
Polyaniline–Carboxymethyl Cellulose Nanocomposite for Cholesterol Detection

Deficiency of the INCL protein Ppt1 resultsin changes in ectopic F1-ATP synthaseand altered cholesterol metabolism
Annina Lyly1, Sanna K. Marjavaara2, Aija Kyttala1, Kristiina Uusi-Rauva1, Kaisu Luiro1,
Outi Kopra3, Laurent O. Martinez4, Kimmo Tanhuanpaa5, Nisse Kalkkinen6,
Anu Suomalainen2, Matti Jauhiainen1 and Anu Jalanko1,�
1National Public Health Institute and FIMM, Institute for Molecular Medicine, Biomedicum Helsinki, PO Box 104,
FIN-00251 Helsinki, Finland, 2Research Program of Molecular Neurology and 3Folkhalsan Institute of Genetics and
the Neuroscience Center, University of Helsinki, Biomedicum Helsinki, FIN-00014 Helsinki, Finland, 4INSERM, U563,
Universite Toulouse III Paul Sabatier, IFR30, Toulouse, France, 5Light Microscopy Unit, Institute of Biotechnology,
University of Helsinki, PO Box 56, FIN-00014 Helsinki, Finland and 6Protein Chemistry Research Group and Core
Facility, Institute of Biotechnology, University of Helsinki, PO Box 65, FIN-00014 Helsinki, Finland
Received December 7, 2007; Revised and Accepted January 24, 2008
Infantile neuronal ceroid lipofuscinosis (INCL) is a severe neurodegenerative disease caused by deficiency ofpalmitoyl protein thioesterase 1 (PPT1). INCL results in dramatic loss of thalamocortical neurons, but the dis-ease mechanism has remained elusive. In the present work we describe the first interaction partner of PPT1,the F1-complex of the mitochondrial ATP synthase, by co-purification and in vitro-binding assays. In additionto mitochondria, subunits of F1-complex have been reported to localize in the plasma membrane, and to becapable of acting as receptors for various ligands such as apolipoprotein A-1. We verified here the plasmamembrane localization of F1-subunits on mouse primary neurons and fibroblasts by cell surface biotinylationand TIRF-microscopy. To gain further insight into the Ppt1-mediated properties of the F1-complex, we utilizedthe Ppt1-deficient Ppt1Dex4 mice. While no changes in the mitochondrial function could be detected in thebrain of the Ppt1Dex4 mice, the levels of F1-subunits a and b on the plasma membrane were specificallyincreased in the Ppt1Dex4 neurons. Significant changes were also detected in the apolipoprotein A-I uptakeby the Ppt1Dex4 neurons and the serum lipid composition in the Ppt1Dex4 mice. These data indicate neuron-specific changes for F1-complex in the Ppt1-deficient cells and give clues for a possible link between lipidmetabolism and neurodegeneration in INCL.
INTRODUCTION
Neuronal ceroid lipofuscinoses (NCLs) constitute the mostcommon group of recessively inherited encephalopathies inchildren (reviewed in 1). Up to eight different genes havebeen identified underlying NCLs (2,3), and the disordersshare similar clinical and pathological findings includingmental and motor deterioration, visual failure, epilepsy,reduced lifespan and autofluorescent lipopigment storage inpatients’ tissues (4). In most NCL diseases the storage materialconsists mainly of subunit C of ATP synthase, while in the
infantile and congenital NCL the main storage material issphingolipid activator proteins (SAPs) A and D (5,6). Thestorage material accumulates in most tissue types, but the hall-mark of NCLs is the selective and devastating loss of neuronsin the central nervous system.
Palmitoyl protein thioesterase 1, PPT1, enzyme deficient inthe infantile form of NCL (INCL) (7), has the structure of ana/b serine hydrolase with three N-linked glycosylation sitesand a fatty acid binding groove (8). PPT1 removes palmiticacid from S-acylated proteins in vitro (9), but the exact phys-iological function and the in vivo substrates have remained
�To whom correspondence should be addressed. Tel: þ358 947448392; Fax: þ358 947448480; Email: [email protected]
# The Author 2008. Published by Oxford University Press. All rights reserved.For Permissions, please email: [email protected]
Human Molecular Genetics, 2008, Vol. 17, No. 10 1406–1417doi:10.1093/hmg/ddn028Advance Access published on February 1, 2008
by guest on Decem
ber 21, 2015http://hm
g.oxfordjournals.org/D
ownloaded from

unknown. In most cell types PPT1 localizes to lysosomes (10),but in neurons it is found also in the presynaptic areas(11–13). The expression of PPT1 in human and mouse brainis developmentally regulated and coincides with corticalsynaptogenesis (14,15). PPT1 has been suggested to beinvolved in various cellular processes, including apoptosis,endocytosis and synaptic function (16,17). Analyses ofPPT1/Ppt1 deficient human and mouse brain have revealedlocal neuroinflammation and changes in lipid profiles(reviewed in 18,19).
PPT1-associated metabolic pathways in neurons haveremained unclear, and the interaction partners of PPT1 havebeen unknown. Here we report an interaction between PPT1and the F1-ATP synthase. We studied this interaction and itsconsequences utilizing Ppt1-deficient Ppt1Dex4 mice. Thesemice mimic the INCL disease with the typical tissue pathologyand early onset neurodegeneration, leading to premature death(20). We show here that Ppt1-deficiency results in alterationsin the amount of F1-ATP synthase on the plasma membrane,but not in mitochondria, and that Ppt1-deficiency is alsomanifested at the level of serum lipids/lipoproteins.
RESULTS
PPT1 interacts with the F1-complex of mitochondrialATP synthase
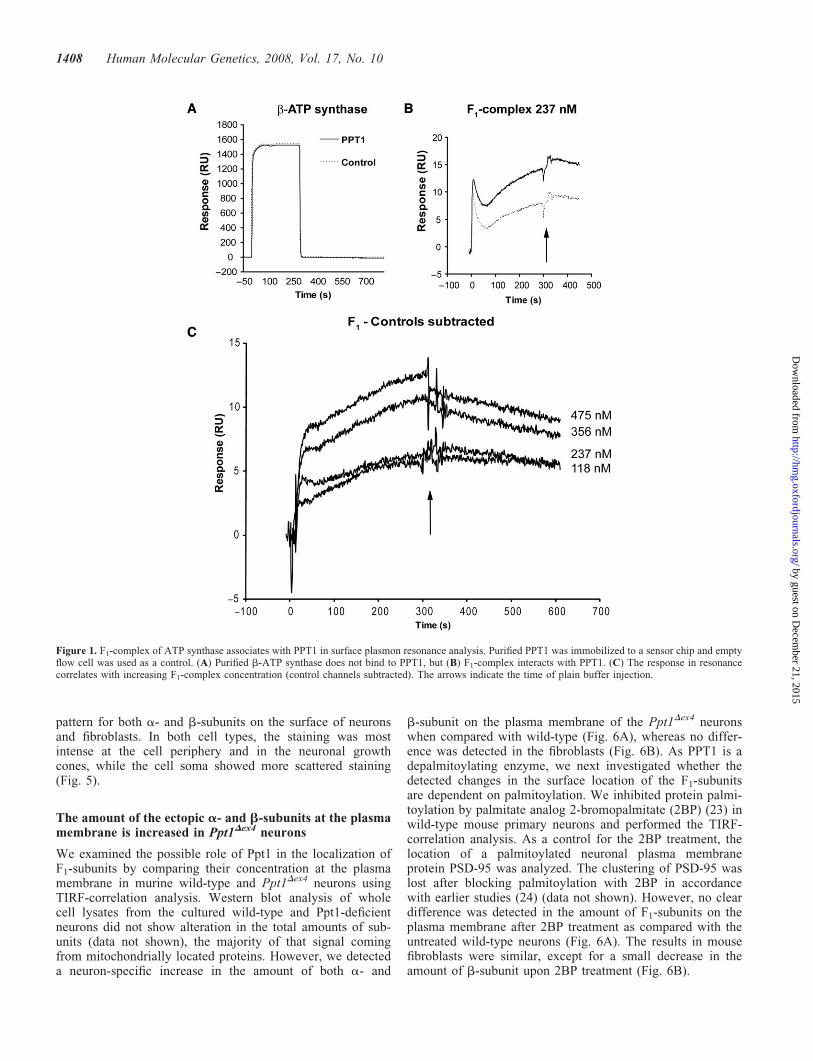
Lysosomal enzymes are secreted upon overexpression, andthis phenomenon was utilized to purify PPT1 from the con-ditioned media of CHO cell line stably expressing PPT1(8,13) by hydrophobic interaction chromatography andsize-exclusion chromatography. During purification, a50 kDa protein was repeatedly co-purified with the recombi-nant PPT1. MALDI-TOF mass spectrometric peptide massfingerprint analysis of the 50 kDa protein band identified thisprotein as the b-subunit of mitochondrial F1-ATP synthase(Supplementary Material), responsible for the final step of oxi-dative ATP production in mitochondria (21). The potentialinteraction between PPT1 and b-subunit was investigated bysurface plasmon resonance assay (Biacore) using purifiedPPT1 immobilized on a sensor chip. To test the chip, polyclo-nal PPT1 antibody was added in the fluid phase and it wasfound to bind the immobilized PPT1 (data not shown). Wethen tested the binding of purified b-subunit and the wholeF1-complex consisting of five different protein subunits(a3b3gDe) to PPT1. PPT1 showed interaction with the purifiedF1-complex, but not with the b-subunit alone (Fig. 1A). Whenthe surface plasmon resonance assay was repeated withincreasing concentrations of F1-complex, the results confirmeda concentration-dependent interaction between PPT1 and theF1-complex (Fig. 1B and C).
To verify this novel interaction, GST pull-down was uti-lized. Recombinant full-length PPT1 lacking the signalsequence was expressed as GST-fusion protein, solubilized,purified and then incubated with mouse brain extract. The pre-cipitated proteins were analyzed by western blot with mono-clonal antibodies against subunits a and b from theF1-complex. GST-PPT1 bound both of these subunits frommouse brain extract, and neither of them interacted withGST alone (Fig. 2). The interaction between PPT1 and
F1-complex but not between PPT1 and b-subunit alone inBiacore-assay allows two possible interpretations of theresult: (i) The interaction is dependent on the conformationof the b-subunit and possible only when it is in the F1
-bound form, or (ii) PPT1 interacts with some other subunitin the F1-complex.
Ppt1Dex4 mice brain do not show alterations inmitochondrial function or assembly of mitochondrialrespiratory complexes
To determine the possible effect of the Ppt1-deficiency on theassembly or steady-state levels of respiratory complexes, andespecially that of the mitochondrial F1-Fo-ATPase, mitochon-dria were isolated from the cerebrum and cerebellum of2-month-old control and Ppt1Dex4 animals, and analyzed byblue native (BN) electrophoresis followed by western blot(Fig. 3A) or Coomassie staining (data not shown). The relativeamounts of the ATP synthase were compared with complex II,and no reduction in the amount or abnormality in the assemblyof the mitochondrial ATP synthase was present in these mice(Fig. 3A). In order to test the mitochondrial function inthe Ppt1Dex4 mice brain, the mitochondria were isolated andthe oxygen consumption was measured at baseline and inresponse to the following components or inhibitors of themitochondrial energy production: (i) pyruvate and malate,(ii) succinate and rotenone, (iii) ascorbate and TMPD, (iv)palmitoyl CoA and (v) a-ketoglutarate. No difference in theoxygen consumption between wild-type and Ppt1Dex4 micewas detected (Fig. 3B).
The a- and b-subunits of the F1-complex localize to the cellsurface in neurons and fibroblasts
To explore the tissue distribution of the F1-subunits in mousebrain, immunohistochemical staining patterns of the a- andb-subunits were analyzed. They were similar to that of Ppt1in the wild-type mouse brain, especially the large pyramidalneurons of cortical layers II–IV were positive for both sub-units and Ppt1 (Fig. 4A–C). Ppt1 staining was absent in thePpt1Dex4 mice, indicating a complete knock-out of the gene(Fig. 4D). Thorough investigation of the brain sections ofthe Ppt1Dex4 mice demonstrated a slightly more intense a-and b-subunit staining in the large neurons of cortical layersII–IV of the mutant mice compared with wild-type controls(Fig. 4E and F).
Several studies have reported ATP-synthase subunits tolocalize in lipid rafts on the plasma membrane in addition tomitochondria (reviewed in 22). No phenotype in the mitochon-drial ATP synthase was identified in the Ppt1 mutant mice, andthus we examined the putative plasma membrane localizationof the a- and b-subunits in the wild-type mouse primaryneurons and fibroblasts. In neurons, both the endogenous a-and b-subunit demonstrated a strong signal in the biotinylatedprotein specimens, indicating their cell surface localizationand supporting an extra-mitochondrial role for these subunitsin mouse neurons as well (Supplementary Material). Toanalyze the pattern of these proteins in the plasma membrane,total internal reflection fluorescence (TIRF) microscopy analy-sis was utilized. TIRF revealed a distinct punctuated staining
Human Molecular Genetics, 2008, Vol. 17, No. 10 1407
by guest on Decem
ber 21, 2015http://hm
g.oxfordjournals.org/D
ownloaded from
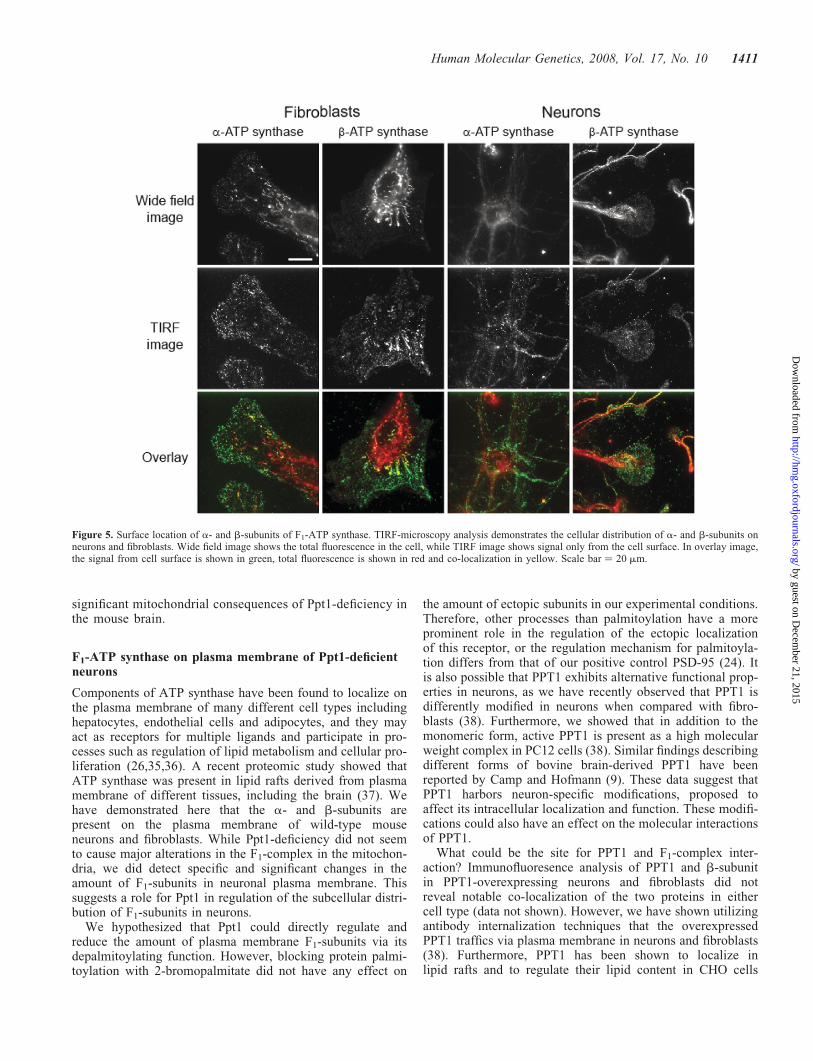
pattern for both a- and b-subunits on the surface of neuronsand fibroblasts. In both cell types, the staining was mostintense at the cell periphery and in the neuronal growthcones, while the cell soma showed more scattered staining(Fig. 5).
The amount of the ectopic a- and b-subunits at the plasmamembrane is increased in Ppt1Dex4 neurons
We examined the possible role of Ppt1 in the localization ofF1-subunits by comparing their concentration at the plasmamembrane in murine wild-type and Ppt1Dex4 neurons usingTIRF-correlation analysis. Western blot analysis of wholecell lysates from the cultured wild-type and Ppt1-deficientneurons did not show alteration in the total amounts of sub-units (data not shown), the majority of that signal comingfrom mitochondrially located proteins. However, we detecteda neuron-specific increase in the amount of both a- and
b-subunit on the plasma membrane of the Ppt1Dex4 neuronswhen compared with wild-type (Fig. 6A), whereas no differ-ence was detected in the fibroblasts (Fig. 6B). As PPT1 is adepalmitoylating enzyme, we next investigated whether thedetected changes in the surface location of the F1-subunitsare dependent on palmitoylation. We inhibited protein palmi-toylation by palmitate analog 2-bromopalmitate (2BP) (23) inwild-type mouse primary neurons and performed the TIRF-correlation analysis. As a control for the 2BP treatment, thelocation of a palmitoylated neuronal plasma membraneprotein PSD-95 was analyzed. The clustering of PSD-95 waslost after blocking palmitoylation with 2BP in accordancewith earlier studies (24) (data not shown). However, no cleardifference was detected in the amount of F1-subunits on theplasma membrane after 2BP treatment as compared with theuntreated wild-type neurons (Fig. 6A). The results in mousefibroblasts were similar, except for a small decrease in theamount of b-subunit upon 2BP treatment (Fig. 6B).
Figure 1. F1-complex of ATP synthase associates with PPT1 in surface plasmon resonance analysis. Purified PPT1 was immobilized to a sensor chip and emptyflow cell was used as a control. (A) Purified b-ATP synthase does not bind to PPT1, but (B) F1-complex interacts with PPT1. (C) The response in resonancecorrelates with increasing F1-complex concentration (control channels subtracted). The arrows indicate the time of plain buffer injection.
1408 Human Molecular Genetics, 2008, Vol. 17, No. 10
by guest on Decem
ber 21, 2015http://hm
g.oxfordjournals.org/D
ownloaded from

Apolipoprotein A-I internalization is increased in Ppt1Dex4
neurons and glia
Our recent transcript profiling studies with neuron cultureshave demonstrated severe disturbances in lipid metabolic path-ways, specifically in cholesterol metabolism, in thePpt1-deficient neurons (25). The b-subunit of F1-ATPsynthase has been linked to lipoprotein metabolism andsuggested to act as an apoA-I –sensing receptor in hepatocytes(26), in which its stimulation activates a P2Y13-mediatedHDL endocytosis pathway (27). Although the function of theplasma membrane F1-complex is not known in neuronalcells, we examined whether the b-subunit operates as anapoA-I-sensing receptor also in neurons and glial cells. Thecells were incubated with an excess of radiolabeled lipid-freeapoA-I. In TCA precipitation, the degree of apoA-I degra-dation was similar between the Ppt1Dex4 and wild-type cellsor culture media (data not shown). A significant increase inthe levels of internalized apoA-I was detected both inPpt1-deficient glial cells (118 versus 100%) and neurons(140 versus 100%), suggesting that the elevated amount ofthe b-subunit on the plasma membrane could mediateenhanced apoA-I uptake (Fig. 7).
Altered serum lipid and lipoprotein profiles in Ppt1Dex4
mice
ApoA-I is the major apolipoprotein in HDL, and thereforeit was of interest to investigate possible changes in lipidmetabolism in the Ppt1Dex4 mice. In serum samples from1-month-old mutant and control mice, the levels of apoA-I,apoE, cholesterol and triglycerides as well as the activity ofplasma phospholipid transfer protein (PLTP) were measured.In the mutant animals, the cholesterol and apoA-I concen-trations as well as the PLTP activity were significantlyreduced when compared with the control mice (Fig. 8A).The observed perturbations are logical since decreased HDLcholesterol levels are associated with reduced apoA-I. Thedecrease in PLTP activity followed the decreased triglyceridelevels. PLTP is a factor suggested to maintain serum HDLlevels (28). Therefore the observed change in PLTP activityis consistent with the drop of cholesterol and apoA-I concen-trations. No significant difference could be detected in serumapoE levels in the Ppt1Dex4 and wild-type mice, but the distri-bution of apoE between different sized lipoprotein particleswas dramatically changed in the Ppt1Dex4 mice. Size-exclusion
chromatography analysis demonstrated that high molecularweight apoE-containing lipoprotein particles were almosttotally absent from the Ppt1Dex4 mice sera. In contrast, par-ticles of smaller size appeared to be more abundant(Fig. 8B). This observation may indicate functional changesin the reverse cholesterol transport pathway, as the largerapoE-containing HDL particles are more efficient in choles-terol removal from macrophage-foam cells when comparedwith smaller HDL particles (29). Taken together, theseresults indicate a change in the systemic lipid homeostasisof Ppt1 deficient mice, and emphasize a novel role for Ppt1/PPT1 in the regulation of lipoprotein metabolism.
DISCUSSION
INCL and mitochondria
In this study, we have characterized the first interaction partnerfor PPT1, the F1-complex of the mitochondrial ATP synthase.This finding was intriguing, since ATP synthase has been pre-
Figure 2. GST pull-down assay. GST fusion proteins were used to pull downinteracting proteins from mouse brain extract. Bound proteins were eluted andanalyzed by western blot using monoclonal antibodies for a- and b-ATPsynthase. Load ¼ 10 mg of total protein from the brain extract.
Figure 3. Mitochondrial function in control and Ppt1Dex4 mouse brain. (A)Steady-state levels of respiratory complexes. Respiratory complexes of mito-chondria, isolated from the cerebrum (Cbr) and cerebellum (Cbl) of wild-typeand Ppt1-deficient mice, were separated by BN-PAGE and analyzed by immu-noblotting with antibodies against b-ATP synthase (complex V) and subunit70 kDa Fp (complex II). The relative intensity of complex V and complexII were calculated with Scion Image software (Scion Corporation). (B) Rela-tive oxygen consumption of mitochondria in response to the followingreagents was determined: (1) pyruvate and malate, (2) succinate and rotenone,(3) ascorbate and TDMP, (4) palmitoyl CoA and (5) a-ketoglutarate.
Human Molecular Genetics, 2008, Vol. 17, No. 10 1409
by guest on Decem
ber 21, 2015http://hm
g.oxfordjournals.org/D
ownloaded from

viously linked with various NCL diseases: a hallmark ofdisease pathology has been the accumulation of storagematerial containing the Fo-subunit C of the mitochondrialATP synthase, and functional and structural abnormalities ofmitochondria have been reported in NCL patients andanimal models (30–33). INCL patient fibroblasts are knownto have reduced basal ATP synthase activity and a defect inthe regulation of the enzyme (34). Furthermore, the myocyticand neuronal mitochondria of ppt-1-deficient Caenorhabditiselegans show abnormal morphology, including fewer cristaeand whorling of the inner membrane (30). However, previousultrastructural analysis of the brain of Ppt1Dex4 mice didnot indicate any abnormalities in the mitochondrial shapeor size, although the storage material of typical GROD
ultrastructure was present in the mutant animals (20 andunpublished data).
We studied here the nature of the detected interactionand its importance in INCL pathogenesis, and first exploredthe mitochondrial function in Ppt1-deficient mouse brain. Nosignificant alterations could be detected in the steady-statelevels or the assembly of respiratory complexes inmitochondria, speaking for normal transportation andstructure of nuclear-encoded subunits in the organelle.Neither was the mitochondrial respiratory function affectedin Ppt1-deficient mouse brain, indicated by the unalteredenzyme activities of the respiratory chain complexes I–IV(data not shown) and mitochondrial oxygen consumption.In the light of the current data, it seems that there are no
Figure 4. Immunohistochemical staining. Sagittal brain sections derived from 1-month-old control and Ppt1Dex4 mice were immunostained with Ppt1 (A and D)a-ATP synthase (B and E) and b-ATP synthase (C and F). Ppt1 staining shows virtually no protein in Ppt1Dex4 cortex, while in the wild-type brain the staining isstrong. The staining for F1-subunits a and b appears stronger in Ppt1Dex4 sections; especially the staining of cell periphery is pronounced (arrows). Images rep-resent cortical layers III-IV. Scale bar ¼ 100mm.
1410 Human Molecular Genetics, 2008, Vol. 17, No. 10
by guest on Decem
ber 21, 2015http://hm
g.oxfordjournals.org/D
ownloaded from
significant mitochondrial consequences of Ppt1-deficiency inthe mouse brain.
F1-ATP synthase on plasma membrane of Ppt1-deficientneurons
Components of ATP synthase have been found to localize onthe plasma membrane of many different cell types includinghepatocytes, endothelial cells and adipocytes, and they mayact as receptors for multiple ligands and participate in pro-cesses such as regulation of lipid metabolism and cellular pro-liferation (26,35,36). A recent proteomic study showed thatATP synthase was present in lipid rafts derived from plasmamembrane of different tissues, including the brain (37). Wehave demonstrated here that the a- and b-subunits arepresent on the plasma membrane of wild-type mouseneurons and fibroblasts. While Ppt1-deficiency did not seemto cause major alterations in the F1-complex in the mitochon-dria, we did detect specific and significant changes in theamount of F1-subunits in neuronal plasma membrane. Thissuggests a role for Ppt1 in regulation of the subcellular distri-bution of F1-subunits in neurons.
We hypothesized that Ppt1 could directly regulate andreduce the amount of plasma membrane F1-subunits via itsdepalmitoylating function. However, blocking protein palmi-toylation with 2-bromopalmitate did not have any effect on
the amount of ectopic subunits in our experimental conditions.Therefore, other processes than palmitoylation have a moreprominent role in the regulation of the ectopic localizationof this receptor, or the regulation mechanism for palmitoyla-tion differs from that of our positive control PSD-95 (24). Itis also possible that PPT1 exhibits alternative functional prop-erties in neurons, as we have recently observed that PPT1 isdifferently modified in neurons when compared with fibro-blasts (38). Furthermore, we showed that in addition to themonomeric form, active PPT1 is present as a high molecularweight complex in PC12 cells (38). Similar findings describingdifferent forms of bovine brain-derived PPT1 have beenreported by Camp and Hofmann (9). These data suggest thatPPT1 harbors neuron-specific modifications, proposed toaffect its intracellular localization and function. These modifi-cations could also have an effect on the molecular interactionsof PPT1.
What could be the site for PPT1 and F1-complex inter-action? Immunofluoresence analysis of PPT1 and b-subunitin PPT1-overexpressing neurons and fibroblasts did notreveal notable co-localization of the two proteins in eithercell type (data not shown). However, we have shown utilizingantibody internalization techniques that the overexpressedPPT1 traffics via plasma membrane in neurons and fibroblasts(38). Furthermore, PPT1 has been shown to localize inlipid rafts and to regulate their lipid content in CHO cells
Figure 5. Surface location of a- and b-subunits of F1-ATP synthase. TIRF-microscopy analysis demonstrates the cellular distribution of a- and b-subunits onneurons and fibroblasts. Wide field image shows the total fluorescence in the cell, while TIRF image shows signal only from the cell surface. In overlay image,the signal from cell surface is shown in green, total fluorescence is shown in red and co-localization in yellow. Scale bar ¼ 20 mm.
Human Molecular Genetics, 2008, Vol. 17, No. 10 1411
by guest on Decem
ber 21, 2015http://hm
g.oxfordjournals.org/D
ownloaded from

(39). Here, the interaction partner was also co-secreted withoverexpressed PPT1 during the purification process,supporting their interaction to occur somewhere along thesecretory pathway.
ApoA-I and the altered neuronal cholesterol metabolismin Ppt1-deficient cells
We have previously shown by transcript profiling studies ofneuronal cultures that Ppt1-deficient neurons have severe dis-turbances in cholesterol metabolism pathways (25). Therefore,it was of interest to investigate whether the increased receptoramount had a role in the same pathway. Indeed, we demon-strated that the cellular uptake of the lipid-free apoA-I wasincreased. In addition to being one of the ligands for the
ectopic b-subunit, apoA-I is also a major apolipoprotein inthe lipoproteins of cerebrospinal fluid (CSF) (26,40). Outsidethe CNS, apoA-I promotes atheroprotective functions, includ-ing facilitation of cholesterol removal from macrophage-foamcells in a process of reverse cholesterol transport that ulti-mately leads to cholesterol removal by the liver (reviewed in41). In the brain, its function seems to be similar, as apoA-Itogether with the activation of Liver X receptor (LXR) wasreported to induce the cholesterol efflux from both neuronsand astrocytes (42).
So far, there is no evidence for apoA-I synthesis in thebrain, but brain endothelial cells have been shown to syn-thesize and secrete apoA-I and abundant amounts of apoA-Ipresent in the CNS are transported across the blood brainbarrier (43). In vitro studies have suggested that vascularendothelial cells also transcytose apoA-I via the SR-BI orABCA1 receptors in the brain or in the aorta, respectively(44,45). Interestingly, another NCL-protein cathepsin D wasrecently found to regulate and promote ABCA1 andapoA-I-mediated cholesterol efflux from macrophages, andcathepsin D inactivation resulted in a lysosomal accumulationof cholesterol (46). Previous data also suggest that whenvascular endothelial cells are exposed to cholesterol, theamount of b-subunit in the plasma membrane increases (47).Our recent study demonstrated that while cholesterol biosyn-thesis was strongly upregulated in the embryonic Ppt1-deficient neurons, no cholesterol accumulation could bedetected (25). In future studies it will be of great interest toinvestigate the relationship of the increased amount ofF1-subunits and the increased uptake of apoA-I with thedetected alteration of cholesterol biosynthesis in thePpt1-deficient neurons.
The cross-talk between astrocytes and neurons
ApoE is the major apolipoprotein expressed in the CNS, and theapoE4 protein, encoded by the e4 allele, has been genetically
Figure 6. Correlation between the ectopic and total amount of a- andb-subunits in mouse primary neurons and fibroblasts. Co-localization of a-and b-subunits between wide field and TIRF images in wild-type (WT),Ppt1Dex4 (Cln12/2) and 2-bromopalmitate-treated (2BP) wild-type neurons(A) and fibroblasts (B). Protein levels were compared using Image Pro Plussoftware’s co-localization analysis tool and Pearson’s correlation value. Thedata from three individual experiments were combined. Error bars representSEM. P-values were calculated using Student’s t-test (�,0.05, ��,0.001).
Figure 7. Intake of radiolabelled apolipoprotein A-1. Neurons and glial cellswere incubated with radiolabelled apoA-I. The ratios of radiolabelled apoA-Iin cells and in culture media were compared between wild-type and Ppt1Dex4
neurons and glial cells [Ppt1Dex4 (counts/mg protein) / WT (counts/mgprotein)]. Bars represent average ratios of three individual experiments andthe error bars are standard deviations. P-values for the ratios were calculatedusing Student’s t-test (�,0.05, ��,0.001).
1412 Human Molecular Genetics, 2008, Vol. 17, No. 10
by guest on Decem
ber 21, 2015http://hm
g.oxfordjournals.org/D
ownloaded from

linked to neurodegenerative Alzheimer’s disease (48). Duringtheir maturation and synaptogenesis, neurons are dependenton the cholesterol provided by astrocytes (reviewed in 49).On the other hand, neurons convert cholesterol to24(S)-hydroxycholesterol, which induces ABCA1-regulated,apoE-mediated cholesterol efflux specifically from astrocytes(42). Astrocytic activation has been shown to be importantfor the pathogenesis of INCL (reviewed in 1,18). Althoughfurther studies are needed to clarify the neuron-astrocyteinterplay during the disease process, it is possible that apoEcontributes to INCL pathogenesis even more than apoA-I.
In this study, we could not observe major alterations inapoA-I or apoE immunostaining in the brain of Ppt1Dex4
mice (data not shown) nor in apoE serum levels. However,the reduced amount of large apoE-containing HDL-particlesin the sera of Ppt1Dex4 mice suggests a disturbance oflipoprotein homeostasis. This is an interesting observationalso in the light of previous studies describing apoE binding
to both a- and b-subunits of F1-complex (50,51). Besidesdecreased levels of cholesterol, apoA-I and abnormal distri-bution of apoE-containing particles, we also detecteddecreased PLTP activity in Ppt1Dex4 mouse sera. Significantly,we have recently demonstrated that active PLTP remodelsHDL particles to apoE-enriched HDL with larger particlesize (52). Moreover, PLTP activity in the CSF was reportedlyreduced in various neurological diseases (53). Finally, a directfunctional interaction between PLTP and apoE was shownwhereby PLTP induced apoE secretion from primary humanastrocytes (53).
CONCLUSIONS
In this study we have described a novel interaction betweenPPT1 and F1-ATP synthase. Although mitochondrial abnorm-alities have been reported in other NCL models, we could not
Figure 8. Lipid profile analysis from mouse serum. (A) Cholesterol, triglycerides, PLTP activity and apoA-I and E levels were determined from individual wild-type and Ppt1Dex4 mouse serum samples. Bars represent an average value, error bars are standard deviations. Student’s t-test was used for calculating theP-values (�,0.05). (B) Pooled serum samples from wild-type and Ppt1Dex4 mice were fractionated with FPLC size-exclusion chromatography and apoElevel was measured from the fractions.
Human Molecular Genetics, 2008, Vol. 17, No. 10 1413
by guest on Decem
ber 21, 2015http://hm
g.oxfordjournals.org/D
ownloaded from

detect any substantial defects in the amount, assembly or func-tion of mitochondrial respiratory complexes in Ppt1-deficientmouse brains. The most striking finding was the increasedamount of F1-complex in the plasma membrane ofPpt1-deficient neurons and their increased uptake of apoA-1.An intriguing previous link suggests a role for plasma mem-brane F1-complex as a receptor for the lipoprotein. Undis-turbed lipid homeostasis is vital for the development andmaintenance of the normal CNS function (49). Braincholesterol homeostasis is still poorly understood, but anincreasing number of studies link it with neurodegenerativediseases, including the NCL disorders. These monogenic dis-eases could provide powerful tools for future studies on regu-latory pathways that connect neuronal homeostasis andsystemic lipid metabolism.
MATERIALS AND METHODS
Plasmids
Expression plasmid for GST-PPT128-306 fusion protein wasprepared as follows: the PPT1 coding sequence without thesignal sequence was multiplied with PCR using primers50CAGGAATTCGACCCGCCGGCGCCGCTGCC and 30GTCCTCGAGTCCAAGGAATGGTATGATGTG. The resultingPCR fragments were digested with EcoRI and XhoI andligated to the corresponding sites in pGEX-4T-1 vector. Thereading frame was confirmed with ABI3730 AutomaticDNA Sequencer using the BigDyeTM Terminator CycleSequencing Kit v3.1 (Applied Biosystems).
PPT1 purification and protein identification
Recombinant PPT1 was purified according to previous proto-col (38). To identify the protein associated with PPT1 duringpurification, the corresponding protein band was cut out fromsilver-stained SDS-PAGE gel, reduced with 20 mM DTT,alkylated with 55 mM iodoacetamide and digested withtrypsin overnight at þ378C. The resulting peptides wereeluted from the gel, desalted with a mC18 ZipTip reversedphase tip (Millipore) and subjected to MALDI-TOF massspectrometric (Ultraflex TOF/TOF, Bruker Daltonics)peptide mass fingerprint analysis. Protein identification wasperformed using the MASCOT Peptide Mass Fingerprintprogram (http://www.matrixscience.com).
Surface plasmon resonance analysis
Binding of PPT1 and F1-complex or b-subunit of ATPsynthase was studied with surface plasmon resonance analysisin Biacore 2000 biosensor (Biacore). Saturating amount ofpurified PPT1 was covalently attached to a flow cell of a bio-sensor chip using standard amine coupling according to themanufacturer’s protocol. Recombinant full-length humanb-ATP synthase [LGC Promochem, France, ATCC number59118 (54)], subcloned in pPROexTMHTb (Invitrogen), wastransformed into E. coli BL21 for protein expression. Aftersolubilization, the proteins were separated on 3 mM-thickness10% (w/v) Glycine-SDS–PAGE. One part of the gel wasstained by Coomassie brilliant blue R250 and a non-stained
band corresponding to the �50kDa b-ATP synthase proteinwas cut off and recovered from the gel by microelectroelutionas previously described (55). The eluted protein fraction wasthen dialyzed in water, quantified and lyophilized. Purity ofthe b-ATP synthase preparation was confirmed by Coomassiestaining and western blot detection with b-subunit monoclonalantibody (Molecular Probes). F1-ATPase sector of theATP synthase complex was purified from bovine heartmitochondria as previously described (56) and kindly providedby J. Walker (MRC Dunn Human Nutrition Unit, Cambridge,UK). Varying concentrations of purified human b-subunit andbovine F1-ATPase were diluted in Hepes-buffered saline andused as analytes. An empty flow cell was used as a negativecontrol.
GST pull-down assay
E. coli BL21 cells were transformed with GST-vector andGST-fusion construct GST-PPT128-306. The GST fusionprotein expression, purification and immobilization toGlutathione-agarose beads were performed according to man-ufacturer’s instructions (Amersham Biosciences). Mouse brainextracts were obtained by homogenizing the brain from adultC57BL mouse in 3 ml extraction buffer (50 mM Hepes pH 7.4,100 mM NaCl, 5 mM MgCl2, 0.5% Triton X-100 and proteaseinhibitor cocktail) and centrifuging for 10 min at 48C at 1000gfollowed by ultracentrifugation of the supernatant at 100 000gat 48C for 40 min. Immobilized GST-fusion proteins wereincubated with mouse brain extract (2 mg/ml) in extractionbuffer for 3 h at 48C with gentle rotation. Samples werewashed, boiled with Laemmli buffer and 70 mM DTT andanalyzed by western blot to detect a- and b-subunits (mono-clonal antibodies from Molecular Probes).
Animals and cell culture
Ppt1Dex4 mice (20) were maintained on a congenic C57/BL6Jstrain background. Animals used in this study were a result often generations of backcrosses onto C57/BL6J. C57/BL6Jmice were used as wild-type controls. The study was approvedby the Laboratory Animal Care and Use Committee of theNational Public health Institute, Helsinki. The study hasbeen carried out following good practice in laboratoryanimal handling and the regulations for handling geneticallymodified organisms. Mouse cortical neurons were preparedfrom C57BL WT and Ppt1Dex4 mouse embryos (E14–E16)and maintained as previously described (11). The glial cellcultures were prepared from mouse cubs (P2–P5) accordingto the protocol of Harkke et al. (57).
Immunohistochemical staining
Brains of three 1-month-old Ppt1Dex4 knock-out mice andthree wild-type controls were immersion fixed (4% PFA)and paraffin embedded. The tissue was cut into 5 mm sectionsand mounted on a Super Frost (Menzel Glazer) objectiveglass. Immunostaining with polyclonal PPT1 (1:200) (38)and monoclonal a- and b-subunit antibodies (1:500, Molecu-lar Probes) was performed using a standard protocol (20).
1414 Human Molecular Genetics, 2008, Vol. 17, No. 10
by guest on Decem
ber 21, 2015http://hm
g.oxfordjournals.org/D
ownloaded from

Examination of mitochondrial function
The mitochondrial function was examined according to theprotocol of Luiro et al. (32). In brief, two Ppt1Dex4 mice andwild-type controls were sacrificed and the brains were hom-ogenized in SETH buffer (0.25 M sucrose, 2 mM EDTA,10 mM Tris–HCl, 50 U/ml heparin, pH 7.4) and centrifugedtwice at 400 g at 48C for 10 min. The postnuclear supernatantwas further centrifuged twice at 9300 g at 48C for 10 min topellet the mitochondria. The pellet was resuspended inBuffer I (0.25 M mannitol, 0.2 M EDTA, 1 mM MgCl2,10 mM KCl, 10 mM KPi) and the oxygen consumption wasmeasured at baseline and systemically in response to the com-ponents of the citric acid cycle and other regulatory com-ponents: ADP, (i) pyruvate and malate, (ii) succinate androtenone, (iii) ascorbate and TMPD (N,N,N’,N’ –tetramethyl-p-phenyleneamine dihydrochloride), (iv) palmi-toyl CoA (b-oxidation of fatty acids) and (v) a-ketoglutarate(58). The results were normalized relative to activity ofcitrate synthase, which has been traditionally used as an indi-cator of mitochondrial mass.
Mitochondrial isolation and blue native gel electrophoresis
Cerebrum and cerebellum were taken from 2-month-old malemice for mitochondrial isolation. Tissues were homogenizedwith a zero-clearance Teflon pestle in ice-cold HIM buffer(200 mM mannitol, 70 mM sucrose, 10 mM HEPES, 1 mMEGTA, adjusted to pH 7.5 with KOH) (59) and centrifugedat 600g for 20 min. The supernatant was further centrifugedat 600g for 20 min and at 10 000g for 10 min. The resultingmitochondrial pellet was washed with HIM buffer and centri-fuged again at 10 000g for 10 min. The pellet was resuspendedin PBS containing protease inhibitor cocktail, and the proteinconcentration was determined. Native gradient gels (5–12%)were casted and run according to the protocol of Antonickaet al. (60). Equal amounts of protein (10–20 mg) fromcontrol and Ppt1Dex4 mouse tissues were analyzed with Coo-massie staining and western blot using monoclonal antibodiesagainst b-subunit of complex V (Molecular Probes) andcomplex II subunit 70 kDa Fp (Mitosciences). They werecompared with reveal possible differences caused by altera-tions in usability of nuclear- and mitochondrial-encodedproteins.
TIRF analysis
Mouse primary neurons and fibroblasts were seeded on 35 mMglass bottom dishes (MatTek) and grown for 8 and 1 days,respectively. Cells were fixed with 4% PFA and permeabilizedwith 0.1% Triton X-100 in PBS for 30 min in 48C. Immuno-fluorescence staining was performed in room temperatureusing monoclonal antibodies against a- and b-subunits andsecondary Alexa 488-fluorescent antibody (MolecularProbes). Images were taken with TILL imaging system includ-ing TILL TIRF condenser, Polychrome IV monochromator,Omnichrome series 43 krypton–argon laser (TILL Photonics)and Olympus NA 1.45 objective on Olympus IX-71 micro-scope including Andor DV885 EMCCD camera (Andor Tech-nology) with Z488BP filter set (Chroma Technology). The
images were further processed with Adobe Photoshop CSand Adobe Illustrator CS software. The correlation analysiswas performed with Image Pro Plus software’s co-localizationtool (Media Cybernetics) by comparing the co-localization ofthe total fluorescence and the fluorescence from the cellsurface. For co-localization analysis a fixed value determinedas the intensity on a cell free area in the image has been sub-tracted. In short, the higher the co-localization correlationvalue (Pearson’s correlation), the more protein is located onthe cell surface. Neuronal analysis included total of 20–30analyzed images per condition from three individual exper-iments. The correlation was calculated for the whole imagefor neurons. Fibroblast analysis included total of 13–24 ana-lyzed images per condition from at least two individual exper-iments. The mitochondria were excluded from the fibroblastanalysis to reduce the variation.
Apolipoprotein A–I internalization to glial cells andneurons
The internalization of apolipoprotein A-1 was performedaccording to the protocol of Siggins et al. (61) with minormodifications. Briefly, purified human apoA-I (kindly pro-vided by Swiss Red Cross, Dr. Peter Lerch) was radiolabeledaccording to previous protocol (62). Neuronal and glial cellcultures were grown on 58 mM dishes with two coverslipsfor 8 days. The specificity of the cells was confirmed byimmunostaining with neuron-specific mouse anti-tubulin anti-body (b-tubulin III, Chemicon) and astrocyte-specific rabbitanti-cow glial fibrillary acidic protein antibody (GFAP,DAKO). Meanwhile, the cells were washed gently with PBSand incubated in serum-free media containing 5 mg/ml of[35S]-apo-A-1 (specific activity �20,000 cpm/mg A-I) for24 h. The cells internalized 1–2% of the available radio-labeled lipid-free apoA-I. Culture media was collected andcells from two dishes were combined and lysed with 1 ml oflysis buffer (0.1 M Tris–HCl pH 7.8, 1% SDS, proteaseinhibitor cocktail, Roche). Samples from culture media andcell lysates were measured for protein content and precipitatedwith 5% trichloroacetic acid. The radioactivity of the precipi-tated (native) and non-precipitated (degraded) samples wasmeasured with liquid scintillation counting, normalized toprotein concentration and the radioactivity of culture mediaand cells were compared.
Mouse serum lipid analysis
One-month-old female mice were fasted for a 4-h period andblood for serum lipid and lipoprotein analysis was collected.Iodoacetic acid (1 mM final) was added to samples toprevent LCAT activity. Wild-type mice (n ¼ 6) andPpt1Dex4 mice (n ¼ 15) were used for the study. Triglycerides(GPO-PAP 1488872 kit, Roche Diagnostics) and total choles-terol (CHOD-PAP 1489232 kit, Roche Diagnostics) weremeasured using fully enzymatic methods. PLTP activity wasmeasured using radiometric assay (63). Mouse apoA-I wasquantified by a sandwich ELISA (64). Mouse apoE was ana-lyzed by ELISA method using specific antibodies against pur-ified mouse apoE. For the lipoprotein analysis, serum sampleswere pooled (final pool size: 120–200 ml) and fractionated by
Human Molecular Genetics, 2008, Vol. 17, No. 10 1415
by guest on Decem
ber 21, 2015http://hm
g.oxfordjournals.org/D
ownloaded from

fast-performance liquid chromatography (FPLC) using aSuperose 6HR 10/30 size-exclusion chromatography column(Amersham Pharmacia Biotech). The column was equilibratedwith Tris-buffered saline (10 mM Tris, 150 mM NaCl, pH 7.4)and 0.5 ml fractions were collected at flow rate 0.5 ml/min.Column fractions were analyzed immediately for lipids andapoE.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
ACKNOWLEDGEMENTS
We are grateful to Auli Toivola for excellent technical assist-ance. Kaija Antila and Tuula Manninen are thanked for thehelp with the immunohistochemical stainings and mouseserum sample collection, respectively. Gunilla Ronnholm isthanked for the mass spectrometric analyses. Tomas Strandinis thanked for the Biacore-analysis. Jari Metso, Sari Nuutinenand Mikko Muilu are thanked for the help and expertise inserum assays. We thank the Molecular Imaging Unit (MIU,Biomedicum Helsinki), where the image analysis was partlydone, for providing the facilities.
Conflict of Interest statement. None declared.
FUNDING
This study was financially supported by the EuropeanCommission 6th Framework Research Grant LSHM-CT-2003-503051, Academy of Finland Centre of Excellence inComplex Disease Genetics Grant 213506 and Centre ofExcellence in Mitochondrial Disease and Aging, as well asby the Sigrid Juselius Foundation and Arvo and Lea YlppoFoundation, which are gratefully acknowledged.
REFERENCES
1. Haltia, M. (2006) The neuronal ceroid-lipofuscinoses: from past topresent. Biochim. Biophys. Acta, 1762, 850–856.
2. Siintola, E., Lehesjoki, A.E. and Mole, S.E. (2006) Molecular genetics ofthe NCLs—status and perspectives. Biochim. Biophys. Acta, 1762,857–864.
3. Siintola, E., Topcu, M., Aula, N., Lohi, H., Minassian, B.A., Paterson,A.D., Liu, X.Q., Wilson, C., Lahtinen, U., Anttonen, A.K. et al. (2007)The novel neuronal ceroid lipofuscinosis gene mfsd8 encodes a putativelysosomal transporter. Am. J. Hum. Genet., 81, 136–146.
4. Santavuori, P., Vanhanen, S.L. and Autti, T. (2001) Clinical andneuroradiological diagnostic aspects of neuronal ceroid lipofuscinosesdisorders. Eur. J. Paediatr. Neurol., 5(Suppl. A), 157–161.
5. Tyynela, J., Palmer, D.N., Baumann, M. and Haltia, M. (1993) Storage ofsaposins A and D in infantile neuronal ceroid-lipofuscinosis. FEBS Lett.,330, 8–12.
6. Siintola, E., Partanen, S., Stromme, P., Haapanen, A., Haltia, M.,Maehlen, J., Lehesjoki, A.E. and Tyynela, J. (2006) Cathepsin Ddeficiency underlies congenital human neuronal ceroid-lipofuscinosis.Brain, 129, 1438–1445.
7. Vesa, J., Hellsten, E., Verkruyse, L.A., Camp, L.A., Rapola, J.,Santavuori, P., Hofmann, S.L. and Peltonen, L. (1995) Mutations in thepalmitoyl protein thioesterase gene causing infantile neuronal ceroidlipofuscinosis. Nature, 376, 584–587.
8. Bellizzi, J.J., III, Widom, J., Kemp, C., Lu, J.Y., Das, A.K., Hofmann,S.L. and Clardy, J. (2000) The crystal structure of palmitoyl proteinthioesterase 1 and the molecular basis of infantile neuronal ceroidlipofuscinosis. Proc. Natl Acad. Sci. USA, 97, 4573–4578.
9. Camp, L.A. and Hofmann, S.L. (1993) Purification and properties of apalmitoyl-protein thioesterase that cleaves palmitate from H-Ras. J. Biol.
Chem., 268, 22566–22574.
10. Hellsten, E., Vesa, J., Olkkonen, V.M., Jalanko, A. and Peltonen, L.(1996) Human palmitoyl protein thioesterase: evidence for lysosomaltargeting of the enzyme and disturbed cellular routing in infantile neuronalceroid lipofuscinosis. EMBO J., 15, 5240–5245.
11. Ahtiainen, L., Van Diggelen, O.P., Jalanko, A. and Kopra, O. (2003)Palmitoyl protein thioesterase 1 is targeted to the axons in neurons.J. Comp. Neurol., 455, 368–377.
12. Heinonen, O., Kyttala, A., Lehmus, E., Paunio, T., Peltonen, L. andJalanko, A. (2000) Expression of palmitoyl protein thioesterase inneurons. Mol. Genet. Metab., 69, 123–129.
13. Lehtovirta, M., Kyttala, A., Eskelinen, E.L., Hess, M., Heinonen, O. andJalanko, A. (2001) Palmitoyl protein thioesterase (PPT) localizes intosynaptosomes and synaptic vesicles in neurons: implications for infantileneuronal ceroid lipofuscinosis (INCL). Hum. Mol. Genet., 10, 69–75.
14. Heinonen, O., Salonen, T., Jalanko, A., Peltonen, L. and Copp, A. (2000)CLN-1 and CLN-5, genes for infantile and variant late infantile neuronalceroid lipofuscinoses, are expressed in the embryonic human brain.J. Comp. Neurol., 426, 406–412.
15. Isosomppi, J., Heinonen, O., Hiltunen, J.O., Greene, N.D., Vesa, J.,Uusitalo, A., Mitchison, H.M., Saarma, M., Jalanko, A. and Peltonen, L.(1999) Developmental expression of palmitoyl protein thioesterase innormal mice. Brain Res. Dev. Brain Res., 118, 1–11.
16. Ahtiainen, L., Luiro, K., Kauppi, M., Tyynela, J., Kopra, O. and Jalanko,A. (2006) Palmitoyl protein thioesterase 1 (PPT1) deficiency causesendocytic defects connected to abnormal saposin processing. Exp. Cell
Res., 312, 1540–1553.
17. Kyttala, A., Lahtinen, U., Braulke, T. and Hofmann, S.L. (2006)Functional biology of the neuronal ceroid lipofuscinoses (NCL) proteins.Biochim. Biophys. Acta, 1762, 920–933.
18. Cooper, J.D., Russell, C. and Mitchison, H.M. (2006) Progress towardsunderstanding disease mechanisms in small vertebrate models of neuronalceroid lipofuscinosis. Biochim. Biophys. Acta, 1762, 873–889.
19. Jalanko, A., Tyynela, J. and Peltonen, L. (2006) From genes to systems:new global strategies for the characterization of NCL biology. Biochim.
Biophys. Acta, 1762, 934–944.
20. Jalanko, A., Vesa, J., Manninen, T., von Schantz, C., Minye, H., Fabritius,A.L., Salonen, T., Rapola, J., Gentile, M., Kopra, O. et al. (2005) Micewith Ppt1(Deltaex4) mutation replicate the INCL phenotype and showan inflammation-associated loss of interneurons. Neurobiol. Dis., 18,226–241.
21. Boyer, P.D. (2002) A research journey with ATP synthase. J. Biol. Chem.,277, 39045–39061.
22. Champagne, E., Martinez, L.O., Collet, X. and Barbaras, R. (2006)Ecto-F1Fo ATP synthase/F1 ATPase: metabolic and immunologicalfunctions. Curr. Opin. Lipidol., 17, 279–284.
23. Webb, Y., Hermida-Matsumoto, L. and Resh, M.D. (2000) Inhibition ofprotein palmitoylation, raft localization, and T cell signaling by2-bromopalmitate and polyunsaturated fatty acids. J. Biol. Chem., 275,261–270.
24. El-Husseini Ael, D., Schnell, E., Dakoji, S., Sweeney, N., Zhou, Q.,Prange, O., Gauthier-Campbell, C., Aguilera-Moreno, A., Nicoll, R.A.and Bredt, D.S. (2002) Synaptic strength regulated by palmitate cyclingon PSD-95. Cell, 108, 849–863.
25. Ahtiainen, L., Kolikova, J., Mutka, A.L., Luiro, K., Gentile, M., Ikonen,E., Khiroug, L., Jalanko, A. and Kopra, O. (2007) Palmitoyl proteinthioesterase 1 (Ppt1)-deficient mouse neurons show alterations incholesterol metabolism and calcium homeostasis prior to synapticdysfunction. Neurobiol. Dis., 28, 52–64.
26. Martinez, L.O., Jacquet, S., Esteve, J.P., Rolland, C., Cabezon, E.,Champagne, E., Pineau, T., Georgeaud, V., Walker, J.E., Terce, F. et al.
(2003) Ectopic beta-chain of ATP synthase is an apolipoprotein A-Ireceptor in hepatic HDL endocytosis. Nature, 421, 75–79.
27. Fabre, A.C., Vantourout, P., Champagne, E., Terce, F., Rolland, C.,Perret, B., Collet, X., Barbaras, R. and Martinez, L.O. (2006) Cell surfaceadenylate kinase activity regulates the F(1)-ATPase/P2Y (13)-mediated
1416 Human Molecular Genetics, 2008, Vol. 17, No. 10
by guest on Decem
ber 21, 2015http://hm
g.oxfordjournals.org/D
ownloaded from

HDL endocytosis pathway on human hepatocytes. Cell Mol. Life Sci., 63,2829–2837.
28. Qin, S., Kawano, K., Bruce, C., Lin, M., Bisgaier, C., Tall, A.R. and Jiang,X. (2000) Phospholipid transfer protein gene knock-out mice have lowhigh density lipoprotein levels, due to hypercatabolism, and accumulateapoA-IV-rich lamellar lipoproteins. J. Lipid Res., 41, 269–276.
29. Matsuura, F., Wang, N., Chen, W., Jiang, X.C. and Tall, A.R. (2006) HDLfrom CETP-deficient subjects shows enhanced ability to promotecholesterol efflux from macrophages in an apoE- and ABCG1-dependentpathway. J. Clin. Invest., 116, 1435–1442.
30. Porter, M.Y., Turmaine, M. and Mole, S.E. (2005) Identification andcharacterization of Caenorhabditis elegans palmitoyl proteinthioesterase1. J. Neurosci. Res., 79, 836–848.
31. Jolly, R.D., Brown, S., Das, A.M. and Walkley, S.U. (2002)Mitochondrial dysfunction in the neuronal ceroid-lipofuscinoses (Battendisease). Neurochem. Int., 40, 565–571.
32. Luiro, K., Kopra, O., Blom, T., Gentile, M., Mitchison, H.M., Hovatta, I.,Tornquist, K. and Jalanko, A. (2006) Batten disease (JNCL) is linked todisturbances in mitochondrial, cytoskeletal, and synaptic compartments.J. Neurosci. Res., 84, 1124–1138.
33. Fossale, E., Wolf, P., Espinola, J.A., Lubicz-Nawrocka, T., Teed, A.M.,Gao, H., Rigamonti, D., Cattaneo, E., MacDonald, M.E. and Cotman, S.L.(2004) Membrane trafficking and mitochondrial abnormalities precedesubunit c deposition in a cerebellar cell model of juvenile neuronal ceroidlipofuscinosis. BMC Neurosci., 5, 57.
34. Das, A.M., Jolly, R.D. and Kohlschutter, A. (1999) Anomalies ofmitochondrial ATP synthase regulation in four different types of neuronalceroid lipofuscinosis. Mol. Genet. Metab., 66, 349–355.
35. Arakaki, N., Nagao, T., Niki, R., Toyofuku, A., Tanaka, H., Kuramoto, Y.,Emoto, Y., Shibata, H., Magota, K. and Higuti, T. (2003) Possible role ofcell surface Hþ -ATP synthase in the extracellular ATP synthesis andproliferation of human umbilical vein endothelial cells. Mol. Cancer Res.,1, 931–939.
36. Kim, B.W., Choo, H.J., Lee, J.W., Kim, J.H. and Ko, Y.G. (2004)Extracellular ATP is generated by ATP synthase complex in adipocytelipid rafts. Exp. Mol. Med., 36, 476–485.
37. Kim, K.B., Lee, J.W., Lee, C.S., Kim, B.W., Choo, H.J., Jung, S.Y., Chi,S.G., Yoon, Y.S., Yoon, G. and Ko, Y.G. (2006) Oxidation-reductionrespiratory chains and ATP synthase complex are localized indetergent-resistant lipid rafts. Proteomics, 6, 2444–2453.
38. Lyly, A., von Schantz, C., Salonen, T., Kopra, O., Saarela, J., Jauhiainen,M., Kyttala, A. and Jalanko, A. (2007) Glycosylation, transport, andcomplex formation of palmitoyl protein thioesterase 1 (PPT1) - distinctcharacteristics in neurons. BMC Cell Biol., 8, 22.
39. Goswami, R., Ahmed, M., Kilkus, J., Han, T., Dawson, S.A. and Dawson,G. (2005) Differential regulation of ceramide in lipid-rich microdomains(rafts): antagonistic role of palmitoyl:protein thioesterase and neutralsphingomyelinase 2. J. Neurosci. Res., 81, 208–217.
40. Koch, S., Donarski, N., Goetze, K., Kreckel, M., Stuerenburg, H.J.,Buhmann, C. and Beisiegel, U. (2001) Characterization of four lipoproteinclasses in human cerebrospinal fluid. J. Lipid Res., 42, 1143–1151.
41. Rader, D.J. (2006) Molecular regulation of HDL metabolism andfunction: implications for novel therapies. J. Clin. Invest., 116,3090–3100.
42. Abildayeva, K., Jansen, P.J., Hirsch-Reinshagen, V., Bloks, V.W.,Bakker, A.H., Ramaekers, F.C., de Vente, J., Groen, A.K., Wellington,C.L., Kuipers, F. et al. (2006) 24(S)-hydroxycholesterol participates in aliver X receptor-controlled pathway in astrocytes that regulatesapolipoprotein E-mediated cholesterol efflux. J. Biol. Chem., 281,12799–12808.
43. Panzenboeck, U., Balazs, Z., Sovic, A., Hrzenjak, A., Levak-Frank, S.,Wintersperger, A., Malle, E. and Sattler, W. (2002) ABCA1 andscavenger receptor class B, type I, are modulators of reverse steroltransport at an in vitro blood-brain barrier constituted of porcine braincapillary endothelial cells. J. Biol. Chem., 277, 42781–42789.
44. Cavelier, C., Rohrer, L. and von Eckardstein, A. (2006) ATP-Bindingcassette transporter A1 modulates apolipoprotein A-I transcytosis throughaortic endothelial cells. Circ. Res., 99, 1060–1066.
45. Kratzer, I., Wernig, K., Panzenboeck, U., Bernhart, E., Reicher, H.,Wronski, R., Windisch, M., Hammer, A., Malle, E., Zimmer, A. et al.(2007) Apolipoprotein A-I coating of protamine-oligonucleotidenanoparticles increases particle uptake and transcytosis in an in vitromodel of the blood-brain barrier. J. Control. Release, 117, 301–311.
46. Haidar, B., Kiss, R.S., Sarov-Blat, L., Brunet, R., Harder, C., McPherson,R. and Marcel, Y.L. (2006) Cathepsin D, a Lysosomal Protease, RegulatesABCA1-mediated Lipid Efflux. J. Biol. Chem., 281, 39971–39981.
47. Wang, T., Chen, Z., Wang, X., Shyy, J.Y. and Zhu, Y. (2006) Cholesterolloading increases the translocation of ATP synthase beta chain intomembrane caveolae in vascular endothelial cells. Biochim. Biophys. Acta,1761, 1182–1190.
48. Farrer, L.A., Cupples, L.A., Haines, J.L., Hyman, B., Kukull, W.A.,Mayeux, R., Myers, R.H., Pericak-Vance, M.A., Risch, N. and van Duijn,C.M. (1997) Effects of age, sex, and ethnicity on the association betweenapolipoprotein E genotype and Alzheimer disease. A meta-analysis.APOE and Alzheimer Disease Meta Analysis Consortium. JAMA, 278,1349–1356.
49. Dietschy, J.M. and Turley, S.D. (2004) Thematic review series: brainLipids. Cholesterol metabolism in the central nervous system during earlydevelopment and in the mature animal. J. Lipid Res., 45, 1375–1397.
50. Beisiegel, U., Weber, W., Havinga, J.R., Ihrke, G., Hui, D.Y.,Wernette-Hammond, M.E., Turck, C.W., Innerarity, T.L. and Mahley,R.W. (1988) Apolipoprotein E-binding proteins isolated from dog andhuman liver. Arteriosclerosis, 8, 288–297.
51. Hui, D.Y., Brecht, W.J., Hall, E.A., Friedman, G., Innerarity, T.L. andMahley, R.W. (1986) Isolation and characterization of the apolipoproteinE receptor from canine and human liver. J. Biol. Chem., 261, 4256–4267.
52. Vikstedt, R., Metso, J., Hakala, J., Olkkonen, V.M., Ehnholm, C. andJauhiainen, M. (2007) Cholesterol efflux from macrophage foam cells isenhanced by active phospholipid transfer protein through generation oftwo types of acceptor particles. Biochemistry, 46, 11979–11986.
53. Vuletic, S., Peskind, E.R., Marcovina, S.M., Quinn, J.F., Cheung, M.C.,Kennedy, H., Kaye, J.A., Jin, L.W. and Albers, J.J. (2005) Reduced CSFPLTP activity in Alzheimer’s disease and other neurologic diseases; PLTPinduces ApoE secretion in primary human astrocytes in vitro. J. Neurosci.
Res., 80, 406–413.54. Ohta, S. and Kagawa, Y. (1986) Human F1-ATPase: molecular cloning of
cDNA for the beta subunit. J. Biochem., 99, 135–141.55. Clarke, N.J., Li, F., Tomlinson, A.J. and Naylor, S. (1998) One step
microelectroelution concentration method for efficient coupling of sodiumdodecylsulfate gel electrophoresis and matrix-assisted laser desorptiontime-of-flight mass spectrometry for protein analysis. J. Am. Soc. Mass
Spectr., 9, 88–91.56. Lutter, R., Abrahams, J.P., van Raaij, M.J., Todd, R.J., Lundqvist, T.,
Buchanan, S.K., Leslie, A.G. and Walker, J.E. (1993) Crystallization ofF1-ATPase from bovine heart mitochondria. J. Mol. Biol., 229, 787–790.
57. Harkke, S., Laine, M. and Jalanko, A. (2003) Aspartylglucosaminidase(AGA) is efficiently produced and endocytosed by glial cells: implicationfor the therapy of a lysosomal storage disorder. J. Gene Med., 5, 472–482.
58. Majander, A., Rapola, J., Sariola, H., Suomalainen, A., Pohjavuori, M.and Pihko, H. (1995) Diagnosis of fatal infantile defects of themitochondrial respiratory chain: age dependence and postmortem analysisof enzyme activities. J. Neurol. Sci., 134, 95–102.
59. Steenaart, N.A. and Shore, G.C. (1997) Mitochondrial cytochrome coxidase subunit IV is phosphorylated by an endogenous kinase. FEBS
Lett., 415, 294–298.60. Antonicka, H., Sasarman, F., Kennaway, N.G. and Shoubridge, E.A.
(2006) The molecular basis for tissue specificity of the oxidativephosphorylation deficiencies in patients with mutations in themitochondrial translation factor EFG1. Hum. Mol. Genet., 15,1835–1846.
61. Siggins, S., Bykov, I., Hermansson, M., Somerharju, P., Lindros, K.,Miettinen, T.A., Jauhiainen, M., Olkkonen, V.M. and Ehnholm, C. (2007)Altered hepatic lipid status and apolipoprotein A-I metabolism in micelacking phospholipid transfer protein. Atherosclerosis, 190, 114–123.
62. Jauhiainen, M., Huuskonen, J., Baumann, M., Metso, J., Oka, T.,Egashira, T., Hattori, H., Olkkonen, V.M. and Ehnholm, C. (1999)Phospholipid transfer protein (PLTP) causes proteolytic cleavage ofapolipoprotein A-I. J. Lipid Res., 40, 654–664.
63. Jauhiainen, M. and Ehnholm, C. (2005) Determination of human plasmaphospholipid transfer protein mass and activity. Methods, 36, 97–101.
64. van Haperen, R., van Tol, A., Vermeulen, P., Jauhiainen, M., van Gent, T.,van den Berg, P., Ehnholm, S., Grosveld, F., van der Kamp, A. and deCrom, R. (2000) Human plasma phospholipid transfer protein increasesthe antiatherogenic potential of high density lipoproteins in transgenicmice. Arterioscler. Thromb. Vasc. Biol., 20, 1082–1088.
Human Molecular Genetics, 2008, Vol. 17, No. 10 1417
by guest on Decem
ber 21, 2015http://hm
g.oxfordjournals.org/D
ownloaded from
Copyright © 2022 FDOKUMEN




















