
Bacterial lipopolysaccharide both renders resistant mice susceptible to mercury-induced autoimmunity...
10
Clinical and Experimental Immunology 238 © 2005 British Society for Immunology, Clinical and Experimental Immunology , 141: 238–247 doi:10.1111/j.1365-2249.2005.02849.x et al. Accepted for publication 25 April 2005 Correspondence: Manuchehr Abedi-Valugerdi, Department Biochemistry and Biophysics, Arrhenius Laboratories for the Natural Sciences, Stockholm University, S-106 91 Stockholm, Sweden. E-mail: address: [email protected] ORIGINAL ARTICLE Bacterial lipopolysaccharide both renders resistant mice susceptible to mercury-induced autoimmunity and exacerbates such autoimmunity in susceptible mice M. Abedi-Valugerdi,* C. Nilsson, † A. Zargari, ‡ F. Gharibdoost, § J. W. DePierre* and M. Hassan † *Department of Biochemistry and Biophysics, Arrhenius Laboratories for the Natural Sciences, Stockholm University, Stockholm, Sweden, † Department of Medicine, Huddinge University Hospital, Karolinska Institute, Stockholm, Sweden, ‡ Department of Medicine, Clinical Allergy Research Unit, Karolinska Institute and Hospital, Stockholm, Sweden, and § Rheumatology Research Center, Tehran University of Medical Sciences, Tehran, Iran Summary The initiation and severity of systemic autoimmune diseases are influenced by a variety of genetic and environmental factors, in particular bacterial infec- tions and products. Here, we have employed bacterial lipopolysaccharide (LPS), which non-specifically activates the immune system, to explore the involvement of innate immunity in mercury-induced autoimmunity in mice. Following treatment of mouse strains resistant [DBA/2 (H-2 d )] or susceptible [SJL(H-2 s )] to such autoimmunity with mercuric chloride and/or LPS or with physiological saline alone (control), their immune/autoimmune responses were monitored. Resistant DBA/2 mice were rendered susceptible to mercury- induced autoimmunity by co-administration of LPS, exhibiting pronounced increases in the synthesis of IgG1 and IgE, high titres of IgG1 deposits in the kidneys and elevated circulating levels of IgG1 antibodies of different speci- ficities. Furthermore, the percentages of the T cells isolated from the spleens of DBA/2 mice exposed to both mercury and LPS that produced pro-inflamma- tory cytokines were markedly increased by in vitro stimulation with phorbol myristate acetate (PMA) and ionomycin, which was not the case for splenic T cells isolated from mice receiving mercuric chloride, LPS or saline alone. In addition, exposure of susceptible SJL mice to mercury in combination with LPS aggravated the characteristic features of mercury-induced autoimmunity, including increased synthesis of IgG1 and IgE, the production of IgG1 anti- nucleolar antibodies (ANolA) and the formation of renal deposits of IgG1. In summary, our findings indicate that activation of the innate immune system plays a key role in both the induction and severity of chemically induced autoimmunity. Keywords: autoimmunity, innate immunity, lipopolysaccharide, mercuric chloride, pro- and non-inflammatory cytokines Introduction The underlying aetiologies of many systemic autoimmune diseases, including systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), multiple sclerosis (MS) and sys- temic sclerosis (SS) or scleroderma, are still unknown. It has been proposed that the aetiology, severity and recurrence of such diseases are multifactorial, involving both exogenous factors, such as infectious organisms and environmental tox- ins/pollutants, and endogenous hormonal, immunological and genetic factors [1,2]. In agreement with this suggestion is the observation that flare-ups of SLE in patients undergo- ing remission can often be triggered by infections, UV light, ingestion of certain food-stuffs and/or physical, mental or hormonal stress (reviewed in [3]). Although such phenom- ena are poorly understood, it is conceivable that infectious organisms induce autoimmune responses by molecular mimicry and disruption of immunoregulation, whereas tox- ins and/or drugs alter cellular responsiveness and the immu- nogenicity of self-antigens [3–6]. Among the environmental factors that may contribute to the development and/or exacerbation of autoimmune dis- eases is the bacterial endotoxin/lipopolysaccharide (LPS), a component of the cell wall of Gram-negative bacteria dis- tributed widely in the environment, as well as inhabiting the digestive tracts of humans and other animals [7–9]. It is now
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Bacterial lipopolysaccharide both renders resistant mice susceptible to mercury-induced autoimmunity...

Clinical and Experimental Immunology
238
© 2005 British Society for Immunology,
Clinical and Experimental Immunology
,
141:
238–247
doi:10.1111/j.1365-2249.2005.02849.x
1412
238247Original Article
Exacerbation of murine mercury-induced autoimmunity by LPSM. Abedi-Valugerdi et al.
Accepted for publication 25 April 2005
Correspondence: Manuchehr Abedi-Valugerdi,
Department Biochemistry and Biophysics,
Arrhenius Laboratories for the Natural Sciences,
Stockholm University, S-106 91 Stockholm,
Sweden.
E-mail: address: [email protected]
OR IG INAL ART I C L E
Bacterial lipopolysaccharide both renders resistant mice susceptible to mercury-induced autoimmunity and exacerbates such autoimmunity in susceptible mice
M. Abedi-Valugerdi,* C. Nilsson,
†
A. Zargari,
‡
F. Gharibdoost,
§
J. W. DePierre* and M. Hassan
†
*Department of Biochemistry and Biophysics,
Arrhenius Laboratories for the Natural Sciences,
Stockholm University, Stockholm, Sweden,
†
Department of Medicine, Huddinge University
Hospital, Karolinska Institute, Stockholm, Sweden,
‡
Department of Medicine, Clinical Allergy
Research Unit, Karolinska Institute and Hospital,
Stockholm, Sweden, and
§
Rheumatology Research
Center, Tehran University of Medical Sciences,
Tehran, Iran
Summary
The initiation and severity of systemic autoimmune diseases are influenced bya variety of genetic and environmental factors, in particular bacterial infec-tions and products. Here, we have employed bacterial lipopolysaccharide(LPS), which non-specifically activates the immune system, to explore theinvolvement of innate immunity in mercury-induced autoimmunity in mice.Following treatment of mouse strains resistant [DBA/2 (H-2
d
)] or susceptible[SJL(H-2
s
)] to such autoimmunity with mercuric chloride and/or LPS or withphysiological saline alone (control), their immune/autoimmune responseswere monitored. Resistant DBA/2 mice were rendered susceptible to mercury-induced autoimmunity by co-administration of LPS, exhibiting pronouncedincreases in the synthesis of IgG1 and IgE, high titres of IgG1 deposits in thekidneys and elevated circulating levels of IgG1 antibodies of different speci-ficities. Furthermore, the percentages of the T cells isolated from the spleens ofDBA/2 mice exposed to both mercury and LPS that produced pro-inflamma-tory cytokines were markedly increased by
in vitro
stimulation with phorbolmyristate acetate (PMA) and ionomycin, which was not the case for splenic Tcells isolated from mice receiving mercuric chloride, LPS or saline alone. Inaddition, exposure of susceptible SJL mice to mercury in combination withLPS aggravated the characteristic features of mercury-induced autoimmunity,including increased synthesis of IgG1 and IgE, the production of IgG1 anti-nucleolar antibodies (ANolA) and the formation of renal deposits of IgG1. Insummary, our findings indicate that activation of the innate immune systemplays a key role in both the induction and severity of chemically inducedautoimmunity.
Keywords:
autoimmunity, innate immunity, lipopolysaccharide, mercuricchloride, pro- and non-inflammatory cytokines
Introduction
The underlying aetiologies of many systemic autoimmunediseases, including systemic lupus erythematosus (SLE),rheumatoid arthritis (RA), multiple sclerosis (MS) and sys-temic sclerosis (SS) or scleroderma, are still unknown. It hasbeen proposed that the aetiology, severity and recurrence ofsuch diseases are multifactorial, involving both exogenousfactors, such as infectious organisms and environmental tox-ins/pollutants, and endogenous hormonal, immunologicaland genetic factors [1,2]. In agreement with this suggestionis the observation that flare-ups of SLE in patients undergo-ing remission can often be triggered by infections, UV light,
ingestion of certain food-stuffs and/or physical, mental orhormonal stress (reviewed in [3]). Although such phenom-ena are poorly understood, it is conceivable that infectiousorganisms induce autoimmune responses by molecularmimicry and disruption of immunoregulation, whereas tox-ins and/or drugs alter cellular responsiveness and the immu-nogenicity of self-antigens [3–6].
Among the environmental factors that may contribute tothe development and/or exacerbation of autoimmune dis-eases is the bacterial endotoxin/lipopolysaccharide (LPS), acomponent of the cell wall of Gram-negative bacteria dis-tributed widely in the environment, as well as inhabiting thedigestive tracts of humans and other animals [7–9]. It is now

Exacerbation of murine mercury-induced autoimmunity by LPS
© 2005 British Society for Immunology,
Clinical and Experimental Immunology
,
141:
238–247
239
well established that interaction of LPS with the LPS-bindingprotein (LBP) causes the membrane-bound or soluble formof CD14 and the recently identified Toll-like receptor 4(TLR4)-MD-2 complex to activate the innate immunesystem non-specifically, resulting in the production of proin-flammatory cytokines together with up-regulation of co-stimulatory molecules [10–12]. Moreover, probably via thissame receptor complex, LPS is capable of inducing poly-clonal activation of B cells, which leads to the production ofantibodies of different specificities, e.g. the production ofautoantibodies by murine splenic cells, both
in vitro
and
invivo
[13–16].It has been proposed that such non-specific activation of
innate immunity by LPS enhances the responses of the adap-tive immune system to both non-self (i.e. pathogen-related)and self (host-related)-antigens [12,17,18]. Thus, severalstudies indicate that under certain conditions LPS might ini-tiate and/or accelerate autoimmune processes. For example,immunization of susceptible mouse strains with mouse thy-roglobulin in combination with LPS can induce an autoim-mune thyroiditis which resembles the analogous syndrome(Hashimoto’s thyroiditis) in humans [19]. Moreover, LPSenhances polyclonal activation of B cells and exacerbatesnephritis in mouse strains prone to develop lupus, includingMRL/lpr/lpr (NZB
¥
NZW)F1 and BXSB mice [20–22].Mercurial compounds are widely spread environmental
pollutants that have been shown to induce a systemicautoimmune reaction in various mammals, and particularlyin rodents [23–26]. In susceptible mice, mercury-inducedautoimmunity is characterized by CD4
+
T cell-dependentpolyclonal activation of B cells, increased serum levels ofimmunoglobulin (Ig) G1 and IgE, production of autoanti-bodies of different specificities (especially anti-nucleolarautoantibodies (ANolA) and formation of deposits of IgG inthe kidney [25–27]. Indeed, in most of these respects thisautoimmunity resembles the lupus developed by(NZB
¥
NZW)F1 hybrids and BXSB mice [28,29], as well ashuman systemic lupus erythematosus [30]. In addition, as isthe case for the development of lupus in susceptible animalsand for human SLE, genetic susceptibility to mercury-induced autoimmunity is determined by both H-2 and non-H-2 background genes [31–35].
Because LPS is known to exacerbate the autoimmunemanifestations of lupus in mouse strains prone to developthis condition spontaneously [20–22], the present investiga-tion was designed to test the hypothesis that LPS also influ-ences the frequency and/or severity of mercury-inducedautoimmunity in both resistant and susceptible strains ofmice. Monitoring of various immunological parameters
-
including activation of B cells
in vivo
and T cells
in vitro
, pro-duction of specific autoantibodies, synthesis of variouscytokines and formation of renal IgG deposits revealed thatLPS is able to render resistant animals susceptible to mer-cury-induced autoimmunity, as well as to exacerbate suchautoimmunity in susceptible mice.
Materials and methods
Animals and treatment with mercuric chloride and/or LPS
Female SJL (H-2
s
) and DBA/2 (H-2
d
) mice (6–8 weeks old atthe beginning of each experiment) were obtained fromM&B A/S (M&B A/S, Ry, Denmark) and housed in the ani-mal facilities either at the Department of Immunology,Stockholm University, or at the Department of Medicine,Karolinska University Hospital at Huddinge, with a 12-hdark/12-h light cycle and access to standard laboratory chowand tap water
ad libitum
. Four groups of each of thesestrains, each group containing four to five mice, wereemployed. Groups 1 and 2 were injected subcutaneously(s.c.) with 0·1 ml of a solution containing 0·4 mg HgCl
2
(Merck, Darmstedt, Germany) per ml sterile physiologicalsaline (to give a dose of 1·6 mg/kg body weight) every thirdday for 4 weeks. Group 2 also received intravenous (i.v.)injections of 50
m
g LPS (
Escherichia coli
, 055:B5; Sigma-Aldrich Sweden, Stockholm) in 0·1 ml sterile physiologicalsaline through their tail veins on the first day of mercurytreatment and once again 2 weeks later. The animals ingroup 3 received LPS alone in the same manner, while themice in group 4 (the control group) were injected s.c. with0·1 ml physiological saline alone according to the sameschedule employed for mercury treatment. These experi-ments were approved by the Northern Stockholm EthicalCommittee for Animal Experimentation.
Collection of blood, spleens and kidneys and preparation of serum and cell suspensions
Following the 4-week treatment period, the mice were bledby retroorbital puncture under light isofluorane anaesthesiaand thereafter killed by cervical dislocation and their spleensand kidneys dissected out aseptically. The blood sampleswere allowed to clot at 4
∞
C and then centrifuged to obtainthe serum, which was stored at
-
20
∞
C until being assayed forantibody/autoantibody levels. Approximately three-quartersof each spleen was teased apart with forceps in Earle’s bal-anced salt solution (EBSS) in order to obtain single cell sus-pensions, which were washed three times and resuspended in5 ml of this same solution for performance of the protein Aplaque assay.
The protein A plaque assay
The numbers of splenic cells secreting antibodies belongingto different Ig classes and subclasses were quantified utilizingthe protein A plaque assay described by Gronowicz
et al
.[36], employing rabbit anti-mouse IgM, IgG1, IgG3 (Orga-non Teknika, Durham, NC, USA) and IgG2b (Nordic Immu-nological Laboratories, Tillburg, the Netherlands) as thedeveloping reagents.

M. Abedi-Valugerdi
et al.
240
© 2005 British Society for Immunology,
Clinical and Experimental Immunology
,
141:
238–247
Quantification of mouse IgE by enzyme-linked immunosorbent assay (ELISA)
Total serum levels of IgE were determined with a sandwichELISA procedure, as described previously [37]. A ratanti-mouse IgE monoclonal antibody (mAb) (R35-72;Pharmingen, San Diego, CA, USA) was utilized as the ‘cap-ture’ antibody and a biotinylated rat anti-mouse IgE mAb(R35-92; Pharmingen) as the ‘detection’ antibody.
Detection of ANolA by indirect immunofluorescence (IIF)
The levels of IgG1- and IgG2a-type ANolAs in serum sampleswere determined employing indirect immunofluorescence.For this purpose, HEp-2 cells grown as monolayers on slides(Immuno Concepts, Sacramento, CA, USA) served as thesubstrate and FITC-conjugated goat anti-mouse IgG1 andIgG2a (Southern Biotechnology, Birmingham, AL, USA) asthe visualizing antibodies. The pattern and titres of differentANolAs were subsequently assessed under a Reichard–JungPolyvar microscope (Vienna, Austria), with serum samplesthat exhibited no specific green fluorescence at a dilution of1 : 50 being assigned a value of zero. The highest dilution atwhich nucleolar fluorescence could still be detected wasdefined as the titre of the IgG1 and/or IgG2a ANolA.
Activation of T cells
in vitro
The remaining quarter of each spleen (see above) was pooledwith the corresponding samples from the mice in the samegroup and single cell suspensions subsequently prepared incomplementary RPMI-1640 (cRPMI) (Invitrogen AB,Stockholm, Sweden). These spleen samples had to be pooledin order to obtain adequate numbers of cells for intracellularstaining of cytokines and flow cytometry. Erythrocytes inthese cell suspensions were lysed with a solution of NH
3
Cl
(StemCell Technologies Inc. Vancouver, Canada) and theother cells then washed twice in phosphate-buffered saline(PBS)-Dulbecco’s medium, re-suspended in 5 ml cRPMIand adjusted to a density of 1
¥
10
6
cells/ml.To stimulate the T cells, 10
6
cells from this suspension wereplated in duplicates in each well of six-well flat-bottomedplates (BD Labware, NJ, USA) containing 4-ml medium sup-plemented either with phorbol 12-myristate 13-acetate(PMA) (10 ng/ml, final concentration), ionomycin (1
m
g/ml) and brefeldin A (10
m
g/ml) (all were purchased fromSigma, Stenheim, Germany) or with brefeldin A alone (con-trol wells). Following incubation for 6 h at 37
∞
C in a humid-ified atmosphere containing 5% CO
2
, the cells wereharvested and centrifuged at 510
g
for 5 min at 10
∞
C and thepellets thus obtained washed in PBS-Dulbecco’s medium(Invitrogen AB, Stockholm, Sweden) by centrifugation. Theresulting pellet was fixed by incubation at room temperaturefor 10–20 min in 1 ml 4% paraformaldehyde. Thereafter, thefixed cells were washed twice in PBS, re-suspended in 1 mlPharMingenStain buffer (FBS) and stored at 4
∞
C until thestaining procedure was performed.
Intracellular staining of cytokines and flow cytometry
The mAb employed for detection of surface markers andintracellular cytokines (all purchased from PharMingen, SanDiego, CA, USA) are documented in Table 1. Intracellularstaining for interleukin (IL)-2, interferon (IFN)-
g
andtumour necrosis factor (TNF)-
a
was performed according tothe standard protocols recommended by the BD Pharmingencompany. Briefly, the cells were first stained (for 30 min onice in the dark) for CD4, CD8 and CD19 using the appro-priate mAb conjugated with phycoerythrin (PE). Thereafter,the cells were washed twice with staining buffer, permeabi-lized Perm/Wash
™
solution, and stained for intracellular IL-2, IFN-
g
and TNF-
a
using the PE-conjugated mAbs directlyspecifically against these cytokines (see Table 1).
Table 1.
The monoclonal antibodies used for detection of surface markers and intracellular cytokines.
Raised in Specificity Conjugated with Clone number Isotype form
Rat Mouse CD4 FITC
1
H129·19 IgG2a
Rat Mouse CD8 PE
2
53–6·7 IgG2a
Rat Mouse CD4 PerCp
3
RM4-5 IgG2a
Rat Mouse CD3 FITC 17A2 IgG2b
Rat Mouse CD19 PE 1D3 IgG2a
Rat Mouse IL-2 PE S4B6 IgG2a
Rat Mouse IL-4 PE BVD4–1D11 IgG2b
Rat Mouse IDN-
g
PE XMG1·2 IgG1
Rat Mouse TNF-
a
PE MP6-XT22 IgG1
Rat Unknown
4
FITC R3-34 IgG1
Rat Unknown PE and FITC R35-95 IgG2a
Rat Unknown PE R3-34 IgG1
Rat Unknown FITC R35-38 IgG2b
All these monoclonal antibodies were purchased from PharMingen (San Diego, CA).
1
Fluorescein isothiocyanate;
2
phycoerythrin;
3
peridinin
chlorophyll protein;
4
negative isotype control.

Exacerbation of murine mercury-induced autoimmunity by LPS
© 2005 British Society for Immunology,
Clinical and Experimental Immunology
,
141:
238–247
241
All samples were analysed on a FACSCalibur flow cytom-eter with CellQuest software (BD Immunocytometry Sys-tems). The percentages of cells producing IL-2, IFN-
g
and/orTNF-
a
was calculated as 100
¥
(the number of cells express-ing both the cytokine and CD3/the total number of cellsexpressing CD3) and the fold-increase caused by
in vitro
stimulation calculated by dividing this percentage for stim-ulated cells by the corresponding value for the cells priorstimulation.
Detection of renal deposits of IgG1
The presence of glomerular deposits of IgG1 antibodies wasdetected by direct immunofluorescence (DIF), as describedpreviously [37]. Briefly, 5
m
m-thick cryostat sections of kid-ney tissue were fixed in acetone and incubated with serialdilutions of fluorescein isothiocyanate (FITC)-conjugatedgoat anti-mouse IgG1 antibodies (Southern Biotechnology),starting with an initial dilution of 1: 40. When no specificgreen fluorescence was detected at this initial dilution, thesample was assigned a value of zero. The highest dilution ofthe antibody at which specific fluorescence could still be seenwas defined as the end-point titre for the glomerulardeposits.
Enzyme immunoassays
For quantification of serum IgG1 antibodies directed againstboth self and non-self antigens, micro-ELISA plates (Costar,Cambridge, MA, USA) were first coated with 50
m
l single-stranded (ss)-DNA (10
m
g/ml in phosphate-buffered saline(PBS); Serva, Heidelberg, Germany), chicken collagen typeII (10
m
g/ml; Sigma Chemical Co, St Louis, MO, USA) orbovine thyroglobulin (10
m
g/ml in 0·035
M
carbonate–bicar-bonate buffer, pH 9·8; Sigma Chemical Co, St Louis, MO,USA) (self-antigens) or with the non-self antigen trini-trobenzene sulphonic acid coupled to bovine serum albumin(TNP-BSA) (10
m
g/ml in 0·035
M
carbonate-bicarbonatebuffer, pH 9·8; a kind gift from Dr Carmen Fernandez,Department of Immunology, Stockholm University) andthen incubated overnight at 4
∞
C. Thereafter, the plates werewashed three times with PBS containing 1% Tween-20 (PBS-Tween) and unoccupied binding sites blocked by incubationwith 1% BSA in PBS for 2 h at room temperature. Sub-sequently, the plates were washed again three times withPBS-Tween and 50
m
l serially diluted serum samples (in PBS-Tween, starting with a dilution of 1 : 10) added to the wellsand the plates again incubated overnight at 4
∞
C. Followingthree more washes with PBS-Tween, the plates were incu-bated with alkaline phosphatase-labelled goat anti-mouseIgG1 (diluted 1 : 2000 in PBS-Tween; Southern Biotechnol-ogy) for 2 h at 37
∞
C, followed by addition of 50
m
l of thephosphatase substrate p-nitrophenyl phosphate (1 mg/mldissolved in 100 mM diethanolamine-HCl, pH 9·8; SigmaChemical Co). After a 40-min incubation at room tempera-
ture, the OD
405 nm
was quantified using an ELISA reader(Anthos, Labtec Instrument, Salzburg, Austria).
Expression and statistical analysis of the data
The numbers of cells secreting IgG1 antibody, serum levels ofIgE, the titres of IgG1 ANolAs and titres of renal IgG1 depos-its are all expressed as means
±
standard error (s.e.). Differ-ences between these parameters for the control and treatedgroups were analysed for statistical significance using analy-sis of variance (
anova
), with a
P
-value of
<
0·05 being con-sidered statistically significant. All statistical analyses wereperformed utilizing WinSTAT software (R. Fitch Software,Medina AB, Vänerborg, Sweden). The experiments shown inFigs 1 and 4 are representative of three and those in Fig. 3and Table 2 are representative of two independentexperiments.
Results
Administration of LPS during treatment with mercuric chloride renders resistant DBA/2 mice susceptible to mercury-induced autoimmunity
We and other investigators have demonstrated that DBA/2(H-2
d
) is the only inbred strain of mice that exhibits noimmune/autoimmune response at all upon exposure to mer-cury [31–34]. This was confirmed here by the observationsthat the percentages of splenic cells producing IgG1 antibod-ies, serum levels of IgE and renal deposits of IgG1 (Figs 1 and2), as well as the production of circulating IgG1 antibodiesdirected against various antigens (Fig. 3) were not signifi-cantly different in DBA/2 mice treated with mercuric chlo-ride or saline alone. Nor did treatment of these same animalswith LPS alone exert any statistically significant effect on anyof these parameters (Figs 1, 2 and 3).
In sharp contrast, DBA/2 mice treated with mercury incombination with LPS exhibited increased percentages ofsplenic cells secreting IgG1 antibodies (Fig. 1a), elevatedserum levels of IgE (Fig. 1b) and high titres of IgG1 depositsin their renal glomeruli (Figs 1c and 2d). Moreover, thiscombined treatment also enhanced serum levels of specificIgG1 antibodies in DBA/2 mice exposed to both exogenous(TNP) and endogenous (ssDNA, collagen and thyroglobu-lin) antigens (Fig. 3a–d). However, administration of mer-cury and LPS to these mice did not result in a significantincrease in the serum levels of ANolA (not shown).
Cytokine production by splenic T cells isolated from DBA/2 mice treated with mercuric chloride and/or LPS
T cells, and in particular CD4
+
T-cells, together with thecytokines they produce have been shown to play crucial rolesin the development of mercury-induced autoimmunity [24–27,38]. Therefore, in attempt to elucidate the possible mech-

M. Abedi-Valugerdi
et al.
242
© 2005 British Society for Immunology,
Clinical and Experimental Immunology
,
141:
238–247
Fig. 1.
Administration of bacterial lipopolysaccharide (LPS) during treatment with mercuric chloride renders DBA/2 mice susceptible to mercury-
induced autoimmunity. Group 1 (five mice per group) (filled hexagons) received repeated subcutaneous injections of subtoxic doses of mercuric
chloride (Hg) for a period of 4 weeks. In addition to similar administration of mercuric chloride, group 2 (filled circles) was injected i.v. with LPS
(50
m
g per mouse) on the 1st and 15th days of mercury treatment. The two control groups 3 (filled squares) and 4 (open squares) received LPS alone
on two occasions or repeated injections of physiological saline, according to the corresponding schedules used for groups 2 and 1, respectively. At the
end of the period of treatment (a) the percentages of splenic cells secreting IgG1 antibodies (PFC) were determined employing the protein A plaque
assay. In addition (b) the levels of IgE in the serum samples were determined employing a sandwich enzyme-linked immunosorbent assay (ELISA)
assay and (c) the kidneys were analysed for the presence of glomerular deposits of IgG1 utilizing a direct immunofluorescence technique, as described
in the Materials and methods. The mean
±
s.e. values are presented as thin vertical lines. *
P
<
0·05 for the difference between the two groups indicated.
These results are representative of three independent experiments.
0
20
40
60
80
100
120
Sal
ine
Hg
alon
e
Hg+
LPS
LPS
alo
ne
Spl
enic
IgG
1 P
FC
¥ 1
0–3
0
10
20
30
40
50
60
70
Sal
ine
Hg
alon
e
Hg+
LPS
LPS
alo
ne
Ser
um le
vels
of I
gE (mg
/ml)
0123456789
10111213
Sal
ine
Hg
alon
e
Hg+
LPS
LPS
alo
ne
Rec
ipro
cal t
itres
of r
enal
IgG
1 de
posi
ts ¥
10–
2
(a) (b) (c)
* *** **
* **
Fig. 2.
Immunofluorescent detection of glomer-
ular deposits of IgG1 in the kidneys of DBA/2
mice administered mercuric chloride in combi-
nation with lipopolysaccharide (LPS). The kid-
neys from the same animals presented in Fig. 1
were tested for the presence of glomular deposits
of IgG1 employing the direct immunofluores-
cence procedure described in the Materials and
methods. Representative kidney sections from
animals administered saline (a), LPS (b) or mer-
cury (c) alone show no green fluorescence inten-
sity; whereas the kidney of a DBA/2 mouse
treated with mercury in combination with LPS
(d) displays intense green fluorescence in the
glomeruli.
(a) (b)
(c) (d)
Saline LPS alone
Hg alone Hg + LPS

Exacerbation of murine mercury-induced autoimmunity by LPS
© 2005 British Society for Immunology,
Clinical and Experimental Immunology
,
141:
238–247
243
anism(s) by which LPS renders DBA/2 mice susceptible tomercury-induced autoimmunity, a separate experiment wasperformed to analyse the
in vitro
cytokine production bysplenic T cells (with and without stimulation) isolated fromDBA/2 mice subjected to different treatments. As shown inTable 2, the spleens of animals treated with saline or mercuryalone contained intermediate and comparable numbers of Tcells producing IL-2, TNF-
a
and/or IFN-
g
. In these twocases,
in vitro
stimulation caused a similar increase in thepercentage of cells producing IL-2, but more pronouncedincreases in the percentages of cells producing TNF-
a
and/orIFN-
g
following treatment with mercuric chloride alone
(Table 2). On the other hand, the spleens of animals admin-istered LPS alone contained a higher percentage of T cellsproducing IL-2, TNF-
a
and/or IFN-g than did the spleens ofthe mice in the other groups (Table 2). However, a 6-h invitro exposure of these cells to PMA/ionomycin enhancedonly the percentages of cells producing IL-2 and/or TNF-aand this effect was slight (Table 2).
In contrast, the spleens of mice treated with Hg in com-bination with LPS exhibited the lowest percentages of T cellsproducing all of the cytokines examined here (Table 2). Atthe same time, in vitro stimulation of these cells led to a morepotent elevation in the percentages of cells producing IL-2,TNF-a and/or IFN-g than was observed in the case of any ofthe other groups (Table 2).
Treatment with LPS potentiates mercury-induced autoimmunity in susceptible SJL mice
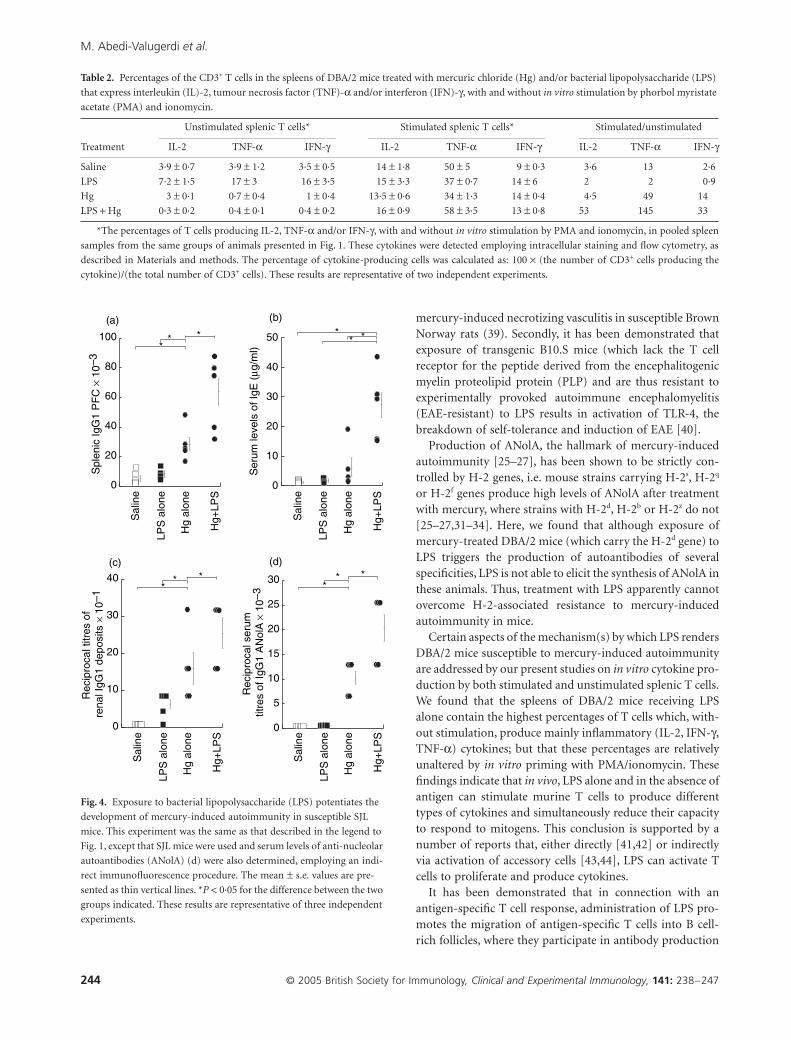
As expected, 4 weeks of treatment with mercury aloneinduced an immune/autoimmune response in susceptibleSJL mice, characterized by increased percentages of spleniccells secreting IgG1 antibodies (Fig. 4a), elevated serum lev-els of IgE (Fig. 4b), increased titres of renal IgG1 deposits(Fig. 4c) and high serum titres of IgG1 ANolA (Fig. 4d).Treatment with LPS alone did not alter these parameters incomparison with control administration of saline alone(Fig. 4a–d). However, the SJL mice that received both mer-cury and LPS exhibited even higher levels of all four of thesemanifestations of autoimmunity than did animals injectedwith mercuric chloride alone (Fig. 4a–d). In contrast, LPShad no influence on the elevation in the percentages ofsplenic cells secreting IgM, IgG2b and/or IgG3 antibodies orthe enhancement of serum levels of IgG2a ANolA elicited byadministration of mercury alone (data not shown).
Discussion
In the present investigation we have demonstrated thatwhile, as expected, treatment with mercuric chloride or LPSalone did not elicit an immune/autoimmune response inDBA/2 mice, combined treatment renders these animalshighly susceptible to mercury-induced autoimmunity.Indeed, our observation that most of the characteristics ofmercury-induced autoimmune developed potently in themice subjected to combined treatment indicates that undercertain circumstances two or more environmental factorscan act in concert to break self-tolerance and initiate thedevelopment of autoimmunity, even in resistant individuals.Our findings also suggest that activation of the innate branchof immune responses by bacteria or their products plays acrucial role with regard to this synergistic effect. Interest-ingly, two types of observations support this suggestion.First, it has been shown that pretreatment with broad-spectrum anti-microbial drugs including tylosin, ivermectinand metronidazole is able, to a large extent, to prevent
Fig. 3. Administration of bacterial lipopolysaccharide (LPS) together
with mercuric chloride to DBA/2 mice increases their production of
IgG1 antibodies to both exogenous and endogenous antigens. The levels
of IgG1 antibodies directed against TNP (a), ssDNA (b), collagen (c)
and thyroglobulin (d) in serum samples from the same animals pre-
sented in Fig. 1 were determined employing sandwich enzyme-linked
immunosorbent assay (ELISA) assays, as described in detail in the Mate-
rials and methods. In all cases a serum dilution of 1 : 40 was utilized and
the values obtained were within the linear range of the dilution curve.
The mean ± s.e. values are presented as thin vertical lines. *P < 0·05 for
the difference between the two groups indicated. These results are rep-
resentative of two independent experiments.
0.0
0.2
0.4
0.6
0.8
1.0
Sal
ine
Hg
alon
e
Hg+
LPS
LPS
alo
ne
0.0
0.2
0.4
0.6
0.8
1.0
Sal
ine
Hg
alon
e
Hg+
LPS
LPS
alo
ne
0.0
0.2
0.4
0.6
0.8
1.0
Sal
ine
Hg
alon
e
Hg+
LPS
LPS
alo
ne
* **
0.0
0.2
0.4
0.6
0.8
1.0
Sal
ine
Hg
alon
e
Hg+
LPS
LPS
alo
ne
Ser
um le
vels
of I
gG1
anti-
TN
P (
OD
405
nm)
Ser
um le
vels
of I
gG1
anti-
ssD
NA
(O
D40
5 nm
)
Ser
um le
vels
of I
gG1
anti-
colla
gen
(OD
405
nm)
(a) (b)
(c) (d)
Ser
um le
vels
of I
gG1
anti-
TG
(O
D40
5 nm
)
* **
* *** **
M. Abedi-Valugerdi et al.
244 © 2005 British Society for Immunology, Clinical and Experimental Immunology, 141: 238–247
mercury-induced necrotizing vasculitis in susceptible BrownNorway rats (39). Secondly, it has been demonstrated thatexposure of transgenic B10.S mice (which lack the T cellreceptor for the peptide derived from the encephalitogenicmyelin proteolipid protein (PLP) and are thus resistant toexperimentally provoked autoimmune encephalomyelitis(EAE-resistant) to LPS results in activation of TLR-4, thebreakdown of self-tolerance and induction of EAE [40].
Production of ANolA, the hallmark of mercury-inducedautoimmunity [25–27], has been shown to be strictly con-trolled by H-2 genes, i.e. mouse strains carrying H-2s, H-2q
or H-2f genes produce high levels of ANolA after treatmentwith mercury, where strains with H-2d, H-2b or H-2z do not[25–27,31–34]. Here, we found that although exposure ofmercury-treated DBA/2 mice (which carry the H-2d gene) toLPS triggers the production of autoantibodies of severalspecificities, LPS is not able to elicit the synthesis of ANolA inthese animals. Thus, treatment with LPS apparently cannotovercome H-2-associated resistance to mercury-inducedautoimmunity in mice.
Certain aspects of the mechanism(s) by which LPS rendersDBA/2 mice susceptible to mercury-induced autoimmunityare addressed by our present studies on in vitro cytokine pro-duction by both stimulated and unstimulated splenic T cells.We found that the spleens of DBA/2 mice receiving LPSalone contain the highest percentages of T cells which, with-out stimulation, produce mainly inflammatory (IL-2, IFN-g,TNF-a) cytokines; but that these percentages are relativelyunaltered by in vitro priming with PMA/ionomycin. Thesefindings indicate that in vivo, LPS alone and in the absence ofantigen can stimulate murine T cells to produce differenttypes of cytokines and simultaneously reduce their capacityto respond to mitogens. This conclusion is supported by anumber of reports that, either directly [41,42] or indirectlyvia activation of accessory cells [43,44], LPS can activate Tcells to proliferate and produce cytokines.
It has been demonstrated that in connection with anantigen-specific T cell response, administration of LPS pro-motes the migration of antigen-specific T cells into B cell-rich follicles, where they participate in antibody production
Table 2. Percentages of the CD3+ T cells in the spleens of DBA/2 mice treated with mercuric chloride (Hg) and/or bacterial lipopolysaccharide (LPS)
that express interleukin (IL)-2, tumour necrosis factor (TNF)-a and/or interferon (IFN)-g, with and without in vitro stimulation by phorbol myristate
acetate (PMA) and ionomycin.
Treatment
Unstimulated splenic T cells* Stimulated splenic T cells* Stimulated/unstimulated
IL-2 TNF-a IFN-g IL-2 TNF-a IFN-g IL-2 TNF-a IFN-g
Saline 3·9 ± 0·7 3·9 ± 1·2 3·5 ± 0·5 14 ± 1·8 50 ± 5 9 ± 0·3 3·6 13 2·6
LPS 7·2 ± 1·5 17 ± 3 16 ± 3·5 15 ± 3·3 37 ± 0·7 14 ± 6 2 2 0·9
Hg 3 ± 0·1 0·7 ± 0·4 1 ± 0·4 13·5 ± 0·6 34 ± 1·3 14 ± 0·4 4·5 49 14
LPS + Hg 0·3 ± 0·2 0·4 ± 0·1 0·4 ± 0·2 16 ± 0·9 58 ± 3·5 13 ± 0·8 53 145 33
*The percentages of T cells producing IL-2, TNF-a and/or IFN-g, with and without in vitro stimulation by PMA and ionomycin, in pooled spleen
samples from the same groups of animals presented in Fig. 1. These cytokines were detected employing intracellular staining and flow cytometry, as
described in Materials and methods. The percentage of cytokine-producing cells was calculated as: 100 ¥ (the number of CD3+ cells producing the
cytokine)/(the total number of CD3+ cells). These results are representative of two independent experiments.
Fig. 4. Exposure to bacterial lipopolysaccharide (LPS) potentiates the
development of mercury-induced autoimmunity in susceptible SJL
mice. This experiment was the same as that described in the legend to
Fig. 1, except that SJL mice were used and serum levels of anti-nucleolar
autoantibodies (ANolA) (d) were also determined, employing an indi-
rect immunofluorescence procedure. The mean ± s.e. values are pre-
sented as thin vertical lines. *P < 0·05 for the difference between the two
groups indicated. These results are representative of three independent
experiments.
0
20
40
60
80
100
Sal
ine
Hg
alon
e
Hg+
LPS
LPS
alo
ne
0
10
20
30
40
50
Sal
ine
Hg
alon
e
Hg+
LPS
LPS
alo
ne
Ser
um le
vels
of I
gE (mg
/ml)
0
10
20
30
40
Sal
ine
Hg
alon
e
Hg+
LPS
LPS
alo
ne
Rec
ipro
cal t
itres
of
rena
l IgG
1 de
posi
ts ¥
10–
1
0
5
10
15
20
25
30
Sal
ine
Hg
alon
e
Hg+
LPS
LPS
alo
ne
Rec
ipro
cal s
erum
tit
res
of Ig
G1
AN
olA
¥ 1
0–3
* ** * **
* **
Spl
enic
IgG
1 P
FC
¥ 1
0–3
* **
(a) (b)
(c) (d)

Exacerbation of murine mercury-induced autoimmunity by LPS
© 2005 British Society for Immunology, Clinical and Experimental Immunology, 141: 238–247 245
[45,46]. Moreover, the antigen-experienced T cells thatremain in the lymphoid tissues for several weeks followingexposure to an antigen and an adjuvant such as LPS exhibitthe functional characteristics of memory T cells [47], i.e.upon activation in vitro, these cells rapidly produce high lev-els of a broad spectrum of both pro- and anti-inflammatorycytokines [47,48]. Similarly, our present observation thatsplenic T cells isolated from DBA/2 mice treated with mer-cury and LPS and subsequently primed in vitro producelarge amounts of different types of cytokines, including IL-2,TNF-a, and IFN-g, suggests that these T cells also demon-strate functional characteristics reminiscent of those ofmemory T cells. Therefore, it appears likely that co-admin-istration of LPS with mercury enhances the generation andactivation of T cells specific for self-antigens altered by mer-cury and that, after aiding autoreactive B cells, some of thesecells are transformed into memory T cells. Clearly, furtherstudy is required to test this hypothesis.
Furthermore, we observed that in vitro priming of splenicT cells isolated from DBA/2 mice treated with mercury alonecauses larger increases in the percentages of these cellsexpressing TNF-a and/or IFN-g than was the case for splenicT cells from untreated mice, but that the increase in IL-2 wassimilar. These results suggest that even in these resistantmice, mercury alone can activate certain T cells, which candevelop further into memory-type T cells with a low capacityfor production of IL-2. This proposal receives strong supportfrom the findings that exposure of mice to antigen alone, inthe absence of adjuvant, leads to the development of a fewantigen-specific CD4+ T cells that remain in the lymphoidtissue for a long period of time and exhibit a phenotype char-acteristic of memory cells [49,50]. However, unlike thememory cells produced in response to administration ofantigen together with adjuvant, these antigen-experienced Tcells do not produce IL-2 or effector lymphokines [49,50].
Thus, it is possible that in the resistant mice and in theabsence of non-specific stimuli (here, LPS) alteration of thepresentation of self-antigens by mercury is inefficient,because this presentation is performed by dendritic cells thatdo not express optimal levels of co-stimulatory molecules.This situation, together with a lack of inflammatory cytok-ines, leads to the activation of specific T cells with low pro-liferative capacity, pronounced susceptibility to apoptosisand the ability to develop into functionally defective mem-ory cells. In contrast, the presence of LPS converts tolero-genic dendritic cells into autoimmune dendritic cells, whichare capable of activating nontolerant CD4+ T cells.
Interestingly, it has recently been shown that non-specificstimuli, including LPS, are not able to break cross-toleranceto self-antigens in autoreactive cytotoxic T (CD8+) cells,unless help from specific autoreactive CD4+ T cells is pro-vided [51]. Although, as the authors have suggested, theexact underlying mechanisms for this co-operation between‘autoreactive CD8+ and CD4+ T cells and non-specific stim-uli require further investigations’ [51], this interesting
finding, together with our present observations, suggest thatautoreactive CD4+ T cells are more prone than autoreactiveCD8+ T cells to activation by non-specific stimuli.
Finally, we also show here that exposure of SJL mice(which are susceptible to mercury-induced autoimmunity)to LPS twice during the course of mercury treatment poten-tiates most, if not all the immune/autoimmune responseselicited by mercury, including the formation of IgG1, syn-thesis of IgE, development of renal deposits of IgG1 and pro-duction of IgG1 ANolA. Such LPS-induced exacerbation ofmercury-induced autoimmunity in SJL was not strain-dependent, as we have made identical observations inanother mercury-susceptible mouse strain, A.SW (H-2s)(data not shown). Thus, these findings are in agreement withother reports that LPS exacerbates autoimmune manifesta-tions in mouse strains genetically prone to develop a lupus-like autoimmune disease [20–22].
Although the mechanism(s) underlying this potentiationof mercury-induced autoimmunity by LPS remains to beelucidated, several lines of evidence indicate that signals pro-vided by co-stimulatory surface molecules play a key role inthis connection. For instance, like any other adaptiveimmune response, mercury-induced autoimmunity hasbeen shown to be dependent on the expression of co-stimulatory molecules such as CD40, CD80 (B7-1) andCD86 (B7-2) on professional antigen-presenting cells,including macrophages, dendritic cells and activated B cells[52–54]. On the other hand, it is now well established that byinteracting with the TLR-4-MD-2 complex and CD14 onprofessional antigen-presenting cells, LPS initiates TLR sig-nalling, which exerts adjuvant effects on adaptive immuneresponses [55,56].
This signalling involves various specific adaptor proteins,including MyD88 (myeloid differentiation factor 88), MAL(MyD88 adaptor-like), Trif (Toll receptor-associated activa-tor of interferon) and TEAM (Toll receptor-associated mol-ecule), as well as recruitment of the Tank-binding proteinkinase-1 ((TBK-1), which leads eventually to the productionof IFN-a/b [55,56]. Synthesis of IFN-a/b would then up-regulate the expression of co-stimulatory molecules onantigen-presenting cells [57], thereby generating robust anti-gen-specific T and B cells [55,56]. Therefore, it is conceivablethat in SJL mice treated with mercury alone, antigen-specificT cells are activated primarily by dendritic cells, which con-stitutively express both MHC and co-stimulatory molecules;whereas in animals administered mercury in combinationwith LPS, other antigen-presenting cells such as macrophagesand B cells also express co-stimulatory molecules and arethus capable of activating T cells and thereby potentiating theimmune/autoimmune responses induced by mercury.
Considered together, our present findings reveal that acti-vation of the innate immune system plays a key role ininduction and exacerbation of mercury-induced autoimmu-nity. This conclusion supports the hypothesis that environ-mental risk factors, such as infections, can either initiate and/

M. Abedi-Valugerdi et al.
246 © 2005 British Society for Immunology, Clinical and Experimental Immunology, 141: 238–247
or accelerate autoimmune processes. Accordingly, we suggestthat simultaneous exposure to certain environmental factorscan cause individuals who are normally genetically or immu-nologically resistant to become susceptible to autoimmunediseases.
Acknowledgements
This study was financed by grants from Karolinska Institute’sResearch Foundations and the Rab Rashidi Institute for Bio-science in Tabriz, Iran. We would also like to thank CarlottaKuylenstierna, Monika Hansson and Mounira Djerbi fortheir skilful technical assistance in connection with thebeginning of this study.
References
1 Brickman CM, Shoenfeld Y. The mosaic of autoimmunity. Scand J
Clin Lab Invest Suppl 2001; 235:3–15.
2 Luppi P, Rossiello MR, Faas S, Trucco M. Genetic background and
environment contribute synergistically to the onset of autoim-
mune diseases. J Mol Med 1995; 73:381–93.
3 Mok CC, Lau CS. Pathogenesis of systemic lupus erythematosus. J
Clin Pathol 2003; 56:481–90.
4 Zandman-Goddard G, Shoenfeld Y. SLE and infections. Clin Rev
Allergy Immunol 2003; 25:29–40.
5 Adelman MK, Marchalonis JJ. Endogenous retroviruses in sys-
temic lupus erythematosus: candidate lupus viruses. Clin Immunol
2002; 102:107–16.
6 Antonov D, Kazandjieva J, Etugov D, Gospodinov D, Tsankov N.
Drug-induced lupus erythematosus. Clin Dermatol 2004; 22:157–
66.
7 Di Luzio NR, Friedmann TJ. Bacterial endotoxins in the environ-
ment [Letter]. Nature 1973; 244:49–51.
8 Anderson WB, Slawson RM, Mayfield CI. A review of drinking-
water-associated endotoxin, including potential routes of human
exposure. Can J Microbiol 2002; 48:567–87.
9 van Deventer SJ, Knepper A, Landsman J et al. Endotoxins in portal
blood. Hepatogastroenterology 1988; 35:223–5.
10 Heumann D, Roger T. Initial responses to endotoxins and Gram-
negative bacteria. Clin Chim Acta 2002; 323:59–72.
11 Triantafilou M, Triantafilou K. Lipopolysaccharide recognition:
CD14, TLRs and the LPS-activation cluster. Trends Immunol 2002;
23:301–4.
12 Kaisho T, Akira S. Toll-like receptors as adjuvant receptors. Bio-
chim Biophys Acta 2002; 1589:1–13.
13 Moller G. Receptors for innate pathogen defence in insects are nor-
mal activation receptors for specific immune responses in mam-
mals. Scand J Immunol 1999; 50:341–7.
14 Dziarski R. Preferential induction of autoantibody secretion in
polyclonal activation by peptidoglycan and lipopolysaccharide. I.
In vitro studies. J Immunol 1982; 128:1018–25.
15 Dziarski R. Preferential induction of autoantibody secretion in
polyclonal activation by peptidoglycan and lipopolysaccharide. II.
In vivo studies. J Immunol 1982; 128:1026–30.
16 Moller G. Lipopolysaccharide as a tool to reveal autoreactive B
cells. APMIS 1988; 96:93–100.
17 Barton GM, Medzhitov R. Control of adaptive immune responses
by Toll-like receptors. Curr Opin Immunol 2002; 14:380–3.
18 Dabbagh K, Lewis DB. Toll-like receptors and T-helper-1/T-helper-
2 responses. Curr Opin Infect Dis 2003; 16:199–204.
19 Charreire J. Immune mechanisms in autoimmune thyroiditis. Adv
Immunol 1989; 46:263–334.
20 Cavallo T, Granholm NA. Lipopolysaccharide from Gram-negative
bacteria enhances polyclonal B cell activation and exacerbates
nephritis in MRL/lpr mice. Clin Exp Immunol 1990; 82:515–21.
21 Granholm NA, Cavallo T. Long-lasting effects of bacterial
lipopolysaccharide promote progression of lupus nephritis in
NZB/W mice. Lupus 1994; 3:507–14.
22 Granholm NA, Cavallo T. Enhancement of renal disease in BXSB
lupus prone mice after prior exposure to bacterial lipopolysaccha-
ride. Lupus 1995; 4:339–47.
23 Sweet LI, Zelikoff JT. Toxicology and immunotoxicology of mer-
cury: a comparative review in fish and humans. J Toxicol Environ
Health B Crit Rev 2001; 4:161–205.
24 Moszczynski P. Mercury compounds and the immune system: a
review. Int J Occup Med Environ Health 1997; 10:247–58.
25 Bagenstose LM, Salgame P, Monestier M. Murine mercury-
induced autoimmunity: a model of chemically related autoimmu-
nity in humans. Immunol Res 1999; 20:67–78.
26 Griem P, Gleichmann E. Metal ion induced autoimmunity. Curr
Opin Immunol 1995; 7:831–8.
27 Enestrom S, Hultman P. Does amalgam affect the immune system?
A controversial issue. Int Arch Allergy Immunol 1995; 106:180–
203.
28 Borchers A, Ansari AA, Hsu T, Kono DH, Gershwin ME. The
pathogenesis of autoimmunity in New Zealand mice. Semin
Arthritis Rheum 2000; 29:385–99.
29 Izui S, Ibnou-Zekri N, Fossati-Jimack L, Iwamoto M. Lessons from
BXSB and related mouse models. Int Rev Immunol 2000; 19:447–
72.
30 Kotzin BL. Systemic lupus erythematosus. Cell 1996; 85:303–6.
31 Hultman P, Bell LJ, Enestrom S, Pollard KM. Murine susceptibility
to mercury. I. Autoantibody profiles and systemic immune depos-
its in inbred, congenic, and intra-H-2 recombinant strains. Clin
Immunol Immunopathol 1992; 65:98–109.
32 Hultman P, Bell LJ, Enestrom S, Pollard KM. Murine susceptibility
to mercury. II. autoantibody profiles and renal immune deposits in
hybrid, backcross, and H-2d congenic mice. Clin Immunol Immu-
nopathol 1993; 68:9–20.
33 Abedi-Valugerdi M, Moller G. Contribution of H-2 and non-H-2
genes in the control of mercury-induced autoimmunity. Int
Immunol 2000; 12:1425–30.
34 Abedi-Valugerdi M, Hansson M, Moller G. Genetic control of
resistance to mercury-induced immune/autoimmune activation.
Scand J Immunol 2001; 54:190–7.
35 Kono DH, Park MS, Szydlik A et al. Resistance to xenobiotic-
induced autoimmunity maps to chromosome 1. J Immunol 2001;
167:2396–403.
36 Gronowicz E, Coutinho A, Melchers F. A plaque assay for all cells
secreting Ig of a given type or class. Eur J Immunol 1976; 6:588–
90.
37 al-Balaghi S, Moller E, Moller G, Abedi-Valugerdi M. Mercury
induces polyclonal B cell activation, autoantibody production and
renal immune complex deposits in young (NZB ¥ NZW) F1
hybrids. Eur J Immunol 1996; 26:1519–26.
38 Hultman P, Johansson U, Dagnaes-Hansen F. Murine mercury-
induced autoimmunity. the role of T-helper cells. J Autoimmun
1995; 8:809–23.

Exacerbation of murine mercury-induced autoimmunity by LPS
© 2005 British Society for Immunology, Clinical and Experimental Immunology, 141: 238–247 247
39 Mathieson PW, Thiru S, Oliveira DB. Mercuric chloride-treated
brown Norway rats develop widespread tissue injury including
necrotizing vasculitis. Lab Invest 1992; 67:121–9.
40 Waldner H, Collins M, Kuchroo VK. Activation of antigen-
presenting cells by microbial products breaks self tolerance and
induces autoimmune disease. J Clin Invest 2004; 113:990–7.
41 Vogel SN, Hilfiker ML, Caulfield MJ. Endotoxin-induced T lym-
phocyte proliferation. J Immunol 1983; 130:1774–9.
42 Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M,
Demengeot J. Regulatory T cells selectively express toll-like recep-
tors and are activated by lipopolysaccharide. J Exp Med 2003;
197:403–11.
43 Tough DF, Sun S, Sprent J. T cell stimulation in vivo by
lipopolysaccharide (LPS). J Exp Med 1997; 185:2089–94.
44 Castro A, Bemer V, Nobrega A, Coutinho A, Truffa-Bachi P.
Administration to mouse of endotoxin from Gram-negative bac-
teria leads to activation and apoptosis of T lymphocytes. Eur J
Immunol 1998; 28:488–95.
45 Pape KA, Khoruts A, Mondino A, Jenkins MK. Inflammatory
cytokines enhance the in vivo clonal expansion and differentiation
of antigen-activated CD4+ T cells. J Immunol 1997; 159:591–8.
46 Jenkins MK, Khoruts A, Ingulli E et al. In vivo activation of
antigen-specific CD4 T cells. Annu Rev Immunol 2001; 19:23–45.
47 Dutton RW, Bradley LM, Swain SL. T cell memory. Annu Rev
Immunol 1998; 16:201–23.
48 Cerwenka A, Carter LL, Reome JB, Swain SL, Dutton RW. In vivo
persistence of CD8 polarized T cell subsets producing type 1 or
type 2 cytokines. J Immunol 1998; 161:97–105.
49 Pape KA, Merica R, Mondino A, Khoruts A, Jenkins MK. Direct
evidence that functionally impaired CD4+ T cells persist in vivo
following induction of peripheral tolerance. J Immunol 1998;
160:4719–29.
50 Perez VL, Van Parijs L, Biuckians A, Zheng XX, Strom TB, Abbas
AK. Induction of peripheral T cell tolerance in vivo requires CTLA-
4 engagement. Immunity 1997; 6:411–7.
51 Hamilton-Williams EE, Lang A, Benke D, Davey GM, Wiesmuller
KH, Kurts C. Cutting edge: TLR ligands are not sufficient to break
cross-tolerance to self-antigens. J Immunol 2005; 174:1159–63.
52 Biancone L, Andres G, Ahn H et al. Distinct regulatory roles of
lymphocyte costimulatory pathways on T helper type-2 mediated
autoimmune disease. J Exp Med 1996; 183:1473–81.
53 Bagenstose LM, Class R, Salgame P, Monestier M. B7-1 and B7-2
co-stimulatory molecules are required for mercury-induced
autoimmunity. Clin Exp Immunol 2002; 127:12–9.
54 Pollard KM, Arnush M, Hultman P, Kono DH. Costimulation
requirements of induced murine systemic autoimmune disease. J
Immunol 2004; 173:5880–7.
55 Hoebe K, Janssen E, Beutler B. The interface between innate and
adaptive immunity. Nat Immunol 2004; 5:971–4.
56 Hoebe K, Beutler B. LPS, dsRNA and the interferon bridge to adap-
tive immune responses: Trif, Tram, and other TIR adaptor pro-
teins. J Endotoxin Res 2004; 10:130–6.
57 Hoebe K, Janssen EM, Kim SO et al. Upregulation of costimulatory
molecules induced by lipopolysaccharide and double-stranded
RNA occurs by Trif-dependent and Trif-independent pathways.
Nat Immunol 2003; 4:1223–9.




















