
Cytotoxic Effect of p-Coumaric Acid on Neuroblastoma, N2a Cell via Generation of Reactive Oxygen...
12
Cytotoxic Effect of p-Coumaric Acid on Neuroblastoma, N2a Cell via Generation of Reactive Oxygen Species Leading to Dysfunction of Mitochondria Inducing Apoptosis and Autophagy S. Shailasree & M. Venkataramana & S. R. Niranjana & H. S. Prakash Received: 22 January 2014 /Accepted: 25 March 2014 /Published online: 24 April 2014 # Springer Science+Business Media New York 2014 Abstract p-Coumaric acid (p-CA), an ubiquitous plant phe- nolic acid, has been proven to render protection against path- ological conditions. In the present study, p-CA was evaluated for its capacity to induce cytotoxic effect to neuroblastoma N2a cells and we report here the possible mechanism of its action. p-CA at a concentration of 150 μmol/L, upon exposure for 72 h, stimulated 81.23 % of cells to apoptosis, as evi- denced by flow cytometer studies mediated through elevated levels of ROS (7.5-fold over control). Excess ROS production activated structural injury to mitochondrial membrane, ob- served as dissipation of its membrane potential and followed by the release of cytochrome c (8.73-fold). Enhanced gener- ation of intracellular ROS correlated well with the decreased levels (~60 %) of intracellular GSH. Sensitizing neuroblasto- ma cells for induction of apoptosis by p-CA identified p53- mediated upregulated accumulation of caspase-8 messenger Highlights 1. We have used in vitro model to investigate the cytotoxic effects of p-coumaric acid (p-CA) on neuroblastoma N2a cells. 2. p-Coumaric acid-induced cytotoxicity by 72 h at 150 μmol/L stimu- lated 12.06 % of cells to early apoptosis and 81.23 % of cells to apoptotic or necrotic cells, as evidenced by flow cytometer studies. 3. Cytotoxic effect was mediated by excessive production of reactive oxygen species (7.5-fold over control) causing mitochondrial dysfunction and followed by release of cytochrome c. 4. Sensitizing N2a cells for induction of apoptosis by p-CA identified p53-mediated upregulation of caspase-8 messenger RNA (2.8-fold) as target gene. 5. Our data report on autophagy, representing an additional mechanism of p-CA to induce growth arrest, detected by immunoblotting and fluores- cence, correlated with accumulation of elevated levels (1.2-fold) of the LC3-II protein and acridine orange-stained autophagosomes, both au- tophagy markers. 6. Detection of lactate dehydrogenase in supernatants of p-CA-treated N2a cells relative to control cells leads to interpretation that a few cells died by necrosis. S. Shailasree Institution of Excellence, Vijnana Bhavana, University of Mysore, Mysore 570006, India e-mail: [email protected] S. R. Niranjana : H. S. Prakash (*) Department of Studies in Biotechnology, University of Mysore, Mysore 570006, India e-mail: [email protected] H. S. Prakash e-mail: [email protected] S. R. Niranjana e-mail: [email protected] M. Venkataramana Defence Research Development Organization, Bharathiar University, Coimbatore, Tamil Nadu, India Mol Neurobiol (2015) 51:119–130 DOI 10.1007/s12035-014-8700-2
-
Upload
uni-mysore -
Category
Documents
-
view
6 -
download
0
Transcript of Cytotoxic Effect of p-Coumaric Acid on Neuroblastoma, N2a Cell via Generation of Reactive Oxygen...

Cytotoxic Effect of p-Coumaric Acid on Neuroblastoma, N2a Cellvia Generation of Reactive Oxygen Species Leadingto Dysfunction of Mitochondria Inducing Apoptosisand Autophagy
S. Shailasree & M. Venkataramana & S. R. Niranjana &
H. S. Prakash
Received: 22 January 2014 /Accepted: 25 March 2014 /Published online: 24 April 2014# Springer Science+Business Media New York 2014
Abstract p-Coumaric acid (p-CA), an ubiquitous plant phe-nolic acid, has been proven to render protection against path-ological conditions. In the present study, p-CAwas evaluatedfor its capacity to induce cytotoxic effect to neuroblastomaN2a cells and we report here the possible mechanism of itsaction. p-CA at a concentration of 150 μmol/L, upon exposurefor 72 h, stimulated 81.23 % of cells to apoptosis, as evi-denced by flow cytometer studies mediated through elevated
levels of ROS (7.5-fold over control). Excess ROS productionactivated structural injury to mitochondrial membrane, ob-served as dissipation of its membrane potential and followedby the release of cytochrome c (8.73-fold). Enhanced gener-ation of intracellular ROS correlated well with the decreasedlevels (~60 %) of intracellular GSH. Sensitizing neuroblasto-ma cells for induction of apoptosis by p-CA identified p53-mediated upregulated accumulation of caspase-8 messenger
Highlights 1. We have used in vitro model to investigate the cytotoxiceffects of p-coumaric acid (p-CA) on neuroblastoma N2a cells.2. p-Coumaric acid-induced cytotoxicity by 72 h at 150 μmol/L stimu-lated 12.06 % of cells to early apoptosis and 81.23 % of cells to apoptoticor necrotic cells, as evidenced by flow cytometer studies.3. Cytotoxic effect was mediated by excessive production of reactiveoxygen species (7.5-fold over control) causing mitochondrial dysfunctionand followed by release of cytochrome c.4. Sensitizing N2a cells for induction of apoptosis by p-CA identifiedp53-mediated upregulation of caspase-8 messenger RNA (2.8-fold) astarget gene.5. Our data report on autophagy, representing an additional mechanism ofp-CA to induce growth arrest, detected by immunoblotting and fluores-cence, correlated with accumulation of elevated levels (1.2-fold) of theLC3-II protein and acridine orange-stained autophagosomes, both au-tophagy markers.6. Detection of lactate dehydrogenase in supernatants of p-CA-treatedN2a cells relative to control cells leads to interpretation that a few cellsdied by necrosis.
S. ShailasreeInstitution of Excellence, Vijnana Bhavana, University of Mysore,Mysore 570006, Indiae-mail: [email protected]
S. R. Niranjana :H. S. Prakash (*)Department of Studies in Biotechnology, University of Mysore,Mysore 570006, Indiae-mail: [email protected]
H. S. Prakashe-mail: [email protected]
S. R. Niranjanae-mail: [email protected]
M. VenkataramanaDefence Research Development Organization, BharathiarUniversity, Coimbatore, Tamil Nadu, India
Mol Neurobiol (2015) 51:119–130DOI 10.1007/s12035-014-8700-2

RNA (2.8-fold). Our data report on autophagy, representingan additional mechanism of p-CA to induce growth arrest,detected by immunoblotting and fluorescence, correlated withaccumulation of elevated levels (1.2-fold) of the LC3-II pro-tein and acridine orange-stained autophagosomes, both au-tophagy markers. The present study indicates p-CA was ef-fective in production of ROS-dependent mitochondrialdamage-induced cytotoxicity in N2a cells.
Keywords p-Coumaric acid . NeuroblastomaN2a cells .
Apoptosis . p53 . Caspase-8 . Autophagy . LC3-II
Introduction
Mitochondria are an intracellular source of reactive oxygenspecies (ROS) [1, 2], continuously generated as byproduct ofthe respiratory chain and involved in the signaling pathwayfrom the mitochondria to the nucleus [3]. Any imbalance inconcentration of ROS, beyond the threshold, is reported tocontribute to the “cumulative oxidative stress” resulting toalterations in abundance of mitochondria, indiscriminate dam-age to copy number, and integrity of mitochondrial DNA.Further, ROS-induced stress response initiates altered expres-sion of specific nuclear genes requisite for normal physiologicalfunction of cells, a cause maybe resulting in diminished anti-oxidant defense, thiol-reducing buffer of small proteins withredox active sulfhydryl moieties like glutathione (GSH),thioredoxin (TRX), and enzymatic systems (superoxide dis-mutase, catalase, and glutathione peroxidase [4]. In the affectedcells, ROS damage could lead to initiation of dissemination ofmitochondrial membrane potential, with release of mitochon-drial proteins, like, cytochrome c. Thus, oxidative stress, eitherto an increase in reactive oxygen species (ROS) and/or adiminished antioxidant defense in the cells, leads to patholog-ical processes, causing cancer, diabetes, and neurodegenerativediseases. These cells under oxidative stress, even at rest, have ametabolic rate higher than normal cells, resulting in elevatedconcentrations of ROS, thus rendering themmore vulnerable toROS-mediated events, like, apoptosis [5] and autophagy [6, 7].
Apoptosis, defined as a controlled suicide program to removedefective cells, without damage to neighboring cells, acts viadeath receptor (extrinsic) and the mitochondrial (intrinsic) path-way [8, 9]. The mitochondrial pathway, involving loss of mito-chondrial membrane potential (Δψm), is regulated through ahighly intricate cross talk, which includes caspases 8, 9, and 3situated at pivotal points of the signaling process [10]. Caspase-8studies represent a yet unknown proapoptotic pathway activatedby cytotoxic drugs, and further, it was identified as an importantp53 target gene in drug-induced death of cancerous cells [11, 12].
Autophagy, type-II cell death, a catabolic process, is pheno-typically characterized by formation of large intracellular vac-uoles, termed autophagosomes. It includes sequestration of
cellular proteins and organelles in a specific sequence prior tothe destruction of the nucleus and has been recognized invarious cancer cell types as a response to anticancer therapies[13, 14]. Induction of autophagy results in posttranslationalprocessing of cytosolic-associated microtubule-associatedprotein-1A/1B light chain 3 (LC3-I; ~17 kDa) to autophagosomemembrane-bound LC3-phosphatidylethanolamine conjugate(LC3-II; ~19 kDa), leading to formation of autophagosomes[15].
Neuroblastoma and solid extracranial cancer of childhoodwith an annual incidence of ~9.1 cases per million children isreported [16]. Standard treatments include radiation alongwith chemotherapy, however results in side effects to childrendue to its acute toxicity. Advances in technology has allowedfor search of new nontoxic drugs to be used as a single ormultidrug therapy in natural products [17–20] with almost60 % of drugs approved for cancer treatment being fromnatural origin [21].
p-Coumaric acid (p-CA), a ubiquitous plant phenolic acidand an abundant isomer of cinnamic acid, has been reportedfrom cereals, fruits, and vegetables. The phenolic hydroxylgroup provides the hydrogen atoms for scavenging the ROS.Numerous reports on use of p-CA for therapeutic benefitsagainst cancer cell lines include antiproliferative effect of p-CA against colonic epithelial cells (Caco-2) by p-CA(1,500 μmol/L) by 24–72 h treatment [22]. A study relatedto benefits of honey, reported on p-CA, credited for thebenefits, inhibited proliferation of colon cancer cells.Inhibiting its growth through mitochondrial malfunction byROS generated during the treatment and apoptosis as themechanism accompanied by blebbing and shrinkage of cellsis reported [23]. The inhibition of mitogen-activated proteinkinase (MAPK) signaling, leading to blocking of nuclear-factor kappa B and activator protein-1 was speculated. Pro-tective effects of p-CA include a study involving combinationof p-CA and hydrocaffeic acid in reducing UV-B oxidativedamage on both in vivo (cornea and sclera) and in vitro(conjunctiva cells) [24]. Neutralization of doxorubicin-induced oxidative stress for protection of rat’s heart withreduced serum levels of lactic dehydrogenase and creatinephosphokinase is recorded [25].
Further reports on p-CA include protection of mouse cor-tical neurons against neuronal injury induced by 5-S-cysteinyl-dopamine neurotoxicity [26], a capacity to reducerisk on stomach cancer by suppressing formation of nitrosa-mines [27], and as an antimelanogenic agent [28] with antiul-cer [29] and antiplatelet activities [30]. Although numerousstudies have described arrest of cancerous cells by p-CA, littleis known regarding the mechanism of action of p-CA leadingto suppression of tumor cell growth through its ability toinduce apoptosis and autophagy.
In the present study, neuroblastoma N2a cell, a subcultureof mouse C1300, has been used for testing neurotoxicity of
120 Mol Neurobiol (2015) 51:119–130

putative drugs [31], elucidating cellular and molecular mech-anisms [32] were included. There have been very few reportson the effect of p-CA in neuroblastoma. Using N2a cells, inthe present study, we show exposure to p-CA augments ROSlevels and the loss of mitochondrial membrane potential as ap-CA-induced event. p-CA treatment results in the release ofcytochrome c with subsequent activation of mitochondrialintrinsic apoptotic pathway and eventually leading to celldeath in vitro. p-CA engender autophagic cell death associatedwith ROS [33] leading to formation of autophagosomes withaccumulation of LC3-II, an autophagy-related unique proteininvolved in formation of autophagosomal membranes in can-cer cells during autophagy.
Materials and Methods
Neuroblastoma Cell Cultures
Mouse neuroblastoma, N2a cell line procured from the Na-tional Center for Cell Science (NCCS, Pune, India) was usedfor the studies. The cells were cultured in Dulbecco’s modifiedEagle’s medium (DMEM; Sigma-Aldrich, St. Louis, MO,U.S.A), supplemented with 5 % fetal calf serum (Sigma-Aldrich, St. Louis, MO, U.S.A) and streptomycin-penicillin(Sigma-Aldrich, St. Louis, MO, U.S.A). The cells were platedat 3×104 cells/cm2, grown in humidified 5 % CO2 with 95 %air atmosphere at 37 °C, and experiments were initiated 48 hafter plating. The culture medium was replaced two times aweek. For the experiments, confluent cells were trypsinizedand plated in 96-, 6-well plates, or into tissue culture dishes(6 mm).
Evaluation of p-Coumaric Acid (p-CA) Induced Cytotoxicity
p-CA (Sigma-Aldrich, St. Louis, MO, U.S.A) was tested atconcentrations between 1 and 200 μmol/L for its cytotoxiceffect. Replicates were maintained for each concentration in96-well plates. N2a cells were subjected to p-CA exposure for72 h. The cell viability was tested using 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich, St. Louis, MO, U.S.A). MTT (5 mg/mL) was dis-solved in sterile phosphate buffer saline (PBS; 0.05 M phos-phate buffer, pH 7.2, 0.8 % NaCl) at room temperature. Thesolution was further sterilized by passing through a 0.2-μmfilter and stored at 4 °C in the dark. For cell viability assay,MTT (10 μL; 1 mg/mL) was added to each well and the plateswere incubated at 37 °C for 4 h in the dark. The supernatantswere aspirated from the wells and washed thrice with PBS.DMSO (100 μL) and 25 μL of 0.1 M glycine buffer (pH 10.5)were added to each well. After an incubation period of 15min,the absorbance was measured at 570 nm using multimodeplate reader (Varioskan Flash Top, Thermo Fisher Scientific,
Germany). Controls consisting of same concentration of cellswithout p-CAwere maintained. Any absorbance due to reac-tion of p-CAwithMTT in wells devoid of cells was subtractedfrom the readings. Time course of p-CA-induced cytotoxicitywas studied under similar conditions wherein the N2a cellswere subjected for 24, 48, and 72 h of exposure to 150 μmol/Lp-CA. Respective controls were maintained in 96-wells foreach time period under similar conditions. Cell viability wasmeasured as described above. The absorbance was measuredat 595 nm as described above.
Lactate dehydrogenase (LDH) release has been correlatedto the extent of damage caused by the drug. N2a cells grown in6-well plate were exposed to different concentrations of p-CA(1–200 μmol/L p-CA), and cytotoxicity was assessed bymeasurement of LDH activity in the medium 72 h after p-CA exposure. LDH activity was determined using LDH kit(Agappe Diagnostics Ltd, Bangalore, India) as per manufac-turers’ protocol. Briefly, an aliquot of the supernatant (controland p-CA-treated cell culture) was mixed with nicotinamideadenine dinucleotide-lactate solution and absorbance wasmeasured at 450 nm using multimode plate reader (VarioskanFlash Top, Thermo Fisher Scientific, Germany).
Morphological Analysis by Phase Contrast Microscopy
Cell morphology was evaluated using phase contrast invertedmicroscope with digital imaging (Axiovert A1, Zeiss,Germany).
Involvement of Intracellular Reactive Oxygen Species (ROS)in p-CA-Induced Cell Cytotoxicity
Accumulation of intracellular ROS was monitored using fluo-rescent probe 2′,7′-dichlorodihydrofluorescein diacetate(H2DCF-DA; Sigma-Aldrich, St. Louis, MO, U.S.A.). Inbrief, 2×105 cells were plated in serum-containing DMEMin 6 mm culture dishes and allowed to attach. After 72 h andfurther exposure to p-CA (150 and 200 μmol/L), the cellswere washed with warm PBS for 30 min at 37 °C and furtherexposed to H2DCF-DA (10 μmol/L) for 30 min in the darkunder 5 % CO2 in the incubator to label intracellular ROS.Controls consisted of cells with the cell culture medium (mi-nus p-CA). The fluorescence was measured using multimodeplate reader (Varioskan Flash Top, Thermo Fisher Scientific,Germany) and photomicrographs were captured using ad-vanced fluorescence research microscope with digital imaging(AxioImager A2, Zeiss Germany).
Estimation of Intracellular Glutathione (GSH)
Intracellular level of GSH in N2a cells was estimated byortho-phthalaldehyde (5 mg/mL) solution, a fluorescent dye.N2a cells were exposed to treatment with p-CA as described
Mol Neurobiol (2015) 51:119–130 121

for ROS. Controls consisted of cells with cell culture medium(minus p-CA). Cells were collected and lyzed with sonicationand centrifuged at 103×g for 3 min. The lysate obtained aftercentrifugation (25 μl) was treated with 5 μM (5 μl) of ortho-phthaldehyde in PBS. The fluorescence due to GSH wasmeasured using multimode plate reader with excitation at365 nm and emission 530 nm (Varioskan Flash Top, ThermoFisher Scientific, Germany).
Mitochondrial Membrane Potential (Δψm) Measurement
The mitochondrial membrane potential was measured usinglipophilic cationic probe, 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbcyanine iodide (JC-1; MolecularProbes, Eugene, OR, U.S.A.) most widely applied for de-tecting mitochondrial depolarization occurring in the earlystages of apoptosis. JC-1 exists as a green fluorescent mono-mer at depolarized membrane potentials or as a red fluores-cent J-aggregate at hyperpolarized membrane potentialsexhibiting a potential-dependent accumulation in the mito-chondria with emission shift from green (530 nm) to red(590 nm). After exposure to p-CA (150 and 200 μmol/L) for72 h, the cells were washed with PBS and subjected to JC-1(10 μg/mL). After 20 min incubation at 37 °C, changes inmitochondrial membrane potential was measured as fluores-cent intensity by multimode plate reader (Varioskan FlashTop, Thermo Fisher Scientific, Germany) [34]. Alternatively,cells were grown on poly-l-lysine slides (Sigma-Aldrich,M.O., USA) and exposed to p-CA (150 and 200 μmol/L)for 72 h in serum-containing DMEM as described above.Subsequently, they were stained for fluorescence with JC-1(10 μg/mL) for 20 min. After incubation, cells were visual-ized and analyzed by fluorescence microscopy (Advancedfluorescence research microscope with digital imaging,AxioImager A2, Zeiss, Germany).
Annexin V FITC, Propidium (PI) Labeling and FlowCytometer
After exposure to p-CA (150μmol/L) for 72 h, these N2a cellsand control (minus p-CA) were trypsinized and centrifuged at103×g for 3 min. Cells were resuspended in 200 μL of PBSand incubated with annexin V FITC and PI for 15 min ac-cording to manufacturers’ protocol (Beckman Coulter,U.S.A.). Theoretically, cells stained by annexin V were earlyapoptotic cells (lower right) and cells stained by PI wereconsidered as necrotic (dead) cells (upper left). Viable cellswere not stained (lower left). Late apoptotic cells were doublestained (upper right). A fluorescent activated flow cytometer(Cell Lab QuantaTM, SC, Beckman Coulter, U.S.A.) was usedto examine N2a cells (3×104).
Quantitative Real-Time PCR
N2a cells were cultured in 75 cm2 flasks (1×105), treated withp-CA (50, 150, and 200 μmol/L) for 72 h. Total cellular RNAwas isolated from the above samples and controls using com-mercially available RNA-isolation kit as per the manufac-turer’s protocol (Sigma, St. Louis, MO, USA). RNA (2 μg)were primed to oligo (dT) primers and reverse transcribedwith HS-RT-PCR kit (Sigma, St. Louis, MO, USA). The geneexpression of caspase-8, p53, and β-2 myoglobulin (house-keeping gene) were analyzed by quantitative real-time PCR(RT-PCR) with SYBR Green I Mastermix (Qiagen, Gambh,Germany) using a total reaction volume of 30 μl. Appropriatetarget primers: caspase-8: fw, 5′-CACTAGAAAGGAGGAGATGGAAAG-3′; rev, 5′-CTATCCTGTTCTCTTGGAGAGTCC-3′; p53: fw, 5′-AGAGTCTATAGGCCCACCCC-3′; rev,5 ′-GCTCGACGCTAGGATCTGAC-3 ′, and the β-2myoglobulin were used and reactions were run followingmanufacturer’s instructions, Roche Light Cycler 480. Eachsample was assayed, and the relative gene expression wasquantified using ΔΔCT method by normalizing with β-2myoglobulin.
Evaluation of Autophagic Vacuoles by Ethidium Bromideand Acridine Orange
N2a cells were grown to subconfluence in six-well plates,incubated with p-CA (150 μmol/L) for 72 h, and control cellsconsisted of cells minus p-CA. At the end of the incubationperiod, cells were stained with ethidium bromide and acridineorange (5 μg/mL; Sigma) in serum-free medium for 15 min[35] and were subsequently washed three times in PBS (clearsexcess of staining components). The fluorescence was record-ed using confocal laser scanning microscope, LSM 710 (CarlZeiss, Germany).
Western Blot Analysis: LC3-I and -II and of Cytochrome c
Cytosolic fractions of N2a cells treated with p-CA (50, 150,and 200 μmol/L) for 72 h and control cells (minus p-CA) werecompared for release of cytochrome c by Western blot analy-sis. Cell cultures (1×107 cells) were washed, and cytosolicfractions were prepared as reported earlier [34]. Briefly, cell,p-CA treated, and controls were lyzed by cold lysis 50 mM,Tris–HCl buffer (pH 7.5) containing NaCl (150mM), NonidetP-40 (0.5 %), phenylmethylsulfonyl fluoride (PMSF; 1 mM),NaF (1 mM), dithiothreitol (DTT; 1 mM), and completeprotease inhibitor cocktail (4 mg/ml). Protein content in thecell lysates were determined using Bradford method (Bio-Radprotein assay kit, Bio-Rad Hercules, CA). Total protein(30 μg) was separated by sodium docecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followingthe method of Laemmli [36] in a 1-mm thick, 12 %
122 Mol Neurobiol (2015) 51:119–130
polyacrylamide gel. Immediately after SDS-PAGE, the sepa-rated proteins were blotted [37] onto nitrocellulose membrane(Millipore) using Multiphor II (LKB, Pharmacia) electropho-retic transfer apparatus. The blots were blocked in fat-freemilk [2 % in Tris-buffered saline; TBS (10 mM Tris–HCl,pH 8.0 containing 125 mMNaCl)]. They were incubated withprimary antibodies:
a. LC3—anti LC3B antibody (ab48394) at 2 μg/ml abcam®,USA
b. Cytochrome c, β-actin, and GAPDH—at 2 μg/ml BDPharMingen, San Diego, CA; Biogenesis Inc., Poole, UK
for 2 h at 37 °C, diluted in TBS. After three washes with TBS,the blots were incubated with secondary antibodies (anti-goatIgG for cytochrome c; goat anti-rabbit for LC3-I, −II, and goat
anti-mouse IgG for GAPDH and β-actin; Upstate Cell signal-ing, Charlottesville, VA) conjugated with horseradish peroxi-dase for 1 h at room temperature. After three washes, the blotswere subjected to 3,3′-diaminobenzedine and hydrogen per-oxide treatment staining peroxidase activity to visualize im-munoreactive proteins.
Statistical Analysis
All experiments/measurements were made in triplicate, andall the values are expressed as the mean±S.E.M. Theresults were subjected to an analysis of variance followedby Tukey’s post hoc to analyze differences between p-CAand control conditions. A value of p<0.05 was consideredsignificant.
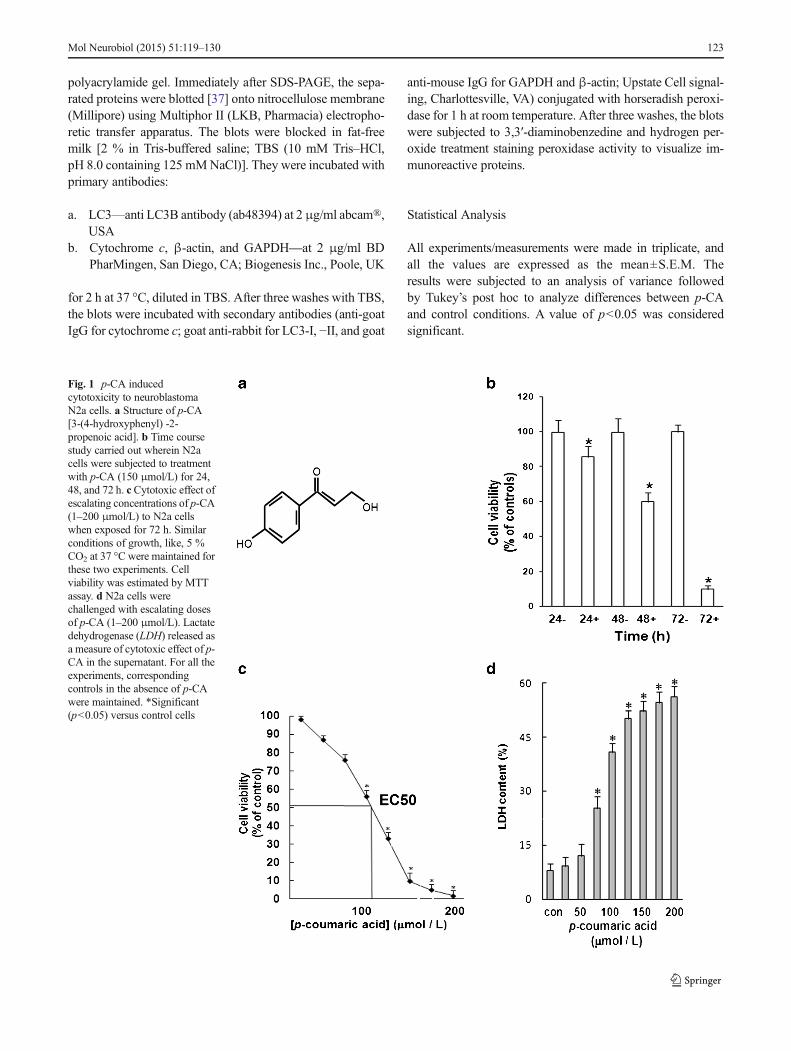
Fig. 1 p-CA inducedcytotoxicity to neuroblastomaN2a cells. a Structure of p-CA[3-(4-hydroxyphenyl) -2-propenoic acid]. b Time coursestudy carried out wherein N2acells were subjected to treatmentwith p-CA (150 μmol/L) for 24,48, and 72 h. c Cytotoxic effect ofescalating concentrations of p-CA(1–200 μmol/L) to N2a cellswhen exposed for 72 h. Similarconditions of growth, like, 5 %CO2 at 37 °C were maintained forthese two experiments. Cellviability was estimated by MTTassay. d N2a cells werechallenged with escalating dosesof p-CA (1–200 μmol/L). Lactatedehydrogenase (LDH) released asa measure of cytotoxic effect of p-CA in the supernatant. For all theexperiments, correspondingcontrols in the absence of p-CAwere maintained. *Significant(p<0.05) versus control cells
Mol Neurobiol (2015) 51:119–130 123
Results
p-Coumaric Acid (p-CA) Induced Cytotoxicityto Neuroblastoma, N2a Cells
A time course study on the effect of p-CA (Fig. 1a) to inducecytotoxicity to N2a cells was investigated. A decrease in cellviability between control and p-CA (150 μmol/L) at 24 and48 h was recorded with maximum cytotoxicity recorded by72 h (Fig. 1b). The viability of these cells decreased signifi-cantly with increase in p-CA concentrations (1–200 μmol/L)exposed for 72 h (Fig. 1c). The EC50 was calculated as104 μmol/L. The cytotoxic effect was also confirmed byLDH assay (Fig. 1d). Compared with the control, cells ex-posed to p-CA exhibited a significant (p<0.05) increase in theLDH release.
N2a cells were visualized under inverted microscope formorphological changes induced by exposure to p-CA. In con-trast to clear neural bodywith emerging neuritis observed inN2acells of the control group (Fig. 2a), cells exposed to p-CAexhibited the loss of these structural characteristics (Fig. 2b, c).
p-CATreatment Caused ROS Production in NeuroblastomaCells
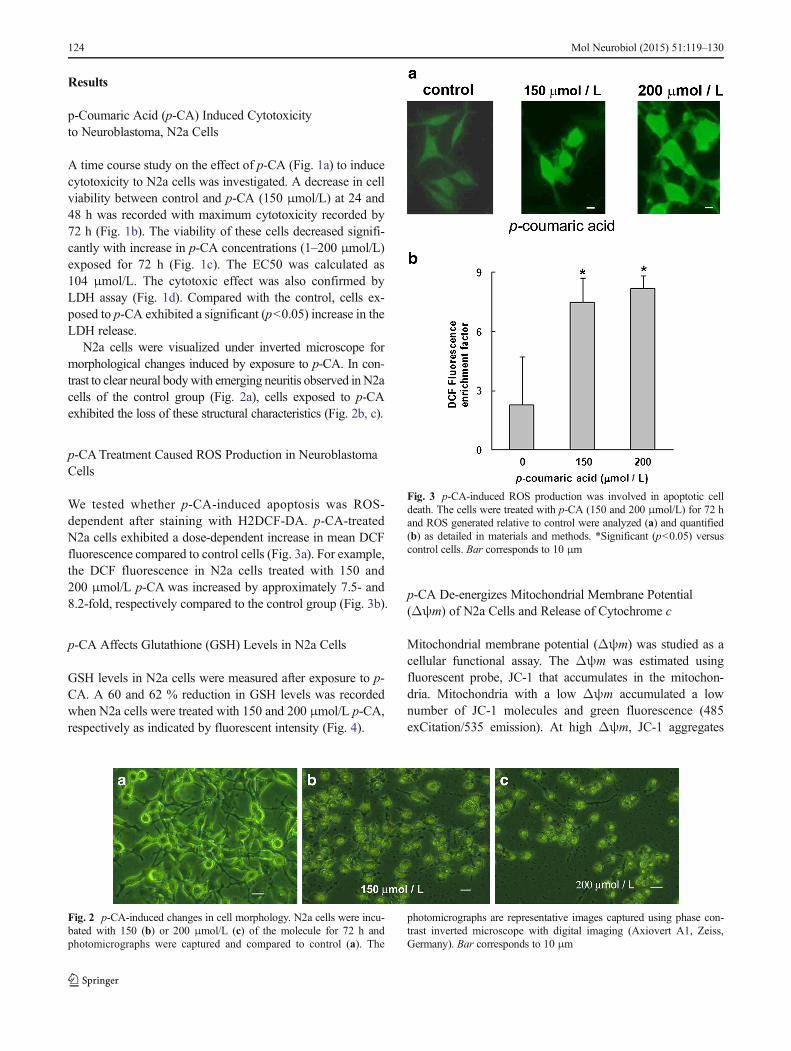
We tested whether p-CA-induced apoptosis was ROS-dependent after staining with H2DCF-DA. p-CA-treatedN2a cells exhibited a dose-dependent increase in mean DCFfluorescence compared to control cells (Fig. 3a). For example,the DCF fluorescence in N2a cells treated with 150 and200 μmol/L p-CA was increased by approximately 7.5- and8.2-fold, respectively compared to the control group (Fig. 3b).
p-CA Affects Glutathione (GSH) Levels in N2a Cells
GSH levels in N2a cells were measured after exposure to p-CA. A 60 and 62 % reduction in GSH levels was recordedwhen N2a cells were treated with 150 and 200 μmol/L p-CA,respectively as indicated by fluorescent intensity (Fig. 4).
p-CA De-energizes Mitochondrial Membrane Potential(Δψm) of N2a Cells and Release of Cytochrome c
Mitochondrial membrane potential (Δψm) was studied as acellular functional assay. The Δψm was estimated usingfluorescent probe, JC-1 that accumulates in the mitochon-dria. Mitochondria with a low Δψm accumulated a lownumber of JC-1 molecules and green fluorescence (485exCitation/535 emission). At high Δψm, JC-1 aggregates
Fig. 2 p-CA-induced changes in cell morphology. N2a cells were incu-bated with 150 (b) or 200 μmol/L (c) of the molecule for 72 h andphotomicrographs were captured and compared to control (a). The
photomicrographs are representative images captured using phase con-trast inverted microscope with digital imaging (Axiovert A1, Zeiss,Germany). Bar corresponds to 10 μm
Fig. 3 p-CA-induced ROS production was involved in apoptotic celldeath. The cells were treated with p-CA (150 and 200 μmol/L) for 72 hand ROS generated relative to control were analyzed (a) and quantified(b) as detailed in materials and methods. *Significant (p<0.05) versuscontrol cells. Bar corresponds to 10 μm
124 Mol Neurobiol (2015) 51:119–130
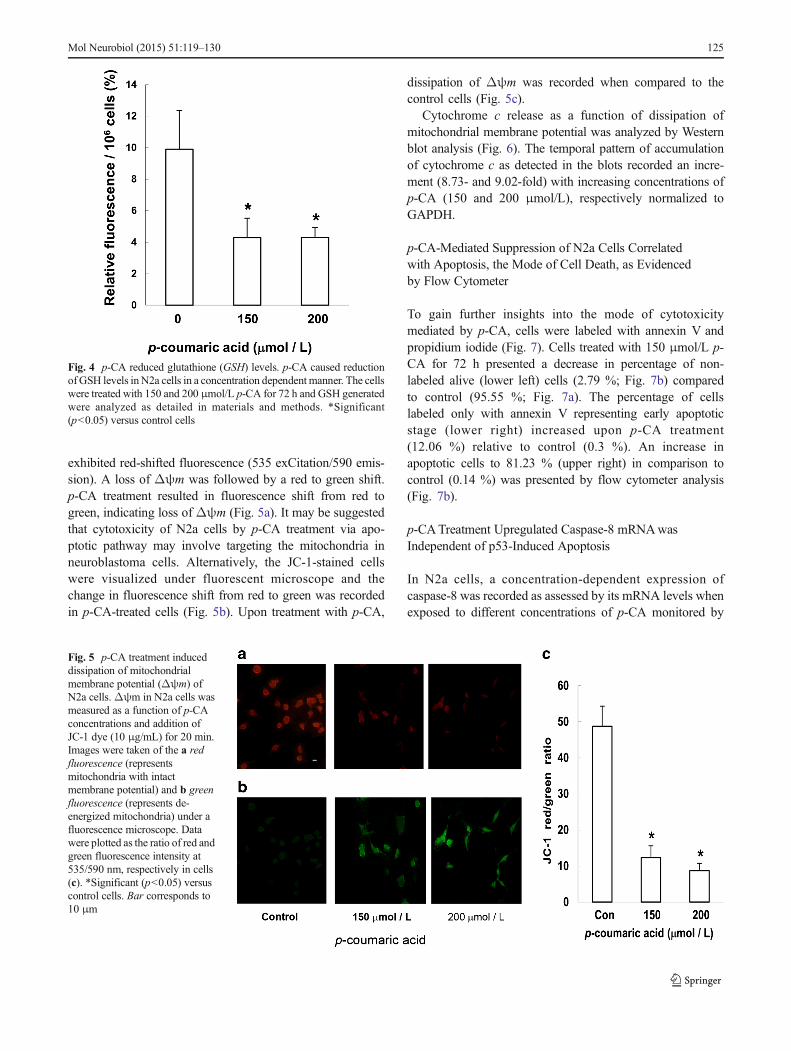
exhibited red-shifted fluorescence (535 exCitation/590 emis-sion). A loss of Δψm was followed by a red to green shift.p-CA treatment resulted in fluorescence shift from red togreen, indicating loss of Δψm (Fig. 5a). It may be suggestedthat cytotoxicity of N2a cells by p-CA treatment via apo-ptotic pathway may involve targeting the mitochondria inneuroblastoma cells. Alternatively, the JC-1-stained cellswere visualized under fluorescent microscope and thechange in fluorescence shift from red to green was recordedin p-CA-treated cells (Fig. 5b). Upon treatment with p-CA,
dissipation of Δψm was recorded when compared to thecontrol cells (Fig. 5c).
Cytochrome c release as a function of dissipation ofmitochondrial membrane potential was analyzed by Westernblot analysis (Fig. 6). The temporal pattern of accumulationof cytochrome c as detected in the blots recorded an incre-ment (8.73- and 9.02-fold) with increasing concentrations ofp-CA (150 and 200 μmol/L), respectively normalized toGAPDH.
p-CA-Mediated Suppression of N2a Cells Correlatedwith Apoptosis, the Mode of Cell Death, as Evidencedby Flow Cytometer
To gain further insights into the mode of cytotoxicitymediated by p-CA, cells were labeled with annexin V andpropidium iodide (Fig. 7). Cells treated with 150 μmol/L p-CA for 72 h presented a decrease in percentage of non-labeled alive (lower left) cells (2.79 %; Fig. 7b) comparedto control (95.55 %; Fig. 7a). The percentage of cellslabeled only with annexin V representing early apoptoticstage (lower right) increased upon p-CA treatment(12.06 %) relative to control (0.3 %). An increase inapoptotic cells to 81.23 % (upper right) in comparison tocontrol (0.14 %) was presented by flow cytometer analysis(Fig. 7b).
p-CATreatment Upregulated Caspase-8 mRNAwasIndependent of p53-Induced Apoptosis
In N2a cells, a concentration-dependent expression ofcaspase-8 was recorded as assessed by its mRNA levels whenexposed to different concentrations of p-CA monitored by
Fig. 5 p-CA treatment induceddissipation of mitochondrialmembrane potential (Δψm) ofN2a cells.Δψm in N2a cells wasmeasured as a function of p-CAconcentrations and addition ofJC-1 dye (10 μg/mL) for 20 min.Images were taken of the a redfluorescence (representsmitochondria with intactmembrane potential) and b greenfluorescence (represents de-energized mitochondria) under afluorescence microscope. Datawere plotted as the ratio of red andgreen fluorescence intensity at535/590 nm, respectively in cells(c). *Significant (p<0.05) versuscontrol cells. Bar corresponds to10 μm
Fig. 4 p-CA reduced glutathione (GSH) levels. p-CA caused reductionof GSH levels inN2a cells in a concentration dependent manner. The cellswere treated with 150 and 200 μmol/L p-CA for 72 h and GSH generatedwere analyzed as detailed in materials and methods. *Significant(p<0.05) versus control cells
Mol Neurobiol (2015) 51:119–130 125
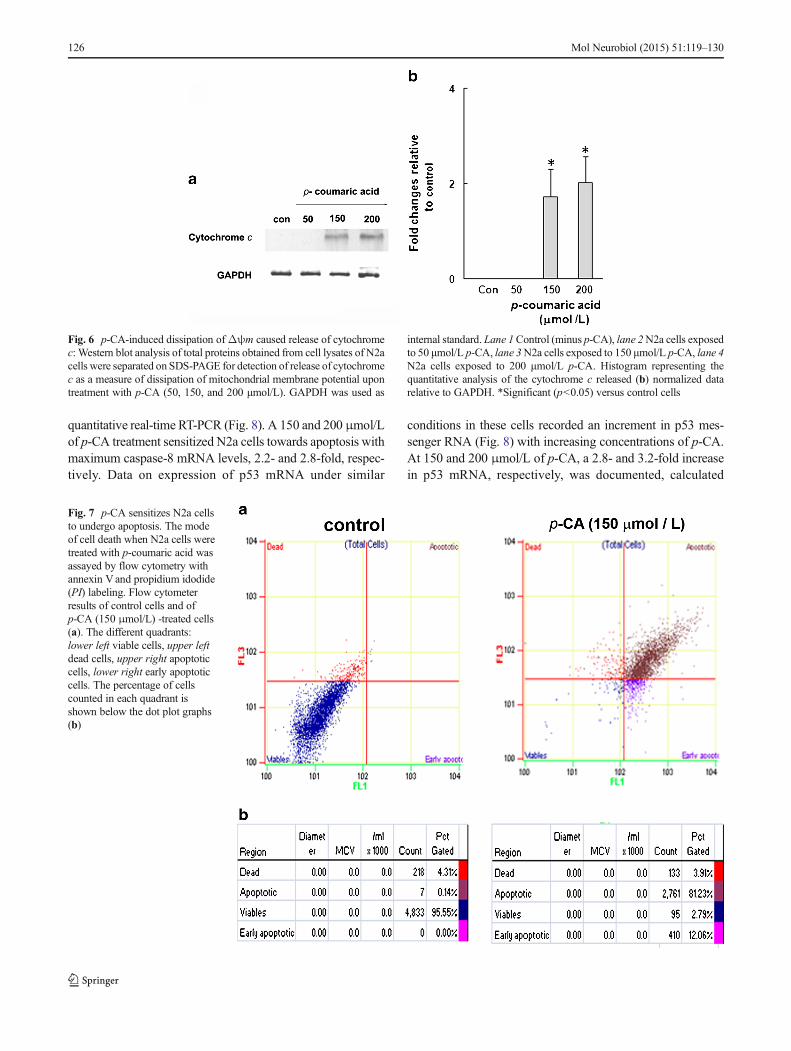
quantitative real-time RT-PCR (Fig. 8). A 150 and 200 μmol/Lof p-CA treatment sensitized N2a cells towards apoptosis withmaximum caspase-8 mRNA levels, 2.2- and 2.8-fold, respec-tively. Data on expression of p53 mRNA under similar
conditions in these cells recorded an increment in p53 mes-senger RNA (Fig. 8) with increasing concentrations of p-CA.At 150 and 200 μmol/L of p-CA, a 2.8- and 3.2-fold increasein p53 mRNA, respectively, was documented, calculated
Fig. 7 p-CA sensitizes N2a cellsto undergo apoptosis. The modeof cell death when N2a cells weretreated with p-coumaric acid wasassayed by flow cytometry withannexin Vand propidium idodide(PI) labeling. Flow cytometerresults of control cells and ofp-CA (150 μmol/L) -treated cells(a). The different quadrants:lower left viable cells, upper leftdead cells, upper right apoptoticcells, lower right early apoptoticcells. The percentage of cellscounted in each quadrant isshown below the dot plot graphs(b)
Fig. 6 p-CA-induced dissipation ofΔψm caused release of cytochromec: Western blot analysis of total proteins obtained from cell lysates of N2acells were separated on SDS-PAGE for detection of release of cytochromec as a measure of dissipation of mitochondrial membrane potential upontreatment with p-CA (50, 150, and 200 μmol/L). GAPDH was used as
internal standard. Lane 1Control (minus p-CA), lane 2N2a cells exposedto 50 μmol/L p-CA, lane 3N2a cells exposed to 150 μmol/L p-CA, lane 4N2a cells exposed to 200 μmol/L p-CA. Histogram representing thequantitative analysis of the cytochrome c released (b) normalized datarelative to GAPDH. *Significant (p<0.05) versus control cells
126 Mol Neurobiol (2015) 51:119–130
based on normalization with β-2myoglobin expression(housekeeping gene).
p-CATreatment Promotes Autophagy
The cumulative data of experiments are shown in Fig. 9. Thep-CA-treated N2a cells exhibit severe change in membranestructure. It triggered the accumulation of vacuolated N2acells. The vacuoles were positively stained with the lysosomemarker dye, acridine orange (Fig. 9b) identified asautophagosomes [35]. However, in control N2a cells, suchmembrane damage or presence of autophagosomes were ab-sent (Fig. 9a).
p-CA Induces a Dose-Dependent Formation of LC3-II,an Autophagy Marker
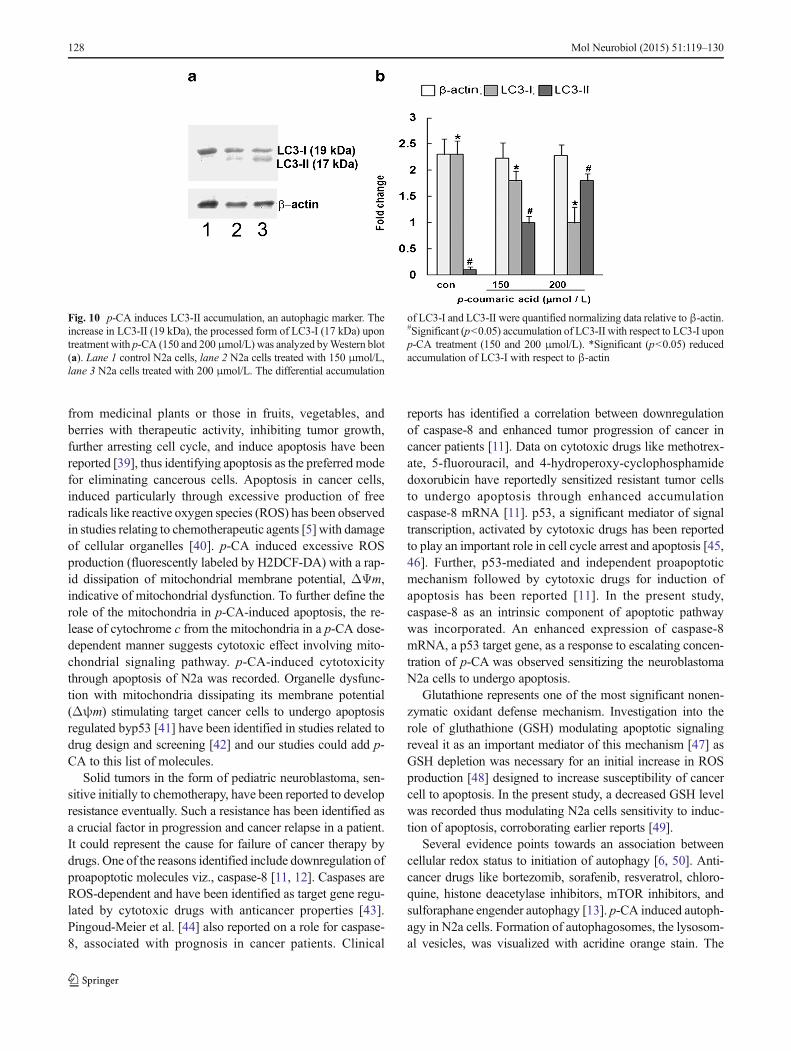
The Western blots identified LC3-I (17 kDa), the cytosolicmitrotubule-associated protein-1A/1B light chain 3, and theLC3-II (19 kDa) autophagosome membrane-bound LC3-phosphatidylethanolamine (processed form of LC3-I) conju-gate (Fig. 10a). Densitometry analysis indicated a gradualdecrease in LC3-I with a concomitant increase in LC3-II(1.2- and 1.8-fold) with increase in p-CA concentration from150 to 200 μmol/L, respectively (Fig. 10b).
Discussion
Cytotoxicity of cancer cells upon exposure to drugs byblocking the cell cycle programming resulting in apoptosis,autophagy, and programmed necrosis has been reported whiledocumenting mechanism of their action in cancer treatment[38]. In the present study, p-coumaric acid (p-CA) treatmentresulted in cytotoxicity to N2a cells by 72 h. Several bioactive
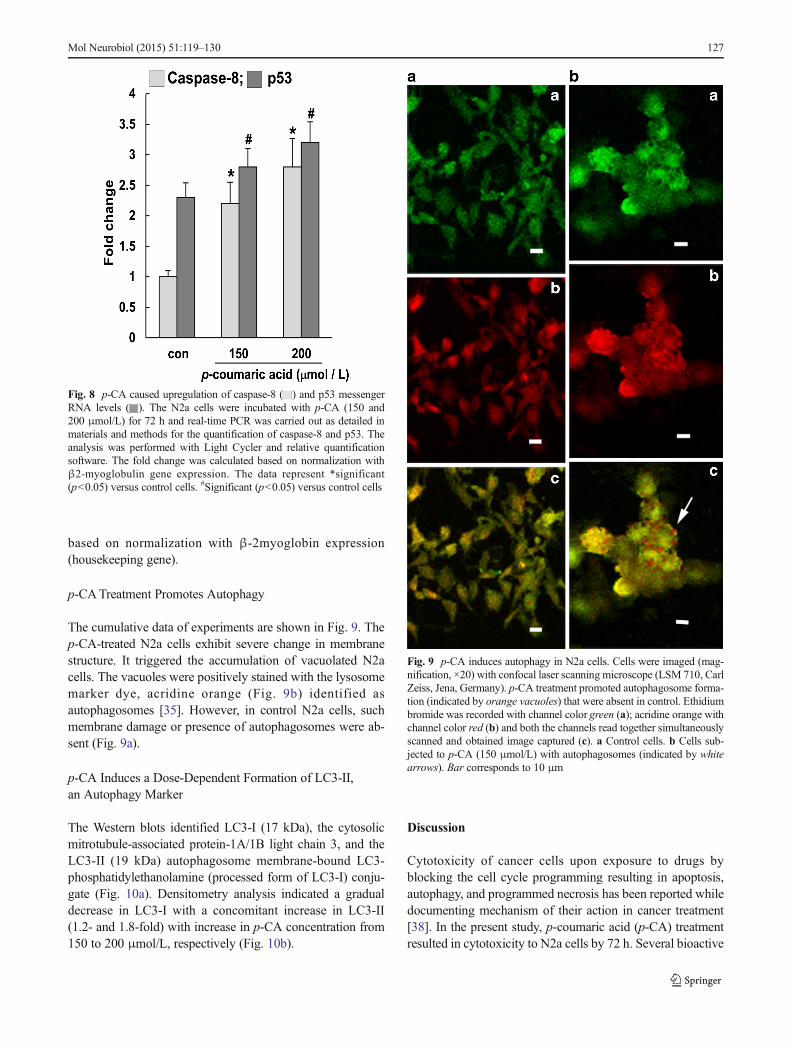
Fig. 9 p-CA induces autophagy in N2a cells. Cells were imaged (mag-nification, ×20) with confocal laser scanning microscope (LSM 710, CarlZeiss, Jena, Germany). p-CA treatment promoted autophagosome forma-tion (indicated by orange vacuoles) that were absent in control. Ethidiumbromide was recorded with channel color green (a); acridine orange withchannel color red (b) and both the channels read together simultaneouslyscanned and obtained image captured (c). a Control cells. b Cells sub-jected to p-CA (150 μmol/L) with autophagosomes (indicated by whitearrows). Bar corresponds to 10 μm
Fig. 8 p-CA caused upregulation of caspase-8 ( ) and p53 messengerRNA levels ( ). The N2a cells were incubated with p-CA (150 and200 μmol/L) for 72 h and real-time PCR was carried out as detailed inmaterials and methods for the quantification of caspase-8 and p53. Theanalysis was performed with Light Cycler and relative quantificationsoftware. The fold change was calculated based on normalization withβ2-myoglobulin gene expression. The data represent *significant(p<0.05) versus control cells. #Significant (p<0.05) versus control cells
Mol Neurobiol (2015) 51:119–130 127
from medicinal plants or those in fruits, vegetables, andberries with therapeutic activity, inhibiting tumor growth,further arresting cell cycle, and induce apoptosis have beenreported [39], thus identifying apoptosis as the preferred modefor eliminating cancerous cells. Apoptosis in cancer cells,induced particularly through excessive production of freeradicals like reactive oxygen species (ROS) has been observedin studies relating to chemotherapeutic agents [5] with damageof cellular organelles [40]. p-CA induced excessive ROSproduction (fluorescently labeled by H2DCF-DA) with a rap-id dissipation of mitochondrial membrane potential, ΔΨm,indicative of mitochondrial dysfunction. To further define therole of the mitochondria in p-CA-induced apoptosis, the re-lease of cytochrome c from the mitochondria in a p-CA dose-dependent manner suggests cytotoxic effect involving mito-chondrial signaling pathway. p-CA-induced cytotoxicitythrough apoptosis of N2a was recorded. Organelle dysfunc-tion with mitochondria dissipating its membrane potential(Δψm) stimulating target cancer cells to undergo apoptosisregulated byp53 [41] have been identified in studies related todrug design and screening [42] and our studies could add p-CA to this list of molecules.
Solid tumors in the form of pediatric neuroblastoma, sen-sitive initially to chemotherapy, have been reported to developresistance eventually. Such a resistance has been identified asa crucial factor in progression and cancer relapse in a patient.It could represent the cause for failure of cancer therapy bydrugs. One of the reasons identified include downregulation ofproapoptotic molecules viz., caspase-8 [11, 12]. Caspases areROS-dependent and have been identified as target gene regu-lated by cytotoxic drugs with anticancer properties [43].Pingoud-Meier et al. [44] also reported on a role for caspase-8, associated with prognosis in cancer patients. Clinical
reports has identified a correlation between downregulationof caspase-8 and enhanced tumor progression of cancer incancer patients [11]. Data on cytotoxic drugs like methotrex-ate, 5-fluorouracil, and 4-hydroperoxy-cyclophosphamidedoxorubicin have reportedly sensitized resistant tumor cellsto undergo apoptosis through enhanced accumulationcaspase-8 mRNA [11]. p53, a significant mediator of signaltranscription, activated by cytotoxic drugs has been reportedto play an important role in cell cycle arrest and apoptosis [45,46]. Further, p53-mediated and independent proapoptoticmechanism followed by cytotoxic drugs for induction ofapoptosis has been reported [11]. In the present study,caspase-8 as an intrinsic component of apoptotic pathwaywas incorporated. An enhanced expression of caspase-8mRNA, a p53 target gene, as a response to escalating concen-tration of p-CA was observed sensitizing the neuroblastomaN2a cells to undergo apoptosis.
Glutathione represents one of the most significant nonen-zymatic oxidant defense mechanism. Investigation into therole of gluthathione (GSH) modulating apoptotic signalingreveal it as an important mediator of this mechanism [47] asGSH depletion was necessary for an initial increase in ROSproduction [48] designed to increase susceptibility of cancercell to apoptosis. In the present study, a decreased GSH levelwas recorded thus modulating N2a cells sensitivity to induc-tion of apoptosis, corroborating earlier reports [49].
Several evidence points towards an association betweencellular redox status to initiation of autophagy [6, 50]. Anti-cancer drugs like bortezomib, sorafenib, resveratrol, chloro-quine, histone deacetylase inhibitors, mTOR inhibitors, andsulforaphane engender autophagy [13]. p-CA induced autoph-agy in N2a cells. Formation of autophagosomes, the lysosom-al vesicles, was visualized with acridine orange stain. The
Fig. 10 p-CA induces LC3-II accumulation, an autophagic marker. Theincrease in LC3-II (19 kDa), the processed form of LC3-I (17 kDa) upontreatment with p-CA (150 and 200μmol/L) was analyzed byWestern blot(a). Lane 1 control N2a cells, lane 2 N2a cells treated with 150 μmol/L,lane 3 N2a cells treated with 200 μmol/L. The differential accumulation
of LC3-I and LC3-II were quantified normalizing data relative to β-actin.#Significant (p<0.05) accumulation of LC3-II with respect to LC3-I uponp-CA treatment (150 and 200 μmol/L). *Significant (p<0.05) reducedaccumulation of LC3-I with respect to β-actin
128 Mol Neurobiol (2015) 51:119–130

enhanced conversion of LC3-I to LC3-II (autophagic marker)was detected by Western blot analysis of cell lysate, an au-tophagic marker [33].
Necrosis has always been considered to be almost “acci-dental” cell death, a random, uncontrolled process. Intracellu-lar lactate dehydrogenase (LDH), as an index of the cellmembranes integrity, associated with programmed necrosisin response to the oxidative stress is known [51]. An incre-ment in the release of intracellular LDH from N2a cells as afunction of increasing concentration of this molecule definesp-CA with activity against proliferation of these cancer cells,supporting the idea that few cells died by necrosis as identifiedby flow cytometer analysis.
The ultimate determinant preferred for cancer cell fate is itsdeath, which could be by apoptosis, autophagy, or pro-grammed necrosis. The presence of an intricate interrelation-ship between them has been postulated. Several reports accorda synergistic effect between apoptosis and autophagy; how-ever, others incur situations wherein autophagy is triggered intreating resistant cancer cells with high apoptotic thresholds[38, 52]. Necrosis is envisaged to be triggered as a pro-grammed backup mechanism [53] for apoptosis wherein thedeath mode switch could be due to the status of mitochondrialmembrane permeability (MOMP) [54]. Necrosis accompa-nied by autophagy [55] also finds mention. Thus, knowledgeof the signaling pathways along with physiological responsesto therapeutic drugs defines the essentials to understandingtheir mechanism of toxicity.
Acknowledgments The authors acknowledge the recognition of theUniversity ofMysore as an Institution of Excellence and financial supportfrom the Ministry of Human Resource Development, Govt. of Indiathrough UGC under UOM/IOE/RESEARCH/1/2010-11, dt 22-04-2010project.
Conflict of Interest The authors declare that they have no conflict ofinterest.
References
1. Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ(2003) Production of reactive oxygen species by mitochondria. JBiol Chem 278:36027–36031
2. Turrens JF (2003) Mitochondrial formation of reactive oxygen spe-cies. J Physiol 552:335–344
3. Laura AS, Chandel NS (2012) Physiological roles of mitochondrialreactive oxygen species. Mol Cells 48:158–167
4. Nakamura H, Nakamura K, Yodoi J (1997) Redox regulation ofcellular activation. Annu Rev Immunol 15:351–369
5. Ozben T (2007) Oxidative stress and apoptosis: impact on cancertherapy. J Pharm Sci 96:2181–2196
6. Xie CM, ChanWY, Yu S, Zhao J, Cheng CH (2011) Bufalin inducesautophagy-mediated cell death in human colon cancer cells throughreactive oxygen species generation and JNK activation. Free RadicBiol Med 51:1365–1375
7. Gao M, Yeh PY, Lu YS, Hsu CH, Chen KF, Lee WC, Feng WC,Chen CS, KuoML, Cheng AL (2008) OSU-03012, a novel celecoxibderivative, induces reactive oxygen species-related autophagy inhepatocellular carcinoma. Cancer Res 68:9348–9357
8. Fulda S, Debatin KM (2006) Extrinsic versus intrinsic apoptosispathways in anticancer chemotherapy. Oncogene 25:4798–4811
9. Mehta SS, Manhas N, Raghubir R (2007) Molecular targets incerebral ischemia for developing novel therapeutics. Brain Res Rev54:34–66
10. Ashkenazi A, Dixit VM (1998) Death receptors: signaling and mod-ulation. Science 281:1305–1308
11. Ehrhardt H, Hacker S, Wittmann S, Maurer M, Borkhardt A,Toloczko A, Debatin KM, Fulda S, Jeremias I (2008) Cytotoxicdrug-induced, p53-mediated upregulation of caspase-8 in tumorcells. Oncogene 27:783–793
12. Ehrhardt H, Wachter F, Maurer M, Stahnke K, Jeremias I (2011)Important role of caspase-8 for chemo-sensitivity of ALL cells. ClinCancer Res 17:7605–7613
13. Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ (2007)Potential therapeutic applications of autophagy. Nat Rev DrugDiscov 6:304–312
14. Periyasamy-Thandavan S, Jiang M, Schoenlein P, Dong Z (2009)Autophagy: molecular machinery, regulation and implications forrenal pathophysiology. Am J Physiol Ren Physiol 297:F244–F256
15. Tanida I, Ueno T, Kominami E (2008) LC3 and autophagy. MethodsMol Biol 445:77–88
16. Gurney JG, Smith MA, Ross JA (1999) Cancer among infants. In:Gloeckler Ries LA (ed) Cancer incidence and survival among chil-dren and adolescents: United States SEER Program, 1975–1995.National Cancer Institute, Bethesda (MD), pp 149–156
17. Surh YJ, Chun KS, Cha HH, Han SS, Keum YS, Park KK, Lee SS(2001) Molecular mechanisms underlying chemopreventive activi-ties of anti-inflammatory phytochemicals: down-regulation of COX-2 and iNOS through suppression of NF-kappa B activation. MutatRes 480–481:243–268
18. Dragsted LO (2003) Antioxidant actions of polyphenols in humans.Int J Vitam Nutr Res 73:112–119
19. Dore S (2005) Unique properties of polyphenol stilbenes in the brain:more than direct antioxidant actions; gene/protein regulatory activity.Neurosignals 14:61–70
20. Sang S, Hou Z, Lambert JD, Yang CS (2005) Redox properties of teapolyphenols and related biological activities. Antioxid Redox Signal7:1704–1714
21. Grever MCB (2001) Cancer drug discovery and development. In: DeVita VHS, Rosenberg SA (eds) Cancer: principles and practice ofoncology. Lippincott Raven, Philadelphia, pp 328–339
22. Janicke B, Onning G, Oredsson SM (2005) Differential effects offerulic acid and p-coumaric acid on S phase distribution and length ofS phase in the human colonic cell line Caco-2. J Agric Food Chem53:6658–6665
23. Jaganathan SK, Supriyanto E, Mandal M (2013) Events associatedwith apoptotic effect of p-coumaric acid in HCT-15 colon cancercells. World J Gastroenterol 19:7726–7734
24. Larrosa M, Lodovici M, Morbidelli L, Dolara P (2008) Hydrocaffeicand p-coumaric acids, natural phenolic compounds, inhibit UV-Bdamage in WKD human conjunctival cells in vitro and rabbit eyein vivo. Free Radic Res 42:903–910
25. Abdel-WahabMH, El-MahdyMA, Abd-EllahMF, Helal GK, KhalifaF, Hamada FM (2003) Influence of p-coumaric acid on doxorubicin-induced oxidative stress in rat’s heart. Pharmacol Res 48:461–465
26. Vauzour D, Corona G, Spencer JP (2010) Caffeic acid, tyrosol and p-coumaric acid are potent inhibitors of 5-S-cysteinyldopamine in-duced neurotoxicity. Arch Biochem Biophys 501:106–111
27. Ferguson LR, Zhu ST, Harris PJ (2005) Antioxidant andantigenotoxic effects of plant cell wall hydroxycinnamic acids incultured HT-29 cells. Mol Nutr Food Res 49:585–593
Mol Neurobiol (2015) 51:119–130 129

28. Seo YK, Kim SJ, Boo YC, Baek JH, Lee SH, Koh JS (2010) Effectsof p-coumaric acid on erythema and pigmentation of human skinexposed to ultraviolet radiation. Clin Exp Dermatol 36:260–266
29. Barros MP, Lemos M, Maistro EL, Leite MF, Sousa JP, Bastos JK,Andrade SF (2008) Evaluation of antiulcer activity of the mainphenolic acids found in Brazilian green propolis. J Ethnopharmacol120:372–377
30. Luceri CL, Lodovici GM, Antonucci E, Abbate R, Masini E, DolaraP (2007) p-Coumaric acid, a common dietary phenol, inhibits plateletactivity in vitro and in vivo. Br J Nutr 97:458–463
31. Shastry P, Basu A, Rajadhyaksha MS (2001) Neuroblastoma celllines—a versatile in vitro model in neurobiology. Int J Neurosci 108:109–126
32. Radio NM, Mundy WR (2008) Developmental neurotoxicity testingin vitro: models for assessing chemical effects on neurite outgrowth.Neurotoxicology 29:361–376
33. Azad MB, Chen Y, Gibson SB (2009) Regulation of autophagy byreactive oxygen species (ROS): implications for cancer progressionand treatment. Antioxid Redox Signal 11:777–790
34. Sareen D, van Ginkel PR, Takach JC, Mohiuddin A, Darjatmoko SR,Albert DM, Polans AS (2006) Mitochondria as the primary target ofresveratrol-induced apoptosis in human retinoblastoma cells. InvestOphthalmol Vis Sci 47:3708–3716
35. Yang C, Kaushal V, Shah SV, Kaushal GP (2008) Autophagy isassociated with apoptosis in cisplatin injury to renal tubular epithelialcells. Am J Physiol Ren Physiol 294:777–787
36. Laemmli UK (1970) Cleavage of structural proteins during assemblyof the head of the bacteriophage T4. Nature 227:680–685
37. Shailasree S, Kini KR, Deepak S, Kumudini BS, Shetty HS (2004)Accumulation of hydroxyproline-rich glycoproteins in pearl milletseedlings in response to Sclerospora graminicola infection. Plant Sci167:1227–1234
38. Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B, Hao JK (2012)Programmed cell death pathways in cancer: a review of apoptosis,autophagy and programmed necrosis. Cell Prolif 45:487–498
39. Kerr JF, Winterford CM, Harmon BV (1994) Apoptosis, its signifi-cance in cancer and cancer therapy. Cancer 73:2013–2026
40. Petrosillo G, Matera M, Moro N, Ruggiero FM, Paradies G (2009)Mitochondrial complex I dysfunction in rat heart with aging: criticalrole of reactive oxygen species and cardiolipin. Free Radic Biol Med46:88–94
41. Li PF, Dietz R, von Harsdorf R (1999) p53 regulates mitochondrialmembrane potential through reactive oxygen species and inducescytochrome c-independent apoptosis blocked by Bcl-1. EMBO J18:6027–6036
42. Smith RAJ, Porteous CM,Gane AM,MurphyMP (2003)Delivery ofbioactive molecules to mitochondria in vivo. Proc Natl Acad Sci U SA 100:5407–5412
43. Von Harsdorf R, Li P, Dietz R (1999) Signaling pathways in reactiveoxygen species-induced cardiomyocyte apoptosis. Circulation 99:2934–2941
44. Pingoud-Meier C, Lang D, Janss AJ, Rorke LB, Phillips PC, ShalabyT (2003) Downregulation of caspase-8 protein expression correlateswith unfavorable survival outcome in childhood medulloblastoma.Clin Cancer Res 9:6401–6409
45. Blagosklonny MV (2002) p53: an ubiquitous target of anticancerdrugs. Int J Cancer 98:161–166
46. Fuster JJ, Sanz-Gonzalez SM, Moll UM, Andres V (2007) Classicaland novel roles of p53: prospects for anticancer therapy. Trends MolMed 13:192–199
47. Hug H, Enari M, Nagata S (1994) No requirement of reactiveoxygen intermediates in Fas-mediated apoptosis. FEBS Lett 351:311–313
48. Langer C, Jurgensmeier JM, Bauer G (1996) Reactive oxygen spe-cies act at both TGF-beta-dependent and -independent steps duringinduction of apoptosis of transformed cells by normal cells. Exp CellRes 222:117–124
49. Armstrong JS, Steinauer KK, Hornung B, Irish JM, Lecane P, BirrellGW, Peehl DM, Knox SJ (2002) Role of glutathione depletion andreactive oxygen species generation in apoptotic signaling in a humanB lymphoma cell line. Cell Death Differ 9:252–263
50. Ling LU, Tan KB, Lin H, Chiu GN (2011) The role of reactiveoxygen species and autophagy in safingol-induced cell death. CellDeath Dis 2:e129
51. Mi YL, Zhang CQ (2005) Protective effect of quercetin on aroclor1254-induced oxidative damage in cultured chicken spermatogonialcells. Toxicol Sci 88:545–550
52. Amelio I, Melino G, Knight RA (2011) Cell death pathology: cross-talk with autophagy and its clinical implications. Biochem BiophysRes Commun 414:277–281
53. Cho YS, Park SY, Shin HS, Chan FK (2010) Physiological conse-quences of programmed necrosis, an alternative form of cell demise.Mol Cells 29:327–332
54. Han W, Xie J, Li L, Liu Z, Hu X (2009) Necrostatin-1 revertsshikonin-induced necroptosis to apoptosis. Apoptosis 14:674–686
55. ChaabaneW,User SD, El-GazzahM, Jaksik R, Sajjadi E, Joanna R-W,Los MJ (2013) Autophagy, apoptosis, mitoptosis and necrosis: inter-dependence between those pathways and effects on cancer. ArchImmunol Ther Exp 61:43–58
130 Mol Neurobiol (2015) 51:119–130




















