
Using generative adversarial networks for genome variant ...
Upload
independentCategory
view
2download
0
Molecular & Biochemical Parasitology 113 (2001) 67–78
Conservation of metacyclic variant surface glycoprotein expressionsites among different trypanosome isolates�
Frederic Bringaud a,*, Nicolas Biteau a, John E. Donelson b, Theo Baltz a
a Laboratoire de Parasitologie Moleculaire, Uni6ersite Victor Segalen Bordeaux II, UMR-5016 CNRS, 146 rue Leo Saignat,33076 Bordeaux cedex, France
b Department of Biochemistry, Uni6ersity of Iowa, Iowa City, IA 52242, USA
Received 10 May 2000; received in revised form 28 November 2000; accepted 30 November 2000
Abstract
We identified in a Trypanosoma brucei brucei strain (AnTat 1) an expression site for a metacyclic variant surface glycoprotein(MVSG) gene (MVSG) that was previously characterized in a T. b. rhodesiense strain (WRATat 1.1). The 3.4 kb sequences of thetwo expression sites are 99.6% identical, with no differences in the sequence of the 1.5 kb MVSG. Two other MVSGs in theWRATat 1.1 genome are not present in the AnTat 1 genome. In addition, five other T. b. brucei and T. b. rhodesiense strains,isolated in the same geographic region as the two former strains, do not contain any of these three MVSGs. Two of these fivestrains, however, appear to possess a very similar MVSG expression site, but with different MVSGs in it. Thus, the presence ofthe same MVSG in the same expression site in two different isolates is unusual and may be the result of genetic exchange in thefield between T. b. brucei and T. b. rhodesiense isolates. Analysis of other African trypanosome strains for the presence of the threeWRATat 1.1 MVSG expression sites demonstrated that the expression sites’ promoter sequences are much more likely to bepresent than are specific MVSGs, suggesting that loss of MVSGs is the result of replacement by other VSGs. The promoter regionof the MVSG expression site active in the WRATat 1.1 MVAT7 variant was found to be highly conserved among T. b. brucei,T. b. rhodesiense and T. b. gambiense group 2 isolates, whereas it does not occur in the T. b. gambiense group 1 isolates tested.A phylogenetic analysis of this promoter region sequence shows that the T. b. gambiense group 2 isolates form a monophyleticclade well separated from the T. b. brucei/T. b. rhodesiense isolates. Thus, whilst the T. b. brucei, T. b. rhodesiense and T. b.gambiense group 2 isolates are closely related but heterogenous, molecular tools may be developed to distinguish T. b. gambiensegroup 2 isolates from the others. © 2001 Elsevier Science B.V. All rights reserved.
Keywords: Expression site; Metacyclic VSG gene repertoire; Phylogeny; Trypanosoma brucei gambiense typing; Trypanosoma brucei group
www.parasitology-online.com.
1. Introduction
African trypanosomes, which are protozoan parasitesthat cause potentially fatal diseases in humans anddomestic livestock, have a life cycle that alternates
between the tsetse fly and the mammal. In the blood-stream of their mammalian hosts, the parasites evadethe immune response by antigenic variation, a contin-ual switching of the variant surface glycoprotein (VSG)that constitutes the surface coat. Although each blood-stream trypanosome has a single VSG species on itssurface, the parasite genome has a repertoire of severalhundred different VSG genes (VSGs), which are ex-pressed in a mutually exclusive manner from about 20potential bloodstream expression sites (BESs) that areinvariantly located near telomeres [1,2]. Only one of theapproximately 20 BESs is activated at a time by amechanism that is still unknown. These expression sitesare long polycistronic transcription units in which theVSG is co-transcribed with several intervening expres-
Abbre6iations: BES, bloodstream expression site; BVAT, blood-stream variant antigen type; BVSG, bloodstream variable surfaceglycoprotein; ES, expression site; MES, metacyclic expression site;MVAT, metacyclic variant antigen type; MVSG, metacyclic variablesurface glycoprotein; VSG, variable surface glycoprotein.� Note : Nucleotide data reported in this paper have been submitted
to the GenBank™ database with the accession number AF259553.* Corresponding author. Tel.: +33-5-57571010; fax: +33-5-
57571015.E-mail address: [email protected] (F. Bringaud).
0166-6851/01/$ - see front matter © 2001 Elsevier Science B.V. All rights reserved.PII: S 0 1 6 6 -6851 (00 )00381 -9

F. Bringaud et al. / Molecular & Biochemical Parasitology 113 (2001) 67–7868
sion site associated genes (ESAGs) from a promoterabout 45–60 kb upstream [3,4].
VSGs are first expressed during the metacyclic stage(MVSGs) of the life cycle in the salivary glands of thetsetse fly as a pre-adaptation to life in the mammals.During its blood meal, the fly transmits to the mammala metacyclic trypanosome population that is antigeni-cally heterogeneous, but with each individual expressinga single MVSG. The metacyclic parasites as a wholeexpress a limited repertoire of approximately 20–30variant antigen types (MVATs), compared to the sev-eral hundred that can be potentially expressed in thebloodstream form [5]. The few metacyclic variants stud-ied in detail were derived from the WRATat 1.1(MVAT4, 5 and 7) and EATRO 795 (1.22 and 1.61)strains of T. b. rhodesiense and T. b. brucei, respectively[6,7]. All the MVSG genes (MVSGs) characterized sofar are located in telomere-linked metacyclic expressionsites (MESs) where they are transcribed into mono-cistronic precursor RNAs from a proximal promoterlocated within 2 kb of the MVSG [8–13]. The MVSGsare activated transcriptionally in the metacyclic formand continue to be expressed in the bloodstream formsduring the first 6–7 days following fly transmission.
Here, we have examined different strains of the T.brucei group for the presence of MVSGs and promoterregions contained in the active MESs previously char-acterized in three variants of the WRATat 1.1 strain(MVAT4, 5 and 7). The analyses revealed that thepromoter regions of MESs are much more conservedthan MVSGs, suggesting that gene conversion ofMVSGs occurs in MESs. The MES active in MVAT7could be a tool for trypanosome isolate typing, since itis present in all the strains tested, except the T. b.gambiense group 1 strains, and a phylogenetic analysisof MVAT7-ES promoter region shows that the T. b.gambiense group 2 isolates can be distinguished fromthe T. b. brucei/T. b. rhodesiense isolates.
2. Materials and methods
2.1. Trypanosomes and nucleic acids isolation
The origins of all the trypanosome stocks used werepreviously detailed by Biteau et al. [14], except theWRATat 1.1 strain [15] (see also Fig. 4 for the originsof the WRATat 1.1, AnTat 1, EATRO 427, STIB 345,ETat 1.1, UTat 1/8 and STIB 859 strains). These stockswere either grown as procyclic cells in axenic liquidculture medium or maintained as bloodstream form inexperimental animals and purified by anion exchangechromatography [16]. The parasites (109–1010) werecollected, and standard genomic DNA preparationswere performed [17].
2.2. Construction and screening of genomic library
Cosmid vector c2X75 [18] was used to generate a T.b. brucei genomic DNA library, as described previously[19]. Large DNA fragments generated by partial Sau3Adigestion of genomic DNA were inserted into theBamHI site of the vector as previously described [20].The cosmid library (2000 clones) was screened with an[a-32P]dCTP-labeled PCR fragment corresponding tothe 5% coding region of the MVSG expressed in theMVAT5 expression site (MVSG5). This PCR fragmentwas amplified from WRATat 1.1 genomic DNA usingVSG5.3 and VSG5.4 primers.
2.3. Polymerase chain reaction and detection
All PCR amplifications were carried out with theGeneAmp® PCR System 2400 (Perkin Elmer) in 25 mlreaction mixtures containing 100 mM of each dATP,dTTP, dCTP, dGTP, 0.1 mM of each primer, 10 ng ofpurified genomic DNA, 1× reaction buffer containing3 mM MgCl2 and 0.2 unit GoldStar Red DNA poly-merase (Eurogentec). The PCR reaction was conductedby ‘‘touch down’’ as follows: denaturation 5 min at96°C, 5 cycles (10 s at 96°C, 30 s at 60°C and up to 5min at 72°C), 25 cycles (10 s at 96°C, 30 s at 55°C andup to 5 min at 72°C) and 10 min at 72°C. The PCRproducts were separated on an ethidium bromide-stained, 0.8 or 2% agarose gel together with molecularweight standards. The Sequences of the primers are(5%–3%): ES1.1, GTTAGCGGGCGCTGTCAGCA;VSG1.1, CGCAGCGGCCAACGTCAAC; VSG1.2,CTAGTCCTGCCGCTTTAAGC; ES4.1, CAATCG-TTCCTATTTGTCGC; ES4.3, CTTTGAAGAGGA-ACCGCA; ES4.4, TGATACTCAGCAGTTGGTC;ES4.5, CAGATATATAGCCATTCAC; VSG4.1, TC-TATATGACCGGCTTGCTG; VSG4.3, CAAGTTT-CGCGGCAACCA; ES5.3, TACAGAAC-CAGTG-TGCTTCG; ES5.5, CGTTCTGTATATGCACGAC;ES5.6, TAAGATGGAAACAGTGCG; VSG5.3, AG-AATCCGTTTATCACGCTG; VSG5.4, CGAGCT-GCCAAGCCCGAC; ES7.1, GCCGGTGTCAAA-CCTTAGCT; ES7.3, CTGTACTGAAAGCTGTCC;VSG7.2, GTGCACAAACTATGCAGCAA; VSG7.3,TTTCGGTCGGCGTGAATG.
2.4. Southern blotting analysis
Digestion of 3 mg of parasite genomic DNA by therestriction enzymes EcoRV and SmaI/HpaI and sepa-ration on 0.8 and 2% agarose gels were performed asdescribed by Sambrook et al. [21]. After electrophore-sis, DNA was transferred onto neutral nylon mem-branes (Appligene®, Oncor®) and hybridized with an

F. Bringaud et al. / Molecular & Biochemical Parasitology 113 (2001) 67–78 69
[a-32P]dCTP-labeled (Rediprime™ II kit -AmershamPharmacia Biotech) DNA fragment in 0.1% SDS and6× SSPE (1× SSPE is 0.18 M NaCl; 10 mMNaH2PO4; 1 mM EDTA [pH 7.0]). Membranes werewashed at low stringency (50°C in 0.5× SSPE and0.5% SDS) before autoradiography. Before rehybridiz-ing blots, probes were removed by boiling in a solutionof 0.5% SDS.
2.5. Sequencing
PCR fragments purified using the GFX™ kit (Amer-sham Pharmacia Biotech), and cosmid and plasmidDNAs purified using the Wizard™ plus kits (Promega),were sequenced with unlabeled primers on an auto-mated ABI 377 DNA sequencer (PE, Applied Biosys-tems) using the PRISM™ Dye Terminator CycleSequencing Ready Reaction kit (Applied Biosystems)as described by the manufacturer.
2.6. Alignments and phylogenetic analysis
DNA and amino acid sequences were analysed usingthe DNA STRIDER program, and database searcheswere conducted using BLAST. Multiple alignment ofthe 264 bp nucleotide sequences containing theMVAT7-ES promoter and flanking regions was ob-tained using CLUSTAL W version 1.6 [22], and thephylogenetic tree was constructed using version 3.5c ofthe PHYLIP program package of J. Felsenstein(BLAST, CLUSTAL W and PHYLIP were obtainedthrough Bisance and Infobiogen facilities). The matrixof pairwise sequence distances was calculated by Day-hoff’s method using DNADIST. The unrooted phylo-genetic trees were constructed from the distance matrixusing the neighbor or Fitch methods. The phylogenetictree was drawn with TREEVIEW version 1.3 [23]. Thestatistical robustness of the resulting phylogenetic treeswas assessed with the SEQBOOT program by bootstrapresampling analysis generating 100 reiterated data sets.The resulting bootstrap values were added manually ateach corresponding node.
3. Results
3.1. T. b. brucei and T. b. rhodesiense strains possessthe same MVAT-ES with the same MVSG
In the course of analysing a T. b. brucei (strainAnTat 1) cosmid DNA library by sequencing both endsof individual clones, we identified a clone (Cos.x35)containing at one end a VSG homologous to thatexpressed in the MVAT5 clone of T. b. rhodesiensestrain WRATat 1.1 [24] and a retroposon (ingi) at theother end. To obtain the full-length MVSG5, a second
cosmid clone (Cos.x42) overlapping the Cos.x35 clonewas isolated using a MVSG5 probe (Fig. 1A). A physi-cal map of both cosmids was constructed, and bothends of some internal fragments were sequenced toconfirm the locations of the ESAG1 that was previouslydetected upstream of the MVAT5 expression site(MVAT5-ES) of T. b. rhodesiense strain WRATat 1.1[24]. The restriction map and localisation of ESAG1and MVSG5 appeared to be identical in the Cos.x35/x42 clones and the MVAT5-ES of T. b. rhodesiense.The MVAT5-ES of the WRATat 1.1 strain contains,about 1.6 kb upstream of MVSG5, a transcriptionpromoter that is functional in transient transfections ofprocyclic organisms [6,8], and 740 bp downstream ofthis site is a sequence homologous to the metacyclicpromoter identified in the MVAT1.61-ES of theEATRO 795 strain [12]. For convenience, these se-quences will be called promoter 1 and promoter 2,respectively. A 3.4 kb region of the clone Cos.x42starting 242 bp upstream of the promoter 1 [6,8] andextending to the end of MVSG5 was sequenced.MVSG5 and a 500 bp segment containing the promoter1 in Cos.x42 were found to be completely identical tothe T. b. rhodesiense MVAT5-ES. The only differencesbetween these 3.4 kb sequences, which are 99.6% identi-cal, reside in the 1.3 kb region between the promoter 1and MVSG5, where 10 nucleotide substitutions and 10single-nucleotide deletions or insertions occur (Fig. 1B).This result suggests that the MVAT5-ES is present inboth the WRATat 1.1 strain of T. b. rhodesiense andthe AnTat 1 strain of T. b. brucei.
To confirm the presence of the MVAT5-ES in theAnTat 1 strain, a PCR analysis was conducted usingoligonucleotide primers deduced from the promoter 1and the N-terminal half of MVSG5 (Fig. 2A). A PCRproduct with the expected size is generated from theDNAs of WRATat 1.1 (lane 1) and AnTat 1 (lane 2)strains. In addition, the gene copy number of MVSG5was determined by Southern blot analysis of genomicDNAs using as probe a DNA fragment correspondingto the N-terminal coding half of MVSG5. A single 3.2kb EcoRV fragment is detected with this specific probein both strains, indicating that MVSG5 is encoded by asingle copy gene located in the MVAT5-ES (Fig. 3A).Furthermore, to confirm that the T. b. brucei and T. b.rhodesiense isolates are distinct strains, the length poly-morphism of four microsatellites was determined usinga PCR approach, as previously described [14] (Fig. 4).None of the eight alleles analysed are shared by the twostrains, indicating that they are very distant strains.Taken together, these data clearly show that theMVAT5-ES originally characterized in the T. b. rhode-siense WRATat 1.1 strain is also present with very fewsingle-nucleotide changes in the T. b. brucei AnTat 1strain.

F. Bringaud et al. / Molecular & Biochemical Parasitology 113 (2001) 67–7870
3.2. Analysis of MVAT5-ES in other strains
The T. b. brucei AnTat 1 and T. b. rhodesienseWRATat 1.1 strains were isolated in Uganda andKenya, respectively, both in the vicinity of Lake Victo-ria (see Fig. 4). To analyse the presence of MVSG5 inother trypanosome stocks, the same PCR and Southernblotting analyses were conducted on DNAs from twoother T. b. brucei stocks and three other T. b. rhode-siense stocks isolated in the same geographic region.The length polymorphism analysis of four microsatel-lites indicates that all seven stocks analysed, which wereisolated from different hosts at different times and indifferent regions of Kenya and Uganda, are differentstrains (Fig. 4). The PCR (Fig. 2A) and Southern blot(Fig. 3A) analyses showed that MVSG5 is not presentin the genomes of these five isolates.
The presence of the MVAT5-ES was tested in thegenomes of all these isolates by a PCR approach usingoligonucleotide primers deduced from promoter 2(ES5.6 primer) and a region 40 bp upstream of pro-
moter 1 (ES5.3 primer). A PCR product of the expected750 bp length could be amplified from the DNAs of theWRATat 1.1, AnTat 1, STIB 345 and ETat 1.1 strains(Fig. 2A). The sequences of the 750 bp fragmentscontaining both promoters were 89–100% identicalamong these four strains (data not shown). No PCRproduct was amplified from the DNAs of the otherthree strains. Southern blot analysis showed that thestrains whose genomic DNAs were templates for PCRamplification of the 750 bp DNA fragment were theonly strains to be recognized by a probe correspondingto the MVAT5-ES promoter region (Fig. 3C). How-ever, we failed to obtain a PCR product from theDNAs of the STIB 345 and ETat 1.1 strains when theES5.6 primer was replaced with the ES5.5 primer de-duced from a region 260 bp downstream of the pro-moter 2 (Fig. 2A). Taken together, these data suggestthat the STIB 345 and ETat 1.1 strains possess theMVAT5-ES containing a different MVSG and thathomology between these ESs ceases between the ES5.6and ES5.5 primers. Recently, Graham et al. showed
Fig. 1. Comparison of the MVAT5-ES in T. b. brucei AnTat 1 and T. b. rhodesiense WRATat 1.1 strains. (A) The top of the panel representsthe physical map of the MVAT5-ES deduced from analysis of the Cos.x35 and Cos.x42 clones isolated from a T. b. brucei AnTat 1 cosmid library.The MVSG5, ESAG1 and ingi are indicated by arrows, and the positions of the 2 cosmid clones are shown under the map. The lower maprepresents the region containing the MVAT5-ES previously sequenced in T. b. rhodesiense WRATat 1.1 [24]. The black arrowhead represents thetranscription start site identified by transient transfections of procyclic cells (promoter 1), as shown for the WRATat 1.1 strain [6,8], and the whitearrowhead represents the transcription start site in the metacyclic form (promoter 2), as shown in the EATRO 795 strain [12]. (B) The differencesobserved between the sequence of the MVAT5-ES from the two strains are indicated under the physical map. The positions of the differences arebased on the WRATat 1.1 sequence with the position +1 arbitrarily assigned to the beginning of the promoter 1 sequence. The nature of thesubstitutions, deletions (*) or insertions is indicated under the nucleotide positions. Abbreviations for restriction sites: B, BamHI; E, EcoRV; H,HpaI.
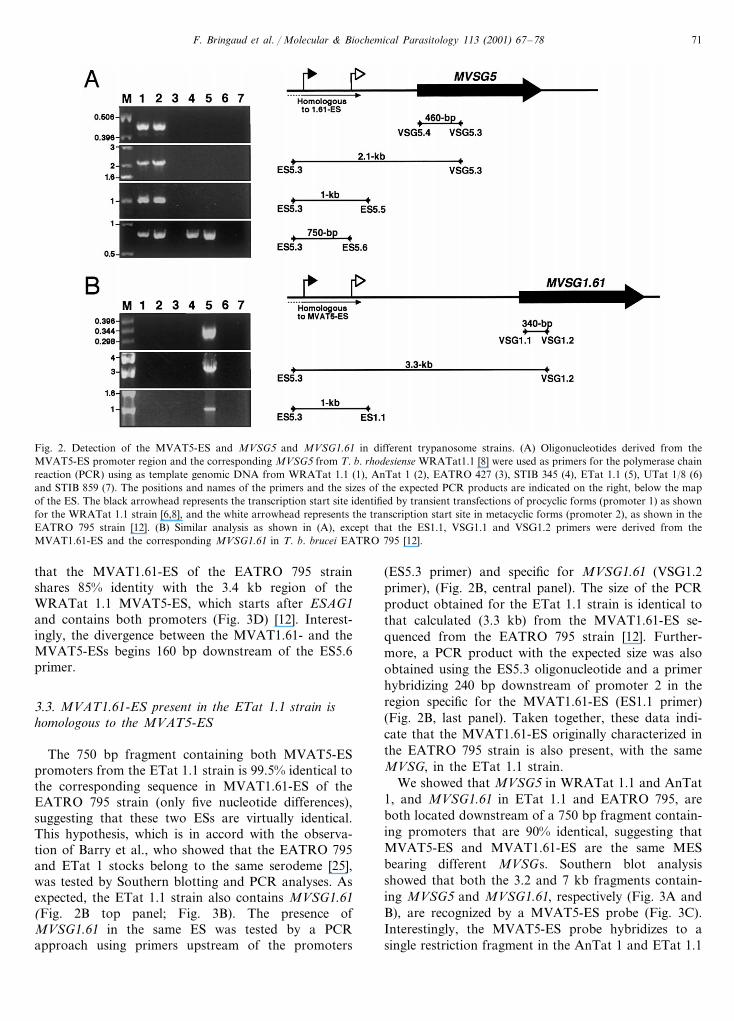
F. Bringaud et al. / Molecular & Biochemical Parasitology 113 (2001) 67–78 71
Fig. 2. Detection of the MVAT5-ES and MVSG5 and MVSG1.61 in different trypanosome strains. (A) Oligonucleotides derived from theMVAT5-ES promoter region and the corresponding MVSG5 from T. b. rhodesiense WRATat1.1 [8] were used as primers for the polymerase chainreaction (PCR) using as template genomic DNA from WRATat 1.1 (1), AnTat 1 (2), EATRO 427 (3), STIB 345 (4), ETat 1.1 (5), UTat 1/8 (6)and STIB 859 (7). The positions and names of the primers and the sizes of the expected PCR products are indicated on the right, below the mapof the ES. The black arrowhead represents the transcription start site identified by transient transfections of procyclic forms (promoter 1) as shownfor the WRATat 1.1 strain [6,8], and the white arrowhead represents the transcription start site in metacyclic forms (promoter 2), as shown in theEATRO 795 strain [12]. (B) Similar analysis as shown in (A), except that the ES1.1, VSG1.1 and VSG1.2 primers were derived from theMVAT1.61-ES and the corresponding MVSG1.61 in T. b. brucei EATRO 795 [12].
that the MVAT1.61-ES of the EATRO 795 strainshares 85% identity with the 3.4 kb region of theWRATat 1.1 MVAT5-ES, which starts after ESAG1and contains both promoters (Fig. 3D) [12]. Interest-ingly, the divergence between the MVAT1.61- and theMVAT5-ESs begins 160 bp downstream of the ES5.6primer.
3.3. MVAT1.61-ES present in the ETat 1.1 strain ishomologous to the MVAT5-ES
The 750 bp fragment containing both MVAT5-ESpromoters from the ETat 1.1 strain is 99.5% identical tothe corresponding sequence in MVAT1.61-ES of theEATRO 795 strain (only five nucleotide differences),suggesting that these two ESs are virtually identical.This hypothesis, which is in accord with the observa-tion of Barry et al., who showed that the EATRO 795and ETat 1 stocks belong to the same serodeme [25],was tested by Southern blotting and PCR analyses. Asexpected, the ETat 1.1 strain also contains MVSG1.61(Fig. 2B top panel; Fig. 3B). The presence ofMVSG1.61 in the same ES was tested by a PCRapproach using primers upstream of the promoters
(ES5.3 primer) and specific for MVSG1.61 (VSG1.2primer), (Fig. 2B, central panel). The size of the PCRproduct obtained for the ETat 1.1 strain is identical tothat calculated (3.3 kb) from the MVAT1.61-ES se-quenced from the EATRO 795 strain [12]. Further-more, a PCR product with the expected size was alsoobtained using the ES5.3 oligonucleotide and a primerhybridizing 240 bp downstream of promoter 2 in theregion specific for the MVAT1.61-ES (ES1.1 primer)(Fig. 2B, last panel). Taken together, these data indi-cate that the MVAT1.61-ES originally characterized inthe EATRO 795 strain is also present, with the sameMVSG, in the ETat 1.1 strain.
We showed that MVSG5 in WRATat 1.1 and AnTat1, and MVSG1.61 in ETat 1.1 and EATRO 795, areboth located downstream of a 750 bp fragment contain-ing promoters that are 90% identical, suggesting thatMVAT5-ES and MVAT1.61-ES are the same MESbearing different MVSGs. Southern blot analysisshowed that both the 3.2 and 7 kb fragments contain-ing MVSG5 and MVSG1.61, respectively (Fig. 3A andB), are recognized by a MVAT5-ES probe (Fig. 3C).Interestingly, the MVAT5-ES probe hybridizes to asingle restriction fragment in the AnTat 1 and ETat 1.1
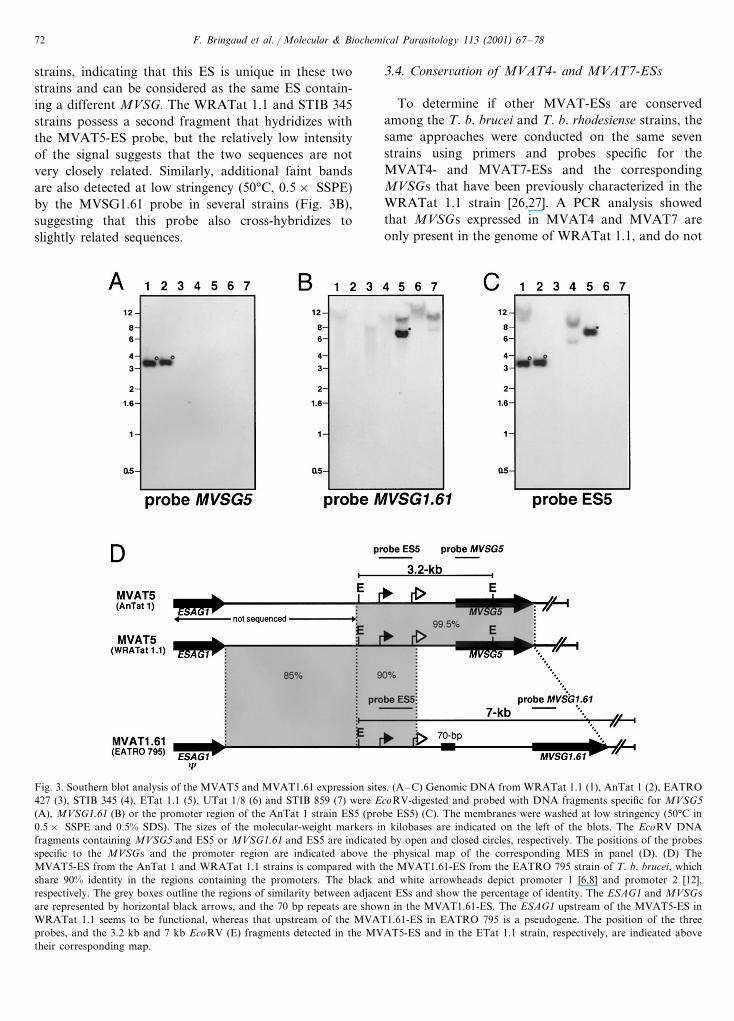
F. Bringaud et al. / Molecular & Biochemical Parasitology 113 (2001) 67–7872
strains, indicating that this ES is unique in these twostrains and can be considered as the same ES contain-ing a different MVSG. The WRATat 1.1 and STIB 345strains possess a second fragment that hydridizes withthe MVAT5-ES probe, but the relatively low intensityof the signal suggests that the two sequences are notvery closely related. Similarly, additional faint bandsare also detected at low stringency (50°C, 0.5× SSPE)by the MVSG1.61 probe in several strains (Fig. 3B),suggesting that this probe also cross-hybridizes toslightly related sequences.
3.4. Conser6ation of MVAT4- and MVAT7-ESs
To determine if other MVAT-ESs are conservedamong the T. b. brucei and T. b. rhodesiense strains, thesame approaches were conducted on the same sevenstrains using primers and probes specific for theMVAT4- and MVAT7-ESs and the correspondingMVSGs that have been previously characterized in theWRATat 1.1 strain [26,27]. A PCR analysis showedthat MVSGs expressed in MVAT4 and MVAT7 areonly present in the genome of WRATat 1.1, and do not
Fig. 3. Southern blot analysis of the MVAT5 and MVAT1.61 expression sites. (A–C) Genomic DNA from WRATat 1.1 (1), AnTat 1 (2), EATRO427 (3), STIB 345 (4), ETat 1.1 (5), UTat 1/8 (6) and STIB 859 (7) were EcoRV-digested and probed with DNA fragments specific for MVSG5(A), MVSG1.61 (B) or the promoter region of the AnTat 1 strain ES5 (probe ES5) (C). The membranes were washed at low stringency (50°C in0.5× SSPE and 0.5% SDS). The sizes of the molecular-weight markers in kilobases are indicated on the left of the blots. The EcoRV DNAfragments containing MVSG5 and ES5 or MVSG1.61 and ES5 are indicated by open and closed circles, respectively. The positions of the probesspecific to the MVSGs and the promoter region are indicated above the physical map of the corresponding MES in panel (D). (D) TheMVAT5-ES from the AnTat 1 and WRATat 1.1 strains is compared with the MVAT1.61-ES from the EATRO 795 strain of T. b. brucei, whichshare 90% identity in the regions containing the promoters. The black and white arrowheads depict promoter 1 [6,8] and promoter 2 [12],respectively. The grey boxes outline the regions of similarity between adjacent ESs and show the percentage of identity. The ESAG1 and MVSGsare represented by horizontal black arrows, and the 70 bp repeats are shown in the MVAT1.61-ES. The ESAG1 upstream of the MVAT5-ES inWRATat 1.1 seems to be functional, whereas that upstream of the MVAT1.61-ES in EATRO 795 is a pseudogene. The position of the threeprobes, and the 3.2 kb and 7 kb EcoRV (E) fragments detected in the MVAT5-ES and in the ETat 1.1 strain, respectively, are indicated abovetheir corresponding map.
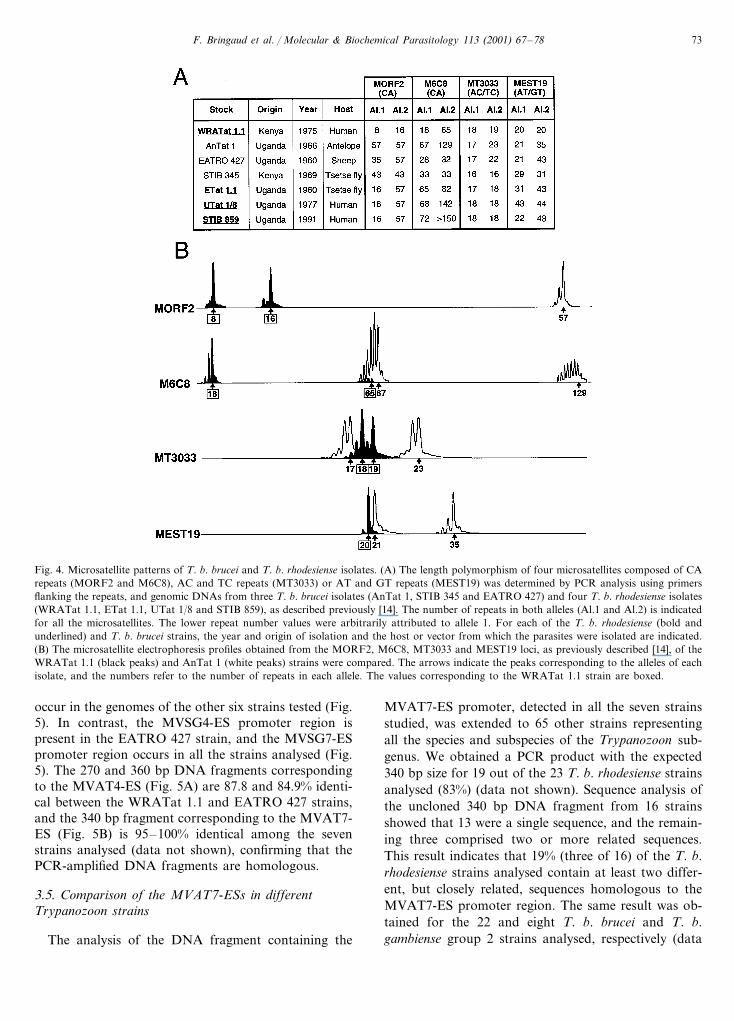
F. Bringaud et al. / Molecular & Biochemical Parasitology 113 (2001) 67–78 73
Fig. 4. Microsatellite patterns of T. b. brucei and T. b. rhodesiense isolates. (A) The length polymorphism of four microsatellites composed of CArepeats (MORF2 and M6C8), AC and TC repeats (MT3033) or AT and GT repeats (MEST19) was determined by PCR analysis using primersflanking the repeats, and genomic DNAs from three T. b. brucei isolates (AnTat 1, STIB 345 and EATRO 427) and four T. b. rhodesiense isolates(WRATat 1.1, ETat 1.1, UTat 1/8 and STIB 859), as described previously [14]. The number of repeats in both alleles (Al.1 and Al.2) is indicatedfor all the microsatellites. The lower repeat number values were arbitrarily attributed to allele 1. For each of the T. b. rhodesiense (bold andunderlined) and T. b. brucei strains, the year and origin of isolation and the host or vector from which the parasites were isolated are indicated.(B) The microsatellite electrophoresis profiles obtained from the MORF2, M6C8, MT3033 and MEST19 loci, as previously described [14], of theWRATat 1.1 (black peaks) and AnTat 1 (white peaks) strains were compared. The arrows indicate the peaks corresponding to the alleles of eachisolate, and the numbers refer to the number of repeats in each allele. The values corresponding to the WRATat 1.1 strain are boxed.
occur in the genomes of the other six strains tested (Fig.5). In contrast, the MVSG4-ES promoter region ispresent in the EATRO 427 strain, and the MVSG7-ESpromoter region occurs in all the strains analysed (Fig.5). The 270 and 360 bp DNA fragments correspondingto the MVAT4-ES (Fig. 5A) are 87.8 and 84.9% identi-cal between the WRATat 1.1 and EATRO 427 strains,and the 340 bp fragment corresponding to the MVAT7-ES (Fig. 5B) is 95–100% identical among the sevenstrains analysed (data not shown), confirming that thePCR-amplified DNA fragments are homologous.
3.5. Comparison of the MVAT7-ESs in differentTrypanozoon strains
The analysis of the DNA fragment containing the
MVAT7-ES promoter, detected in all the seven strainsstudied, was extended to 65 other strains representingall the species and subspecies of the Trypanozoon sub-genus. We obtained a PCR product with the expected340 bp size for 19 out of the 23 T. b. rhodesiense strainsanalysed (83%) (data not shown). Sequence analysis ofthe uncloned 340 bp DNA fragment from 16 strainsshowed that 13 were a single sequence, and the remain-ing three comprised two or more related sequences.This result indicates that 19% (three of 16) of the T. b.rhodesiense strains analysed contain at least two differ-ent, but closely related, sequences homologous to theMVAT7-ES promoter region. The same result was ob-tained for the 22 and eight T. b. brucei and T. b.gambiense group 2 strains analysed, respectively (data
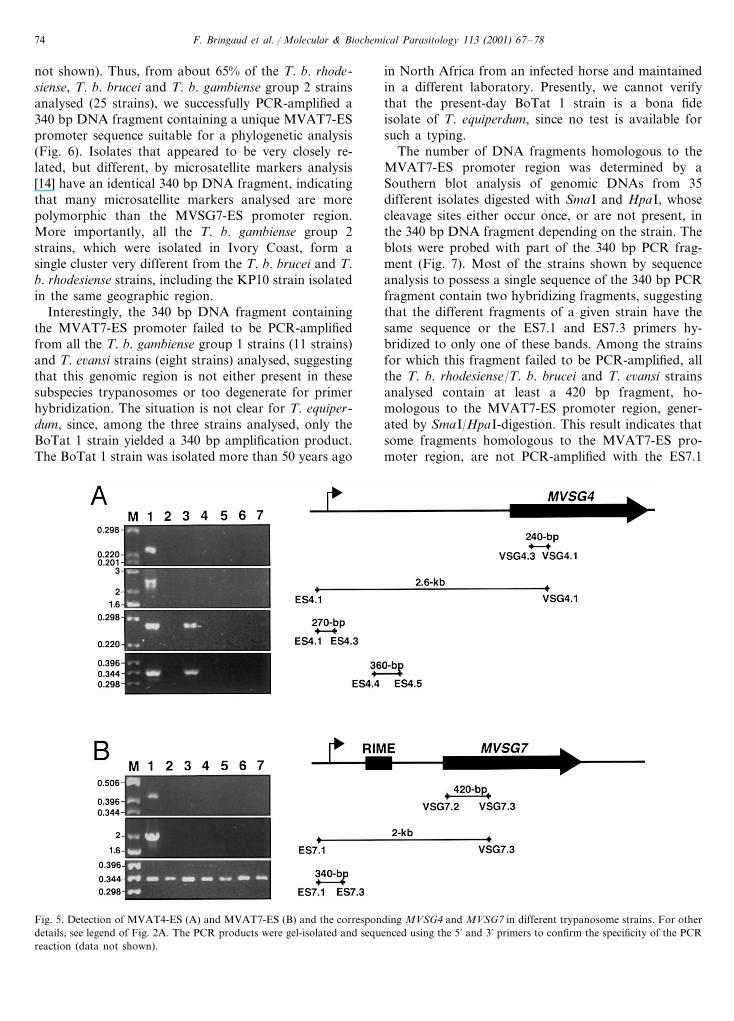
F. Bringaud et al. / Molecular & Biochemical Parasitology 113 (2001) 67–7874
not shown). Thus, from about 65% of the T. b. rhode-siense, T. b. brucei and T. b. gambiense group 2 strainsanalysed (25 strains), we successfully PCR-amplified a340 bp DNA fragment containing a unique MVAT7-ESpromoter sequence suitable for a phylogenetic analysis(Fig. 6). Isolates that appeared to be very closely re-lated, but different, by microsatellite markers analysis[14] have an identical 340 bp DNA fragment, indicatingthat many microsatellite markers analysed are morepolymorphic than the MVSG7-ES promoter region.More importantly, all the T. b. gambiense group 2strains, which were isolated in Ivory Coast, form asingle cluster very different from the T. b. brucei and T.b. rhodesiense strains, including the KP10 strain isolatedin the same geographic region.
Interestingly, the 340 bp DNA fragment containingthe MVAT7-ES promoter failed to be PCR-amplifiedfrom all the T. b. gambiense group 1 strains (11 strains)and T. e6ansi strains (eight strains) analysed, suggestingthat this genomic region is not either present in thesesubspecies trypanosomes or too degenerate for primerhybridization. The situation is not clear for T. equiper-dum, since, among the three strains analysed, only theBoTat 1 strain yielded a 340 bp amplification product.The BoTat 1 strain was isolated more than 50 years ago
in North Africa from an infected horse and maintainedin a different laboratory. Presently, we cannot verifythat the present-day BoTat 1 strain is a bona fideisolate of T. equiperdum, since no test is available forsuch a typing.
The number of DNA fragments homologous to theMVAT7-ES promoter region was determined by aSouthern blot analysis of genomic DNAs from 35different isolates digested with SmaI and HpaI, whosecleavage sites either occur once, or are not present, inthe 340 bp DNA fragment depending on the strain. Theblots were probed with part of the 340 bp PCR frag-ment (Fig. 7). Most of the strains shown by sequenceanalysis to possess a single sequence of the 340 bp PCRfragment contain two hybridizing fragments, suggestingthat the different fragments of a given strain have thesame sequence or the ES7.1 and ES7.3 primers hy-bridized to only one of these bands. Among the strainsfor which this fragment failed to be PCR-amplified, allthe T. b. rhodesiense/T. b. brucei and T. e6ansi strainsanalysed contain at least a 420 bp fragment, ho-mologous to the MVAT7-ES promoter region, gener-ated by SmaI/HpaI-digestion. This result indicates thatsome fragments homologous to the MVAT7-ES pro-moter region, are not PCR-amplified with the ES7.1
Fig. 5. Detection of MVAT4-ES (A) and MVAT7-ES (B) and the corresponding MVSG4 and MVSG7 in different trypanosome strains. For otherdetails, see legend of Fig. 2A. The PCR products were gel-isolated and sequenced using the 5% and 3% primers to confirm the specificity of the PCRreaction (data not shown).

F. Bringaud et al. / Molecular & Biochemical Parasitology 113 (2001) 67–78 75
Fig. 6. Evolutionary relationships among subgenus Trypanozoon strains based on comparisons of the 340 bp fragment sequences containing theMVAT7-ES promoter. The tree was obtained by distance-based analyses (neighbour joining and Fitch–Margoliash methods), using 264 bp of the340 bp fragment (Fig. 7B). The asterisk (*) indicates that the sequence of the WRATat 1.1 strain used for this analysis is not the 340 bpPCR-fragment, which generated at least two different sequences, but corresponds to the previously published sequence [27]. The tree wasarbitrarily rooted with the T. b. brucei EATRO 427 sequence. The scale bar represents a genetic distance of 0.01 nucleotide substitution per site,and the numbers given on the nodes represent the percentage of bootstrap replicas yielding this tree. A number is arbitrarily given to each strainfor which the subspecies, name, year and place of isolation are indicated on the right of the tree. Abbreviations: Tbb, Trypanosoma brucei brucei ;Tbr, Trypanosoma brucei rhodesiense ; Tbg2, Trypanosoma brucei gambiense group 2.
and ES7.3 primers. In contrast, none of the T. b.gambiense group 1 strains analysed contain theMVAT7-ES promoter region, since no hybridizationwas observed (Fig. 7A).
4. Discussion
To study the stability of the MVAT repertoire in thebrucei subspecies of trypanosomes, we examined sixdifferent T. b. brucei and T. b. rhodesiense stocks for thepresence of the three MVSGs and MVAT-ES pro-moters (MVAT4, 5 and 7) previously characterized inthe T. b. rhodesiense WRATat 1.1 strain. All sevenstrains were originally isolated in the vicinity of LakeVictoria in either Uganda or Kenya. The T. b. bruceiAnTat 1 strain was found to share one MVSG with theT. b. rhodesiense WRATat 1.1 strain, while the genomesof the five other stocks analysed do not contain any ofthe three MVSGs. As far as we know, the only previousstudy addressing this issue of MVSG stability reportedsubstantial differences in the MVAT repertoire of dif-ferent stocks isolated along the north-east shore ofLake Victoria between 1958 and 1979 [25]. In addition,a T. b. brucei clone transmitted sequentially through
tsetse flies 10 times lost the expression of three MVATs[25]. However, both of these analyses were based on animmunological approach, so it was not possible todetermine if the drift in MVAT expression was due todeletion of MVSGs or modification of the expressioncontrol. We observed here that the three MVSGs areonly present in one out of four T. b. rhodesiense stocksisolated around Lake Victoria between 1960 and 1991,indicating that deletion of MVSGs contributes to theinstability of MVAT repertoires.
A question to consider is how are MVSGs deletedand what, if anything, are they replaced by? VSGsexpressed in the bloodstream form (BVSGs) are typi-cally encoded by the active VSG copy located in anexpression site (BES) and a minimum of one silent VSGcopy elsewhere in the genome. During antigenic varia-tion of BVSGs, an active VSG is often destroyed andreplaced by duplication and transposition of anothersilent VSG into the same BES [1,2]. Thus, conservationof the VAT repertoire in the bloodstream form (BVATrepertoire) is ensured by maintenance of the silent VSGdonors. In contrast, many MVSGs are encoded by asingle copy MVSG invariably located in the telomere-linked MESs [6,7], and eventual duplication/transposi-tion of a silent VSG in a MES may lead to the definitive

F. Bringaud et al. / Molecular & Biochemical Parasitology 113 (2001) 67–7876
loss of a MVSG. Such a scenario on the molecular levelhas not yet been identified. We observed that thepromoter regions of MESs active in three variants ofWRATat 1.1 are more highly conserved in try-panosome isolates than MVSGs present in these MESs.Furthermore, we observed that the AnTat 1 andWRATat 1.1 strains, on one hand, and the EATRO795 and ETat 1.1 strains, on the other hand, expressunrelated MVSGs (MVSG5 and MVSG1.61, respec-tively) located in the same MES (the 750 bp DNAfragments containing the transcription promoters are90% identical). Interestingly, a Southern blot analysisshowed that this particular MES is a single copy in theAnTat 1 and ETat 1.1 genomes, indicating that thesame MES contains a different MVSG depending onthe strain. These observations suggest that MVSGslocated in MESs can be replaced by other VSGs,probably by duplication and transposition, i.e. geneconversion. This conclusion is supported by reportsshowing that MVSGs located in MESs can serve as
donor genes in homologous recombination events lead-ing to antigenic variation [6,24,28,29]. Since replace-ments of VSGs present in BESs by copies of MVSGslocated in MESs have been observed several times, it isreasonable to consider that the reverse is also possible.Nevertheless, the relative stability of the MVAT reper-toire compared to that of the BVAT repertoire suggeststhat gene conversion of MVSGs in MESs is much lessfrequent than for the VSGs located in BESs. Therelative stability of the MVAT repertoire is probablydue to the lack [13,24] or the presence of only a fewrepeats [6,27–29], of the 70 bp repeats located upstreamof the MVSGs, which are known to be recombinationhot spots for VSG gene conversion.
None of the MVSG4, 5 or 7 originally characterizedin the WRATat 1.1 strain are present in the genomes ofsix T. b. brucei and T. b. rhodesiense isolates, with theexception of MVSG5 in T. b. brucei AnTat 1. Surpris-ingly, the ES containing MVSG5 is virtually identical inT. b. rhodesiense WRATat 1.1 and T. b. brucei AnTat 1
Fig. 7. Southern blot analysis of the promoter region in the MVAT7 expression site. (A) Genomic DNA from T. b. brucei (not specified), T. b.rhodesiense (not specified), T. e6ansi (T.e6.), T. b. gambiense group 2 (T.g.2) and T. b. gambiense group 1 (T.g.1) was SmaI/HpaI-digested andprobed with a 175 bp DNA fragment generated by HpaI-digestion of pooled 340 bp PCR fragments obtained from five different strains. Themembranes were washed at low stringency (50°C in 0.5× SSPE and 0.5% SDS). The sizes of the molecular-weight markers in kilobases areindicated on the left of the blot. The numbers at the top of the first 18 lanes (T. b. brucei, T. b. rhodesiense and T. b. gambiense group 2 strainspossessing a single sequence for the 340 bp PCR fragment) correspond to the numbers used in the phylogenetic tree (Fig. 6). Strains possessingidentical sequences for the 340 bp PCR fragment are boxed. The names of the strains that either possess multiple sequences for the 340 bp PCRfragment or were negative for PCR-amplification of the 340 bp DNA fragment are indicated. Under the blot, the eventual presence of the HpaI(H) and SmaI (S) restriction sites in the sequenced 340 bp PCR fragments is indicated. The arrow on both sides of the blot indicates the 420 bpfragment present in 90% of the strains, except the T. b. gambiense group 1 isolates. (B) Schematic representation of the 340 bp PCR fragmentshowing the putative MVAT7-ES promoter (white box), the 264 bp DNA region used for the phylogenetic analysis (black box), the ES7.1 andES7.3 primers (arrows), the position of the eventual HpaI (H) and SmaI (S) restriction sites and the probe used for the Southern blot analysis.

F. Bringaud et al. / Molecular & Biochemical Parasitology 113 (2001) 67–78 77
with only 20 substitutions, deletions or insertions ofsingle nucleotides in the 3451 bp region containing thepromoters and MVSG5. Furthermore, none of the dif-ferences are located in MVSG5 or in promoter 1. Thislow rate of divergence is comparable to that observedfor housekeeping genes or intergenic regions flankinghousekeeping genes, which was not expected for theESs since they are thought to be highly variable due totheir frequent participation in VSG rearrangements.One possibility is that conservation of MVSG5 mayreflect a high selection pressure to keep this particularMVSG intact, but the absence of MVSG5 in five otherstrains isolated in the same area does not favour thishypothesis. Alternatively, the AnTat 1 or WRATat 1.1strains may have recently acquired the MVAT5-ESfrom each other by genetic exchange in the tsetse fly,one of the strains being parental, the other a hybrid.Genetic exchange in the tsetse fly has been demon-strated unequivocally in the laboratory, particularlybetween human-infective (T. b. rhodesiense) and non-in-fective (T. b. brucei ) strains, indicating that suchcrosses may occur in the field [30].
T. b. gambiense isolates are divided into two groups,groups 1 and 2, which differ in their virulence torodents, isoenzyme patterns and RFLPs in VSGs [31].The T. b. gambiense group 1 isolates appear to be muchmore homogenous compared to the other Trypanozoonsubspecies [31]. This property was used to developseveral specific markers that efficiently differentiate T.b. gambiense group 1 isolates from all the other Try-panozoon isolates [14,32–36]. The absence of MVAT7-ES in all the T. b.gambiense group 1 isolates tested sofar is in accord with the homogeneity of this group. Incontrast, T. b. gambiense group 2 isolates, which haveso far only been found in Burkina Faso and IvoryCoast, are very heterogenous and are still indistinguish-able from T. b. brucei and T. b. rhodesiense isolatesusing molecular tools. By PCR analysis of the genomicregion containing the MVAT7-ES promoter, we ob-served that T. b. gambiense group 2 isolates behave likeT. b. brucei and T. b. rhodesiense isolates, as opposed tothe T. b. gambiense group 1 isolates, as previouslyobserved [31]. More interestingly, a phylogenetic analy-sis of isolates, from which the 340 bp DNA fragmentcontaining the MVAT7-ES promoter was PCR-am-plified and sequenced, showed that all the T. b. gambi-ense group 2 isolates studied form a monophyletic cladewell separated from T. b. brucei and T. b. rhodesienseisolates. Furthermore, T. b. brucei and the T. b. gambi-ense group 2 stocks, isolated in the same geographicregion, are very different. These preliminary resultsindicate that molecular tools may be developed todistinguish T. b. gambiense group 2 isolates, such asthose isolated from domestic or wild animals that areknown to constitute potential reservoirs for T. b. gam-biense [33].
Acknowledgements
We are grateful to C. Giroud for cell cultures andmanipulation of experimental animals. This work wassupported by CNRS, grants from the Conseil Regionald’Aquitaine, GDR/CNRS and the Ministere de l’Edu-cation Nationale de la Recherche et de la Technologie(Action Microbiologie).
References
[1] Pays E, Vanhamme L, Berberof M. Genetic controls for theexpression of surface antigens in African trypanosomes. AnnuRev Microbiol 1994;48:25–52.
[2] Borst P, Fairlamb AH. Surface receptors and transporters ofTrypanosoma brucei. Annu Rev Microbiol 1998;52:745–78.
[3] Kooter JM, Van der Spek HJ, Wagter R, D’Oliveira CE, Vander Hoeven F, Johnson P, Borst P. The anatomy and transcrip-tion of a telomeric expression site for variant-specific surfaceantigen in Trypanosoma brucei. Cell 1987;51:261–72.
[4] Pays E, Tebabi P, Pays A, Coquelet H, Salmon D, Steinert M.The genes and transcripts of an antigen gene expression site fromT. brucei. Cell 1989;57:835–45.
[5] Turner CM, Barry JD, Maudlin I, Vickerman K. An estimate ofthe size of the metacyclic variable antigen repertoire of Try-panosoma brucei rhodesiense. Parasitology 1988;97:269–76.
[6] Donelson JE, Hill KL, El-Sayed NMA. Multiple mechanisms ofimmune evasion by African trypanosomes. Mol Biochem Para-sitol 1998;91:51–66.
[7] Barry JD, Graham SV, Fotheringham M, Graham VS, KobrynK, Wymer B. VSG gene control and infectivity strategy ofmetacyclic stage Trypanosoma brucei. Mol Biochem Parasitol1998;91:93–105.
[8] Nagoshi YL, Alarcon CM, Donelson JE. The putative promoterfor a metacyclic VSG gene in African trypanosomes. MolBiochem Parasitol 1995;72:33–45.
[9] Graham SV, Barry JD. Trancriptional regulation of metacyclicvariant surface glycoprotein gene expression during the life cycleof Trypanosoma brucei. Mol Cell Biol 1995;15:5945–56.
[10] Graham SV, Wymer B, Barry JD. A trypanosome metacyclicVSG gene promoter with two functionally distinct, life cyclestage-specific activities. Nucleic Acids Res 1998;26:1985–90.
[11] Alarcon CM, Pedram M, Donelson JE. Leaky transcription ofvariant surface glycoprotein gene expression sites in bloodstreamtrypanosomes. J Biol Chem 1999;274:16884–93.
[12] Graham SV, Terry S, Barry JD. A structural and transcriptionpattern for variant surface glycoprotein gene expression sitesused in metacyclic stages Trypanosoma brucei. Mol BiochemParasitol 1999;103:141–54.
[13] Pedram M, Donelson JE. The anatomy and transcription of amonocistronic expression site for metacyclic variant surface gly-coprotein gene in Trypanosoma brucei. J Biol Chem1999;274:16876–83.
[14] Biteau N, Bringaud F, Gibson W, Truc P, Baltz T. Characteriza-tion of Trypanozoon isolates using a repeated coding sequenceand microsatellite markers. Mol Biochem Parasitol2000;105:185–201.
[15] Campbell GH, Esser KM, Wellde BT, Diggs CL. Isolation andcharacterization of a new serodeme of a Trypanosoma rhode-siense. Am J Trop Med Hyg 1979;28:974–83.
[16] Lanham SM, Godfrey DG. Isolation of salivarian trypanosomesfrom man and other mammals using DEAE-cellulose. Exp Para-sitol 1970;28:521–34.

F. Bringaud et al. / Molecular & Biochemical Parasitology 113 (2001) 67–7878
[17] Van der Ploeg LH, Bernards A, Rijsewijk FA, Borst P. Charac-terization of the DNA duplication-transposition that controlsthe expression of two genes for variant surface glycoproteins inTrypanosoma brucei. Nucleic Acids Res 1982;10:593–609.
[18] Campbell DA. c2X75, a derivative of the cosmid vector c2XB.Nucleic Acids Res 1989;17:458.
[19] Bringaud F, Vedrenne C, Cuvillier A, Parzy D, Baltz D, TetaudE, Pays E, Venegas J, Merlin G, Baltz T. Conserved organiza-tion of genes in trypanosomatids. Mol Biochem Parasitol1998;94:249–64.
[20] Bates PF, Swift RA. Double cos site vectors: simplified cosmidcloning. Gene 1983;26:137–46.
[21] Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: aLaboratory Manual, second ed. Cold Spring Harbor, NY: ColdSpring Harbor Laboratory Press, 1989.
[22] Thompson JD, Higgins DG, Gibson TJ. Clustal W: Improvingthe sensitivity of progressive multiple sequence alignmentthrough sequence weighting, position-specific gap penalties andweight matrix choice. Nucleic Acids Res 1994;22:4673–80.
[23] Page RDM. TREEVIEW: An application to display phyloge-netic trees on personal computers. Comput Appl Biosci1996;12:357–8.
[24] Lu Y, Hall T, Gay LS, Donelson JE. Point mutations areassociated with a gene duplication leading to the bloodstreamreexpression of a trypanosome metacyclic VSG. Cell1993;72:397–406.
[25] Barry JD, Crowe JS, Vickerman K. Instability of the Try-panosoma brucei rhodesiense metacyclic variable antigen reper-toire. Nature 1983;306:699–701.
[26] Alarcon CM, Son HJ, Hall T, Donelson JE. A monocistronictranscript for a trypanosome VSG. Mol Cell Biol 1994;14:5579–91.
[27] Kim KS, Donelson JE. Co-duplication of a variant surface
glycoprotein gene and its promoter to an expression site inAfrican trypanosomes. J Biol Chem 1997;272:24637–45.
[28] Graham SV, Matthews KR, Shiels PG, Barry JD. Distinct,developmental stage-specific activation mechanisms of try-panosome VSG genes. Parasitology 1990;101:361–7.
[29] Matthews KR, Shiels PG, Graham SV, Cowan C, Barry JD.Duplicative activation mechanisms of two trypanosome telom-eric VSG genes with structurally simple 5% flanks. Nucleic AcidsRes 1990;18:7219–27.
[30] Gibson W, Stevens J. Genetic exchange in the Trypanosomatidae.Adv Parasitol 1999;9:1–46.
[31] Gibson W. Will the real Trypanosoma b. gambiense please standup. Parasitol Today 1986;43:255–7.
[32] Mehlitz D, Zillman U, Scott CM, Godfrey DG. Epidemiologicalstudies on the animal reservoir of gambiense sleeping sickness.III. Characterization of Trypanozoon stocks by isoenzymes andsensitivity to human serum. Trop Parasitol 1982;33:113–8.
[33] Paindavoine P, Pays E, Laurent M, Geltmeyer Y, Le Ray D,Mehlitz D, Steinert M. The use of DNA hybridization andnumerical taxonomy in determining relationships between Try-panosoma brucei stocks and subspecies. Parasitology 1986;92:31–50.
[34] Bromidge T, Gibson W, Hudson K, Dukes P. Identification ofTrypanosoma brucei gambiense by PCR amplification of variantsurface glycoprotein genes. Act Trop 1993;53:107–19.
[35] Mathieu-Daude F, Bicart-See A, Bosseno M-F, Breniere SF,Tibayrenc M. Identification of Trypanosoma brucei gambiensegroup I by a specific kinetoplast DNA probe. Am J Trop MedHyg 1994;50:13–9.
[36] Mathieu-Daude F, Stevens J, Welsh J, Tibayrenc M, McClellandM. Genetic diversity and population structure of Trypanosomabrucei : clonality versus sexuality. Mol Biochem Parasitol1995;72:89–101.
.
Copyright © 2022 FDOKUMEN




















