
Calibration of the response of 9-amino acridine fluorescence to transmembrane pH differences in...
10
ARCHIVES OF BIOCHEMISTRY AND BIOPHYSICS Vol. 238, No. 1, April, pp. 219-228, 1985 Calibration of the Response of g-Amino Acridine Fluorescence to Transmembrane pH Differences in Bacterial Chromatophores RITA CASADIO’ AND B. ANDREA MELANDRI* Institute of Botany ad *Chair of Molecular Biology, University of Bologna, Via Irnerio 42, 401% Bologna, Italy Received June 20, 1984, and in revised form November 29, 1984 The spectral characteristics of absorption and fluorescence emission of g-amino acridine are not altered by the interaction with bacterial chromatophores, except for the attenuation of both the absorption and emission following the formation of a protonic gradient. The lifetime of fluorescence of the dye is significantly affected in the presence of membranes, and even more following illumination. The shortening of the lifetime induced by light is reversible and prevented by nigericin and K+. The onset kinetics of the fluorescence quenching following the generation of an artificial transmembrane pH difference is temperature dependent, with an activation energy of 17 f 3 kcal/mol. The effect of pH on the rate constants is consistent with a model assuming that the diffusion of the unprotonated species is the limiting step in the quenching phenomenon. The response of g-amino acridine to artificially imposed ApH’s has been utilized as a calibration method for the measurements of the light- induced protonic gradient. The apparent inner volume of chromatophores, evaluated from the extraplation of the response at ApH = 0, was found to be much larger (15 to 40-fold) than the true osmotic volume, indicating that most of the dye is bound to the membrane when accumulated into the inner lumen. o 1985 Academic press, I~~. g-Amino acridine is a probe commonly utilized for the evaluation of transmem- brane pH differences in acidic-inside membrane vesicles or organelles (l-3). The model, originally proposed by Schul- diner et al. (2) for an interpretation of energy-dependent fluorescence changes, assumes that the dye is redistributed across the membrane in response to a proton activity difference, and that the percentage quenching reflects the distri- bution ratio of the probe between the inner and outer compartments. According to this model g-amino acri- dine behaves as a weak amine (4), being in the unprotonated form freely permeable and in diffusional equilibrium across the membrane, whereas the charged form is completely impermeable. Consequently the ’ To whom correspondence should be addressed. dye distributes according to ApH, as a result of diffusion and protonation coupled equilibria. The quantitative equilibrium relation for this situation is [Ain1 ApH = pH,“t - pHi, = log - [&utl Q V = log (1oo _ &) + log $ PI where A is the total concentration (activ- ity) of the dye in the inner and outer compartments, and V,,, and D,,, are the volumes of these two phases. In Eq. [l] Q is the percentage quenching of fluores- cence; the use of this parameter, in rela- tion to the concentration of g-amino ac- ridine, implies the notion that the inten- sity of fluorescence is proportional to the concentration of the dye only in the outer 219 0003-9861/85 $3.00 Copyright 0 1985 by Academic Press, Inc. All rights of reproduction in any form reserved.
Transcript of Calibration of the response of 9-amino acridine fluorescence to transmembrane pH differences in...

ARCHIVES OF BIOCHEMISTRY AND BIOPHYSICS
Vol. 238, No. 1, April, pp. 219-228, 1985
Calibration of the Response of g-Amino Acridine Fluorescence to Transmembrane pH Differences in Bacterial Chromatophores
RITA CASADIO’ AND B. ANDREA MELANDRI*
Institute of Botany ad *Chair of Molecular Biology, University of Bologna, Via Irnerio 42, 401% Bologna, Italy
Received June 20, 1984, and in revised form November 29, 1984
The spectral characteristics of absorption and fluorescence emission of g-amino acridine are not altered by the interaction with bacterial chromatophores, except for the attenuation of both the absorption and emission following the formation of a protonic gradient. The lifetime of fluorescence of the dye is significantly affected in the presence of membranes, and even more following illumination. The shortening of the lifetime induced by light is reversible and prevented by nigericin and K+. The onset kinetics of the fluorescence quenching following the generation of an artificial transmembrane pH difference is temperature dependent, with an activation energy of 17 f 3 kcal/mol. The effect of pH on the rate constants is consistent with a model assuming that the diffusion of the unprotonated species is the limiting step in the quenching phenomenon. The response of g-amino acridine to artificially imposed ApH’s has been utilized as a calibration method for the measurements of the light- induced protonic gradient. The apparent inner volume of chromatophores, evaluated from the extraplation of the response at ApH = 0, was found to be much larger (15 to 40-fold) than the true osmotic volume, indicating that most of the dye is bound to the membrane when accumulated into the inner lumen. o 1985 Academic press, I~~.
g-Amino acridine is a probe commonly utilized for the evaluation of transmem- brane pH differences in acidic-inside membrane vesicles or organelles (l-3). The model, originally proposed by Schul- diner et al. (2) for an interpretation of energy-dependent fluorescence changes, assumes that the dye is redistributed across the membrane in response to a proton activity difference, and that the percentage quenching reflects the distri- bution ratio of the probe between the inner and outer compartments.
According to this model g-amino acri- dine behaves as a weak amine (4), being in the unprotonated form freely permeable and in diffusional equilibrium across the membrane, whereas the charged form is completely impermeable. Consequently the
’ To whom correspondence should be addressed.
dye distributes according to ApH, as a result of diffusion and protonation coupled equilibria. The quantitative equilibrium relation for this situation is
[Ain1 ApH = pH,“t - pHi, = log - [&utl
Q V = log (1oo _ &) + log $ PI
where A is the total concentration (activ- ity) of the dye in the inner and outer compartments, and V,,, and D,,, are the volumes of these two phases. In Eq. [l] Q is the percentage quenching of fluores- cence; the use of this parameter, in rela- tion to the concentration of g-amino ac- ridine, implies the notion that the inten- sity of fluorescence is proportional to the concentration of the dye only in the outer
219 0003-9861/85 $3.00 Copyright 0 1985 by Academic Press, Inc. All rights of reproduction in any form reserved.

220 CASADIO AND MELANDRI
phase, without any contribution from the inner compartment. Equation [l] holds only at pH’s, both in the inner and outer phase, much lower than the pK of the amine (9.99) (2, 5).
Quantitative evaluations of the validity of this model have been attempted both in natural and model membrane systems. In chloroplasts a good agreement between ApH evaluated with g-amino acridine and [14C]methylamine or NH: has been re- ported (2, 6-8). A linear relation between the log Q/(100 - Q) and ApH, with an ideal slope equal to 1, was described in vesides of soybean or bacterial phospho- lipids (9-11). The linearity was indepen- dent of the absolute value of the inner pH and extended up to ApH as high as 2- 2.5 units. A similar linearity was also found between log Q/(100 - Q) and log V,,JV,,, as expected from Eq. [l]. Fur- thermore, the quenching was uniform for the entire wavelength range of the emis- sion spectrum (3) and was completely prevented, or reversed, by any condition inhibiting the formation of ApH (NH: > 1 mM, nigericin + K+, detergents) (2). According to Eq. [l], the extrapolated value of log Q/(100 - Q) to ApH = 0 should give a measure of log Vo”,/ Vi,; following this rationale, the apparent in- ner volume of the acidic compartment can be evaluated. The values obtained with this method, however, were found to be unreasonably high, both in liposomes and in chromatophores, as compared to the volumes expected or measured (12, 13). This discrepancy was taken as an indica- tion of the nonideal behavior of the probe within the inner compartment, bringing about a large overestimation of the actual ApH (12).
The problem of the interaction of 9- amino acridine (and of other acridines as well) with the membrane, its relevance for the activity of the probe in the inner compartment, and the evaluation of ApH is still largely unsolved. Nonideal behavior in g-amino acridine distribution, and con- sequent overestimation of ApH by this technique, has been inferred by many researchers and in many membrane sys- tems (14-16). In these studies, the values of ApH obtained with g-amino acridine
were generally compared with those eval- uated from the distribution of isotope- labeled organic amines and considered, somewhat arbitrarily, as correct. It can be considered well established, in any case, that g-amino acridine yields values of ApH systematically higher than those obtained with other amines: this can also be evidenced by the very large value of log V,,,/ I$, observed during empirical cal- ibration with artificially imposed pH dif- ferences (12, 13).
Models for the interpretation of these interactions have been proposed. Forma- tion of dimers and multimers on the membrane, promoted by the interaction of acridine cations with fixed electrically charged groups, has been suggested by Massari et al. (1’7) as an interpretation both for fluorescence quenching of acridine dyes and for ApH overestimation. Alter- natively, a hydrophobic partition of the dye between the aqueous buffer and the membrane lipids has been considered (18); this partition will also be influenced by the electric potential difference at the interface and/or across the membrane. Large deviations from the ideal distribu- tion ratio of Eq. [l] were calculated ac- cording to the latter model (18). All these approaches, however, are largely specu- lative and do not allow, in general, a quantitative evaluation of the interaction of the dye with the membrane.
In this paper we describe an empirical calibration method for assessing the re- sponse of g-amino acridine fluorescence to transmembrane pH differences artificially imposed on bacterial chromatophores. This calibration is utilized for an evalua- tion of the apparent internal volume of chromatophores and, consequently, for testing the relevance of membrane-dye interactions in determining ApH. Data are also presented on the fluorescence lifetime of the dye both in the presence and absence of chromatophores, and on the kinetics of the fluorescence response to artificial pH changes. The results in- dicate that the fluorescent dye behaves as expected for a freely rotating molecule in aqueous solution and distributes rapidly between the inner (acidic) and the outer

CALIBRATION OF B-AMINO ACRIDINE RESPONSE TO ApH 221
(alkaline) compartments, according to the model of Schuldiner et al. (2), except that its accumulation ratio largely exceeds that expected for an ideal monoamine. Allow- ance for this deviation from an ideal behavior should be made during ApH measurements, and corrections applied on the basis of empirical calibration curves.
MATERIALS AND METHODS
Rho&pseudomonas sphaeroides GA was grown photoheterotrophycally on a synthetic medium con- taining malate as a carbon source (18), and was harvested at an early stationary phase of growth; chromatophores were prepared by French press (SLM-Aminco, Urbana) disruption and differential centrifugation, as previously described (19). The preparation was stored at -16°C in 20 mM glycyl- glycine, pH 7.5, 5 mM MgClz and 60% glycerol. Bacteriochlorophyll and proteins were determined according to Clayton (20) and Lowry et al. (211, respectively.
Static fluorescence of g-amino acridine was mea- sured using either a Jasco Model FP-550 or a Perkin- Elmer Model MPF-3 spectrofluorimeter, equipped, when specified, with excitation and emission light polarizers. A standard assay contained, in a final volume of 2 ml, 20 ?IIM glycylglycine, pH 7.5 (or other, when specified), 5 mM MgClz, 0.2 mM sodium succinate, 50 mM KCl, chromatophores corresponding to 15 to 25 pM bacterioehlorophyll, and 5 FM g-amino acridine.
Quenching of fluorescence was induced by an ar- tificially imposed ApH as follows: Chromatophores were equilibrated at pH 5.5 (or other, when specified) with a 20 mM succinate-Trieine’ buffer. After 1 h of incubation in the dark, 2 ml of the sample was transferred into a fluorescence cuvette, equipped with magnetic stirring, and supplemented with 5 pM
g-amino acridine (as a 2 IIIM ethanolic solution). The fluorescence transition was recorded while the pH was brought to alkaline values by addition of 5-30 ~1 of 1 M NaOH. The final pH of the sample was determined immediately after the addition of NaOH, utilizing a Radiometer Model PHM 62 pH meter. When necessary, additions of ionic solutions, sucrose, or ionophores were made before the acid-base tra- sition. At a transmembrane ApH = 0, 100% fluores- cence was recorded after addition of nigericin and KC1 to the sample. Excitation and emission wave- lengths were 400 and 454 nm, respectively; actinic
’ Abbreviation used: Tricine, N-[2-hydroxy-l,l- bis(hydroxymethyl)ethyllglycine.
light, produced by a 55-W quartz-iodine lamp, was screened through an 88A Wratten Kodak gelatin filter.
Stopped-flow fluorescence experiments were per- formed, adopting the same conditions of buffering and ApH as for the static experiments. In this case a Biochem Sigma-ZWII spectrophotometer, equipped with stopped-flow apparatus and fluorescence at- tachment, was used. The mixing chamber had a volume of 0.2 ml, and the minimal mixing time was better than 5 ms; the overall time resolution of the instrument was less than 10 ms. The excitation wavelength was 400 nm; the total emission band was detected in this case.
Lifetime measurements were performed under the same conditions, as static experiments, utilizing for excitation a Lambda Physics Model 100 A nitrogen laser (pulse half-width, 3.5 ns; 1 MW). Fluorescence transients were detected by a lP28 RCA photomul- tiplier, operating at 1.25 kV in a 5-dynode configu- ration, through a Bausch & Lomb high intensity monochromator. The signal was fed into a Tektronix Model R712 transient digitizer, equipped with a ‘IA19 vertical amplifier, and interfaced with a Z80- based Cromemco microcomputer. The time resolution was better than 1 ns, but was limited by the emission profile of the excitation laser.
All chemicals utilized were reagent grade. g-Amino acridine was purchased from Fluka AG, Buchs, Switzerland.
RESULTS
Spectral Characteristics of g-Amino Acm’dine Fluorescence
The emission spectrum of 5 PM g-amino acridine solutions in 20 mM glycylglycine buffer, pH 8.0, containing 5 mM MgCla, 50 mM KCl, and 0.2 mM sodium succinate, is presented in Fig. 1 (spectrum A). Upon addition of chromatophores the spectrum appears diminished in intensity and largely distorted (spectrum B). Correction for internal filter effects (23) due to the absorption of the membrane pigments can, however, account completely for these ef- fects (spectrum C). Any possible interac- tion with the membrane, if even present, does not appear to affect the quantum yield or the spectral shape of the emission. Upon excitation of the membranes with infrared actinic light, the fluorescence emission is quenched, in an energy-depen- dent manner, with kinetics that have been described previously (3); addition of ni- gericin, in the presence of 50 mM K+,
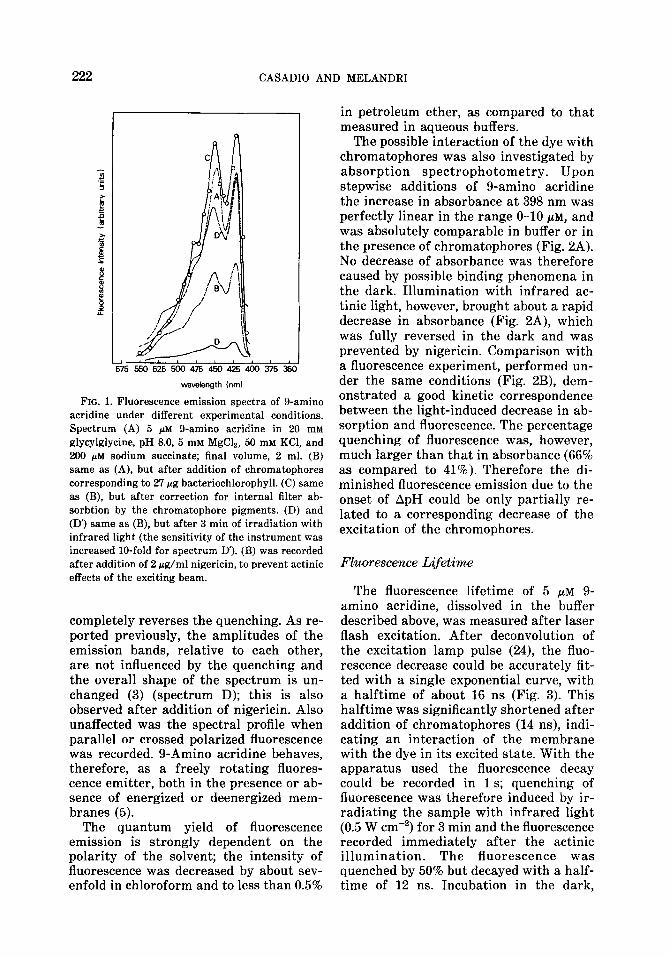
CASADIO AND MELANDRI
wavelength (nmJ
FIG. 1. Fluorescence emission spectra of g-amino acridine under different experimental conditions. Spectrum (A) 5 @M g-amino acridine in 20 mM glycylglycine, pH 8.0, 5 mM MgCla, 50 mM KCl, and 200 pM sodium succinate; final volume, 2 ml. (B) same as (A), but after addition of chromatophores corresponding to 27 Fg bacteriochlorophyll. (C) same as (B), but after correction for internal filter ab- sorbtion by the chromatophore pigments. (D) and (D’) same as (B), but after 3 min of irradiation with infrared light (the sensitivity of the instrument was increased lo-fold for spectrum D’). (B) was recorded after addition of 2 rg/ml nigericin, to prevent actinic effects of the exciting beam.
completely reverses the quenching. As re- ported previously, the amplitudes of the emission bands, relative to each other, are not influenced by the quenching and the overall shape of the spectrum is un- changed (3) (spectrum D); this is also observed after addition of nigericin. Also unaffected was the spectral profile when parallel or crossed polarized fluorescence was recorded. g-Amino acridine behaves, therefore, as a freely rotating fluores- cence emitter, both in the presence or ab- sence of energized or deenergized mem- branes (5).
The quantum yield of fluorescence emission is strongly dependent on the polarity of the solvent; the intensity of fluorescence was decreased by about sev- enfold in chloroform and to less than 0.5%
in petroleum ether, as compared to that measured in aqueous buffers.
The possible interaction of the dye with chromatophores was also investigated by absorption spectrophotometry. Upon stepwise additions of g-amino acridine the increase in absorbance at 398 nm was perfectly linear in the range O-10 PM, and was absolutely comparable in buffer or in the presence of chromatophores (Fig. 2A). No decrease of absorbance was therefore caused by possible binding phenomena in the dark. Illumination with infrared ac- tinic light, however, brought about a rapid decrease in absorbance (Fig. 2A), which was fully reversed in the dark and was prevented by nigericin. Comparison with a fluorescence experiment, performed un- der the same conditions (Fig. 2B), dem- onstrated a good kinetic correspondence between the light-induced decrease in ab- sorption and fluorescence. The percentage quenching of fluorescence was, however, much larger than that in absorbance (66% as compared to 41%). Therefore the di- minished fluorescence emission due to the onset of ApH could be only partially re- lated to a corresponding decrease of the excitation of the chromophores.
Fluorescence Lifetime
The fluorescence lifetime of 5 PM 9- amino acridine, dissolved in the buffer described above, was measured after laser flash excitation. After deconvolution of the excitation lamp pulse (24), the fluo- rescence decrease could be accurately fit- ted with a single exponential curve, with a halftime of about 16 ns (Fig. 3). This halftime was significantly shortened after addition of chromatophores (14 ns), indi- cating an interaction of the membrane with the dye in its excited state. With the apparatus used the fluorescence decay could be recorded in 1 s; quenching of fluorescence was therefore induced by ir- radiating the sample with infrared light (0.5 W cm-‘) for 3 min and the fluorescence recorded immediately after the actinic illumination. The fluorescence was quenched by 50% but decayed with a half- time of 12 ns. Incubation in the dark,

CALIBRATION OF O-AMINO ACRIDINE RESPONSE TO ApH
1 Light on A
1
Light on B
I 5AA * 103
t off
7 10 pM SAA
20 1 mm
FIG. 2. (A) Reversible decrease of the absorbance of O-amino acridine in the presence of chromatophores upon energization with infrared actinic light. Conditions were as in Fig. 1, except that 10 pM O-amino acridine was added. (B) Reversible decrease in fluorescence of g-amino acridine induced by energization with infrared actinic light in a sample identical to that of (A).
223
after illumination, for 1.5 or 3 min com- pletely restored the original intensity and lifetime. The decrease was prevented by nigericin addition. These halftimes were, however, only indicative, since the x2 for single exponential fitting was not as sat- isfactory as in buffer alone. The fluores- cence emission decay was possibly the result of several nearly overlapping com-
0 10 20 30 40 50 60 70 80 SO
mle (“S,
FIG. 3. Lifetime of fluorescence of g-amino acridine under different experimental conditions. (A) 5 pM 9- amino acridine in the same buffer as spectrum (1A). (B) as (A), but after addition of chromatophores (28 pg bateriochlorophyWm1) and readjustment of the instrumental gain. (C) as (B), but after addition of 2 pg/ml nigericin. (D) as (B), but immediately after irradiation for 3 min with infrared light.
ponents which could not be resolved by the apparatus used, but were indicative of some interaction with the membrane. This interaction was seemingly enhanced following accumulation of the probe, as indicated by the shortened apparent half- time. The light-induced decrease of 9- amino acridine fluorescence, in any case, showed properties of a dynamic quenching, which was completely reversible in the dark and inhibited by nigericin and K+.
Kinetics of the Fluorescence Response to Arti$cially Imposed Transmembrane pH Differences
As previously described for liposomes (9, 10, 12), when chromatophores were suspended at acidic pH (usually 20 mM
succinate-Tricine buffer, pH 5.5), allowed to equilibrate, and then suddenly exposed to a pH shift caused by addition of NaOH, the fluorescence of g-amino acridine was transitorily decreased (Fig. 4). The extent of the quenching was a function of the amount of the vesicles added, of the pH, and also of the parameters described be- low. It was not, however, dependent on the absolute values of the inner or outer pH’s, but only on ApH. The quenching was completely prevented by detergents or by nigericin and K+. The kinetics of

224 CASADIO AND MELANDRI
fluorescence recovery was very similar when the quenching was induced either by an acid-base transition or by light (Fig. 4).
The kinetics of the fluorescence decrease upon an acid-base transition was time resolved with stopped-flow experiments. When a fixed ApH was applied, the flue- rescence was observed to decrease with first-order kinetics, fitting the whole tran- sition (Fig. 5A) according to the equation I - 1, = (I0 - 1,) e@ (where 1, and I,, are the emission intensities at the end of the transition and at t = 0, respectively). The time constant of the fluorescence quenching was markedly dependent on the temperature (Fig. 5B); the activation energy between 5 and 30°C was constant and equal to 1’7 + 3 kcal/mol. This value did not change when the ionic strength of the buffer was increased to 300 mM KCl.
The increase of the final pH of the acid- base transition had profound kinetic ef- fects, drastically increasing the rate con- stants but leaving the activation energy practically unaffected. This stimulation would be in agreement with Schuldiner’s model, where the diffusion of the unpro- tonated species from the outer phase was the limiting step for the onset of the quenching process. Quantitatively, how- ever, the agreement was not complete; an
9.
NH&I
1 mn
FIG. 4. Trasitory quenching of fluorescence of 9- amino acridine induced by an artifically imposed ApH, or by illumination. Conditions were as in Fig. 2D. The formation of a small protonic gradient by the measuring beam, before addition of NH&l, is apparent.
increase of the final pH from 7.6 to 9.4, that raises the concentration of the un- charged amine about 40 times, caused an increase in rate constants of about ll- fold. It is clear, therefore, that the onset of the quenching is not entirely controlled kinetically by the concentration of the unprotonated amine in the outer bulk phase.
Calibration with Artificially Imposed ApH
The transitory quenching of fluorescence caused by an artificially imposed ApH was exploited for the calibration of the fluo- rescence response (13). The kinetics of the quenching onset were, in general, fast enough to prevent any underestimation of the degree of quenching due to the backflow of protons into the external buffer; an accurate recording of the tran- sient quenching was, therefore, also pos- sible when ApH exceeded 3 units and the external pH approached the value of 9.0, all conditions in which a marked deviation from a linear response was detected.
The main diagnostic feature for testing the accuracy of Schuldiner’s model is the linearity of log Q/(100 - Q) vs ApH. This was generally observed when ApH was smaller than about 2 to 2.5 units; at higher ApH a progressive decrease in slope and, consequently, a decrease in the ap- parent inner volume (see below), was ob- served. Critical for this evaluation was the choice of the baseline of fluorescence in the presence of deenergized vesicles. We routinely selected the trace after ad- dition of nigericin as 100% fluorescence. This was particularly important for pH’s close to the pK of g-amino acridine (pH > 8.5), since the emission intensity of the unprotonated species was about 20% higher than that of the protonated one [but cf. (5)]. As p reviously observed for liposomes, the extent of the quenching was independent of the absolute values of the external and internal pH’s, and was related only to the extent of the protonic gradient.
Also consistent with the model was the response of the quenching to changes in

CALIBRATION OF g-AMINO ACRIDINE RESPONSE TO ApH 225
11 1 1 * a h 1 0 06 12 18 24 30 36 42
lime (s)
32 34 36
l/T IK-'X 103)
FIG. 5. (A) Time course of the onset of quenching induced by artificial ApH’s, as a function of temperature, and resolved by stopped-flow fluorescence. Conditions were as detailed under Materials and Methods. (B) Activation energy of the rate of quenching under conditions of low (0) or high (&) ionic strength.
the amount of chromatophore added. As expected the experimental values of logQ/ (100 - Q) were linear with the log win, win being the total inner volume in the sample. The apparent volume was, therefore, in- dependent of the concentration of chro- matophores added in the assay when nor- malized per milligram of bacteriochloro- phyll.
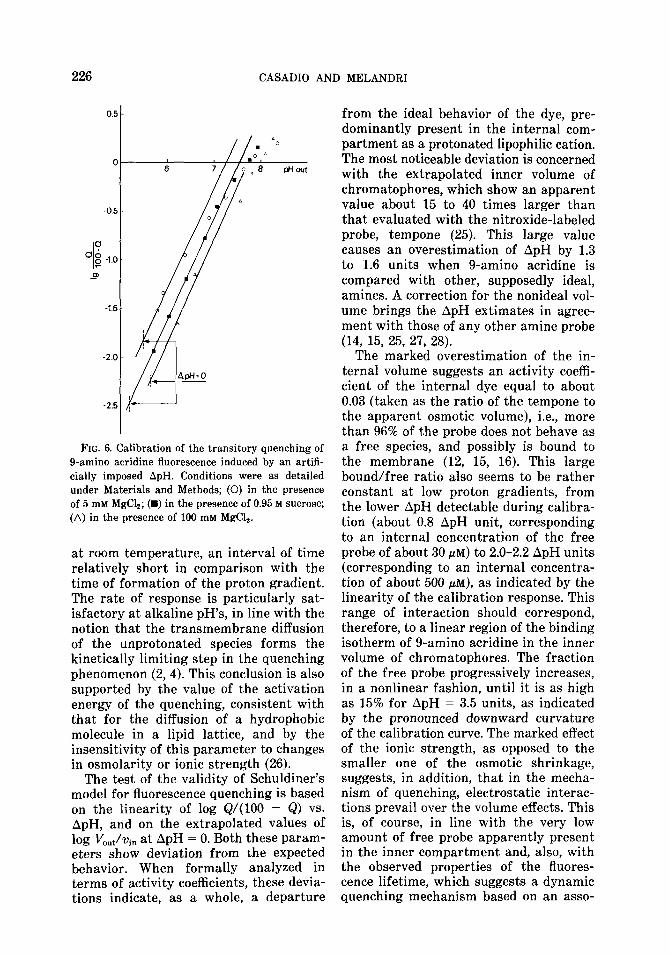
The composition of the buffer was changed in order to test the effect of ionic strength and osmolarity. Changes in os- molarity from 0 to 0.95 M sucrose, a set of conditions shown to cause a marked osmotic shrinkage of chromatophores (25), altered the extrapolated value at ApH = 0 of the log Q/(100 - Q) curve. Similarly, an increase in ionic strength (MgClz in- creased from 5 to 100 mM) decreased markedly the extrapolated value without changing the slope. The extrapolated val- ues of the apparent volumes in three sets of conditions, 5 mM MgClz, 100 mM MgCl,, and 5 mM MgC12 plus 0.95 M sucrose were, respectively, 0.67, 0.15, and 0.29 ml/mg bacteriochlorophyll (Fig. 6). It is evident, therefore, that the effect of MgClz could not be entirely attributed to an osmotic action, although an effect of osmotic shrinkage in low ionic strength was also
apparent. Addition of KC1 affected both the slope, increasing the curvature at high pH’s, and the intercept of the calibration curve. This effect seemed to be specific for K+, however, since it could not be mim- icked by any other univalent salt tested. It is possible that addition of KC1 at high concentrations accelerated the decay of the proton gradient through a K+/H+ antiporter mechanism. This possibility was also supported by a faster decay rate of the quenching in the presence of KC1 and deserves further investigation.
DISCUSSION
The main point of interest in the present study is the possibility of obtaining a direct calibration of the fluorescence re- sponse of g-amino acridine to transmem- brane ApH’s. If one accepts the concept that an artificially imposed pH difference essentially simulates the formation of a proton gradient driven by electron trans- fer, this calibration allows an evaluation, in real time, of the ApH existing across a coupling membrane. This conclusion is strongly supported by the excellent kinetic response of this dye, which equilibrates across the membrane in less than 100 ms,
226 CASADIO AND MELANDRI
FIG. 6. Calibration of the transitory quenching of O-amino acridine fluorescence induced by an artifi- cially imposed ApH. Conditions were as detailed under Materials and Methods; (0) in the presence of 5 miv Mg&; (B) in the presence of 0.95 M sucrose; (A) in the presence of 100 mM MgC12.
at room temperature, an interval of time relatively short in comparison with the time of formation of the proton gradient. The rate of response is particularly sat- isfactory at alkaline pH’s, in line with the notion that the transmembrane diffusion of the unprotonated species forms the kinetically limiting step in the quenching phenomenon (2,4). This conclusion is also supported by the value of the activation energy of the quenching, consistent with that for the diffusion of a hydrophobic molecule in a lipid lattice, and by the insensitivity of this parameter to changes in osmolarity or ionic strength (26).
The test of the validity of Schuldiner’s model for fluorescence quenching is based on the linearity of log Q/(100 - Q) vs. ApH, and on the extrapolated values of log Vout/qn at ApH = 0. Both these param- eters show deviation from the expected behavior. When formally analyzed in terms of activity coefficients, these devia- tions indicate, as a whole, a departure
from the ideal behavior of the dye, pre- dominantly present in the internal com- partment as a protonated lipophilic cation. The most noticeable deviation is concerned with the extrapolated inner volume of chromatophores, which show an apparent value about 15 to 40 times larger than that evaluated with the nitroxide-labeled probe, tempone (25). This large value causes an overestimation of ApH by 1.3 to 1.6 units when g-amino acridine is compared with other, supposedly ideal, amines. A correction for the nonideal vol- ume brings the ApH extimates in agree- ment with those of any other amine probe (14, 15, 25, 27, 28).
The marked overestimation of the in- ternal volume suggests an activity coefi- cient of the internal dye equal to about 0.03 (taken as the ratio of the tempone to the apparent osmotic volume), i.e., more than 96% of the probe does not behave as a free species, and possibly is bound to the membrane (12, 15, 16). This large bound/free ratio also seems to be rather constant at low proton gradients, from the lower ApH detectable during calibra- tion (about 0.8 ApH unit, corresponding to an internal concentration of the free probe of about 30 PM) to 2.0-2.2 ApH units (corresponding to an internal concentra- tion of about 500 PM), as indicated by the linearity of the calibration response. This range of interaction should correspond, therefore, to a linear region of the binding isotherm of g-amino acridine in the inner volume of chromatophores. The fraction of the free probe progressively increases, in a nonlinear fashion, until it is as high as 15% for ApH = 3.5 units, as indicated by the pronounced downward curvature of the calibration curve. The marked effect of the ionic strength, as opposed to the smaller one of the osmotic shrinkage, suggests, in addition, that in the mecha- nism of quenching, electrostatic interac- tions prevail over the volume effects. This is, of course, in line with the very low amount of free probe apparently present in the inner compartment and, also, with the observed properties of the tluores- cence lifetime, which suggests a dynamic quenching mechanism based on an asso-

CALIBRATION OF g-AMINO ACRIDINE RESPONSE TO ApH 227
ciation between a cationic dye and mem- brane binding groups. In this respect, the increasing fraction of free dye observable at high accumulation ratios may indicate that the number of binding groups on the membrane is limited, and approaches sat- uration at high concentrations (as high as 1.6 to 15 mM for 5 PM external probe and ApH = 2.5 to 3.5 units, respectively). This model also offers a rationale and quantitative interpretation for the quenching mechanism, through the for- mation of dimers and multimers of the cationic dye on an array of descrete, neg- atively charged groups (29). This mecha- nism corresponds to the membrane-di- rected dye stacking and consequent fluo- rescence quenching previously suggested by Massari et al. (17) for other acridines.
The accumulation and stacking of the dye in the inner lumen and/or on the membrane causes also a severe and re- versible hypochromic effect on the ab- sorption bands of g-amino acridine. This effect is likely to occur following the ac- cumulation of the dye in a very small fraction of the total sample volume, and, possibly the formation of ordered struc- tures of the chromophores on the mem- brane, causing mutual shielding and a marked decrease in their cross-section for the interaction with the incident photons (30). The lack of excitation, together with the increased efficiency of any quenching mechanism on ordered arrays of chro- mophores, and with the drastically re- duced quantum yield of fluorescence of the dye partitioned into the lipid phase, is probably the main cause of the complete quenching of the fluorescence emission of the accumulated g-amino acridine.
In conclusion, the calibration procedure described here allows the utilization, with a good degree of confidence, of g-amino acridine as a probe for ApH in bacterial chromatophores. The fast kinetic response of the probe and the convenience of the fluorescence measurements make this ap- proach quite suitable for studies of energy transduction on bacterial photosynthetic systems, yielding direct information on the extent and rate of formation of the protonic gradient. An extension of this
type of calibration to other energy-trans- ducing vesicular systems, acidic inside, should not present large difficulties.
ACKNOWLEDGMENTS
We are grateful to Professor F. Bolletta, Institute of Chemistry, University of Bologna, for his kind help during the measurements of fluorescence life time, and to the FRAE Laboratory of the Consiglio Nazionale delle Ricerche for making the instrumen- tation available to us. Many thanks to Dr. B. Ferrari and to Professor L. Masotti, Institute of Biochem- istry, University of Parma, for the deconvolution calculations, and to Dr. G. Venturoli, of this Institute, for general advice and criticism. This work was supported by Consiglio Nazionale delle Ricerche Grant 82.02028.04.
REFERENCES
1. KRAAYENHOF, R. (1970) FEBS Lett. 13, 161-165. 2. SCHULDINER, S., ROSENBERG, H., AND AVRON,
M. (1972) Eur. J. Biochem. 25, 64-70. 3. CASADIO, R., BACCARINI-MELANDRI, A., AND ME-
LANDRI, B. A. (1974) Eur. J. Biochem. 47, 121- 128.
4. CROFTS, A. R. (1963) J. Biol Chem. 242, 3352- 3359.
5. HUANG, C.-S., KOPACZ, S. J., AND LEE, C.-P. (1983) Biochim. Biophys. Acta 722, 107-115.
6. ROTPENBERG, H., GRUNWALD, T., AND AVRON, M. (1972) Eur. J. B&hem 25, 54-63.
7. ROSENBERG, H., AND GRUNWALD, T. (1972) EUT. J. Biochem 25, 71-74.
8. CASADIO, R. (1982) in Transport in Biomemhranes Model Systems and Reconstitution (Antolini, R., Gliozzi, A., and Govio, A., eds.), pp. 201- 214, Raven Press, New York.
9. DEAMER, D. W., PRINCE, R. C., AND CROFTS, R. A. (1972) B&him. Biophys. Aeta 274, 323- 335.
10. CASADIO, R., AND MELANDRI, B. A. (1977) J. Bioenerg. Bknnembr. 9, 17-29.
11. LEE, H. C., AND FORTE, J. (1978) B&him Biophys. Acta 508, 339-356.
12. FIOLET, J. W. T., BAKKER, E. P., AND VAN DAM, K. (1974) Biochim. Biophys. Acta 368.432-445.
13. MELANDRI, B. A., DE SANTIS, A., VENTUROLI, G., AND BACCARINI-MELANDRI, A. (1978) FEBS I.&t. 95, 130-139.
14. LEISER, M., AND GROMET-ELHANAN, Z. (1977) Arch Biochem Biophys. 178,79-88.
15. CIRILLO, V. P., AND GROMET-ELHANAN, Z. (1981) Biochim. Biophys. Ada 636, 244-253.
16. MICHELS, P. A. M., AND KONINGS, W. N. (1978) Eur. J. B&hem. 85. 147-155.

228 CASADIO AND MELANDRI
17. MASSARI, S., DELL’ANTONE, P., COLONNA, R., AND AZZONE, G. F. (1974) Biochemistry 13, 103% 1043.
18. HARAUX, F., AND DE KOUCHKOVSKY, Y. (1980) Biochim. Biophys. Acta 592,153-168.
19. ORMEROD, J. G., ORMEROD, K. S., AND GEST, H. (1961) Arch Biochem. Biophys. 94,449-463.
20. BACCARINI-MELANDRI, A., AND MELANDRI, B. A. (1977) FEBS I&t. 80, 459-464.
21. CLAYTON, R. K. (1963) Biochim Biophys. Acta 75,312-323.
22. LOWRY, 0. H., ROSEBROUGH, N. J., FARR, A. L., AND RANDALL, R. J. (1951) J. Biol. Chem 193, 265-275.
23. BRAND, L., AND WITHOLD, B. (1967) in Methods in Enzymology (Hirs, C. H. W., ed.), Vol. 11, pp. 776-856, Academic Press, New York.
24. GRINWALD, A., AND STEINBERG, I. Z. (1974) And B&&em. 59, 583498.
25. MELANDRI, B. A., MELHORN, R. J., AND PACKER, L. (1984) Arch Biochem Biophys. 235,97-105.
26. STEIN, W. D. (1967) The Movement of Molecules across Biomembranes, p. 81, Academic Press, New York.
27. ELEMA, R. P., MICHELS, P. A. M., AND KONINGS, W. N. (1978) Eur. J. Biochem. 92, 381-387.
28. DE BENEDETTI, E., AND GARLASCHI, F. M. (1977) J. Bioewget. Bicnnembr. 9,195-201.
29. MATSUURA, K., MASAMOTO, K., ITOH, S., AND NISHIMURA, M. (1980) B&him. Biophys. Acta 592, 121-12s.
30. DUYSENS, L. M. N. (1956) Biochim. Biophys. Acta 19, l-22.




















