
Genetic analysis of lobular carcinoma in situ and associated invasive lobular cancer

Clonality of Lobular Carcinoma in Situ andSynchronous Invasive Lobular Carcinoma
E. Shelley Hwang, M.D.1
Sarah J. Nyante, B.S.1
Yunn Yi Chen, M.D., Ph.D.2
Dan Moore, Ph.D.3
Sandy DeVries, B.S., M.S.3
James E. Korkola, Ph.D.3
Laura J. Esserman, M.D., M.B.A.1
Frederic M. Waldman, M.D., Ph.D.3–5
1 Department of Surgery, University of California–San Francisco, San Francisco, California.
2 Department of Pathology, University of Califor-nia–San Francisco, San Francisco, California.
3 University of California–San Francisco Compre-hensive Cancer Center, San Francisco, California.
4 Department of Laboratory Medicine, University ofCalifornia–San Francisco, San Francisco, Califor-nia.
5 Department of Urology, University of California–San Francisco, San Francisco, California.
Supported by grants from the National Institutes ofHealth (CA44768 and CA58207) and by the Uni-versity of California–San Francisco Clinical Inves-tigator Research Program Award (to Dr. Hwang).
The authors thank the University of California–SanFrancisco Cancer Center Array Core, directed byDr. Donna Albertson and Dr. Dan Pinkel, for pro-viding Human Array 2.0 genomic arrays.
Address for reprints: E. Shelley Hwang, M.D., De-partment of Pathology, University of California–SanFrancisco Cancer Center, 1600 Divisadero Avenue,B606, San Francisco, CA 94115; Fax: (415) 353-9571; E-mail: [email protected]
Received January 8, 2004; revision receivedMarch 2, 2004; accepted March 9, 2004.
BACKGROUND. Lobular carcinoma in situ (LCIS) of the breast is considered a
marker for an increased risk of carcinoma in both breasts. However, the frequent
association of LCIS with invasive lobular carcinoma (ILC) suggests a precursor-
product relation. The possible genomic relation between synchronous LCIS and
ILC was analyzed using the technique of array-based comparative genomic hy-
bridization (CGH).
METHODS. Twenty-four samples from the University of California–San Francisco
pathology archives that contained synchronous LCIS and ILC were identified.
Array CGH was performed using random primer–amplified microdissected DNA.
Samples were hybridized onto bacterial artificial chromosome arrays composed of
approximately 2400 clones. Patterns of alterations within synchronous LCIS and
ILC were compared.
RESULTS. A substantial proportion of the genome was altered in samples of both
LCIS and ILC. The most frequent alterations were gain of 1q and loss of 16q, both
of which usually occurred as whole-arm changes. Smaller regions of gain and loss
were seen on other chromosome arms. Fourteen samples of LCIS were related
more to their paired samples of ILC than to any other ILC, as demonstrated by a
weighted similarity score.
CONCLUSIONS. LCIS and ILC are neoplastic lesions that demonstrate a range of
genomic alterations. In the current study, the genetic relation between synchro-
nous LCIS and ILC suggested clonality in a majority of the paired specimens. These
data were consistent with a progression pathway from LCIS to ILC. The authors
conclude that LCIS, which is known to be a marker for an environment that is
permissive of neoplasia, may itself represent a precursor to invasive carcinoma.
Cancer 2004;100:2562–72. © 2004 American Cancer Society.
KEYWORDS: lobular carcinoma in situ, ductal carcinoma in situ, comparativegenomic hybridization, breast neoplasms.
Our clinical understanding of lobular carcinoma in situ (LCIS) ofthe breast has undergone a gradual evolution since the term was
coined by Foote and Stewart in 1941.1 Although, histologically aneoplastic lesion, LCIS has long been considered a marker for in-creased breast carcinoma risk rather than a precursor for invasivecarcinoma, because it appears to confer an equally increased risk ofbreast carcinoma in both the contralateral and ipsilateral breasts.2–5
Furthermore, over half of invasive carcinomas that develop after adiagnosis of LCIS develop � 10 years subsequent to the index lesion,with most of these invasive carcinomas exhibiting ductal histology.2– 4
Because of the concern regarding increased bilateral risk, thehistorically recommended treatment for LCIS included bilateral mas-tectomy. However, most clinicians now choose to follow their pa-tients with expectant management, sometimes in conjunction with
2562
© 2004 American Cancer SocietyDOI 10.1002/cncr.20273Published online 6 May 2004 in Wiley InterScience (www.interscience.wiley.com).
tamoxifen chemoprophylaxis. However, both the clin-ical significance and the biologic significance of LCISremain uncertain. Over the past 2 decades, the greateruse of screening mammography has resulted in morefrequent incidental diagnoses of LCIS.6 In addition,more meticulous histologic evaluation has shown thatLCIS often can be found adjacent to invasive carci-noma, particularly invasive lobular carcinoma (ILC).Epidemiologic studies conducted after the establish-ment of widespread mammographic screening havesupported a model in which both LCIS and ductalcarcinoma in situ (DCIS) confer an increased risk ofcontralateral breast carcinoma.6,7 These findings againhave raised the question of whether LCIS, like DCIS,may act as a precursor to invasive carcinoma.
Recently, advances in molecular techniques havefostered an unprecedented ability to identify discretechanges in the genome. This has allowed examinationat high resolution of possible genomic relations be-tween tumors. Using array-based comparativegenomic hybridization (CGH), we explored the asso-ciation between LCIS and synchronous ILC to deter-mine whether they share a clonal relation.
MATERIALS AND METHODSPatientsThe current study was evaluated and approved by theUniversity of California–San Francisco (UCSF; SanFrancisco, CA) Institutional Review Board. Twenty-four samples from the pathology archives at our insti-tution that showed concurrent ILC and LCIS diag-nosed between 1994 and 2001 were selected. Eachspecimen was reviewed to validate the diagnosis andto select regions for microdissection. Altered immu-nohistochemical staining for E-cadherin was used toconfirm the diagnosis of ILC in all specimens.
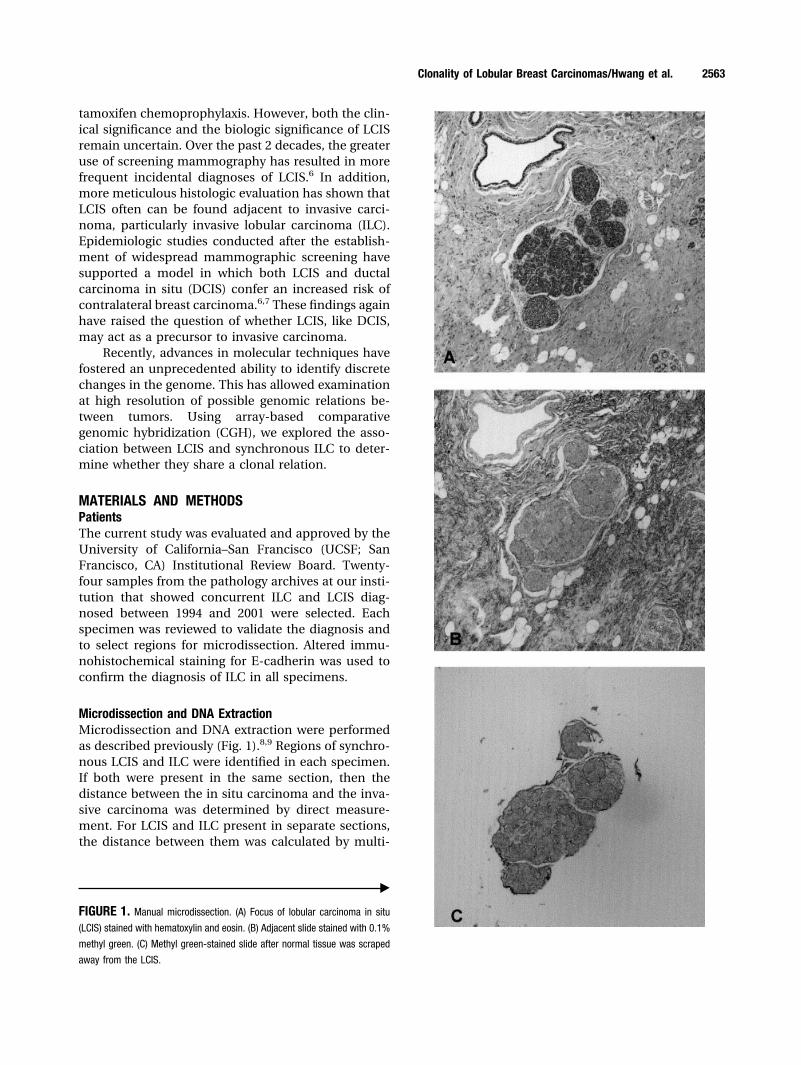
Microdissection and DNA ExtractionMicrodissection and DNA extraction were performedas described previously (Fig. 1).8,9 Regions of synchro-nous LCIS and ILC were identified in each specimen.If both were present in the same section, then thedistance between the in situ carcinoma and the inva-sive carcinoma was determined by direct measure-ment. For LCIS and ILC present in separate sections,the distance between them was calculated by multi-
‹
FIGURE 1. Manual microdissection. (A) Focus of lobular carcinoma in situ
(LCIS) stained with hematoxylin and eosin. (B) Adjacent slide stained with 0.1%
methyl green. (C) Methyl green-stained slide after normal tissue was scraped
away from the LCIS.
Clonality of Lobular Breast Carcinomas/Hwang et al. 2563

plying the number of intervening blocks with the av-erage thickness of all blocks (4 mm).
Nine slides were cut from each block selected forthe study. Six slides were stained with 0.1% methylgreen for microdissection. Using an adjacent hema-toxylin and eosin (H&E)-stained section as a guide,normal cells were scraped away from the cells of in-terest. A drop of extraction buffer (10 mM Tris, 1.5 mMMgCl2, 50 mM KCl, and 0.5% Tween-20 (Fisher Scien-tific, Tustin, CA) buffer with 4 mg/mL proteinase K)was placed on the cells, which were then removed anddeposited in approximately 15 �L extraction buffer per1000 cells. In situ and invasive lesions were isolatedseparately. Cells were incubated in a shaking waterbath at 55 °C for 3 days, with the addition of 0.3 �L of20 mg/mL proteinase K per 1000 cells on Days 2 and 3.At the end of the incubation, proteinase K was inacti-vated by heating at 95 °C for 15 minutes. Samples wereconcentrated using a Microcon YM-30 column (Ami-con Millipore, Bedford, MA), after which DNA wasquantitated using the TaqMan (Applied Biosystems,Foster City, CA) real-time polymerase chain reaction(PCR).10
DNA Amplification and LabelingDNA was random primer–amplified using compo-nents from the BioPrime DNA Labeling System (In-vitrogen, Carlsbad, CA). The reaction consisted of 50ng tumor DNA, 300 ng/�L random oligodeoxyribo-nucleotide primers, 20 U Klenow enzyme, 200 �Mdeoxyadinosine triphosphate (dATP), 200 �M deoxy-cytidine triphosphate (dCTP), 200 �M deoxythymi-dine triphosphate (dTTP), and 200 �M deoxyguaninetriphosphate (dGTP). Tumor DNA and the randomprimers were denatured at 100 °C for 10 minutes andthen immediately cooled on ice. The remaining reac-tants were then added, and the reaction was incu-bated at 37 °C for 2 hours. The final reaction volumewas 25 �L. Excess nucleotides were removed using theQIAquick PCR Purification Kit (Qiagen Inc., Valencia,CA).
The labeling reaction mixture consisted of ampli-fied DNA, 370 ng/�L random primers, 64 U Klenowenzyme, 200 �M dATP, 200 �M dCTP, 200 �M dGTP,100 �M dTTP, and 75 �M Cy dye– conjugated de-oxyuridine triphosphate (dUTP). DNA and randomprimers were denatured at 100 °C for 10 minutes andcooled on ice immediately. The remaining reactantswere then added, and the reaction was incubated for 2hours at 37 °C. Tumor DNA was labeled with Flu-oroLink indocarbocyanine (Cy3)-dUTP, and referencegenomic male DNA was labeled with FluoroLinkindodicarbocyanine (Cy5)-dUTP (Amersham Pharma-cia, Piscataway, NJ). Excess primers and nucleotides
were removed using a Sephadex G-50 column (Amer-sham Pharmacia).
HybridizationArray CGH was performed according to protocols de-scribed previously.11–13 Human Array 2.0 chromiumsurface arrays were obtained from the UCSF CancerCenter Array Core. Each array was composed of 2464bacterial artificial chromosomes (BACs) printed intriplicate, representing the entire human genome. Be-fore use, arrays were ultraviolet (UV) cross-linked at asetting of 1300 �J using a UV Stratalinker (La Jolla,CA).
Labeled tumor DNA and reference DNA werecombined with 100 �g CotI DNA (Invitrogen), and themixture was precipitated using 0.1 volumes of 3 Msodium acetate and 2.5 volumes of 100% ethanol. DNAwas dissolved in a solution of 33 �g/�L yeast tRNAwith 9% sodium dodecyl sulfate and then mixed thor-oughly with hybridization solution to yield a finalcomposition of 50% formamide, 10% dextran sulfate,and 2X standard saline citrate (SSC). DNA was dena-tured at 73 °C for 15 minutes and then incubated at 37°C for 30 – 60 minutes. The DNA probe was applied tothe array, the slide was sealed inside a slide box hu-midified with 50% formamide/2X SSC, and the slidebox was incubated at 37 °C for 48 hours.
After hybridization, the array was washed twice in50% formamide/2X SSC at 45 °C for 10 minutes andthen twice in phosphate buffer containing 0.1% NP-40, pH 8.0, (PN buffer) at room temperature for 10minutes. While the slide was still wet with PN buffer,100 �L of 3 �g/mL 4,6-diamidino-2-phenylindole(DAPI) in a 10% solution of phosphate-buffered salinein glycerol was added to the slide, which was thencoverslipped and sealed.
Image Capture and ProcessingArrays were imaged using a charged coupled devicecamera as described previously.11 Intensity data wereacquired through the DAPI, Cy3, and Cy5 channels.The SPOT 2.0 software program (available from URL:http://cc.ucsf.edu/jain/public [accessed October 12,2003]) was used to process the image data.14 For seg-mentation purposes, the DAPI image field was leftblank, and combined test and reference images wereused to define spot size and location. Test-to-refer-ence ratios for each spot were calculated using abso-lute intensities, which were determined by subtractingthe local background intensity from the foregroundintensity. Post-SPOT data processing included dis-carding spots that were less than 15 pixels, spots forwhich correlations were less than 0.9 or in the bottom10th percentile of correlations, spots with a test plus
2564 CANCER June 15, 2004 / Volume 100 / Number 12

reference intensity of less than 200 or in the bottom20th percentile, and spots with a (reference intensity/[sum reference and test intensities]) value less than0.1. For each clone, a single centered log2 ratio of testintensity over reference intensity was calculated fromthe three replicate spots on the array. Clones for whichthe ratio was derived from only 1 of 3 replicates andclones for which replicate ratios had a log2 standarddeviation � 0.15 were discarded. Clones that did notyield data in at least 80% of hybridizations were ex-cluded from further analysis, as were suspected poly-morphic clones (28 clones; detailed information avail-able from URL: http://cc.ucsf.edu/people/waldman/breast/hwang.polymorphisms.xls [accessed October12, 2003]). An average of 1900 clones per case wereincluded in the final data analysis. A human DNAsequence draft obtained from the University of Cali-fornia–Santa Cruz Genome Browser (freeze date, Au-gust 2001; available from URL: http://genome.ucsc.edu [accessed October 12, 2003]) was used to mapclones.
Statistical AnalysisA global threshold of � 0.20 defined gains and lossesfor all log2 ratios. High-level gains and losses werescored for clones with log2 intensity ratios � 1.0 or� �0.75, respectively. Differences in continuous vari-ables between LCIS and ILC were evaluated using atwo-tailed unpaired t test. To allow comparison ofarray CGH results with the results of previous studiesof lobular carcinoma that were performed using chro-mosomal CGH, genomic alterations were expressed aswhole-arm changes by calculating the mean log2 ratiofor all clones mapping to each chromosome arm. Athreshold of � 0.14 was then applied to the averagelog2 ratio to define chromosomal arm gains or lossesrelative to the reference sample.
The concordance between the paired in situ andinvasive components was calculated for whole-armaveraged data (i.e., the log2 ratio averaged over anentire chromosome arm), as described previously,9
using the following formula:
Number of changes in common for LCIS and ILC�number of changes in common for LCIS and ILC�
�12
� �number in LCIS only � number in ILC only�
Because 1q gain and 16q loss were observed inalmost every tumor, concordance was calculated afterexcluding these changes and those on the X chromo-some and weighting for less common changes. A logsimilarity score comparing each synchronous LCIS-ILC pairing with all other possible LCIS-ILC pairings
also was calculated. The individual terms of the simi-larity score were negative when alterations were dis-cordant (i.e., when there was a genomic alteration at aparticular locus in one lesion but not at the corre-sponding locus in the other lesion in the pair). Incontrast, similarity scores were increasingly positivewhen set members were increasingly alike (i.e., whenboth had alterations of the same type at the samelocus or when neither lesion had any alteration). Prob-ability weighting ensured that agreement at rare alter-ation sites was weighted more heavily than was agree-ment at common sites.
RESULTSClinical CharacteristicsClinical characteristics of the study population aresummarized in Table 1. None of the patients hadbilateral disease, and all patients were female. Elevenof 24 patients (46%) had pathologic confirmation oflymph node metastasis. No patient had distant metas-tasis at initial diagnosis. The LCIS and ILC were in thesame breast in all patients, although the distance be-tween the two components varied (Table 1). Immuno-histochemical staining of the invasive tumor compo-nent showed loss of E-cadherin in all but twospecimens; weak, variable membrane staining was ob-served in both of these tumors, one of which was amixed ductolobular carcinoma. All ILCs were positivefor estrogen receptor staining.
Genomic Alterations in LCIS and ILCTwenty-four pairs of concurrent LCIS and ILC lesionswere analyzed by array CGH (log2 data are available athttp://cc.ucsf.edu/people/waldman/breast/hwang-.data.xls). A substantial proportion of the genome wasaltered in both LCIS and ILC lesions; 310 (� 105 SD)and 403 (� 122 SD) of 1945 BACs were altered in LCISand ILC, respectively (P � 0.01), representing 16% and21% of the genome (P � 0.04). For both LCIS and ILC,the most frequently altered individual BACs were partsof larger regions of gain or loss (Fig. 2A,B). The mostcommon alterations usually involved the entire chro-mosome arm. Loss of the entire 16q arm, which in-cludes the locus for E-cadherin (CDH1), was seen in100% of samples. The second most common genomicchange was gain of 1q, which was noted in almost 90%of both LCIS and ILC lesions. Nearly the entire 8p armwas lost in � 20% of LCIS and ILC lesions; however, itis noteworthy that the most proximal portion (8p11.1–11.2; 48.1–52.9 megabases [Mb]), which included 2clones that mapped to the locus for fibroblast growthfactor receptor 1 (FGFR1), was gained in � 10% ofsamples. Similarly, although 40% of LCIS samples and60% ILC samples exhibited loss of the distal portion of
Clonality of Lobular Breast Carcinomas/Hwang et al. 2565
11q (11q14 –11qter; 94.1–160 Mb), the proximal por-tion of 11q (11q11–11q13; 62.3–76.2 Mb) was lost lessfrequently, with the exception of the region that con-tained the CCND1 gene (79.2–79.9 Mb), which wasgained in approximately 30% of all samples.
Loss of the distal portion of chromosome 1p(1p36 –1p33; 1.0 – 40.6 Mb) was observed in 25% ofLCIS lesions and in 40% of ILC lesions. This regionincluded a clone for E2F2 (1p36.12; 26.2 Mb), whichwas lost in almost 30% of lesions. A second distinctregion of loss was noted at 1p22–1p13 (92.5–128.7Mb), with almost 40% of lesions exhibiting loss at thislocus.
Overall, 64 clones had high-level gains (log2 inten-sity ratio � 1.0) in at least 1 instance. Ten LCIS lesionsand 7 ILC lesions had at least 1 high-level gain, with amean of 2.8 gains (� 4.8 gains) per sample for LCISand 2.2 gains (� 5.5 gains) per sample for ILC. Thehighest gene copy numbers (9.2-fold increase) wereobserved at 11q13, which includes the loci for thegenes CCND1, FGF19, FGF4, and FGF3.
One hundred thirty clones showed high-levellosses (log2 intensity ratio � �0.75) in at least 1 ILC
lesion or 1 LCIS lesion. Ten LCIS lesions (mean, 6.4� 14.7 high-level losses) and 10 ILC lesions (mean, 4.0� 9.9 high-level losses) had at least 1 such loss. Theregions that most commonly contained BACs withhigh-level losses generally belonged to larger loci ofgenomic loss, primarily on 11q, 8p, and 16q, as de-scribed above.
In addition to the region that included the locusfor E2F2, which was lost in almost 30% of lesions,several other loci that contained genes known to beassociated with disease progression were frequentlyaltered. Epidermal growth factor receptor (EGFR;7p11.2; 59.7 Mb) was gained in � 70% of LCIS lesionsand almost 60% of ILC lesions. Cyclin D1 (CCND1;11q13; 79.2 Mb) was gained in almost 40% of bothLCIS and ILC lesions. p53 (17p13.1; 75.1 Mb) was lostin 30% of LCIS lesions and in � 50% of ILC lesions,and MDM2 (12q15; 79.8 Mb) was gained in 20% ofboth LCIS and ILC lesions. Only 10% of LCIS lesionsand 15% of ILC lesions gained ERBB2 (17q21.1; 41.9Mb). E-cadherin (CDH1; 16q22.1; 81.6 Mb) was lost innearly every lesion.
Chromosomal arm changes were calculated for
TABLE 1Clinical Characteristics
Patient no.Age(yrs)
LICSextenta
Invasivesize (cm)
Invasivegradeb
Lymph nodestatus
E-cadherinexpression (ILC)c
Approximate distancebetween LCIS and ILC (cm)
LC02 50 2 3.5 2 � �/� 1.6LC03 49 2 1.7 2 � � � 0.1LC04 50 2 4.5 1 � � � 0.1LC06 78 2 0.7 2 � � 0.3LC07 47 2 2.5 1 � � 0.7LC12 51 2 0.9 1 � �/� 0.2LC17 55 1 1.2 1 � � 0.8LC19 48 2 1.8 2 � � 0.7LC23 40 2 1.9 1 � � 2.8LC28 46 2 4.5 1 � � 2.4LC31 68 2 2.1 1 � � � 0.1LC33 46 2 3.0 3 � � � 0.1LC38 55 2 4.0 2 � � 2.4LC39 55 2 3.0 2 � � 1.8LC42 45 2 1.2 2 � � 2.4LC43 79 2 3.5 1 � � 0.4LC45 39 2 1.6 2 � � 0.5LC48 53 2 8.2 1 � � 0.4LC53 53 2 12.5 2 � � � 0.1LC55 77 1 2.5 2 � � � 0.1LC56 53 2 1.6 2 � � � 0.1LC57 50 2 5.4 1 � � 0.6LC59 40 2 2.4 2 � � 0.2LC69 73 1 1.3 1 � � � 0.1
LCIS: lobular carcinoma in situ; ILC: invasive lobular carcinoma; �: positive; �: negative.a For LCIS extent, 1 corresponds to � 10 lobules, and 2 corresponds to � 10 lobules.b Tumor grade was based on modified Scarff–Bloom–Richardson scoring.c �: circumferential cell membrane staining, and �/�: partial membrane staining; �: no membrane staining.
2566 CANCER June 15, 2004 / Volume 100 / Number 12

each lesion based on thresholded averages of copynumber ratios along the chromosome so that arraydata could be compared with data previously obtainedusing chromosomal CGH (see Materials and Meth-ods). The mean number of chromosome arms alteredin ILC was 8.9, compared with 5.0 in LCIS (P � 0.004).The most common changes in LCIS and ILC were 1qgain and 16q loss, which were present according towhole-arm averaged analysis in 87.5% and 100% oflesions, respectively (Table 2).
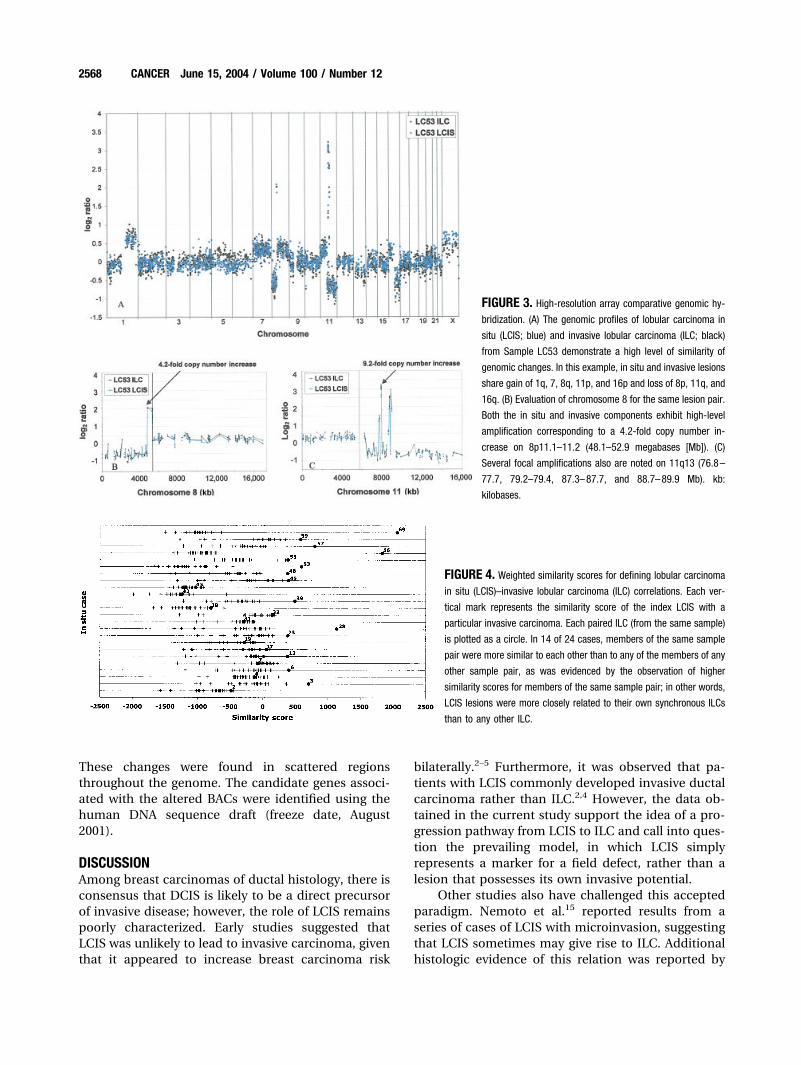
Relation between Synchronous in Situ and InvasiveTumorsArray-based CGH allowed discrimination of uniquealteration patterns shared by synchronous in situ andinvasive carcinomas at high resolution. Figure 3 showsexamples of the pattern of alteration seen in twomatched pairs of LCIS and ILC lesions. The strikingsimilarity in genomic changes between the in situ andinvasive components of these lesions clearly demon-strated the common clonality of the two lesions.
Two approaches were used to quantify the degreeof similarity between LCIS and its associated ILC. Aweighted similarity score was calculated from high-resolution data (Fig. 4); strong evidence for the com-mon clonality of synchronous specimens was indi-cated when the similarity score for the pair ofspecimens was higher than the score for any otherdifferent-case pairing of invasive and in situ carci-noma. In 14 of 24 cases, the synchronous lesions weremore similar to each another than to any of the otherlesion pairings. In addition, concordance of whole-armalterations was calculated for each LCIS-ILC pair;these calculations revealed clear similarities betweensynchronous pairs in a large subset of cases (Table 3).
To determine whether specific candidate genesmay play a role in the progression of in situ carcinomato invasive carcinoma, we searched for BACs that wereeither gained or lost in invasive tumors, but not in thematched in situ tumors, in at least 4 of 14 lesions thatwere related clonally according to the weighted simi-larity score (Table 4). For each BAC, the mean log2
ratio for the ILC that exhibited this change was calcu-lated. Most BACs that were altered only in ILC lesionsand not in LCIS lesions exhibited low-level alterations.
FIGURE 2. Frequency of changes in bacterial artificial chromosome (BAC)
arrays in (A) 4 cases of lobular carcinoma in situ and (B) 24 cases of
synchronous invasive lobular carcinoma. Log2 ratios from each hybridization
were scored as being indicative of gain or loss using a threshold of � 0.20. The
frequency with which each BAC was gained is reported as a positive frequency,
and the frequency of loss is reported as a negative frequency.
TABLE 2Frequency of Whole-Arm Alterations
EventFrequency inin LCIS (%)a
Frequency inin ILC (%)
16q� 100 1001q� 88 8811q� 25 388p� 25 2916p� 21 5417p� 21 5413� 21 3822� 17 4618q� 13 218q� 13 2112p� 13 131p� 8 256q� 8 217p� 8 177q� 8 1320p� 4 175p� 4 1717q� 4 1318p� 4 1320q� 4 133p� 4 1311p� 0 176p� 0 1719q� 0 13
LCIS: lobular carcinoma in situ; ILC: invasive lobular carcinoma; �: gain; �: loss.a Frequencies based on average log2 ratios of bacterial artificial chromosome arrays on specified
chromosome arms.
Clonality of Lobular Breast Carcinomas/Hwang et al. 2567
These changes were found in scattered regionsthroughout the genome. The candidate genes associ-ated with the altered BACs were identified using thehuman DNA sequence draft (freeze date, August2001).
DISCUSSIONAmong breast carcinomas of ductal histology, there isconsensus that DCIS is likely to be a direct precursorof invasive disease; however, the role of LCIS remainspoorly characterized. Early studies suggested thatLCIS was unlikely to lead to invasive carcinoma, giventhat it appeared to increase breast carcinoma risk
bilaterally.2–5 Furthermore, it was observed that pa-tients with LCIS commonly developed invasive ductalcarcinoma rather than ILC.2,4 However, the data ob-tained in the current study support the idea of a pro-gression pathway from LCIS to ILC and call into ques-tion the prevailing model, in which LCIS simplyrepresents a marker for a field defect, rather than alesion that possesses its own invasive potential.
Other studies also have challenged this acceptedparadigm. Nemoto et al.15 reported results from aseries of cases of LCIS with microinvasion, suggestingthat LCIS sometimes may give rise to ILC. Additionalhistologic evidence of this relation was reported by
FIGURE 3. High-resolution array comparative genomic hy-
bridization. (A) The genomic profiles of lobular carcinoma in
situ (LCIS; blue) and invasive lobular carcinoma (ILC; black)
from Sample LC53 demonstrate a high level of similarity of
genomic changes. In this example, in situ and invasive lesions
share gain of 1q, 7, 8q, 11p, and 16p and loss of 8p, 11q, and
16q. (B) Evaluation of chromosome 8 for the same lesion pair.
Both the in situ and invasive components exhibit high-level
amplification corresponding to a 4.2-fold copy number in-
crease on 8p11.1–11.2 (48.1–52.9 megabases [Mb]). (C)
Several focal amplifications also are noted on 11q13 (76.8–
77.7, 79.2–79.4, 87.3–87.7, and 88.7–89.9 Mb). kb:
kilobases.
FIGURE 4. Weighted similarity scores for defining lobular carcinoma
in situ (LCIS)–invasive lobular carcinoma (ILC) correlations. Each ver-
tical mark represents the similarity score of the index LCIS with a
particular invasive carcinoma. Each paired ILC (from the same sample)
is plotted as a circle. In 14 of 24 cases, members of the same sample
pair were more similar to each other than to any of the members of any
other sample pair, as was evidenced by the observation of higher
similarity scores for members of the same sample pair; in other words,
LCIS lesions were more closely related to their own synchronous ILCs
than to any other ILC.
2568 CANCER June 15, 2004 / Volume 100 / Number 12
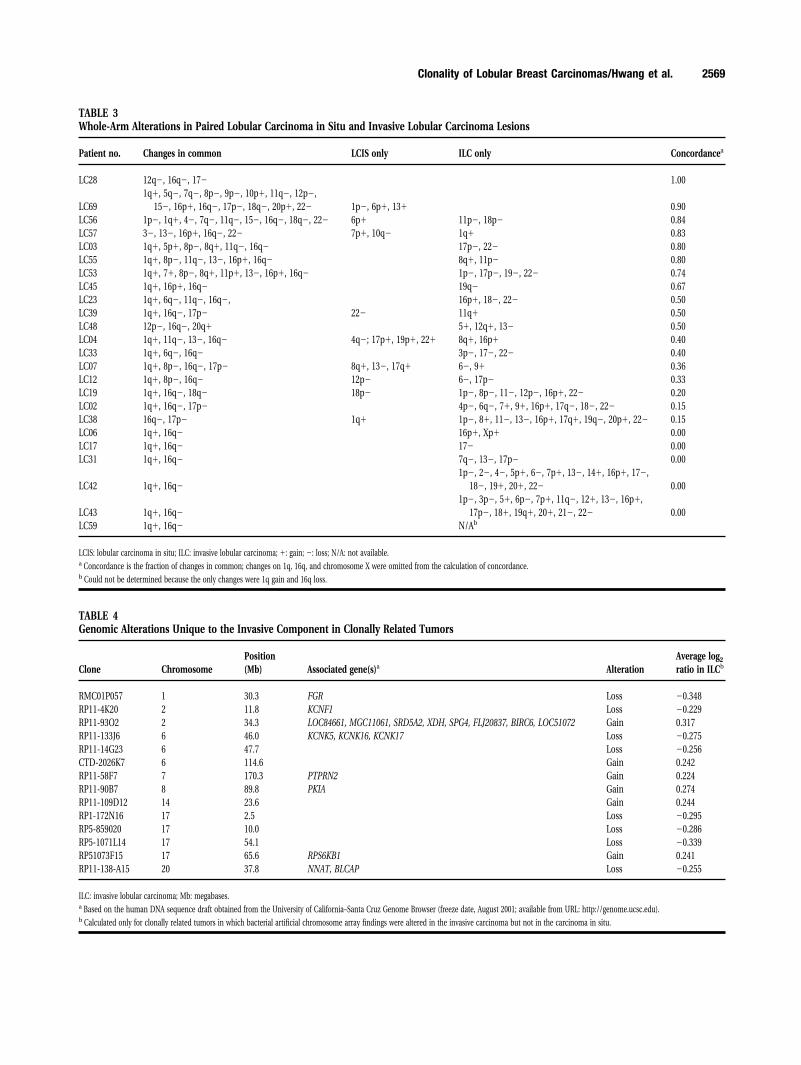
TABLE 3Whole-Arm Alterations in Paired Lobular Carcinoma in Situ and Invasive Lobular Carcinoma Lesions
Patient no. Changes in common LCIS only ILC only Concordancea
LC28 12q�, 16q�, 17� 1.00
LC691q�, 5q�, 7q�, 8p�, 9p�, 10p�, 11q�, 12p�,
15�, 16p�, 16q�, 17p�, 18q�, 20p�, 22� 1p�, 6p�, 13� 0.90LC56 1p�, 1q�, 4�, 7q�, 11q�, 15�, 16q�, 18q�, 22� 6p� 11p�, 18p� 0.84LC57 3�, 13�, 16p�, 16q�, 22� 7p�, 10q� 1q� 0.83LC03 1q�, 5p�, 8p�, 8q�, 11q�, 16q� 17p�, 22� 0.80LC55 1q�, 8p�, 11q�, 13�, 16p�, 16q� 8q�, 11p� 0.80LC53 1q�, 7�, 8p�, 8q�, 11p�, 13�, 16p�, 16q� 1p�, 17p�, 19�, 22� 0.74LC45 1q�, 16p�, 16q� 19q� 0.67LC23 1q�, 6q�, 11q�, 16q�, 16p�, 18�, 22� 0.50LC39 1q�, 16q�, 17p� 22� 11q� 0.50LC48 12p�, 16q�, 20q� 5�, 12q�, 13� 0.50LC04 1q�, 11q�, 13�, 16q� 4q�; 17p�, 19p�, 22� 8q�, 16p� 0.40LC33 1q�, 6q�, 16q� 3p�, 17�, 22� 0.40LC07 1q�, 8p�, 16q�, 17p� 8q�, 13�, 17q� 6�, 9� 0.36LC12 1q�, 8p�, 16q� 12p� 6�, 17p� 0.33LC19 1q�, 16q�, 18q� 18p� 1p�, 8p�, 11�, 12p�, 16p�, 22� 0.20LC02 1q�, 16q�, 17p� 4p�, 6q�, 7�, 9�, 16p�, 17q�, 18�, 22� 0.15LC38 16q�, 17p� 1q� 1p�, 8�, 11�, 13�, 16p�, 17q�, 19q�, 20p�, 22� 0.15LC06 1q�, 16q� 16p�, Xp� 0.00LC17 1q�, 16q� 17� 0.00LC31 1q�, 16q� 7q�, 13�, 17p� 0.00
LC42 1q�, 16q�1p�, 2�, 4�, 5p�, 6�, 7p�, 13�, 14�, 16p�, 17�,
18�, 19�, 20�, 22� 0.00
LC43 1q�, 16q�1p�, 3p�, 5�, 6p�, 7p�, 11q�, 12�, 13�, 16p�,
17p�, 18�, 19q�, 20�, 21�, 22� 0.00LC59 1q�, 16q� N/Ab
LCIS: lobular carcinoma in situ; ILC: invasive lobular carcinoma; �: gain; �: loss; N/A: not available.a Concordance is the fraction of changes in common; changes on 1q, 16q, and chromosome X were omitted from the calculation of concordance.b Could not be determined because the only changes were 1q gain and 16q loss.
TABLE 4Genomic Alterations Unique to the Invasive Component in Clonally Related Tumors
Clone ChromosomePosition(Mb) Associated gene(s)a Alteration
Average log2
ratio in ILCb
RMC01P057 1 30.3 FGR Loss �0.348RP11-4K20 2 11.8 KCNF1 Loss �0.229RP11-93O2 2 34.3 LOC84661, MGC11061, SRD5A2, XDH, SPG4, FLJ20837, BIRC6, LOC51072 Gain 0.317RP11-133J6 6 46.0 KCNK5, KCNK16, KCNK17 Loss �0.275RP11-14G23 6 47.7 Loss �0.256CTD-2026K7 6 114.6 Gain 0.242RP11-58F7 7 170.3 PTPRN2 Gain 0.224RP11-90B7 8 89.8 PKIA Gain 0.274RP11-109D12 14 23.6 Gain 0.244RP1-172N16 17 2.5 Loss �0.295RP5-859020 17 10.0 Loss �0.286RP5-1071L14 17 54.1 Loss �0.339RP51073F15 17 65.6 RPS6KB1 Gain 0.241RP11-138-A15 20 37.8 NNAT, BLCAP Loss �0.255
ILC: invasive lobular carcinoma; Mb: megabases.a Based on the human DNA sequence draft obtained from the University of California–Santa Cruz Genome Browser (freeze date, August 2001; available from URL: http://genome.ucsc.edu).b Calculated only for clonally related tumors in which bacterial artificial chromosome array findings were altered in the invasive carcinoma but not in the carcinoma in situ.
Clonality of Lobular Breast Carcinomas/Hwang et al. 2569

DeLeeuw et al.16 as well as our own group,17 whichnoted a loss of E-cadherin and catenins in both LCISand ILC, but not in ductal lesions. Moran and Haffty18
reported on a series of 1096 patients who underwentbreast-conserving surgery before 1992. In specimensthat contained LCIS, those investigators found a 53%prevalence of ILC, compared with a 5% prevalence ofILC in patients without LCIS (P � 0.001).
We have observed that LCIS is an advanced neo-plastic state with alterations that, on average, involve� 20% of the genome. Whole-arm averaging of thelog2 ratio across each chromosome arm was used tocompare our array CGH data with results reportedpreviously using chromosomal CGH; this analysisshowed that the average number of chromosomal al-terations in LCIS (5.0) was less than the average num-ber of alterations in ILC (8.9), DCIS (8.8), or invasiveductal carcinoma (8.6), but greater than the corre-sponding figure in atypical ductal hyperplasia (3.6).8,19
These findings suggest that LCIS has greater genomicstability compared with other breast neoplasms andmay indicate that LCIS has a lower propensity fordisease progression than does DCIS.
Loss of chromosome arm 16q was seen in 100% ofboth ILC and LCIS lesions. Genomic alteration of 16qis an early change that also has been described in avariety of other types of breast neoplasms, such asatypical lobular hyperplasia, atypical ductal hyperpla-sia, tubular carcinoma, invasive papillary carcinoma,and low-grade DCIS.8,9,20 –22 In these typically well dif-ferentiated, low-grade tumors, this loss frequently isdue to a 1;16 fusion (as detected by fluorescence insitu hybridization), which results in loss of the distalarm of 16q combined with a translocation and gain of1q.23 For low-grade breast carcinomas, including LCISand ILC, this translocation may be an important earlyevent in carcinogenesis.
The ubiquitous loss of 16q in both LCIS and ILCincludes the locus for E-cadherin (16q22.1) and isassociated with the loss of E-cadherin immunohisto-chemical staining. Promoter methylation, mutation,and allelic loss all have been proposed as mechanismsto explain loss of E-cadherin staining in breast carci-nomas. Our data support a model in which it is likelythat regional genomic loss plays an important role.The finding that 16q is lost in other tumors that none-theless have normal E-cadherin staining21,22 suggeststhat genomic loss is an early event. This genomicalteration may be followed by a “second hit” throughmutation or methylation, resulting in the reduced E-cadherin staining phenotype seen in both LCIS andILC. E-cadherin is central to cell adhesion and tissuemorphogenesis and recently has been reported to playa role in the suppression of invasion.24 Therefore, its
loss may play an important role in tumorigenesis forlobular carcinomas.
The region that contains the p53 gene (17p13.1)was lost in one-third of LCIS lesions and one-half ofILC lesions, whereas cyclin D1 (11q13) was gained inalmost 40% of lesions. Also noteworthy are the differ-ences between our own findings and the genomicalterations described in invasive ductal carcinomas.The region that codes for EGFR (7p11.2) is gainedmore frequently in lobular carcinomas than in ductalcarcinomas (� 60% in the current study vs. 27% re-ported in a series of 1029 invasive ductal breast carci-nomas25). Gains in the locus coding for ERBB2(17q21.1) were conspicuously rare in the current dataset (occurring in 10% of LCIS lesions and 15% of ILClesions), whereas it has been shown that � 30% ofinvasive ductal carcinomas amplify and overexpressthis gene. This finding is consistent with previous re-ports, which also have shown that ERBB2 amplifica-tion is uncommon in lobular carcinomas,26,27 and in-dicates that genomic alteration of ERBB2 is not criticalfor lobular carcinoma progression.
The relation between LCIS and synchronous ILCwas evaluated with two different metrics—a weightedsimilarity score and a concordance score (see Materi-als and Methods). Both analyses confirmed that manypaired LCIS and ILC lesions were genomically similar.We have referred to synchronous LCIS/ILC lesionswith the highest similarity scores as clonal, becausethe extent of shared genomic alterations was highlyindicative that these lesions shared a common cell oforigin. However, a possible alternate interpretation ofthe data may be that the presence of unrecognizedpolymorphisms may cause lesions from the same pa-tient to appear more closely related to one anotherthan to other lesions on the basis of these polymor-phisms alone. We minimized this potential confound-ing influence by excluding all known polymorphicBACs from the analysis. After excluding a total of 28BACs, the majority of LCIS/ILC pairs still were relatedmore closely to one another than to all other lesions.Another potential confounder in our study was possi-ble cross-contamination of in situ and invasive carci-nomas. However, clonality was observed even in casesin which the LCIS and ILC lesions were separated by� 2 cm or were present in separate tissue blocks; thisis the most compelling evidence to date of a precur-sor-product relation between LCIS and ILC.
Among the clonal pairs, there were several regionsthat were altered in ILC but not in LCIS (Table 4).Although some of these genomic changes may reflectonly the overall increased genomic instability associ-ated with disease progression, it is possible that geneslocated within these loci may be important to lobular
2570 CANCER June 15, 2004 / Volume 100 / Number 12

carcinoma progression. Protein tyrosine phosphatasereceptor type N, which is located at 7q36.3 (170.3 Mb),is a member of the protein tyrosine phosphatase fam-ily, which is known to play an important role in theregulation of cell growth and oncogenic differentia-tion. It has been shown that the protein kinase inhib-itor � gene, which is located at 8q21.11 (89.8 Kb), is apotent competitive inhibitor of cyclic AMP– depen-dent protein kinase activity.28 It also has been re-ported that a group of ion channel genes, KCNF1(2p25; 11.8 Mb), KCNK5, and KCNK17 (6p21; 46.0 Mb),were gained in invasive carcinomas but not in pairedin situ carcinomas. It is known that some of thesegenes affect electrolyte metabolism, whereas othersplay a role in cell cycle regulation.
The clinical implications of these findings meritdiscussion. Although complete surgical excision ofLCIS to clear margins is not the current standard ofcare, do our data support such a shift in practice? Inthe National Surgical Adjuvant Breast Project (NSABP)data set, only 4 of 182 women with LCIS developedinvasive carcinoma at 5 years.29 Thus, at best, fourincidents of ILC may have been prevented by moreaggressive treatment for LCIS. However, the impact onlong-term outcome remains uncertain, because it isunknown how many of those four patients would havehad a different outcome as a result of earlier treat-ment. Furthermore, aggressive local treatment wouldnot be expected to affect the contralateral risk of car-cinoma conferred by LCIS. Fisher et al. showed that at4 years of follow-up, women with LCIS were affordeda 56% overall reduction in invasive carcinoma riskwith tamoxifen compared with placebo.30 Thus, forthe majority of patients who are diagnosed with LCIS,current epidemiologic and clinical evidence supportsthe established approach of close expectant manage-ment and consideration of prophylactic tamoxifentherapy for bilateral risk reduction.
Nevertheless, although we do not advocate anabrupt change in the surgical management of LCIS,our data suggest a reevaluation of the role that LCISplays in the breast carcinoma progression model. Forpatients with preinvasive lobular carcinoma, diseaseprogression can lead to ILC. Previous reports indi-cated that any ipsilateral invasive carcinoma appear-ing after a diagnosis of LCIS was more likely to haveductal histology rather than lobular histology.2,3 Theapparently increased incidence of ipsilateral invasiveductal carcinoma after LCIS in earlier studies may beattributable to the absence of standardized criteria forLCIS, with the possibility that many cases of DCISwere incorrectly diagnosed as LCIS. Studies that wereconducted after more comprehensive histologic crite-ria for lobular neoplasias were established indicate
that ipsilateral ILC is more common after LCIS thanafter DCIS. In the NSABP-B17 data set, all four invasiveipsilateral carcinomas that developed after LCIS wereof the lobular type.29 It is noteworthy that the onlypredictor of invasive carcinoma was the degree oflobular distortion (a possible marker of tumor grade)in the LCIS. Others have found that when LCIS ispresent in the setting of invasive carcinoma, the his-tology of the invasive component is more than twiceas likely to be lobular than it is to be ductal.2,4,18 Theseclinical observations are entirely consistent with ourgenomic findings.
We propose that in addition to histologic type, thegrade of preinvasive disease also may be an importantdeterminant of both the rate and the likelihood ofdisease progression (Fig. 5). It is probable that somesubtypes of in situ disease have a limited likelihood ofever progressing to invasion. Other subtypes, such ashigh-grade DCIS and pleomorphic LCIS, may be muchmore likely to progress. The samples of LCIS evaluatedin the current study were selected specifically becauseof the accompanying ILC; thus, our data may charac-terize a subset of LCIS with an elevated invasive po-tential. However, even in the current data set, we haveshown that LCIS exhibits significantly fewer genomicaberrations than have been reported in DCIS. Over thelong term, this relative genomic stability may be man-ifested as both a decreased likelihood and a slowerrate of disease progression (Fig. 5). Because most pub-lished studies cover relatively short follow-up periods,there may be an ascertainment bias in favor of in situlesions that progress at a faster rate. It is certain thataggressive treatment is warranted for any in situ lesionthat has a high propensity for progression. However,for an in situ lesion with limited invasive potential(such as most LCIS lesions and low-grade DCIS le-sions), prevention strategies aimed at global risk re-duction may represent the optimal long-term ap-proach.
We conclude that genomic alterations, particu-larly 16q loss and 1q gain, are highly prevalent in both
FIGURE 5. Proposed progression pathway from carcinoma in situ to invasive
carcinoma. The propensity for disease progression is indicated by the relative
thickness of the arrows. The length of the arrow indicates the time course over
which such progression can develop; it is likely that the time course for lobular
carcinoma in situ (LCIS) is longer than that for ductal carcinoma in situ (DCIS).
IDC: invasive ductal carcinoma; ILC: invasive lobular carcinoma.
Clonality of Lobular Breast Carcinomas/Hwang et al. 2571

LCIS and ILC, with invasive lesions exhibiting morealterations overall compared with in situ lesions.High-resolution data were particularly powerful inidentifying the genomic regions that may be impor-tant in the progression from in situ disease to invasivedisease. Despite the overall greater frequency ofgenomic changes in ILC versus LCIS, our data suggestthat progression to the invasive phenotype does notrequire a specific genomic change and is not depen-dent on the cumulative burden of genomic changes,thus implicating multiple potential pathways for pro-gression. The clonal relation exhibited by most ofthese matched pairs supports the idea of a precursor-product relation between LCIS and ILC and indicatesthat LCIS is more than simply a marker for an in-creased risk of breast carcinoma.
REFERENCES1. Foote FW Jr., Stewart FW. Lobular carcinoma in situ. Am J
Pathol. 1941;17:491– 499.2. Rosen PP, Kosloff C, Lieberman PH, Adair F, Braun DW Jr.
Lobular carcinoma in situ of the breast. Detailed analysis of99 patients with average follow-up of 24 years. Am J SurgPathol. 1978;2:225–251.
3. Page DL, Kidd TE Jr., Dupont WD, Simpson JF, Rogers LW.Lobular neoplasia of the breast: higher risk for subsequentinvasive cancer predicted by more extensive disease. HumPathol. 1991;22:1232–1239.
4. Wheeler JE, Enterline HT, Roseman JM, et al. Lobular car-cinoma in situ of the breast. Long-term follow up. Cancer.1974;34:554 –563.
5. Frykberg ER, Bland KI. Management of in situ and mini-mally invasive breast carcinoma. World J Surg. 1994;18:45–57.
6. Habel LA, Moe RE, Daling JR, Holte S, Rossing MA, WeissNS. Risk of contralateral breast cancer among women withcarcinoma in situ of the breast. Ann Surg. 1997;225:69 –75.
7. Ottesen GL, Graversen HP, Blichert-Toft M, Christensen IJ,Andersen JA. Carcinoma in situ of the female breast. 10 yearfollow-up results of a prospective nationwide study. BreastCancer Res Treat. 2000;62:197–210.
8. Gong G, DeVries S, Chew KL, Cha I, Ljung BM, WaldmanFM. Genetic changes in paired atypical and usual ductalhyperplasia of the breast by comparative genomic hybrid-ization. Clin Cancer Res. 2001;7:2410 –2414.
9. Waldman FM, DeVries S, Chew KL, Moore DH II, Ker-likowske K, Ljung BM. Chromosomal alterations in ductalcarcinomas in situ and their in situ recurrences. J NatlCancer Inst. 2000;92:313–320.
10. Ginzinger DG, Godfrey TE, Nigro J, et al. Measurement ofDNA copy number at microsatellite loci using quantitativePCR analysis. Cancer Res. 2000;60:5405–5409.
11. Pinkel D, Segraves R, Sudar D, et al. High resolution analysisof DNA copy number variation using comparative genomichybridization to microarrays. Nat Genet. 1998;20:207–211.
12. Wilhelm M, Veltman JA, Olshen AB, et al. Array-based com-parative genomic hybridization for the differential diagnosisof renal cell cancer. Cancer Res. 2002;62:957–960.
13. Veltman JA, Fridlyand J, Pejavar S, et al. Array-based com-parative genomic hybridization for genome-wide screening
of DNA copy number in bladder tumors. Cancer Res. 2003;63:2872–2880.
14. Jain AN, Tokuyasu TA, Snijders AM, Segraves R, AlbertsonDG, Pinkel D. Fully automatic quantification of microarrayimage data. Genome Res. 2002;12:325–332.
15. Nemoto T, Castillo N, Tsukada Y, Koul A, Eckhert KH Jr.,Bauer RL. Lobular carcinoma in situ with microinvasion.J Surg Oncol. 1998;67:41– 46.
16. De Leeuw WJ, Berx G, Vos CB, et al. Simultaneous loss ofE-cadherin and catenins in invasive lobular breast cancerand lobular carcinoma in situ. J Pathol. 1997;183:404 – 411.
17. Etzell JE, Devries S, Chew K, et al. Loss of chromosome 16qin lobular carcinoma in situ. Hum Pathol. 2001;32:292–296.
18. Moran M, Haffty BG. Lobular carcinoma in situ as a com-ponent of breast cancer: the long-term outcome in patientstreated with breast-conservation therapy. Int J Radiat OncolBiol Phys. 1998;40:353–358.
19. Isola JJ, Kallioniemi OP, Chu LW, et al. Genetic aberrationsdetected by comparative genomic hybridization predict out-come in node-negative breast cancer. Am J Pathol. 1995;147:905–911.
20. Lu YJ, Osin P, Lakhani SR, Di Palma S, Gusterson BA, ShipleyJM. Comparative genomic hybridization analysis of lobularcarcinoma in situ and atypical lobular hyperplasia and po-tential roles for gains and losses of genetic material in breastneoplasia. Cancer Res. 1998;58:4721– 4727.
21. Waldman FM, Hwang ES, Etzell J, et al. Genomic alterationsin tubular breast carcinomas. Hum Pathol. 2001;32:222–226.
22. Thor AD, Eng C, Devries S, et al. Invasive micropapillarycarcinoma of the breast is associated with chromosome 8abnormalities detected by comparative genomic hybridiza-tion. Hum Pathol. 2002;33:628 – 631.
23. Tsuda H, Takarabe T, Hirohashi S. Correlation of numericaland structural status of chromosome 16 with histologicaltype and grade of non-invasive and invasive breast carcino-mas. Int J Cancer. 1999;84:381–387.
24. Berx G, Cleton-Jansen AM, Nollet F, et al. E-cadherin is atumour/invasion suppressor gene mutated in human lobu-lar breast cancers. EMBO J. 1995;14:6107– 6115.
25. Tsutsui S, Ohno S, Murakami S, Hachitanda Y, Oda S. Prog-nostic value of epidermal growth factor receptor (EGFR) andits relationship to the estrogen receptor status in 1029 pa-tients with breast cancer. Breast Cancer Res Treat. 2002;71:67–75.
26. Porter PL, Garcia R, Moe R, Corwin DJ, Gown AM. C-erbB-2oncogene protein in in situ and invasive lobular breastneoplasia. Cancer. 1991;68:331–334.
27. Mercapide J, Zhang SY, Fan X, et al. CCND1- and ERBB2-gene deregulation and PTEN mutation analyses in invasivelobular carcinoma of the breast. Mol Carcinog. 2002;35:6 –12.
28. Olsen SR, Uhler MD. Inhibition of protein kinase-A by over-expression of the cloned human protein kinase inhibitor.Mol Endocrinol. 1991;5:1246 –1256.
29. Fisher ER, Costantino J, Fisher B, et al. Pathologic findingsfrom the National Surgical Adjuvant Breast Project (NSABP)Protocol B-17. Five-year observations concerning lobularcarcinoma in situ. Cancer. 1996;78:1403–1416.
30. Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen forprevention of breast cancer: report of the National SurgicalAdjuvant Breast and Bowel Project P-1 study. J Natl CancerInst. 1998;90:1371–1388.
2572 CANCER June 15, 2004 / Volume 100 / Number 12
Copyright © 2022 FDOKUMEN




















