
ChemInform Abstract: Transfer Hydrogenation of Carbonyl Compounds Catalyzed by Ruthenium...
8
Transfer hydrogenation of ketones catalyzed by new rhodium and iridium complexes of aminophosphine containing cyclohexyl moiety and photosensing behaviors of rhodium and iridium based devices Khadichakhan Rafikova a , Nurzhamal Kystaubayeva a , Murat Aydemir b, * , Cezmi Kayan b , Yusuf Selim Ocak c , Hamdi Temel d , Alexey Zazybin a , Nevin Gürbüz e , _ Ismail Özdemir e a Department of Chemical Engineering, Kazakh-British Technical University, 050000 Almaty, Kazakhstan b Department of Chemistry, Faculty of Science, University of Dicle, 21280 Diyarbakir, Turkey c Department of Science, Faculty of Education, University of Dicle, 21280 Diyarbakir, Turkey d Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Dicle University, Diyarbakir 21280, Turkey e Department of Chemistry, Faculty of Science and Art, University of _ Inönü, 44280 Malatya, Turkey article info Article history: Received 6 December 2013 Received in revised form 14 January 2014 Accepted 20 January 2014 Keywords: Aminophosphine Iridium Transfer hydrogenation Rectifying contact Electrical properties Photoconductor abstract The reaction of [Rh(m-Cl)(cod)] 2 and Ir(h 5 -C 5 Me 5 )(m-Cl)Cl] 2 with aminophosphine ligands Cy 2 PNHCH 2 eC 4 H 3 X (X: O; S) gave a range of new monodendate [Rh(Cy 2 PNHCH 2 eC 4 H 3 O)(cod)Cl], (1), [Rh(Cy 2 PNHCH 2 eC 4 H 3 S)(cod)Cl], (2), [Ir(Cy 2 PNHCH 2 eC 4 H 3 O)(h 5 -C 5 Me 5 )Cl 2 ], (3) and [Ir(Cy 2 PNHCH 2 eC 4 H 3 S)(h 5 -C 5 Me 5 )Cl 2 ], (4) complexes, which were characterized by analytical and spectroscopic methods. The new rhodium(I) and iridium(III) catalysts were applied to transfer hydrogenation of ace- tophenone derivatives using 2-propanol as a hydrogen source. The results showed that the corre- sponding alcohols could be obtained with high activity (up to 99%) under mild conditions. Notably, [Rh(Cy 2 PNHCH 2 eC 4 H 3 O)(cod)Cl] complex (1) is much more active than the other analogous complexes in the transfer hydrogenation. Moreover, organiceinorganic rectifying contacts were fabricated forming rhodium(I) and iridium(III) complex thin films on n-Si semiconductors and evaporating Au metal on the structures. Electrical properties of the contacts including ideality factor, barrier height and series resis- tance were determined using their currentevoltage (IeV) data. The photoelectrical characteristics of the devices were examined under the light with 40e100 mW/cm 2 illumination conditions. It was seen that light had strong effects on IeV characteristics of the devices and the ones fabricated using 3 and 4 complexes had unusually forward and reverse bias photoconducting behavior. Ó 2014 Elsevier B.V. All rights reserved. 1. Introduction Transition metal complexes are powerful catalysts for organic transformations and when suitable ligands are bound to a metal center, they can offer chemio, regio or stereo selectivity under mild conditions [1]. However, the appropriate choice of metal precursors and the reaction conditions are crucial for catalytic properties [2].A number of transition metal complexes are known to catalyze hydrogen transfer from an alcohol to a ketone [3e5]. Over the last three decades, most effort on hydrogenation has been focused on the use of ruthenium, rhodium and iridium catalysts [6e10]. Rhodium and iridium complexes have been proven to lead to very efficient processes along with potential industrial applications [11e 14]. To date, a number of such systems with a variety of backbone frameworks have been synthesized and their transition metal chemistry has been explored [15e17]. Phosphorusenitrogen con- taining ligands have particular use in catalysis where it is necessary for part of ligand to dissociate to allow an organic fragment to co- ordinate and undergo transformations [18,19]. The presence of PeN ligands enables many different and important catalytic processes to occur [7,20,21]. Especially, aminophosphines are able to stabilize many different metals in various oxidation states, controlling the performance of metals in a large variety of useful transformations. Synthesis of new aminophosphines to stabilize transition metals in * Corresponding author. Department of Chemistry, Dicle University, TR-21280 Diyarbakır, Turkey. Tel.: þ90 412 248 8550; fax: þ90 412 248 8300. E-mail address: [email protected] (M. Aydemir). Contents lists available at ScienceDirect Journal of Organometallic Chemistry journal homepage: www.elsevier.com/locate/jorganchem http://dx.doi.org/10.1016/j.jorganchem.2014.01.025 0022-328X/Ó 2014 Elsevier B.V. All rights reserved. Journal of Organometallic Chemistry 758 (2014) 1e8
Transcript of ChemInform Abstract: Transfer Hydrogenation of Carbonyl Compounds Catalyzed by Ruthenium...

lable at ScienceDirect
Journal of Organometallic Chemistry 758 (2014) 1e8
Contents lists avai
Journal of Organometallic Chemistry
journal homepage: www.elsevier .com/locate/ jorganchem
Transfer hydrogenation of ketones catalyzed by new rhodium andiridium complexes of aminophosphine containing cyclohexyl moietyand photosensing behaviors of rhodium and iridium based devices
Khadichakhan Rafikova a, Nurzhamal Kystaubayeva a, Murat Aydemir b,*, Cezmi Kayan b,Yusuf Selim Ocak c, Hamdi Temel d, Alexey Zazybin a, Nevin Gürbüz e, _Ismail Özdemir e
aDepartment of Chemical Engineering, Kazakh-British Technical University, 050000 Almaty, KazakhstanbDepartment of Chemistry, Faculty of Science, University of Dicle, 21280 Diyarbakir, TurkeycDepartment of Science, Faculty of Education, University of Dicle, 21280 Diyarbakir, TurkeydDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Dicle University, Diyarbakir 21280, TurkeyeDepartment of Chemistry, Faculty of Science and Art, University of _Inönü, 44280 Malatya, Turkey
a r t i c l e i n f o
Article history:Received 6 December 2013Received in revised form14 January 2014Accepted 20 January 2014
Keywords:AminophosphineIridiumTransfer hydrogenationRectifying contactElectrical propertiesPhotoconductor
* Corresponding author. Department of ChemistryDiyarbakır, Turkey. Tel.: þ90 412 248 8550; fax: þ90
E-mail address: [email protected] (M. Aydemir
http://dx.doi.org/10.1016/j.jorganchem.2014.01.0250022-328X/� 2014 Elsevier B.V. All rights reserved.
a b s t r a c t
The reaction of [Rh(m-Cl)(cod)]2 and Ir(h5-C5Me5)(m-Cl)Cl]2 with aminophosphine ligands Cy2PNHCH2
eC4H3X (X: O; S) gave a range of new monodendate [Rh(Cy2PNHCH2eC4H3O)(cod)Cl], (1),[Rh(Cy2PNHCH2eC4H3S)(cod)Cl], (2), [Ir(Cy2PNHCH2eC4H3O)(h5-C5Me5)Cl2], (3) and [Ir(Cy2PNHCH2
eC4H3S)(h5-C5Me5)Cl2], (4) complexes, which were characterized by analytical and spectroscopic
methods. The new rhodium(I) and iridium(III) catalysts were applied to transfer hydrogenation of ace-tophenone derivatives using 2-propanol as a hydrogen source. The results showed that the corre-sponding alcohols could be obtained with high activity (up to 99%) under mild conditions. Notably,[Rh(Cy2PNHCH2eC4H3O)(cod)Cl] complex (1) is much more active than the other analogous complexesin the transfer hydrogenation. Moreover, organiceinorganic rectifying contacts were fabricated formingrhodium(I) and iridium(III) complex thin films on n-Si semiconductors and evaporating Au metal on thestructures. Electrical properties of the contacts including ideality factor, barrier height and series resis-tance were determined using their currentevoltage (IeV) data. The photoelectrical characteristics of thedevices were examined under the light with 40e100 mW/cm2 illumination conditions. It was seen thatlight had strong effects on IeV characteristics of the devices and the ones fabricated using 3 and 4complexes had unusually forward and reverse bias photoconducting behavior.
� 2014 Elsevier B.V. All rights reserved.
1. Introduction
Transition metal complexes are powerful catalysts for organictransformations and when suitable ligands are bound to a metalcenter, they can offer chemio, regio or stereo selectivity under mildconditions [1]. However, the appropriate choice of metal precursorsand the reaction conditions are crucial for catalytic properties [2]. Anumber of transition metal complexes are known to catalyzehydrogen transfer from an alcohol to a ketone [3e5]. Over the lastthree decades, most effort on hydrogenation has been focused on
, Dicle University, TR-21280412 248 8300.).
the use of ruthenium, rhodium and iridium catalysts [6e10].Rhodium and iridium complexes have been proven to lead to veryefficient processes alongwith potential industrial applications [11e14].
To date, a number of such systems with a variety of backboneframeworks have been synthesized and their transition metalchemistry has been explored [15e17]. Phosphorusenitrogen con-taining ligands have particular use in catalysis where it is necessaryfor part of ligand to dissociate to allow an organic fragment to co-ordinate and undergo transformations [18,19]. The presence of PeNligands enables many different and important catalytic processes tooccur [7,20,21]. Especially, aminophosphines are able to stabilizemany different metals in various oxidation states, controlling theperformance of metals in a large variety of useful transformations.Synthesis of new aminophosphines to stabilize transition metals in

Scheme 1. Synthesis of the [Rh(Cy2PNHCH2eC4H3O)(cod)Cl], (1), [Rh(Cy2PNHCH2e
C4H3S)(cod)Cl], (2), [Ir(Cy2PNHCH2eC4H3O)(h5-C5Me5)Cl2], (3) and [Ir(Cy2PNHCH2e
C4H3S)(h5-C5Me5)Cl2], (4) complexes (i) 1/2 equiv. [Rh(m-Cl)(cod)]2, thf; (ii) 1/2 equiv.[Ir(h5-C5Me5)(m-Cl)Cl]2, thf.
K. Rafikova et al. / Journal of Organometallic Chemistry 758 (2014) 1e82
low valent states is considered to be amost challenging task in viewof their potential utility in a variety of metal-mediated organictransformations [22].
Hydrogen transfer reactions are mild methodologies forreduction of ketones or imines and oxidation of alcohols or aminesin which a substrate-selective catalyst transfers hydrogen be-tween the substrate and a hydrogen donor or acceptor, respec-tively [23e25]. From an industrial point of view, catalytic transferhydrogenation is an attractive alternative for high pressure cata-lytic hydrogenations with molecular hydrogen [26]. Here,hydrogen donors such as secondary alcohols (e.g., 2-propanol) areapplied to convert carbonyl compounds to alcohols. The riskassociated with the use of molecular hydrogen at high pressures isthereby eliminated [27,28]. Furthermore, there are several otheradvantages for use of 2-propanol such as, it is inexpensive, readilyavailable and has an appropriate boiling point. It is also a goodsolvent for many organic compounds. Upon dehydrogenation, 2-propanol is converted to acetone, which can be easily removedfrom the reaction mixture, and thus simplifies the reaction process[29,30].
Organic materials have obtained an important place in the fieldsof electrical and optical device industry. They have been preferredin device applications because of their key advantages includinglarge area coating, using on flexible substrates and low cost [31,32].Among the other organic compounds, there is a growing interest onmetal complexes because of their mechanical and chemical sta-bilities [33]. While some studies have concentrated on findingsuitable metal complexes for device technology [34,35], othershave focused on the usage of these compounds in the fabrication ofdevices including solar cells, Schottky diodes and light emittingdiodes [36e39]. Studies on metalesemiconductor (MS) deviceswith organic semiconductor have shown that these structurespresented photovoltaic and photoconductivity influences whenthey are exposed to light because of electron and hole production atthe interface [40].
To the best our knowledge, there is no report on the use of thesecomplexes including aminophosphines having cyclohexyl moietyon phosphorus atom in rhodium and iridium catalyzed transferhydrogenation reaction. As part of our research program, we reporthere the synthesis and full characterization of four new amino-phosphine complexes [Rh(Cy2PNHCH2eC4H3O)(cod)Cl] (1),[Rh(Cy2PNHCH2eC4H3S)(cod)Cl], (2), [Ir(Cy2PNHCH2eC4H3O)(h5-C5Me5)Cl2], (3) and [Ir(Cy2PNHCH2eC4H3S)(h5-C5Me5)Cl2], (4). Wealso report their catalytic activity in transfer hydrogenation re-actions of ketones with iso-PrOH. Furthermore, up to know, studieshave reported to reverse bias photosensing properties of organicbased devices [38,41,42]. In this study, fabrication of organiceinorganic heterojunction devices, their electrical propertiesincluding ideality factor, barrier height and series resistance valuesand photoelectrical properties are presented. It is also reported thatthe devices obtained using 3 and 4 complexes have reverse andcurrent bias photoconduction behavior which have not reported upto now for organic based devices.
2. Results and discussion
2.1. Synthesis and characterization of the metal complexes
Synthesis and characterization of the ligands, furfuryl-2-(N-dicyclohexylphosphino)methylamine and thiophene-2-(N-dicy-clohexylphosphino)methyla-mine, were mentioned elsewhere[43]. We examined various coordination chemistry of these ami-nophosphines with [Rh(m-Cl)(cod)]2 precursor. Reaction of furfuryl-2-(N-dicyclohexylphosphino)methylamine or thiophene-2-(N-dicyclohexylphosphino) methylamine with [Rh(m-Cl)(cod)]2 in a
molar ratio of 2/1 at room temperature for 45 min afforded[Rh(Cy2PNHCH2eC4H3O)(cod)Cl], (1) and [Rh(Cy2PNHCH2e
C4H3S)(cod)Cl] (2), respectively as crystalline yellow powders(Scheme 1). The complexes of 1 and 2were isolated as indicated bydoublets in the 31Pe{1H} NMR spectra at d 71.98 (d, 1JRhP: 156.5 Hz)and 74.39 (d, 1JRhP: 154.1 Hz) ppm, respectively, (Fig. 1). In their 1HNMR spectra, 1 and 2 are characterized by CH resonances of cod atd w5.35 and 3.55 ppm, whereas in the 13C-{1H} NMR spectra, res-onances at dw70 and 104 ppm correspond to CH resonances of cod(for details see Experimental Section). Furthermore, other 1H and13C-{1H} NMR data are in agreement with the proposed structures.The complexes were also characterized by IR and microanalysis.
We studied coordination chemistry of aminophosphinesincluding cyclohexyl moiety with [Ir(h5-C5Me5)(m-Cl)Cl]2 precursoras well. [Ir(Cy2PNHCH2eC4H3O)(h5-C5Me5)Cl2], 3 and [Ir(Cy2PNHCH2eC4H3S)(h5-C5Me5)Cl2], 4 were obtained by the reaction of li-gands with [Ir(h5-C5Me5)(m-Cl)Cl]2 in a molar ratio of 2/1 at roomtemperature for 1 h (Scheme 1). In the 31Pe{1H} NMR spectra,resonances at d w43 ppm may be attributed to complexes of 3 and4 (Fig. 1). 13C NMR spectra of the complexes display singlets atd w10 ppm attributable to methyl carbons of Cp* and doublets atd w92 due to carbons of Cp* ring. The 1H NMR spectra are consis-tent with the anticipated structures. The structural compositions ofthe complexes 3 and 4 were further confirmed by IR spectroscopyand microanalysis, and found to be in good agreement with thetheoretical values (for details see Experimental section).
2.2. Catalytic transfer hydrogenation of ketones
The brilliant catalytic performance of aminophosphine-basedtransition metal complexes [44], and references therein] promp-ted us to develop new Rh(I) and Ir(III) complexes with well-shaped ligands, since NH unit forms a hydrogen bond with thecarbonyl oxygen atom to stabilize the transition state. Therefore,the presence of an NH moiety in the ligands is crucially importantto determine the catalytic performance of the bifunctional cata-lysts [45e47]. An important and unprecedented aspect is that thecarbonyl compound does not interact directly with the metalcenter for its own activation [48]. To this end, we observed thecatalytic activation of complexes 1e4 in the transfer hydrogena-tion of ketones to the corresponding alcohols. In a typical exper-iment, 0.005 mmol of the complex and 0.5 mmol of ketone wereadded to a solution of NaOH in iso-PrOH (0.025 mmol of NaOH in5 mL iso-PrOH) and refluxed at 82 �C, the reaction being moni-tored by GC. In all reactions, these complexes catalyzed the
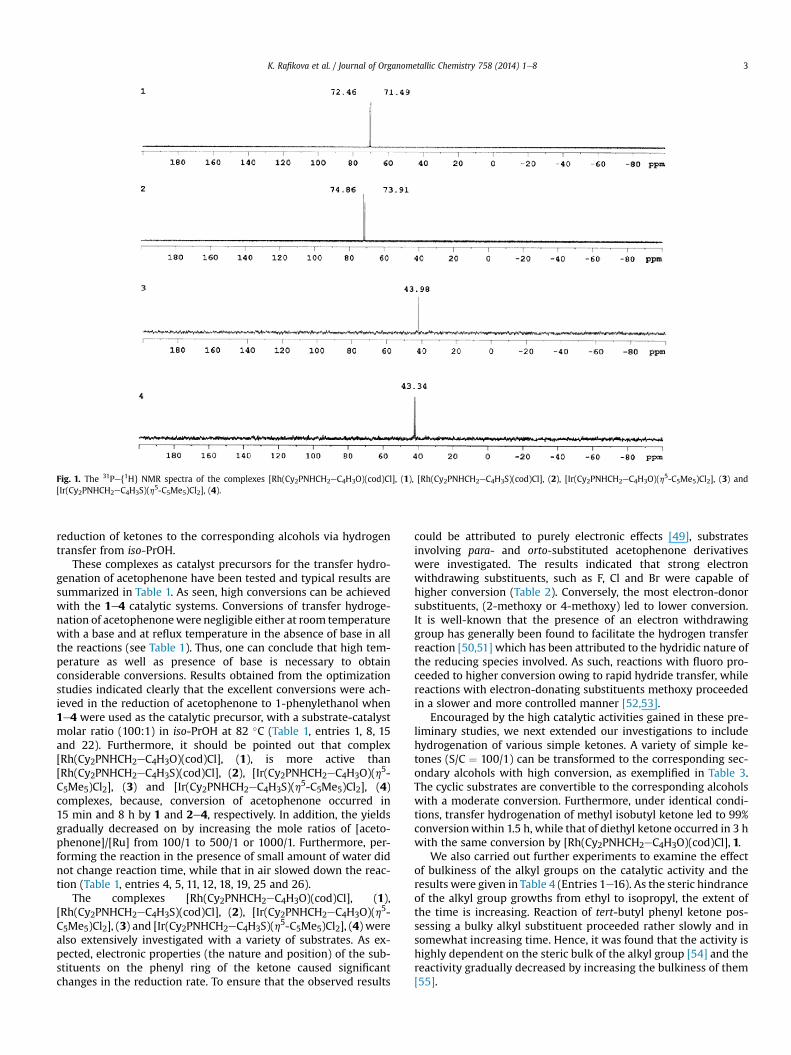
Fig. 1. The 31Pe{1H} NMR spectra of the complexes [Rh(Cy2PNHCH2eC4H3O)(cod)Cl], (1), [Rh(Cy2PNHCH2eC4H3S)(cod)Cl], (2), [Ir(Cy2PNHCH2eC4H3O)(h5-C5Me5)Cl2], (3) and[Ir(Cy2PNHCH2eC4H3S)(h5-C5Me5)Cl2], (4).
K. Rafikova et al. / Journal of Organometallic Chemistry 758 (2014) 1e8 3
reduction of ketones to the corresponding alcohols via hydrogentransfer from iso-PrOH.
These complexes as catalyst precursors for the transfer hydro-genation of acetophenone have been tested and typical results aresummarized in Table 1. As seen, high conversions can be achievedwith the 1e4 catalytic systems. Conversions of transfer hydroge-nation of acetophenonewere negligible either at room temperaturewith a base and at reflux temperature in the absence of base in allthe reactions (see Table 1). Thus, one can conclude that high tem-perature as well as presence of base is necessary to obtainconsiderable conversions. Results obtained from the optimizationstudies indicated clearly that the excellent conversions were ach-ieved in the reduction of acetophenone to 1-phenylethanol when1e4 were used as the catalytic precursor, with a substrate-catalystmolar ratio (100:1) in iso-PrOH at 82 �C (Table 1, entries 1, 8, 15and 22). Furthermore, it should be pointed out that complex[Rh(Cy2PNHCH2eC4H3O)(cod)Cl], (1), is more active than[Rh(Cy2PNHCH2eC4H3S)(cod)Cl], (2), [Ir(Cy2PNHCH2eC4H3O)(h5-C5Me5)Cl2], (3) and [Ir(Cy2PNHCH2eC4H3S)(h5-C5Me5)Cl2], (4)complexes, because, conversion of acetophenone occurred in15 min and 8 h by 1 and 2e4, respectively. In addition, the yieldsgradually decreased on by increasing the mole ratios of [aceto-phenone]/[Ru] from 100/1 to 500/1 or 1000/1. Furthermore, per-forming the reaction in the presence of small amount of water didnot change reaction time, while that in air slowed down the reac-tion (Table 1, entries 4, 5, 11, 12, 18, 19, 25 and 26).
The complexes [Rh(Cy2PNHCH2eC4H3O)(cod)Cl], (1),[Rh(Cy2PNHCH2eC4H3S)(cod)Cl], (2), [Ir(Cy2PNHCH2eC4H3O)(h5-C5Me5)Cl2], (3) and [Ir(Cy2PNHCH2eC4H3S)(h5-C5Me5)Cl2], (4) werealso extensively investigated with a variety of substrates. As ex-pected, electronic properties (the nature and position) of the sub-stituents on the phenyl ring of the ketone caused significantchanges in the reduction rate. To ensure that the observed results
could be attributed to purely electronic effects [49], substratesinvolving para- and orto-substituted acetophenone derivativeswere investigated. The results indicated that strong electronwithdrawing substituents, such as F, Cl and Br were capable ofhigher conversion (Table 2). Conversely, the most electron-donorsubstituents, (2-methoxy or 4-methoxy) led to lower conversion.It is well-known that the presence of an electron withdrawinggroup has generally been found to facilitate the hydrogen transferreaction [50,51] which has been attributed to the hydridic nature ofthe reducing species involved. As such, reactions with fluoro pro-ceeded to higher conversion owing to rapid hydride transfer, whilereactions with electron-donating substituents methoxy proceededin a slower and more controlled manner [52,53].
Encouraged by the high catalytic activities gained in these pre-liminary studies, we next extended our investigations to includehydrogenation of various simple ketones. A variety of simple ke-tones (S/C ¼ 100/1) can be transformed to the corresponding sec-ondary alcohols with high conversion, as exemplified in Table 3.The cyclic substrates are convertible to the corresponding alcoholswith a moderate conversion. Furthermore, under identical condi-tions, transfer hydrogenation of methyl isobutyl ketone led to 99%conversionwithin 1.5 h, while that of diethyl ketone occurred in 3 hwith the same conversion by [Rh(Cy2PNHCH2eC4H3O)(cod)Cl], 1.
We also carried out further experiments to examine the effectof bulkiness of the alkyl groups on the catalytic activity and theresults were given in Table 4 (Entries 1e16). As the steric hindranceof the alkyl group growths from ethyl to isopropyl, the extent ofthe time is increasing. Reaction of tert-butyl phenyl ketone pos-sessing a bulky alkyl substituent proceeded rather slowly and insomewhat increasing time. Hence, it was found that the activity ishighly dependent on the steric bulk of the alkyl group [54] and thereactivity gradually decreased by increasing the bulkiness of them[55].

Table 1Transfer hydrogenation of acetophenone with iso-PrOH catalyzed [Rh(Cy2PNHCH2e
C4H3O)(cod)Cl], (1), [Rh(Cy2PNHCH2eC4H3S)(cod)Cl], (2), [Ir(Cy2PNHCH2e
C4H3O)(h5-C5Me5)Cl2], (3) and [Ir(Cy2PNHCH2eC4H3S)(h5-C5Me5)Cl2],(4).
Entry Catalyst S/C/NaOH Time Conversion (%)i TOF (h�1)j
1 1a 100:1:5 15 min 98 3922 1b 100:1:5 24 h Trace e
3 1c 100:1 24 h Trace e
4 1d 100:1:5 15 min 96 3845 1e 100:1:5 3 h 98 336 1f 500:1:5 45 min 99 1327 1g 1000:1:5 1.5 h 97 658 2a 100:1:5 8 h 98 129 2b 100:1:5 24 h Trace e
10 2c 100:1 24 h Trace e
11 2d 100:1:5 8 h 96 1212 2e 100:1:5 30 h 97 <513 2f 500:1:5 20 h 98 <514 2g 1000:1:5 40 h 97 <515 3a 100:1:5 8 h 97 1216 3b 100:1:5 24 h Trace e
17 3c 100:1 24 h Trace e
18 3d 100:1:5 8 h 97 1219 3e 100:1:5 30 h 98 <520 3f 500:1:5 20 h 97 <521 3g 1000:1:5 40 h 96 <522 4a 100:1:5 8 h 96 1223 4b 100:1:5 24 h Trace e
24 4c 100:1 24 h Trace e
25 4d 100:1:5 8 h 98 1226 4e 100:1:5 30 h 98 <527 4f 500:1:5 20 h 98 <528 4g 1000:1:5 40 h 96 <5
Reaction conditions:a Refluxing in iso-PrOH; acetophenone/Ru/NaOH, 100:1:5.b At room temperature; acetophenone/Ru/NaOH, 100:1:5.c Refluxing in iso-PrOH; acetophenone/Ru, 100:1, in the absence of base.d Added 0.1 mL of H2O.e Refluxing the reaction in air.f Refluxing in iso-PrOH; acetophenone/Ru/NaOH, 500:1:5.g Refluxing in iso-PrOH; acetophenone/Ru/NaOH, 1000:1:5.i Determined by GC (three independent catalytic experiments).j Referred at the reaction time indicated in column; TOF ¼ (mol product/mol
Ru(II)Cat.) � h�1.
Table 2Transfer hydrogenation results for substituted acetophenones with the catalystsystems, [Rh(Cy2PNHCH2eC4H3O)(cod)Cl], (1), [Rh(Cy2PNHCH2eC4H3S)(cod)Cl], (2),[Ir(Cy2PNHCH2eC4H3O)(h5-C5Me5)Cl2], (3) and [Ir(Cy2PNHCH2eC4H3S)(h5-C5Me5)Cl2], (4).a
Entry R Time Conversion (%)b TOF (h�1)c
Cat:(I) complex 11 4-F 10 min 96 5762 4-Cl 10 min 98 5883 4-Br 15 min 98 3924 2-MeO 25 min 97 2335 4-MeO 20 min 96 288Cat:(I) complex 26 4-F 6 h 96 167 4-Cl 6 h 98 168 4-Br 8 h 97 129 2-MeO 12 h 98 810 4-MeO 10 h 98 10Cat:(III) complex 311 4-F 6 h 96 1612 4-Cl 6 h 95 1613 4-Br 8 h 95 1214 2-MeO 12 h 98 815 4-MeO 10 h 97 10Cat:(III) complex 416 4-F 6 h 97 1617 4-Cl 6 h 97 1618 4-Br 8 h 95 1219 2-MeO 12 h 97 820 4-MeO 10 h 96 10
a Catalyst (0.005 mmol), substrate (0.5 mmol), iso-PrOH (5 mL), NaOH(0.025 mmol %), 82 �C, the concentration of acetophenone derivatives is 0.1 M.
b Purity of compounds is checked by 1H NMR and GC (three independent catalyticexperiments), yields are based on methyl aryl ketone.
c TOF ¼ (mol product/mol Cat.) � h�1.
Table 3Transfer hydrogenation of various simple ketones with iso-PrOH catalyzed by[Rh(Cy2PNHCH2eC4H3O)(cod)Cl], (1), [Rh(Cy2PNHCH2eC4H3S)(cod)Cl], (2),[Ir(Cy2PNHCH2eC4H3O)(h5-C5Me5)Cl2], (3) and [Ir(Cy2PNHCH2eC4H3S)(h5-C5Me5)Cl2],(4).a
Entry Cat. Substrate Time Conversion (%)b
1 1 a 30 min 962 2 b 10 h 983 3 c 10 h 974 4 d 10 h 985 1 a 30 min 966 2 b 10 h 987 3 c 10 h 958 4 d 10 h 979 1 a 3/2 h 9910 2 b 20 h 9711 3 c 20 h 9812 4 d 20 h 9713 1 a 3 h 9914 2 b 30 h 9615 3 c 30 h 9916 4 d 30 h 95
a Refluxing in iso-PrOH; ketone/Ru/NaOH, 100:1:5.b Determined by GC (three independent catalytic experiments). Purity of com-
pounds is checked by 1H NMR and GC (three independent catalytic experiments),yields are based on methyl aryl ketone.
K. Rafikova et al. / Journal of Organometallic Chemistry 758 (2014) 1e84
2.3. Electrical and photoelectrical properties of Au/metal complex/n-Si devices
IeV measurements of a structure give important informationabout electrical characteristics of the device. Main electrical pa-rameters of a diode such as ideality factor, barrier height and seriesresistance can be determined using its IeV data in dark. In addition,photoelectrical parameters of a structure including open circuitvoltage (VOC) closed circuit current (ISC) can be extracted using its IeV data under light. Fig. 2 presents IeV measurements of Au/metalcomplex/n-Si structures in dark and under a solar simulator withvarious illumination conditions. As seen from the figures, allstructures have excellent rectification behavior. When a rectifyingdiode is taken into account, IeV can be analyzed using thermionicemission theory.
According to the theory, the ideality factor and the barrierheight values of a device can be extracted from the slope and thecurrent axis intercept of the linear regions of the forward bias IeVplots, respectively. The calculated ideality factor and the barrierheight values are given in Table 5. As seen from the table, theideality factors of the junctions are greater than unity. According tothe theory, ideality factor should be very close to unity (1.03) whenimage force lowering is considered. The deviation from ideal diodecan be attributed the effects of series resistance of the structure,existence of interface states at organiceinorganic interface,recombinationegeneration at the interface and tunneling [56,57].

Table 4Transfer hydrogenation results for substituted alkyl phenyl ketones with the catalystsystems [Rh(Cy2PNHCH2eC4H3O)(cod)Cl], (1), [Rh(Cy2PNHCH2eC4H3S)(cod)Cl], (2),[Ir(Cy2PNHCH2eC4H3O)(h5-C5Me5)Cl2], (3) and [Ir(Cy2PNHCH2eC4H3S)(h5-C5Me5)Cl2],(4).a
Entry R Time Conversion (%)b TOF (h�1)c
Cat:(I) complex 11 Ethyl 20 min 97 2912 Propyl 30 min 96 1923 iso-Propyl 40 min 96 1444 ter-Butyl 1.5 h 95 63Cat:(I) complex 25 Ethyl 16 h 96 <106 Propyl 32 h 98 <57 iso-Propyl 40 h 95 <58 ter-Butyl 50 h 97 <5Cat:(III) complex 39 Ethyl 16 h 95 <1010 Propyl 32 h 97 <511 iso-Propyl 40 h 98 <512 ter-Butyl 50 h 97 <5Cat:(III) complex 413 Ethyl 16 h 97 <1014 Propyl 32 h 97 <515 iso-Propyl 40 h 97 <516 ter-Butyl 50 h 96 <5
a Catalyst (0.005 mmol), substrate (0.5 mmol), iso-PrOH (5 mL), NaOH(0.025 mmol %),82 �C, respectively, the concentration of alkyl phenyl ketones is0.1 M.
b Purity of compounds is checked by 1H NMR and GC (three independent catalyticexperiments), yields are based on methyl aryl ketone.
c TOF ¼ (mol product/mol Cat.) � h�1.
K. Rafikova et al. / Journal of Organometallic Chemistry 758 (2014) 1e8 5
The barrier height values of the structures are very close to eachother and vary between 0.76 and 0.80 eV. Up to now, many kinds ofmolecules including small organic compounds, metal complexes,polymers and biological compounds have been used in the fabri-cation of organiceinorganic devices. For instance, several re-searchers [39] have synthesized and characterized Cu(II), Co(III),Ni(II) and Pd(II) complexes of N2S2O2 thio Schiff base ligand andshowed their usage in the fabrication of organiceinorganic hybriddevices. The calculated barrier heights of the devices were between0.75 and 0.88 eV. Other researchers [58] have reported the IeV andcapacitanceevoltage (CeV) characteristics of an Al/p-Si/organicsemiconductor formed using CoPc thin film on p-Si semi-conductor. They have showed the strong effects of CoPc interlayeron electrical properties of metal/semiconductor (MS) contact.Therefore, it can be easily said that the organic thin films havestrong impacts on the electrical performance of the organiceinor-ganic devices and they behave as an active layer.
The curvature in the higher voltage values of the IeV mea-surements are because of the series resistance of the structures. Theseries resistance of the devices may be caused by contact wires orbulk resistance of the organic material and semiconductor [59].Series resistance of a diode can be calculated by the help of Nordefunctions [60]. It is also possible to determine barrier height valuesusing Norde Functions. The obtained barrier height and seriesresistance values are also given in Table 5. As seen from the table,the devices obtained using [Ir(Cy2PNHCH2eC4H3O)(h5-C5Me5)Cl2],(3), and [Ir(Cy2PNHCH2eC4H3S)(h5-C5Me5)Cl2], (4), have lower se-ries resistance compared with ones fabricated using[Rh(Cy2PNHCH2eC4H3O)(cod)Cl], (1) and [Rh(Cy2PNHCH2e
C4H3S)(cod)Cl], (2). The large differences in series resistance be-tween the compounds can be attributed to the higher conductivity
values of compounds 3 and 4. As it is well known, conjugation isone of the most important properties increasing the conductivity oforganic compounds. Thus, the obtained results are in consistentwith the theory.
Fig. 2 also presents the influences of light intensity on IeVproperties of the structures. Although all structures have sensitivityto the light, the devices obtained using 3 and 4 have reverse biasand forward bias photosensing properties. As seen from the figures,current values of Au/3/n-Si and Au/4/n-Si are nearly fixed after theseries resistance curvatures in forward bias. The reverse and for-ward bias photosensing properties of the structures imply thephotoconductivity of [Ir(Cy2PNHCH2eC4H3O)(h5-C5Me5)Cl2] and[Ir(Cy2PNHCH2eC4H3S)(h5-C5Me5)Cl2], compounds. There aremany papers on the increase of reverse bias current with the in-crease of light intensity [38,42,43]. To the best our knowledge, thereis no report on the forward bias photosensing properties of a de-vice. The photovoltaic parameters of all structures determinedunder the light with 100 mW/cm2 illumination intensity called onesun are given in Table 6. The table presents the superior photo-voltaic properties of the junctions formed using compounds 3 and4. By taking Fig. 1 and Table 6 into account, one can easily say thatthe [Ir(Cy2PNHCH2eC4H3O)(h5-C5Me5)Cl2] and [Ir(Cy2PNHCH2e
C4H3S)(h5-C5Me5)Cl2] might be used in the fabrication reverse biasor forward bias photosensor applications.
3. Conclusion
In conclusion, new rhodium(I) and iridium(III) complexes con-taining furfuryl-2-(N-dicyclohexylphosphino)methylamine andthiophene-2-(N-dicyclohexylphosphino)methyl amine ligandshave been synthesized with high yield. All complexes were char-acterized usingmulti nuclear NMR, IR andmicroanalysis. The use ofthe new complexes for the reduction of the ketonic C]O bond ofacetophenone derivatives under hydrogen transfer conditions wasinvestigated. Surprisingly, [Rh(Cy2PNHCH2eC4H3O)(cod)Cl] was amore efficient catalyst in the transfer hydrogenation reaction thanthe other complexes. It was also found that these catalysts con-taining cyclohexyl moiety exhibited promising catalytic activitycompared to those of containing phenyl moiety. The construction ofthe catalysts containing cyclohexyl moiety and their flexibility to-ward transfer hydrogenation make these encouraging systems tofollow. In addition, organiceinorganic rectifying contacts wereformed by spin coating of complexes on n-Si semiconductor andevaporating Au metal on thin films. The electrical properties ofthe devices were analyzed in dark and under a solar simulatorwith various illumination conditions and the results showedexcellent reverse and forward bias photosensing behaviorsfor [Ir(Cy2PNHCH2eC4H3O)(h5-C5Me5)Cl2] and [Ir(Cy2PNHCH2e
C4H3S)(h5-C5Me5)Cl2] complexes, which were attributed the pho-toconducting properties of the complexes.
4. Experimental
4.1. Materials and methods
Unless otherwise stated, all reactions were carried out under anatmosphere of argon using conventional Schlenk glassware, sol-vents were dried using established procedures and distilled underargon immediately prior to use. Analytical grade and deuteratedsolvents were purchased from Merck. [Rh(m-Cl)(cod)]2 and Ir(h5-C5Me5)(m-Cl)Cl]2 are purchased from Fluka and were used asreceived. The IR spectra were recorded on a Mattson 1000 ATIUNICAM FT-IR spectrometer as KBr pellets. 1H (400.1 MHz), 13CNMR (100.6 MHz) and 31Pe{1H} NMR spectra (162.0 MHz) wererecorded on a Bruker AV400 spectrometer, with d referenced to

Fig. 2. IeV measurements of Au/metal complex/n-Si structures fabricated using a) 1 b) 2 c) 3 and d) 4 compounds.
K. Rafikova et al. / Journal of Organometallic Chemistry 758 (2014) 1e86
external TMS and 85% H3PO4 respectively. Elemental analysis wascarried out on a Fisons EA 1108 CHNS-O instrument. Melting pointswere recorded by Gallenkamp Model apparatus with open capil-laries. GC analyses were performed on a Shimadzu 2010 Plus Gas
Table 5Electrical parameters of the devices obtained using metal complexes.
Device ln IeV Norde
n fb (eV) fb (eV) RS (W)
Au/1/n-Si 1.65 0.79 0.83 17,080Au/2/n-Si 1.67 0.80 0.83 165,800Au/3/n-Si 1.82 0.76 0.85 313Au/4/n-Si 1.41 0.78 0.83 3518
Chromatograph equipped with capillary column (5% biphenyl, 95%dimethylsiloxane) (30m� 0.32mm� 0.25 mm). The GC parametersfor transfer hydrogenation of ketones were as follows; initialtemperature, 110 �C; initial time, 1 min; solvent delay, 4.48 min;
Table 6Photovoltaic parameters of the devices under the light with 100 mW/cm2 illumi-nation intensity.
Device ISC (mA) VOC (mV)
Au/1/n-Si 5.40 125Au/2/n-Si 0.16 95Au/3/n-Si 196 226Au/4/n-Si 114 206

K. Rafikova et al. / Journal of Organometallic Chemistry 758 (2014) 1e8 7
temperature ramp 80 �C/min; final temperature, 200 �C, 19 min;final time, 21.13 min; injector port temperature, 200 �C; detectortemperature, 200 �C, injection volume, 2.0 mL.
4.2. General procedure for the transfer hydrogenation of ketones
Typical procedure for the catalytic hydrogen transfer reaction: asolution of complexes [Rh(Cy2PNHCH2eC4H3O)(cod)Cl], (1),[Rh(Cy2PNHCH2eC4H3S)(cod)Cl], (2), [Ir(Cy2PNHCH2eC4H3O)(h5-C5Me5)Cl2], (3) and [Ir(Cy2PNHCH2eC4H3S)(h5-C5Me5)Cl2], (4)(0.005 mmol), NaOH (0.025 mmol) and the corresponding ketone(0.5 mmol) in degassed iso-PrOH (5 mL) were refluxed until thereactions were completed. After this period a sample of the reactionmixture was taken off, diluted with acetone and analyzed imme-diately by GC. Conversions obtained are related to the residualunreacted ketone.
4.3. Synthesis of rhodium and iridium complexes
4.3.1. [Rh(Cy2PNHCH2eC4H3O)(cod)Cl], (1)A mixture of [Rh(m-Cl)(cod)]2 (0.235 g, 0.48 mmol) and
[Cy2PNHCH2eC4H3O] (0.279 g, 0.95 mmol) in 15 mL of tetrahy-drofuran was stirred at room temperature for 45 min. The volumeof the solvent was then reduced to 0.5 mL before addition of pe-troleum ether (10 mL). The precipitated product was filtered anddried in vacuo yielding 1 as a yellow microcrystalline solid. Yield0.472 g, 91.8%, m.p. ¼ 148e150 �C. 1H NMR (400.1 MHz, CDCl3):d 7.36 (d, 1H, 3J¼ 1.9 Hz,H-5), 6.32 (dd,1H, 3J¼ 1.9 and 3.0 Hz,H-4),6.19 (d, 1H, 3J ¼ 3.0 Hz, H-3), 5.34 (br, 2H, CH of cod), 4.03 (dd, 2H,3J ¼ 5.7 and 5.8 Hz, CH2e), 3.55 (br, 2H, CH of cod), 2.78 (dt, 1H,3J(HH)¼ 5.7 and 2J(HP)¼ 12.6 Hz), NHe), 1.23e2.42 (m, 30H, CH2 ofcodþ protons of cyclohexyl); 13C NMR (100.6MHz, CDCl3): d (26.43,27.14, 27.23, 27.27, 27.33, 27.86, 28.38, 28.81, 29.01, 29.05) (CH2 ofcyclohexylþ cod), 35.69 (d, 1J¼ 25.2 Hz, CH of cyclohexyl), 40.49 (d,2J ¼ 11.1 Hz, CH2), 68.79 (d, 1J ¼ 15.1 Hz, CH of cod (a)), 103.88 (d,1J ¼ 6.4 Hz, CH of cod (b)), 106.23 (C-3), 110.26 (C-4), 141.86 (C-5),153.80 (d, 3J ¼ 5.0 Hz, C-2); assignment was based on the 1He13CHETCOR, DEPT and 1He1H COSY spectra; 31Pe{1H} NMR(162.0 MHz, CDCl3): d 71.98 (d, 1J (103Rhe31P)¼ 156.5 Hz); IR, (KBr):y 3320 (NeH), 1076 (PeN) cm�1. C25H40NOPRhCl (539.9 g/mol):calcd. C 55.61, H 7.47, N 2.59; found C 55.42, H 7.31, N 2.45.
4.3.2. [Rh(Cy2PNHCH2eC4H3S)(cod)Cl], (2)A mixture of [Rh(m-Cl)(cod)]2 (0.208 g, 0.42 mmol) and
[Cy2PNHCH2eC4H3S] (0.261 g, 0.84 mmol) in 15 mL of tetrahy-drofuran was stirred at room temperature for 45 min. The volumeof the solvent was then reduced to 0.5 mL before addition of pe-troleum ether (10 mL). The precipitated product was filtered anddried in vacuo yielding 2 as a yellow microcrystalline solid. Yield0.418 g, 89.1%, m.p. ¼ 150e152 �C. 1H NMR (400.1 MHz, CDCl3):d 7.23 (dd, 1H, 3J ¼ 1.2, 4.9 Hz, H-5), 6.94e6.97 (m, 2H, H-3 þ H-4),5.35 (br, 2H, CH of cod), 4.25 (dd, 2H, 3J ¼ 6.7 and 6.8 Hz, CH2e),3.56 (br, 2H, CH of cod), 3.08 (dt, 1H, 3J HH) ¼ 6.7 and 2JHP)¼ 12.8 Hz, NH), 1.39e2.46 (m, 30H, protons of cyclohexylþ CH2of cod); 13C NMR (100.6 MHz, CDCl3): d (25.98, 26.43, 27.14, 27.23,27.27, 27.33, 28.03, 28.40, 29.01, 29.10) (CH2 of cyclohexyls þ cod),35.89 (d, 1J¼ 25.2 Hz, CH of cyclohexyls), 42.75 (d, 2J¼ 9.1 Hz, CH2e
), 68.88 (d, 1J ¼ 13.1 Hz, CH of cod, (a)), 104.04 (d, 1J ¼ 9.1 Hz, CH ofcod, (b)), 126.81, 124.55, 124.38 (C-3, C-4 and C-5), 144.40 (d,3J ¼ 5.0 Hz, C-2); assignment was based on the 1He13C HETCOR,DEPT and 1He1H COSY spectra; 31Pe{1H} NMR (162.0 MHz, CDCl3):d 74.39 (d, 1J (103Rhe31P) ¼ 154.1 Hz); IR, (KBr): y 3322 (NeH), 849(PeN) cm�1. C25H40NSPRhCl (556.0 g/mol): calcd. C 54.00, H 7.25, N2.52; found C 53.82, H 7.19, N 2.41.
4.3.3. [Ir(Cy2PNHCH2eC4H3O)(h5-C5Me5)Cl2], (3)
A mixture of [Ir(h5-C5Me5)(m-Cl)Cl]2 (0.379 g, 0.48 mmol) and[Cy2PNHCH2eC4H3O] (0.279 g, 0.95 mmol) in 15 mL of tetrahy-drofuranwas stirred at room temperature for 1 h. The volume of thesolvent was then reduced to 0.5 mL before addition of petroleumether (20 mL). The precipitated product was filtered and dried invacuo yielding 3 as an orange microcrystalline solid. Yield 0.583 g,88.6%, m.p.¼189e191 �C. 1H NMR (400.1MHz, CDCl3) d: 7.31 (d,1H,3J ¼ 2.6 Hz H-5), 6.43 (dd, 1H, 3J ¼ 1,7 and 2.6 Hz, H-4), 6.30 (d, 1H,3J ¼ 1.7 Hz, H-3), 4.15 (dd, 2H, 3J ¼ 6.2 and 6.4 Hz, CH2e), 3.30 (dt,1H, 3J(HH) ¼ 6.2 and 2J(HP) ¼ 12.2 Hz, NH), 1.19e2.24 (m, 37H,protons of cyclohexyls þ CH3 of Cp* (C5Me5); 13C NMR (100.6 MHz,CDCl3): d 9.58 (C5Me5), 26.44, 27.20, 27.32, 27.41, 27.97, 28.37 (CH2of cyclohexyls), 39.07 (d, 1J ¼ 34.2 Hz, CH of cyclohexyls), 41.48 (d,2J ¼ 7.0 Hz, CH2e), 91.92 (d, 2J ¼ 12.0 Hz, C5Me5), 106.53 (C-4),110.40 (C-3), 141.21 (C-5), 154.51 (C-2); assignment was based onthe 1He13C HETCOR, DEPT and 1He1H COSY spectra; 31Pe{1H} NMR(162.0 MHz, CDCl3): d 43.98 (s); IR, (KBr): y 3341 (NeH), 852 (PeN)cm�1; C27H43NOPIrCl2 (691.7 g/mol): calcd. C 46.88, H 6.27, N 2.02;found C 46.73, H 6.21, N 1.98.
4.3.4. [Ir(Cy2PNHCH2eC4H3S)(h5-C5Me5)Cl2], 4
A mixture of [Ir(h5-C5Me5)(m-Cl)Cl]2 (0.336 g, 0.42 mmol) and[Cy2PNHCH2eC4H3S] (0.261 g, 0.84 mmol) in 15 mL of tetrahy-drofuranwas stirred at room temperature for 1 h. The volume of thesolvent was then reduced to 0.5 mL before addition of petroleumether (10 mL). The precipitated product was filtered and dried invacuo yielding 4 as an orange microcrystalline solid. Yield 0.539 g,90.3%, m.p.¼169e171 �C. 1H NMR (400.1 MHz, CDCl3): d 7.15 (d, 1H,3J ¼ 2.0 Hz, H-5), 7.12 (dd, 1H, 3J ¼ 2.0 and 3.2 Hz, H-4), 6.93 (d, 1H,3J ¼ 3.2 Hz, H-3), 4.36 (dd, 2H, 3J ¼ 5.6 and 5.7 Hz, CH2e), 3.39 (dt,1H, 3J HH) ¼ 5.6 and 2J (HP) ¼ 12.8 Hz, NH), 1.20e2.26 (m, 37H,protons of cyclohexyls þ CH3 of Cp*); 13C NMR (100.6 MHz, CDCl3):d 9.61 (C5Me5), (26.43, 27.21, 27.30, 27.40, 28.05, 28.42) (CH2e ofcyclohexyls), 39.06 (d, 1J ¼ 34.2 Hz, CH of cyclohexyls), 43.50 (d,2J ¼ 8.0 Hz, CH2e), 91.89 (d, 2J ¼ 2.0 Hz, C5Me5), 124.45, 123.74 (C-4and C-5), 126.80 (C-3), 144.79 (C-2); assignment was based on the1He13C HETCOR and 1He1H COSY spectra; 31Pe{1H} NMR(162.0 MHz, CDCl3): d 43.34 (s). IR, (KBr): y 3332 (NeH), 1070 (PeN)cm�1; C27H43NSPIrCl2 (707.8 g/mol): calcd. C 45.82, H 6.12, N 1.98;found C 45.71, H 6.03, N 1.85.
4.4. Fabrication and characterization of Au/metal complex/n-Sidevices
An n-Si semiconductor with (100) orientation and 1e10 Ucmresistivity was used in this study to fabricate Au/metal complex/n-Si devices. The semiconductor was cleaned by boiling in trichlo-roethylene and ultrasonically vibrating in acetone and methanol.The native oxide on n-Si wafer was removed using 0.4% HF:H2Osolution. The wafer was dipped in deionized water and dried underN2 flow. Au ohmic back contact was formed by evaporation of Au onn-Si in high vacuum and annealing the n-Si/Au contact in N2 at-mosphere at 420 �C. The thin films of metal complexes were formedon n-Si wafer bymeans of a SCS G3P-8 spin coater. Finally, Au metalwas evaporated on the structure to obtain the front contact of thedevices. Currentevoltage (IeV) measurements were carried outusing Keithley 2400 sourcemeter in dark and under a solar simu-lator with AM1.5 global filter for various light intensities.
References
[1] M.T. Miller, P.K. Gantzel, T.B. Karpishin, Inorg. Chem. 38 (1999) 3414e3422.[2] G. Venkatachalam, R. Ramesh, Inorg. Chem. Commun. 9 (2006) 703e707.[3] O. Pamies, J.-E. Backvall, Chem. Eur. J. 23 (2001) 5052e5058.

K. Rafikova et al. / Journal of Organometallic Chemistry 758 (2014) 1e88
[4] N. Meriç, C. Kayan, M. Aydemir, Y.S. Ocak, A. Baysal, H. Temel, Dicle Univ. J.Inst. Sci. Technol. 2 (2013) 1e8.
[5] C. Kayan, N. Meriç, M. Aydemir, A. Baysal, H. Temel, Dicle Univ. J. Inst. Sci.Technol. 2 (2013) 20e27.
[6] M. Aydemir, A. Baysal, N. Meric, B. Gümgüm, J. Organomet. Chem. 694 (2009)2488e2492.
[7] M. Aydemir, A. Baysal, Polyhedron 29 (2010) 1219e1224.[8] M. Aydemir, N. Meriç, C. Kayan, F. Ok, A. Baysal, Inorg. Chim. Acta 398 (2013)
1e10.[9] A.N. Ajjou, J.-L. Pinet, J. Mol. Catal. A Chem. 214 (2004) 203e206.
[10] Y.-Y. Li, H. Zhang, J.-S. Chen, X.-L. Liao, Z.-R. Dong, J.-X. Gao, J. Mol. Catal. AChem. 218 (2004) 153e156.
[11] R. Noyori, T. Ohkuma, Angew. Chem. Int. Ed. 40 (2001) 40e73.[12] H.U. Blaser, F. Spindler, M. Studer, Enantioselective catalysis in fine chemicals
production, Appl. Catal. A Gen. 221 (2001) 119e143.[13] S.C. Stinson, Chem. Eng. News 77 (1999) 101e120.[14] V. Turcry, C. Pasquier, F. Agbossou-Niedercorn, Aminophosphine phosphinite
(AMMP) and enantioselective hydrogenation of ketones: further de-velopments, C. R. Chim. 6 (2003) 179e184.
[15] (a) J.T. Mague, J. Krinsky, Inorg. Chem. 40 (2001) 1962e1971;(b) A.M.Z. Slawin, M.B. Smith, J.D. Woollins, J. Chem. Soc. Dalton. Trans. (1998)1537e1539;(c) K.V. Katti, V.S. Reddy, P.R. Singh, Chem. Soc. Rev. 24 (1995) 97e107;(d) M.S. Balakrishna, P.P. George, S.M. Mobin, Polyhedron 24 (2005) 475e480.
[16] M. Aydemir, A. Baysal, F. Durap, B. Gümgüm, S. Özkar, L.-T. Yıldırım, Appl.Organomet. Chem. 23 (2009) 467e475.
[17] A. Kılıç, F. Durap, M. Aydemir, A. Baysal, E. Tas, J. Organomet. Chem. 693(2008) 2835e2842.
[18] P. Bhattacharyya, J.D. Woollins, Polyhedron 14 (1995) 3367e3388.[19] M. Aydemir, A. Baysal, E. Sahin, B. Gümgüm, S. Özkar, Inorg. Chim. Acta 378
(2011) 10e18.[20] M. Aydemir, A. Baysal, N. Gürbüz, _I. Özdemir, B. Gümgüm, S. Özkar, N. Çaylak,
L.-T. Yıldırım, Appl. Organomet. Chem. 24 (2010) 17e24.[21] M. Aydemir, N. Meriç, A. Baysal, J. Organomet. Chem. 720 (2012) 38e45.[22] [a] A. Buhling, P.C.J. Kamer, P.W.N.M. van Leeuwen, Organometallics 16 (1997)
3027e3037;[b] T.J. Kwok, D.J. Wink, Organometallics 12 (1993) 1954e1959;[c] H. Bricout, J.-F. Carpentier, A. Mortreuz, Tetrahedron Lett. 37 (1996) 6105e6108;[d] I. Suisse, H. Bricout, A. Mortreux, Tetrahedron Lett. 35 (1994) 413e416;[e] D. Gleich, R. Schmid, W.A. Herrmann, Organometallics 17 (1998) 2141e2143.
[23] S. Gladiali, E. Alberico, in: M. Beller, C. Bolm (Eds.), Transition Metals forOrganic Synthesis, vol. 2, Wiley-VCH, Weinheim, 2004, pp. 145e166.
[24] S.E. Clapham, A. Hadzovic, R.H. Morris, Coord. Chem. Rev. 248 (2004) 2201e2237.
[25] J.E. Backvall, J. Organomet. Chem. 652 (2002) 105e111.[26] G. Zassinovich, G. Mestroni, S. Gladiali, Chem. Rev. 92 (1992) 1051e1069.[27] R.V. Wisman, J.G. de Vries, B.-J. Deelman, H.J. Heeres, Org. Process Res. Dev. 10
(2006) 423e429.[28] M. Aydemir, F. Durap, A. Baysal, N. Meric, A. Bulda�g, B. Gümgüm, S. Özkar,
L.T. Yıldırım, J. Mol. Catal. A Chem. 326 (2010) 75e81.[29] Y.X. Chen, Syntheses and Applications of Novel Chiral Ligands, Polytechnic
University, Hong Kong, 1999 (Ph.D. Thesis).[30] M. Aydemir, N. Meric, F. Durap, A. Baysal, M. To�grul, J. Organomet. Chem. 695
(2010) 1392e1398.[31] S.E. Habas, H.A.S. Platt, M.F.A.M. van Hest, D.S. Ginley, Chem. Rev. 110 (2010)
6571e6594.[32] R.V. Salvatierra, C.E. Cava, L.S. Roman, A.J.G. Zarbin, Adv. Funct. Mater. 23
(2013) 1490e1499.[33] N. Sekar, V.Y. Gehlot, Resonance 15 (2010) 819.[34] U. Olgun, M. Gulfen, Dyes Pigm. 99 (2013) 1004e1009.
[35] L. Giribabu, V.K. Singh, T. Jella, Y. Soujanya, A. Amat, F. De Angelis, A. Yella,P. Gao, M.K. Nazeeruddin, Dyes Pigm. 98 (2013) 518e529.
[36] C. Dragonetti, A. Valore, A. Colombo, M. Magni, P. Mussini, D. Roberto, R. Ugo,A. Valsecchi, V. Trifiletti, N. Manfredi, A. Abbotto, Inorg. Chim. Acta 405 (2013)98e104.
[37] S. Pasa, Y.S. Ocak, H. Temel, T. Kilicoglu, Inorg. Chim. Acta 405 (2013) 493e504.
[38] H. Temel, S. Pasa, Y.S. Ocak, I. Yilmaz, S. Demir, I. Ozdemir, Synth. Met. 161(2012) 2765e2775.
[39] H. Tang, Y. Li, Q. Chen, B. Chen, Q. Qiao, W. Yang, H. Wu, Y. Cao, Dyes Pigm. 100(2013) 79e86.
[40] M. El-Nahass, K. Abd-El-Rahman, A. Darwish, Microelectron. J. 38 (2007) 91e95.[41] T. Kılıcoglu, Y.S. Ocak, Microelectron. Eng. 88 (2011) 150e154.[42] R.K. Gupta, M. Cavas, A.A. Al-Ghamdi, Z.H. Gafer, F. El-Tantawy,
F. Yakuphanoglu, Sol. Energy 92 (2013) 1e6.[43] Furfuryl-(N-dicyclohexylphosphino)amine: Yield 0.279 g, 93.3%. 1H NMR
(400.1 Hz, CDCl3): d 7.33 (d, 1H, H-5, 3J ¼ 1.6 Hz), 6.29 (dd, 1H, H-4, 3J ¼ 1.6and 3.0 Hz), 6.13 (d, 1H, H-3, 3J ¼ 3.0 Hz), 4.04 (dd, 2H, CH2e, 3J(HP) ¼ 7.7 and3J(HH) ¼ 7.6 Hz), 2.42 (dt, eNHe, 1H, 3J(HH) ¼ 7.6 and 2J(HP) ¼ 7.7 Hz), 1.16e1.77 (m, 22H, protons of cyclohexyls); 13C NMR (100.6 MHz, CDCl3): d 26.55,27.03, 27.16, 27.28, 29.12, 29.30 (CH2- of cyclohexyls), 36.40 (d, CHe ofcyclohexyls, 1J ¼ 11.1 Hz), 45.86 (d, CH2e, 2J ¼ 27.2 Hz), 105.56 (C-3), 110.09(C-4), 141.27 (C-5), 156.23 (d, C-2, 3J ¼ 3.0 Hz), assignment was based on the1He13C HETCOR, DEPT and 1He1H COSY spectra; 31Pe{1H} NMR (162.0 MHz,CDCl3): d 61.61 (s, NHeP(Cy)2); IR, (KBr): y 801 (PeN), 3257 (NeH) cm�1;C17H28NOP (293.39 g/mol): calcd. C 69.60, H 9.62, N 4.77; found C 69.49, H9.54, N 4.61%. Thiophene(N-dicyclohexylphosphino)amine: Yield 0.261 g,95.6%. 1H NMR (400.1 Hz, CDCl3): d 7.18 (d, 1H, H-5, 3J ¼ 5.0 Hz), 6.90e6.94(m, 2H, H-3 and H-4), 4.27 (dd, 2H, CH2e, 3J(HP) ¼ 6.4 and 3J(HH) ¼ 6.3 Hz),2.98 (m, 1H, NH), 1.24e1.81 (m, 22H, protons of cyclohexyls); 13C NMR(100.6 MHz, CDCl3): d 26.01, 26.57, 27.00, 27.70, 29.43, 29.62 (CH2 of cyclo-hexyls), 36.58 (d, CH of cyclohexyls, 1J ¼ 10.1 Hz), 48.16 (d, CH2, 2J ¼ 4.0 Hz),123.46 (C-3), 123.67 (C-5), 126.36 (C-4), 147.40 (C-2), assignment was basedon the 1He13C HETCOR, DEPT and 1He1H COSY spectra; 31Pe{1H} NMR(162.0 MHz, CDCl3): d 61.69 (s, NHeP(Cy)2); IR, (KBr): y 798 (PeN), 3253 (NeH) cm�1; C17H28NSP (309.45 g/mol): calcd. C 65.98, H 9.12, N 4.53; found C65.87, H 8.99, N 4.35%
[44] M. Aydemir, A. Baysal, N. Meriç, B. Gümgüm, J. Organomet. Chem. 694 (2009)2488e2492.
[45] [a] S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya, R. Noyori, J. Am. Chem. Soc.117 (1995) 7562e7563;[b] J. Takehara, S. Hashiguchi, A. Fujii, S. Inoue, T. Ikariya, R. Noyori, J. Chem.Soc. Chem. Commun. (1996) 233e234.
[46] T. Ohkuma, R. Noyori, Angew. Chem. Int. Ed. 40 (2001) 40e73.[47] R. Noyori, Angew. Chem. Int. Ed. 41 (2002) 2008e2022.[48] T. Ikariya, A.J. Blacker, Acc. Chem. Res. 40 (2007) 1300e1308.[49] H.L. Lowry, K.S. Richardson, Mechanism and Theory in Organic Chemistry,
Harper Collins, New York, 1987 (Chapter 2).[50] I. Yamada, R. Noyori, Org. Lett. 2 (2000) 3425e3427.[51] M.J. Palmer, M. Wills, Tetrahedron: Asymmetry 10 (1999) 2045e2061.[52] D.H. McDaniel, H.C. Brown, J. Org. Chem. 23 (1958) 420e427.[53] J.W. Faller, A.R. Lavoie, Organometallics 20 (2001) 5245e5247.[54] J.-X. Gao, X.-D. Yi, P.-P. Xu, C.-L. Tang, H.-L. Wan, T. Ikariya, J. Organomet.
Chem. 592 (1999) 290e295.[55] J.-X. Gao, H. Zhang, X.-D. Yi, P.-P. Xu, C.-L. Tang, H.-L. Wan, K.-R. Tsai, T. Ikariya,
Chirality 12 (2000) 383e388.[56] M. Campos, L.O.S. Bulhoes, C.A. Lindino, Sens. Actuators 87 (2000) 67e71.[57] O. Gullu, A. Turut, S. Asubay, J. Phys. Condens. Matter 20 (2008) 045215.[58] F. Yakuphanoglu, M. Kandaz, B.F. Senkal, Thin Solid Films 516 (2008) 8793e
8796.[59] O. Gullu, A. Turut, Sol. Energy Mater. Sol. Cells 92 (2008) 1205e1210.[60] H. Norde, J. Appl. Phys. 50 (1979) 5052.




















