
Barium and strontium-enriched (Ba 0.5Sr 0.5) 1+ x Co 0.8Fe 0.2O 3− δ oxides as high-performance...
12
Barium- and strontium-enriched (Ba 0.5 Sr 0.5 ) 1+x Co 0.8 Fe 0.2 O 3d oxides as high-performance cathodes for intermediate-temperature solid-oxide fuel cells Wei Zhou, Ran Ran, Zongping Shao * , Wei Zhuang, Jing Jia, Hongxia Gu, Wanqin Jin, Nanping Xu State Key Laboratory of Materials-oriented Chemical Engineering, Nanjing University of Technology, No. 5 Xin Mofan Road, Nanjing, JiangSu 210009, China Received 23 August 2007; received in revised form 1 February 2008; accepted 5 February 2008 Available online 14 March 2008 Abstract (Ba 0.5 Sr 0.5 ) 1+x Co 0.8 Fe 0.2 O 3d , or BSCF(1 + x), (0 6 x 6 0.3) oxides were synthesized and investigated as cathodes for intermediate- temperature solid-oxide fuel cells. The A-site cation excess in BSCF(1 + x) resulted in a lattice expansion and the creation of more active sites for oxygen reduction reaction due to the lowered valence states of the B-site ions and the increased oxygen vacancy concentration, which improved the oxygen adsorption process. On the other hand, the A-site excess could also result in higher resistances for oxygen adsorption (due to the formation of BaO and/or SrO impurities), and oxygen-ion transfer (by facilitating the solid-phase reaction between the cathode and the electrolyte). By taking all these factors into account, we found BSCF1.03 to be the optimal composition, which lead to a peak power density of 1026.2 ± 12.7 mW cm 2 at 650 °C for a single cell. Ó 2008 Acta Materialia Inc. Published by Elsevier Ltd. All rights reserved. Keywords: Perovskites; Fuel cell materials; Electrochemistry; Vacancies; Electrochemical impedance 1. Introduction Fuel cells are promising next-generation energy conver- sion systems reputed for their high efficiency, silent opera- tion and extremely low emissions. Among many types of fuel cells, solid-oxide fuel cell (SOFC) is of particular inter- est due to its capacity for both power/heat co-generation and fuel flexibility. Due to the significant problems associ- ated with high operating temperatures, such as fast degra- dation of performance and stringent requirement of the fuel cell materials, current efforts are devoted to lowering the operating temperature to 800 °C or less for the com- mercialization of SOFCs [1–5]. As the operating tempera- ture decreases, some other problems emerge. Among them, one problem is the significant drop of electro-cata- lytic activity for oxygen reduction over the cathode. For example, the polarization resistance of Sr-doped LaM- nO 3 (LSM), a state-of-the-art cathode material for high- temperature SOFCs, increases dramatically at T < 800 °C [6–10]. Since LSM has a negligible oxygen ionic conductiv- ity, the oxygen reduction process is limited to the electro- lyte–electrode–oxygen triple-phase boundary (TPB). In comparison, mixed ionic and electronic conducting oxides effectively extend the oxygen reduction sites from the TPB to the entire cathode–oxygen dual-phase boundary, which typically results in a significant improvement in cathode performance at intermediate temperatures (IT). Nowadays, they have become the cornerstones of high-per- formance cathodes for IT-SOFCs [11–16]. Very recently, Shao et al. [17,18] reported the applica- tion of a novel mixed-conducting perovskite-type compos- ite oxide, Ba 0.5 Sr 0.5 Co 0.8 Fe 0.2 O 3d (BSCF), as the cathode for IT-SOFCs. An extremely low polarization resistance 1359-6454/$34.00 Ó 2008 Acta Materialia Inc. Published by Elsevier Ltd. All rights reserved. doi:10.1016/j.actamat.2008.02.002 * Corresponding author. Tel.: +86 25 83587722; fax: +86 25 83365813. E-mail address: [email protected] (Z. Shao). www.elsevier.com/locate/actamat Available online at www.sciencedirect.com Acta Materialia 56 (2008) 2687–2698
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Barium and strontium-enriched (Ba 0.5Sr 0.5) 1+ x Co 0.8Fe 0.2O 3− δ oxides as high-performance...

Available online at www.sciencedirect.com
www.elsevier.com/locate/actamat
Acta Materialia 56 (2008) 2687–2698
Barium- and strontium-enriched (Ba0.5Sr0.5)1+xCo0.8Fe0.2O3�d oxidesas high-performance cathodes for intermediate-temperature
solid-oxide fuel cells
Wei Zhou, Ran Ran, Zongping Shao *, Wei Zhuang, Jing Jia, Hongxia Gu,Wanqin Jin, Nanping Xu
State Key Laboratory of Materials-oriented Chemical Engineering, Nanjing University of Technology, No. 5 Xin Mofan Road, Nanjing,
JiangSu 210009, China
Received 23 August 2007; received in revised form 1 February 2008; accepted 5 February 2008Available online 14 March 2008
Abstract
(Ba0.5Sr0.5)1+xCo0.8Fe0.2O3�d, or BSCF(1 + x), (0 6 x 6 0.3) oxides were synthesized and investigated as cathodes for intermediate-temperature solid-oxide fuel cells. The A-site cation excess in BSCF(1 + x) resulted in a lattice expansion and the creation of more activesites for oxygen reduction reaction due to the lowered valence states of the B-site ions and the increased oxygen vacancy concentration,which improved the oxygen adsorption process. On the other hand, the A-site excess could also result in higher resistances for oxygenadsorption (due to the formation of BaO and/or SrO impurities), and oxygen-ion transfer (by facilitating the solid-phase reactionbetween the cathode and the electrolyte). By taking all these factors into account, we found BSCF1.03 to be the optimal composition,which lead to a peak power density of 1026.2 ± 12.7 mW cm�2 at 650 �C for a single cell.� 2008 Acta Materialia Inc. Published by Elsevier Ltd. All rights reserved.
Keywords: Perovskites; Fuel cell materials; Electrochemistry; Vacancies; Electrochemical impedance
1. Introduction
Fuel cells are promising next-generation energy conver-sion systems reputed for their high efficiency, silent opera-tion and extremely low emissions. Among many types offuel cells, solid-oxide fuel cell (SOFC) is of particular inter-est due to its capacity for both power/heat co-generationand fuel flexibility. Due to the significant problems associ-ated with high operating temperatures, such as fast degra-dation of performance and stringent requirement of thefuel cell materials, current efforts are devoted to loweringthe operating temperature to 800 �C or less for the com-mercialization of SOFCs [1–5]. As the operating tempera-ture decreases, some other problems emerge. Amongthem, one problem is the significant drop of electro-cata-
1359-6454/$34.00 � 2008 Acta Materialia Inc. Published by Elsevier Ltd. All
doi:10.1016/j.actamat.2008.02.002
* Corresponding author. Tel.: +86 25 83587722; fax: +86 25 83365813.E-mail address: [email protected] (Z. Shao).
lytic activity for oxygen reduction over the cathode. Forexample, the polarization resistance of Sr-doped LaM-nO3(LSM), a state-of-the-art cathode material for high-temperature SOFCs, increases dramatically at T < 800 �C[6–10]. Since LSM has a negligible oxygen ionic conductiv-ity, the oxygen reduction process is limited to the electro-lyte–electrode–oxygen triple-phase boundary (TPB). Incomparison, mixed ionic and electronic conducting oxideseffectively extend the oxygen reduction sites from theTPB to the entire cathode–oxygen dual-phase boundary,which typically results in a significant improvement incathode performance at intermediate temperatures (IT).Nowadays, they have become the cornerstones of high-per-formance cathodes for IT-SOFCs [11–16].
Very recently, Shao et al. [17,18] reported the applica-tion of a novel mixed-conducting perovskite-type compos-ite oxide, Ba0.5Sr0.5Co0.8Fe0.2O3�d(BSCF), as the cathodefor IT-SOFCs. An extremely low polarization resistance
rights reserved.

2688 W. Zhou et al. / Acta Materialia 56 (2008) 2687–2698
was observed at �600 �C [17]. Baumann et al. demon-strated that the surface exchange rate of BSCF reachedas high as 5 � 10�5 cm s�1 at 750 �C in air [19], whichwas about 100 times faster than La0.6Sr0.4Co0.8Fe0.2O3�d
under the identical conditions [20]. On the other hand, itwas found that many factors might result in a change ofthe bulk diffusion and surface exchange properties, andconsequently the cathode performance of BSCF. Forexample, we observed that both the synthetic route ofBSCF powders and the calcination temperature could havea significant impact on the cathode polarization resistanceof the BSCF-based electrodes [21,22]. Recently Wang et al.[23] also demonstrated that the addition of a small amountof silver to BSCF + GDC (Gd2O3-doped CeO2) improvedthe efficiency of current collector and the cathodeperformance.
It is well known that an idealized perovskite structure(ABO3) contains an equivalent number of A-site and B-sitecations. In reality, some perovskite oxides can exist as atype of unequal molar amounts of A- and B-site cations(i.e. A/B cation mole ratio 6¼ 1), which can be caused bymany factors such as experimental deviations [24–27]. Ithas been observed that this A/B nonstoichiometry mayaffect many properties of the perovskite material, includingconductivity, magnetic, phase transition and microstruc-ture. For example, the electronic conductivity was foundto decrease when A-site ions deficiency was introduced intoLa0.6Sr0.4Co0.2Fe0.8O3�d (LSCF) [28,29]. In these A-site-deficient perovskites, the charge compensation mechanismwas probably explained by the formation of oxygen vacan-cies rather than the oxidation of B3+ to B4+. It wasexpected that these additional oxygen vacancies improvedthe oxide ionic conductivity and catalytic property of thecathode for oxygen reduction [28,29]. The primary resultsof Doshi et al. [30] indeed demonstrated that the A-site-deficient LSCF had an area-specific resistance (ASR) aslow as 0.1 X cm2 at 500 �C. It has also been well-estab-lished that variations in the cationic stoichiometry (i.e.the A/B cation ratio) can also affect the sintering behaviorof the oxide [31]. Varied sintering characteristics can inturn lead to over- or under-sintering of the cathode (at agiven temperature). Simner et al. [32] found that a slightA-site deficiency of La0.8Sr0.2FeO3�d improved its sinteringbehavior and subsequently made it possible to fabricate thecathode layer at lower temperatures to prevent particlecoarsening and iron diffusion into the (Sm2O3-dopedCeO2) SDC layer.
Unlike the A-site-deficient perovskites, the properties ofA-site enriched ones have been addressed by virtually noliterature. In this work, A-site barium- and strontium-enriched BSCF oxides with the composition of(Ba0.5Sr0.5)1+xCo0.8Fe0.2O3�d, or BSCF(1 + x) (0 6 x 6
0.3) were synthesized and systematically characterized,with the major focus being on the effect of A-site cationsexcess on the cathode performance for IT-SOFCs basedon doped-ceria electrolytes. The aim of the present workis to further optimize the cathode performance of BSCF
oxides by clarifying the relationship between the cathodeperformance and the nonstoichiometry of A/B.
2. Experimental procedures
All BSCF(1 + x) (x = 0.0–0.3) oxides were synthesizedby our standard combined EDTA-citrate complexing sol–gel process [33]. The metal nitrates, the starting materialsfor BSCF(1 + x), were first prepared as respective aqueoussolution with its precise concentration determined by thestandard EDTA titration method. A solution was preparedby mixing required amounts of metal nitrates, followed bythe addition of EDTA and citric acid at a pH value of �6with the help of NH4OH. The molar ratio of total metalions, EDTA and citric acid was set at 1:1:2. The final prod-ucts were obtained after several steps: (1) evaporate waterat 90 �C to get a transparent gel; (2) pre-fire the gel at250 �C; (3) further calcine the gel at 1000 �C for 5 h inair. In order to evaluate the effect of impurities on the cath-ode performance of BSCF(1 + x), the BSCF(1 + x) pow-der (0.005 mol) was boiled in deionized water (200 ml)and filtrated several times. Since the solubility of Ba(OH)2
and Sr(OH)2 in water reaches 0.067 and 0.20 mol l�1 at293 K, respectively, the concentrations of Ba2+ and/orSr2+ in the water during the washing process were farbelow saturation level. The pH value of the filtrated solu-tion was detected by a pH meter with an accuracy of±0.001.
A symmetrical cell with the configuration of: elec-trodejSDCjelectrode was used for the a.c. impedance stud-ies. Dense SDC pellets 12 mm in diameter and 0.8 mm inthickness were prepared by dry-pressing and sintering at1350 �C for 5 h. The cathode powders were first dispersedinto a pre-mixed solution of glycerol, ethylene glycol andisopropyl alcohol to form a colloidal suspension by high-energy ball-mill (Fritsch, Pulverisettle 6) at the rotationalspeed of 400 rpm for 0.5 h. The slurry thus obtained waspainted symmetrically on both surfaces of the SDC pellet,followed by calcining at 1000 �C for 2 h in air. Anode pow-ders consisting of 60 wt.% NiO and 40 wt.% SDC were pre-pared by mixing NiO and SDC in an agate mortar. A dry-press method was used to fabricate anode-supported SDCbi-layer pellets [34]. The bi-layer pellet was then fired at1450 �C for 5 h in air for the densification of the electrolytelayer. The thicknesses of the electrolyte and anode are �30and �350 lm, respectively. The porosity of the anode isabout 30% after the reduction of NiO by hydrogen. Thecathode slurry was painted onto the central surface of theelectrolyte and then fired at 1000 �C for 2 h in air, whichresulted in a coin-shaped cathode with a thickness of 10–20 lm, an effective geometric area of 0.42 cm2, and a poros-ity of �35%.
The I–V polarization curves were collected using aKeithley 2420 source meter based on the four-terminal con-figuration. Hydrogen humidified by 3% H2O was fed intothe anode chamber as fuel at a flow rate of 80 ml min�1
(STP) and air was fed into the cathode chamber as oxidant

W. Zhou et al. / Acta Materialia 56 (2008) 2687–2698 2689
gas at a flow rate of 200 ml min�1 (STP). The gas flow rateswere controlled by mass flow controllers. Silver paste wasadopted as the current collector. The electrode perfor-mance was investigated with a symmetrical cell or completecell configuration by the a.c. impedance method using anelectrochemical workstation based on a Solartron 1287potentiostat and a 1260 A frequency response analyzer.The frequency range applied was from 0.01 Hz to100 kHz with signal amplitude of 10 mV under the open-circuit condition. The overall impedance data were fittedby a complex non-linear least square (CNLS) fitting pro-gram in the Z-View 2.9b software.
In order to investigate the chemical compatibilitybetween the cathode and the electrolyte, the BSCF(1 + x)(x = 0.00–0.20) (70 wt.%) and SDC (30 wt.%) powderswere mixed and calcined at 1000 �C for 2 h in air. The reac-tion products were analyzed by X-ray diffraction (XRD,Bruker D8 Advance) at room temperature. Fourier trans-form infrared spectroscopy (FT-IR, AVATAR-360) ofthe as-prepared powders was recorded from 4000 to400 cm�1 by the KBr pellet method. Oxygen nonstoichiom-etry (d) and a mean valence of B-site cations (n) in the sam-ples were evaluated by iodometry as described in Ref. [35].Briefly, the samples with the weight of 0.1000 g were dis-solved in HCl (6.0 mol l�1) under an inert gas atmospherein order to prevent the oxidation of I� ions by air, and thenthe solution was titrated by standard thiosulfate solution.The microscopic features of the prepared electrodes werecharacterized by an environmental scanning electronmicroscopy (ESEM, QUANTA-2000). Meanwhile, thecomposition of the cathode material was also analyzed byenergy-dispersive X-ray analysis (EDXA). The specific sur-face area of the samples was characterized by N2 adsorp-tion using a BELSORP II instrument at the temperatureof liquid nitrogen. Electrical conductivity was measuredby the four-terminal DC technique in air, using Ag pasteas electrodes. The current and the voltage were detectedby Keithley 2420 source meter at intervals of 5 �C withina temperature range of 300–900 �C. Oxygen desorptionproperties of the powders were studied by oxygen temper-ature-programmed desorption technique (O2-TPD). About50 mg of BSCF(1 + x) powder was loaded in a quartz tube.The assembly was placed in a single-zone furnace equippedwith a temperature controller. Argon was used as the car-rier gas with a flow rate of 20 ml min�1 (STP). The temper-ature was increased from 50 to 950 �C at the rate of
Table 1Compositions of the (Ba0.5Sr0.5)1+xCo0.8Fe0.2O3�d (0 6 x 6 0.2) powders acco
Composition Abbreviation (n) Ratio A/Bmeasured (d
Ba0.5Sr0.5Co0.8Fe0.2O3�d BSCF1.00 1.01(Ba0.5Sr0.5)1.03Co0.8Fe0.2O3�d BSCF1.03 1.04(Ba0.5Sr0.5)1.09Co0.8Fe0.2O3�d BSCF1.09 1.10(Ba0.5Sr0.5)1.15Co0.8Fe0.2O3�d BSCF1.15 1.16(Ba0.5Sr0.5)1.20Co0.8Fe0.2O3�d BSCF1.20 1.21
10 �C min�1 and the effluent gases were monitored by themass spectrometer (MS, Hiden, QIC-20).
3. Results and discussion
3.1. Elemental composition and crystal structure
It is well known that small variations in the compositionof perovskite oxides can lead to a significant change in theirphysical and chemical properties. In order to ensure theaimed cation stoichiometry for the synthesized materials,all starting materials were first prepared in their respectiveaqueous solutions with their accurate concentrations deter-mined by the EDTA titration method [33]. The exact A/Bcation ratios of the as-prepared materials were analyzed bythe EDX technique. As shown in Table 1, they match theaimed stoichiometry well within the experimental error.Fig. 1 shows the X-ray diffraction patterns of BSCF(1 + x)powders calcined at 1000 �C for 5 h in air. All the oxidesexhibit the cubic perovskite structure with a space groupof Pm3m. No secondary phase was detected by XRD untilthe A-site excess was over 20 mol.% (x = 0.2). The calcu-lated lattice constants are also listed in Table 1. BSCF1.00(for consensus from previous publications, it is also abbre-viated as BSCF) has a lattice constant of 3.9891(2) A,which agrees with the reported value in Ref. [36]. The lat-tice constant for BSCF(1 + x) first increases with x andthen reaches a plateau at around 4.1384(3) A at x > 0.09.It indicates that the upper-limit of the A-site excess isaround 9 mol.% for sustaining the pure-phase perovskitestructure of BSCF(1 + x).
It was reported that La(OH)3, which originated fromLa2O3, could be found in La excessive (La,Sr)MnO3 and(La,Sr)CrO3 [37,38]. Such hydroxide impurities might havea detrimental effect on the oxygen surface exchange processfor the oxygen reduction reaction (ORR) on cathode. Jiangand Love [39] reported that the surface impurities of LSMcathode could be removed by diluted acid, which resultedin an improvement of the cathode performance. Fig. 2shows the FT–IR spectra of BSCF1.03, BSCF1.09 andBSCF1.20 powders. The band at around 3548 cm�1 canbe attributed to the H–O–H bending mode of coordinatedwater in Ba(OH)2 � nH2O and/or Sr(OH)2 � nH2O, whilethe smaller but sharper band at around 3500 cm�1 isassigned to the stretching vibration for the lattice hydroxylgroup. The larger sharp band at around 3400 cm�1 is the
rding to EDX and XRD results
)System (m) Lattice constant (A)
Before washing After washing
Cubic 3.9891(2) 3.9897(1)Cubic 4.1112(7) 4.1107(3)Cubic 4.1384(3) 4.1323(7)Cubic 4.1401(9) 4.1348(2)Cubic 4.1410(2) 4.1371(4)

Fig. 1. XRD patterns of (Ba0.5Sr0.5)1+xCo0.8Fe0.2O3�d before washing treatment (a), and after washing treatment (b).
Fig. 2. FTIR spectra of BSCF1.03, BSCF1.09 and BSCF1.20 beforewashing treatment (a), and after washing treatment (b).
Fig. 3. pH Values of filtrated solution from (Ba0.5Sr0.5)1+xCo0.8Fe0.2O3�d
(0 6 x 6 0.2) after the first washing treatment and the third treatment.
2690 W. Zhou et al. / Acta Materialia 56 (2008) 2687–2698
characteristic peak for the vibration of surface-adsorbedhydroxyl group [40,41]. Only a very weak stretching vibra-tion for the lattice and surface hydroxyl groups is detectedin BSCF1.03 sample, which suggests that a negligibleamount of Ba(OH)2 � nH2O and/or Sr(OH)2 � nH2O isformed. The appearance of the stretching vibration forcoordinated water, the increased stretching vibration forthe lattice and surface hydroxyl groups both demonstratethat a small amount of Ba(OH)2 � nH2O and/or Sr(OH)2 �nH2O is formed in BSCF1.09. The fact that all the threevibration peaks strengthened significantly in the case ofBSCF1.20 suggests that the amount of Ba(OH)2 � nH2Oand/or Sr(OH)2 � nH2O impurities increased obviously.
As we mentioned above, BSCF(1 + x) powders wereboiled in deionized water and filtrated for several timesfor the comparison with the unwashed one to evaluatethe effect of impurities on the cathode performance. Afterthis treatment, the pH value of the filtrated solution fromeach experiment was recorded and shown in Fig. 3. ThepH value of the solution is very similar for BSCF1.00(7.33(1)), BSCF1.03 (7.36(0)), BSCF1.06 (7.38(2)) andBSCF1.09 (7.39(9)), while a sharp increase can be observedfor the case of BSCF1.15 (9.12(2)). These phenomenastrongly support the formation of hydrates in BSCF(1 + x)
at x > 0.09. The samples after the washing treatment werethen calcined at 500 �C for 5 h to get rid of the waterresidual. The X-ray diffraction patterns and FT–IR spectraof the corresponding samples are shown in Fig. 1b andFig. 2b, respectively. According to the XRD results, thewashing treatment has a negligible impact on the crystalstructure of the materials. BSCF1.09 and BSCF1.20have similar characteristic vibration peaks to that ofBSCF1.03 in the range of 3300–3600 cm�1 after the wash.It suggests that the impurities can be removed by water.The EDX examination demonstrates the compositionof Ba0.542Sr0.542Co0.792Fe0.208O3�d for BSCF1.20 afterthe washing treatment (x = 0.084). It further confirmsthat the maximum A-site excess amount, x, for(Ba0.5Sr0.5)1+xCo0.8Fe0.2O3�d is around 0.09.
For the ABO3-structured perovskite oxide with fixed A-site cations, its cell volume is mainly determined by the BO6
octahedron. Generally speaking, the lower the valence stateof the B cation, the longer the distance of the B–O bond,and thus the larger lattice volume is. Therefore, theincreased cell volume with the A-site excess of BSCF(1 + x)may be partly related to the decrease of the valence statesof the B-site cations. The B-site mean valences (n) andthe oxygen contents (3 � d) of the samples were then

W. Zhou et al. / Acta Materialia 56 (2008) 2687–2698 2691
determined by the iodometry method with the results sum-marized in Table 2. The n value as well as the oxygen non-stoichiometry (d) indeed first decreases with the A-siteexcess up to x = 0.09, and then remains constant for higherx values. It is interesting that BSCF can incorporate excessBa into its lattice and the A-site excess (or called as B-sitedeficiency) does not promote the charge compensation byelevating the valence state of the B-site cations. These phe-nomenons can be explained by the defect chemical incorpo-ration reactions as follows:
BaO! BaA þ V0000
B þOþO2V��O ð1Þ
Table 2Oxygen content (3 � d) and B-site mean valence (n) at room temperature
Sample 3 � da n
BSCF1.00 2.65 3.30BSCF1.03 2.57 3.23BSCF1.09 2.44 3.14BSCF1.15b 2.43 3.12BSCF1.20b 2.45 3.16
a The 3 � d is calculated by regarding the molecular formula as theB-site-deficient form. For example, the molecular formula of BSCF1.03 isregarded as Ba0.5Sr0.5(Co0.8Fe0.2)0.971O3�d and the 3�d = (0.50 � 2 +0.50 � 2 + 3.23 � 0.97)/2 = 2.57.
b The composition for oxygen content calculation is Ba0.5Sr0.5(Co0.8-Fe0.2)0.917O3�d + 0.028BaO + 0.028SrO for BSCF1.15 and Ba0.5Sr0.5-(Co0.8Fe0.2)0.917O3�d + 0.050BaO + 0.050SrO for BSCF1.20.
Fig. 4. Electrical conductivities of: fresh (Ba0.5Sr0.5)1+xCo0.8Fe0.2O3�d (0 6Co0.8Fe0.2O3�d (0 6 x 6 0.2) after washing treatment (c).
The increase in lattice constant in such a case would result,in part, from the fact that Ba has an ionic radius that islarger than the average of Ba and Sr. On the other hand,since the initial oxidation state on the B-site is not actually4+, one vacancy on the B-site would not exactly providecharge balance for the two oxygen vacancies. Therefore,a complete charge balance would require that some ofthe B-site cations lower their valences, as observedexperimentally.
3.2. Electrical conductivity
Fig. 4a shows the temperature dependence of the electri-cal conductivity of BSCF(1 + x) (0 6 x 6 0.2) before thewashing treatment. It shows that the electrical conductivityof all samples first increases linearly with temperature to atransition temperature (Ts) of about 450–550 �C, and thenit almost remains constant afterwards. The creation ofadditional oxygen vacancies, accompanied by a reductionof Fe4+ to Fe3+ or Co4+ to Co3+ at higher temperatures,causes a decrease in the charge carrier concentration anda covalent interaction because of a perturbation of theO–(Fe,Co)–O periodic potential, which may account forsuch a transition [42–44].
At the temperatures below Ts, the electron-conductingmechanism can be attributed to the hopping of the p-typesmall polarons, which are associated with the behavior of
x 6 0.2) (a); the corresponding Arrhenius plots (b); (Ba0.5Sr0.5)1+x-

2692 W. Zhou et al. / Acta Materialia 56 (2008) 2687–2698
triple- and tetra-valent states of cobalt and iron cations.The charge transfer is thermally activated and its tempera-ture dependence is given by the equation:
r ¼ ðA=T Þ expð�Ea=kT Þ ð2Þwhere r is the electrical conductivity, A is a material con-stant containing the carrier concentration term, T is theabsolute temperature, Ea is the activation energy for smallpolaron hopping, and k is the Boltzmann constant. Plots oflogrT versus 1/T are shown in Fig. 4b, from which theapparent activation energy (Ea) can be calculated fromthe slopes of the linear correlations. The values of Ea arevery close for all the samples (about 30 kJ mol�1), whichindicates that the conduction mechanism is the same attemperatures lower than Ts. However, the electronic con-ductivity decreases steadily with the amount of A-site ex-cess at a given temperature. The maximum conductivityis 43.0(4) S cm�1 for BSCF1.00, while it is only 32.9(1)and 25.0(2) S cm�1 for BSCF1.03 and BSCF1.09, respec-tively, which indicates that the oxygen vacancy concentra-tion increases with the A-site excess. The valence state ofCo and Fe also has a significant effect on the electronicconductivity. Based on the iodometry results, at room tem-perature, the mean valence state of the B-site cations de-creases from 3.3(0) to 3.1(2) with the A-site excessconcentration. The decrease of Co4+ and Fe4+ concentra-tions leads to the decrease of Co�co (hole) and Fe�Fe (hole)concentrations and consequently that of the hole conduc-tivity. For example, Zhou et al. found that La0.6Sr0.4FeO3
showed the lowest electronic conductivity when all theFecations were in 3+ valence state [45]. The Ts ofBSCF(1 + x) decreases with the A-site excess because thetemperature at which oxygen loss began to occur drops.This fact reveals that the Co–O and Fe–O bond energy isweakened due to the expansion of the lattice structure.
In the case of BSCF1.15 and BSCF1.20, since the oxy-gen vacancy concentration and mean valence states of themetal ions are very similar to those of BSCF1.09, the low-ered electronic conductivities can be related to the forma-tion of Ba(OH)2 � nH2O and Sr(OH)2 � H2O impuritiesover the material surface. To verify this hypothesis, thefour-terminal DC test was performed on all the samplesafter the washing treatment, as shown in Fig. 4c. It indi-cates that the washing treatment does not result in an obvi-ous change in the conductivity of BSCF1.00, BSCF1.03and BSCF1.09, while the conductivities of BSCF1.15 andBSCF1.20 are improved significantly. Their temperature–conductivity curves overlap with that of BSCF1.09 between300 and 900 �C. Therefore, the impurities in BSCF1.15 andBSCF1.20 probably block the electronic conductance tosome extent; fortunately, such impurities can be removedby a simple washing treatment.
3.3. Microstructure
Fig. 5 shows the surface morphology of theBSCF(1 + x) cathodes with various A-site excess concen-
tration sintered at 1000 �C for 2 h. The A-site excess con-centration has an obvious influence on the particle size,the connection between the particles, and the porosity ofthe cathode. The particle size decreases and the porosityincreases with the A-site excess concentration. However,worse adhesion between particles was observed forBSCF1.09 and BSCF1.20 because of the under-sinteringbehavior of them. It is well known that the sintering behav-ior can be controlled by adjusting the concentration of theA-site cations. Stevenson et al. found that a slight defi-ciency of the A-site cations improved the densification ofdoped lanthanum and yttrium manganite substantially[30]. Such a behavior was attributed to an increased con-centration of the A-site vacancies which enhanced the dif-fusion of the A-site cations during the sintering. On thecontrary, the excess of the A-site cations may decreasethe diffusion rate of A-site cations, and consequentlydecrease the sintering activity of the oxide. It was foundthat for BaTiO3, a 2 mol.% excess of Ba (A-site excess)resulted in a fine-grained (1 lm) microstructure, while theformation of a liquid phase and a coarse-grained micro-structure was observed for a 1 mol.% excess in Ti (A-sitedeficiency) [46]. Therefore, the microstructure of the cath-ode can be optimized by tailoring the concentration ofthe A-site cations. The specific surface area of the BSCFpowder calcined at 1000 �C is only 0.25(3) m2 g�1 becausethe sintering behavior of BSCF1.00 is extremely good.The introduction of the A-site deficiency in barium stron-tium cobalt ferrites results in an over-sintering of the oxide[47], and therefore the A-site excess ones may be an advan-tage. The corresponding specific surface area of BSCF1.03is 0.28(1) m2 g�1, which indicates that the enhancement ofthe surface area is very little. Therefore, the microstructurehas a negligible contribution to the differences betweenthese two cathodes. On the contrary, BSCF1.09 andBSCF1.20 are under-sintered. It is very likely that theimpurity phases, such as BaO and SrO, pin the grainboundaries of BSCF1.09 and BSCF1.20. Different fromBSCF1.09, there are some bright particles in BSCF1.20,which are proved by the EDX as the Ba- and Sr-richphase(s).
3.4. A.c. impedance studies
The cathode performance for BSCF(1 + x), x = 0,0.03, 0.09 and 0.20 calcined at 1000 �C was then investi-gated by the ac impedance spectroscopy based on asymmetrical cell configuration in air. The Nyquist andBode plots for the cell measured at 800 �C under theopen-circuit condition are given in Fig. 6. As shown inthe Bode plots, there are two obvious peaks for each ofthe BSCF1.00, BSCF1.09 and BSCF1.20-based cathodes.It suggests that the oxygen reduction reaction is limitedby at least two different electrode processes during themolecular oxygen reduction. However, the peak at lowfrequencies is not visible for the BSCF1.03 cathode at800 �C.

Fig. 5. SEM micrographs of the cross section and the surface of BSCF1.03 (a, b), BSCF1.09 (c, d) and BSCF1.20 (e, f) cathodes, fired at 1000 �C for 2 h inair before washing treatment, the arrow-indexed domains are the Ba and Sr enriched phases.
Fig. 6. Nyquist- (a) and bode-type (b) impedance curves for BSCF, BSCF1.00, BSCF1.03, BSCF1.09 and BSCF1.20 cathodes before washing treatmentmeasured at 800 �C and open circuit.
W. Zhou et al. / Acta Materialia 56 (2008) 2687–2698 2693

2694 W. Zhou et al. / Acta Materialia 56 (2008) 2687–2698
To separate these two process, we fitted the impedancespectra data to an equivalent circuit with a configurationof LRohm(RE1 � CPE1)(RE2 � CPE2), where Rohm is theoverall ohmic resistance including the electrolyte resistance,electrode ohmic resistance and lead resistance. ParameterL is the inductance, which could be due to the Ag cur-rent/voltage probes or the high-frequency phase shift ofthe electrochemical equipment. Parameters CPE1 andCPE2 are constant phase elements. The resistance at thehigh frequency is probably associated with the charge-transfer processes (RE1), which include the ion-transferprocess occurring at the electrode/electrolyte interfacesand the electron-transfer process accompanying with theoxygen reduction reaction. The low-frequency arc can beattributed to the diffusion processes (RE2), which includethe adsorption–desorption of oxygen, oxygen diffusion atthe gas-cathode interface and surface diffusion of interme-diate oxygen species [48–53]. The difference between realaxes intercepts of the impedance plot is considered to bethe electrode polarization resistance (Rp = RE1 + RE2).The symbols in Fig. 6 represent the measured data andthe solid lines are the fitted curves. Good agreement isfound between the equivalent circuit and the observedimpedance data, which implies that the equivalent circuitshould be a reasonable choice. The fitted impedanceparameters are given in Table 3. The effect of the A-siteexcess on the overall electrode polarization resistance(Rp) and the high- and low-frequency arcs (RE1 and RE2)at different temperatures is shown in Fig. 7 and the corre-sponding activation energy is listed in Table 4. As can beseen in Fig. 7a, the BSCF1.03 cathode exhibits a verylow polarization resistance, with values of 0.068(9),0.014(9) and 0.004(4) X cm2 at 600, 700 and 800 �C, respec-tively. The activation energy of oxygen reduction is alsoreduced by using BSCF1.03 as the cathode as comparedto BSCF1.00, which suggests that the surface exchangeprocess is optimized with the slight A-site excess. However,the polarization resistance increases dramatically as the A-site excess further increases. Fig. 7a also compares theASRs of the BSCF1.09 and BSCF1.20 cathodes beforeand after the washing treatment. The interfacial resistancesof BSCF1.09 before and after the treatment were very sim-ilar, while the cathode performance of BSCF1.20 isenhanced substantially with the ASR values comparable
Table 3Fitted impedance results for O2 reduction reaction on BSCF1.00, BSCF1.03,temperatures
T (�C) BSCF1.00 BSCF1.03
RE1 RE2 RE1 RE2
550 0.024(1) 0.301(2) 0.112(9) 0.078(600 0.016(1) 0.077(9) 0.048(3) 0.020(650 0.013(0) 0.027(5) 0.023(4) 0.009(700 0.010(6) 0.008(6) 0.011(6) 0.003(750 0.007(8) 0.003(0) 0.006(7) 0.001(800 0.005(0) 0.001(4) 0.004(4) –
to that of BSCF1.09. It suggests that the impurities havea negative effect on the cathode performance. Fortunately,the impurities can be simply removed by the washing treat-ment without destroying other properties of the material.
As shown in Fig. 7b, the resistance RE2 of the diffusionprocesses for BSCF1.03 is 0.020(6) X cm2 at 600 �C, only26.4% of that for BSCF1.00. The creation of additionaloxygen vacancies by introducing excessive A-site cationshas a positive effect on the adsorption of oxygen and thesurface diffusion of intermediate oxygen species. Generallyfor a given chemical potential driving force, the morevacancies there are, the faster would oxygen adsorb andmove through the lattice. However, with the furtherincrease of the A-site excess, the oxygen adsorption resis-tance increases steadily. For BSCF1.20, the resistancereaches 0.809(2) X cm2 that is much higher than that ofBSCF1.00. It is likely that the active sites on the surfaceof the BSCF1.20 cathode are blocked by the impurities,which is highly detrimental to the oxygen adsorption andthe surface diffusion of the intermediate oxygen species.
Fig. 8 shows the O2–TPD curves of BSCF1.00,BSCF1.03, BSCF1.09 and BSCF1.20. There are two mainareas of oxygen loss, with onset temperatures being about250 and 750 �C, respectively. These two regions of oxygenloss are generally referred to as being from so-called a- andb-oxygen, respectively [54]. The low-temperature peak (a-oxygen) can be originating from two sources. One isascribed to the reduction of the high valence state Co4+
and/or Fe4+ to the tri-valence state Co3+ and/or Fe3+
[55], and it can also be attributed to the desorption of theadsorbed oxygen on the surface of the oxides [56]. Theonset temperature for a-oxygen desorption decreases withthe A-site excess concentration, which indicates that theCo–O and Fe–O bond energy is weakened because of theenlarged cell volume. Additionally, the peak area reflectsthe amount of O2 desorbing from the material. As can beseen from Fig. 8, the a-oxygen desorbing from BSCF1.03has a higher and broader peak than that of BSCF1.00,which indicates that more active sites are created on thesurface of BSCF1.03 for the adsorption of oxygen. How-ever, the amount of desorbed oxygen from theBSCF(1 + x) oxides decreases as the A-site excess concen-tration further increases, which suggests that the impuritiesoccupying the active sites block the oxygen adsorption and
BSCF1.09 and BSCF1.20 cathodes before washing treatment at various
BSCF1.09 BSCF1.20
RE1 RE2 RE1 RE2
4) 0.422(1) 0.526(5) 2.812(1) 2.921(1)6) 0.185(9) 0.110(3) 1.274(2) 0.809(2)1) 0.081(8) 0.034(8) 0.550(5) 0.254(1)3) 0.036(0) 0.016(4) 0.266(6) 0.047(3)2) 0.020(8) 0.004(5) 0.113(1) 0.018(2)
0.012(0) 0.003(1) 0.053(1) 0.012(3)

Fig. 7. Temperature dependence of the Rp (a) before and after washing treatment, RE1 (b) and RE2 (c) for BSCF1.00, BSCF1.03, BSCF1.09 andBSCF1.20.
Table 4Activation energy for RE1, RE2 and Rp of BSCF1.00, BSCF1.03,BSCF1.09 and BSCF1.20 cathodes before washing treatment
Sample Activation energy (kJ mol�1)
RE1 RE2 Rp
BSCF1.00 43 ± 1 158 ± 7 109 ± 2BSCF1.03 104 ± 8 142 ± 12 107 ± 5BSCF1.09 106 ± 3 152 ± 13 127 ± 11BSCF1.20 116 ± 7 170 ± 4 126 ± 2
Fig. 8. O2-TPD profiles for BSCF1.00, BSCF1.03, BSCF1.09 andBSCF1.20 before washing treatment.
W. Zhou et al. / Acta Materialia 56 (2008) 2687–2698 2695
also decrease the mean valences of the B-site ions. As awhole, the oxygen adsorption resistance is reduced dramat-ically for BSCF1.03, while it is unfavorably high forBSCF1.20.
The charge-transfer processes are also significantlyaffected by introducing the excessive Ba and Sr into theA-sites. At the cathode, the charge-transfer reactioninvolves the conversion of an oxygen molecule to oxideions. The creation of additional oxygen vacancies is benefi-cial to the oxygen adsorption process over the cathode, butthe decreased electronic conductivity is detrimental to thecharge-transfer process for oxygen reduction reaction. Onthe other hand, the solid-state reaction between the cath-ode and the electrolyte also contributes to the increase ofthe RE1. In our previous study [57], it was found that theSDC could react with BSCF1.00 to form BaCeO3 and(Ba,Sr,Sm,Ce)(Co,Fe)O3 at temperatures higher than1000 �C, which would block the migration of the electronsand the oxygen ions between the BSCF1.00 phase and theSDC phase, so that it might increase the interfacial resis-tance between cathode and electrolyte. In this study, thecathode materials (70 wt.%) and the SDC (30 wt.%) pow-ders were mixed and calcined at 1000 �C for 2 h in air forthe investigation of the phase reaction between them, andthe XRD results are shown in Fig. 9. For the BSCF1.03 +SDC mixture, an additional diffraction peak appears nearto the right side of the main BSCF1.03 peak at around

Fig. 9. XRD patterns of SDC, BSCF1.00, and BSCF(1 + x) + SDCmixture calcined at 1000 �C in air for 5 h.
2696 W. Zhou et al. / Acta Materialia 56 (2008) 2687–2698
2h = 31.7�, suggesting the formation of a new perovskiteoxide with a smaller lattice parameter than BSCF1.03.Based on our previous study, this can be identified as thediffraction peak of (Ba,Sr,Sm,Ce)(Co,Fe)O3 [57]. Further-more, additional diffractions at 2h values around 28.8�,41.1� and 50.8� are also observed, which can be ascribedto the new phase of BaCeO3 perovskite. With the furtherincrease of the A-site excess concentration, the intensityof the BSCF(1 + x) and the SDC diffraction peaksdecreases while that of the BaCeO3 and the (Ba,Sr,Sm,-Ce)(Co,Fe)O3 increased, indicating that the increase ofthe A-site excess greatly aggravates the reaction betweenthe BSCF(1 + x) and the SDC. Therefore, both the resis-tance and the activation energy of the charge-transfer pro-cess increase dramatically for the A-site excess bariumstrontium cobalt ferrites.
Based on the aforementioned results, the oxygen reduc-tion reaction mechanism on the A-site excessive bariumstrontium cobalt ferrites cathode is proposed and shownin Fig. 10. For BSCF1.00, based on the activation energy
Fig. 10. Mechanism for A-site excess barium
of RE1 and RE2, the diffusion process (RE2) is a rate-deter-mining step as compared with the charge-transfer process(RE1). The excess of the A-site cations creates additionaloxygen vacancies, which supplies more active sites for oxy-gen adsorption, so that the RE2 decreases dramatically forBSCF1.03. On the other hand, the creation of vacanciesalso sacrifices the electronic conductivity, which deterio-rates the electron-transfer process. Moreover, the solid-state reaction between the A-site excess barium strontiumcobalt ferrites and the SDC becomes more significant thanthat between the BSCF1.00 and the SDC. The formedBaCeO3 blocks not only the transfer of oxygen ions fromthe cathode to the electrolyte but also the incorporationof oxygen ions from the three-phase boundary into theSDC lattice, so that the RE1 increased for BSCF1.03. Withthe further increase of the A-site excess amount (x > 0.09),the BaO and/or SrO impurities formed (in the formation ofBa(OH)2 � nH2O and Sr(OH)2 � nH2O) on the surface of thecathode, which occupies the active sites for the ORR, lead-ing to an increase of RE2 to as high as 0.809(2) X cm2 at600 �C for BSCF1.20. By comparison with the oxygenadsorption process, the deterioration of the electron-trans-fer and the ion-transfer processes is even more substantialdue to the very low electronic conductivity and the moredramatic solid-phase reaction. By taking all these factorsinto account, the BSCF1.03 is found to be the optimalcathode material in this study.
It should be noted that silver is often added into theceramic cathode to improve the cathode performance byfacilitating the oxygen reduction reaction [58,59]. In ourprevious study, the effect of the silver paste on the BSCFcathode performance has been investigated [59]. We foundthat the silver can migrate into the cathode and improvethe cathode performance only at the temperature higherthan 850 �C. Therefore, in this study, considering the rela-tively lower operating temperature (<800 �C), we confirmthat the influence of silver paste can be ignored.
strontium cobalt ferrites as the cathode.
W. Zhou et al. / Acta Materialia 56 (2008) 2687–2698 2697
3.5. Cell performance
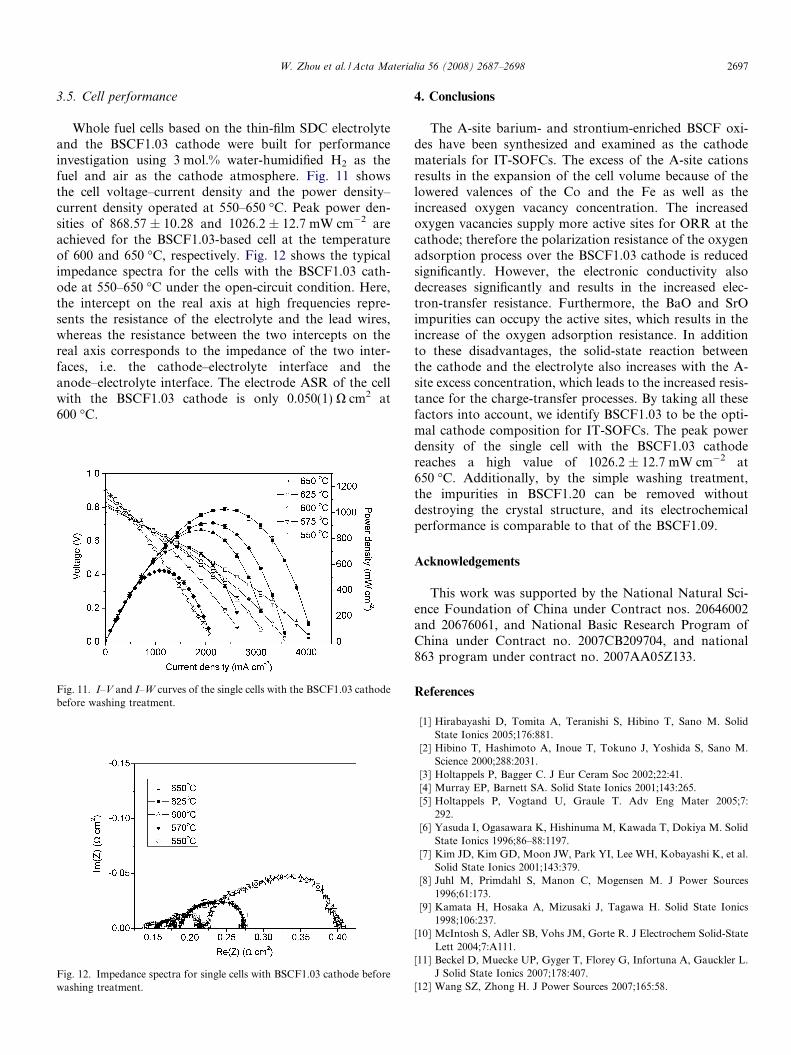
Whole fuel cells based on the thin-film SDC electrolyteand the BSCF1.03 cathode were built for performanceinvestigation using 3 mol.% water-humidified H2 as thefuel and air as the cathode atmosphere. Fig. 11 showsthe cell voltage–current density and the power density–current density operated at 550–650 �C. Peak power den-sities of 868.57 ± 10.28 and 1026.2 ± 12.7 mW cm�2 areachieved for the BSCF1.03-based cell at the temperatureof 600 and 650 �C, respectively. Fig. 12 shows the typicalimpedance spectra for the cells with the BSCF1.03 cath-ode at 550–650 �C under the open-circuit condition. Here,the intercept on the real axis at high frequencies repre-sents the resistance of the electrolyte and the lead wires,whereas the resistance between the two intercepts on thereal axis corresponds to the impedance of the two inter-faces, i.e. the cathode–electrolyte interface and theanode–electrolyte interface. The electrode ASR of the cellwith the BSCF1.03 cathode is only 0.050(1) X cm2 at600 �C.
Fig. 11. I–V and I–W curves of the single cells with the BSCF1.03 cathodebefore washing treatment.
Fig. 12. Impedance spectra for single cells with BSCF1.03 cathode beforewashing treatment.
4. Conclusions
The A-site barium- and strontium-enriched BSCF oxi-des have been synthesized and examined as the cathodematerials for IT-SOFCs. The excess of the A-site cationsresults in the expansion of the cell volume because of thelowered valences of the Co and the Fe as well as theincreased oxygen vacancy concentration. The increasedoxygen vacancies supply more active sites for ORR at thecathode; therefore the polarization resistance of the oxygenadsorption process over the BSCF1.03 cathode is reducedsignificantly. However, the electronic conductivity alsodecreases significantly and results in the increased elec-tron-transfer resistance. Furthermore, the BaO and SrOimpurities can occupy the active sites, which results in theincrease of the oxygen adsorption resistance. In additionto these disadvantages, the solid-state reaction betweenthe cathode and the electrolyte also increases with the A-site excess concentration, which leads to the increased resis-tance for the charge-transfer processes. By taking all thesefactors into account, we identify BSCF1.03 to be the opti-mal cathode composition for IT-SOFCs. The peak powerdensity of the single cell with the BSCF1.03 cathodereaches a high value of 1026.2 ± 12.7 mW cm�2 at650 �C. Additionally, by the simple washing treatment,the impurities in BSCF1.20 can be removed withoutdestroying the crystal structure, and its electrochemicalperformance is comparable to that of the BSCF1.09.
Acknowledgements
This work was supported by the National Natural Sci-ence Foundation of China under Contract nos. 20646002and 20676061, and National Basic Research Program ofChina under Contract no. 2007CB209704, and national863 program under contract no. 2007AA05Z133.
References
[1] Hirabayashi D, Tomita A, Teranishi S, Hibino T, Sano M. SolidState Ionics 2005;176:881.
[2] Hibino T, Hashimoto A, Inoue T, Tokuno J, Yoshida S, Sano M.Science 2000;288:2031.
[3] Holtappels P, Bagger C. J Eur Ceram Soc 2002;22:41.[4] Murray EP, Barnett SA. Solid State Ionics 2001;143:265.[5] Holtappels P, Vogtand U, Graule T. Adv Eng Mater 2005;7:
292.[6] Yasuda I, Ogasawara K, Hishinuma M, Kawada T, Dokiya M. Solid
State Ionics 1996;86–88:1197.[7] Kim JD, Kim GD, Moon JW, Park YI, Lee WH, Kobayashi K, et al.
Solid State Ionics 2001;143:379.[8] Juhl M, Primdahl S, Manon C, Mogensen M. J Power Sources
1996;61:173.[9] Kamata H, Hosaka A, Mizusaki J, Tagawa H. Solid State Ionics
1998;106:237.[10] McIntosh S, Adler SB, Vohs JM, Gorte R. J Electrochem Solid-State
Lett 2004;7:A111.[11] Beckel D, Muecke UP, Gyger T, Florey G, Infortuna A, Gauckler L.
J Solid State Ionics 2007;178:407.[12] Wang SZ, Zhong H. J Power Sources 2007;165:58.

2698 W. Zhou et al. / Acta Materialia 56 (2008) 2687–2698
[13] Xia CR, Liu ML. Solid State Ionics 2001;144:249.[14] Zhang X, Robertson M, Yick S, Deces-Petit C, Styles E, Qu W, et al.
J Power Sources 2006;160:1211.[15] Zhang YL, Zha SW, Liu ML. Adv Mater 2005;17:487.[16] Bellino MG, Sacanell JG, Lamas DG, Leyva AG, Walsoe de Reca
NE. J Am Chem Soc 2007;129:3066.[17] Shao ZP, Haile SM. Nature 2004;431:170.[18] Shao ZP, Haile SM, Ahn J, Ronney PD, Zhan ZL, Barnett SA.
Nature 2005;435:795.[19] Baumann FS, Fleig J, Habermeier HU, Maier J. Solid State Ionics
2006;177:3187.[20] Baumann FS, Fleig J, Habermeier HU, Maier J. Solid State Ionics
2006;177:1071.[21] Zhou W, Ran R, Shao ZP, Gu HX, Jin WQ, Xu NP. J Power Sources
2007;174:237.[22] Zhou W, Shao ZP, Ran R, Zeng PY, Gu HX, Jin WQ, et al. J Power
Sources 2007;168:330.[23] Wang YS, Wang SR, Wang ZR, Wen TL, Wen ZY. J Alloys Compd
2007;428:286.[24] Yakovlev SO, Kharton VV, Naumovich EN, Zekonyte J, Zap-
orojtchenko V, Kovalevsky AV, et al. Solid State Sci 2006;8:1302.[25] Otoshi S, Sasaki H, Ohnishi H, Hase M, Ishimaru K, Ippommatsu M.
J Electrochem Soc 1991;138:1519.[26] Hatchwell C, Bonanos N, Mogensen M. Solid State Ionics
2004;167:349.[27] Stevenson JW, Armstrong TR, Pederson LR, Li J, Lewinsohn CA,
Baskaran S. Solid State Ionics 1998;113–115:571.[28] Mineshige A, Izutsu J, Nakamura M, Nigaki K, Abe J, Kobune M,
et al. Solid State Ionics 2005;176:1145.[29] Kostogloudis GCh, Ftikos Ch. Solid State Ionics 1999;126:143.[30] Doshi R, Richards VL, Carter JD, Wang XP, Krumpelta M. J
Electrochem Soc 1999;146:1273.[31] Stevenson JW, Hallman PE, Armstrong TR, Chick LA. J Am Ceram
Soc 1995;78:507.[32] Simner SP, Bonnett JF, Canfield NL, Meinhardt KD, Shelton JP,
Sprenkle VL, et al. J Power Sources 2003;113:1.[33] Zhou W, Shao ZP, Jin WQ. J Alloys Compd 2006;426:368.[34] Xia CR, Liu ML. J Am Ceram Soc 2001;84:1903.[35] Jonker GH, Van Santen JH. Physica 1953;19:120.
[36] Kostera H, Mertins FHB. J Powder Diff 2003;18:56.[37] Bergsmark E, Furuseth S, Dyrlie O, Norby T, Kofstad P. In: Grosz F,
Zegers P, Singhal SC, Yamamoto O, editors. Proceedings of thesecond international symposium on solid oxide fuel cells. Luxem-bourg: Commission of European Communities; 1991. p. 473.
[38] Khattak CP, Cox DE. Mater Res Bull 1977;12:463.[39] Jiang SP, Love JG. Solid State Ionics 2001;138:183.[40] Yoon S, Baik S, Kim MG, Shin N, Kim I. J Am Ceram Soc
2007;90:311.[41] Khollam YB, Potdar HS, Deshpande SB, Gaikwad AB. Mater Chem
Phys 2006;97:295.[42] Lee KT, Manthiram A. J Power Sources 2006;158:1202.[43] Lee KT, Manthiram A. Chem Mater 2006;18:1621.[44] Takahashi H, Munakata F, Yamanaka M. Phys Rev B
1998;57:15211.[45] Zhou XD, Anderson HU. In: Singhal SC, Mizusaki J, editors. SOFC-
IX, PV2005-07. Pennington (NJ): The Electrochemical Society; 2005.p. 1479.
[46] Jaffe B, Cook WR, Jaffe H. Piezoelectric ceramics. New York: Aca-demic Press; 1971.
[47] Ge L, Zhou W, Ran R, Liu SM, Shao ZP, Jin WQ, et al. J Membr Sci2007;306:318.
[48] Adler SB, Lane JA, Steele BCH. J Electrochem Soc 1996;143:3554.[49] Chen XJ, Chan SH, Khor KA. Electrochim Acta 2004;49:1851.[50] Qiang F, Sun KN, Zhang NQ, Zhu XD, Le SR, Zhou DR. J Power
Sources 2007;168:338.[51] Adler SB. Solid State Ionics 1998;111:125.[52] Jørgensen MJ, Mogensen M. J Electrochem Soc 2001;148:A433.[53] Jiang SP, Wang W. J Electrochem Soc 2005;152:A1398.[54] Pena MA, Fierro JLG. Chem Rev 2001;101:1981.[55] Shao ZP, Yang WS, Cong Y, Dong H, Tong JH, Xiong GX. J
Membr Sci 2000;172:177.[56] Russo N, Fino D, Saracco G, Specchia V. J Catal 2005;229:459.[57] Wang K, Ran R, Zhou W, Gu HX, Shao ZP, Ahn J. J Power Sources,
in press, doi:10.1016/j.powsour.2007.12.051.[58] Sholklapper TZ, Radmilovic V, Jacobson CP, Visco SJ, De Jonghe
LC. J Power Sources 2008;175:206.[59] Zhou W, Ran R, Shao ZP, Cai R, Jin WQ, Xu NP, Ahn J.
Electrochim Acta, in press, doi:10.1016/j.electacta.2008.01.058.




















