
Cell-to-Cell Contact Results in a Selective Translocation of Maternal Human Immunodeficiency Virus...
13
10.1128/JVI.75.10.4780-4791.2001. 2001, 75(10):4780. DOI: J. Virol. Transmission of HIV-1 Bomsel and the European Network for the Study of In Utero Mauclère, G. Scarlatti, G. Chaouat, F. Barré-Sinoussi, M. S. Lagaye, M. Derrien, E. Menu, C. Coi?to, E. Tresoldi, P. Barrier by both Transcytosis and Infection Quasispecies across a Trophoblastic Immunodeficiency Virus Type 1 Translocation of Maternal Human Cell-to-Cell Contact Results in a Selective http://jvi.asm.org/content/75/10/4780 Updated information and services can be found at: These include: REFERENCES http://jvi.asm.org/content/75/10/4780#ref-list-1 at: This article cites 63 articles, 27 of which can be accessed free CONTENT ALERTS more» articles cite this article), Receive: RSS Feeds, eTOCs, free email alerts (when new http://journals.asm.org/site/misc/reprints.xhtml Information about commercial reprint orders: http://journals.asm.org/site/subscriptions/ To subscribe to to another ASM Journal go to: on May 22, 2013 by 00308377 http://jvi.asm.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Cell-to-Cell Contact Results in a Selective Translocation of Maternal Human Immunodeficiency Virus...

10.1128/JVI.75.10.4780-4791.2001.
2001, 75(10):4780. DOI:J. Virol. Transmission of HIV-1Bomsel and the European Network for the Study of In UteroMauclère, G. Scarlatti, G. Chaouat, F. Barré-Sinoussi, M. S. Lagaye, M. Derrien, E. Menu, C. Coi?to, E. Tresoldi, P. Barrier by both Transcytosis and InfectionQuasispecies across a Trophoblastic Immunodeficiency Virus Type 1Translocation of Maternal Human Cell-to-Cell Contact Results in a Selective
http://jvi.asm.org/content/75/10/4780Updated information and services can be found at:
These include:
REFERENCEShttp://jvi.asm.org/content/75/10/4780#ref-list-1at:
This article cites 63 articles, 27 of which can be accessed free
CONTENT ALERTS more»articles cite this article),
Receive: RSS Feeds, eTOCs, free email alerts (when new
http://journals.asm.org/site/misc/reprints.xhtmlInformation about commercial reprint orders: http://journals.asm.org/site/subscriptions/To subscribe to to another ASM Journal go to:
on May 22, 2013 by 00308377
http://jvi.asm.org/
Dow
nloaded from

JOURNAL OF VIROLOGY,0022-538X/01/$04.0010 DOI: 10.1128/JVI.75.10.4780–4791.2001
May 2001, p. 4780–4791 Vol. 75, No. 10
Copyright © 2001, American Society for Microbiology. All Rights Reserved.
Cell-to-Cell Contact Results in a Selective Translocation of MaternalHuman Immunodeficiency Virus Type 1 Quasispecies across a
Trophoblastic Barrier by both Transcytosis and InfectionS. LAGAYE,1* M. DERRIEN,1,2 E. MENU,1,3 C. COITO,2 E. TRESOLDI,4 P. MAUCLERE,5 G. SCARLATTI,4
G. CHAOUAT,3 F. BARRE-SINOUSSI,1 M. BOMSEL,2 AND THE EUROPEAN NETWORKFOR THE STUDY OF IN UTERO TRANSMISSION OF HIV-1†
Institut Pasteur, Unite de Biologie des Retrovirus, 75 724 Paris Cedex 15,1 INSERM U 131, Hopital Antoine Beclere,92 140 Clamart,3 and INSERM U 332, Institut Cochin de Genetique Moleculaire, 75014 Paris,2 France;
Unit of Immunology, DIBIT, San Raffaele Scientific Institute, 20 132 Milan, Italy4; andCentre Pasteur du Cameroun, Yaounde, Cameroon5
Received 23 October 2000/Accepted 22 February 2001
Mother-to-child transmission can occur in utero, mainly intrapartum and postpartum in case of breast-feeding. In utero transmission is highly restricted and results in selection of viral variant from the mother tothe child. We have developed an in vitro system that mimics the interaction between viruses, infected cellspresent in maternal blood, and the trophoblast, the first barrier protecting the fetus. Trophoblastic BeWo cellswere grown as a tight polarized monolayer in a two-chamber system. Cell-free virions applied to the apical poleneither crossed the barrier nor productively infected BeWo cells. In contrast, apical contact with humanimmunodeficiency virus (HIV)-infected peripheral blood mononuclear cells (PBMCs) resulted in transcytosisof infectious virus across the trophoblastic monolayer and in productive infection correlating with the fusionof HIV-infected PBMCs with trophoblasts. We showed that viral variants are selected during these two stepsand that in one case of in utero transmission, the predominant maternal viral variant characterized aftertranscytosis was phylogenetically indistinguishable from the predominant child’s virus. Hence, the first stepsof transmission of HIV-1 in utero appear to involve the interaction between HIV type 1-infected cells and thetrophoblastic layer, resulting in the passage of infectious HIV by transcytosis and by fusion/infection, bothleading to a selection of virus quasispecies.
The transmission of human immunodeficiency virus type 1(HIV-1) from mother to child is believed to occur mainlyduring delivery (intrapartum) and later through breast feeding(14). The percentage of infected children decreased markedly,from 15 to 35% to 0.8 to 3%, when pregnant women weregiven zidovudine (AZT) early in pregnancy and when theirchildren were delivered by cesarean section (33, 34, 53). Earlyin utero transmission is a rare event (2). The evidence for inutero transmission of HIV includes identification of the virusin fetuses aborted in the first, second, and even third trimesters(35, 37, 40, 54). Comparison of maternal HIV-1 variants andvirus populations detected in neonates at birth indicated thatonly some viruses are transmitted to the fetus and suggestedthat a selection occurs (1, 48, 52, 61). Although maternal HIVmay enter the fetal blood directly through microbreaches of
the placenta (9), studies on blood and placenta specimensobtained from a cohort of untreated, HIV-1-positive pregnantwomen and infants have shown HIV-1 sequences within theplacenta of all seropositive pregnant women, irrespective ofthe virological status of the infants (20, 41, 64). In addition,only a limited number of maternal viruses were detected in theplacenta (41), suggesting that in utero transmission is con-trolled by subtle regulatory mechanisms related to character-istics (structural and/or functional) of the viral variants and/orrelated to the development/organization of placenta cell sub-populations.
The placental chorionic villi are constituted by several cellsubpopulations, including a trophoblast layer (cyto- and syn-cytiotrophoblast) forming the interface between maternal andfetal blood. These epithelium-like cells are organized as a tightmonolayer (39). Cytotrophoblasts lying inside the villus grad-ually form a syncytium during pregnancy, which becomes ex-posed to maternal blood. Neighboring cells are connected bytight junctional complexes that actively participate in the tro-phoblast barrier function by precluding the passive paracellu-lar transport of even small macromolecules. Exchanges be-tween the mother and the fetus occur by transcytosis, apathway of membrane trafficking specific to polarized epithe-lial cells (44). During this vesicular transcellular transport, thetranscytosed cargo remains enclosed in transcytotic vesicles,precluding contact with the host cell cytoplasm, and is releasedintact at the opposite pole (29, 44). Maternal blood immuno-globulins G (IgGs) are carried across the placenta by receptor-
* Corresponding author. Present address: INSERM U342, Hopitalsaint Vincent de Paul, 82, Avenue Denfert-Rochereau, 75014 Paris,France. Phone: 33-1-40-48-82-49. Fax: 33-1-40-48-83-52. E-mail: [email protected].
† Participants of the European Network for In Utero Transmissionof HIV-1 who contributed to this study include the following: B.Mognetti and M. Moussa (INSERM U 131, Clamart, France); P.Martin, A. Ayouba, and E. Tina (Centre Pasteur, Yaounde, Cam-eroon); E. M. Fenyo and C. Tscherning (Karolinska Institute—Micro-biology and Tumorbiology Center, Stockholm, Sweden); D. Dormont,P. Roques, and C. Depienne (CEA/CENFAR, Fontenay aux Roses,France); P. Gounon and A. Topilko (Institut Pasteur, Paris, France);and I. Athanassakis (Department of Biology, Division of Immunology,University of Crete, Heraklion, Greece).
4780
on May 22, 2013 by 00308377
http://jvi.asm.org/
Dow
nloaded from

mediated transcytosis into fetal blood (24, 36). Trophoblasticcells also form the first efficient barrier to protect the fetusfrom maternal infection, but its efficiency depends on thepathogen (28, 38, 55, 56). Data concerning the permissivity ofpurified primary trophoblasts or trophoblastic cell lines to cell-free HIV infection remain controversial (18, 32, 43, 47, 62, 63).All these studies were done using trophoblast target cells thatwere not organized as a functional, polarized trophoblast bar-rier.
We have therefore established an in vitro experimentalmodel, based on our previous study on HIV interaction withsimple epithelia, that mimics the interaction of primary HIVisolates from infected mothers and the trophoblastic barrier.The barrier is represented by BeWo cells (46) that were ini-tially isolated from a choriocarcinoma and are now widely usedas a model for trophoblast cells. These cells form a polarized,tight monolayer when cultured on polycarbonate filters in atwo-chamber system (12, 24).
We found that cell-free virus does not cross the BeWomonolayer either by transcytosis or following BeWo cell infec-tion. In contrast, the contact between HIV-infected peripheralblood mononuclear cells (PBMCs) and the apical side of thetrophoblast layer leads to two events. The first and rapid eventconsists of HIV-1 particles budding at the contact site (19),followed by their transcytosis across the trophoblastic barrierwithout viral replication. The second event is the fusion ofHIV-1-infected PBMCs with trophoblastic cells, resulting invirus replication and the production of virus progeny towardthe basal pole of the trophoblastic monolayer. Maternal virusvariants are selected following both transcytosis and fusion-induced virus replication. The comparison of the in vitro-trans-located virus quasispecies derived from infected mothers withthose transmitted to their children in vivo evidenced that in thecase of in utero transmission, the child’s virus was indistin-guishable from the maternal, transcytosed, predominant vari-ant. Our data suggest therefore that our in vitro system is atleast partially reflecting the initial steps of in vivo transmissionof HIV-1 in utero.
MATERIALS AND METHODS
Cells. The human choriocarcinoma BeWo cell line (ATCC CCL 98) (46),obtained from the American Type Culture Collection (Rockville, Md.), wasmaintained in Dulbecco’s modified Eagle medium containing 25 mM glucose,supplemented with 20% heat-inactivated fetal calf serum (GIBCO BRL; Gaith-ersburg, Md.), 20 mM glutamine, 25 mM HEPES (pH 7.4), and antibiotics (50IU of penicillin/ml and 50 mg of streptomycin [GIBCO BRL]/ml in 5% CO2–95%air) (12). Venous blood samples were taken from healthy blood donors, andPBMCs were isolated by Ficoll-Paque (Pharmacia Biotech, Uppsala, Sweden)centrifugation and used either as effector cells or as indicator cells or to amplifyviral isolates. Phytohemagglutinin (PHA)-stimulated PBMCs were cultivated inRPMI 1640 medium supplemented with 10% heat-inactived fetal calf serum(GIBCO BRL) and 10 U of interleukin-2/ml. T-cell (CD31/CD142) and mono-cyte/macrophage (CD32/CD141) populations were separated by negative selec-tion using Dynabead columns as recommended by the manufacturer (Dynal,S.A., Oslo, Norway).
Viral isolates and HIV-1-infected PBMCs. Primary virus isolates were recov-ered from PBMCs of HIV-1-infected, pregnant women after their informedconsent to participate in the study (41, 51). None of them was under antiretro-viral therapy. Viral stocks were obtained after two passages of the primaryisolates on PHA-stimulated PBMCs. Two HIV-1 subtype A isolates (1329 and1379) were both of the non-syncytium-inducing (NSI) (R5) phenotype derivedfrom Cameroonian nontransmitting mothers. Subtype B viruses were from Ital-ian mothers: two were NSI (R5) (A113 and 115) and three were syncytiuminducing (SI) (X4) (A245, A196, and A204). A204, A115, and A196 were from
mothers who transmitted the virus to their child, either in utero (A204) orintrapartum (A115 and A196). The other viruses were from nontransmittingmothers (1329, 1379, A113, and A245). Virus stocks were snap frozen in aliquotsand were stored at 280°C. The infectious titer of each virus stock was deter-mined by limiting dilution assays on PHA-activated PBMCs, as previously de-scribed (49), and was expressed as 50% tissue culture infective dose (TCID50)per milliliter. Either cell-free viruses or infected PBMCs were used in the in vitromodel. Infected PBMCs used as effector cells were prepared as follows: 106
PBMCs were inoculated with a dose of 100 TCID50 of virus previously treatedwith 200 U of RNase-free DNase per ml (15 min at room temperature). Theinfected PBMCs were extensively washed and maintained at 37°C for 7 to 10additional days, until virus production was maximal, as determined by HIV-1 p24antigen content in cell culture supernatant. Uninfected PBMCs or T-cell-lineCEM cells chronically infected with laboratory-adapted HIV-1 strains (HIV-1LAI subtype B and HIV-1 NDK subtype D) or HIV-2 strain (HIV-2 RODsubtype type A) (59) were used as controls.
In vitro model to study HIV translocation across polarized BeWo trophoblas-tic monolayers. We investigated the mechanisms responsible for the passage ofHIV across the trophoblastic barrier, using the in vitro model shown in Fig. 1.This model involves three distinct cell types: (i) effector cells consisting ofHIV-1-infected PBMCs (replaced in some experiments by cell-free virus); (ii)target cells, a tight, polarized monolayer of BeWo trophoblastic cells; and (iii)indicator cells, uninfected PBMCs supporting replication of infectious HIV-1particles that had been transcytosed through or were released from infectedBeWo monolayers.
Target BeWo cells (5 3 105 cells) were grown on permeable supports (12-mm-diameter Transwell polycarbonate filters, 0.45-mm porosity; Costar Corp.,Cambridge, Mass.) in a two-chamber system for 5 days. Under these conditions,BeWo cells form a tight, polarized monolayer that mimicked the trophoblasticbarrier in vivo (12, 13). We checked the integrity of target BeWo cell monolayersat the onset of the experiments and after contact with infectious material by usingthree parameters. The integrity and tightness of BeWo monolayers were ana-lyzed by measuring the stability of the volume of medium in the upper chamber.The electrical transepithelial resistance was measured prior to addition of infec-tious materials and after inoculation; it remained stable as previously reported(6, 12). Lastly, morphological tests were performed to confirm the integrity of theBeWo monolayer and to preclude the translocation of even a single effectorHIV-infected PBMC (paracellular passage) across the trophoblastic layer duringthe experiments (6). Briefly, effector HIV-infected PBMCs were loaded with afluorescent dye (Cell Tracker-AM [CT] green) before the start of the assay andwere added to the apical chamber. No fluorescent cells were detected in thebasolateral chamber at the end of the 2-h-30-min inoculation period by directobservation under an epifluorescent microscope. Similarly, CT green added tothe basolateral medium at the end of the inoculation period revealed no trans-location of effector cells from the apical chamber to the basolateral chamber. Incontrast, opening tight junctions with EGTA (10 mM) resulted in the detectionof HIV-infected fluorescent cells in the basolateral medium, presumably due toa paracellular transport. We also used extensive washing and examination of themonolayer by confocal microscopy—a series of 30 optical sections, 0.4 mm apartand using an (x, z) resolution of 0.6 mm—to ensure that effector HIV-infectedcells did not bypass tight junctions and remained in the trophoblastic monolayerabove the filter, where they could have produced HIV. We thus demonstratedthe integrity of the trophoblastic barrier at the onset of and throughout theexperiment and precluded a rupture of the monolayer in contact with infectiousmaterial and a paracellular transport of effector HIV-infected PBMCs. Thisculture system is widely employed to study polarized epithelial cell monolayers(6, 11, 24) and offers independent access to both apical and basolateral fluids,making it suitable for studying the transport of viruses or virus-infected cellsacross the trophoblastic barrier.
Nonpolarized BeWo cells. To study the effects of HIV-1 on unpolarized tro-phoblastic cells, BeWo (5 3 105 cells/well) were cultured on plastic support(6-well plates [Costar Corp.]) for 36 h to reach 50 to 70 percent confluency.
Inoculation of BeWo target cells and virus detection. The experimental pro-tocol used to study the interaction of HIV (cell-free particles or HIV-infectedeffector PBMCs) with the BeWo cell monolayer is shown in Fig. 1. We studiedthe interaction of HIV-1-infected effector PBMCs with the apical surface ofBeWo cell monolayers by adding 2 3 106 effector PBMCs infected with HIV-1primary maternal isolates (clade A or B) to the apical chamber where they werein contact with trophoblastic BeWo target cells (6, 7). To study cell-free virusinteraction with BeWo, these target cell monolayers were inoculated with cell-free HIV-1 by adding virus isolates (100 TCID50) to the apical or basolateralchamber or to unpolarized BeWo cells. Fresh media were added into the oppo-site chamber. In some experiments, BeWo cells were treated with AZT (10 mM;
VOL. 75, 2001 TROPHOBLASTIC BARRIER AND HIV-1 INFECTION 4781
on May 22, 2013 by 00308377
http://jvi.asm.org/
Dow
nloaded from

Sigma); cells were incubated for 2 h before contact with cell-free virus and duringall the following steps. As a control, PBMCs (106 cells) infected with cell-freeHIV-1 were treated with AZT under similar conditions.
HIV-infected PBMCs or cell-free virus was carefully removed from BeWo cellmonolayers immediately after the 2-h-30-min contact. The medium in the apicaland basolateral chambers was collected and immediately frozen. IndicatorPBMCs (1.5 3 106) were added to the basolateral chamber during the apicalcontact between the inoculum and BeWo cells to allow propagation of anytranscytosed virus. Then indicator PBMCs were cultured separately from filter-grown BeWo cells, and their virus content was determined immediately and atdifferent times after culture, up to 5 days later. Cell-free supernatants wereanalyzed for HIV-1 p24 antigen quantification.
Infectious material was removed from the apical and basolateral sides of theBeWo cell monolayer, and the cells were extensively washed, including one washwith prewarmed trypsin for 1 min at 37°C to remove any adherent effectorPBMCs or virus particles. The last of the six washes was negative for p24 viralantigen when cell-free virus was used as inoculum. Unpolarized BeWo cellscultured on plastic support were washed as polarized BeWo cell monolayers.BeWo cell monolayers were then cultured for five additional days in the presenceof 2 3 106 uninfected indicator PBMCs plated in the basolateral chamber tomonitor trophoblastic cell infection and the production of infectious virus prog-eny. At the end of this period, monolayers and basolateral indicator PBMCs werecollected for DNA extraction and PCR amplification of HIV-1 sequences. Theviral content of supernatants from the different compartments was analyzed byHIV-1 p24 antigen enzyme-linked immunosorbent assay (Sanofi DiagnosticsPasteur, Marnes la Coquette, France) as indicated by the manufacturer.
Morphological detection of fusion between HIV-1-infected PBMCs and tro-phoblastic cells. Effector HIV-1-infected PBMCs were loaded with a fluorescentprobe, CT green (0.5 mM), for 30 min at 37°C and were placed in the apicalchamber in contact with the trophoblastic BeWo target cell monolayer for 2 h 30min. The BeWo cell monolayers were fixed, and nuclei were fluorescently labeledwith Hoechst 3345 (5 mg/ml) for 15 min. The cell monolayers were then mountedin Mowiol (7). The redistribution of the CT green from effector HIV-infectedPBMCs to BeWo target cells indicated fusion. This was analyzed by confocalmicroscopy (Bio-Rad 1024 with a 603 optical lens [Nikon] using argon/kryptonand UV lasers) as previously described (7). Consecutive sections were 0.5 mmapart. Images corresponding to the vertical projection of at least 20 consecutiveoptical sections were processed using the double-labeling laser Sharp software.CT green appears yellow and nuclei appear red after merging of two sectionsrecorded at the same level in each channel.
Quantitation of fusion. Cell fusion was measured by fluorescence resonanceenergy transfer using compatible dyes, CT green (excitation wavelength, 488 nm;emission wavelength, 524 nm) and CT red (excitation wavelength, 540 nm;emission wavelength, 572 nm) (31, 60). Briefly, each of the fusion partners wasloaded with one of the two dyes, effector HIV-1-infected PBMCs with CT green
and target trophoblastic BeWo cells with CT red (0.5 mM) by incubation for 30min at 37°C. Effectors were added to the targets as described above. Fusioncaused the formation of a syncytium and the mixing of the two dyes in sufficientlyclose contact to allow resonance energy transfer to occur from CT green to CTred following CT green excitation. Cells were then detached from their supportby using trypsin, washed in phosphate-buffered saline, and fusion quantified byflow cytometry after CT green excitation at 488 nm and collection of the CT redsignal at 572 nm (31, 60), using the Elite flow cytometer and Elite software(Coulter, Marseilles, France). Results are expressed as the percentage of targetcells (index of fusion) that underwent fusion.
Analysis of virus phenotype and coreceptor usage. The phenotypes of mater-nal viruses were analyzed before and after transcytosis or replication. The NSI/SIphenotype was identified using the standard test involving MT2 T-cell lines (30,42, 50). Briefly, MT2 cells (5 3 105) were incubated for 1 h with the sample foranalysis, washed extensively, and cultured for up to 15 days. The cultures wereanalyzed by visual inspection for cytopathic effect (syncytium formation), andinfection was quantified by measuring HIV-1 p24 antigen in culture supernatantsby enzyme-linked immunosorbent assay. The chemokine receptor usage of asample was determined by using human glioma CD41 U87 cell lines stablyexpressing CCR5 (R5) or CXCR4 (X4) (22, 23). Briefly, cells were seeded at alow concentration (104/well) in 96-well plates, incubated for 48 h, and inoculatedwith 200 ml of the test sample for 1 h. The plates were washed extensively toremove the virus inoculum and cultured in fresh medium. Cultures were ob-served daily for cytopathic effect (syncytium formation). Supernatants were col-lected from each well and tested for HIV-1 p24 antigen.
DNA extraction and PCR amplification of HIV-1 tat and V3-V5 env fragments.Total DNA was extracted from the three cell types involved in the in vitro model:(i) effector-infected PBMCs, (ii) infected target BeWo cells immediately or at theindicated times after contact with effector-infected PBMCs (or with cell-freevirus) and extensive washes, and (iii) from infected indicator PBMCs. The cellpellets were resuspended in a lysis buffer (0.01 M Tris-HCl [pH 7.4], 0.1 M NaCl,0.01 M EDTA, 1% sodium dodecyl sulfate) containing RNase (100 mg/ml) andproteinase K (100 mg/ml). DNA was then extracted with phenol, phenol-chloro-form, and chloroform-isoamylalcohol; precipitated with ethanol; and suspendedin sterile water for PCR amplification.
HIV-1 sequences from PBMCs and BeWo DNA were PCR amplified using tatand env specific primers. The tat primers used were tat A1, tat A11, and tat A2;tat A1/tat A2 were outer primers, and tat A11/tat A2 were inner primers. tat A20was used as a probe for hybridization (41). The env primers used were ED3 andED14; ED5 and ED12 were outer primers, and ES7 and ES8 were inner primers(41). The sensitivity of the PCRs was one copy for 105 cells or for 1 mg of DNA.Three PCR amplifications were performed for each DNA sample tested. Am-plification with primer pairs specific for the b-actin gene was also performed oneach sample to control the quality and quantity of DNA samples. The primersused were b-actin 1, 59-GTGGGGCGCCCCAGGCACCA 39 (nucleotides 1260
FIG. 1. Experimental system for monitoring HIV translocation across the polarized trophoblastic monolayer. The inoculum, cell-free HIV orHIV-infected PBMCs (effectors), is applied at the apical pole of target trophoblastic BeWo cells. Both transcytosis across and productive infectionof target cells are monitored in the basolateral chamber after addition of indicator PBMCs.
4782 LAGAYE ET AL. J. VIROL.
on May 22, 2013 by 00308377
http://jvi.asm.org/
Dow
nloaded from

to 1280); and b-actin 2, 59 CTCCTTAATGTCACGCACGATTTC 39 (nucleo-tides 1350 to 1375). HIV-1 V3-V5 env and tat fragments were PCR amplified aspreviously described (21, 41). The expected size of the final PCR products wasvisualized after 2% agarose gel electrophoresis, ethidium bromide staining (0.5mg/ml), and Southern hybridization (17).
HMA. Comparative genetic analysis of HIV samples was performed by het-eroduplex mobility assay (HMA) (21). Each amplified gene product in the V3-V5env region was mixed with a homologous PCR fragment from a reference virusDNA corresponding to maternal HIV-1 subtype A or B. Heteroduplexes wereseparated by electrophoresis on a 5% polyacrylamide gel (using a 30% acryl-amide–0.8% bisacrylamide stock), stained with ethidium bromide (1 mg/ml), andexamined. Patterns of HIV-1 env quasispecies detected in the different fractions(Ap, apical inoculum [representative of maternal virus]; BW, BeWo monolayerafter 2-h-30-min contact with infected PBMCs; PI, produced by BeWo cells afterinfection; Tr, transcytosed; or Ch, pair child viral isolate) were analyzed togetherfor direct comparison.
PCR and direct sequencing of gp120-V3 region. Sequences specific for HIV inthe V3 region of the gp120 gene were PCR amplified (49, 52), first with the outerprimers (JA 9-12). One-tenth of the product was further amplified using a set ofinner primers (JA 10-53). The amplified product was directly used for solid-phase sequencing. Briefly, DNA fragments obtained by amplification with innerprimers were purified by immobilization of biotinylated primers on streptavidin-coated magnetic beads (Dynabeads M280-streptavidin; Dynal). The sequencingreaction was performed with fluorescein-labeled primers using a commercial kit(AutoRead; Pharmacia). The reaction products were then loaded on a 6%polyacrylamide gel in an automated laser fluorescent sequencing apparatus(ALF; Pharmacia LKB).
Sequence analysis. V3 nucleotide sequences were aligned using the CLUSTALW program with minor manual adjustments. Phylogenetic trees were generatedby the neighbor-joining and Fitch-Margoliash distance methods implemented inthe PHYLIP package, and their reliability was estimated from 100 bootstrapreplicates. Phylogenetic trees were also constructed by maximum parsimony andmaximum likelihood.
RESULTS
BeWo cells are resistant to cell-free virus infection andtranscytosis, regardless of their polarity. We first investigated
the permissivity of target BeWo trophoblastic cells to cell-freeHIV, as cell-free HIV is generally assumed to be the mainvector of transmission and is present in maternal blood at thetrophoblast interface. Trophoblast monolayers were infectedwith cell-free HIV stocks (100 TCID50). No evidence of trans-cytosis from the apical to the basolateral side or productiveinfection by cell-free HIV was observed, since no HIV-1 p24antigen was detectable in the basolateral medium when ana-lyzed directly or when indicator PBMCs were inoculated withthese HIV-1 p24 antigen-negative supernatants and culturedup to five additional days (Table 1). These results were similarregardless of the virus subtype, coreceptor usage, SI/NSI phe-notype, or transmitted or nontransmitted status. There was nodetectable HIV-1 p24 antigen in the supernatant from theapical chamber when the polarity of the infection was reversedand when virus isolates were added to the basolateral side (notshown). Similarly, no significant HIV-1 p24 antigen was de-tected in the culture supernatant of unpolarized BeWo cellsinoculated with cell-free virus (not shown). In contrast, BeWomonolayers treated with EGTA (10 mM) to alter the integrityof their tight junctions showed a paracellular transport of thevirus, together with the leveling of medium in both chambers(not shown) (6).
We investigated the presence of HIV-1 in the target cells byPCR amplification of tat sequences. Except in rare cases, neg-ative signals were obtained with DNA from the trophoblastmonolayer, polarized or not, using tat primers (Table 2 andFig. 2), and indicator PBMCs were always negative (Table 2).Similar data were obtained irrespective of the viral isolatesused or the polarity of the inoculation (apical or basolateral)(Fig. 2A and B). Moreover, no viral sequences were amplifiedin BeWo cells after AZT treatment (Fig. 2C) at a dose that
TABLE 1. Kinetics of detection of transcytosed virus in the basolateral chamber or after one passage on indicator PBMCsa
Chamber or cell type
p24 antigen level (pg/ml) found by inoculating HIV1 PBMCs or cell-free virus after:
0 h 2 h 30 min 5 h 5 days
HIV1 PBMCs Cell-free virus HIV1 PBMCs Cell-free virus HIV1 PBMCs Cell-free virus HIV1 PBMCs Cell-free virus
Basolateral chamberb 10 6 0.003 — 32 6 0.004 — 43 6 0.011 — 1,350 6 0.19 —Indicator PBMCsc — — — — — — 6,300 6 0.82 —
a BeWo target cells were inoculated apically with either cell-free HIV-1 or HIV-1-infected PBMCs for 2 h 30 min. Inoculum was then removed. Detection oftrancytosed virus was performed by HIV-1 p24 antigen measurement in the basolateral chamber at different times after removal of the apical inoculum. Results aregiven as amount of p24 antigen detected in the supernatants.
b HIV p24 antigen was measured directly in the basolateral medium at various times after the end of the apical contact.c Indicator PBMCs were placed into the basolateral chamber during the contact period and were then cultured separately from BeWo monolayer for five additional
days. HIV p24 antigen was measured in the supernatant. —, p24 antigen level of ,10 pg/ml. Data are expressed as mean values 6 standard deviations of fourindependent experiments performed in duplicates.
TABLE 2. PCR-amplified tat and/or env sequences in BeWo cells or in indicator PBMCs located in the basolateral chamberat different times after removal of the apical inoculuma,b
Cell type
Results found with inoculation of HIV-1-infected PBMCs or of cell-free HIV-1
0 h 2 h 30 min 5 h 5 days
HIV1 PBMCs Cell-free virus HIV1 PBMCs Cell-free virus HIV1 PBMCs Cell-free virus HIV1 PBMCs Cell-free virus
BeWo 1 2 1 2 1 2 1 6Indicator PBMCs 2 2 2 2 2 2 1 2
a Cell-free HIV-1 or HIV-1-infected PBMCs were inoculated apically onto BeWo cells. The assay was conducted as for Table 1.b Indicator PBMCs were placed into the basolateral chamber during the contact period and were then cultured separately from the BeWo monolayer for five
additional days. PCR amplification was performed on BeWo and PBMC DNA. Results are representative of at least four independent experiments. 1, always positive;2, always negative; 6, rarely positive.
VOL. 75, 2001 TROPHOBLASTIC BARRIER AND HIV-1 INFECTION 4783
on May 22, 2013 by 00308377
http://jvi.asm.org/
Dow
nloaded from

reduced by 50% the infection of PBMCs. Indeed, in AZT-treated PBMCs, HIV tat sequences were detected in three outof six PCR amplifications performed, whereas in cells culturedwithout AZT, all DNA samples tested were positive for theHIV tat sequence with the same viral isolates (Fig. 2E). Theoccasional detection of HIV-1 DNA sequences in BeWo mightbe either related to remaining particles or to the entry of veryfew viral particles into BeWo cells which could reverse tran-scribe as suggested by AZT treatment.
Thus, contact between cell-free virus and BeWo resultedneither in translocation across the monolayer nor in a detect-able production of virion by BeWo cells, regardless of thedegree of polarization of the target BeWo cell.
Cell-to-cell contact between HIV-infected PBMCs and BeWocell monolayers induces both transcytosis of infectious virusand HIV replication. Since cell-free viruses did not cross theBeWo monolayer, we investigated whether cell-to-cell contactbetween effector HIV-1-infected PBMCs and polarized BeWotarget cells could result in virus transcytosis, as was previouslyreported when epithelial cells were used as the target (6). Incontrast to cell-free virus, HIV-1 p24 antigen was detected inthe supernatant of the indicator PBMCs cultured for 5 daysafter being in the basolateral chamber during inoculation (Ta-ble 1). As described in detail in Materials and Methods, theparacellular passage of virus could be excluded since no effec-tor apical HIV-infected PBMCs loaded with a fluorescent dye(CT green) were found in the basolateral chamber throughout
the experiment. In contrast, opening tight junctions withEGTA resulted in the detection of HIV-infected fluorescentcells in the basolateral medium, presumably due to a paracel-lular transport. Furthermore, examination of the monolayer byconfocal microscopy has confirmed that effector HIV-infectedcells did not bypass tight junctions.
The HIV-1 p24 antigen released by the indicator PBMCsindicated that the transcytosed virus was infectious. HIV-1 tat-and env-specific sequences were PCR amplified from DNAsamples recovered from both BeWo cells and indicatorPBMCs (Table 2).
We then investigated whether in addition to rapid transcy-tosis of infectious HIV, there was replication in filter-grownBeWo trophoblastic cells after apical contact with PBMCs.The challenged trophoblastic monolayer was extensivelywashed to remove effector cells and further cultured for fiveadditional days with indicator PBMCs in the basolateral cham-ber. HIV-1 tat and env sequences were PCR amplified fromtrophoblast DNA, and HIV-1 p24 antigen was detected in thebasolateral supernatant derived from indicator cells as soon as3 days after incubation in the basolateral chamber (Table 3).
Thus, apical cell-to-cell contact of effector HIV-infectedPBMCs with trophoblastic target cells leads to the transport ofinfectious HIV-1 across the placental barrier by two indepen-dent pathways within different time frames: a rapid transcytosisof HIV and a slower process of viral replication.
HIV-1 replication correlates with fusion between infectedPBMCs and BeWo cells. To study whether fusion betweeninfected PBMCs and BeWo cells contributes to HIV-1 repli-cation, we used HIV-1-infected PBMCs loaded with a fluores-cent probe (CT green) placed on the apical side of trophoblas-tic BeWo target cells. At the end of the cell-to-cell-contactfollowed by extensive washes, the incubated monolayers werefixed and nuclei were fluorescently labeled to visualize thecells, and the two fluorescences were analyzed simultaneouslyby confocal microscopy. We observed a redistribution of thefluorescent CT green dye from the apical effector HIV-in-fected PBMCs to trophoblastic BeWo target cell cytoplasm(Fig. 3A, panels b to c), indicating a fusion between effectorand target cells. The fusion occurred with HIV-infected effec-tor cells, regardless of the isolate subtype (A or B) or the SI orNSI phenotype (Fig. 3C, panels a to d). Furthermore, thefusion was observed with total PBMCs (Fig. 3A and C), as well
FIG. 2. Infection of polarized and unpolarized BeWo trophoblasticcells by cell-free virus. Media conditioned with uninfected PBMCs(Mock), viral isolate 115 (A), or 204 (B to E) were inoculated either tothe apical (Ap) or to the basolateral (BL) side of a trophoblastic BeWocell monolayer (as indicated), to unpolarized BeWo cells (D), or touninfected PBMCs (E). Cells were treated (1 AZT) or not treated (2AZT) with AZT. Five days after contact, DNA was extracted andHIV-1 tat sequences were PCR amplified, resolved on an agarose gelnext to a PCR-amplified HIV-1 tat sequence from HIV-1-infected-PBMCs used as a positive control (1), and transferred onto mem-branes before hybridization with a 32P-labeled tat A20 probe. Mem-branes were exposed to Kodak film for autoradiographic visualization.b-Actin PCR-amplified sequences are represented in panel F. Eachbar represents a triplicate (or a duplicate for HIV-1-infected PBMCs)of PCR-amplified sequence for each sample.
TABLE 3. Detection of HIV-1 p24 antigen in the supernatant ofindicator PBMCs placed into the basolateral chamber after
removal of the inoculum
Inoculuma
Level of p24 antigen (pg/ml) found by placement ofindicator PBMCsb
1day
2days 3 days 5 days
HIV1 PBMCs — — 1,780 6 0.08 5,450 6 0.25Cell-free virus — — — —
a Cell-free HIV-1 or HIV-1-infected PBMCs were inoculated apically ontoBeWo cells. The assay was conducted as for Table 1.
b Indicator PBMCs were placed into the basolateral chamber after removal ofthe inoculum. HIV p24 antigen was measured in the supernatant at differenttimes. Results are levels of p24 antigen detected in the supernatants. —, p24antigen level of ,20 pg/ml. Data are expressed as means 6 standard deviationsof four independent experiments performed in duplicate.
4784 LAGAYE ET AL. J. VIROL.
on May 22, 2013 by 00308377
http://jvi.asm.org/
Dow
nloaded from
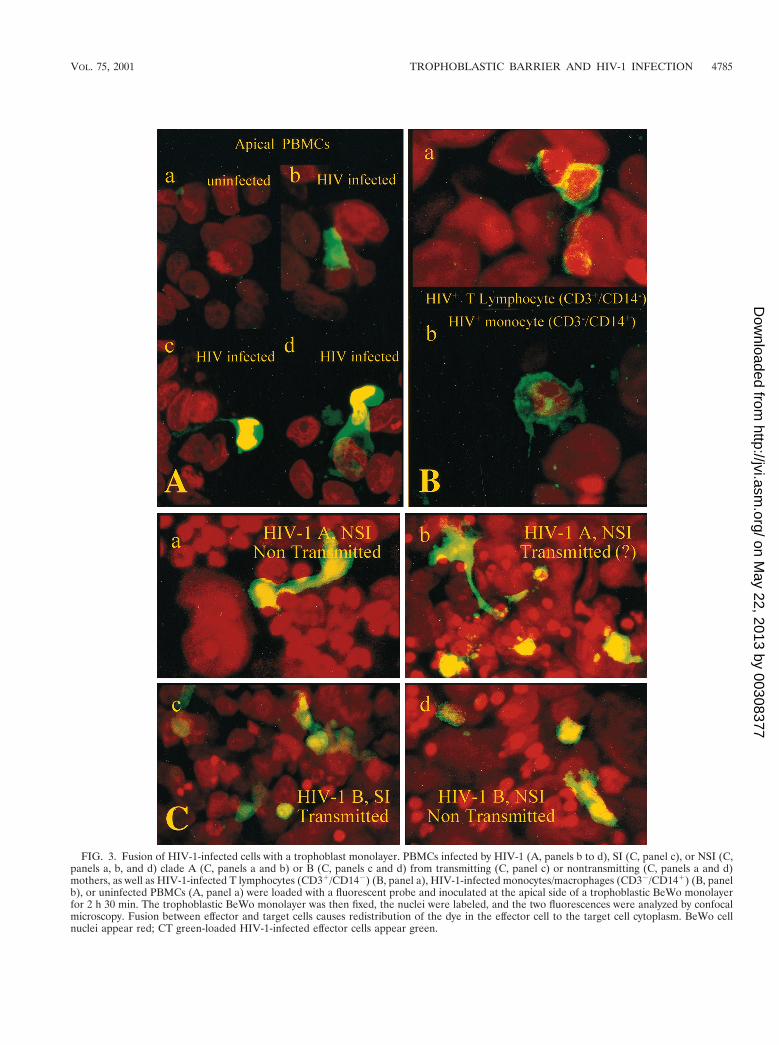
FIG. 3. Fusion of HIV-1-infected cells with a trophoblast monolayer. PBMCs infected by HIV-1 (A, panels b to d), SI (C, panel c), or NSI (C,panels a, b, and d) clade A (C, panels a and b) or B (C, panels c and d) from transmitting (C, panel c) or nontransmitting (C, panels a and d)mothers, as well as HIV-1-infected T lymphocytes (CD31/CD142) (B, panel a), HIV-1-infected monocytes/macrophages (CD32/CD141) (B, panelb), or uninfected PBMCs (A, panel a) were loaded with a fluorescent probe and inoculated at the apical side of a trophoblastic BeWo monolayerfor 2 h 30 min. The trophoblastic BeWo monolayer was then fixed, the nuclei were labeled, and the two fluorescences were analyzed by confocalmicroscopy. Fusion between effector and target cells causes redistribution of the dye in the effector cell to the target cell cytoplasm. BeWo cellnuclei appear red; CT green-loaded HIV-1-infected effector cells appear green.
VOL. 75, 2001 TROPHOBLASTIC BARRIER AND HIV-1 INFECTION 4785
on May 22, 2013 by 00308377
http://jvi.asm.org/
Dow
nloaded from
as with CD31/CD142 lymphocytes (Fig. 3B, panel a) or CD32/CD141 monocytes (Fig. 3B, panel b) but in all cases only wheninfected (Fig. 3A, panel a).
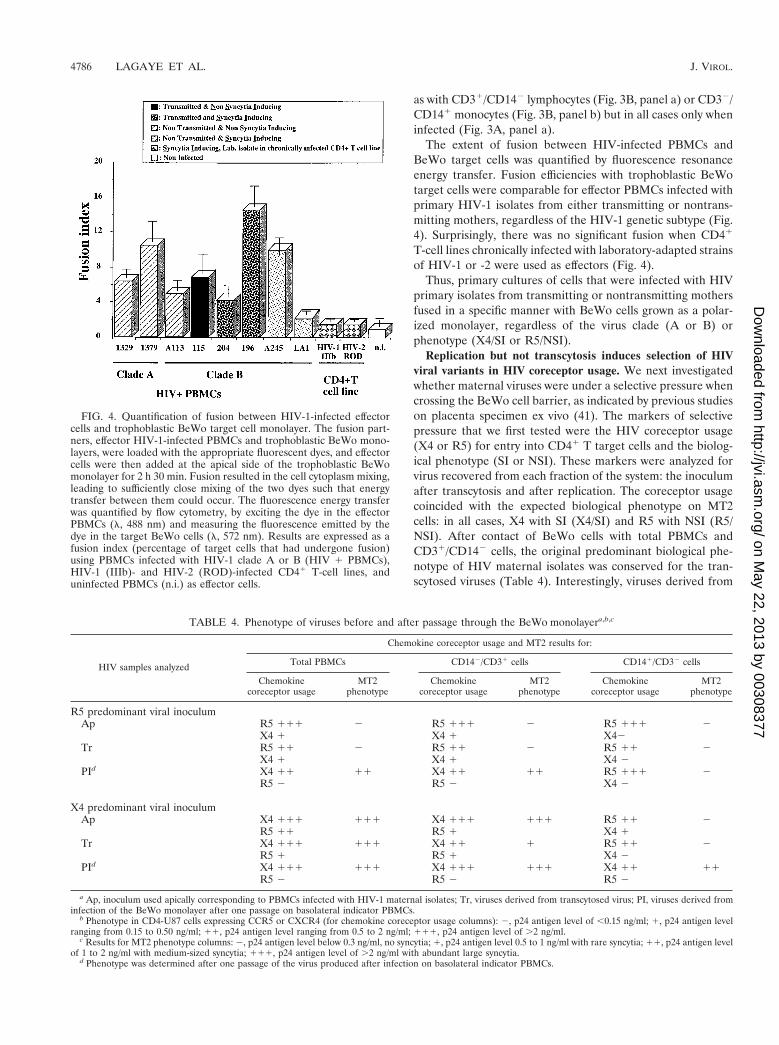
The extent of fusion between HIV-infected PBMCs andBeWo target cells was quantified by fluorescence resonanceenergy transfer. Fusion efficiencies with trophoblastic BeWotarget cells were comparable for effector PBMCs infected withprimary HIV-1 isolates from either transmitting or nontrans-mitting mothers, regardless of the HIV-1 genetic subtype (Fig.4). Surprisingly, there was no significant fusion when CD41
T-cell lines chronically infected with laboratory-adapted strainsof HIV-1 or -2 were used as effectors (Fig. 4).
Thus, primary cultures of cells that were infected with HIVprimary isolates from transmitting or nontransmitting mothersfused in a specific manner with BeWo cells grown as a polar-ized monolayer, regardless of the virus clade (A or B) orphenotype (X4/SI or R5/NSI).
Replication but not transcytosis induces selection of HIVviral variants in HIV coreceptor usage. We next investigatedwhether maternal viruses were under a selective pressure whencrossing the BeWo cell barrier, as indicated by previous studieson placenta specimen ex vivo (41). The markers of selectivepressure that we first tested were the HIV coreceptor usage(X4 or R5) for entry into CD41 T target cells and the biolog-ical phenotype (SI or NSI). These markers were analyzed forvirus recovered from each fraction of the system: the inoculumafter transcytosis and after replication. The coreceptor usagecoincided with the expected biological phenotype on MT2cells: in all cases, X4 with SI (X4/SI) and R5 with NSI (R5/NSI). After contact of BeWo cells with total PBMCs andCD31/CD142 cells, the original predominant biological phe-notype of HIV maternal isolates was conserved for the tran-scytosed viruses (Table 4). Interestingly, viruses derived from
FIG. 4. Quantification of fusion between HIV-1-infected effectorcells and trophoblastic BeWo target cell monolayer. The fusion part-ners, effector HIV-1-infected PBMCs and trophoblastic BeWo mono-layers, were loaded with the appropriate fluorescent dyes, and effectorcells were then added at the apical side of the trophoblastic BeWomonolayer for 2 h 30 min. Fusion resulted in the cell cytoplasm mixing,leading to sufficiently close mixing of the two dyes such that energytransfer between them could occur. The fluorescence energy transferwas quantified by flow cytometry, by exciting the dye in the effectorPBMCs (l, 488 nm) and measuring the fluorescence emitted by thedye in the target BeWo cells (l, 572 nm). Results are expressed as afusion index (percentage of target cells that had undergone fusion)using PBMCs infected with HIV-1 clade A or B (HIV 1 PBMCs),HIV-1 (IIIb)- and HIV-2 (ROD)-infected CD41 T-cell lines, anduninfected PBMCs (n.i.) as effector cells.
TABLE 4. Phenotype of viruses before and after passage through the BeWo monolayera,b,c
HIV samples analyzed
Chemokine coreceptor usage and MT2 results for:
Total PBMCs CD142/CD31 cells CD141/CD32 cells
Chemokinecoreceptor usage
MT2phenotype
Chemokinecoreceptor usage
MT2phenotype
Chemokinecoreceptor usage
MT2phenotype
R5 predominant viral inoculumAp R5 111 2 R5 111 2 R5 111 2
X4 1 X4 1 X42Tr R5 11 2 R5 11 2 R5 11 2
X4 1 X4 1 X4 2PId X4 11 11 X4 11 11 R5 111 2
R5 2 R5 2 X4 2
X4 predominant viral inoculumAp X4 111 111 X4 111 111 R5 11 2
R5 11 R5 1 X4 1Tr X4 111 111 X4 11 1 R5 11 2
R5 1 R5 1 X4 2PId X4 111 111 X4 111 111 X4 11 11
R5 2 R5 2 R5 2
a Ap, inoculum used apically corresponding to PBMCs infected with HIV-1 maternal isolates; Tr, viruses derived from transcytosed virus; PI, viruses derived frominfection of the BeWo monolayer after one passage on basolateral indicator PBMCs.
b Phenotype in CD4-U87 cells expressing CCR5 or CXCR4 (for chemokine coreceptor usage columns): 2, p24 antigen level of ,0.15 ng/ml; 1, p24 antigen levelranging from 0.15 to 0.50 ng/ml; 11, p24 antigen level ranging from 0.5 to 2 ng/ml; 111, p24 antigen level of .2 ng/ml.
c Results for MT2 phenotype columns: 2, p24 antigen level below 0.3 ng/ml, no syncytia; 1, p24 antigen level 0.5 to 1 ng/ml with rare syncytia; 11, p24 antigen levelof 1 to 2 ng/ml with medium-sized syncytia; 111, p24 antigen level of .2 ng/ml with abundant large syncytia.
d Phenotype was determined after one passage of the virus produced after infection on basolateral indicator PBMCs.
4786 LAGAYE ET AL. J. VIROL.
on May 22, 2013 by 00308377
http://jvi.asm.org/
Dow
nloaded from
the fusion of effector cells with BeWo target cells alwaysshowed a strict X4/SI phenotype, even when effector PBMCswere infected with viruses with a predominant R5/NSI pheno-type. In contrast, the contact with BeWo cells of CD141/CD32
cells (mostly monocytes and macrophages) purified from X4/SI- or R5/NSI-infected effector PBMCs preferentially selectedand propagated viruses with the R5/NSI phenotype (Table 4,apical results). As for unfractionated effector PBMCs, the fu-sion of CD141/CD32 cells that were isolated from effectorPBMCs infected with predominant X4/SI viruses resulted inthe production of X4/SI viruses, whereas the fusion of CD141/CD32 cells purified from predominant R5/NSI-infected effec-tor PBMCs resulted in the production of R5/NSI viruses (Ta-ble 4).
HIV translocation across trophoblastic BeWo monolayerresults in enrichment of HIV minor variants. To further ex-amine whether maternal viruses were under selective pressureduring passage across BeWo monolayers, the genetic diversityof viruses isolated from the different compartments was ana-lyzed by HMA. We used viral isolates from a nontransmitting
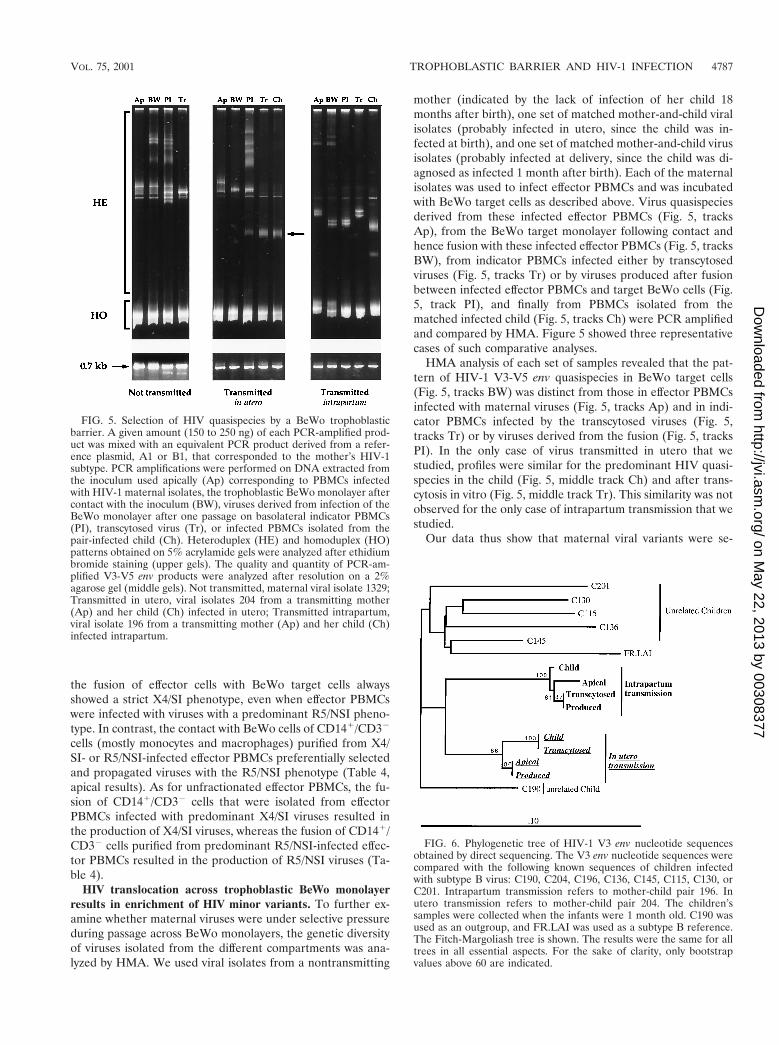
mother (indicated by the lack of infection of her child 18months after birth), one set of matched mother-and-child viralisolates (probably infected in utero, since the child was in-fected at birth), and one set of matched mother-and-child virusisolates (probably infected at delivery, since the child was di-agnosed as infected 1 month after birth). Each of the maternalisolates was used to infect effector PBMCs and was incubatedwith BeWo target cells as described above. Virus quasispeciesderived from these infected effector PBMCs (Fig. 5, tracksAp), from the BeWo target monolayer following contact andhence fusion with these infected effector PBMCs (Fig. 5, tracksBW), from indicator PBMCs infected either by transcytosedviruses (Fig. 5, tracks Tr) or by viruses produced after fusionbetween infected effector PBMCs and target BeWo cells (Fig.5, track PI), and finally from PBMCs isolated from thematched infected child (Fig. 5, tracks Ch) were PCR amplifiedand compared by HMA. Figure 5 showed three representativecases of such comparative analyses.
HMA analysis of each set of samples revealed that the pat-tern of HIV-1 V3-V5 env quasispecies in BeWo target cells(Fig. 5, tracks BW) was distinct from those in effector PBMCsinfected with maternal viruses (Fig. 5, tracks Ap) and in indi-cator PBMCs infected by the transcytosed viruses (Fig. 5,tracks Tr) or by viruses derived from the fusion (Fig. 5, tracksPI). In the only case of virus transmitted in utero that westudied, profiles were similar for the predominant HIV quasi-species in the child (Fig. 5, middle track Ch) and after trans-cytosis in vitro (Fig. 5, middle track Tr). This similarity was notobserved for the only case of intrapartum transmission that westudied.
Our data thus show that maternal viral variants were se-
FIG. 5. Selection of HIV quasispecies by a BeWo trophoblasticbarrier. A given amount (150 to 250 ng) of each PCR-amplified prod-uct was mixed with an equivalent PCR product derived from a refer-ence plasmid, A1 or B1, that corresponded to the mother’s HIV-1subtype. PCR amplifications were performed on DNA extracted fromthe inoculum used apically (Ap) corresponding to PBMCs infectedwith HIV-1 maternal isolates, the trophoblastic BeWo monolayer aftercontact with the inoculum (BW), viruses derived from infection of theBeWo monolayer after one passage on basolateral indicator PBMCs(PI), transcytosed virus (Tr), or infected PBMCs isolated from thepair-infected child (Ch). Heteroduplex (HE) and homoduplex (HO)patterns obtained on 5% acrylamide gels were analyzed after ethidiumbromide staining (upper gels). The quality and quantity of PCR-am-plified V3-V5 env products were analyzed after resolution on a 2%agarose gel (middle gels). Not transmitted, maternal viral isolate 1329;Transmitted in utero, viral isolates 204 from a transmitting mother(Ap) and her child (Ch) infected in utero; Transmitted intrapartum,viral isolate 196 from a transmitting mother (Ap) and her child (Ch)infected intrapartum.
FIG. 6. Phylogenetic tree of HIV-1 V3 env nucleotide sequencesobtained by direct sequencing. The V3 env nucleotide sequences werecompared with the following known sequences of children infectedwith subtype B virus: C190, C204, C196, C136, C145, C115, C130, orC201. Intrapartum transmission refers to mother-child pair 196. Inutero transmission refers to mother-child pair 204. The children’ssamples were collected when the infants were 1 month old. C190 wasused as an outgroup, and FR.LAI was used as a subtype B reference.The Fitch-Margoliash tree is shown. The results were the same for alltrees in all essential aspects. For the sake of clarity, only bootstrapvalues above 60 are indicated.
VOL. 75, 2001 TROPHOBLASTIC BARRIER AND HIV-1 INFECTION 4787
on May 22, 2013 by 00308377
http://jvi.asm.org/
Dow
nloaded from

lected during both virus translocation by transcytosis acrossand after replication of maternal HIV in the trophoblast-likebarrier.
Viral variants selected during transcytosis are closely re-lated to in utero-transmitted viruses. We next analyzed the V3gp120 env sequences of the HIV-1 variants in the fractionsanalyzed in Fig. 5. The major V3 gp120 env sequence detectedin the PBMCs of the only in utero-infected child studied wasclosely related, if not identical, to the major V3 sequences oftranscytosis-derived virus. In contrast, the major V3 env se-quence detected in the fusion-derived virus was closer to thematernal PBMC-derived virus (Fig. 6). Such homology was notobserved in the case of intrapartum transmission, where themajor V3 sequences from the child were related to but distinctfrom those derived from both transcytosis and fusion.
DISCUSSION
The human syncytiotrophoblast is a highly polarized epithe-lium-like layer responsible for regulating maternal-fetal ex-changes (39). During pregnancy, the apical side of this barrieris in contact with infected maternal blood which contains bothcell-free HIV and HIV-infected cells. We have used an in vitromodel to examine the first steps that could restrict the in uterotransmission of HIV-1. This model uses trophoblast-like BeWocells that form a tight, polarized monolayer in a two-chamberculture system. The apical side of the trophoblastic barrier,when in contact with HIV-1-infected PBMCs or cell-freeHIV-1 isolated from seropositive mothers, mimics the mater-nal blood-placenta interface, whereas the basolateral side maybe considered the first compartment of the fetal side. We firstdemonstrated that cell-free viruses cannot cross the BeWomonolayer by transcytosis from the apical to the basolateralside or by target cell infection and replication. This is consis-tent with previous results showing that intestinal or endome-trial epithelial cells grown as a tight monolayer do not allowtranscytosis of cell-free virus (6, 7), as well as with studies withprimary trophoblasts or trophoblastic cells that appeared to beresistant to HIV infection (18, 19), although the cells used inthese previous studies were not polarized. The reason for thisresistance is not yet clear. The study reporting that BeWo cellscould be infected by cell-free virus showed a poor productiveinfection limited to few HIV-adapted laboratory strains (43).In the present study, primary HIV isolates were used ratherthan laboratory strains. Virus sequences were occasionally de-tected in BeWo cells after contact between cell-free virus andthe apical or basolateral surface of trophoblastic cells, but thisdetection was never coupled with a detectable production ofvirions. No viral sequence was detected in AZT-treated cells,which is consistent with a recent study showing that tropho-blasts isolated from the placentas of AZT-treated, pregnant,HIV-positive women are not infected (57). The inability ofcell-free HIV to transcytose or to infect BeWo cells cannot beascribed to factors related to cell polarity, as identical resultswere obtained when cell-free virus was applied either apicallyor basolaterally or when we attempted to infect unpolarizedBeWo cells. This contrasts with findings for other viruses thatinfect and replicate in epithelia in a polarized manner, such asvesicular stomatitis virus or canine parvovirus (infecting pre-dominantly through the basolateral membrane) (3, 27) and
measles virus or simian virus 40, which preferentially infects viathe apical membrane (5, 15).
In contrast to cell-free HIV, HIV-infected PBMCs inducedHIV translocation across the trophoblastic barrier. Contactbetween HIV-infected PBMCs and trophoblastic cells is re-quired for the release of infectious viruses into the basolateralchamber both by transcytosis and after fusion between effectorPBMCs and target trophoblastic cells. Trancytosis of infectiousHIV across the trophoblastic barrier occurs shortly after cell-to-cell contact, as expected for such a process (6). In contrast,replicating viruses were detected later in the basolateral side, 3days after effector-target cell contact and fusion of the two cellpartners.
The molecular mechanisms governing HIV binding to, entryinto (47, 62, 63), and transcytosis across trophoblastic cellsremain unclear. Some authors have suggested that entry isindependent on any cell surface molecules known to be in-volved in HIV-1 entry, including CD4 (62, 63). Supporting thistheory, CD4 is not detected on BeWo cells by immunofluores-cence assay when they are cultured as a polarized monolayer(M. Bomsel, unpublished data), even though CD4 has beendetected on fresh primary trophoblastic cells from early pla-centa (63) and to a certain extent from term placenta (43).Alternative receptors for HIV envelope glycoproteins, such asthe glycolipid galactosyl ceramide, a marker of the apical sur-face of epithelial cells, might be involved in HIV transcytosisand/or entry into trophoblastic cells. The observations thatHIV transcytosis across epithelial cells is impaired by a mono-clonal antibody specific for galactosyl ceramide (6) and thatgalactosyl ceramide can be detected at the BeWo apical pole(Bomsel, unpublished) are in favor of this possibility.
As adhesion molecules may act as a cofactor for HIV fusion(4, 45, 47, 58), they may also be involved in the fusion ofprimary infected cells with trophoblasts and/or the transmis-sion of the virus. Previous reports on the impact of adhesionmolecules (26) and of human leukocyte antigen (HLA) class I(16) and class II molecules (10) on the infectivity of HIVparticles are consistent with this hypothesis. We find that bothlymphocyte (CD31/CD142) and monocyte/macrophage(CD32/CD141) effectors infected with SI or NSI viruses un-dergo fusion with trophoblastic target cells. In contrast, notonly uninfected PBMCs but also chronically infected T-celllines do not significantly fuse with trophoblastic cells. Thus, inaddition to viral envelope glycoproteins, host cell molecules,including adhesion molecules, may participate in fusion bybeing recruited into the plasma membrane microdomain whereHIV envelope glycoproteins bind (25). Such host cell HIVfusion cofactors may be absent or inactive in the chronicallyinfected cell lines that we used. In support of this hypothesis, aprevious study showed that choriocarcinoma-derived tropho-blasts cannot be infected by chronically infected HIV-1 mono-cytic and lymphocytic cell lines with impaired adhesion capac-ity (8). However, the lack of infection by cell-free HIV suggeststhat the capacity of infected cells to fuse with and infect targetBeWo cells is mediated by host molecules distinct from thoseinvolved in virus-cell fusion.
Virus transmission resulting from cell-to-cell contact mightalso be independent of cell fusion. It might also be a fusionprocess between budding particles released from the surface ofinfected PBMCs and BeWo.
4788 LAGAYE ET AL. J. VIROL.
on May 22, 2013 by 00308377
http://jvi.asm.org/
Dow
nloaded from

Both CCR5 and CXCR4 coreceptors are detected by immu-nohistochemistry at the apical surface of BeWo monolayers(Bomsel, unpublished), as reported for early primary tropho-blasts (63). Viruses derived from cell-to-cell contact exhibitedan X4 phenotype, irrespective of the initial predominant R5 orX4 phenotype of the virus propagated in effector PBMCs. Thismay be due to either a preferential fusion of X4-infectedPBMCs with the BeWo monolayer or by a preferential repli-cation of X4 viruses after fusion.
As expected, R5 viruses are preferentially selected inCD141/CD32 cells derived from X4-infected PBMCs (Table4). However, the fusion of these cells with BeWo cells resultedin the release of X4 virus. These data indicate that, despite thepreferential tropism of R5 viruses for CD141/CD32 cells, X4minor variants are preferentially selected after cell-to-cell con-tact and fusion of the two cell types. These results furthersupport the hypothesis proposed above of either a preferentialfusion of X4-infected cells with BeWo cells or a preferentialreplication of X4 viruses in target cells. Finally, our results alsodemonstrated that both fusion of R5-infected cells (Fig. 3) andreplication of R5 viruses (Table 4) are not restricted in BeWocells.
Previous studies have shown that maternal HIV-1 variantsare selected during mother-to-child infection (1, 48, 52). Thedata presented here suggest that a selection may first occur attwo levels: during transcytosis and after trophoblast fusion withthe effector cells resulting in productive infection. In the onlycase of in utero transmission that we studied up to now, thepredominant transcytosed viral variant detected was geneti-
cally identical to the predominant env variant found in theinfected child. This suggests that transcytosis may be partlyinvolved in controlling a complex series of events which mayrestrict the transmission of HIV in utero. However, furtherinvestigations of additional cases of in utero and intrapartumtransmissions are needed to confirm this hypothesis.
In summary, we presented several lines of evidence that ourreconstitution system of a trophoblast barrier in vitro is asuitable model for dissecting the first steps leading to a re-stricted passage of HIV-1 in utero. The trophoblast barrier isnot permeable to cell-free virus, but the contact between thisbarrier and infected cells results in the transcytosis of infec-tious virus and in the fusion between effector and target cells.Infected cells, either T cells or monocytes, fuse efficiently withthe trophoblastic barrier and lead to the release of infectiousvirus into the basolateral side. The fusion process clearly in-duces a selection of viral variants, as reported in studies on inutero transmission in vivo (41). In addition, in vitro transcytosisof infectious virus may be relevant to the processes leading toin utero transmission of HIV.
Our genetic and phenotypic analysis of viruses crossing thetrophoblast barrier by transcytosis or replication after fusionsuggests that the selection of maternal viruses within the pla-centa may indeed involve multiple steps. The initial selectionwithin trophoblastic cells is likely to be regulated by factors inthe placental microenvironment, such as cytokines, chemo-kines, hormones, and maternal antibodies (as illustrated in Fig.7). Other placental cells, such as macrophages (Hofbauercells), which are present underneath the trophoblast in vivo,
FIG. 7. Model to approach the first steps of in utero transmission of HIV. Step 1: HIV-1-infected mononuclear effector cells representative ofmaternal blood cells are in contact with the apical side of the syncytiotrophoblast. This contact can initiate two events (steps 2 to 3 or steps 4 to5). The first one is a fusion of infected effector target cells (step 2a) or a fusion between virions freshly released from infected effector cells (step2b) and syncytiotrophoblast target cells, resulting in both cases in a productive infection (step 3), with viral progeny released into the basolateralfetal side (step 6). The second event is transcytosis of the virus (steps 4 and 5) and release of infectious virus into the fetal side (step 6). Maternalviruses translocated to the fetal side by either pathway (step 2 or 4) undergo a first series of selection followed by a further selective process (step7) involving other placental cells, such as macrophages (Hofbauer cells), monocytes, or CD41 T cells in the fetal side. Such selective forces maylead to the emergence of viral species that may or may not be transmissible to the fetus. Hormones, chemokines, and cytokines, in the placentalmicroenvironment, could also regulate the two translocation pathways of cell-associated virus (step 8). In contrast, no translocation of infectiouscell-free virus across the trophoblast barrier is detectable (step 9). However, infectious cell-free virus complexed with maternal specific IgG in thematernal blood should also be considered for a potential IgG receptor-mediated transcytosis pathway to reach the fetal side of the trophoblast layer(step 10).
VOL. 75, 2001 TROPHOBLASTIC BARRIER AND HIV-1 INFECTION 4789
on May 22, 2013 by 00308377
http://jvi.asm.org/
Dow
nloaded from

may also play a role in the selective process. Our in vitro modelis a very useful tool to approach the selective forces within theplacenta that lead to control of HIV-1 transmission in utero.
ACKNOWLEDGMENTS
We thank all of the medical and counseling staff at the CentralHospital and at the Child Welfare Center in Yaounde, Cameroon, andat the First Departments of Gynecology and Pediatrics, University ofMilan, Milan, Italy, for their dedicated cooperation. We also thank thewomen who participated in this study, as well as the members of thePasteur Center in Yaounde for their technical assistance. We espe-cially thank Emmanuel Tina for his considerable technical help. Wethank D. R. Littman (Skirball Institute of Biomolecular Medicine andHoward Hughes Medical Institute, New York University School Uni-versity of Medicine, New York, N.Y.) for providing the human gliomacell line U87.CD4 expressing the chemokine receptors R5 and X4. Wealso thank M. C. Muller-Trutwin for advice on phylogenetic sequenceanalysis and P. Versmisse for his technical assistance. We are verygrateful to Robert Bassin for his critical reading of the manuscript.
This work was supported by the French National Agency for AIDSResearch (ANRS), the European Program Biomed 2 (BMH4-CT96–1509) on the study of in utero transmission of HIV-1, and the InstitutoSuperior di Sanita (ISS). E.M. was a recipient of a SIDAction fellow-ship; C.C. and M.D. were recipients of an ANRS fellowship.
REFERENCES
1. Ahmad, N., B. M. Baroudy, R. C. Baker, and C. Chappey. 1995. Geneticanalysis of human immunodeficiency virus type 1 envelope V3 region isolatesfrom mothers and infants after perinatal transmission. J. Virol. 69:1001–1012.
2. Amirhessami-Aghili, N., and S. A. Spector. 1991. Human immunodeficiencyvirus type 1 infection of human placenta: potential route for fetal infection.J. Virol. 65:2231–2236.
3. Basak, S., and R. W. Compans. 1989. Polarized entry of canine parvovirus inan epithelial cell line. J. Virol. 63:3164–3167.
4. Berman, P. W., and G. R. Nakamura. 1994. Adhesion mediated by intercel-lular adhesion molecule 1 attenuates the potency of antibodies that blockHIV-1 gp160-dependent syncytium formation. AIDS Res. Hum. Retrovir.10:585–593.
5. Blau, D. M., and R. W. Compans. 1995. Entry and release of measles virusare polarized in epithelial cells. Virology 210:91–99.
6. Bomsel, M. 1997. Transcytosis of infectious human immunodeficiency virusacross a tight human epithelial cell line barrier. Nat. Med. 3:42–47.
7. Bomsel, M., M. Heyman, H. Hocini, S. Lagaye, L. Belec, C. Dupont, and C.Desgranges. 1998. Intracellular neutralization of HIV transcytosis acrosstight epithelial barriers by anti-HIV envelope protein dIgA or IgM. Immu-nity 9:277–287.
8. Bourinbaiar, A. S., and R. Nagorny. 1993. Human immunodeficiency virustype-1 infection of choriocarcinoma-derived trophoblasts. Acta Virol. 37:21–28.
9. Burton, G. J., S. O’Shea, T. Rostron, J. E. Mullen, S. Aiyer, J. N. Skepper,R. Smith, and J. Banatvala. 1996. Physical breaks in the placental tropho-blastic surface: significance in vertical transmission of HIV. AIDS 10:1294–1296.
10. Cantin, R., J. F. Fortin, G. Lamontagne, and M. Tremblay. 1997. Theacquisition of host-derived major histocompatibility complex class II glyco-proteins by human immunodeficiency virus type 1 accelerates the process ofvirus entry and infection in human T-lymphoid cells. Blood 90:1091–1100.
11. Caplan, M. J., H. C. Anderson, G. E. Palade, and J. D. Jamieson. 1986.Intracellular sorting and polarized cell surface delivery of (Na1,K1)ATPase, an endogenous component of MDCK cell basolateral plasma mem-branes. Cell 46:623–631.
12. Cerneus, D. P., and A. van der Ende. 1991. Apical and basolateral transferrinreceptors in polarized BeWo cells recycle through separate endosomes.J. Cell Biol. 114:1149–1158.
13. Cerneus, D. P., G. J. Strous, and A. van der Ende. 1993. Bidirectionaltranscytosis determines the steady state distribution of the transferrin recep-tor at opposite plasma membrane domains of BeWo cells. J. Cell Biol.122:1223–1230.
14. Chouquet, C., M. Burgard, S. Richardson, C. Rouzioux, and D. Costagliola.1997. Timing of mother-to-child HIV-1 transmission and diagnosis of infec-tion based on polymerase chain reaction in the neonatal period by a non-parametric method. AIDS 11:1183–1199.
15. Clayson, E. T., and R. W. Compans. 1988. Entry of simian virus 40 isrestricted to apical surfaces of polarized epithelial cells. Mol. Cell. Biol.8:3391–3396.
16. Cosma, A., D. Blanc, J. Braun, C. Quillent, C. Barassi, C. Moog, S. Klasen,
B. Spire, G. Scarlatti, E. Pesenti, A. G. Siccardi, and A. Beretta. 1999.Enhanced HIV infectivity and changes in GP120 conformation associatedwith viral incorporation of human leucocyte antigen class I molecules. AIDS13:2033–2042.
17. Courcoul, M., C. Patience, F. Rey, D. Blanc, A. Harmache, J. Sire, R. Vigne,and B. Spire. 1995. Peripheral blood mononuclear cells produce normalamounts of defective Vif2 human immunodeficiency virus type 1 particleswhich are restricted for the preretrotranscription steps. J. Virol. 69:2068–2074.
18. David, F. J., B. Autran, H. C. Tran, E. Menu, M. Raphael, P. Debre, B. L.Hsi, T. G. Wegmann, F. Barre-Sinoussi, and G. Chaouat. 1992. Humantrophoblast cells express CD4 and are permissive for productive infectionwith HIV-1. Clin. Exp. Immunol. 88:10–16.
19. David, F. J., H. C. Tran, N. Serpente, B. Autran, C. Vaquero, V. Djian, E.Menu, F. Barre-Sinoussi, and G. Chaouat. 1995. HIV infection of chorio-carcinoma cell lines derived from human placenta: the role of membraneCD4 and Fc-Rs into HIV entry. Virology 208:784–788.
20. De Andreis, C., G. Simoni, F. Rossella, C. Castagna, E. Pesenti, G. Porta, G.Colucci, S. Giuntelli, G. Pardi, and A. E. Semprini. 1996. HIV-1 proviralDNA polymerase chain reaction detection in chorionic villi after exclusion ofmaternal contamination by variable number of tandem repeats analysis.AIDS 10:711–715.
21. Delwart, E. L., E. G. Shpaer, J. Louwagie, F. E. McCutchan, M. Grez, H.Rubsamen-Waigmann, and J. I. Mullins. 1993. Genetic relationships deter-mined by a DNA heteroduplex mobility assay: analysis of HIV-1 env genes.Science 262:1257–1261.
22. Deng, H., R. Liu, W. Ellmeier, S. Choe, D. Unutmaz, M. Burkhart, P. DiMarzio, S. Marmon., R. E. Sutton, M. C. Hill, C. B. Davis, S. C. Peiper, T. J.Schall, D. R. Littman, and N. R. Landau. 1996. Identification of a majorco-receptor for primary isolates of HIV-1. Nature 381:661–666.
23. Deng, H. K., D. Unutmaz, V. N. Kewalramani, and D. R. Littman. 1997.Expression cloning of new receptors used by simian and human immunode-ficiency viruses. Nature 388:296–300.
24. Ellinger, I., M. Schwab, A. Stefanescu, W. Hunziker, and R. Fuchs. 1999.IgG transport across trophoblast-derived BeWo cells: a model system tostudy IgG transport in the placenta. Eur. J. Immunol. 29:733–744.
25. Fais, S., M. R. Capobianchi, I. Abbate, C. Castilletti, M. Gentile, P. CordialiFei, F. Ameglio, and F. Dianzani. 1995. Unidirectional budding of HIV-1 atthe site of cell-to-cell contact is associated with co-polarization of intercel-lular adhesion molecules and HIV-1 viral matrix protein. AIDS 9:329–335.
26. Fortin, J. F., R. Cantin, G. Lamontagne, and M. Tremblay. 1997. Host-derived ICAM-1 glycoproteins incorporated on human immunodeficiencyvirus type 1 are biologically active and enhance viral infectivity. J. Virol.71:3588–3596.
27. Fuller, S., C. H. von Bonsdorff, and K. Simons. 1984. Vesicular stomatitisvirus infects and matures only through the basolateral surface of the polar-ized epithelial cell line, MDCK. Cell 38:65–77.
28. Halwachs-Baumann, G., M. Wilders-Truschnig, G. Desoye, T. Hahn, L.Kiesel, K. Klingel, G. Jahn, and C. Sinzger. 1998. Human trophoblast cellsare permissive to the complete replicative cycle of human cytomegalovirus.J. Virol. 72:7598–7602.
29. Hamad, A. R., P. Marrack, and J. W. Kappler. 1997. Transcytosis of staph-ylococcal superantigen toxins. J. Exp. Med. 185:1447–1454.
30. Harada, S., Y. Koyanagi, and N. Yamamoto. 1985. Infection of HTLV-III/LAV in HTLV-I-carrying cells MT-2 and MT-4 and application in a plaqueassay. Science 229:563–566.
31. Jaroszeski, M. J., R. Gilbert, P. G. Fallon, and R. Heller. 1994. Mechanicallyfacilitated cell-cell electrofusion. Biophys. J. 67:1574–1581.
32. Kilani, R. T., L. J. Chang, M. I. Garcia-Lloret, D. Hemmings, B. Winkler-Lowen, and L. J. Guilbert. 1997. Placental trophoblasts resist infection bymultiple human immunodeficiency virus (HIV) type 1 variants even withcytomegalovirus coinfection but support HIV replication after provirustransfection. J. Virol. 71:6359–6372.
33. Kind, C., C. Rudin, C. A. Siegrist, C. A. Wyler, K. Biedermann, U. Lauper,O. Irion, J. Schupbach, and D. Nadal. 1998. Prevention of vertical HIVtransmission: additive protective effect of elective cesarean section andzidovudine prophylaxis. AIDS 12:205–210.
34. Lansky, A., J. L. Jones, P. C. Wan, M. L. Lindegren, and P. Wortley. 1998.Trends in zidovudine prescription for pregnant women infected with HIV. J.Acquir. Immune Defic. Syndr. Hum. Retrovirol. 18:289–292.
35. Lapointe, N., J. Michaud, D. Pekovic, J. P. Chausseau, and J. M. Dupuy.1985. Transplacental transmission of HTLV-III virus. N. Engl. J. Med. 312:1325–1326.
36. Leach, J. L., D. D. Sedmak, J. M. Osborne, B. Rahill, M. D. Lairmore, andC. L. Anderson. 1996. Isolation from human placenta of the IgG transporter,FcRn, and localization to the syncytiotrophoblast: implications for maternal-fetal antibody transport. J. Immunol. 157:3317–3322.
37. Lewis, S. H., C. Reynolds-Kohler, H. E. Fox, and J. A. Nelson. 1990. HIV-1in trophoblastic and villous Hofbauer cells, and haematological precursors ineight-week fetuses. Lancet 335:565–568.
38. Liu, X., V. Zachar, H. Hager, U. Koppelhus, and P. Ebbesen. 1996. Transfer
4790 LAGAYE ET AL. J. VIROL.
on May 22, 2013 by 00308377
http://jvi.asm.org/
Dow
nloaded from

of human T cell lymphotropic virus type I to human term trophoblast cells invitro. J. Gen. Virol. 77:369–374.
39. Loke, Y. W., and A. King. 1996. Human implantation: cell biology andimmunology. Cambridge University Press, Cambridge, England.
40. Mano, H., and J. C. Chermann. 1991. Fetal human immunodeficiency virustype 1 infection of different organs in the second trimester. AIDS Res. Hum.Retrovir. 7:83–88.
41. Menu, E., F. X. Mbopi Keou, S. Lagaye, S. Pissard, P. Mauclere, G. Scar-latti, J. Martin, M. Goossens, G. F. Chaouat, F. Barre-Sinoussi, and theEuropean Network for in utero transmission of HIV-1. 1999. Selection ofmaternal HIV-1 variants in the human placenta. J. Infect. Dis. 179:44–51.
42. Miyoshi, I., I. Kubonishi, S. Yoshimoto, T. Akagi, Y. Ohtsuki, Y. Shiraishi,K. Nagata, and Y. Hinuma. 1981. Type C virus particules in a cord T-cell linederived by co-cultivating normal human cord leukocytes and leukaemic Tcells. Nature 294:770–771.
43. Mognetti, B., M. Moussa, J. Croitoru, M. Menu, D. Dormont, P. Roques,and G. Chaouat. 2000. HIV-1 co-receptor expression on trophoblastic cellsfrom early placentas and permissivity to infection by several HIV-1 primaryisolates. Clin. Exp. Immunol. 119:486–492.
44. Mostov, K. E., and N. E. Simister. 1985. Transcytosis. Cell 43:389–390.45. Pantaleo, G., G. Poli, L. Butini, C. Fox, A. I. Dayton, and A. S. Fauci. 1991.
Dissociation between syncytia formation and HIV spreading. Suppression ofsyncytia formation does not necessarily reflect inhibition of HIV infection.Eur. J. Immunol. 21:1771–1774.
46. Pattillo, R. A., and G. O. Gey. 1968. The establishment of a cell line of humanhormone-synthesizing trophoblastic cells in vitro. Cancer Res. 28:1231–1236.
47. Phillips, D. M., and X. Tan. 1992. HIV-1 infection of the trophoblast cell lineBeWo: a study of virus uptake. AIDS Res. Hum. Retrovir. 8:1683–1691.
48. Roth, W. W., J. A. Zuberi, H. G. Stringer, S. K. Davidson, and V. C. Bond.1996. Examination of HIV type 1 variants in mother-child pairs. AIDS Res.Hum. Retrovir. 12:925–930.
49. Scarlatti, G., J. Albert, P. Rossi, V. Hodara, P. Biraghi, L. Muggiasca, andE. M. Fenyo. 1993. Mother-to-child transmision of human immunodeficiencyvirus type1: correlation with neutralizing antibodies against primary isolates.J. Infect. Dis. 168:207–210.
50. Scarlatti, G., E. Tresoldi, A. Bjorndal, R. Fredriksson, C. Colognesi, H. K.Den, M. S. Malnati, A. Plebani, A. G. Siccardi, D. R. Littman, E. M. Fenyo,and P. Lusso. 1997. In vivo evolution of HIV-1 co-receptor usage andsensitivity to chemokine-mediated suppression. Nat. Med. 3:1259–1265.
51. Scarlatti, G., V. Hodara, P. Rossi, L. Muggiasca, A. Bucceri, J. Albert, andE. M. Fenyo. 1993. Transmission of human immunodeficiency virus type1(HIV-1) from mother to child correlates with viral phenotype. Virology197:624–629.
52. Scarlatti, G., T. Leitner, E. Halapi, J. Wahlberg, P. Marchisio, M. A. Clerici-Schoeller, H. Wigzell, E. M. Fenyo, J. Albert, M. Uhlen, and P. Rossi. 1993.Comparison of variable region 3 sequences of human immunodeficiencyvirus type-1 from infected children with the RNA and DNA sequences of thevirus populations of their mothers. Proc. Natl. Acad. Sci. USA 90:1721–1725.
53. Sperling, R. S., D. E. Shapiro, R. W. Coombs, J. A. Todd, S. A. Herman,
G. D. McSherry, M. J. O’Sullivan, R. B. Van Dyke, E. Jimenez, C. Rouzioux,P. M. Flynn, and J. L. Sullivan. 1996. Maternal viral load, zidovudinetreatment, and the risk of transmission of human immunodeficiency virustype 1 from mother to infant. N. Engl. J. Med. 335:1621–1629.
54. Sprecher, S, G. Soumenkoff, F. Puissant, and M. Degueldre. 1986. Verticaltransmission of HIV in 15-week fetus. Lancet ii:288–289.
55. Toth, F. D., G. Aboagye-Mathiesen, J. Szabo, X. Liu, P. Mosborg-Petersen,J. Kiss, H. Hager, M. Zdravkovic, I. Andirko, J. Aranyosi, and P. Ebbesen.1995. Bidirectional enhancing activities between human T cell leukemia-lymphoma virus type I and human cytomegalovirus in human term syncy-tiotrophoblast cells cultured in vitro. AIDS Res. Hum. Retrovir. 11:1495–1507.
56. Toth, F. D., P. Mosborg-Petersen, J. Kiss, G. Aboagye-Mathiesen, H. Hager,C. B. Juhl, L. Gergely, M. Zdravkovic, J. Aranyosi, L. Lampe, and P.Ebbesen. 1995. Interactions between human immunodeficiency virus type 1and human cytomegalovirus in human term syncytiotrophoblast cells coin-fected with both viruses. J. Virol. 69:2223–2232.
57. Tscherning-Casper, C., N. Papadogiannakis, M. Anvret, L. Stolpe, S.Lindgren, A. Britt Bohlin, J. Albert, and E. M. Fenyo. 1999. The tropho-blastic epithelial barrier is not infected in full-term placentae of humanimmunodeficiency virus-seropositive mothers undergoing antiretroviral ther-apy. J. Virol. 73:9673–9678.
58. Valentin, A., K. Lundin, M. Patarroyo, and B. Asjo. 1990. The leukocyteadhesion glycoprotein CD18 participates in HIV-1-induced syncytia forma-tion in monocytoid and T cells. J. Immunol. 144:934–937.
59. Valentin, H., M. T. Nugeyre, F. Vuillier, L. Boumsell, M. Schmid, F. Barre-Sinoussi, and R. Pereira. 1994. Two subpopulations of human triple-negativethymic cells are susceptible to infection by human immunodeficiency virustype 1 in vitro. J. Virol. 68:3041–3050.
60. Van Amersfoort, E. S., and J. A. Van Strijp. 1994. Evaluation of a flowcytometric fluorescence quenching assay of phagocytosis of sensitized sheeperythrocytes by polymorphonuclear leukocytes. Cytometry 17:294–301.
61. Wolinsky, S. M., C. M. Wike, B. T. Korber, C. Hutto, W. P. Parks, L. L.Rosenblum, K. J. Kunstman, M. R. Furtado, and J. L. Munoz. 1992. Selec-tive transmission of human immunodeficiency virus type-1 variants frommothers to infants. Science 255:1134–1137.
62. Zachar, V., B. Spire, I. Hirsch, J. C. Chermann, and P. Ebbesen. 1991.Human transformed trophoblast-derived cells lacking CD4 receptor exhibitrestricted permissiveness for human immunodeficiency virus type 1. J. Virol.65:2102–2107.
63. Zachar, V., N. Norskov-Lauristen, C. Juhl, B. Spire, J. C. Chermann, and P.Ebbesen. 1991. Susceptibility of cultured human trophoblasts cells to infec-tion with human immunodeficiency virus type 1. J. Gen. Virol. 72:1253–1260.
64. Zachar, V., V. Zacharova, T. Fink, R. A. Thomas, B. R. King, P. Ebbesen,T. B. Jones, and A. S. Goustin. 1999. Genetic analysis reveals ongoing HIVtype 1 evolution in infected human placental trophoblast. AIDS Res. Hum.Retrovir. 15:1673–1683.
VOL. 75, 2001 TROPHOBLASTIC BARRIER AND HIV-1 INFECTION 4791
on May 22, 2013 by 00308377
http://jvi.asm.org/
Dow
nloaded from




















