
CCR5 and CXCR3 Are Dispensable for Liver Infiltration, but CCR5 Protects against Virus-Induced...
12
JOURNAL OF VIROLOGY, Sept. 2007, p. 10101–10112 Vol. 81, No. 18 0022-538X/07/$08.000 doi:10.1128/JVI.01242-07 Copyright © 2007, American Society for Microbiology. All Rights Reserved. CCR5 and CXCR3 Are Dispensable for Liver Infiltration, but CCR5 Protects against Virus-Induced T-Cell-Mediated Hepatic Steatosis P. J. Holst, 1 C. Orskov, 2 K. Qvortrup, 2 J. P. Christensen, 1 and A. R. Thomsen 1 * Institute of Medical Microbiology and Immunology, 1 and Institute of Medical Anatomy, 2 University of Copenhagen, the Panum Institute, 3C Blegdamsvej, DK-2200 N, Copenhagen, Denmark Received 7 June 2007/Accepted 29 June 2007 CCR5 and CXCR3 are important molecules in regulating the migration of activated lymphocytes. Thus, the majority of tissue-infiltrating T cells found in the context of autoimmune conditions and viral infections express CCR5 and CXCR3, and the principal chemokine ligands are expressed within inflamed tissues. Accordingly, intervention studies have pointed to nonredundant roles of these receptors in models of allograft rejection, viral infection, and autoimmunity. In spite of this, considerable controversy exists, with many studies failing to support a role for CCR5 or CXCR3 in disease pathogenesis. One possible explanation is that different chemokine receptors may take over in the absence of any individual receptor, thus rendering individual receptors redundant. We have attempted to address this issue by analyzing CCR5 / , CXCR3 / , and CCR5/CXCR3 / mice with regard to virus-induced liver inflammation, generation and recruitment of effector cells, virus control, and immunopathology. Our results indicate that CCR5 and CXCR3 are largely dispensable for tissue infiltration and virus control. In contrast, the T-cell response is accelerated in CCR5 / and CCR5/CXCR3 / mice and the absence of CCR5 is associated with the induction of CD8 T-cell-mediated immunopathology consisting of marked hepatic microvesicular steatosis. Chemokines play a key role in virus-induced inflammation and virus clearance, acting as regulators of leukocyte trafficking (18, 49). Among the chemokine receptors, CCR5 has drawn considerable attention as the main coreceptor for human im- munodeficiency virus and CXCR3 is a promising target for antiinflammatory therapeutics (41, 52). Notably, both recep- tors have been associated with type 1 cytokine T-cell responses and the majority of tissue-infiltrating T cells in several human inflammatory diseases are found to coexpress these receptors (7, 42, 48). During viral infection, CCR5 and CXCR3 are expressed on NK cells and activated CD4 and CD8 T cells and the ligands are generally expressed in virus-infected tissues (30, 38, 50). Further support for the important role of CCR5 and CXCR3 comes from the existence of virally encoded che- mokine receptor antagonists targeting these receptors (23, 27). In this context, it is not surprising that CXCR3 or its ligands have been experimentally demonstrated to be important for immune-mediated virus control in the central nervous system (CNS), the lungs, the ovaries, and the islet of Langerhans in the pancreas (9, 15, 20, 26, 51). In contrast, despite the sug- gestive expression patterns, several studies have indicated that CCR5 or CCR5 ligands are less important in antiviral immu- nity (30, 37). The positive findings suggest a role for CCR5 in macrophage and CD4 T-cell recruitment, but the receptor appears not to be required for CD8 T-cell migration except in young mice, in which the immune system has not yet reached full maturity (3, 17). The reason for this difference between the two receptors could lie in a difference in biological redundancy. Thus, several CCR5 ligands can be expressed simultaneously and these can also signal through CCR1, which can be ex- pressed on CD8 T cells. On the other hand, CXCR3 is the only lymphocyte receptor for CXCL9 through CXCL11 and this may explain why the requirement for this receptor is less easily circumvented (31, 36). Another factor clouding the interpretation of chemokine function in vivo remains organ-specific effects (8). Several re- ports have presented convincing, yet different conclusions re- garding the requirement for these receptors in liver inflamma- tion. For example, during chronic infections of hepatitis C, a disease entity affecting more than 200 million people world- wide (43), CCR5, CXCR3, and CXCR6 are expressed on liver- infiltrating lymphocytes (LILs) and the expression correlates with disease severity (53). Yet, contrary to expectations, the CCR532 human chemokine defect is associated with sponta- neous hepatitis C clearance and lower inflammatory scores (19). In contrast, the CCR532 polymorphism or genetically engineered deficiency of CCR5 results in accelerated graft- versus-host disease in mice and reduced graft-versus-host dis- ease in humans. Notably, the data are not unequivocal and the effect seems to depend on the regimen of pretransplant con- ditioning (35, 54, 55). The strongest evidence for a role of CCR5 in hepatic immunity in humans comes from the discov- ery that homozygosity for the CCR532 chemokine defect is associated with primary sclerosing cholangitis and an increased risk of ischemic-type billiary lesion following liver transplanta- tion (13, 33). Additionally, the CCR5 ligand CCL3 has been suggested to play an essential role in virus control through the induction of gamma interferon (IFN-) and the production of CXCR3 ligand during acute murine cytomegalovirus (MCMV) infection (45, 46). We have recently taken the rational approach and attempted to unravel redundancy within the chemokine system by gener- * Corresponding author. Mailing address: Institute of Medical Micro- biology and Immunology, University of Copenhagen, The Panum Insti- tute, building 22.5.16, 3C Blegdamsvej, DK-2200 Copenhagen N, Den- mark. Phone: 45-35-32-78-71. Fax: 45-35-32-78-74. E-mail: a.r.thomsen @immi.ku.dk. Published ahead of print on 11 July 2007. 10101
Transcript of CCR5 and CXCR3 Are Dispensable for Liver Infiltration, but CCR5 Protects against Virus-Induced...

JOURNAL OF VIROLOGY, Sept. 2007, p. 10101–10112 Vol. 81, No. 180022-538X/07/$08.00�0 doi:10.1128/JVI.01242-07Copyright © 2007, American Society for Microbiology. All Rights Reserved.
CCR5 and CXCR3 Are Dispensable for Liver Infiltration, but CCR5Protects against Virus-Induced T-Cell-Mediated Hepatic Steatosis�
P. J. Holst,1 C. Orskov,2 K. Qvortrup,2 J. P. Christensen,1 and A. R. Thomsen1*Institute of Medical Microbiology and Immunology,1 and Institute of Medical Anatomy,2 University of Copenhagen,
the Panum Institute, 3C Blegdamsvej, DK-2200 N, Copenhagen, Denmark
Received 7 June 2007/Accepted 29 June 2007
CCR5 and CXCR3 are important molecules in regulating the migration of activated lymphocytes. Thus, themajority of tissue-infiltrating T cells found in the context of autoimmune conditions and viral infectionsexpress CCR5 and CXCR3, and the principal chemokine ligands are expressed within inflamed tissues.Accordingly, intervention studies have pointed to nonredundant roles of these receptors in models of allograftrejection, viral infection, and autoimmunity. In spite of this, considerable controversy exists, with many studiesfailing to support a role for CCR5 or CXCR3 in disease pathogenesis. One possible explanation is that differentchemokine receptors may take over in the absence of any individual receptor, thus rendering individualreceptors redundant. We have attempted to address this issue by analyzing CCR5�/�, CXCR3�/�, andCCR5/CXCR3�/� mice with regard to virus-induced liver inflammation, generation and recruitment of effectorcells, virus control, and immunopathology. Our results indicate that CCR5 and CXCR3 are largely dispensablefor tissue infiltration and virus control. In contrast, the T-cell response is accelerated in CCR5�/� andCCR5/CXCR3�/� mice and the absence of CCR5 is associated with the induction of CD8� T-cell-mediatedimmunopathology consisting of marked hepatic microvesicular steatosis.
Chemokines play a key role in virus-induced inflammationand virus clearance, acting as regulators of leukocyte trafficking(18, 49). Among the chemokine receptors, CCR5 has drawnconsiderable attention as the main coreceptor for human im-munodeficiency virus and CXCR3 is a promising target forantiinflammatory therapeutics (41, 52). Notably, both recep-tors have been associated with type 1 cytokine T-cell responsesand the majority of tissue-infiltrating T cells in several humaninflammatory diseases are found to coexpress these receptors(7, 42, 48). During viral infection, CCR5 and CXCR3 areexpressed on NK cells and activated CD4� and CD8� T cellsand the ligands are generally expressed in virus-infected tissues(30, 38, 50). Further support for the important role of CCR5and CXCR3 comes from the existence of virally encoded che-mokine receptor antagonists targeting these receptors (23, 27).In this context, it is not surprising that CXCR3 or its ligandshave been experimentally demonstrated to be important forimmune-mediated virus control in the central nervous system(CNS), the lungs, the ovaries, and the islet of Langerhans inthe pancreas (9, 15, 20, 26, 51). In contrast, despite the sug-gestive expression patterns, several studies have indicated thatCCR5 or CCR5 ligands are less important in antiviral immu-nity (30, 37). The positive findings suggest a role for CCR5 inmacrophage and CD4� T-cell recruitment, but the receptorappears not to be required for CD8� T-cell migration except inyoung mice, in which the immune system has not yet reachedfull maturity (3, 17). The reason for this difference between thetwo receptors could lie in a difference in biological redundancy.
Thus, several CCR5 ligands can be expressed simultaneouslyand these can also signal through CCR1, which can be ex-pressed on CD8� T cells. On the other hand, CXCR3 is theonly lymphocyte receptor for CXCL9 through CXCL11 andthis may explain why the requirement for this receptor is lesseasily circumvented (31, 36).
Another factor clouding the interpretation of chemokinefunction in vivo remains organ-specific effects (8). Several re-ports have presented convincing, yet different conclusions re-garding the requirement for these receptors in liver inflamma-tion. For example, during chronic infections of hepatitis C, adisease entity affecting more than 200 million people world-wide (43), CCR5, CXCR3, and CXCR6 are expressed on liver-infiltrating lymphocytes (LILs) and the expression correlateswith disease severity (53). Yet, contrary to expectations, theCCR5�32 human chemokine defect is associated with sponta-neous hepatitis C clearance and lower inflammatory scores(19). In contrast, the CCR5�32 polymorphism or geneticallyengineered deficiency of CCR5 results in accelerated graft-versus-host disease in mice and reduced graft-versus-host dis-ease in humans. Notably, the data are not unequivocal and theeffect seems to depend on the regimen of pretransplant con-ditioning (35, 54, 55). The strongest evidence for a role ofCCR5 in hepatic immunity in humans comes from the discov-ery that homozygosity for the CCR5�32 chemokine defect isassociated with primary sclerosing cholangitis and an increasedrisk of ischemic-type billiary lesion following liver transplanta-tion (13, 33). Additionally, the CCR5 ligand CCL3 has beensuggested to play an essential role in virus control through theinduction of gamma interferon (IFN-�) and the production ofCXCR3 ligand during acute murine cytomegalovirus (MCMV)infection (45, 46).
We have recently taken the rational approach and attemptedto unravel redundancy within the chemokine system by gener-
* Corresponding author. Mailing address: Institute of Medical Micro-biology and Immunology, University of Copenhagen, The Panum Insti-tute, building 22.5.16, 3C Blegdamsvej, DK-2200 Copenhagen N, Den-mark. Phone: 45-35-32-78-71. Fax: 45-35-32-78-74. E-mail: [email protected].
� Published ahead of print on 11 July 2007.
10101

ating CCR5/CXCR3 double-deficient (CCR5/CXCR3�/�)mice. In a prior study, we infected these mice intracerebrallywith the model virus, lymphocytic choriomeningitis virus(LCMV), and in that case, we found opposing actions of CCR5and CXCR3 in relation to T-cell-mediated inflammation in theCNS (10). We have now taken the system further to evaluatechemokine receptor requirements for liver inflammation fol-lowing intraperitoneal (i.p.) infection with a rapidly invasivestrain of LCMV. This model system is characterized by dra-matic expansion of virus-specific cytotoxic T lymphocytes thatmediate nearly all virus-associated pathology as well as viruscontrol during the acute phase of infection. Surprisingly, wefind that both chemokine receptors are redundant for the elim-ination of virus infection in the liver but not for the regulationof virus-induced immunopathology.
MATERIALS AND METHODS
Mice. The generation of CXCR3-deficient (CXCR3�/�) mice has been de-scribed before (21). The animals used in these experiments were the progeny ofbreeder pairs kept at the Panum Institute, University of Copenhagen. CCR5-deficient (CCR5�/�) mice (B6; 129P-CmKbr5�tm/Kn2�) were bred locallyfrom breeder pairs obtained from the Jackson Laboratory (Bar Harbor, ME).CCR5/CXCR3�/� mice were produced as recently described (10). Wild-type(WT) C57BL/6 mice were purchased from Taconic M & B (Ry, Denmark). Micefrom outside sources were always allowed to rest for at least a week before beingentered into experiments; by that time, the animals were about 7 to 9 weeks old.Animals were housed under controlled (specific-pathogen-free) conditions, andexperiments were conducted according to national guidelines.
Virus infection. Mice were infected i.p. with a virus dose of 105 PFU of LCMVclone 13 in a volume of 0.3 ml.
Organ virus titers. To determine virus titers in organs, the organs were firsthomogenized in phosphate-buffered saline (PBS) to yield 10% (vol/wt) organsuspensions and then serial 10-fold dilutions were prepared. Each dilution wasthen plated in duplicate onto MC57G cells. Forty-eight hours after infection,infected cell clusters were detected using monoclonal rat anti-LCMV (VL-4)antibody, peroxidase-labeled goat anti-rat antibody, and o-phenylenediamine(substrate) (6). The numbers of PFU were counted, and results were expressedas PFU per gram of tissue.
Splenocyte preparation. Single cell suspensions of splenocytes were obtainedby pressing the organs through a fine steel mesh, and cells were counted in ahemocytometer.
Preparation of LILs. The protocol for liver leukocyte isolation was modifiedfrom that of Liu et al. (28). Mice were sacrificed by careful cervical dislocation.The abdomen was opened, and the portal vein was cut to create an outlet. Thecaval vein was then cannulated and perfused with 5 to 10 ml of PBS. Perfusedlivers were homogenized in 20 ml of Hank’s balanced salt solution (HBSS) bypassing the tissue through a fine steel mesh, and cells were pelleted by centrif-ugation at 500 � g. Pellets were resuspended by vortexing in gradient buffer (92.5ml Percoll, 3.6 ml 20� PBS, 750 ml of 50 ku/�l of heparin, 2 ml of 4.16%NaHCO3, and 152 ml of HBSS for each 250 ml). Leukocytes were then pelletedby 20 min of centrifugation at 800 � g, and the pellets were resuspended in 0.83%NH4Cl. Cell suspensions were incubated for 10 min to allow erythrocytes to lyse,and cells were washed twice in HBSS, counted in a hemocytometer, and sub-jected to fluorescence-activated cell sorter analysis.
Antibodies and dextramers for flow cytometry. The following monoclonal an-tibodies were purchased from BD Pharmingen (San Diego, CA) as rat anti-mouseantibody: phycoerythrin (PE)-conjugated and Cy-chrome-conjugated anti-CD8, al-lophycocyanin-conjugated anti-CD4, fluorescein isothiocyanate (FITC)-conjugatedanti-CD44, FITC-conjugated anti-Mac-1 (CD11b), FITC-conjugated CD11c, PE-conjugated anti-B220 (CD45R), PE-conjugated anti-NK1.1, Alexa 488-conjugatedanti-T-cell receptor alpha/beta (TCR-/), PE-conjugated anti-IFN-�, and PE-con-jugated immunoglobulin G1 isotype standard. The detection of LCMV-specificCD8� T cells was performed with PE-conjugated H-2Db/gp33-41 or H-2Db/np396-404
dextramers kindly provided by Dako (Glostrup, Denmark).Flow cytometric analysis. Surface staining of cells for flow cytometry was
performed according to standard laboratory procedure (2). For the evaluation ofcytokine production by LCMV-specific CD8� T cells, splenocytes were incu-bated in vitro at 37°C in 5% CO2 with or without gp33-41 peptide (0.1 �g/ml) inthe presence of monensin (3 �M; Sigma Chemicals Co., St. Louis, MO), and
murine recombinant interleukin-2 (10 U/well; R&D Systems Europe Ltd.,Abingdon, United Kingdom). After incubation, cells were surface stained,washed, and permeabilized using 0.5% saponin. Cells were then stained withanti-IFN-� or immunoglobulin G1 isotype control for 20 min at 4°C. Sampleswere analyzed using a Becton Dickinson FACSCalibur, and at least 104 mono-nuclear cells were gated using a combination of low angle and side scatter toexclude dead cells and debris. Data analysis was conducted using Cell Quest Pro(B&D Biosciences).
In vivo CD8� T-cell depletion. The depletion of CD8� T cells was obtained byi.p. injection on day �1 and �1 relative to virus infection with the ascites frommice carrying the 2.43 hybridoma. The efficiency of the depletion was verified byflow cytometric analysis of splenocytes from the treated animals.
Detection of mRNA in the liver. Livers from mice deeply anesthetized andexsanguinated were immediately removed, snap frozen on dry ice, and stored at�80°C until processed. Total RNA was extracted by homogenizing livers in 4 Mguanidine isothiocyanate (GITC), followed by centrifugation to pellet debris withdouble extraction of RNA from the supernatant by phenol and chloroformisoamylalcohol. The aqueous supernatant was then precipitated in isopropanol,and the pellet was resuspended in 4 M GITC, precipitated again using isopro-panol, pelleted, and subsequently washed twice in ethanol. Pellets were resus-pended in diethyl pyrocarbonate-treated water. Transcription levels were thenstudied using the RiboQuant multiprobe RNase protection assay (RPA) system(Pharmingen, San Diego, CA). The following templates sets (from Pharmingen)were used: cytokine marker mRNA (tumor necrosis factor beta [TNF-], LT,TNF-, interleukin-6, IFN-�, IFN-, transforming growth factor 1 [TGF-1] toTGF-3, migration inhibition factor [MIF]), and chemokine marker mRNA(XCL1 [lymphotactin], CCL5 [RANTES], CCL4 [MIP-1], CCL3 [MIP-1],CXCL1 and CXCL2 [MIP-2], CXCL10 [IP-10], and CCL2 [MCP-1]). All sets ofprobes included templates for the housekeeping genes L-32 and GAPDH(glyceraldehyde-3-phosphate dehydrogenase) to serve as loading controls. TheRPA was performed according to the manufacturer’s instructions. Briefly,[-32P]UTP-labeled antisense RNA transcript was generated from the templatesets using T7 RNA polymerase. RNA from each sample was allowed to hybridizeto the labeled probe for 16 to 20 h at 56°C. Single-stranded RNA was digestedwith an RNase/T1 mixture, and the hybrids were analyzed on a denaturingurea-polyacrylamide gel. For qualitative and quantitative results, gels were sub-jected to PhosphorImager analysis (Amersham Pharmacia Biotech) and the datawere subsequently analyzed using ImageMaster TotalLab software with L-32 asthe normalization control (Amersham Pharmacia Biotech).
Quantification of cytokine and chemokine levels in the liver. The amounts ofcytokine or chemokine protein in the liver were evaluated using tissue homog-enates containing protein inhibitor. Levels of IFN-� were measured using astandard sandwich enzyme-linked immunosorbent assay, and chemokine levelswere assessed using a multiplex fluorescent bead assay from Biosource and aLuminex analyzer. All analyses were carried out according to the manufacturers’instructions.
CD8 staining. Frozen liver sections were fixed in acetone and stained for CD8immunoreactivity by using the rat anti-mouse CD8 antibody (catalog no. 550281;BD Biosciences, San Jose, CA) diluted 1:1,000. Immunoreactive cells were vi-sualized using biotin-labeled anti-rat antibody, the tyramide signal amplificationkit as instructed by the manufacturer (PerkinElmer, Boston, MA), and diamino-benzidine. No staining was seen in the absence of primary antibody. The tissuesections were lightly counterstained with hematoxylin.
Oil Red staining. Frozen liver sections were fixed in paraformaldehyde andstained with Oil Red O solution for 10 min at room temperature, followed by alight counterstain with hematoxylin. The percentage of Oil Red O staining wasmeasured in four random visual fields from each slide using Image Pro, and themean percent � standard error of the mean (SEM) was then calculated for eachslide.
Transmission electron microscopy. WT and CCR5/CXCR3�/� mice werefixed by vascular perfusion through the left ventricle of the heart with 2%glutaraldehyde in 0.05 M sodium phosphate buffer (pH 7.2) for 2 min. Followingfixation, the abdomen was opened and the liver was removed and stored in thesame fixative. Following isolation of suitable specimen blocks, the samples wererinsed three times in 0.15 M sodium cacodylate buffer (pH 7.2) and subsequentlypostfixed in 1% OsO4 in 0.12 M sodium cacodylate buffer (pH 7.2) for 2 h. Thespecimens were dehydrated in graded series of ethanol, transferred to propyleneoxide, and embedded in Epon according to standard procedures. Ultrathinsections were cut with a Reichert-Jung Ultracut E microtome and collected on200 mesh copper grids with Formvar supporting membranes. The sections werestained with uranyl acetate and lead citrate and examined with a Philips CM 100transmission electron microscope operated at an accelerating voltage of 80 kV
10102 HOLST ET AL. J. VIROL.

and equipped with a SIS MegaView II camera. Digital images were recordedwith the analySIS software package.
Statistical analysis. Quantitative results were compared using the Mann-Whitney U test. A P value of �0.05 was considered evidence of statisticalsignificance.
RESULTS
Characterization of LCMV-induced hepatitis following i.p.infection with clone 13. To define the basic parameters of ourhepatitis model, we analyzed the kinetics of leukocyte accumu-lation in the livers of mice infected i.p. with 105 PFU of LCMVclone 13. Mice were sacrificed either before infection or 2, 5,and 7 days after infection, and the numbers of CD4� andCD8� T cells, NK cells, NKT cells, B cells, and Mac-1� cellsisolated from the perfused liver were determined (Fig. 1A).Two days after the infection, we did not find any increase in thenumber of LILs. Substantial cellular infiltration was noted 5days after infection, with the most marked increases in num-bers of NK and CD8� T cells. Interesting also is that thenumber of B220� cells increased significantly, raising the pos-sibility that some of these cells were plasmacytoid dendriticcells. However, additional phenotypic characterization re-vealed that none of these cells were CD11c� cells, demonstrat-ing that few if any were dendritic cells. At day 7 after infection,the number of infiltrating CD8� T cells had increased evenfurther, whereas numbers of other cell types were either un-changed or had begun to decline.
To determine how the T-cell influx correlated with viruscontrol and clearance, we quantified the viral load within theliver at days 2, 3, 4, 5, and 7 after infection (Fig. 1B). Viraltiters were found to peak between days 3 and 5 after infection,and virus was present, but only at a very low level on day 7postinfection.
To map the chemokines that might be involved in effectorcell recruitment, we next screened broadly for likely candidatesby using an RPA to look for relevant gene transcripts in thelivers of uninfected mice and of mice infected 2, 5, and 7 dayspreviously (Fig. 1C). We found that the expression of all of thechemokines studied here was increased in the livers of infectedmice. Notably, the CXCR3 ligand CXCL10 was found to bemaximally expressed on day 5 after infection, coinciding withpeak viral titers, while the expression levels of the CCR5 li-gands CCL3, CCL4, and CCL5 were stable between days 5 and7 after infection. These correlations seem to match previousreports suggesting CXCL10 to be produced predominantlyfrom virally infected cells and the CCR5 ligands to be pro-duced from activated NK and T cells (4, 30). Based on theseresults, we selected days 5 and 7 postinfection as the mostoptimal time points for studying NK cell recruitment, the earlyphase of the CD8� T-cell infiltration, and the resulting viruscontrol.
Unimpaired leukocyte infiltration in CCR5�/�, CXCR3�/�,and CCR5/CXCR3�/� mice. To define the actual chemokine/receptor interactions involved in effector cell recruitment, weinfected chemokine receptor-deficient mice and WT mice inparallel. First, we investigated whether antiviral effector T cellswere efficiently generated and capable of homing to the liver inthe CCR5�/�, CXCR3�/�, and CCR5/CXCR3�/� mice. Tothis end, we quantitated leukocyte subsets in the spleen andliver on days 5 and 7 after i.p. LCMV infection and compared
the response of the chemokine receptor-deficient mice to thatof WT mice.
All in all, there were few substantial differences in the cel-lular response patterns of the four different genotypes; how-
FIG. 1. Kinetics of the intrahepatic immune response in WT miceinfected with 105 PFU of LCMV clone 13 i.p. (A) Leukocytes werepurified from uninfected and infected animals at the indicated timesafter infection and were stained with monoclonal antibodies to revealthe total numbers of each of the main hepatic leukocyte subsets de-fined by CD4�/NK1.1� (CD4� T cells), CD8�/CD8� (CD8� Tcells), TCR-/�/NK1.1� (NKT), TCR-/�/NK1.1� (NK), B220� (Bcells), and Mac-1� (macrophages). (B) Following virus infection, ani-mals were sacrificed at the indicated times and viral titers in the liverwere determined by plaque assay on homogenates. (C) Chemokineexpression within infected and uninfected livers as determined by RPAassay and normalized to L32 expression. All data shown are averagesfor four to eight mice. Bars represent standard deviations.
VOL. 81, 2007 CCR5 AND CXCR3 IN INTRAHEPATIC ANTIVIRAL IMMUNITY 10103

ever, a few trends could be discerned. On day 7 after infection,both CXCR3�/� strains had significantly more CD8� T cells inthe spleen (Fig. 2A) and this was true also for cells with knownspecificity for LCMV (Fig. 2B). In contrast, both CCR5�/�
strains had higher numbers of liver-infiltrating CD8� T cells,including LCMV-specific cells, on day 7 after infection thandid CCR5-replete mice (Fig. 2A and B).
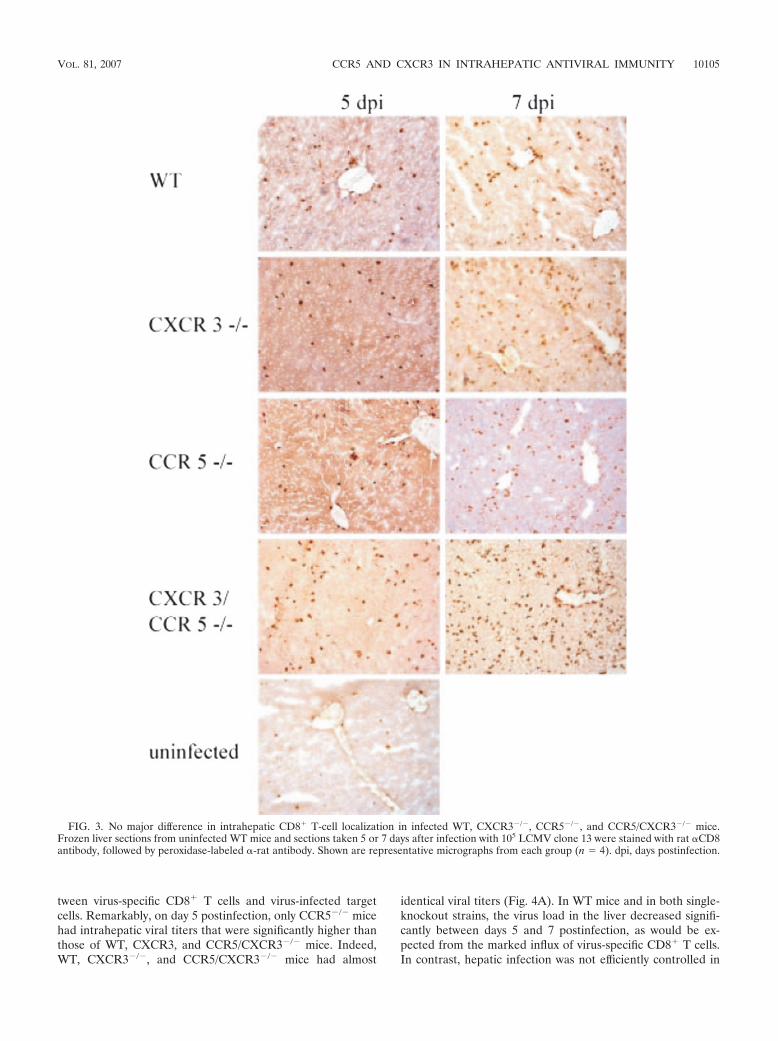
Immunohistochemical visualization of CD8� cells failed toreveal any obvious differences between the different mousestrains in the intrahepatic localization of the infiltrating cells(Fig. 3). In summary, these results clearly revealed that all ofthe tested mouse strains efficiently support the generation ofantigen-specific CD8� T cells following LCMV infection andthat the generated cells are capable of infiltrating the liver.
The number of NK and NKT cells also increased in thespleen, but no differences were observed between the geno-types, neither here nor in the liver, indicating that both of thesecell types could infiltrate the LCMV-infected liver in the ab-sence of CCR5 and CXCR3. Notably, no differences werefound if uninfected WT and CCR5/CXCR3�/� mice werecompared (data not shown).
Slightly impaired virus control in CCR5 knockout mice,similar parenchymal liver damage, and similar inflammatoryresponses in WT, CXCR3, CCR5, and CCR5/CXCR3 double-knockout mice. In addition to studying the hepatic cell infil-trates, we also evaluated the ability of chemokine receptor-deficient mice to control and eliminate infectious virus fromthe liver, a process which requires direct cellular contact be-
FIG. 2. Liver and splenic lymphocyte subsets following infection of WT, CXCR3�/�, CCR5�/�, and CCR5/CXCR3�/� mice with 105 PFU ofLCMV clone 13 i.p. Animals infected either 5 or 7 days previously were sacrificed, and spleen and liver leukocytes were collected. (A) Totalnumbers of NK cells (NK1.1�/TCR-/�), NKT cells (NK1.1�/TCR-/�), CD4� T cells (CD4�/NK1.1�), and CD8� T cells (CD8�/CD8�)(n � 8 mice/group). (B) Total numbers of LCMV-specific CD8� T cells defined by CD8 staining and binding of either gp33-41 or np396-404dextramers. All results shown are averages � standard deviations (error bars) of four animals. *, P was less than 0.05, Mann-Whitney rank sumtest versus WT mice.
10104 HOLST ET AL. J. VIROL.
tween virus-specific CD8� T cells and virus-infected targetcells. Remarkably, on day 5 postinfection, only CCR5�/� micehad intrahepatic viral titers that were significantly higher thanthose of WT, CXCR3, and CCR5/CXCR3�/� mice. Indeed,WT, CXCR3�/�, and CCR5/CXCR3�/� mice had almost
identical viral titers (Fig. 4A). In WT mice and in both single-knockout strains, the virus load in the liver decreased signifi-cantly between days 5 and 7 postinfection, as would be ex-pected from the marked influx of virus-specific CD8� T cells.In contrast, hepatic infection was not efficiently controlled in
FIG. 3. No major difference in intrahepatic CD8� T-cell localization in infected WT, CXCR3�/�, CCR5�/�, and CCR5/CXCR3�/� mice.Frozen liver sections from uninfected WT mice and sections taken 5 or 7 days after infection with 105 LCMV clone 13 were stained with rat CD8antibody, followed by peroxidase-labeled -rat antibody. Shown are representative micrographs from each group (n � 4). dpi, days postinfection.
VOL. 81, 2007 CCR5 AND CXCR3 IN INTRAHEPATIC ANTIVIRAL IMMUNITY 10105
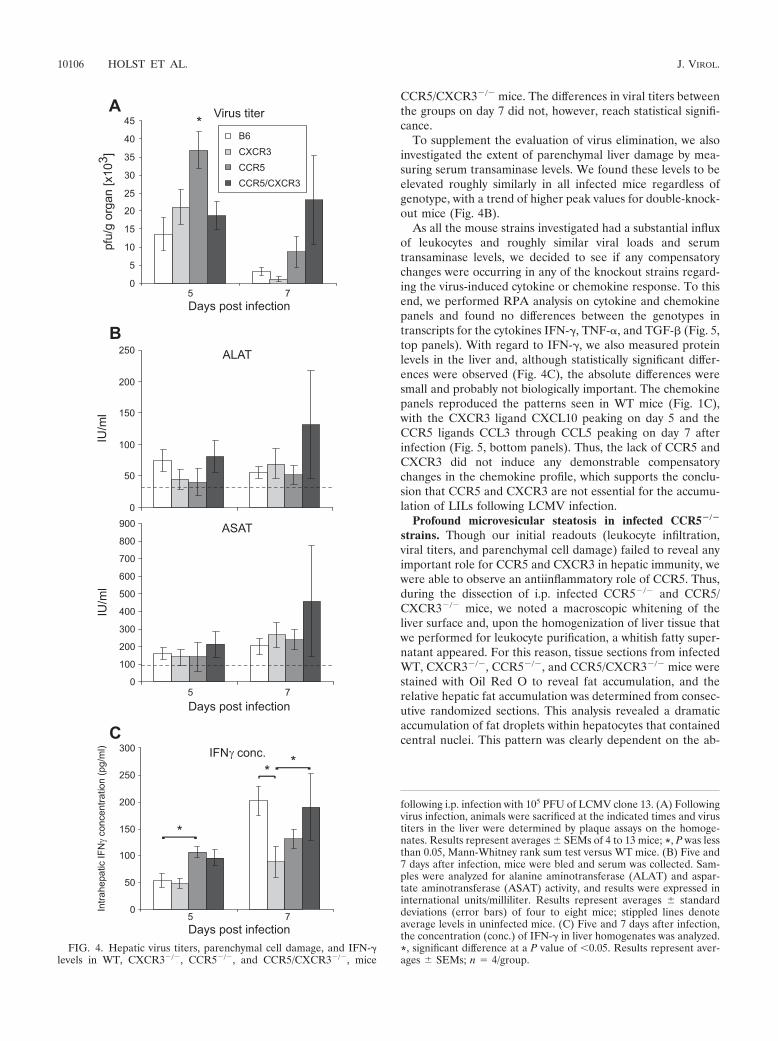
CCR5/CXCR3�/� mice. The differences in viral titers betweenthe groups on day 7 did not, however, reach statistical signifi-cance.
To supplement the evaluation of virus elimination, we alsoinvestigated the extent of parenchymal liver damage by mea-suring serum transaminase levels. We found these levels to beelevated roughly similarly in all infected mice regardless ofgenotype, with a trend of higher peak values for double-knock-out mice (Fig. 4B).
As all the mouse strains investigated had a substantial influxof leukocytes and roughly similar viral loads and serumtransaminase levels, we decided to see if any compensatorychanges were occurring in any of the knockout strains regard-ing the virus-induced cytokine or chemokine response. To thisend, we performed RPA analysis on cytokine and chemokinepanels and found no differences between the genotypes intranscripts for the cytokines IFN-�, TNF-, and TGF- (Fig. 5,top panels). With regard to IFN-�, we also measured proteinlevels in the liver and, although statistically significant differ-ences were observed (Fig. 4C), the absolute differences weresmall and probably not biologically important. The chemokinepanels reproduced the patterns seen in WT mice (Fig. 1C),with the CXCR3 ligand CXCL10 peaking on day 5 and theCCR5 ligands CCL3 through CCL5 peaking on day 7 afterinfection (Fig. 5, bottom panels). Thus, the lack of CCR5 andCXCR3 did not induce any demonstrable compensatorychanges in the chemokine profile, which supports the conclu-sion that CCR5 and CXCR3 are not essential for the accumu-lation of LILs following LCMV infection.
Profound microvesicular steatosis in infected CCR5�/�
strains. Though our initial readouts (leukocyte infiltration,viral titers, and parenchymal cell damage) failed to reveal anyimportant role for CCR5 and CXCR3 in hepatic immunity, wewere able to observe an antiinflammatory role of CCR5. Thus,during the dissection of i.p. infected CCR5�/� and CCR5/CXCR3�/� mice, we noted a macroscopic whitening of theliver surface and, upon the homogenization of liver tissue thatwe performed for leukocyte purification, a whitish fatty super-natant appeared. For this reason, tissue sections from infectedWT, CXCR3�/�, CCR5�/�, and CCR5/CXCR3�/� mice werestained with Oil Red O to reveal fat accumulation, and therelative hepatic fat accumulation was determined from consec-utive randomized sections. This analysis revealed a dramaticaccumulation of fat droplets within hepatocytes that containedcentral nuclei. This pattern was clearly dependent on the ab-
FIG. 4. Hepatic virus titers, parenchymal cell damage, and IFN-�levels in WT, CXCR3�/�, CCR5�/�, and CCR5/CXCR3�/�, mice
following i.p. infection with 105 PFU of LCMV clone 13. (A) Followingvirus infection, animals were sacrificed at the indicated times and virustiters in the liver were determined by plaque assays on the homoge-nates. Results represent averages � SEMs of 4 to 13 mice; *, P was lessthan 0.05, Mann-Whitney rank sum test versus WT mice. (B) Five and7 days after infection, mice were bled and serum was collected. Sam-ples were analyzed for alanine aminotransferase (ALAT) and aspar-tate aminotransferase (ASAT) activity, and results were expressed ininternational units/milliliter. Results represent averages � standarddeviations (error bars) of four to eight mice; stippled lines denoteaverage levels in uninfected mice. (C) Five and 7 days after infection,the concentration (conc.) of IFN-� in liver homogenates was analyzed.*, significant difference at a P value of �0.05. Results represent aver-ages � SEMs; n � 4/group.
10106 HOLST ET AL. J. VIROL.

sence of CCR5 and protracted in CCR5/CXCR3�/� mice (Fig.6A). Steatosis was markedly reduced in all groups on day 9after infection, and little if any fat accumulation was found at15 and 30 days after infection (percentages of liver sectionsstaining with Oil Red O were below 2% for both CCR5 andCCR5/CXCR3�/� mice).
To further characterize the nature of the apparent microve-sicular steatosis, tissue sections from perfusion-fixed livers ofWT and CCR5/CXCR3�/� mice were investigated by trans-mission electron microscopy. This analysis supported the diag-nosis of microvesicular steatosis and documented disorganizedchristae in the mitochondria of CCR5/CXCR3�/� mice (Fig.6B). This sign of mitochondrial dysfunction is often foundunder conditions of microvesicular steatosis associated withinflammation (16, 24, 25).
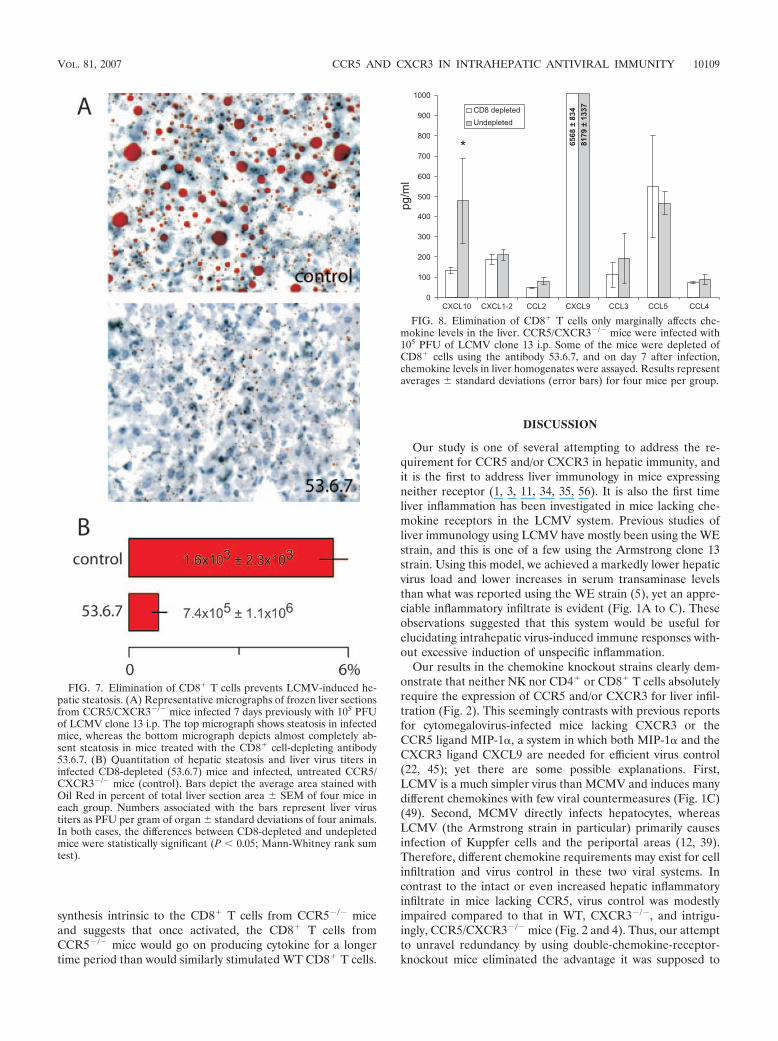
LCMV-induced hepatic steatosis is dependent on CD8� Tcells. The virus-induced hepatic steatosis in CCR5-deficientmice either could reflect direct virus-induced cell damage con-fined to mice with this genotype or could be the indirect resultof a modified inflammatory response. As virus-specific CD8� Tcells cause most known LCMV-induced pathology, we per-formed a depletion of CD8� T cells before and during LCMV
infection in CCR5/CXCR3�/� mice. The effect of this deple-tion was analyzed in CCR5/CXCR3�/� mice on day 7 afterinfection, as this was the setting of the most consistent anddramatic steatosis. As can be seen in Fig. 7, virus-inducedsteatosis was almost completely abolished in CD8� T-cell-depleted CCR5/CXCR3�/� mice, despite increased hepaticinfection in these mice. Notably, supplementary analysis ofchemokine levels in the liver revealed that except for a signif-icant reduction in the concentration of CXCL10, levels of allrelevant chemokines, including the CCR5 ligands CCL3through CCL5, were unaffected by the CD8� T-cell depletion(Fig. 8).
Absence of CCR5 leads to a T-cell-intrinsic defect in theregulation of IFN-� synthesis. Although the analysis of whole-organ homogenates for content of IFN-� did not point to thiscytokine as the crucial factor in mediating liver steatosis, theextent and distribution of the morphological changes did pointto a T-cell-derived soluble factor as the underlying mechanism.Therefore, to study the regulation of cytokine synthesis inCD8� T cells from CCR5�/� mice more directly, we comparedex vivo production of IFN-� in CD8� T cells from these miceand matched wild types in the presence or absence of overt
FIG. 5. Lack of compensatory changes in cytokine or chemokine expression in CXCR3�/�, CCR5�/�, or CCR5/CXCR3�/� mice infected with105 PFU of LCMV clone 13 i.p. RNA was purified from livers taken 5 or 7 days after infection in each of the above strains and from livers fromWT mice, and relevant transcripts were quantified using RNase protection assays. Shown are the signals from the most highly expressed cytokinesand chemokines normalized to the expression of the housekeeping gene L32; for levels in uninfected mice, see Fig. 1C. Shown are averages �standard deviations (error bars); n � 3 in each group.
VOL. 81, 2007 CCR5 AND CXCR3 IN INTRAHEPATIC ANTIVIRAL IMMUNITY 10107

stimulation (plus/minus relevant peptide). The cells were in-cubated in vitro for 1, 3, or 5 h, and monensin was added forthe last hour before analysis.
Expanding on our own published results, the most remark-able difference was an increased and sustained synthesis ofIFN-� in CD8� T cells from CCR5�/� mice in the absence ofadded peptide (Fig. 9, top panel). Given that previous analysisfailed to reveal substantial differences between CCR5�/� miceand matched wild types in the total number of gp33-specificsplenic CD8� T cells (Fig. 2B), this pattern could reflect eitheran increased frequency of cells expressing virus naturally in the
splenocyte cultures from CCR5�/� mice or a less-stringentregulation of cytokine synthesis intrinsic to CD8� T cells fromthese mice. In order to discriminate between these possibili-ties, we separated splenocytes from both types of mice intoCD8� and non-T cells and subsequently cultured the cells ofeither type with syngeneic cells or cells of the opposite geno-type for 5 h. As can be seen clearly in Fig. 9, bottom panel, thecapacity of the CD8� T cells to “spontaneously” synthesizeIFN-� followed the origin of the T cells and not the genotypeof the cells with which they were cocultured. This finding ismost consistent with a less-stringent regulation of cytokine
FIG. 6. Hepatic steatosis and mitochondrial abnormalities in infected CCR5�/� strains. (A) Shown are representative images of frozen liver sectionstaken from mice infected either 5 or 7 days previously with 105 PFU of LCMV clone 13 i.p. The sections have been stained with Oil Red solution to revealintracellular fat content. Numbers in the lower part of each picture show average area stained with Oil Red in percent of total liver section area � SEMof four mice in each group. dpi, days postinfection. (B) Representative transmission electron micrographs from WT and CCR5/CXCR3�/� mice infected5 days previously. Disrupted mitochondrial architecture with loss of clearly definable christae is evident in CCR5/CXCR3�/� mice. Bar � 1 �m.
10108 HOLST ET AL. J. VIROL.
synthesis intrinsic to the CD8� T cells from CCR5�/� miceand suggests that once activated, the CD8� T cells fromCCR5�/� mice would go on producing cytokine for a longertime period than would similarly stimulated WT CD8� T cells.
DISCUSSION
Our study is one of several attempting to address the re-quirement for CCR5 and/or CXCR3 in hepatic immunity, andit is the first to address liver immunology in mice expressingneither receptor (1, 3, 11, 34, 35, 56). It is also the first timeliver inflammation has been investigated in mice lacking che-mokine receptors in the LCMV system. Previous studies ofliver immunology using LCMV have mostly been using the WEstrain, and this is one of a few using the Armstrong clone 13strain. Using this model, we achieved a markedly lower hepaticvirus load and lower increases in serum transaminase levelsthan what was reported using the WE strain (5), yet an appre-ciable inflammatory infiltrate is evident (Fig. 1A to C). Theseobservations suggested that this system would be useful forelucidating intrahepatic virus-induced immune responses with-out excessive induction of unspecific inflammation.
Our results in the chemokine knockout strains clearly dem-onstrate that neither NK nor CD4� or CD8� T cells absolutelyrequire the expression of CCR5 and/or CXCR3 for liver infil-tration (Fig. 2). This seemingly contrasts with previous reportsfor cytomegalovirus-infected mice lacking CXCR3 or theCCR5 ligand MIP-1, a system in which both MIP-1 and theCXCR3 ligand CXCL9 are needed for efficient virus control(22, 45); yet there are some possible explanations. First,LCMV is a much simpler virus than MCMV and induces manydifferent chemokines with few viral countermeasures (Fig. 1C)(49). Second, MCMV directly infects hepatocytes, whereasLCMV (the Armstrong strain in particular) primarily causesinfection of Kuppfer cells and the periportal areas (12, 39).Therefore, different chemokine requirements may exist for cellinfiltration and virus control in these two viral systems. Incontrast to the intact or even increased hepatic inflammatoryinfiltrate in mice lacking CCR5, virus control was modestlyimpaired compared to that in WT, CXCR3�/�, and intrigu-ingly, CCR5/CXCR3�/� mice (Fig. 2 and 4). Thus, our attemptto unravel redundancy by using double-chemokine-receptor-knockout mice eliminated the advantage it was supposed to
FIG. 7. Elimination of CD8� T cells prevents LCMV-induced he-patic steatosis. (A) Representative micrographs of frozen liver sectionsfrom CCR5/CXCR3�/� mice infected 7 days previously with 105 PFUof LCMV clone 13 i.p. The top micrograph shows steatosis in infectedmice, whereas the bottom micrograph depicts almost completely ab-sent steatosis in mice treated with the CD8� cell-depleting antibody53.6.7. (B) Quantitation of hepatic steatosis and liver virus titers ininfected CD8-depleted (53.6.7) mice and infected, untreated CCR5/CXCR3�/� mice (control). Bars depict the average area stained withOil Red in percent of total liver section area � SEM of four mice ineach group. Numbers associated with the bars represent liver virustiters as PFU per gram of organ � standard deviations of four animals.In both cases, the differences between CD8-depleted and undepletedmice were statistically significant (P � 0.05; Mann-Whitney rank sumtest).
FIG. 8. Elimination of CD8� T cells only marginally affects che-mokine levels in the liver. CCR5/CXCR3�/� mice were infected with105 PFU of LCMV clone 13 i.p. Some of the mice were depleted ofCD8� cells using the antibody 53.6.7, and on day 7 after infection,chemokine levels in liver homogenates were assayed. Results representaverages � standard deviations (error bars) for four mice per group.
VOL. 81, 2007 CCR5 AND CXCR3 IN INTRAHEPATIC ANTIVIRAL IMMUNITY 10109

confer. This result is, however, similar to what we recentlyreported for LCMV-induced inflammation in the CNS (10). Itmust be stressed that the mechanism by which CXCR3 defi-ciency improves initial virus control in CCR5�/� mice remainsundetermined, yet the literature offers a possible explanation.Shields et al. (47) studied chemokine expression during hepa-titis C virus infection in the liver and found CCR5 ligands to be
expressed in the periportal areas, whereas the CXCR3 ligandwas expressed from the liver sinusoids and hepatocytes. Ac-cordingly, the balance between different stimuli would deter-mine the localization of CCR5/CXCR3 double-positive cells (asimilar mechanism is in fact determining splenic leukocytepositioning) (44). When applied to our system, a lack of CCR5might drive the CD8� T cells away from the periportal areas,where most of the virus is located, deeper into the liver paren-chyma, where CXCR3 ligands would be present. In this situa-tion, concomitant loss of CXCR3 stimulation might restoreperiportal localization through other signals. Unfortunately,the majority of our LILs are not antigen specific and we havenot been able to costain sections with both antigen-specificmajor histocompatibility complex dextramers and portal tis-sue-specific stains. Thus, a direct demonstration of an alteredintrahepatic localization remains elusive. In any case, the effectof selective CCR5 deficiency on viral titers is transient andmodest and any differences in intrahepatic localization couldlikewise be subtle.
Remarkably and quite unexpected, despite almost normalviral control, morphological analysis demonstrated a markedmicrovesicular steatosis in both strains of CCR5-deficient mice(Fig. 6). In our system, the presence of any appreciable fattychange within the livers of infected mice clearly depended onthe absence of CCR5, yet similar steatotic changes were ob-served previously in WT mice infected with high doses of therapidly invasive WE strain of LCMV and indeed in the contextof viral infections other than LCMV (29, 32). We are, however,the first to demonstrate that such pathological changes can beCD8� T-cell dependent in a system where other features ofvirus-associated pathology are rather limited (Fig. 7). Indeed,our observation of specific inflammatory changes in the ab-sence of CCR5 may add to the understanding of our ownrecent report of opposing effects of CCR5 and CXCR3 onLCMV-induced meningitis (10). LCMV-induced hepatic ste-atosis was found to be pronounced and short lived in CCR5�/�
mice, yet it was maintained at increasing levels until day 7 inCCR5/CXCR3�/� mice. We suggest that this pattern may re-flect the fact that only double-knockout mice failed to reducethe viral load between days 5 and 7 postinfection.
We noticed that the clinical history of LCMV-induced ste-atosis has several features in common with virus-induced mi-crovesicular steatosis in humans, i.e., classical Reye’s syn-drome, an apparently virus-triggered, acute liver failure andencephalopathy in children hallmarked by similar light andelectron microscopical findings in the liver (16, 24). Indeed, therequirement for CCR5 also seems to vary according to age ina herpes simplex model of liver inflammation (3). However,aspirin administration, the use of which is associated withReye’s syndrome (14) in humans, failed to increase the pathol-ogy in our mice (data not shown). If the human CCR5 poly-morphism plays a similar role in classical Reye’s syndrome,other predisposing factors are likely to vary considerably. Thus,although identical mechanisms at the genetic level between ourmodel and Reye’s syndrome remains elusive, we have demon-strated that microvesicular steatosis can be triggered by CD8�
T-cell activities even when viral replication in the liver is quitelimited, and this could very well be a mimic to the situation ininfluenza-infected humans.
Although the observation of CCR5-dependent protection
FIG. 9. Absence of CCR5 leads to impaired regulation of IFN-�synthesis. (A) Splenocytes from WT and CCR5�/� mice infected with105 PFU of LCMV clone 13 i.p. 7 days earlier were incubated in vitrowith or without LCMV-derived peptide (gp33-41). At the indicated timepoints, monensin was added to the cultures, and CD8� cells wereanalyzed for cytokine production by intracellular staining 1 h later;cells from uninfected mice were included as a control. Means of du-plicate samples are presented; results are representative of two exper-iments. (B) Splenocytes from infected WT and CCR5�/� mice wereseparated into CD8� cells and non-T cells, and each population wasincubated with syngeneic or allogeneic cells of the opposite phenotype.After 5 h of incubation, CD8� cells were analyzed for intracellularcytokine. Representative plots are depicted (n � 2/group).
10110 HOLST ET AL. J. VIROL.

from virus-induced hepatic steatosis is unique, recent reportshave demonstrated that CCR5 protects mice against the hep-atotoxic effects of systemic concanavalin A administration (1,34). Taken together with our study, it seems that evidence foran important role of CCR5 in controlling hepatic immunopa-thology is accumulating.
Precisely how the CD8� T cells induce liver steatosis is notclear. Given the extent of the observed steatosis, a solublefactor seems most likely to be the underlying mechanism. Yet,variation in neither whole-organ levels of mRNA for severalproinflammatory mediators nor protein levels of IFN-� orCCR5 ligands readily explains the association between steato-sis and the lack of CCR5 expression. On the other hand, our invitro analysis of CD8� T cells from CCR5�/� mice points to aprolonged synthesis of IFN-� by the activated T cells in thesemice, which could support the assumption that this cytokineand/or other associated secreted molecules may play an im-portant role in the observed pathology. As to the reason for thediscrepancy between our in vivo and in vitro results, it could beargued that the in vitro assay much more precisely detectsdifferences in cytokine synthesis by activated CD8� T cells,whereas analysis of the whole organ simply gives a snapshot ofthe accumulated potential of these cells, many of which maynot realize this normally.
In summary, we have shown that neither CCR5 nor CXCR3is necessary for virus-induced NK cell or CD4� and CD8�
T-cell infiltration into the liver. Lack of CCR5 resulted in avery modest impairment of virus control that was initially coun-teracted by a concomitant lack of CXCR3. Lack of CCR5 orboth CCR5 and CXCR3 resulted in marked virus-inducedCD8� T-cell-mediated steatosis. Our findings may shed lighton studies showing adverse effects of CCR5 deficiency in stud-ies of human immunopathology (13, 33, 40).
ACKNOWLEDGMENTS
This work was supported in part by the Novo Nordisk Foundation,the Danish Cancer Society, the Lundbeck Foundation, the SophusC. E. Friis Foundation, the Foundation for the Advancement of Med-ical Sciences, the Aase and Ejnar Danielsen Foundation, the HedeNielsen Family Foundation, the Leo Nielsen Foundation, MerchantFoght’s Foundation, the Christian and Ellen Larsen Foundation, andthe Leo Pharma Research Foundation. P.J.H. is the recipient of aresearch fellowship from the Faculty of Health Science, University ofCopenhagen, Copenhagen, Denmark.
We thank Grethe Thorner Andersen and Lone Malte for expertlaboratory assistance and Grazyna Hahn for technical assistance. Ma-jor histocompatibility complex dextramers were kindly donated byJørgen Schøller, Dako, Glostrup, Denmark.
REFERENCES
1. Ajuebor, M. N., A. I. Aspinall, F. Zhou, T. Le, Y. Yang, S. J. Urbanski, S.Sidobre, M. Kronenberg, C. M. Hogaboam, and M. G. Swain. 2005. Lack ofchemokine receptor CCR5 promotes murine fulminant liver failure by pre-venting the apoptosis of activated CD1d-restricted NKT cells. J. Immunol.174:8027–8037.
2. Andreasen, S. O., J. P. Christensen, O. Marker, and A. R. Thomsen. 1999.Virus-induced non-specific signals cause cell cycle progression of primedCD8(�) T cells but do not induce cell differentiation. Int. Immunol. 11:1463–1473.
3. Ank, N., K. Petersen, L. Malmgaard, S. C. Mogensen, and S. R. Paludan.2005. Age-dependent role for CCR5 in antiviral host defense against herpessimplex virus type 2. J. Virol. 79:9831–9841.
4. Asensio, V. C., C. Kincaid, and I. L. Campbell. 1999. Chemokines and theinflammatory response to viral infection in the central nervous system with afocus on lymphocytic choriomeningitis virus. J. Neurovirol. 5:65–75.
5. Balkow, S., A. Kersten, T. T. Tran, T. Stehle, P. Grosse, C. Museteanu, O.
Utermohlen, H. Pircher, F. von Weizsacker, R. Wallich, A. Mullbacher, andM. M. Simon. 2001. Concerted action of the FasL/Fas and perforin/gran-zyme A and B pathways is mandatory for the development of early viralhepatitis but not for recovery from viral infection. J. Virol. 75:8781–8791.
6. Battegay, M., S. Cooper, A. Althage, J. Banziger, H. Hengartner, and R. M.Zinkernagel. 1991. Quantification of lymphocytic choriomeningitis virus withan immunological focus assay in 24- or 96-well plates. J. Virol. Methods33:191–198.
7. Burns, W. R., Y. Wang, P. C. Tang, H. Ranjbaran, A. Iakimov, J. Kim, M.Cuffy, Y. Bai, J. S. Pober, and G. Tellides. 2005. Recruitment of CXCR3�
and CCR5� T cells and production of interferon-gamma-inducible chemo-kines in rejecting human arteries. Am. J. Transplant. 5:1226–1236.
8. Chen, S. C., G. Vassileva, D. Kinsley, S. Holzmann, D. Manfra, M. T.Wiekowski, N. Romani, and S. A. Lira. 2002. Ectopic expression of themurine chemokines CCL21a and CCL21b induces the formation of lymphnode-like structures in pancreas, but not skin, of transgenic mice. J. Immu-nol. 168:1001–1008.
9. Christensen, J. E., A. Nansen, T. Moos, B. Lu, C. Gerard, J. P. Christensen,and A. R. Thomsen. 2004. Efficient T-cell surveillance of the CNS requiresexpression of the CXC chemokine receptor 3. J. Neurosci. 24:4849–4858.
10. de Lemos, C., J. E. Christensen, A. Nansen, T. Moos, B. Lu, C. Gerard, J. P.Christensen, and A. R. Thomsen. 2005. Opposing effects of CXCR3 andCCR5 deficiency on CD8� T cell-mediated inflammation in the centralnervous system of virus-infected mice. J. Immunol. 175:1767–1775.
11. Duffner, U., B. Lu, G. C. Hildebrandt, T. Teshima, D. L. Williams, P. Reddy,R. Ordemann, S. G. Clouthier, K. Lowler, C. Liu, C. Gerard, K. R. Cooke,and J. L. Ferrara. 2003. Role of CXCR3-induced donor T-cell migration inacute GVHD. Exp. Hematol. 31:897–902.
12. Dutko, F. J., and M. B. Oldstone. 1983. Genomic and biological variationamong commonly used lymphocytic choriomeningitis virus strains. J. Gen.Virol. 64:1689–1698.
13. Eri, R., J. R. Jonsson, N. Pandeya, D. M. Purdie, A. D. Clouston, N. Martin,D. Duffy, E. E. Powell, J. Fawcett, T. H. Florin, and G. L. Radford-Smith.2004. CCR5-Delta32 mutation is strongly associated with primary sclerosingcholangitis. Genes Immun. 5:444–450.
14. Forsyth, B. W., R. I. Horwitz, D. Acampora, E. D. Shapiro, C. M. Viscoli,A. R. Feinstein, R. Henner, N. B. Holabird, B. A. Jones, and A. D. Karabelas.1989. New epidemiologic evidence confirming that bias does not explain theaspirin/Reye’s syndrome association. JAMA 261:2517–2524.
15. Frigerio, S., T. Junt, B. Lu, C. Gerard, U. Zumsteg, G. A. Hollander, and L.Piali. 2002. Beta cells are responsible for CXCR3-mediated T-cell infiltra-tion in insulitis. Nat. Med. 8:1414–1420.
16. Fromenty, B., and D. Pessayre. 1997. Impaired mitochondrial function inmicrovesicular steatosis. Effects of drugs, ethanol, hormones and cytokines.J. Hepatol. 26(Suppl. 2):43–53.
17. Glass, W. G., M. T. Liu, W. A. Kuziel, and T. E. Lane. 2001. Reducedmacrophage infiltration and demyelination in mice lacking the chemokinereceptor CCR5 following infection with a neurotropic coronavirus. Virology288:8–17.
18. Glass, W. G., H. F. Rosenberg, and P. M. Murphy. 2003. Chemokine regu-lation of inflammation during acute viral infection. Curr. Opin. Allergy Clin.Immunol. 3:467–473.
19. Goulding, C., A. Murphy, G. MacDonald, S. Barrett, J. Crowe, J. Hegarty, S.McKiernan, and D. Kelleher. 2005. The CCR5-delta32 mutation: impact ondisease outcome in individuals with hepatitis C infection from a singlesource. Gut 54:1157–1161.
20. Hamilton, N. H., S. Mahalingam, J. L. Banyer, I. A. Ramshaw, and S. A.Thomson. 2004. A recombinant vaccinia virus encoding the interferon-in-ducible T-cell alpha chemoattractant is attenuated in vivo. Scand. J. Immu-nol. 59:246–254.
21. Hancock, W. W., B. Lu, W. Gao, V. Csizmadia, K. Faia, J. A. King, S. T.Smiley, M. Ling, N. P. Gerard, and C. Gerard. 2000. Requirement of thechemokine receptor CXCR3 for acute allograft rejection. J. Exp. Med.192:1515–1520.
22. Hokeness, K. L., E. S. Deweerd, M. W. Munks, C. A. Lewis, R. P. Gladue, andT. P. Salazar-Mather. 2007. CXCR3-dependent recruitment of antigen-specific T lymphocytes to the liver during murine cytomegalovirus infection.J. Virol. 81:1241–1250.
23. Holst, P. J., H. R. Luttichau, T. W. Schwartz, and M. M. Rosenkilde. 2003.Virally encoded chemokines and chemokine receptors in the role of viralinfections. Contrib. Microbiol. 10:232–252.
24. Hou, J. W., S. P. Chou, and T. R. Wang. 1996. Metabolic function and liverhistopathology in Reye-like illnesses. Acta Paediatr. 85:1053–1057.
25. Kaneda, M., S. Kashiwamura, H. Ueda, K. Sawada, A. Sugihara, N. Terada,A. Kimura-Shimmyo, Y. Fukuda, T. Shimoyama, and H. Okamura. 2003.Inflammatory liver steatosis caused by IL-12 and IL-18. J. Interferon Cyto-kine Res. 23:155–162.
26. Lee, B. J., F. Giannoni, A. Lyon, S. Yada, B. Lu, C. Gerard, and S. R.Sarawar. 2005. Role of CXCR3 in the immune response to murine gamma-herpesvirus 68. J. Virol. 79:9351–9355.
27. Lindow, M., A. Nansen, C. Bartholdy, A. Stryhn, N. J. Hansen, T. P. Boesen,T. N. Wells, T. W. Schwartz, and A. R. Thomsen. 2003. The virus-encoded
VOL. 81, 2007 CCR5 AND CXCR3 IN INTRAHEPATIC ANTIVIRAL IMMUNITY 10111

chemokine vMIP-II inhibits virus-induced Tc1-driven inflammation. J. Virol.77:7393–7400.
28. Liu, Z. X., S. Govindarajan, S. Okamoto, and G. Dennert. 2001. Fas-medi-ated apoptosis causes elimination of virus-specific cytotoxic T cells in thevirus-infected liver. J. Immunol. 166:3035–3041.
29. Lohler, J., J. Gossmann, T. Kratzberg, and F. Lehmann-Grube. 1994. Mu-rine hepatitis caused by lymphocytic choriomeningitis virus. I. The hepaticlesions. Lab. Investig. 70:263–278.
30. Madsen, A. N., A. Nansen, J. P. Christensen, and A. R. Thomsen. 2003. Roleof macrophage inflammatory protein-1 in T-cell-mediated immunity toviral infection. J. Virol. 77:12378–12384.
31. Mantovani, A. 1999. The chemokine system: redundancy for robust outputs.Immunol. Today 20:254–257.
32. Martin, J. P., A. Bingen, F. Koehren, J. P. Gut, and A. Kirn. 1991. Alteredpathogenicity in the liver induced by a mouse hepatitis virus type 3 thermo-sensitive mutant. J. Hepatol. 13:61–70.
33. Moench, C., A. Uhrig, A. W. Lohse, and G. Otto. 2004. CC chemokinereceptor 5�32 polymorphism—a risk factor for ischemic-type biliary lesionsfollowing orthotopic liver transplantation. Liver Transpl. 10:434–439.
34. Moreno, C., T. Gustot, C. Nicaise, E. Quertinmont, N. Nagy, M. Parmentier,O. Le Moine, J. Deviere, and H. Louis. 2005. CCR5 deficiency exacerbatesT-cell-mediated hepatitis in mice. Hepatology 42:854–862.
35. Murai, M., H. Yoneyama, A. Harada, Z. Yi, C. Vestergaard, B. Guo, K.Suzuki, H. Asakura, and K. Matsushima. 1999. Active participation ofCCR5� CD8� T lymphocytes in the pathogenesis of liver injury in graft-versus-host disease. J. Clin. Investig. 104:49–57.
36. Murphy, P. M., M. Baggiolini, I. F. Charo, C. A. Hebert, R. Horuk, K.Matsushima, L. H. Miller, J. J. Oppenheim, and C. A. Power. 2000. Inter-national union of pharmacology. XXII. Nomenclature for chemokine recep-tors. Pharmacol. Rev. 52:145–176.
37. Nansen, A., J. P. Christensen, S. O. Andreasen, C. Bartholdy, J. E. Christensen,and A. R. Thomsen. 2002. The role of CC chemokine receptor 5 in antiviralimmunity. Blood 99:1237–1245.
38. Nansen, A., O. Marker, C. Bartholdy, and A. R. Thomsen. 2000. CCR2� andCCR5� CD8� T cells increase during viral infection and migrate to sites ofinfection. Eur. J. Immunol. 30:1797–1806.
39. Papadimitriou, J. M., G. R. Shellam, and T. A. Robertson. 1984. An ultra-structural investigation of cytomegalovirus replication in murine hepato-cytes. J. Gen. Virol. 65:1979–1990.
40. Petrek, M., J. Drabek, V. Kolek, J. Zlamal, K. I. Welsh, M. Bunce, E. Weigl,and B. R. Du. 2000. CC chemokine receptor gene polymorphisms in Czechpatients with pulmonary sarcoidosis. Am. J. Respir. Crit. Care Med. 162:1000–1003.
41. Pierson, T. C., R. W. Doms, and S. Pohlmann. 2004. Prospects of HIV-1entry inhibitors as novel therapeutics. Rev. Med. Virol. 14:255–270.
42. Qin, S., J. B. Rottman, P. Myers, N. Kassam, M. Weinblatt, M. Loetscher,A. E. Koch, B. Moser, and C. R. Mackay. 1998. The chemokine receptorsCXCR3 and CCR5 mark subsets of T cells associated with certain inflam-matory reactions. J. Clin. Investig. 101:746–754.
43. Rehermann, B., and M. Nascimbeni. 2005. Immunology of hepatitis B virusand hepatitis C virus infection. Nat. Rev. Immunol. 5:215–229.
44. Reif, K., E. H. Ekland, L. Ohl, H. Nakano, M. Lipp, R. Forster, and J. G.Cyster. 2002. Balanced responsiveness to chemoattractants from adjacentzones determines B-cell position. Nature 416:94–99.
45. Salazar-Mather, T. P., T. A. Hamilton, and C. A. Biron. 2000. A chemokine-to-cytokine-to-chemokine cascade critical in antiviral defense. J. Clin. Inves-tig. 105:985–993.
46. Salazar-Mather, T. P., C. A. Lewis, and C. A. Biron. 2002. Type I interferonsregulate inflammatory cell trafficking and macrophage inflammatory protein1 delivery to the liver. J. Clin. Investig. 110:321–330.
47. Shields, P. L., C. M. Morland, M. Salmon, S. Qin, S. G. Hubscher, and D. H.Adams. 1999. Chemokine and chemokine receptor interactions provide amechanism for selective T cell recruitment to specific liver compartmentswithin hepatitis C-infected liver. J. Immunol. 163:6236–6243.
48. Sørensen, T. L., M. Tani, J. Jensen, V. Pierce, C. Lucchinetti, V. A. Folcik,S. Qin, J. Rottman, F. Sellebjerg, R. M. Strieter, J. L. Frederiksen, and R. M.Ransohoff. 1999. Expression of specific chemokines and chemokine recep-tors in the central nervous system of multiple sclerosis patients. J. Clin.Investig. 103:807–815.
49. Thomsen, A. R., A. Nansen, A. N. Madsen, C. Bartholdy, and J. P. Christensen.2003. Regulation of T cell migration during viral infection: role of adhesionmolecules and chemokines. Immunol. Lett. 85:119–127.
50. Trebst, C., S. M. Staugaitis, B. Tucky, T. Wei, K. Suzuki, K. D. Aldape, C. A.Pardo, J. Troncoso, H. Lassmann, and R. M. Ransohoff. 2003. Chemokinereceptors on infiltrating leucocytes in inflammatory pathologies of the cen-tral nervous system (CNS). Neuropathol. Appl. Neurobiol. 29:584–595.
51. Tsunoda, I., T. E. Lane, J. Blackett, and R. S. Fujinami. 2004. Distinct rolesfor IP-10/CXCL10 in three animal models, Theiler’s virus infection, EAE,and MHV infection, for multiple sclerosis: implication of differing roles forIP-10. Mult. Scler. 10:26–34.
52. Vincenti, F. 2002. What’s in the pipeline? New immunosuppressive drugs intransplantation. Am. J. Transplant. 2:898–903.
53. Wang, J., T. H. Holmes, R. Cheung, H. B. Greenberg, and X. S. He. 2004.Expression of chemokine receptors on intrahepatic and peripheral lympho-cytes in chronic hepatitis C infection: its relationship to liver inflammation.J. Infect. Dis. 190:989–997.
54. Welniak, L. A., Z. Wang, K. Sun, W. Kuziel, M. R. Anver, B. R. Blazar, andW. J. Murphy. 2004. An absence of CCR5 on donor cells results in acceler-ation of acute graft-vs-host disease. Exp. Hematol. 32:318–324.
55. Wysocki, C. A., S. B. Burkett, A. Panoskaltsis-Mortari, S. L. Kirby, A. D.Luster, K. McKinnon, B. R. Blazar, and J. S. Serody. 2004. Differential rolesfor CCR5 expression on donor T cells during graft-versus-host disease basedon pretransplant conditioning. J. Immunol. 173:845–854.
56. Wysocki, C. A., Q. Jiang, A. Panoskaltsis-Mortari, P. A. Taylor, K. P.McKinnon, L. Su, B. R. Blazar, and J. S. Serody. 2005. Critical role forCCR5 in the function of donor CD4�CD25� regulatory T cells during acutegraft-versus-host disease. Blood 106:3300–3307.
10112 HOLST ET AL. J. VIROL.




















