
Cycloaromatization reactions: the testing ground for theory and experiment
47
Cycloaromatization reactions: the testing ground for theory and experiment IGOR ALABUGIN,BORIS BREINER and MARIAPPAN MANOHARAN Department of Chemistry and Biochemistry, Florida State University, Tallahassee, FL, USA 1 Introduction: bonds lost and bonds formed, or chemical bookkeeping 1 2 Cycloaromatization reactions: breaking p-bonds and breaking the rules 2 3 The diversity of cycloaromatization reactions 3 4 The relative role of s- versus p-effects at the early reaction stage 6 5 s-Effects on reactant stability 10 Strain and antiaromaticity in cyclic enediynes 11 Control of strain through ligand–metal coordination 16 Steric assistance of ortho substituents 17 Control of steric repulsion with positively charged ortho substituents 20 Hybridization effects 21 6 p-Effects on reactant stability 22 7 Transition state effects: communication of orthogonal orbitals in the transition state of radical-anionic cyclizations 23 8 Effects on product stability 27 s-Effects 27 p-Effects 30 9 Conclusion 31 References 31 1 Introduction: bonds lost and bonds formed, or chemical bookkeeping Chemical reactions involve the reorganization of electron density in which bonds are broken and formed in accordance with the reaction mechanism. The energetic con- sequences of such reorganization are accounted for by the reaction thermodynamics. Unless a very weak bond is broken with a significant increase in entropy, bond breaking is thermodynamically unfavorable and leads to the formation of highly reactive intermediates: radicals and diradicals through bond homolysis, anions and cations through heterolysis, and carbenes as a result of a-elimination. On the other hand, bond formation is favorable and when a strong bond is formed, serves as a thermodynamic sink that often terminates a cascade of chemical transformations, thus balancing the thermodynamic checkbook or registering a profit. In a perfect world of balanced checkbooks, the bond breaking and bond forming processes are synchronized, as it happens (albeit not always perfectly) in concerted pericyclic reactions. Even when such synchronization is not perfect, unimolecular 1 ADVANCES IN PHYSICAL ORGANIC CHEMISTRY VOLUME 42 ISSN 0065-3160 DOI: 10.1016/S0065-3160(07)42001-9 r 2008 Elsevier Inc. All rights reserved
Transcript of Cycloaromatization reactions: the testing ground for theory and experiment

Cycloaromatization reactions: the testingground for theory and experiment
IGOR ALABUGIN, BORIS BREINER and MARIAPPAN MANOHARAN
Department of Chemistry and Biochemistry, Florida State University,
Tallahassee, FL, USA
1 Introduction: bonds lost and bonds formed, or chemical bookkeeping 12 Cycloaromatization reactions: breaking p-bonds and breaking
the rules 23 The diversity of cycloaromatization reactions 34 The relative role of s- versus p-effects at the early reaction stage 65 s-Effects on reactant stability 10
Strain and antiaromaticity in cyclic enediynes 11Control of strain through ligand–metal coordination 16Steric assistance of ortho substituents 17Control of steric repulsion with positively charged ortho substituents 20Hybridization effects 21
6 p-Effects on reactant stability 227 Transition state effects: communication of orthogonal orbitals in the transition state of
radical-anionic cyclizations 238 Effects on product stability 27
s-Effects 27p-Effects 30
9 Conclusion 31References 31
1 Introduction: bonds lost and bonds formed, or chemicalbookkeeping
Chemical reactions involve the reorganization of electron density in which bonds arebroken and formed in accordance with the reaction mechanism. The energetic con-sequences of such reorganization are accounted for by the reaction thermodynamics.Unless a very weak bond is broken with a significant increase in entropy, bondbreaking is thermodynamically unfavorable and leads to the formation of highlyreactive intermediates: radicals and diradicals through bond homolysis, anions andcations through heterolysis, and carbenes as a result of a-elimination. On the otherhand, bond formation is favorable and when a strong bond is formed, serves as athermodynamic sink that often terminates a cascade of chemical transformations,thus balancing the thermodynamic checkbook or registering a profit.
In a perfect world of balanced checkbooks, the bond breaking and bond formingprocesses are synchronized, as it happens (albeit not always perfectly) in concertedpericyclic reactions. Even when such synchronization is not perfect, unimolecular
1
ADVANCES IN PHYSICAL ORGANIC CHEMISTRYVOLUME 42 ISSN 0065-3160 DOI: 10.1016/S0065-3160(07)42001-9
r 2008 Elsevier Inc.All rights reserved

rearrangements are still rarely accompanied with a change in the number of chemicalbonds. In other words, the number of chemical bonds generally is conserved.
2 Cycloaromatization reactions: breaking p-bonds and breakingthe rules
An interesting breakdown of the tendency for the conservation of chemical bondsoccurs in the so-called cycloaromatization reactions1 (Scheme 1).
In these reactions, a s-bond is formed at the expense of two p-bonds and, thus, theprocess leads to a net loss of one chemical bond that is intrinsically unfavorablethermodynamically. Formation of the new s-bond leads to ring closure, whereas thenet loss of a bond leads to the formation of two radical centers, which can be eitherinside (the ‘‘endo’’ pattern in Scheme 1) or outside of the newly formed cycle(the ‘‘exo’’ pattern). Note that s-radicals are formed through the endo path, whileexo-closures may produce either a s-radical when a triple bond is involved or aconjugated p-radical when the new bond is formed at the central carbon of an allene.The parent version of this process is the transformation of enediyne 1 into p-benzynediradical2 (the Bergman cyclization), shown in Scheme 2.
This transformation requires significant thermal energy to overcome the activa-tion barrier and is endergonic. The endergonicity of the Bergman cyclization is notsurprising and stems logically from the net loss of a bond in the product. What is
surprising is that the energy penalty (DH) for this intrinsically unfavorable
Xa
b
bX X
endo, exo endo, endoc
a
c
X
exo, exo bis-allene
X
c′
exo, exo bis-alkyne
Scheme 1 Schematic representation of cycloaromatization reactions. Double lines corre-spond to the out-of-plane p-systems of a bis-alkyne reagent. Only orbitals of the in-planep-system in the reactant and of new s-bond and radical centers in the product are shownexplicitly.
I. ALABUGIN ET AL.2

transformation is only 8–9 kcal/mol.3 This low value results from the aromaticstabilization of the product provided by the completely conjugated system of out-of-plane p-orbitals at each carbon of the enediyne system. Thus, even though the out-of-plane p-array does not directly participate in the bond forming/bond breakingevent, it is not a simple bystander in the overall process. Developing aromaticstabilization in the conjugated out-of-plane system plays an important role in deep-ening the potential energy well for the diradical product to the extent that thisspecies can be trapped by external reactions such as H-atom abstraction or additionto a p-system.
The goal of this review is to dissect the relative contributions of the twoorthogonal p-systems in the most general sense and provide a clear and generalanalysis of electronic effects in the Bergman cyclization as they apply to and orig-inate from these two different sources. We will outline the relative role of the twosystems in defining the kinetics and thermodynamics of the parent Bergmancycloaromatization pathway, delineate factors that affect these systems separatelyand discuss ways to provide communication between the two orthogonal p-systems.This review will be selective rather than comprehensive since a number of excellentand detailed reviews are available on various aspects of the chemistry (and bio-chemical application) of cycloaromatization reactions in the recent literature: Rawatand Zaleski4 compared and contrasted geometric and electronic effects in thermalBergman cyclizations to those of the recently developed metalloenediynes, whileBasak and coworkers5 concentrated even more closely on the use of enediynylligands in chelation-controlled Bergman cyclizations. The Schreiner group6 reviewedthe application of computational methods toward a variety of cycloaromatizationreactions and Konig and coworkers7 provided a compendium of experimentalresults related to the electronic effects on the Bergman cyclization. Intricate detailsof biological activity of natural enediynes were most recently reviewed by Shen andcoworkers,8 whereas medicinal utility of designed enediynes was expertly discussedby Jones and Fouad.9
3 The diversity of cycloaromatization reactions
Fig. 1 summarizes cyclizations of the two prototype molecules, (Z) hex-3-ene-1,5-diyne and (Z) hept-3,5,6-triene-1-yne. In all of these processes, bonds are formedfrom in-plane p-orbitals in the presence of an orthogonal p-system. However, it isclear that the properties of the newly formed cyclic conjugated systems can be quite
H
H
.
.
2RH
1 2
Scheme 2 Bergman cyclization of (Z)-3-ene-1,5-diyne.
CYCLOAROMATIZATION REACTIONS 3

different. A new aromatic system is formed only in the Bergman and Myers-Saitocyclizations, and thus, in a sense, only these two processes in Fig. 1 are bona fidecycloaromatization reactions. Note also that s,s-diradicals are produced fromenediynes, whereas s,p-diradicals are formed from enyne allenes.
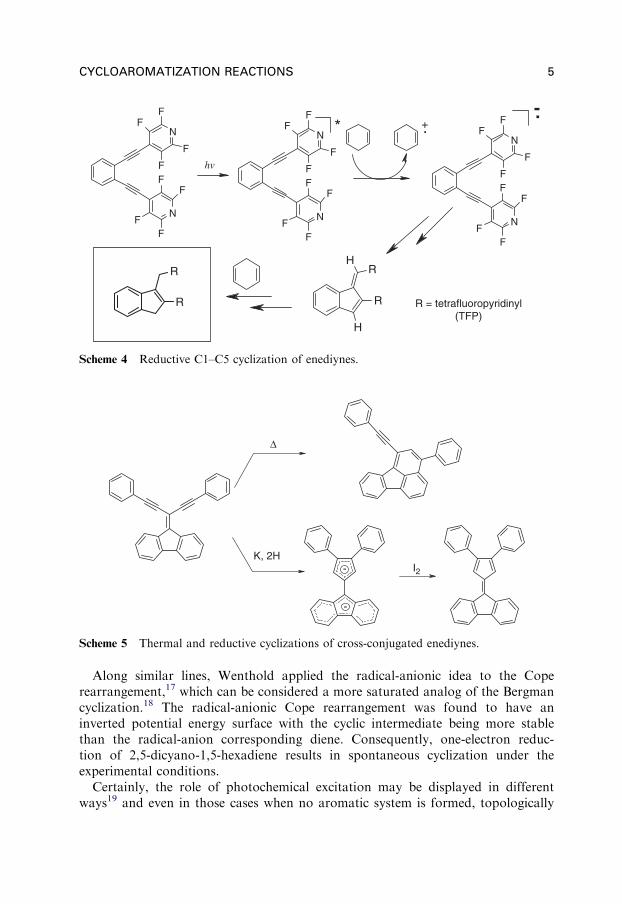
The benzene ring is not the only aromatic moiety that can be formed in acycloaromatization process and formation of other aromatic systems was consideredas the driving force of new cycloaromatization processes. For example, Matzger(X ¼ S)10 and Schreiner11 investigated whether the 5-membered aromatic hetero-cyclic systems of pyrrole, thiophene, and furan can be formed in these processes(Scheme 3), while Alabugin12 (Scheme 4) and Rabinovitz13 (Scheme 5) investigatedanionic species where cycloaromatization benefited (albeit to a different extent) fromthe aromaticity of the cyclopentadienyl anion.
Whitlock et al.14 discovered a reductive cyclization of enediynes promoted bylithium naphthalenide that provides substituted fulvenes and suggested a dianionicmechanism (Scheme 6). However, even now it is still unclear whether the enediynedianion is indeed the cyclizing species or whether the initially formed acyclic radical-anion cyclizes first to give a fulvene radical-anion which is further reduced bylithium to give the cyclic dianion.
Aromaticity of the products is only one of the factors accounting for the efficiencyof these cyclizations as evidenced by the discovery of a dianionic synthesis of non-aromatic 5-membered heterocycles by Tamao and coworkers who found that re-duction of bis(phenylethynyl)dialkylsilanes with lithium naphthalenide resulted information of a cyclized product by endo–endo cyclization15 (Scheme 7).
Under analogous conditions, cyclic 1,2-bis(silylethynyl)benzenes afford 1,4-di-lithio-2,3-disylylnaphthalenes via dianionic endo–endo cyclization leading to theformation of a 6-membered ring (Scheme 8).16
Schreiner Bergman
Schmittel Myers-Saito
a ba b
c d
cd
Fig. 1 Cycloaromatization reactions of (Z) hex-3-ene-1,5-diyne and (Z) hept-3,5,6-triene-1-yne.
X
R′R
X
R′R
X = NH, O, S, PH, NH2
+, OH+, SH+, CH-, etc.
Scheme 3 Formation of 5-membered heterocycles through cyclization reaction.
I. ALABUGIN ET AL.4
Along similar lines, Wenthold applied the radical-anionic idea to the Coperearrangement,17 which can be considered a more saturated analog of the Bergmancyclization.18 The radical-anionic Cope rearrangement was found to have aninverted potential energy surface with the cyclic intermediate being more stablethan the radical-anion corresponding diene. Consequently, one-electron reduc-tion of 2,5-dicyano-1,5-hexadiene results in spontaneous cyclization under theexperimental conditions.
Certainly, the role of photochemical excitation may be displayed in differentways19 and even in those cases when no aromatic system is formed, topologically
N
FF
FF
NF
F
F
F
N
FF
FF
NF
F
F
F
R
RH
H
R
R
N
FF
FF
NF
F
F
F
.-
hv
*
R = tetrafluoropyridinyl (TFP)
+.
Scheme 4 Reductive C1–C5 cyclization of enediynes.
I2K, 2H
Δ
Scheme 5 Thermal and reductive cyclizations of cross-conjugated enediynes.
CYCLOAROMATIZATION REACTIONS 5

similar cyclization photoreactions are still possible. A classic example involves thephotochemical cyclization of diethynylmethanes discovered by Zimmerman andPincock20 around the same time as the thermal Bergman cyclization was reported byBergman. The reaction proceeded upon direct irradiation of the diyne in isopropylalcohol or, alternatively, when triplet-sensitized by acetophenone or xanthone.As in the Bergman cycloaromatization, two hydrogen atoms are abstracted from asuitable donor (Scheme 9).
Taken together these data suggest that the family of cycloaromatization reactionswill continue to grow and new interesting transformations may be discovered in thefuture.21 Thus, understanding of general factors which can be used to controlcycloaromatization process should be of significant value for organic chemistry.
4 The relative role of r- versus p-effects at the early reaction stage
Although data presented in the previous section illustrate the diversity ofcycloaromatization reactions, most of the following discussion will concentrate onthe Bergman cyclization – a reaction that has been studied intensively in recentdecades due to its role in the mechanism of biological activity of natural anticancerantibiotics.8,9 We will take advantage of the large body of data produced by this
D
D
1. Li / naphthalene
2. D2O
Scheme 6 Dianionic cyclization of enediynes promoted by double lithium naphthalenidereduction.
SiR R
Ph Ph
Si
PhPh
Li
R R
Li
lithium naphthalenide
(4 equivalents)
Scheme 7 Dianionic endo–endo cyclization of diethynylsilanes.
Si
Si
Li
Li
Si
Si
lithium
naphthalenide
Scheme 8 Endo–endo reductive cyclization of cyclic 1,2-bis(silylethynyl)benzenes.
I. ALABUGIN ET AL.6

research and use this archetypal reaction to illustrate the general electronic factorsinvolved in a typical cycloaromatization process.
Since both in-plane and out-of-plane p-systems are involved, a clear dissection oftheir respective roles in the cycloaromatization process is beneficial for the analysisof electronic effects on reactivity. Galbraith et al.22 approached this problem byutilizing valence bond (VB) theory, which provided insight into the structural andelectronic nature of the transition state and illustrated the role of the s- andp-frameworks. The dominant VB configuration in the enediyne was found to playonly a small role in the p-benzyne diradical, while the role of the dominant VBconfiguration (accounting for the biradical) in the product was seemingly insignifi-cant in the starting material. The avoided crossing of the energy curves of these twoVB configurations leads to the transition state, but the changes in the electronicwave function and in geometry are non-synchronous. The transition state (TS) wasfound to be �80% product-like geometrically and �70% reactant-like electronicallyin an apparent violation of the Hammond postulate.23 This is a direct consequenceof the high stabilization energy provided by the formation of the aromatic system,the development of which only has significance once the reaction approaches theproduct.
Independently, Alabugin and coworkers utilized classic molecular orbital (MO)correlation diagrams for understanding cycloaromatization processes. The advan-tage of this approach is that it clearly describes changes accompanying the cycliza-tion process at the level of individual MOs and allows comprehensive analysis ofelectronic reorganization that accompanies the cyclization processes including therelative roles of the two orthogonal p-arrays.
As an example of the latter approach, let us analyze the MO correlation diagramfor the Bergman cyclization of the parent enediyne given in Fig. 2. This diagramshows the transformation of MOs of the starting material to MOs of TS and theproduct. The most important change at the TS stage is the dramatic increase in theenergy of HOMO-1 of the enediyne, which is paralleled by the decrease in the energyof LUMO+1 due to the transformation of these in-plane orbitals (bonding andantibonding respectively) into non-bonding MOs (radical centers). Destabilizationof the HOMO-1 is the most dramatic energy penalty that the molecule has to pay toreach the TS. This effect is partially offset by the s-bond forming interaction thatstabilizes HOMO-2 (in phase combination of the in-plane enediyne p-orbitals).Developing aromatic stabilization of the out-of-plane p-system lags behind the
hν
2-propanol
Scheme 9 Photochemical cyclization of diethynylmethanes. Although this reaction is topo-logically analogous to the cyclization reactions discussed earlier, it does not lead to an ar-omatic product.
CYCLOAROMATIZATION REACTIONS 7
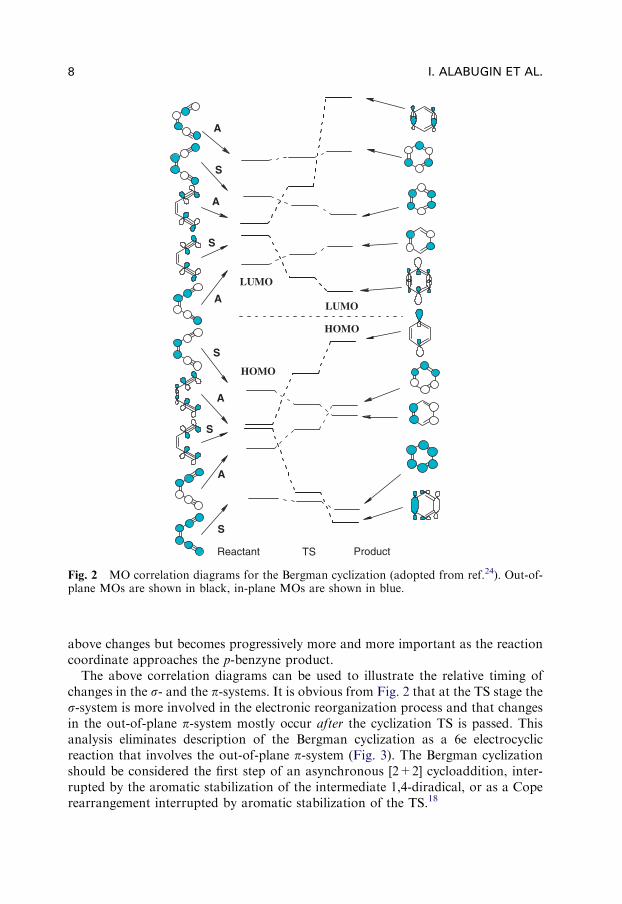
above changes but becomes progressively more and more important as the reactioncoordinate approaches the p-benzyne product.
The above correlation diagrams can be used to illustrate the relative timing ofchanges in the s- and the p-systems. It is obvious from Fig. 2 that at the TS stage thes-system is more involved in the electronic reorganization process and that changesin the out-of-plane p-system mostly occur after the cyclization TS is passed. Thisanalysis eliminates description of the Bergman cyclization as a 6e electrocyclicreaction that involves the out-of-plane p-system (Fig. 3). The Bergman cyclizationshould be considered the first step of an asynchronous [2+2] cycloaddition, inter-rupted by the aromatic stabilization of the intermediate 1,4-diradical, or as a Coperearrangement interrupted by aromatic stabilization of the TS.18
LUMO
HOMO
LUMO
S
A
S
A
S
S
A
S
A
HOMO
Reactant TS Product
A
Fig. 2 MO correlation diagrams for the Bergman cyclization (adopted from ref.24). Out-of-plane MOs are shown in black, in-plane MOs are shown in blue.
I. ALABUGIN ET AL.8

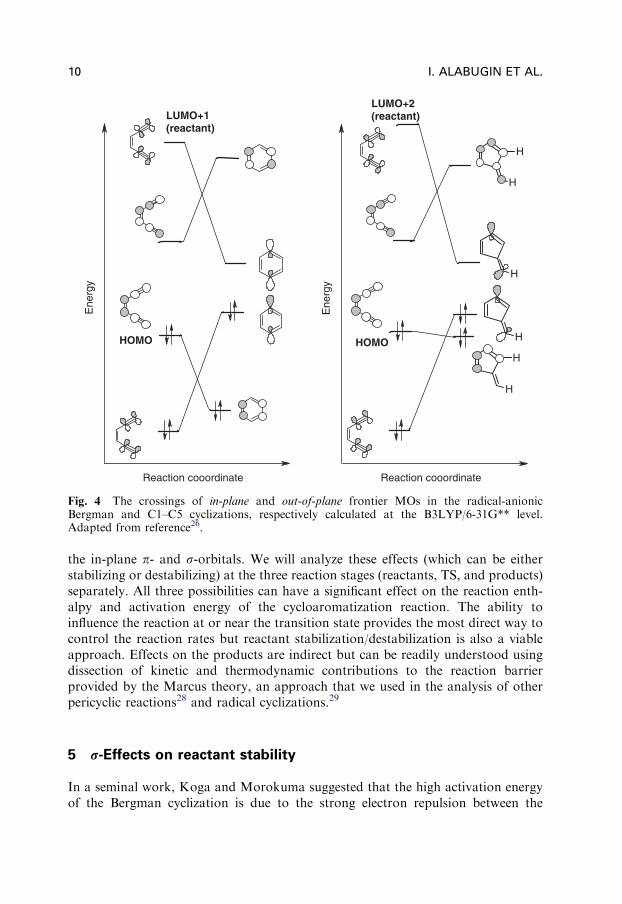
It is interesting to use MO correlation diagrams for a comparison of the twoalternative cyclizations of enediynes (Fig. 4): the Bergman cyclization and theC1–C5 cyclization (Fig. 1b,a), investigated first computationally by Schreiner.25
In both these processes, a s bond is formed at the expense of the same two in-planep-bonds in either an endo–endo (Bergman) or an endo–exo (C1–C5) fashion (Fig. 4).Not surprisingly, the changes in in-plane MOs are comparable in the two diagramsand consistent with these p-bonds being broken in order to form the new s-bond.In contrast, changes in the out-of-plane MOs are significantly different. In contrastto the Bergman cyclization where the enediyne highest occupied molecular orbital(HOMO) is strongly stabilized, such stabilization is small in the non-aromaticdidehydrofulvene product of the C1–C5 cyclization. Another interesting difference isthat the LUMO+2 of the reactant correlates with the lowest unoccupied molecularorbital (LUMO) of the product in the C1–C5 cyclization instead of LUMO+1.Such changes illustrate the different properties of the out-of-plane conjugatedsystems formed in the two processes (aromatic for the Bergman and non-aromaticfor the C1–C5 cyclization).
The above analysis illustrates why it is helpful to consider the enediyne moiety astwo independent p-systems. As discussed above, the conjugated out-of-planep-system of the reactant is smoothly transformed into the conjugate p-system of theproduct (e.g., the aromatic system of benzene) without an overall change in thenumber of bonds. We will refer to this group of electrons as ‘‘out-of-plane MOs’’ orsimply as ‘‘the p-system’’. In contrast, the two in-plane p-bonds are transformed ina more drastic way to the new s-bond and a pair of radical centers. We will refer tothis system of orbitals as ‘‘in-plane p-bonds’’ or as ‘‘the s system’’.
Since the breaking p-bonds and the developing radical centers are orthogonal tothe out-of-plane p-system and the aromatic system develops mostly after the systemproceeds through the transition state, the activation barrier for the Bergmancyclization is relatively insensitive to effects in the p-system. This provides achallenge for the substituent control of such reactions: it is not always clear whetherhyperconjugative and inductive s-effects are sufficiently strong to control thesereactions efficiently. On the other hand, the well understood p-effects are usuallyindirect and they display themselves mostly in reaction thermodynamics.27
Due to the different role of s- and p-effects, we will separate the generaldescription of electronic effects into s- and p-effects in this study. s-Effects include
Fig. 3 Incorrect description of the Bergman cycloaromatization as a 6p-electrocyclic ringclosure (top). A better description shown for comparison (below).
CYCLOAROMATIZATION REACTIONS 9
the in-plane p- and s-orbitals. We will analyze these effects (which can be eitherstabilizing or destabilizing) at the three reaction stages (reactants, TS, and products)separately. All three possibilities can have a significant effect on the reaction enth-alpy and activation energy of the cycloaromatization reaction. The ability toinfluence the reaction at or near the transition state provides the most direct way tocontrol the reaction rates but reactant stabilization/destabilization is also a viableapproach. Effects on the products are indirect but can be readily understood usingdissection of kinetic and thermodynamic contributions to the reaction barrierprovided by the Marcus theory, an approach that we used in the analysis of otherpericyclic reactions28 and radical cyclizations.29
5 r-Effects on reactant stability
In a seminal work, Koga and Morokuma suggested that the high activation energyof the Bergman cyclization is due to the strong electron repulsion between the
H
H
H
H
H
H
HOMO
Ene
rgy
Reaction cooordinate
Ene
rgy
Reaction cooordinate
LUMO+2 (reactant)LUMO+1
(reactant)
HOMO
Fig. 4 The crossings of in-plane and out-of-plane frontier MOs in the radical-anionicBergman and C1–C5 cyclizations, respectively calculated at the B3LYP/6-31G** level.Adapted from reference26.
I. ALABUGIN ET AL.10

in-plane occupied acetylene p-orbitals.30 These authors also pointed out that thelower activation energy of the Myers-Saito cyclization can be attributed to thedecrease in electron repulsion due to the transoid orientation of the interactingp-orbitals.31
Recently, we analyzed the role of electron repulsion relative to bond breaking andantiaromaticity effects on a quantitative basis using Natural Bond Orbital (NBO)analysis.24 Two other destabilizing factors were considered at the initial stage of thecyclization in addition to four-electron repulsion between the filled in-plane
acetylenic p-orbitals – distortion/breaking of the acetylenic bonds as a result oftheir bending, and the fact that, at a distance of ca. 3 A, the in-plane p-orbitalsbecome parallel and reach a geometry that resembles the antiaromatic TS of thesymmetry forbidden [2s+2s] cycloaddition (vide infra).
STRAIN AND ANTIAROMATICITY IN CYCLIC ENEDIYNES
The effect of cyclic constraints that bring the two ends of the enediyne system closertogether is extensively studied. This is not surprising because nature utilizes thehigher reactivity of cyclic enediynes in the naturally occurring enediyne antibiotics.When the enediyne moiety is incorporated in a large cycle (more than 10 carbonatoms), the Bergman cyclization is not fast enough to proceed at physiologicallyrelevant temperatures. In contract, enediynes in 9-membered cycles are too unstableto be isolated (unless stabilized by additional electronic effects, vide infra), whereas10-membered enediynes cyclize within hours (Fig. 5, Table 1).32 Nicolaou andcoworkers33 suggested a simple empirical criterion based on ‘‘the c–d distance’’ (theproximity of the terminal carbons of the enediyne moiety) as a measure of increasedring torsion, which is relieved when the compounds undergo the Bergman cycliza-tion. Only below the ‘‘critical upper limit’’ (the c–d distance of 3.2–3.3 A), thecyclization is expected to proceed at a measurable rate at ambient temperature.More elaborate analysis incorporates the relative strain energies of the ground and
(CH2)n
c
d
OH
OH
c
d
I
OH
OH
c
d
II
OH
OH
c
d
III
O
OO
c
d
IV
Fig. 5 Cyclic model enediynes used by Nicolaou to establish the critical distance model.
CYCLOAROMATIZATION REACTIONS 11

transition states.34,35 In the 11-membered system, the distance is only slightly larger(ca. 3.5 A), yet such enediynes are rather unreactive.36
Overall, 9-membered enediynes are usually too unstable to be utilized in thedesign of new anticancer drugs. An interesting way to overcome this limitation wasshown recently by Jones and coworkers who utilized an electronic effect in a rareexample of intentional deceleration of the Bergman cyclization (Fig. 6). Substitutionof a hydrogen atom with a chlorine atom at the vinylic position of a 9-memberedcyclic enediyne37 decreased the cyclization rate and allowed the first enediynewith a 9-membered ring to be isolated. Nevertheless, the half-life of this compoundwas found to be only 6min at 313K, corresponding to an activation barrier ofo18 kcal/mol.
An extensive computational analysis expanded the range of the c–d distances forreactive cyclic enediynes to 2.9–3.4 A.38 By comparing unsubstituted enediynes withdialkyl-substituted enediynes, it was found that the activation enthalpy is dependenton factors other than the c–d distance and that reactivity hinges on a subtle interplayof steric and electronic effects that accompany distortion caused by incorpora-tion into a macrocycle. For example, since alkyl substituents stabilize acetylenicbonds to a greater extend than olefinic bonds,39 such substituents stabilize thestarting material, thus increasing both the activation barrier and the reactionendothermicity.
Despite the contribution of the above factors, it is clear that bending of thetwo alkyne moieties toward each other increases the energy of the system asillustrated by the three plots in Fig. 7.24 These plots illustrate the relation betweenthe ring size and calculated cyclization parameters in more detail and dependenceof the total energy of the system from the c–d distance. Interestingly, simplebending of alkyne moiety reproduced the effect of cyclic restraints reasonably well.A similar conclusion has been reached even earlier by Kraka and Cremer.40
Table 1 Calculated c–d distances and stabilities of cyclic enediynes
Compound n Ring Size c–d Distance (A) Stability
1 9 2.84 Unknown2 10 3.25 t1/2 ¼ 18 h at 251C3 11 3.61 Stable at 251C
I – 10 3.20 t1/2 ¼ 11.8 h at 371CII – 10 3.29 t1/2 ¼ 4 h at 501CIII – 10 3.34 t1/2 ¼ 2 h at 501CIV – 10 3.42 Stable at 251C
Cl Cl
Fig. 6 Cyclization of the first isolable a 9-membered enediyne.
I. ALABUGIN ET AL.12
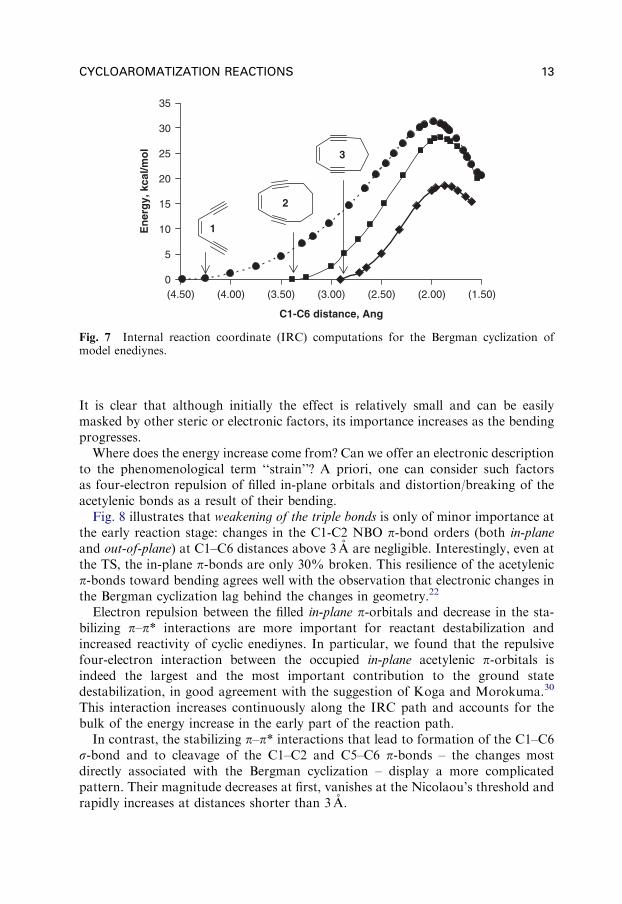
It is clear that although initially the effect is relatively small and can be easilymasked by other steric or electronic factors, its importance increases as the bendingprogresses.
Where does the energy increase come from? Can we offer an electronic descriptionto the phenomenological term ‘‘strain’’? A priori, one can consider such factorsas four-electron repulsion of filled in-plane orbitals and distortion/breaking of theacetylenic bonds as a result of their bending.
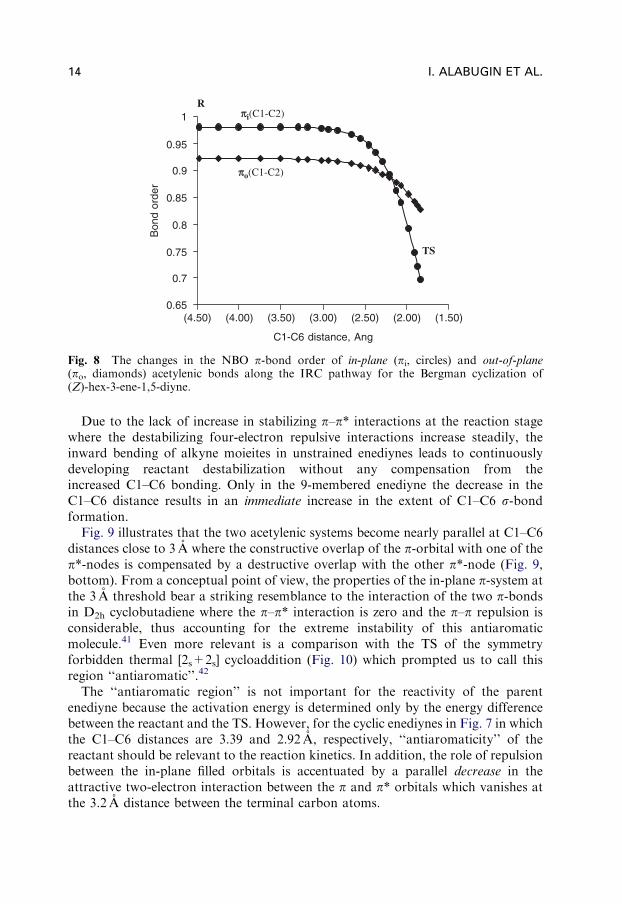
Fig. 8 illustrates that weakening of the triple bonds is only of minor importance atthe early reaction stage: changes in the C1-C2 NBO p-bond orders (both in-plane
and out-of-plane) at C1–C6 distances above 3 A are negligible. Interestingly, even atthe TS, the in-plane p-bonds are only 30% broken. This resilience of the acetylenicp-bonds toward bending agrees well with the observation that electronic changes inthe Bergman cyclization lag behind the changes in geometry.22
Electron repulsion between the filled in-plane p-orbitals and decrease in the sta-bilizing p–p* interactions are more important for reactant destabilization andincreased reactivity of cyclic enediynes. In particular, we found that the repulsivefour-electron interaction between the occupied in-plane acetylenic p-orbitals isindeed the largest and the most important contribution to the ground statedestabilization, in good agreement with the suggestion of Koga and Morokuma.30
This interaction increases continuously along the IRC path and accounts for thebulk of the energy increase in the early part of the reaction path.
In contrast, the stabilizing p–p* interactions that lead to formation of the C1–C6s-bond and to cleavage of the C1–C2 and C5–C6 p-bonds – the changes mostdirectly associated with the Bergman cyclization – display a more complicatedpattern. Their magnitude decreases at first, vanishes at the Nicolaou’s threshold andrapidly increases at distances shorter than 3 A.
0
5
10
15
20
25
30
35
(4.50) (4.00) (3.50) (3.00) (2.50) (2.00) (1.50)
C1-C6 distance, Ang
En
erg
y, k
cal/m
ol
1
2
3
Fig. 7 Internal reaction coordinate (IRC) computations for the Bergman cyclization ofmodel enediynes.
CYCLOAROMATIZATION REACTIONS 13
Due to the lack of increase in stabilizing p–p* interactions at the reaction stagewhere the destabilizing four-electron repulsive interactions increase steadily, theinward bending of alkyne moieites in unstrained enediynes leads to continuouslydeveloping reactant destabilization without any compensation from theincreased C1–C6 bonding. Only in the 9-membered enediyne the decrease in theC1–C6 distance results in an immediate increase in the extent of C1–C6 s-bondformation.
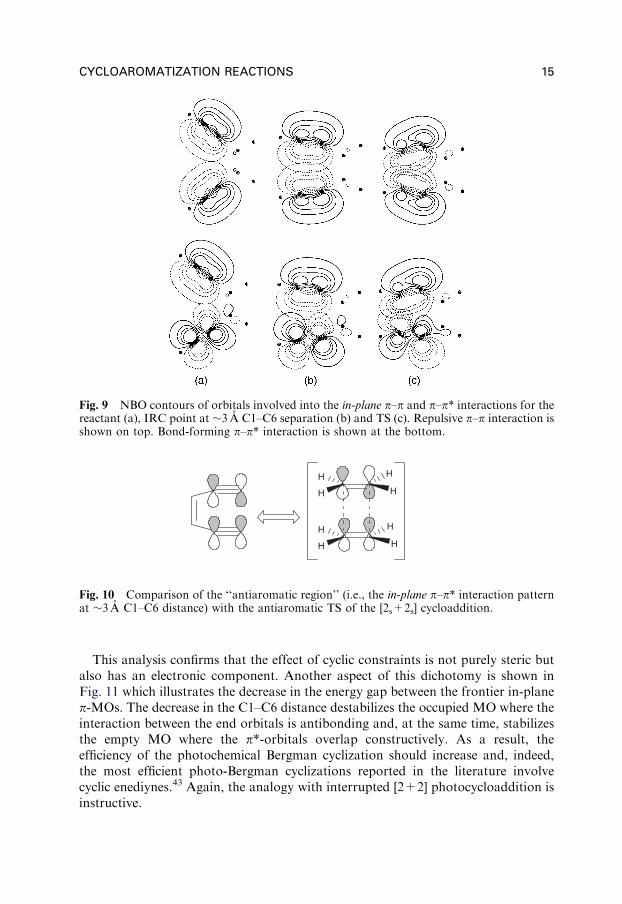
Fig. 9 illustrates that the two acetylenic systems become nearly parallel at C1–C6distances close to 3 A where the constructive overlap of the p-orbital with one of thep*-nodes is compensated by a destructive overlap with the other p*-node (Fig. 9,bottom). From a conceptual point of view, the properties of the in-plane p-system atthe 3 A threshold bear a striking resemblance to the interaction of the two p-bondsin D2h cyclobutadiene where the p–p* interaction is zero and the p–p repulsion isconsiderable, thus accounting for the extreme instability of this antiaromaticmolecule.41 Even more relevant is a comparison with the TS of the symmetryforbidden thermal [2s+2s] cycloaddition (Fig. 10) which prompted us to call thisregion ‘‘antiaromatic’’.42
The ‘‘antiaromatic region’’ is not important for the reactivity of the parentenediyne because the activation energy is determined only by the energy differencebetween the reactant and the TS. However, for the cyclic enediynes in Fig. 7 in whichthe C1–C6 distances are 3.39 and 2.92 A, respectively, ‘‘antiaromaticity’’ of thereactant should be relevant to the reaction kinetics. In addition, the role of repulsionbetween the in-plane filled orbitals is accentuated by a parallel decrease in theattractive two-electron interaction between the p and p* orbitals which vanishes atthe 3.2 A distance between the terminal carbon atoms.
0.65
0.7
0.75
0.8
0.85
0.9
0.95
1
(4.50) (4.00) (3.50) (3.00) (2.50) (2.00) (1.50)
C1-C6 distance, Ang
Bon
d or
der
TS
R π πi(C1-C2)
π πo(C1-C2)
Fig. 8 The changes in the NBO p-bond order of in-plane (pi, circles) and out-of-plane(po, diamonds) acetylenic bonds along the IRC pathway for the Bergman cyclization of(Z)-hex-3-ene-1,5-diyne.
I. ALABUGIN ET AL.14
This analysis confirms that the effect of cyclic constraints is not purely steric butalso has an electronic component. Another aspect of this dichotomy is shown inFig. 11 which illustrates the decrease in the energy gap between the frontier in-planep-MOs. The decrease in the C1–C6 distance destabilizes the occupied MO where theinteraction between the end orbitals is antibonding and, at the same time, stabilizesthe empty MO where the p*-orbitals overlap constructively. As a result, theefficiency of the photochemical Bergman cyclization should increase and, indeed,the most efficient photo-Bergman cyclizations reported in the literature involvecyclic enediynes.43 Again, the analogy with interrupted [2+2] photocycloaddition isinstructive.
HH
HH
HH
HH
Fig. 10 Comparison of the ‘‘antiaromatic region’’ (i.e., the in-plane p–p* interaction patternat �3 A C1–C6 distance) with the antiaromatic TS of the [2s+2s] cycloaddition.
Fig. 9 NBO contours of orbitals involved into the in-plane p–p and p–p* interactions for thereactant (a), IRC point at �3 A C1–C6 separation (b) and TS (c). Repulsive p–p interaction isshown on top. Bond-forming p–p* interaction is shown at the bottom.
CYCLOAROMATIZATION REACTIONS 15

CONTROL OF STRAIN THROUGH LIGAND–METAL COORDINATION
An elegant way to control both strain and electronics is to take advantage of metalcoordination to an enediynyl ligand – a topic that has been reviewed intensively44
and, thus, will be discussed rather briefly here. The influence of metal complexationon enediynes can be divided into several aspects: It can work through either s-donorcoordination or p-complexation, resulting in any combination of geometric changes(analogous to the change in c–d distance proposed by Nicolaou) or by their influenceon the electronic environment of the enediyne moiety.
Strain-based systems work the same way as the cyclic enediynes: they reduce thec–d distance in the molecule. The first example was provided by Buchwald andcoworkers,45 who used metal complexation of a PPh2 substituted enediyne toproduce a species with considerably lower activation barrier (compared to the non-coordinated enediyne) (Scheme 10).
While in the unbound enediyne the c–d distance is 4.1 A, this distance is dimini-shed upon metal complexation: �3.3 A for Pt(II) and Pd(II), and �3.4 A for Hg(II).The Pt and Pd species cyclize in the solid state at only slightly elevated temperatures,and give Bergman products below ambient temperature in solution. While thechange in reactivity was attributed to the change in distance between the alkynetermini, an accelerating influence of the metal cannot be ruled out.
Since both s acceptors and p donors at the alkyne termini are known to facilitatethe Bergman cyclization, Zaleski and coworkers established a model46 in which thecoordination of a Lewis acid (metal ion) would change the electronic environment infavor of diradical formation (Scheme 11).
Metal coordination was assumed to accelerate the reaction by inductive effects.A comparison of the reactivity of the benzylated compound in presence of Mo wasused to rule out any p influence on the acetylenes.
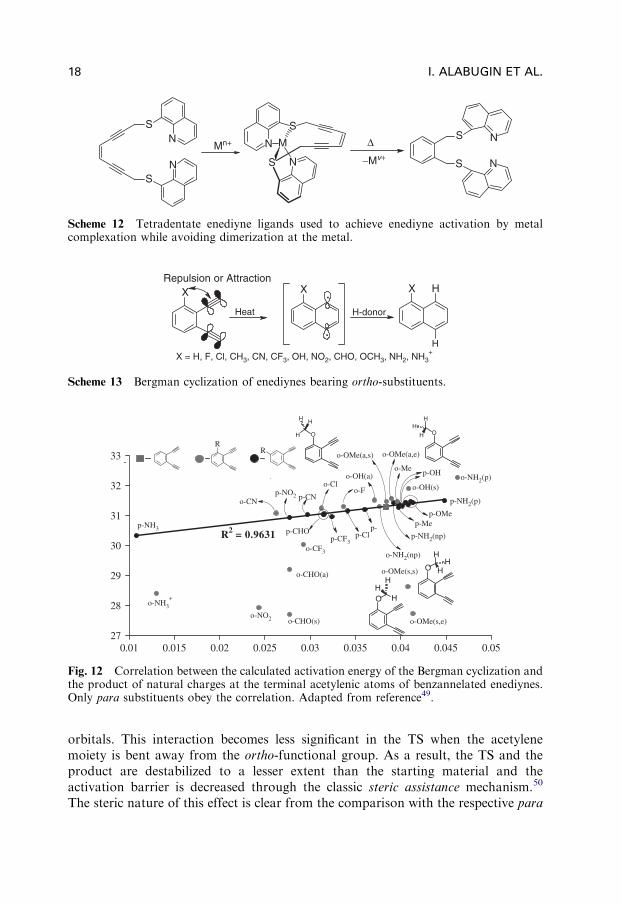
Zaleski and coworkers47 later expanded upon this line of work by using tetra-dentate enediyne ligands, in which the reactivity could be modulated by metalcomplexation (Scheme 12).
The advantage of tetradentate ligand systems is the fact that they help to avoiddimerization, and that with the right choice of metal (and therefore the right
Fig. 11 Comparison of the energy gap between highest occupied in-plane MO and lowestunoccupied in-plane MO in acyclic (left) and cyclic (right) benzannelated enediynes. Incor-poration of the enediyne moiety into a cyclic structure simultaneously increases the energy ofthe occupied MO and lowers the energy of the unoccupied MO.
I. ALABUGIN ET AL.16

coordination geometry), tuning of both the c–d distance and electronic effects iseasily accomplished.
STERIC ASSISTANCE OF ORTHO SUBSTITUENTS
An additional way to use strain for control of reactivity is steric assistance. Thiseffect can be easily introduced through a choice of appropriate ortho substituentsin benzannelated enediynes (Scheme 13). Whereas activation energies for theneutral para substituents lie within a range of only 0.6 kcal/mol,48 the presence ofortho-NO2, NH2, CHO, CF3, and syn-OMe groups results in large changes in theactivation energy (Fig. 12).49 Depending on the substituent, three different factorsaccount for these changes: steric assistance (decrease in steric destabilization in TS),extra stabilization of the TS, and decrease in TS stabilization.
In particular, the NO2, CF3, syn-CHO, and syn-OMe groups were predicted todecrease the activation energy for Bergman cyclization by destabilizing reactantsthrough steric repulsion between the ortho-substituent and the in-plane acetylenic
PPh2
PPh2
P
P PMCl2
PMCl2 MCl2
Ph2
Ph2
Ph2
Ph2
Pt: 61°C Pd: 81°C
Δ
Scheme 10 Metal coordination activates enediynes by drastically reducing the distancebetween the acetylenic carbons.
S
S SK
SK
S
S SMoS Cp
Cp
[MoCp2Cl2], CHD, 60°C
30 min, 40 %
S
S S
S
Ph
Ph
[MoCp2Cl2], CHD
60°C, THF/MeOHno reaction
S
S S
S
Ph
Ph
S
S
S
S
Ph
Ph
DMSO, CHD, 180 °C
24 h, 60 %
S
S S
S
Ph
Ph
DMSO, CHD
180 °C, 24 hno reaction
S
S
SMoS
CpCp
S
S SMoS Cp
CpDMSO, CHD, 120°C
5 h, 15%
Scheme 11 Model systems illustrating enediyne activation through metal complexation.
CYCLOAROMATIZATION REACTIONS 17
orbitals. This interaction becomes less significant in the TS when the acetylenemoiety is bent away from the ortho-functional group. As a result, the TS and theproduct are destabilized to a lesser extent than the starting material and theactivation barrier is decreased through the classic steric assistance mechanism.50
The steric nature of this effect is clear from the comparison with the respective para
S
S
N
N N
N
S
S
M N
N
S
S
Mn+
−Mv+
Δ
Scheme 12 Tetradentate enediyne ligands used to achieve enediyne activation by metalcomplexation while avoiding dimerization at the metal.
XX X H
H
Heat H-donor
X = H, F, Cl, CH3, CN, CF3, OH, NO2, CHO, OCH3, NH2, NH3+
Repulsion or Attraction
.
.
Scheme 13 Bergman cyclization of enediynes bearing ortho-substituents.
R2 = 0.9631
27
28
29
30
31
32
33
0.01 0.015 0.02 0.025 0.03 0.035 0.04 0.045 0.05
RR
O
H H
H OH
H
H
OH
H
H
O
H
HH
o-NH3+
p-NH3
o-NO2 o-CHO(s)
o-CHO(a) o-OMe(s,s)
o-OMe(s,e)
o-CN
o-Clo-F
p-NH2(np)
o-OMe(a,s)
p-OMe
o-Meo-NH2(p)
p-Me
p-CNp-NO2
p-CHO p-Clp-
o-OMe(a,e)
o-NH2(np)
p-OH
p-CF3o-CF3
o-OH(s)
p-NH2(p)
o-OH(a)
Fig. 12 Correlation between the calculated activation energy of the Bergman cyclization andthe product of natural charges at the terminal acetylenic atoms of benzannelated enediynes.Only para substituents obey the correlation. Adapted from reference49.
I. ALABUGIN ET AL.18
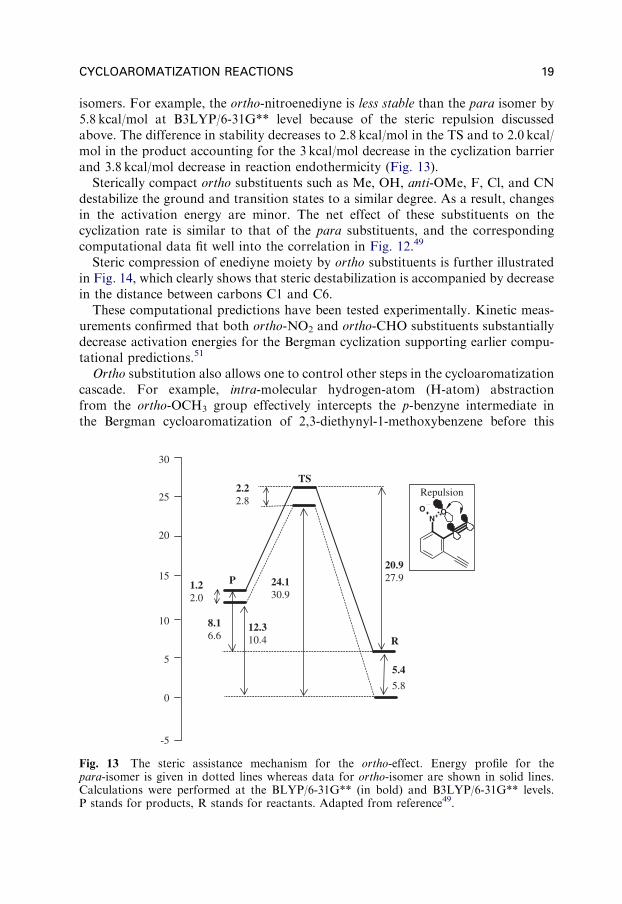
isomers. For example, the ortho-nitroenediyne is less stable than the para isomer by5.8 kcal/mol at B3LYP/6-31G** level because of the steric repulsion discussedabove. The difference in stability decreases to 2.8 kcal/mol in the TS and to 2.0 kcal/mol in the product accounting for the 3 kcal/mol decrease in the cyclization barrierand 3.8 kcal/mol decrease in reaction endothermicity (Fig. 13).
Sterically compact ortho substituents such as Me, OH, anti-OMe, F, Cl, and CNdestabilize the ground and transition states to a similar degree. As a result, changesin the activation energy are minor. The net effect of these substituents on thecyclization rate is similar to that of the para substituents, and the correspondingcomputational data fit well into the correlation in Fig. 12.49
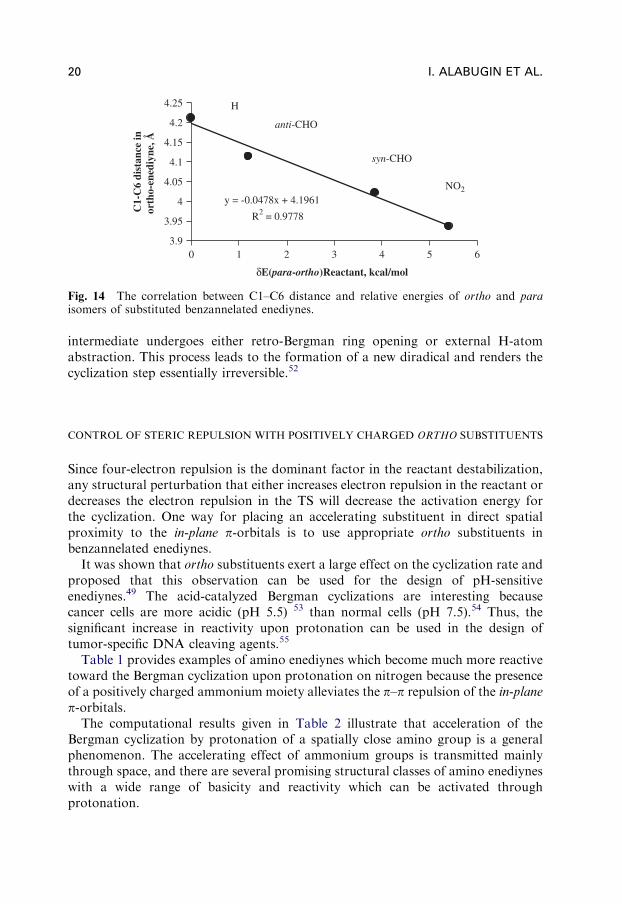
Steric compression of enediyne moiety by ortho substituents is further illustratedin Fig. 14, which clearly shows that steric destabilization is accompanied by decreasein the distance between carbons C1 and C6.
These computational predictions have been tested experimentally. Kinetic meas-urements confirmed that both ortho-NO2 and ortho-CHO substituents substantiallydecrease activation energies for the Bergman cyclization supporting earlier compu-tational predictions.51
Ortho substitution also allows one to control other steps in the cycloaromatizationcascade. For example, intra-molecular hydrogen-atom (H-atom) abstractionfrom the ortho-OCH3 group effectively intercepts the p-benzyne intermediate inthe Bergman cycloaromatization of 2,3-diethynyl-1-methoxybenzene before this
N+ OO
30
25
20
15
10
5
0
-5
8.1 6.6
12.3 10.4
5.4
5.8
R
24.1 30.9
2.2 2.8
1.2 2.0
20.9 27.9
TS
P
Repulsion
Fig. 13 The steric assistance mechanism for the ortho-effect. Energy profile for thepara-isomer is given in dotted lines whereas data for ortho-isomer are shown in solid lines.Calculations were performed at the BLYP/6-31G** (in bold) and B3LYP/6-31G** levels.P stands for products, R stands for reactants. Adapted from reference49.
CYCLOAROMATIZATION REACTIONS 19
intermediate undergoes either retro-Bergman ring opening or external H-atomabstraction. This process leads to the formation of a new diradical and renders thecyclization step essentially irreversible.52
CONTROL OF STERIC REPULSION WITH POSITIVELY CHARGED ORTHO SUBSTITUENTS
Since four-electron repulsion is the dominant factor in the reactant destabilization,any structural perturbation that either increases electron repulsion in the reactant ordecreases the electron repulsion in the TS will decrease the activation energy forthe cyclization. One way for placing an accelerating substituent in direct spatialproximity to the in-plane p-orbitals is to use appropriate ortho substituents inbenzannelated enediynes.
It was shown that ortho substituents exert a large effect on the cyclization rate andproposed that this observation can be used for the design of pH-sensitiveenediynes.49 The acid-catalyzed Bergman cyclizations are interesting becausecancer cells are more acidic (pH 5.5) 53 than normal cells (pH 7.5).54 Thus, thesignificant increase in reactivity upon protonation can be used in the design oftumor-specific DNA cleaving agents.55
Table 1 provides examples of amino enediynes which become much more reactivetoward the Bergman cyclization upon protonation on nitrogen because the presenceof a positively charged ammonium moiety alleviates the p–p repulsion of the in-plane
p-orbitals.The computational results given in Table 2 illustrate that acceleration of the
Bergman cyclization by protonation of a spatially close amino group is a generalphenomenon. The accelerating effect of ammonium groups is transmitted mainlythrough space, and there are several promising structural classes of amino enediyneswith a wide range of basicity and reactivity which can be activated throughprotonation.
C1-
C6
dist
ance
in
orth
o-en
ediy
ne, Å
y = -0.0478x + 4.1961
R2 = 0.9778
3.9
3.95
4
4.05
4.1
4.15
4.2
4.25
0 1 4 5 62 3
NO2
anti-CHO
syn-CHO
H
δE(para-ortho)Reactant, kcal/mol
Fig. 14 The correlation between C1–C6 distance and relative energies of ortho and paraisomers of substituted benzannelated enediynes.
I. ALABUGIN ET AL.20

Interestingly, both donor (syn-OMe) and acceptor (NO2) substitution can increasethe accelerating effect of an ortho ammonium group when the steric interferencewith the above substituents ‘‘pushes’’ the adjacent acetylene moiety toward the otheracetylene group, increasing the C1–C6 bonding. This is an example of an interestingcooperating effect between two ortho-substituents further illustrated in Fig. 15.
HYBRIDIZATION EFFECTS
As a direct consequence of Bent’s rule,56 carbon compounds with a significantamount of s-character in their bonds to an electronegative element, e.g., fluoro-alkynes, are unstable. Thus, reactions which involve change in hybridization at acarbon atom bearing such an acceptor can be promoted when the amount ofs-character at the reactive atom decreases. The role of rehybridization in a variety ofchemical and supramolecular processes has been extensively analyzed and reviewedrecently.55,58
An illustrative example of how rehybridization can be used to control theBergman cyclization is provided by substituent effects at the alkyne termini ofenediynes. This effect in cycloaromatization chemistry was first studied by Schreinerand coworkers, who found dramatic acceleration of the Bergman cyclization upon
Table 2 Calculated energetics in kcal/mol for the Bergman cyclizations of protonated andunprotonated aminoenediynes
Protonated EnediynesaNeutral Amines
DE 6¼ DH 6¼ DG6¼ DEr DE6¼ a
NH3+
st ec
29.2 28.6 29.7 10.6 32.328.4 27.1 29.1 11.7
NH3+ st
ec30.3 29.6 31.3 9.6 31.530.2 29.3 31.1 9.2
NH3+
syn anti
25.5 24.6 26.2 10.0 27.229.9 28.7 30.4 10.2
st, staggered; ec, eclipsed.aThe reaction barriers for the Bergman cyclization of corresponding neutral amino enediynes.
CYCLOAROMATIZATION REACTIONS 21

introduction of s acceptors at the terminal carbons of (Z)-1,5-hexadiyne-3-ene.59
In particular, cyclization of the enediynes with terminal fluoro-substituents waspredicted to have the lowest barrier and be significantly exothermic. A subsequentexperimental study found that the reverse reaction (retro-Bergman ring opening) inthis system is endothermic, thus confirming the earlier computational predictions.60
In contrast, the computational work of Jones and Warner found that acceptorsubstituents positioned at the ene part of the enediyne moiety decelerate the reac-tion61 in full accord with the earlier experimental results of Jones and coworkers.
Rehybridization provides a unified explanation for these two sets of results.57
Since the transition state is reactant-like22 and TS and reactant have the samedominant Lewis structures, application of the NBO method for the analysis of thetransition state is straightforward. The hybrid orbital ‘‘h’’ that connects the terminalacetylene carbon to the substituent undergoes the most dramatic rehybridization(sp-sp2 or 48%-33% s-character) in the Bergman cyclization of enediyne I inFig. 16. According to Bent’s rule, terminal fluorine substitution destabilizes thereagent by preventing this hybrid orbital from attaining its ‘‘natural’’ sp-hybridi-zation (in other words, it is unfavorable to direct a hybrid orbital with 50% ofs-character toward a strong acceptor). As a result, the hybrid h has only 36–37% ofs-character in the reactant – a dramatic effect of F substitution! However, thedifferences in hybridization decrease in the TS, illustrating that the destabilizing effectof electron acceptor in the reactant is removed by rehybridization in the TS (Fig. 17).
6 p-Effects on reactant stability
As mentioned in the preceding section, p-effects on the stability of the reactants aregoing to be rather subtle in thermal cyclizations, since the determining factor in theactivation barrier for this reaction is the formation of the bond between in-planeorbitals. One way to accelerate this reaction would be to destabilize the reactantp-system. The challenge is in designing a system where the reactant destabilization isnot transferred to the transition state and product as well. An elegant approach to
NH3+
NH3+
NH3+
OMe
NH3+
NO2
Ea=30.8 Ea=22.6 Ea=25.7
Push-push Pull-pull Push-pull
NO2
NO2
O
O
Me
Me
Ea=25.5Ea=25.6
Fig. 15 Predicted cooperative effects on activation energies (in kcal/mol) at the B3LYP/6-31G** level for model enediynes (‘‘push’’ and ‘‘pull’’ denote through-space repulsive (steric)and attractive (H-bonding) interactions of ortho-substituents with in-plane p-orbitals of anadjacent acetylene moeity).
I. ALABUGIN ET AL.22

achieve such selective reactant destabilization is described in the next sectionwhich describes systems with selective destabilization of reactants and efficientcommunication between the p and s arrays.
7 Transition state effects: communication of orthogonal orbitals inthe transition state of radical-anionic cyclizations
In order to activate remote substituent effects in cycloaromatization reactions, twoorthogonal p-systems need to find a way to communicate. At first glance, this task
X
X
X
X
X
X
35.6
31.2
41.0
27.9 17.1
3.93.3 3.3
Reaction coordinate
Ene
rgy,
kca
l/mol
X = HX = F
TSBC
TSRBC
0
21.0
ED1 BZY ED2
s-ch
ar o
f C, %
ED1 ED2BZYTSBC TSRBC
22.9423.22
23.88
30.07
36.59
Reaction coordinate
F
F
F
F
(a) (b)
Fig. 16 (a) Comparison of potential energy profile for the formal Cope rearrangement of3,4-difluorohexa-1,5-diyne-3-ene with that of (Z)-hexa-1,5-diyne-3-ene, (b) Rehybridization in theC(F) bond along the reaction path. ED1 ¼ 3,4-difluoro-hex- 3-ene-1,5-diyne; ED2 ¼ 1,6-di-fluoro-hex-3-ene-1,5-diyne; BZY ¼ difluoro-1,4-didehydrobenzezne; TSBC ¼ the transition statefor the Bergman cyclization; TSRBC ¼ the transition state for the retro Bergman cyclization.
F
F
F
F
F
F
Ea = 31.2
Ea = 24.0
4.480
4.452
1.212
1.210
1.978
2.052
1.265
1.268
I
I
ΔEr = 3.3
ΔEr = -17.1
Fig. 17 The Bergman cyclizations of parent and fluoro-substituted enediynes with thetriple bond and the incipient bond lengths and the activation energies calculated at theBS-UB3LYP/6-31G** level.
CYCLOAROMATIZATION REACTIONS 23
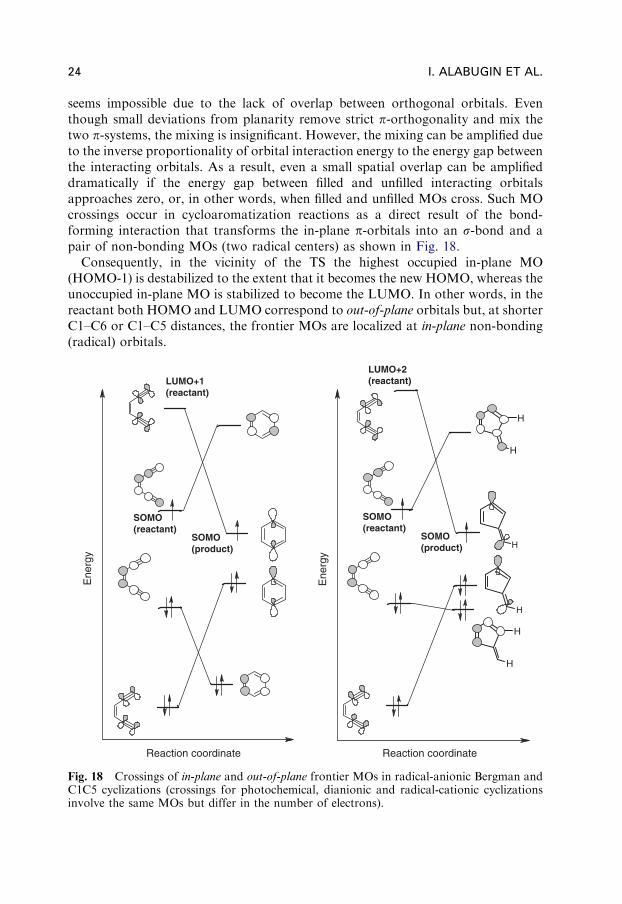
seems impossible due to the lack of overlap between orthogonal orbitals. Eventhough small deviations from planarity remove strict p-orthogonality and mix thetwo p-systems, the mixing is insignificant. However, the mixing can be amplified dueto the inverse proportionality of orbital interaction energy to the energy gap betweenthe interacting orbitals. As a result, even a small spatial overlap can be amplifieddramatically if the energy gap between filled and unfilled interacting orbitalsapproaches zero, or, in other words, when filled and unfilled MOs cross. Such MOcrossings occur in cycloaromatization reactions as a direct result of the bond-forming interaction that transforms the in-plane p-orbitals into an s-bond and apair of non-bonding MOs (two radical centers) as shown in Fig. 18.
Consequently, in the vicinity of the TS the highest occupied in-plane MO(HOMO-1) is destabilized to the extent that it becomes the new HOMO, whereas theunoccupied in-plane MO is stabilized to become the LUMO. In other words, in thereactant both HOMO and LUMO correspond to out-of-plane orbitals but, at shorterC1–C6 or C1–C5 distances, the frontier MOs are localized at in-plane non-bonding(radical) orbitals.
H
H
H
H
H
H
SOMO (reactant)
SOMO (product)
Ene
rgy
SOMO (reactant)
SOMO (product)
Ene
rgy
Reaction coordinateReaction coordinate
LUMO+2 (reactant)LUMO+1
(reactant)
Fig. 18 Crossings of in-plane and out-of-plane frontier MOs in radical-anionic Bergman andC1C5 cyclizations (crossings for photochemical, dianionic and radical-cationic cyclizationsinvolve the same MOs but differ in the number of electrons).
I. ALABUGIN ET AL.24
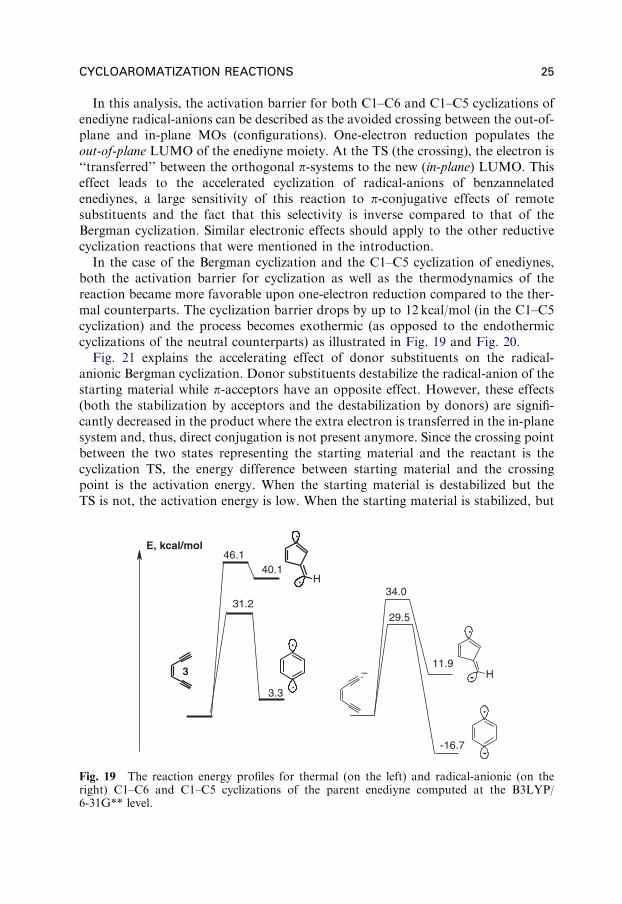
In this analysis, the activation barrier for both C1–C6 and C1–C5 cyclizations ofenediyne radical-anions can be described as the avoided crossing between the out-of-plane and in-plane MOs (configurations). One-electron reduction populates theout-of-plane LUMO of the enediyne moiety. At the TS (the crossing), the electron is‘‘transferred’’ between the orthogonal p-systems to the new (in-plane) LUMO. Thiseffect leads to the accelerated cyclization of radical-anions of benzannelatedenediynes, a large sensitivity of this reaction to p-conjugative effects of remotesubstituents and the fact that this selectivity is inverse compared to that of theBergman cyclization. Similar electronic effects should apply to the other reductivecyclization reactions that were mentioned in the introduction.
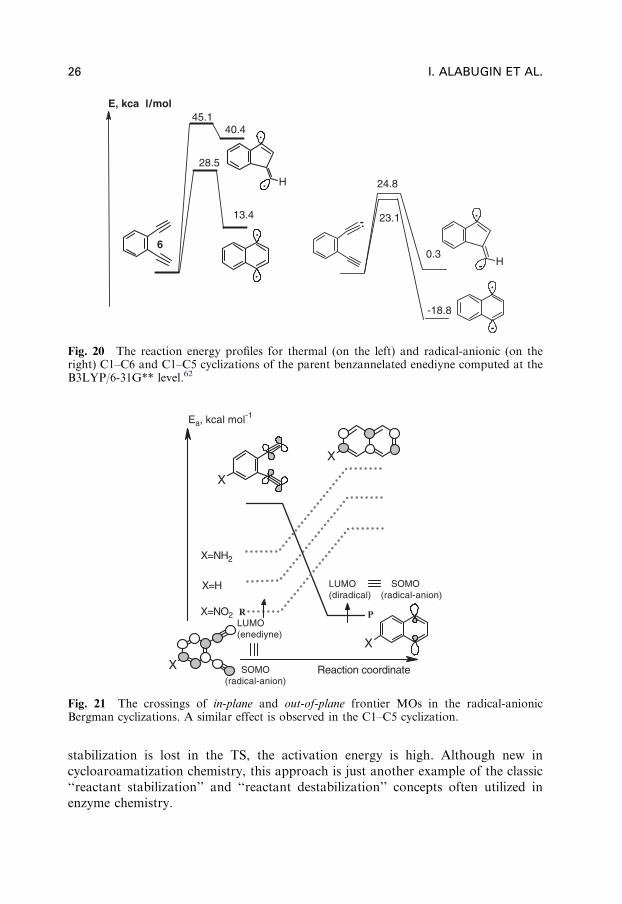
In the case of the Bergman cyclization and the C1–C5 cyclization of enediynes,both the activation barrier for cyclization as well as the thermodynamics of thereaction became more favorable upon one-electron reduction compared to the ther-mal counterparts. The cyclization barrier drops by up to 12 kcal/mol (in the C1–C5cyclization) and the process becomes exothermic (as opposed to the endothermiccyclizations of the neutral counterparts) as illustrated in Fig. 19 and Fig. 20.
Fig. 21 explains the accelerating effect of donor substituents on the radical-anionic Bergman cyclization. Donor substituents destabilize the radical-anion of thestarting material while p-acceptors have an opposite effect. However, these effects(both the stabilization by acceptors and the destabilization by donors) are signifi-cantly decreased in the product where the extra electron is transferred in the in-planesystem and, thus, direct conjugation is not present anymore. Since the crossing pointbetween the two states representing the starting material and the reactant is thecyclization TS, the energy difference between starting material and the crossingpoint is the activation energy. When the starting material is destabilized but theTS is not, the activation energy is low. When the starting material is stabilized, but
H-
.
H
.
.34.0
11.9
E, kcal/mol46.1
40.1
-
.
.
.
31.2
3.3
-16.7
29.5
3 −.
Fig. 19 The reaction energy profiles for thermal (on the left) and radical-anionic (on theright) C1–C6 and C1–C5 cyclizations of the parent enediyne computed at the B3LYP/6-31G** level.
CYCLOAROMATIZATION REACTIONS 25
stabilization is lost in the TS, the activation energy is high. Although new incycloaroamatization chemistry, this approach is just another example of the classic‘‘reactant stabilization’’ and ‘‘reactant destabilization’’ concepts often utilized inenzyme chemistry.
H
-
.
H
.
-
24.8
0.3
E, kca l/mol45.1
40.4
28.5
13.4
-18.8
23.1
6
.
.
-..
.
Fig. 20 The reaction energy profiles for thermal (on the left) and radical-anionic (on theright) C1–C6 and C1–C5 cyclizations of the parent benzannelated enediyne computed at theB3LYP/6-31G** level.62
X
X
X
X
Ea, kcal mol-1
R
X=H
X=NO2
X=NH2
LUMO (diradical)
P
Reaction coordinate
LUMO (enediyne)
SOMO (radical-anion)
SOMO (radical-anion)
Fig. 21 The crossings of in-plane and out-of-plane frontier MOs in the radical-anionicBergman cyclizations. A similar effect is observed in the C1–C5 cyclization.
I. ALABUGIN ET AL.26

8 Effects on product stability
At this stage, both s- and p-effects are important. However, the influence ofthe p-effects dramatically increases. In fact, it is the formation of a p-aromaticsystem in the cycloaromatization reactions that makes these processes energeticallyfeasible.
s-EFFECTS
Interaction of non-bonding electrons
The non-bonding electrons can be coupled either directly through space (TS) orindirectly through antibonding (s*) bridge orbitals (through bond (TB) coupling).The most well-recognized of the s-effects is the TB interaction of the two radicalcenters in p-benzynes and related molecules.63 A well-recognized effect of thisinteraction is displayed in the lower reactivity of p-benzyne in H-abstractionreactions relative to that of the phenyl radical.55 TB interaction, which is absentin monoradicals,54 provides an additional 3–5 kcal/mol of stabilization energy to thep-benzyne-type diradicals. Since this stabilizing energy is lost with the first H-atomabstraction, the p-benzyne diradicals are less reactive and more selective thansimple phenyl radicals. Interestingly, coupling between the non-bonding orbitalsis dramatically enhanced upon one-electron reduction of p-benzynes ordidehydrofulvenes, possibly because the TS interaction adds to the TB coupling.12b
Zwitterionic products
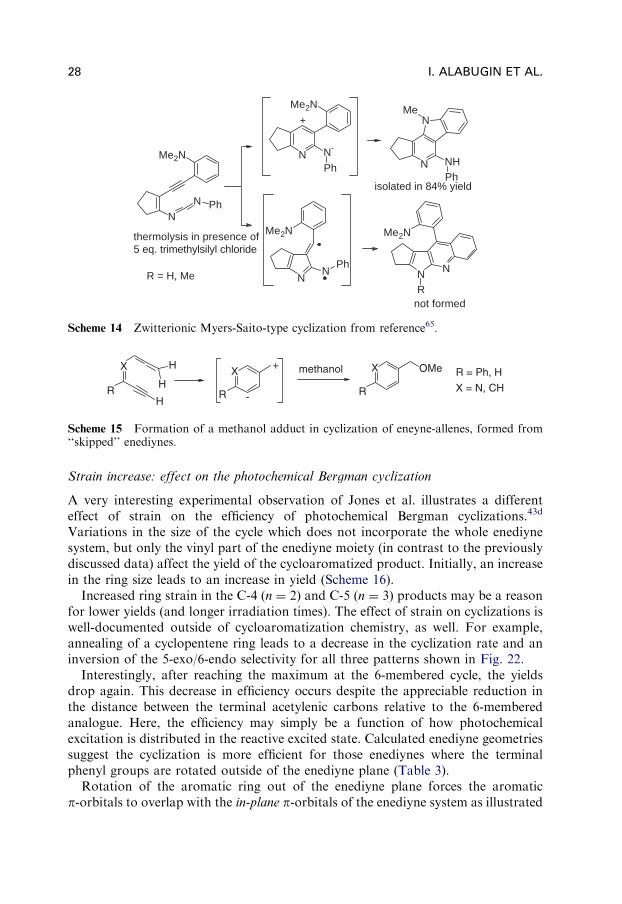
Full -polarization in diradicals can give rise to zwitterionic products. First exampleswere studied in detail by Carpenter and coworker who investigated solvent effects onrates and product distribution in Myers-Saito cyclizations.64 Polar solvents andsubstitution patterns that stabilize either positive or negative charges (or both) favorthe zwitterionic products. For example, the presence of a dimethylamino groupleads to stabilization of cations and isolation of pyrrolo-quinolines, rather thanpyrido-indoles from eneyne-carbodiimides, as reported by Wang and coworkers(Scheme 14).65
Even though these products are not formed by a diradical mechanism, these polarspecies may still have relevance to DNA damage, because of the potential alkylatingability of their electrophilic (cationic) sites.
Another example of a zwitterionic product of a cycloaromatization reactionwas given by Kerwin and coworkers. Their ‘‘skipped’’ (aza)enediynes rearranged toyield (aza)eneyne-allenes that subsequently cyclized under addition of methanol(in a byproduct), which is consistent with a partitioning between a diradical and azwitterionic reaction pathway (Scheme 15).66
CYCLOAROMATIZATION REACTIONS 27
Strain increase: effect on the photochemical Bergman cyclization
A very interesting experimental observation of Jones et al. illustrates a differenteffect of strain on the efficiency of photochemical Bergman cyclizations.43d
Variations in the size of the cycle which does not incorporate the whole enediynesystem, but only the vinyl part of the enediyne moiety (in contrast to the previouslydiscussed data) affect the yield of the cycloaromatized product. Initially, an increasein the ring size leads to an increase in yield (Scheme 16).
Increased ring strain in the C-4 (n ¼ 2) and C-5 (n ¼ 3) products may be a reasonfor lower yields (and longer irradiation times). The effect of strain on cyclizations iswell-documented outside of cycloaromatization chemistry, as well. For example,annealing of a cyclopentene ring leads to a decrease in the cyclization rate and aninversion of the 5-exo/6-endo selectivity for all three patterns shown in Fig. 22.
Interestingly, after reaching the maximum at the 6-membered cycle, the yieldsdrop again. This decrease in efficiency occurs despite the appreciable reduction inthe distance between the terminal acetylenic carbons relative to the 6-memberedanalogue. Here, the efficiency may simply be a function of how photochemicalexcitation is distributed in the reactive excited state. Calculated enediyne geometriessuggest the cyclization is more efficient for those enediynes where the terminalphenyl groups are rotated outside of the enediyne plane (Table 3).
Rotation of the aromatic ring out of the enediyne plane forces the aromaticp-orbitals to overlap with the in-plane p-orbitals of the enediyne system as illustrated
+X
RH
H
H
X
R
OMeX
R
R = Ph, H
X = N, CH-
methanol
Scheme 15 Formation of a methanol adduct in cyclization of eneyne-allenes, formed from‘‘skipped’’ enediynes.
Me2N
N
N
N
N
NH
Ph
Me
NN
Me2N
R
N N-
Me2N
Ph
+
NN
Me2N
Ph
Ph
thermolysis in presence of
5 eq. trimethylsilyl chloride
R = H, Me
isolated in 84% yield
not formed
Scheme 14 Zwitterionic Myers-Saito-type cyclization from reference65.
I. ALABUGIN ET AL.28
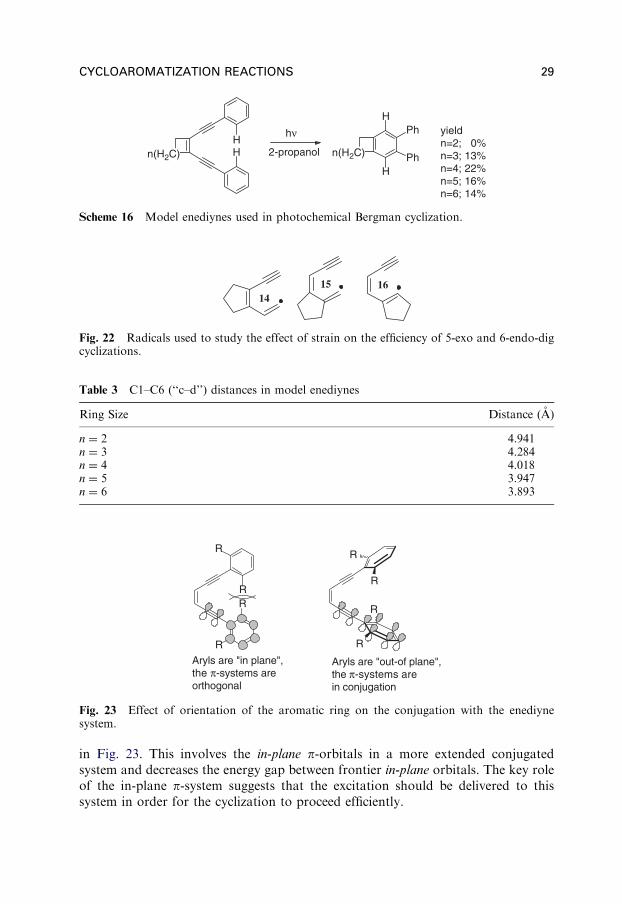
in Fig. 23. This involves the in-plane p-orbitals in a more extended conjugatedsystem and decreases the energy gap between frontier in-plane orbitals. The key roleof the in-plane p-system suggests that the excitation should be delivered to thissystem in order for the cyclization to proceed efficiently.
HH
Ph
Ph
H
H
n(H2C)
hν
2-propanol n(H2C)
yield n=2; 0% n=3; 13% n=4; 22% n=5; 16% n=6; 14%
Scheme 16 Model enediynes used in photochemical Bergman cyclization.
1415 16
Fig. 22 Radicals used to study the effect of strain on the efficiency of 5-exo and 6-endo-digcyclizations.
Table 3 C1–C6 (‘‘c–d’’) distances in model enediynes
Ring Size Distance (A)
n ¼ 2 4.941n ¼ 3 4.284n ¼ 4 4.018n ¼ 5 3.947n ¼ 6 3.893
RR
R
R R
R
R
R
Aryls are "in plane", the π-systems are orthogonal
Aryls are "out-of plane", the π-systems are in conjugation
Fig. 23 Effect of orientation of the aromatic ring on the conjugation with the enediynesystem.
CYCLOAROMATIZATION REACTIONS 29

p-EFFECTS
The most obvious effect on cycloaromatization, as the name implies, is the forma-tion of an aromatic system. By delocalizing electrons in an aromatic ring, the prod-uct gains a high degree of stability, which is reflected in the small endothermicity ofthe Bergman cyclization and the exothermicity of the Myers-Saito cyclization. Sincethe Schmittel and Schreiner cyclizations are not true cycloaromatization reactionsper se, they do not have the beneficial effect of the formation of an aromatic systemand are therefore much are more endothermic than their counterparts.
Product stabilization is much more pronounced when the enediyne or ene-yne-allene starting materials are not already part of an aromatic system, since forming anaromatic system constitutes a much higher degree of stabilization than expanding anaromatic system (Fig. 24). Conjugation of the radical center provides additionalstabilization to the p-radical formed by the Myers-Saito and Schmittel cyclizations.
Both the Schmittel and the Schreiner cyclization do not formally producean aromatic system, Schleyer, Schreiner and co-workers compared these reactionsto the Bergman and Myers-Saito cyclization using standard aromaticity criteria,such as magnetic susceptibility exaltations, aromatic stabilization energy (ASE),and nucleus independent chemical shift (NICS). Interestingly, the degree ofcyclic electron delocalization found in these systems is comparable to that foundin benzene. The effect of benzannelation however is smaller in magnitude, and forSchreiner cyclization it has an opposite effect leading to a slight decrease in reactionendothermicity.
Bergman
Schreiner
Erel.: 25.2 (Erel.: 24.6)
Erel.: 41.0 (Erel.: 37.2)
Erel.: 8.5 (Erel.: 14.4)
Erel.: 41.3 (Erel.: 37.4)
Myers-Saito
Schmittel
Erel.: 18.7 (Erel.: 19.5)
Erel.: 30.0 (Erel.: 25.2)
Erel.: -9.7 (Erel.: -4.5)
Erel.: 12.9 (Erel.: 11.9)
Fig. 24 Relative energies for the transition states and radical products for the differentcyclization pathways (benzannelated systems in parentheses).
I. ALABUGIN ET AL.30

These results, of course, contrast the significant effect of benzannelation inradical-anionic cycloaromatization reactions discussed above12 where not just theformation of a new aromatic cycle but also restoration of aromaticity in the pre-viously existing cycle occurs at the same time in the cyclorearomatization process.
9 Conclusion
The great potential utility of cycloaromatization reactions is matched by theinherent complexity of electronic changes that accompany these processes. Thiscomplexity stems from the presence of two orthogonal p-arrays, both of whichundergo significant changes during the reactions. The asynchronous nature of s- andp-effects opens numerous opportunities to control these processes and will continueto provide challenges for experimentalists and theoreticians alike. Even though someof the general factors controlling the Bergman cyclization are applicable to newlydiscovered cycloaromatization reactions, new phenomena such as communicationof orthogonal orbitals in radical-anionic, radical-cationic and dianioniccycloaromatizaton and cyclorearomatization will continue to stimulate futuredevelopment of this interesting field.
References
1. We are omitting reactions such as the trimerization of acetylene on transition metalcatalysts, even though they could be considered cycloaromatization reactions
2. Bergman, R.G. (1973). Acc. Chem. Res. 6, 253. Roth, W.R., Hopf, H. and Horn, C. (1994). C. Chem. Ber. 127, 1765; Wenthold, P.G.
and Squires, R.R. (1994). J. Am. Chem. Soc. 116, 6401; Davico, G.E., Bierbaum, V.M.,De Puy, C.H., Ellison, G.B. and Squires, R.R. (1995). J. Am. Chem. Soc. 117, 2590
4. Rawat, D.S. and Zaleski, J.M. (2004). Synlett 393; Bhattacharyya, S. and Zaleski, J.M.(2004). Curr. Topics Med. Chem. 4, 1637
5. Basak, A., Mandal, S. and Bag, S.S. (2003). Chem. Rev. 103, 40776. Schreiner, P.R., Navarro-Vazquez, A. and Prall, M. (2005). Acc. Chem. Res. 38, 297. Klein, M., Walenzyk, T. and Konig, B. (2004). Collect. Czech. Chem. Commun. 69, 9458. Galm, U., Hager, M.H., Van Lanen, S.G., Ju, J., Thorson, J.S. and Shen, B. (2005).
Chem. Rev. 105, 7399. Jones, G.B. and Fouad, F.S. (2002). Curr. Pharm. Design 8, 241510. Lewis, K.D., Wenzler, D.L. and Matzger, A.J. (2003). Org. Lett. 5, 2195; Lewis, K.D.,
Rowe, M.P. and Matzger, A.J. (2004). Tetrahedron 60, 719111. Kawatkar, S.P. and Schreiner, P.R. (2002). Org. Lett. 4, 364312. (a) Alabugin, I.V. and Kovalenko, S.V. (2002). J. Am. Chem. Soc. 124, 9052;
(b) Alabugin, I.V. and Manoharan, M. (2003). J. Am. Chem. Soc. 125, 449513. Eshdat, L., Berger, H., Hopf, H. and Rabinovitz, M. (2002). J. Am. Chem. Soc. 124,
3822; Treitel, N., Eshdat, L., Sheradsky, T., Donovan, P.M., Tykwinski, R.R., Scott,L.T., Hopf, H. and Rabinovitz, M. (2006). J. Am. Chem. Soc. 128, 4703
14. Whitlock Jr., H.W., Sandvick, P.E., Overman, L.E. and Reichardt, P.B. (1969). J. Org.Chem. 34, 879
15. Tamao, K., Yamaguchi, S. and Shiro, M. (1994). J. Am. Chem. Soc. 116, 1171516. Yamaguchi, S., Miyasato, M. and Tamao, K. (2003). Chem. Lett. 32, 1104
CYCLOAROMATIZATION REACTIONS 31

17. Wenthold, P.G. (1999). J. Chem. Soc., Perkin Trans. 2, 2357; Hammad, L.A. andWenthold, P.G. (2003). J. Am. Chem. Soc. 125, 10796
18. Navarro-Vazquez, A., Prall, M. and Schreiner, P.R. (2004). Org. Lett. 6, 298119. Clark, A.E., Davidson, E.R. and Zaleski, J.M. (2001). Am. Chem. Soc. 123, 265020. Zimmerman, H.E. and Pincock, J.A. (1973). J. Am. Chem. Soc. 95, 324621. Schreiner, P.R. and Bui, B.H. (2006). Eur. J. Org. Chem. 5, 116222. Galbraith, J.M., Schreiner, P.R., Harris, N.R., Wei, W., Wittkopp, A. and Shaik, S.
(2000). Chem. Eur. J. 6, 144623. Leffler, J.E. (1953). Science 117, 340; Hammond, G.S. (1995). J. Am. Chem. Soc. 77,
33424. Alabugin, I.V. and Manoharan, M. (2003). J. Phys. Chem. A 107, 336325. Prall, M., Wittkopp, A. and Schreiner, P.R. (2001). J. Phys. Chem. 105, 926526. Alabugin, I.V. and Manoharan, M. (2003). J. Am. Chem. Soc. 125, 449527. The only known exception is the fascinating class of radical-anionic and dianionic
cyclizations where p-effects can be activated through MO crossings28. Alabugin, I.V., Manoharan, M., Breiner, B. and Lewis, F. (2003). J. Am. Chem. Soc.
125, 932929. Alabugin, I.V. and Manoharan, M. (2005). J. Am. Chem. Soc. 127, 12583; Alabugin,
I.V. and Manoharan, M. (2005). J. Am. Chem. Soc. 127, 953430. Koga, N. and Morokuma, K. (1991). J. Am. Chem. Soc. 113, 1907–191131. For theoretical analysis of the Myers-Saito cyclization, see: (a) Engel, B. and Hanrath,
M. (1998). J. Am. Chem. Soc. 120, 6356–6361; (b) Schreiner, P.R. and Prall, M. (1999).J. Am. Chem. Soc. 121, 8615–8627
32. In very small macrocycles (7- and 8-membered rings), cyclization is not expected, despitethe very short c–d distance of of 2.5 and 2.6 A, respectively. The ring strain in thecyclized product would simply be too high
33. Nicolaou, K.C., Smith, A.L. and Yue, E.W. (1993). Proc. Natl. Acad. Sci. USA 90, 588134. Snyder, J.P. (1989). J. Am. Chem. Soc. 111, 763035. Magnus, P., Carter, P., Elliott, J., Lewis, R., Harling, R., Pitterna, T., Bauta, W.E. and
Fortt, S. (1992). J. Am. Chem. Soc. 114, 254436. Wandel, H. and Wiest, O. (2002). J. Org. Chem. 67, 38837. Plourde, G.W., Warner, P.M., Parrish, D.A. and Jones, G.B. (2002). J. Org. Chem. 67,
536938. Schreiner, P.R. (1998). J. Am. Chem. Soc. 120, 418439. This notion has been the subject of a recent controversy, which was resolved40. Kraka, E. and Cremer, D. (1994). J. Am. Chem. Soc. 116, 492941. Jafri, J.A. and Newton, M.D. (1978). J. Am. Chem. Soc. 100, 501242. One can argue that the presence of the in-plane C3-C4 bond and the 6-electron out-of-
plane p-system renders the whole molecule non-antiaromatic. Therefore, we use thequotation marks for the term ‘‘antiaromatic region’’. However, use of this term is jus-tified because the lack of the p–p* stabilization along the strong p–p repulsion providesan appealing analogy to antiaromatic molecules
43. (a) Funk, R.L., Young, E.R.R., Williams, R.M., Flanagan, M.F. and Cecil, T.L. (1996).J. Amer. Chem. Soc. 118, 3291; (b) Kaneko, T., Takanashi, M. and Hirama, M. (1999).Angew. Chem. Int. Ed. 38, 1267; (c) Choy, N., Blanco, B., Wen, J., Krishan, A. andRussell, K.C. (2000). Org. Lett. 2, 3761; (d) Jones, G.B., Wright, J.M., Plourde II, G.,Purohit, A.D., Wyatt, J.K., Hynd, G. and Fouad, F. (2000). J. Amer. Chem. Soc. 122,9872
44. Rawat, D.S. and Zaleski, J.M. (2004). Synlett 393; Bhattacharyya, S. and Zaleski, J.M.(2004). Curr. Topics Med. Chem. 4, 1637; Basak, A., Mandal, S. and Bag, S.S. (2003).Chem. Rev. 103, 4077; as wel l asKonig, B. (2000). Eur. J. Org. Chem. 12, 381
45. Warner, B.P., Millar, S.P., Broene, R.D. and Buchwald, S.L. (1995). Science 269, 814
I. ALABUGIN ET AL.32

46. Bhattacharyya, S., Pink, M., Baik, M. and Zaleski, J.M. (2005). Angew. Chem. Int. Ed.44, 592
47. Bhattacharyya, S., Dye, D.F., Pink, M. and Zaleski, J.M. (2005). Chem. Commun. 5295;For a review on metal-bound enediynes, see: Rawat, D.S. and Zaleski, J.M. (2004).Synlett 3, 393
48. Kim, C.-S. and Russell, K.C. (1998). J. Org. Chem. 63, 8229; Choy, N. and Russell,K.C. (1999). Heterocycles 51, 13; Kim, C.-S. and Russell, K.C. (1999). Tetrahedron Lett.40, 3835; Kim, C.-S., Diez, C. and Russell, K.C. (2000). Chem. Eur. J. 6, 1555
49. Alabugin, I.V., Manoharan, M. and Kovalenko, S.V. (2002). Org. Lett. 4, 111950. Eliel, E.L., Wilen, S.H. and Doyle, M.P. (2001). Basic Organic Stereochemistry, p. 459,
Wiley-Interscience, New York51. Zeidan, T., Kovalenko, S.V., Manoharan, M. and Alabugin, I.V. (2006). J. Org. Chem.
71, 96252. Zeidan, T., Manoharan, M. and Alabugin, I.V. (2006). J. Org. Chem. 71, 95453. Osinsky, S.P., Levitin, I.Y., Bubnovskaya, L.N., Ganusevich II, Sigan, A.L., Tsykalova,
M.V. and Zagorujko, L.I. (1999). Exp. Oncol. 21, 216; Tannock, I.F. and Rotin, D.(1989). Cancer Res. 49, 4373
54. Kraka, E. and Cremer, D. (2000). J. Am. Chem. Soc. 122, 824555. Hoffner, J., Schottelius, J., Feichtinger, D. and Chen, P. (1998). J. Am. Chem. Soc. 120,
376–38556. Bent, H.A. (1961). Chem. Rev. 61, 27557. (a) Alabugin, I.V. and Manoharan, M. (2007). J. Comp. Chem. 28, 373; (b) Additional
examples of rehybridization are analyzed in Alabugin, I.V., Manoharan, M., Buck, M.and Clark, R.J. (2007) Theochem, 813, 21–27
58. For an analysis of rehybridization effects in supramolecular chemistry, see: Alabugin,I.V., Manoharan, M., Peabody, S. and Weinhold, F. (2003). J. Am. Chem. Soc. 125,5973; Alabugin, I.V., Manoharan, M. and Weinhold, F. (2004). J. Phys. Chem. A 108,4720
59. Prall, M., Wittkopp, A., Fokin, A.A. and Schreiner, P.R. (2001). J. Comp. Chem. 22,1605
60. Wenk, H.H., Balster, A., Sander, W., Hrovat, D.A. and Borden, W.T. (2001). Angew.Chem. Int. Ed. Engl. 40, 2295
61. Jones, G.B. and Warner, P.M. (2001). J. Am. Chem Soc. 66, 213462. The starting points for both C1–C5 and C1–C6 cyclizations are characterized using
C1–C6 distances to stress that starting point for both cyclizations is the same enediyneradical-anion. However, for transition states and products of C1–C5 and C1–C6cyclizations, the respective incipient bond length (C1–C5 or C1–C6) was used as thereaction coordinate
63. (a) Hoffman, R., Imamura, A. and Hehre, W.J. (1968). J. Am. Chem. Soc. 90, 1499;Hoffman, R. (1971). Acc. Chem. Res. 4, 1; (b) Paddon-Row, M.N. (1982). Acc. Chem.Res. 15, 245; (c) Gleiter, R. and Schafer, W. (1990). Chem. Res. 23, 369–375;(d) Brodskaya, E.I., Ratovskii, G.V. and Voronkov, M.G. (1993). Russ. Chem. Rev. 62,975
64. Hughes, T.S. and Carpenter, B.K. (1999). J. Chem. Soc., Perkin Trans. 2, 229165. Li, H., Petersen, J.L. and Wang, K.K. (2003). J. Org. Chem. 68, 551266. Feng, L., Kumar, D., Birney, D.M. and Kerwin, S.M. (2004). Org. Lett. 6, 2059
CYCLOAROMATIZATION REACTIONS 33

This page intentionally left blank

N-Acyloxy-N-alkoxyamides – structure,properties, reactivity and biological activity
STEPHEN A. GLOVER
School of Science and Technology, University of New England, Armidale, NSW 2350,
Australia
1 Introduction 35General 35Background 37
2 Synthesis 393 Structure 43
General 43Theoretical structures 44X-ray structures 47Spectroscopic properties 51
4 Chemical reactivity 59Solvolysis studies – AAl1 reactivity 60Nucleophilic substitution reactions – SN2 reactivity 70Thermal decomposition reactions 90
5 Biological activity of N-acyloxy-N-alkoxyamides 97Mutagenicity in the Ames Salmonella/microsome assay 97Anticancer activity of N-acyloxy-N-alkoxyamides 115
6 Conclusions 115Acknowledgements 116References 117
1 Introduction
GENERAL
The properties of amides are determinants of the structure and characteristics of awide range of molecules and particularly those of peptides and proteins.1 Univer-sally, amides and amide linkages are characterised by a nitrogen that is largely sp2
hybridised and in which the lone pair resides in a 2pz orbital. As a consequence thereis a strong interaction between the amide nitrogen and the carbonyl. While this hastraditionally been described as a resonance delocalisation involving I and II inFig. 1a, contemporary views strongly favour a third resonance contributor III inwhich there is s back donation from carbon to the sp2 hybridised nitrogen.2,3
35
ADVANCES IN PHYSICAL ORGANIC CHEMISTRYVOLUME 42 ISSN 0065-3160 DOI: 10.1016/S0065-3160(07)42002-0
r 2008 Elsevier Inc.All rights reserved

According to Wiberg, the p donation from nitrogen is best described as a HighestOccupied Molecular Orbital (HOMO)—Lowest Unoccupied Molecular Orbital(LUMO) interaction in which case there is little charge transfer to oxygen since thiscontributes weakly to the LUMO (Fig. 1b).2 Either way, the predominant inter-action is between the nitrogen lone pair and the carbon of the carbonyl bond and thedouble bond character is the prime cause of both planarity at nitrogen and therestricted rotation about C–N bonds in amides. Properties of hydroxamic esterslargely mirror those of amides. The attachment of one oxygen to the amide nitrogendoes not dramatically alter the degree of pyramidality at nitrogen or the extent oflone pair delocalisation/interaction with the carbonyl.4–8
A good number of amides are not planar at nitrogen owing to twistingdue to steric interactions (Fig. 2a),9–17 configurational properties that close theangles at nitrogen (Fig. 2b)11,18–20 or lactams that lock the nitrogen lone pair outof alignment with the carbonyl 2pz orbital (Fig. 2c).21–24 The result in all casesis a disconnection between the nitrogen lone pair and the amide carbonyl. Inthese cases, the amide nitrogen tends towards sp3 hybridisation and the C–Nbonds assume single bond character. All these amides constitute the class of‘‘twisted amides’’ although in Fig. 2b, twisting may be the result of lone pair dis-connection rather than the cause thereof. However, they can all best be regarded asN-acylamines.
Kirby’s configurationally rigid 1-azaadamantan-2-one 1 is the extreme of thisclass.21–23,25 The nitrogen properties of this lactam are clearly amine-like while thecarbonyl is ketonic in all respects. Twisted amides undergo rapid hydrolysis orreduction22,25–30 and exhibit enhanced reactivity.31
C
O
NC
O
N
I II
R CO
N
(b)(a)
C
O
N
III
NHOMO
COLUMO
Fig. 1 (a) Resonance and (b) HOMO–LUMO interaction in simple amides.
CO
NR2
R1
(a)
R3R3C
O
N
(b)
RN
(c)
O
Fig. 2 (a) A sterically twisted amide; (b) an angularly constrained amide; (c) a twistedlactam.
S.A. GLOVER36

N O
1
H
O
N
XY
a X=Y=ORb X=Cl,Y=ORc X=OR,Y=NR2d X=Y=Halogen
2
R3 CO
NO OC
O
R2
R1
3
The author has identified another class of amides that possess pyramidal nitrogensbut are devoid of any steric or configurational imposition that results in enforcedtwisting about the amide linkage. ‘‘Anomeric amides’’, are defined as amidesthat bear two heteroatoms at the amide nitrogen 2.32 There is ample evidence ofpyramidality in N,N-dioxoamides 2a, N-halo-N-oxoamides 2b and N-amino-N-oxoamides 2c. N,N-dihaloamides 2d may well fall into the same class. In all ofthese, the amide nitrogen responds to the collective electronegativity of the subs-tituents by rehybridising from sp2 to sp3. This enables an electron density distri-bution that better satisfies the electron demand of the nitrogen substituents, X andY. The configurational change results in smaller angles at nitrogen and reducedp-character of the lone pair orbital with attendant disconnection from the amidecarbonyl as evidenced by spectroscopic properties, radically reduced amide iso-merisation barriers, reactivity patterns and theoretical attributes.6,32–36
This review will cover the structural, physical, spectroscopic, chemical and bio-logical properties of the most widely studied class of these anomeric amides, theN-acyloxy-N-alkoxyamides 3.
These not only exhibit all the structural and spectroscopic characteristics of an-omeric amides 2, their reactivity patterns embrace a range of reactions at the amidenitrogen that in many respects parallel those at a saturated carbon.37–46 In addition,they are mutagenic as well as DNA damaging agents that have anticancer capa-bility.37,38,40,46–50 To date nearly a hundred of these have been synthesised in ourlaboratory while, recently, a range of urea and urethane analogues have also beenreported.51–53
BACKGROUND
Aromatic amines 4 are metabolised in vivo by cytochrome P450 mediated oxidationto phenolic and hydroxylamine derivatives 5 and 6. Phase II conjugation of thelatter with PAPS or acyl transferase results in formation of the sulfuric or acetic acidesters 7. Nitrogen conjugation to give the N-acetyl analogues is also possible(Scheme 1).54–65
These metabolites have been shown to produce arylnitrenium ions 8 undersolvolytic conditions and McClelland and Novak have demonstrated, respectively
N-ACYLOXY-N-ALKOXYAMIDES 37
by Laser Flash Photolysis (LFP) and azide clock methods, that the lifetimes of thesenitrenium ions are discriminating; those aromatic amines that produce long-livedarylnitrenium ions are generally more carcinogenic and the rates of hydration ofthese ions in water are critical.66–85 Arylnitrenium ions are highly delocalised andsteric hindrance to hydration at the 4-position seems to be critical in determiningthe lifetimes in solution. Nitrenium ions from carcinogenic 4-biphenylamine 9,N-acetyl-2-aminofluorene 10, 2-naphthylamine 11 and benzidine 12 have beenshown to have much longer lifetimes than other, less harmful arylamines and theirreaction with guanine to afford C-8 (G-C8) adducts 13 is the primary cause ofmutations of DNA.86–89
NHAcNH2
NH2NH2NH2
9 10
11 12
NH
N
NO
NH2
N
Deoxyribose
NAr
(Ac)H
13
In 1984, we demonstrated that N-alkoxy-N-acyl nitrenium ions 15 could be gen-erated by the reaction of N-alkoxy-N-chloroamides 14 with Lewis acids such as Ag+
and Zn2+ and used these to form heterocycles by intramolecular aromatic substi-tution reactions (Scheme 2).90 In this manner, several novel N-acyl-3,4-dihydro-2,1-benzoxazines 16a and N-acyl-4,5-dihydro-(1H,3H)-2,1-benzoxazepines 16b weremade. Subsequent work91,92 and that of Kikugawa93–96 produced numerous syn-theses involving alkoxynitrenium ions including formation of natural products.97–99
In a seminal theoretical paper, we showed that, at the Modified Neglect ofDifferential Overlap (MNDO) level, phenylnitrenium ions 17 as well as NH2, PH2,
NH
OH
NH2
Acetylase(PAPS)
P450 NH2
Ar Ar Ar
HO4 5 6
N
OAc
H (Ac)NH (Ac)
Ar Ar
(SO3–)
78
Scheme 1.
S.A. GLOVER38

SH and OH-substituted nitrenium ions 19a–d and their N-formyl analogues havesinglet ground states with a similar degree of p-overlap with the electron-deficientnitrogen (p-bond orders 40.9). Ab initio calculations (HF/6-31G*) on heteroatom-substituted nitrenium ions showed that all the heteroatoms strongly stabilised thesinglet state relative to their triplet state.100 Extensive double bond character inhydroxynitrenium ion was also determined by Schwarz and coworkers.101
NH (CHO)
17
XN
H (CHO)
19
a X=NH2b X=PH2c X=SHd X=OH
NR
18
For phenylnitrenium ions, we85 and others 72,78,102–104 have computed that there isextensive positive charge delocalisation into the aromatic ring and arylnitreniumions are best described as 4-imino-2,5-cyclohexadienyl-1-yl carbenium ions 18.
From appraisal of their respective resonance stabilisation, arylnitrenium ions andalkoxynitrenium ions should form with similar facility. On account of the fact thatN-acetoxy-N-acetyl arylamines 7 are penultimate carcinogens in the metabolism ofaromatic amines, N-acyloxy-N-alkoxyamides 3 were designed to test their potentialas DNA-damaging agents.
2 Synthesis
N-Acyloxy-N-alkoxyamides 20 are synthesised from N-chlorohydroxamic esters 22by replacement of chlorine by a carboxyl group (Scheme 3). Initially we employedsilver acetate in anhydrous ether to make N-acetoxy derivatives.47 However, mosthave been made using sodium carboxylates in dry acetone by analogy withFinkelstein chemistry.5,38–40,42,43,46,48,49,105 The reactions can be monitored conven-iently by thin layer chromatography and N-acyloxy-N-alkoxyamides generally
(CH2)nO
NCl
O
R'
(CH2)nO
N
O
R'
(CH2)nO
N
O
R'
Lewis acids
14 15 16a n=2b n=3
Scheme 2.
N-ACYLOXY-N-ALKOXYAMIDES 39

oxidise to brown spots on silica plates. Yields vary depending upon the stability ofthe N-acyloxy-N-alkoxyamide to hydrolysis but in most instances, conversion isclean, or minor impurities (such as the corresponding alkyl esters) can be removedusing centrifugal chromatography.
Scheme 3 outlines synthetic strategies for the introduction of a range of subs-tituents on the amide, alkoxyl and acyloxyl side chains. Hydroxamic esters 23
are readily synthesised from potassium salts of hydroxamic acids 21 according toCooley et al.7 or by condensation of the corresponding acid chloride 24 with analkoxyamine.
This group has synthesised a wide range of N-acyloxy-N-alkoxyamides that canbe categorised as follows:
1. N-Acetoxy-N-alkoxybenzamides 25, with variation in the alkoxyl side chain2. N-Acetoxy-N-butoxyarylamides 26, with variation on the benzamide ring3. N-Acetoxy-N-arylmethyloxybenzamides 27, with variation on the benzyloxyl side
chain4. N-Aroyloxy-N-benzyloxybenzamides 28, with variation on the ester side chain5. N-Acyloxy-N-butoxyamides 29, with variable amide and acyloxyl side chains6. N-Acyloxy-N-alkoxyacetamides 30, with variable alkoxyl and acyloxyl side
chains7. N-Benzoyloxy- and N-acetoxy-N-benzyloxybenzamides 31–33, bearing one or
more para-tert-butyl substituents8. N-Acyloxy-N-alkoxyamides 34 and 35 where two amide groups are tethered by
alkoxyl or acyl chains9. N-Acyloxy-N-alkoxyamides 36 and 37 of a specialised nature
N
OAc (COR2)
O
R3
R1O
HN O
R3R1O
N O
R3R1O
ClButOCl
HN O
R3KO
R1Briii
i AgOAc anhydrous diethyl etherii NaOAc (NaOCOR2) anhydrous acetone
20 21
22 23
Cl O
R3
24
R1ONH2
Et3N
Scheme 3.
S.A. GLOVER40
O
N OR
AcO
O
N OBu
AcO
XO
N OAcO
Y
25 26 27
a R=Etb R=Prc R=Bud R=Pentyle R=Octylf R=Pri
g R=Bui
h R=Penti
i R=But
j R=2-butyl
a X=Hb X=MeOc X=Phd X=Mee X=Clf X=Brg X=NO2h X=m-NO2i X=But
a Y=Hb Y=MeOc Y=PhOd Y=Phe Y=Mef Y=Clg Y=Brh Y=NO2i Y=But
O
N OO
O
Z
R3O
N OBu
OO
R2
28 29
a Z=Hb Z=MeOc Z=Phd Z=Mee Z=Clf Z=CHO
g Z=CNh Z=NO2i Z=But
j Z=CF3k Z=m-NO2l Z=m-MeO
a R2=Ph, R3=Etb R2=Ph, R3=Pri
c R2=Ph, R3=But
d R2=Ph, R3=neopentyle R2=Ph, R3=1-Adamantylf R2=R3=Phg R2=Pr, R3=Phh R2=Pri, R3=Phi R2=(S)-2-butyl, R3=Phj R2=Neopentyl, R3=Ph
k R2=1-Ad, R3=Phl R2=But, R3=Ph
m R2=Me, R3=2-naphthyln R2=Me, R3=fluoren-1-ylo R2=Me, R3=9,10-anthra-
quinone-2-ylp R2=Me, R3=pyren-1-ylr R2=2-naphthyl, R3=Phs R2=2,6-dimethylphenyl, R3=Pht R2=3,5-dimethylphenyl, R3=Phu R2=Me, R3=3,5-dimethylphenylv R2=hexyl, R3=But
w R2=hexyl, R3=Phx R2=5-hexen-1-yl, R3=But
y R2=5-hexen-1-yl, R3=Ph
CH3
O
N OR1
OO
R2
a R1=Bu, R2=Meb R1=Bu, R2=Phc R1=Bn, R2=Med R1=Bn, R2=Phe R1=Bu, R2=2-Npf R1=CH2(1-Np),R2=Meg R1=CH2(2-Np),R2=Meh R1=(CH2)2(2-Np),R2=Mei R1=(CH2)3(2-Np),R2=Mej R1=Bu, R2=fluoren-1-ylk R1=Bu, R2=pyren-1-yll R1=Bu, R2=CH2(pyren-1-yl)
m R1=Bu, R2=CH2(2-naphthyl)n R1=Bu, R2=(CH2)2(2-naphthyl)
30
N-ACYLOXY-N-ALKOXYAMIDES 41
O
NO
O
CH3
OR3
a R1=R2=Buta R1=R2=H, R3=But
b R1=But , R2=R3=Hc R1=R2=But, R3=H;d R1=H, R2=R3=But;e R1=R3=But, R2=H;f R1=R2=R3=But
O O
But But
a Y=H;b Y=But,
R1O
NO
Y
31
O
N OO
O
R3R1
R2
32 33
O
N O(CH2)3
AcO34
2
35
O
NO
OAc
Ph(CH2)n
O
NO
OAc
Ph
a n=7b n=8
But
O
NO
O
CH3
O
R1
36
a R1=Hb R1=Cl
O
NO
O
CH3
O
a
b
c
d
37
a a,b,=Me, c,d=Hb a,b=H, c,d=Mec a,b,d=H, c=Med a=Me, b,c,d=H
Recently, Shtamburg et al. synthesised a variety of N-acyloxy-N-alkoxyureas 38,N-acyloxy-N-alkoxycarbamates 39 as well as N-acetoxy-N-ethoxybenzamide 25a byan analogous procedure using appropriate sodium carboxylates in CH3CN.51–53
Compound 39h was synthesised from ethyl N-butoxy-N-chlorocarbamate using ourNaOAc in acetone method.106
S.A. GLOVER42

N O
NR32
R1O
38
N
OCOR2OCOR2
O
OR3R1O
39
aR1=R2=R3=MebR1=R3=Me, R2=EtcR1=R3=Me, R2=Pri
dR1=R3=Me, R2=PheR1=Prn, R2=R3=Mef R1=Prn, R2=Me, R3=Me,HgR1=Prn, R2=Ph, R3=Me,HhR1=Et, R2=Me, R3=CH2(1-Np),Hi R1=Et, R2=Me, R3=Hj R1=Bu,R2=Me, R3=HkR1=n-dodecyl, R2=Me, R3=H
a R1=R2=R3=Meb R1=R3=Me, R2=Etc R1=n-octyl,R2=R3=Med R1=R2=Me, R3=Ete R1=Me, R2=Ph, R3=Etf R1=Pri, R2=Me, R3=Etg R1=Me, R2=4-ClC6H4, R3=Meh R1=Bu, R2=C6H4, R3=Et
3 Structure
GENERAL
N-Acyloxy-N-alkoxyamides are archetypal anomeric amides. The combined electro-negativity of the alkoxyl and acyloxyl oxygens strongly alters the geometry at theamide nitrogen and the attendant pyramidality manifests itself in radically reducedor negligible amide conjugation; the nitrogen lone pair is in an sp3 rather than a 2pzorbital (Fig. 3a). As a consequence, relative to amides and hydroxamic esters, theyshould have much longer N–C(O) bonds, low barriers to E–Z isomerisation andhigher carbonyl stretch frequencies in their IR spectra. However the barrier tonitrogen inversion is likely to be low since, in the planar transition state for inver-sion, the lone pair can interact with the carbonyl (Fig. 3b). In contrast to N-acyloxy-N-alkoxyamides, dialkoxyamines, which are also pyramidal at nitrogen, haveabnormally high nitrogen inversion barriers. Here the planar inversion transitionstate is destabilised by sp2 hybridisation, which necessitates shorter N–O bonds, aswell as lone pair repulsion.107–111
OR
N
OAc
CRO
NAcO
CR
O
OR
sp3N
poor overlap low barrier
(b)
C N
OAc
OR
‡O
R
(a)
Fig. 3 (a) Poor p overlap in N-acyloxy-N-alkoxyamides; (b) stabilisation of the inversiontransition state.
N-ACYLOXY-N-ALKOXYAMIDES 43

In addition to these unusual amide characteristics, the bisoxo substitution and thesp3 hybridisation at nitrogen results in a strong anomeric interaction between nO, thealkoxyl oxygen lone pair and the s*N–OAc bond (Fig. 4a). This is a common featureof all bisheteroatom-substituted amides that we have studied.32 In the case ofN-acyloxy-N-alkoxyamides this anomeric interaction is likely to be strongest of thetwo possible anomeric effects; the s*N–OAc is lower in energy than s*N–OR while thealkoxyl oxygen p-type lone pair would be higher in energy than that of the acyloxylether oxygen (Fig. 4b). As illustrated in Fig. 4c, to maximise such interactions, theoptimum geometry would require twist angles about the donor oxygen–nitrogenbond of around 901.
Computed structures for model N-acyloxy-N-alkoxyamides, X-ray diffractiondata for a number of congeners, as well as spectroscopic evidence fully support thesequalitative arguments.
THEORETICAL STRUCTURES
Both AM1 calculations on N-acetoxy-N-methoxybenzamide38 and ab initio 6-31G*calculations on N-formyloxy-N-methoxyformamide 405,45 predict a stronglypyramidal nitrogen. Its lowest energy conformation at HF/6-31G* is depicted inFig. 5a while structural data are provided in Table 1 together with that forN-methoxyformamide 41.
H
O
NH2H
O
N H
O
NHOCH3
CHO
40 41 42
OCH3
The conformation in which the amide carbonyl and the formloxy groups are syn
and both methyl and formyloxy carbonyl groups are exo to the nitrogen pyramid islowest in energy. The average angle at nitrogen was 110.31 while dihedral angles givea Winkler–Dunitz amide distortion index of wN ¼ 58.5 (Table 1).112,113 The N–CO(1.405 A) and amide CQO (1.178 A) bond lengths are respectively longer and
RCOR
90°
OAc
ON
AcO
ORO
σ*N—OAc
nO
σ*N—OAc
nO
σN—OAc
E
(b)(a) (c)
nOAc
σN—OR
σ*N—OR
Fig. 4 (a) Anomeric overlap in N-acyloxy-N-alkoxyamides; (b) lone pair stabilisationthrough an nO–s*N–OAc interaction; (c) optimum conformation for anomeric overlap in N-acyloxy-N-alkoxyamides.
S.A. GLOVER44
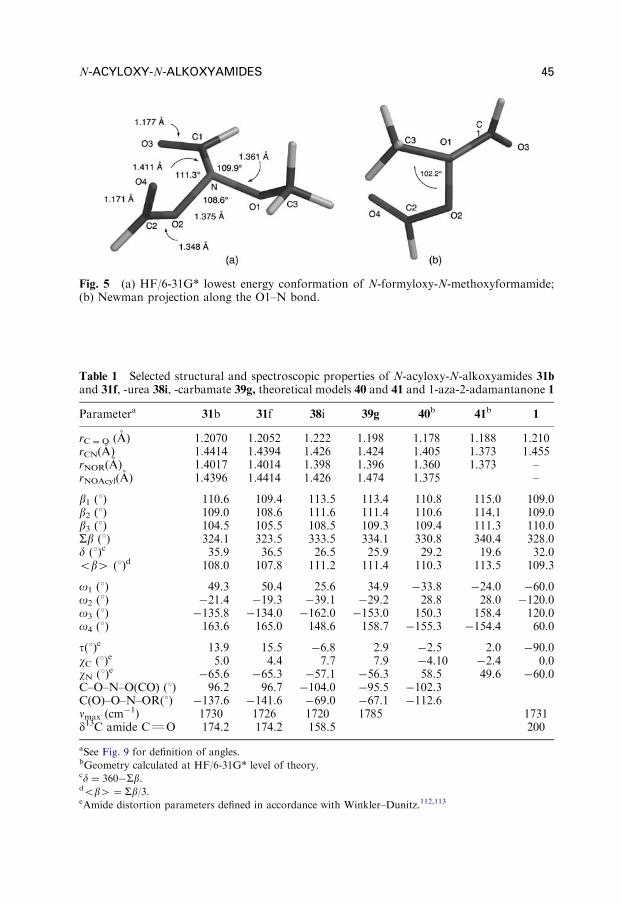
Table 1 Selected structural and spectroscopic properties of N-acyloxy-N-alkoxyamides 31band 31f, -urea 38i, -carbamate 39g, theoretical models 40 and 41 and 1-aza-2-adamantanone 1
Parametera 31b 31f 38i 39g 40b 41b 1
rC ¼ O (A) 1.2070 1.2052 1.222 1.198 1.178 1.188 1.210rCN(A) 1.4414 1.4394 1.426 1.424 1.405 1.373 1.455rNOR(A) 1.4017 1.4014 1.398 1.396 1.360 1.373 –rNOAcyl(A) 1.4396 1.4414 1.426 1.474 1.375 –
b1 (1) 110.6 109.4 113.5 113.4 110.8 115.0 109.0b2 (1) 109.0 108.6 111.6 111.4 110.6 114.1 109.0b3 (1) 104.5 105.5 108.5 109.3 109.4 111.3 110.0Sb (1) 324.1 323.5 333.5 334.1 330.8 340.4 328.0d (1)c 35.9 36.5 26.5 25.9 29.2 19.6 32.0ob4 (1)d 108.0 107.8 111.2 111.4 110.3 113.5 109.3
o1 (1) 49.3 50.4 25.6 34.9 �33.8 �24.0 �60.0o2 (1) �21.4 �19.3 �39.1 �29.2 28.8 28.0 �120.0o3 (1) �135.8 �134.0 �162.0 �153.0 150.3 158.4 120.0o4 (1) 163.6 165.0 148.6 158.7 �155.3 �154.4 60.0
t(1)e 13.9 15.5 �6.8 2.9 �2.5 2.0 �90.0wC (1)e 5.0 4.4 7.7 7.9 �4.10 �2.4 0.0wN (1)e �65.6 �65.3 �57.1 �56.3 58.5 49.6 �60.0C–O–N–O(CO) (1) 96.2 96.7 �104.0 �95.5 �102.3C(O)–O–N–OR(1) �137.6 �141.6 �69.0 �67.1 �112.6nmax (cm
�1) 1730 1726 1720 1785 1731d13C amide CQO 174.2 174.2 158.5 200
aSee Fig. 9 for definition of angles.bGeometry calculated at HF/6-31G* level of theory.cd ¼ 360�Sb.dob4 ¼ Sb/3.eAmide distortion parameters defined in accordance with Winkler–Dunitz.112,113
Fig. 5 (a) HF/6-31G* lowest energy conformation of N-formyloxy-N-methoxyformamide;(b) Newman projection along the O1–N bond.
N-ACYLOXY-N-ALKOXYAMIDES 45

shorter than those computed for N-methoxyformamide (1.373 A and 1.188 A)reflecting significantly decreased interaction with the amide carbonyl. TheCH3–O–N–OCHO dihedral angle was 102.31 at the HF/6-31G* level reflecting astrong nO–s*N–OCHO anomeric overlap in this direction (Fig. 5b). The dihedral angleHCO–O–N–OMe of 112.61 reflects a poorer alignment for the alternativenOCHO–s*N–O interaction. The anomeric overlap in the preferred conformationalso results in a shorter CH3O–N bond in 40 (1.360 A) than that found formethoxyformamide 41 (1.373 A) despite the reduced sp3 character in the latter(ob4 ¼ 113.5, wN ¼ 49.61). However, a more meaningful comparison would bewith O-methylhydroxylamine, which like 40 at the HF/6-31G* level, is sp3 hybrid-ised at nitrogen with an N–O bond length of 1.399 A.
Gas-phase vibrational frequencies can be computed using molecular orbitalmethods,114,115 which can also provide structural data about transition states orunstable conformations. Fig. 6 gives HF/6-31G* gas-phase carbonyl and C–Nstretch frequencies for ground-state and fully twisted conformations of N-formyl-oxy-N-methoxyformamide (Fig. 6a), N-methoxyformamide (Fig. 6b) and form-amide (Fig. 6c). Fully twisted formamide and N-methoxyformamide have carbonylstretch frequencies that are 29 and 47 cm�1, respectively, higher than the planar ornear-planar ground state structures.� The C–N stretch frequencies are however re-duced concomitantly by 147 and 208 cm�1, respectively, indicating a significant lossof lone pair overlap in the twisted forms. The carbonyl vibrational frequencies for
NH HOH
N
H
H
H O
NH OMe
HO
N
H
MeO
H O
N
MeO O2CH
OH
N
HCO2
MeO
O H
CO 1784 (1.193)CN 1231 (1348)t=0˚; <b> =120°-169.887003
CO 1796 (1.188)CN 1229 (1.373)t=2.0°; <b> = 113.5°-284.349123
CO1838(s)/1819(a) (1.178)CN 1232 (1.405)t = 0.5°; <b> =110.3°-472.845200
CO 1813 (1.183)CN 1083 (1.427)t = 90°; <b> =107.5°-169.858270
CO 1843 (1.178)CN 1020 (1.436)t = 90°; <b> =105.2°-284.324215
CO 1852(s)/1818(a) (1.171)CN 1182 (1.462)t = 90°; <b> = 105.8°-472.824551
(c)(b)
N
HCO2
MeO
H O
CO 1861(s)/1824(a) (1.172)CN 1190 (1.452)t = 90°; <b> = 104.2°-472.833170
(a)
Fig. 6 Amide CQO and C–N vibrational frequencies (cm�1) and bond lengths (A; inparentheses), twist angles (t), average angles at nitrogen ob4 computed at HF/6-31G* levelfor ground state and orthogonal conformations of (a) N-formyloxy-N-methoxyformamide 40,(b) N-methoxyformamide 41 and (c) formamide 42. Energies (Hartrees) at B3LYP/6-31G*//HF/6-31G* level.
�The frequency change is small when compared to the difference between CQO (1700 cm�1) and C–O
(1100 cm�1) stretches in line with the prevailing view that in planar amides relatively little charge is
transferred to oxygen. The changes in frequency represent a small stiffening of the carbonyl bond at best.
S.A. GLOVER46

both twisted forms of N-formyloxy-N-methoxyformamide 40 are strongly coupledbut the averages of both the symmetrical and asymmetrical stretch frequencies (1835and 1842 cm�1) are comparatively similar to that of the ground state conformation(1828 cm�1) (Fig. 6a). In this case, the C–N stretch frequency is reduced by only 50or 42 cm�1. Thus, when compared with formamide or N-methoxyformamide, boththe carbonyl stretch and the C–N bond stretch frequencies are much less sensitive tothe orientation of the sp3 lone pair. The small changes in the frequencies and bondlengths upon twisting the lone pair of N-formyloxy-N-methoxyformamide out ofconjugation (Fig. 6a) would suggest that, at HF/6-31G* level in the gas phase atleast, some residual interaction is lost. However, it should be borne in mind thatchanges in the degree of pyramidality will also affect bond lengths. While the C–Nbond length in the ground state of 40 (1.405 A) is of the same order as twistedformamide 42 (1.427 A), the bond lengthens by 0.005–0.006 A in the twisted forms of40. To a large degree this must be due to the tighter angles at nitrogen, which resultin orbitals with greater p character.
The extent of nitrogen lone pair-carbonyl overlap is also reflected in the barriersto E–Z isomerisation. B3LYP/6-31G*//HF/6-31G* barriers to rotation in form-amide (18.0 kcalmol�1), methoxyformamide (15.6 kcalmol�1) are similar in con-currence with our results in an earlier study using B3LYP/6-31G(D) optimisedgeometries.6 However both barriers are significantly higher than the smallest barrierfor isomerisation of formyloxymethoxyformamide (7.5 kcalmol�1; nitrogen lonepair and carbonyl syn). Though the nitrogen of the hydroxamic ester is computed tobe significantly pyramidal (wN ¼ 49.6), the similarity in IR stretch frequencies andisomerisation barriers to those of the formamide confirm that the impact of oneoxygen substituent upon amide characteristics is far smaller than that brought aboutby bisoxo-substitution.
The calculated variations in CQO and C–N vibrational frequencies for form-amide (+29 and �147 cm�1, respectively) upon twisting the lone pair into the OCNplane are in accordance with changes in bond lengths (�0.001 and+0.008 Arespectively) and reflect the contemporary view that p-donation to the carbonylcarbon in the planar form does not result in an equivalent loss in CQO pi-bondcharacter.2 Similar results are found for N-methoxyformamide. The small differ-ences in CQO bond lengths between planar (�sp2 N) and twisted (sp3 N) form-amide and methoxyformamide, as well as the similarity in CQO bond length of allthree amides in the ground state (sp2 N in 42, sp3 N in 40) also lend support toWiberg’s HOMO–LUMO theory of amide resonance.
X-RAY STRUCTURES
X-ray data are available for several N-acyloxy-N-alkoxyamides 31b and 31f,5 anN-acyloxy-N-alkoxyurea 38i and an N-acyloxy-N-alkoxycarbamate 39g.51 Thestructure of all four is dominated by pyramidality at the amide nitrogen. Whileamides are typically planar or close to planar with average angles at nitrogen close
N-ACYLOXY-N-ALKOXYAMIDES 47




















