
Bidirectional diffusion of ammonium and sodium cations in forward osmosis: role of membrane active...
8
Bidirectional Diffusion of Ammonium and Sodium Cations in Forward Osmosis: Role of Membrane Active Layer Surface Chemistry and Charge Xinglin Lu, † Chanhee Boo, ‡ Jun Ma,* ,† and Menachem Elimelech* ,‡ † State Key Laboratory of Urban Water Resource and Environment, Harbin Institute of Technology, Harbin 150090, China ‡ Department of Chemical and Environmental Engineering, Yale University, New Haven, Connecticut 06520-8286, United States * S Supporting Information ABSTRACT: Systematic fundamental understanding of mass trans- port in osmosis-driven membrane processes is important for further development of this emerging technology. In this work, we investigate the role of membrane surface chemistry and charge on bidirectional solute diffusion in forward osmosis (FO). In particular, bidirectional diffusion of ammonium (NH 4 + ) and sodium (Na + ) is examined using FO membranes with different materials and surface charge character- istics. Using an ammonium bicarbonate (NH 4 HCO 3 ) draw solution, we observe dramatically enhanced cation fluxes with sodium chloride feed solution compared to that with deionized water feed solution for thin- film composite (TFC) FO membrane. However, the bidirectional diffusion of cations does not change, regardless of the type of feed solution, for cellulose triacetate (CTA) FO membrane. We relate this phenomenon to the membrane fixed surface charge by employing different feed solution pH to foster different protonation conditions for the carboxyl groups on the TFC membrane surface. Membrane surface modification is also carried out with the TFC membrane using ethylenediamine to alter carboxyl groups into amine groups. The modified TFC membrane, with less negatively charged groups, exhibits a significant decrease in the bidirectional diffusion of cations under the same conditions employed with the pristine TFC membrane. Based on our experimental observations, we propose Donnan dialysis as a mechanism responsible for enhanced bidirectional diffusion of cations in TFC membranes. ■ INTRODUCTION Membrane technologies provide promising solutions to address the global challenge for adequate and safe water. 1−3 As an emerging membrane technology, osmosis-driven membrane processes (ODMP), such as forward osmosis (FO) and pressure retarded osmosis (PRO), have been gradually developed in the past decade. Because of their low fouling propensity, 4 ODMP have potential applications in treatment of a variety of high fouling potential source waters, including desalination of high salinity brines from shale gas produced water, 5−9 municipal wastewater reclamation, 10−16 and dilution of brine from reverse osmosis (RO) plants. 17−19 Previous efforts on the development of ODMP have mainly focused on two technical obstacles: fabrication of high performance membranes and selection of appropriate draw solutes to meet the special needs of ODMP. 20 Early studies on ODMP used a commercial cellulose triacetate (CTA) FO membrane fabricated on a polyester woven mesh support, which sparked resurgence in ODMP research and development in the past decade. 21 More recently, thin-film composite (TFC) polyamide FO membranes have been successfully fabricated through a carefully tailored support layer fabrication process. 22−24 TFC FO membranes showed higher water permeability and salt rejection as well as a wider pH operation range than the CTA FO membrane, thus further accelerating the growth and applications of ODMP. 25−27 At the same time, significant efforts have been made in the development of draw solutes for various potential applications that can produce high osmotic pressure with low reverse draw permeation. 28 Among those potential draw solutes, the thermolytic ammonia−carbon dioxide (NH 3 −CO 2 ) draw solution has been investigated at the lab scale 29,30 and more recently demonstrated in a pilot-scale desalination system. 5 As a unique property of ODMP, bidirectional diffusion of solutesreverse diffusion of draw solutes and forward diffusion of feed solutesis detrimental to OMDP due to loss of draw solutes, 31 acceleration of water flux decline by fouling, 32 and decrease in product water quality. 33,34 Previous studies 35−39 investigated bidirectional ion diffusion in ODMP with the Received: August 24, 2014 Revised: November 19, 2014 Accepted: November 24, 2014 Article pubs.acs.org/est © XXXX American Chemical Society A dx.doi.org/10.1021/es504162v | Environ. Sci. Technol. XXXX, XXX, XXX−XXX
Transcript of Bidirectional diffusion of ammonium and sodium cations in forward osmosis: role of membrane active...

Bidirectional Diffusion of Ammonium and Sodium Cations inForward Osmosis: Role of Membrane Active Layer Surface Chemistryand ChargeXinglin Lu,† Chanhee Boo,‡ Jun Ma,*,† and Menachem Elimelech*,‡
†State Key Laboratory of Urban Water Resource and Environment, Harbin Institute of Technology, Harbin 150090, China‡Department of Chemical and Environmental Engineering, Yale University, New Haven, Connecticut 06520-8286, United States
*S Supporting Information
ABSTRACT: Systematic fundamental understanding of mass trans-port in osmosis-driven membrane processes is important for furtherdevelopment of this emerging technology. In this work, we investigatethe role of membrane surface chemistry and charge on bidirectionalsolute diffusion in forward osmosis (FO). In particular, bidirectionaldiffusion of ammonium (NH4
+) and sodium (Na+) is examined usingFO membranes with different materials and surface charge character-istics. Using an ammonium bicarbonate (NH4HCO3) draw solution, weobserve dramatically enhanced cation fluxes with sodium chloride feedsolution compared to that with deionized water feed solution for thin-film composite (TFC) FO membrane. However, the bidirectionaldiffusion of cations does not change, regardless of the type of feedsolution, for cellulose triacetate (CTA) FO membrane. We relate thisphenomenon to the membrane fixed surface charge by employingdifferent feed solution pH to foster different protonation conditions for the carboxyl groups on the TFC membrane surface.Membrane surface modification is also carried out with the TFC membrane using ethylenediamine to alter carboxyl groups intoamine groups. The modified TFC membrane, with less negatively charged groups, exhibits a significant decrease in thebidirectional diffusion of cations under the same conditions employed with the pristine TFC membrane. Based on ourexperimental observations, we propose Donnan dialysis as a mechanism responsible for enhanced bidirectional diffusion ofcations in TFC membranes.
■ INTRODUCTION
Membrane technologies provide promising solutions to addressthe global challenge for adequate and safe water.1−3 As anemerging membrane technology, osmosis-driven membraneprocesses (ODMP), such as forward osmosis (FO) andpressure retarded osmosis (PRO), have been graduallydeveloped in the past decade. Because of their low foulingpropensity,4 ODMP have potential applications in treatment ofa variety of high fouling potential source waters, includingdesalination of high salinity brines from shale gas producedwater,5−9 municipal wastewater reclamation,10−16 and dilutionof brine from reverse osmosis (RO) plants.17−19
Previous efforts on the development of ODMP have mainlyfocused on two technical obstacles: fabrication of highperformance membranes and selection of appropriate drawsolutes to meet the special needs of ODMP.20 Early studies onODMP used a commercial cellulose triacetate (CTA) FOmembrane fabricated on a polyester woven mesh support,which sparked resurgence in ODMP research and developmentin the past decade.21 More recently, thin-film composite (TFC)polyamide FO membranes have been successfully fabricatedthrough a carefully tailored support layer fabrication
process.22−24 TFC FO membranes showed higher waterpermeability and salt rejection as well as a wider pH operationrange than the CTA FO membrane, thus further acceleratingthe growth and applications of ODMP.25−27 At the same time,significant efforts have been made in the development of drawsolutes for various potential applications that can produce highosmotic pressure with low reverse draw permeation.28 Amongthose potential draw solutes, the thermolytic ammonia−carbondioxide (NH3−CO2) draw solution has been investigated at thelab scale29,30 and more recently demonstrated in a pilot-scaledesalination system.5
As a unique property of ODMP, bidirectional diffusion ofsolutesreverse diffusion of draw solutes and forward diffusionof feed solutesis detrimental to OMDP due to loss of drawsolutes,31 acceleration of water flux decline by fouling,32 anddecrease in product water quality.33,34 Previous studies35−39
investigated bidirectional ion diffusion in ODMP with the
Received: August 24, 2014Revised: November 19, 2014Accepted: November 24, 2014
Article
pubs.acs.org/est
© XXXX American Chemical Society A dx.doi.org/10.1021/es504162v | Environ. Sci. Technol. XXXX, XXX, XXX−XXX

commercial CTA FO membrane. These studies found that ionvalence and hydration radius influence the bidirectionaldiffusion rate of different ions36,37 and suggested electrostaticinteractions can impact bidirectional diffusion when nitrate ispresent in the system.38,39 However, all observed phenomenaand proposed mechanisms of bidirectional diffusion in thosestudies were based on the CTA FO membrane.Using commercial TFC and CTA FO membranes, Coday et
al.38 indicated a higher diffusion of cations relative to anions forthe TFC membranes, attributing this phenemenon to differentsurface charge of the support layers of TFC and CTAmembranes. More recently, Arena et al.,40 using a TFC ROmembrane in FO, have reported enhanced reverse diffusion ofammonium (NH4
+) through the membrane when a strongelectrolyte (i.e., NaCl) was used as a feed solution. They furtherobserved substantially lower rejection of sodium than chloridein FO, attributing the result to cation exchange between thedraw and feed solutions. Given the significance of TFCmembranes in ODMP and the advantages of NH4
+ based drawsolutes, such as NH3−CO2 and concentrated fertilizers,41 amore fundamental understanding of the transport mechanismsin FO with TFC membranes and NH4
+ based draw solutions isof significant importance for the future development of theODMP. Such fundamental understanding can lead to thedevelopment of TFC membranes with reduced reversediffusion of ammonium, which will significantly increase theefficiency of OMDP.The objective of this study is to investigate the bidirectional
diffusion of ammonium (NH4+) and sodium (Na+) in FO and
to relate this phenomenon to membrane material properties.Water and cation (NH4
+ and Na+) fluxes are determined withdifferent membrane materials at different solution pH toconfirm the role of membrane surface chemistry and charge inbidirectional diffusion of NH4
+ and Na+. Surface chemicalmodification of the TFC membrane active layer to alter itssurface charge characteristics is also carried out to betterunderstand the role of surface charge in bidirectional diffusionof NH4
+ and Na+. The results of our experiments are used toelucidate the mechanism for the enhanced reverse transport ofNH4
+ and the bidirectional diffusion of cations in TFC FOmembranes.
■ MATERIALS AND METHODS
FO Membranes and Membrane Characterization. Acommercial asymmetric cellulose triacetate (CTA) FOmembrane was obtained from Hydration Technology In-novation (Albany, OR). This proprietary FO membrane,denoted as CTA in this paper, is thought to be prepared byphase inversion using the same polymer material for the entiremembrane. CTA membranes have negligible or no surfacecharge because the polymer functional groups are converted toester during the fabrication process.42,43
A high performance thin-film composite (TFC) polyamideFO membrane was acquired from Oasys Water (Oasys WaterInc., Boston, MA). The membrane consists of a dense selectivepolyamide active layer on top of a porous polysulfone supportlayer. The surface of the polyamide active layer is abundantwith carboxyl groups,43 which renders the membrane surfacenegatively charged. To better understand the role of membranesurface charge in bidirectional ion diffusion, we modified thefunctional groups on the TFC membrane surface as describedin the next subsection. We denote the TFC FO membrane
before and after surface modification as “pristine TFC” and“modified TFC”, respectively.Prior to the experiments, all membranes were immersed in
25% isopropanol (Fisher Scientific) for 30 min to allowcomplete wetting.44 The pure water permeability coefficient, A,salt (NaCl) permeability coefficient, B, and structure parameter,S, were determined using an RO and FO characterizationmethod as described in our previous study.22 The determinedproperties (A, B, and S) of the FO membranes are provided inTable S1 of the Supporting Information (SI).An electrokinetic analyzer (EKA, Brookhaven Instruments,
Holtsville, NY) was used to determine the membrane surfacezeta potential. Carboxyl group density was measured usingtoluidine blue O (TBO, technical grade, Sigma-Aldrich) asreported in our earlier publication.43 More details of thesemembrane surface characterization methods are given in the SI.
Draw Solutions. Ammonia−carbon dioxide (NH3−CO2)draw solution was prepared by mixing 1.8 M ammoniumbicarbonate (NH4HCO3, Sigma-Aldrich) and 0.2 M ammo-nium hydroxide (NH4OH, Sigma-Aldrich) in deionized (DI)water (Milli-Q ultrapure water purification system, Millipore,Billerica, MA). A total ammonium salt concentration of 2.0 Mwas achieved with an ammonia to carbon dioxide molar ratio of1.1. The measured pH of the NH3−CO2 draw solution was 7.8.Ammonium chloride (NH4Cl) draw solution was also used
to investigate the effect of solution pH on the bidirectionalcation diffusion. ACS reagent grade ammonium chloride(NH4Cl, Sigma-Aldrich) was dissolved in DI water at aconcentration of 2.0 M. The measured pH of the 2.0 M NH4Cldraw solution was 4.5.
Measurement of Water Flux and Bidirectional IonFlux. A laboratory scale crossflow FO unit with channeldimensions of 77 mm × 26 mm × 3 mm was used to measurethe water flux and solute flux across the membrane. Theexperiments were performed using DI water or 0.2 M NaCl asfeed solution and NH3−CO2 or NH4Cl as draw solution in FOmode (i.e., draw solution facing the membrane support layerand no applied hydraulic pressure on either side of the cell). Allexperiments were conducted for 1 h with a crossflow velocity of21.4 cm/s. Temperatures of both draw and feed solutions weremaintained at 25 ± 0.5 °C.Water flux, Jw, across the membrane was measured by
recording the increase in draw solution weight at 1 minintervals. The data collected after the water flux had stabilized(always within 10 min) was averaged and used for thecalculation of ion flux. Forward ion fluxes, JS
forward, weredetermined by measuring the total concentrations of feedions (i.e., Na+ and Cl−) in the draw solution after 1 h using
+ =C V J A t J A t( )D Dinitial
w m Sforward
m (1)
where CD is the feed ion concentration in the draw solutionafter 1 h, VD
initial is the initial volume of the draw solution, Jw isthe average water flux, Am is the membrane area, and t is thetime. Similarly, reverse ion fluxes, JS
reverse, were determined bymeasuring the total concentrations of draw ions (i.e., NH4
+ andHCO3
−) in the feed solution using
− =C V J A t J A t( )F Finitial
w m Sreverse
m (2)
where CF is the draw ion concentration in the feed solutionafter 1 h and VF
initial is the initial volume of the feed solution.Concentrations of sodium and chloride ions in the draw
solution (i.e., NH3−CO2 or NH4Cl solutions) were quantified
Environmental Science & Technology Article
dx.doi.org/10.1021/es504162v | Environ. Sci. Technol. XXXX, XXX, XXX−XXXB
using inductively coupled plasma mass spectrometry (ICP-MS;Optima 8000, PerkinElmer, Fremont, CA) and ion chromatog-raphy (IC; ICS-3000, Dionex, Sunnyvale, CA), respectively. A10 mL sample of draw solution was collected after a 1 h FOexperiment and boiled until all water evaporated. During theboiling, the dissolved thermolytic ammonium salts decomposeinto NH3 and CO2 gases. Next, the remaining NaCl wasdissolved in a known volume of DI water and the Na+ and Cl−
concentrations were measured.29 Note that concentration ofCl− ion was analyzed only for experiments with NH3−CO2draw solution. Concentrations of total ammonia and totalcarbon species in the feed solution (DI water or NaCl solution)were measured using the phenate method and total organiccarbon (TOC) analyzer (TOC-V, Shimadzu Corp., Japan),respectively. We note that the detectable concentration rangesof Na+ by ICP-MS and Cl− by IC are several orders ofmagnitude lower than the measured values in this work. Also,the sensitivities of the phenate method for ammonia and theTOC analyzer for carbonate species are less than 1 ppm,indicating the accuracy and reliability of our analytical methods.FO Membrane Surface Modification. In order to alter
the surface charge of the pristine TFC membrane, surfacemodification was carried out using a custom-made mold thatisolated the polyamide active layer side of the membrane forthe desired reaction. After membrane characterization throughthe experimental procedures described above, the samemembrane coupon was placed on a glass plate with thepolyamide active layer facing up. A soft rubber spacer andrectangular frame were then clamped on top of the membraneto isolate the polyamide active surface while in contact with thesurface modification solution. Both the rubber spacer andrectangular frame have open windows of 77 mm × 26 mm toensure that the modification takes place on the same membranearea which was previously characterized in the FO system. Themodification reaction was carried out on an orbital shaker(Cole-Parmer, IL) at 60 rpm.For surface modification, the polyamide active layer was first
contacted with an aqueous solution of 4 mM 1-ethyl-3-(3-dimethlaminopropyl) carbodiimide (EDC, 98%, ThermoScientific), 10 mM N-hydroxysuccinimide (NHS, 98%, Sigma-Aldrich), and 0.5 M NaCl (ACS grade, J.T. Baker) buffered atpH 5 using 10 mM MES monohydrate (BioXtra, 99%, Sigma-Aldrich) for 1 h. This step converted the native carboxyl groupson the polyamide surface into amine-reactive esters. After thisstep, the polyamide was washed with DI water for three timesand subsequently contacted with an ethylenediamine (BioXtra,Sigma-Aldrich) aqueous solution containing 0.15 M NaCl andbuffered at pH 7.5 with 10 mM HEPES (Sigma-Aldrich) for 30min. In this step, ethylenediamine readily binds to the activelayer by the formation of amide links with the amine-reactiveesters. The membrane surface was then washed three timeswith DI water to remove unreacted ethylenediamine on thesurface. Ethylenediamine concentration was varied from 10 to100 mM to examine its influence on membrane transportproperties and a concentration of 100 mM was finally chosenfor further solute transport experiments.
■ RESULTS AND DISCUSSIONEffect of Membrane Surface Chemistry on Bidirec-
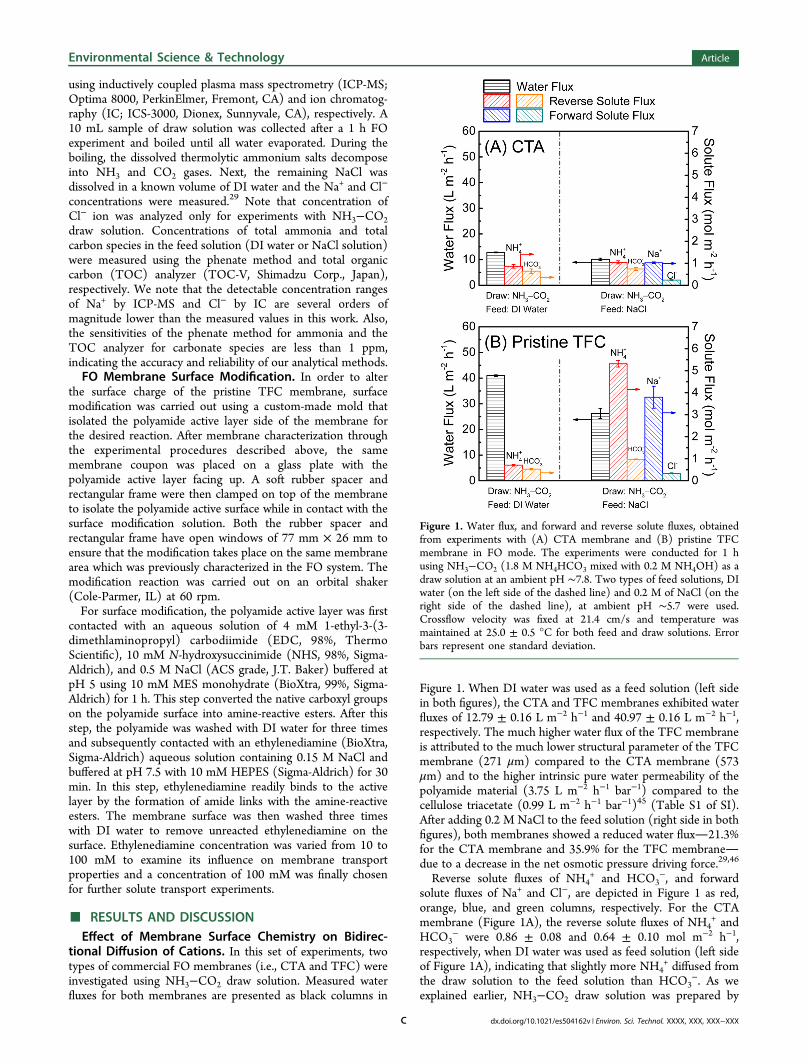
tional Diffusion of Cations. In this set of experiments, twotypes of commercial FO membranes (i.e., CTA and TFC) wereinvestigated using NH3−CO2 draw solution. Measured waterfluxes for both membranes are presented as black columns in
Figure 1. When DI water was used as a feed solution (left sidein both figures), the CTA and TFC membranes exhibited waterfluxes of 12.79 ± 0.16 L m−2 h−1 and 40.97 ± 0.16 L m−2 h−1,respectively. The much higher water flux of the TFC membraneis attributed to the much lower structural parameter of the TFCmembrane (271 μm) compared to the CTA membrane (573μm) and to the higher intrinsic pure water permeability of thepolyamide material (3.75 L m−2 h−1 bar−1) compared to thecellulose triacetate (0.99 L m−2 h−1 bar−1)45 (Table S1 of SI).After adding 0.2 M NaCl to the feed solution (right side in bothfigures), both membranes showed a reduced water flux21.3%for the CTA membrane and 35.9% for the TFC membranedue to a decrease in the net osmotic pressure driving force.29,46
Reverse solute fluxes of NH4+ and HCO3
−, and forwardsolute fluxes of Na+ and Cl−, are depicted in Figure 1 as red,orange, blue, and green columns, respectively. For the CTAmembrane (Figure 1A), the reverse solute fluxes of NH4
+ andHCO3
− were 0.86 ± 0.08 and 0.64 ± 0.10 mol m−2 h−1,respectively, when DI water was used as feed solution (left sideof Figure 1A), indicating that slightly more NH4
+ diffused fromthe draw solution to the feed solution than HCO3
−. As weexplained earlier, NH3−CO2 draw solution was prepared by
Figure 1. Water flux, and forward and reverse solute fluxes, obtainedfrom experiments with (A) CTA membrane and (B) pristine TFCmembrane in FO mode. The experiments were conducted for 1 husing NH3−CO2 (1.8 M NH4HCO3 mixed with 0.2 M NH4OH) as adraw solution at an ambient pH ∼7.8. Two types of feed solutions, DIwater (on the left side of the dashed line) and 0.2 M of NaCl (on theright side of the dashed line), at ambient pH ∼5.7 were used.Crossflow velocity was fixed at 21.4 cm/s and temperature wasmaintained at 25.0 ± 0.5 °C for both feed and draw solutions. Errorbars represent one standard deviation.
Environmental Science & Technology Article
dx.doi.org/10.1021/es504162v | Environ. Sci. Technol. XXXX, XXX, XXX−XXXC

mixing 1.8 M NH4HCO3 and 0.2 M NH4OH. The slightlyhigher reverse NH4
+ diffusion is attributed to the higher totalammonia concentration (2.0 M) than bicarbonate species (1.8M) in the draw solution. When 0.2 M NaCl was used as a feedsolution, we observed slight increases in reverse diffusion ofboth NH4
+ (1.04 ± 0.07 mol m−2 h−1) and HCO3− (0.74 ±
0.07 mol m−2 h−1) compared to those obtained fromexperiments with DI water feed solution. We note that ourdata are comparable to previous studies using the same type ofmembrane.47
Concomitantly, for the CTA membrane, the forward fluxesof Na+ and Cl− in the experiment with 0.2 M NaCl feedsolution were 1.02 ± 0.02 and 0.24 ± 0.02 mol m−2 h−1,respectively. We attribute this observation to the higher reversediffusion of NH4
+, which is balanced by the forward diffusion ofcations from the feed solution to the draw solution. In our case,two cations (i.e., Na+ and H+) are present in the feed solution.However, due to the much higher Na+ concentration than H+
in the feed solution, transport of Na+ plays a dominant role inbalancing the solution charge, which is manifested as enhancedNa+ transport.Reverse and forward solute fluxes obtained from FO
experiments with the TFC membrane are presented in Figure1B. Although the TFC membrane exhibited a much higherwater flux than the CTA membrane, the reverse solute fluxes ofNH4
+ and HCO3− were comparable for both the CTA and
TFC membranes when DI water was used as a feed solution.However, a substantial increase in reverse NH4
+flux was
observed with the TFC membrane when a 0.2 M NaCl feedsolution was employed (right side of Figure 1B). We alsoobserved a significantly higher forward Na+ flux compared tothat obtained with the CTA membrane. Interestingly, therewere only slight changes in the rates of anion diffusion,regardless of the types of feed solution and membranes used.The results of bidirectional ion diffusion with the TFC
membrane indicate that with NaCl feed solution the cationsand anions diffused through the membrane with an unequal-molar proportion. In order to evaluate the effect of feedsolution on draw solute reverse diffusion in our study, we define
an enhanced diffusion ratio (EDR) of ions (i.e., NH4+ and
HCO3−):
=EDRRSFRSFion
NaCl
DI (3)
where RSFDI and RSFNaCl are the reverse solute fluxes of aspecific ion when DI water and 0.2 M NaCl were used as feedsolutions, respectively. The CTA membrane exhibited EDRNH4
+
and EDRHCO3− of 1.22 ± 0.06 and 1.17 ± 0.09, respectively,
indicating that NaCl feed solution has a slight influence on thetransport of NH4
+ and HCO3− for the CTA membrane. On the
other hand, EDRNH4+ and EDRHCO3
− were 7.49 ± 0.24 and 1.85± 0.24 for the TFC membrane, implying a dramatic increase inNH4
+ diffusion, but a relatively mild enhancement in HCO3−.
We note that previous publications38,40 also reported a similarphenomenon with commercial TFC membranes underconditions similar to those used in this study. Taken together,these results confirm that the presence of Na+ in the feedsolution could significantly enhance the reverse transport ofNH4
+ for TFC polyamide membranes.The most important difference between the CTA and TFC
membranes relevant to our study is the active layer surfacechemistry. The CTA membrane is made of cellulose triacetate,which contains acetyl and hydroxyl functional groups. Sincethese functional groups do not dissociate under the pH rangeinvestigated, the membrane active layer does not possess fixedsurface charge.48,49 On the other hand, the TFC membraneconsists of a thin polyamide active layer, which is formed viainterfacial polymerization between amine containing monomerand acyl chloride on top of a microporous support. After thefabrication, unreacted acyl chloride groups hydrolyze intocarboxyl groups, which dominate the surface functional groupsof the TFC membrane.48,50 Therefore, the enhanced bidirec-tional diffusion of NH4
+ and Na+ with the TFC membranemight be attributed to the presence of carboxyl groups.
Role of Membrane Surface Charge in BidirectionalDiffusion of Cations. To further unravel the mechanism forthe enhanced bidirectional diffusion of cations with the TFCmembrane, we performed a set of experiments using 2.0 M
Figure 2. Effect of feed solution pH on bidirectional diffusion of cations with pristine TFC membrane. Water flux, and forward and reverse solutefluxes, were obtained from 1 h FO experiments using 2 M NH4Cl (at ambient solution pH 4.5) as a draw solution and DI water or 0.2 M NaCl attwo different solution pH (pH 6.0 and 3.0) as feed solutions. All experiments were conducted with a crossflow velocity of 21.4 cm/s and a constanttemperature of 25.0 ± 0.5 °C. Error bars represent one standard deviation.
Environmental Science & Technology Article
dx.doi.org/10.1021/es504162v | Environ. Sci. Technol. XXXX, XXX, XXX−XXXD
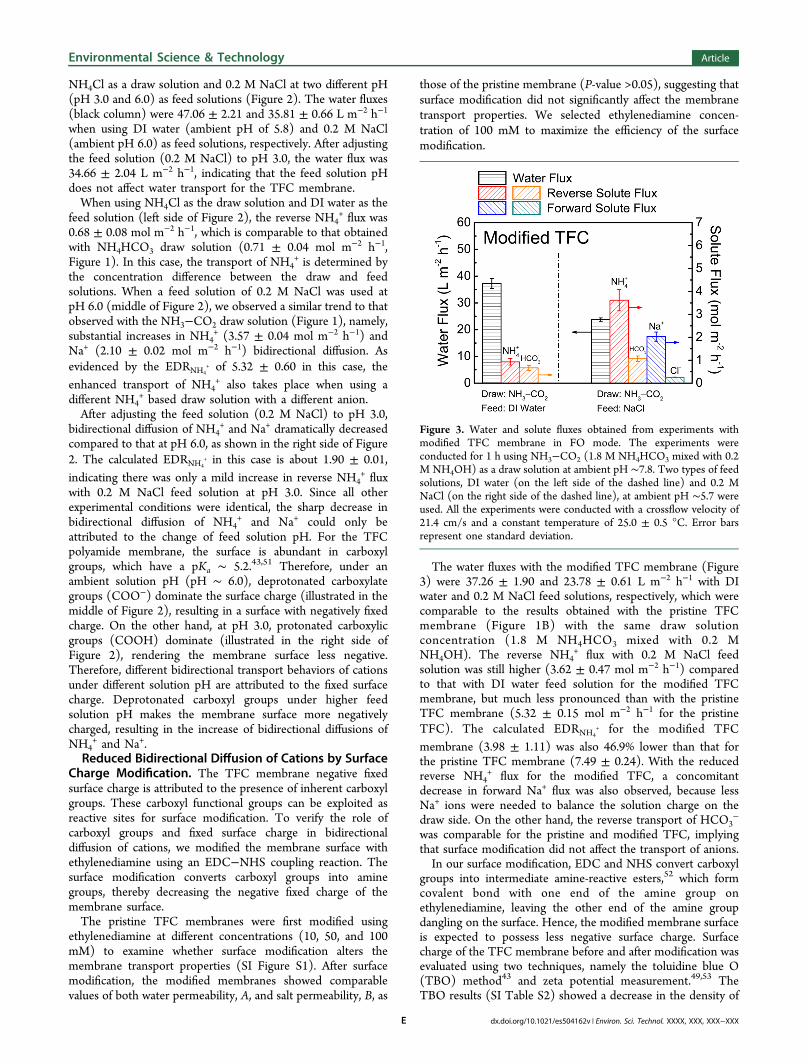
NH4Cl as a draw solution and 0.2 M NaCl at two different pH(pH 3.0 and 6.0) as feed solutions (Figure 2). The water fluxes(black column) were 47.06 ± 2.21 and 35.81 ± 0.66 L m−2 h−1
when using DI water (ambient pH of 5.8) and 0.2 M NaCl(ambient pH 6.0) as feed solutions, respectively. After adjustingthe feed solution (0.2 M NaCl) to pH 3.0, the water flux was34.66 ± 2.04 L m−2 h−1, indicating that the feed solution pHdoes not affect water transport for the TFC membrane.When using NH4Cl as the draw solution and DI water as the
feed solution (left side of Figure 2), the reverse NH4+flux was
0.68 ± 0.08 mol m−2 h−1, which is comparable to that obtainedwith NH4HCO3 draw solution (0.71 ± 0.04 mol m−2 h−1,Figure 1). In this case, the transport of NH4
+ is determined bythe concentration difference between the draw and feedsolutions. When a feed solution of 0.2 M NaCl was used atpH 6.0 (middle of Figure 2), we observed a similar trend to thatobserved with the NH3−CO2 draw solution (Figure 1), namely,substantial increases in NH4
+ (3.57 ± 0.04 mol m−2 h−1) andNa+ (2.10 ± 0.02 mol m−2 h−1) bidirectional diffusion. Asevidenced by the EDRNH4
+ of 5.32 ± 0.60 in this case, theenhanced transport of NH4
+ also takes place when using adifferent NH4
+ based draw solution with a different anion.After adjusting the feed solution (0.2 M NaCl) to pH 3.0,
bidirectional diffusion of NH4+ and Na+ dramatically decreased
compared to that at pH 6.0, as shown in the right side of Figure2. The calculated EDRNH4
+ in this case is about 1.90 ± 0.01,indicating there was only a mild increase in reverse NH4
+flux
with 0.2 M NaCl feed solution at pH 3.0. Since all otherexperimental conditions were identical, the sharp decrease inbidirectional diffusion of NH4
+ and Na+ could only beattributed to the change of feed solution pH. For the TFCpolyamide membrane, the surface is abundant in carboxylgroups, which have a pKa ∼ 5.2.43,51 Therefore, under anambient solution pH (pH ∼ 6.0), deprotonated carboxylategroups (COO−) dominate the surface charge (illustrated in themiddle of Figure 2), resulting in a surface with negatively fixedcharge. On the other hand, at pH 3.0, protonated carboxylicgroups (COOH) dominate (illustrated in the right side ofFigure 2), rendering the membrane surface less negative.Therefore, different bidirectional transport behaviors of cationsunder different solution pH are attributed to the fixed surfacecharge. Deprotonated carboxyl groups under higher feedsolution pH makes the membrane surface more negativelycharged, resulting in the increase of bidirectional diffusions ofNH4
+ and Na+.Reduced Bidirectional Diffusion of Cations by Surface
Charge Modification. The TFC membrane negative fixedsurface charge is attributed to the presence of inherent carboxylgroups. These carboxyl functional groups can be exploited asreactive sites for surface modification. To verify the role ofcarboxyl groups and fixed surface charge in bidirectionaldiffusion of cations, we modified the membrane surface withethylenediamine using an EDC−NHS coupling reaction. Thesurface modification converts carboxyl groups into aminegroups, thereby decreasing the negative fixed charge of themembrane surface.The pristine TFC membranes were first modified using
ethylenediamine at different concentrations (10, 50, and 100mM) to examine whether surface modification alters themembrane transport properties (SI Figure S1). After surfacemodification, the modified membranes showed comparablevalues of both water permeability, A, and salt permeability, B, as
those of the pristine membrane (P-value >0.05), suggesting thatsurface modification did not significantly affect the membranetransport properties. We selected ethylenediamine concen-tration of 100 mM to maximize the efficiency of the surfacemodification.
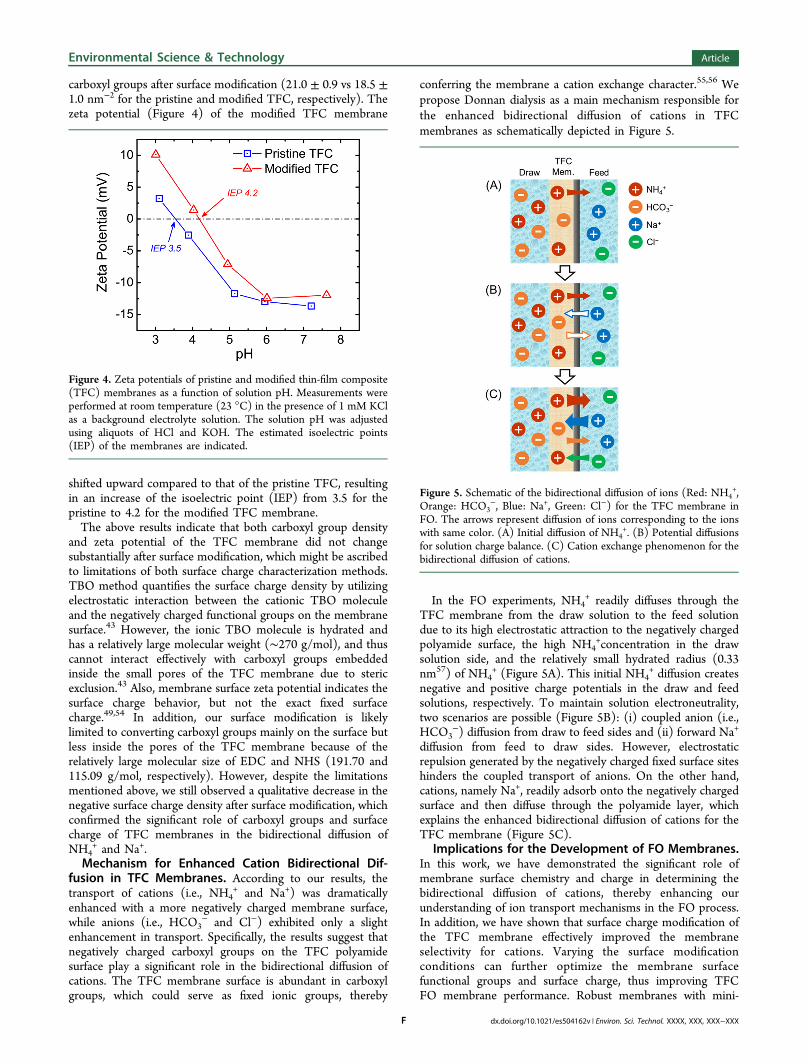
The water fluxes with the modified TFC membrane (Figure3) were 37.26 ± 1.90 and 23.78 ± 0.61 L m−2 h−1 with DIwater and 0.2 M NaCl feed solutions, respectively, which werecomparable to the results obtained with the pristine TFCmembrane (Figure 1B) with the same draw solutionconcentration (1.8 M NH4HCO3 mixed with 0.2 MNH4OH). The reverse NH4
+flux with 0.2 M NaCl feed
solution was still higher (3.62 ± 0.47 mol m−2 h−1) comparedto that with DI water feed solution for the modified TFCmembrane, but much less pronounced than with the pristineTFC membrane (5.32 ± 0.15 mol m−2 h−1 for the pristineTFC). The calculated EDRNH4
+ for the modified TFCmembrane (3.98 ± 1.11) was also 46.9% lower than that forthe pristine TFC membrane (7.49 ± 0.24). With the reducedreverse NH4
+flux for the modified TFC, a concomitant
decrease in forward Na+ flux was also observed, because lessNa+ ions were needed to balance the solution charge on thedraw side. On the other hand, the reverse transport of HCO3
−
was comparable for the pristine and modified TFC, implyingthat surface modification did not affect the transport of anions.In our surface modification, EDC and NHS convert carboxyl
groups into intermediate amine-reactive esters,52 which formcovalent bond with one end of the amine group onethylenediamine, leaving the other end of the amine groupdangling on the surface. Hence, the modified membrane surfaceis expected to possess less negative surface charge. Surfacecharge of the TFC membrane before and after modification wasevaluated using two techniques, namely the toluidine blue O(TBO) method43 and zeta potential measurement.49,53 TheTBO results (SI Table S2) showed a decrease in the density of
Figure 3. Water and solute fluxes obtained from experiments withmodified TFC membrane in FO mode. The experiments wereconducted for 1 h using NH3−CO2 (1.8 M NH4HCO3 mixed with 0.2M NH4OH) as a draw solution at ambient pH ∼7.8. Two types of feedsolutions, DI water (on the left side of the dashed line) and 0.2 MNaCl (on the right side of the dashed line), at ambient pH ∼5.7 wereused. All the experiments were conducted with a crossflow velocity of21.4 cm/s and a constant temperature of 25.0 ± 0.5 °C. Error barsrepresent one standard deviation.
Environmental Science & Technology Article
dx.doi.org/10.1021/es504162v | Environ. Sci. Technol. XXXX, XXX, XXX−XXXE
carboxyl groups after surface modification (21.0 ± 0.9 vs 18.5 ±1.0 nm−2 for the pristine and modified TFC, respectively). Thezeta potential (Figure 4) of the modified TFC membrane
shifted upward compared to that of the pristine TFC, resultingin an increase of the isoelectric point (IEP) from 3.5 for thepristine to 4.2 for the modified TFC membrane.The above results indicate that both carboxyl group density
and zeta potential of the TFC membrane did not changesubstantially after surface modification, which might be ascribedto limitations of both surface charge characterization methods.TBO method quantifies the surface charge density by utilizingelectrostatic interaction between the cationic TBO moleculeand the negatively charged functional groups on the membranesurface.43 However, the ionic TBO molecule is hydrated andhas a relatively large molecular weight (∼270 g/mol), and thuscannot interact effectively with carboxyl groups embeddedinside the small pores of the TFC membrane due to stericexclusion.43 Also, membrane surface zeta potential indicates thesurface charge behavior, but not the exact fixed surfacecharge.49,54 In addition, our surface modification is likelylimited to converting carboxyl groups mainly on the surface butless inside the pores of the TFC membrane because of therelatively large molecular size of EDC and NHS (191.70 and115.09 g/mol, respectively). However, despite the limitationsmentioned above, we still observed a qualitative decrease in thenegative surface charge density after surface modification, whichconfirmed the significant role of carboxyl groups and surfacecharge of TFC membranes in the bidirectional diffusion ofNH4
+ and Na+.Mechanism for Enhanced Cation Bidirectional Dif-
fusion in TFC Membranes. According to our results, thetransport of cations (i.e., NH4
+ and Na+) was dramaticallyenhanced with a more negatively charged membrane surface,while anions (i.e., HCO3
− and Cl−) exhibited only a slightenhancement in transport. Specifically, the results suggest thatnegatively charged carboxyl groups on the TFC polyamidesurface play a significant role in the bidirectional diffusion ofcations. The TFC membrane surface is abundant in carboxylgroups, which could serve as fixed ionic groups, thereby
conferring the membrane a cation exchange character.55,56 Wepropose Donnan dialysis as a main mechanism responsible forthe enhanced bidirectional diffusion of cations in TFCmembranes as schematically depicted in Figure 5.
In the FO experiments, NH4+ readily diffuses through the
TFC membrane from the draw solution to the feed solutiondue to its high electrostatic attraction to the negatively chargedpolyamide surface, the high NH4
+concentration in the drawsolution side, and the relatively small hydrated radius (0.33nm57) of NH4
+ (Figure 5A). This initial NH4+ diffusion creates
negative and positive charge potentials in the draw and feedsolutions, respectively. To maintain solution electroneutrality,two scenarios are possible (Figure 5B): (i) coupled anion (i.e.,HCO3
−) diffusion from draw to feed sides and (ii) forward Na+
diffusion from feed to draw sides. However, electrostaticrepulsion generated by the negatively charged fixed surface siteshinders the coupled transport of anions. On the other hand,cations, namely Na+, readily adsorb onto the negatively chargedsurface and then diffuse through the polyamide layer, whichexplains the enhanced bidirectional diffusion of cations for theTFC membrane (Figure 5C).
Implications for the Development of FO Membranes.In this work, we have demonstrated the significant role ofmembrane surface chemistry and charge in determining thebidirectional diffusion of cations, thereby enhancing ourunderstanding of ion transport mechanisms in the FO process.In addition, we have shown that surface charge modification ofthe TFC membrane effectively improved the membraneselectivity for cations. Varying the surface modificationconditions can further optimize the membrane surfacefunctional groups and surface charge, thus improving TFCFO membrane performance. Robust membranes with mini-
Figure 4. Zeta potentials of pristine and modified thin-film composite(TFC) membranes as a function of solution pH. Measurements wereperformed at room temperature (23 °C) in the presence of 1 mM KClas a background electrolyte solution. The solution pH was adjustedusing aliquots of HCl and KOH. The estimated isoelectric points(IEP) of the membranes are indicated.
Figure 5. Schematic of the bidirectional diffusion of ions (Red: NH4+,
Orange: HCO3−, Blue: Na+, Green: Cl−) for the TFC membrane in
FO. The arrows represent diffusion of ions corresponding to the ionswith same color. (A) Initial diffusion of NH4
+. (B) Potential diffusionsfor solution charge balance. (C) Cation exchange phenomenon for thebidirectional diffusion of cations.
Environmental Science & Technology Article
dx.doi.org/10.1021/es504162v | Environ. Sci. Technol. XXXX, XXX, XXX−XXXF

mized reverse and forward transport of ions are critical for theefficient application of the FO process in treating a variety ofsource waters. Our results further underscore the uniquerequirements for TFC membranes for the FO processcompared to TFC membranes for pressure-driven membraneprocesses (i.e., RO and nanofiltration).
■ ASSOCIATED CONTENT*S Supporting InformationDetails on the membrane surface characterization (zetapotential and TBO method) (S1); effect of ethylenediamineconcentration on membrane transport properties (Figure S1);transport properties of pristine membranes (Table S1);carboxyl group density of TFC membranes (Table S2). Thismaterial is available free of charge via the Internet at http://pubs.acs.org/.
■ AUTHOR INFORMATIONCorresponding Authors*(J.M.) Phone: +86 451 86283010; fax: +86 451 86283010; e-mail: [email protected].*(M.E.) Phone: +1 203 432 2789; fax: +1 203 432 4387; e-mail: [email protected] authors declare no competing financial interest.
■ ACKNOWLEDGMENTSWe acknowledge the support received from the NationalScience Foundation under Award Number CBET 1232619 andthe Funds for Creative Research Groups of China (Grant No.51121062). We thank Oasys Water Inc. for providing thepristine TFC membranes and Dr. Helmut Ernstberger (YaleUniversity) for assistance with use of the ion chromatographyand inductively coupled plasma mass spectrometry. We alsoacknowledge a graduate fellowship (to X.L.) made possible bythe China Scholarship Council (CSC), and the use of facilitiessupported by YINQE and NSF MRSEC DMR 1119826.
■ REFERENCES(1) Shannon, M. A.; Bohn, P. W.; Elimelech, M.; Georgiadis, J. G.;Marinas, B. J.; Mayes, A. M. Science and technology for waterpurification in the coming decades. Nature 2008, 452 (7185), 301−310.(2) Elimelech, M., Special Paper: The global challenge for adequateand safe water. J. Water Supply: Res. Technol.Aqua 2006, 55, (1).(3) Sima, L. C.; Elimelech, M. More than a drop in the bucket:Decentralized Membrane-based drinking water refill stations inSoutheast Asia. Environ. Sci. Technol. 2013, 47 (14), 7580−7588.(4) Mi, B. X.; Elimelech, M. Organic fouling of forward osmosismembranes: Fouling reversibility and cleaning without chemicalreagents. J. Membr. Sci. 2010, 348 (1−2), 337−345.(5) McGinnis, R. L.; Hancock, N. T.; Nowosielski-Slepowron, M. S.;McGurgan, G. D. Pilot demonstration of the NH3/CO2 forwardosmosis desalination process on high salinity brines. Desalination 2013,312, 67−74.(6) Shaffer, D. L.; Arias Chavez, L. H.; Ben-Sasson, M.; Romero-Vargas Castrillon, S.; Yip, N. Y.; Elimelech, M. Desalination and reuseof high-salinity shale gas produced water: Drivers, technologies, andfuture directions. Environ. Sci. Technol. 2013, 47 (17), 9569−9583.(7) Hickenbottom, K. L.; Hancock, N. T.; Hutchings, N. R.;Appleton, E. W.; Beaudry, E. G.; Xu, P.; Cath, T. Y. Forward osmosistreatment of drilling mud and fracturing wastewater from oil and gasoperations. Desalination 2013, 312, 60−66.(8) Hutchings, N. R.; Appleton, E. W.; McGinnis, R. A. Making highquality frac water out of oilfield waste. In The International Society of
Petrolume Engineers 2010 Annual Technical Conference and Exhibition,Florence, Italy, 2010.(9) Li, X.-M.; Zhao, B.; Wang, Z.; Xie, M.; Song, J.; Nghiem, L.; He,T.; Yang, C.; Li, C.; Chen, G. Water reclamation from shale gas drillingflow-back fluid using a novel forward osmosis-vacuum membranedistillation hybrid system. Water Sci. Technol. 2014, 69, 1036−1044.(10) Achilli, A.; Cath, T. Y.; Marchand, E. A.; Childress, A. E. Theforward osmosis membrane bioreactor: A low fouling alternative toMBR processes. Desalination 2009, 239 (1−3), 10−21.(11) Cornelissen, E. R.; Harmsen, D.; Beerendonk, E. F.; Qin, J. J.;Oo, H.; de Korte, K. F.; Kappelhof, J. W. M. N. The innovativeOsmotic Membrane Bioreactor (OMBR) for reuse of wastewater.Water Sci. Technol. 2011, 63 (8), 1557−1565.(12) Holloway, R. W.; Regnery, J.; Nghiem, L. D.; Cath, T. Y.Removal of trace organic chemicals and performance of a novel hybridultrafiltration-osmotic membrane bioreactor. Environ. Sci. Technol.2014, 48 (18), 10859−10868.(13) Nguyen, N. C.; Chen, S.-S.; Yang, H.-Y.; Hau, N. T. Applicationof forward osmosis on dewatering of high nutrient sludge. Bioresour.Technol. 2013, 132, 224−229.(14) Zhang, J.; Loong, W. L. C.; Chou, S.; Tang, C.; Wang, R.; Fane,A. G. Membrane biofouling and scaling in forward osmosis membranebioreactor. J. Membr. Sci. 2012, 403−404, 8−14.(15) Chen, L.; Gu, Y.; Cao, C.; Zhang, J.; Ng, J.-W.; Tang, C.Performance of a submerged anaerobic membrane bioreactor withforward osmosis membrane for low-strength wastewater treatment.Water Res. 2014, 50, 114−123.(16) Holloway, R. W.; Wait, A. S.; Fernandes da Silva, A.; Herron, J.;Schutter, M. D.; Lampi, K.; Cath, T. Y. Long-term pilot scaleinvestigation of novel hybrid ultrafiltration-osmotic membranebioreactors. Desalination 2014, http://dx.doi.org/10.1016/j.desal.2014.05.040.(17) Cath, T. Y.; Hancock, N. T.; Lundin, C. D.; Hoppe-Jones, C.;Drewes, J. E. A multi-barrier osmotic dilution process for simultaneousdesalination and purification of impaired water. J. Membr. Sci. 2010,362 (1−2), 417−426.(18) Hancock, N. T.; Black, N. D.; Cath, T. Y. A comparative lifecycle assessment of hybrid osmotic dilution desalination andestablished seawater desalination and wastewater reclamationprocesses. Water Res. 2012, 46 (4), 1145−1154.(19) Hancock, N. T.; Xu, P.; Roby, M. J.; Gomez, J. D.; Cath, T. Y.Towards direct potable reuse with forward osmosis: Technicalassessment of long-term process performance at the pilot scale. J.Membr. Sci. 2013, 445, 34−46.(20) Shaffer, D. L.; Werber, J. R.; Jaramillo, H.; Lin, S.; Elimelech, M.,Forward osmosis: Where are we now? Desalination 2014, http://dx.doi.org/10.1016/j.desal.2014.10.031.(21) Cath, T. Y.; Childress, A. E.; Elimelech, M. Forward osmosis:Principles, applications, and recent developments. J. Membr. Sci. 2006,281 (1−2), 70−87.(22) Yip, N. Y.; Tiraferri, A.; Phillip, W. A.; Schiffman, J. D.;Elimelech, M. High performance thin-film composite forward osmosismembrane. Environ. Sci. Technol. 2010, 44 (10), 3812−3818.(23) Wei, J.; Qiu, C. Q.; Tang, C. Y. Y.; Wang, R.; Fane, A. G.Synthesis and characterization of flat-sheet thin film compositeforward osmosis membranes. J. Membr. Sci. 2011, 372 (1−2), 292−302.(24) Han, G.; Chung, T. S.; Toriida, M.; Tamai, S. Thin-filmcomposite forward osmosis membranes with novel hydrophilicsupports for desalination. J. Membr. Sci. 2012, 423, 543−555.(25) Zhao, S. F.; Zou, L.; Tang, C. Y. Y.; Mulcahy, D. Recentdevelopments in forward osmosis: Opportunities and challenges. J.Membr. Sci. 2012, 396, 1−21.(26) Lu, X.; Romero-Vargas Castrillon, S.; Shaffer, D. L.; Ma, J.;Elimelech, M. In situ surface chemical modification of thin-filmcomposite forward osmosis membranes for enhanced organic foulingresistance. Environ. Sci. Technol. 2013, 47 (21), 12219−12228.(27) Romero-Vargas Castrillon, S.; Lu, X.; Shaffer, D. L.; Elimelech,M. Amine enrichment and poly(ethylene glycol) (PEG) surface
Environmental Science & Technology Article
dx.doi.org/10.1021/es504162v | Environ. Sci. Technol. XXXX, XXX, XXX−XXXG

modification of thin-film composite forward osmosis membranes fororganic fouling control. J. Membr. Sci. 2014, 450, 331−339.(28) Chekli, L.; Phuntsho, S.; Shon, H. K.; Vigneswaran, S.;Kandasamy, J.; Chanan, A. A review of draw solutes in forwardosmosis process and their use in modern applications. Desalin. WaterTreat. 2012, 43 (1−3), 167−184.(29) McCutcheon, J. R.; McGinnis, R. L.; Elimelech, M. Desalinationby ammonia-carbon dioxide forward osmosis: Influence of draw andfeed solution concentrations on process performance. J. Membr. Sci.2006, 278 (1−2), 114−123.(30) McGinnis, R. L.; McCutcheon, J. R.; Elimelech, M. A novelammonia-carbon dioxide osmotic heat engine for power generation. J.Membr. Sci. 2007, 305 (1−2), 13−19.(31) Achilli, A.; Cath, T. Y.; Childress, A. E. Selection of inorganic-based draw solutions for forward osmosis applications. J. Membr. Sci.2010, 364 (1−2), 233−241.(32) Lee, S.; Boo, C.; Elimelech, M.; Hong, S. Comparison of foulingbehavior in forward osmosis (FO) and reverse osmosis (RO). J.Membr. Sci. 2010, 365 (1−2), 34−39.(33) Jin, X.; Tang, C. Y.; Gu, Y.; She, Q.; Qi, S. Boric acid permeationin forward osmosis membrane processes: Modeling, experiments, andimplications. Environ. Sci. Technol. 2011, 45 (6), 2323−30.(34) Xie, M.; Nghiem, L. D.; Price, W. E.; Elimelech, M. Comparisonof the removal of hydrophobic trace organic contaminants by forwardosmosis and reverse osmosis. Water Res. 2012, 46 (8), 2683−2692.(35) Phillip, W. A.; Yong, J. S.; Elimelech, M. Reverse draw solutepermeation in forward osmosis: Modeling and experiments. Environ.Sci. Technol. 2010, 44 (13), 5170−5176.(36) Hancock, N. T.; Cath, T. Y. Solute coupled diffusion inosmotically driven membrane processes. Environ. Sci. Technol. 2009, 43(17), 6769−6775.(37) Hancock, N. T.; Phillip, W. A.; Elimelech, M.; Cath, T. Y.Bidirectional permeation of electrolytes in osmotically drivenmembrane processes. Environ. Sci. Technol. 2011, 45 (24), 10642−10651.(38) Coday, B. D.; Heil, D. M.; Xu, P.; Cath, T. Y. Effects oftransmembrane hydraulic pressure on performance of forward osmosismembranes. Environ. Sci. Technol. 2013, 47 (5), 2386−2393.(39) Irvine, G. J.; Rajesh, S.; Georgiadis, M.; Phillip, W. A. Ionselective permeation through cellulose acetate membranes in forwardosmosis. Environ. Sci. Technol. 2013, 47 (23), 13745−13753.(40) Arena, J. T.; Manickam, S. S.; Reimund, K. K.; Freeman, B. D.;McCutcheon, J. R. Solute and water transport in forward osmosisusing polydopamine modified thin film composite membranes.Desalination 2014, 343, 8−16.(41) Phuntsho, S.; Shon, H. K.; Hong, S.; Lee, S.; Vigneswaran, S. Anovel low energy fertilizer driven forward osmosis desalination fordirect fertigation: Evaluating the performance of fertilizer drawsolutions. J. Membr. Sci. 2011, 375 (1−2), 172−181.(42) Boo, C.; Lee, S.; Elimelech, M.; Meng, Z. Y.; Hong, S. Colloidalfouling in forward osmosis: Role of reverse salt diffusion. J. Membr. Sci.2012, 390, 277−284.(43) Tiraferri, A.; Elimelech, M. Direct quantification of negativelycharged functional groups on membrane surfaces. J. Membr. Sci. 2012,389, 499−508.(44) Tiraferri, A.; Yip, N. Y.; Straub, A. P.; Romero-Vargas Castrillon,S.; Elimelech, M. A method for the simultaneous determination oftransport and structural parameters of forward osmosis membranes. J.Membr. Sci. 2013, 444, 523−538.(45) Mulder, M. Basic Principles of Membrane Technology, 2nd ed.;Kluwer Academic Pulisher: The Netherlands, 1996.(46) McCutcheon, J. R.; Elimelech, M. Influence of concentrativeand dilutive internal concentration polarization on flux behavior inforward osmosis. J. Membr. Sci. 2006, 284 (1−2), 237−247.(47) Boo, C.; Khalil, Y. F.; Elimelech, M. Performance evaluation oftrimethylamine−carbon dioxide thermolytic draw solution forengineered osmosis. J. Membr. Sci. 2015, 473, 302−309.
(48) Elimelech, M.; Chen, W. H.; Waypa, J. J. Measuring the zeta(electrokinetic) potential of reverse-osmosis membranes by astreaming potential analyzer. Desalination 1994, 95 (3), 269−286.(49) Childress, A. E.; Elimelech, M. Effect of solution chemistry onthe surface charge of polymeric reverse osmosis and nanofiltrationmembranes. J. Membr. Sci. 1996, 119 (2), 253−268.(50) Petersen, R. J. Composite reverse-osmosis and nanofiltrationmembranes. J. Membr. Sci. 1993, 83 (1), 81−150.(51) Coronell, O.; Marinas, B. J.; Zhang, X. J.; Cahill, D. G.Quantification of functional groups and modeling of their ionizationbehavior in the active layer of FT30 reverse osmosis membrane.Environ. Sci. Technol. 2008, 42 (14), 5260−5266.(52) Staros, J. V.; Wright, R. W.; Swingle, D. M. Enhancement by N-hydroxysulfosuccinimide of water-soluble carbodiimide-mediatedcoupling reactions. Anal. Biochem. 1986, 156 (1), 220−222.(53) Walker, S. L.; Bhattacharjee, S.; Hoek, E. M. V.; Elimelech, M. Anovel asymmetric clamping cell for measuring streaming potential offlat surfaces. Langmuir 2002, 18 (6), 2193−2198.(54) Elimelech, M.; Gregory, J.; Jia, X.; Williams, R. A. ParticleDeposition and Aggregation: Measurement, Modelling, and Simulation;Butterworth-Heinemann Ltd: Oxford, England, 1995.(55) Nasef, M. M.; Hegazy, E. S. A. Preparation and applications ofion exchange membranes by radiation-induced graft copolymerizationof polar monomers onto non-polar films. Prog. Polym. Sci. 2004, 29(6), 499−561.(56) Xu, T. W. Ion exchange membranes: State of their developmentand perspective. J. Membr. Sci. 2005, 263 (1−2), 1−29.(57) Nightingale, E. R. Phenomenological theory of ion solvationEffective radii of hydrated ions. J. Phys. Chem-Us 1959, 63 (9), 1381−1387.
Environmental Science & Technology Article
dx.doi.org/10.1021/es504162v | Environ. Sci. Technol. XXXX, XXX, XXX−XXXH




















