
BBAMEM-19-17R1 Title: Molecular basis f - Digital CSIC
89
Elsevier Editorial System(tm) for BBA - Biomembranes Manuscript Draft Manuscript Number: BBAMEM-19-17R1 Title: Molecular basis for the protective effects of low-density lipoprotein receptor-related protein 1 (LRP1)-derived peptides against LDL aggregation Article Type: Regular Paper Keywords: Lipoprotein aggregation; LRP1-derived peptides; ApoB-100; SMase, PLA2, atherosclerosis Corresponding Author: Dr. Concepción Vicenta Llorente, Corresponding Author's Institution: First Author: Aleyda Benitez Amaro, PhD student Order of Authors: Aleyda Benitez Amaro, PhD student; Chiara Pallara, PhD; Laura Nasarre; Andrea Rivas Urbina, PhD student; Sonia Benitez, PhD; Angela Vea, PhD student; Olga Bornachea, PhD; David de Gonzalo Calvo, PhD; Gabriel Serra Mir, PhD; Sandra Villegas, PhD; Roger Prades, PhD; Jose Luis Sanchez Quesada, Phd; Cristina Chiva, PhD; Eduard Sabido, PhD; Teresa Tarragó, PhD; Concepción Vicenta Llorente Abstract: Aggregated LDL is the first ligand reported to interact with the cluster II CR9 domain of low-density lipoprotein receptor-related protein 1 (LRP1). In particular, the C-terminal half of domain CR9, comprising the region Gly1127-Cys1140 exclusively recognizes aggregated LDL and it is crucial for aggregated LDL binding. Our aim was to study the effect of the sequence Gly1127-Cys1140 (named peptide LP3 and its retro-enantio version, named peptide DP3) on the structural characteristics of sphingomyelinase- (SMase) and phospholipase 2 (PLA2)- modified LDL particles. Turbidimetry, gel filtration chromatography (GFC) and tomography electronic microscopy (TEM) analysis showed that LP3 and DP3 peptides strongly inhibited SMase- and PLA2-induced LDL aggregation. Nondenaturing polyacrylamide gradient gel electrophoresis (GGE), agarose gel electrophoresis and high-performance thin-layer chromatography (HPTLC) indicated that LP3 and DP3 prevented SMase-induced alterations in LDL particle size, electric charge and phospholipid content, respectively, but not those induced by PLA2. Western blot analysis showed that LP3 and DP3 counteracted changes in ApoB-100 conformation induced by the two enzymes. LDL proteomics (LDL trypsin digestion followed by mass spectroscopy) and computational modeling methods evidenced that peptides protect ApoB-100 conformation due to their electrostatic interactions with a basic region of ApoB-100. These results demonstrate that LRP1- derived peptides are protective against LDL aggregation, even in conditions of extreme lipolysis, through their capacity to bind to ApoB- 100 regions critical to preserve ApoB-100 conformation. These results suggests that these LRP1(CR9) derived peptides could be promising tools to prevent LDL aggregation induced by the main proteolytic enzymes acting in the arterial intima. Response to Reviewers: ANSWERS TO REVIEWERS
-
Upload
khangminh22 -
Category
Documents
-
view
2 -
download
0
Transcript of BBAMEM-19-17R1 Title: Molecular basis f - Digital CSIC

Elsevier Editorial System(tm) for BBA -
Biomembranes
Manuscript Draft
Manuscript Number: BBAMEM-19-17R1
Title: Molecular basis for the protective effects of low-density
lipoprotein receptor-related protein 1 (LRP1)-derived peptides against
LDL aggregation
Article Type: Regular Paper
Keywords: Lipoprotein aggregation; LRP1-derived peptides; ApoB-100;
SMase, PLA2, atherosclerosis
Corresponding Author: Dr. Concepción Vicenta Llorente,
Corresponding Author's Institution:
First Author: Aleyda Benitez Amaro, PhD student
Order of Authors: Aleyda Benitez Amaro, PhD student; Chiara Pallara, PhD;
Laura Nasarre; Andrea Rivas Urbina, PhD student; Sonia Benitez, PhD;
Angela Vea, PhD student; Olga Bornachea, PhD; David de Gonzalo Calvo,
PhD; Gabriel Serra Mir, PhD; Sandra Villegas, PhD; Roger Prades, PhD;
Jose Luis Sanchez Quesada, Phd; Cristina Chiva, PhD; Eduard Sabido, PhD;
Teresa Tarragó, PhD; Concepción Vicenta Llorente
Abstract: Aggregated LDL is the first ligand reported to interact with
the cluster II CR9 domain of low-density lipoprotein receptor-related
protein 1 (LRP1). In particular, the C-terminal half of domain CR9,
comprising the region Gly1127-Cys1140 exclusively recognizes aggregated
LDL and it is crucial for aggregated LDL binding. Our aim was to study
the effect of the sequence Gly1127-Cys1140 (named peptide LP3 and its
retro-enantio version, named peptide DP3) on the structural
characteristics of sphingomyelinase- (SMase) and phospholipase 2 (PLA2)-
modified LDL particles. Turbidimetry, gel filtration chromatography (GFC)
and tomography electronic microscopy (TEM) analysis showed that LP3 and
DP3 peptides strongly inhibited SMase- and PLA2-induced LDL aggregation.
Nondenaturing polyacrylamide gradient gel electrophoresis (GGE), agarose
gel electrophoresis and high-performance thin-layer chromatography
(HPTLC) indicated that LP3 and DP3 prevented SMase-induced alterations in
LDL particle size, electric charge and phospholipid content,
respectively, but not those induced by PLA2. Western blot analysis showed
that LP3 and DP3 counteracted changes in ApoB-100 conformation induced by
the two enzymes. LDL proteomics (LDL trypsin digestion followed by mass
spectroscopy) and computational modeling methods evidenced that peptides
protect ApoB-100 conformation due to their electrostatic interactions
with a basic region of ApoB-100. These results demonstrate that LRP1-
derived peptides are protective against LDL aggregation, even in
conditions of extreme lipolysis, through their capacity to bind to ApoB-
100 regions critical to preserve ApoB-100 conformation. These results
suggests that these LRP1(CR9) derived peptides could be promising tools
to prevent LDL aggregation induced by the main proteolytic enzymes acting
in the arterial intima.
Response to Reviewers: ANSWERS TO REVIEWERS

Manuscript No.: BBAMEM-19-17
Title: Molecular basis for the protective effects of low-density
lipoprotein receptor-related protein 1 (LRP1)-derived peptides against
LDL aggregation
Article Type: Regular Paper
Journal Title: BBA - Biomembranes
Corresponding Author: Dr. Vicenta Llorente Cortés
Submit Date: Jan 20, 2019
We thank the Reviewers for their valuable suggestions and critical
comments that have enhanced the quality of the manuscript.
Reviewer #1: The manuscript is a well-written report on the effect of two
peptides derived from LRP1 on the inhibition of enzyme-induced
aggregation of LDL. Overall the experiments are well designed and
executed, and the results are presented clearly showing that DP3 and LP3
peptides inhibit LDL aggregation.
It would help if the authors provide more detail on the rational of using
the retro-enantio peptides, it would help if this is included in the
first paragraph of the result section. This is relevant as the authors
clearly show that DP3 is more active than LP3, and an explanation in the
discussion would be welcome. Further, provide detail about the blocking
of the termini in the methods section (in particular explain "H" on the
N-terminus). This is important as DP3 is more active than LP3. This can
also be discussed in the discussion.
We thank the Reviewer by this comment that will help to understand the
rational to use retro-enantio peptides. Peptide drugs offer high
activity, high specificity and low toxicity, but are susceptible to
proteolytic degradation. A strategy to overcome this problem is to
produce peptides using non-natural or D-amino acids, which usually are
not recognized by plasma proteases. However, in most of the cases, the
all-D peptide version (enantio) of an active L-peptide is not active or
show very low activity because the opposite orientation of the amino
acids side-chains compared to the parent peptide, which challenges the
recognition of the enantio peptide by its target receptor. Nevertheless,
in the literature there are some examples with enantio versions of
peptides that do show activity (Soto C et al, Biochem Biophys Res Commun
1996; Chalifour RJ et al, J Biol Chem 2003). A more conservative strategy
to preserve peptide activity using D-amino acids consists in applying the
retro-enantio approach. The idea behind this approach is to
simultaneously invert the sequence and chirality of the peptide (Chorev M
et al, Trends Biotechnol 1995; Jameson BA et al, Nature 1994) In this
way, the amine bone of the retro-enantio version is shift one unit but
the orientation of the side chains remain the same:
Usually, using this approach the retro-enantio peptide retains the
activity of the parent peptide (Prades R et al, Angew Chem Int Ed 2015).
This explanation has been summarized in the first paragraph of Results
(part 3.1) and expanded in the Discussion section of the revised ms.
In the first paragraph of the result (part 3.1)_“Peptide drugs offer high
activity, high specificity and low toxicity, but are susceptible to
proteolytic degradation. A strategy to overcome this problem is to
produce peptides using non-natural or D-amino acids, which shows improved
stability. Although all-D peptides are not recognized by plasma
proteases, they may be less active than their corresponding all-L
peptides because of differences in the orientation of amino acid side-
chains. To preserve the orientation of the side-chains of the parent
peptide in a D-analogue, the D-amino acid residues can be linked in the
reverse order to that of the parent peptide in order to produce a retro-

enantio peptide. This approach emphasizes the maintenance of the
topological native orientation of the side-chains but not of the
backbone, where the amide bond is shift one unit (Chorev M et al, Trends
Biotechnol 1995). By using this approach, peptides usually preserve their
activity while notably improve their plasma stability. In the literature
there are examples where this approach has been successfully used (Prades
R et al, Angew Chem Int Ed 2015)”.
In the Discussion section we have added the following explanation (first
paragraph)_“we tested both the original LP3 version and its retro-enantio
version DP3. Although all-D peptides are not recognised by proteases, and
thus have higher stability, they may be less active than their
corresponding all-L peptides because of differences in the orientation of
amino acid side-chains. To preserve the orientation of the side-chains of
the parent L-peptide in a D-analogue, the retro-enantio approach (Soto C
et al, Biochem Biophys Res Commun 1996; Chalifour RJ et al, J Biol Chem
2003) has been used in this study. Here, we demonstrate that LP3 and its
derived retro-enantio version (DP3) per se have a high capacity to
inhibit LDL aggregation induced by SMase and PLA2. In this regard, the
preservation of the topological side-chain orientation of DP3 together
with a high stability in biological samples account for its notable
performance in the inhibition assays.”
In the reference section we have added three new references
Further, provide detail about the blocking of the termini in the methods
section (in particular explain "H" on the N-terminus). Peptides are
always written from N to C terminus (N->C). The peptides described in
this manuscript have an amide at their C-terminus and we have indicated
so by typing –NH2 at this termini. The way of indicating this could be
misleading for readers not familiar with peptide nomenclature, since
these could think that the peptide sequences is typed from C to N. To
clearly indicate the N and C terminus of the peptide we added the H- at
the N-terminal part of the sequence to indicate that it finalizes as
amine.
To avoid any confusion in this regard, this explanation has been included
in the Methods section of the revised ms_“Peptide sequences are written
from N to C, H- at the end terminus indicate amine ending, whereas –NH2
at the C terminus indicates amide”.
It is evidently shown that the two peptides are able to inhibit the SMase
and PLA2 induced LDL aggregation. Can the authors show that the peptides
associate with the LDL surface prior or after enzyme treatment?
Thanks to the Reviewer by this interesting question. Limited trypsin
proteolysis combined with Mass spectrometry-based proteomics was the
strategy used to analyze this key aspect mentioned by the Reviewer. This
strategy allows to identify the sequences of ApoB-100 in native LDL
protected by the peptide against trypsin-induced proteolysis. Five
sequences of ApoB-100 in native LDL were protected from proteolysis. The
peptide protection was obviously temporal, it was observed at 1 hour and
disappeared at 3 hours (Figure 7A). These results evidenced that the
peptide interacts with the LDL surface prior enzymatic treatment,
protecting certain sequences against proteolysis.
We have remarked this interesting point in the discussion section of the
revised manuscript _ “Limited trypsin proteolysis combined with Mass
spectrometry-based proteomics of ApoB-100/DP3 complexes evidenced that
the peptide interacts with the LDL surface prior enzymatic treatment”.
Is it possible that the peptides bind directly to the enzyme thereby
preventing LDL aggregation? This possibility should be discussed.

We have tested this possibility by exploring the effect of peptides on
SMase activity in the current incubation conditions. As shown in
Supplemental Figure 6, SMase activity, measured in relation to the amount
of phosphorylcholine generated, was similar in absence or presence of the
peptides, indicating that, although we cannot discard a potential
interaction between peptide and SMase, it is clear that this interaction
has not effect on SMase activity. This comment has been included in the
Discussion of the revised ms. as following: “Although we can not discard
a potential interaction between peptides and SMase, SMase activity assays
clearly show that peptides does not exert any effect on SMase functional
activity”.
The authors discuss the appearance of non-polar spots on the surface that
trigger aggregation (page 20). Is it possible that the peptides associate
with these spots (hydrophobic defects?), as has been shown for
amphipathic helices of apolipoproteins? (4F is an amphipathic peptide).
This has been reported in a manuscript by Liu et al, in which PL-C
treated LDL resulted in the recruitment of apolipoproteins, thereby
offering protection against aggregation. (Liu, H., Scraba, D.G., and
Ryan, R.O. 1993 Prevention of phospholipase-C induced aggregation of low
density lipoprotein by amphipathic apolipoproteins, FEBS Lett. 316, 27-
33.)
Thanks to the Reviewer for this interesting observation. As the Reviewer
comments, several peptides and proteins bearing amphipathic properties
have been reported to be able to block LDL aggregation by a direct
interaction with LDL phospholipid surface. In this respect, common
examples are 4F peptide (Nguyen SD et al, J Lipid Res 2015) or different
apolipoproteins (Liu H et al, FEBS Lett 1993). Despite the lack of
amphipathic properties in any of the LRP1-derived peptides, this
hypothesis was even considered as possible mechanism of action for the
peptides described herein.Thus, as a preliminary test, molecular ligand
docking was performed using a phospholipidic monolayer mimicking LDL
surface (Hevonoja T et al, Biochem Biophys Acta 2000) as receptor and DP3
peptide as ligand. The top ranked docking poses showed rather low binding
energy values (around -4 kcal/mol) and mostly unreasonable binding modes.
Despite their theoretical character, these results support that there is
not a direct association between LRP1-derived peptides and LDL
phospholipid surface. This explanation has been summarized in the Results
(part 3.7) and expanded in the Discussion section of the revised ms.
In Result (part 3.7)_”Molecular docking calculations were perfomed using
a phospholipidic monolayer mimicking LDL surface (Hevonoja T et al,
Biochem Biophys Acta 2000) as receptor and DP3 peptide as ligand. The top
ranked docking poses showed rather low binding energy values (around -4
kcal/mol) and mostly unreasonable binding modes. This theoretical
calculations support that peptides do not associate with LDL phospholipid
surface”.
This argumentation has been expanded in the Discussion section_ Two
different explanations about the protective effect of peptides against
SMase-induced LDL lipolysis have been explored. By one side, we have
explored whether there is a direct effect of peptides on SMase activity.
This possibility has been discarded since SMase activity assays clearly
show that peptides does not exert any effect on SMase functional
activity. Other possibility that has been explored is a potential
interaction of peptides with the LDL phospholipid surface. Several
peptides and proteins bearing amphipathic properties have been reported
to be able to block LDL aggregation by a direct interaction with LDL
phospholipid surface. In this respect, common examples are 4F peptide
(Nguyen SD et al, J Lipid Res 2015) or different apolipoproteins (Liu H

et al, FEBS Lett 1993). Despite the lack of amphipathic properties in
LRP1-derived peptides, this hypothesis has been considered as possible
mechanism of action for the peptides described herein. Theoretical
molecular docking calculations performed in the present study indicate a
lack of association between peptides and LDL phospholipid surface”
New references about this issue have been included in the Reference
section_ One new reference has been added to the reference section of the
revised ms.
Fig 1 legend. Indicate that turbidity was measured by the absorbance at
405 nm.
We have included this indication in the Figure 1 legend of the revised
ms.
Table S3, indicate that this is a molar ratio.
We have included this indication in Table S3 of the revised ms.
Reviewer #3: The manuscript is a comparative study among different
peptides and their effects on the aggregation of sphingomyelinase-
(SMase) and phospholipase 2 (PLA2)-modified LDL particles.
The manuscript is well-written. The study is interesting. The peptides
were succesfully synthetized by solid-phase peptide synthesis as reported
in table S1.
The manuscript is ready for publication.
We thank the Reviewer for his/her positive evaluation of our manuscript
Reviewer #4: In the manuscript entitled "Molecular basis for the
protective effects of low-density lipoprotein receptor-related protein 1
(LRP1)-derived peptides against LDL aggregation » the authors have
investigate the effect of two peptide (LP3 and DP3) on the structural
characteristics of SMase and PLA2 modified LDL particles. They showed by
turbidimetry, gel filtration chromatography and tomography electronic
microscopy (TEM) analysis that LP3 and DP3 peptides strongly inhibited
SMase- and PLA2-induced LDL aggregation. They showed that :
LRP1-derived peptides prevent SMase-induced alterations in LDL particle
size, electric charge and phospholipid content but do not prevent PLA2-
induced alterations in LDL particle size, electric charge and
phospholipid content. Their data demonstrate that LRP1-derived peptides
are protective against LDL aggregation, through their capacity to bind to
ApoB-100 regions critical to preserve ApoB-100 conformation.
The article is well written and easy to read. The authors have convincing
data demonstrating mechanism involved by these both peptides and provide
evidence that these peptides could be promising tools to prevent LDL

aggregation in the arterial intima. This work present interesting data
that could be published with some minor complementary informations listed
below:
1- In the discussion section, the authors should better explain why both
peptides exerted protection against conformational alterations of poB-100
induced by both SMases and PLA2 whereas they didn't counteract particle
size charge and phospholipids induced by PLA2.
We thank the Reviewer by this interesting comment. LRP1-derived peptides
preserved ApoB-100 conformation, sphyngomyelin (SM) content and LDL size
in SMase-LDL while in PLA2-LDL, peptides preserve only ApoB-100
conformation but not phosphatidylcholine (PC) content or LDL size. We
propose that the differential effect of LRP1-derived peptides in these
two modified LDLs could be explained by topological reasons. Although
sphingomyelin (SM), compared to phosphatidylcholine (PC), is a minor
phospholipid in LDL (185 versus 450 molecules/LDL), SM, the target
phospholipid for SMase, is localized in cholesterol-enriched
environments, while PC is associated with poor-cholesterol environment.
Surface cholesterol seems to be a key regulator of the process of
phospholipolysis (Hevonoja T et al, Biochem Biophys Acta 2000) and thus,
could modulate SM but not PC lipolysis. We propose that peptide
preservation of ApoB-100 conformation directly impacts on the maintenance
of the cholesterol-enriched environment structure since, according to our
molecular modeling studies, ApoB-100 sequences interacting with peptide
are those close to cholesterol in the LDL surface. Therefore, SM would be
protected against SMase phospholipolysis by its privilege of being part
of cholesterol-enriched domains, whose structure is likely dependent on
the maintenance of ApoB-100 conformation. In contrast, PC, belonging to
cholesterol-poor domains, seems to be unprotected against PLA2
independently of the status of ApoB-100 conformation.
These comments, complementary to those performed by Reviewer 1, have been
jointly integrated in the Discussion as following_ “Two different
explanations about the protective effect of peptides against SMase-
induced LDL lipolysis have been explored. By one side, we have explored
whether there is a direct effect of peptides on SMase activity. This
possibility has been discarded since SMase activity assays clearly show
that peptides does not exert any effect on SMase functional activity.
Other possibility that has been explored is a potential interaction of
peptides with the LDL phospholipid surface. Several peptides and proteins
bearing amphipathic properties have been reported to be able to block LDL
aggregation by a direct interaction with LDL phospholipid surface. In
this respect, common examples are 4F peptide (Nguyen SD et al, J Lipid
Res 2015) or different apolipoproteins (Liu H et al, FEBS Lett 1993).
Despite the lack of amphipathic properties in LRP1-derived peptides, this
hypothesis has been considered as possible mechanism of action for the
peptides described herein. Theoretical molecular docking calculations
performed in the present study indicate a lack of association between
peptides and LDL phospholipid surface. A key point that offer clues about
how peptides protect SM against SMase-induced hydrolysis is their
unability to protect PC against PLA2-induced hydrolysis. We propose that
the differential anti-phospholypolytic effects of the peptide on SM and
PC phospholipids could be explained by the particular topological
distribution of SM and PC on the LDL surface. Although SM, compared to
PC, is a minor phospholipid in LDL (185 versus 450 molecules/LDL), SM is
localized in cholesterol-enriched environments, key regulators of
phospholipolysis (Hevonoja T et al, Biochem Biophys Acta 2000). We
propose that peptide preservation of ApoB-100 conformation directly
impacts on the maintenance of the cholesterol-enriched environment

structure since, according to our molecular modeling studies, ApoB-100
sequences complexed with peptide are those close to cholesterol in the
LDL surface. Therefore, SM would be peptide-protected against
phospholipolysis by its privilege of being part of cholesterol-enriched
domains, whose structure would be preserved through peptide-ApoB-100
conformational stabilization. In contrast, PC, belonging to cholesterol-
poor domains, is unprotected against PLA2, independently of the status of
ApoB-100 conformational stabilization”.
2-The authors indicated at the end of the discussion that "LRP1-derived
peptides emerge as tools potentially useful to prevent atherosclerosis
and cardiovascular complications, in particular in individuals with
aggregation-prone LDL such as obese and diabetic patients. » This is a
very interesting point but the authors should discuss a little bit more
the possibility to use these peptides in a therapeutic way. How do they
envisage to deliver these peptides in the future? (Are these peptides
stable? Do they consider local delivery after a first cardiovascular
event to decrease recurrence risk as a possible strategy? Or systemic
delivery…?
We thank the Reviewer for these interesting comments that will indeed
increase the potential translational impact of our manuscript.
In the future, we are going to test the effect of the retro-enantio
peptide (increased stability) in in vivo models of atherosclerosis. We
are mainly interested in the capacity of DP3 to reduce LDL retention and
LDL-induced smooth muscle cell (SMC)-foam cell formation in the vascular
wall.
Our first option will be to inject DP3 subcutaneously (not focal delivery
at the moment) taking advantage that atherosclerotic-prone areas of the
vasculature in in vivo models of angioplasty-induced atherosclerosis have
increased endothelial permeability.
These comments have been included in the Discussion (part conclusions) of
the revised ms._“In the future, we are going to test the efficacy of the
retro-enantio peptide version (DP3) to reduce vascular LDL retention and
LDL-induced smooth muscle cell (SMC)-foam cell formation in in vivo
models of atherosclerosis. Our first option in the near future will be to
inject the peptide DP3 subcutaneously (not focal delivery at the moment)
in a murine model of angioplasty-induced atherosclerosis taking
advantage of the increased endothelial permeability the vasculature of in
vivo models of angioplasty-induced atherosclerosis. In a not too distant
future, we expect LRP1-derived peptides to emerge as tools potentially
useful to prevent atherosclerosis and cardiovascular complications, in
particular in individuals with aggregation-prone LDL as obese and
diabetic patients”.

Editor-in-Chief
BBA_Biomembranes
Editorial Office
Barcelona, 2019, April 9th
Dear Editor,
Enclose please find our article BBAMEM-19-17 (R1) entitled “Molecular basis for the protective effects of
low-density lipoprotein receptor-related protein 1 (LRP1)-derived peptides against LDL aggregation”
to be considered for publication in BBA_Biomembranes.
We have addressed, point-by-point, all the issues raised by the Reviewers. According to their suggestions,
we have modified Methods, Results and Discussion sections of the manuscript. As a result, we think that the
manuscript has been deeply improved. We have changed each section of the manuscript by including new
information that has highly increased the impact of reported results.
We hope that the manuscript will be now acceptable for publication in BBA Biomembranes.
All authors have read and approved submission of the manuscript and the manuscript has not been published
and is not being considered for publication elsewhere in whole or part in any language except as an abstract.
Roger Prades, Chiara Pallara and Teresa Tarragó are employees of Iproteos SL. Teresa Tarragó is
cofounder of Iproteos SL and member of its board of directors.
None of the rest of authors have any conflict of interest with the publication of the manuscript.
We thank you for your time and effort.
Sincerely
Dr. Vicenta Llorente Cortes, Lipids and Cardiovascular Pathology group, Biomedical Research Institute IIB
Sant Pau, IIBB-CSIC, Hospital de la Santa Creu i Sant Pau, Sant Antoni Mª Claret, 167, 08025 Barcelona.
Phone: +34 935565888, Fax: +34 935565559, E-mail: [email protected]; [email protected]
Cover Letter

BBAMEM-19-17 (R1)
1
ANSWERS TO REVIEWERS
Manuscript No.: BBAMEM-19-17
Title: Molecular basis for the protective effects of low-density lipoprotein receptor-
related protein 1 (LRP1)-derived peptides against LDL aggregation
Article Type: Regular Paper
Journal Title: BBA - Biomembranes
Corresponding Author: Dr. Vicenta Llorente Cortés
Submit Date: Jan 20, 2019
Editor-in-Chief
BBA_Biomembranes
Editorial Office
Barcelona, 2019, April 9th
Dear Editor,
Enclose please find our article BBAMEM-19-17 (R1) entitled “Molecular basis for the
protective effects of low-density lipoprotein receptor-related protein 1 (LRP1)-derived
peptides against LDL aggregation” to be considered for publication in BBA_Biomembranes.
We have addressed, point-by-point, all the issues raised by the Reviewers. According to
their suggestions, we have modified Methods, Results and Discussion sections of the
manuscript. As a result, we think that the manuscript has been deeply improved. We have
changed each section of the manuscript by including new information that has highly increased
the impact of reported results.
We hope that the manuscript will be now acceptable for publication in BBA Biomembranes.
All authors have read and approved submission of the manuscript and the manuscript has not
been published and is not being considered for publication elsewhere in whole or part in any
language except as an abstract.
Roger Prades, Chiara Pallara and Teresa Tarragó are employees of Iproteos SL. Teresa Tarragó
is cofounder of Iproteos SL and member of its board of directors.
None of the rest of authors have any conflict of interest with the publication of the manuscript.
We thank you for your time and effort.
Response to Reviewers

BBAMEM-19-17 (R1)
2
Sincerely
Dr. Vicenta Llorente Cortes, Lipids and Cardiovascular Pathology group, Biomedical Research
Institute IIB Sant Pau, IIBB-CSIC, Hospital de la Santa Creu i Sant Pau, Sant Antoni Mª Claret,
167, 08025 Barcelona. Phone: +34 935565888, Fax: +34 935565559, E-mail:

BBAMEM-19-17 (R1)
3
We thank the reviewer for the valuable suggestions and critical comments that have
enhanced the quality of the manuscript.
Reviewer #1: The manuscript is a well-written report on the effect of two peptides
derived from LRP1 on the inhibition of enzyme-induced aggregation of LDL. Overall the
experiments are well designed and executed, and the results are presented clearly
showing that DP3 and LP3 peptides inhibit LDL aggregation.
It would help if the authors provide more detail on the rational of using the retro-enantio
peptides, it would help if this is included in the first paragraph of the result section. This
is relevant as the authors clearly show that DP3 is more active than LP3, and an
explanation in the discussion would be welcome. Further, provide detail about the
blocking of the termini in the methods section (in particular explain "H" on the N-
terminus). This is important as DP3 is more active than LP3. This can also be
discussed in the discussion.
We thank the Reviewer by this comment that will help to understand the rational to use
retro-enantio peptides. Peptide drugs offer high activity, high specificity and low toxicity,
but are susceptible to proteolytic degradation. A strategy to overcome this problem is to
produce peptides using non-natural or D-amino acids, which usually are not recognized
by plasma proteases. However, in most of the cases, the all-D peptide version
(enantio) of an active L-peptide is not active or show very low activity because the
opposite orientation of the amino acids side-chains compared to the parent peptide,
which challenges the recognition of the enantio peptide by its target receptor.
Nevertheless, in the literature there are some examples with enantio versions of
peptides that do show activity (Soto C et al, Biochem Biophys Res Commun 1996;
Chalifour RJ et al, J Biol Chem 2003). A more conservative strategy to preserve
peptide activity using D-amino acids consists in applying the retro-enantio approach.
The idea behind this approach is to simultaneously invert the sequence and chirality of
the peptide (Chorev M et al, Trends Biotechnol 1995; Jameson BA et al, Nature 1994)
In this way, the amine bone of the retro-enantio version is shift one unit but the
orientation of the side chains remain the same:
Usually, using this approach the retro-enantio peptide retains the activity of the parent
peptide (Prades R et al, Angew Chem Int Ed 2015). This explanation has been
summarized in the first paragraph of Results (part 3.1) and expanded in the Discussion
section of the revised ms.
In the first paragraph of the result (part 3.1)_“Peptide drugs offer high activity, high
specificity and low toxicity, but are susceptible to proteolytic degradation. A
strategy to overcome this problem is to produce peptides using non-natural or
D-amino acids, which shows improved stability. Although all-D peptides are not
recognized by plasma proteases, they may be less active than their
corresponding all-L peptides because of differences in the orientation of amino
acid side-chains. To preserve the orientation of the side-chains of the parent
peptide in a D-analogue, the D-amino acid residues can be linked in the reverse
order to that of the parent peptide in order to produce a retro-enantio peptide.
This approach emphasizes the maintenance of the topological native orientation
of the side-chains but not of the backbone, where the amide bond is shift one

BBAMEM-19-17 (R1)
4
unit (Chorev M et al, Trends Biotechnol 1995). By using this approach, peptides
usually preserve their activity while notably improve their plasma stability. In the
literature there are examples where this approach has been successfully used
(Prades R et al, Angew Chem Int Ed 2015)”.
In the Discussion section we have added the following explanation (first
paragraph)_“we tested both the original LP3 version and its retro-enantio
version DP3. Although all-D peptides are not recognised by proteases, and thus
have higher stability, they may be less active than their corresponding all-L
peptides because of differences in the orientation of amino acid side-chains. To
preserve the orientation of the side-chains of the parent L-peptide in a D-
analogue, the retro-enantio approach (Soto C et al, Biochem Biophys Res
Commun 1996; Chalifour RJ et al, J Biol Chem 2003) has been used in this
study. Here, we demonstrate that LP3 and its derived retro-enantio version
(DP3) per se have a high capacity to inhibit LDL aggregation induced by SMase
and PLA2. In this regard, the preservation of the topological side-chain
orientation of DP3 together with a high stability in biological samples account for
its notable performance in the inhibition assays.”
In the reference section we have added three new references
Further, provide detail about the blocking of the termini in the methods section (in
particular explain "H" on the N-terminus). Peptides are always written from N to C
terminus (N->C). The peptides described in this manuscript have an amide at their C-
terminus and we have indicated so by typing –NH2 at this termini. The way of indicating
this could be misleading for readers not familiar with peptide nomenclature, since these
could think that the peptide sequences is typed from C to N. To clearly indicate the N
and C terminus of the peptide we added the H- at the N-terminal part of the sequence
to indicate that it finalizes as amine.
To avoid any confusion in this regard, this explanation has been included in the
Methods section of the revised ms_“Peptide sequences are written from N to C, H-
at the end terminus indicate amine ending, whereas –NH2 at the C terminus indicates
amide”.
It is evidently shown that the two peptides are able to inhibit the SMase and PLA2
induced LDL aggregation. Can the authors show that the peptides associate with the
LDL surface prior or after enzyme treatment?
Thanks to the Reviewer by this interesting question. Limited trypsin proteolysis
combined with Mass spectrometry-based proteomics was the strategy used to analyze
this key aspect mentioned by the Reviewer. This strategy allows to identify the
sequences of ApoB-100 in native LDL protected by the peptide against trypsin-induced
proteolysis. Five sequences of ApoB-100 in native LDL were protected from
proteolysis. The peptide protection was obviously temporal, it was observed at 1 hour
and disappeared at 3 hours (Figure 7A). These results evidenced that the peptide
interacts with the LDL surface prior enzymatic treatment, protecting certain sequences
against proteolysis.
We have remarked this interesting point in the discussion section of the revised
manuscript _ “Limited trypsin proteolysis combined with Mass spectrometry-based

BBAMEM-19-17 (R1)
5
proteomics of ApoB-100/DP3 complexes evidenced that the peptide interacts with the
LDL surface prior enzymatic treatment”.
Is it possible that the peptides bind directly to the enzyme thereby preventing LDL
aggregation? This possibility should be discussed.
We have tested this possibility by exploring the effect of peptides on SMase activity in
the current incubation conditions. As shown in Supplemental Figure 6, SMase activity,
measured in relation to the amount of phosphorylcholine generated, was similar in
absence or presence of the peptides, indicating that, although we cannot discard a
potential interaction between peptide and SMase, it is clear that this interaction has not
effect on SMase activity. This comment has been included in the Discussion of the
revised ms. as following: “Although we can not discard a potential interaction between
peptides and SMase, SMase activity assays clearly show that peptides does not exert
any effect on SMase functional activity”.
The authors discuss the appearance of non-polar spots on the surface that trigger
aggregation (page 20). Is it possible that the peptides associate with these spots
(hydrophobic defects?), as has been shown for amphipathic helices of apolipoproteins?
(4F is an amphipathic peptide). This has been reported in a manuscript by Liu et al, in
which PL-C treated LDL resulted in the recruitment of apolipoproteins, thereby offering
protection against aggregation. (Liu, H., Scraba, D.G., and Ryan, R.O. 1993 Prevention
of phospholipase-C induced aggregation of low density lipoprotein by amphipathic
apolipoproteins, FEBS Lett. 316, 27-33.)
Thanks to the Reviewer for this interesting observation. As the Reviewer comments,
several peptides and proteins bearing amphipathic properties have been reported to be
able to block LDL aggregation by a direct interaction with LDL phospholipid surface. In
this respect, common examples are 4F peptide (Nguyen SD et al, J Lipid Res 2015) or
different apolipoproteins (Liu H et al, FEBS Lett 1993). Despite the lack of amphipathic
properties in any of the LRP1-derived peptides, this hypothesis was even considered
as possible mechanism of action for the peptides described herein.Thus, as a
preliminary test, molecular ligand docking was performed using a phospholipidic
monolayer mimicking LDL surface (Hevonoja T et al, Biochem Biophys Acta 2000) as
receptor and DP3 peptide as ligand. The top ranked docking poses showed rather low
binding energy values (around -4 kcal/mol) and mostly unreasonable binding modes.
Despite their theoretical character, these results support that there is not a direct
association between LRP1-derived peptides and LDL phospholipid surface. This
explanation has been summarized in the Results (part 3.7) and expanded in the
Discussion section of the revised ms.
In Result (part 3.7)_”Molecular docking calculations were perfomed using a
phospholipidic monolayer mimicking LDL surface (Hevonoja T et al, Biochem Biophys
Acta 2000) as receptor and DP3 peptide as ligand. The top ranked docking poses
showed rather low binding energy values (around -4 kcal/mol) and mostly
unreasonable binding modes. This theoretical calculations support that peptides do not
associate with LDL phospholipid surface”.
This argumentation has been expanded in the Discussion section_ Two different
explanations about the protective effect of peptides against SMase-induced LDL
lipolysis have been explored. By one side, we have explored whether there is a direct
effect of peptides on SMase activity. This possibility has been discarded since SMase
activity assays clearly show that peptides does not exert any effect on SMase
functional activity. Other possibility that has been explored is a potential interaction of

BBAMEM-19-17 (R1)
6
peptides with the LDL phospholipid surface. Several peptides and proteins bearing
amphipathic properties have been reported to be able to block LDL aggregation by a
direct interaction with LDL phospholipid surface. In this respect, common examples are
4F peptide (Nguyen SD et al, J Lipid Res 2015) or different apolipoproteins (Liu H et al,
FEBS Lett 1993). Despite the lack of amphipathic properties in LRP1-derived peptides,
this hypothesis has been considered as possible mechanism of action for the peptides
described herein. Theoretical molecular docking calculations performed in the present
study indicate a lack of association between peptides and LDL phospholipid surface”
New references about this issue have been included in the Reference section_
One new reference has been added to the reference section of the revised ms.
Fig 1 legend. Indicate that turbidity was measured by the absorbance at 405 nm.
We have included this indication in the Figure 1 legend of the revised ms.
Table S3, indicate that this is a molar ratio.
We have included this indication in Table S3 of the revised ms.
Reviewer #3: The manuscript is a comparative study among different peptides and
their effects on the aggregation of sphingomyelinase- (SMase) and phospholipase 2
(PLA2)-modified LDL particles.
The manuscript is well-written. The study is interesting. The peptides were succesfully
synthetized by solid-phase peptide synthesis as reported in table S1.
The manuscript is ready for publication.
We thank the Reviewer for his/her positive evaluation of our manuscript

BBAMEM-19-17 (R1)
7
Reviewer #4: In the manuscript entitled "Molecular basis for the protective effects of
low-density lipoprotein receptor-related protein 1 (LRP1)-derived peptides against LDL
aggregation » the authors have investigate the effect of two peptide (LP3 and DP3) on
the structural characteristics of SMase and PLA2 modified LDL particles. They showed
by turbidimetry, gel filtration chromatography and tomography electronic microscopy
(TEM) analysis that LP3 and DP3 peptides strongly inhibited SMase- and PLA2-
induced LDL aggregation. They showed that :
LRP1-derived peptides prevent SMase-induced alterations in LDL particle size, electric
charge and phospholipid content but do not prevent PLA2-induced alterations in LDL
particle size, electric charge and phospholipid content. Their data demonstrate that
LRP1-derived peptides are protective against LDL aggregation, through their capacity
to bind to ApoB-100 regions critical to preserve ApoB-100 conformation.
The article is well written and easy to read. The authors have convincing data
demonstrating mechanism involved by these both peptides and provide evidence that
these peptides could be promising tools to prevent LDL aggregation in the arterial
intima. This work present interesting data that could be published with some minor
complementary informations listed below:
1- In the discussion section, the authors should better explain why both peptides
exerted protection against conformational alterations of poB-100 induced by both
SMases and PLA2 whereas they didn't counteract particle size charge and
phospholipids induced by PLA2.
We thank the Reviewer by this interesting comment. LRP1-derived peptides preserved
ApoB-100 conformation, sphyngomyelin (SM) content and LDL size in SMase-LDL
while in PLA2-LDL, peptides preserve only ApoB-100 conformation but not
phosphatidylcholine (PC) content or LDL size. We propose that the differential effect of
LRP1-derived peptides in these two modified LDLs could be explained by topological
reasons. Although sphingomyelin (SM), compared to phosphatidylcholine (PC), is a
minor phospholipid in LDL (185 versus 450 molecules/LDL), SM, the target
phospholipid for SMase, is localized in cholesterol-enriched environments, while PC is
associated with poor-cholesterol environment. Surface cholesterol seems to be a key
regulator of the process of phospholipolysis (Hevonoja T et al, Biochem Biophys Acta
2000) and thus, could modulate SM but not PC lipolysis. We propose that peptide
preservation of ApoB-100 conformation directly impacts on the maintenance of the
cholesterol-enriched environment structure since, according to our molecular modeling
studies, ApoB-100 sequences interacting with peptide are those close to cholesterol in
the LDL surface. Therefore, SM would be protected against SMase phospholipolysis by
its privilege of being part of cholesterol-enriched domains, whose structure is likely
dependent on the maintenance of ApoB-100 conformation. In contrast, PC, belonging
to cholesterol-poor domains, seems to be unprotected against PLA2 independently of
the status of ApoB-100 conformation.
These comments, complementary to those performed by Reviewer 1, have been
jointly integrated in the Discussion as following_ “Two different explanations about
the protective effect of peptides against SMase-induced LDL lipolysis have been
explored. By one side, we have explored whether there is a direct effect of peptides on
SMase activity. This possibility has been discarded since SMase activity assays clearly
show that peptides does not exert any effect on SMase functional activity. Other
possibility that has been explored is a potential interaction of peptides with the LDL
phospholipid surface. Several peptides and proteins bearing amphipathic properties
have been reported to be able to block LDL aggregation by a direct interaction with

BBAMEM-19-17 (R1)
8
LDL phospholipid surface. In this respect, common examples are 4F peptide (Nguyen
SD et al, J Lipid Res 2015) or different apolipoproteins (Liu H et al, FEBS Lett 1993).
Despite the lack of amphipathic properties in LRP1-derived peptides, this hypothesis
has been considered as possible mechanism of action for the peptides described
herein. Theoretical molecular docking calculations performed in the present study
indicate a lack of association between peptides and LDL phospholipid surface. A key
point that offer clues about how peptides protect SM against SMase-induced hydrolysis
is their unability to protect PC against PLA2-induced hydrolysis. We propose that the
differential anti-phospholypolytic effects of the peptide on SM and PC phospholipids
could be explained by the particular topological distribution of SM and PC on the LDL
surface. Although SM, compared to PC, is a minor phospholipid in LDL (185 versus
450 molecules/LDL), SM is localized in cholesterol-enriched environments, key
regulators of phospholipolysis (Hevonoja T et al, Biochem Biophys Acta 2000). We
propose that peptide preservation of ApoB-100 conformation directly impacts on the
maintenance of the cholesterol-enriched environment structure since, according to our
molecular modeling studies, ApoB-100 sequences complexed with peptide are those
close to cholesterol in the LDL surface. Therefore, SM would be peptide-protected
against phospholipolysis by its privilege of being part of cholesterol-enriched domains,
whose structure would be preserved through peptide-ApoB-100 conformational
stabilization. In contrast, PC, belonging to cholesterol-poor domains, is unprotected
against PLA2, independently of the status of ApoB-100 conformational stabilization”.
2-The authors indicated at the end of the discussion that "LRP1-derived peptides
emerge as tools potentially useful to prevent atherosclerosis and cardiovascular
complications, in particular in individuals with aggregation-prone LDL such as obese
and diabetic patients. » This is a very interesting point but the authors should discuss a
little bit more the possibility to use these peptides in a therapeutic way. How do they
envisage to deliver these peptides in the future? (Are these peptides stable? Do they
consider local delivery after a first cardiovascular event to decrease recurrence risk as
a possible strategy? Or systemic delivery…?
We thank the Reviewer for these interesting comments that will indeed increase the
potential translational impact of our manuscript.
In the future, we are going to test the effect of the retro-enantio peptide (increased
stability) in in vivo models of atherosclerosis. We are mainly interested in the capacity
of DP3 to reduce LDL retention and LDL-induced smooth muscle cell (SMC)-foam cell
formation in the vascular wall.
Our first option will be to inject DP3 subcutaneously (not focal delivery at the moment)
taking advantage that atherosclerotic-prone areas of the vasculature in in vivo models
of angioplasty-induced atherosclerosis have increased endothelial permeability.
These comments have been included in the Discussion (part conclusions) of the
revised ms._“In the future, we are going to test the efficacy of the retro-enantio peptide
version (DP3) to reduce vascular LDL retention and LDL-induced smooth muscle cell
(SMC)-foam cell formation in in vivo models of atherosclerosis. Our first option in the
near future will be to inject the peptide DP3 subcutaneously (not focal delivery at the
moment) in a murine model of angioplasty-induced atherosclerosis taking advantage
of the increased endothelial permeability the vasculature of in vivo models of
angioplasty-induced atherosclerosis. In a not too distant future, we expect LRP1-
derived peptides to emerge as tools potentially useful to prevent atherosclerosis and
cardiovascular complications, in particular in individuals with aggregation-prone LDL as
obese and diabetic patients”.
ApoB-100/DP3 complex
STABILIZATION OF ApoB-100 CONFORMATION
SMase PLA2
STABILIZATION OF CHOLESTEROL/SM
enriched environment
Unaltered PL Surface Unaltered LDL size
NO EFFECT ON PC-enriched/colesterol-poor
environment
Altered PL Surface Decreased LDL size
LDL aggregation
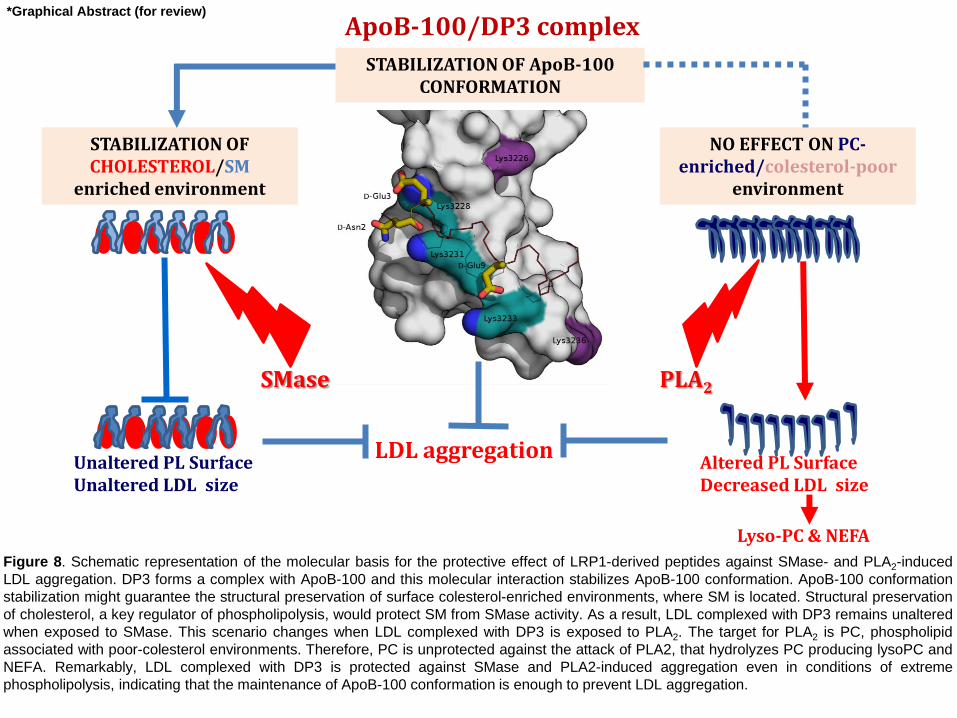
Figure 8. Schematic representation of the molecular basis for the protective effect of LRP1-derived peptides against SMase- and PLA2-induced
LDL aggregation. DP3 forms a complex with ApoB-100 and this molecular interaction stabilizes ApoB-100 conformation. ApoB-100 conformation
stabilization might guarantee the structural preservation of surface colesterol-enriched environments, where SM is located. Structural preservation
of cholesterol, a key regulator of phospholipolysis, would protect SM from SMase activity. As a result, LDL complexed with DP3 remains unaltered
when exposed to SMase. This scenario changes when LDL complexed with DP3 is exposed to PLA2. The target for PLA2 is PC, phospholipid
associated with poor-colesterol environments. Therefore, PC is unprotected against the attack of PLA2, that hydrolyzes PC producing lysoPC and
NEFA. Remarkably, LDL complexed with DP3 is protected against SMase and PLA2-induced aggregation even in conditions of extreme
phospholipolysis, indicating that the maintenance of ApoB-100 conformation is enough to prevent LDL aggregation.
Lyso-PC & NEFA
*Graphical Abstract (for review)

Highlights
LRP1-derived peptides prevent SMase-induced alterations in LDL particle size,
electric charge and phospholipid content.
LRP1-derived peptides do not prevent PLA2-induced alterations in LDL particle
size, electric charge and phospholipid content.
LRP1-derived peptides prevent lipolytic-induced LDL aggregation through
electrostatical interaction with basic ApoB-100 sequences key for preservation
of ApoB-100 conformation.
Highlights (for review)

Manuscript BBAMEM-19-17 (R1)
1
Molecular basis for the protective effects of low-density lipoprotein receptor-related protein 1
(LRP1)-derived peptides against LDL aggregation
Short title: LRP1-derived peptides and LDL aggregation
Aleyda Benitez-Amaroa,b, Chiara Pallarac, Laura Nasarrea, Andrea Rivas-Urbinad, Sonia Benitezd,
Angela Veaa, Olga Bornacheaa,b, David de Gonzalo-Calvoa,b,e, Gabriel Serra-Mirf, Sandra Villegasf,
Roger Pradesc, José Luís Sanchez-Quesadad,g, Cristina Chivah,i, Eduard Sabidoh,i, Teresa
Tarragóc, and Vicenta Llorente-Cortésa,b,e**
aGroup of Lipids and Cardiovascular Pathology. Biomedical Research Institute Sant Pau (IIB Sant
Pau), Hospital de la Santa Creu i Sant Pau. Barcelona. Spain.
bInstitute of Biomedical Research of Barcelona (IIBB). Spanish National Research Council (CSIC),
Barcelona, Spain.
cIproteos S.L., Barcelona Science Park (PCB), Barcelona, Spain.
dCardiovascular Biochemistry Group, Research Institute of the Hospital de Sant Pau (IIB Sant Pau),
Barcelona, Spain.
eCIBER enfermedades cardiovasculares (CIBERcv).
fProtein Design and Immunotherapy Group. Departament de Bioquímica i Biologia Molecular,
Facultat de Biociències, Universitat Autònoma de Barcelona, Bellaterra, Barcelona, Spain.
gCIBER diabetes y enfermedades metabólicas asociadas (CIBERdem)
hProteomics Unit, Centre de Regulació Genòmica, Barcelona Institute of Science and Technology,
Barcelona, Spain.
iUniversitat Pompeu Fabra, Barcelona, Spain.
**To whom correspondence should be addressed: Vicenta Llorente-Cortés, Lipids and
Cardiovascular Pathology group leader, ICCC Program, Biomedical Research Institute IIB Sant
Pau, Hospital de la Santa Creu i Sant Pau, Sant Antoni Mª Claret, 167, 08025 Barcelona. Phone:
+34 935565888, Fax: +34 935565559, E-mail: [email protected]; [email protected]
*REVISED Manuscript (text UNmarked)Click here to view linked References

Manuscript BBAMEM-19-17 (R1)
2
Abstract
Aggregated LDL is the first ligand reported to interact with the cluster II CR9 domain of low-density
lipoprotein receptor-related protein 1 (LRP1). In particular, the C-terminal half of domain CR9,
comprising the region Gly1127-Cys1140 exclusively recognizes aggregated LDL and it is crucial for
aggregated LDL binding. Our aim was to study the effect of the sequence Gly1127-Cys1140 (named
peptide LP3 and its retro-enantio version, named peptide DP3) on the structural characteristics of
sphingomyelinase- (SMase) and phospholipase 2 (PLA2)-modified LDL particles. Turbidimetry, gel
filtration chromatography (GFC) and transmission electronic microscopy (TEM) analysis showed
that LP3 and DP3 peptides strongly inhibited SMase- and PLA2-induced LDL aggregation.
Nondenaturing polyacrylamide gradient gel electrophoresis (GGE), agarose gel electrophoresis and
high-performance thin-layer chromatography (HPTLC) indicated that LP3 and DP3 prevented
SMase-induced alterations in LDL particle size, electric charge and phospholipid content,
respectively, but not those induced by PLA2. Western blot analysis showed that LP3 and DP3
counteracted changes in ApoB-100 conformation induced by the two enzymes. LDL proteomics
(LDL trypsin digestion followed by mass spectroscopy) and computational modeling methods
evidenced that peptides preserve ApoB-100 conformation due to their electrostatic interactions with
a basic region of ApoB-100. These results demonstrate that LRP1-derived peptides are protective
against LDL aggregation, even in conditions of extreme lipolysis, through their capacity to bind to
ApoB-100 regions critical for ApoB-100 conformational preservation. These results suggests that
these LRP1(CR9) derived peptides could be promising tools to prevent LDL aggregation induced by
the main proteolytic enzymes acting in the arterial intima.
Key words: Lipoprotein aggregation; LRP1-derived peptides; ApoB-100; SMase, PLA2,
atherosclerosis

Manuscript BBAMEM-19-17 (R1)
3
Abbreviations: agLDL, aggregated LDL; apo, apolipoprotein; CE, cholesteryl esters; FC, free
cholesterol; hcVSMC, human coronary vascular smooth muscle cells; GFC, gel filtration
chromatography; GGE, nondenaturing polyacrylamide gradient gel electrophoresis; HPTLC, high-
performance thin-layer chromatography; LC-MS/MS, liquid chromatography tandem-mass
spectrometry; LDL, low-density lipoprotein; LysoPC, lysophosphatidylcholine; LRP1, low-density
lipoprotein receptor-related protein 1; NEFA, nonesterified fatty acids; PC, phosphatidylcholine;
PLA2, phospholipase A2; PL, phospholipids; REMD, Replica Exchange Molecular Dynamics; SM,
sphingomyelin; SMase, sphingomyelinase; TEM, transmission electron microscopy

Manuscript BBAMEM-19-17 (R1)
4
1. Introduction
Ischemic heart disease is the first cause of death in Western countries, and myocardial infarction
accounts for about 50% of deaths from this disease. The Framingham study showed that
cardiovascular risk positively correlates with low-density lipoprotein (LDL)-cholesterol and inversely
with high-density lipoprotein (HDL)-cholesterol 1,2. High levels of LDL-cholesterol induce
alterations in the endothelium permeability, which in turn promote LDL influx into the arterial intima
3. Intimal LDL-cholesterol accumulation is a critical step in vascular cholesteryl ester (CE)
deposition, a process that increases the tendency of the atherosclerotic plaque to rupture, triggering
thrombosis and the development of ischemic cardiomyopathy 4-6. CEs in atherosclerotic plaques
are deposited both extra- and intra-cellularly. Extracellular deposition of LDL-CEs, a crucial initiating
event in atherosclerosis 7, is mediated by proteoglycans in the extracellular matrix of the arterial
intima. The electrostatic interaction between proteoglycans and LDL and the proteolytic/lipolytic
actions of enzymes on LDL are enhanced in the arterial intima and promote LDL retention and
aggregation 8,9. Two of the main enzymes acting on LDL are sphingomyelinase (SMase),
secreted by endothelial cells and macrophages 10,11 and phospholipase A2 (PLA2) 12,13. These
two enzymes play key roles in LDL aggregation in the arterial intima during atherogenesis 12-15.
Aggregated LDL (agLDL) has been detected and isolated from atherosclerotic plaques from animal
models and humans 11,16. Unlike native LDL, agLDL is a potent inducer of massive intracellular
CE accumulation both in macrophages 17-19 and human coronary vascular smooth muscle cells
(hcVSMCs) 20. In particular, in hcVSMCs, agLDL is actively taken up through the low-density
lipoprotein receptor-related protein 1 (LRP1) 21,22. The internalization of agLDL, in turn, induces
LRP1 expression 23,24, promoting a positive feedback loop that efficiently transforms hcVSMCs
into foam cells. We have also demonstrated that hcVSMC-foam cells synthesize and release high
amounts of tissue factor, which is crucial for the pro-thrombotic transformation of the vascular wall
and thus for the progression of atherosclerosis to thrombosis 25,26. The relevance of this
mechanism in atherosclerosis is evident since VSMCs are the main components of the vascular wall

Manuscript BBAMEM-19-17 (R1)
5
and more than 50% of foam cells previously considered to be monocyte-derived macrophages in
human atherosclerotic plaques originate from VSMCs 27. Together, these findings support the
notion that the generation of hVSMC-derived foam cells through LRP1-mediated agLDL uptake is a
key mechanism underlying cholesterol accumulation in vasculature susceptible to atherosclerosis.
Our group has identified LRP1 cluster II, in particular the region Gly1127-Cys1140 (peptide LP3: H-
GDNDSEDNSDEENC-NH2) that spans the C-terminal half of domain CR9, as pivotal for binding to
agLDL and subsequent internalization of this agLDL into human VSMCs 28. We hypothesize that
peptide LP3 or its retro-enantio version, peptide DP3, would counteract LDL aggregation. Using a
range of biochemical, biophysical and molecular experiments, here we demonstrate that LP3 and
DP3 peptides efficiently prevent LDL aggregation induced by the lipolytic enzymes SMase and
PLA2. Although LP3 and DP3 efficiently counteracted alterations in LDL particle size, charge and
SM loss induced by SMase, they were unable to prevent the alterations induced in the same
parameters by PLA2. However, both peptides protected ApoB-100 from conformational alterations
induced by SMase and PLA2. Proteomics, ab initio modeling and molecular dynamics showed that
LRP1-derived peptides establish stable electrostatic interactions with basic sequences of ApoB-100.
2. Material and Methods
2.1. Peptide sequences and synthesis
Peptide sequences are written from N to C, H- at the end terminus indicate amine ending, whereas
–NH2 at the C terminus indicates amide. The amino acid sequence of LP3 peptide (parent) is H-
GDNDSEDNSDEENC-NH2, DP3 is the retro-enantio version of LP3 without cysteine: H-
needsndesdndh-NH2, where the amino acid letter code in lowercase stands for amino acids with D-
chirality. As a negative control, we used an ApoB-100-based peptide coded as IP321 with the
sequence: H-RLTRKRGLK-NH2. We also compared the efficacy of LP3, DP3 and IP321 with the
previously described anti-atherosclerotic apoA-I mimetic, 4F. The latter is 18 amino acids long and
has the following sequence: Ac-DWFKAFYDKVAEKFKEAF-NH2 VSMCs 29. All peptides were

Manuscript BBAMEM-19-17 (R1)
6
synthesized by Iproteos by standard 9-fluorenylmethoxycarbonyl/tert-butyl (Fmoc/tBu) solid-phase
peptide synthesis. Syntheses were performed manually on a 100-µmols scale/each using Rink
amide ChemMatrix® resin. Peptide elongation and other solid-phase manipulations were done
manually in polypropylene syringes, each fitted with a polyethylene porous disk. Solvents and
soluble reagents were removed by suction. Washings between synthetic steps were done with
dimethylformamide and dichloromethane using 5 mL of solvents/g resin each time. Nα-Fmoc-
protected amino acids (4 equivalents) were coupled using 2-(1H-benzotriazole-1-yl)-1,1,3,3-
tetramethyluronium (TBTU, 4 equivalents), and N,N-diisopropylethylamine (8 equivalents). The
extent of the reaction was monitored using the Kaiser test (primary amines). During couplings, the
mixture was allowed to react with intermittent manual stirring. Fmoc group was removed by treating
the resin with 20% piperidine in DMF (3 mL/g resin). The peptides were cleaved from the resin using
a mixture of trifluoroacetic acid, triisopropylsilane and water (95:2.5:2.5). The crude products
obtained were purified in reverse phase using a semi-prep HPLC instrument equipped with a C18
column. The purity and identity of the synthesized peptides was assessed by HPLC, HPLC-MS and
MALDI-TOF analysis (Table S1).
2.2. Peptide stability in human serum and cell culture medium
The stability of the peptides was studied in human serum (Sigma-Aldrich). For the stability assay,
the peptides were dissolved in a mixture of 10% Hanks’ Balanced Solution Salt (HBSSx1) and
human serum. The final concentration of peptide was 200 mM VSMCs 30. The samples were
incubated at 37°C with orbital agitation for 24 h. At selected time points, serum aliquots were
extracted and the serum proteins of the mixture were then precipitated by adding methanol.
Samples were then centrifuged at 4°C, and the supernatant was filtered out and analyzed by HPLC.
Peptide stability during the assays was monitored using HPLC (UV detection at 220 nm).
2.3. LDL isolation and purification
Human LDL (d1.019-d1.063 g/mL) was obtained from pooled normolipemic human plasma by
sequential ultracentrifugation in a KBr density gradient. Briefly, very low density lipoproteins (VLDLs)

Manuscript BBAMEM-19-17 (R1)
7
were first discarded after spinning plasma at 36,000 rpm for 18 h at 4ºC using a fixed-angle rotor
(50.2 Ti, Beckman) mounted on an Optima L-100 XP ultracentrifuge (Beckman). Subsequently,
VLDL-free plasma was layered with 1.063 g/mL KBr solution and centrifuged at 36,000 rpm for 18 h
at 4ºC. LDLs were dialyzed against 0.02 M Trizma, 0.15 M NaCl, 1 mM EDTA, pH 7.5 for 18 h, and
then against normal saline for 2 h. Finally, isolated LDLs were filter-sterilized (0.22 μm Millex-GV
filter unit, Millipore). Protein concentration was determined using the BCA protein assay (Thermo
Scientific) and the cholesterol concentration with a commercial kit (IL test Cholesterol, Izasa).
2.4. Analysis of SMase activity in the presence and absence of peptides
The capacity of LP3 and DP3, IP321 and manumycin A (positive control) to inhibit
sphingomyelinase activity was measured using the Amplex Red Sphingomyelinase Assay Kit
(Molecular Probes Invitrogen Detection Technologies, #A12220), an assay that measures the
formation of phosphorylcholine. To measure SMase activity in the presence and absence of
peptides, DMSO volume and peptide concentrations were kept constant [10 M (molar ratio
peptide/ApoB= 4.7/1)] while SMase concentration was increased (0, 0.2, 0.4, 0.6, 0.8 and 1 mU/ml).
Each reaction contained 50 μM Amplex Red reagent, 1 U/mL HRP, 0,1 U/ml choline oxidase, 4 U/ml
of alkaline phosphatase, 0,25 mM sphingomyelin (diluted with Triton X-100 or with the Reaction
Buffer 1x). Reactions were incubated at 37ºC for 30 min. Fluorescence intensity was measured
using a fluorescence microplate reader (Victor 1420 multilabel counter) using excitation at 530 nm
and fluorescent detection at 590nm.
2.5. LDL modification by Sphingomyelinase (SMase) and Phospholipase A2 (PLA2) in
the presence and absence of peptides
Human LDL (1.44 mg/mL) were incubated with 40 U/L of Bacillus cereus SMase (Sigma-Aldrich,
Schnelldorf, Germany) or with 50 µg/L of type II secretory PLA2 from honey bee venom (Sigma-
Aldrich, Schnelldorf, Germany) in 20 mM Tris buffer (pH 7.0) containing 150 mM NaCl, 2 mM CaCl2,
and 2 mM MgCl2 at 37ºC. LDL incubation with SMase and PLA2 was performed at peptide
concentrations of 0, 2.5, 5, 7.7, 8.8, 10 µM peptide/ApoB-100 ratio :0/1, 1.2/1, 2.4/1, 3.5/1, 4.1/1,

Manuscript BBAMEM-19-17 (R1)
8
4.7/1) and 0, 4.2, 6.3, 10.0, 14.6, 20.8 µM peptide/ApoB-100 ratio (0/1, 2.0/1, 2.9/1, 4.7/1, 6.8/1,
9.8/1), respectively, for increased time periods. LDL lipolysis was stopped by addition of EDTA
(final concentration 10 mM).
2.6. Characterization of SMase- and PLA2-induced LDL alterations in the presence and
absence of peptides. A detailed information of the different procedures
2.6.1.LDL Composition_The LDL composition was measured by commercial methods adapted to a
Cobas 6000/c501 autoanalyzer (Roche Diagnostics, Indianapolis, IN, USA). Total cholesterol,
triglycerides and ApoB reagents were obtained from Roche Diagnostics. PLs, NEFAs, and free
cholesterol reagents were obtained from Wako Pure Chemicals (Tokyo Japan).
2.6.2.Turbidity_Sample turbidity (LDL aggregation monitorization) was determined by measuring
absorbance of 100 µl of LDLs (1.44 mg/mL ApoB) in a 96-well microplates at 405 nm.
2.6.3.Gel Filtration Chromatography (GFC)_To separate aggregated from non-aggregated LDL,
samples (50 µL at 1.44 mg/mL) were subjected to gel filtration chromatography (GFC) in two online
connected Superose 6 Increase 5/150 GL columns with an AKTA-FPLC system (GE Healthcare) at
a flow of 0.3 mL/min 31,32. The area under the curve of each peak was calculated using the
Unicorn 4.0 software (GE Healthcare).
2.6.4. Agarose gel electrophoresis_LDL (1.5 µL of LDL (1.44 mg/mL) was electrophoresed on 0.5%
agarose gels at 65V for 40 min followed by 90V for 15 min. Fixed gels were dehydrated and stained
with Coomassie Blue. Decolored gels were captured with the Bio-Rad GS-800 scanner.
2.6.5. SDS-PAGE and Western blot
ApoB-100 was detected by Western blot analysis after electrophoresis of LDL in 6% SDS-PAGE
gels. Nitrocellulose membranes were incubated with anti-ApoB-100 polyclonal antibodies (Roche
Diagnostics S.L., Ref: 3032574122, polyclonal anti-human ApoB-100, dilution 1: 5000) for 1 h at

Manuscript BBAMEM-19-17 (R1)
9
room temperature. After several washes, membranes were incubated with secondary anti-sheep
antibodies (dilution 1: 10000, Dako; Glostrup, Denmark). Bands were detected using ECL Prime
Western Blotting Detection Reagent (Amersham) and quantified by densitometry using a ChemiDoc
system and Quantity One software (Bio‐Rad, Hercules, CA, USA). The results are expressed as
arbitrary units of intensity.
2.6.6.TEM
TEM was performed as previously described 32. LDL was placed on a 400 mesh glow-discharge
carbon-coated grid, negatively stained with 2% potassium phosphotungstate, pH 7.0, and air dried.
Grids were placed in a Jeol 120-kV JEM-1400 (Jeol, Japan) transmission electron microscope. TEM
images were processed using ImageJ (Fiji) software. Briefly, contrast was enhanced, followed by
background subtraction.
2.6.7. Nondenaturing acrylamide gradient gel electrophoresis (GGE)
GGE was performed following Nichols et al., with small modifications 32. Two solutions at 2% and
16% were prepared by using a stock solution of acrylamide and bis-acrylamide (30% total, 5%
cross-linker) and mixed using 2 P-1 peristaltic pumps (Pharmacia). The different samples (5 μL at
1.44 mg/mL) were preincubated for 15 min with 10 μL Sudan black (0.1% [wt/vol]) in ethylene glycol
and 5 μL saccharose (50% [wt/vol]). Ten microliters of this mixture was electrophoresed at 4°C for
30 min at 20 V, 30 min at 70 V, and 4 h at 100 V. Bands were scanned by densitometry.
2.6.8. HPTLC
LDL (15 µg) was lipid-extracted with organic solvents as previously described by our group with
minimal modifications 25. Samples were applied to horizontal, one-dimensional high-performance
thin-layer chromatography (HPTLC) (Merck, HPTLC Silica Gel 60, 1.05641.0001). Phospholipids
were separated on HPTLC by chloroform:ethyl
acetate:acetone:isopropanol:ethanol:methanol:water:acetic acid (30:6:6:6:16:28:6:2); visualized by

Manuscript BBAMEM-19-17 (R1)
10
staining with 7.5% Cu-acetate (w/v), 2.5% CuSO4 (w/v), and 8% H3PO4 (v/v) in water. A mixture of
phospholipids (Sigma Ref pH-7) were also run as standards. The spots corresponding to the
different phospholipids were quantified by densitometry against the standard curve using a GS-800
Calibrated Densitometer (Bio-Rad).
2.7. Proteomics analyses of LDL samples
2.7.1. Sample preparation_LDL, LDL unexposed or exposed to SMase in the absence of peptide or
in the presence of DP3 or IP321) were directly subjected to limited trypsin digestion (1 µg) for 1 h or
3 h. In both cases, tryptic peptides were desalted using a C18 column and evaporated to dryness.
2.7.2. Mass spectrometry data acquisition_ LDL samples were analyzed using a LTQ-Orbitrap Velos
Pro mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) coupled to an EasyLC
(Thermo Fisher Scientific (Proxeon), Odense, Denmark). Peptides were separated by reverse-
phase chromatography using a 25-cm column with an inner diameter of 75 μm, packed with 5 μm
C18 particles (Nikkyo Technos Co., Ltd. Japan). Chromatographic gradients started at 93% buffer A
and 7% buffer B and gradually increased to 65% buffer A / 35% buffer B in 60 min at a flow rate of
250 nl/min. The mass spectrometer was operated in positive ionization mode with nanospray
voltage set at 2.2 kV and source temperature at 250°C. Ultramark 1621 for the FT mass analyzer
was used for external calibration prior to the analyses, and an internal calibration was performed
using the background polysiloxane ion signal at m/z 445.1200. The acquisition was performed in
data-dependent acquisition mode, and full MS scans with 1 micro scans at a resolution of 60,000
were used over a mass range of m/z 350-2000 with detection in the Orbitrap. Auto gain control
(AGC) was set to 1E6, dynamic exclusion (60 seconds) and charge state filtering disqualifying singly
charged peptides was activated in each cycle of data-dependent acquisition analysis. After each
survey scan, the top ten most intense ions with multiple charged ions above a threshold ion count of
5000 were selected for fragmentation at normalized collision energy of 35%. Fragment ion spectra
produced via collision-induced dissociation (CID) were acquired in an Ion Trap apparatus. AGC set

Manuscript BBAMEM-19-17 (R1)
11
to 5e4, an isolation window of 2.0 m/z, activation time of 0.1ms and maximum injection time of 100
ms were used. All data were acquired with Xcalibur software v2.2.
2.7.3. Mass spectrometry data analysis
The spectra acquired for the LDL samples were analyzed using the Proteome Discoverer software
suite (v2.0, Thermo Fisher Scientific) and the Mascot search engine (v2.5, Matrix Science). The
data were searched against the Swiss-Prot human database (v.2017/10). At the MS1 level, a
precursor ion mass tolerance of 7 ppm was used, and up to three missed cleavages were allowed.
The fragment ion mass tolerance was set to 0.5 Da for MS2 spectra. Methionine oxidation and N-
terminal protein acetylation were defined as variable modifications, whereas carbamidomethylation
on cysteine residues was set as a fixed modification. False discovery rate (FDR) in peptide
identification was limited to a maximum of 5% using a decoy database. Quantitation data were
retrieved from the “Precursor ion area detector” node of Proteome Discoverer (v2.0) using 2 ppm
mass tolerance for the peptide extracted ion current (XIC). Finally, for each LDL sample, Skyline
software v4.1 was used to build a spectral library for the ApoB100 protein with the corresponding
identified spectra and to extract all its MS1 areas. Extracted ion chromatograms for the different LDL
samples have been deposited to Panorama with identified PXD011261; URL
(https://panoramaweb.org/j8CaUy.url).
2.8. Molecular modeling of peptide-LDL interactions
The structural model of ApoB-100 in complex with DP3 peptide was built using rigid body molecular
docking simulation and starting from the isolated ab initio models of both docking partners. First of
all, an exhaustive conformational sampling was performed on the peptide in order to identify the
most stable and populated conformational states, which were assumed to be the most favorable to
bind ApoB-100 protein. For that, on one hand, a 3D structure of DP3 was designed from scratch
and consequently used as input for Replica Exchange Molecular Dynamics (REMD) simulations in
implicit water using the AMBER ff12SB force field 33 and NAMD software 34. In each REMD
scheme, 16 independent 100 ns-long simulations (replicas) were performed simultaneously at

Manuscript BBAMEM-19-17 (R1)
12
temperatures ranging from 300 to 613 K. Finally, the representative structure of the most populated
cluster was extracted and used as input structure for the docking simulation. On the other hand, the
3D structure of ApoB-100 protein was built by ab initio protein modeling, since no crystal structure of
protein was available and very low sequence identity templates (ranging from 8 to 13%) were found
in the PDB. Indeed, this computational technique tends to require vast resources, thus only a small
domain of roughly 100 residues around the basic region believed to be responsible for LDL receptor
binding (3189-3261 residues) was selected for modeling. Consequently, a total of 10 ab initio
models were built using ROBETTA server 35 and finally used as input structures for the docking
simulation. Starting from each ApoB-100 protein structure and DP3 peptide previously obtained, 10
independent rigid-body docking simulations were performed using AutoDock Vina software fork
Smina 36 and setting the docking grid box around Apo-B100 3227IKFDKYKAEK3236 region. Finally,
all the docking results were merged and the lowest-energy poses were visually inspected.
2.9. Statistical analysis
Data are presented as mean±SD. One-way ANOVA followed by the Bonferroni or the Storey (q-
value) correction was performed for comparison of >2 groups. Student’s t test was performed for
comparison of 2 groups. P<0.05 was considered significant.
3. Results
3.1. DP3 shows higher stability than the parent peptide LP3 in human serum
Peptide drugs offer high activity, high specificity and low toxicity, but are susceptible to proteolytic
degradation. A strategy to overcome this problem is to produce peptides using non-natural or D-
amino acids, which shows improved stability. Although all-D peptides are not recognized by plasma
proteases, they may be less active than their corresponding all-L peptides because of differences in
the orientation of amino acid side-chains. To preserve the orientation of the side-chains of the
parent peptide in a D-analogue, the D-amino acid residues can be linked in the reverse order to that
of the parent peptide in order to produce a retro-enantio peptide. This approach emphasizes the

Manuscript BBAMEM-19-17 (R1)
13
maintenance of the topological native orientation of the side-chains but not of the backbone, where
the amide bond is shift one unit 37. By using this approach, peptides usually preserve their activity
while notably improve their plasma stability. In the literature there are examples where this approach
has been successfully used 30. LP3 and DP3 were synthesized by means of solid-phase peptide
synthesis using a Fmoc/tBu strategy. The purity of both peptides (≥98%) was assessed by HPLC
analysis (Supplemental Fig. S1). Other parameters indicative of the purity and identity of the
synthesized peptides were obtained by HPLC-MS and MALDI-TOF analysis (Supplemental Table
S1). To study the stability of the parent peptide (LP3), it was dissolved in a mixture of HBSS (x1)
and human serum. LP3 showed moderate stability with a calculated half-life of 8 h (Supplemental
Fig. S2A). To achieve an increase in the stability of LP3 for the purpose of in vivo studies, a retro-
enantio version of the peptide (DP3) was synthesized. This approach consisted of using all D-amino
acids and reversing the sequence of the parent peptide. This synthetic strategy has been shown to
be useful 38. As the retro-enantio version of the peptide is synthesized using D-amino acids, the
sequence is not recognized by serum proteases, thus extending its half-life considerably
(Supplemental Fig. S2B).
3.2. Time- and dose-response blocking effects of LP3 and DP3 on SMase- and PLA2- induced
LDL turbidity
To study the effects of the peptides on SMase-induced LDL aggregation, LDL was incubated with
SMase and PLA2 in the presence and absence of LP3, DP3 and IP321 (negative control) under the
experimental conditions detailed in Supplemental Fig. S3. The peptide concentrations and
peptide/ApoB-100 molar ratios tested in the aggregation of SMase-modified LDL (SMase-LDL) are
detailed in Supplemental Table S2. LP3 and DP3 reduced SMase-LDL aggregation at all the times
tested (4, 6 and 18 h) in a dose-dependent manner (Fig. 1A). DP3 showed a higher inhibitory
capacity than LP3 at concentrations of 5.0, 7.7 and 8.8 µM, and SMase-LDL aggregation was fully
inhibited by LP3 at 10 µM (peptide/ApoB-100 ratio: 4.7/1) and by DP3 at 7.7 µM (peptide/ApoB-100
ratio: 3.5/1). To study the effects of the peptidomimetics on the aggregation of PLA2-modified LDL,

Manuscript BBAMEM-19-17 (R1)
14
LDL was incubated with PLA2 in the presence or absence of LP3, DP3 and IP321 at the doses and
peptide/ApoB-100 ratios detailed in Supplemental Table S3. We did not detect any effect of PLA2 on
LDL aggregation at time points lower than 24 h (data not shown). LP3 and DP3 showed high
inhibitory activity on PLA2-LDL aggregation, although, as in the case of SMase, DP3 showed
outperformed LP3 at concentrations of 10 µM and 14.6 µM at 24, 30 and 36 h (Fig. 1B). PLA2-LDL
aggregation was fully inhibited by LP3 at a concentration of 20.83 µM (peptide/ApoB-100 ratio:
9.8/1) and by DP3 at 14.6 µM (peptide/ApoB-100 ratio: 6.8/1) at all tested times. IP321 did not
inhibit SMase- or PLA2-LDL aggregation at any dose or time tested (Fig. 1A,B). Given that
Apolipoprotein A-I peptidomimetic 4F has been reported to block SMase-LDL aggregation 29, we
compared the capacity of LP3, DP3 and 4F (10 M, ratio 4.7/1, 24 h) to inhibit SMase- and PLA2-
LDL aggregation. SMase-induced aggregation was almost fully blocked by LP3 (86.2%±1.8) and
DP3 (98.1%±1.4), but only partially blocked by 4F (55.7%±2.3). PLA2-LDL aggregation was partially
blocked by LP3 (59.6%±0.8) and 4F (45.8%±1.1) and almost completely inhibited by DP3
(84.4%±1.9) (Table 1). IP321 did not have any significant effect on SMase- or PLA2-LDL
aggregation. None of the peptides alter the turbidimetry values of untreated LDL (Supplemental Fig.
S4).
3.3. LP3 and DP3 decrease the percentage of aggregated particles in SMase-LDL and PLA2-
LDL
To separate aggregated from monomeric LDL particles, LDL samples were subjected to gel filtration
chromatography (GFC). GFC showed that LDL comprised mainly monomeric LDL (Fig. 2a). A small
percentage of residual LDL aggregates was detected in untreated LDL due to the fact that both
untreated and treated LDL were exposed to 37ºC for the same period (Supplemental Fig. S3). In
contrast to LDL, SMase-LDL (Fig. 2b) was composed mainly of LDL aggregates. LP3 (Fig. 2c) and
DP3 (Fig. 2d) partially prevented the formation of LDL aggregates, DP3 showing higher efficiency
than LP3. IP321 did not alter the GFC pattern observed in SMase-LDL (Fig. 2e). As observed with
SMase-LDL, PLA2-LDL (Fig. 2f) showed a large peak of LDL aggregates. LP3 (Fig. 2g), but

Manuscript BBAMEM-19-17 (R1)
15
specially DP3 (Fig. 2h), increased the percentage of monomeric LDL and decreased that of LDL
aggregates. IP321 did not cause any significant alteration in the pattern of PLA2-LDL (Fig. 2i).
LRP1-derived peptides did not alter per se the GFC pattern of LDL (Supplemental Fig. S5).
Quantification of peak areas showed that LP3 and DP3, but not IP321, conferred SMase-LDL and
PLA2-LDL with a GFC profile closer to that of LDL (Fig. 2j,k). TEM images of LDL size distribution
(Fig. 3) showed that LDL comprised mainly individual LDL particles (Fig. 3A). In contrast, SMase-
LDL (Fig. 3B) and PLA2-LDL (Fig. 3C) generated in the presence of IP321 or in the absence of
peptides were formed mostly by aggregated and fused LDL. In the presence of LP3 and DP3,
SMase-LDL (Fig. 3B) and PLA2-LDL (Fig. 3C) were composed of LDL particles with a wide variety
of diameter sizes, although most were in the range present in LDL.
3.4. LP3 and DP3 block the alterations in LDL phospholipid composition in SMase-LDL but
not in PLA2-LDL
Lipoprotein composition analysis (Table 2) revealed that exposure of LDL to SMase or PLA2 did not
alter cholesterol, triglyceride or ApoB content of LDL, either in the presence or absence of the
peptides. In contrast, phospholipid (PL) content of LDL was significantly reduced by both enzymes
(P<0.005). LP3 and DP3 efficiently prevented PL reduction in SMase-LDL (P<0.005). However,
peptides did not show any effect on PL depletion or on non-esterified fatty acid (NEFA) increase
induced by PLA2 treatment. To identify the PL species influenced by enzymatic treatment, HPTLC
was used to separate LDL PLs. This procedure revealed that the main PLs in the LDLs were L--
phosphatidylcholine (L--PC) and sphingomyelin (SM). L--PC levels were unaltered by SMase
treatment (Fig. 4A,B). In contrast, SM was removed from LDL by this treatment (Fig. 4A,C). LP3 and
DP3, but not IP321, efficiently protected LDL-SM against SMase-induced SM degradation (Fig.
4A,C). To examine whether the protective action of LRP1-derived peptides on LDL was due to their
capacity to inhibit SMase activity, we tested the effect of these compounds on the capacity of
SMase to promote SM lipolysis. SMase activity, measured in relation to the amount of
phosphorylcholine generated, was similar in the presence and absence peptides, thereby indicating

Manuscript BBAMEM-19-17 (R1)
16
that none of the peptides exerted direct effects on SMase activity (Supplemental Fig. S6). PLA2
completely removed L--PC from LDL (Fig. 4D,E), leading to a sharp increase in the L--LysoPC
content (Fig. 4D,F). PLA2 did not have any effect on the LDL-SM content (Fig. 4D,G). The peptides
were unable to counteract L--PC decay and the increase in L--LysoPC induced by PLA2 in LDL.
3.5. LP3 and DP3 block the alterations in the LDL electrophoretic profile of SMase-LDL but
not those of PLA2-LDL
To study the effects of the peptides on the electrophoretic mobility of SMase- and PLA2-LDL, we
used gradient gel electrophoresis (GGE) and agarose gels to analyze the modification of LDL in the
presence and absence of the peptides. In native GGE gels (4 h of electrophoresis), the position of
the LDL particle was observed to be determined by particle size. GGE gels showed that SMase
caused a strong loss of the band corresponding to LDL. The lack of this band in SMase-LDL is
attributed to the fact that the pore size of GGE gels precludes the entrance of the largest LDL
aggregates. The band loss induced by SMase was efficiently counteracted by LP3 and DP3 but not
by IP321 (Fig. 5A). In PLA2-LDL (Fig. 5B), GGE showed a minor band corresponding to LDL
(marked as band 1), an intensive band (marked as band 2) with higher electrophoretic mobility
(smaller size) than band 1, and a third band (marked as band 3), which was a minor band with lower
electrophoretic mobility (higher size) than band 1. LP3 and DP3 did not have any effect on the GGE
pattern of PLA2-LDL.
Agarose gels, where LDL runs according to its negative charge, showed that SMase-LDL had a
more diffuse band compared to the well-defined LDL band. SMase-LDL in the presence of LP3 and
DP3 also showed a defined LDL band (Fig. 5C). In contrast to SMase-LDL, PLA2-LDL showed a
single band with a higher negative electric charge than LDL combined with a degradation smear
pattern (Fig. 5D). LP3, and in particular DP3, slightly increased the electrophoretic mobility and
band intensity of PLA2-LDL while they diminished the degradation smear pattern. The peptides did
not cause any change in the electrophoretic mobility of untreated LDL (Supplemental Fig. S7).

Manuscript BBAMEM-19-17 (R1)
17
3.6. DP3 blocks the conformational changes of ApoB-100 in SMase-LDL and PLA2-LDL
We studied the potential conformational modifications of ApoB-100 by Western blot analysis.
Treatment of LDL with SMase (Fig. 6A) or PLA2 (Fig. 6B) increased the exposure of reactive ApoB-
100 epitopes to anti-ApoB-100 antibodies. Both LP3 and DP3 protected ApoB-100 against the
higher reactivity to anti-ApoB-100 antibodies induced by exposure to SMase and PLA2 (Fig. 6A,B).
IP321 had no effect on the increased ApoB-100 reactivity induced by the two enzymatic treatments.
3.7. LRP1-derived peptides electrostatically interact with specific lysine-enriched sequences
in ApoB-100 protein
Limited proteolysis combined with mass spectrometry-based proteomics (LC-MS/MS) evidenced
that DP3 protects specific sequences of ApoB-100 in LDL either by direct interaction or by
remodeling the protein structure (Fig. 7A). The protein/peptide complexes were subjected to limited
proteolysis with trypsin for 1 h and 3 h. The protection of protein sequences by the peptide was
observed by shotgun LC-MSMS analysis at short times (The whole data concerning LDL is available
via PanoramaPublic with identifier PXD011261). At 3-hours digestions, the effect disappeared. The
transient protection of the peptide can be explained by the proteolytic action that trypsin exerted on
the protein complex. Remarkably, most of the protected sequences were also protected in SMase-
LDL unexposed to peptides, suggesting that the aggregation process safeguards them from trypsin
digestion and that these specific sequences are likely to be those that play a major role in LDL
aggregation. The high coincidence between sequences protected by DP3 and those protected by
aggregation indicates that most DP3-protected sequences participate in LDL aggregation, thereby
explaining the high efficiency of LRP1-derived peptides to prevent LDL aggregation.
To further study the molecular basis for ApoB-100 recognition by DP3, peptide homology modeling
and molecular docking methods were used. First, to identify a potential binding site of DP3 peptide
on ApoB-100 protein structure, each ApoB-100 fragment protected by DP3 binding during trypsin
digestion was mapped on the overall ApoB-100 sequence. Virtually all the fragments corresponded
to lipid-associating domains 39, except for a highly positively charged sequence
(3227IKFDKYKAEK3236) located in the ApoB-100 C-terminal region and adjacent to the LDL receptor

Manuscript BBAMEM-19-17 (R1)
18
binding domain (3385RLTRKRGLK3393) 40-42. This sequence was thus assumed to be responsible
for DP3 peptide recognition and used as the binding site for the molecular docking calculations.
Finally, an energy-based rigid body docking experiment was performed between ApoB-100 protein
and DP3 peptide structures previously modeled alone by ab initio methods. Interestingly, we found
that one of the lowest-energy poses of the docking simulations accommodated DP3 in such an
orientation that ApoB-100 Lys3228, Lys3231 and Lys3233 (but not Lys3226 nor Lys3236) were deeply buried
upon peptide binding. This finding is in strong agreement with the trypsin digestion data described
above. Additionally, strong electrostatic interactions involving same specific key residues of the two
docking partners were also found. In this regard, two of the acidic residues in DP3 correspond to
those assumed to be responsible for the specific recognition between LRP1 module CR9 and
agLDL particles [PMID 25918169] (namely D-Glu3 and D-Glu9) established salt bridge contacts with
two positively charged ApoB-100 residues (namely Lys3229 and Lys3234), while the oxygen atom of
the DP3 D-Asn2 backbone and ApoB-100 Lys3232 were found to be involved in a hydrogen bond
favored by the positive ionic charge carried by the lysine side chain amine group (Fig. 7B).
4. Discussion
A previous study from our group demonstrated that LRP1, in particular the C-terminal half of domain
CR9 (from Gly1127 to Cys1140) named LP3, interacts with agLDL 28. In the current study, we tested
the effect of both the original LP3 version and its retro-enantio version DP3 on SMase and PLA2-
induced LDL aggregation.
In the present study, we observed that PLA2 required larger times and doses than SMase to induce
LDL aggregation. This finding is consistent with previous studies showing that LDL hydrolysis by
PLA2 is slower than that by SMase 43-45. The latter cleaves SM into phosphocholine and
ceramide, whereas PLA2 hydrolyzes the fatty acids in the sn-2 position of PC, generating free fatty
acids and lyso-PC molecules. When SM is lost, ceramide-enriched microdomains have been
proposed to act as non-polar spots at the LDL surface, thereby facilitating LDL aggregation through
hydrophobic associations between domains 44. Here, PLA2-induced PC degradation induces a

Manuscript BBAMEM-19-17 (R1)
19
dramatic increase in Lyso-L--PC and a strong decrease in LDL surface area, in line with previous
studies 44,46,47. In addition, SMase and PLA2 provoked higher accessibility of ApoB-100 epitopes
to anti-ApoB-100 antibodies, as showed by Western blot analysis of LDL. This observation would
suggest conformational alterations in ApoB-100 linked to phospholipolysis, in line with previous
results from other groups 48. GFC followed by TEM analysis evidenced that LDL treatment with
SMase induced aggregated and fused LDL particles, in agreement with previous studies 44and
that PLA2-LDL comprises a high variety of aggregate sizes, as previously shown 13.
Here, we demonstrate that LP3 and its derived retro-enantio version (DP3) per se have a high
capacity to inhibit LDL aggregation induced by SMase and PLA2. However, all experimental
approaches (turbidimetry, GFC, and TEM) used in this work demonstrate that DP3 outperforms
LP3. Although all-D peptides are not recognised by proteases, and thus have higher stability, they
may be less active than their corresponding all-L peptides because of differences in the orientation
of amino acid side-chains. To preserve the orientation of the side-chains of the parent L-peptide in a
D-analogue, the retro-enantio approach 49,50 has been used in this study. In this regard, the
preservation of the topological side-chain orientation of DP3 together with a high stability in
biological samples account for its notable performance in the inhibition assays. In addition,
comparison of the capacity of LRP1 (LP3 and DP3) and ApoA-1 (4F) peptides to inhibit LDL
aggregation revealed that at 10 µM (peptide/ApoB-100 ratio: 4.7:1), LP3 and DP3 almost completely
repressed SMase-induced LDL turbidimetry, while L-4F exerted only a partial effect. Previous
studies addressing the efficacy of 4F to inhibit LDL aggregation showed that a molar ratio of 20:1 (4-
fold higher) is required to fully block SMase-induced LDL aggregation 29. However, the
experimental conditions used for LDL aggregation by Nguyen SD et al. (shorter times, and different
procedures for incubation of LDL with L-4F) differed from those applied in our study and thus hinder
a direct comparison between results. To the best of our knowledge, the present study is the first to
report data on the efficacy of 4F to inhibit PLA2-induced LDL aggregation. Here we found that
comparing the inhibitory capacities of L-4F, DP3 and LP3 at 4.7:1 peptide:ApoB-100 ratio, L-4F

Manuscript BBAMEM-19-17 (R1)
20
showed a similar capacity than LP3 to inhibit PLA2-induced LDL aggregation but lower capacity than
DP3.
One key question is how LRP1-derived peptides inhibit LDL aggregation. An important observation
in this regard is that these peptides counteracted alterations in LDL particle size, charge and
phospholipids induced by SMase but not those induced by PLA2. Despite of this, LRP1-derived
peptides exerted protection against SMase and PLA2-induced conformational alterations of ApoB-
100 and against LDL aggregation. These results indicate that 1) peptide protection of ApoB-100
conformation occurrs even in the context of extreme lipolysis and, 2) preservation of ApoB-100
conformation seems to be a common mechanisms underlying LDL protection against SMase and
PLA2-induced LDL aggregation. But, how LRP1-derived peptides preserve ApoB-100
conformation???. In the current study, limited trypsin proteolysis combined with Mass spectrometry-
based proteomic studies of LDL/DP3 complexes evidenced that peptide DP3 interacts with the LDL
surface prior to enzymatic treatment, protecting certain ApoB-100 sequences against trypsin-
induced proteolysis. Most of the peptide-protected sequences in ApoB-100, correspond to lipid-
associated ApoB-100 domains in LDL. The protection that peptide confers to ApoB-100 can be
induced by direct interaction or by remodeling the protein structure. Moreover, a highly positively
charged sequence (3227IKFDKYKAEK3236) located in the ApoB-100 C-terminal region and adjacent to
the LDL receptor binding domain (3385RLTRKRGLK3393) 51-54 was assumed to be responsible for
DP3 peptide recognition and used as the binding site for the molecular docking calculations. In
addition, homology modeling and molecular dynamics methods combined with molecular docking
provided a crucial first insight into the mechanistic basis of LRP1 peptide recognition by ApoB-100
protein. We propose a structural model of DP3 peptide in complex with ApoB-100. This complex is
mainly formed by salt bridge contacts between two of the acidic residues in DP3 (namely D-Glu3
and D-Glu9) and two positively charged ApoB-100 residues (namely Lys3229 and Lys3234), while the
oxygen atom of the DP3 D-Asn2 backbone and ApoB-100 Lys3232 were found to be involved in a
hydrogen bond favored by the positive ionic charge carried by the lysine side chain amine group.
This model is in complete agreement with the trypsin-limited proteolysis data reported herein, as
well as with the study of the recognition between LRP1 module CR9 and aggregated LDL particles

Manuscript BBAMEM-19-17 (R1)
21
recently published 28. Previous studies have consistently demonstrated that conformational
alterations of ApoB-100 lead to ApoB-100 degradation, a key process for LDL aggregation
48,55,56. In this context, the capacity of LRP1-derived peptides to preserve ApoB-100
conformation explain the high potential of these peptides to protect LDL against LDL aggregation.
Other crucial question is the mechanism by which LRP1-derived peptides protect SM but not PC
from enzymatic hydrolysis. Two different mechanistical options for the protective effect of peptides
against SMase-induced LDL lipolysis have been explored. By one side, we have explored whether
there is a direct effect of peptides on SMase activity. This possibility has been discarded since
SMase activity assays clearly show that peptides does not exert any effect on SMase functional
activity. Other possibility that has been explored is a potential interaction of peptides with the LDL
phospholipid surface. Several peptides and proteins bearing amphipathic properties have been
reported to be able to block LDL aggregation by a direct interaction with LDL phospholipid surface.
In this respect, common examples are 4F peptide 29 or different apolipoproteins 57. Despite the
lack of amphipathic properties in LRP1-derived peptides, this hypothesis has been considered as
possible mechanism of action for the peptides described herein. Theoretical molecular docking
calculations performed in the present study indicate a lack of association between peptides and LDL
phospholipid surface. A key point offering clues about how peptides protect SM is their unability to
protect PC hydrolysis. On the basis of the differential topological distribution of SM and PC in the
LDL surface 46, we propose that the differential anti-phospholypolytic effects of the peptide on SM
and PC phospholipolysis may be determined, at least in part, by the particular imbrication of SM and
PC with free cholesterol on the LDL surface. Although SM, compared to PC, is a minor phospholipid
in LDL (185 versus 450 molecules/LDL), SM is localized in cholesterol-enriched environments,
considered key regulators of phospholipolysis 46. ApoB-100 conformational stabilization could
directly impact the maintenance of the cholesterol-enriched environment structure since, according
to our molecular modeling studies, ApoB-100 sequences complexed with peptide are those close to
cholesterol in the LDL surface. Therefore, SM could be protected by LRP1-derived peptides against
phospholipolysis by its privilege of being part of cholesterol-enriched domains, whose structure

Manuscript BBAMEM-19-17 (R1)
22
would be preserved through the formation of peptide-ApoB-100 complexes. In contrast, PC,
belonging to cholesterol-poor domains, would be unprotected against PLA2-induced lipolysis,
independently of the status of ApoB-100 conformation.
5. Conclusion
Here, we show that LRP1-derived peptides, LP3 and, in particular, its retro-enantio version (DP3),
efficiently preserve LDL against SMase or PLA2-induced LDL aggregation. LP3 and DP3 maintain
LDL particle size, charge, phospholipid composition and ApoB-100 conformation in LDL treated with
SMase, but only ApoB-100 conformation in LDL exposed to PLA2 (summarized in Figure 8,
Graphical abstract). LP3 and DP3 keep ApoB-100 conformation even under extreme lipolysis
through key electrostatic interactions with a highly positively charged sequence
(3227IKFDKYKAEK3236) located in the ApoB-100 C-terminal region. A recent study has shown that the
individual susceptibility of LDL to aggregation is associated with future cardiac deaths independently
of other classical risk factors 58. LRP(CR9) derived peptides protect LDL against the main
proteolytic enzymes contributing to LDL retention and aggregation in the arterial intima. In the next
future, we are going to test the efficacy of the retro-enantio peptide version (DP3) to reduce
vascular LDL retention and LDL-induced smooth muscle cell (SMC)-foam cell formation in in vivo
models of atherosclerosis. Our first option in the near future will be to inject the peptide DP3
subcutaneously (not focal delivery at the moment) in a murine model of angioplasty-induced
atherosclerosis. This model offers the advantage of increased endothelial permeability, crucial to
favor peptide arrival to vasculature. In a not too distant future, we expect LRP1-derived peptides to
emerge as tools potentially useful to prevent atherosclerosis and cardiovascular complications, in
particular in individuals with aggregation-prone LDL as obese and diabetic patients
DATA AVAILABILITY
Mass spectroscopy proteomics of LDL. Panorama Public PXD011261
https://panoramaweb.org/j8CaUy.url

Manuscript BBAMEM-19-17 (R1)
23
Conflict of interest
R.P., C.P., and T.T. are employees of Iproteos SL. T.T. is cofounder of Iproteos SL and member of
its board of directors.
Acknowledgments
The Ministry of Science and Innovation of Spain, in the framework of the State Plan of Scientific
and Technical Innovation Investigation 2013-2016, awarded funding to the project
“DEVELOPMENT OF AN INNOVATIVE THERAPY FOR THE TREATMENT OF THE
ATHEROSCLEROSIS THROUGH INHIBITION OF CHOLESTEROL VASCULAR
ACCUMULATION”, led by IPROTEOS SL with file number RTC-2016-5078-1. This project was also
financed by the Ministry of Economy, Industry and Competitiveness (MINECO) in the framework of
the Subprogram RETOS-COLABORACIÓN, 2016 call. The project is also co-financed by the
European Union with the objective to promoting the technological development, innovation and
quality research. Support was also received from SAF2017-89613R (to SV), co-financed by the
European Regional Development Fund (ERDF), the Fundació MARATÓ TV3 Project 201521-10 (to
VLl-C), FIS PI13/00364 and PI16/00471 (to JSQ) and FIS PI18/01584 (to VLl-C) from the Instituto
de Salud Carlos III (ISCIII) and co-financed with ERDFs. Support was also received from Ministerio
de Economía y Competitividad to DdG-C (IJCI-2016-29393). CIBER Diabetes y Enfermedades
Metabólicas Asociadas (CIBERDEM; CB07/08/0016 (JSQ) and CIBER Enfermedades
Cardiovasculares (CIBERCV; CB16/1100403 (DdG-C, VLl-C) are projects run by the Instituto de
Salud Carlos III (ISCIII). The CRG/UPF Proteomics Unit is member of ProteoRed PRB3 consortium
which is supported by grant PT17/0019 of the PE I+D+i 2013-2016 from the Instituto de Salud
Carlos III (ISCIII) and ERDF. We also acknowledge support from the Spanish Ministry of Economy
and Competitiveness, “Centro de Excelencia Severo Ochoa 2013-2017”, SEV-2012-0208, and
“Secretaria d’Universitats i Recerca del Departament d’Economia I Coneixement de la Generalitat
de Catalunya” (2017SGR595 to ES and 2017SGR946 to VLl-C. SB, JLS-Q, AR-U, DdG-C and VL-
C are members of the Group of Vascular Biology of the Spanish Society of Atherosclerosis (SEA).

Manuscript BBAMEM-19-17 (R1)
24
References
[1] Castelli WP, Anderson K, Wilson PWF, Levy D. Lipids and risk of coronary heart disease The Framingham Study. Ann Epidemiol. 1992; 2:23–28.
[2] Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR, High density lipoprotein as a protective factor against coronary heart disease. Am J Med. 1977; 62:707–714.
[3] Guretzki H-J, Gerbitz K-D, Olgemöller B, Schleicher E. Atherogenic levels of low density lipoprotein alter the permeability and composition of the endothelial barrier. Atherosclerosis. 1994; 107:15–24.
[4] Aikawa M, Libby P. The vulnerable atherosclerotic plaque. Cardiovasc Pathol. 2004; 13 :125–138.
[5] Mauriello A, Sangiorgi GM, Virmani R, Trimarchi S, Holmes Jr DR, Kolodgie FD, Piepgras DG, Piperno G, Liotti D, Narula J, Righini P, Ippoliti A, Spagnoli LG. A pathobiologic link between risk factors profile and morphological markers of carotid instability. Atherosclerosis. 2010; 208: 572–580.
[6] Puri R, Nissen SE, Shao M, Elshazly MB, Kataoka Y, Kapadia SR, Tuzcu EM, Nicholls SJ. Non-HDL Cholesterol and Triglycerides: Implications for Coronary Atheroma Progression and Clinical Events., Arterioscler Thromb Vasc Biol. 2016; 36:2220-2228.
[7] Skålén K, Gustafsson M, Knutsen Rydberg E, Hultén LM, Wiklund, Innerarity TL, Boren J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002; 417:750-754.
[8] Öörni K, Pentikäinen MO, Ala-Korpela M, Kovanen PT. Aggregation, fusion, and vesicle formation of modified low density lipoprotein particles: molecular mechanisms and effects on matrix interactions. J Lipid Res. 2000; 41:1703–1714.
[9] Öörni K, Posio P, Ala-Korpela M, Jauhiainen M, Kovanen PT. Sphingomyelinase induces aggregation and fusion of small very low-density lipoprotein and intermediate-density lipoprotein particles and increases their retention to human arterial proteoglycans, Arterioscler Thromb Vasc Biol. 2005; 25:1678-1683
[10] Marathe S, Schissel SL, Yellin MJ, Beatini N, Mintzer R, Williams KJ, Tabas I. Human vascular endothelial cells are a rich and regulatable source of secretory sphingomyelinase. Implications for early atherogenesis and ceramide-mediated cell signaling. J Biol Chem. 1998; 273:4081-4088.
[11] Schissel SL, Schuchman EH, Williams KJ, Tabas I. Zn2+-stimulated Sphingomyelinase Is Secreted by Many Cell Types and Is a Product of the Acid Sphingomyelinase Gene. J Biol Chem. 1996;. 271:18431–18436.
[12] Aviram M, Maor I. Phospholipase A2-modified LDL is taken up at enhanced rate by macrophages. Biochem Biophys Res Commun. 1992; 185:465–472.
[13] Öörni K, Hakala JK, Annila A, Ala-Korpela M, Kovanen PT. Sphingomyelinase induces aggregation and fusion, but phospholipase A2 only aggregation, of low density lipoprotein (LDL) particles: Two distinct mechanisms leading to increased binding strength of LDL to human aortic proteoglycans. J Biol Chem. 1998; 273:29127-29134.
[14] Hakala JK, Öörni K, Pentikäinen MO, Hurt-Camejo E, Kovanen PT. Lipolysis of LDL by Human Secretory Phospholipase A Induces Particle Fusion and Enhances the Retention of LDL to Human Aortic Proteoglycans. Arterioscler Thromb Vasc Biol. 2001; 21:1053-1058.
[15] Tabas I, Li Y, Brocia RW, Xu SW, Swenson TL, Williams KJ. Lipoprotein lipase and sphingomyelinase synergistically enhance the association of atherogenic lipoproteins with smooth muscle cells and extracellular matrix. A possible mechanism for low density lipoprotein and lipoprotein(a) retention and macrophage foam cell formation, J Biol Chem. 1993; 268:20419-20432.
[16] Von Ballmoos MW, Dubler D, Mirlacher M, Cathomas G, Muser J, Biedermann BC. Increased apolipoprotein deposits in early atherosclerotic lesions distinguish symptomatic from

Manuscript BBAMEM-19-17 (R1)
25
asymptomatic patients. Arterioscler Thromb Vasc Biol. 2006; 26:359-364. [17] Khoo JC, Miller E, McLoughlin P, Steinberg D. Enhanced macrophage uptake of low density
lipoprotein after self-aggregation. Arterioscler Thromb Vasc Biol. 1988; 8:348-358. [18] Zhang WY, Ishii I, Kruth HS. Plasmin-mediated macrophage reversal of low density
lipoprotein aggregation. J Biol Chem. 2000; 275:33176-33183. [19] Kruth HS. Sequestration of aggregated low-density lipoproteins by macrophages. Curr Opin
Lipidol. 2002; 13:483-488. [20] Llorente-Cortés V, Martínez-González J, Badimon L. Esterified Cholesterol Accumulation
Induced by Aggregated LDL Uptake in Human Vascular Smooth Muscle Cells Is Reduced by HMG-CoA Reductase Inhibitors, Arterioscler Thromb Vasc Biol. 1998; 18:738-746.
[21] Llorente-Cortés V, Martínez-González J, Badimon L. LDL Receptor–Related Protein Mediates Uptake of Aggregated LDL in Human Vascular Smooth Muscle Cells. Arterioscler Thromb Vasc Biol. 2000; 20:1572-1579.
[22] Llorente-Cortés V, Otero-Viñas M, Hurt-Camejo E, Martínez-González J, Badimon L. Human coronary smooth muscle cells internalize versican-modified LDL through LDL receptor-related protein and LDL receptors. Arterioscler Thromb Vasc Biol. 2002; 22:387-393.
[23] Llorente-Cortés V, Otero-Viñas M, Sánchez S, Rodríguez C, Badimon L. Low-Density Lipoprotein Upregulates Low-Density Lipoprotein Receptor-Related Protein Expression in Vascular Smooth Muscle Cells. Circulation. 2002; 106:3104-3110
[24] Costales P, Aledo R, Vérnia S, Das A, Shah VH, Casado M, Badimon L, Llorente-Cortés V. Selective role of sterol regulatory element binding protein isoforms in aggregated LDL-induced vascular low density lipoprotein receptor-related protein-1 expression, Atherosclerosis. 2010; 213:458–468.
[25] Llorente-Cortés V, Otero-Viñas M, Camino-López S, Llampayas O, Badimon L. Aggregated low-density lipoprotein uptake induces membrane tissue factor procoagulant activity and microparticle release in human vascular smooth muscle cells, Circulation. 2004; 110:452-459.
[26] Camino‐ López S, Badimon L, GonzáLez A, Canals D, Peña E, Llorente‐ Cortés V. Aggregated low density lipoprotein induces tissue factor by inhibiting sphingomyelinase activity in human vascular smooth muscle cells. J Thromb Haemost. 2009; 7:2137–2146.
[27] Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis., Circulation.2014; 129:1551-1559.
[28] Costales P, Fuentes-Prior P, Castellano J, Revuelta-Lopez E, Corral-Rodríguez MÁ, Nasarre L, Badimon L, Llorente-Cortes V. Domain CR9 of Low Density Lipoprotein (LDL) Receptor-related Protein 1 (LRP1) Is Critical for Aggregated LDL-induced Foam Cell Formation from Human Vascular Smooth Muscle Cells. J Biol Chem. 2015; 290:14852–14865.
[29] Nguyen SD, Javanainen M, Rissanen S, Zhao H, Huusko J, Kivelä AM, Ylä-Herttuala S, Navab M, Fogelman AM, Vattulainen I, Kovanen PT, Öörni K. Apolipoprotein A-I mimetic peptide 4F blocks sphingomyelinase-induced LDL aggregation J Lipid Res. 2015; 56:1206–1221.
[30] Pujals S, Fernández-Carneado J, Ludevid MD, Giralt E. D-SAP: A new, noncytotoxic, and fully protease resistant cell-penetrating peptide. ChemMedChem. 2008; 3:296-301.
[31] Bancells C, Villegas S, Blanco FJ, Benítez S, Gállego I, Beloki L, Pérez-Cuellar M, Ordóñez-Llanos J, Sánchez-Quesada JL. Aggregated Electronegative Low Density Lipoprotein in Human Plasma Shows a High Tendency toward Phospholipolysis and Particle Fusion. J Biol Chem. 2010; 285:32425–32435.
[32] Martínez-Bujidos M, Rull A, González-Cura B, Pérez-Cuéllar M, Montoliu-Gaya L, Villegas S, Ordóñez-Llanos J, Sánchez-Quesada J.L. Clusterin/apolipoprotein J binds to aggregated LDL in human plasma and plays a protective role against LDL aggregation. FASEB J. 2014; 29:1688–1700.
[33] Case D, Cheatham TE 3rd, Darden T, Gohlke H, Luo R, Merz KM Jr, Inufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668-1688.
[34] Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé

Manuscript BBAMEM-19-17 (R1)
26
L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781-1802.
[35] Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004;32:W526-W531.
[36] Koes DR, Baumgartner MP, Camacho CJ. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J Chem Inf Model. 2013; 53:1893-1904.
[37] Chorev M, Goodman M. Recent developments in retropeptides and proteins_An ongoing topochemical exploration. Trends Biotechnol. 1995;13:438-445.
[38] Prades R, Oller-Salvia B, Schwarzmaier SM, Selva J, Moros M, Balbi M, Grazú V, De La Fuente JM, Egea G, Plesnila N, Teixidõ M, Giralt E. Applying the retro-enantio approach to obtain a peptide capable of overcoming the blood-brain barrier. Angew. Chemie - Int. Ed. 2015; 54:3967-3972.
[39] Prassl R, Laggner P. Molecular structure of low density lipoprotein: current status and future challenges. Eur Biophys J. 2009;38:145-158.
[40] Segrest JP, Jones MK, De Loof H, Dashti N. Structure of Apolipoprotein B-100 in low density lipoproteins. J Lipid Res. 2001; 42:1346-1367.
[41] Yang CY, Chen SH, Gianturco SH, Bradley WA, Sparrow JT, Tanimura M, Li WH, Sparrow DA, DeLoof H, Rosseneu M, et al. Sequence, structure, receptor-binding domains and internal repeats of human apolipoprotein B-100. Nature. 1986;323:738-742.
[42] Hospattankar AV, Law SW, Lackner K, Brewer HB Jr. Identification of low density lipoprotein receptor binding domains of human apolipoprotein B-100: a proposed consensus LDL receptor binding sequence of apoB-100. Biochem Biophys Res Commun. 1986;139:1078-1085.[43] Schissel SL, Tweedie-Hardman J, Rapp JH, Graham G, Williams KJ, Tabas I. Rabbit aorta and human atherosclerotic lesions hydrolyze the sphingomyelin of retained low-density lipoprotein. Proposed role for arterial-wall sphingomyelinase in subendothelial retention and aggregation of atherogenic lipoproteins. J Clin Invest. 1996; 98:1455–1464.
[44] Sartipy P, Camejo G, Svensson L, Hurt-Camejo E. Phospholipase A2 modification of low density lipoproteins forms small high density particles with increased affinity for proteoglycans and glycosaminoglycans, J Biol Chem. 1999; 274:25913-25920.
[45] Oestvang J, Bonnefont-Rousselot D, Ninio E, Hakala JK, Johansen B, Anthonsen MW, Modification of LDL with human secretory phospholipase A2 or sphingomyelinase promotes its arachidonic acid-releasing propensity. J Lipid Res. 2004; 45:831–838.
[46] Hevonoja T, Pentikäinen MO, Hyvönen MT, Kovanen PT, Ala-Korpela M. Structure of low density lipoprotein (LDL) particles: Basis for understanding molecular changes in modified LDL. Biochim Biophys Acta - Mol. Cell Biol. Lipids. 2000; 1488:189–210.
[47] Benítez S, Camacho M, Arcelus R, Vila L, Bancells C, Ordóñez-Llanos J, Sánchez-Quesada JL. Increased lysophosphatidylcholine and non-esterified fatty acid content in LDL induces chemokine release in endothelial cells. Atherosclerosis. 2004; 177:299–305.
[48] Kleinman Y, Krul ES, Burnes M, Aronson W, Pfleger B, Schonfeld G. Lipolysis of LDL with phospholipase A2 alters the expression of selected apoB-100 epitopes and the interaction of LDL with cells, J Lipid Res. 1988; 29:729-743.
[49] Soto C, Kindy MS, Baumann M, Frangione B. Inhibition of Alzheimer amyloidosis by peptides that prevent beta-sheet conformation. Biochem Biophys Res Commun. 1996; 226:672–680.
[50] Chalifour RJ, McLaughlin RW, Lavoie L, Morissette C, Tremblay N, Boulé M, Sarazin P, Stéa D, Lacombe D, Tremblay P, Gervais F. Stereoselective interactions of peptide inhibitors with the beta-amyloid peptide. J Biol Chem. 2003;278:34874-34881.
[51] Sneck M, Nguyen SD, Pihlajamaa T, Yohannes G, Riekkola M-L, Milne R, Kovanen PT, Öörni K. Conformational changes of apoB-100 in SMase-modified LDL mediate formation of large aggregates at acidic pH, J. Lipid Res. 2012; 53:1832-1839.
[52] Bancells C, Benítez S, Ordóñez-Llanos J, Öörni K, Kovanen PT, Milne RW, Sánchez-Quesada JL. Immunochemical Analysis of the Electronegative LDL Subfraction Shows That Abnormal N-terminal Apolipoprotein B Conformation Is Involved in Increased Binding to Proteoglycans, J Biol Chem. 2011; 286:1125–1133.
[53] Liu H, Scraba DG, Ryan RO. Prevention of phospholipase-C induced aggregation of LDL by amphipathic apolipoproteins. FEBS Lett. 1993; 316:27–33.

Manuscript BBAMEM-19-17 (R1)
27
[54] Ruuth M, Nguyen SD, Vihervaara T, Hilvo M, Laajala TD, Kondadi PK, Gisterå A, Lähteenmäki H, Kittilä T, Huusko J, Uusitupa M, Schwab U, Savolainen MJ, Sinisalo J, Lokki M-L, Nieminen MS, Jula A, Perola M, Ylä-Herttula S, Rudel L, Öörni A, Baumann M, Baruch A, Laaksonen R, Ketelhuth DFJ, Aittokallio T, Jauhiainen M, Käkelä R, Borén J, Williams KJ, Kovanen PT, Öörni K. Susceptibility of low-density lipoprotein particles to aggregate depends on particle lipidome, is modifiable, and associates with future cardiovascular deaths. Eur Heart J. 2018; 39:2562-2573
Figure Legends
Fig.1. LRP1-derived peptides (LP3 and DP3) decrease SMase- and PLA2-induced LDL aggregation in a dose-dependent manner. LDL was used at 1.44 mg/mL ApoB. Peptides were tested at
increasing concentrations (from 2.5 to 10 M for SMase-LDL and from 4.16 to 20.83 M for PLA2-LDL). Aggregation of LDL was induced by SMase (A) or PLA2 (B) and monitored at the indicated times by measuring turbidimetry (absorbance at 405 nm). Data are mean ± SD of 2 experiments performed in duplicate. Unsighted bar errors are minor than symbol size.*P<0.005 versus SMase-
LDL + IP321; P<0.005 versus SMase-LDL + LP3.
Fig. 2. LRP1-derived peptides (LP3 and DP3) reduce the AUC of agLDL peak in SMase-LDL and PLA2-LDL. LDL was untreated (LDL) or treated with SMase (40 U/L) and PLA2 (50 µg/L) in the absence (w/o peptide) or presence of LP3, DP3 or IP321 (10 µM, peptide/ApoB-100 ratio: 4.7:1, 24 h). LDLs (0.5 mL at 0.4 mg/mL ApoB) were chromatographed at a flow rate of 0.5 ml/min using a Superose 6 column in a AKTA-FPLC system (GE Healthcare) as described in Material and Methods. Representative GFC of: a) non enzymatically-treated LDL; b) LDL incubated with SMase; c) LDL incubated with SMase in the presence of LP3; d) LDL incubated with SMase in the presence of DP3; e) LDL incubated with SMase in the presence of IP321; f) LDL incubated with PLA2; g) LDL incubated with PLA2 in the presence of LP3; h) LDL incubated with PLA2 in the presence of DP3; and i) LDL incubated with PLA2 in the presence of IP321. j) and k) Bar graphs showing the percentage of aggregated versus monomeric LDL in SMase-LDL and PLA2-LDL, respectively, calculated from the chromatograms as area under the curve as explained in Methods.
Fig. 3. LRP1-derived peptides (LP3 and DP3) diminish the proportion of large size LDL aggregates in SMase-LDL and PLA2-LDL. Untreated LDL (A) or LDL treated with SMase (40 U/L) (B) and PLA2 (50 µg/L) (C) in the presence of LP3, DP3 or IP321 (10 µM, peptide/ApoB-100 ratio: 4.7:1, 24 h) or in absence of peptides (w/o peptide). TEM was performed in a Jeol 120 kV JEM-1400 microscope in the LDL preparations treated as explained above for 24 h at 37ºC. Black bars, 500 nm.
Fig. 4. LRP1-derived peptides (LP3 and DP3) prevent SM depletion in SMase-LDL but not L--PC depletion in PLA2-LDL. LDL was untreated (LDL) or treated with SMase (40 U/L) and PLA2 (50 µg/L) in the absence (w/o peptide) or presence of LP3, DP3 or IP321 (10 µM, peptide/ApoB-100 ratio: 4.7:1, 24 h) and the phospholipid pattern was analyzed by HPTLC after lipid extraction. Representative HPTLC of LDL (A and D) and bar graphs (B, C, E, F and G) showing quantification of phospholipid bands. Results are expressed as percentage of PL relative to untreated LDL and
shown as mean ± SD of 3 independent experiments performed in triplicate. L--PC indicates L--
phosphatidylcholine; SM: sphingomyelin; L--lyso-PC: L--lysophosphatidylcholine. *P<0.005 versus LDL.
Fig. 5. LRP1-derived peptides (LP3 and DP3) counteract LDL alterations in size and electric charge induced by SMase but not those induced by PLA2. LDL was untreated (LDL) or treated with SMase (40 U/L) and PLA2 (50 µg/L) in the absence (w/o peptide) or presence of LP3, DP3 o IP321 (10 µM,

Manuscript BBAMEM-19-17 (R1)
28
ratio peptide/ApoB-100: 4.7:1, 24 hours). LDL electrophoretic mobility was analyzed by GGE (A and B) and agarose gels (C and D). In GGE, LDL (5 µg/well) was electrophoresed in 2-16% acrylamide native gradient gels. LDLs were prestained with 1% Sudan black. In agarose gel electrophoresis, LDL (2 µg/well) was electrophoresed in 0.5% agarose gels, which were dehydrated and stained with Coomassie Blue. Peptides were assayed at 10 µM (Ratio Peptide/ApoB= 4.7/1)
Fig. 6. LRP1-derived peptides (LP3 and DP3) counteract ApoB-100 conformational alterations induced by SMase and PLA2 in LDL. LDL was untreated (LDL) or treated with SMase (40 U/L) and PLA2 (50 µg/L) in the absence (w/o peptide) or presence of LP3, DP3 or IP321 (10 µM, peptide/ApoB-100 ratio: 4.7:1, 24 h). Representative western blot analysis of ApoB-100 in LDL exposed to SMase (A) or PLA2 (B). LDL (5 µg/well) was electrophoresed in 10% acrylamide gels under non-reducing conditions for 2 h at 100 V and transferred to nitrocellulose membranes, which were then incubated with anti-ApoB-100 antibodies. Fig. 7. DP3 protect ApoB-100 sequences against tryptic-limited proteolysis A) List of ApoB100 peptides that are protected by DP3 and by LDL aggregation as shown by tryptic-limited proteolysis coupled to mass spectrometry analysis. Green indicates signal detected, Yellow a poor signal, and Red no MS signal. B) Structural details of the molecular interaction between Apo-B100 protein and DP3 peptide. View of the ApoB-100 protein surface contacting the DP3 peptide: the basic residues of ApoB-100 exposed to trypsin digestion and buried deeply upon peptide binding highlighted in teal and violet, respectively. DP3 peptide acid residues responsible for ApoB-100 binding highlighted in yellow sticks.
Fig. 8. Schematic representation of the molecular basis for the protective effect of LRP1-derived peptides against SMase- and PLA
2-induced LDL aggregation. DP3 forms a complex with ApoB-100
and this molecular interaction stabilizes ApoB-100 conformation. ApoB-100 conformation stabilization could guarantee the structural preservation of surface colesterol-enriched environments, where SM is located. Structural preservation of cholesterol, a key regulator of phospholipolysis, would protect SM from SMase activity. As a result, LDL complexed with DP3 remains unaltered when exposed to SMase. This scenario changes when LDL complexed with DP3 is exposed to PLA
2. The target for PLA
2 is PC, phospholipid associated with poor-colesterol
environments. Therefore, PC is unprotected against the attack of PLA2, that hydrolyzes PC producing lyso-PC and NEFA. Remarkably, LDL complexed with DP3 is protected against SMase and PLA2-induced aggregation even in conditions of extreme phospholipolysis, indicating that the maintenance of ApoB-100 conformation is key in the prevention of LDL aggregation.

Manuscript BBAMEM-19-17 (R1)
1
Molecular basis for the protective effects of low-density lipoprotein receptor-related protein 1
(LRP1)-derived peptides against LDL aggregation
Short title: LRP1-derived peptides and LDL aggregation
Aleyda Benitez-Amaroa,b, Chiara Pallarac, Laura Nasarrea, Andrea Rivas-Urbinad, Sonia Benitezd,
Angela Veaa, Olga Bornacheaa,b, David de Gonzalo-Calvoa,b,e, Gabriel Serra-Mirf, Sandra Villegasf,
Roger Pradesc, José Luís Sanchez-Quesadad,g, Cristina Chivah,i, Eduard Sabidoh,i, Teresa
Tarragóc, and Vicenta Llorente-Cortésa,b,e**
aGroup of Lipids and Cardiovascular Pathology. Biomedical Research Institute Sant Pau (IIB Sant
Pau), Hospital de la Santa Creu i Sant Pau. Barcelona. Spain.
bInstitute of Biomedical Research of Barcelona (IIBB). Spanish National Research Council (CSIC),
Barcelona, Spain.
cIproteos S.L., Barcelona Science Park (PCB), Barcelona, Spain.
dCardiovascular Biochemistry Group, Research Institute of the Hospital de Sant Pau (IIB Sant Pau),
Barcelona, Spain.
eCIBER enfermedades cardiovasculares (CIBERcv).
fProtein Design and Immunotherapy Group. Departament de Bioquímica i Biologia Molecular,
Facultat de Biociències, Universitat Autònoma de Barcelona, Bellaterra, Barcelona, Spain.
gCIBER diabetes y enfermedades metabólicas asociadas (CIBERdem)
hProteomics Unit, Centre de Regulació Genòmica, Barcelona Institute of Science and Technology,
Barcelona, Spain.
iUniversitat Pompeu Fabra, Barcelona, Spain.
**To whom correspondence should be addressed: Vicenta Llorente-Cortés, Lipids and
Cardiovascular Pathology group leader, ICCC Program, Biomedical Research Institute IIB Sant
Pau, Hospital de la Santa Creu i Sant Pau, Sant Antoni Mª Claret, 167, 08025 Barcelona. Phone:
+34 935565888, Fax: +34 935565559, E-mail: [email protected]; [email protected]
Formatted: Spanish (Spain,
International Sort)
Formatted: English (United States)
*REVISED Manuscript (text with changes Marked)Click here to view linked References

Manuscript BBAMEM-19-17 (R1)
2
Abstract
Aggregated LDL is the first ligand reported to interact with the cluster II CR9 domain of low-density
lipoprotein receptor-related protein 1 (LRP1). In particular, the C-terminal half of domain CR9,
comprising the region Gly1127-Cys1140 exclusively recognizes aggregated LDL and it is crucial for
aggregated LDL binding. Our aim was to study the effect of the sequence Gly1127-Cys1140 (named
peptide LP3 and its retro-enantio version, named peptide DP3) on the structural characteristics of
sphingomyelinase- (SMase) and phospholipase 2 (PLA2)-modified LDL particles. Turbidimetry, gel
filtration chromatography (GFC) and transmission electronic microscopy (TEM) analysis showed
that LP3 and DP3 peptides strongly inhibited SMase- and PLA2-induced LDL aggregation.
Nondenaturing polyacrylamide gradient gel electrophoresis (GGE), agarose gel electrophoresis and
high-performance thin-layer chromatography (HPTLC) indicated that LP3 and DP3 prevented
SMase-induced alterations in LDL particle size, electric charge and phospholipid content,
respectively, but not those induced by PLA2. Western blot analysis showed that LP3 and DP3
counteracted changes in ApoB-100 conformation induced by the two enzymes. LDL proteomics
(LDL trypsin digestion followed by mass spectroscopy) and computational modeling methods
evidenced that peptides preserve ApoB-100 conformation due to their electrostatic interactions with
a basic region of ApoB-100. These results demonstrate that LRP1-derived peptides are protective
against LDL aggregation, even in conditions of extreme lipolysis, through their capacity to bind to
ApoB-100 regions critical for ApoB-100 conformational preservation. These results suggests that
these LRP1(CR9) derived peptides could be promising tools to prevent LDL aggregation induced by
the main proteolytic enzymes acting in the arterial intima.
Key words: Lipoprotein aggregation; LRP1-derived peptides; ApoB-100; SMase, PLA2,
atherosclerosis

Manuscript BBAMEM-19-17 (R1)
3
Abbreviations: agLDL, aggregated LDL; apo, apolipoprotein; CE, cholesteryl esters; FC, free
cholesterol; hcVSMC, human coronary vascular smooth muscle cells; GFC, gel filtration
chromatography; GGE, nondenaturing polyacrylamide gradient gel electrophoresis; HPTLC, high-
performance thin-layer chromatography; LC-MS/MS, liquid chromatography tandem-mass
spectrometry; LDL, low-density lipoprotein; LysoPC, lysophosphatidylcholine; LRP1, low-density
lipoprotein receptor-related protein 1; NEFA, nonesterified fatty acids; PC, phosphatidylcholine;
PLA2, phospholipase A2; PL, phospholipids; REMD, Replica Exchange Molecular Dynamics; SM,
sphingomyelin; SMase, sphingomyelinase; TEM, transmission electron microscopy

Manuscript BBAMEM-19-17 (R1)
4
1. Introduction
Ischemic heart disease is the first cause of death in Western countries, and myocardial infarction
accounts for about 50% of deaths from this disease. The Framingham study showed that
cardiovascular risk positively correlates with low-density lipoprotein (LDL)-cholesterol and inversely
with high-density lipoprotein (HDL)-cholesterol 1,2. High levels of LDL-cholesterol induce
alterations in the endothelium permeability, which in turn promote LDL influx into the arterial intima
3. Intimal LDL-cholesterol accumulation is a critical step in vascular cholesteryl ester (CE)
deposition, a process that increases the tendency of the atherosclerotic plaque to rupture, triggering
thrombosis and the development of ischemic cardiomyopathy 4-6. CEs in atherosclerotic plaques
are deposited both extra- and intra-cellularly. Extracellular deposition of LDL-CEs, a crucial initiating
event in atherosclerosis 7, is mediated by proteoglycans in the extracellular matrix of the arterial
intima. The electrostatic interaction between proteoglycans and LDL and the proteolytic/lipolytic
actions of enzymes on LDL are enhanced in the arterial intima and promote LDL retention and
aggregation 8,9. Two of the main enzymes acting on LDL are sphingomyelinase (SMase),
secreted by endothelial cells and macrophages 10,11 and phospholipase A2 (PLA2) 12,13. These
two enzymes play key roles in LDL aggregation in the arterial intima during atherogenesis 12-15.
Aggregated LDL (agLDL) has been detected and isolated from atherosclerotic plaques from animal
models and humans 11,16. Unlike native LDL, agLDL is a potent inducer of massive intracellular
CE accumulation both in macrophages 17-19 and human coronary vascular smooth muscle cells
(hcVSMCs) 20. In particular, in hcVSMCs, agLDL is actively taken up through the low-density
lipoprotein receptor-related protein 1 (LRP1) 21,22. The internalization of agLDL, in turn, induces
LRP1 expression 23,24, promoting a positive feedback loop that efficiently transforms hcVSMCs
into foam cells. We have also demonstrated that hcVSMC-foam cells synthesize and release high
amounts of tissue factor, which is crucial for the pro-thrombotic transformation of the vascular wall
and thus for the progression of atherosclerosis to thrombosis 25,26. The relevance of this
mechanism in atherosclerosis is evident since VSMCs are the main components of the vascular wall

Manuscript BBAMEM-19-17 (R1)
5
and more than 50% of foam cells previously considered to be monocyte-derived macrophages in
human atherosclerotic plaques originate from VSMCs 27. Together, these findings support the
notion that the generation of hVSMC-derived foam cells through LRP1-mediated agLDL uptake is a
key mechanism underlying cholesterol accumulation in vasculature susceptible to atherosclerosis.
Our group has identified LRP1 cluster II, in particular the region Gly1127-Cys1140 (peptide LP3: H-
GDNDSEDNSDEENC-NH2) that spans the C-terminal half of domain CR9, as pivotal for binding to
agLDL and subsequent internalization of this agLDL into human VSMCs 28. We hypothesize that
peptide LP3 or its retro-enantio version, peptide DP3, would counteract LDL aggregation. Using a
range of biochemical, biophysical and molecular experiments, here we demonstrate that LP3 and
DP3 peptides efficiently prevent LDL aggregation induced by the lipolytic enzymes SMase and
PLA2. Although LP3 and DP3 efficiently counteracted alterations in LDL particle size, charge and
SM loss induced by SMase, they were unable to prevent the alterations induced in the same
parameters by PLA2. However, both peptides protected ApoB-100 from conformational alterations
induced by SMase and PLA2. Proteomics, ab initio modeling and molecular dynamics showed that
LRP1-derived peptides establish stable electrostatic interactions with basic sequences of ApoB-100.
2. Material and Methods
2.1. Peptide sequences and synthesis
Peptide sequences are written from N to C, H- at the end terminus indicate amine ending, whereas
–NH2 at the C terminus indicates amide. The amino acid sequence of LP3 peptide (parent) is H-
GDNDSEDNSDEENC-NH2, DP3 is the retro-enantio version of LP3 without cysteine: H-
needsndesdndh-NH2, where the amino acid letter code in lowercase stands for amino acids with D-
chirality. As a negative control, we used an ApoB-100-based peptide coded as IP321 with the
sequence: H-RLTRKRGLK-NH2. We also compared the efficacy of LP3, DP3 and IP321 with the
previously described anti-atherosclerotic apoA-I mimetic, 4F. The latter is 18 amino acids long and
has the following sequence: Ac-DWFKAFYDKVAEKFKEAF-NH2 VSMCs 29. All peptides were

Manuscript BBAMEM-19-17 (R1)
6
synthesized by Iproteos by standard 9-fluorenylmethoxycarbonyl/tert-butyl (Fmoc/tBu) solid-phase
peptide synthesis. Syntheses were performed manually on a 100-µmols scale/each using Rink
amide ChemMatrix® resin. Peptide elongation and other solid-phase manipulations were done
manually in polypropylene syringes, each fitted with a polyethylene porous disk. Solvents and
soluble reagents were removed by suction. Washings between synthetic steps were done with
dimethylformamide and dichloromethane using 5 mL of solvents/g resin each time. Nα-Fmoc-
protected amino acids (4 equivalents) were coupled using 2-(1H-benzotriazole-1-yl)-1,1,3,3-
tetramethyluronium (TBTU, 4 equivalents), and N,N-diisopropylethylamine (8 equivalents). The
extent of the reaction was monitored using the Kaiser test (primary amines). During couplings, the
mixture was allowed to react with intermittent manual stirring. Fmoc group was removed by treating
the resin with 20% piperidine in DMF (3 mL/g resin). The peptides were cleaved from the resin using
a mixture of trifluoroacetic acid, triisopropylsilane and water (95:2.5:2.5). The crude products
obtained were purified in reverse phase using a semi-prep HPLC instrument equipped with a C18
column. The purity and identity of the synthesized peptides was assessed by HPLC, HPLC-MS and
MALDI-TOF analysis (Table S1).
2.2. Peptide stability in human serum and cell culture medium
The stability of the peptides was studied in human serum (Sigma-Aldrich). For the stability assay,
the peptides were dissolved in a mixture of 10% Hanks’ Balanced Solution Salt (HBSSx1) and
human serum. The final concentration of peptide was 200 mM VSMCs 30. The samples were
incubated at 37°C with orbital agitation for 24 h. At selected time points, serum aliquots were
extracted and the serum proteins of the mixture were then precipitated by adding methanol.
Samples were then centrifuged at 4°C, and the supernatant was filtered out and analyzed by HPLC.
Peptide stability during the assays was monitored using HPLC (UV detection at 220 nm).
2.3. LDL isolation and purification
Human LDL (d1.019-d1.063 g/mL) was obtained from pooled normolipemic human plasma by
sequential ultracentrifugation in a KBr density gradient. Briefly, very low density lipoproteins (VLDLs)

Manuscript BBAMEM-19-17 (R1)
7
were first discarded after spinning plasma at 36,000 rpm for 18 h at 4ºC using a fixed-angle rotor
(50.2 Ti, Beckman) mounted on an Optima L-100 XP ultracentrifuge (Beckman). Subsequently,
VLDL-free plasma was layered with 1.063 g/mL KBr solution and centrifuged at 36,000 rpm for 18 h
at 4ºC. LDLs were dialyzed against 0.02 M Trizma, 0.15 M NaCl, 1 mM EDTA, pH 7.5 for 18 h, and
then against normal saline for 2 h. Finally, isolated LDLs were filter-sterilized (0.22 μm Millex-GV
filter unit, Millipore). Protein concentration was determined using the BCA protein assay (Thermo
Scientific) and the cholesterol concentration with a commercial kit (IL test Cholesterol, Izasa).
2.4. Analysis of SMase activity in the presence and absence of peptides
The capacity of LP3 and DP3, IP321 and manumycin A (positive control) to inhibit
sphingomyelinase activity was measured using the Amplex Red Sphingomyelinase Assay Kit
(Molecular Probes Invitrogen Detection Technologies, #A12220), an assay that measures the
formation of phosphorylcholine. To measure SMase activity in the presence and absence of
peptides, DMSO volume and peptide concentrations were kept constant [10 M (molar ratio
peptide/ApoB= 4.7/1)] while SMase concentration was increased (0, 0.2, 0.4, 0.6, 0.8 and 1 mU/ml).
Each reaction contained 50 μM Amplex Red reagent, 1 U/mL HRP, 0,1 U/ml choline oxidase, 4 U/ml
of alkaline phosphatase, 0,25 mM sphingomyelin (diluted with Triton X-100 or with the Reaction
Buffer 1x). Reactions were incubated at 37ºC for 30 min. Fluorescence intensity was measured
using a fluorescence microplate reader (Victor 1420 multilabel counter) using excitation at 530 nm
and fluorescent detection at 590nm.
2.5. LDL modification by Sphingomyelinase (SMase) and Phospholipase A2 (PLA2) in
the presence and absence of peptides
Human LDL (1.44 mg/mL) were incubated with 40 U/L of Bacillus cereus SMase (Sigma-Aldrich,
Schnelldorf, Germany) or with 50 µg/L of type II secretory PLA2 from honey bee venom (Sigma-
Aldrich, Schnelldorf, Germany) in 20 mM Tris buffer (pH 7.0) containing 150 mM NaCl, 2 mM CaCl2,
and 2 mM MgCl2 at 37ºC. LDL incubation with SMase and PLA2 was performed at peptide
concentrations of 0, 2.5, 5, 7.7, 8.8, 10 µM peptide/ApoB-100 ratio :0/1, 1.2/1, 2.4/1, 3.5/1, 4.1/1,

Manuscript BBAMEM-19-17 (R1)
8
4.7/1) and 0, 4.2, 6.3, 10.0, 14.6, 20.8 µM peptide/ApoB-100 ratio (0/1, 2.0/1, 2.9/1, 4.7/1, 6.8/1,
9.8/1), respectively, for increased time periods. LDL lipolysis was stopped by addition of EDTA
(final concentration 10 mM).
2.6. Characterization of SMase- and PLA2-induced LDL alterations in the presence and
absence of peptides. A detailed information of the different procedures
2.6.1.LDL Composition_The LDL composition was measured by commercial methods adapted to a
Cobas 6000/c501 autoanalyzer (Roche Diagnostics, Indianapolis, IN, USA). Total cholesterol,
triglycerides and ApoB reagents were obtained from Roche Diagnostics. PLs, NEFAs, and free
cholesterol reagents were obtained from Wako Pure Chemicals (Tokyo Japan).
2.6.2.Turbidity_Sample turbidity (LDL aggregation monitorization) was determined by measuring
absorbance of 100 µl of LDLs (1.44 mg/mL ApoB) in a 96-well microplates at 405 nm.
2.6.3.Gel Filtration Chromatography (GFC)_To separate aggregated from non-aggregated LDL,
samples (50 µL at 1.44 mg/mL) were subjected to gel filtration chromatography (GFC) in two online
connected Superose 6 Increase 5/150 GL columns with an AKTA-FPLC system (GE Healthcare) at
a flow of 0.3 mL/min 31,32. The area under the curve of each peak was calculated using the
Unicorn 4.0 software (GE Healthcare).
2.6.4. Agarose gel electrophoresis_LDL (1.5 µL of LDL (1.44 mg/mL) was electrophoresed on 0.5%
agarose gels at 65V for 40 min followed by 90V for 15 min. Fixed gels were dehydrated and stained
with Coomassie Blue. Decolored gels were captured with the Bio-Rad GS-800 scanner.
2.6.5. SDS-PAGE and Western blot
ApoB-100 was detected by Western blot analysis after electrophoresis of LDL in 6% SDS-PAGE
gels. Nitrocellulose membranes were incubated with anti-ApoB-100 polyclonal antibodies (Roche
Diagnostics S.L., Ref: 3032574122, polyclonal anti-human ApoB-100, dilution 1: 5000) for 1 h at

Manuscript BBAMEM-19-17 (R1)
9
room temperature. After several washes, membranes were incubated with secondary anti-sheep
antibodies (dilution 1: 10000, Dako; Glostrup, Denmark). Bands were detected using ECL Prime
Western Blotting Detection Reagent (Amersham) and quantified by densitometry using a ChemiDoc
system and Quantity One software (Bio‐Rad, Hercules, CA, USA). The results are expressed as
arbitrary units of intensity.
2.6.6.TEM
TEM was performed as previously described 32. LDL was placed on a 400 mesh glow-discharge
carbon-coated grid, negatively stained with 2% potassium phosphotungstate, pH 7.0, and air dried.
Grids were placed in a Jeol 120-kV JEM-1400 (Jeol, Japan) transmission electron microscope. TEM
images were processed using ImageJ (Fiji) software. Briefly, contrast was enhanced, followed by
background subtraction.
2.6.7. Nondenaturing acrylamide gradient gel electrophoresis (GGE)
GGE was performed following Nichols et al., with small modifications 32. Two solutions at 2% and
16% were prepared by using a stock solution of acrylamide and bis-acrylamide (30% total, 5%
cross-linker) and mixed using 2 P-1 peristaltic pumps (Pharmacia). The different samples (5 μL at
1.44 mg/mL) were preincubated for 15 min with 10 μL Sudan black (0.1% [wt/vol]) in ethylene glycol
and 5 μL saccharose (50% [wt/vol]). Ten microliters of this mixture was electrophoresed at 4°C for
30 min at 20 V, 30 min at 70 V, and 4 h at 100 V. Bands were scanned by densitometry.
2.6.8. HPTLC
LDL (15 µg) was lipid-extracted with organic solvents as previously described by our group with
minimal modifications 25. Samples were applied to horizontal, one-dimensional high-performance
thin-layer chromatography (HPTLC) (Merck, HPTLC Silica Gel 60, 1.05641.0001). Phospholipids
were separated on HPTLC by chloroform:ethyl
acetate:acetone:isopropanol:ethanol:methanol:water:acetic acid (30:6:6:6:16:28:6:2); visualized by

Manuscript BBAMEM-19-17 (R1)
10
staining with 7.5% Cu-acetate (w/v), 2.5% CuSO4 (w/v), and 8% H3PO4 (v/v) in water. A mixture of
phospholipids (Sigma Ref pH-7) were also run as standards. The spots corresponding to the
different phospholipids were quantified by densitometry against the standard curve using a GS-800
Calibrated Densitometer (Bio-Rad).
2.7. Proteomics analyses of LDL samples
2.7.1. Sample preparation_LDL, LDL unexposed or exposed to SMase in the absence of peptide or
in the presence of DP3 or IP321) were directly subjected to limited trypsin digestion (1 µg) for 1 h or
3 h. In both cases, tryptic peptides were desalted using a C18 column and evaporated to dryness.
2.7.2. Mass spectrometry data acquisition_ LDL samples were analyzed using a LTQ-Orbitrap Velos
Pro mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) coupled to an EasyLC
(Thermo Fisher Scientific (Proxeon), Odense, Denmark). Peptides were separated by reverse-
phase chromatography using a 25-cm column with an inner diameter of 75 μm, packed with 5 μm
C18 particles (Nikkyo Technos Co., Ltd. Japan). Chromatographic gradients started at 93% buffer A
and 7% buffer B and gradually increased to 65% buffer A / 35% buffer B in 60 min at a flow rate of
250 nl/min. The mass spectrometer was operated in positive ionization mode with nanospray
voltage set at 2.2 kV and source temperature at 250°C. Ultramark 1621 for the FT mass analyzer
was used for external calibration prior to the analyses, and an internal calibration was performed
using the background polysiloxane ion signal at m/z 445.1200. The acquisition was performed in
data-dependent acquisition mode, and full MS scans with 1 micro scans at a resolution of 60,000
were used over a mass range of m/z 350-2000 with detection in the Orbitrap. Auto gain control
(AGC) was set to 1E6, dynamic exclusion (60 seconds) and charge state filtering disqualifying singly
charged peptides was activated in each cycle of data-dependent acquisition analysis. After each
survey scan, the top ten most intense ions with multiple charged ions above a threshold ion count of
5000 were selected for fragmentation at normalized collision energy of 35%. Fragment ion spectra
produced via collision-induced dissociation (CID) were acquired in an Ion Trap apparatus. AGC set

Manuscript BBAMEM-19-17 (R1)
11
to 5e4, an isolation window of 2.0 m/z, activation time of 0.1ms and maximum injection time of 100
ms were used. All data were acquired with Xcalibur software v2.2.
2.7.3. Mass spectrometry data analysis
The spectra acquired for the LDL samples were analyzed using the Proteome Discoverer software
suite (v2.0, Thermo Fisher Scientific) and the Mascot search engine (v2.5, Matrix Science). The
data were searched against the Swiss-Prot human database (v.2017/10). At the MS1 level, a
precursor ion mass tolerance of 7 ppm was used, and up to three missed cleavages were allowed.
The fragment ion mass tolerance was set to 0.5 Da for MS2 spectra. Methionine oxidation and N-
terminal protein acetylation were defined as variable modifications, whereas carbamidomethylation
on cysteine residues was set as a fixed modification. False discovery rate (FDR) in peptide
identification was limited to a maximum of 5% using a decoy database. Quantitation data were
retrieved from the “Precursor ion area detector” node of Proteome Discoverer (v2.0) using 2 ppm
mass tolerance for the peptide extracted ion current (XIC). Finally, for each LDL sample, Skyline
software v4.1 was used to build a spectral library for the ApoB100 protein with the corresponding
identified spectra and to extract all its MS1 areas. Extracted ion chromatograms for the different LDL
samples have been deposited to Panorama with identified PXD011261; URL
(https://panoramaweb.org/j8CaUy.url).
2.8. Molecular modeling of peptide-LDL interactions
The structural model of ApoB-100 in complex with DP3 peptide was built using rigid body molecular
docking simulation and starting from the isolated ab initio models of both docking partners. First of
all, an exhaustive conformational sampling was performed on the peptide in order to identify the
most stable and populated conformational states, which were assumed to be the most favorable to
bind ApoB-100 protein. For that, on one hand, a 3D structure of DP3 was designed from scratch
and consequently used as input for Replica Exchange Molecular Dynamics (REMD) simulations in
implicit water using the AMBER ff12SB force field 33 and NAMD software 34. In each REMD
scheme, 16 independent 100 ns-long simulations (replicas) were performed simultaneously at

Manuscript BBAMEM-19-17 (R1)
12
temperatures ranging from 300 to 613 K. Finally, the representative structure of the most populated
cluster was extracted and used as input structure for the docking simulation. On the other hand, the
3D structure of ApoB-100 protein was built by ab initio protein modeling, since no crystal structure of
protein was available and very low sequence identity templates (ranging from 8 to 13%) were found
in the PDB. Indeed, this computational technique tends to require vast resources, thus only a small
domain of roughly 100 residues around the basic region believed to be responsible for LDL receptor
binding (3189-3261 residues) was selected for modeling. Consequently, a total of 10 ab initio
models were built using ROBETTA server 35 and finally used as input structures for the docking
simulation. Starting from each ApoB-100 protein structure and DP3 peptide previously obtained, 10
independent rigid-body docking simulations were performed using AutoDock Vina software fork
Smina 36 and setting the docking grid box around Apo-B100 3227IKFDKYKAEK3236 region. Finally,
all the docking results were merged and the lowest-energy poses were visually inspected.
2.9. Statistical analysis
Data are presented as mean±SD. One-way ANOVA followed by the Bonferroni or the Storey (q-
value) correction was performed for comparison of >2 groups. Student’s t test was performed for
comparison of 2 groups. P<0.05 was considered significant.
3. Results
3.1. DP3 shows higher stability than the parent peptide LP3 in human serum
Peptide drugs offer high activity, high specificity and low toxicity, but are susceptible to proteolytic
degradation. A strategy to overcome this problem is to produce peptides using non-natural or D-
amino acids, which shows improved stability. Although all-D peptides are not recognized by plasma
proteases, they may be less active than their corresponding all-L peptides because of differences in
the orientation of amino acid side-chains. To preserve the orientation of the side-chains of the
parent peptide in a D-analogue, the D-amino acid residues can be linked in the reverse order to that
of the parent peptide in order to produce a retro-enantio peptide. This approach emphasizes the
Formatted: Font: 11 pt

Manuscript BBAMEM-19-17 (R1)
13
maintenance of the topological native orientation of the side-chains but not of the backbone, where
the amide bond is shift one unit 37. By using this approach, peptides usually preserve their activity
while notably improve their plasma stability. In the literature there are examples where this approach
has been successfully used 30. LP3 and DP3 were synthesized by means of solid-phase peptide
synthesis using a Fmoc/tBu strategy. The purity of both peptides (≥98%) was assessed by HPLC
analysis (Supplemental Fig. S1). Other parameters indicative of the purity and identity of the
synthesized peptides were obtained by HPLC-MS and MALDI-TOF analysis (Supplemental Table
S1). To study the stability of the parent peptide (LP3), it was dissolved in a mixture of HBSS (x1)
and human serum. LP3 showed moderate stability with a calculated half-life of 8 h (Supplemental
Fig. S2A). To achieve an increase in the stability of LP3 for the purpose of in vivo studies, a retro-
enantio version of the peptide (DP3) was synthesized. This approach consisted of using all D-amino
acids and reversing the sequence of the parent peptide. This synthetic strategy has been shown to
be useful 3833. As the retro-enantio version of the peptide is synthesized using D-amino acids, the
sequence is not recognized by serum proteases, thus extending its half-life considerably
(Supplemental Fig. S2B).
3.2. Time- and dose-response blocking effects of LP3 and DP3 on SMase- and PLA2- induced
LDL turbidity
To study the effects of the peptides on SMase-induced LDL aggregation, LDL was incubated with
SMase and PLA2 in the presence and absence of LP3, DP3 and IP321 (negative control) under the
experimental conditions detailed in Supplemental Fig. S3. The peptide concentrations and
peptide/ApoB-100 molar ratios tested in the aggregation of SMase-modified LDL (SMase-LDL) are
detailed in Supplemental Table S2. LP3 and DP3 reduced SMase-LDL aggregation at all the times
tested (4, 6 and 18 h) in a dose-dependent manner (Fig. 1A). DP3 showed a higher inhibitory
capacity than LP3 at concentrations of 5.0, 7.7 and 8.8 µM, and SMase-LDL aggregation was fully
inhibited by LP3 at 10 µM (peptide/ApoB-100 ratio: 4.7/1) and by DP3 at 7.7 µM (peptide/ApoB-100
ratio: 3.5/1). To study the effects of the peptidomimetics on the aggregation of PLA2-modified LDL,
Formatted: English (United States)
Formatted: Font: 11 pt
Formatted: English (United States)

Manuscript BBAMEM-19-17 (R1)
14
LDL was incubated with PLA2 in the presence or absence of LP3, DP3 and IP321 at the doses and
peptide/ApoB-100 ratios detailed in Supplemental Table S3. We did not detect any effect of PLA2 on
LDL aggregation at time points lower than 24 h (data not shown). LP3 and DP3 showed high
inhibitory activity on PLA2-LDL aggregation, although, as in the case of SMase, DP3 showed
outperformed LP3 at concentrations of 10 µM and 14.6 µM at 24, 30 and 36 h (Fig. 1B). PLA2-LDL
aggregation was fully inhibited by LP3 at a concentration of 20.83 µM (peptide/ApoB-100 ratio:
9.8/1) and by DP3 at 14.6 µM (peptide/ApoB-100 ratio: 6.8/1) at all tested times. IP321 did not
inhibit SMase- or PLA2-LDL aggregation at any dose or time tested (Fig. 1A,B). Given that
Apolipoprotein A-I peptidomimetic 4F has been reported to block SMase-LDL aggregation 29, we
compared the capacity of LP3, DP3 and 4F (10 M, ratio 4.7/1, 24 h) to inhibit SMase- and PLA2-
LDL aggregation. SMase-induced aggregation was almost fully blocked by LP3 (86.2%±1.8) and
DP3 (98.1%±1.4), but only partially blocked by 4F (55.7%±2.3). PLA2-LDL aggregation was partially
blocked by LP3 (59.6%±0.8) and 4F (45.8%±1.1) and almost completely inhibited by DP3
(84.4%±1.9) (Table 1). IP321 did not have any significant effect on SMase- or PLA2-LDL
aggregation. None of the peptides alter the turbidimetry values of untreated LDL (Supplemental Fig.
S4).
3.3. LP3 and DP3 decrease the percentage of aggregated particles in SMase-LDL and PLA2-
LDL
To separate aggregated from monomeric LDL particles, LDL samples were subjected to gel filtration
chromatography (GFC). GFC showed that LDL comprised mainly monomeric LDL (Fig. 2a). A small
percentage of residual LDL aggregates was detected in untreated LDL due to the fact that both
untreated and treated LDL were exposed to 37ºC for the same period (Supplemental Fig. S3). In
contrast to LDL, SMase-LDL (Fig. 2b) was composed mainly of LDL aggregates. LP3 (Fig. 2c) and
DP3 (Fig. 2d) partially prevented the formation of LDL aggregates, DP3 showing higher efficiency
than LP3. IP321 did not alter the GFC pattern observed in SMase-LDL (Fig. 2e). As observed with
SMase-LDL, PLA2-LDL (Fig. 2f) showed a large peak of LDL aggregates. LP3 (Fig. 2g), but

Manuscript BBAMEM-19-17 (R1)
15
specially DP3 (Fig. 2h), increased the percentage of monomeric LDL and decreased that of LDL
aggregates. IP321 did not cause any significant alteration in the pattern of PLA2-LDL (Fig. 2i).
LRP1-derived peptides did not alter per se the GFC pattern of LDL (Supplemental Fig. S5).
Quantification of peak areas showed that LP3 and DP3, but not IP321, conferred SMase-LDL and
PLA2-LDL with a GFC profile closer to that of LDL (Fig. 2j,k). TEM images of LDL size distribution
(Fig. 3) showed that LDL comprised mainly individual LDL particles (Fig. 3A). In contrast, SMase-
LDL (Fig. 3B) and PLA2-LDL (Fig. 3C) generated in the presence of IP321 or in the absence of
peptides were formed mostly by aggregated and fused LDL. In the presence of LP3 and DP3,
SMase-LDL (Fig. 3B) and PLA2-LDL (Fig. 3C) were composed of LDL particles with a wide variety
of diameter sizes, although most were in the range present in LDL.
3.4. LP3 and DP3 block the alterations in LDL phospholipid composition in SMase-LDL but
not in PLA2-LDL
Lipoprotein composition analysis (Table 2) revealed that exposure of LDL to SMase or PLA2 did not
alter cholesterol, triglyceride or ApoB content of LDL, either in the presence or absence of the
peptides. In contrast, phospholipid (PL) content of LDL was significantly reduced by both enzymes
(P<0.005). LP3 and DP3 efficiently prevented PL reduction in SMase-LDL (P<0.005). However,
peptides did not show any effect on PL depletion or on non-esterified fatty acid (NEFA) increase
induced by PLA2 treatment. To identify the PL species influenced by enzymatic treatment, HPTLC
was used to separate LDL PLs. This procedure revealed that the main PLs in the LDLs were L--
phosphatidylcholine (L--PC) and sphingomyelin (SM). L--PC levels were unaltered by SMase
treatment (Fig. 4A,B). In contrast, SM was removed from LDL by this treatment (Fig. 4A,C). LP3 and
DP3, but not IP321, efficiently protected LDL-SM against SMase-induced SM degradation (Fig.
4A,C). To examine whether the protective action of LRP1-derived peptides on LDL was due to their
capacity to inhibit SMase activity, we tested the effect of these compounds on the capacity of
SMase to promote SM lipolysis. SMase activity, measured in relation to the amount of
phosphorylcholine generated, was similar in the presence and absence peptides, thereby indicating

Manuscript BBAMEM-19-17 (R1)
16
that none of the peptides exerted direct effects on SMase activity (Supplemental Fig. S6). PLA2
completely removed L--PC from LDL (Fig. 4D,E), leading to a sharp increase in the L--LysoPC
content (Fig. 4D,F). PLA2 did not have any effect on the LDL-SM content (Fig. 4D,G). The peptides
were unable to counteract L--PC decay and the increase in L--LysoPC induced by PLA2 in LDL.
3.5. LP3 and DP3 block the alterations in the LDL electrophoretic profile of SMase-LDL but
not those of PLA2-LDL
To study the effects of the peptides on the electrophoretic mobility of SMase- and PLA2-LDL, we
used gradient gel electrophoresis (GGE) and agarose gels to analyze the modification of LDL in the
presence and absence of the peptides. In native GGE gels (4 h of electrophoresis), the position of
the LDL particle was observed to be determined by particle size. GGE gels showed that SMase
caused a strong loss of the band corresponding to LDL. The lack of this band in SMase-LDL is
attributed to the fact that the pore size of GGE gels precludes the entrance of the largest LDL
aggregates. The band loss induced by SMase was efficiently counteracted by LP3 and DP3 but not
by IP321 (Fig. 5A). In PLA2-LDL (Fig. 5B), GGE showed a minor band corresponding to LDL
(marked as band 1), an intensive band (marked as band 2) with higher electrophoretic mobility
(smaller size) than band 1, and a third band (marked as band 3), which was a minor band with lower
electrophoretic mobility (higher size) than band 1. LP3 and DP3 did not have any effect on the GGE
pattern of PLA2-LDL.
Agarose gels, where LDL runs according to its negative charge, showed that SMase-LDL had a
more diffuse band compared to the well-defined LDL band. SMase-LDL in the presence of LP3 and
DP3 also showed a defined LDL band (Fig. 5C). In contrast to SMase-LDL, PLA2-LDL showed a
single band with a higher negative electric charge than LDL combined with a degradation smear
pattern (Fig. 5D). LP3, and in particular DP3, slightly increased the electrophoretic mobility and
band intensity of PLA2-LDL while they diminished the degradation smear pattern. The peptides did
not cause any change in the electrophoretic mobility of untreated LDL (Supplemental Fig. S7).

Manuscript BBAMEM-19-17 (R1)
17
3.6. DP3 blocks the conformational changes of ApoB-100 in SMase-LDL and PLA2-LDL
We studied the potential conformational modifications of ApoB-100 by Western blot analysis.
Treatment of LDL with SMase (Fig. 6A) or PLA2 (Fig. 6B) increased the exposure of reactive ApoB-
100 epitopes to anti-ApoB-100 antibodies. Both LP3 and DP3 protected ApoB-100 against the
higher reactivity to anti-ApoB-100 antibodies induced by exposure to SMase and PLA2 (Fig. 6A,B).
IP321 had no effect on the increased ApoB-100 reactivity induced by the two enzymatic treatments.
3.7. LRP1-derived peptides electrostatically interact with specific lysine-enriched sequences
in ApoB-100 protein
Limited proteolysis combined with mass spectrometry-based proteomics (LC-MS/MS) evidenced
that DP3 protects specific sequences of ApoB-100 in LDL either by direct interaction or by
remodeling the protein structure (Fig. 7A). The protein/peptide complexes were subjected to limited
proteolysis with trypsin for 1 h and 3 h. The protection of protein sequences by the peptide was
observed by shotgun LC-MSMS analysis at short times (The whole data concerning LDL is available
via PanoramaPublic with identifier PXD011261). At 3-hours digestions, the effect disappeared. The
transient protection of the peptide can be explained by the proteolytic action that trypsin exerted on
the protein complex. Remarkably, most of the protected sequences were also protected in SMase-
LDL unexposed to peptides, suggesting that the aggregation process safeguards them from trypsin
digestion and that these specific sequences are likely to be those that play a major role in LDL
aggregation. The high coincidence between sequences protected by DP3 and those protected by
aggregation indicates that most DP3-protected sequences participate in LDL aggregation, thereby
explaining the high efficiency of LRP1-derived peptides to prevent LDL aggregation.
To further study the molecular basis for ApoB-100 recognition by DP3, peptide homology modeling
and molecular docking methods were used. First, to identify a potential binding site of DP3 peptide
on ApoB-100 protein structure, each ApoB-100 fragment protected by DP3 binding during trypsin
digestion was mapped on the overall ApoB-100 sequence. Virtually all the fragments corresponded
to lipid-associating domains [PMID 18797861] 39, except for a highly positively charged sequence
(3227IKFDKYKAEK3236) located in the ApoB-100 C-terminal region and adjacent to the LDL receptor
Formatted: Highlight

Manuscript BBAMEM-19-17 (R1)
18
binding domain (3385RLTRKRGLK3393) 40-42. This sequence was thus assumed to be responsible
for DP3 peptide recognition and used as the binding site for the molecular docking calculations.
Finally, an energy-based rigid body docking experiment was performed between ApoB-100 protein
and DP3 peptide structures previously modeled alone by ab initio methods. Interestingly, we found
that one of the lowest-energy poses of the docking simulations accommodated DP3 in such an
orientation that ApoB-100 Lys3228, Lys3231 and Lys3233 (but not Lys3226 nor Lys3236) were deeply buried
upon peptide binding. This finding is in strong agreement with the trypsin digestion data described
above. Additionally, strong electrostatic interactions involving same specific key residues of the two
docking partners were also found. In this regard, two of the acidic residues in DP3 correspond to
those assumed to be responsible for the specific recognition between LRP1 module CR9 and
agLDL particles [PMID 25918169] (namely D-Glu3 and D-Glu9) established salt bridge contacts with
two positively charged ApoB-100 residues (namely Lys3229 and Lys3234), while the oxygen atom of
the DP3 D-Asn2 backbone and ApoB-100 Lys3232 were found to be involved in a hydrogen bond
favored by the positive ionic charge carried by the lysine side chain amine group (Fig. 7B).
4. Discussion
A previous study from our group demonstrated that LRP1, in particular the C-terminal half of domain
CR9 (from Gly1127 to Cys1140) named LP3, interacts with agLDL 28. In the current study, we tested
the effect of both the original LP3 version and its retro-enantio version DP3 on SMase and PLA2-
induced LDL aggregation.
In the present study, we observed that PLA2 required larger times and doses than SMase to induce
LDL aggregation. This finding is consistent with previous studies showing that LDL hydrolysis by
PLA2 is slower than that by SMase 34-3643-45. The latter cleaves SM into phosphocholine and
ceramide, whereas PLA2 hydrolyzes the fatty acids in the sn-2 position of PC, generating free fatty
acids and lyso-PC molecules. When SM is lost, ceramide-enriched microdomains have been
proposed to act as non-polar spots at the LDL surface, thereby facilitating LDL aggregation through
hydrophobic associations between domains 44. Here, PLA2-induced PC degradation induces a
Formatted: Subscript

Manuscript BBAMEM-19-17 (R1)
19
dramatic increase in Lyso-L--PC and a strong decrease in LDL surface area, in line with previous
studies 44,46,47. In addition, Here, Both SMase and PLA2 inducedprovoked higher accessibility of
ApoB-100 epitopes to anti-ApoB-100 antibodies, as showed by Western blot analysis of LDL. This
observation would suggest conformational alterations in ApoB-100 linked to phospholipolysis, in line
with previous results from other groups 48. GFC followed by TEM analysis evidenced that LDL
treatment with SMase induced aggregated and fused LDL particles, in agreement with previous
studies 3544. TEM also revealedand that PLA2-LDL comprises a high variety of aggregate sizes,
as previously shown 13.
Here, we demonstrate that LP3 and its derived retro-enantio version (DP3) per se have a high
capacity to inhibit LDL aggregation induced by SMase and PLA2. However, all experimental
approaches (turbidimetry, GFC, and TEM) used in this work demonstrate that DP3 outperforms
LP3. Although all-D peptides are not recognised by proteases, and thus have higher stability, they
may be less active than their corresponding all-L peptides because of differences in the orientation
of amino acid side-chains. To preserve the orientation of the side-chains of the parent L-peptide in a
D-analogue, the retro-enantio approach 49,50 has been used in this study. In this regard, the
preservation of the topological side-chain orientation of DP3 together with a high stability in
biological samples account for its notable performance in the inhibition assays. In addition,
comparison of the capacity of LRP1 (LP3 and DP3) and ApoA-1 (4F) peptides to inhibit LDL
aggregation revealed that at 10 µM (peptide/ApoB-100 ratio: 4.7:1), LP3 and DP3 almost completely
repressed SMase-induced LDL turbidimetry, while L-4F exerted only a partial effect. Previous
studies addressing the efficacy of 4F to inhibit LDL aggregation showed that a molar ratio of 20:1 (4-
fold higher) is required to fully block SMase-induced LDL aggregation 29. However, the
experimental conditions used for LDL aggregation by Nguyen SD et al. (shorter times, and different
procedures for incubation of LDL with L-4F) differed from those applied in our study and thus hinder
a direct comparison between results. To the best of our knowledge, the present study is the first to
report data on the efficacy of 4F to inhibit PLA2-induced LDL aggregation. Here we found that
comparing the inhibitory capacities of L-4F, DP3 and LP3 at 4.7:1 peptide:ApoB-100 ratio, L-4F
Formatted: Font: Italic
Manuscript BBAMEM-19-17 (R1)
20
showed a similar capacity than LP3 to inhibit PLA2-induced LDL aggregation but lower capacity than
DP3.
One key question is how LRP1-derived peptides inhibit LDL aggregation. An important observation
in this regard is that these peptides counteracted alterations in LDL particle size, charge and
phospholipids induced by SMase but not those induced by PLA2. Despite of this, LRP1-derived
peptides exerted protection against SMase and PLA2-induced conformational alterations of ApoB-
100 and against LDL aggregation. These results indicate that 1) peptide protection of ApoB-100
conformation occurreds even in the context of extreme lipolysis and, 2) preservation of ApoB-100
conformation seems to be a common mechanisms underlying LDL protection against SMase and
PLA2-induced LDL aggregation. But, how LRP1-derived peptides preserve ApoB-100
conformation???. In the current study, limited trypsin proteolysis combined with Mass spectrometry-
based proteomic studies of LDL/DP3 complexes evidenced that peptide DP3 interacts with the LDL
surface prior to enzymatic treatment, protecting certain ApoB-100 sequences against trypsin-
induced proteolysis. Most of the peptide-protected sequences in ApoB-100, correspond to lipid-
associated ApoB-100 domains in LDL. The protection that peptide confers to ApoB-100 can be
induced by direct interaction or by remodeling the protein structure. Moreover, a highly positively
charged sequence (3227IKFDKYKAEK3236) located in the ApoB-100 C-terminal region and adjacent to
the LDL receptor binding domain (3385RLTRKRGLK3393) 51-54 was assumed to be responsible for
DP3 peptide recognition and used as the binding site for the molecular docking calculations. In
addition, homology modeling and molecular dynamics methods combined with molecular docking
provided a crucial first insight into the mechanistic basis of LRP1 peptide recognition by ApoB-100
protein. We propose a structural model of DP3 peptide in complex with ApoB-100. This complex is
mainly formed by salt bridge contacts between two of the acidic residues in DP3 (namely D-Glu3
and D-Glu9) and two positively charged ApoB-100 residues (namely Lys3229 and Lys3234), while the
oxygen atom of the DP3 D-Asn2 backbone and ApoB-100 Lys3232 were found to be involved in a
hydrogen bond favored by the positive ionic charge carried by the lysine side chain amine group.
This model is in complete agreement with the trypsin-limited proteolysis data reported herein, as
well as with the study of the recognition between LRP1 module CR9 and aggregated LDL particles
Formatted: Not Superscript/ Subscript
Formatted: Font: Not Italic
Formatted: Font: Not Italic
Formatted: Font: Not Italic
Formatted: Font: Not Italic
Formatted: Font: Not Italic

Manuscript BBAMEM-19-17 (R1)
21
recently published 28. Previous studies have consistently demonstrated that conformational
alterations of ApoB-100 lead to ApoB-100 degradation, a key process for LDL aggregation
48,55,56. In this context, the capacity of LRP1-derived peptides to preserve ApoB-100
conformation explain the high potential of these peptides to protect LDL against LDL aggregation.
Other crucial question is the mechanism by which LRP1-derived peptides protect SM but not PC
from enzymatic hydrolysis. Two different mechanistical options for the protective effect of peptides
against SMase-induced LDL lipolysis have been explored. By one side, we have explored whether
there is a direct effect of peptides on SMase activity. This possibility has been discarded since
SMase activity assays clearly show that peptides does not exert any effect on SMase functional
activity. Other possibility that has been explored is a potential interaction of peptides with the LDL
phospholipid surface. Several peptides and proteins bearing amphipathic properties have been
reported to be able to block LDL aggregation by a direct interaction with LDL phospholipid surface.
In this respect, common examples are 4F peptide 29 or different apolipoproteins 57. Despite the
lack of amphipathic properties in LRP1-derived peptides, this hypothesis has been considered as
possible mechanism of action for the peptides described herein. Theoretical molecular docking
calculations performed in the present study indicate a lack of association between peptides and LDL
phospholipid surface. A key point offering clues about how peptides protect SM is their unability to
protect PC hydrolysis. On the basis of the differential topological distribution of SM and PC in the
LDL surface 46, we propose that the differential anti-phospholypolytic effects of the peptide on SM
and PC phospholipolysis may be determined, at least in part, by the particular imbrication of SM and
PC with free cholesterol on the LDL surface. Although SM, compared to PC, is a minor phospholipid
in LDL (185 versus 450 molecules/LDL), SM is localized in cholesterol-enriched environments,
considered key regulators of phospholipolysis 46. ApoB-100 conformational stabilization could
directly impact the maintenance of the cholesterol-enriched environment structure since, according
to our molecular modeling studies, ApoB-100 sequences complexed with peptide are those close to
cholesterol in the LDL surface. Therefore, SM could be protected by LRP1-derived peptides against
phospholipolysis by its privilege of being part of cholesterol-enriched domains, whose structure

Manuscript BBAMEM-19-17 (R1)
22
would be preserved through the formation of peptide-ApoB-100 complexes. In contrast, PC,
belonging to cholesterol-poor domains, would be unprotected against PLA2-induced lipolysis,
independently of the status of ApoB-100 conformation.
5. Conclusion
Here, we show that LRP1-derived peptides, LP3 and, in particular, its retro-enantio version (DP3),
efficiently preserve LDL against SMase or PLA2-induced LDL aggregation. LP3 and DP3 maintain
LDL particle size, charge, phospholipid composition and ApoB-100 conformation in LDL treated with
SMase, but only ApoB-100 conformation in LDL exposed to PLA2 (summarized in Figure 8,
Graphical abstract). LP3 and DP3 keep ApoB-100 conformation even under extreme lipolysis
through key electrostatic interactions with a highly positively charged sequence
(3227IKFDKYKAEK3236) located in the ApoB-100 C-terminal region. A recent study has shown that the
individual susceptibility of LDL to aggregation is associated with future cardiac deaths independently
of other classical risk factors 4258. LRP(CR9) derived peptides protect LDL against the main
proteolytic enzymes contributing to LDL retention and aggregation in the arterial intima. In the next
future, we are going to test the efficacy of the retro-enantio peptide version (DP3) to reduce
vascular LDL retention and LDL-induced smooth muscle cell (SMC)-foam cell formation in in vivo
models of atherosclerosis. Our first option in the near future will be to inject the peptide DP3
subcutaneously (not focal delivery at the moment) in a murine model of angioplasty-induced
atherosclerosis. This model offers the taking advantage of the increased endothelial permeability,
crucial to favor peptide arrival to vasculature. In a not too distant future, we expect LRP1-derived
peptides to emerge as tools potentially useful to prevent atherosclerosis and cardiovascular
complications, in particular in individuals with aggregation-prone LDL as obese and diabetic patients
DATA AVAILABILITY
Mass spectroscopy proteomics of LDL. Panorama Public PXD011261
https://panoramaweb.org/j8CaUy.url

Manuscript BBAMEM-19-17 (R1)
23
Conflict of interest
R.P., C.P., and T.T. are employees of Iproteos SL. T.T. is cofounder of Iproteos SL and member of
its board of directors.
Acknowledgments
The Ministry of Science and Innovation of Spain, in the framework of the State Plan of Scientific
and Technical Innovation Investigation 2013-2016, awarded funding to the project
“DEVELOPMENT OF AN INNOVATIVE THERAPY FOR THE TREATMENT OF THE
ATHEROSCLEROSIS THROUGH INHIBITION OF CHOLESTEROL VASCULAR
ACCUMULATION”, led by IPROTEOS SL with file number RTC-2016-5078-1. This project was also
financed by the Ministry of Economy, Industry and Competitiveness (MINECO) in the framework of
the Subprogram RETOS-COLABORACIÓN, 2016 call. The project is also co-financed by the
European Union with the objective to promoting the technological development, innovation and
quality research. Support was also received from SAF2017-89613R (to SV), co-financed by the
European Regional Development Fund (ERDF), the Fundació MARATÓ TV3 Project 201521-10 (to
VLl-C), FIS PI13/00364 and PI16/00471 (to JSQ) and FIS PI18/01584 (to VLl-C) from the Instituto
de Salud Carlos III (ISCIII) and co-financed with ERDFs. Support was also received from Ministerio
de Economía y Competitividad to DdG-C (IJCI-2016-29393). CIBER Diabetes y Enfermedades
Metabólicas Asociadas (CIBERDEM; CB07/08/0016 (JSQ) and CIBER Enfermedades
Cardiovasculares (CIBERCV; CB16/1100403 (DdG-C, VLl-C) are projects run by the Instituto de
Salud Carlos III (ISCIII). The CRG/UPF Proteomics Unit is member of ProteoRed PRB3 consortium
which is supported by grant PT17/0019 of the PE I+D+i 2013-2016 from the Instituto de Salud
Carlos III (ISCIII) and ERDF. We also acknowledge support from the Spanish Ministry of Economy
and Competitiveness, “Centro de Excelencia Severo Ochoa 2013-2017”, SEV-2012-0208, and
“Secretaria d’Universitats i Recerca del Departament d’Economia I Coneixement de la Generalitat
de Catalunya” (2017SGR595 to ES and 2017SGR946 to VLl-C. SB, JLS-Q, AR-U, DdG-C and VL-
C are members of the Group of Vascular Biology of the Spanish Society of Atherosclerosis (SEA).

Manuscript BBAMEM-19-17 (R1)
24
References
[1] Castelli WP, Anderson K, Wilson PWF, Levy D. Lipids and risk of coronary heart disease The Framingham Study. Ann Epidemiol. 1992; 2:23–28.
[2] Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR, High density lipoprotein as a protective factor against coronary heart disease. Am J Med. 1977; 62:707–714.
[3] Guretzki H-J, Gerbitz K-D, Olgemöller B, Schleicher E. Atherogenic levels of low density lipoprotein alter the permeability and composition of the endothelial barrier. Atherosclerosis. 1994; 107:15–24.
[4] Aikawa M, Libby P. The vulnerable atherosclerotic plaque. Cardiovasc Pathol. 2004; 13 :125–138.
[5] Mauriello A, Sangiorgi GM, Virmani R, Trimarchi S, Holmes Jr DR, Kolodgie FD, Piepgras DG, Piperno G, Liotti D, Narula J, Righini P, Ippoliti A, Spagnoli LG. A pathobiologic link between risk factors profile and morphological markers of carotid instability. Atherosclerosis. 2010; 208: 572–580.
[6] Puri R, Nissen SE, Shao M, Elshazly MB, Kataoka Y, Kapadia SR, Tuzcu EM, Nicholls SJ. Non-HDL Cholesterol and Triglycerides: Implications for Coronary Atheroma Progression and Clinical Events., Arterioscler Thromb Vasc Biol. 2016; 36:2220-2228.
[7] Skålén K, Gustafsson M, Knutsen Rydberg E, Hultén LM, Wiklund, Innerarity TL, Boren J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002; 417:750-754.
[8] Öörni K, Pentikäinen MO, Ala-Korpela M, Kovanen PT. Aggregation, fusion, and vesicle formation of modified low density lipoprotein particles: molecular mechanisms and effects on matrix interactions. J Lipid Res. 2000; 41:1703–1714.
[9] Öörni K, Posio P, Ala-Korpela M, Jauhiainen M, Kovanen PT. Sphingomyelinase induces aggregation and fusion of small very low-density lipoprotein and intermediate-density lipoprotein particles and increases their retention to human arterial proteoglycans, Arterioscler Thromb Vasc Biol. 2005; 25:1678-1683
[10] Marathe S, Schissel SL, Yellin MJ, Beatini N, Mintzer R, Williams KJ, Tabas I. Human vascular endothelial cells are a rich and regulatable source of secretory sphingomyelinase. Implications for early atherogenesis and ceramide-mediated cell signaling. J Biol Chem. 1998; 273:4081-4088.
[11] Schissel SL, Schuchman EH, Williams KJ, Tabas I. Zn2+-stimulated Sphingomyelinase Is Secreted by Many Cell Types and Is a Product of the Acid Sphingomyelinase Gene. J Biol Chem. 1996;. 271:18431–18436.
[12] Aviram M, Maor I. Phospholipase A2-modified LDL is taken up at enhanced rate by macrophages. Biochem Biophys Res Commun. 1992; 185:465–472.
[13] Öörni K, Hakala JK, Annila A, Ala-Korpela M, Kovanen PT. Sphingomyelinase induces aggregation and fusion, but phospholipase A2 only aggregation, of low density lipoprotein (LDL) particles: Two distinct mechanisms leading to increased binding strength of LDL to human aortic proteoglycans. J Biol Chem. 1998; 273:29127-29134.
[14] Hakala JK, Öörni K, Pentikäinen MO, Hurt-Camejo E, Kovanen PT. Lipolysis of LDL by Human Secretory Phospholipase A Induces Particle Fusion and Enhances the Retention of LDL to Human Aortic Proteoglycans. Arterioscler Thromb Vasc Biol. 2001; 21:1053-1058.
[15] Tabas I, Li Y, Brocia RW, Xu SW, Swenson TL, Williams KJ. Lipoprotein lipase and sphingomyelinase synergistically enhance the association of atherogenic lipoproteins with smooth muscle cells and extracellular matrix. A possible mechanism for low density lipoprotein and lipoprotein(a) retention and macrophage foam cell formation, J Biol Chem. 1993; 268:20419-20432.
[16] Von Ballmoos MW, Dubler D, Mirlacher M, Cathomas G, Muser J, Biedermann BC. Increased apolipoprotein deposits in early atherosclerotic lesions distinguish symptomatic from
Formatted: Spanish (Spain,
International Sort)
Formatted: Spanish (Spain,
International Sort)

Manuscript BBAMEM-19-17 (R1)
25
asymptomatic patients. Arterioscler Thromb Vasc Biol. 2006; 26:359-364. [17] Khoo JC, Miller E, McLoughlin P, Steinberg D. Enhanced macrophage uptake of low density
lipoprotein after self-aggregation. Arterioscler Thromb Vasc Biol. 1988; 8:348-358. [18] Zhang WY, Ishii I, Kruth HS. Plasmin-mediated macrophage reversal of low density
lipoprotein aggregation. J Biol Chem. 2000; 275:33176-33183. [19] Kruth HS. Sequestration of aggregated low-density lipoproteins by macrophages. Curr Opin
Lipidol. 2002; 13:483-488. [20] Llorente-Cortés V, Martínez-González J, Badimon L. Esterified Cholesterol Accumulation
Induced by Aggregated LDL Uptake in Human Vascular Smooth Muscle Cells Is Reduced by HMG-CoA Reductase Inhibitors, Arterioscler Thromb Vasc Biol. 1998; 18:738-746.
[21] Llorente-Cortés V, Martínez-González J, Badimon L. LDL Receptor–Related Protein Mediates Uptake of Aggregated LDL in Human Vascular Smooth Muscle Cells. Arterioscler Thromb Vasc Biol. 2000; 20:1572-1579.
[22] Llorente-Cortés V, Otero-Viñas M, Hurt-Camejo E, Martínez-González J, Badimon L. Human coronary smooth muscle cells internalize versican-modified LDL through LDL receptor-related protein and LDL receptors. Arterioscler Thromb Vasc Biol. 2002; 22:387-393.
[23] Llorente-Cortés V, Otero-Viñas M, Sánchez S, Rodríguez C, Badimon L. Low-Density Lipoprotein Upregulates Low-Density Lipoprotein Receptor-Related Protein Expression in Vascular Smooth Muscle Cells. Circulation. 2002; 106:3104-3110
[24] Costales P, Aledo R, Vérnia S, Das A, Shah VH, Casado M, Badimon L, Llorente-Cortés V. Selective role of sterol regulatory element binding protein isoforms in aggregated LDL-induced vascular low density lipoprotein receptor-related protein-1 expression, Atherosclerosis. 2010; 213:458–468.
[25] Llorente-Cortés V, Otero-Viñas M, Camino-López S, Llampayas O, Badimon L. Aggregated low-density lipoprotein uptake induces membrane tissue factor procoagulant activity and microparticle release in human vascular smooth muscle cells, Circulation. 2004; 110:452-459.
[26] Camino‐ López S, Badimon L, GonzáLez A, Canals D, Peña E, Llorente‐ Cortés V. Aggregated low density lipoprotein induces tissue factor by inhibiting sphingomyelinase activity in human vascular smooth muscle cells. J Thromb Haemost. 2009; 7:2137–2146.
[27] Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis., Circulation.2014; 129:1551-1559.
[28] Costales P, Fuentes-Prior P, Castellano J, Revuelta-Lopez E, Corral-Rodríguez MÁ, Nasarre L, Badimon L, Llorente-Cortes V. Domain CR9 of Low Density Lipoprotein (LDL) Receptor-related Protein 1 (LRP1) Is Critical for Aggregated LDL-induced Foam Cell Formation from Human Vascular Smooth Muscle Cells. J Biol Chem. 2015; 290:14852–14865.
[29] Nguyen SD, Javanainen M, Rissanen S, Zhao H, Huusko J, Kivelä AM, Ylä-Herttuala S, Navab M, Fogelman AM, Vattulainen I, Kovanen PT, Öörni K. Apolipoprotein A-I mimetic peptide 4F blocks sphingomyelinase-induced LDL aggregation J Lipid Res. 2015; 56:1206–1221.
[30] Pujals S, Fernández-Carneado J, Ludevid MD, Giralt E. D-SAP: A new, noncytotoxic, and fully protease resistant cell-penetrating peptide. ChemMedChem. 2008; 3:296-301.
[31] Bancells C, Villegas S, Blanco FJ, Benítez S, Gállego I, Beloki L, Pérez-Cuellar M, Ordóñez-Llanos J, Sánchez-Quesada JL. Aggregated Electronegative Low Density Lipoprotein in Human Plasma Shows a High Tendency toward Phospholipolysis and Particle Fusion. J Biol Chem. 2010; 285:32425–32435.
[32] Martínez-Bujidos M, Rull A, González-Cura B, Pérez-Cuéllar M, Montoliu-Gaya L, Villegas S, Ordóñez-Llanos J, Sánchez-Quesada J.L. Clusterin/apolipoprotein J binds to aggregated LDL in human plasma and plays a protective role against LDL aggregation. FASEB J. 2014; 29:1688–1700.
[33] Case D, Cheatham TE 3rd, Darden T, Gohlke H, Luo R, Merz KM Jr, Inufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668-1688.
[34] Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé
Formatted: English (United States)

Manuscript BBAMEM-19-17 (R1)
26
L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781-1802.
[35] Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004;32:W526-W531.
[36] Koes DR, Baumgartner MP, Camacho CJ. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J Chem Inf Model. 2013; 53:1893-1904.
[37] Chorev M, Goodman M. Recent developments in retropeptides and proteins_An ongoing topochemical exploration. Trends Biotechnol. 1995;13:438-445.
[383] Prades R, Oller-Salvia B, Schwarzmaier SM, Selva J, Moros M, Balbi M, Grazú V, De La Fuente JM, Egea G, Plesnila N, Teixidõ M, Giralt E. Applying the retro-enantio approach to obtain a peptide capable of overcoming the blood-brain barrier. Angew. Chemie - Int. Ed. 2015; 54:3967-3972.
[39] Prassl R, Laggner P. Molecular structure of low density lipoprotein: current status and future challenges. Eur Biophys J. 2009;38:145-158.
[40] Segrest JP, Jones MK, De Loof H, Dashti N. Structure of Apolipoprotein B-100 in low density lipoproteins. J Lipid Res. 2001; 42:1346-1367.
[41] Yang CY, Chen SH, Gianturco SH, Bradley WA, Sparrow JT, Tanimura M, Li WH, Sparrow DA, DeLoof H, Rosseneu M, et al. Sequence, structure, receptor-binding domains and internal repeats of human apolipoprotein B-100. Nature. 1986;323:738-742.
[42] Hospattankar AV, Law SW, Lackner K, Brewer HB Jr. Identification of low density lipoprotein receptor binding domains of human apolipoprotein B-100: a proposed consensus LDL receptor binding sequence of apoB-100. Biochem Biophys Res Commun. 1986;139:1078-1085.
[3443] Schissel SL, Tweedie-Hardman J, Rapp JH, Graham G, Williams KJ, Tabas I. Rabbit aorta and human atherosclerotic lesions hydrolyze the sphingomyelin of retained low-density lipoprotein. Proposed role for arterial-wall sphingomyelinase in subendothelial retention and aggregation of atherogenic lipoproteins. J Clin Invest. 1996; 98:1455–1464.
[3544] Sartipy P, Camejo G, Svensson L, Hurt-Camejo E. Phospholipase A2 modification of low density lipoproteins forms small high density particles with increased affinity for proteoglycans and glycosaminoglycans, J Biol Chem. 1999; 274:25913-25920.
[3645] Oestvang J, Bonnefont-Rousselot D, Ninio E, Hakala JK, Johansen B, Anthonsen MW, Modification of LDL with human secretory phospholipase A2 or sphingomyelinase promotes its arachidonic acid-releasing propensity. J Lipid Res. 2004; 45:831–838.
[3746] Hevonoja T, Pentikäinen MO, Hyvönen MT, Kovanen PT, Ala-Korpela M. Structure of low density lipoprotein (LDL) particles: Basis for understanding molecular changes in modified LDL. Biochim Biophys Acta - Mol. Cell Biol. Lipids. 2000; 1488:189–210.
[3847] Benítez S, Camacho M, Arcelus R, Vila L, Bancells C, Ordóñez-Llanos J, Sánchez-Quesada JL. Increased lysophosphatidylcholine and non-esterified fatty acid content in LDL induces chemokine release in endothelial cells. Atherosclerosis. 2004; 177:299–305.
[3948] Kleinman Y, Krul ES, Burnes M, Aronson W, Pfleger B, Schonfeld G. Lipolysis of LDL with phospholipase A2 alters the expression of selected apoB-100 epitopes and the interaction of LDL with cells, J Lipid Res. 1988; 29:729-743.
[49] Soto C, Kindy MS, Baumann M, Frangione B. Inhibition of Alzheimer amyloidosis by peptides that prevent beta-sheet conformation. Biochem Biophys Res Commun. 1996; 226:672–680.
[50] Chalifour RJ, McLaughlin RW, Lavoie L, Morissette C, Tremblay N, Boulé M, Sarazin P, Stéa D, Lacombe D, Tremblay P, Gervais F. Stereoselective interactions of peptide inhibitors with the beta-amyloid peptide. J Biol Chem. 2003;278:34874-34881.
[4051] Sneck M, Nguyen SD, Pihlajamaa T, Yohannes G, Riekkola M-L, Milne R, Kovanen PT, Öörni K. Conformational changes of apoB-100 in SMase-modified LDL mediate formation of large aggregates at acidic pH, J. Lipid Res. 2012; 53:1832-1839.
[5241] Bancells C, Benítez S, Ordóñez-Llanos J, Öörni K, Kovanen PT, Milne RW, Sánchez-Quesada JL. Immunochemical Analysis of the Electronegative LDL Subfraction Shows That Abnormal N-terminal Apolipoprotein B Conformation Is Involved in Increased Binding to Proteoglycans, J Biol Chem. 2011; 286:1125–1133.
[53] Liu H, Scraba DG, Ryan RO. Prevention of phospholipase-C induced aggregation of LDL by
Formatted: Indent: Left: 0 cm,
Hanging: 1.13 cm
Formatted: Font: (Default) Arial, 11
Formatted: Font: (Default) Arial
Formatted: Font: (Default) Arial
Formatted: Font: (Default) Arial, 11
Formatted: Check spelling and
grammar
Formatted: English (United States)
Formatted: Font: (Default) Arial
Formatted: English (United States)
Formatted: Font: (Default) Arial
Formatted: Check spelling and
grammar
Formatted: Spanish (Spain,
International Sort)
Formatted: Indent: Left: 0 cm,
Hanging: 1.13 cm
Formatted: Font: (Default) Arial,
Spanish (Spain, International Sort)
Formatted: Spanish (Spain,
International Sort)

Manuscript BBAMEM-19-17 (R1)
27
amphipathic apolipoproteins. FEBS Lett. 1993; 316:27–33. [4254] Ruuth M, Nguyen SD, Vihervaara T, Hilvo M, Laajala TD, Kondadi PK, Gisterå A,
Lähteenmäki H, Kittilä T, Huusko J, Uusitupa M, Schwab U, Savolainen MJ, Sinisalo J, Lokki M-L, Nieminen MS, Jula A, Perola M, Ylä-Herttula S, Rudel L, Öörni A, Baumann M, Baruch A, Laaksonen R, Ketelhuth DFJ, Aittokallio T, Jauhiainen M, Käkelä R, Borén J, Williams KJ, Kovanen PT, Öörni K. Susceptibility of low-density lipoprotein particles to aggregate depends on particle lipidome, is modifiable, and associates with future cardiovascular deaths. Eur Heart J. 2018; 39:2562-2573
Figure Legends
Fig.1. LRP1-derived peptides (LP3 and DP3) decrease SMase- and PLA2-induced LDL aggregation in a dose-dependent manner. LDL was used at 1.44 mg/mL ApoB. Peptides were tested at
increasing concentrations (from 2.5 to 10 M for SMase-LDL and from 4.16 to 20.83 M for PLA2-LDL). Aggregation of LDL was induced by SMase (A) or PLA2 (B) and monitored at the indicated times by measuring turbidimetry (absorbance at 405 nm). Data are mean ± SD of 2 experiments performed in duplicate. Unsighted bar errors are minor than symbol size.*P<0.005 versus SMase-
LDL + IP321; P<0.005 versus SMase-LDL + LP3.
Fig. 2. LRP1-derived peptides (LP3 and DP3) reduce the AUC of agLDL peak in SMase-LDL and PLA2-LDL. LDL was untreated (LDL) or treated with SMase (40 U/L) and PLA2 (50 µg/L) in the absence (w/o peptide) or presence of LP3, DP3 or IP321 (10 µM, peptide/ApoB-100 ratio: 4.7:1, 24 h). LDLs (0.5 mL at 0.4 mg/mL ApoB) were chromatographed at a flow rate of 0.5 ml/min using a Superose 6 column in a AKTA-FPLC system (GE Healthcare) as described in Material and Methods. Representative GFC of: a) non enzymatically-treated LDL; b) LDL incubated with SMase; c) LDL incubated with SMase in the presence of LP3; d) LDL incubated with SMase in the presence of DP3; e) LDL incubated with SMase in the presence of IP321; f) LDL incubated with PLA2; g) LDL incubated with PLA2 in the presence of LP3; h) LDL incubated with PLA2 in the presence of DP3; and i) LDL incubated with PLA2 in the presence of IP321. j) and k) Bar graphs showing the percentage of aggregated versus monomeric LDL in SMase-LDL and PLA2-LDL, respectively, calculated from the chromatograms as area under the curve as explained in Methods.
Fig. 3. LRP1-derived peptides (LP3 and DP3) diminish the proportion of large size LDL aggregates in SMase-LDL and PLA2-LDL. Untreated LDL (A) or LDL treated with SMase (40 U/L) (B) and PLA2 (50 µg/L) (C) in the presence of LP3, DP3 or IP321 (10 µM, peptide/ApoB-100 ratio: 4.7:1, 24 h) or in absence of peptides (w/o peptide). TEM was performed in a Jeol 120 kV JEM-1400 microscope in the LDL preparations treated as explained above for 24 h at 37ºC. Black bars, 500 nm.
Fig. 4. LRP1-derived peptides (LP3 and DP3) prevent SM depletion in SMase-LDL but not L--PC depletion in PLA2-LDL. LDL was untreated (LDL) or treated with SMase (40 U/L) and PLA2 (50 µg/L) in the absence (w/o peptide) or presence of LP3, DP3 or IP321 (10 µM, peptide/ApoB-100 ratio: 4.7:1, 24 h) and the phospholipid pattern was analyzed by HPTLC after lipid extraction. Representative HPTLC of LDL (A and D) and bar graphs (B, C, E, F and G) showing quantification of phospholipid bands. Results are expressed as percentage of PL relative to untreated LDL and
shown as mean ± SD of 3 independent experiments performed in triplicate. L--PC indicates L--
phosphatidylcholine; SM: sphingomyelin; L--lyso-PC: L--lysophosphatidylcholine. *P<0.005 versus LDL.
Fig. 5. LRP1-derived peptides (LP3 and DP3) counteract LDL alterations in size and electric charge induced by SMase but not those induced by PLA2. LDL was untreated (LDL) or treated with SMase

Manuscript BBAMEM-19-17 (R1)
28
(40 U/L) and PLA2 (50 µg/L) in the absence (w/o peptide) or presence of LP3, DP3 o IP321 (10 µM, ratio peptide/ApoB-100: 4.7:1, 24 hours). LDL electrophoretic mobility was analyzed by GGE (A and B) and agarose gels (C and D). In GGE, LDL (5 µg/well) was electrophoresed in 2-16% acrylamide native gradient gels. LDLs were prestained with 1% Sudan black. In agarose gel electrophoresis, LDL (2 µg/well) was electrophoresed in 0.5% agarose gels, which were dehydrated and stained with Coomassie Blue. Peptides were assayed at 10 µM (Ratio Peptide/ApoB= 4.7/1)
Fig. 6. LRP1-derived peptides (LP3 and DP3) counteract ApoB-100 conformational alterations induced by SMase and PLA2 in LDL. LDL was untreated (LDL) or treated with SMase (40 U/L) and PLA2 (50 µg/L) in the absence (w/o peptide) or presence of LP3, DP3 or IP321 (10 µM, peptide/ApoB-100 ratio: 4.7:1, 24 h). Representative western blot analysis of ApoB-100 in LDL exposed to SMase (A) or PLA2 (B). LDL (5 µg/well) was electrophoresed in 10% acrylamide gels under non-reducing conditions for 2 h at 100 V and transferred to nitrocellulose membranes, which were then incubated with anti-ApoB-100 antibodies. Fig. 7. DP3 protect ApoB-100 sequences against tryptic-limited proteolysis A) List of ApoB100 peptides that are protected by DP3 and by LDL aggregation as shown by tryptic-limited proteolysis coupled to mass spectrometry analysis. Green indicates signal detected, Yellow a poor signal, and Red no MS signal. B) Structural details of the molecular interaction between Apo-B100 protein and DP3 peptide. View of the ApoB-100 protein surface contacting the DP3 peptide: the basic residues of ApoB-100 exposed to trypsin digestion and buried deeply upon peptide binding highlighted in teal and violet, respectively. DP3 peptide acid residues responsible for ApoB-100 binding highlighted in yellow sticks.
Fig. 8. Fig. 8. Schematic representation of the molecular basis for the protective effect of LRP1-derived peptides against SMase- and PLA
2-induced LDL aggregation. DP3 forms a complex with
ApoB-100 and this molecular interaction stabilizes ApoB-100 conformation. ApoB-100 conformation stabilization could guarantee the structural preservation of surface colesterol-enriched environments, where SM is located. Structural preservation of cholesterol, a key regulator of phospholipolysis, would protect SM from SMase activity. As a result, LDL complexed with DP3 remains unaltered when exposed to SMase. This scenario changes when LDL complexed with DP3 is exposed to PLA
2. The target for PLA
2 is PC, phospholipid associated with poor-colesterol
environments. Therefore, PC is unprotected against the attack of PLA2, that hydrolyzes PC producing lyso-PC and NEFA. Remarkably, LDL complexed with DP3 is protected against SMase and PLA2-induced aggregation even in conditions of extreme phospholipolysis, indicating that the maintenance of ApoB-100 conformation is key in the prevention of LDL aggregation.
Formatted: Subscript
Formatted: English (United States)
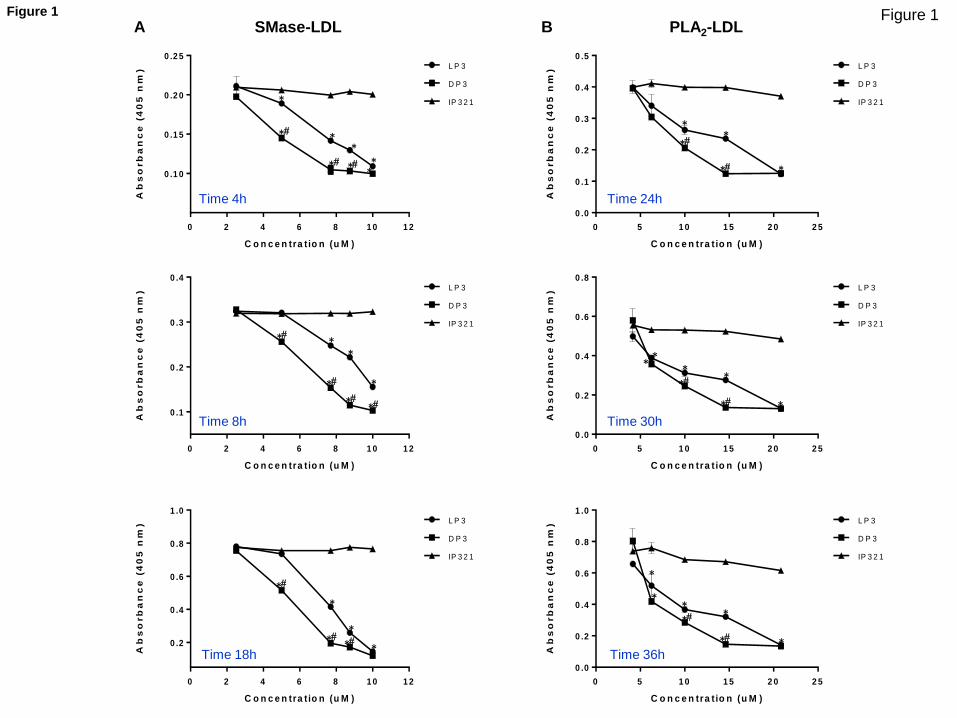
Figure 1
C o n c e n tra t io n (u M )
Ab
so
rb
an
ce
(4
05
nm
)
0 2 4 6 8 1 0 1 2
0 .1 0
0 .1 5
0 .2 0
0 .2 5L P 3
D P 3
IP 3 2 1
C o n c e n tra t io n (u M )
Ab
so
rb
an
ce
(4
05
nm
)
0 2 4 6 8 1 0 1 2
0 .1
0 .2
0 .3
0 .4L P 3
D P 3
IP 3 2 1
C o n c e n tra t io n (u M )
Ab
so
rb
an
ce
(4
05
nm
)
0 2 4 6 8 1 0 1 2
0 .2
0 .4
0 .6
0 .8
1 .0L P 3
D P 3
IP 3 2 1
Time 4h
Time 8h
Time 18h
SMase-LDL
C o n c e n tra t io n (u M )
Ab
so
rb
an
ce
(4
05
nm
)
0 5 1 0 1 5 2 0 2 5
0 .0
0 .1
0 .2
0 .3
0 .4
0 .5L P 3
D P 3
IP 3 2 1
C o n c e n tra t io n (u M )
Ab
so
rb
an
ce
(4
05
nm
)
0 5 1 0 1 5 2 0 2 5
0 .0
0 .2
0 .4
0 .6
0 .8L P 3
D P 3
IP 3 2 1
C o n c e n tra t io n (u M )
Ab
so
rb
an
ce
(4
05
nm
)
0 5 1 0 1 5 2 0 2 5
0 .0
0 .2
0 .4
0 .6
0 .8
1 .0L P 3
D P 3
IP 3 2 1
PLA2-LDL
Time 24h
Time 30h
Time 36h
A B
*
*
*
* *
* *
* *
*
*
* * *
*
*
*
*
*
* *
* *
*
*
*
* *
* *
*
* *
*
* *
*
*
* *
Figure 1

Figure 2
1a001:1_UV
0.0
10.0
20.0
30.0
40.0
50.0
60.0
70.0
80.0
mAU
0.0 2.0 4.0 6.0 8.0 10.0 ml
Aggregated LDL
Monomeric LDL
3a001:1_UV
0
50
100
150
200
250
mAU
0.0 2.0 4.0 6.0 8.0 10.0 ml
Aggregated
Monomeric
4a001:1_UV
0
20
40
60
80
mAU
0.0 2.0 4.0 6.0 8.0 10.0 ml
Aggregated
Monomeric
5a001:1_UV
0.0
10.0
20.0
30.0
40.0
50.0
60.0
70.0
mAU
0.0 2.0 4.0 6.0 8.0 10.0 ml
Aggregated
Monomeric 7a001:1_UV
0
50
100
150
200
250
300
mAU
0.0 2.0 4.0 6.0 8.0 10.0 ml
Aggregated
Monomeric
8a001:1_UV
0
50
100
150
mAU
0.0 2.0 4.0 6.0 8.0 10.0 ml
Aggregated
Monomeric
9a001:1_UV
0.0
20.0
40.0
60.0
80.0
mAU
0.0 2.0 4.0 6.0 8.0 10.0 ml
Aggregated
Monomeric
10a001:1_UV
0.0
10.0
20.0
30.0
40.0
50.0
60.0
mAU
0.0 2.0 4.0 6.0 8.0 10.0 ml
Aggregated
Monomeric
12a001:1_UV
0
50
100
150
200
250
mAU
0.0 2.0 4.0 6.0 8.0 10.0 ml
Aggregated
Monomeric
w/o peptide LP3
DP3 IP321
SMase-LDL
LDL
DP3 IP321
PLA2-LDL
w/o peptide LP3
SMas
e-i
nd
uce
d L
DL
aggr
ega
tio
n
(%)
PLA
2-in
du
ced
LD
L ag
gre
gati
on
(
%)
a
b c
d e
f g
h i
SMase-LDL PLA2-LDL
0
20
40
60
80
100
Aggregated
Monomeric
0
20
40
60
80
100
Aggregated
Monomeric
j K
Figure 2
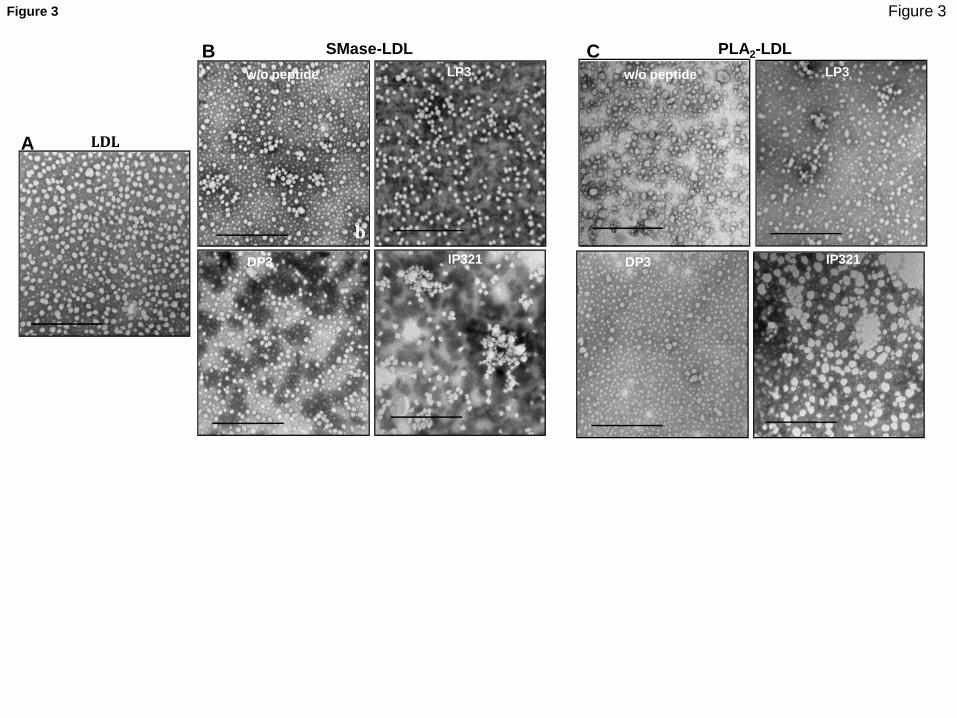
Figure 3
b
c d e
f
h
w/o peptide
LDL
LP3
SMase-LDL PLA2-LDL
DP3 IP321
w/o peptide LP3
DP3 IP321
A
B C
Figure 3
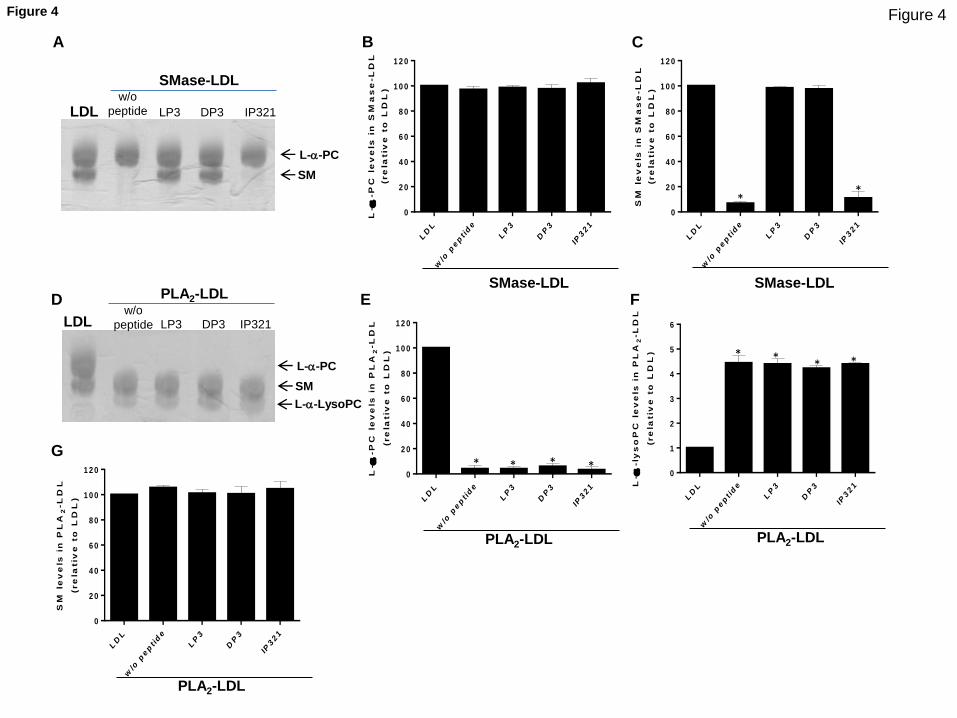
Figure 4
LDL LP3 DP3 IP321
SMase-LDL
LP3 DP3 IP321
PLA2-LDL
SM
L-a-PC
LDL
w/o
peptide
w/o
peptide
L-a-LysoPC
SM
le
ve
ls i
n P
LA
2-L
DL
(re
lativ
e t
o L
DL
)
LD
L
w/o
pept ide
LP
3
DP
3
IP321
0
2 0
4 0
6 0
8 0
1 0 0
1 2 0 L-a
-P
C l
ev
els
in
PL
A2
-L
DL
(re
lativ
e t
o L
DL
)
LD
L
w/o
pept ide
LP
3
DP
3
IP321
0
2 0
4 0
6 0
8 0
1 0 0
1 2 0
L-a
-ly
so
PC
le
ve
ls i
n P
LA
2-L
DL
(re
lativ
e t
o L
DL
)
LD
L
w/o
pept ide
LP
3
DP
3
IP321
0
1
2
3
4
5
6
L-a
-P
C l
ev
els
in
SM
as
e-L
DL
(re
lativ
e t
o L
DL
)
LD
L
w/o
pept ide
LP
3
DP
3
IP321
0
2 0
4 0
6 0
8 0
1 0 0
1 2 0
SM
le
ve
ls i
n S
Ma
se
-L
DL
(re
lativ
e t
o L
DL
)
LD
L
w/o
pept ide
LP
3
DP
3
IP321
0
2 0
4 0
6 0
8 0
1 0 0
1 2 0
SM
L-a-PC
SMase-LDL SMase-LDL
PLA2-LDL
PLA2-LDL
PLA2-LDL
* *
* * * *
* * * *
A B C
D E F
G
Figure 4
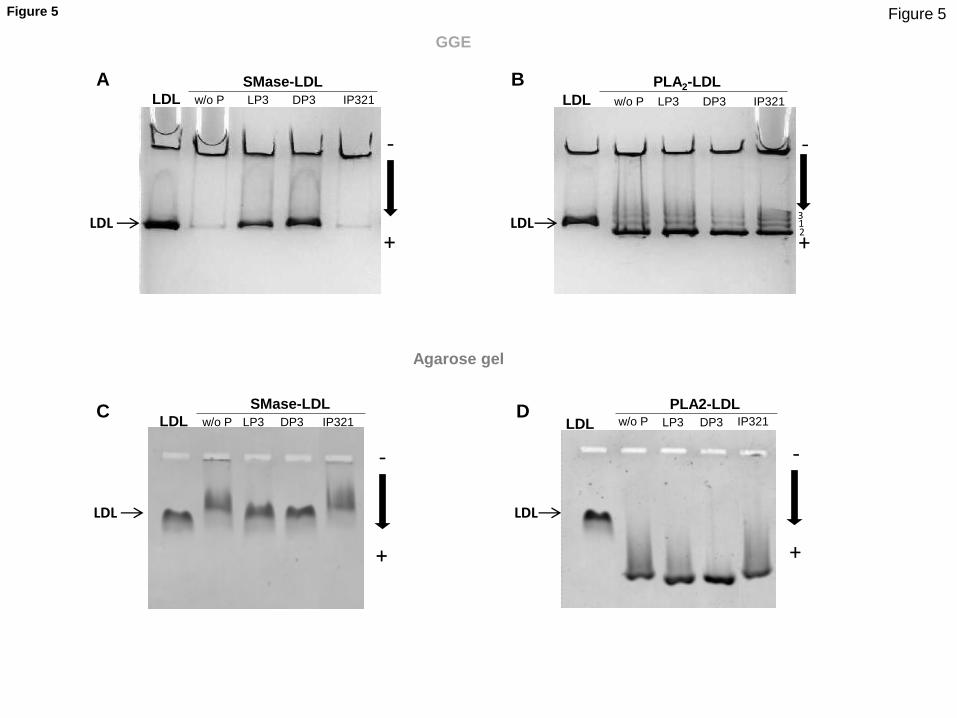
Figure 5
GGE
Agarose gel
w/o P LP3 DP3 IP321
SMase-LDL
LP3 DP3 IP321
PLA2-LDL
-
+
LDL LDL
w/o P LP3 DP3 IP321
SMase-LDL
LDL LP3 DP3 IP321
PLA2-LDL
-
+
-
+
-
+
LDL LDL
LDL LDL
1 2
3
w/o P
w/o P LDL
A B
C D
Figure 5

Figure 6
LDL LP3 DP3 IP321
SMase-LDL
LDL LP3 DP3 IP321
PLA2-LDL
ApoB-100 ApoB-100
250 kDa
250 kDa
Mw Mw
A B
w/o
peptide w/o
peptide
Figure 6

Figure 7 A
B
Peptide Start End Status Protein nat
iveL
DL
nat
iveL
DL
+ D
P3
Smas
e-LD
L
Smas
e-LD
L +
DP
3
YELKLAIPEGK 128 138 protected sp|P04114|APOB_HUMAN 1h
3h
YGMVAQVTQTLKLEDTPK 275 292 protected sp|P04114|APOB_HUMAN 1h
3h
LEDTPKINSR 287 296 protected sp|P04114|APOB_HUMAN 1h
3h
IKFDKYKAEK 3227 3236 protected sp|P04114|APOB_HUMAN 1h
3h
FDKYKAEK 3229 3236 protected sp|P04114|APOB_HUMAN 1h
3h
Figure 7

ApoB-100/DP3 complex
STABILIZATION OF ApoB-100 CONFORMATION
SMase PLA2
STABILIZATION OF CHOLESTEROL/SM
enriched environment
Unaltered PL Surface Unaltered LDL size
NO EFFECT ON PC-enriched/colesterol-poor
environment
Altered PL Surface Decreased LDL size
LDL aggregation
Lyso-PC & NEFA
Figure 8 Figure 8

Supplemental Tables
Table S1. HPLC chromatography peptide characteristics
Peptide Retention time (min) Purity (%) Calculated mass (Da) Found Mass (Da)
LP3 4,9 99 1542 1543 [M+H]+
DP3 3,1 98 1481 1482 [M+H]+
IP321 3,3 98 1126 1127 [M+H]+
HPLC spectra were recorded in Waters Alliance 2695 separation module equipped with a 2998
photodiode array detector, a Sunfire C18 column (100 mm x 4.6 mm, 3.5 µm, 100 Å, Waters)
and Empower software (Waters); flow rate = 1 mL/min; gradient 0-100% B in 8 min (A = 0.045%
trifuloroacetic acid in water, and B = 0.036% trifluoroacetic acid in acetonitrile), detection at 220
nm. HPLC-MS were recorded in a Waters Alliance 2695 equipped with a 2998 photodiode array
detector, ESI-MS micromass ZQ, a Sunfire C18 column (2.1 mm x 100 mm, 3.5 µm, 100 Å,
Waters) and Masslynx software (Waters); flow rate = 0.3 mL/min; gradient 0-100% B in 8 min (A
= 0.1% formic acid in water, and B = 0.07% formic acid in acetonitrile).
Table S2. Concentration and ratio of peptides used in SMase-induced LDL
turbidimetry experiments
Concentration (M) Peptide/ApoB-100 Molar Ratio
10 4.7/1
8.75 4.1/1
7.7 3.5/1
5 2.4/1
2.5 1.2/1
Table S3. Concentration and ratio of peptides used in PLA2-induced LDL
turbidimetry experiments
Concentration (uM) Peptide/ApoB-100 Molar Ratio
20.83 9.8/1
14.60 6.8/1
10 4.7/1
6.25 2.9/1
4.16 2.0/1
Supplementary Material (for online publication)Click here to download Supplementary Material (for online publication): BBA Supplemental Tables and Figures R1.docx

Figure S1
Figure S1. HPLC trace of LP3 (A) and DP3 (B). Each compound was analyzed using a C18 analytical HPLC column (Sunfire, Waters).
Flow rate 1 mL/min; Gradient 0-100% a-b in 8 min (a= 0.045% trifluoroacetic acid in water and b 0.036% trifluoroacetic acid in acetonitrile).
Detection wavelength 220 nm. The peptide shows purity above 98%. The observed mass (HPLC-MS, not shown) conforms to the mass of
the target compound.
A B
Figure S2
Figure S2. Percentage of peptide LP3 (A) and of DP3 (B) versus incubation time in human serum. Experiment was performed in triplicate. Data are shown
as the mean ± SD
1) Generation of Normolipemic Pool of humanplasma
2) LDL isolation
3) LDL modification by SMase and PLA2 with and w/o peptides
4) hVSMCc are exposed for 2h with modified LDL
Turbidimetry(Absorbance 405 nm)
37ºC18 hours
Lipid extraction and TLC to quantify
intracelular CE/FC ratio
Confocal MicroscopyBODIPY
Figure S3
Figure S3. Schematic representation of the experimental procedure

Figure S4
Figure S4. LRP1-derived peptides does not increase LDL turbidimetry. LDL at 1.44 mg/ml apoB was exposed to peptides (10 M,
Ratio Peptide/ApoB= 4.7/1) for 24 hours. Aggregation of LDL was monitored by measuring absorbance at 405 nm. Data are mean ±
SD of 4 experiments performed in duplicate. 5
Ab
so
rba
nc
e(4
05
nm
)
0
0,1
0,2
0,3
0,4
0,5
w/opeptide
LP3 DP3 IP321
1a001:1_UV
0.0
10.0
20.0
30.0
40.0
50.0
60.0
70.0
80.0
mAU
0.0 2.0 4.0 6.0 8.0 10.0 ml
LDL + buffer
2a001:1_UV
0.0
10.0
20.0
30.0
40.0
50.0
60.0
70.0
mAU
0.0 2.0 4.0 6.0 8.0 10.0 ml
LDL + DP3
Figure S5
Figure S5. Gel filtration chromatography (GFC) of LDL (0.5 mL at 0.4 mg/mL ApoB) unexposed or exposed to DP3 (10 M, Ratio
Peptide/ApoB= 4.7/1) for 24 hours. LDLs were chromatographed at a flow rate of 0.5 ml/min using a Superose 6 column in a AKTA-FPLC
system (GE Healthcare) as described in Methods.

Figure S6
Figure S6. Effect of LP3, DP3, IP321 and Manumycin A on SMase activity. SMase activity was measured by the Amplex Red assay
based on the SMase-induced formation of phosphorylcholine. The results are shown as mean ± SD of three independent experiments
performed in triplicate. *P<0.005 control (w/o peptide)
5 3 0 -5 9 0 n m
S M a s e (m U /m L )
SM
as
e a
cti
vit
y (
a.u
.)
0 .0 0 .2 0 .4 0 .6 0 .8 1 .0 1 .2
0
2 0 0 0 0
4 0 0 0 0
6 0 0 0 0
8 0 0 0 0w /o p e p tid e
L P 3
D P 3
IP 3 2 1
M a n u m y c in
* * * * *
Figure S7
LDL
-
+
LP3 DP3 IP321
Figure S7. Agarose gel electrophoresis of LDL (2 g/well) exposed to LP3, DP3 or IP321 (10 M, Ratio Peptide/ApoB= 4.7/1) for 18 hours.
LDLs were electrophoresed in 0.5% agarose gels that were dehydrated and stained with Coomassie Blue.

AUTHOR DECLARATION TEMPLATE
We wish to confirm that there are no known conflicts of interest associated with this publication
entitled “Molecular basis for the protective effects of low-density lipoprotein receptor-related
protein 1 (LRP1)-derived peptides against LDL aggregation” and there has been no significant
financial support for this work that could have influenced its outcome.
We state that Roger Prades, Chiara Pallara and Teresa Tarragó are employees of Iproteos SL.
Teresa Tarragó is cofounder of Iproteos SL and member of its board of directors.
We confirm that the manuscript has been read and approved by all named authors and that there
are no other persons who satisfied the criteria for authorship but are not listed.
We further confirm that the order of authors listed in the manuscript has been approved by all of
us.
We confirm that we have given due consideration to the protection of intellectual property
associated with this work and that there are no impediments to publication, including the timing of
publication, with respect to intellectual property. In so doing we confirm that we have followed the
regulations of our institutions concerning intellectual property.
We further confirm that any aspect of the work covered in this manuscript that has involved human
patients has been conducted with the ethical approval of all relevant bodies.
We understand that the Corresponding Author is the sole contact for the Editorial process
(including Editorial Manager and direct communications with the office). He/she is responsible for
communicating with the other authors about progress, submissions of revisions and final approval
of proofs.
We confirm that we have provided a current, correct email address which is accessible by the
Corresponding Author and which has been configured to accept email from VICENTA LLORENTE
CORTES ([email protected])
Signed by the corresponding author in the name of the rest of authors
Vicenta Llorente Cortes
*Conflict of Interest





















