
Natural variation in CDC28 underlies morphological phenotypes in an environmental yeast isolate

www.elsevier.com/locate/yviro
Virology 338 (20
Generation and properties of a human immunodeficiency virus
type 1 isolate resistant to the small molecule CCR5 inhibitor,
SCH-417690 (SCH-D)
Andre J. Marozsana, Shawn E. Kuhmanna, Thomas Morgana, Carolina Herreraa,
Enid Rivera-Trochea, Serena Xub, Bahige M. Baroudyb,1, Julie Strizkib, John P. Moorea,*
aDepartment of Microbiology and Immunology, Joan and Sanford I. Weill Medical College of Cornell University,
1300 York Avenue, W-805, New York, NY 10021, USAbSchering Plough Research Institute, Kenilworth, NJ 07033, USA
Received 19 April 2005; accepted 21 April 2005
Available online 1 June 2005
Abstract
We describe the generation of two genetically related human immunodeficiency virus type 1 (HIV-1) isolates highly (>20,000-fold)
resistant to the small molecule CCR5 inhibitor, SCH-417690 (formerly SCH-D). Both viruses were cross-resistant to other small molecules
targeting entry via CCR5, but they were inhibited by some MAbs against the same coreceptor on primary CD4+ T-cells. The resistant isolates
remained sensitive to inhibitors of other stages of virus entry, and to replication inhibitors acting post-entry. Neither escape mutant could
replicate detectably in peripheral blood mononuclear cells (PBMC) from two donors homozygous for the CCR5-D32 allele and both were
insensitive to the CXCR4-specific inhibitor, AMD3100. Hence, the SCH-D escape mutants retained the R5 phenotype. One of the resistant
isolates was, however, capable of replication in U87.CD4.CXCR4 cells and, after expansion in those cells, was sensitive to AMD3100 in
primary CD4+ T-cells. Hence, some X4 variants may be present in this escape mutant swarm. A notable observation was that the SCH-D
escape mutants were also cross-resistant to PSC-RANTES and AOP-RANTES, chemokine derivatives that are reported to down-regulate cell
surface CCR5 almost completely. However, the extent to which CCR5 is down-regulated was dependent upon the detection MAb. Hence, the
escape mutants may be using a CCR5 configuration that is only detected by some anti-CCR5 MAbs. Finally, two SCH-D-resistant clonal
viruses revealed no amino acid changes in the gp120 V3 region relative to the parental viruses, in marked contrast to clones resistant to the
AD101 small molecule CCR5 inhibitor that possess 4 such sequence changes. Several sequence changes elsewhere in gp120 (V2, C3 and
V4) were present in the SCH-D-resistant clones. Their influence on the resistant phenotype remains to be determined.
D 2005 Elsevier Inc. All rights reserved.
Keywords: HIV-1; CCR5; CCR5 inhibitors; SCH-D; Drug resistance; Escape mutants
Introduction
Entry inhibitors constitute a new class of drugs to treat
infection by human immunodeficiency virus type 1 (HIV-1)
(Baldwin et al., 2003; De Clercq, 2003; Kazmierski et al.,
2003; Kilby and Eron, 2003; Moore and Doms, 2003;
0042-6822/$ - see front matter D 2005 Elsevier Inc. All rights reserved.
doi:10.1016/j.virol.2005.04.035
* Corresponding author. Fax: +1 212 746 8340.
E-mail address: [email protected] (J.P. Moore).1 Present affiliation: Avance Pharma Inc., Laval, Quebec, Canada
H7V 5B7.
Moore and Stevenson, 2000; O’Hara and Olson, 2002). The
first member of this class, enfuvirtide, previously known as
T-20, has now been licensed for therapeutic use (Lalezari et
al., 2003; Lazzarin et al., 2003). Several other entry
inhibitors are in various stages of pre-clinical or clinical
development. They target any of several stages in the multi-
stage process by which the HIV-1 envelope glycoprotein
(Env) complex attaches to receptors via the gp120 glyco-
protein, then undergoes the conformational changes that
drive the fusion of the viral and cellular membranes (Kilby
and Eron, 2003; Moore and Doms, 2003). One particular
05) 182 – 199

A.J. Marozsan et al. / Virology 338 (2005) 182–199 183
step that is being actively pursued as a drug target is the
CD4-dependent binding of gp120 to the CCR5 coreceptor
(Trkola et al., 1996; Wu et al., 1996).
Various CCR5-reactive inhibitors of HIV-1 Env-medi-
ated membrane fusion have now been described, including
small molecules, chemokines and their derivatives, and
monoclonal antibodies (MAbs) (Kazmierski et al., 2003;
Olson et al., 1999; Schwarz and Wells, 2002; Seibert and
Sakmar, 2004). The small molecule CCR5 inhibitor SCH-D
(SCH-417690) is a receptor antagonist that has improved
potency and better pharmacology properties compared to its
predecessor, SCH-C (Strizki et al., 2001; Tagat et al., 2004).
SCH-D is in clinical development, and both it and SCH-C
have antiviral activity in HIV-1-infected humans (Maeda et
al., 2004b; Reynes et al., 2002; Schurmann et al., 2004).
It is an unfortunate but well-recognized aspect of HIV-1
drug development that resistance to any single inhibitor
frequently develops in vivo, and often occurs with multiple
inhibitors used in combination (Hammer, 2002; Larder,
2001). Entry inhibitors are highly unlikely, however, to share
resistance pathways with protease inhibitors (PI) or reverse
transcriptase inhibitors (RTI). In addition, inhibitors that
block different stages of the fusion process will probably not
suffer from cross-resistance, as their molecular targets are
different (Moore and Doms, 2003; Wei et al., 2002). Indeed,
countering otherwise drug-resistant strains is an attractive
rationale for the clinical use of entry inhibitors (Kilby and
Eron, 2003; Moore and Doms, 2003). We have, therefore,
been studying the development of resistance to CCR5
inhibitors in vitro, to gain insights into what might happen
if and when these compounds are used extensively in vivo.
We previously described how a primary R5 isolate, CC1/85,
became resistant to AD101, a prototypic CCR5 inhibitor,
when cultured in primary human T-cells (Kuhmann et al.,
2004; Trkola et al., 2002). We concluded that the resistance
of CC1/85 to AD101 involved the retention of CCR5 use in
an inhibitor-insensitive manner, rather than switching to the
use of the CXCR4 coreceptor. The fully AD101-resistant
virus contained 4 amino acid changes in the V3 region of
gp120, which were necessary and sufficient to confer
resistance when engineered into clonal viruses derived from
CC1/85 (Kuhmann et al., 2004). Escape of a different R5
primary virus, the subtype G strain JV1083, from SCH-C
also involves the sequential evolution of multiple amino acid
changes in V3 that create an inhibitor-insensitive R5 variant
(Riley et al., submitted for publication).
We now describe the generation of two different CC1/85
escape mutants under the selection pressure of SCH-D in
vitro. The input virus for one of the escape cultures was a
variant of CC1/85 that had been selected for limited
resistance to AD101; the other was CC1/85 itself. The fully
SCH-D-resistant (>20,000-fold) viruses emerged over 12
and 16 passages, respectively. Like the fully AD101-
resistant virus, the SCH-D escape mutant remained depend-
ent upon CCR5 for entry into primary CD4+ T-cells, rather
than switching to CXCR4 usage, although some variants
capable of CXCR4 use in T-cell lines were detected at low
frequency. However, unlike what was seen in the AD101
escape study, the development of resistance to SCH-D was
not associated with amino acid substitutions in V3. We also
show that the SCH-D escape mutants are cross-resistant to
several other CCR5 inhibitors, including the modified
chemokine PSC-RANTES. In contrast, their sensitivity to
other entry inhibitors, and to PI and RTI, is unchanged.
Results
Generation of an HIV-1 escape mutant to SCH-D
SCH-D is a piperazino-piperidine-based compound that
binds to the CCR5 receptor with high affinity and is an
antagonist of the natural CC-chemokine ligand, RANTES,
MIP-a and MIP-h. The derivation of SCH-D, its chemical
synthesis and its antiviral properties are all described
elsewhere (Tagat et al., 2001, 2004). In brief, SCH-D inhibits
a wide range of HIV-1 strains from various genetic subtypes,
with inhibitory concentrations in the low nM range (Tagat et
al., 2001, 2004). In comparative studies, SCH-D is typically
10-fold more potent than the earlier generation CCR5
inhibitor, SCH-C (Tagat et al., 2004). SCH-D has comparable
potency in vitro to the AD101 compound used in one of our
earlier escape studies although its pharmaceutic and pharma-
cokinetic properties are markedly superior (Strizki et al.,
submitted for publication; Tagat et al., 2004). SCH-D is now
entering Phase 3 clinical trials after demonstrating significant
antiviral activity in earlier trials (Maeda et al., 2004b).
To generate HIV-1 variants resistant to SCH-D, we used
mitogen-stimulated primary human CD4+ T-cells as the
target for virus replication. Two separate, but related, escape
cultures were initiated. In the first study, the input virus was
HIV-1 CC1/85, a subtype B, R5 primary isolate of USA
origin (Connor et al., 1997). When CC1/85 was isolated
from patient Case C in January 1985, he had a CD4+ T-cell
count of 1038 cells/mm3 and only viruses able to use CCR5
were detectable in his blood. However, R5X4 viruses had
evolved within Case C by February 1986 (Connor et al.,
1997). CC1/85 was the input isolate in our earlier CCR5
inhibitor-resistance selection experiment using AD101
(Kuhmann et al., 2004; Trkola et al., 2002).
The second, simultaneously performed escape experi-
ment used CC101.6 as the input virus. CC101.6 had been
isolated from the passage 6 culture of CC1/85 with AD101
(Kuhmann et al., 2004; Trkola et al., 2002). It is partially (3-
fold) resistant to AD101 (and also to SCH-D, see below).
CC101.6 is also much more genetically homogenous than
CC1/85, reflecting a rapid selection for naturally AD101-
resistant variants from among the initial swarm present in
the CC1/85 isolate (Kuhmann et al., 2004). In particular,
CC101.6 contains a single amino acid change in the V3
region, H308P, which was selected for very early in the
CC1/85 escape culture. We considered it worthwhile to
A.J. Marozsan et al. / Virology 338 (2005) 182–199184
investigate whether the escape process would differ for
these two genetically related input viruses.
The CC1/85 and CC101.6 escape cultures were initiated
by culturing the input viruses in the presence of 500 nM of
SCH-D, a concentration that was approximately 500 and
150 times the previously determined IC50 values of 0.95 nM
and 3.0 nM respectively. The PC cultures were maintained
in exactly the same way as the SCH-D-treated cultures,
except that no inhibitor was present. The use of PC cultures
allows the identification and analysis of genotypic and
phenotypic changes that can occur upon prolonged passage
of HIV-1 in vitro, even in PBMC (Kuhmann et al., 2004;
Pugach et al., 2004). The culture supernatants and cells were
passaged weekly and monitored for HIV-1 replication by
measuring p24 antigen production. The SCH-D concen-
tration was increased whenever virus replication in an
inhibitor-treated culture approached that in the correspond-
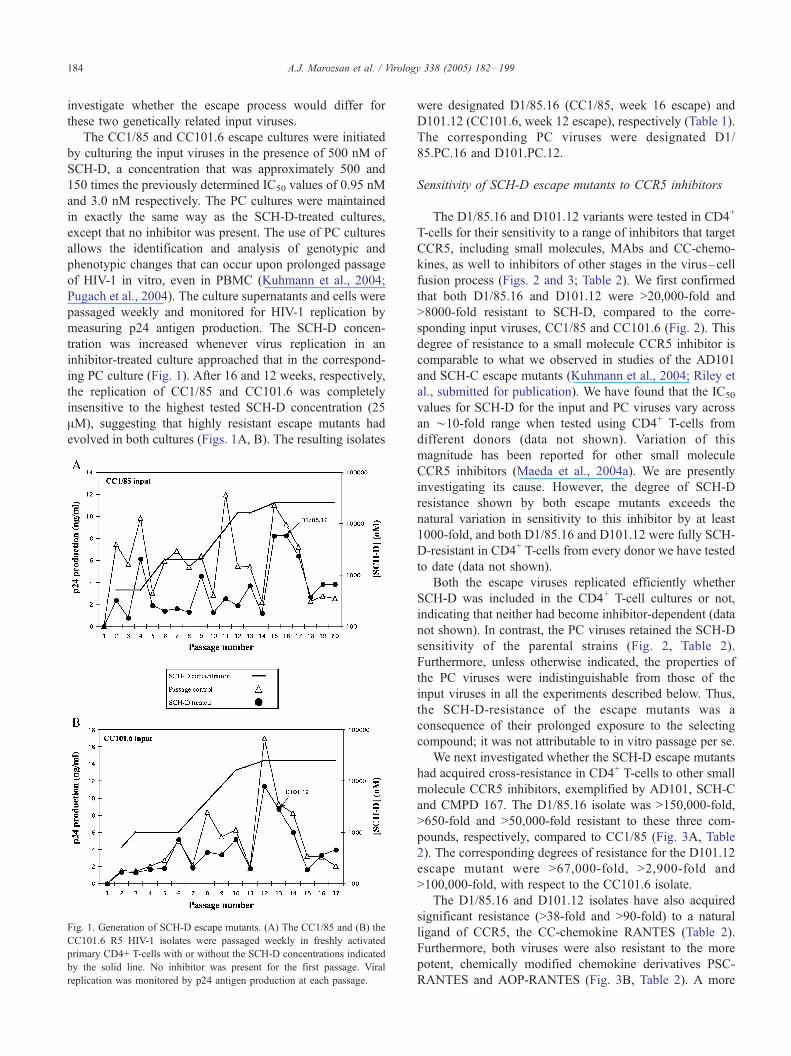
ing PC culture (Fig. 1). After 16 and 12 weeks, respectively,
the replication of CC1/85 and CC101.6 was completely
insensitive to the highest tested SCH-D concentration (25
AM), suggesting that highly resistant escape mutants had
evolved in both cultures (Figs. 1A, B). The resulting isolates
Fig. 1. Generation of SCH-D escape mutants. (A) The CC1/85 and (B) the
CC101.6 R5 HIV-1 isolates were passaged weekly in freshly activated
primary CD4+ T-cells with or without the SCH-D concentrations indicated
by the solid line. No inhibitor was present for the first passage. Viral
replication was monitored by p24 antigen production at each passage.
were designated D1/85.16 (CC1/85, week 16 escape) and
D101.12 (CC101.6, week 12 escape), respectively (Table 1).
The corresponding PC viruses were designated D1/
85.PC.16 and D101.PC.12.
Sensitivity of SCH-D escape mutants to CCR5 inhibitors
The D1/85.16 and D101.12 variants were tested in CD4+
T-cells for their sensitivity to a range of inhibitors that target
CCR5, including small molecules, MAbs and CC-chemo-
kines, as well to inhibitors of other stages in the virus–cell
fusion process (Figs. 2 and 3; Table 2). We first confirmed
that both D1/85.16 and D101.12 were >20,000-fold and
>8000-fold resistant to SCH-D, compared to the corre-
sponding input viruses, CC1/85 and CC101.6 (Fig. 2). This
degree of resistance to a small molecule CCR5 inhibitor is
comparable to what we observed in studies of the AD101
and SCH-C escape mutants (Kuhmann et al., 2004; Riley et
al., submitted for publication). We have found that the IC50
values for SCH-D for the input and PC viruses vary across
an ¨10-fold range when tested using CD4+ T-cells from
different donors (data not shown). Variation of this
magnitude has been reported for other small molecule
CCR5 inhibitors (Maeda et al., 2004a). We are presently
investigating its cause. However, the degree of SCH-D
resistance shown by both escape mutants exceeds the
natural variation in sensitivity to this inhibitor by at least
1000-fold, and both D1/85.16 and D101.12 were fully SCH-
D-resistant in CD4+ T-cells from every donor we have tested
to date (data not shown).
Both the escape viruses replicated efficiently whether
SCH-D was included in the CD4+ T-cell cultures or not,
indicating that neither had become inhibitor-dependent (data
not shown). In contrast, the PC viruses retained the SCH-D
sensitivity of the parental strains (Fig. 2, Table 2).
Furthermore, unless otherwise indicated, the properties of
the PC viruses were indistinguishable from those of the
input viruses in all the experiments described below. Thus,
the SCH-D-resistance of the escape mutants was a
consequence of their prolonged exposure to the selecting
compound; it was not attributable to in vitro passage per se.
We next investigated whether the SCH-D escape mutants
had acquired cross-resistance in CD4+ T-cells to other small
molecule CCR5 inhibitors, exemplified by AD101, SCH-C
and CMPD 167. The D1/85.16 isolate was >150,000-fold,
>650-fold and >50,000-fold resistant to these three com-
pounds, respectively, compared to CC1/85 (Fig. 3A, Table
2). The corresponding degrees of resistance for the D101.12
escape mutant were >67,000-fold, >2,900-fold and
>100,000-fold, with respect to the CC101.6 isolate.
The D1/85.16 and D101.12 isolates have also acquired
significant resistance (>38-fold and >90-fold) to a natural
ligand of CCR5, the CC-chemokine RANTES (Table 2).
Furthermore, both viruses were also resistant to the more
potent, chemically modified chemokine derivatives PSC-
RANTES and AOP-RANTES (Fig. 3B, Table 2). A more
Table 1
Virus nomenclature
Virus Classification Lineage Phenotype
CC1/85 Parental/input Patient
isolate
R5
D1/85.16 Escape CC1/85 R5
D1/85.PC.16 Passage control CC1/85 R5
D1/85.X4 U87.CD4.CXCR4
expanded escape
D1/85.16 R5/X4 or
R5+X4
CC101.6 Input CC1/85 R5
D101.12 Escape CC101.6 R5
D101.PC.12 Passage control CC101.6 R5
Fig. 2. The escape mutants are resistant to SCH-D. Viruses CC1/85, D1/
85.PC, D1/85.16, CC101.6, D101.PC and D101.12 were tested for their
sensitivity to SCH-D in primary CD4+ T-cells. The extent of HIV-1
replication is represented relative to p24 antigen production in the absence
of any inhibitor, which is defined as 100%. Individual data points were the
averages of values derived from 4 separate experiments, each performed
using duplicate wells.
A.J. Marozsan et al. / Virology 338 (2005) 182–199 185
detailed analysis of the resistance to these compounds is
described below.
In contrast to what was observed with the chemokines,
both SCH-D escape mutants were inhibited by the anti-
CCR5 MAbs PA14 and 2D7 that bind to epitopes formed by
the extracellular domains of CCR5 (Figs. 3C, D, Table 2).
Indeed, D101.12 was substantially more sensitive than the
other isolates to 2D7, including the input and PC viruses. It
was completely (100%) inhibited by 2D7 concentrations
above ¨10 Ag/ml (Fig. 3D). The D1/85.16 escape mutant
was only partially inhibited by this concentration of 2D7.
Thus although 50% inhibition was achieved (Table 2), the
extent of inhibition did not exceed ¨60% at the highest 2D7
concentration tested (20 Ag/ml) (Fig. 3D). The reasons for
the hypersensitivity of D101.12 to 2D7, the partial
sensitivity of D1/85.16, and the differences in the inhibitory
activities of PA14 and 2D7 are all now under investigation.
Neither MAb 45523 or 45531 had inhibitory activity against
any of the parental, PC and escape mutant isolates in CD4+
T-cells (data not shown).
Overall, when replicating in CD4+ T-cells, the SCH-D
escape mutants are resistant to some, but not all, agents that
target CCR5.
The SCH-D escape mutants remain sensitive to other classes
of entry and post-entry inhibitors
The SCH-D-resistant viruses also remained sensitive to
compounds targeting other stages of the virus–cell fusion
process. Thus, the IC50 values of D1/85.16 and D101.12 for
CD4-IgG2, a compound that blocks gp120-CD4 binding,
were not significantly different for the escape mutants and
the PC viruses (Fig. 3E, Table 2). A similar observation was
made using T20 and T1249, peptide inhibitors of gp41-
mediated conformational changes that take place after
receptor binding has occurred (Table 2).
Both the SCH-D escape mutants, as well as the
corresponding PC viruses, were comparable to the parental
isolates in their sensitivities to the NRTI zidovudine, the
NNRTI nevirapine, the PI indinavir and the integrase
inhibitor 118-D-24 (Table 2). This implies, not unexpect-
edly, that adaptation to the presence of SCH-D has primarily
involved the HIV-1 env gene, and not other elements of the
viral genome that influence sensitivity to drugs that act post-
entry. Moreover, if any compensatory mutations have arisen
in the pol gene as a result of SCH-D resistance, they have
not overtly affected sensitivity to the RT, PI or integrase
inhibitors that target the pol-encoded enzymes.
The SCH-D escape mutants continue to use CCR5 for entry
into human CD4+ T-cells, with no evidence of coreceptor
switching
The sensitivity of D1/85.16 and D101.12 to the anti-
CCR5 MAb PA14 suggested that both viruses still required
CCR5 to enter CD4+ T-cells, despite their acquired
resistance to small molecule CCR5 inhibitors. To confirm
this supposition, we evaluated their ability to replicate in
PBMC from two donors who are homozygous for the CCR5
32 allele and hence lack functional CCR5 expression. We
also investigated whether the CXCR4-specific small mole-
cule inhibitor, AMD3100, affected replication of the escape
variants.
The two SCH-D escape mutants were unable to replicate
to significant levels in PBMC from either CCR5-D32
homozygous donor under conditions in which they did so
efficiently in PBMC from a normal (i.e., CCR5 wild-type)
donor. In contrast, the reference viruses NL4-3 (X4), DH123
(R5X4) and CC2/86 (R5X4) all replicated to high levels in
cells from both the CCR5 wild type and CCR5-D32
homozygous donors (Fig. 4A). The escape mutants and
the input and PC viruses were all insensitive to AMD3100
in normal donor PBMC (Fig. 4B). Furthermore, the addition
of a fixed, saturating concentration of AMD3100 (1 AM) did
not significantly affect the replication of D1/85.16 or
D101.12 to PA14 in normal donor PBMC (data not shown).
Under the same experimental conditions, the various X4 and

Fig. 3. Sensitivity of the SCH-D escape mutants to other entry inhibitors. Viruses CC1/85, D1/85.PC, D1/85.16, CC101.6, D101.PC and D101.12 were tested
for their sensitivity to (A) SCH-C, (B) PSC-RANTES, (C) PA14, (D) 2D7, (E) CD4-IgG2 in primary CD4+ T-cells. The extent of viral replication is represented
relative to p24 antigen production in the absence of any inhibitor, which is defined as 100%. Individual data points were the averages of values derived from 4
separate experiments, each performed using duplicate wells.
A.J. Marozsan et al. / Virology 338 (2005) 182–199186
R5X4 reference viruses were all inhibited in the expected
manner by AMD3100 in PBMC from both CCR5 wild-type
and CCR5-D32 donors (Figs. 4A, B).
Taken together, these various experiments show that the
SCH-D escape mutants are both completely dependent upon
CCR5 for entry into human PBMC, and that neither virus
has acquired the ability to use CXCR4 (or any other
coreceptor) to enter these cells.
Replication of SCH-D escape mutants in
coreceptor-expressing cell lines
The coreceptor usage profiles of the SCH-D escape
mutants were further analyzed using GHOST cell lines that
each expressed CD4 and a different putative HIV-1
coreceptor. The D101.12 and D1/85.16 escape mutants,
the CC101.6 input isolate and the D101.PC.12 virus all
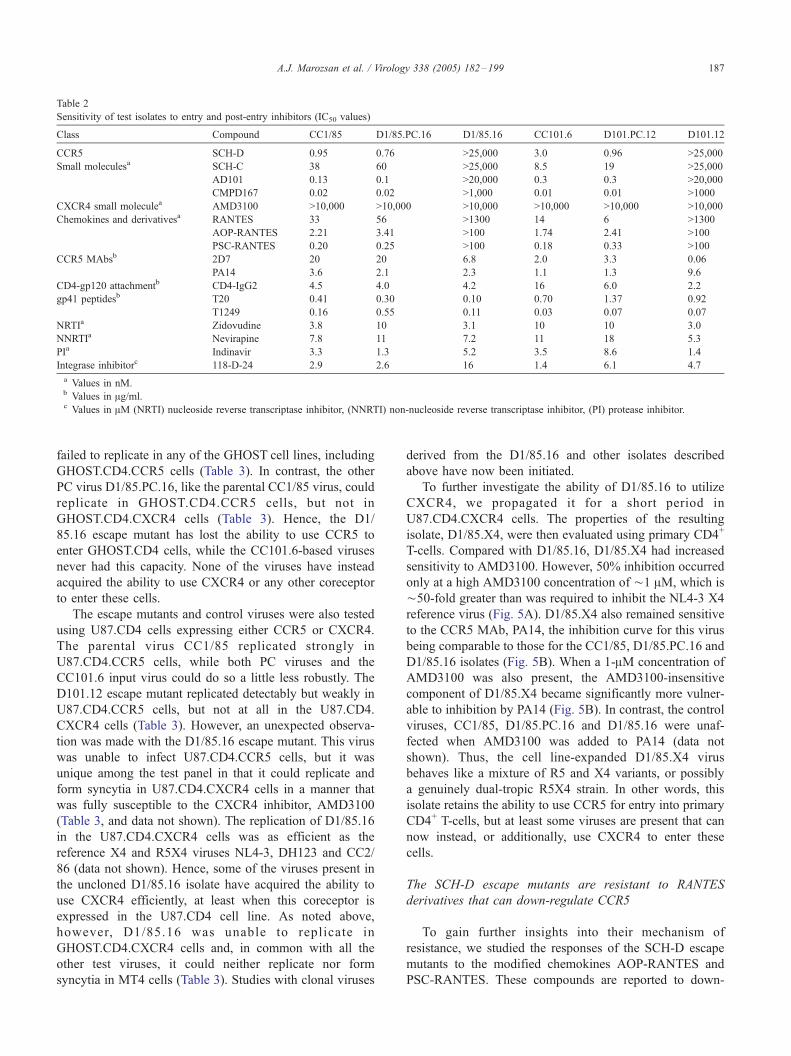
Table 2
Sensitivity of test isolates to entry and post-entry inhibitors (IC50 values)
Class Compound CC1/85 D1/85.PC.16 D1/85.16 CC101.6 D101.PC.12 D101.12
CCR5 SCH-D 0.95 0.76 >25,000 3.0 0.96 >25,000
Small moleculesa SCH-C 38 60 >25,000 8.5 19 >25,000
AD101 0.13 0.1 >20,000 0.3 0.3 >20,000
CMPD167 0.02 0.02 >1,000 0.01 0.01 >1000
CXCR4 small moleculea AMD3100 >10,000 >10,000 >10,000 >10,000 >10,000 >10,000
Chemokines and derivativesa RANTES 33 56 >1300 14 6 >1300
AOP-RANTES 2.21 3.41 >100 1.74 2.41 >100
PSC-RANTES 0.20 0.25 >100 0.18 0.33 >100
CCR5 MAbsb 2D7 20 20 6.8 2.0 3.3 0.06
PA14 3.6 2.1 2.3 1.1 1.3 9.6
CD4-gp120 attachmentb CD4-IgG2 4.5 4.0 4.2 16 6.0 2.2
gp41 peptidesb T20 0.41 0.30 0.10 0.70 1.37 0.92
T1249 0.16 0.55 0.11 0.03 0.07 0.07
NRTIa Zidovudine 3.8 10 3.1 10 10 3.0
NNRTIa Nevirapine 7.8 11 7.2 11 18 5.3
PIa Indinavir 3.3 1.3 5.2 3.5 8.6 1.4
Integrase inhibitorc 118-D-24 2.9 2.6 16 1.4 6.1 4.7
a Values in nM.b Values in Ag/ml.c Values in AM (NRTI) nucleoside reverse transcriptase inhibitor, (NNRTI) non-nucleoside reverse transcriptase inhibitor, (PI) protease inhibitor.
A.J. Marozsan et al. / Virology 338 (2005) 182–199 187
failed to replicate in any of the GHOST cell lines, including
GHOST.CD4.CCR5 cells (Table 3). In contrast, the other
PC virus D1/85.PC.16, like the parental CC1/85 virus, could
replicate in GHOST.CD4.CCR5 cells, but not in
GHOST.CD4.CXCR4 cells (Table 3). Hence, the D1/
85.16 escape mutant has lost the ability to use CCR5 to
enter GHOST.CD4 cells, while the CC101.6-based viruses
never had this capacity. None of the viruses have instead
acquired the ability to use CXCR4 or any other coreceptor
to enter these cells.
The escape mutants and control viruses were also tested
using U87.CD4 cells expressing either CCR5 or CXCR4.
The parental virus CC1/85 replicated strongly in
U87.CD4.CCR5 cells, while both PC viruses and the
CC101.6 input virus could do so a little less robustly. The
D101.12 escape mutant replicated detectably but weakly in
U87.CD4.CCR5 cells, but not at all in the U87.CD4.
CXCR4 cells (Table 3). However, an unexpected observa-
tion was made with the D1/85.16 escape mutant. This virus
was unable to infect U87.CD4.CCR5 cells, but it was
unique among the test panel in that it could replicate and
form syncytia in U87.CD4.CXCR4 cells in a manner that
was fully susceptible to the CXCR4 inhibitor, AMD3100
(Table 3, and data not shown). The replication of D1/85.16
in the U87.CD4.CXCR4 cells was as efficient as the
reference X4 and R5X4 viruses NL4-3, DH123 and CC2/
86 (data not shown). Hence, some of the viruses present in
the uncloned D1/85.16 isolate have acquired the ability to
use CXCR4 efficiently, at least when this coreceptor is
expressed in the U87.CD4 cell line. As noted above,
however, D1/85.16 was unable to replicate in
GHOST.CD4.CXCR4 cells and, in common with all the
other test viruses, it could neither replicate nor form
syncytia in MT4 cells (Table 3). Studies with clonal viruses
derived from the D1/85.16 and other isolates described
above have now been initiated.
To further investigate the ability of D1/85.16 to utilize
CXCR4, we propagated it for a short period in
U87.CD4.CXCR4 cells. The properties of the resulting
isolate, D1/85.X4, were then evaluated using primary CD4+
T-cells. Compared with D1/85.16, D1/85.X4 had increased
sensitivity to AMD3100. However, 50% inhibition occurred
only at a high AMD3100 concentration of ¨1 AM, which is
¨50-fold greater than was required to inhibit the NL4-3 X4
reference virus (Fig. 5A). D1/85.X4 also remained sensitive
to the CCR5 MAb, PA14, the inhibition curve for this virus
being comparable to those for the CC1/85, D1/85.PC.16 and
D1/85.16 isolates (Fig. 5B). When a 1-AM concentration of
AMD3100 was also present, the AMD3100-insensitive
component of D1/85.X4 became significantly more vulner-
able to inhibition by PA14 (Fig. 5B). In contrast, the control
viruses, CC1/85, D1/85.PC.16 and D1/85.16 were unaf-
fected when AMD3100 was added to PA14 (data not
shown). Thus, the cell line-expanded D1/85.X4 virus
behaves like a mixture of R5 and X4 variants, or possibly
a genuinely dual-tropic R5X4 strain. In other words, this
isolate retains the ability to use CCR5 for entry into primary
CD4+ T-cells, but at least some viruses are present that can
now instead, or additionally, use CXCR4 to enter these
cells.
The SCH-D escape mutants are resistant to RANTES
derivatives that can down-regulate CCR5
To gain further insights into their mechanism of
resistance, we studied the responses of the SCH-D escape
mutants to the modified chemokines AOP-RANTES and
PSC-RANTES. These compounds are reported to down-

Fig. 4. The SCH-D escape mutants do not use CXCR4 for entry into
PBMC. (A) The escape mutants and the PC viruses were assessed for their
abilities to replicate in PBMC from CCR5-D32 homozygous and CCR5
wild-type donors, in the presence and absence of the CXCR4 inhibitor
AMD3100 (1 AM). The reference viruses NL4-3 (X4), CC1/85 (parental
strain, R5), DH123 (R5X4) and CC2/86 (R5X4) were also tested. The
extent of HIV-1 replication was determined by the amount of p24 antigen
production on day 14. Similar results were obtained using PBMC from a
second CCR5-D32 homozygous donor (not shown). (B) The SCH-D escape
mutants, the PC viruses, the parental virus CC1/85 and the X4 reference
virus NL4-3 were tested for their abilities to replicate in normal donor
primary CD4+ T-cells in the presence of the AMD3100 concentrations
indicated. The extent of HIV-1 replication is expressed relative to what
occurred in the absence of AMD3100 (=100%).
A.J. Marozsan et al. / Virology 338 (2005) 182–199188
regulate a substantial fraction of cell surface CCR5, as well
as binding the receptor with greater affinity than RANTES.
Hence, the chemokine derivatives are significantly more
Table 3
Replication of SCH-D escape mutants and control isolates in coreceptor-expressi
U87.CD4 GHOST.CD4
Virus CCR5 CXCR4 CCR5 CXCR4 BOB BONZO
CC1/85 +++ – ++ – – –
D1/85.PC ++ – + – – –
D1/85.16 – ++* – – – –
CC101.6 ++ – – – – –
D101.PC ++ – – – – –
D101.12 + – – – – –
+ Weak replication.
++ Strong replication.
* Replication blocked by 1 AM AMD3100.
effective than the natural RANTES molecule at inhibiting
R5 virus replication (Hartley et al., 2004; Kawamura et al.,
2004; Simmons et al., 1997; Torre et al., 2000). However,
we noted above that both the D1/85.16 and D101.12 escape
mutants were substantially resistant to both AOP-RANTES
and PSC-RANTES when replicating in primary CD4+ T-
cells (Fig. 3B, Table 2). Thus, D1/85.16 replication was not
affected detectably by PSC-RANTES, whereas D101.12
was only partially inhibited at the highest PSC-RANTES
concentration (Fig. 3B). The resistance displayed by the two
SCH-D escape mutants stands in contrast to the PSC-
RANTES sensitivity of not just the parental and PC viruses,
but also of the AD101 escape isolate, CC101.19 (Fig. 3B,
Table 2 and data not shown). Hence, the SCH-D and AD101
escape mutants differ significantly in their sensitivities to the
modified chemokines, despite their shared genetic origin
and otherwise similar phenotypes, and despite the general
chemical similarity of SCH-D and AD101. Again, we could
find no evidence that the SCH-D escape mutants were using
CXCR4 to enter CD4+ T-cells after PSC-RANTES treat-
ment because AMD3100 (1 AM) did not inhibit their
replication even in the presence of 100 nM PSC-RANTES
(data not shown).
We concluded above that D1/85.16 and D101.12 were
still predominantly or exclusively using CCR5 for repli-
cation in primary CD4+ T-cells. Yet, they are resistant to
compounds that apparently down-regulate a very substantial
fraction of CCR5 from the cell surface (Hartley et al., 2004;
Kawamura et al., 2004; Simmons et al., 1997; Torre et al.,
2000). To investigate this apparent paradox, we studied the
effect of RANTES and its derivatives on the cell surface
exposure of CCR5 on normal donor PBMC, using flow
cytometry and several MAbs previously described as being
CCR5-specific. Nine days after mitogen activation, the
staining of CCR5 on PBMC by the 2D7 MAb was reduced
after 1 h of incubation at 3 -C with 640 nM of RANTES,
100 nM of AOP-RANTES or PSC-RANTES (Fig. 6). In the
case of PSC-RANTES, the level of 2D7 staining was almost
undetectable (<2% positive cells).
We then repeated the above experiment but using two
other anti-CCR5 MAbs, 45531 or 45523, in place of 2D7.
MAb 45523 differs from 45531 and 2D7 in that it only
ng cell lines
MT4
US28 GPR1 CCR1 CCR2 CCR3 CCR8 CXCR4
– – – – – – –
– – – – – – –
– – – – – – –
– – – – – – –
– – – – – – –
– – – – – – –
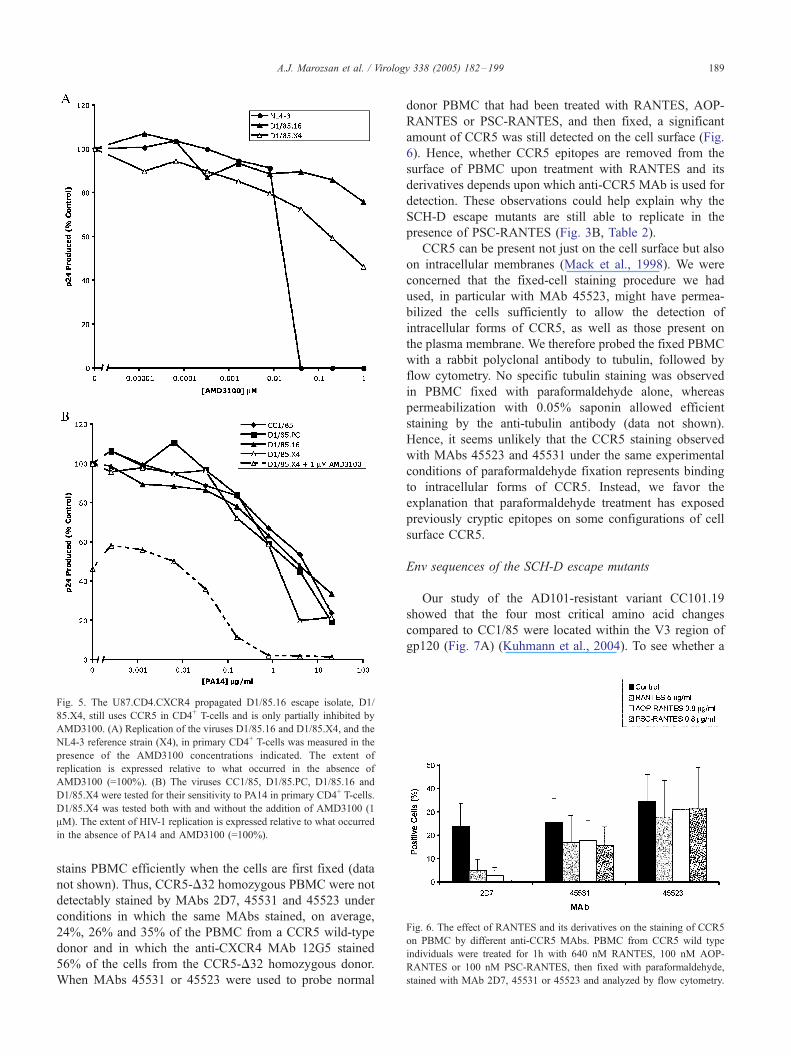
Fig. 5. The U87.CD4.CXCR4 propagated D1/85.16 escape isolate, D1/
85.X4, still uses CCR5 in CD4+ T-cells and is only partially inhibited by
AMD3100. (A) Replication of the viruses D1/85.16 and D1/85.X4, and the
NL4-3 reference strain (X4), in primary CD4+ T-cells was measured in the
presence of the AMD3100 concentrations indicated. The extent of
replication is expressed relative to what occurred in the absence of
AMD3100 (=100%). (B) The viruses CC1/85, D1/85.PC, D1/85.16 and
D1/85.X4 were tested for their sensitivity to PA14 in primary CD4+ T-cells.
D1/85.X4 was tested both with and without the addition of AMD3100 (1
AM). The extent of HIV-1 replication is expressed relative to what occurred
in the absence of PA14 and AMD3100 (=100%).
Fig. 6. The effect of RANTES and its derivatives on the staining of CCR5
on PBMC by different anti-CCR5 MAbs. PBMC from CCR5 wild type
individuals were treated for 1h with 640 nM RANTES, 100 nM AOP-
RANTES or 100 nM PSC-RANTES, then fixed with paraformaldehyde,
stained with MAb 2D7, 45531 or 45523 and analyzed by flow cytometry.
A.J. Marozsan et al. / Virology 338 (2005) 182–199 189
stains PBMC efficiently when the cells are first fixed (data
not shown). Thus, CCR5-D32 homozygous PBMC were not
detectably stained by MAbs 2D7, 45531 and 45523 under
conditions in which the same MAbs stained, on average,
24%, 26% and 35% of the PBMC from a CCR5 wild-type
donor and in which the anti-CXCR4 MAb 12G5 stained
56% of the cells from the CCR5-D32 homozygous donor.
When MAbs 45531 or 45523 were used to probe normal
donor PBMC that had been treated with RANTES, AOP-
RANTES or PSC-RANTES, and then fixed, a significant
amount of CCR5 was still detected on the cell surface (Fig.
6). Hence, whether CCR5 epitopes are removed from the
surface of PBMC upon treatment with RANTES and its
derivatives depends upon which anti-CCR5 MAb is used for
detection. These observations could help explain why the
SCH-D escape mutants are still able to replicate in the
presence of PSC-RANTES (Fig. 3B, Table 2).
CCR5 can be present not just on the cell surface but also
on intracellular membranes (Mack et al., 1998). We were
concerned that the fixed-cell staining procedure we had
used, in particular with MAb 45523, might have permea-
bilized the cells sufficiently to allow the detection of
intracellular forms of CCR5, as well as those present on
the plasma membrane. We therefore probed the fixed PBMC
with a rabbit polyclonal antibody to tubulin, followed by
flow cytometry. No specific tubulin staining was observed
in PBMC fixed with paraformaldehyde alone, whereas
permeabilization with 0.05% saponin allowed efficient
staining by the anti-tubulin antibody (data not shown).
Hence, it seems unlikely that the CCR5 staining observed
with MAbs 45523 and 45531 under the same experimental
conditions of paraformaldehyde fixation represents binding
to intracellular forms of CCR5. Instead, we favor the
explanation that paraformaldehyde treatment has exposed
previously cryptic epitopes on some configurations of cell
surface CCR5.
Env sequences of the SCH-D escape mutants
Our study of the AD101-resistant variant CC101.19
showed that the four most critical amino acid changes
compared to CC1/85 were located within the V3 region of
gp120 (Fig. 7A) (Kuhmann et al., 2004). To see whether a

A.J. Marozsan et al. / Virology 338 (2005) 182–199190
similar pattern of genetic changes had occurred during the
development of SCH-D resistance by the same virus, we
made clonal, SCH-D-resistant viruses by cassetting env
genes from the resistant isolates into the NL4-3 background
(Kuhmann et al., 2004; Marozsan and Arts, 2003; Marozsan
et al., 2005). The same procedure was used to make clonal,
chimeric viruses from the PC isolates (Marozsan and Arts,
2003; Marozsan et al., 2005). The V3 sequences of the

A.J. Marozsan et al. / Virology 338 (2005) 182–199 191
chimeric viruses were determined, as were IC50 values for
SCH-D inhibition of the replication of some of the chimeras
in primary CD4+ T-cells (Fig. 7A).
The SCH-D resistant clone of D1/85.16 had an identical
V3 sequence to the parental CC1/85 virus, but was
essentially completely resistant to SCH-D (Fig. 7A). A
single change, T319A, was present in the V3 region of the
clonal PC virus, D1/85.PC.16. This same change was
observed in 2/12 clones derived from the control virus
from passage 20 in the AD101 escape experiment, and may
represent an adaptation to long-term culture in PBMC
(Kuhmann et al., 2004). The H308P change in the CC101.6
virus that conferred partial resistance to AD101 was retained
in the SCH-D-resistant clone D101.12, but the additional 3
substitutions that conferred full AD101 resistance, shown in
the CC101.19 cl.7 sequence, had not arisen (Fig. 7A). The
AD101-resistant clone was found to also be resistant to
SCH-D (Fig. 7A), presumably due to these 4 changes in V3.
The H308P substitution had reverted back to the wild-type
sequence in the passage control clone from D101.PC.12,
suggesting that the original change was not always stable
during prolonged culture (Fig. 7A).
The clonal viruses derived from both D1/85.16 and
D101.12 were fully resistant to SCH-D (IC50 > 25 AM) in
primary CD4+ T-cells (Fig. 7A). In contrast, the clone
derived from CC1/85 was unusually sensitive to SCH-D,
something we have previously observed in studies of the
AD101 sensitivity of clonal viruses (Kuhmann et al., 2004).
Replication of the presently available D1/85.PC.16 and
CC101.6 clones was too limited for determination of IC50
values (Fig. 7A).
To obtain a fuller understanding of why the D1/85.16 and
D101.12 env clones were resistant to SCH-D, we sequenced
the region from the C1 domain of gp120 through end of the
ectodomain of gp41 (Fig. 7B). It is reasonable to assume
that resistance to SCH-D maps to this surface-exposed
region of Env.
In total, there were 17 amino acid differences between
the depicted CC1/85 and D1/85.16 clones (Fig. 7B). Of
these, 9 occur in gp120 and are shown in Fig. 7C. Position
146 in the V1 region is variable in wild-type CC1/85 clones,
2/8 clones having a glutamic acid residue at this position, 6/
Fig. 7. Sequences and SCH-D sensitivity of clonal viruses. (A) The amino acid sequ
previously sequenced from the CC1/85 isolate, CC1/85 cl.6 (GenBank accession n
this study. Of the CC1/85 clones sequenced in our previous study, 7/8 had identica
the CC101.6 isolate, CC101.6 cl.2 (GenBank accession number AY357371) is th
CC101.6 clones sequenced in our previous study, 9/11 had identical V3 sequence
derived from the AD101-resistant isolate CC101.19. The sequences are recorded
sequence identity. The IC50 values in CD4+ T-cells for SCH-D inhibition of clonal
the right of the sequences. ND=not done. (B) The amino acid sequence of gp120 a
As in (A), CC1/85 cl.6 is used as a reference, dashes indicate identity in the aligne
beginning and end of gp120, the beginning of gp41, and the beginning and end of
numbers indicate the amino acid number, which by convention is based on that o
amino acid to which it refers. The sequences for the D1/85.16 and D101.12
recombination in the yeast system used to create these clones. The alignment ends i
from (B) within the gp120 subunit that either differ between CC1/85 cl.6 and the D
along with the amino acid number within Env (again, based on HXBc2 numberi
8 a lysine (Kuhmann et al., 2004). The selection of K146 in
the D1/85.16 clone may then represent a founder effect.
Glutamic acid at position 146 became dominant at early
passages in the generation of the AD101 escape mutant, and
so the CC101.6 cl.2 and D101.12 clones contain this residue
(Figs. 7B, C) (Kuhmann et al., 2004). Likewise, position
167 in V2 is variable in the CC1/85 clones. 2/8 CC1/85
clones have asparagine at amino acid 167, the remainder
have aspartic acid, and asparagine was found to be present
in the D1/85.16 clone. The remaining substitutions in V2,
V169M and K171R, were not found in the wild-type or PC
viruses in this or our previous study (Figs. 7B, C)
(Kuhmann et al., 2004). Residue 351 in C3 is also variable
in CC1/85 clones. Indeed, CC1/85 cl.6 is the only clone
containing lysine, the other 7 having glutamic acid. The
other changes in C3, G354P and N355D (deletion of N355),
were not seen in any of the sequenced CC1/85 clones (Figs.
7B, C) (Kuhmann et al., 2004). The remaining 8 sequence
differences between D1/85.16 and CC1/85 cl.6 are in the
ectodomain of gp41 (Fig. 7B). Three of these are
conservative substitutions of hydrophobic residues in the
gp41 fusion peptide, the remaining 5 are at locations that are
variable in CC1/85 and represent alternative amino acids
found at those positions in other CC1/85 clones (Fig. 7B)
(Kuhmann et al., 2004).
The CC101.6 and D101.12 clones differ by only 2 amino
acids in gp120 and 4 in gp41 (Figs. 7B,C). The N406K
substitution in the V4 loop results in the loss of a potential
N-linked glycosylation site. The G474D substitution in
D101.12 relative to CC101.6 cl.2 occurs because CC101.6
cl.2 contains glycine at amino acid 474, whereas all the
other sequenced CC101.6 clones, as well as all CC1/85
clones, have aspartic acid (Fig. 7B) (Kuhmann et al., 2004).
Two hydrophobic substitutions are found in the gp41 fusion
peptide and two additional changes are at amino acids 534
and 535. Serine is found at position 534 in the D101.12
clone and 7/8 CC1/85 clones but is alanine is all of the
available CC101.6 clones. The V535M substitution is not
found in any of the CC1/85 or CC101.6 clones (Fig. 7B)
(Kuhmann et al., 2004). Which of the amino acid changes in
the SCH-D-resistant clones account for the escape mutant
phenotype has not yet been determined.
ences of the V3 region from 7 env clones are shown. Among the env clones
umber AY357338) is the most closely related to the D1/85.16 clone used in
l V3 sequences. Likewise, among the env clones previously sequenced from
e one most closely related to the D101.12 clone used in this study. Of the
s. CC101.19 cl.7 (GenBank accession number AY357465) is an env clone
relative to that of CC1/85 cl.6 (top line) with dashes indicating amino acid
viruses containing these env genes in the NL4-3 background are recorded on
nd the ectodomain of gp41 from four of the env clones from (A) are aligned.
d sequences and a dot indicates a gap in the alignment. The positions of the
each conserved and variable region are indicated above the alignment. The
f the prototypic strain HXBc2; the last digit of the number aligns with the
clones begin at amino acid 42 because this is the point of homologous
mmediately before the transmembrane domain in gp41. (C) The amino acids
1/85.16 clone or differ between CC101.6 and the D101.12 clone are shown,
ng) and the region in which that amino acid is found.

A.J. Marozsan et al. / Virology 338 (2005) 182–199192
Discussion
In our previous reports on how HIV-1 escapes from the
selection pressure exerted by small molecule CCR5
inhibitors in vitro, we concluded that the predominant
pathway involved the generation of viral variants that
retained the ability to use CCR5, but in an inhibitor-
insensitive manner (Kuhmann et al., 2004; Riley et al.,
submitted for publication; Trkola et al., 2002). Thus, the
AD101-resistant CC1/85 variants were unable to replicate in
PBMC from CCR5-D32 homozygous donors; inhibited by
CCR5-specific MAbs; insensitive to CXCR4 inhibitors; and
unable to use any coreceptor other than CCR5 to enter
indicator cell lines (Kuhmann et al., 2004; Trkola et al.,
2002). We have since made similar findings using another
primary isolate, JV1083 and SCH-C, albeit with CCR5-
expressing PM1 cells substituting for PBMC (Strizki et al.,
submitted for publication).
The present study, again performed using the CC1/85
primary isolate and PBMC, but with a different inhibitor,
SCH-D, has generated some findings that are consistent
with the previous experiments, but also some unexpected
ones. Two escape mutants were made, one from CC1/85, the
second from a partially (¨3-fold) resistant variant,
CC101.6. The latter virus is more genetically homogenous
than CC1/85, reflecting a rapid selection for naturally
resistant variants during the initial stages of the AD101
resistance-selection process (Kuhmann et al., 2004; Trkola
et al., 2002). SCH-D-resistant viruses were generated from
both input isolates over a comparable number of passages,
16 for CC1/85, 12 for CC101.6. The escape mutants were
completely resistant to SCH-D and also to other small
molecule CCR5 inhibitors: SCH-C, AD101 and CMPD 167.
These compounds are from different chemical families
(Shen et al., 2004). However, all the small molecule
inhibitors probably bind to a broadly similar site within
CCR5’s transmembrane helices (Tsamis et al., 2003). Cross-
resistance within the drug class would be the expected
outcome if these compounds were used clinically, although
it is not necessarily inevitable. In contrast, the SCH-D
escape mutants retained the sensitivity of the parental
isolates to other entry inhibitors, CD4-IgG2 (PRO 542), T-
20 and T-1249, with different mechanisms of action. There
was also no change in their susceptibility to RT, protease or
integrase inhibitors. Hence, as expected, the small molecule
CCR5 inhibitors are unlikely to cause cross-resistance to
other classes of licensed drugs (Moore and Doms, 2003).
Both the SCH-D escape mutants are R5 viruses. Thus,
they lack any ability to replicate in PBMC from CCR5-D32
homozygous individuals; they are inhibited by CCR5-
specific MAbs in CD4+ T-cells from normal donors; and
they have no detectable sensitivity to the CXCR4-specific
inhibitor AMD3100 in the same cells. In this regard, they
resemble the AD101- and SCH-C-escape mutants (Kuh-
mann et al., 2004; Riley et al., submitted for publication;
Trkola et al., 2002). However, the SCH-D-resistant variants
did possess some unusual properties when evaluated in
cell lines expressing various putative coreceptors. For
example, D1/85.16 and D101.12 could not replicate in
GHOST.CD4.CCR5 cells, nor could the PC virus,
D101.PC.12. In contrast, the other PC virus, D1/85.PC.16,
resembled the input CC1/85 isolate by retaining the ability
to replicate in GHOST.CD4.CCR5 cells. The reasons
underlying this pattern of viral tropism remain uncertain
pending additional studies.
Of particular note is that none of the escape mutants or PC
viruses could replicate in any other GHOST.CD4 cell line
that we tested, including GHOST.CD4.CXCR4 cells. The
D101.12 escape mutant was also unable to replicate in
U87.CD4.CXCR4 cells, although it could do so in
U87.CD4.CCR5 cells, albeit only at low levels. However,
the D1/85.16 escape mutant behaved unexpectedly, in that it
could replicate and form syncytia in U87.CD4.CXCR4 cells,
but not in U87.CD4.CCR5 cells. Replication in the
U87.CD4.CXCR4 cells was blocked by AMD3100, suggest-
ing that D1/85.16 was using CXCR4 to enter these cells, and
not any other coreceptor. When the D1/85.16 variant was
propagated for a short period in U87.CD4.CXCR4 cells, then
tested on primary CD4+ T-cells, its properties resembled an
R5X4 virus or a mixture of R5 and X4 variants.
It seems likely then that the swarm of viruses in the D1/
85.16 isolate includes at least some with atypical coreceptor
usage properties compared to the input and PC viruses. The
presence of these variants allows low-level replication in
U87.CD4.CXCR4 cells, but not in GHOST.CD4.CXCR4
cells. Furthermore, the D1/85.16 isolate could not replicate
or form syncytia in MT4 cells, so by the classical definition,
it would not be a syncytium-inducing or SI virus (Moore et
al., 2004; Schuitemaker et al., 1991) Presumably, the
amount or configuration of CXCR4 on the GHOST cells
differs critically from what is present on the U87 cells. The
replication of primary isolates, whether of the R5 or X4
phenotype, in coreceptor-transfected cell lines is frequently
discordant with their usage of CCR5, CXCR4 or other
coreceptors in primary cells (McKnight et al., 1997; Trkola
et al., 1999; Yi et al., 2005; Zhang et al., 1998, 2000). Such
replication restrictions may also apply to different cell lines.
Without deliberate propagation of the D1/85.16 isolate
on U87.CD4.CXCR4 cells, the CXCR4-using variants were
either present at too low a frequency to permit a detectable
level of replication in primary CD4+ T-cells, or they were
unable to use CXCR4 in the configuration or quantity
present on these cells. Genetic sequencing revealed an
unusual amino acid change, G314R, in the crown of the V3
region from some of the clones from the U87.CD4.CXCR4
cell expanded stock (D1/85.X4) (data not shown). The
functional significance of this change is now under
investigation. The selection pressure of SCH-D was, then,
able to drive the emergence of at least some viruses able to
use CXCR4 under some conditions, in vitro, an outcome
that needs to be considered in relation to ongoing clinical
trials of CR5 inhibitors for HIV-1 therapy. However, we did

A.J. Marozsan et al. / Virology 338 (2005) 182–199 193
not observe any usage of CXCR4 by viruses generated in
the other SCH-D resistance culture or by the AD101 or
SCH-C escape mutants, suggesting that switching to
CXCR4 use is not a common pathway for escape from
CCR5 inhibitors (Kuhmann et al., 2004; Riley et al.,
submitted for publication; Trkola et al., 2002).
The paramount property of both SCH-D escape mutants
was, however, their retention of CCR5 usage in primary
CD4+ T-cells. It still remains uncertain exactly how this was
achieved. We initially hypothesized that a CCR5-dependent
escape mutant could either have acquired an increased
affinity for CCR5 that enabled it to out-compete the inhibitor,
or else the ability to use the coreceptor despite the presence of
the inhibitor (Trkola et al., 2002). We obtained evidence that
an increased affinity for CCR5 could occur in the early stages
of the AD101 escape process, but this was associated with
only a modest degree of resistance (¨3-fold). Intuitively, it
seems unlikely that resistance as profound as we have
observed (i.e., >20,000-fold) could arise by this mechanism;
the affinity gain required would be colossal.
Recognition of the inhibitor-bound form of CCR5
remains a plausible, but unconfirmed, escape mechanism.
We have, however, shown that SCH-C can interact with the
macaque form of CCR5 without inhibiting SIV or SHIV
entry, an event explained by a single amino acid difference,
I198M, between the human and macaque coreceptors that is
located outside the inhibitor-binding site (Billick et al.,
2004). On the basis of this and related structure–function
studies, we suggested that small molecule CCR5 inhibitors
either prevent the coreceptor from adopting a configuration
that can be recognized by HIV-1, or stabilize a CCR5
conformation that the virus cannot use (Billick et al., 2004;
Tsamis et al., 2003). Perhaps, the SCH-D escape mutants
have acquired the ability to use the latter configuration.
An alternative escape process is that the mutant virus has
evolved to use a particular configuration of CCR5 to which
SCH-D does not bind (Kuhmann et al., 2004). This CCR5
isoform would either not be recognized by the parental
virus or else it is present at only very low levels because
otherwise SCH-D would have no activity against the
parental virus (or R5 viruses in general). However, some
rare R5 HIV-1 isolates are intrinsically relatively resistant to
small molecule CCR5 inhibitors (Strizki et al., 2001), and
there is variation among SIV-infected macaques in their
response to CCR5 inhibitor therapy (Veazey et al., 2003;
Wolinsky et al., 2004). Viruses, and perhaps also hosts, that
are naturally resistant to CCR5 inhibitors might utilize a
mechanism for Env-CCR5 interaction that is analogous to
the one(s) acquired by the in vitro-selected SCH-D escape
mutants. There is, in fact, evidence based on MAb-
reactivity patterns for the presence of multiple CCR5
configurations on primary cells (Blanpain et al., 2002; Hill
et al., 1998; Lee et al., 1999; Olson et al., 1999). This
evidence also applies to CXCR4 (Baribaud et al., 2001;
Carnec et al., 2005). Studies of G-protein-coupled receptors
in general provide strong support for the existence of these
proteins in alternative allosteric conformations (Kenakin,
2004; Seibert and Sakmar, 2004). Soluble gp120 from the
AD101 escape mutant was unable to bind to human CCR5
expressed on transfected murine fibroblasts, an observation
that might be attributable to the presence of different CCR5
configurations on different cell types, although other
possibilities exist (Kuhmann et al., 2004). We have not
yet performed such studies with gp120 from the SCH-D
escape variants.
The resistance to AOP-RANTES and PSC-RANTES
displayed by the SCH-D escape mutants, but curiously not
by the AD101-resistant virus, might also be consistent with
the hypothesis that cell surface CCR5 exists in multiple
antigenic forms that are recognized differently not just by
various anti-CCR5 MAbs, but also by HIV-1 phenotypic
isolates. PSC-RANTES treatment of PBMC reduced the
staining of cell surface CCR5 proteins by MAb 2D7 by
over 98% within 1 h, yet this treatment did not prevent the
replication of the SCH-D escape mutants. However, MAbs
45523 and 45531 still bound significantly to the surface of
PSC-RANTES-treated cells, at ¨40–60% of the baseline
level. All three MAbs are specific for CCR5; they did not
stain PBMC from CCR5-D32 homozygous individuals.
Perhaps, then, the escape mutants recognize a CCR5
configuration that is also bound by MAbs 45523 and
45531 and which is not affected by PSC-RANTES? At
present, we do not know what such a CCR5 conformation
might be, but one clue for future studies is provided by the
observation that MAb 45523 only binds to CCR5 wild
type PBMC after fixation of the cells. Hence, the 45523
epitope may normally be cryptic, perhaps occluded by
another cell surface molecule or buried within a CCR5
hetero- or homo-dimer. This epitope is formed from
multiple CCR5 domains (Lee et al., 1999). MAb 2D7,
on the other hand, binds to live cells and fixation does not
affect its epitope, which is located principally within the
second extracellular (2-A) loop of CCR5 (Lee et al., 1999;
Olson et al., 1999). The 45531 epitope is also located in
this loop, but in a different area of it (2-B) (Lee et al.,
1999). Thus, the epitopes for the above three MAbs may
partially overlap but they are not identical, allowing the
possibility that they can each recognize different CCR5
configurations to various extents.
The above scenario needs to be reconciled with the
inhibitory effect of MAb 2D7 on the SCH-D escape
mutants. While 2D7 only partially inhibits D1/85.16
(¨60%), the D101.12 isolate is unusually sensitive to this
MAb and is completely blocked by it. How, then, can 2D7
inhibit the replication of D101.12 when the same virus is
little affected by the essentially complete removal of the
2D7 epitope from the cell-surface by PSC-RANTES treat-
ment? AOP-RANTES has been reported to efficiently
down-regulate CCR5 (i.e., physically remove it) from the
surface of various CCR5-transfected cell lines, but it also
occludes the 2D7 epitope (Sabbe et al., 2001; Signoret et al.,
2000). The same is true for PSC-RANTES (data not shown).

A.J. Marozsan et al. / Virology 338 (2005) 182–199194
The inability of 2D7 to stain CCR5 on PSC-RANTES-
treated PBMC does not, therefore, necessarily mean that
CCR5 is completely absent from the PBMC surface. It
could mean that some CCR5 proteins remain on the surface,
but with their 2D7 epitope obscured by the modified
chemokine, but with their epitopes for 45523 and 45531
unaffected. D101.12 might be able to utilize these chemo-
kine-bound CCR5 proteins. However, the efficiency of this
interaction may be diminished compared to the unbound
form of CCR5 because D101.12 was partially sensitive to
the highest concentration of PSC-RANTES.
As noted above, the potency of CCR5 inhibitors in vitro
and in vivo does vary between individuals (Kuhmann et al.,
2004; Moore and Doms, 2003). Additional studies may
reveal whether HIV-1 variants use a CCR5 configuration(s)
that is expressed to different extents between individuals.
Donor-dependent variation in the secretion of chemokines
by PBMC, either basally or in response to treatment with a
chemokine antagonist like SCH-D, could be another
relevant factor. Yet another might be SCH-D-induced up-
regulation of CCR5 over the duration of the 14-day
replication assays, a phenomenon that has been reported
in studies using SCH-C (Xu et al., 2002). An isoform(s) of
CCR5 that is unaffected by PSC-RANTES but recognized
by the SCH-D escape mutants might be upregulated
disproportionately under these conditions.
In genetic studies, we found that escape of CC1/85 from
AD101 was caused by four sequential amino acid changes
in the gp120 V3 region (Kuhmann et al., 2004). A different
R5 primary isolate also escaped from SCH-C in PM1 cells
via V3 sequence changes (Strizki et al., submitted for
publication). In contrast, the V3 sequences of two clones
fully resistant to SCH-D were identical to those of the
corresponding input isolates (Fig. 7). Hence, viruses
resistant to small molecule CCR5 inhibitors can emerge
without any changes in V3. Presumably, genetic changes
elsewhere in the envelope glycoproteins have the same
phenotypic effect.
In the SCH-D-resistant D1/85.16 clone, amino acid
changes in two regions stand out as the ones most likely
to influence SCH-D sensitivity. These changes, V169M and
K171R in the V2 region and G354P and N355D in C3, were
present in the D1/85.16 clone, but in none of the clones
from the parental CC1/85 isolate. The two V2 changes are
proximal to another alteration, D167N, that was a minor
polymorphism in the CC1/85 parental isolate. The D167N
change was previously found to be slowly selected for in the
PC viruses from the AD101 escape cultures, and it is
enriched in the later passages of the AD101-resistant viruses
(Kuhmann et al., 2004; Pugach et al., 2004). The D167N
polymorphism, along with two other changes in V2, I165K
and the deletion of residues 188 and 189, are associated with
increased sensitivity to neutralization by soluble CD4 and
the CD4 binding site MAb b12, and probably cause an
increase in the affinity of Env for cell surface CD4 (Pugach
et al., 2004). Other changes in V2 have also been implicated
in affecting CD4 and coreceptor binding (Platt et al., 2005;
Walter et al., 2005), and the V1/V2 structure is thought to
functionally interact with V3 (Carrillo and Ratner, 1996). In
addition, the V1/V2 structure from one gp120 subunit in the
native Env trimer may physically interact with V3 from a
neighboring subunit (Chen et al., 2005). Thus, it seems
reasonable to assume that the V2 changes in the SCH-D-
resistant virus could have a significant impact on the
receptor interactions of Env, a hypothesis that is now under
investigation.
The C3 changes G354P and N355D are also unique to
the SCH-D-resistant clone D1/85.16 compared to CC1/85
clones (Fig. 7). However, these changes are present in 2/9
clones from the CC2/86 isolate and 1/12 clones from the
CC12/86 isolate, which are R5X4 viruses generated from
the same individual as CC1/85, but at later time points
(Kuhmann et al., 2004). The region containing these
changes is present in the structures of the CD4-liganded
HIV-1 gp120 core and the unliganded SIV gp120 core
(Chen et al., 2005; Kwong et al., 1998). This region is
located in the surface exposed loop E, proximal to the V4
loop and the virion surface (Chen et al., 2005; Kwong et al.,
1998). Whether changes in this region could affect
coreceptor binding, either CCR5 vs. CXCR4 or different
usage of CCR5, is not clear, but it is now under
investigation.
The changes outside of V3 that might influence the SCH-
D resistance of the D101.12 clone are striking by their
scarcity. Within gp120, only one difference between the
SCH-D-resistant D101.12 clone and the clone from the
input CC101.6 virus was unique to this study (i.e., had not
previously been observed). This change, N406K, is found in
the V4 loop and results in the deletion of a potential N-
linked glycosylation site. Of note is that a similar loss of a
glycosylation site in the C-terminal portion of the V4 loop in
the JR-CSF isolate has recently been shown to be associated
with the more efficient use of a CCR5 mutant from which
the N-terminus was deleted (Platt et al., 2005). Relative to
the parental CC1/85 virus, CC101.6 has an H308P
substitution in V3, a change maintained in the D101.12
SCH-D resistant clone. This substitution has been shown to
confer partial resistance to AD101 (Kuhmann et al., 2004),
and the CC101.6 isolate has a similar level of resistance to
SCH-D (Fig. 2 and Table 2). It is possible that the loss of the
V4 glycan acts in concert with P308 to confer the high level
SCH-D resistance of the D101.12 isolate and clone, perhaps
by altering how the resistant virus interacts with CCR5
(Platt et al., 2005). Again, this hypothesis is now under
investigation.
The finding that V3 changes are not involved in the
resistance of the tested D1/85.16 and D101.12 clones was
unexpected given that we had used the same primary isolate,
CC1/85, in both the AD101 and SCH-D resistance experi-
ments, and that both these small molecule CCR5 inhibitors
are of comparable size and potency. However, in each
escape study, we used PBMC from different donors for each

A.J. Marozsan et al. / Virology 338 (2005) 182–199 195
passage, which creates a source of variation even when the
same input virus and similar inhibitors are studied. This
factor might account for why we observed the emergence of
some viruses able to use CXCR4 in some cell lines in the
present study, but not in those where the selection pressure
was from AD101 or SCH-C.
It appears, then, that HIV-1 can follow multiple genetic
pathways to escape from small molecule CCR5 inhibitors.
As we have noted previously, the plasticity of the HIV-1
envelope glycoproteins adds an extra dimension to the drug-
resistance process, compared to what has been experienced
with RT and protease inhibitors (Kuhmann et al., 2004;
Moore and Doms, 2003). Another important variable may
be donor-dependent differences in the expression of CCR5
isoforms. Quasispecies variation in HIV-1-infected individ-
uals creates the potential for even more complexity. An
additional factor to consider in vivo is the presence of
humoral immunity, which also acts against the viral
envelope glycoproteins. Any virus resistant to CCR5
inhibitors, indeed to any entry inhibitor, that emerges in
vivo must also retain its resistance to neutralizing anti-
bodies. Hence, some escape pathways that are favored in
vitro may be disfavored in vivo. We should expect the
unexpected when studying escape from CCR5 inhibitors
under clinical conditions.
Methods
Reagents
The small molecule CCR5 inhibitors, AD101, SCH-C
and SCH-D, have all been described previously and were
synthesized by Schering-Plough Research Institute (Kenil-
worth, NJ) (Strizki et al., 2001; Tagat et al., 2004; Trkola et
al., 2002). SCH-D has is also known as SCH-417690, but
we have retained the SCH-D nomenclature in this paper, for
clarity and convenience. The small molecule CCR5
inhibitor CMPD 167 was a gift from Dr. Marty Springer
(Merck Research Labs, Rahway, NJ) (Veazey et al., 2003;
Wolinsky et al., 2004). The CC-chemokine RANTES was
purchased from RD Systems (Minneapolis, MN). The
chemically modified chemokines PSC-RANTES and
AOP-RANTES were donated by Dr. Oliver Hartley (Centre
Medicale Universitaire, Geneva) (Hartley et al., 2004;
Kawamura et al., 2004; Simmons et al., 1997). The CD4-
IgG2 (PRO 542) molecule (Aarons et al., 2001), the murine
anti-CCR5 MAb PA14 (Olson et al., 1999) and the gp41-
based peptides T20 and T1249 were all gifts from Dr.
William Olson (Progenics Pharmaceuticals Inc, Tarrytown,
NY). The murine anti-CCR5 MAbs 2D7, and 45523 and
45531, were purchased from Pharmingen (San Diego, CA)
and from RD Systems Inc., respectively. The following
reagents were obtained through the NIH AIDS Research and
Reference Reagent Program, Division of AIDS, NIAID,
NIH: the PI, indinavir sulfate; the non-nucleoside reverse
transcriptase inhibitor (NNRTI) nevirapine; the nucleoside
reverse transcriptase inhibitor (NRTI) zidovudine; the
integrase inhibitor 118-D-24; the CXCR4 inhibitor
AMD3100.
Selection of escape mutants in PBMC and assessment of
HIV-1 replication in primary cells
The procedure used to generate SCH-D escape mutants
was closely based on that used to make a virus resistant to
AD101, an earlier generation small molecule CCR5
inhibitor (Kuhmann et al., 2004; Trkola et al., 2002). Two
separate cultures were initiated and carried out simulta-
neously, the input viruses being either CC1/85, an R5
primary isolate (Connor et al., 1993; Trkola et al., 2002), or
CC101.6, a virus isolated from the passage 6 culture of
CC1/85 with AD101 (Kuhmann et al., 2004; Trkola et al.,
2002). CC101.6 is partially (3-fold) resistant to AD101 (and
also to SCH-D, see below). In both cases, passage control
cultures were also established in which the cells and viruses
were treated exactly as in the cultures containing SCH-D,
but without the addition of any inhibitor. Our nomenclature
for isolates and env clones is based on the parental virus, the
selecting drug used and the passage history. Thus, CC1/85
and CC101.6 are the parental isolates, and D1/85.XX or
D101.XX refer to the isolates made after XX passages under
selection pressure from SCH-D. Similarly, D1/85.PC.XX
and D101.PC.XX refer to the control viruses that were
cultured for XX passages in the same PBMC, but in the
absence of SCH-D (Table 1).
In brief, CC1/85 or CC101.6 (1000 TCID50/ml) was
added to 20 ml of PBMC (2 � 106/ml), prepared from
leukopacks pooled from the blood of 4 healthy volunteers,
with sufficient SCH-D (500 nM) to cause >90% inhibition.
The pooled PBMC were depleted of CD8+ T-cells,
fractionated on a Ficoll density gradient, then mitogen-
activated for 3 days, as described previously (Kuhmann et
al., 2004; Trkola et al., 2002). The use of a PBMC pool
minimizes the influence of donor-dependent variation in
CCR5 expression (or in other parameters) on the rate of
HIV-1 replication and, conceivably, on how escape mutants
develop. The CD8 + T-cell-depleted PBMC preparations
are referred to hereafter as primary CD4+ T-cells. The
cultures were passaged weekly, maintaining a constant
density of cells throughout the experiment. The production
of p24 antigen was monitored to ensure that virus
replication was occurring and to determine the extent of
inhibition by SCH-D. Because PBMC from different
donors were used at each passage, p24 production varies
in both the SCH-D-treated and control cultures. SCH-D
(500 nM) was first added after passage 1, then its
concentration was escalated over sequential passages when
viral replication began to increase in the inhibitor-treated
cultures (Trkola et al., 2002). Culture supernatants were
frozen periodically for later inhibitor-sensitivity and cor-
eceptor-utilization studies.

A.J. Marozsan et al. / Virology 338 (2005) 182–199196
The assessment of HIV-1 sensitivity to SCH-D and other
replication inhibitors was performed using primary CD4+ T-
cells as described previously, the assay endpoint being the
production of p24 antigen after 7 and 14 days of culture
(Kuhmann et al., 2004; Pugach et al., 2004). HIV-1
replication in PBMC from CCR5-D32 homozygous donors
(or from CCR5 wild type donors in comparative studies)
was measured as described elsewhere (Kuhmann et al.,
2004; Trkola et al., 2002). Note that these preparations were
not depleted of CD8+ T-cells.
HIV-1 replication in cell lines
The U87.CD4.CCR5 and U87.CD4.CXCR4 cell lines
were obtained through the AIDS Research and Reference
Reagent Program, Division of AIDS, NIAID, NIH: from Dr.
HongKui Deng and Dr. Dan R. Littman (Bjorndal et al.,
1997). The U87 cells were cultured in Dulbecco’s Mod-
ification of Eagle’s Medium (DMEM) with 10% fetal
bovine serum (FBS), 300 Ag/ml G418, 1 Ag/ml puromycin
and penicillin/streptomycin.
GHOST cell lines were obtained through the AIDS
Research and Reference Reagent Program, Division of
AIDS, NIAID, NIH: from Dr. Vineet N. KewalRamani and
Dr. Dan R. Littman (Morner et al., 1999). GHOST cells
were cultured in DMEM with 10% FBS, 500 Ag/ml G418, 1
Ag/ml Puromycin, 100 Ag/ml hygromycin and penicillin/
streptomycin.
Cells from the T-lymphoblast line MT4 were a gift from
Dr. Robert Doms (University of Pennsylvania) and were
cultured in RPMI 1640 medium supplemented with l-
glutamine, 10% FBS and penicillin/streptomycin (Larder
et al., 1989).
HIV-1 replication in the various cell lines was measured
essentially as described previously (Kuhmann et al., 2004;
Trkola et al., 2002). The assay endpoint was the measure-
ment of p24 antigen concentration using an in-house
immunoassay (Kuhmann et al., 2004; Trkola et al., 2002).
Flow cytometry analysis of CCR5 expression
Primary CD4+ T-cells were prepared as described above;
cells from the 4 donors were then pooled and incubated for
6 more days. The cells were harvested, washed with PBS
and incubated for 1 h at 37-C with or without RANTES
(640 nM), AOP-RANTES (100 nM) or PSC-RANTES
(100 nM). After washing with ice-cold PBS, the cells were
fixed in a PBS solution containing 2% paraformaldehyde,
60 mM sucrose, pH 7.4, for 15 min at room temperature.
Fixation is necessary for MAb 45523 to bind to cell surface
CCR5 on primary CD4+ T-cells. In some experiments, cells
were fixed and permeabilized simultaneously by the
addition of 0.05% saponin during the preceding incubation.
The fixed cells were washed twice with PBS containing 20
mM glycine (buffer A), then incubated for 15 min at room
temperature in buffer A supplemented with 1% BSA and
0.05% NaN3 (buffer B). A 50-Al volume of buffer B
containing an anti-CCR5 MAb, either APC-conjugated
2D7 (10 Al/106 cells), FITC-labeled 45531 (20 Ag/ml) or
FITC-labeled 45523 (20 Ag/ml), was then added for 1 h at
room temperature. In some experiments, the cells were
incubated for 45 min in 50 Al buffer B containing rabbit
anti-tubulin (AB935, Chemicon International, Temecula,
CA), washed three times in buffer B and incubated a
further 45 min in 50 Al buffer B containing FITC-
conjugated anti-rabbit IgG (Jackson ImmunoResearch,
West Grove, PA). After 3 washes in buffer B, the cells
were resuspended for analysis using a BD LSR II flow
cytometer (BD Biosciences; San Jose, CA). The parameters
used to select cell populations for analysis were forward-
and side-light scatter, with a total of 10,000 events being
collected for analysis.
Env chimeras and V3 sequence analysis
The cloning of full-length D1/85.PC.16, D1/85.16,
D101.PC.12 and D101.12 env genes from infected PBMC,
and a partial analysis of their sequences, were accomplished
by using a newly developed yeast recombination system that
offers several advantages over previous methods (Marozsan
and Arts, 2003). Briefly, the env gene was PCR-amplified
from the respective virus isolates using the Env-Start and
Env-End primers (Marozsan and Arts, 2003). The product
was then recombined into the pRec D env vector as described
previously (Marozsan and Arts, 2003; Marozsan et al.,
2005). The V3 regions of env clones of the parental, passage
control and SCH-D-resistant viruses were sequenced at the
Cornell Biotechnology Resource Center, making use of
primers CC01 (TTAACCCCACTCTGTGTTAC), CC02
(TAAACCTACCAAGCCTCC), CC03 (CAGTCTAGCA-
GAAGAAGAGG) and CC04 (TGCGCCCATAGTGCTTC).
Acknowledgments
We thank Dr. Robert Doms for providing MT4 cells, Dr.
Ruth Connor for providing the CC1/85 and CC2/86 HIV-1
isolates, Drs. Jim Hoxie and Nelson Michael for blood from
CCR5D32/D 32 homozygous donors, and Dr. William
Olson for providing PA14, T20 and T1249. We are also
grateful to Dr. Oliver Hartley for AOP- and PSC-RANTES.
We thank Tom Ketas, Ashley Palmer, Christopher Kibler,
Janice Riley and Kudzai Chikwava for technical support.
Finally, we thank Dr. Per Johan Klasse for his helpful
critical comments. This work was supported by NIH grant
R01 AI41420 (JPM), NIH Immunology training grant T32
AI07621 (AJM), NIH Fellowship grant F32 AI062664
(SEK), and by Schering Plough Research Institute. JPM is a
Stavros S. Niarchos Scholar. The Department of Micro-
biology and Immunology at the Weill Medical College
gratefully acknowledges the support of the William Ran-
dolph Hearst Foundation.

A.J. Marozsan et al. / Virology 338 (2005) 182–199 197
References
Aarons, E.J., Beddows, S., Willingham, T., Wu, L., Koup, R.A., 2001.
Adaptation to blockade of human immunodeficiency virus type 1 entry
imposed by the anti-CCR5 monoclonal antibody 2D7. Virology 287 (2),
382–390.
Baldwin, C.E., Sanders, R.W., Berkhout, B., 2003. Inhibiting HIV-1 entry
with fusion inhibitors. Curr. Med. Chem. 10 (17), 1633–1642.
Baribaud, F., Edwards, T.G., Sharron, M., Brelot, A., Heveker, N., Price,
K., Mortari, F., Alizon, M., Tsang, M., Doms, R.W., 2001. Antigeni-
cally distinct conformations of CXCR4. J. Virol. 75 (19), 8957–8967.
Billick, E., Seibert, C., Pugach, P., Ketas, T., Trkola, A., Endres, M.J.,
Murgolo, N.J., Coates, E., Reyes, G.R., Baroudy, B.M., Sakmar, T.P.,
Moore, J.P., Kuhmann, S.E., 2004. The differential sensitivity of human
and rhesus macaque CCR5 to small-molecule inhibitors of human
immunodeficiency virus type 1 entry is explained by a single amino
acid difference and suggests a mechanism of action for these inhibitors.
J. Virol. 78 (8), 4134–4144.
Bjorndal, A., Deng, H., Jansson, M., Fiore, J.R., Colognesi, C., Karlsson,
A., Albert, J., Scarlatti, G., Littman, D.R., Fenyo, E.M., 1997.
Coreceptor usage of primary human immunodeficiency virus type 1
isolates varies according to biological phenotype. J. Virol. 71 (10),
7478–7487.
Blanpain, C., Vanderwinden, J.M., Cihak, J., Wittamer, V., Le Poul, E.,
Issafras, H., Stangassinger, M., Vassart, G., Marullo, S., Schlndorff, D.,
Parmentier, M., Mack, M., 2002. Multiple active states and oligome-
rization of CCR5 revealed by functional properties of monoclonal
antibodies. Mol. Biol. Cell 13 (2), 723–737.
Carnec, X., Quan, L., Olson, W.C., Hazan, U., Dragic, T., 2005. Anti-
CXCR4 monoclonal antibodies recognizing overlapping epitopes differ
significantly in their ability to inhibit entry of human immunodeficiency
virus type 1. J. Virol. 79 (3), 1930–1933.
Carrillo, A., Ratner, L., 1996. Cooperative effects of the human
immunodeficiency virus type 1 envelope variable loops V1 and V3 in
mediating infectivity for T cells. J. Virol. 70 (2), 1310–1316.
Chen, B., Vogan, E.M., Gong, H., Skehel, J.J., Wiley, D.C., Harrison, S.C.,
2005. Structure of an unliganded simian immunodeficiency virus gp120
core. Nature 433 (7028), 834–841.
Connor, R.I., Mohri, H., Cao, Y., Ho, D.D., 1993. Increased viral burden
and cytopathicity correlate temporally with CD4+ T-lymphocyte decline
and clinical progression in human immunodeficiency virus type 1-
infected individuals. J. Virol. 67 (4), 1772–1777.
Connor, R.I., Sheridan, K.E., Ceradini, D., Choe, S., Landau, N.R., 1997.
Change in coreceptor use coreceptor use correlates with disease
progression in HIV-1-infected individuals. J. Exp. Med. 185 (4),
621–628.
De Clercq, E., 2003. The bicyclam AMD3100 story. Nat. Rev., Drug
Discov. 2 (7), 581–587.
Hammer, S.M., 2002. HIV drug resistance: implications for management.
Top. HIV Med. 10 (5), 10–15.
Hartley, O., Gaertner, H., Wilken, J., Thompson, D., Fish, R., Ramos, A.,
Pastore, C., Dufour, B., Cerini, F., Melotti, A., Heveker, N., Picard, L.,
Alizon, M., Mosier, D., Kent, S., Offord, R., 2004. Medicinal chemistry
applied to a synthetic protein: development of highly potent HIV entry
inhibitors. Proc. Natl. Acad. Sci. U.S.A. 101 (47), 16460–16465.
Hill, C.M., Kwon, D., Jones, M., Davis, C.B., Marmon, S., Daugherty,
B.L., DeMartino, J.A., Springer, M.S., Unutmaz, D., Littman, D.R.,
1998. The amino terminus of human CCR5 is required for its function
as a receptor for diverse human and simian immunodeficiency virus
envelope glycoproteins. Virology 248 (2), 357–371.
Kawamura, T., Bruse, S.E., Abraha, A., Sugaya, M., Hartley, O., Offord,
R.E., Arts, E.J., Zimmerman, P.A., Blauvelt, A., 2004. PSC-RANTES
blocks R5 human immunodeficiency virus infection of Langerhans cells
isolated from individuals with a variety of CCR5 diplotypes. J. Virol. 78
(14), 7602–7609.
Kazmierski, W., Bifulco, N., Yang, H., Boone, L., DeAnda, F., Watson, C.,
Kenakin, T., 2003. Recent progress in discovery of small-molecule
CCR5 chemokine receptor ligands as HIV-1 inhibitors. Bioorg. Med.
Chem. 11 (13), 2663–2676.
Kenakin, T., 2004. G-protein coupled receptors as allosteric machines.
Recept. Channels 10 (2), 51–60.
Kilby, J.M., Eron, J.J., 2003. Novel therapies based on mechanisms of HIV-
1 cell entry. N. Engl. J. Med. 348 (22), 2228–2238.
Kuhmann, S.E., Pugach, P., Kunstman, K.J., Taylor, J., Stanfield, R.L.,
Snyder, A., Strizki, J.M., Riley, J., Baroudy, B.M., Wilson, I.A., Korber,
B.T., Wolinsky, S.M., Moore, J.P., 2004. Genetic and phenotypic
analyses of human immunodeficiency virus type 1 escape from a small-
molecule CCR5 inhibitor. J. Virol. 78 (6), 2790–2807.
Kwong, P.D., Wyatt, R., Robinson, J., Sweet, R.W., Sodroski, J.,
Hendrickson, W.A., 1998. Structure of an HIV gp120 envelope
glycoprotein in complex with the CD4 receptor and a neutralizing
human antibody. Nature 393 (6686), 648–659.
Lalezari, J.P., Henry, K., O’Hearn, M., Montaner, J.S., Piliero, P.J.,
Trottier, B., Walmsley, S., Cohen, C., Kuritzkes, D.R., Eron Jr., J.J.,
Chung, J., DeMasi, R., Donatacci, L., Drobnes, C., Delehanty, J.,
Salgo, M., 2003. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-
resistant HIV infection in North and South America. N. Engl. J. Med.
348 (22), 2175–2185.
Larder, B., 2001. Mechanisms of HIV-1 drug resistance. Aids 15 (Suppl. 5),
S27–S34.
Larder, B.A., Darby, G., Richman, D.D., 1989. HIV with reduced
sensitivity to zidovudine (AZT) isolated during prolonged therapy.
Science 243 (4899), 1731–1734.
Lazzarin, A., Clotet, B., Cooper, D., Reynes, J., Arasteh, K., Nelson, M.,
Katlama, C., Stellbrink, H.J., Delfraissy, J.F., Lange, J., Huson, L.,
DeMasi, R., Wat, C., Delehanty, J., Drobnes, C., Salgo, M., 2003.
Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in
Europe and Australia. N. Engl. J. Med. 348 (22), 2186–2195.
Lee, B., Sharron, M., Blanpain, C., Doranz, B.J., Vakili, J., Setoh, P., Berg,
E., Liu, G., Guy, H.R., Durell, S.R., Parmentier, M., Chang, C.N., Price,
K., Tsang, M., Doms, R.W., 1999. Epitope mapping of CCR5 reveals
multiple conformational states and distinct but overlapping structures
involved in chemokine and coreceptor function. J. Biol. Chem. 274
(14), 9617–9626.
Mack, M., Luckow, B., Nelson, P.J., Cihak, J., Simmons, G., Clapham,
P.R., Signoret, N., Marsh, M., Stangassinger, M., Borlat, F., Wells,
T.N., Schlondorff, D., Proudfoot, A.E., 1998. Aminooxypentane-
RANTES induces CCR5 internalization but inhibits recycling: a novel
inhibitory mechanism of HIV infectivity. J. Exp. Med. 187 (8),
1215–1224.
Maeda, K., Nakata, H., Koh, Y., Miyakawa, T., Ogata, H., Takaoka, Y.,
Shibayama, S., Sagawa, K., Fukushima, D., Moravek, J., Koyanagi, Y.,
Mitsuya, H., 2004a. Spirodiketopiperazine-based CCR5 inhibitor which
preserves CC- chemokine/CCR5 interactions and exerts potent activity
against R5 human immunodeficiency virus type 1 in vitro. J. Virol. 78
(16), 8654–8662.
Maeda, K., Nakata, H., Ogata, H., Koh, Y., Miyakawa, T., Mitsuya, H.,
2004b. The current status of, and challenges in, the development of
CCR5 inhibitors as therapeutics for HIV-1 infection. Curr. Opin.
Pharmacol. 4 (5), 447–452.
Marozsan, A.J., Arts, E.J., 2003. Development of a yeast-based recombi-
nation cloning/system for the analysis of gene products from diverse
human immunodeficiency virus type 1 isolates. J. Virol. Methods 111
(2), 111–120.
Marozsan, A.J., Moore, D., Lobritz, M., Fraundorf, E., Abraha, A., Reeves,
J.D., Arts, E.J., 2005. Differences in the fitness of two diverse wild-type
human immunodeficiency virus type 1 isolates are related to the
efficiency of cell binding and entry. J. Virol. 79, 7121–7134.
McKnight, A., Wilkinson, D., Simmons, G., Talbot, S., Picard, L., Ahuja,
M., Marsh, M., Hoxie, J.A., Clapham, P.R., 1997. Inhibition of human
immunodeficiency virus fusion by a monoclonal antibody to a coreceptor
(CXCR4) is both cell type and virus strain dependent. J. Virol. 71 (2),
1692–1696.
Moore, J.P., Doms, R.W., 2003. The entry of entry inhibitors: a fusion

A.J. Marozsan et al. / Virology 338 (2005) 182–199198
of science and medicine. Proc. Natl. Acad. Sci. U.S.A. 100 (19),
10598–10602.
Moore, J.P., Stevenson, M., 2000. New targets for inhibitors of HIV-1
replication. Nat. Rev., Mol. Cell Biol. 1 (1), 40–49.
Moore, J.P., Kitchen, S.G., Pugach, P., Zack, J.A., 2004. The CCR5 and
CXCR4 coreceptors-central to understanding the transmission and
pathogenesis of human immunodeficiency virus type 1 infection. AIDS
Res. Hum. Retroviruses 20 (1), 111–126.
Morner, A., Bjorndal, A., Albert, J., Kewalramani, V.N., Littman, D.R.,
Inoue, R., Thorstensson, R., Fenyo, E.M., Bjorling, E., 1999. Primary
human immunodeficiency virus type 2 (HIV-2) isolates, like HIV-1
isolates, frequently use CCR5 but show promiscuity in coreceptor
usage. J. Virol. 73 (3), 2343–2349.
O’Hara, B.M., Olson, W.C., 2002. HIV entry inhibitors in clinical
development. Curr. Opin. Pharmacol. 2 (5), 523–528.
Olson, W.C., Rabut, G.E., Nagashima, K.A., Tran, D.N., Anselma, D.J.,
Monard, S.P., Segal, J.P., Thompson, D.A., Kajumo, F., Guo, Y., Moore,
J.P., Maddon, P.J., Dragic, T., 1999. Differential inhibition of human
immunodeficiency virus type 1 fusion, gp120 binding, and CC-
chemokine activity by monoclonal antibodies to CCR5. J. Virol. 73
(5), 4145–4155.
Platt, E.J., Shea, D.M., Rose, P.P., Kabat, D., 2005. Variants of human
immunodeficiency virus type 1 that efficiently use CCR5 lacking the
tyrosine-sulfated amino terminus have adaptive mutations in gp120,
including loss of a functional N-glycan. J. Virol. 79 (7), 4357–4368.
Pugach, P., Kuhmann, S.E., Taylor, J., Marozsan, A.J., Snyder, A., Ketas,
T., Wolinsky, S.M., Korber, B.T., Moore, J.P., 2004. The prolonged
culture of human immunodeficiency virus type 1 in primary lympho-
cytes increases its sensitivity to neutralization by soluble CD4. Virology
321 (1), 8–22.
Reynes, J., Rouzier, R., Kanouni, T., Baillat, V., Baroudy, B., Keung, A.,
Hogan, C., Markowitz, M., Laughlin, M., 2002. 9th Conference on
Retroviruses and Opportunistic Infections, Seattle, WA.
Riley, J., Wojcik, L., Wei, H., Xu, S., Kuhmann, S., Marozsan, A.J.,
Whitcomb, J.M., Petropoulos, C.J., Moore, J.P., Strizki, J.M., submitted
for publication. Genotypic and phenotypic analysis of Human Immu-
nodeficiency Virus Type 1 escape variants generated under the selection
pressure of the CCR5 antagonist SCH-C (SCH-351125). In Vitro.
Sabbe, R., Picchio, G.R., Pastore, C., Chaloin, O., Hartley, O., Offord, R.,
Mosier, D.E., 2001. Donor- and ligand-dependent differences in C-C
chemokine receptor 5 reexpression. J. Virol. 75 (2), 661–671.
Schuitemaker, H., Kootstra, N.A., de Goede, R.E., de Wolf, F., Miedema,
F., Tersmette, M., 1991. Monocytotropic human immunodeficiency
virus type 1 (HIV-1) variants detectable in all stages of HIV-1 infection
lack T-cell line tropism and syncytium-inducing ability in primary T-cell
culture. J. Virol. 65 (1), 356–363.
Schurmann, D., Rouzier, R., Nougarede, R., Reynes, J., Fatkenheuer, G.,
Raffi, F., Michelet, C., Tarral, A., Hoffmann, C., Kiunke, J., Sprenger,
H., vanLier, J., Sansone, A., Jackson, M., Laughlin, M., 2004. 11th
Conference on Retroviruses and Opportunistic Infections, San
Francisco, CA.
Schwarz, M.K., Wells, T.N., 2002. New therapeutics that modulate
chemokine networks. Nat. Rev., Drug Discov. 1 (5), 347–358.
Seibert, C., Sakmar, T.P., 2004. Small-molecule antagonists of CCR5 and
CXCR4: a promising new class of anti-HIV-1 drugs. Curr. Pharm. Des.
10 (17), 2041–2062.
Shen, D.M., Shu, M., Willoughby, C.A., Shah, S., Lynch, C.L., Hale, J.J.,
Mills, S.G., Chapman, K.T., Malkowitz, L., Springer, M.S., Gould,
S.L., DeMartino, J.A., Siciliano, S.J., Lyons, K., Pivnichny, J.V., Kwei,
G.Y., Carella, A., Carver, G., Holmes, K., Schleif, W.A., Danzeisen, R.,
Hazuda, D., Kessler, J., Lineberger, J., Miller, M.D., Emini, E.A., 2004.
Antagonists of human CCR5 receptor containing 4-(pyrazolyl)piper-
idine side chains: Part 2. discovery of potent, selective, and orally
bioavailable compounds. Bioorg. Med. Chem. Lett. 14 (4), 941–945.
Signoret, N., Pelchen-Matthews, A., Mack, M., Proudfoot, A.E., Marsh, M.,
2000. Endocytosis and recycling of the HIV coreceptor CCR5. J. Cell
Biol. 151 (6), 1281–1294.
Simmons, G., Clapham, P.R., Picard, L., Offord, R.E., Rosenkilde, M.M.,
Schwartz, T.W., Buser, R., Wells, T.N., Proudfoot, A.E., 1997. Potent
inhibition of HIV-1 infectivity in macrophages and lymphocytes by a
novel CCR5 antagonist. Science 276 (5310), 276–279.
Strizki, J.M., Xu, S., Wagner, N.E., Wojcik, L., Liu, J., Hou, Y., Endres, M.,
Palani, A., Shapiro, S., Clader, J.W., Greenlee, W.J., Tagat, J.R.,
McCombie, S., Cox, K., Fawzi, A.B., Chou, C.C., Pugliese-Sivo, C.,
Davies, L., Moreno, M.E., Ho, D.D., Trkola, A., Stoddart, C.A., Moore,
J.P., Reyes, G.R., Baroudy, B.M., 2001. SCH-C (SCH 351125), an
orally bioavailable, small molecule antagonist of the chemokine
receptor CCR5, is a potent inhibitor of HIV-1 infection in vitro and in
vivo. Proc. Natl. Acad. Sci. U.S.A. 98 (22), 12718–12723.
Strizki, J.M., Tremblay, C., Xu, S., Wojcik, L., Wagner, N., Gonsiorek,
W., Hipkin, R.W., Chou, C.C., Pugliese-Sivo, C., Xiao, Y., Tagat, J.,
Cox, K., Priestly, T., Sorota, S., Huang, W., Hirsch, M.S., Reyes,
G.R., Baroudy, B.M., submitted for publication. Discovery and
characterization of SCH-417690 (SCH-D), a second generation
CCR5 antagonist.
Tagat, J.R., McCombie, S.W., Steensma, R.W., Lin, S., Nazareno, D.V.,
Baroudy, B., Vantuno, N., Xu, S., Liu, J., 2001. Piperazine-based
CCR5 antagonists as HIV-1 inhibitors: I. 2 (S)-methyl piperazine as a
key pharmacophore element. Bioorg. Med. Chem. Lett. 11 (16),
2143–2146.
Tagat, J.R., McCombie, S.W., Nazareno, D., Labroli, M.A., Xiao, Y.,
Steensma, R.W., Strizki, J.M., Baroudy, B.M., Cox, K., Lachowicz, J.,
Varty, G., Watkins, R., 2004. Piperazine-based CCR5 antagonists as
HIV-1 inhibitors: IV. Discovery of 1-[(4,6-dimethyl-5-pyrimidinyl)
carbonyl]-4-[4-[2-methoxy-1(R)-4-(trifluoromethyl)phenyl]ethyl-3(S)-
methyl-1-piperaz inyl]-4-methylpiperidine (Sch-417690/Sch-D), a
potent, highly selective, and orally bioavailable CCR5 antagonist.
J. Med. Chem. 47 (10), 2405–2408.
Torre, V.S., Marozsan, A.J., Albright, J.L., Collins, K.R., Hartley, O.,
Offord, R.E., Quinones-Mateu, M.E., Arts, E.J., 2000. Variable
sensitivity of CCR5—tropic human immunodeficiency virus type 1
isolates to inhibition by RANTES analogs. J. Virol. 74 (10),
4868–4876.
Trkola, A., Dragic, T., Arthos, J., Binley, J.M., Olson, W.C., Allaway, G.P.,
Cheng- Mayer, C., Robinson, J., Maddon, P.J., Moore, J.P., 1996. CD4-
dependent, antibody-sensitive interactions between HIV-1 and its co-
receptor CCR-5. Nature 384 (6605), 184–187.
Trkola, A., Matthews, J., Gordon, C., Ketas, T., Moore, J.P., 1999. A cell
line-based neutralization assay for primary human immunodeficiency
virus type 1 isolates that use either the CCR5 or the CXCR4 coreceptor.
J. Virol. 73 (11), 8966–8974.
Trkola, A., Kuhmann, S.E., Strizki, J.M., Maxwell, E., Ketas, T., Morgan,
T., Pugach, P., Xu, S., Wojcik, L., Tagat, J., Palani, A., Shapiro, S.,
Clader, J.W., McCombie, S., Reyes, G.R., Baroudy, B.M., Moore, J.P.,
2002. HIV-1 escape from a small molecule, CCR5-specific entry
inhibitor does not involve CXCR4 use. Proc. Natl. Acad. Sci. U.S.A.
99 (1), 395–400.
Tsamis, F., Gavrilov, S., Kajumo, F., Seibert, C., Kuhmann, S., Ketas, T.,
Trkola, A., Palani, A., Clader, J.W., Tagat, J.R., McCombie, S.,
Baroudy, B., Moore, J.P., Sakmar, T.P., Dragic, T., 2003. Analysis of
the mechanism by which the small-molecule CCR5 antagonists SCH-
351125 and SCH-350581 inhibit human immunodeficiency virus type 1
entry. J. Virol. 77 (9), 5201–5208.
Veazey, R.S., Klasse, P.J., Ketas, T.J., Reeves, J.D., Piatak Jr., M.,
Kunstman, K., Kuhmann, S.E., Marx, P.A., Lifson, J.D., Dufour, J.,
Mefford, M., Pandrea, I., Wolinsky, S.M., Doms, R.W., DeMartino,
J.A., Siciliano, S.J., Lyons, K., Springer, M.S., Moore, J.P., 2003. Use
of a small molecule CCR5 inhibitor in macaques to treat simian
immunodeficiency virus infection or prevent simian-human immuno-
deficiency virus infection. J. Exp. Med. 198 (10), 1551–1562.
Walter, B.L., Wehrly, K., Swanstrom, R., Platt, E., Kabat, D., Chesebro, B.,
2005. Role of low CD4 levels in the influence of human immunode-
ficiency virus type 1 envelope v1 and v2 regions on entry and spread in
macrophages. J. Virol. 79 (8), 4828–4837.

A.J. Marozsan et al. / Virology 338 (2005) 182–199 199
Wei, X., Decker, J.M., Liu, H., Zhang, Z., Arani, R.B., Kilby, J.M., Saag,
M.S., Wu, X., Shaw, G.M., Kappes, J.C., 2002. Emergence of resistant
human immunodeficiency virus type 1 in patients receiving fusion
inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46 (6),
1896–1905.
Wolinsky, S.M., Veazey, R.S., Kunstman, K.J., Klasse, P.J., Dufour, J.,
Marozsan, A.J., Springer, M.S., Moore, J.P., 2004. Effect of a CCR5
inhibitor on viral loads in macaques dual-infected with R5 and X4
primate immunodeficiency viruses. Virology 328 (1), 19–29.
Wu, L., Gerard, N.P., Wyatt, R., Choe, H., Parolin, C., Ruffing, N.,
Borsetti, A., Cardoso, A.A., Desjardin, E., Newman, W., Gerard, C.,
Sodroski, J., 1996. CD4-induced interaction of primary HIV-1 gp120
glycoproteins with the chemokine receptor CCR-5. Nature 384 (6605),
179–183.
Xu, S., Wojcik, L., Strizki, J., 2002. 9th Conference on Retroviruses and
Opportunistic Infection, Seattle, WA.
Yi, Y., Shaheen, F., Collman, R.G., 2005. Preferential use of CXCR4 by
R5X4 human immunodeficiency virus type 1 isolates for infection of
primary lymphocytes. J. Virol. 79 (3), 1480–14806.
Zhang, Y.J., Dragic, T., Cao, Y., Kostrikis, L., Kwon, D.S., Littman, D.R.,
KewalRamani, V.N., Moore, J.P., 1998. Use of coreceptors other than
CCR5 by non-syncytium-inducing adult and pediatric isolates of
human immunodeficiency virus type 1 is rare in vitro. J. Virol. 72
(11), 9337–9344.
Zhang, Y., Lou, B., Lal, R.B., Gettie, A., Marx, P.A., Moore, J.P., 2000. Use
of inhibitors to evaluate coreceptor usage by simian and simian/human
immunodeficiency viruses and human immunodeficiency virus type 2
in primary cells. J. Virol. 74 (15), 6893–6910.
Copyright © 2022 FDOKUMEN




















