
Strategies and tools for studying microglial-mediated synapse ...
Upload
independentCategory
view
2download
0
U.S. government work not protected by U.S. copyright The FASEB Journal express article 10.1096/fj.05-4578fje. Published online October 11, 2005. CpG-containing oligodeoxynucleotide promotes microglial cell uptake of amyloid β 1-42 peptide by up-regulating the expression of the G-protein-coupled receptor mFPR2 Pablo Iribarren,* Keqiang Chen,* Jinyue Hu,* Wanghua Gong,† Edward H. Cho,‡ Stephen Lockett,‡ Badarch Uranchimeg,§ and Ji Ming Wang*
*Laboratory of Molecular Immunoregulation, Center for Cancer Research, National Cancer Institute at Frederick, Frederick, Maryland 21702 USA; †Basic Research Program, ‡Image Analysis Laboratory and §Developmental Therapeutics Program, Tumor Hypoxia Laboratory, SAIC-Frederick, Frederick, Maryland 21702 USA
Corresponding author: Ji Ming Wang, Laboratory of Molecular Immunoregulation, Center for Cancer Research, National Cancer Institute at Frederick, Building 560, Room 31-40, Frederick, MD 21702-1201. E-mail: [email protected]
ABSTRACT
Human G protein-coupled formyl peptide receptor like 1 (FPRL1) and its mouse homologue murine formyl peptide receptor 2 (mFPR2) mediate the chemotactic activity of amyloid β 1-42 (Aβ42), a key pathogenic peptide in Alzheimer’s disease (AD). Since mFPR2 is up-regulated in mouse microglia by lipopolysaccharide (LPS), a Toll-like receptor 4 ligand, we investigated the capacity of CpG-containing oligodeoxynucleotide (ODN), a Toll-like receptor (TLR) 9 ligand, to regulate the expression of mFPR2 in mouse microglia. CpG ODN markedly enhanced the expression and function of mFPR2 in microglial cells, which exhibited increased chemotactic responses to mFPR2 agonists, including Aβ42. The effect of CpG ODN is dependent on activation of p38 MAPK. Further studies showed that CpG ODN-treated microglia increased their capacity to endocytose Aβ42 through mFPR2, as this process was abrogated by pertussis toxin, a Gi protein inhibitor, and W peptide, another potent mFPR2 agonist. Our results suggest that TLR9 may play an important role in promoting microglial recognition of Aβ42, thus affecting the pathogenic process of AD.
Key words: Alzheimer’s disease • chemotaxis • inflammation • phagocytosis • TLR9
lzheimer’s disease (AD) is the most common cause of dementia characterized by progressive neurodegeneration (1). Pathological features in the brain parenchyma of AD patients include focal deposits of fibrillary amyloid β (Aβ) peptides in blood vessel wall
and in neuritic plaques, accumulation of abnormal tau filaments as neurofibrillary tangles, reactive gliosis, and inflammation (2–6). Neuritic plaques consisting of Aβ deposits, in particular those formed by the 42 amino acid peptide (Aβ42) (7), are considered the foci of local inflammatory responses in AD brain, as evidenced by increased levels of acute phase proteins,
A
Page 1 of 19(page number not for citation purposes)

proinflammatory cytokines, complement components, and proteases (8–10). Of note is the association of activated microglia with neuritic plaques containing high levels of Aβ42 (11, 12), which have been shown to be directly toxic to neurons and also act as a proinflammatory stimulant of microglial cells of the myeloid lineage. Aβ42 has been reported to interact with microglial cells through several cell surface receptors (13). Among those, the G protein-coupled formyl peptide receptor like 1 (FPRL1) and its mouse homologue murine formyl peptide receptor 2 (mFPR2) have been shown to mediate the chemotactic activity and uptake of Aβ42 for cells of the myeloid lineage, including microglia, thus may be involved in the recruitment of microglial cells in AD brain. In human macrophages, FPRL1 has been shown to be crucial for Aβ42 uptake, a process that results in intracellular Aβ42 aggregation (13–18). Moreover, FPRL1, on cells of the neuroblastoma origin, appears to be responsible for the cytotoxicity of Aβ42 (18). These results suggest that FPRL1 may play an important role in the inflammatory amyloidogenic and neurodegenerative processes in AD.
The expression of the FPRL1 counterpart mFPR2 by mouse microglial cells is subject to regulation by proinflammatory stimulants. We recently reported that microglial cells isolated from newborn mice expressed markedly enhanced levels of mFPR2 upon activation by TNF-α and lipopolysaccharide (LPS), supporting the notion that microbial infection and proinflammatory stress conditions may profoundly affect the pathogenic process of AD via the up-regulation mFPR2 in microglial cells (19, 20). The aim of the present study was to examine whether CpG oligodeoxynucleotide (ODN), a Toll-like receptor (TLR) 9 ligand, which is contained in bacteria and virus (21) but may also be released by damaged host cells (22), also activates microglial cells and enhances the expression and function of mFPR2. We show that CpG ODN is a potent inducer of mFPR2 in microglia by using a p38 MAPK-dependent signaling pathway. Furthermore, microglial cells activated by CpG exhibited markedly increased capacity to endocytose Aβ42 through mFPR2.
MATERIAL AND METHODS
Reagents and cells
Mouse-specific stimulatory CpG ODN 1826 and inactive control CpG ODN were purchased from InvivoGen (San Diego, CA). fMLF and LPS were purchased Sigma-Aldrich (St. Louis, MO). Mouse SDF-1α was from PeproTech (Rocky Hill, NJ). The chemotactic peptide WKYMVm (designated W peptide) was synthesized and purified by the Department of Biochemistry, Colorado State University (Fort Collins, CO). The antibodies against phospho-ERK1/2, total ERK1/2, phospho-p38 MAPK, and total p38 MAPK were from Cell Signaling Technology, Inc. (Beverly, MA). Primary murine microglial cells were isolated from 1-day-old newborn C57BL/6 (WT) mice. The murine microglial cell line N9 was a kind gift from Dr. P. Ricciardi-Castagnoli (Universita Degli Studi di Milano-Bicocca, Milan, Italy). The cells were grown in IMDM supplemented with 5% heat-inactivated FCS, 2 mM glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin, and 50 μM 2-mercaptoethanol.
Chemotaxis assays
Microglial cell chemotaxis assays were performed with 48-well chemotaxis chambers (NeuroProbe, Cabin John, MD) as previously described (11). The results are expressed as the
Page 2 of 19(page number not for citation purposes)

mean ± SE of chemotaxis index (CI), which represents the fold increase in the number of migrated cells counted in three high-power fields (×400) in response to chemoattractants over spontaneous cell migration (to control medium).
RT-PCR and real-time PCR
Total RNA was extracted from cells with an RNeasy Mini kit and depleted of contaminating DNA with RNase-free DNase (Qiagen, Valencia, CA). For amplification of mFPR2 gene, primers 5′-TCT ACC ATC TCC AGA GTT CTG TGG-3′ (sense) and 5′-TTA CAT CTA CCA CAA TGT GAA CTA-3′ (antisense) were designed to generate a 268-bp product. Primers for mouse CXCR4 were 5′-GGC TGT AGA GCG AGT GTT GC-3′ (sense) and 5′-GTA GAG GTT GAC AGT GTA GAT-3′ (antisense), which generate a product of 390 bp. RT-PCR was performed with 0.5 µg of total RNA for each sample (High Fidelity ProSTAR System; Stratagene, La Jolla, CA), consisting of a 15 min reverse transcription at 37°C, 1 min inactivation of Moloney murine leukemia virus reverse transcriptase at 95°C, 40 cycles of denaturing at 95°C (45 s), annealing at 55°C (52°C for CXCR4; 45 s), and extension at 72°C (1 min), with a final extension for 10 min at 72°C. Primers for murine β-actin gene were used as controls (Stratagene). Real-time PCR was performed by using an ABI-Prism 7700 Sequence Detector (Applied Biosystems, Foster City, CA). Briefly, 5 ng of reverse-transcribed cDNA were used in triplicate samples. The assays were initiated with 2 min at 50°C, 10 min at 95°C, and then 40 cycles of 15 s at 95°C and 1 min at 60°C. Primers for mFPR2 and a specific probe were obtained from Applied Biosystems and consisted of the following: 5′-CCTTA TAGTC TTGAG AGAGC CCTGA-3′ (sense), 5′-TGCAG GAGGT GAAGT AGAAC TGG-3′ (antisense), and the probe 5′-FAM-TGAGG ATTCT GGTCA AACCA GTGAT TCAAG C-TAMRA-3′. Detection of mFPR2 and control β-actin were performed using TaqMan Universal PCR Master Mix (Applied Biosystems).
Flow cytometry
Murine microglia were examined for expression of CD14, TLR4, and TLR9 by labeling with PE- or FITC-conjugated monoclonal antibodies (PharMingen, San Diego, CA). All staining procedures were completed at 4°C in DPBS containing 5 mM EDTA and 1% FCS. After extensive washing, the cells were analyzed using a FACScan flow cytometer (BD Biosciences, Mountain View, CA).
Western immunoblotting
N9 cells were grown in 60 mm dishes until subconfluency and then were cultured overnight in FCS-free medium. After treatment with stimulants at the indicated time points, the cells were lysed with 150 μl ice-cold lysis buffer. The cell lysate was centrifuged at 14,000 rpm (4°C) for 5 min and the protein concentration of the supernatant was measured by using a BCA Protein Assay System (Pierce, Rockfold, IL). Western blotting of phosphorylated ERK1/2 or p38 MAPK was performed according to the manufacturer’s instructions using phospho protein-specific antibodies. Briefly, proteins were electrophoresed on a 10% SDS-PAGE precast gel (Invitrogen, Carlsbad, CA) under reducing conditions and were transferred onto Immun-Blot PVDF Membrane (Bio-Rad, Hercules, CA). The membranes were blocked with 5% nonfat milk (0.1% Tween-20) in TBS overnight at 4°C and then were incubated with primary antibodies for 3 h at
Page 3 of 19(page number not for citation purposes)

room temperature. After incubation with a horseradish-peroxidase conjugated secondary antibody, the protein bands were detected with a Super Signal Chemiluminescent Substrate (Pierce) and BIOMAX-MR film (Eastman Kodak, Rochester, NY). For detection of total Akt the membranes were stripped with Restore Western blot stripping buffer (Pierce) followed by incubation with specific antibodies.
Fluorescence confocal microscopy
CpG ODN-activated N9 cells grown on chamber slides were treated in the presence or absence of G-protein receptor deactivator pertussis toxin or mFPR2 agonist W peptide, followed by incubation with Aβ42 for different time points. The cells were fixed in 4% paraformaldehyde for 10 min at room temperature and washed with PBS. The slides were then incubated with 5% normal goat serum (Sigma) in PBS, 0.05% Tween-20 (PBS-T-NGS), for 1 h to reduce nonspecific binding of antibodies to the cell surface and for cell permeabilization. An anti-Aβ42 antibody (Sigma) was applied to the slides, which were further incubated for 1 h at room temperature. After three rinses with PBS, the slides were incubated with FITC-conjugated goat anti-mouse IgG (PharMingen) in TBS containing 1% BSA for 60 min. After three washes with PBS, the slides were stained with propidium iodide (PI) for 20 min at room temperature. The slides were mounted with an antifade, water-based mounting medium and analyzed under a laser scanning confocal fluorescence microscope (Zeiss LSM410 NLO Meta, Jena, Germany). Excitation wavelengths of 488 (for FITC) and 568 (for PI) nm were used to generate fluorescence emission in green (for Aβ42) and red (for nuclei), respectively.
Statistical analysis
All experiments were performed at least three times, and the results presented are from representative experiments. For cell migration, the significance of the difference between test and control groups was analyzed by using a computer-aided t test (GraphPad Prism, GraphPad Software, Inc., San Diego, CA). P ≤ 0.05 was considered statistically significant.
RESULTS
Expression of TLR9 in microglia and the effect of CpG ODN on mFPR2 expression
We first investigated the interaction of CpG ODN with TLR9 in microglial cells. Figure 1A shows that the mouse microglial cell line N9 expressed TLR9 on the surface as assessed by confocal microscopy with a TLR9-specific monoclonal antibody. After stimulation of N9 cells for 24 h with CpG ODN, TLR9 on the surface (Fig. 1A) was enhanced, in agreement with the reported studies with other cells types (23). The effect of CpG ODN on microglial cell TLR9 is specific, since it did not down-regulate TLR4, in contrast to LPS (Fig. 1B).
We next evaluated whether CpG ODN, by interacting with TLR9, was able to regulate the expression of mFPR2 in microglial cells. RT-PCR using total RNA from the mouse microglial cell line N9 shows that CpG ODN in a dose-dependent manner induced an increase in the levels of mRNA for mFPR2 (Fig. 2A). Real-time PCR (Fig. 2B) confirmed an enhanced mFPR2 transcription in CpG ODN-treated N9 cells. In contrast to its effect on mFPR2 mRNA expression, CpG ODN did not significantly modify the levels of mRNA for CXCR4, a receptor
Page 4 of 19(page number not for citation purposes)

for chemokine SDF-1α (CXCL12) (Fig. 2A). The induction of increased mFPR2 gene expression by CpG ODN in microglial cells was associated with the acquirement of potent chemotactic responses of N9 and primary mouse microglial cells to mFPR2 agonists, including the bacterial formylated peptide fMLF (24), a synthetic hexapeptide W peptide (25), and Aβ42, a key pathogenic molecule in AD (16) (Fig. 3A–C). The effect of CpG ODN on microglial responses to mFPR2 agonist peptides was dose- and time-dependent, reaching the maximum at 24 h after stimulation (Fig. 3A, 3D). A control ODN, devoid of the capacity to activate TLR9, failed to up-regulate the expression of mFPR2 mRNA and mFPR2-mediated cell chemotaxis (Fig. 2 and 3A). In contrast to its effect on mFPR2, CpG ODN treatment inhibited the chemotactic responses of microglia to SDF-1α (Fig. 3), suggesting that CpG ODN selectively regulates microglial responses to different chemoattractants. Since CpG ODN-induced signaling via TLR9 requires acidification and maturation of endosomes (25, 26, 27), and chloroquine by causing perturbation of endosomal and lysosomal pH prevents the activation of TLR9 (26, 28), we tested chloroquine on TLR9 signaling in microglial cells. Microglial cells treated with chloroquine failed to respond to CpG ODN by increased mFPR2 gene expression and chemotactic responses to mFPR2 agonists (Fig. 4A, 4B), indicating that TLR9 is required for CpG ODN to up-regulate mFPR2 in microglia.
Involvement of p38 MAPK activation in CpG ODN induction of mFPR2
Activation of TLR9 recruits the adaptor protein MyD88 followed by phosphorylation of MAPKs and NF-κB translocation to stimulate gene transcription (29). We found that CpG ODN induced a rapid and potent phosphorylation of p38, with a relatively weaker phosphorylation of ERK MAPK (Fig. 5A). This is in contrast to the effect of LPS, which stimulated a markedly increased phosphorylation of both ERK and p38 MAPK in microglial cells (Fig. 5A). CpG ODN also induced phosphorylation of IκB and Akt, an effect that was similar to LPS in microglia (Fig. 5A). Interestingly, although CpG ODN induced the phosphorylation of both p38 and ERK1/2 in microglia, its effect on p38 appeared to be critical for induction of mFPR2 transcription, since the p38 MAPK inhibitor SB202190, but not the MEK1/2 inhibitor PD98059, decreased CpG ODN-induced expression of mFPR2 mRNA (Fig. 5B). However, the chemotactic responses of CpG ODN-activated microglia to mFPR2 agonists were also inhibited by PD98059, suggesting that ERK activation is required for mFPR2 signaling in activated microglia (data not shown). In addition, we observed that a selective inhibitor of IκBα phosphorylation, BAY 11-7082, decreased CpG ODN-induced expression of mFPR2 mRNA (Fig. 5B) in microglial cells. These results suggest that p38 and NF-κB are critical molecules coupled to TLR9 signaling pathway that resulted in enhanced mFPR2 expression in microglial cells.
Increased Aβ42 uptake by CpG ODN-activated microglia
Microglia are key immune effector and phagocytic cells in the central nervous system (CNS) (9, 11). Microglial cells have been shown to ingest Aβ peptides and this process may contribute to the clearance of Aβ42 or to its fibrillary deposition in AD brain, which may profoundly affect the progression of AD (30). Since in our previous study we found that interaction of Aβ42 with FPRL1 in human macrophages resulted in internalization of the ligand/receptor complexes (17, 18), we evaluated the effect of CpG ODN on Aβ42 internalization in microglia. Stimulation of microglial cells with CpG ODN for 24 h significantly increased the levels of Aβ42 uptake as
Page 5 of 19(page number not for citation purposes)

compared with unstimulated cells (Fig. 6A). After 5 min of incubation of CpG ODN-treated microglia with Aβ42, the peptide was mostly associated with the cell membrane. After 24 h, Aβ42 was detected at markedly increased levels in the cytoplasmic compartment of microglial cells, indicating that Aβ42 was internalized (Fig. 6A). The internalization of Aβ42 was apparently mediated by mFPR2 because preincubation of microglial cells with pertussis toxin (PTX) (Fig. 6B), an inhibitor of Giα protein-coupled receptor, abrogated Aβ42 internalization by CpG ODN-activated microglia (Fig. 6B). Moreover, an mFPR2-specific agonist, W peptide (24), also reduced Aβ42 internalization in CpG ODN-activated microglial cells. Note that a low level of Aβ42 internalization was visible in PTX- or W peptide-treated microglial cells, which was comparable to the level shown in unstimulated microglia (Fig. 6B). These results suggest that mFPR2 induced by CpG ODN in microglial cells plays a prominent role in mediating Aβ42 internalization.
DISCUSSION
In this study, we have shown that CpG ODN enhances mFPR2 expression and function in microglial cells, through TLR9-mediated activation of p38 MAPK. The expression of mFPR2 in CpG ODN-activated microglial cells is associated with a markedly increased Aβ42 internalization, which is inhibited by PTX and another mFPR2 agonist, W peptide. To our knowledge, this is the first demonstration of the capacity of CpG ODN to promote the interaction of microglia with Aβ42, a key pathogenic factor in AD, via the induction of the G protein-coupled receptor mFPR2.
Microglia plays a critical role in many CNS diseases (31). In fact, activation of microglia is an essential component in the pathogenesis of AD, Parkinson’s disease (30), multiple sclerosis, AIDS dementia (32), and brain trauma caused by stroke (33). Our previous studies showed that murine microglial cells in resting state express very low levels of mFPR2 (19), a receptor that recognizes a diverse array of chemotactic agonists, including not only Aβ42 but also the bacterial formyl peptide fMLF, HIV-1 envelope protein-derived peptides, and a neuroprotective peptide, humanin (16, 18, 19). When stimulated with LPS, microglial cells expressed higher levels of mFPR2 transcripts and became responsive to mFPR2-specific agonists (19, 20, 34), suggesting that activation of TLR4 may promote microglial responses in CNS diseases in which agonists for mFPR2 are elevated (35, 36). Our present study further reveals that TLR9 engagement by CpG containing DNA is also able to induce the expression of mFPR2 by microglia. Thus, TLRs on microglial cells may act as sensors for proinflammatory signals and orchestrate the host responses in the CNS.
TLR9 is expressed by resting microglia, suggesting the potential of such cells in host defense against microbial infection and in the maintenance of homeostasis in the CNS (37). Although bacterial infection is the most common cause of CNS inflammation (38, 39), viruses are also important sources that cause inflammatory responses in the CNS. For example, herpes simplex virus causes encephalitis in children and young adults, and cytomegalovirus infects the CNS of immunocompromised individuals such as AIDS patients (40). In addition, HIV infection may cause lethal AIDS dementia. Recently, the genomes of herpes simplex virus type 1 (HSV-1), HSV-2, and murine cytomegalovirus (MCMV) have been found to contain a large quantity of CpG DNA motifs, which activate TLR9 in host cells to increase secretion of inflammatory
Page 6 of 19(page number not for citation purposes)

cytokines and IFN-α (41–43). In addition, CpG ODN may also be produced by mammalian cells and act as host-derived agonists for TLR9. For instance, cross-linking of autoimmune B cell receptor (BCR) with chromatin and antichromatin complexes induces potent autoimmune responses that involve the participation of TLR9 (44, 45). The heavy- and light-chain genes from one of the rheumatoid factor positive hybridoma lines were used to construct a AM14 BCR transgenic mouse line, which was activated by self-chromatin-containing immune complex (IC) (46). Studies have clearly demonstrated that chromatin-containing ICs were much more effective in activating the rheumatoid factor positive AM14 B cells than ICs alone. On the other hand, both MyD88-deficient (47) and TLR9-deficient (48) AM14 cells were resistent to activation by chromatin-containing immune complexes; furthermore, cloroquine blocked AM14 responses to chromatin ICs, indicating that TLR9 is activated by CpG ODN contained in the chromatin.
The evidence for presence of TLR9 agonists in mammalian cells is also provided by the observation that DNA fragments isolated from ICs in the sera of systemic lupus erythematosus patients contain CpG dinucleotides at high frequencies, and anti-DNA autoantibodies isolated from lupus sera preferentially bound to CG-rich DNA (48). This is an important finding because mammalian chromatin was originally considered a weak adjuvant (49); nevertheless, the adjuvant efficacy is significantly increased for apoptotic chromatin derived from stressed cells or in the form of immune complex (44, 45). Thus, endogenous ligands for TLR9 can be generated and the recognition of self-DNA by TLR9 may occur in pathogenic conditions such as neurodegenerative diseases in which increased host cell death is a hallmark (38).
It has been postulated that in AD brain, microglia accumulate at lesions containing high levels of Aβ (50). This process was thought to be an important host response to an aberrantly produced noxious agent. In fact, serial three-dimensional reconstruction of Aβ plaques in an animal model of AD revealed that Aβ deposits were enveloped by microglial processes (51). In addition, microglial cells have been documented to phagocytose fibrillary Aβ (52–54) and to break isolated senile plaques in vitro (55). Furthermore, active immunization with Aβ peptide (56, 57) and passive immunization with antibodies to Aβ (58) both result in substantial removal of Aβ deposits, which apparently is mediated by microglia.
It is relevant that Aβ has been reported to bind several cell surface receptors (16), including type A (SR-A) and type B (CD36) scavenger receptors (16, 59) as well as the receptor for advanced glycation end products (RAGE) (60). However, our results suggest that in CpG ODN-activated microglia, the G protein-coupled receptor mFPR2 plays a major role in mediating Aβ42 internalization. This is supported by inhibition of Aβ42 internalization with PTX and another mFPR2 agonist peptide. In addition, in our experiments, CD36 and RAGE agonists failed to affect Aβ42 internalization by CpG ODN-activated microglia (data not shown), indicating a prominent role of mFPR2.
Targeted delivery of genes and transplantation of suitable cell types into adult nervous system for the treatment of neurodegenerative diseases have raised considerable interest (61). Although gene delivery vehicles are designed to reduce or avoid toxicity and immunogenicity, viral proteins and genes as well as CpG DNA contained in plasmids (62) may affect host immune responses during transduction of foreign genes into target tissues (62). It is especially important when CpG containing vectors are to be considered in the context of AD treatment because such
Page 7 of 19(page number not for citation purposes)

treatment may increase the microglial expression of mFPR2, which upon interaction with Aβ42, may mediate proinflammatory responses, such as chemotaxis and release of superoxide (16). On the other hand, mFPR2-mediated internalization of Aβ42 may also facilitate the degradation and clearance of Aβ42. This was evidenced by our observation with immunoblotting of the lysates of CpG-activated microglia, which showed a prominent increase in Aβ42 antigenic activity 3 h after cell exposure to Aβ42 and the Aβ42 antigenic activity markedly decreased after 24 h exposure (data not shown). Further research is merited to delineate the beneficial vs. detrimental effects of FPRL1/mFPR2 in the pathogenic process of AD, thus to develop novel therapeutic agents.
ACKNOWLEDGMENTS
We thank Dr. Joost J. Oppenheim for reviewing the manuscript, Ms. Nancy Dunlop for technical support, and Ms. Cheryl Fogle and Ms. Cheryl Nolan for secretarial assistance. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government. The publisher or recipient acknowledges right of the U.S. government to retain a nonexclusive, royalty-free license in and to any copyright covering the article. This research was supported in part by the Intramural Research Program of the National Cancer Institute, and in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract no. NO1-CO-12400. Animal care was provided in accordance with the procedures outlined in the “Guide for the Care and Use of Laboratory Animals” (NIH publication no. 86-23, 1985). REFERENCES 1. Sadik, K., and Wilcock, G. (2003) The increasing burden of Alzheimer disease. Alzheimer
Dis. Assoc. Disord. 3, S75–S79
2. Dickson, D. W. (1997) The pathogenesis of senile plaques. J. Neuropathol. Exp. Neurol. 56, 321–339
3. Scheff, S. W., and Price, D. A. (1998) Synaptic density in the inner molecular layer of the hippocampal dentate gyrus in Alzheimer disease. J. Neuropathol. Exp. Neurol. 57, 1146–1153
4. Selkoe, D. (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev. 81, 741–766
5. Wegiel, J., Wisniewski, H. M., Dziewiatkowski, J., Tarnawski, M., Nowakowski, J., Dziewiatkowska, A., and Soltysiak, Z. (1995) The origin of amyloid in cerebral vessels of aged dogs. Brain Res. 705, 225–234
6. Wisniewski, K. E., Wisniewski, H. M., and Wen, G. Y. (1985) Occurrance of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Ann. Neurol. 17, 278–282
Page 8 of 19(page number not for citation purposes)

7. Citron, M., Diehl, T. S., Gordon, G., Biere, A. L., Seubert, P., and Selkoe, D. J. (1996) Evidence that the 42- and 40-amino acid forms of amyloid β protein are generated from the β-amyloid precursor protein by different protease activities. Proc. Natl. Acad. Sci. USA 93, 13,170–13,175
8. Eikelenboom, P., Rozemuller, J. M., and van Muiswinkel, F. L. (1998) Inflammation and Alzheimer’s disease: relationships between pathogenic mechanisms and clinical expression. Exp. Neurol. 154, 89–98
9. Cotter, R. L., Burke, W. J., Thomas, V. S., Potter, J. F., Zheng, J., and Gendelman, H. E. (1999) Insights into the neurodegenerative process of Alzheimer’s disease: a role for mononuclear phagocyte-associated inflammation and neurotoxicity. J. Leukoc. Biol. 65, 416–427
10. McGeer, P. L., and McGeer, E. G. (1999) Inflammation of the brain in Alzheimer’s disease: implications for therapy. J. Leukoc. Biol. 65, 409–415
11. McGeer, P. L., Itagaki, S., Boyes, B. E., and McGeer, E. G. (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38, 1285–1291
12. Uchihara, T., Akiyama, H., Kondo, H., and Ikeda, K. (1997) Activated microglial cells are colocalized with perivascular deposits of amyloid-β protein in Alzheimer’s disease brain. Stroke 28, 1948–1950
13. Iribarren, P., Zhou, Y., Hu, J., Le, Y., and Wang, J. M. (2005) The role of formyl peptide receptor like 1 (FPRL1/MFPR2) in mononuclear phagocyte responses in alzheimer’s disease. Immunol. Res. 31, 165–176
14. Giulian, D. (1999) Microglia and the immune pathology of Alzheimer disease. Am. J. Hum. Genet. 65, 13–18
15. Cui, Y., Le, Y., Yazawa, H., Gong, W., and Wang, J. M. (2002) Potential role of the formyl peptide receptor-like 1 (FPRL1) in inflammatory aspects of Alzheimer’s disease. J. Leukoc. Biol. 72, 628–635
16. Tiffany, H. L., Lavigne, M. C., Cui, Y. H., Wang, J. M., Leto, T. L., Gao, J. L., and Murphy, P. M. (2001) Amyloid-beta induces chemotaxis and oxidant stress by acting at formylpeptide receptor 2, a G protein-coupled receptor expressed in phagocytes and brain. J. Biol. Chem. 276, 23,645–23,652
17. Yazawa, H., Yu, Z. X., Takeda, Le, Y., Gong, W., Ferrans, V. J., Oppenheim, J. J., Li, C. C., and Wang, J. M. (2001) Beta amyloid peptide (Abeta42) is internalized via the G-protein-coupled receptor FPRL1 and forms fibrillar aggregates in macrophages. FASEB J. 15, 2454−2462
Page 9 of 19(page number not for citation purposes)

18. Ying, G., Iribarren, P., Zhou, Y., Gong, W., Zhang, N., Yu, Z. X., Le, Y., Cui, Y., and Wang, J. M. (2004) Humanin, a newly identified neuroprotective factor, uses the G protein-coupled formylpeptide receptor-like-1 as a functional receptor. J. Immunol. 172, 7078–7085
19. Cui, Y. H., Le, Y., Gong, W., Proost, P., Van Damme, J., Murphy, W. J., and Wang, J. M. (2002) Bacterial lipopolysaccharide selectively up-regulates the function of the chemotactic peptide receptor formyl peptide receptor 2 in murine microglial cells. J. Immunol. 168, 434–442
20. Cui, Y. H., Le, Y., Zhang, X., Gong, W., Abe, K., Sun, R., Van Damme, J., Proost, P., and Wang, J. M. (2002) Up-regulation of FPR2, a chemotactic receptor for amyloid beta 1-42 (A beta 42), in murine microglial cells by TNF alpha. Neurobiol. Dis. 10, 366–377
21. Boehme, K. W., and Compton, T. (2004) Innate sensing of viruses by toll-like receptors. J. Virol. 78, 7867–7873
22. Marshak-Rothstein, A., Busconi, L., Rifkin, I. R., and Viglianti, G. A. (2004) The stimulation of Toll-like receptors by nuclear antigens: a link between apoptosis and autoimmunity. Rheum. Dis. Clin. North Am. 30, 559–574
23. Liu, L., Zhou, X., Shi, J., Xie, X., and Yuan, Z. (2003) Toll-like receptor-9 induced by physical trauma mediates release of cytokines following exposure to CpG motif in mouse skin. Immunology 110, 341–347
24. Hartt, J. K., Barish, G., Murphy, P. M., and Gao, J. L. (1999) N-formylpeptides induce two distinct concentration optima for mouse neutrophil chemotaxis by differential interaction with two N-formylpeptide receptor (FPR) subtypes. Molecular characterization of FPR2, a second mouse neutrophil FPR. J. Exp. Med. 190, 741–747
25. Le, Y., Gong, W., Li, B., Dunlop, N. M., Shen, W., Su, S. B., Ye, R. D., and Wang J, M. (1999) Utilization of two seven-transmembrane, G protein-coupled receptors, formyl peptide receptor-like 1 and formyl peptide receptor, by the synthetic hexapeptide WKYMVm for human phagocyte activation. J. Immunol. 163, 6777-6784
26. Hacker, H., Mischak, H., Miethke, T., Liptay, S., Schmid, R., Sparwasser, T., Heeg, K., Lipford, G. B., and Wagner, H. (1998) CpGDNA-specific activation of antigen-presenting cells requires stress kinase activity and is preceded by non-specific endocytosis and endosomal maturation. EMBO J. 17, 6230–6240
27. Yi, A. K., Tuetken, R., Redford, T., Waldschmidt, M., Kirsch, J., and Krieg, A. M. (1998) CpG motifs in bacterial DNA activate leukocytes through the pH-dependent generation of reactive oxygen species. J. Immunol. 160, 4755–4761
28. Macfarlane, D. E., and Manzel, L. (1998) Antagonism of immunostimulatory CpG-oligodeoxynucleotides by quinacrine, chloroquine, and structurally related compounds. J. Immunol. 160, 1122–1131
Page 10 of 19(page number not for citation purposes)

29. Krieg, A. M. (2002) CpG motifs in bacterial DNA and their immune effects. Annu. Rev. Immunol. 20, 709–760
30. Monsonego, A., and Weiner, H. L. (2003) Immunotherapeutic approaches to Alzheimer's disease. Science 302, 834–838
31. Hirsch, E. C., Breidert, T., Rousselet, E., Hunot, S., Hartmann, A., and Michel, P. P. (2003) The role of glial reaction and inflammation in Parkinson's disease. Ann. N. Y. Acad. Sci. 991, 214–228
32. Minagar, A., Shapshak, P., Fujimura, R., Ownby, R., Heyes, M., and Eisdorfer, C. (2002) The role of macrophage/microglia and astrocytes in the pathogenesis of three neurologic disorders: HIV-associated dementia, Alzheimer disease, and multiple sclerosis. J. Neurol. Sci. 202, 13–23
33. Pocock, J. M., Liddle, A. C., Hooper, C., Taylor, D. L., Davenport, C. M., and Morgan, S. C. (2002) Activated microglia in Alzheimer's disease and stroke. Ernst Schering Res. Found. Workshop 39, 105–132
34. Iribarren, P., Cui, Y. H., Le, Y., Ying, G., Zhang, X., Gong, W., and Wang, J. M. (2003) IL-4 down-regulates lipopolysaccharide-induced formyl peptide receptor 2 in murine microglial cells by inhibiting the activation of mitogen-activated protein kinases. J. Immunol. 171, 5482–5488
35. Kalaria, R. N. (1999) Microglia and Alzheimer's disease. Curr. Opin. Hematol. 6, 15–24
36. Akiyama, H., Barger, S., Barnum, S., Bradt, B., Bauer, J., Cole, G. M., Cooper, N. R., Eikelenboom, P., Emmerling, M., Fiebich, B. L., et al. (2000) Inflammation and Alzheimer's disease. Neurobiol. Aging 21, 383–421
37. Dalpke, A. H., Schafer, M. K., Frey, M., Zimmermann, S., Tebbe, J., Weihe, E., and Heeg, K. (2002) Immunostimulatory CpG-DNA activates murine microglia. J. Immunol. 168, 4854–4863
38. Lee, S. J., and Lee, S. (2002) Toll-like receptors and inflammation in the CNS. Curr. Drug Targets Inflamm. Allergy 1, 181–191
39. Durand, M. L., Calderwood, S. B., Weber, D. J., Miller, S. I., Southwick, F. S., Caviness, V. S., Jr., and Swartz, M. N. (1993) Acute bacterial meningitis in adults. A review of 493 episodes. N. Engl. J. Med. 328, 21–28
40. Whitley, R. J. Viral encephalitis. (1990) N. Engl. J. Med. 323, 242−250.
41. Krug, A., Luker, G. D., Barchet, W., Leib, D. A., Akira, S., and Colonna, M. (2004) Herpes simplex virus type 1 activates murine natural interferonproducing cells through toll-like receptor 9. Blood 103, 1433–1437
Page 11 of 19(page number not for citation purposes)

42. Lund, J., Sato, A., Akira, S., Medzhitov, R., and Iwasaki, A. (2003) Toll-like receptor 9-mediated recognition of herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 198, 513–520
43. Tabeta, K., Georgel, P., Janssen, E., Du, X., Hoebe, K., Crozat, K., Mudd, S., Shamel, L., Sovath, S., Goode, J., et al. (2004) Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc. Natl. Acad. Sci. USA 101, 3516–3521
44. Leadbetter, E. A., Rifkin, I. R., Hohlbaum, A. M., Beaudette, B. C., Shlomchik, M. J., and Marshak-Rothstein, A. (2002) Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 416, 603–607
45. Viglianti, G. A., Lau, C. M., Hanley, T. M., Miko, B. A., Shlomchik, M. J., and Marshak-Rothstein, A. (2003) Activation of autoreactive B cells by CpG dsDNA. Immunity 19, 837–847
46. Shlomchik, M. J., Zharhary, D., Camper, S., Saunders, T., and Weigert, M. (1993) A rheumatoid factor transgenic mouse model for autoantibody regulation. Int. Immunol. 5, 1329–1341
47. Leadbetter, E. A., Rifkin, I. R., Hohlbaum, A. H., Beaudette, B., Shlomchik, M. J., and Marshak-Rothstein, A. (2002) Immune complexes activate autoreactive B cells by co-engagement of sIgM and Toll-like receptors. Nature 419, 603–607
48. Sano, H., Takai, O., Harata, N., Yoshinaga, K., Kodama-Kamada, I., and Sasaki, T. (1989) Binding properties of human anti-DNA antibodies to cloned human DNA fragments. Scand. J. Immunol. 30, 51–63
49. Pisetsky, D. S. (1992) Anti-DNA antibodies in systemic lupus erythematosus. Rheum. Dis. Clin. North Am. 18, 437–454
50. Rogers, J., and Lue, L. F. (2001) Microglial chemotaxis, activation, and phagocytosis of amyloid beta-peptide as linked phenomena in Alzheimer’s disease. Neurochem. Int. 39, 333–340
51. Stalder, M., Deller, T., Staufenbiel, M., and Jucker, M. (2001) 3D-Reconstruction of microglia and amyloid in APP23 transgenic mice: no evidence of intracellular amyloid. Neurobiol. Aging 22, 427–434
52. Ard, M. D., Cole, G. M., Wei, J., Mehrle, A. P., and Fratkin, J. D. (1996) Scavenging of Alzheimer’s amyloid beta-protein by microglia in culture. J. Neurosci. Res. 43, 190–202
53. Paresce, D. M., Ghosh, R. N., and Maxfield, F. R. (1996) Microglial cells internalize aggregates of the Alzheimer’s disease amyloid beta-protein via a scavenger receptor. Neuron 17, 553–565
Page 12 of 19(page number not for citation purposes)

54. Bard, F., Cannon, C., Barbour, R., Burke, R. L., Games, D., Grajeda, H., Guido, T., Hu, K., Huang, J., Johnson-Wood, K., et al. (2000) Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 6, 916–919
55. DeWitt, D. A., Perry, G., Cohen, M., Doller, C., and Silver, J. (1998) Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer’s disease. Exp. Neurol. 149, 329–340
56. Schenk, D., Barbour, R., Dunn, W., Gordon, G., Grajeda, H., Guido, T., Hu, K., Huang, J., Johnson-Wood, K., Khan, K., et al. (1999) Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173–177
57. Lombardo, J. A., Stern, E. A., McLellan, M. E., Kajdasz, S. T., Hickey, G. A., Bacskai, B. J., and Hyman, B. T. (2003) Amyloid-beta antibody treatment leads to rapid normalization of plaque-induced neuritic alterations. J. Neurosci. 23, 10,879–10883
58. Wilcock, D. M., DiCarlo, G., Henderson, D., Jackson, J., Clarke, K., Ugen, K. E., Gordon, M. N., and Morgan, D. (2003) Intracranially administered anti-Aβ antibodies reduce β-amyloid deposition by mechanisms both independent of and associated with microglial activation. J. Neurosci. 23, 3745–3751
59. El Khoury, J., Hickman, S. E., Thomas, C. A., Cao, L., Silverstein, S. C., and Loike, J. D. (1996) Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature 382, 716–719
60. Yan, S. D., Chen, X., Fu, J., Chen, M., Zhu, H., Roher, A., Slattery, T., Zhao, L., Nagashima, M., Morser, J., et al. (1996) RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 382, 685–691
61. Blesch, A., and Tuszynski, M. H. (2004) Gene therapy and cell transplantation for Alzheimer's disease and spinal cord injury. Yonsei Med. J. 45, Suppl, 28–31
62. Bessis, N., GarciaCozar, F. J., and Boissier, M. C. (2004) Immune responses to gene therapy vectors: influence on vector function and effector mechanisms. Gene Ther. 11 Suppl 1, S10–S17
Received July 4, 2005; accepted August 10, 2005.
Page 13 of 19(page number not for citation purposes)
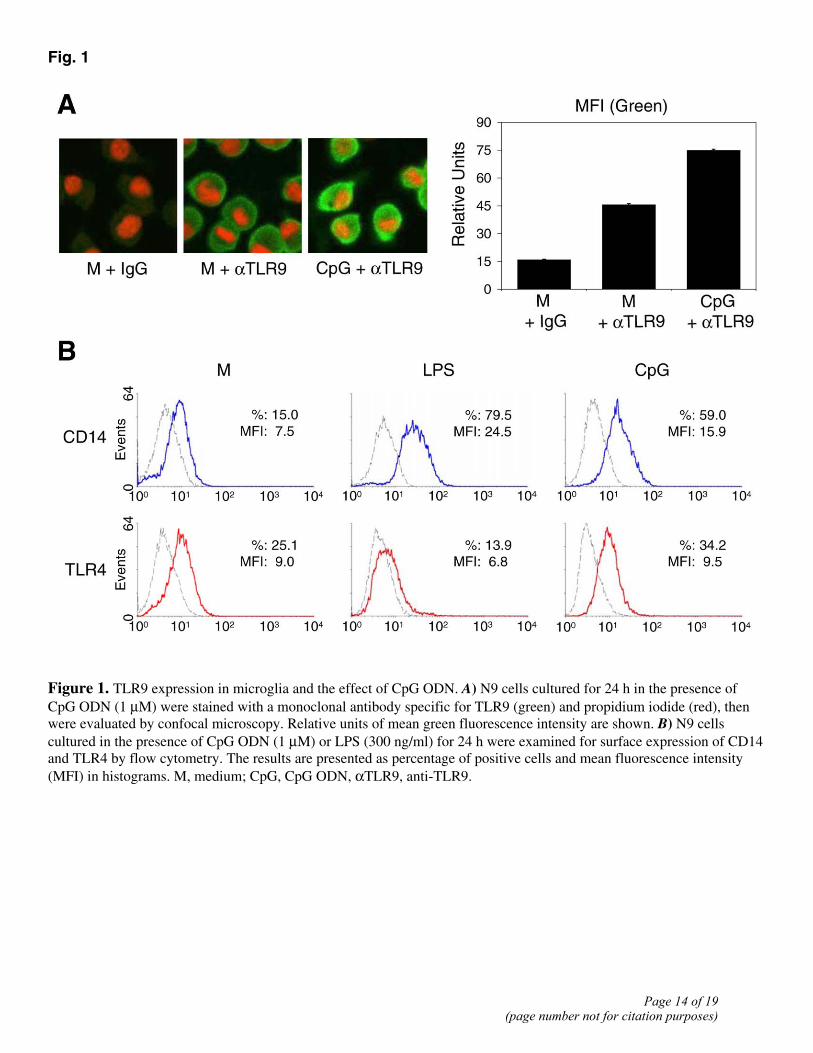
Fig. 1
Figure 1. TLR9 expression in microglia and the effect of CpG ODN. A) N9 cells cultured for 24 h in the presence of CpG ODN (1 µM) were stained with a monoclonal antibody specific for TLR9 (green) and propidium iodide (red), then were evaluated by confocal microscopy. Relative units of mean green fluorescence intensity are shown. B) N9 cells cultured in the presence of CpG ODN (1 µM) or LPS (300 ng/ml) for 24 h were examined for surface expression of CD14 and TLR4 by flow cytometry. The results are presented as percentage of positive cells and mean fluorescence intensity (MFI) in histograms. M, medium; CpG, CpG ODN, αTLR9, anti-TLR9.
Page 14 of 19(page number not for citation purposes)
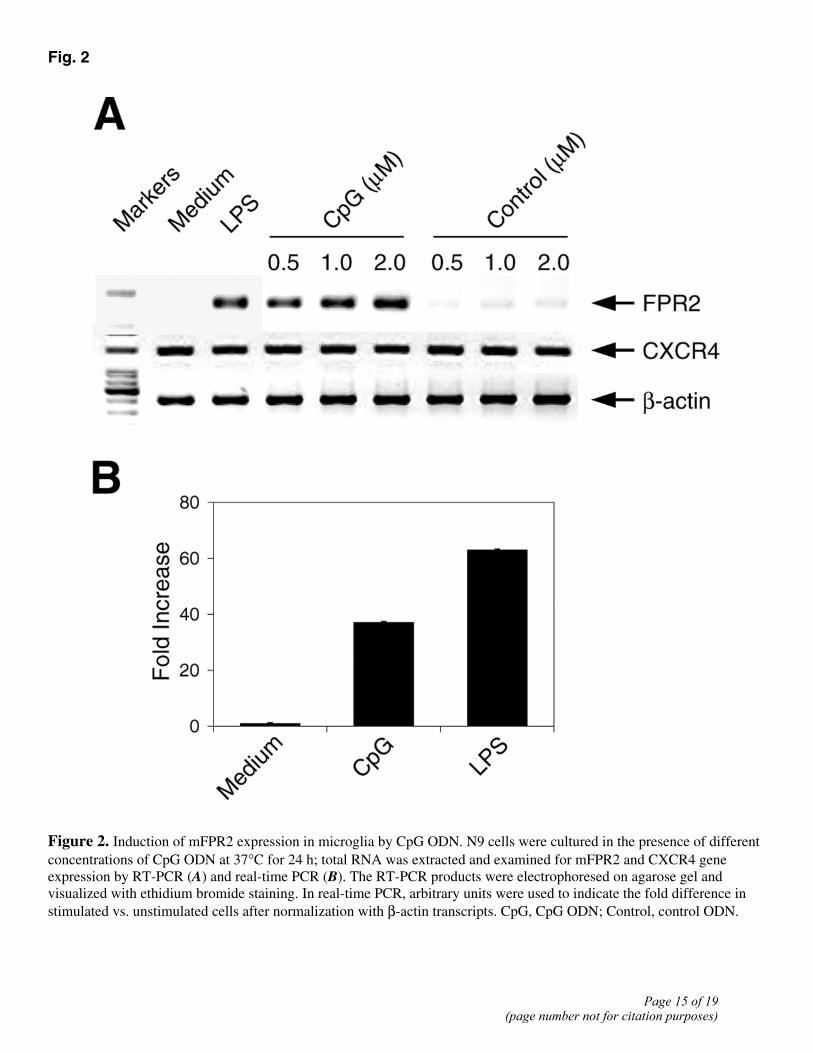
Fig. 2
Figure 2. Induction of mFPR2 expression in microglia by CpG ODN. N9 cells were cultured in the presence of different concentrations of CpG ODN at 37°C for 24 h; total RNA was extracted and examined for mFPR2 and CXCR4 gene expression by RT-PCR (A) and real-time PCR (B). The RT-PCR products were electrophoresed on agarose gel and visualized with ethidium bromide staining. In real-time PCR, arbitrary units were used to indicate the fold difference in stimulated vs. unstimulated cells after normalization with β-actin transcripts. CpG, CpG ODN; Control, control ODN.
Page 15 of 19(page number not for citation purposes)

Fig. 3
Figure 3. mFPR2 function in CpG ODN-activated microglia. N9 cells (A) and primary mouse microglia (B) were incubated with different concentrations of CpG ODN or a control ODN for 24 h. The cells were then examined for migration in response to the mFPR2 agonists W peptide (1 μM) and fMLF (10 μM), as well as the chemokine SDF-1α (10 nM). C) Primary microglia were cultured in the presence of CpG ODN for 24 h. The cells were then examined for migration in response to Aβ42 (10 μM) and SDF-1α (10 nM). D) N9 cells were cultured in the presence of CpG ODN (1 μM) for different time points and then were examined for migration in response to W peptide (1 μM), fMLF (10 μM), and SDF-1α (10 nM). The results are expressed as chemotaxis index (CI) representing fold increase in cell migration in response to chemoattractants over the baseline migration (to medium). *Statistically significant (P<0.01) increase in cell migration compared with unstimulated cells. CpG, CpG ODN; Control, control ODN.
Page 16 of 19(page number not for citation purposes)
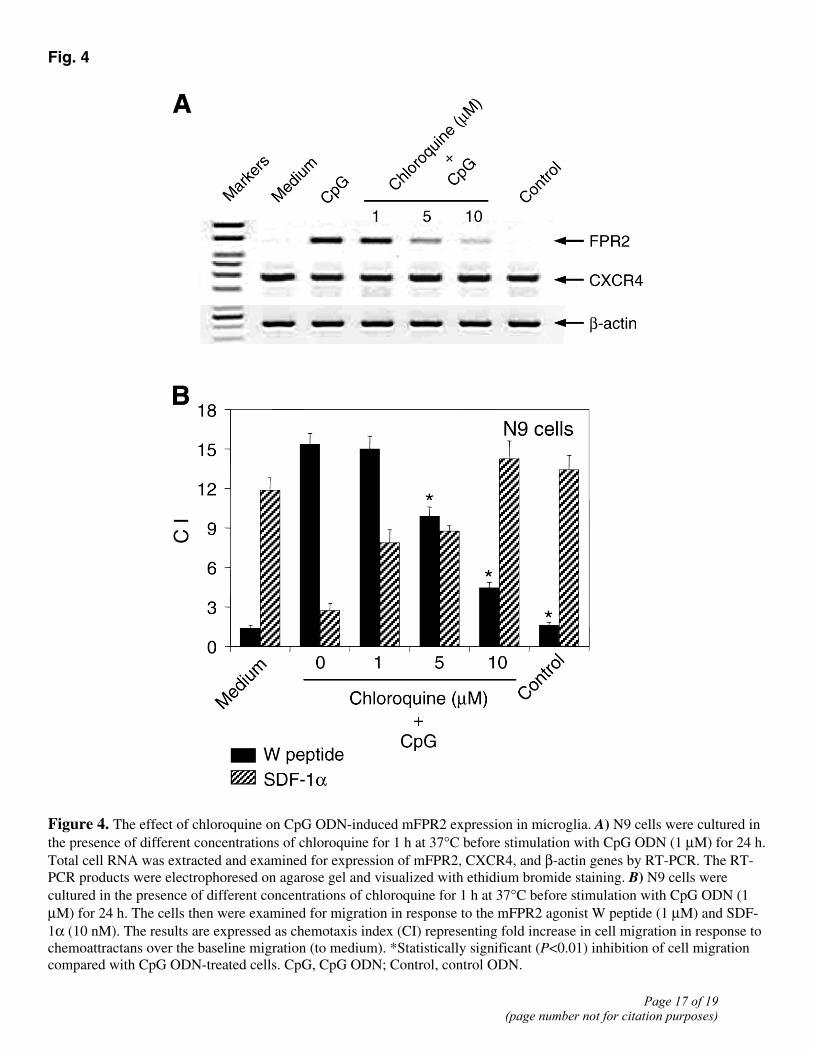
Fig. 4
Figure 4. The effect of chloroquine on CpG ODN-induced mFPR2 expression in microglia. A) N9 cells were cultured in the presence of different concentrations of chloroquine for 1 h at 37°C before stimulation with CpG ODN (1 µM) for 24 h. Total cell RNA was extracted and examined for expression of mFPR2, CXCR4, and β-actin genes by RT-PCR. The RT-PCR products were electrophoresed on agarose gel and visualized with ethidium bromide staining. B) N9 cells were cultured in the presence of different concentrations of chloroquine for 1 h at 37°C before stimulation with CpG ODN (1 µM) for 24 h. The cells then were examined for migration in response to the mFPR2 agonist W peptide (1 µM) and SDF-1α (10 nM). The results are expressed as chemotaxis index (CI) representing fold increase in cell migration in response to chemoattractans over the baseline migration (to medium). *Statistically significant (P<0.01) inhibition of cell migration compared with CpG ODN-treated cells. CpG, CpG ODN; Control, control ODN.
Page 17 of 19(page number not for citation purposes)

Fig. 5
Figure 5. Involvement of p38 MAPK activation in CpG ODN induction of mFPR2. A) N9 cells were cultured in the presence of CpG ODN (1 µM) or LPS (300 ng/ml) at 37°C and lysed at the indicated time points. Equal amounts of total proteins were electrophoresed and blotted, and protein bands were detected with anti-phospho-p38, anti-phospho-ERK1/2, anti-phospho-IκB, and anti-phospho-Akt antibodies, respectively. The membranes were stripped and examined for total Akt. B) N9 cells were cultured in the presence of different concentrations of SB202190, PD98059, or BAY 11-7082 for 1 h at 37°C before stimulation with CpG ODN (1 µM) and for an additional 24 h at 37°C. Total cell RNA was extracted and examined for expression of mFPR2, CXCR4, and β-actin genes by RT-PCR. The RT-PCR products were electrophoresed on agarose gel and visualized with ethidium bromide staining. CpG, CpG ODN; Control, control ODN; SB, SB202190; PD, PD98059; BAY, BAY 11-7082.
Page 18 of 19(page number not for citation purposes)
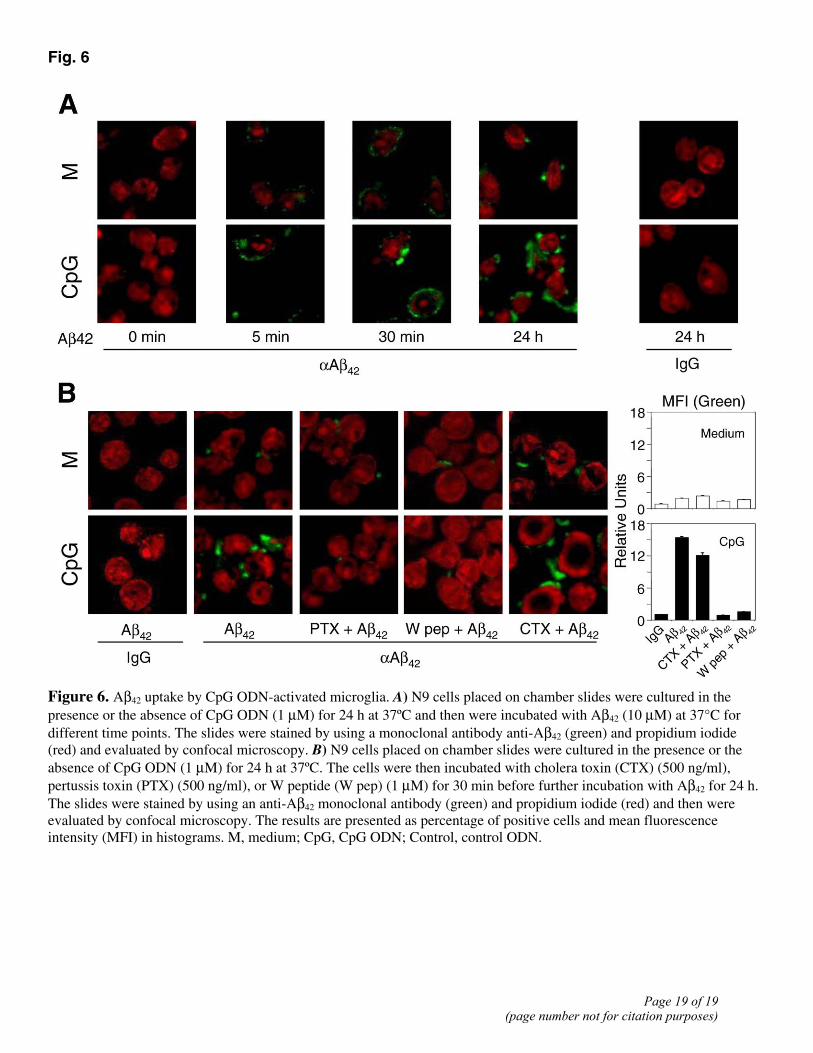
Fig. 6
Figure 6. Aβ42 uptake by CpG ODN-activated microglia. A) N9 cells placed on chamber slides were cultured in the presence or the absence of CpG ODN (1 µM) for 24 h at 37ºC and then were incubated with Aβ42 (10 µM) at 37°C for different time points. The slides were stained by using a monoclonal antibody anti-Aβ42 (green) and propidium iodide (red) and evaluated by confocal microscopy. B) N9 cells placed on chamber slides were cultured in the presence or the absence of CpG ODN (1 µM) for 24 h at 37ºC. The cells were then incubated with cholera toxin (CTX) (500 ng/ml), pertussis toxin (PTX) (500 ng/ml), or W peptide (W pep) (1 µM) for 30 min before further incubation with Aβ42 for 24 h. The slides were stained by using an anti-Aβ42 monoclonal antibody (green) and propidium iodide (red) and then were evaluated by confocal microscopy. The results are presented as percentage of positive cells and mean fluorescence intensity (MFI) in histograms. M, medium; CpG, CpG ODN; Control, control ODN.
Page 19 of 19(page number not for citation purposes)
Copyright © 2022 FDOKUMEN




















