
Eletrochemical synthesis of CdTe and CdSe quantum dots TGA-capped
Upload
univ-montpellierCategory
view
0download
0
Characterization of the unstability of 4-mercaptoaniline capped
platinum nanoparticles solution by combining LB technique
and X-ray photoelectron spectroscopy
Frederic Raynal a, Arnaud Etcheberry a, Sara Cavaliere a,b,Vincent Noel b, Henri Perez b,*
a Institut Lavoisier (IREM, UMR 8637 CNRS), Universite de Versailles-Saint Quentin, 45 ave des Etats Unis, 78035 Versailles, Franceb CEA-Saclay, DSM/DRECAM/SPAM-LFP, Bat. 522, 91191 Gif-sur-Yvette, France
Received 26 April 2005; received in revised form 24 May 2005; accepted 25 May 2005
Available online 1 July 2005
Abstract
This paper reports on the study of the evolution of 4-mercaptoaniline ( p-HSC6H4NH2) functionalized platinum nanoparticles
in solution by coupling the Langmuir–Blodgett technique and X-ray photoelectron spectroscopy (XPS). The spectra are
recorded on mixed LB films containing fatty acid and platinum particles in proportion 50/50. Several samples built from fresh
and aged solutions of particles are analyzed. Comparison of the Pt 4f, S 2p and N 1s regions in each case points to the time
dependant chemical evolution of the functionalized particles involving at once platinum, thiolate and amine components. The
particle aging in solution is reported during several months, until the complete flocculation of the functionalized platinum
nanoparticles. Using the compared XPS analysis of the LB layers obtained from the different particle solutions, unstability of the
storage looks then clearly related to the chemical evolution of the bifunctional organic crown.
# 2005 Elsevier B.V. All rights reserved.
Keywords: Platinum nanoparticles; Functionalization; Langmuir–Blodgett technique; XPS
www.elsevier.com/locate/apsusc
Applied Surface Science 252 (2006) 2422–2431
1. Introduction
Self-assembled monolayers (SAMs) formed on
metals provide an elegant route to the preparation of
well-defined organic assemblies on solid surfaces.
These SAMs consist essentially in thiolate derivatives
* Corresponding author. Tel.: +33 1 69 08 41 83;
fax: +33 1 69 08 12 13.
E-mail address: [email protected] (H. Perez).
0169-4332/$ – see front matter # 2005 Elsevier B.V. All rights reserved
doi:10.1016/j.apsusc.2005.05.042
adsorbed on flat surfaces of gold [1–3], silver [1],
copper [1], iron [4], nickel [5] or platinum [6].
Recently, an electrochemical way has been described
to form SAMs on flat surfaces of gold [7].
The strong interaction involving sulfur and metal
atoms has been exploited to control the formation of
noble metal nanoparticles of gold [8–13], platinum
[14–20], silver [14,21], or gold/silver alloy [22]. An
efficient approach in the elaboration of nanoparticle
thin film is the Langmuir–Blodgett (LB) technique
.

F. Raynal et al. / Applied Surface Science 252 (2006) 2422–2431 2423
[23–26]. In a previous study, we described the
elaboration of LB thin films of 4-mercaptoaniline
( p-HSC6H4NH2) functionalized platinum nanoparti-
cles [25].
The aging of the structures involving self-
assembled thiolate monolayer is presumably related
to the stability of the chemical bond between thiolate
and metal surfaces. Oxidation of thiolate to sulfonate
species has been evidenced on flat surfaces of silver,
copper, as well as gold, and related to air oxidation
processes [1,2,27]. In a previous paper, we reported
similar degradation of 4-mercaptoaniline functiona-
lized platinum nanoparticles in air atmosphere. The
degradation was evidenced by infrared spectroscopy
recorded on the powder stored in air several months
after the synthesis [16]. The spectrum exhibits new
absorption attributed to sulfonates species. Moreover,
we also pointed out the slow aggregation process of
the nanoparticles in DMSO solution. This process was
suspected to be closely related to the chemical
evolution of the 4-mercaptoaniline capping molecules.
In the current work, we characterize the step by step
chemical evolution of the organic crown of the particle
stored in solution by combining Langmuir–Blodgett
technique and X-ray photoelectron spectroscopy
(XPS). Solutions prepared just after the synthesis of
the nanoparticles are referred as fresh solutions
whereas solutions which have been kept in flask for
at least ten days are referred as aged solutions. To
follow the aging process aliquots of an initially fresh
solution are taken and used to build up LB films,
which are then analyzed by XPS.
2. Experimental
All products were obtained from Aldrich and were
used as received without further purification.
2.1. Synthesis
Platinum nanoparticles capped with mercaptoani-
line molecules were prepared by reduction of PtCl4 in
hexylamine by sodium borohydride. Just after the
reduction 4-mercaptoaniline is added to the reaction
media resulting in the functionalisation of the
platinum nanoparticles [16]. This procedure provides
particles with an average diameter of 2 nm that can be
dissolved in dimethyl sulfoxide (DMSO). The solu-
tions are prepared in sealed flask that are stored under
air until further utilization.
2.2. LB films elaboration
The formation of Langmuir–Blodgett (LB) thin
films of 4-mercaptoaniline functionalized nanoparti-
cles has been already described [25]. The films are
transferred on glass slides provided with gold
electrodes deposit using a laboratory-made LB trough
filled with Millipore-grade water (resistivity higher
than 18 MV cm). The transfer is carried out vertically
at a surface pressure of 28 mN/m with a transfer speed
of 0.3–0.5 cm/min. The substrate being hydrophilic an
odd number of monolayers can be deposited. In this
work the samples consist in three monolayers thick LB
films containing behenic acid (CH3-(CH2)20-COOH)
and platinum nanoparticles. The proportion of fatty
acid to nanoparticles is defined by the ratio of the area
associated respectively to both species at the air water-
interface which is set at 50/50 [25]. In the three
monolayers configuration, the Au 4f signal provided
by the buried interface, can be detected and used as an
internal reference.
2.3. XPS characterization
XPS analysis is performed using a VG Esca-
lab_220i XL system. High-resolution XPS conditions
have been fixed, i.e. constant analyzer energy mode
with 20 eV as pass energy, and a monochromatic Al
Ka X-ray excitation giving high-resolution spectra.
The analyzed area is 1 mm2 and the contact is taken on
the inter-digital gold electrodes part where none LB
deposition is present. Binding energies (BE) of all core
levels are referenced to the Au 4f 7/2 core level
(BE = 84 eV). Curve fitting is performed using the
Eclipse data system developed by VG Scientific.
3. Results
3.1. XPS spectra of samples built from fresh
nanoparticle solutions
The XPS spectra are recorded first on freshly
synthesized particles from which a fresh DSMO

F. Raynal et al. / Applied Surface Science 252 (2006) 2422–24312424
solution is prepared. The period of time between the
isolation of the powder, the preparation of the DMSO
solution and the LB film elaboration is approximately
two hours. The LB sample is then analyzed within
twelve hours after elaboration.
Spectrum analysis is performed on several core
levels: N 1s, S 2p, O 1s, C 1s, Pt 4f and Au 4f that are
characteristic of the particles deposited on substrate
provided with gold electrodes. The most interesting
feature is observed for S 2p region, which is shown in
Fig. 1. Several contributions are distributed between
161 and 171 eV. The main contribution at low binding
energy exhibits an apparent spin orbit splitting related
to the 2p character of the core level. For comparison
the S 2p region of a CdS oxide free layer [28], is also
proposed in Fig. 1 with the same XPS resolution as for
the functionalized particles. It appears that this
reference S 2p region presents an only one contribu-
tion with a perfectly defined spin orbit splitting.
According to the S 2p region of a CdS sample, easily
Fig. 1. S 2p region of sulfur in freshly 4-mercaptoaniline functio-
nalized platinum nanoparticles compared to S 2p region of sulfur in
a CdS structure [28].
fitted with one split contribution [28], the modified
shape of the S 2p region of functionalized nanoparti-
cules at low BE cannot be fitted, in its mean feature,
with only one contribution, but with at least two. So,
for the nanoparticles the S 2p peak fitting must be
considered with attention. It is presented in Fig. 2. The
S 2p fitting parameters are deduced from the ones used
for the CdS signals for which we have taken into
account only the effect of the spin-orbit splitting that
gives rise to a doublet. Three specific parameters of the
2p doublet have been fixed: the branching ratio 3/2:1/2
equal to 2, the distance between S 2p3/2 and S 2p1/2
taken equal to 1.2 eV, and the FWHM equal to 1.2 eV
for each sub level. These parameters are then injected
to fit the mean feature of the signal of nanoparticles.
Clearly a double contribution is detected on the lowest
binding energy region. For the S 2p contributions at
higher BE, where the 2p3/2 and 2p1/2 sub levels cannot
be obviously differentiated a simple component must
be used. Finally, with a rather good reproducibility,
a four component structure can be always proposed
for describing the S 2p spectra associated to the
nanoparticles associated to fresh solution.
The other core levels can be easily fitted, with one
(Pt 4f, Au 4f, N 1s) or two (C 1s, O 1s) contributions.
The Pt 4f fitting is also given in Fig. 2. The only
parameter fixed for the Pt 4f doublet, fitted in
asymmetric mode, is the branching ratio 7/2:5/2 equal
to 1.3.
Finally fitted BE positions and FWHMs for the
different components of each region investigated are
reported in Table 1. We note that all the FWHM are
Table 1
Binding energy (BE) and full width at half maximum (FWHM) of
the core levels investigated in LB films built using freshly 4-
mercaptoaniline functionalized platinum nanoparticles
Region BEa (eV) FWHM (eV)
C 1s 285.1 1.1
284.5 1.1
O 1s 532.6 1.9
531.2 1.5
N 1s 399.5 1.6
Pt 4f7/2 71.3 1
S 2p3/2 162.3 1.2b
162.7 1.2b
S 2p 164.9 2.6
167.7 2.5a Values are determined with respect to the Au 4f7/2 BE at 84 eV.b Fixed value in the fit.

F. Raynal et al. / Applied Surface Science 252 (2006) 2422–2431 2425
Fig. 2. Fitted S 2p and Pt 4f XPS spectra of LB films elaborated from freshly 4-mercaptoaniline functionalized platinum nanoparticles. Arrows
in the S 2p region show the S 2p3/2 and S 2p1/2 contributions at lower energy.
included between 1 and 1.9 eV except for the S 2p
contributions at higher BE, where the 2p3/2 and 2p1/2
sub levels cannot be differentiated.
3.2. LB films built from aged particles solutions
The LB thin films were elaborated from aged
nanoparticle solutions of respectively 3 weeks and 3
months. The corresponding XPS spectra were
recorded in each case. The BE shifts of the XPS
signals are reported in Table 2. Compared to those of
Table 2
Binding energy (BE) and relative peak area of N 1s, Pt 4f7/2, S 2p3/2 and S
mercaptoaniline functionalized platinum nanoparticles
Sample Fresh solution Three wee
BE (eV) Peak area (%) BE (eV)
S 2p3/2 162.3 38 162.3
162.7 40 162.8
S 2p 164.9 16 165.6
167.7 6 167.8
Pt 4f7/2 71.3 100 71.4
N 1s 399.5 100 399.5
400.3
Values are determined with respect to the Au 4f7/2 BE at 84 eV.
the fresh sample, the Pt 4f, S 2p and N 1s regions of the
sample prepared from 3 weeks aged solution exhibit
more or less important evolution. The general
tendency is a peak shift to higher energy which is
tiny for Pt 4f, more pronounced for S 2p and quite
important for N 1s. The latter shows a clear
broadening.
The Pt 4f, S 2p and N 1s regions of the sample built
from 3 months aged solution are presented in Fig. 4.
Whereas N 1s region shows similar features than after
3 weeks the changes become clearly marked for S 2p
2p regions investigated in LB films built using freshly and aged 4-
ks aged solution Three months aged solution
Peak area (%) BE (eV) Peak area (%)
21 162.3 14
50 163.2 60
15 166.2 13
14 168.2 13
100 71.4 44
72.3 33
73.2 23
51 399.4 46
49 400.5 54

F. Raynal et al. / Applied Surface Science 252 (2006) 2422–24312426
and Pt 4f. The shift of the S 2p signal to higher
BE is increasing and the Pt 4f region is characterized
by the emergence of a new contribution at higher
BE.
Several modifications are therefore observed with
samples built from aged particles solutions. The peak
fitting allows to precise these evolutions which are
reported in Table 2. It contains the BE energies and the
relative area of the peaks when several BE are
identified for a given element.
4. Discussion
4.1. XPS spectra of samples built from fresh
nanoparticle solutions
The comments concern essentially the S 2p region,
which exhibits four different contributions. The
main signal below 165 eV was fitted with two
S 2p contributions, related to sulfur in two different
chemical environments. The S 2p3/2 at low BE, located
at 162.3 � 0.05 eV, is in perfect agreement with
results from 4-mercaptoaniline functionalized gold
nanoparticles reported by Johnson et al. (BE of S 2p3/2
at 162.5 eV and BE of Au 4f7/2 at 84.2 eV) [10].
Data from literature for SAMs on metal flat surfaces
[1,2,5,6,29,30], and more recently on organically
capped nanoparticles [10,19,21,22,31,32], indicate that
S 2p3/2 at low BE (BE = 162.3 � 0.05 eV) results
from the dissociative adsorption of the S–H bond (or
S–S bond when disulfide instead of thiol is involved) on
the metal (Eq. (1)):
RSH þ Mð0Þ ! RS-MðIÞ þ 1=2H2 (1)
The S 2p3/2 BE is related to the charge density on the
sulfur atom, as was demonstrated in the well known
work of Siegbahn and co-workers [33]. This work,
based on 132 organic sulfur compounds ranging from
thiolates to sulfonium ions, presented a linear free
energy relationship (LFER) between the S 2p3/2 BE
and the calculated charge of the sulfur. In our case,
interpolation of S 2p3/2 into the Siegbahn LFER
suggests that the sulfur bears a charge of (�0.2e),
which is consistent with a thiolate like Pt–S bond with
covalent character.
A second S 2p3/2 contribution is located at
162.7 � 0.05 eV. Interpolation of S 2p3/2 into the
Siegbahn LFER suggests that this sulfur bears a charge
of (�0.1e). It is likely that such a BE does not involve
oxidized sulfur. Considering that Pt nanoparticles have
a cuboctahedral shape [25] and taking into account the
small size of the core (ca. 2 nm) the fraction of
platinum atoms that belongs to Pt(1 0 0) faces is quite
low compared to Pt(1 1 1) faces [34]. Therefore, the
two sulfur contributions cannot be related to Pt–S
bonds corresponding to different platinum faces
because both sulfurs are found in equal quantities
(Table 2). The attribution of this second S 2p structure
to free alkylthiols or disulfides cannot be completely
excluded because the expected binding energies are
�163.2 eV [6,17,29,30,33]. However, the fact that the
nanoparticles undergoes several washing after synth-
esis conflicts with such a conclusion. A third point that
could explain two thiolate like peaks regards the
binding modes which can be expected for a thiolate
species on a given crystal face. Indeed, theoretical
studies on alkanethiolates suggested that there were
different binding sites for thiolates on Au(1 1 1)
surfaces referred as top, bridge and hollow sites
[35]. The relative strength of the Au–S bond in the
various sites was in the order hollow > bridge > top.
Although quite difficult to prove such different
binding sites could be tentatively proposed to be
responsible for the two different environments
corresponding to Pt–S bonds at 162.3 and 162.7 eV.
The S 2p peak fitting shows also the presence of
two sulfurs exhibiting BE higher than 164 eV. The
following attributions can be done in agreement with
literature: sulfinate and sulfonate species respectively
at 164.9 � 0.05 and 167.7 � 0.05 eV. These struc-
tures correspond therefore to oxidized species of 4-
mercaptoaniline, consistent with previous observa-
tions [16]. In contrast to other para substituted
arylthiolate, 4-mercaptoaniline looks actually easily
oxidizable, as suggested by similar XPS results
recorded with 4-mercaptoaniline functionalized gold
nanoparticles [10].
Even if the small size of the nanoparticle makes the
surface atoms a significant part of the total atoms
number, numerous data reported in literature
[10,19,21,22,31,32] do not give clear evidence on
the possibility to distinguish between the metal
surface atoms which are combined to the capping
molecule, and those from the internal part of the
particle. With that respect, our results are consistent

F. Raynal et al. / Applied Surface Science 252 (2006) 2422–2431 2427
with most of those reported in literature. The Pt 4f7/2
signal of fresh sample, reported in Fig. 1, is located at
71.2 � 0.05 eV, a value consistent with platinum
nanoparticles in the metallic Pt(0) state. The shape and
FMWH of the Pt 4f7/2 peak (Table 1) do not allow to
distinguish between different platinum environments.
The N 1s region of fresh sample, reported in Fig. 1, is
located at 399.5 � 0.05 eV, a value consistent with a
primary amine function. As we previously shown [25],
this point confirms that there is no interaction between
the carboxylic function of the fatty acid, which is used
to build up LB films containing nanoparticles, and the
amine function of the latter. At least for freshly
prepared solution the question of a competitive
binding between amine and thiolate function with
respect to platinum surface can be reasonably ruled
out for two reasons. First, the synthesis proceeds in
hexylamine as temporary stabilizing agent before
disulfide introduction [16], if disulfide is omitted then
the reaction media flocculates irreversibly suggesting
that the interaction with amine function with platinum
is weaker than with thiolate. Second, if the interaction
of the amine function with platinum was efficient then
the nanoparticles would be crosslinked through amine
and thiolate links and could not be solubilized
spontaneously.
4.2. LB films built from 3 weeks aged particles
solution
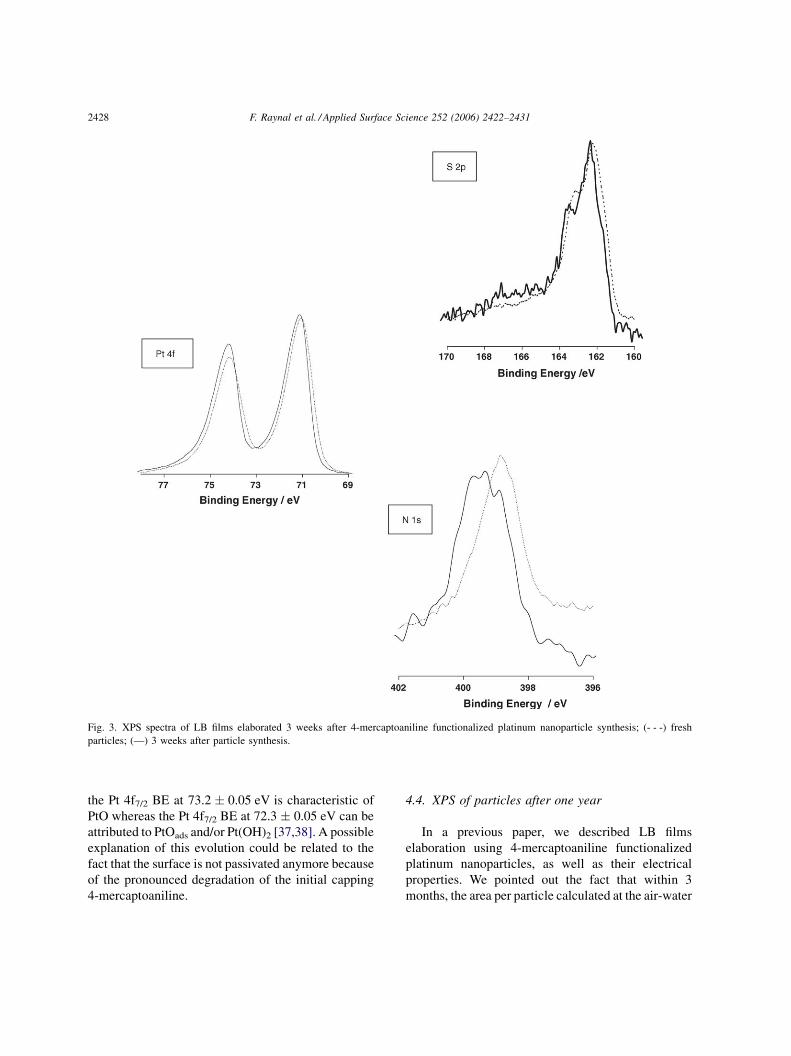
The S 2p region of LB films elaborated from 3
weeks aged solution is presented in Fig. 3. At first
view this S 2p region looks rather similar to the fresh
sample ones. However, peak fitting and relative peak
area point to a real evolution characterized by an
increasing proportion of sulfur S 2p at higher binding
energies compared to the lowest BE of 162.3 eV. This
evolution suggests a possible reorganization of Pt–S
bonds for which the component at 163.8 eV became
majoritary with respect to one at 162.3 eV. Possibly
related to this evolution, the same tendency is
observed for S 2p signals at BE higher than 164 eV.
S 2p located at 167.8 eV appears now with an
equivalent relative weight than the one pointing at
lower energy (165.6 eV).
Besides the evolution observed on the S 2p signals,
the N 1s contribution exhibits a strong evolution. The
peak, given in Fig. 3, cannot be fitted anymore with a
unique contribution attributed to primary amine
function (BE N 1s at 399.5 � 0.05 eV), but a second
contribution centered at 400.3 eV has to be introduced
and is found to be in an equivalent proportion
(Table 2). The BE of the second N 1s contribution
looks quite low to reflect an oxydized or quaternized
species [33]. It has to be mentionned that a double
structure of N 1s peak is reported in 4-aminothio-
phenol monolayers on gold in pristine and oxidized
form [36]. In this paper a complex oxidation
mechanism of 4-mercaptoaniline is proposed that
finally results in 40-mercapto-N-phenyl quinone. It
cannot be excluded that such a process occurs on
platinum nanoparticles. In contrast to sulfur and
nitrogen evolution the Pt 4f peak barely evolves except
for a very small displacement (0.1 eV) towards higher
BE.
4.3. LB films built from 3 months aged particles
solution
In Fig. 4, we report the S 2p region for samples built
from 3 months aged solution. The evolution in term of
binding energy shift is now obvious. The evolution
observed after 3 weeks looks more pronounced after 3
months, the Pt–S bond at lower energy (162.3 eV)
appears to decrease in favour of a new peak that points
at 163.2 eV (Table 2). This BE could be now attributed
to disulfide species [6,17,29,30,33]. The S 2p signals
attributed to oxydized sulfur are located at higher
energies (166.2 and 168.2 eV) but their relative
intensities do not increase in a significant way. This
point could be related to the elimination of oxydized
species (the interaction of which with platinum
surface is supposed to be weak) during the LB
procedure. However, this explanation is not conforted
by a significant evolution of the Pt/S ratio between the
different samples showing the quite complex pro-
cesses taking place in the chemical evolution reported
here. Whereas the N 1s signal does not exhibit
important evolution compared to those observed on
samples built from 3 weeks aged solution the Pt 4f
signal is characterized by a strong modification. The
signal cannot be fitted with one contribution but
requires three contributions located at 71.4, 72.3 and
73.2 eV. The lowest energy contribution is still
attributed to Pt(0) whereas the new ones correspond
to Ptd+ forms of platinum. Referring to the literature,
F. Raynal et al. / Applied Surface Science 252 (2006) 2422–24312428
Fig. 3. XPS spectra of LB films elaborated 3 weeks after 4-mercaptoaniline functionalized platinum nanoparticle synthesis; (- - -) fresh
particles; (—) 3 weeks after particle synthesis.
the Pt 4f7/2 BE at 73.2 � 0.05 eV is characteristic of
PtO whereas the Pt 4f7/2 BE at 72.3 � 0.05 eV can be
attributed to PtOads and/or Pt(OH)2 [37,38]. A possible
explanation of this evolution could be related to the
fact that the surface is not passivated anymore because
of the pronounced degradation of the initial capping
4-mercaptoaniline.
4.4. XPS of particles after one year
In a previous paper, we described LB films
elaboration using 4-mercaptoaniline functionalized
platinum nanoparticles, as well as their electrical
properties. We pointed out the fact that within 3
months, the area per particle calculated at the air-water

F. Raynal et al. / Applied Surface Science 252 (2006) 2422–2431 2429
Fig. 4. XPS spectra of LB films elaborated 3 months after 4-mercaptoaniline functionalized platinum nanoparticle synthesis; (- - -) fresh
particles; (—) 3 months after particle synthesis.
interface decreases by ca. 50% [25]. This behavior is
now clearly related to the chemical degradation of the
organic crown surrounding the particles than we report
here. The ultimate step of this process is the
macroscopic flocculation of the solution.
In a last set of experiment, we recorded the XPS
spectra of the platinum precipitate resulting from the
complete flocculation of a one year old solution. This
precipitate was briefly washed with acetone and the
cast powder was then analyzed (Fig. 5). On this
sample, about 80% of the sulfur detected is into
sulfinate and sulfonate forms, whereas only 30% of the
sulfur was oxidized in the sample built from 3 months
aged solution.
The peak fitting of the Pt 4f region shows the same
contributions already observed after 3 months with
one difference: the Pt 4f7/2 contribution at higher
BE is shifted at 74.4 � 0.05 eV instead of 73.2 �0.05 eV. This Pt 4f signal can be attributed to PtO2,
involving a platinum evolution after several months in
DMSO from PtO to PtO2. The XPS quantitative
analysis gives a Pt(0)/Ptd+ ratio close to 36/64, close
to the sample built 3 months after particle synthesis
(Pt(0)/Ptd+ = 44/56).

F. Raynal et al. / Applied Surface Science 252 (2006) 2422–24312430
Fig. 5. XPS spectra of the precipitate collected in a 1-year aged solution of 4-mercaptoaniline functionalized platinum.
Along this paper we focussed on the spectroscopic
study to characterize the functionalized nanoparticle
and its chemical evolution. In spite of the good
reproducibility of the shape and BE of the XPS regions
analyzed, the correlation between these chemical
evolutions and the quantitative analysis gave surpris-
ing results. Except for a consistent general tendency
showing an increase of the oxygen contents in samples
from aged solution, we noticed that the various atomic
ratio where moving into a large range from one sample
to another. This point contrasts with the consistent
quantitative analysis recently we reported on such LB
films bearing increasing number of layers and built
from fresh solutions [39].
5. Conclusion
X-ray photoelectron spectroscopy measurements
have been conducted on Langmuir–Blodgett films of
4-mercaptoaniline functionalized platinum nanopar-
ticles built from fresh and aged solutions. For fresh
solutions the S 2p region binding energy confirms the
expected formation of a thiolate-like bond between

F. Raynal et al. / Applied Surface Science 252 (2006) 2422–2431 2431
sulfur and platinum. Reasonable fitting implies the
introduction of two different thiolates binding sites
corresponding to different environments.
We have pointed out the step-by-step degradation
of the organic crown in DMSO solution (storage
solution) and found a good correlation with XPS
spectroscopic characterization. This process looks
quite complex and seems to involve reorganization of
Pt–S bonds and sulfur environment. These observa-
tions look related to the chemical stability of 4-
mercaptoaniline itself, which is oxydizable and
explain the time unstability of the solution of
nanoparticles. Further publication will report on the
stabilization of the solution of platinum nanoparticles
by chemical reaction of the amine function of 4-
mercaptoaniline crown.
References
[1] P.E. Laibinis, G.M. Whitesides, D.L. Allara, Y.-T. Tao, A.N.
Parikh, R.G. Nuzzo, J. Am. Chem. Soc. 113 (1991) 7152–
7167.
[2] P.E. Laibinis, G.M. Whitesides, J. Am. Chem. Soc. 114 (1992)
9022–9028.
[3] A. Ulman, Chem. Rev. 96 (1996) 1533–1554.
[4] M. Stratmann, Adv. Mater. 2 (1990) 191–195.
[5] Z. Mekhalif, J. Riga, J.-J. Pireaux, J. Delhalle, Langmuir 13
(1997) 2285–2290.
[6] R. Brito, V.A. Rodriguez, J. Figueroa, C.R. Cabrera, J. Elec-
troanal. Chem. 520 (2002) 47–52.
[7] D. Qu, M. Morin, J. Electroanal. Chem. 524–525 (2002) 77–
80.
[8] M. Brust, M. Walker, D. Bethell, D.J. Schiffrin, R. Whyman, J.
Chem. Soc., Chem. Commun. (1994) 801–802.
[9] D. Bethell, M. Brust, D.J. Schiffrin, C. Kiely, J. Electroanal.
Chem. 409 (1996) 137–143.
[10] S.R. Johnson, S.D. Evans, R. Brydson, Langmuir 14 (1998)
6639–6647.
[11] F.L. Leibowitz, W. Zheng, M.M. Maye, C.-J. Zhong, Anal.
Chem. 71 (1999) 5076–5083.
[12] S. Chen, R.W. Murray, Langmuir 15 (1999) 682–689.
[13] A.C. Templeton, W.P. Wuelfing, R.W. Murray, Acc. Chem.
Res. 33 (2000) 27–36.
[14] K.V. Sarathy, G. Raina, R.T. Yadav, G.U. Kulkarni, C.N.R.
Rao, J. Phys. Chem. B 101 (1997) 9876–9880.
[15] C. Yee, M. Scotti, A. Ulman, H. White, M. Rafailovich, J.
Sokolov, Langmuir 15 (1999) 4314–4316.
[16] H. Perez, J.-P. Pradeau, P.-A. Albouy, J. Perez-Omil, Chem.
Mater. 11 (1999) 3460–3463.
[17] K.V. Sarathy, P.J. Thomas, G.U. Kulkarni, C.N.R. Rao, J. Phys.
Chem. B 103 (1999) 399–401.
[18] S. Chen, K. Kimura, J. Phys. Chem. B 105 (2001) 5397–
5403.
[19] X. Fu, Y. Wang, N. Wu, L. Gui, Y. Tang, J. Colloid Interface
Sci. 243 (2001) 326–330.
[20] J.E. Martin, J.P. Wilcoxon, J. Odinek, P. Provencio, J. Phys.
Chem. B 106 (2002) 971–978.
[21] S.Y. Kang, K. Kim, Langmuir 14 (1998) 226–230.
[22] S.W. Han, Y. Kim, K. Kim, J. Colloid Interface Sci. 208 (2001)
272–278.
[23] J.R. Heath, C.M. Knobler, D.V. Leff, J. Phys. Chem. B 101
(1997) 189–197.
[24] J.-P. Bourgoin, C. Kergueris, E. Lefevre, S. Palacin, Thin Solid
Films 327–329 (1998) 515–519.
[25] H. Perez, R.M. Lisboa de Sousa, J.-P. Pradeau, P.-A. Albouy,
Chem. Mater. 13 (2001) 1512–1517.
[26] S. Chen, Langmuir 17 (2001) 2878–2884.
[27] J. Huang, J.C. Hemminger, J. Am. Chem. Soc. 115 (1993)
3342–3343.
[28] B. Canava, J. Vigneron, A. Etcheberry, D. Guimard, P.P. Grand,
J.-F. Guillemoles, D. Lincot, S. Ould Saad Hamatly, Z. Djeb-
bour, D. Mencaraglia, Thin Solid Films 431 (2003) 289–295.
[29] D.G. Castner, K. Hinds, D.W. Grainger, Langmuir 12 (1996)
5083–5086.
[30] R.G. Nuzzo, B.R. Zegarski, L.H. Dubois, J. Am. Chem. Soc.
109 (1987) 733–740.
[31] M.-C. Bourg, A. Badia, R.B. Lennox, J. Phys. Chem. B 104
(2000) 6562–6567.
[32] M.J. Hostetler, J.E. Wingate, C.-J. Zhong, J.E. Harris, R.W.
Vachet, M.R. Clark, J.D. Londono, S.J. Green, J.J. Stokes, G.D.
Wignall, G.L. Glish, M.D. Porter, N.D. Evans, R.W. Murray,
Langmuir 14 (1998) 17–30.
[33] B.J. Lindberg, K. Hamrin, G. Johansson, U. Gelius, A. Fahl-
man, C. Nordling, K. Siegbahn, Phys. Scr. 1 (1970) 286–298.
[34] K. Kinoshita, J. Electrochem. Soc. 137 (1990) 845–848.
[35] M. Tachibana, K. Yoshizawa, A. Ogawa, H. Fujimoto, R.
Hoffmann, J. Phys. Chem. B 106 (2002) 12727–12736.
[36] J. Lukkari, K. Kleemola, M. Meretoja, T. Ollonqvist, J.
Kankare, Langmuir 14 (1998) 1705–1715.
[37] S.K. Shaikhutdinov, M. Schildenberger, M. Noeske, G. Mestl,
React. Kinet. Catal. Lett. 67 (1999) 129.
[38] K.S. Kim, N. Winograd, R.E. Davis, J. Am. Chem. Soc. 93
(1971) 6296.
[39] F. Raynal, A. Etcheberry, C. Reynaud, H. Perez, Appl. Surf.
Sci. 236 (2004) 198–207.
Copyright © 2022 FDOKUMEN




















