
Blockage of dopaminergic D 2 receptors produces decrease of REM but not of slow wave sleep in rats...
6
Available online at www.sciencedirect.com Behavioural Brain Research 188 (2008) 406–411 Research report Blockage of dopaminergic D 2 receptors produces decrease of REM but not of slow wave sleep in rats after REM sleep deprivation Marcelo M.S. Lima a,∗ , Monica L. Andersen a , Angela B. Reksidler b , Andressa Silva a , Adriano Zager a , S´ ılvio M. Zanata c , Maria A.B.F. Vital b , Sergio Tufik a a Departamento de Psicobiologia, Universidade Federal de S˜ ao Paulo, S˜ ao Paulo, SP, Brazil b Departamento de Farmacologia, Universidade Federal do Paran´ a, Curitiba, PR, Brazil c Departamento de Patologia B´ asica, Universidade Federal do Paran´ a, Curitiba, PR, Brazil Received 12 September 2007; received in revised form 26 November 2007; accepted 30 November 2007 Available online 8 December 2007 Abstract Dopamine (DA) has, as of late, become singled out from the profusion of other neurotransmitters as what could be called a key substance, in the regulation of the sleep–wake states. We have hypothesized that dopaminergic D 2 receptor blockage induced by haloperidol could generate a reduction or even an ablation of rapid eye movement (REM) sleep. Otherwise, the use of the selective D 2 agonist, piribedil, could potentiate REM sleep. Electrophysiological findings demonstrate that D 2 blockage produced a dramatic reduction of REM sleep during the rebound (REB) period after 96 h of REM sleep deprivation (RSD). This reduction of REM sleep was accompanied by an increment in SWS, which is possibly accounted for the observed increase in the sleep efficiency. Conversely, our findings also demonstrate that the administration of piribedil did not generate additional increase of REM sleep. Additionally, D 2 receptors were found down-regulated, in the haloperidol group, after RSD, and subsequently up-regulated after REB group, contrasting to the D 1 down-regulation at the same period. In this sense, the current data indicate a participation of the D 2 receptor for REM sleep regulation and consequently in the REM sleep/SWS balance. Herein, we propose that the mechanism underlying the striatal D 2 up-regulation is due to an effect as consequence of RSD which originally produces selective D 2 supersensitivity, and after its period probably generates a surge in D 2 expression. In conclusion we report a particular action of the dopaminergic neurotransmission in REM sleep relying on D 2 activation. © 2007 Elsevier B.V. All rights reserved. Keywords: Dopamine; REM sleep; Haloperidol; Piribedil; Rat; D 2 receptor 1. Introduction From the plethora of neurotransmitters that orchestrate neu- ral function dopamine has, as of late, become singled out as what can be referred to as a key substance in the regulation of the sleep–wake states [10,11,14,27]. Recently, dopaminergic neurons present in the ventral tegmental area (VTA) and in the substantia nigra pars compacta (SNpc) have been reported to be strongly associated to the regulation and probably with the gen- eration of electrophysiological states of sleep, especially rapid eye movement sleep [4,11]. ∗ Corresponding author at: Departamento de Psicobiologia, Universidade Fed- eral de S˜ ao Paulo, Escola Paulista de Medicina, Rua Napole˜ ao de Barros, 925 Vila Clementino, 04024-002 S˜ ao Paulo, SP, Brazil. Fax: +55 11 5572 5092. E-mail address: [email protected] (M.M.S. Lima). Clinically, it is observed that patients with Parkinson’s dis- ease (PD) who have extensive loss of dopaminergic cells within the SNpc, and less so within the VTA, often have increased sleepiness, which is aggravated by the presence of dopaminergic D 2 receptor agonists [2,3,9]. Otherwise, treatment with the D 2 antagonist and antipsychotic agent haloperidol attenuates hip- pocampal theta and gamma oscillations which is characteristic of REM sleep [5]. For instance, REM sleep could be recovered in the dopaminergic transporter knockout (DAT-KO) mice by selective activation of the D 2 , but not the D 1 , suggesting a par- ticular role of this receptor in the regulation of REM sleep [5]. Such involvement of DA has been previously reported subse- quent to sleep deprivation protocols, as being directly involved in the generation of burly dopaminergic D 2 supersensitivity [17,25,26]. To evaluate the participation of dopaminergic D 2 receptors in the regulation of REM sleep of rats we resorted to record 0166-4328/$ – see front matter © 2007 Elsevier B.V. All rights reserved. doi:10.1016/j.bbr.2007.11.025
Transcript of Blockage of dopaminergic D 2 receptors produces decrease of REM but not of slow wave sleep in rats...

A
trsafauttpr©
K
1
rwonssee
eV
0d
Available online at www.sciencedirect.com
Behavioural Brain Research 188 (2008) 406–411
Research report
Blockage of dopaminergic D2 receptors produces decrease of REMbut not of slow wave sleep in rats after REM sleep deprivation
Marcelo M.S. Lima a,∗, Monica L. Andersen a, Angela B. Reksidler b, Andressa Silva a,Adriano Zager a, Sılvio M. Zanata c, Maria A.B.F. Vital b, Sergio Tufik a
a Departamento de Psicobiologia, Universidade Federal de Sao Paulo, Sao Paulo, SP, Brazilb Departamento de Farmacologia, Universidade Federal do Parana, Curitiba, PR, Brazil
c Departamento de Patologia Basica, Universidade Federal do Parana, Curitiba, PR, Brazil
Received 12 September 2007; received in revised form 26 November 2007; accepted 30 November 2007Available online 8 December 2007
bstract
Dopamine (DA) has, as of late, become singled out from the profusion of other neurotransmitters as what could be called a key substance, inhe regulation of the sleep–wake states. We have hypothesized that dopaminergic D2 receptor blockage induced by haloperidol could generate aeduction or even an ablation of rapid eye movement (REM) sleep. Otherwise, the use of the selective D2 agonist, piribedil, could potentiate REMleep. Electrophysiological findings demonstrate that D2 blockage produced a dramatic reduction of REM sleep during the rebound (REB) periodfter 96 h of REM sleep deprivation (RSD). This reduction of REM sleep was accompanied by an increment in SWS, which is possibly accountedor the observed increase in the sleep efficiency. Conversely, our findings also demonstrate that the administration of piribedil did not generatedditional increase of REM sleep. Additionally, D2 receptors were found down-regulated, in the haloperidol group, after RSD, and subsequentlyp-regulated after REB group, contrasting to the D1 down-regulation at the same period. In this sense, the current data indicate a participation of
he D2 receptor for REM sleep regulation and consequently in the REM sleep/SWS balance. Herein, we propose that the mechanism underlyinghe striatal D2 up-regulation is due to an effect as consequence of RSD which originally produces selective D2 supersensitivity, and after its periodrobably generates a surge in D2 expression. In conclusion we report a particular action of the dopaminergic neurotransmission in REM sleepelying on D2 activation.2007 Elsevier B.V. All rights reserved.
etsDapo
eywords: Dopamine; REM sleep; Haloperidol; Piribedil; Rat; D2 receptor
. Introduction
From the plethora of neurotransmitters that orchestrate neu-al function dopamine has, as of late, become singled out ashat can be referred to as a key substance in the regulationf the sleep–wake states [10,11,14,27]. Recently, dopaminergiceurons present in the ventral tegmental area (VTA) and in theubstantia nigra pars compacta (SNpc) have been reported to be
trongly associated to the regulation and probably with the gen-ration of electrophysiological states of sleep, especially rapidye movement sleep [4,11].∗ Corresponding author at: Departamento de Psicobiologia, Universidade Fed-ral de Sao Paulo, Escola Paulista de Medicina, Rua Napoleao de Barros, 925ila Clementino, 04024-002 Sao Paulo, SP, Brazil. Fax: +55 11 5572 5092.
E-mail address: [email protected] (M.M.S. Lima).
istSqi[
i
166-4328/$ – see front matter © 2007 Elsevier B.V. All rights reserved.oi:10.1016/j.bbr.2007.11.025
Clinically, it is observed that patients with Parkinson’s dis-ase (PD) who have extensive loss of dopaminergic cells withinhe SNpc, and less so within the VTA, often have increasedleepiness, which is aggravated by the presence of dopaminergic2 receptor agonists [2,3,9]. Otherwise, treatment with the D2
ntagonist and antipsychotic agent haloperidol attenuates hip-ocampal theta and gamma oscillations which is characteristicf REM sleep [5]. For instance, REM sleep could be recoveredn the dopaminergic transporter knockout (DAT-KO) mice byelective activation of the D2, but not the D1, suggesting a par-icular role of this receptor in the regulation of REM sleep [5].uch involvement of DA has been previously reported subse-uent to sleep deprivation protocols, as being directly involved
n the generation of burly dopaminergic D2 supersensitivity17,25,26].To evaluate the participation of dopaminergic D2 receptorsn the regulation of REM sleep of rats we resorted to record

Brai
tiithatOpecucp
2
2
oamti(vas
2
R
t(etcN
(I(pfiw
tTad
2
dk(lsbpTt
Ftd
M.M.S. Lima et al. / Behavioural
he electrocorticographic activity of the Brattleboro rat, whichs a lineage consistent with elevated density of D2 receptorsn both the dorsal and ventral striatum [23]. We hypothesizedhat dopaminergic D2 blockage induced pharmacologically byaloperidol would most likely generate a reduction or even anblation of REM sleep, considering that physiological func-ion is dependent on dopaminergic D2 autoreceptor activation.therwise, the use of the selective D2 agonist, piribedil, couldotentiate REM sleep. In view of the influence of the dopamin-rgic system over sleep patterns it becomes fundamental toomprehend the physiology underlying the systems that makese of this very important neurotransmitter so that improvementsan be made in treatment protocols of sleep disturbances in PDatients.
. Experimental procedures
.1. Subjects
All experiments were conducted in accordance with National Institutesf Health (USA) guidelines for the care and use of animals and abided byn approved animal protocol from our university’s ethical committee for ani-al experimentation (#0737/06). Male Brattleboro rats weighing 300–350 g at
he beginning of the experiments were used. These were housed individuallyn transparent acrylic cages and maintained in standard laboratory conditions22 ± 2 ◦C, 12 h light/dark cycle, lights on 7:00 a.m.) with food and water pro-ided ad libitum. Maximal efforts were employed to reduce the number ofnimals used in the experiments yet enough to ensure unambiguous and reliabletatistical analysis and data interpretation.
.2. Experimental design
In order to assess the influence of dopaminergic D2 receptors in regulatingEM sleep we adopted haloperidol as a D2 antagonist and piribedil as a selec-
w(dwa
ig. 1. Schematic representation of the experimental design. Panel A depicts a time-he experiments. Panel B illustrates the subdivisions of groups, with the respective ameprived, NSD; rebound, REB; Western blotting, WB.
n Research 188 (2008) 406–411 407
ive D2 agonist. Two distinct groups of animals, named REM sleep deprivedRSD) and non-sleep deprived (NSD), were surgically implanted electrodes forlectrophysiological recording of sleep–wake states. After 1 week of recoveryhe animals underwent the basal recording. Subsequently, the animals were allo-ated for a 96 h of REM sleep deprivation protocol, and the animals from theSD group were conducted to their home cages, being allowed to sleep freely.
Subsequent to the RSD procedure groups received a single intraperitoneali.p.) injection of saline 0.9% or haloperidol (3.0 mg/kg) or piribedil (8.0 mg/kg).mmediately afterwards the distinct groups initiated the sleep–wake evaluationinitiated between 7:30 and 8:30 a.m.) for a period up to 48 h, called rebounderiod. The first 24 h of recording concentrated all the alterations reported. Theollowing 24 h did not present statistical differences among the groups tested,ndicating a clearance of the effects of the RSD and of the drugs to which subjectsere exposure.
In parallel, a different set of animals underwent the same experimental designo determine the protein expressions of D1, D2 and tyrosine hydroxylase (TH).his set of animals was decapitated (between 7:30 and 9:00 a.m.) immediatelyfter: drug administrations, RSD and REB. (For more details of the experimentalesign see Fig. 1.)
.3. Stereotaxic surgery for sleep–wake cycle recording
Rats were distributed at random into the three groups named saline, haloperi-ol and piribedil. Animals were anesthetized with diazepam (10 mg/kg, i.p.) andetamine (90 mg/kg, i.p.) and they were mounted in a classical stereotaxic frameInsight Instruments). Body temperature was maintained at 37 ◦C with a regu-ated electric heating pad (Harvard Apparatus). Two bipolar electrodes with fourtainless-steel screws (Ø 0.9 mm) were placed into the skull through small holesored into the right lateral fronto-parietal (one pair) and in the left medial fronto-arietal (another pair) in order to monitor bipolar electroencephalogram (EEG).he free ends of the electrodes were soldered to a socket that was attached
o the skull with acrylic dental cement. Two nickel–chromium flexible wires
ere inserted into the neck muscles in order to record the electromyogramEMG). All the rats received penicillin (20,000 U in 0.1 ml, i.m.) and sodiumiclofenac (25 mg/ml, i.p.) after surgery. One week after surgery, the socketsere connected via flexible recording cables and a commutator to a polygraph
nd computer. After 3 days with the cables the rats progressively habituated
line illustration (not represented in scale) of the sequence of events throughoutostral numbers for each examination. REM sleep deprivation, RSD; non-sleep
4 Brain Research 188 (2008) 406–411
tr
2
abswictl
Fb
2
QwwcthDpoaa
2
bstb8smR
fsnaaSl(ToDfhbt
2
Aem
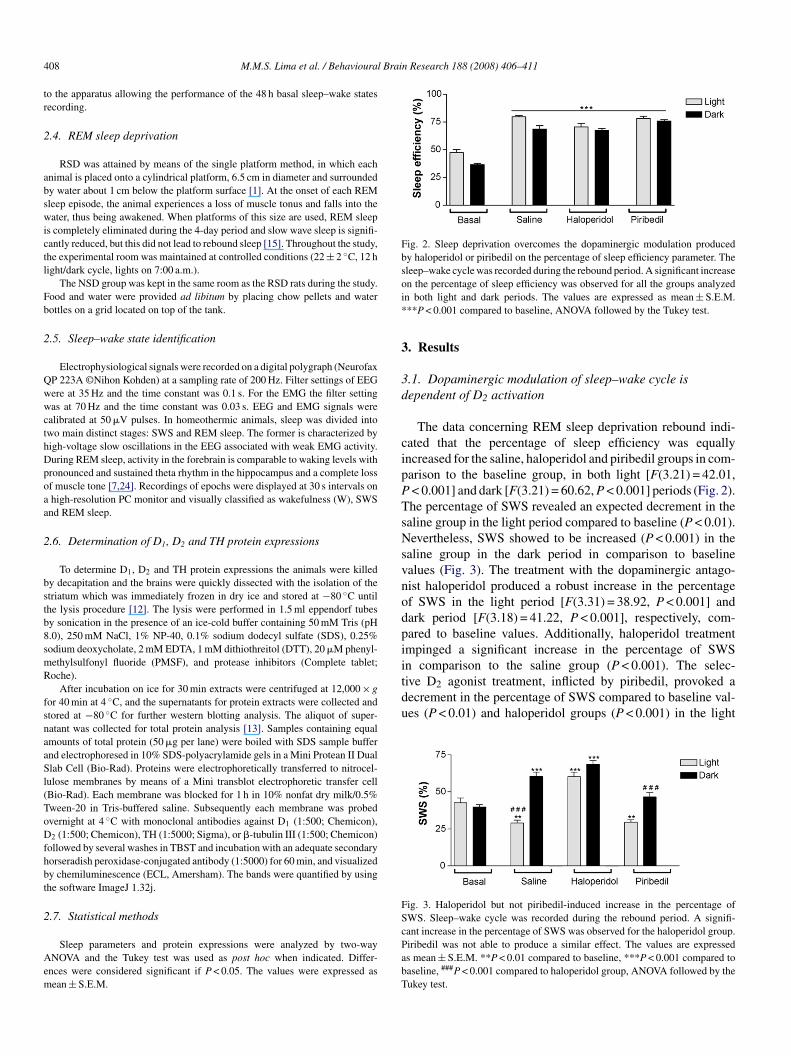
Fig. 2. Sleep deprivation overcomes the dopaminergic modulation producedby haloperidol or piribedil on the percentage of sleep efficiency parameter. Thesleep–wake cycle was recorded during the rebound period. A significant increaseoi*
3
3d
cipPTsNsvnodpiitive D2 agonist treatment, inflicted by piribedil, provoked adecrement in the percentage of SWS compared to baseline val-ues (P < 0.01) and haloperidol groups (P < 0.001) in the light
Fig. 3. Haloperidol but not piribedil-induced increase in the percentage ofSWS. Sleep–wake cycle was recorded during the rebound period. A signifi-
08 M.M.S. Lima et al. / Behavioural
o the apparatus allowing the performance of the 48 h basal sleep–wake statesecording.
.4. REM sleep deprivation
RSD was attained by means of the single platform method, in which eachnimal is placed onto a cylindrical platform, 6.5 cm in diameter and surroundedy water about 1 cm below the platform surface [1]. At the onset of each REMleep episode, the animal experiences a loss of muscle tonus and falls into theater, thus being awakened. When platforms of this size are used, REM sleep
s completely eliminated during the 4-day period and slow wave sleep is signifi-antly reduced, but this did not lead to rebound sleep [15]. Throughout the study,he experimental room was maintained at controlled conditions (22 ± 2 ◦C, 12 hight/dark cycle, lights on 7:00 a.m.).
The NSD group was kept in the same room as the RSD rats during the study.ood and water were provided ad libitum by placing chow pellets and waterottles on a grid located on top of the tank.
.5. Sleep–wake state identification
Electrophysiological signals were recorded on a digital polygraph (NeurofaxP 223A ©Nihon Kohden) at a sampling rate of 200 Hz. Filter settings of EEGere at 35 Hz and the time constant was 0.1 s. For the EMG the filter settingas at 70 Hz and the time constant was 0.03 s. EEG and EMG signals were
alibrated at 50 �V pulses. In homeothermic animals, sleep was divided intowo main distinct stages: SWS and REM sleep. The former is characterized byigh-voltage slow oscillations in the EEG associated with weak EMG activity.uring REM sleep, activity in the forebrain is comparable to waking levels withronounced and sustained theta rhythm in the hippocampus and a complete lossf muscle tone [7,24]. Recordings of epochs were displayed at 30 s intervals onhigh-resolution PC monitor and visually classified as wakefulness (W), SWS
nd REM sleep.
.6. Determination of D1, D2 and TH protein expressions
To determine D1, D2 and TH protein expressions the animals were killedy decapitation and the brains were quickly dissected with the isolation of thetriatum which was immediately frozen in dry ice and stored at −80 ◦C untilhe lysis procedure [12]. The lysis were performed in 1.5 ml eppendorf tubesy sonication in the presence of an ice-cold buffer containing 50 mM Tris (pH.0), 250 mM NaCl, 1% NP-40, 0.1% sodium dodecyl sulfate (SDS), 0.25%odium deoxycholate, 2 mM EDTA, 1 mM dithiothreitol (DTT), 20 �M phenyl-ethylsulfonyl fluoride (PMSF), and protease inhibitors (Complete tablet;oche).
After incubation on ice for 30 min extracts were centrifuged at 12,000 × gor 40 min at 4 ◦C, and the supernatants for protein extracts were collected andtored at −80 ◦C for further western blotting analysis. The aliquot of super-atant was collected for total protein analysis [13]. Samples containing equalmounts of total protein (50 �g per lane) were boiled with SDS sample buffernd electrophoresed in 10% SDS-polyacrylamide gels in a Mini Protean II Duallab Cell (Bio-Rad). Proteins were electrophoretically transferred to nitrocel-
ulose membranes by means of a Mini transblot electrophoretic transfer cellBio-Rad). Each membrane was blocked for 1 h in 10% nonfat dry milk/0.5%ween-20 in Tris-buffered saline. Subsequently each membrane was probedvernight at 4 ◦C with monoclonal antibodies against D1 (1:500; Chemicon),
2 (1:500; Chemicon), TH (1:5000; Sigma), or �-tubulin III (1:500; Chemicon)ollowed by several washes in TBST and incubation with an adequate secondaryorseradish peroxidase-conjugated antibody (1:5000) for 60 min, and visualizedy chemiluminescence (ECL, Amersham). The bands were quantified by usinghe software ImageJ 1.32j.
.7. Statistical methods
Sleep parameters and protein expressions were analyzed by two-wayNOVA and the Tukey test was used as post hoc when indicated. Differ-
nces were considered significant if P < 0.05. The values were expressed asean ± S.E.M.
cPabT
n the percentage of sleep efficiency was observed for all the groups analyzedn both light and dark periods. The values are expressed as mean ± S.E.M.**P < 0.001 compared to baseline, ANOVA followed by the Tukey test.
. Results
.1. Dopaminergic modulation of sleep–wake cycle isependent of D2 activation
The data concerning REM sleep deprivation rebound indi-ated that the percentage of sleep efficiency was equallyncreased for the saline, haloperidol and piribedil groups in com-arison to the baseline group, in both light [F(3.21) = 42.01,< 0.001] and dark [F(3.21) = 60.62, P < 0.001] periods (Fig. 2).he percentage of SWS revealed an expected decrement in thealine group in the light period compared to baseline (P < 0.01).evertheless, SWS showed to be increased (P < 0.001) in the
aline group in the dark period in comparison to baselinealues (Fig. 3). The treatment with the dopaminergic antago-ist haloperidol produced a robust increase in the percentagef SWS in the light period [F(3.31) = 38.92, P < 0.001] andark period [F(3.18) = 41.22, P < 0.001], respectively, com-ared to baseline values. Additionally, haloperidol treatmentmpinged a significant increase in the percentage of SWSn comparison to the saline group (P < 0.001). The selec-
ant increase in the percentage of SWS was observed for the haloperidol group.iribedil was not able to produce a similar effect. The values are expresseds mean ± S.E.M. **P < 0.01 compared to baseline, ***P < 0.001 compared toaseline, ###P < 0.001 compared to haloperidol group, ANOVA followed by theukey test.
M.M.S. Lima et al. / Behavioural Brain Research 188 (2008) 406–411 409
Fig. 4. REM sleep reduction mediated by haloperidol. Sleep–wake cycle wasrecorded during the rebound period. A significant decrease in the percentage ofRat
pSp(
oiv[tpt
3om
ot5tttaoowpa
pSc
segppMn
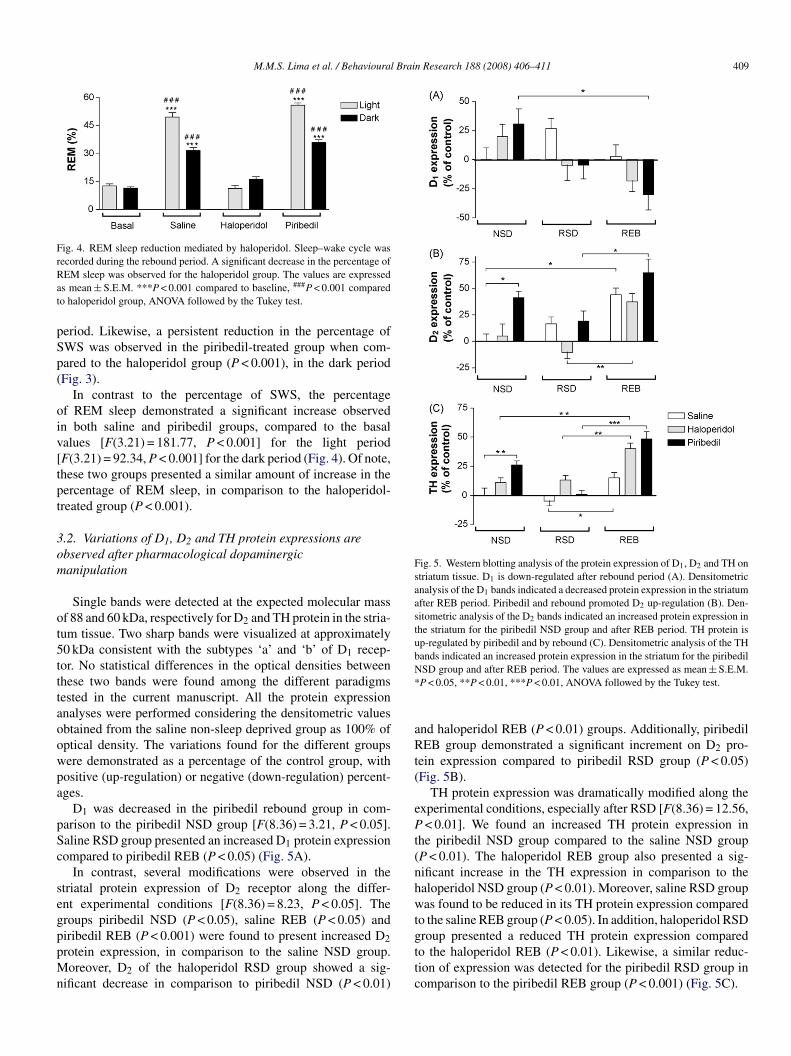
Fig. 5. Western blotting analysis of the protein expression of D1, D2 and TH onstriatum tissue. D1 is down-regulated after rebound period (A). Densitometricanalysis of the D1 bands indicated a decreased protein expression in the striatumafter REB period. Piribedil and rebound promoted D2 up-regulation (B). Den-sitometric analysis of the D2 bands indicated an increased protein expression inthe striatum for the piribedil NSD group and after REB period. TH protein isup-regulated by piribedil and by rebound (C). Densitometric analysis of the THbands indicated an increased protein expression in the striatum for the piribedilN*
aRt(
ePt(nhwt
EM sleep was observed for the haloperidol group. The values are expresseds mean ± S.E.M. ***P < 0.001 compared to baseline, ###P < 0.001 comparedo haloperidol group, ANOVA followed by the Tukey test.
eriod. Likewise, a persistent reduction in the percentage ofWS was observed in the piribedil-treated group when com-ared to the haloperidol group (P < 0.001), in the dark periodFig. 3).
In contrast to the percentage of SWS, the percentagef REM sleep demonstrated a significant increase observedn both saline and piribedil groups, compared to the basalalues [F(3.21) = 181.77, P < 0.001] for the light periodF(3.21) = 92.34, P < 0.001] for the dark period (Fig. 4). Of note,hese two groups presented a similar amount of increase in theercentage of REM sleep, in comparison to the haloperidol-reated group (P < 0.001).
.2. Variations of D1, D2 and TH protein expressions arebserved after pharmacological dopaminergicanipulation
Single bands were detected at the expected molecular massf 88 and 60 kDa, respectively for D2 and TH protein in the stria-um tissue. Two sharp bands were visualized at approximately0 kDa consistent with the subtypes ‘a’ and ‘b’ of D1 recep-or. No statistical differences in the optical densities betweenhese two bands were found among the different paradigmsested in the current manuscript. All the protein expressionnalyses were performed considering the densitometric valuesbtained from the saline non-sleep deprived group as 100% ofptical density. The variations found for the different groupsere demonstrated as a percentage of the control group, withositive (up-regulation) or negative (down-regulation) percent-ges.
D1 was decreased in the piribedil rebound group in com-arison to the piribedil NSD group [F(8.36) = 3.21, P < 0.05].aline RSD group presented an increased D1 protein expressionompared to piribedil REB (P < 0.05) (Fig. 5A).
In contrast, several modifications were observed in thetriatal protein expression of D2 receptor along the differ-nt experimental conditions [F(8.36) = 8.23, P < 0.05]. Theroups piribedil NSD (P < 0.05), saline REB (P < 0.05) and
iribedil REB (P < 0.001) were found to present increased D2rotein expression, in comparison to the saline NSD group.oreover, D2 of the haloperidol RSD group showed a sig-ificant decrease in comparison to piribedil NSD (P < 0.01)
gttc
SD group and after REB period. The values are expressed as mean ± S.E.M.P < 0.05, **P < 0.01, ***P < 0.01, ANOVA followed by the Tukey test.
nd haloperidol REB (P < 0.01) groups. Additionally, piribedilEB group demonstrated a significant increment on D2 pro-
ein expression compared to piribedil RSD group (P < 0.05)Fig. 5B).
TH protein expression was dramatically modified along thexperimental conditions, especially after RSD [F(8.36) = 12.56,< 0.01]. We found an increased TH protein expression in
he piribedil NSD group compared to the saline NSD groupP < 0.01). The haloperidol REB group also presented a sig-ificant increase in the TH expression in comparison to thealoperidol NSD group (P < 0.01). Moreover, saline RSD groupas found to be reduced in its TH protein expression compared
o the saline REB group (P < 0.05). In addition, haloperidol RSD
roup presented a reduced TH protein expression comparedo the haloperidol REB (P < 0.01). Likewise, a similar reduc-ion of expression was detected for the piribedil RSD group inomparison to the piribedil REB group (P < 0.001) (Fig. 5C).
4 Brai
4
pawaCtaatddsdia
tupaespitlPd[aa
ca[dripias
aootadptspt
riadhdtpttnwp
poaspcogwdromtdpttTtipb
msdFipm
arisy
10 M.M.S. Lima et al. / Behavioural
. Discussion
Our main findings demonstrate that a selective D2 blockageroduced a dramatic reduction of REM sleep during a REB situ-tion after a period of 96 h of RSD. Such decrease of REM sleepas accompanied by an increment in SWS, which is possibly
ccounted for in the observed increase in the sleep efficiency.onversely, our findings also demonstrate that the adminis-
ration of a selective D2 agonist (piribedil) did not generatedditional increase in the percentage of REM sleep. Addition-lly, SWS was slightly reduced compared to baseline (duringhe light period) and to the haloperidol RSD group (during theark period). Besides, the dopaminergic D2 receptor was foundown-regulated, in the haloperidol group, after RSD, and sub-equently up-regulated after REB group, contrasting to the D1own-regulation at the same period. In this sense, the current datandicate a participation of D2 receptor for REM sleep regulationnd consequently in the REM sleep/SWS balance.
As of late, such involvement of the dopaminergic system inhe regulation of sleep, particularly of REM sleep, has beennder scrutiny [4,5,11,14,16]. In a prospective study in PDatients, cabergoline (a D1 and D2 agonist) treatment increasedrousals, stage shifts, and awakenings, although quantitativelectrophysiological measures of sleep were maintained, andubjective measures of sleep quality even improved [6]. Therevention of sleep and enhancement of waking by DA reuptakenhibitors and DA receptors agonists are the basis for their use inhe treatment of other conditions such as narcolepsy and somno-ence associated with hypodopaminergic states [19]. Moreover,D patients present several disruptions of sleep, which areeteriorated by the use of dopaminergic D2 receptor agonists2,3,9]. Opposing to that, treatment with the D2 antagonist andntipsychotic agent haloperidol attenuates hippocampal thetand gamma oscillations which is characteristic of REM sleep [5].
Previous studies have related the administration of antipsy-hotic drugs with the tendency to induce sedation, which isproperty commonly observed with the use of these drugs
18]. Analogously, the clinical antipsychotic potency of theserugs is correlated with their ability to block dopaminergiceceptors [20]. The strongest evidence for the DA hypothesiss the observation that all medications efficacious for positivesychotic symptoms block D2 receptors [21] and drugs thatncrease mesolimbic dopamine transmission, such as cocaine ormphetamine, can induce schizophrenia-like positive psychoticymptoms [22].
In the present study our hypothesis contemplates the possiblebility of D2 blockage to produce reduction or even suppressionf REM sleep, based on the recently identified role of dopaminen sleep [4,5,11]. In this context, our findings indicated that thisheory is plausible according to the D2 antagonism, although, thedministration of D2 agonist did not show the inverse. Such evi-ence dovetails with a possible ceiling effect of REM inductionreviously promoted by RSD. Additionally, it is possible that
he agonist-induced increase of REM sleep (and of slow waveleep) depends upon the selective activation of dopaminergic D2resynaptic autoreceptors located in DA neurons of the ventralegmental area or in the substantia nigra pars compacta.A
M
n Research 188 (2008) 406–411
Biochemical data, concerning the receptors expressions,evealed that after RSD, D2 was initially down-regulatedn the haloperidol group, although it became up-regulatedfter REB period. Opposing to that, D1 showed a persistentown-regulation, after REB, for the groups administered withaloperidol and piribedil. It is suggested that the dramaticecrease in the percentage of REM sleep is possibly related tohe down-regulation of striatal D2 receptors, found after RSD,articularly in the haloperidol group. This is in accordance withhe evidence that the D2 antagonist agent haloperidol attenuateshe REM-like hippocampal oscillations, indicating that D2 isecessary to generate REM sleep [5]. Nevertheless, D1 receptoras down-regulated at the same time-point, suggesting somearticipation in this regulatory event.
The mechanism underlying the striatal D2 up-regulation isroposed to be an effect came as consequence of RSD whichriginally produces selective D2 supersensitivity [17,25,26],nd after its period probably generates a surge in D2 expres-ion. A similar effect was observed, during the RSD and REBeriods, after the administration of the D2 agonist. Anotheromplementary theory to explain this D2 up-regulation reliesn the importance of this receptor for the REM sleep trig-ering. While haloperidol blocked D2 receptors REM sleepas barely detected, generating a biological pressure to pro-uce a D2 up-regulation and consequently the expected REMebound in the subsequent 24 h. Still regarding the etiologyf the D2 up-regulation, it is possible that it is a pri-ary abnormality or, conceivably, a compensatory response
o the decreased DA concentrations. Indeed, there is evi-ence that DA levels in Brattleboro rats are decreased [8]ossibly to compensate for increased D2 receptors. Never-heless, striatal TH protein expression was incremented afterhe administration of piribedil to the NSD group. Likewise,H was up-regulated from RSD to REB period of all groups
ested, suggesting that the RSD manipulation reverberatedn this enzyme expression more dominantly than did theharmacological intervention, possibly exacerbating the DAiosynthesis.
Taken together our findings demonstrate that D2 is instru-ental for sleep homeostasis and, in particular, for REM
leep regulation, once haloperidol severely depleted REM sleepuring the REB period without modifying sleep efficiency.urthermore, haloperidol and piribedil administrations induced
ncreased expressions of D2 and TH proteins after the REBeriod, suggesting that D2 supersensitivity overcame the phar-acological effect produced by the drugs.In conclusion, the present investigation reported a particular
ction of the dopaminergic neurotransmission on REM sleepelying on D2 activation which is consistent with recent findingsn the literature that indicate a novel role of DA in regulatingleep which will only be determined after further investigationsield the data needed in that comprehension.
cknowledgements
The authors would like to thank Waldermaks Aires Leite andarilde Aires Costa for their capable technical assistance.

Brai
Fa
R
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
M.M.S. Lima et al. / Behavioural
Funding: This work was supported by AFIP, CAPES,APESP-CEPID (98/14.303-3 to ST). ST, MABFV and SMZre recipients of CNPq fellowships.
eferences
[1] Andersen ML, Perry JC, Bignotto M, Perez-Mendes P, Cinini SM, MelloLE, et al. Influence of chronic cocaine treatment and sleep deprivation onsexual behavior and neurogenesis of the male rat. Prog Neuropsychophar-macol Biol Psychiatry 2007;31:1224–9.
[2] Arnulf I. Sleep and wakefulness disturbances in Parkinson’s disease. JNeural Transm Suppl 2006:357–60.
[3] Arnulf I, Konofal E, Merino-Andreu M, Houeto JL, Mesnage V, Welter ML,et al. Parkinson’s disease and sleepiness: an integral part of PD. Neurology2002;58:1019–24.
[4] Dahan L, Astier B, Vautrelle N, Urbain N, Kocsis B, Chouvet G. Prominentburst firing of dopaminergic neurons in the ventral tegmental area duringparadoxical sleep. Neuropsychopharmacology 2007;32:1232–41.
[5] Dzirasa K, Ribeiro S, Costa R, Santos LM, Lin SC, Grosmark A, et al.Dopaminergic control of sleep–wake states. J Neurosci 2006;26:10577–89.
[6] Hogl B, Rothdach A, Wetter TC, Trenkwalder C. The effect of cabergo-line on sleep, periodic leg movements in sleep, and early morning motorfunction in patients with Parkinson’s disease. Neuropsychopharmacology2003;28:1866–70.
[7] Jouvet M. Neurophysiology of the states of sleep. Physiol Rev 1967;47:117–77.
[8] Kovacs GL, Szabo G, Szontagh L, Medve L, Telegdy G, Laszlo FA.Hereditary diabetes insipidus in rats. Altered cerebral indolamine and cat-echolamine metabolism. Neuroendocrinology 1980;31:189–93.
[9] Larsen JP, Tandberg E. Sleep disorders in patients with Parkinson’s disease:epidemiology and management. CNS Drugs 2001;15:267–75.
10] Lena I, Parrot S, Deschaux O, Muffat-Joly S, Sauvinet V, Renaud B, etal. Variations in extracellular levels of dopamine, noradrenaline, gluta-mate, and aspartate across the sleep–wake cycle in the medial prefrontalcortex and nucleus accumbens of freely moving rats. J Neurosci Res2005;81:891–9.
11] Lima MMS, Andersen ML, Reksidler AB, Vital MA, Tufik S. The role ofthe substantia nigra pars compacta in regulating sleep patterns in rats. PLoSONE 2007;2:e513.
12] Lima MMS, Braga Reksidler A, Marques Zanata S, Bueno MachadoH, Tufik S, Vital MA. Different parkinsonism models produce a time-
[
[
n Research 188 (2008) 406–411 411
dependent induction of COX-2 in the substantia nigra of rats. Brain Res2006;1101:117–25.
13] Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurementwith the Folin protein reagent. J Bio Chem 1951;193:265–75.
14] Lu J, Jhou TC, Saper CB. Identification of wake-active dopaminergic neu-rons in the ventral periaqueductal gray matter. J Neurosci 2006;26:193–202.
15] Machado RB, Suchecki D, Tufik S. Sleep homeostasis in rats assessed by along-term intermittent paradoxical sleep deprivation protocol. Behav BrainRes 2005;160:356–64.
16] Monti JM, Monti D. The involvement of dopamine in the modulation ofsleep and waking. Sleep Med Rev 2007;11:113–33.
17] Nunes GP, Tufik S, Nobrega JN. Autoradiographic analysis of D1 and D2dopaminergic receptors in rat brain after paradoxical sleep deprivation.Brain Res Bull 1994;34:453–6.
18] Ongini E, Bonizzoni E, Ferri N, Milani S, Trampus M. Differential effectsof dopamine D-1 and D-2 receptor antagonist antipsychotics on sleep–wakepatterns in the rat. J Pharmacol Exp Ther 1993;266:726–31.
19] Pace-Schott EF, Hobson JA. The neurobiology of sleep: genetics, cellu-lar physiology and subcortical networks. Nat Rev Neurosci 2002;3:591–605.
20] Seeman P. Atypical neuroleptics: role of multiple receptor, endogenousdopamine, and receptor linkage. Acta Phychiatry Scand 1990;82:14–20.
21] Seeman P. Dopamine receptor sequences. Therapeutic levels of neuro-leptics occupy D2 receptors, clozapine occupies D4. Neuropsychophar-macology 1992;7:261–84.
22] Segal DS, Schuckit MA. Animal models of stimulant-induced psychosis.In: Creese I, editor. Stimulants: neurochemical, behavioral and clinicalperspectives. New York: Raven Press; 1983. p. 131–67.
23] Shilling PD, Kinkead B, Murray T, Melendez G, Nemeroff CB, FeifelD. Upregulation of striatal dopamine-2 receptors in Brattleboro rats withprepulse inhibition deficits. Biol Psychiatry 2006;60:1278–81.
24] Timo-Iaria C, Negrao N, Schmidek WR, Hoshino K, Lobato de MenezesCE, Leme da Rocha T. Phases and states of sleep in the rat. Physiol Behav1970;5:1057–62.
25] Tufik S. Changes of response to dopaminergic drugs in rats submitted toREM-sleep deprivation. Psychopharmacology (Berl) 1981;72:257–60.
26] Tufik S, Lindsey CJ, Carlini EA. Does REM sleep deprivation induce asupersensitivity of dopaminergic receptors in the rat brain? Pharmacology1978;16:98–105.
27] Wisor JP, Nishino S, Sora I, Uhl GH, Mignot E, Edgar DM. Dopaminergicrole in stimulant-induced wakefulness. J Neurosci 2001;21:1787–94.




















