
Sex differences in corticotropin-releasing factor signaling and trafficking
Upload
independentCategory
view
0download
0
Neuropharmacology 45 (2003) 623–636www.elsevier.com/locate/neuropharm
Corticotropin-releasing factor (CRF) and related peptides conferneuroprotection via type 1 CRF receptors
L. Faccia, D.A. Stevensa, M. Pangallob, D. Franceschinia, S.D. Skapera,P.J.L.M. Strijbosa,∗
a Neurology and GI Centre of Excellence for Drug Discovery, GlaxoSmithKline Research and Development Limited, New Frontiers SciencePark, Third Avenue, Harlow CM19 5AW, UK
b Department of Pharmacology, University of Florence, Viale Pieraccini 6, 50139 Florence, Italy
Received 21 November 2002; received in revised form 25 April 2003; accepted 23 May 2003
Abstract
Corticotropin-releasing factor (CRF) receptors are members of the superfamily of G-protein coupled receptors that utilise adenylatecyclase and subsequent production of cAMP for signal transduction in many tissues. Activation of cAMP-dependent pathways,through elevation of intracellular cAMP levels is known to promote survival of a large variety of central and peripheral neuronalpopulations. Utilising cultured primary rat central nervous system neurons, we show that stimulation of endogenous cAMP signallingpathways by forskolin confers neuroprotection, whilst inhibition of this pathway triggers neuronal death. CRF and the related CRFfamily peptides urotensin I, urocortin, and sauvagine, which also induced cAMP production, prevented the apoptotic death ofcerebellar granule neurons triggered by inhibition of phosphatidylinositol kinase-3 pathway activity with LY294002. These effectswere negated by the highly selective CRF-R1 antagonist CP154,526. CRF even conferred neuroprotection when its application wasdelayed by up to 8 h following LY294002 addition. The CRF peptides also protected cortical and hippocampal neurons againstdeath induced byβ-amyloid peptide (1-42), in a CRF-R1 dependent manner. In separate experiments, LY294002 reduced neuronalprotein kinase B activity while increasing glycogen synthase kinase-3, whilst CRF (and related peptides) promoted phosphorylationof glycogen synthase kinase-3 without protein kinase B activation. Taken together, these results suggest that the neuroprotectiveactivity of CRF may involve cAMP-dependent phosphorylation of glycogen synthase kinase-3. 2003 Elsevier Ltd. All rights reserved.
Keywords: Central nervous system neurons; Apoptosis; Phosphatidylinositol 3-kinase;β-Amyloid; Neuroprotection; Glycogen synthase kinase-3
1. Introduction
Corticotropin-releasing factor (CRF) is a key mediatorof the stress response and evokes the release of adreno-corticotrophic hormone from the pituitary (Arborelius etal., 1999). CRF is expressed throughout the central ner-vous system (CNS) and has a wide spectrum of auto-nomic, electrophysiological and behavioural actions inthe brain (Owens and Nemeroff, 1991). As predicted,deficits in this pathway lead to aberrant stress responses.Abnormal CRF physiology has also been linked withcertain neurodegenerative diseases including Hunt-
∗ Corresponding author. Tel.:+44-1279-622681; fax:+44-1279-622555.
E-mail address: [email protected] (P.J.L.M. Strijbos).
0028-3908/$ - see front matter 2003 Elsevier Ltd. All rights reserved.doi:10.1016/S0028-3908(03)00211-9
ington’s chorea (Heuser et al., 1991), olivopontocerebel-lar atrophy and spinocerebellar degeneration (Mizuno etal., 1995; Suemaru et al., 1995), and Alzheimer’s disease(De Souza, 1995). However, the precise role of CRF,and its down-stream effectors, in these diseases ispresently unclear.
CRF receptors belong to a superfamily of Gs-proteincoupled receptors which show considerable homology tothe vasoactive intestinal polypeptide/calcitonin family ofGs-protein coupled receptors, which are also positivelycoupled to adenylate cyclase and cAMP production(Perrin and Vale, 1999). Members of the CRF peptidefamily (CRF, urocortin, urotensin 1 and sauvagine)themselves are potent activators of adenylate cyclase andcAMP production (Chen et al., 1997; Battaglia et al.,1987). Two types of CRF receptors, namely CRF-R1 andCRF-R2 exist, of which CRF-R2 can additionally occur

624 L. Facci et al. / Neuropharmacology 45 (2003) 623–636
as three alternative splice variant forms, CRF-R2α,CRF-R2β, CRF-R2γ (Chang et al., 1993; Kishimoto etal., 1995; Lovenberg et al., 1995; Perrin et al., 1995;Chalmers et al., 1996). In analogy to their ligands, bothtypes of CRF receptors are expressed in the brain, withCRF-R1 predominating in the cerebral cortex, sensoryrelay nuclei, and in the cerebellum and its major affer-ents (van Pett et al., 2000). CRF-R2 is additionally foundin peripheral tissues like heart and skeletal muscle.
Apoptotic cell death has been suggested to underlievarious human brain disorders, such as cerebralischemia, Alzheimer’s disease (Loo et al., 1993), Hunt-ington’s disease (Portera-Cailliau et al., 1995), and amy-trophic lateral sclerosis (Rabizadeh et al., 1995). Thesearch for novel neuroprotective therapies to alleviatethese devastating conditions has identified a multitudeof molecules and pathways that may serve as putativetargets. Elevating intracellular cAMP levels promotessurvival of cultured sympathetic and sensory neurons(Rydel and Greene, 1988), dopamine neurons (Mena etal., 1995), developing septal cholinergic neurons (Kewet al., 1996), spinal cord motor neurons (Hanson et al.,1998), and cerebellar granule neurons (D’Mello et al.,1993). Furthermore, activation of cAMP-dependent thirdmessenger pathways can substitute for the loss of neuro-trophic activity following removal of growth factors(Rydel and Greene, 1988; D’Mello et al., 1993; Hansonet al., 1998).
In the present study, we have evaluated the neuropro-tective efficacy of CRF, and related family members, inrat primary cerebellar granule neurons undergoingapoptosis. We report that CRF (peptides) are extremelypotent neuroprotectants in this paradigm and that thisactivity is mediated entirely by type 1 CRF receptorsand, partially, by cAMP and subsequent inhibition ofglycogen synthase kinase-3 (GSK-3). Furthermore, CRF(peptides) protected cortical and hippocampal neuronsfrom injury caused by β-amyloid peptide (Aβ) 1-42,which constitutes the mature form of amyloid peptidefound in Alzheimer’s disease brain plaques. Parts of thiswork have been reported in abstract form (Facci et al.,2000).
2. Methods
2.1. Primary neuronal cell cultures
Cultures of cerebellar granule neurons were preparedfrom 8-day-old Sprague-Dawley rats (Charles River,Margate, UK) as described previously (Skaper et al.,1990). Dissociated cells were plated on poly-d-lysinecoated 48-well plates (Nunclon) (3.5 × 105 cells /well)for survival and cAMP experiments or 6-well plates (3× 106 cells /well) for western blot analysis. Neurons weremaintained in Basal Medium Eagle’s (BME) sup-
plemented with 10% heat-inactivated fetal bovine serum,2 mM l-glutamine, 50 µg/ml gentamicin, and 25 mMKCl, and incubated at 37 °C in a humidified atmospherewith 5% CO2. Cytosine arabinoside (10 µM) was added18–24 h after plating to inhibit growth of non-neuronalcells. Cultures prepared in this manner typically contain�5% non-neuronal cells (Boje et al., 1993; Gallo et al.,1987). Experiments were performed on granule neuronsgrown in culture for 8 days.
Hippocampal and cortical neurons were prepared fromembryonic day 17.5 to 18 day gestation Sprague-Dawleyrat fetuses, as described (Skaper et al., 1990). Cells weremaintained in Neurobasal medium supplemented withB27 additives with antioxidants, 1 mM sodium pyruvate,2 mM glutamine, 25 µM glutamate, 50 U/ml penicillin,and 50 µg/ml streptomycin. Half of the medium wasreplaced every 3–4 days with fresh maintenance medium(Neurobasal supplemented with 50 U/ml penicillin, 50µg/ml streptomycin, 2 mM glutamine and B27 withoutantioxidants). Cultures were utilised after 7–9 days, andcomprised �95% neuronal cells at this time.
2.2. Neuronal survival assays
At 8 days in vitro, apoptosis was triggered by shiftingcerebellar granule neuron cultures to serum-free BMEcontaining 25 mM KCl, 50 U/ml penicillin, 50 µg/mlstreptomycin and the selective phosphatidylinositol 3-kinase (PI-3-K) inhibitor LY294002. Under these con-ditions maximal granule neuron cell death is achievedwith 50–75 µM LY294002 (Miller et al., 1997; ourunpublished observations). Test agent was added at thistime, unless indicated otherwise. Neuron survival wasquantified 48 h after start of LY294002 treatment by thecolorimetric MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay (Cross et al., 2001;Manthorpe et al., 1986; Skaper et al., 1990), which reactswith living cells but not with dead cells or their lyticdebris. Absolute MTT values were normalised by scalingto the mean of cultures in serum-free medium with 25mM KCl (defined as 100%). Release of lactate dehydro-genase (LDH) into the culture medium was also utilisedas an index of cell vitality, with a commercially availablekit (Sigma #228-10), following the manufacturer’sinstructions. The MTT technique is equivalent to LDHrelease in the measurement of neurotoxin-mediated neu-ronal death in vitro (Patel et al., 1990). Cell vitality wasroutinely confirmed by morphological observation.
For β-amyloid peptide toxicity experiments, Aβ1-42was reconstituted in 1 mM NH3, left at 37 °C for 1 hand then diluted into culture medium. Cultures weretreated with CRF peptide ± β-amyloid peptide for 3days, then fixed with 4% paraformaldehyde and stainedwith 0.4% trypan blue. Neurons with intact neurites ofuniform diameter and soma with a smooth round appear-ance were considered viable, whereas neurons with frag-

625L. Facci et al. / Neuropharmacology 45 (2003) 623–636
mented neurites and a vacuolated or swollen soma wereconsidered nonviable (Mattson et al., 1992).
2.3. Characterisation of apoptosis
Chromatin condensation was analysed in cerebellargranule neurons treated with 75 µM LY294002 ± 30nM CRF for 24 h. Cells were grown on poly-d-lysine-coated coverglasses, at the same plating density used forsurvival measurements. Hoechst 33258 fluorescent dye(Molecular Probes) prepared fresh as a 1 mM stock sol-ution in water was added directly to cell culture medium,to a final concentration of 1 µM. Cells were incubatedfor 10 min at 22 °C and then fixed with 4% paraformal-dehyde (freshly prepared in PBS). Cells were rinsedthree times with PBS and then coverslipped with Citi-fluor to prevent fading. Stained nuclei were photo-graphed with a Leica DMR microscope equipped forfluorescence at 40× magnification.
DNA fragmentation was quantified in granule neuronstreated with 75 µM LY294002 ± test agent for 24 h,using a commercially available ELISA kit (TUNEL) thatdetects the enrichment of mono- and oligonucleosomesthat occurs in apoptotic cells (Cross et al., 2001), accord-ing to the manufacturer’s instructions (Roche Molecu-lar Diagnostics).
2.4. Cyclic AMP assays
Cerebellar granule neuron cultures were shifted toserum-free BME containing 25 mM KCl, 50 U/ml peni-cillin, 50 µg/ml streptomycin, and incubated for 15 min(37 °C) with the phosphodiesterase inhibitor isobutylme-thyl xanthine (IBMX, 0.5 mM) and test agent as indi-cated in the appropriate figure. Cyclic AMP was quant-ified by enzyme immunoassay, using the BIOTRAK kit(Amersham Pharmacia Biotech) and following themanufacturer’s instructions.
2.5. Western blot analysis
Granule neuron cultures were shifted to serum-freeBME containing 25 mM KCl, 50 U/ml penicillin, 50µg/ml streptomycin, and treated for 3 h as indicated inthe appropriate figure. Cell extracts were prepared bylysing cells on ice in 1% Triton X-100, 0.5% SDS,0.75% deoxycholate, 10 mM Tris–Cl (pH 7.4), 75 mMNaCl, 10 mM EDTA, 0.5 mM phenylmethylsulfonylfluoride, 2 mM sodium orthovanadate, 4 mg/ml leupep-tin, 10 mg/ml aprotinin, 4.2 mg/ml NaF, and 17.8 mg/mlsodium pyrophosphate. Lysates were then centrifuged at13,000 rpm for 30 min at 4 °C and the supernatantsstored at �80 °C. Protein lysates were resuspended inLaemmeli buffer, size fractionated by SDS-PAGE (12%)and transferred to PVDF nitrocellulose membranes(BioRad) for western blotting. Membranes were blocked
in TBS-T buffer (20 mM Tris base, pH 7.6, 150 mMNaCl, 0.1% Tween-20) containing 20% w/v non-fat milkpowder (Marvel, Premier Brands, Stafford, UK) at roomtemperature for 2 h. Membranes were agitated at 4 °Covernight with rabbit primary antibodies diluted 1:1000in 5% w/v non-fat milk in TBS/T. Membranes were thenwashed four times, 10 min each, in TBS/T at room tem-perature and incubated for 1 h with horseradish peroxi-dase-conjugated goat anti-rabbit secondary antibodydiluted 1:7500 in TBS/T with 5% non-fat milk. Mem-branes were washed 4 × 10 min in TBS/T and proteinwas detected by enhanced chemiluminescence (ECL,Amersham) after exposure of filters to X-ray film for 30s to 15 min.
2.6. Statistical analysis
Statistical significance was determined using one-wayANOVA followed by Fisher’s post hoc test. Differenceswere considered significant at p � 0.05.
2.7. Materials
Tissue culture media and fetal calf serum were pur-chased from Life Technologies (Paisley, UK); all othertissue culture reagents, LY294002, IBMX, MTT, andbiochemicals were from Sigma (Dorset, UK); forskolin,1,9-dideoxy forskolin, Rp-8-Br-cAMPS, 8-CPT-cAMPand H89 were from Calbiochem (Nottingham, UK);Aβ1-42 and Aβ42-1 were from Bachem (SaffronWalden, UK); CRF (human, rat; lot 055-7180), sauvag-ine (lot 053-7850), urocortin (lot 804075) and urotensinI (lot 054-2374) were from Bachem (Merseyside, UK);tissue culture plasticware was from Nunc (Roskilde,Denmark); cAMP enzyme immunoassay kits(BIOTRAK , RPN 225) were from Amersham Biosci-ences UK Ltd (Buckinghamshire, UK); primary anti-bodies were from Cell Signalling Technology(Hertfordshire, UK), and secondary antibodies andU0126 from Promega (Southampton, UK); polyacrylam-ide gels were from Invitrogen (Paisley, UK). CP154,526(Chen et al., 1997) was synthesised by standard chemi-cal techniques.
3. Results
3.1. Modulation of neuronal cell death by cAMP-dependent signalling
Depolarising concentrations of potassium promote thesurvival of cerebellar granule neurons, and shiftingmature granule neuron cultures from 25 to 5 mM KCltriggers an apoptotic death cascade (D’Mello et al.,1993; Schulz et al., 1996b; Miller et al., 1997). Thisphenomenon is dependent on the PI-3-K pathway (Miller
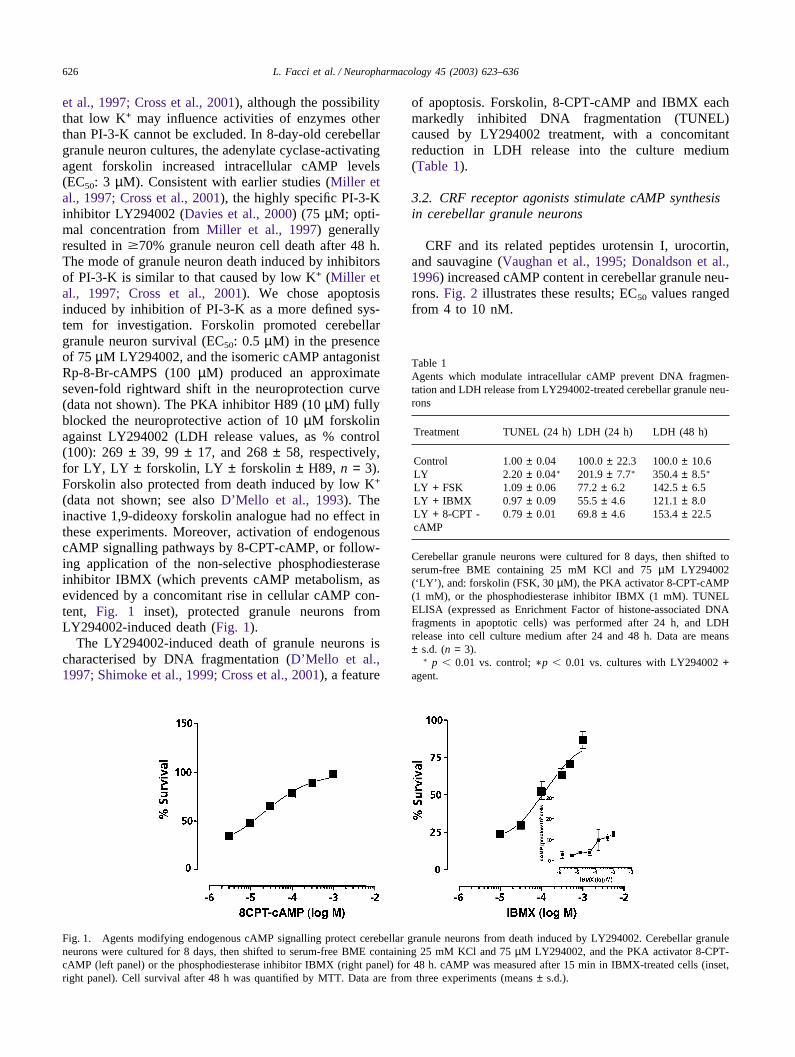
626 L. Facci et al. / Neuropharmacology 45 (2003) 623–636
et al., 1997; Cross et al., 2001), although the possibilitythat low K+ may influence activities of enzymes otherthan PI-3-K cannot be excluded. In 8-day-old cerebellargranule neuron cultures, the adenylate cyclase-activatingagent forskolin increased intracellular cAMP levels(EC50: 3 µM). Consistent with earlier studies (Miller etal., 1997; Cross et al., 2001), the highly specific PI-3-Kinhibitor LY294002 (Davies et al., 2000) (75 µM; opti-mal concentration from Miller et al., 1997) generallyresulted in �70% granule neuron cell death after 48 h.The mode of granule neuron death induced by inhibitorsof PI-3-K is similar to that caused by low K+ (Miller etal., 1997; Cross et al., 2001). We chose apoptosisinduced by inhibition of PI-3-K as a more defined sys-tem for investigation. Forskolin promoted cerebellargranule neuron survival (EC50: 0.5 µM) in the presenceof 75 µM LY294002, and the isomeric cAMP antagonistRp-8-Br-cAMPS (100 µM) produced an approximateseven-fold rightward shift in the neuroprotection curve(data not shown). The PKA inhibitor H89 (10 µM) fullyblocked the neuroprotective action of 10 µM forskolinagainst LY294002 (LDH release values, as % control(100): 269 ± 39, 99 ± 17, and 268 ± 58, respectively,for LY, LY ± forskolin, LY ± forskolin ± H89, n = 3).Forskolin also protected from death induced by low K+
(data not shown; see also D’Mello et al., 1993). Theinactive 1,9-dideoxy forskolin analogue had no effect inthese experiments. Moreover, activation of endogenouscAMP signalling pathways by 8-CPT-cAMP, or follow-ing application of the non-selective phosphodiesteraseinhibitor IBMX (which prevents cAMP metabolism, asevidenced by a concomitant rise in cellular cAMP con-tent, Fig. 1 inset), protected granule neurons fromLY294002-induced death (Fig. 1).
The LY294002-induced death of granule neurons ischaracterised by DNA fragmentation (D’Mello et al.,1997; Shimoke et al., 1999; Cross et al., 2001), a feature
Fig. 1. Agents modifying endogenous cAMP signalling protect cerebellar granule neurons from death induced by LY294002. Cerebellar granuleneurons were cultured for 8 days, then shifted to serum-free BME containing 25 mM KCl and 75 µM LY294002, and the PKA activator 8-CPT-cAMP (left panel) or the phosphodiesterase inhibitor IBMX (right panel) for 48 h. cAMP was measured after 15 min in IBMX-treated cells (inset,right panel). Cell survival after 48 h was quantified by MTT. Data are from three experiments (means ± s.d.).
of apoptosis. Forskolin, 8-CPT-cAMP and IBMX eachmarkedly inhibited DNA fragmentation (TUNEL)caused by LY294002 treatment, with a concomitantreduction in LDH release into the culture medium(Table 1).
3.2. CRF receptor agonists stimulate cAMP synthesisin cerebellar granule neurons
CRF and its related peptides urotensin I, urocortin,and sauvagine (Vaughan et al., 1995; Donaldson et al.,1996) increased cAMP content in cerebellar granule neu-rons. Fig. 2 illustrates these results; EC50 values rangedfrom 4 to 10 nM.
Table 1Agents which modulate intracellular cAMP prevent DNA fragmen-tation and LDH release from LY294002-treated cerebellar granule neu-rons
Treatment TUNEL (24 h) LDH (24 h) LDH (48 h)
Control 1.00 ± 0.04 100.0 ± 22.3 100.0 ± 10.6LY 2.20 ± 0.04∗ 201.9 ± 7.7∗ 350.4 ± 8.5∗
LY + FSK 1.09 ± 0.06 77.2 ± 6.2 142.5 ± 6.5LY + IBMX 0.97 ± 0.09 55.5 ± 4.6 121.1 ± 8.0LY + 8-CPT - 0.79 ± 0.01 69.8 ± 4.6 153.4 ± 22.5cAMP
Cerebellar granule neurons were cultured for 8 days, then shifted toserum-free BME containing 25 mM KCl and 75 µM LY294002(‘LY’ ), and: forskolin (FSK, 30 µM), the PKA activator 8-CPT-cAMP(1 mM), or the phosphodiesterase inhibitor IBMX (1 mM). TUNELELISA (expressed as Enrichment Factor of histone-associated DNAfragments in apoptotic cells) was performed after 24 h, and LDHrelease into cell culture medium after 24 and 48 h. Data are means± s.d. (n = 3).
∗ p � 0.01 vs. control; ∗p � 0.01 vs. cultures with LY294002 +agent.

627L. Facci et al. / Neuropharmacology 45 (2003) 623–636
Fig. 2. CRF and sauvagine elevate cAMP in cerebellar granule neurons. Cerebellar granule neurons were cultured for 8 days, then shifted toserum-free BME containing 25 mM KCl and CRF or sauvagine in the presence of 0.5 mM IBMX. Following incubation for 15 min at 37 °C,cells were lysed and intracellular cAMP content measured. Data are means ± s.d. (n = 3); similar results were obtained in two other experiments.EC50 values were calculated using GraphPad Prism software.
3.3. CRF and related peptides suppress apoptosis andare neuroprotective
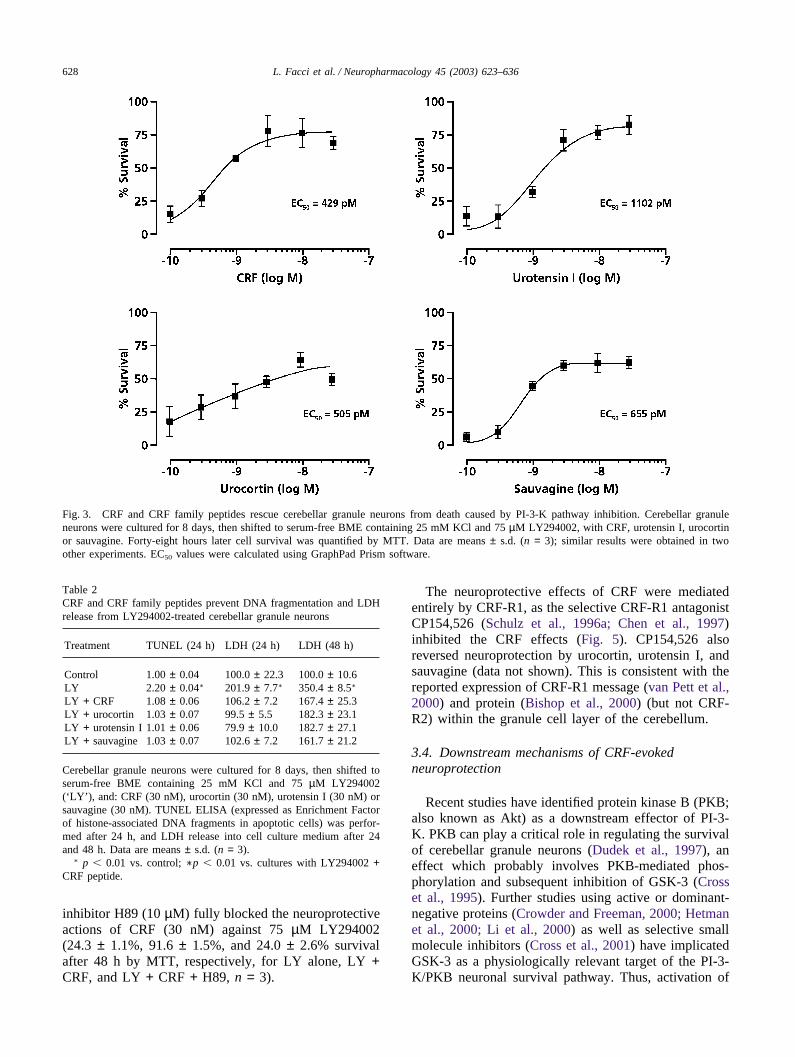
CRF and its related peptides were tested for theirability to protect against apoptotic death caused by inhi-bition of PI-3-K. CRF, urotensin I, urocortin, and sauva-gine all prevented neuronal cell death (Fig. 3; EC50
range: ~0.3–1.2 nM). Injury induced by the structurallydistinct PI-3-K inhibitor wortmannin (0.3 µM) was alsoreduced by CRF (100 nM), as measured by LDH releaseinto the culture medium (190 ± 20% and 115 ± 1% ofcontrol (=100), respectively, n = 6, p � 0.01; comparableresults were obtained using MTT as the index of cellsurvival). Moreover CRF, urotensin I, urocortin, andsauvagine each strongly inhibited DNA fragmentation(TUNEL) caused by LY294002 treatment, with a con-comitant reduction in LDH release into the culturemedium (Table 2). The time-course of LY294002-induced granule neuron death, and rescue by CRF, isshown in Table 3, utilising TUNEL, LDH release, andMTT as readouts. DNA fragmentation (TUNEL assay)preceded both LDH release and MTT, not unexpectedly,and reached plateau by 24 h, while LDH and MTTincreased to 48 h. CRF provided significant neuroprotec-tion over the 48-h incubation period. Delayed applicationof CRF, for up to 8 h following treatment withLY294002 also provided significant neuroprotection
(Table 4). Cerebellar granule neuron death triggered bylowering extracellular K+ to 5 mM was also diminishedby CRF (data not shown).
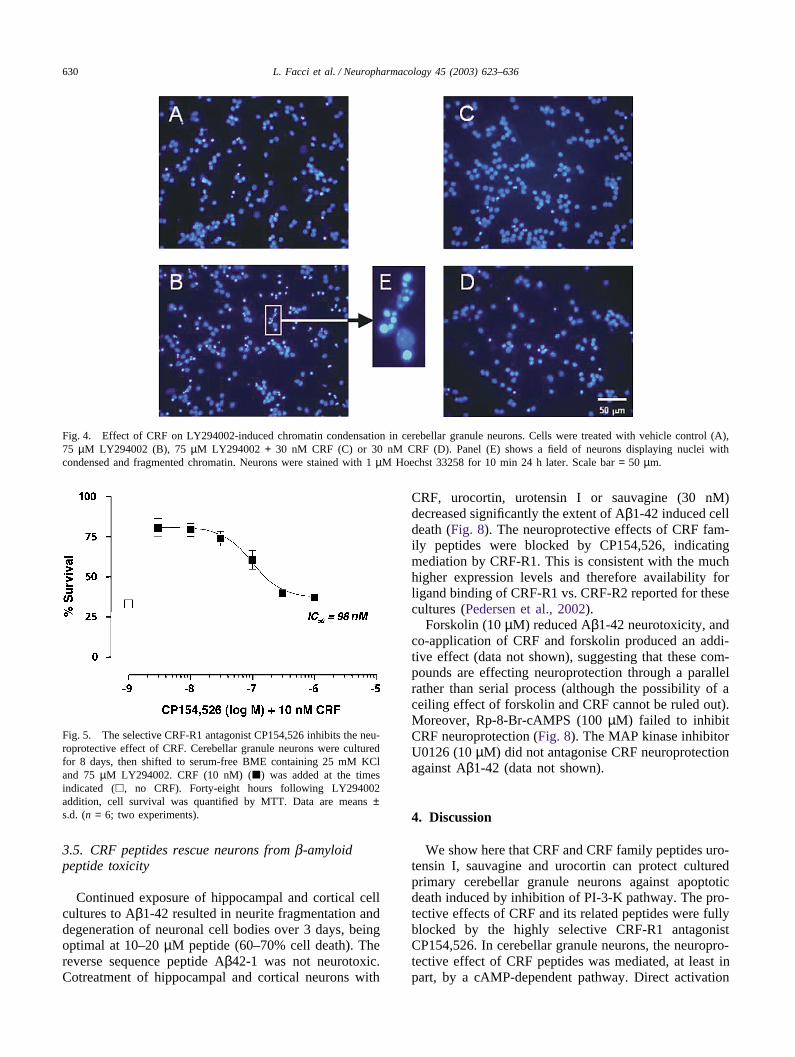
LY294002-induced cerebellar granule neuron celldeath is accompanied by chromatin condensation,another feature of apoptosis (Miller et al., 1997; Shi-moke et al., 1999). Therefore, chromatin condensationwas examined with Hoechst 33258 fluorescent dye. Incontrol cultures, cells showed little fluorescence in thenucleus (Fig. 4A). On the other hand, condensed andfragmented chromatin was clearly evident in LY294002-treated cultures (Fig. 4B). CRF greatly reduced theamount of condensed chromatin in LY294002-treatedneurons (Fig. 4C), and did not influence this parameterin control cultures (Fig. 4D).
The apparent lower concentrations of CRF agonistrequired to obtain neuroprotection (vs. cAMPproduction) may be due to inherent differences in theparameters measured (cAMP vs. cell survival), the timeto assay end-point (15 min vs. 48 h), or to a ‘ threshold’level of cAMP required to promote survival. Interest-ingly, a similar potency pattern was observed with for-skolin and IBMX (Fig. 1). In fact, the non-selective pho-sphodiesterase inhibitor only modestly increased cAMPcontent (less than 10-fold, compared to several hundred-fold by forskolin and CRF), although it was equipotentto CRF and forskolin as a neuroprotectant. The PKA
628 L. Facci et al. / Neuropharmacology 45 (2003) 623–636
Fig. 3. CRF and CRF family peptides rescue cerebellar granule neurons from death caused by PI-3-K pathway inhibition. Cerebellar granuleneurons were cultured for 8 days, then shifted to serum-free BME containing 25 mM KCl and 75 µM LY294002, with CRF, urotensin I, urocortinor sauvagine. Forty-eight hours later cell survival was quantified by MTT. Data are means ± s.d. (n = 3); similar results were obtained in twoother experiments. EC50 values were calculated using GraphPad Prism software.
Table 2CRF and CRF family peptides prevent DNA fragmentation and LDHrelease from LY294002-treated cerebellar granule neurons
Treatment TUNEL (24 h) LDH (24 h) LDH (48 h)
Control 1.00 ± 0.04 100.0 ± 22.3 100.0 ± 10.6LY 2.20 ± 0.04∗ 201.9 ± 7.7∗ 350.4 ± 8.5∗
LY + CRF 1.08 ± 0.06 106.2 ± 7.2 167.4 ± 25.3LY + urocortin 1.03 ± 0.07 99.5 ± 5.5 182.3 ± 23.1LY + urotensin I 1.01 ± 0.06 79.9 ± 10.0 182.7 ± 27.1LY + sauvagine 1.03 ± 0.07 102.6 ± 7.2 161.7 ± 21.2
Cerebellar granule neurons were cultured for 8 days, then shifted toserum-free BME containing 25 mM KCl and 75 µM LY294002(‘LY’ ), and: CRF (30 nM), urocortin (30 nM), urotensin I (30 nM) orsauvagine (30 nM). TUNEL ELISA (expressed as Enrichment Factorof histone-associated DNA fragments in apoptotic cells) was perfor-med after 24 h, and LDH release into cell culture medium after 24and 48 h. Data are means ± s.d. (n = 3).
∗ p � 0.01 vs. control; ∗p � 0.01 vs. cultures with LY294002 +CRF peptide.
inhibitor H89 (10 µM) fully blocked the neuroprotectiveactions of CRF (30 nM) against 75 µM LY294002(24.3 ± 1.1%, 91.6 ± 1.5%, and 24.0 ± 2.6% survivalafter 48 h by MTT, respectively, for LY alone, LY +CRF, and LY + CRF + H89, n = 3).
The neuroprotective effects of CRF were mediatedentirely by CRF-R1, as the selective CRF-R1 antagonistCP154,526 (Schulz et al., 1996a; Chen et al., 1997)inhibited the CRF effects (Fig. 5). CP154,526 alsoreversed neuroprotection by urocortin, urotensin I, andsauvagine (data not shown). This is consistent with thereported expression of CRF-R1 message (van Pett et al.,2000) and protein (Bishop et al., 2000) (but not CRF-R2) within the granule cell layer of the cerebellum.
3.4. Downstream mechanisms of CRF-evokedneuroprotection
Recent studies have identified protein kinase B (PKB;also known as Akt) as a downstream effector of PI-3-K. PKB can play a critical role in regulating the survivalof cerebellar granule neurons (Dudek et al., 1997), aneffect which probably involves PKB-mediated phos-phorylation and subsequent inhibition of GSK-3 (Crosset al., 1995). Further studies using active or dominant-negative proteins (Crowder and Freeman, 2000; Hetmanet al., 2000; Li et al., 2000) as well as selective smallmolecule inhibitors (Cross et al., 2001) have implicatedGSK-3 as a physiologically relevant target of the PI-3-K/PKB neuronal survival pathway. Thus, activation of
629L. Facci et al. / Neuropharmacology 45 (2003) 623–636
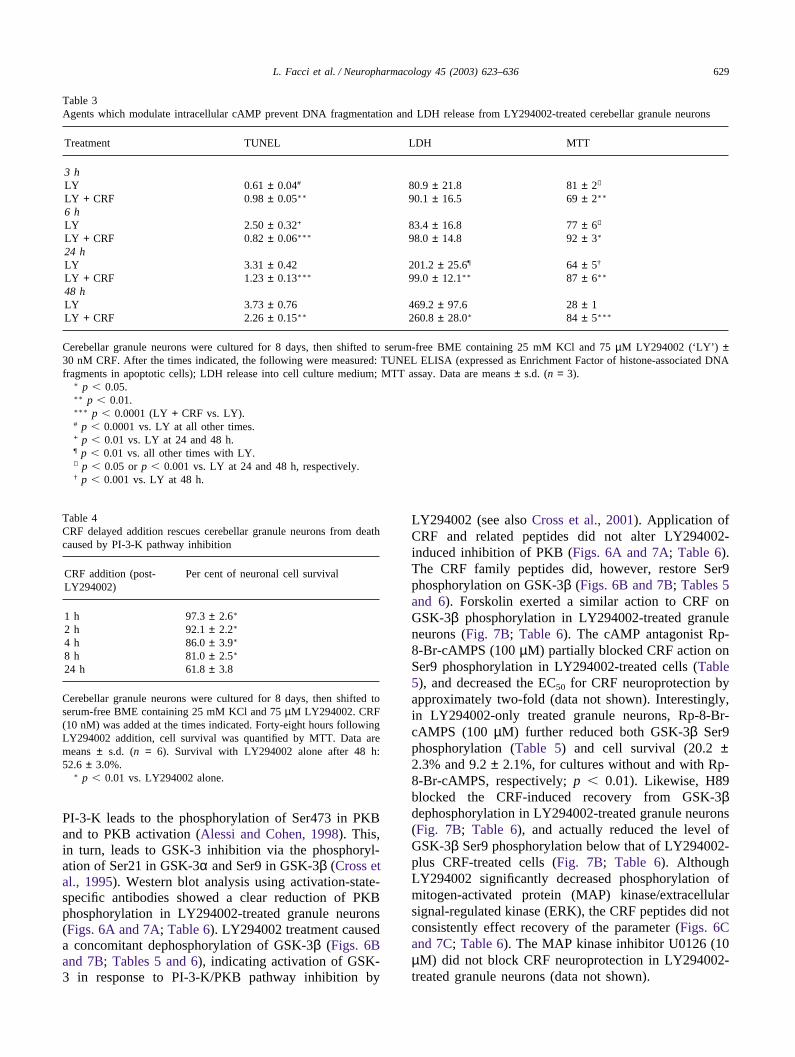
Table 3Agents which modulate intracellular cAMP prevent DNA fragmentation and LDH release from LY294002-treated cerebellar granule neurons
Treatment TUNEL LDH MTT
3 hLY 0.61 ± 0.04# 80.9 ± 21.8 81 ± 2✠
LY + CRF 0.98 ± 0.05∗∗ 90.1 ± 16.5 69 ± 2∗∗
6 hLY 2.50 ± 0.32+ 83.4 ± 16.8 77 ± 6✠
LY + CRF 0.82 ± 0.06∗∗∗ 98.0 ± 14.8 92 ± 3∗
24 hLY 3.31 ± 0.42 201.2 ± 25.6¶ 64 ± 5†
LY + CRF 1.23 ± 0.13∗∗∗ 99.0 ± 12.1∗∗ 87 ± 6∗∗
48 hLY 3.73 ± 0.76 469.2 ± 97.6 28 ± 1LY + CRF 2.26 ± 0.15∗∗ 260.8 ± 28.0∗ 84 ± 5∗∗∗
Cerebellar granule neurons were cultured for 8 days, then shifted to serum-free BME containing 25 mM KCl and 75 µM LY294002 (‘LY’ ) ±30 nM CRF. After the times indicated, the following were measured: TUNEL ELISA (expressed as Enrichment Factor of histone-associated DNAfragments in apoptotic cells); LDH release into cell culture medium; MTT assay. Data are means ± s.d. (n = 3).
∗ p � 0.05.∗∗ p � 0.01.∗∗∗ p � 0.0001 (LY + CRF vs. LY).# p � 0.0001 vs. LY at all other times.+ p � 0.01 vs. LY at 24 and 48 h.¶ p � 0.01 vs. all other times with LY.✠ p � 0.05 or p � 0.001 vs. LY at 24 and 48 h, respectively.† p � 0.001 vs. LY at 48 h.
Table 4CRF delayed addition rescues cerebellar granule neurons from deathcaused by PI-3-K pathway inhibition
CRF addition (post- Per cent of neuronal cell survivalLY294002)
1 h 97.3 ± 2.6∗
2 h 92.1 ± 2.2∗
4 h 86.0 ± 3.9∗
8 h 81.0 ± 2.5∗
24 h 61.8 ± 3.8
Cerebellar granule neurons were cultured for 8 days, then shifted toserum-free BME containing 25 mM KCl and 75 µM LY294002. CRF(10 nM) was added at the times indicated. Forty-eight hours followingLY294002 addition, cell survival was quantified by MTT. Data aremeans ± s.d. (n = 6). Survival with LY294002 alone after 48 h:52.6 ± 3.0%.
∗ p � 0.01 vs. LY294002 alone.
PI-3-K leads to the phosphorylation of Ser473 in PKBand to PKB activation (Alessi and Cohen, 1998). This,in turn, leads to GSK-3 inhibition via the phosphoryl-ation of Ser21 in GSK-3α and Ser9 in GSK-3β (Cross etal., 1995). Western blot analysis using activation-state-specific antibodies showed a clear reduction of PKBphosphorylation in LY294002-treated granule neurons(Figs. 6A and 7A; Table 6). LY294002 treatment causeda concomitant dephosphorylation of GSK-3β (Figs. 6Band 7B; Tables 5 and 6), indicating activation of GSK-3 in response to PI-3-K/PKB pathway inhibition by
LY294002 (see also Cross et al., 2001). Application ofCRF and related peptides did not alter LY294002-induced inhibition of PKB (Figs. 6A and 7A; Table 6).The CRF family peptides did, however, restore Ser9phosphorylation on GSK-3β (Figs. 6B and 7B; Tables 5and 6). Forskolin exerted a similar action to CRF onGSK-3β phosphorylation in LY294002-treated granuleneurons (Fig. 7B; Table 6). The cAMP antagonist Rp-8-Br-cAMPS (100 µM) partially blocked CRF action onSer9 phosphorylation in LY294002-treated cells (Table5), and decreased the EC50 for CRF neuroprotection byapproximately two-fold (data not shown). Interestingly,in LY294002-only treated granule neurons, Rp-8-Br-cAMPS (100 µM) further reduced both GSK-3β Ser9phosphorylation (Table 5) and cell survival (20.2 ±2.3% and 9.2 ± 2.1%, for cultures without and with Rp-8-Br-cAMPS, respectively; p � 0.01). Likewise, H89blocked the CRF-induced recovery from GSK-3βdephosphorylation in LY294002-treated granule neurons(Fig. 7B; Table 6), and actually reduced the level ofGSK-3β Ser9 phosphorylation below that of LY294002-plus CRF-treated cells (Fig. 7B; Table 6). AlthoughLY294002 significantly decreased phosphorylation ofmitogen-activated protein (MAP) kinase/extracellularsignal-regulated kinase (ERK), the CRF peptides did notconsistently effect recovery of the parameter (Figs. 6Cand 7C; Table 6). The MAP kinase inhibitor U0126 (10µM) did not block CRF neuroprotection in LY294002-treated granule neurons (data not shown).
630 L. Facci et al. / Neuropharmacology 45 (2003) 623–636
Fig. 4. Effect of CRF on LY294002-induced chromatin condensation in cerebellar granule neurons. Cells were treated with vehicle control (A),75 µM LY294002 (B), 75 µM LY294002 + 30 nM CRF (C) or 30 nM CRF (D). Panel (E) shows a field of neurons displaying nuclei withcondensed and fragmented chromatin. Neurons were stained with 1 µM Hoechst 33258 for 10 min 24 h later. Scale bar = 50 µm.
Fig. 5. The selective CRF-R1 antagonist CP154,526 inhibits the neu-roprotective effect of CRF. Cerebellar granule neurons were culturedfor 8 days, then shifted to serum-free BME containing 25 mM KCland 75 µM LY294002. CRF (10 nM) (�) was added at the timesindicated (�, no CRF). Forty-eight hours following LY294002addition, cell survival was quantified by MTT. Data are means ±s.d. (n = 6; two experiments).
3.5. CRF peptides rescue neurons from b-amyloidpeptide toxicity
Continued exposure of hippocampal and cortical cellcultures to Aβ1-42 resulted in neurite fragmentation anddegeneration of neuronal cell bodies over 3 days, beingoptimal at 10–20 µM peptide (60–70% cell death). Thereverse sequence peptide Aβ42-1 was not neurotoxic.Cotreatment of hippocampal and cortical neurons with
CRF, urocortin, urotensin I or sauvagine (30 nM)decreased significantly the extent of Aβ1-42 induced celldeath (Fig. 8). The neuroprotective effects of CRF fam-ily peptides were blocked by CP154,526, indicatingmediation by CRF-R1. This is consistent with the muchhigher expression levels and therefore availability forligand binding of CRF-R1 vs. CRF-R2 reported for thesecultures (Pedersen et al., 2002).
Forskolin (10 µM) reduced Aβ1-42 neurotoxicity, andco-application of CRF and forskolin produced an addi-tive effect (data not shown), suggesting that these com-pounds are effecting neuroprotection through a parallelrather than serial process (although the possibility of aceiling effect of forskolin and CRF cannot be ruled out).Moreover, Rp-8-Br-cAMPS (100 µM) failed to inhibitCRF neuroprotection (Fig. 8). The MAP kinase inhibitorU0126 (10 µM) did not antagonise CRF neuroprotectionagainst Aβ1-42 (data not shown).
4. Discussion
We show here that CRF and CRF family peptides uro-tensin I, sauvagine and urocortin can protect culturedprimary cerebellar granule neurons against apoptoticdeath induced by inhibition of PI-3-K pathway. The pro-tective effects of CRF and its related peptides were fullyblocked by the highly selective CRF-R1 antagonistCP154,526. In cerebellar granule neurons, the neuropro-tective effect of CRF peptides was mediated, at least inpart, by a cAMP-dependent pathway. Direct activation
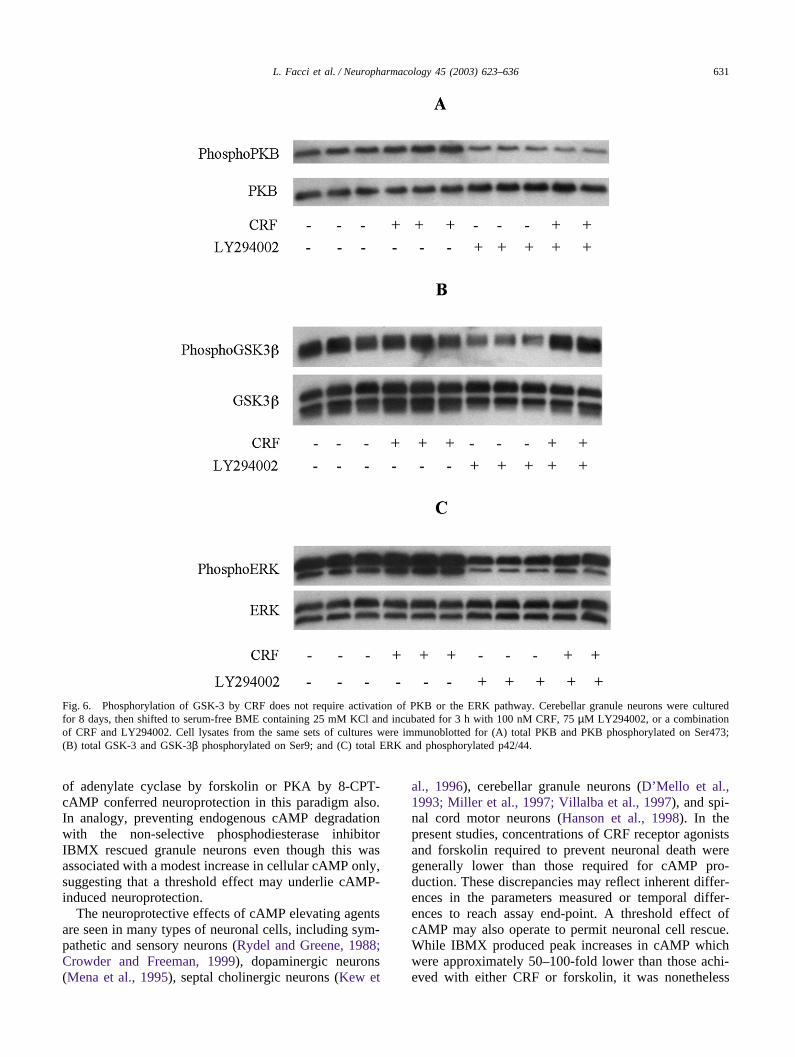
631L. Facci et al. / Neuropharmacology 45 (2003) 623–636
Fig. 6. Phosphorylation of GSK-3 by CRF does not require activation of PKB or the ERK pathway. Cerebellar granule neurons were culturedfor 8 days, then shifted to serum-free BME containing 25 mM KCl and incubated for 3 h with 100 nM CRF, 75 µM LY294002, or a combinationof CRF and LY294002. Cell lysates from the same sets of cultures were immunoblotted for (A) total PKB and PKB phosphorylated on Ser473;(B) total GSK-3 and GSK-3β phosphorylated on Ser9; and (C) total ERK and phosphorylated p42/44.
of adenylate cyclase by forskolin or PKA by 8-CPT-cAMP conferred neuroprotection in this paradigm also.In analogy, preventing endogenous cAMP degradationwith the non-selective phosphodiesterase inhibitorIBMX rescued granule neurons even though this wasassociated with a modest increase in cellular cAMP only,suggesting that a threshold effect may underlie cAMP-induced neuroprotection.
The neuroprotective effects of cAMP elevating agentsare seen in many types of neuronal cells, including sym-pathetic and sensory neurons (Rydel and Greene, 1988;Crowder and Freeman, 1999), dopaminergic neurons(Mena et al., 1995), septal cholinergic neurons (Kew et
al., 1996), cerebellar granule neurons (D’Mello et al.,1993; Miller et al., 1997; Villalba et al., 1997), and spi-nal cord motor neurons (Hanson et al., 1998). In thepresent studies, concentrations of CRF receptor agonistsand forskolin required to prevent neuronal death weregenerally lower than those required for cAMP pro-duction. These discrepancies may reflect inherent differ-ences in the parameters measured or temporal differ-ences to reach assay end-point. A threshold effect ofcAMP may also operate to permit neuronal cell rescue.While IBMX produced peak increases in cAMP whichwere approximately 50–100-fold lower than those achi-eved with either CRF or forskolin, it was nonetheless

632 L. Facci et al. / Neuropharmacology 45 (2003) 623–636
Fig. 7. CRF family peptides and forskolin restore phosphorylation ofGSK-3 in LY294002-treated cerebellar granule neurons. Granule neu-rons were cultured for 8 days, then shifted to serum-free BME contain-ing 25 mM KCl and incubated for 3 h with 75 µM LY294002 ± 30nM CRF or related peptide, 10 µM forskolin (FSK), or 30 nM CRF+ 10 µM H89. Cell lysates from the same sets of cultures were immu-noblotted for (A) total PKB and PKB phosphorylated on Ser473; (B)total GSK-3 and GSK-3β phosphorylated on Ser9; and (C) total ERKand phosphorylated p42/44. Lane 1, control; lanes 2-8, LY294002 plus:none (2); CRF (3); CRF + H89 (4); urotensin I (5); sauvagine (6);urocortin (7); FSK (8).
Table 5Effect of CRF and LY294002 on Ser9 phosphorylation in GSK-3β
Treatment Per cent of control phosphorylation
Control 100LY294002 50.7 ± 8.7 [6]CRF + LY294002 126.9 ± 10.3 [6]CRF + LY294002 + Rp 90.8 ± 12.5∗ [3]LY294002 + Rp 28.4 ± 1.2∗∗ [3]
Cerebellar granule neurons were cultured for 8 days, then shifted toserum-free BME containing 25 mM KCl and incubated for 3 h with100 nM CRF, 75 µM LY294002, and/or 100 µM Rp-8-Br-cAMPS(‘Rp’ ). Cell lysates were immunoblotted for total GSK-3 and GSK-3β phosphorylated on Ser9. Phospho-specific immunoreactivity wasquantified by densitometric analysis and expressed relative to totalGSK-3 protein (means ± s.d.; ‘n’ indicated in brackets).
∗ p � 0.05 vs. CRF + LY294002.∗∗ p � 0.01 vs. LY294002.
neuroprotective in this paradigm. Conversely, inhibitionof endogenous cAMP signalling proved to be injuriousper se, and propose that maintenance of tonic levels ofcAMP may play a role in cerebellar granule neuron sur-vival.
The mechanisms underlying neuroprotection bycAMP are poorly understood. Two putative signallingpathways that could mediate the neuroprotective effectof CRF were explored, namely the MAP kinase/ERKpathway and the PI-3-K/PKB pathway. The ERK path-
way has been implicated in the protection of PC12 cellsfrom nerve growth factor withdrawal-induced apoptosis(Xia et al., 1995). However, other studies with sympath-etic neurons indicate that nerve growth factor and cAMPpromote neuronal survival through ERK-independentpathway(s) (Virdee and Tolkovsky, 1995; Creedon et al.,1996). Activation of the cAMP and MAP kinase signal-ling pathways appears a prerequisite for pituitary adenyl-ate cyclase-activating peptide-mediated protection ofcerebellar granule neurons (Villalba et al., 1997), and forCRF-mediated neuroprotection from excitotoxicity in rathippocampus (Elliott-Hunt et al., 2002). In the presentstudies, CRF family peptides did not appear to consist-ently alter ERK phosphorylation of granule neurons, andMAP kinase inhibitors failed to block the neuroprotec-tive activity of CRF. These results concur with others(Creedon et al., 1996; Li et al., 2000) and suggest that,in rat cerebellar granule neurons, the MAP kinase/ERKpathway does not mediate cAMP-evoked neuroprotec-tion. Thus far, most studies that have examined a roleof PI-3-K/PKB in cAMP-mediated neuroprotection havedemonstrated that the PKB pathway is not involved. Inaccordance, here we shown that CRF family peptides,and forskolin, had no effect on PKB phosphorylation incerebellar granule neurons, and failed to promote recov-ery of PKB activity in the presence of the PI-3-K inhibi-tor LY294002. Thus, PKB is unlikely to mediate theneuroprotective effects of CRF. In contrast, studies usingGSK-3 active or dominant-negative proteins (Crowderand Freeman, 2000; Hetman et al., 2000; Li et al., 2000),as well as selective small molecule inhibitors (Cross etal., 2001) of GSK-3, have provided strong evidence tosuggest that GSK-3 acts as a principal target throughwhich the PI-3-K/PKB pathway regulates neuronalviability. Treatment with CRF and related peptides, aswell as forskolin, restored Ser9 phosphorylation onGSK-3β in cerebellar granule neurons challenged withLY294002. Conversely, the cAMP antagonist Rp-8-Br-cAMPS partially blocked CRF action on Ser9 phos-phorylation in LY294002-treated cells and decreased theEC50 for CRF neuroprotection, and reduced Ser9 phos-phorylation on GSK-3β and cell viability in LY294002-treated granule neurons in the absence of CRF. In a simi-lar vein, the PKA inhibitor H89 blocked fully the neuro-protective effect of CRF (and forskolin) and inhibitedCRF-induced phosphorylation of GSK-3β. Differencesin inhibitor efficacy may relate to their distinct modesof action: Rp-8-Br-cAMPS and H89 compete for thebinding of cAMP and ATP, respectively, to PKA. Acaveat should be noted with regard to the use of H89:although this compound is marketed by Calbiochem as‘a selective and potent inhibitor of PKA’ , H89 (10 µM)is reported to inhibit at least eight protein kinases by 80–100%; three (MSK1, S6K1 and ROCK-II) with apotency similar to or greater than that for PKA (Davieset al., 2000). Our data suggest a novel signalling path-

633L. Facci et al. / Neuropharmacology 45 (2003) 623–636
Table 6Effect of forskolin and CRF family peptides on phosphorylation of PKB, GSK-3β and ERK proteins in LY294002-treated cerebellar granule neurons
Treatment Relative phosphorylation (% control)
PKB GSK-3β ERK
Control 100 100 100LY294002 plusNo addition 32.5 ± 5.0 57.4 ± 11.7 46.8 ± 7.6Forskolin 31.9 ± 11.9 119.3 ± 11.9∗ 88.8 ± 12.9∗
CRF 33.5 ± 5.3 113.6 ± 13.8∗ 67.8 ± 10.0∗
CRF + H89 31.6 ± 5.4 18.1 ± 1.2# 20.4 ± 8.0#
Urotensin I 32.0 ± 4.7 116.8 ± 14.7∗ 73.8 ± 11.7∗
Sauvagine 37.6 ± 8.1 106.7 ± 14.2∗ 51.7 ± 3.9Urocortin 29.4 ± 8.1 90.5 ± 16.5∗ 61.1 ± 6.4
Cerebellar granule neurons were cultured for 8 days, then shifted to serum-free BME containing 25 mM KCl and incubated for 3 h with 75 µMLY294002, and 30 nM of the indicated CRF family peptide or 10 µM forskolin. A second set of cultures with CRF contained also 10 µM H89.Cell lysates were immunoblotted for: PKB phosphorylated on Ser473 and total PKB protein; GSK-3β phosphorylated on Ser9 and total GSK-3protein; phosphorylated p42/44 and total ERK protein. Phospho-specific immunoreactivities were quantified by densitometric analysis and expressedrelative to total protein in each case. Values are means ± s.e.m. (n = 3).
∗ p � 0.05 vs. LY294002 alone.# p � 0.01 vs. LY294002 alone.
Fig. 8. Protective effects of CRF family peptides against Aβ1-42 induced cell death in cultured hippocampal and cortical neurons, and block bythe CRF-R1 antagonist CP154,526. Cultures were exposed for 3 days to CRF, urocortin, urotensin I (Uro I), or sauvagine (each at 30 nM) togetherwith Aβ1-42 at 20 µM. Where indicated, CP154,526 (‘CP’ ) was added at 1 µM. Values are means ± s.d. (n = 3). ∗p � 0.01 vs. Aβ1-42 (‘none’ ).Values for CRF peptide + CP did not differ from Aβ1-42 alone.
way by which CRF promotes neuronal survival throughactivation of type 1 CRF receptors involving, down-stream, a cAMP-dependent mechanism—possibly PKA,but not PKB—that leads to inhibition of GSK-3β, a pro-apoptotic kinase in cerebellar granule neurons (Li et al.,2000). These observations are supported by recent workwhich showed that cAMP activates PKA phosphoryl-ation of Ser9 and subsequent inactivation of GSK-3β (Liet al., 2000).
Excessive activation of the hypothalamic-pituitary–adrenal axis and consequent hypersecretion of glucocort-icoids may underlie a number of conditions, includingdepression and certain anxiety disorders (Arborelius etal., 1999). We, and others, have previously demonstrated
that α-helical CRF (9-41), an antagonist of both CRFreceptor subtypes (Owens and Nemeroff, 1991), reducesbrain damage in a model of focal cerebral ischemia andprotects against excitotoxic damage caused by activationof N-methyl-d-aspartic acid receptors in rat brain (Lyonset al., 1991; Strijbos et al., 1994). In this context, it isworth noting that α-helical CRF (9-41) has recently beenshown to display partial, albeit weak, agonist activity atCRF-R1 (Smart et al., 1999). This raises the possibilitythat the neuroprotective effects seen in vivo with the pre-sumed CRF receptor antagonist (Lyons et al., 1991;Strijbos et al., 1994) may, in fact, be achieved throughan agonist action on CRF type 1 receptors. This notionis supported by a recent report demonstrating that in

634 L. Facci et al. / Neuropharmacology 45 (2003) 623–636
organotypic hippocampal slice cultures, CRF and uro-cortin prevent glutamate-induced neurotoxicity (Elliott-Hunt et al., 2002).
Deficits in CRF signalling may be involved in motordisorders related to the basal ganglia, namely Hunt-ington’s chorea (Heuser et al., 1991), as well as in olivo-pontocerebellar atrophy and spinocerebellar degener-ation (Mizuno et al., 1995; Suemaru et al., 1995), andAlzheimer’s disease (De Souza, 1995). To evaluate apossible role of CRF in the latter disease, we assessedthe neuroprotective efficacy of CRF (peptides) on β-amyloid-evoked neurotoxicity. Our data show that CRFand CRF-like peptides are capable of rescuing corticaland hippocampal neurons from Aβ1-42 induced death, ina PKA and MAP kinase pathway-independent manner.Although these findings are consistent with the reportedability of CRF peptides to protect CNS neurons fromAβ25-35 death (Pedersen et al., 2001, 2002) and oxidat-ive stress (Lezoualc’h et al., 2000), they differ with thesuggested involvement of PKA and MAP kinase signal-ling in the Aβ25-35 effect (Pedersen et al., 2002). Aβ1-42 constitutes the mature form of amyloid peptide foundin Alzheimer’s disease brain plaques and, in contrast toAβ25-35, is a neuropathological hallmark of this neurod-egenerative disorder (Selkoe, 2001). Distinct mech-anisms of neurotoxicity for Aβ1-42 and Aβ25-35 mayaccount for these differences, and will require furtherstudy to clarify this issue. In vivo, urocortin was cardiop-rotective following ischemic and reperfusion injury (Braret al., 2000), and we have recently obtained preliminaryevidence to suggest that urotensin I reduces infarct sizein experimental cerebral ischemia (Strijbos, Skaper andParsons, unpublished observations). Finally, ligandswhich displace CRF from its binding protein improvedspatial learning and memory in rats (Behan et al., 1995).
Taken together, the data strongly suggest that stimul-ating CRF type 1 receptors may confer a number of ben-eficial effects, including direct neuroprotection as shownhere, which could ultimately prove efficacious in pre-venting or reducing the burden of neurodegenerative dis-eases.
References
Alessi, D.R., Cohen, P., 1998. Mechanism of activation and functionof protein kinase B. Current Opinion in Genetics and Development8, 55–62.
Arborelius, L., Owens, M.J., Plotsky, P.M., Nemeroff, C.B., 1999. Therole of corticotrophin-releasing factor in depression and anxietydisorders. Journal of Endocrinology 160, 1–12.
Battaglia, G., Webster, E.L., De Souza, E.B., 1987. Characterization ofcorticotropin-releasing factor receptor-mediated adenylate cyclaseactivity in the rat central nervous system. Synapse 1, 572–581.
Behan, D.P., Heinrichs, S.C., Troncoso, J.C., Liu, X.J., Kawas, C.H.,Liang, N., De Souza, E.B., 1995. Displacement of corticotropinreleasing factor from its binding protein as a possible treatment forAlzheimer’s disease. Nature 378, 284–287.
Bishop, G.A., Seelandt, C.M., King, J.S., 2000. Cellular localizationof corticotrophin releasing factor receptors in the adult mouse cere-bellum. Neuroscience 101, 1083–1092.
Boje, K.M., Wong, G., Skolnick, P., 1993. Desensitization on theNMDA receptor complex by glycinergic ligands in cerebellar gran-ule cell cultures. Brain Research 603, 207–214.
Brar, B.K., Jonassen, A.K., Stephanou, A., Santilli, G., Railson, J.,Knight, R.A., Yellon, D.M., Latchman, D.S., 2000. Urocortin pro-tects against ischemic and reperfusion injury via a MAPK-depen-dent pathway. Journal of Biological Chemistry 275, 8508–8514.
Chalmers, D.T., Lovenberg, T.W., Grigoriadis, D.E., Behan, D.P., DeSouza, E.B., 1996. Corticotrophin-releasing factor receptors: frommolecular biology to drug design. Trends in PharmacologicalSciences 17, 166–172.
Chang, C.-P., Pearse, R.V. II, O’Connell, S., Rosenfeld, M.G., 1993.Identification of a seven transmembrane helix receptor for cortico-trophin-releasing factor and sauvagine in mammalian brain. Neuron11, 1187–1195.
Chen, Y.L., Mansbach, R.S., Winter, S.M., Brooks, E., Collins, J.,Corman, M.L., Dunaiskis, A.R., Faraci, W.S., Gallaschun, R.J.,Schmidt, A., Schulz, D.W., 1997. Synthesis and oral efficacy of a4-(butylethylamino)pyrrolo[2,3-d]pyrimidine: a centrally activecorticotrophin-releasing factor1 receptor antagonist. Journal ofMedicinal Chemistry 40, 1749–1754.
Creedon, D.J., Johnson, E.M., Lawrence, J.C., 1996. Mitogen-activatedprotein kinase-independent pathways mediate the effects of nervegrowth factor and cAMP on neuronal survival. Journal of Biologi-cal Chemistry 271, 20713–20718.
Cross, D.A.E., Alessi, D.R., Cohen, P., Andjelkovich, M., Hemmings,B.A., 1995. Inhibition of glycogen synthase kinase-3 by insulinmediated by protein kinase B. Nature 378, 785–789.
Cross, D.A.E., Culbert, A.A., Chalmers, K.A., Facci, L., Skaper, S.D.,Reith, A.D., 2001. Selective small-molecule inhibitors of glycogensynthase kinase-3 activity protect primary neurones from death.Journal of Neurochemistry 77, 94–102.
Crowder, R.J., Freeman, R.S., 1999. The survival of sympathetic neu-rons promoted by potassium depolarization, but not cAMP, requiresphosphatidylinositol 3-kinase and Akt. Journal of Neurochemistry73, 466–475.
Crowder, R.J., Freeman, R.S., 2000. Glycogen synthase kinase-3βactivity is critical for neuronal death caused by inhibiting phosphat-idylinositol 3-kinase or Akt but not for death caused by nervegrowth factor withdrawal. Journal of Biological Chemistry 275,34266–34271.
Davies, S.P., Reddy, H., Caivano, M., Cohen, P., 2000. Specificityand mechanism of action of some commonly used protein kinaseinhibitors. Biochemical Journal 351, 95–105.
De Souza, E.B., 1995. Corticotrophin-releasing factor receptors: physi-ology, pharmacology, biochemistry and role in central nervous sys-tem and immune disorders. Psychoneuroendocrinology 20, 789–819.
D’Mello, S.R., Galli, C., Ciotti, T., Calissano, P., 1993. Induction ofapoptosis in cerebellar granule neurons by low potassium: inhi-bition of death by insulin-like growth factor I and cAMP. Proceed-ings of the National Academy of Sciences of the United States ofAmerica 90, 10989–10993.
D’Mello, S.R., Borodezt, K., Soltoff, S.P., 1997. Insulin-like growthfactor and potassium depolarization maintain neuronal survival bydistinct pathways: possible involvement of PI 3-kinase in IGF-1signaling. Journal of Neuroscience 17, 1548–1560.
Donaldson, C.J., Sutton, S.W., Perrin, M.H., Corrigan, A.Z., Lewis,K.A., Rivier, J.E., Vaughan, J.M., Vale, W.W., 1996. Cloning andcharacterization of human cerocortin. Endocrinology 137, 3896.
Dudek, H., Datta, S.R., Franke, T.F., Birnbaum, M.J., Yao, R., Cooper,G.M., Segal, R.A., Kaplan, D.R., Greenberg, M.E., 1997. Regu-lation of neuronal survival by the serine-threonine protein kinaseAkt. Science 275, 661–665.

635L. Facci et al. / Neuropharmacology 45 (2003) 623–636
Elliott-Hunt, C.R., Kazlauskaite, J., Wilde, G.J.C., Grammatopoulos,D.K., Hillhouse, E.W., 2002. Potential signalling pathways under-lying corticotrophin-releasing hormone-mediated neuroprotectionfrom excitotoxicity in rat hippocampus. Journal of Neurochemistry80, 416–425.
Facci, L., Stevens, D.L., Skaper, S.D., Strijbos, P.J.L.M., 2000. Cortic-otrophin-releasing factor receptor agonists prevent neuronalapoptosis triggered by phosphatidylinositol 3-kinase pathway inhib-tion. Society for Neuroscience Abstract 27, 2052.
Gallo, V., Kingsbury, A., Balazs, R., Jorgensen, O.S., 1987. The roleof depolarization in the survival and differentiation of cerebellargranule cells in culture. Journal of Neuroscience 7, 2203–2213.
Hanson, M.G. Jr., Shen, S., Wiemelt, A.P., McMorris, F.A., Barres,B.A., 1998. Cyclic AMP elevation is sufficient to promote the sur-vival of spinal motor neurons in vitro. Journal of Neuroscience 18,7361–7371.
Hetman, M., Cavanaugh, J.E., Kimelman, D., Xia, Z., 2000. Role ofglycogen synthase kinase-3β in neuronal apoptosis induced bytrophic withdrawal. Journal of Neuroscience 20, 2567–2574.
Heuser, I.J., Chase, T.N., Mouradian, M.M., 1991. The limbic-hypo-thalamic-pituitary–adrenal axis in Huntington’s disease. BiologicalPsychiatry 30, 943–952.
Kew, J.N., Smith, D.W., Sofroniew, M.V., 1996. Nerve growth factorwithdrawal induces the apoptotic death of developing septal chol-inergic neurons in vitro: protection by cyclic AMP analogue andhigh potassium. Neuroscience 70, 329–339.
Kishimoto, T., Pearse, R.V., Lin, C.R., Rosenfeld, M.G., 1995. Asauvagine/corticotrophin-releasing factor expressed in heart andskeletal muscle. Proceedings of the National Academy of Sciencesof the United States of America 92, 1108–1112.
Lezoualc’h, F., Engert, S., Berning, B., Behl, C., 2000. Corticotrophin-releasing hormone-mediated neuroprotection against oxidativestress is associated with the increased release of nonamyloidogenicamyloid β precursor protein and with suppression of nuclear factor-�B. Molecular Endocrinology 14, 147–159.
Li, M., Wang, X., Meintzer, M.K., Laessig, T., Birnbaum, M.J., Heid-enreich, K.A., 2000. Cyclic AMP promotes neuronal survival byphosphorylation of glycogen synthase kinase 3β. Molecular andCellular Biology 20, 9356–9363.
Loo, D.T., Copani, A., Pike, C.J., Whittemore, E.R., Walencewicz,A.J., Cotman, C.W., 1993. Apoptosis is induced by β-amyloid incultured central nervous system neurons. Proceedings of theNational Academy of Sciences of the United States of America 90,7951–7955.
Lovenberg, T.W., Liaw, C.W., Grigoriadis, D.E., Clevenger, W., Chal-mers, D.T., De Souza, E.B., Oltersdorf, T., 1995. Cloning andcharacterisation of a functionally distinct corticotrophin-releasingfactor receptor subtype from rat brain. Proceedings of the NationalAcademy of Sciences of the United States of America 92, 836–840.
Lyons, M.K., Anderson, R.E., Meyer, F.B., 1991. Corticotropin releas-ing factor antagonist reduces ischemic hippocampal neuronalinjury. Brain Research 545, 339–342.
Manthorpe, M., Fagnani, R., Skaper, S.D., Varon, S., 1986. An auto-mated colorimetric assay for neurotrophic factors. DevelopmentalBrain Research 25, 191–198.
Mattson, M.P., Cheng, B., Davis, D., Bryant, K., Lieberburg, I., Rydel,R.E., 1992. β-Amyloid peptides destabilize calcium homeostasisand render human cortical neurons vulnerable to excitotoxicity.Journal of Neuroscience 12, 376–389.
Mena, M.A., Caserejos, M.J., Bonin, A., Ramos, J.A., Yebenes, J.G.,1995. Effects of dibutyryl cyclic AMP and retinoic acid on thedifferentiation of dopamine neurons: prevention of cell death bydibutyryl cyclic AMP. Journal of Neurochemistry 65, 2612–2620.
Miller, T.M., Tansey, M.G., Johnson, E.M. Jr., Creedon, D.J., 1997.Inhibition of phosphatidylinositol 3-kinase activity blocks depolar-isation- and insulin-like growth factor 1-mediated survival of cer-
ebellar granule cells. Journal of Biological Chemistry 272, 9847–9853.
Mizuno, Y., Takahashi, K., Totsune, K., Ohneda, M., Konno, H.,Murakami, O., Satoh, F., Sone, M., Takase, S., Itoyama, Y., Mouri,T., 1995. Decrease in cerebellin and corticotrophin-releasing hor-mone in the cerebellum of olivopontocerebellar atrophy and Shy-Drager syndrome. Brain Research 686, 115–118.
Owens, M.J., Nemeroff, C.B., 1991. Physiology and pharmacology ofcorticotrophin-releasing factor. Pharmacological Reviews 43,425–473.
Patel, J., Zinkand, W.C., Thompson, C., Keith, R., Salama, A., 1990.Role of glycine in the N-methyl-d-aspartate-mediated neuronalcytotoxicity. Journal of Neurochemistry 54, 849–854.
Pedersen, W.A., McCullers, D., Culmsee, C., Haughey, N.J., Herman,J.P., Mattson, M.P., 2001. Corticotrophin-releasing hormone pro-tects neurons against insults relevant to the pathogenesis of Alzhei-mer’s disease. Neurobiology of Disease 8, 492–503.
Pedersen, W.A., Wan, R., Zhang, P., Mattson, M.P., 2002. Urocortin,but not urocortin II, protects cultured hippocampal neurons fromoxidative and excitotoxic cell death via corticotropin-releasing hor-mone receptor type I. Journal of Neuroscience 22, 404–412.
Perrin, M.H., Vale, W.W., 1999. Corticotrophin releasing factor recep-tors and their ligand family. Annals of the New York Academy ofSciences 885, 312–328.
Perrin, M., Donaldson, C., Chen, R., Blount, A., Berggren, T., Bilezikj-ian, L., Sawchenko, P., Vale, W., 1995. Identification of a secondcorticotrophin-releasing factor receptor gene and characterizationof a cDNA expressed in heart. Proceedings of the National Acad-emy of Sciences of the United States of America 92, 2969–2973.
van Pett, K., Viau, V., Bittencourt, J.C., Chan, R.K.W., Li, H.-Y.,Arias, C., Prins, G.S., Perrin, M., Vale, W., Sawchenko, P.E., 2000.Distribution of mRNAs encoding CRF receptors in brain and pitu-itary of rat and mouse. Journal of Comparative Neurology 428,191–212.
Portera-Cailliau, C., Hedreen, J.C., Price, D.L., Koliatsos, V.E., 1995.Evidence for apoptotic cell death in Huntington disease and excito-toxic animal models. Journal of Neuroscience 15, 3775–3787.
Rabizadeh, S., Butler, G.E., Borchelt, D.R., Gwinn, R., Selverstone,V.J., Sisodia, S., Wong, P., Lee, M., Hahn, H., Bredesen, D.E.,1995. Mutations associated with amyotrophic lateral sclerosis con-vert superoxide dismutase from an antiapoptotic gene to a proapop-totic gene: studies in yeast and neural cells. Proceedings of theNational Academy of Sciences of the United States of America 92,3024–3028.
Rydel, R.E., Greene, L.A., 1988. cAMP analogs promote survival andneurite outgrowth in cultures of rat sympathetic and sensory neu-rons independently of nerve growth factor. Proceedings of theNational Academy of Sciences of the United States of America 85,1257–1261.
Schulz, D.W., Mansbach, R.S., Sprouse, J., Braselton, J.P., Collins, J.,Corman, M., Dunaiskis, A., Faraci, S., Schmidt, A.W., Seeger, T.,Seymour, P., Tingley, F.D. III, Winston, E.N., Chen, Y.L., Heym,J., 1996a. CP-154,526: a potent and selective nonpeptide antagonistof corticotrophin releasing factor receptors. Proceedings of theNational Academy of Sciences of the United States of America 93,10477–10482.
Schulz, J.B., Weller, M., Klockgether, T., 1996b. Potassium depri-vation-induced apoptosis of cerebellar granule neurons: a sequen-tial requirement for new mRNA and protein synthesis, ICE-likeprotease activity, and reactive oxygen species. Journal of Neurosci-ence 16, 4696–4706.
Selkoe, D.J., 2001. Alzheimer’s disease: genes, proteins, and therapy.Physiological Reviews 81, 741–766.
Shimoke, K., Yamagishi, S., Yamada, M., Ikeuchi, T., Hatanaka, H.,1999. Inhibition of phosphatidylinositol 3-kinase activity elevatesc-Jun N-terminal kinase activity in apoptosis of cultured cerebellargranule neurons. Developmental Brain Research 112, 245–253.

636 L. Facci et al. / Neuropharmacology 45 (2003) 623–636
Skaper, S.D., Facci, L., Milani, D., Leon, A., Toffano, G., 1990. Cul-ture and use of primary and clonal neural cells. In: Conn, P.M.(Ed.), Methods in Neurosciences, vol. 2. Academic Press, SanDiego, pp. 17–33.
Smart, D., Coppell, A., Rossant, C., Hall, M., McKnight, A.T., 1999.Characterisation using microphysiometry of CRF receptor pharma-cology. European Journal of Pharmacology 379, 229–235.
Strijbos, P.J.L.M., Relton, J.K., Rothwell, N.J., 1994. Corticotrophinreleasing factor antagonist inhibits neuronal damage induced byfocal cerebral ischemia or activation of NMDA receptors in the ratbrain. Brain Research 656, 405–408.
Suemaru, S., Suemaru, K., Kawai, S., Miyata, S., Nobukuni, K., Ihara,Y., Namba, R., Urakami, K., Hashimoto, K., 1995. Cerebrospinalfluid corticotrophin-releasing hormone in neurodegenerative dis-eases: reduction in spinocerebellar degeneration. Life Sciences 57,2231–2235.
Vaughan, J., Donaldson, J., Bittencourt, J., Perrin, M.H., Lewis, K.,Sutton, S., Chan, R., Turnbull, A.V., Lovejoy, D., Rivier, C., Riv-ier, J., Sawchenko, P.E., Vale, W., 1995. Urocortin, a mammalianneuropeptide related to fish urotensin I and to corticotrophin-releas-ing factor. Nature 378, 287–292.
Villalba, M., Bockaert, J., Journot, L., 1997. Pituitary adenylatecyclase-activating polypeptide (PACAP-38) protects cerebellargranule neurons from apoptosis by activating the mitogen-activatedprotein kinase (MAP kinase) pathway. Journal of Neuroscience 17,83–90.
Virdee, K., Tolkovsky, A.M., 1995. Activation of p44 and p42 MAPkinases is not essential for the survival of rat sympathetic neurons.European Journal of Neuroscience 7, 2159–2169.
Xia, Z., Dickens, M., Raingeaud, J., Davis, R.J., Greenberg, M.E.,1995. Opposing effects of ERK and JNK-p38 MAP kinases onapoptosis. Science 270, 1326–1331.
Copyright © 2022 FDOKUMEN




















