
The magnitude of HIV-1 resistance to the CCR5 antagonist maraviroc may impart a differential...
8
The magnitude of HIV-1 resistance to the CCR5 antagonist maraviroc may impart a differential alteration in HIV-1 tropism for macrophages and T-cell subsets Jacqueline K. Flynn a,d , Geza Paukovics b , Miranda S. Moore a,1 , Anne Ellett a , Lachlan R. Gray a,c , Renee Duncan a , Hamid Salimi a,f , Becky Jubb g , Mike Westby g , Damian F. J. Purcell h , Sharon R. Lewin a,d,i , Benhur Lee j , Melissa J. Churchill a,e,f , Paul R Gorry a,d,h,n , Michael Roche a,d,nn a Center for Virology, Burnet Institute, Melbourne, VIC, Australia b Alfred Medical Research & Education Precinct and Burnet Institute Flow Cytometry Core Facility, Melbourne, VIC, Australia c Department of Biochemistry and Molecular Biology, Monash University, Melbourne, VIC, Australia d Department of Infectious Diseases, Monash University, Melbourne, VIC, Australia e Department of Medicine, Monash University, Melbourne, VIC, Australia f Department of Microbiology, Monash University, Melbourne, VIC, Australia g Pfizer Global Research and Development, Sandwich, UK h Department of Microbiology and Immunology, University of Melbourne, Parkville, VIC, Australia i Infectious Diseases Unit, The Alfred Hospital, Melbourne, VIC, Australia j Department of Microbiology, Immunology, and Molecular Genetics, David Geffen School of Medicine, UCLA, Los Angeles, CA, USA article info Article history: Received 24 December 2012 Returned to author for revisions 31 January 2013 Accepted 26 March 2013 Available online 17 April 2013 Keywords: HIV-1 Maraviroc Resistance T-cell Macrophage Tropism Env gp120 abstract Human immunodeficiency virus type 1 (HIV-1) resistance to CCR5 antagonists, including maraviroc (MVC), results from alterations in the HIV-1 envelope glycoproteins (Env) enabling recognition of antagonist-bound CCR5. Here, we characterized tropism alterations for CD4+ T-cell subsets and macrophages by Envs from two subjects who developed MVC resistance in vivo, which displayed either relatively efficient or inefficient recognition of MVC-bound CCR5. We show that MVC-resistant Env with efficient recognition of drug-bound CCR5 displays a tropism shift for CD4+ T-cell subsets associated with increased infection of central memory T-cells and reduced infection of effector memory and transitional memory T-cells, and no change in macrophage infectivity. In contrast, MVC-resistant Env with inefficient recognition of drug-bound CCR5 displays no change in tropism for CD4+ T-cell subsets, but exhibits a significant reduction in macrophage infectivity. The pattern of HIV-1 tropism alterations for susceptible cells may therefore be variable in subjects with MVC resistance. & 2013 Elsevier Inc. All rights reserved. Introduction The entry of human immunodeficiency virus type 1 (HIV-1) into cells is initiated by the interaction between the gp120 glycoproteins of the HIV-1 envelope (Env) with cellular CD4 and a coreceptor, either CCR5 or CXCR4 [reviewed in (Gorry and Ancuta, 2011)]. Maraviroc (MVC) inhibits HIV-1 entry by binding to a hydrophobic pocket in the transmembrane helices of CCR5, which alters the conformation of the CCR5 extracellular loops (ECLs) rendering them unrecognizable by gp120 (Gorry et al., 2010; Tilton and Doms, 2010). Maraviroc, similar to other CCR5 antagonists such as vicriviroc (VVC) and aplaviroc (APL) (Dragic et al., 2000; Maeda et al., 2008; Seibert et al., 2006), is therefore an allosteric inhibitor of HIV-1 entry. Maraviroc is currently being used as an HIV-1 antiretroviral therapy for both treatment-experienced and antiretroviral therapy (ART)-naïve adults who have no evidence of CXCR4 using virus in their plasma (Gorry et al., 2010). However, similar to other classes of HIV-1 antiretrovirals, drug resistance to CCR5 antagonists can occur. Treatment failure can result from the emergence of CXCR4- using HIV-1 strains that were present at undetectable levels prior to MVC therapy, and also from the development of bona fide resistance (Westby et al., 2006), where adaptive mutations in gp120 enable recognition of- and entry via the drug bound Contents lists available at SciVerse ScienceDirect journal homepage: www.elsevier.com/locate/yviro Virology 0042-6822/$ - see front matter & 2013 Elsevier Inc. All rights reserved. http://dx.doi.org/10.1016/j.virol.2013.03.026 n Corresponding author at: Center for Virology, Burnet Institute, 85 Commercial Rd, Melbourne, 3004, VIC, Australia. nn Corresponding author. E-mail addresses: [email protected] (P. Gorry), [email protected]. au (M. Roche). 1 Fred Hutchinson Cancer Research Center, Seattle, WA, USA. Virology 442 (2013) 51–58
Transcript of The magnitude of HIV-1 resistance to the CCR5 antagonist maraviroc may impart a differential...

Virology 442 (2013) 51–58
Contents lists available at SciVerse ScienceDirect
Virology
0042-68http://d
n CorrRd, Mel
nn CorE-m
au (M. R1 Fr
journal homepage: www.elsevier.com/locate/yviro
The magnitude of HIV-1 resistance to the CCR5 antagonist maravirocmay impart a differential alteration in HIV-1 tropism for macrophagesand T-cell subsets
Jacqueline K. Flynn a,d, Geza Paukovics b, Miranda S. Moore a,1, Anne Ellett a, LachlanR. Gray a,c, Renee Duncan a, Hamid Salimi a,f, Becky Jubb g, Mike Westby g, Damian F.J. Purcell h, Sharon R. Lewin a,d,i, Benhur Lee j, Melissa J. Churchill a,e,f, Paul R Gorry a,d,h,n,Michael Roche a,d,nn
a Center for Virology, Burnet Institute, Melbourne, VIC, Australiab Alfred Medical Research & Education Precinct and Burnet Institute Flow Cytometry Core Facility, Melbourne, VIC, Australiac Department of Biochemistry and Molecular Biology, Monash University, Melbourne, VIC, Australiad Department of Infectious Diseases, Monash University, Melbourne, VIC, Australiae Department of Medicine, Monash University, Melbourne, VIC, Australiaf Department of Microbiology, Monash University, Melbourne, VIC, Australiag Pfizer Global Research and Development, Sandwich, UKh Department of Microbiology and Immunology, University of Melbourne, Parkville, VIC, Australiai Infectious Diseases Unit, The Alfred Hospital, Melbourne, VIC, Australiaj Department of Microbiology, Immunology, and Molecular Genetics, David Geffen School of Medicine, UCLA, Los Angeles, CA, USA
a r t i c l e i n f o
Article history:Received 24 December 2012Returned to author for revisions31 January 2013Accepted 26 March 2013Available online 17 April 2013
Keywords:HIV-1MaravirocResistanceT-cellMacrophageTropismEnvgp120
22/$ - see front matter & 2013 Elsevier Inc. Ax.doi.org/10.1016/j.virol.2013.03.026
esponding author at: Center for Virology, Burbourne, 3004, VIC, Australia.responding author.ail addresses: [email protected] (P. Gorry),oche).ed Hutchinson Cancer Research Center, Seattl
a b s t r a c t
Human immunodeficiency virus type 1 (HIV-1) resistance to CCR5 antagonists, including maraviroc(MVC), results from alterations in the HIV-1 envelope glycoproteins (Env) enabling recognition ofantagonist-bound CCR5. Here, we characterized tropism alterations for CD4+ T-cell subsets andmacrophages by Envs from two subjects who developed MVC resistance in vivo, which displayed eitherrelatively efficient or inefficient recognition of MVC-bound CCR5. We show that MVC-resistant Env withefficient recognition of drug-bound CCR5 displays a tropism shift for CD4+ T-cell subsets associated withincreased infection of central memory T-cells and reduced infection of effector memory and transitionalmemory T-cells, and no change in macrophage infectivity. In contrast, MVC-resistant Env with inefficientrecognition of drug-bound CCR5 displays no change in tropism for CD4+ T-cell subsets, but exhibits asignificant reduction in macrophage infectivity. The pattern of HIV-1 tropism alterations for susceptiblecells may therefore be variable in subjects with MVC resistance.
& 2013 Elsevier Inc. All rights reserved.
Introduction
The entry of human immunodeficiency virus type 1 (HIV-1) intocells is initiated by the interaction between the gp120 glycoproteins ofthe HIV-1 envelope (Env) with cellular CD4 and a coreceptor, eitherCCR5 or CXCR4 [reviewed in (Gorry and Ancuta, 2011)]. Maraviroc(MVC) inhibits HIV-1 entry by binding to a hydrophobic pocket in thetransmembrane helices of CCR5, which alters the conformation of the
ll rights reserved.
net Institute, 85 Commercial
e, WA, USA.
CCR5 extracellular loops (ECLs) rendering them unrecognizable bygp120 (Gorry et al., 2010; Tilton and Doms, 2010). Maraviroc, similar toother CCR5 antagonists such as vicriviroc (VVC) and aplaviroc (APL)(Dragic et al., 2000; Maeda et al., 2008; Seibert et al., 2006), istherefore an allosteric inhibitor of HIV-1 entry.
Maraviroc is currently being used as an HIV-1 antiretroviraltherapy for both treatment-experienced and antiretroviral therapy(ART)-naïve adults who have no evidence of CXCR4 using virus intheir plasma (Gorry et al., 2010). However, similar to other classesof HIV-1 antiretrovirals, drug resistance to CCR5 antagonists canoccur. Treatment failure can result from the emergence of CXCR4-using HIV-1 strains that were present at undetectable levels priorto MVC therapy, and also from the development of bona fideresistance (Westby et al., 2006), where adaptive mutations ingp120 enable recognition of- and entry via the drug bound
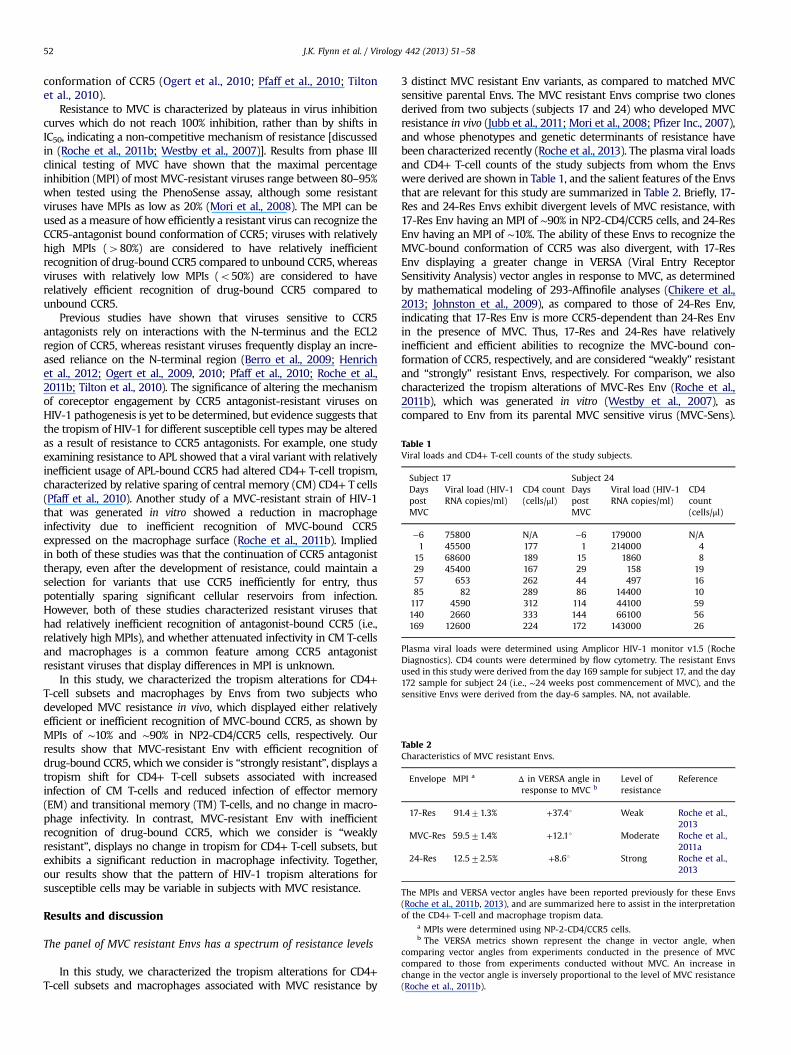
Table 1Viral loads and CD4+ T-cell counts of the study subjects.
Subject 17 Subject 24DayspostMVC
Viral load (HIV-1RNA copies/ml)
CD4 count(cells/μl)
DayspostMVC
Viral load (HIV-1RNA copies/ml)
CD4count(cells/μl)
−6 75800 N/A −6 179000 N/A1 45500 177 1 214000 4
15 68600 189 15 1860 829 45400 167 29 158 1957 653 262 44 497 1685 82 289 86 14400 10117 4590 312 114 44100 59140 2660 333 144 66100 56169 12600 224 172 143000 26
Plasma viral loads were determined using Amplicor HIV-1 monitor v1.5 (RocheDiagnostics). CD4 counts were determined by flow cytometry. The resistant Envsused in this study were derived from the day 169 sample for subject 17, and the day172 sample for subject 24 (i.e., ∼24 weeks post commencement of MVC), and thesensitive Envs were derived from the day-6 samples. NA, not available.
Table 2Characteristics of MVC resistant Envs.
Envelope MPI a Δ in VERSA angle inresponse to MVC b
Level ofresistance
Reference
17-Res 91.471.3% +37.41 Weak Roche et al.,2013
MVC-Res 59.571.4% +12.11 Moderate Roche et al.,2011a
24-Res 12.572.5% +8.61 Strong Roche et al.,2013
The MPIs and VERSA vector angles have been reported previously for these Envs(Roche et al., 2011b, 2013), and are summarized here to assist in the interpretationof the CD4+ T-cell and macrophage tropism data.
a MPIs were determined using NP-2-CD4/CCR5 cells.b The VERSA metrics shown represent the change in vector angle, when
comparing vector angles from experiments conducted in the presence of MVCcompared to those from experiments conducted without MVC. An increase inchange in the vector angle is inversely proportional to the level of MVC resistance(Roche et al., 2011b).
J.K. Flynn et al. / Virology 442 (2013) 51–5852
conformation of CCR5 (Ogert et al., 2010; Pfaff et al., 2010; Tiltonet al., 2010).
Resistance to MVC is characterized by plateaus in virus inhibitioncurves which do not reach 100% inhibition, rather than by shifts inIC50, indicating a non-competitive mechanism of resistance [discussedin (Roche et al., 2011b; Westby et al., 2007)]. Results from phase IIIclinical testing of MVC have shown that the maximal percentageinhibition (MPI) of most MVC-resistant viruses range between 80–95%when tested using the PhenoSense assay, although some resistantviruses have MPIs as low as 20% (Mori et al., 2008). The MPI can beused as a measure of how efficiently a resistant virus can recognize theCCR5-antagonist bound conformation of CCR5; viruses with relativelyhigh MPIs (480%) are considered to have relatively inefficientrecognition of drug-bound CCR5 compared to unbound CCR5, whereasviruses with relatively low MPIs (o50%) are considered to haverelatively efficient recognition of drug-bound CCR5 compared tounbound CCR5.
Previous studies have shown that viruses sensitive to CCR5antagonists rely on interactions with the N-terminus and the ECL2region of CCR5, whereas resistant viruses frequently display an incre-ased reliance on the N-terminal region (Berro et al., 2009; Henrichet al., 2012; Ogert et al., 2009, 2010; Pfaff et al., 2010; Roche et al.,2011b; Tilton et al., 2010). The significance of altering the mechanismof coreceptor engagement by CCR5 antagonist-resistant viruses onHIV-1 pathogenesis is yet to be determined, but evidence suggests thatthe tropism of HIV-1 for different susceptible cell types may be alteredas a result of resistance to CCR5 antagonists. For example, one studyexamining resistance to APL showed that a viral variant with relativelyinefficient usage of APL-bound CCR5 had altered CD4+ T-cell tropism,characterized by relative sparing of central memory (CM) CD4+ T cells(Pfaff et al., 2010). Another study of a MVC-resistant strain of HIV-1that was generated in vitro showed a reduction in macrophageinfectivity due to inefficient recognition of MVC-bound CCR5expressed on the macrophage surface (Roche et al., 2011b). Impliedin both of these studies was that the continuation of CCR5 antagonisttherapy, even after the development of resistance, could maintain aselection for variants that use CCR5 inefficiently for entry, thuspotentially sparing significant cellular reservoirs from infection.However, both of these studies characterized resistant viruses thathad relatively inefficient recognition of antagonist-bound CCR5 (i.e.,relatively high MPIs), and whether attenuated infectivity in CM T-cellsand macrophages is a common feature among CCR5 antagonistresistant viruses that display differences in MPI is unknown.
In this study, we characterized the tropism alterations for CD4+T-cell subsets and macrophages by Envs from two subjects whodeveloped MVC resistance in vivo, which displayed either relativelyefficient or inefficient recognition of MVC-bound CCR5, as shown byMPIs of ∼10% and ∼90% in NP2-CD4/CCR5 cells, respectively. Ourresults show that MVC-resistant Env with efficient recognition ofdrug-bound CCR5, which we consider is “strongly resistant”, displays atropism shift for CD4+ T-cell subsets associated with increasedinfection of CM T-cells and reduced infection of effector memory(EM) and transitional memory (TM) T-cells, and no change in macro-phage infectivity. In contrast, MVC-resistant Env with inefficientrecognition of drug-bound CCR5, which we consider is “weaklyresistant”, displays no change in tropism for CD4+ T-cell subsets, butexhibits a significant reduction in macrophage infectivity. Together,our results show that the pattern of HIV-1 tropism alterations forsusceptible cells may be variable in subjects with MVC resistance.
Results and discussion
The panel of MVC resistant Envs has a spectrum of resistance levels
In this study, we characterized the tropism alterations for CD4+T-cell subsets and macrophages associated with MVC resistance by
3 distinct MVC resistant Env variants, as compared to matched MVCsensitive parental Envs. The MVC resistant Envs comprise two clonesderived from two subjects (subjects 17 and 24) who developed MVCresistance in vivo (Jubb et al., 2011; Mori et al., 2008; Pfizer Inc., 2007),and whose phenotypes and genetic determinants of resistance havebeen characterized recently (Roche et al., 2013). The plasma viral loadsand CD4+ T-cell counts of the study subjects from whom the Envswere derived are shown in Table 1, and the salient features of the Envsthat are relevant for this study are summarized in Table 2. Briefly, 17-Res and 24-Res Envs exhibit divergent levels of MVC resistance, with17-Res Env having an MPI of ∼90% in NP2-CD4/CCR5 cells, and 24-ResEnv having an MPI of ∼10%. The ability of these Envs to recognize theMVC-bound conformation of CCR5 was also divergent, with 17-ResEnv displaying a greater change in VERSA (Viral Entry ReceptorSensitivity Analysis) vector angles in response to MVC, as determinedby mathematical modeling of 293-Affinofile analyses (Chikere et al.,2013; Johnston et al., 2009), as compared to those of 24-Res Env,indicating that 17-Res Env is more CCR5-dependent than 24-Res Envin the presence of MVC. Thus, 17-Res and 24-Res have relativelyinefficient and efficient abilities to recognize the MVC-bound con-formation of CCR5, respectively, and are considered “weakly” resistantand “strongly” resistant Envs, respectively. For comparison, we alsocharacterized the tropism alterations of MVC-Res Env (Roche et al.,2011b), which was generated in vitro (Westby et al., 2007), ascompared to Env from its parental MVC sensitive virus (MVC-Sens).
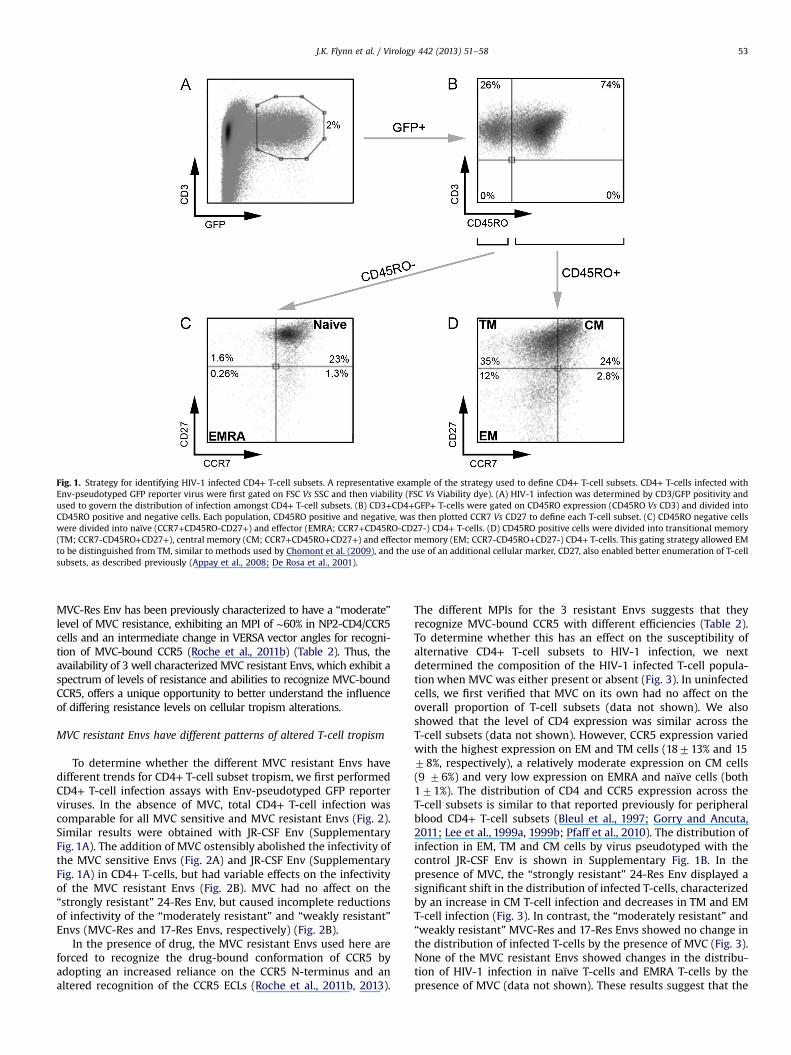
Fig. 1. Strategy for identifying HIV-1 infected CD4+ T-cell subsets. A representative example of the strategy used to define CD4+ T-cell subsets. CD4+ T-cells infected withEnv-pseudotyped GFP reporter virus were first gated on FSC Vs SSC and then viability (FSC Vs Viability dye). (A) HIV-1 infection was determined by CD3/GFP positivity andused to govern the distribution of infection amongst CD4+ T-cell subsets. (B) CD3+CD4+GFP+ T-cells were gated on CD45RO expression (CD45RO Vs CD3) and divided intoCD45RO positive and negative cells. Each population, CD45RO positive and negative, was then plotted CCR7 Vs CD27 to define each T-cell subset. (C) CD45RO negative cellswere divided into naïve (CCR7+CD45RO-CD27+) and effector (EMRA; CCR7+CD45RO-CD27-) CD4+ T-cells. (D) CD45RO positive cells were divided into transitional memory(TM; CCR7-CD45RO+CD27+), central memory (CM; CCR7+CD45RO+CD27+) and effector memory (EM; CCR7-CD45RO+CD27-) CD4+ T-cells. This gating strategy allowed EMto be distinguished from TM, similar to methods used by Chomont et al. (2009), and the use of an additional cellular marker, CD27, also enabled better enumeration of T-cellsubsets, as described previously (Appay et al., 2008; De Rosa et al., 2001).
J.K. Flynn et al. / Virology 442 (2013) 51–58 53
MVC-Res Env has been previously characterized to have a “moderate”level of MVC resistance, exhibiting an MPI of ∼60% in NP2-CD4/CCR5cells and an intermediate change in VERSA vector angles for recogni-tion of MVC-bound CCR5 (Roche et al., 2011b) (Table 2). Thus, theavailability of 3 well characterized MVC resistant Envs, which exhibit aspectrum of levels of resistance and abilities to recognize MVC-boundCCR5, offers a unique opportunity to better understand the influenceof differing resistance levels on cellular tropism alterations.
MVC resistant Envs have different patterns of altered T-cell tropism
To determine whether the different MVC resistant Envs havedifferent trends for CD4+ T-cell subset tropism, we first performedCD4+ T-cell infection assays with Env-pseudotyped GFP reporterviruses. In the absence of MVC, total CD4+ T-cell infection wascomparable for all MVC sensitive and MVC resistant Envs (Fig. 2).Similar results were obtained with JR-CSF Env (SupplementaryFig. 1A). The addition of MVC ostensibly abolished the infectivity ofthe MVC sensitive Envs (Fig. 2A) and JR-CSF Env (SupplementaryFig. 1A) in CD4+ T-cells, but had variable effects on the infectivityof the MVC resistant Envs (Fig. 2B). MVC had no affect on the“strongly resistant” 24-Res Env, but caused incomplete reductionsof infectivity of the “moderately resistant” and “weakly resistant”Envs (MVC-Res and 17-Res Envs, respectively) (Fig. 2B).
In the presence of drug, the MVC resistant Envs used here areforced to recognize the drug-bound conformation of CCR5 byadopting an increased reliance on the CCR5 N-terminus and analtered recognition of the CCR5 ECLs (Roche et al., 2011b, 2013).
The different MPIs for the 3 resistant Envs suggests that theyrecognize MVC-bound CCR5 with different efficiencies (Table 2).To determine whether this has an effect on the susceptibility ofalternative CD4+ T-cell subsets to HIV-1 infection, we nextdetermined the composition of the HIV-1 infected T-cell popula-tion when MVC was either present or absent (Fig. 3). In uninfectedcells, we first verified that MVC on its own had no affect on theoverall proportion of T-cell subsets (data not shown). We alsoshowed that the level of CD4 expression was similar across theT-cell subsets (data not shown). However, CCR5 expression variedwith the highest expression on EM and TM cells (18713% and 1578%, respectively), a relatively moderate expression on CM cells(9 76%) and very low expression on EMRA and naïve cells (both171%). The distribution of CD4 and CCR5 expression across theT-cell subsets is similar to that reported previously for peripheralblood CD4+ T-cell subsets (Bleul et al., 1997; Gorry and Ancuta,2011; Lee et al., 1999a, 1999b; Pfaff et al., 2010). The distribution ofinfection in EM, TM and CM cells by virus pseudotyped with thecontrol JR-CSF Env is shown in Supplementary Fig. 1B. In thepresence of MVC, the “strongly resistant” 24-Res Env displayed asignificant shift in the distribution of infected T-cells, characterizedby an increase in CM T-cell infection and decreases in TM and EMT-cell infection (Fig. 3). In contrast, the “moderately resistant” and“weakly resistant” MVC-Res and 17-Res Envs showed no change inthe distribution of infected T-cells by the presence of MVC (Fig. 3).None of the MVC resistant Envs showed changes in the distribu-tion of HIV-1 infection in naïve T-cells and EMRA T-cells by thepresence of MVC (data not shown). These results suggest that the
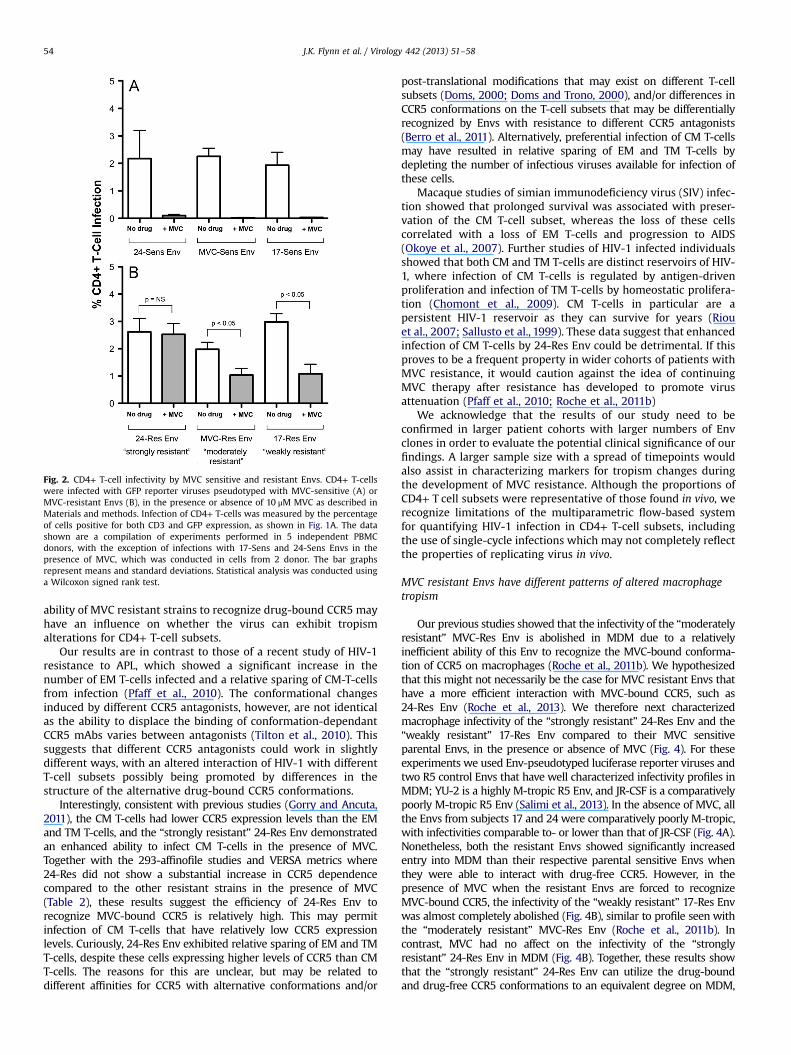
Fig. 2. CD4+ T-cell infectivity by MVC sensitive and resistant Envs. CD4+ T-cellswere infected with GFP reporter viruses pseudotyped with MVC-sensitive (A) orMVC-resistant Envs (B), in the presence or absence of 10 μM MVC as described inMaterials and methods. Infection of CD4+ T-cells was measured by the percentageof cells positive for both CD3 and GFP expression, as shown in Fig. 1A. The datashown are a compilation of experiments performed in 5 independent PBMCdonors, with the exception of infections with 17-Sens and 24-Sens Envs in thepresence of MVC, which was conducted in cells from 2 donor. The bar graphsrepresent means and standard deviations. Statistical analysis was conducted usinga Wilcoxon signed rank test.
J.K. Flynn et al. / Virology 442 (2013) 51–5854
ability of MVC resistant strains to recognize drug-bound CCR5 mayhave an influence on whether the virus can exhibit tropismalterations for CD4+ T-cell subsets.
Our results are in contrast to those of a recent study of HIV-1resistance to APL, which showed a significant increase in thenumber of EM T-cells infected and a relative sparing of CM-T-cellsfrom infection (Pfaff et al., 2010). The conformational changesinduced by different CCR5 antagonists, however, are not identicalas the ability to displace the binding of conformation-dependantCCR5 mAbs varies between antagonists (Tilton et al., 2010). Thissuggests that different CCR5 antagonists could work in slightlydifferent ways, with an altered interaction of HIV-1 with differentT-cell subsets possibly being promoted by differences in thestructure of the alternative drug-bound CCR5 conformations.
Interestingly, consistent with previous studies (Gorry and Ancuta,2011), the CM T-cells had lower CCR5 expression levels than the EMand TM T-cells, and the “strongly resistant” 24-Res Env demonstratedan enhanced ability to infect CM T-cells in the presence of MVC.Together with the 293-affinofile studies and VERSA metrics where24-Res did not show a substantial increase in CCR5 dependencecompared to the other resistant strains in the presence of MVC(Table 2), these results suggest the efficiency of 24-Res Env torecognize MVC-bound CCR5 is relatively high. This may permitinfection of CM T-cells that have relatively low CCR5 expressionlevels. Curiously, 24-Res Env exhibited relative sparing of EM and TMT-cells, despite these cells expressing higher levels of CCR5 than CMT-cells. The reasons for this are unclear, but may be related todifferent affinities for CCR5 with alternative conformations and/or
post-translational modifications that may exist on different T-cellsubsets (Doms, 2000; Doms and Trono, 2000), and/or differences inCCR5 conformations on the T-cell subsets that may be differentiallyrecognized by Envs with resistance to different CCR5 antagonists(Berro et al., 2011). Alternatively, preferential infection of CM T-cellsmay have resulted in relative sparing of EM and TM T-cells bydepleting the number of infectious viruses available for infection ofthese cells.
Macaque studies of simian immunodeficiency virus (SIV) infec-tion showed that prolonged survival was associated with preser-vation of the CM T-cell subset, whereas the loss of these cellscorrelated with a loss of EM T-cells and progression to AIDS(Okoye et al., 2007). Further studies of HIV-1 infected individualsshowed that both CM and TM T-cells are distinct reservoirs of HIV-1, where infection of CM T-cells is regulated by antigen-drivenproliferation and infection of TM T-cells by homeostatic prolifera-tion (Chomont et al., 2009). CM T-cells in particular are apersistent HIV-1 reservoir as they can survive for years (Riouet al., 2007; Sallusto et al., 1999). These data suggest that enhancedinfection of CM T-cells by 24-Res Env could be detrimental. If thisproves to be a frequent property in wider cohorts of patients withMVC resistance, it would caution against the idea of continuingMVC therapy after resistance has developed to promote virusattenuation (Pfaff et al., 2010; Roche et al., 2011b)
We acknowledge that the results of our study need to beconfirmed in larger patient cohorts with larger numbers of Envclones in order to evaluate the potential clinical significance of ourfindings. A larger sample size with a spread of timepoints wouldalso assist in characterizing markers for tropism changes duringthe development of MVC resistance. Although the proportions ofCD4+ T cell subsets were representative of those found in vivo, werecognize limitations of the multiparametric flow-based systemfor quantifying HIV-1 infection in CD4+ T-cell subsets, includingthe use of single-cycle infections which may not completely reflectthe properties of replicating virus in vivo.
MVC resistant Envs have different patterns of altered macrophagetropism
Our previous studies showed that the infectivity of the “moderatelyresistant” MVC-Res Env is abolished in MDM due to a relativelyinefficient ability of this Env to recognize the MVC-bound conforma-tion of CCR5 on macrophages (Roche et al., 2011b). We hypothesizedthat this might not necessarily be the case for MVC resistant Envs thathave a more efficient interaction with MVC-bound CCR5, such as24-Res Env (Roche et al., 2013). We therefore next characterizedmacrophage infectivity of the “strongly resistant” 24-Res Env and the“weakly resistant” 17-Res Env compared to their MVC sensitiveparental Envs, in the presence or absence of MVC (Fig. 4). For theseexperiments we used Env-pseudotyped luciferase reporter viruses andtwo R5 control Envs that have well characterized infectivity profiles inMDM; YU-2 is a highly M-tropic R5 Env, and JR-CSF is a comparativelypoorly M-tropic R5 Env (Salimi et al., 2013). In the absence of MVC, allthe Envs from subjects 17 and 24 were comparatively poorly M-tropic,with infectivities comparable to- or lower than that of JR-CSF (Fig. 4A).Nonetheless, both the resistant Envs showed significantly increasedentry into MDM than their respective parental sensitive Envs whenthey were able to interact with drug-free CCR5. However, in thepresence of MVC when the resistant Envs are forced to recognizeMVC-bound CCR5, the infectivity of the “weakly resistant” 17-Res Envwas almost completely abolished (Fig. 4B), similar to profile seen withthe “moderately resistant” MVC-Res Env (Roche et al., 2011b). Incontrast, MVC had no affect on the infectivity of the “stronglyresistant” 24-Res Env in MDM (Fig. 4B). Together, these results showthat the “strongly resistant” 24-Res Env can utilize the drug-boundand drug-free CCR5 conformations to an equivalent degree on MDM,
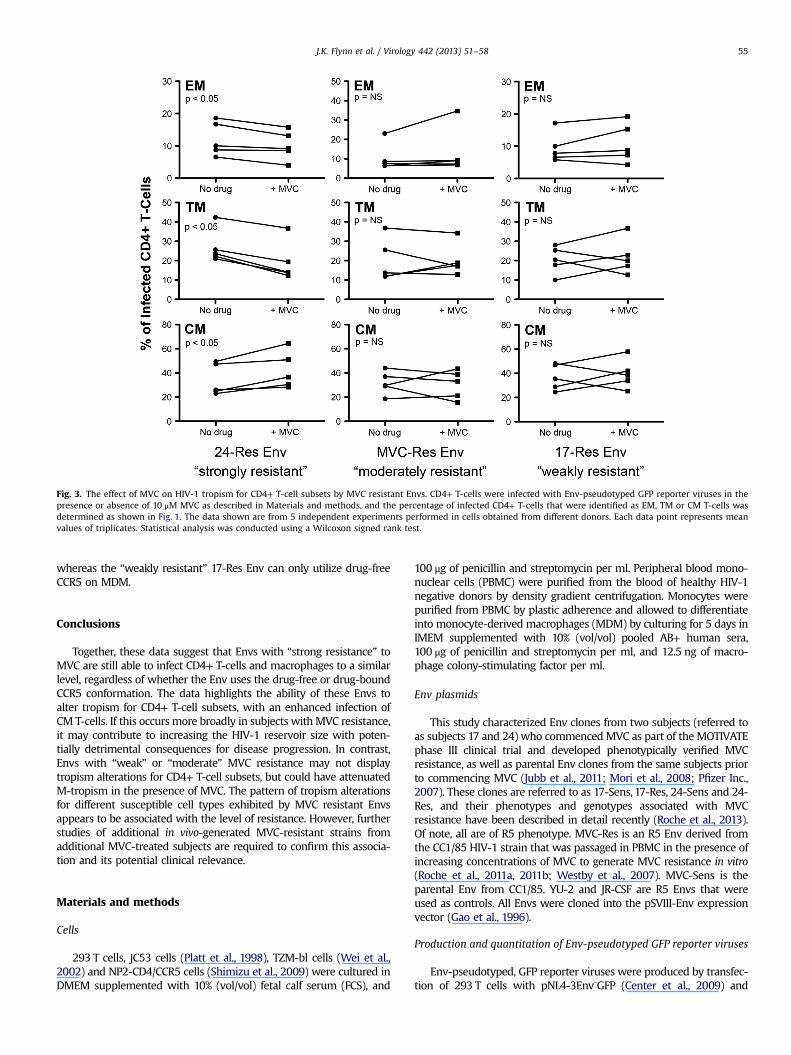
Fig. 3. The effect of MVC on HIV-1 tropism for CD4+ T-cell subsets by MVC resistant Envs. CD4+ T-cells were infected with Env-pseudotyped GFP reporter viruses in thepresence or absence of 10 μM MVC as described in Materials and methods, and the percentage of infected CD4+ T-cells that were identified as EM, TM or CM T-cells wasdetermined as shown in Fig. 1. The data shown are from 5 independent experiments performed in cells obtained from different donors. Each data point represents meanvalues of triplicates. Statistical analysis was conducted using a Wilcoxon signed rank test.
J.K. Flynn et al. / Virology 442 (2013) 51–58 55
whereas the “weakly resistant” 17-Res Env can only utilize drug-freeCCR5 on MDM.
Conclusions
Together, these data suggest that Envs with “strong resistance” toMVC are still able to infect CD4+ T-cells and macrophages to a similarlevel, regardless of whether the Env uses the drug-free or drug-boundCCR5 conformation. The data highlights the ability of these Envs toalter tropism for CD4+ T-cell subsets, with an enhanced infection ofCM T-cells. If this occurs more broadly in subjects withMVC resistance,it may contribute to increasing the HIV-1 reservoir size with poten-tially detrimental consequences for disease progression. In contrast,Envs with “weak” or “moderate” MVC resistance may not displaytropism alterations for CD4+ T-cell subsets, but could have attenuatedM-tropism in the presence of MVC. The pattern of tropism alterationsfor different susceptible cell types exhibited by MVC resistant Envsappears to be associated with the level of resistance. However, furtherstudies of additional in vivo-generated MVC-resistant strains fromadditional MVC-treated subjects are required to confirm this associa-tion and its potential clinical relevance.
Materials and methods
Cells
293 T cells, JC53 cells (Platt et al., 1998), TZM-bl cells (Wei et al.,2002) and NP2-CD4/CCR5 cells (Shimizu et al., 2009) were cultured inDMEM supplemented with 10% (vol/vol) fetal calf serum (FCS), and
100 μg of penicillin and streptomycin per ml. Peripheral blood mono-nuclear cells (PBMC) were purified from the blood of healthy HIV-1negative donors by density gradient centrifugation. Monocytes werepurified from PBMC by plastic adherence and allowed to differentiateinto monocyte-derived macrophages (MDM) by culturing for 5 days inIMEM supplemented with 10% (vol/vol) pooled AB+ human sera,100 μg of penicillin and streptomycin per ml, and 12.5 ng of macro-phage colony-stimulating factor per ml.
Env plasmids
This study characterized Env clones from two subjects (referred toas subjects 17 and 24) who commencedMVC as part of the MOTIVATEphase III clinical trial and developed phenotypically verified MVCresistance, as well as parental Env clones from the same subjects priorto commencing MVC (Jubb et al., 2011; Mori et al., 2008; Pfizer Inc.,2007). These clones are referred to as 17-Sens, 17-Res, 24-Sens and 24-Res, and their phenotypes and genotypes associated with MVCresistance have been described in detail recently (Roche et al., 2013).Of note, all are of R5 phenotype. MVC-Res is an R5 Env derived fromthe CC1/85 HIV-1 strain that was passaged in PBMC in the presence ofincreasing concentrations of MVC to generate MVC resistance in vitro(Roche et al., 2011a, 2011b; Westby et al., 2007). MVC-Sens is theparental Env from CC1/85. YU-2 and JR-CSF are R5 Envs that wereused as controls. All Envs were cloned into the pSVIII-Env expressionvector (Gao et al., 1996).
Production and quantitation of Env-pseudotyped GFP reporter viruses
Env-pseudotyped, GFP reporter viruses were produced by transfec-tion of 293 T cells with pNL4-3Env-GFP (Center et al., 2009) and
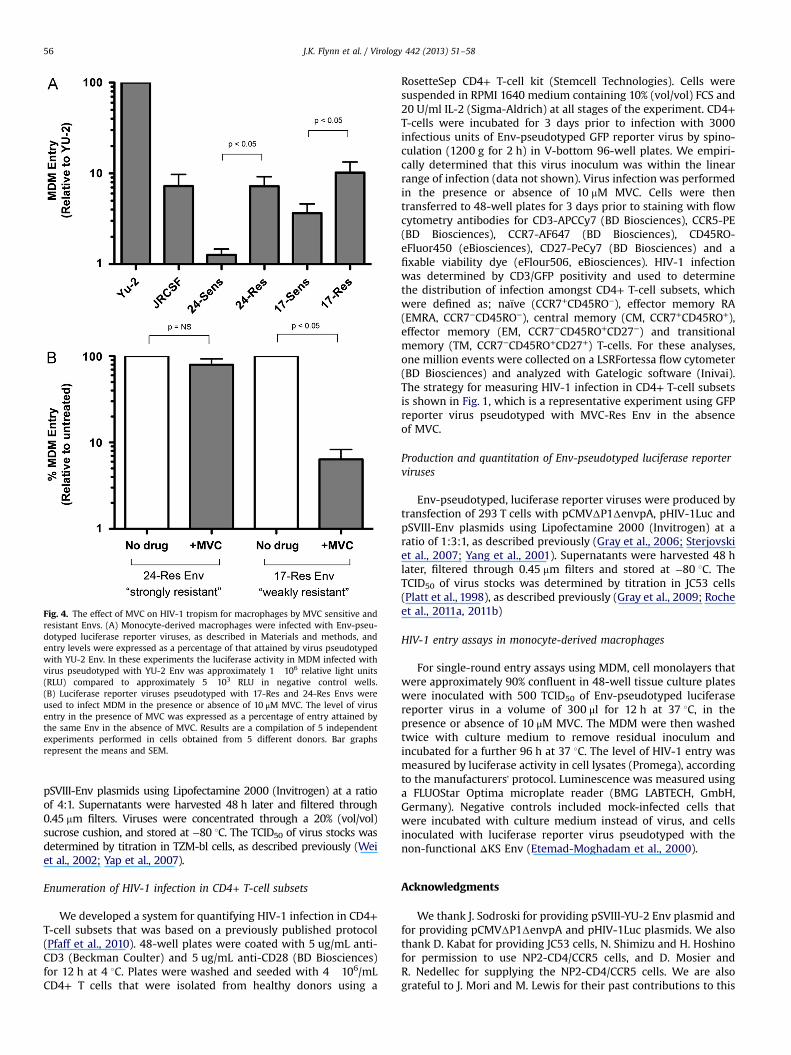
Fig. 4. The effect of MVC on HIV-1 tropism for macrophages by MVC sensitive andresistant Envs. (A) Monocyte-derived macrophages were infected with Env-pseu-dotyped luciferase reporter viruses, as described in Materials and methods, andentry levels were expressed as a percentage of that attained by virus pseudotypedwith YU-2 Env. In these experiments the luciferase activity in MDM infected withvirus pseudotyped with YU-2 Env was approximately 1�106 relative light units(RLU) compared to approximately 5�103 RLU in negative control wells.(B) Luciferase reporter viruses pseudotyped with 17-Res and 24-Res Envs wereused to infect MDM in the presence or absence of 10 μM MVC. The level of virusentry in the presence of MVC was expressed as a percentage of entry attained bythe same Env in the absence of MVC. Results are a compilation of 5 independentexperiments performed in cells obtained from 5 different donors. Bar graphsrepresent the means and SEM.
J.K. Flynn et al. / Virology 442 (2013) 51–5856
pSVIII-Env plasmids using Lipofectamine 2000 (Invitrogen) at a ratioof 4:1. Supernatants were harvested 48 h later and filtered through0.45 mm filters. Viruses were concentrated through a 20% (vol/vol)sucrose cushion, and stored at −80 1C. The TCID50 of virus stocks wasdetermined by titration in TZM-bl cells, as described previously (Weiet al., 2002; Yap et al., 2007).
Enumeration of HIV-1 infection in CD4+ T-cell subsets
We developed a system for quantifying HIV-1 infection in CD4+T-cell subsets that was based on a previously published protocol(Pfaff et al., 2010). 48-well plates were coated with 5 ug/mL anti-CD3 (Beckman Coulter) and 5 ug/mL anti-CD28 (BD Biosciences)for 12 h at 4 1C. Plates were washed and seeded with 4�106/mLCD4+ T cells that were isolated from healthy donors using a
RosetteSep CD4+ T-cell kit (Stemcell Technologies). Cells weresuspended in RPMI 1640 medium containing 10% (vol/vol) FCS and20 U/ml IL-2 (Sigma-Aldrich) at all stages of the experiment. CD4+T-cells were incubated for 3 days prior to infection with 3000infectious units of Env-pseudotyped GFP reporter virus by spino-culation (1200 g for 2 h) in V-bottom 96-well plates. We empiri-cally determined that this virus inoculum was within the linearrange of infection (data not shown). Virus infection was performedin the presence or absence of 10 mM MVC. Cells were thentransferred to 48-well plates for 3 days prior to staining with flowcytometry antibodies for CD3-APCCy7 (BD Biosciences), CCR5-PE(BD Biosciences), CCR7-AF647 (BD Biosciences), CD45RO-eFluor450 (eBiosciences), CD27-PeCy7 (BD Biosciences) and afixable viability dye (eFlour506, eBiosciences). HIV-1 infectionwas determined by CD3/GFP positivity and used to determinethe distribution of infection amongst CD4+ T-cell subsets, whichwere defined as; naïve (CCR7+CD45RO−), effector memory RA(EMRA, CCR7−CD45RO−), central memory (CM, CCR7+CD45RO+),effector memory (EM, CCR7−CD45RO+CD27−) and transitionalmemory (TM, CCR7−CD45RO+CD27+) T-cells. For these analyses,one million events were collected on a LSRFortessa flow cytometer(BD Biosciences) and analyzed with Gatelogic software (Inivai).The strategy for measuring HIV-1 infection in CD4+ T-cell subsetsis shown in Fig. 1, which is a representative experiment using GFPreporter virus pseudotyped with MVC-Res Env in the absenceof MVC.
Production and quantitation of Env-pseudotyped luciferase reporterviruses
Env-pseudotyped, luciferase reporter viruses were produced bytransfection of 293 T cells with pCMVΔP1ΔenvpA, pHIV-1Luc andpSVIII-Env plasmids using Lipofectamine 2000 (Invitrogen) at aratio of 1:3:1, as described previously (Gray et al., 2006; Sterjovskiet al., 2007; Yang et al., 2001). Supernatants were harvested 48 hlater, filtered through 0.45 mm filters and stored at −80 1C. TheTCID50 of virus stocks was determined by titration in JC53 cells(Platt et al., 1998), as described previously (Gray et al., 2009; Rocheet al., 2011a, 2011b)
HIV-1 entry assays in monocyte-derived macrophages
For single-round entry assays using MDM, cell monolayers thatwere approximately 90% confluent in 48-well tissue culture plateswere inoculated with 500 TCID50 of Env-pseudotyped luciferasereporter virus in a volume of 300 μl for 12 h at 37 1C, in thepresence or absence of 10 μM MVC. The MDM were then washedtwice with culture medium to remove residual inoculum andincubated for a further 96 h at 37 1C. The level of HIV-1 entry wasmeasured by luciferase activity in cell lysates (Promega), accordingto the manufacturers' protocol. Luminescence was measured usinga FLUOStar Optima microplate reader (BMG LABTECH, GmbH,Germany). Negative controls included mock-infected cells thatwere incubated with culture medium instead of virus, and cellsinoculated with luciferase reporter virus pseudotyped with thenon-functional ΔKS Env (Etemad-Moghadam et al., 2000).
Acknowledgments
We thank J. Sodroski for providing pSVIII-YU-2 Env plasmid andfor providing pCMVΔP1ΔenvpA and pHIV-1Luc plasmids. We alsothank D. Kabat for providing JC53 cells, N. Shimizu and H. Hoshinofor permission to use NP2-CD4/CCR5 cells, and D. Mosier andR. Nedellec for supplying the NP2-CD4/CCR5 cells. We are alsograteful to J. Mori and M. Lewis for their past contributions to this

J.K. Flynn et al. / Virology 442 (2013) 51–58 57
work, J. Demarest for helpful comments, and T. Spellman for advicewith statistical analysis. Maraviroc was provided by Pfizer. Thisstudy was supported by a grant from the Australian National Healthand Medical Research Council to PRG, MJC and SRL (1006534). PRGwas the recipient of an Australian National Health and MedicalResearch Council (NHMRC) Level 2 Biomedical Career DevelopmentAward, and is now supported by an Australian Research Council(ARC) Future Fellowship (FT2). LRG is the recipient of an AustralianNHMRC Postdoctoral Training Fellowship. HS is supported by aPostgraduate Research Scholarship from the Iranian Ministry ofHealth and Medical Education. SRL is the recipient of a NHMRCPractitioner Fellowship. The authors gratefully acknowledge thecontribution to this work of the Victorian Operational InfrastructureSupport Program received by the Burnet Institute. Pfizer assumesno responsibility for the validation and storage of the data shown inFigs. 1–4 and Table 2.
Appendix A. Supporting information
Supplementary data associated with this article can be found inthe online version at http://dx.doi.org/10.1016/j.virol.2013.03.026.
References
Appay, V., van Lier, R., Sallusto, F., Roederer, M., 2008. Phenotype and function ofhuman T lymphocyte subsets: consensus and issues. Cytometry Part A 73,975–983.
Berro, R., Klasse, P.J., Lascano, D., Flegler, A., Nagashima, K.A., Sanders, R.W., Sakmar,T.P., Hope, T.J., Moore, J.P., 2011. Multiple CCR5 conformations on the cellsurface are used differentially by human immunodeficiency viruses resistant orsensitive to CCR5 inhibitors. J. Virol. 85, 8227–8240.
Berro, R., Sanders, R.W., Lu, M., Klasse, P.J., Moore, J.P., 2009. Two HIV-1 variantsresistant to small molecule CCR5 inhibitors differ in how they use CCR5 forentry. PLoS Pathog. 5, e1000548.
Bleul, C.C., Wu, L., Hoxie, J.A., Springer, T.A., Mackay, C.R., 1997. The HIV coreceptorsCXCR4 and CCR5 are differentially expressed and regulated on humanT lymphocytes. PNAS 94, 1925–1930.
Center, R.J., Wheatley, A.K., Campbell, S.M., Gaeguta, A.J., Peut, V., Alcantara, S.,Siebentritt, C., Kent, S.J., Purcell, D.F., 2009. Induction of HIV-1 subtype B andAE-specific neutralizing antibodies in mice and macaques with DNA prime andrecombinant gp140 protein boost regimens. Vaccine 27, 6605–6612.
Chikere, K., Chou, T., Gorry, P.R., Lee, B., 2013. Affinofile profiling: how efficiency ofCD4/CCR5 usage impacts the biological and pathogenic phenotype of HIV.Virology 435, 81–91.
Chomont, N., El-Far, M., Ancuta, P., Trautmann, L., Procopio, F.A., Yassine-Diab, B.,Boucher, G., Boulassel, M.R., Ghattas, G., Brenchley, J.M., Schacker, T.W., Hill, B.J.,Douek, D.C., Routy, J.P., Haddad, E.K., Sekaly, R.P., 2009. HIV reservoir size andpersistence are driven by T cell survival and homeostatic proliferation. Nat.Med. 15, 893–900.
De Rosa, S., Herzenberg, L., Herzenberg, L., Roederer, M., 2001. 11-color,13-parameter flow cytometry: identification of human naive T cells byphenotype, function, and T-cell receptor diversity. Nat. Med. 7, 245–248.
Doms, R.W., 2000. Beyond receptor expression: the influence of receptor con-formation, density, and affinity in HIV-1 infection. Virology 276, 229–237.
Doms, R.W., Trono, D., 2000. The plasma membrane as a combat zone in the HIVbattlefield. Genes Dev. 14, 2677–2688.
Dragic, T., Trkola, A., Thompson, D.A., Cormier, E.G., Kajumo, F.A., Maxwell, E., Lin, S.W., Ying, W., Smith, S.O., Sakmar, T.P., Moore, J.P., 2000. A binding pocket for asmall molecule inhibitor of HIV-1 entry within the transmembrane helices ofCCR5. PNAS 97, 5639–5644.
Etemad-Moghadam, B., Sun, Y., Nicholson, E.K., Fernandes, M., Liou, K., Gomila, R.,Lee, J., Sodroski, J., 2000. Envelope glycoprotein determinants of increasedfusogenicity in a pathogenic simian-human immunodeficiency virus (SHIV-KB9) passaged in vivo. J. Virol. 74, 4433–4440.
Gao, F., Morrison, S.G., Robertson, D.L., Thornton, C.L., Craig, S., Karlsson, G.,Sodroski, J., Morgado, M., Galvao-Castro, B., von Briesen, H., et al., 1996.Molecular cloning and analysis of functional envelope genes from humanimmunodeficiency virus type 1 sequence subtypes A through G. The WHO andNIAID Networks for HIV isolation and characterization. J. Virol. 70, 1651–1667.
Gorry, P.R., Ancuta, P., 2011. Coreceptors and HIV-1 pathogenesis. Curr. HIV/AIDSRep. 8, 45–53.
Gorry, P.R., Ellett, A., Lewin, S.R., 2010. Maraviroc. In: Grayson, L., Crowe, S.,McCarthy, J., Mills, J., Mouton, J., Norrby, S.R., Paterson, D., Pfaller, M. (Eds.),Kucers' The Use of Antibiotics, sixth ed. Hodder & Stoughton Ltd., London,pp. 2869–2876.
Gray, L., Churchill, M.J., Keane, N., Sterjovski, J., Ellett, A.M., Purcell, D.F., Poumbour-ios, P., Kol, C., Wang, B., Saksena, N.K., Wesselingh, S.L., Price, P., French, M.,Gabuzda, D., Gorry, P.R., 2006. Genetic and functional analysis of R5�4 humanimmunodeficiency virus type 1 envelope glycoproteins derived from twoindividuals homozygous for the CCR5delta32 allele. J. Virol. 80, 3684–3691.
Gray, L., Roche, M., Churchill, M.J., Sterjovski, J., Ellett, A., Poumbourios, P., Sheffief,S., Wang, B., Saksena, N., Purcell, D.F., Wesselingh, S., Cunningham, A.L., Brew, B.J., Gabuzda, D., Gorry, P.R., 2009. Tissue-specific sequence alterations in thehuman immunodeficiency virus type 1 envelope favoring CCR5 usage con-tribute to persistence of dual-tropic virus in the brain. J. Virol. 83, 5430–5441.
Henrich, T.J., Lewine, N.R., Lee, S.H., Rao, S.S., Berro, R., Gulick, R.M., Moore, J.P.,Tsibris, A.M., Kuritzkes, D.R., 2012. Differential use of CCR5 by HIV-1 clinicalisolates resistant to small-molecule CCR5 antagonists. Antimicrob. AgentsChemother. 56, 1931–1935.
Johnston, S.H., Lobriz, M.A., Nguyen, S., Lassen, K., Delair, S., Posta, F., Bryson, Y.J.,Arts, E.J., Chou, T., Lee, B., 2009. A quantitative affinity-profiling system thatreveals distinct CD4/CCR5 usage patterns among human immunodeficiencyvirus type 1 and simian immunodeficiency virus strains. J. Virol. 83,11016–11026.
Jubb, B., Buttler, S., Craig, C., Westby, M., 2011. Maraviroc-resistant viruses continueto use the extracellular loop and N-terminal regions of CCR5 for cell entry. In:Program and Abstracts of the 18th Conference on Retroviruses and Opportu-nistic Infections, Boston, February 27-March 2. Poster P590.
Lee, B., Sharron, M., Blanpain, C., Doranz, B.J., Vakili, J., Setoh, P., Berg, E., Liu, G., Guy,H.R., Durell, S.R., Parmentier, M., Chang, C.N., Price, K., Tsang, M., Doms, R.W.,1999a. Epitope mapping of CCR5 reveals multiple conformational states anddistinct but overlapping structures involved in chemokine and coreceptorfunction. J. Biol. Chem. 274, 9617–9626.
Lee, B., Sharron, M., Montaner, L.J., Weissman, D., Doms, R.W., 1999b. Quantificationof CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, anddifferentially conditioned monocyte-derived macrophages. PNAS 96,5215–5220.
Maeda, K., Das, D., Yin, P.D., Tsuchiya, K., Ogata-Aoki, H., Nakata, H., Norman, R.B.,Hackney, L.A., Takaoka, Y., Mitsuya, H., 2008. Involvement of the secondextracellular loop and transmembrane residues of CCR5 in inhibitor bindingand HIV-1 fusion: insights into the mechanism of allosteric inhibition. J. Mol.Biol. 381, 956–974.
Mori, J., Lewis, M., Simpson, P., Whitcomb, J., Perros, M., van der Ryst, E., Westby, M.,2008. Characterization of Maraviroc Resistance in Patients Failing Treatmentwith CCR5-tropic Virus in MOTIVATE 1 and MOTIVATE 2 (24 week analysis).Program and Abstracts of the Sixth European Drug Resistance Workshop,Budapest, March 26-28. Poster 51.
Ogert, R.A., Ba, L., Hou, Y., Buontempo, C., Qiu, P., Duca, J., Murgolo, N., Buontempo,P., Ralston, R., Howe, J.A., 2009. Structure-function analysis of human immu-nodeficiency virus type 1 gp120 amino acid mutations associated withresistance to the CCR5 coreceptor antagonist vicriviroc. J. Virol. 83,12151–12163.
Ogert, R.A., Hou, Y., Ba, L., Wojcik, L., Qiu, P., Murgolo, N., Duca, J., Dunkle, L.M.,Ralston, R., Howe, J.A., 2010. Clinical resistance to vicriviroc through adaptiveV3 loop mutations in HIV-1 subtype D gp120 that alter interactions with the N-terminus and ECL2 of CCR5. Virology 400, 145–155.
Okoye, A., Meier-Schellersheim, M., Brenchley, J.M., Hagen, S.I., Walker, J.M.,Rohankhedkar, M., Lum, R., Edgar, J.B., Planer, S.I., Legasse, A., Sylwester, A.W., Piatak, M., Lisfson, J.J.D., Maino, V.C., Sodora, D.L., Douck, D.C., Axthelm, M.K., Grossman, Z., Picker, L.J., 2007. Progressive CD4+ central memory T celldecline results in CD4+ effector memory insufficiency and overt disease inchronic SIV infection. J. Exp. Med. 204, 2171–2185.
Pfaff, J.M., Wilen, C.B., Harrison, J.E., Demarest, J.F., Lee, B., Doms, R.W., Tilton, J.C.,2010. HIV-1 resistance to CCR5 antagonists associated with highly efficient useof CCR5 and altered tropism on primary CD4+ T cells. J. Virol. 84, 6505–6514.
Pfizer Inc, 2007. Maraviroc Tablets NDA 22-128: Antiviral Drugs Advisory Commit-tee (AVDAC) Briefing Document. Pfizer Inc, New York, NY.
Platt, E.J., Wehrly, K., Kuhmann, S.E., Chesebro, B., Kabat, D., 1998. Effects of CCR5and CD4 cell surface concentrations on infections by macrophagetropic isolatesof human immunodeficiency virus type 1. J. Virol. 72, 2855–2864.
Riou, C., Yassine-Diab, B., Van grevenynghe, J., Somogyi, R., Greller, L.D., Gagnon, D.,Gimmig, S., Wilkinson, P., Shi, Y., Cameron, M.J., Campos-Gonzalez, R., Balderas,R.S., Kelvin, D., Sekaly, R.P., Haddad, E.K., 2007. Convergence of TCR andcytokine signaling leads to FOXO3a phosphorylation and drives the survivalof CD4+ central memory T cells. J. Exp. Med. 204, 79–91.
Roche, M., Jakobsen, M.R., Ellett, A., Salimiseyedabad, H., Jubb, B., Westby, M., Lee,B., Lewin, S.R., Churchill, M.J., Gorry, P.R., 2011a. HIV-1 predisposed to acquiringresistance to maraviroc (MVC) and other CCR5 antagonists in vitro has aninherent, low-level ability to utilize MVC-bound CCR5 for entry. Retrovirology8, 89.
Roche, M., Jakobsen, M.R., Sterjovski, J., Ellett, A., Posta, F., Lee, B., Jubb, B., Westby,M., Lewin, S.R., Ramsland, P.A., Churchill, M.J., Gorry, P.R., 2011b. HIV-1 escapefrom the CCR5 antagonist maraviroc associated with an altered and lessefficient mechanism of gp120-CCR5 engagement that attenuates macrophage-tropism. J. Virol. 85, 4330–4342.
Roche, M., Salimi, H., Duncan, R., Wilkinson, B., Chikere, K., Moore, M.S., Webb, N.,Zappi, H., Sterjovski, J., Flynn, J., Ellett, A., Gray, L.R., Lee, B., Jubb, B., Westby, M.,Ramsland, P.A., Lewin, S.R., Payne, R., Churchill, M.J., Gorry, P.R., 2013. Acommon mechanism of clinical HIV-1 resistance to Maraviroc despite divergentresistance levels and lack of common gp120 resistance mutations. Retro-virology 10, 43.

J.K. Flynn et al. / Virology 442 (2013) 51–5858
Salimi, H., Roche, M., Webb, N., Gray, L.R., Chikere, K., Sterjovski, J., Ellett, A.,Wesselingh, S.L., Ramsland, P.A., Lee, B., Churchill, M., Gorry, P.R., 2013.Macrophage-tropic HIV-1 variants from brain demonstrate alterations in theway gp120 engages both CD4 and CCR5. J. Leukoc. Biol. 93, 113–126.
Sallusto, F., Lenig, D., Forster, R., Lipp, M., Lanzavecchia, A., 1999. Two subsets ofmemory T lymphocytes with distinct homing potentials and effector functions.Nature 401, 708–712.
Seibert, C., Ying, W., Gavrilov, S., Tsamis, F., Kuhmann, S.E., Palani, A., Tagat, J.R.,Clader, J.W., McCombie, S.W., Baroudy, B.M., Smith, S.O., Dragic, T., Moore, J.P.,Sakmar, T.P., 2006. Interaction of small molecule inhibitors of HIV-1 entry withCCR5. Virology 349, 41–54.
Shimizu, N., Tanaka, A., Oue, A., Mori, T., Ohtsuki, T., Apichartpiyakul, C., Uchiumi,H., Nojima, Y., Hoshino, H., 2009. Broad usage spectrum of G protein-coupledreceptors as coreceptors by primary isolates of HIV. AIDS 23, 761–769.
Sterjovski, J., Churchill, M.J., Ellett, A., Gray, L.R., Roche, M.J., Dunfee, R.L.,Purcell, D.F., Saksena, N., Wang, B., Sonza, S., Wesselingh, S.L., Karlsson, I.,Fenyo, E.M., Gabuzda, D., Cunningham, A.L., Gorry, P.R., 2007. Asn 362 in gp120contributes to enhanced fusogenicity by CCR5-restricted HIV-1 envelopeglycoprotein variants from patients with AIDS. Retrovirology 4, 89.
Tilton, J.C., Doms, R.W., 2010. Entry inhibitors in the treatment of HIV-1 infection.Antiviral Res. 85, 91–100.
Tilton, J.C., Wilen, C.B., Didigu, C.A., Sinha, R., Harrison, J.E., Agrawal-Gamse, C.,Henning, E.A., Bushman, F.D., Martin, J.N., Deeks, S.G., Doms, R.W., 2010.
A maraviroc-resistant HIV-1 with narrow cross-resistance to other CCR5antagonists depends on both N-terminal and extracellular loop domains ofdrug-bound CCR5. J. Virol. 84, 10863–10876.
Wei, X., Decker, J.M., Liu, H., Zhang, Z., Arani, R.B., Kilby, J.M., Saag, M.S., Wu, X.,Shaw, G.M., Kappes, J.C., 2002. Emergence of resistant human immunodefi-ciency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy.Antimicrob. Agents Chemother. 46, 1896–1905.
Westby, M., Lewis, M., Whitcomb, J., Youle, M., Pozniak, A.L., James, I.T., Jenkins, T.M., Perros, M., van der Ryst, E., 2006. Emergence of CXCR4-using humanimmunodeficiency virus type 1 (HIV-1) variants in a minority of HIV-1-infectedpatients following treatment with the CCR5 antagonist maraviroc is from apretreatment CXCR4-using virus reservoir. J. Virol. 80, 4909–4920.
Westby, M., Smith-Burchnell, C., Mori, J., Lewis, M., Mosley, M., Stockdale, M., Dorr,P., Ciaramella, G., Perros, M., 2007. Reduced maximal inhibition in phenotypicsusceptibility assays indicates that viral strains resistant to the CCR5 antagonistmaraviroc utilize inhibitor-bound receptor for entry. J. Virol. 81, 2359–2371.
Yang, X., Wyatt, R., Sodroski, J., 2001. Improved elicitation of neutralizing antibodiesagainst primary human immunodeficiency viruses by soluble stabilized envel-ope glycoprotein trimers. J. Virol. 75, 1165–1171.
Yap, S.H., Sheen, C.W., Fahey, J., Zanin, M., Tyssen, D., Lima, V.D., Wynhoven, B.,Kuiper, M., Sluis-Cremer, N., Harrigan, P.R., Tachedjian, G., 2007. N348I in theconnection domain of HIV-1 reverse transcriptase confers zidovudine andnevirapine resistance. PLoS Med. 4, e335.




















