
CHADS2 score predicts atrial fibrillation following cardiac surgery
Upload
independentCategory
view
0download
0
European Journal of Internal Medicine 20 (2009) 672–681
Contents lists available at ScienceDirect
European Journal of Internal Medicine
j ourna l homepage: www.e lsev ie r.com/ locate /e j im
Review article
Atrial fibrillation: Mechanistic insights and treatment options
Sunil T. Mathew a,⁎, Jigar Patel b, Satheesh Joseph c
a Cardiovascular Section, University of Oklahoma Health Sciences Center, University of Oklahoma School of Medicine, Oklahoma City, OK, USAb Department of Internal Medicine, University of Oklahoma Health Sciences Center, University of Oklahoma School of Medicine, Oklahoma City, OK, USAc Brookhaven Heart, 100 Hospital Rd., Suite 120, East Patchogue, NY 11772, USA
⁎ Corresponding author. Fax: +1 866 384 0496.E-mail address: [email protected] (S.T. Mathew
0953-6205/$ – see front matter © 2009 European Fededoi:10.1016/j.ejim.2009.07.011
a b s t r a c t
a r t i c l e i n f oArticle history:Received 19 December 2008Received in revised form 23 June 2009Accepted 22 July 2009Available online 27 September 2009
Keywords:ArrhythmiaAtrial fibrillationArrhythmia mechanismArrhythmia treatment
Atrial fibrillation (AF) remains the most common clinically encountered arrhythmia. Unlike supraventriculararrhythmias that use a defined mechanism, AF involves a wide spectrum of arrhythmias from lone AF toparoxysmal to chronic AF. AF is an arrhythmia that may develop in several ways. Mechanical remodelingmanifests as decreased atrial contractility and increased atrial compliance which leads to a stretch of theatrial myocardium. Atrial remodeling may also increase in atrial fibrosis which can slow conduction velocityand can shorten the refractory period in atria with long-standing AF. It is still unclear whether initiation of AFactivates direct inflammatory effects or whether the presence of a pre-existing systemic inflammatory statepromotes further persistence of AF.Currently, the patient population undergoing AF ablation has greatly expanded. Patients are older and havelarger left atrial size and are more likely to have persistent/permanent AF. It is likely that AF comprises aspectrum of disease with no single mechanism adequate enough to comprehensively explain AF and itsvariability. The management of patients with AF involves elements of anticoagulation, rate control andrhythm control and such treatment strategies are not necessarily mutually exclusive of each other.
© 2009 European Federation of Internal Medicine. Published by Elsevier B.V. All rights reserved.
1. Introduction
Atrial fibrillation (AF) is the most common clinically encounteredarrhythmia [1]. The arrhythmia is associated with heart failure, se-nescence, and numerous diseases related to senescence and has anincidence of more than 2.3 million in the United States. However, themechanisms underlying AF are only partially understood and recentepidemiologic data indicate that AF prevalence is not explained by agealone [2]. Furthermore, additional risk markers such as obesity [3,4],obstructive sleep apnea [5,6], anger and hostility [7], and long-termalcohol consumption [8–10] only partially account for the increase inAF. Multiple AF descriptors have developed in light of such wide-spread prevalence of the arrhythmia. Various descriptors such as loneAF, paroxysmal AF, and chronic AF had been previously used some-what indiscriminately. Recently however a consensus classificationscheme has been proposed to simplify its description with respect toits clinical relevance (Fig. 1).
Although significant progress in understanding the mechanism ofthis arrhythmia has been accomplished with the advent of catheterablation, the pathophysiology of AF is complex and likely has many
).
ration of Internal Medicine. Publish
possible mechanisms which may be interrelated. This paper reviewsthis clinically widespread arrhythmia. Both traditional and morecontemporary theories responsible for AF as well as standard andevolving treatment modalities are also reviewed herein.
2. Basic science insights on atrial fibrillation mechanism(s)
2.1. Rotors and wavelets
Several theories attempt to explain different aspects of suchmechanism(s), however a unifying theory remains lacking. Threemajor theories are often included in this constituency, including(a) the random reentrymodel; (b) the single or multiple rapidly firingectopic foci; and (c) the multiple reentrant model.
2.1.1. Random reentryMoe and Abildskov's hypothesis [11] of random reentry has been
one of the prevailing concepts. Its basis centers onmultiplewanderingatrial reentrant wavelets colliding with each other to either self-terminate or produce daughter wavelets which sustain atrial excita-tion. Theories describing the genesis of AF typically have originatedfrom animal models describing these random atrial reentry wavelets.Canine models have demonstrated that these AF pathways are vagallymediated and are determined by a critical amount of atrial refrac-toriness and excitability, not on atrial anatomy [11–14].
ed by Elsevier B.V. All rights reserved.

Fig. 1. Updated atrial fibrillation classification schema.(Adapted from Estes NA 3rd, Halperin JL, Calkins H, et al. J Am Coll Cardiol. 2008 Feb26;51(8):865–84.)
Fig. 2. Schematic illustrating the numerous “modulating factors” that influence andmaycontribute to the pathogenesis of atrial fibrillation.(Wyse DG, Gersh BJ. Atrial fibrillation: a perspective: thinking inside and outside thebox. Circulation 2004 Jun 29;109(25):3089–95.)
673S.T. Mathew et al. / European Journal of Internal Medicine 20 (2009) 672–681
2.1.2. Multiple fociSomewhat in contrast to the random reentry model, another
theory suggests single ormultiple rapidly firing ectopic foci. This is notas widely acknowledged, based in part from studies supportingverification of the multiple reentrant concept by Allessie et al. [15,16].
2.1.3. Multiple reentryThough multiple reentrant theory appeared to best explain
persistent AF [12,14], there still lacks a general inability of theseprincipal theories to fully describe AF and this further underscoresboth the difficulty in understanding this arrhythmia.
2.1.4. New dataContemporary investigations have only continued to add complex-
ity. For example, the initiation of AF from anatomic substrates in thearea around the pulmonary veins [17] question the random reentryhypothesis and have resurrected prior theories such as the singlesource of stable reentry. An attempt at understanding the variousputative mechanisms is required in order to adequately utilize someof the state-of-the-art therapies that are now available.
Jalife and colleagues, using optical mapping studies on an ovinemodel, demonstrated that a primary localized generator (or rotor)comprising either an ectopic focus or single small reentry circuit[18,19] in the atria is responsible for AF initiation. In this model, afunctional, anatomic reentry occurs with spiral waves rotating aroundreentrant circuits of about one centimeter diameter and is thought tobe the likely source of AF [20,21]. They have also shown that thesedominant rotors that drive AF consistently arise from the left atriumwith passive activation of the right atrium [18,19]. While such studieshave shown that in some cases the relative roles of focal reentry andabnormal automaticity in the pulmonary vein region may be morerelevant, it is still unclear whether significant rotors exist in humansand what their location may be. There is also ambiguity as to thedifferences between the apparently disparate concepts of multiplewavelet reentry and the primary rotor model: some have hypothe-sized that it is possible that they may have more similarities thandifferences [22,23].
2.2. Modulating factors
Other basic mechanisms include those that modulate arrhythmo-genesis [22] which often occur in conjunction with those mechanismsthat are thought to initiate or perpetuate AF. Thesemodulation factorscan further be described as potentially modifiable or non-modifiable(i.e. age or genetics). As discussed below, there are many modifiablemodulating factors that may maintain or inhibit AF. The majormodulating factors such as endocrine activity, pulmonary vein andatrial function, are typically more familiar to the practicing clinicianand will be described. Those less prevalent clinically, such as mechan-ical and electrical atrial remodeling and inflammatory processes willalso be reviewed, as well as the advanced concept of AFmodulation bythe autonomic nervous system. Wyse and Gersh have proposedschematics to illustrate the presumed relationship amongmodulationfactors to AF (Fig. 2) as well as how these relate to a trigger and/orsubstrate as a fundamental mechanism(s) thought to initiate andsustain AF (Fig. 3). Entities thought to be AF “triggers” are moreassociated with initiating AF and may be more predominant in themechanism of paroxysmal AF. Alternatively, those described as“substrates” are more likely associated with influencing persistent AF.
2.2.1. Mechanical and electrical atrial remodelingOur understanding of AF was broadened by the recognition that
once AF initiated, atrial electrophysiological properties changed and alower threshold induced the arrhythmia. This process explained AFpropagation by a process known as electrical remodeling [24]. Theprimary stimulus needed to induce electrical remodeling was anestimated tenfold increase in the atrial rate caused by AF [25]. Similarchanges were also produced by any form of sufficiently rapid atrialtachycardia [25].
The cellular basis for the observed changes from AF mediatedtachycardia is thought to be due to changes in calcium (Ca2+) loading.Ca2+ enters the cells through inward L-type Ca2+ current (ICa2+) andwith each action potential, there is a substantial increase in cellularCa2+ loading in the presence of the arrhythmia [26]. The ensuingadaptive responses that occur decrease Ca2+ loading by reducing ICa2+,but at the expense of a shorter action potential duration, whichincreases the propensity for atrial fibrillation [25,26]. These AF in-duced electrophysiological changes promote a vicious cycle of ar-rhythmia that enhances the ability of the disorder to sustain itself, andalso create an increasing vulnerability to relapse [24,27–29] (oftendescribed as “AF begets AF”) [24].

Fig. 3. Schematic illustrating how modulating factors relate to the various mechanismsthought to invoke reentry.AF initiation and perpetuation are illustrated as well as howAF can produce modulating factors that further perpetuate it or produce manifestationsof AF.(Wyse DG, Gersh BJ. Atrial fibrillation: a perspective: thinking inside and outside thebox. Circulation 2004 Jun 29;109(25):3089–95.)
674 S.T. Mathew et al. / European Journal of Internal Medicine 20 (2009) 672–681
Although it is well established that the perpetuation of AF causeselectrical remodeling, AF's links to contractile and structural remodel-ing remain controversial. Mechanical remodeling is thought to com-prise both contractile and structural remodeling of the atria as a resultof AF. Whereas electrical and contractile atrial remodeling may bereversible after the cessation of AF, structural remodeling is less likelyto revert [22,30]. Electrical remodeling and constant changes in theL-type Ca2+ channels are both thought to be the primary cause forcontractile remodeling. Mechanical remodeling manifests as de-creased atrial contractility and increased atrial compliance whichcauses the atrial myocardium to stretch. Such decreased atrialcontractility acts as a stimulus for structural remodeling due to theremodeled enlarged atria. This abnormal atrium now becomes anelectro-anatomical substrate for AF by allowing more intra-atrialcircuits to potentially perpetuate due to a reduction in wavelength(shortening of refractoriness and slowing of conduction) and anincrease in non-uniform atrial tissue [30]. Thus, the remodeled atriawith its enlarged chambers act as AF substrate as it promotes the
Fig. 4. Illustration of the potential interplay of electrical, contractile and structuralfeedback loops of atrial remodeling on AF. Increased atrial myocardial stretch ishypothesized to act as a stimulus for structural remodeling of the atria. The resultingenlarged atria allow intra-atrial circuits of small size, due to a reduction in wavelength(shortening of refractoriness and slowing of conduction) and increased non-uniformtissue anisotropy (zigzag conduction).(Allessie M, Ausma J, Schotten U. Electrical, contractile and structural remodelingduring atrial fibrillation. Cardiovasc Res 2002 May;54(2):230–46.)
development of smaller sized intra-atrial circuits [30]. The size of theatrial reentry circuits depends on the circuit's wavelength: thosewith a short wavelength (or smaller atrial circuits) will maintainseveral reentry circuits and thereby promote the continuation of AF(Fig. 4) [28,30]. Larger atria are more likely than smaller atria topromote smaller intra-atrial circuits [30].
2.2.2. Inflammation/fibrosisAtrial remodelingmayalso increase in atrialfibrosis. This process can
slow conduction velocity and may change the atrial refractory periodwith long-standing AF [31,32]. It is still unclear whether initiation of AFactivates direct inflammatory effects or whether the presence of a pre-existing systemic inflammatory state promotes further persistenceof AF[33]. One of the first studies comparing inflammatory markers to atrialremodeling was reported by Frustaci et al. [34]. Nakamura et al. [35]confirmed this association by showing that atrial biopsies from patientswith lone AF had higher amounts of atrial fibrosis, inflammatoryinfiltrates, andmyocyte necrosis compared to those with sinus rhythm.Canine models with sustained AF demonstrated active atrial perimyo-carditis with inflammatory infiltrates, lipid degeneration, and fibrosis[36]. Furthermore, not only have increased C-reactive protein (CRP)levels been demonstrated as early as within the first 24-hours of theonset of AF [37], patients with persistent AF have also shown markedlyincreased CRP levels when compared to patients with paroxysmal AF(independent of other risk factors) [38]. This suggests that inflammationmay also promote the persistence of AF.
Corollaries to these observations came from a positive effect ofcorticosteroid treatment in a randomized study of 216 patients un-dergoing coronary or valvular heart surgery [39]. The study demon-strated that a single dose of dexamethasone was associated with adecreased incidence of new-onset AF in the first 3 days post-surgery.Recently, a similar multi-centered randomized study corroboratedthis by showing that the use of intravenous hydrocortisone reducedthe incidence of AF after cardiac surgery among 241 consecutivepatients without prior AF or flutter [40]. The pleiotropic effects ofHMG-CoA reductase inhibitors have also been shown in studies which
Fig. 5. Illustration of the possible role of oxidative stress and inflammation contributingto electrical remodeling in AF. [ICaL — L-type Ca2+ current; APD — action potentialduration; AERP — atrial effective refractory period; CV — conduction velocity.](Korantzopoulos P, Kolettis T, Siogas K, Goudevenos J. Atrial fibrillation and electricalremodeling: the potential role of inflammation and oxidative stress. Med Sci Monit2003;9(9):RA225-9.)

675S.T. Mathew et al. / European Journal of Internal Medicine 20 (2009) 672–681
investigated the association between statin use and incidence of AF.The most compelling data come from Kumagai et al. [36] whichutilized atorvastatin on AF in a canine pericarditis model. The ator-vastatin group had lower C-reactive protein levels, less pronouncedfibrosis in the atrial myocardium, and a shorter duration of AF. Fibrosisand inflammation are thought to change the anatomical andelectrophysiological substrate of the atria [28] and it is thought thatthe interplay of inflammation may not only be a response to AF butalso an integral part of it [41]. Inflammation and fibrosis in AF meritsfurther study (Fig. 5) [42] since recovery from electrical remodelingby conventional pharmacotherapy remains inadequate.
2.2.3. Endocrine activityUsing a multivariable model based on data from the Framingham
Heart Study (with adjustment for age and other AF risk factors), theodds ratio of AFwith diabetes was 1.4 formen and 1.6 for women [43].Kato et al. hypothesized that endocrine mediated disorders such asdiabetes mellitus may promote the development of atrial fibrosisleading to a predisposition to AF. Using a rat model, they found thatstructural remodeling of the atrium characterized by diffuse inter-stitial fibrosis is amajor substrate for diabetes related AF [44]. Anotherwell-known clinical endocrine modulating factor is the association ofAF with hyperthyroidism whereby changes in thyroid hormoneconcentration directly affects the initiation or inhibition of AF [22].
There is also growing evidence to suggest a role for the renin–angiotensin system (RAS) in the pathogenesis of AF. Inhibition of RASwith captopril or candesartan, yields a reduction in atrial tachycardiaassociated electrical remodeling in dogs subjected to rapid atrialpacing for a relatively short duration. Among dogs subjected to pacingfor a longer duration (i.e. 1–5 weeks), RAS inhibition did not appear toalter atrial remodeling during the relatively early stages of electricalremodeling [45,46]. The role of RAS in atrial remodeling to sustain AFin humans is less clear. Angiotensin II infusion in human subjects hasnot been shown to shorten the atrial refractory period [42] and maynot have a significant acute effect on human atrial electrical re-modeling. It may, however, facilitate the role of structural remodelingand fibrosis of atrial tissue, which suggests a link between RAS andremodeling of atrial tissue [42]. This may be consistent with clinicaldata demonstrating the effectiveness of a RAS blockade in preventingnew-onset or recurrent AF among those with hypertension and leftventricular hypertrophy, congestive heart failure, and those under-going electrical cardioversion for AF [47,48]. However, more clinicaland experimental animal studies are needed in order to define therole of RAS inhibition in patients with AF.
2.2.4. Pulmonary veins and atrial functionHaissaguerre et al. demonstrated that premature depolarizations
originating from the pulmonary veins could contribute to the gen-eration of AF [17]. They showed that the pulmonary vein foci mayinitiate AF by originating as a rapid atrial tachycardia. Subsequentstudies have shown that AF originating from pulmonary vein foci maythen degenerate as a consequence of atrial remodeling which acts tomaintain AF by promoting multiple reentry circuits [49,50]. Otherstudies have also shown this activity and implicate the pulmonaryveins in both the initiation as well as maintenance of AF [51,52].
The pulmonary vein-left atrial junction has also become a targetfor morphological studies in both animal and human hearts indescribing AF arrhythmogenicity. The multilayer left atrial myocar-dium extend in a variable arrangement and distance into each PVresulting in a complex three-dimensional fiber orientation [53,54]].These pulmonary vein-left atrial muscle sleeves consist of specializedconduction cells similar to those found in the sinus node [55]. Thetransition zones of these myocardial extensions were thickest at thevenoatrial junction (mean 1.1 mm) and became thinner movingdistally [56]. Their thickness is variable with the inferior walls of thesuperior veins. In contrast, the superior walls of the inferior veins have
a thicker myocardial extension [55]]. Such observations from the areaof the pulmonary veins with the passive activation of the right atrium[18,19] have lessened the dominance of the multiple wavelethypothesis and renewed interest in concepts of fibrillatory conductionand a dominant rotor as primary drivers of AF [57]. These observationshave provided further pathophysiological insight and have alsohelped expand therapy options for current cutting edge pulmonaryvein ablation procedures.
2.2.5. AutonomicsThe anatomy of the sympathetic and parasympathetic innervation of
the sinus and AV nodes is intricate and each component of the autonomicnervous system follows a different course [58]. Parasympathetic neuronsextend to the heart through the vagus nerves and the ones destined toinnervate the sinus node terminate in the pulmonary vein fat pad,whereas those that predominantly innervate theAVnode terminate in theinferior vena cava–inferior left atrial fat pad [59]]. Parasympatheticdenervation by surgical dissection of the fat pads does not influencesympathetic control of sinus rate and AV-nodal conduction [57]].
The electrophysiologic properties of atrial cells are moderateddifferently by parasympathetic and sympathetic influences. Forexample, vagal mediated influences favor atrial macroreentry phe-nomena and sympathetic influences favor abnormal automaticity andtriggered activity [60]. In normal hearts, vagal influences predomi-nate, which partially explains why the clinical pattern of vagallymediated paroxysmal AF is often found in young males with nodetectable heart disease. These patients usually have an ECG pattern ofatrial flutter alternatingwithfibrillation [61]. The vagal effect is to shortenboth the action potential and refractory period. This is recognized to occurheterogeneously within the atrial wall, suggesting that such inhomo-geneity may contribute to a reentry mechanism rather than hyperexcit-ability [62]]. Conversely, AFmediated by the sympathetic nervous systemis observed in the presence of most any heart disease, of which the firsteffect is often to provoke a vagal withdrawal [59]].
Although studies have demonstrated that the rate of arrhythmo-genicity near the pulmonary vein can be greatly accelerated byadrenergic stimulation [63], little is known about the impact of base-line autonomic tone and the development of new-onset AF in thegeneral population. Despite epidemiological evidence from theFramingham Heart Study subjects suggesting that baseline autonomicdysregulation reflected by altered heart rate variability (HRV) isassociated with risk of AF, the association did not persist afteradjusting for potential confounders. Much of the apparent associationbetween HRV and AF in the Framingham cohort was believed to bemediated by traditional risk factors [64].
Tomita et al. [65] demonstrated a relationship between the time ofparoxysmal AF onset and autonomic tone both before and afterparoxysmal AF and found that the autonomic nervous system plays animportant role in both the initiation and termination of paroxysmalAF. They also found that the onset of paroxysmal AF influences theautonomic tone at the initiation as well as during termination ofparoxysmal AF [65]. However, the role of autonomic tone in theinitiation of paroxysmal atrial flutter has not been reported.
These epidemiologic and clinical observations between AF and theautonomic nervous system gain further credibility with basic scienceassociations. For example, the novel Class III antiarrhythmic agent,NIP-142, shows a link between the autonomic impact on AF in a caninemodel of atrial fibrillation and flutter. As a primary potassium channelblocker, NIP-142 prolonged atrial refractoriness and was effective intreating atrial flutter and vagally-induced AF [66]. Such studies areimportant as they add insight into the autonomic component of thearrhythmia. This understanding forms the basis for some of theemerging therapies aimed at targeting the autonomic nervous systemfor the treatment of refractory AF. Coumel has suggested that in order toconfirm vagally derived initiation of AF, the minutes and hours pre-ceding AF onset need to be recorded in order to demonstrate the

676 S.T. Mathew et al. / European Journal of Internal Medicine 20 (2009) 672–681
relationship between arrhythmia onset and heart rate changes [60]. Thewell-known mechanism of ectopic foci located in the left atrium or thepulmonary veins [17] is compatible with vagal or adrenergic profiles.Such ectopic foci may form the arrhythmia trigger and vagally derivedarrhythmias are often sensitive to focal ablation [67].
3. Clinical science and treatment issues
Although there has been much insight regarding the pathophysiolo-gical triggers of AF, an understanding of itsmechanisms is still inadequate[56]. This manifests by less than optimal treatment of this arrhythmiadespite the development of novel catheter-based ablation therapy andpharmacotherapy. Though these techniquesmay in time lead to a cure forthe disorder in some patients, focal atrial ablation or pulmonary veinisolation is not currently universally effective [68]]. Currently, twofundamental strategies dominate treatment options: (i) ventricular ratecontrol; and (ii) restoration of sinus rhythm to obtain rhythm control.
3.1. Treatment insights from clinical trials: rate or rhythm control
AF management involves three facets: rate control; prevention ofthromboembolism; and the restoration of sinus rhythm. The dataavailable on the clinical relationship between sinus node dysfunctionand AF suggest that the sinoatrial (SA) node is probably passive duringAF. Diseases often affect both the SA node and atria simultaneouslywhich can contribute to persistent AF. This is different from instanceswhere abnormal SA node dysfunction contributes to AF maintenance[28]. Theoretically, the basis for restoration of normal sinus rhythmwasbelieved to have several benefits over rate control. These potentialadvantages include improved cardiac output from the restoration ofatrial systolic function which yields improvement in quality of life;greater exercise capacity; lower risk of stroke; thepossibility of stoppinganticoagulation with maintenance of sinus rhythm; and the theoreticalbenefits of potentially reversingatrial structural or electrical remodeling[28]. Even in light of these potential advantages, it appears that rhythmcontrol is nearly equivocal to rate control for mortality benefit.
Several trials have analyzed the restoration of normal sinus rhythmand demonstrated that not only did rhythm control offer no addedmorbidity and mortality benefit, the quality of life measures did notdiffer significantly during follow-up. Such trials included RACE (RAteControl versus Electrical conversion) [69], AFFIRM (Atrial FibrillationFollow-up of Rhythm Management) [70], PIAF (PharmacologicalIntervention in Atrial Fibrillation) [71], STAF (Strategies of Treatmentof Atrial Fibrillation) [72], and HOT-CAFE (How to Treat Chronic AtrialFibrillation) [73] (the study parameters and key findings aresummarized in Table 1). In summary, these studies showed that arate-control strategy was at least as effective as rhythm control andthat antithrombotic therapy is important evenwhen a rhythm-controlstrategy is employed. Though an important caveat to this was that thetrials were problematic because of the inability to maintain sinusrhythm in a large percentage of the sinus-rhythm arm suggesting thatthe antiarrythmics used proved essentially inadequate for the job andno superiority of sinus control could be demonstrated.
Regardless of how the rate- or rhythm-control strategy is dealtwith, attention must also be directed to antithrombotic therapy forprevention of thromboembolism [74]]. In a meta-analysis, the AtrialFibrillation Investigators [75] reported that warfarin reduced the riskof death by 33% and that warfarin is the only pharmacologic therapythat has been reported to improve survival in AF.
Many patients with AF who are thought to be low risk for AFassociated stroke do not substantially benefit from anticoagulation.These patients can be reliably identified using the CHADS2 stroke riskstratification scheme (an acronym for Congestive heart failure,Hypertension, Age≥75 years, Diabetes and prior Stroke/or transientischemic attack (with S2 to indicate strokewith a weight of 2 points)).Ischemic stroke risk is estimated with the CHADS2 score (one point
for each disease state and 2 points for prior strokes or transientischemic attacks) [69] (see Tables 2 and 3). Anticoagulation withwarfarin is recommended for patients with valvular atrial fibrillationor a CHADS2 score≥2 (INR 2–3 and 2.5–3.5 for patients with me-chanical valves), unless contraindicated or there is an annual majorbleeding risk exceeds 3% [76]. Aspirin or warfarin may be used whenthe CHADS2 score=1. Aspirin, 81–325 mg daily, is recommended inpatients with a CHADS2 score of 0, or if warfarin is contraindicated.Current recommendations include continued anticoagulation withwarfarin in patients with sufficient thromboembolic risk whetherthey have persistent or paroxysmal atrial fibrillation [74,77].
Differing patient selection criteria, endpoints and therapeutic inter-ventions inmanyof theabove studies limit theapplicabilityof thefindingsto all AF populations. For example, AFFIRM, RACE and HOT-CAFE did notevaluate a single drug or type of treatment. Another criticism was thatthese trials were not comparisons of sinus rhythm and AF, but rathercomparisonsof thestrategyof rate control to thatof rhythmcontrolwherethe success rate of the rhythm-control strategy was relatively poor [78].Up to 35% of rhythm-control patients in AFFIRMwere actually using rate-control drugs by the end of the study. In subanalyses,manypatients in therate-control arm were spontaneously in sinus rhythm by the end of thestudy period. The same number was 10% in STAF and RACE to 35% inAFFIRM which suggests that by switching analysis according to thepatient's actual rhythm, the benefit of sinus rhythm over AF becomesobvious [77]]. This is consistent with population-based studies such asSOLVD, DIAMOND and CHF-STAT which have long shown the negativeprognostic impact of AF on survival [79]. With the exception of the PIAFtrial, antithrombotic therapywasdiscontinued in somepatientswhowereassigned to the rhythm-control groups. Withholding antithrombotictherapy is one possible explanation for the higher rate of non-cardiovascular mortality observed in the rhythm-control arm of AFFIRM.The majority of strokes in RACE occurred while receiving inadequateanticoagulation therapy. In RACE, 21 of the 35 thromboemboliccomplications occurred under rhythm control. Six patients had theevent after cessation of oral anticoagulant therapy, with five of them inapparent sinus rhythm. Asymptomatic episodes of AF may add tocontinued stroke risk in rhythm-control patients who are believed to bein persistent sinus rhythm [80]]. Such thromboembolic complicationsdespite apparent sinus rhythm contribute to the benefits of warfarin andits ability to reduce the risk of death by a third [75,81]].
Younger patients who have greater cumulative risk for developingsequelae of chamber remodelingwere underrepresented in these trials.Themeanagewas70 years inAFFIRMand68 years in the RACE study. Inexcluding the younger subgroup, the results of these studiesmay not berepresentative for the full spectrum of AF patients. This youngersymptomatic subgroup is thought to represent at least one third of all AFpatients [82]. With AFFIRM, there was a trend toward a benefit ofrhythm control for patients younger than 65 years which indicated animproved outcome in cases of maintained sinus rhythm [70]. In PIAF,where the mean age was slightly younger (60–61 years), exercisetolerance was better with a rhythm-control strategy. A follow-upanalysis of outcomes in AFFIRM [79] showed that the presence of sinusrhythm was associated with reduced mortality (47% reduction, 99% CI28–61; p≤0.0001). The benefits of sinus-rhythm maintenance overtime have yet to be carefully evaluated [83] and the optimum restingheart rate or heart rate during exercise remains undefined for patientswith permanent AF despite retrospective nonrandomized data ofAFFIRM and the AFFIRM-RACE cooperation [84–87].
4. Additional management options
While the cornerstone of long-term AF management is usuallyanticoagulation with rate and/or rhythm control, there are severaladditional treatment options. The following section will touch onmany of the major management options available and describe someof the benefits and disadvantages of each.
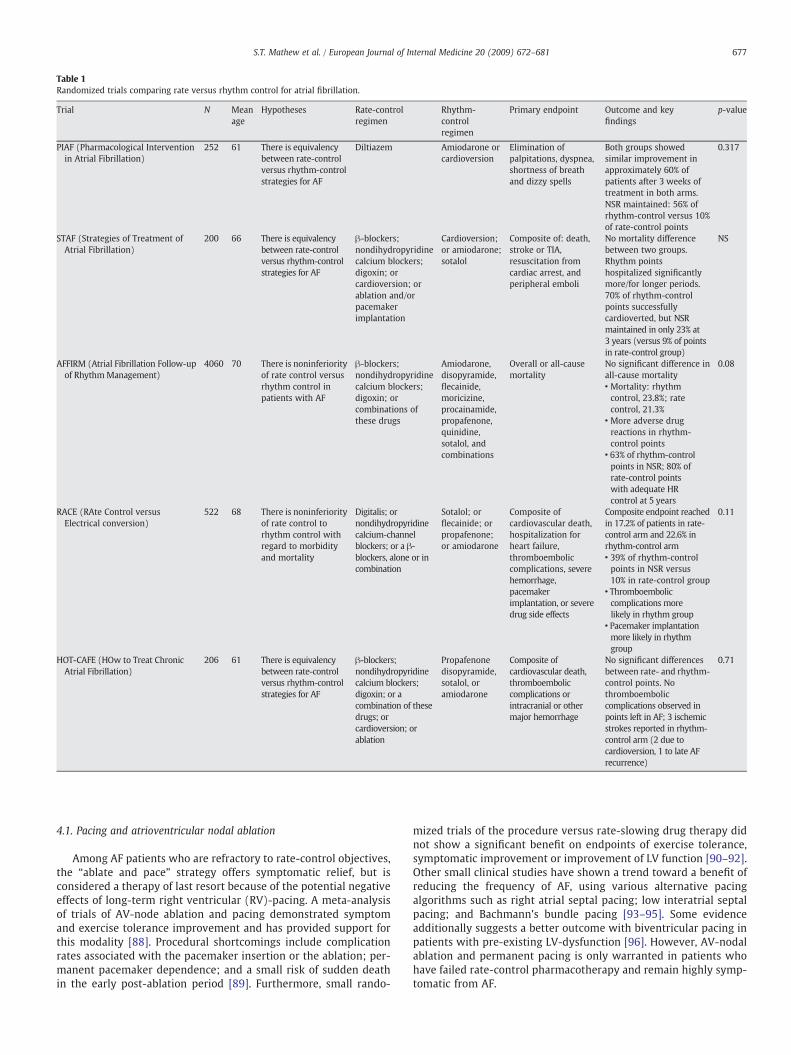
Table 1Randomized trials comparing rate versus rhythm control for atrial fibrillation.
Trial N Meanage
Hypotheses Rate-controlregimen
Rhythm-controlregimen
Primary endpoint Outcome and keyfindings
p-value
PIAF (Pharmacological Interventionin Atrial Fibrillation)
252 61 There is equivalencybetween rate-controlversus rhythm-controlstrategies for AF
Diltiazem Amiodarone orcardioversion
Elimination ofpalpitations, dyspnea,shortness of breathand dizzy spells
Both groups showedsimilar improvement inapproximately 60% ofpatients after 3 weeks oftreatment in both arms.NSR maintained: 56% ofrhythm-control versus 10%of rate-control points
0.317
STAF (Strategies of Treatment ofAtrial Fibrillation)
200 66 There is equivalencybetween rate-controlversus rhythm-controlstrategies for AF
β-blockers;nondihydropyridinecalcium blockers;digoxin; orcardioversion; orablation and/orpacemakerimplantation
Cardioversion;or amiodarone;sotalol
Composite of: death,stroke or TIA,resuscitation fromcardiac arrest, andperipheral emboli
No mortality differencebetween two groups.Rhythm pointshospitalized significantlymore/for longer periods.70% of rhythm-controlpoints successfullycardioverted, but NSRmaintained in only 23% at3 years (versus 9% of pointsin rate-control group)
NS
AFFIRM (Atrial Fibrillation Follow-upof RhythmManagement)
4060 70 There is noninferiorityof rate control versusrhythm control inpatients with AF
β-blockers;nondihydropyridinecalcium blockers;digoxin; orcombinations ofthese drugs
Amiodarone,disopyramide,flecainide,moricizine,procainamide,propafenone,quinidine,sotalol, andcombinations
Overall or all-causemortality
No significant difference inall-cause mortality
0.08
• Mortality: rhythmcontrol, 23.8%; ratecontrol, 21.3%
• More adverse drugreactions in rhythm-control points
• 63% of rhythm-controlpoints in NSR; 80% ofrate-control pointswith adequate HRcontrol at 5 years
RACE (RAte Control versusElectrical conversion)
522 68 There is noninferiorityof rate control torhythm control withregard to morbidityand mortality
Digitalis; ornondihydropyridinecalcium-channelblockers; or a β-blockers, alone or incombination
Sotalol; orflecainide; orpropafenone;or amiodarone
Composite ofcardiovascular death,hospitalization forheart failure,thromboemboliccomplications, severehemorrhage,pacemakerimplantation, or severedrug side effects
Composite endpoint reachedin 17.2% of patients in rate-control arm and 22.6% inrhythm-control arm
0.11
• 39% of rhythm-controlpoints in NSR versus10% in rate-control group
• Thromboemboliccomplications morelikely in rhythm group
• Pacemaker implantationmore likely in rhythmgroup
HOT-CAFE (HOw to Treat ChronicAtrial Fibrillation)
206 61 There is equivalencybetween rate-controlversus rhythm-controlstrategies for AF
β-blockers;nondihydropyridinecalcium blockers;digoxin; or acombination of thesedrugs; orcardioversion; orablation
Propafenonedisopyramide,sotalol, oramiodarone
Composite ofcardiovascular death,thromboemboliccomplications orintracranial or othermajor hemorrhage
No significant differencesbetween rate- and rhythm-control points. Nothromboemboliccomplications observed inpoints left in AF; 3 ischemicstrokes reported in rhythm-control arm (2 due tocardioversion, 1 to late AFrecurrence)
0.71
677S.T. Mathew et al. / European Journal of Internal Medicine 20 (2009) 672–681
4.1. Pacing and atrioventricular nodal ablation
Among AF patients who are refractory to rate-control objectives,the “ablate and pace” strategy offers symptomatic relief, but isconsidered a therapy of last resort because of the potential negativeeffects of long-term right ventricular (RV)-pacing. A meta-analysisof trials of AV-node ablation and pacing demonstrated symptomand exercise tolerance improvement and has provided support forthis modality [88]. Procedural shortcomings include complicationrates associated with the pacemaker insertion or the ablation; per-manent pacemaker dependence; and a small risk of sudden deathin the early post-ablation period [89]. Furthermore, small rando-
mized trials of the procedure versus rate-slowing drug therapy didnot show a significant benefit on endpoints of exercise tolerance,symptomatic improvement or improvement of LV function [90–92].Other small clinical studies have shown a trend toward a benefit ofreducing the frequency of AF, using various alternative pacingalgorithms such as right atrial septal pacing; low interatrial septalpacing; and Bachmann's bundle pacing [93–95]. Some evidenceadditionally suggests a better outcome with biventricular pacing inpatients with pre-existing LV-dysfunction [96]. However, AV-nodalablation and permanent pacing is only warranted in patients whohave failed rate-control pharmacotherapy and remain highly symp-tomatic from AF.

Table 2CHADS2 scoring system for AF.From: Gage, BF, Waterman, AD, Shannon, W, Boechler M, Rich MW, Radford MJ.Validation of clinical classification schemes for predicting stroke: results from theNational Registry of Atrial Fibrillation. JAMA 2001;285:2864–2870.
Risk factor CHADS2 score (points)
Congestive heart failure (EFb35%) 1Hypertension (systolicN160 mm Hg) 1Age greater than 75 years 1Diabetes 1Stroke (prior cerebral ischemia: CVA or TIA) 2
CHADS2 is an acronym derived from the stated risk factors and the scoring of 2 for priorstroke or TIA. The CHADS2 score is calculated by totaling the scores for each risk factorpresent.Abbreviations: CHF, congestive heart failure; EF, ejection fraction; TIA, transient ischemicattack.
678 S.T. Mathew et al. / European Journal of Internal Medicine 20 (2009) 672–681
4.2. Surgical intervention
The goal of AF surgical management is to make atrial incisions atcritical locations that create barriers to conduction and preventpersistent AF preserving sinus node and atrial transport functions[71]. The surgical “maze” or “cut-and-sew” technique has evolvedtremendously since its inception over 25 years ago. The objective is toensure that these transmural incisions isolate the pulmonary veins,while also connecting these incisions to additional surgical incisionsaround the mitral valve annulus, in order to create electrical barrierswithin the atria to prevent macroentrant rhythms—atrial flutter or AF—frombecomingpersistent [97]. Success rates [98] from70% tonearly 95%over 15 years of follow-up have been reported in patients undergoingmitral valve surgery [99]. Despite its reasonably high success rate, thesurgical maze is not routinely performed other than for patientsundergoing cardiac surgery because of the requirement of cardiopul-monary bypass. Several less invasive variations are under investigation,including thoracoscopic and catheter-based epicardial techniques [93].If the efficacy and safety of these surgical adaptations approach that ofthe percutaneous ablative techniques, they may become acceptablealternatives for a larger proportion of patients with AF.
4.3. Percutaneous intervention
Inmost AF patients (94%), the AF focuswas in≥1 of the pulmonaryveins (PVs) which was a landmark observation by Haissaguerre et al.[17]. Extra-PV sites may trigger AF, but is thought to occur in only aminority of patients: no more than 6% to 10% [46]. This facilitatedrecognition of the pathophysiological role played by these “musclesleeves”; or extensions of myocardium within the PV in the initiationof AF. Dominant rotors of AF are localized primarily in the junctionbetween the left atrium (LA) and PVs, have been demonstrated byseveral subsequent investigations [77,100–102]. A recent study hasalso demonstrated that the PV-LA region has heterogeneous electro-physiological properties capable of sustaining reentry (micro ormacro) [103]. Since PV isolation was first described for treating AF in
Table 3Risk of stroke by CHADS2 score.From: Gage, BF, Waterman, AD, Shannon, W, Boechler M, Rich MW, Radford MJ.Validation of clinical classification schemes for predicting stroke: results from theNational Registry of Atrial Fibrillation. JAMA 2001;285:2864–2870.
CHADS2 score Adjusted stroke rate (per 100 person-years)
0 1.91 2.82 4.03 5.94 8.55 12.56 18.2
1998, percutaneous procedures have evolved to replicate andimprove on surgical maze techniques. The former is done by makingcontinuous endocardial lesions to encircle the pulmonary veins withthe goal of contemporary AF ablation to electrically “disconnect” thePVs from the rest of the atrium by ablating around the origin of theveins. These percutaneous procedures are often considered forpatients who have failed antiarrhythmic drug therapy. The current,patient population undergoing AF ablation has expanded significantly.Patients are older and have larger left atrial size and are more likely tohave persistent AF often having been subjected to fewer priorantiarrhythmic agents [104].
Although PV isolation results in a long-term clinically satisfactoryoutcome in an estimated 60–85% [95,105–108] of patients withparoxysmal AF, it is clearly not the ideal approach in all AF patients[98,109]. Histological studies have shown that complex atrialanatomy has marked variations in atrial wall thickness [110,111].Adjacent structures of the atrial wall may vary in depth by severalmillimeters, so applying the same degree of energy at two contiguousspots can result in excessive heating in one spot and non-transmuralheating in the other. Consequently, complete conduction block viaradiofrequency application may not always be achieved [112].Potential complications of catheter ablation for AF include systemicthromboembolism, PV stenosis, pericardial effusion, cardiac tampo-nade, and phrenic nerve paralysis [113–115]]. Packer et al. [116] havesuggested that although the short-term safety of newer ablationtechniques has improved, several serious complications remain areality with long-term safety and ablation efficacy rates are still notclearly defined. Finally, unavoidable procedure-related complicationsmay occur and general skepticism tends to arise which furtherhighlight the need for the development of less complex therapies.
4.4. Targeting ganglionated plexi
Coumel first described adrenergically and vagally derived AF inpatients [60]. Patterson et al. noted that combined parasympatheticand sympathetic nerve stimulation modulate firing in caninepulmonary veins [117]. The human heart contains a variety ofmorphologically distinct nerve terminals concentrated at the gang-lionated plexi (GP). They are distributed more widely than has beendescribed in experimental animals [118]. Other similar arrhythmo-genic foci which may trigger AF have been found in the superior venacava, the right and left atria, the coronary sinus, and the ligament ofMarshall [50]. Focal atrial ablation has a low success rate in patientswith persistent AF and particularly in patients with the vagotonic typeof paroxysmal AF. An approach that involves ablation within the leftatrium is likely to yield better results in these subgroups, as well asthose subgroups with paroxysmal AF that do not respond to focalatrial ablation [81,102]. Multivariate analysis indicated that therewere only two independent predictors of recurrent AF afterpulmonary vein isolation: persistent AF at the time of pulmonaryvein isolation and the vagotonic variety of paroxysmal AF. Theseresults tend to show that the pulmonary veins play a less importantrole in the generation of persistent/permanent AF and in the initiationor maintenance of vagotonic paroxysmal AF than in paroxysmal AFthat is either random or adrenergic in onset [107]. Little is knownabout the impact of baseline autonomic tone and the development ofnew-onset AF in a population-based cohort. Inferences from recentreports have been that stimulation of the GP situated at the PV-atrialjunctions can convert PV focal firing into AF [119] or initiateparoxysmal AF and can provide a basis for potential therapy [120].Radiofrequency ablation of the GP can also prevent AF inducibility[121]. The role of the intrinsic cardiac autonomic nervous system inthe genesis and perpetuation of AF was further emphasized by severalrecent clinical reports. This approach centered on ablation of the GPlocated in proximity to the PV-atrial junction and resulted in a highsuccess rate in eliminating symptomatic AF of the vagotonic variety

679S.T. Mathew et al. / European Journal of Internal Medicine 20 (2009) 672–681
[122–124]. The involvement of local cardiac autonomic elementsrequires further investigation and it remains to be determinedwhether targeting the GPs clinically will result in significantly higherAF ablation success rates.
4.5. Pharmacologic therapy
Pharmacotherapy remains an essential component in the manage-ment of AF since invasive procedures are currently not globallyefficacious nor can they be performed on all patients with AF.Available antiarrhythmic drugs are only modestly effective insuppressing AF. Drug therapy leading to sinus-rhythm maintenancemay be a marker of improved outcome or less serious disease. Or itmay indicate that, while the ability of treatment to maintain sinusrhythm is inherently beneficial, such benefit may come at the expenseof toxic effects and poor efficacy [27]. It is uncommon for patients tohave complete suppression of AF. Successful therapy is often gaugedby reduction in frequency severity and duration of episodes.
Most antiarrhythmic drugs used for AF were never designed totarget the atria but rather developed for ventricular arrhythmiaswhich partially explains their toxicity limitations predominantlyrelated to prolonged QT. Recently, a meta-analysis of over 11,000patients reported that Class IA, IC, and III drugs are effective inmaintaining sinus rhythm but also reported an increase in adverseeffects, as well as an increase in mortality with Class IA drugs [125].Specifically, several Class IA (disopyramide phosphate, quinidinesulfate), Class IC (flecainide acetate, propafenone hydrochloride), andClass III (amiodarone, dofetilide, sotalol hydrochloride) drugs sig-nificantly reduced recurrence of atrial fibrillation, but they also hadassociated increased withdrawals due to adverse effects. Pooledanalysis of Class IA drugs were associated with increased mortalitycompared with controls and with the exception of amiodarone andpropafenone, each showed an increased propensity toward arrhythmia[125]. Investigational drugs with potentially improved efficacy andsafety profiles are under development. Some of these novel agents arenon-selective ion channel blocking drugs or analogs to amiodarone.Other such experimental drugs are designed to target specific ionchannels to provide atrial antiarrhythmic effects without ventriculararrhythmias by preferential action on the atria (inhibition of the ITOchannel) or via selective action on the atria (inhibition of IKUR currents)[126]. Combined pharmacologic and nonpharmacologic strategies mayultimately replace current approaches to the treatment of AF.
5. Conclusion
Unlike other supraventricular arrhythmias, AF ismore complex. AF isan arrhythmia that may develop in several ways but our basic insightinto its mechanism remains too limited to allow reliable classification.Mechanisms operating in AF derived from different animal andexperimental models are often different in genesis and perpetuationfrom clinical models involving humans [127]. It is likely that AFcomprises a spectrum of diseases with no single mechanism adequateenough to comprehensively explain the variability of AF. A unifyingconclusion may be that the AF mechanism is multifactorial and wecannot be certain about such mechanism of AF in most individualpatients. Despite the treatment implications evidenced in clinical trials,it would be an oversimplification to assume that rate control should bethemanagement approach for all patients. Themanagement of patientswith AF involves elements of anticoagulation, rate control and rhythmcontrol and such treatment strategies are not necessarily mutuallyexclusive of each other. The development of novel drugs allows forimproved efficacy, atrial selectivity and reduced toxicity. Evolvingtechnological improvement in ablative procedural devices and theincorporation of real time three-dimensional imaging modalities mayalso improve chances of the termination long-term arrhythmia[14,27,126]. It is hoped that continued research and improved
electrophysiological instrumentation may contribute to better insightinto AF mechanisms for purposes of more effective therapies.
6. Learning points
• Significant progress in understanding the mechanism of thisarrhythmia has been accomplished with the advent of catheterablation, the pathophysiology of AF is complex and likely has manypossible mechanisms whichmay be interrelated. This paper reviewsthis clinically widespread arrhythmia. Both traditional and morecontemporary theories responsible for AF as well as standard andevolving treatment modalities are also reviewed herein.
References
[1] Go AS, Hylek EM, Phillips KA, Chang Y, Henault LE, Selby JV, et al. Prevalence ofdiagnosed atrial fibrillation in adults: national implications for rhythm manage-ment and stroke prevention: the AnTicoagulation and Risk Factors in AtrialFibrillation (ATRIA) Study. JAMA 2001;285:2370–5.
[2] Chen LY, Shen WK. Epidemiology of atrial fibrillation: a current perspective.Heart Rhythm 2007;4:S1–6.
[3] Miyasaka Y, BarnesME, Gersh BJ, Cha SS, Bailey KR, AbhayaratnaWP, et al. Seculartrends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to2000, and implications on the projections for future prevalence. Circulation2006;114:119–25.
[4] Wang TJ, Parise H, Levy D, D'Agostino Sr RB, Wolf PA, Vasan RS, et al. Obesity andthe risk of new-onset atrial fibrillation. JAMA 2004;292:2471–7.
[5] Gami AS, PressmanG, Caples SM, Kanagala R, Gard JJ, DavisonDE, et al. Associationof atrial fibrillation and obstructive sleep apnea. Circulation 2004;110:364–7.
[6] Kanagala R, Murali NS, Friedman PA, Ammash NM, Gersh BJ, Ballman KV, et al.Obstructive sleep apnea and the recurrence of atrial fibrillation. Circulation2003;107:2589–94.
[7] Eaker ED, Sullivan LM, Kelly-Hayes M, D'Agostino Sr RB, Benjamin EJ. Anger andhostility predict the development of atrial fibrillation in men in the FraminghamOffspring Study. Circulation 2004;109:1267–71.
[8] Denison H, Jern S, Jagenburg R, Wendestam C, Wallerstedt S. Influence ofincreased adrenergic activity and magnesium depletion on cardiac rhythm inalcohol withdrawal. Br Heart J 1994;72:554–60.
[9] Maki T, Toivonen L, Koskinen P, Naveri H, Harkonen M, Leinonen H. Effect ofethanol drinking, hangover, and exercise on adrenergic activity and heart ratevariability in patients with a history of alcohol-induced atrial fibrillation. Am JCardiol 1998;82:317–22.
[10] Mukamal KJ, Tolstrup JS, Friberg J, Jensen G, Gronbaek M. Alcohol consumptionand risk of atrial fibrillation in men and women: the Copenhagen City HeartStudy. Circulation 2005;112:1736–42.
[11] Moe GK, Abildskov JA. Atrial fibrillation as a self-sustaining arrhythmiaindependent of focal discharge. Am Heart J 1959;58:59–70.
[12] Waldo AL. Mechanisms of atrial fibrillation. J Cardiovasc Electrophysiol 2003;14:S267–74.
[13] Moe GK, Rheinboldt WC, Abildskov JA. A computer model of atrial fibrillation. AmHeart J 1964;67:200–20.
[14] Moe GK. A conceptual model of atrial fibrillation. J Electrocardiol 1968;1:145–6.
[15] Allessie MALW, Bonke FIM, Hollen J. Experimental evaluation of Moe's multiplewavelet hypothesis of atrial fibrillation. In: Zipes DPJJ, editor. Cardiovascularelectrophysiology and arrhythmias. Orlando: Grune & Stratton; 1985. p. 265–76.
[16] Allessie MALW, Smeets J, Bonke F, Hollen. Total mapping of atrial excitationduring acetylcholine-induced atrial flutter and fibrillation in the isolated canineheart. Molndal: AB Hassle; 1982.
[17] Haissaguerre M, Jais P, Shah DC, Takahashi A, Hocini M, Quiniou G, et al.Spontaneous initiation of atrial fibrillation by ectopic beats originating in thepulmonary veins. N Engl J Med 1998;339:659–66.
[18] Mansour M, Mandapati R, Berenfeld O, Chen J, Samie FH, Jalife J. Left-to-rightgradient of atrial frequencies during acute atrial fibrillation in the isolated sheepheart. Circulation 2001;103:2631–6.
[19] Mandapati R, Skanes A, Chen J, Berenfeld O, Jalife J. Stable microreentrant sourcesas a mechanism of atrial fibrillation in the isolated sheep heart. Circulation2000;101:194–9.
[20] Fynn SP, Kalman JM. Pulmonary veins: anatomy, electrophysiology, tachycardia,and fibrillation. Pacing Clin Electrophysiol 2004;27:1547–59.
[21] Jalife J, Berenfeld O, Mansour M. Mother rotors and fibrillatory conduction: amechanism of atrial fibrillation. Cardiovasc Res 2002;54:204–16.
[22] Wyse DG, Gersh BJ. Atrial fibrillation: a perspective: thinking inside and outsidethe box. Circulation 2004;109:3089–95.
[23] Cosio FG, Castillo E. Left atrial anatomic remodeling in atrial fibrillation.J Cardiovasc Electrophysiol 2007;18:53–4.
[24] Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA. Atrial fibrillation begets atrialfibrillation. A study in awake chronically instrumented goats. Circulation 1995;92:1954–68.
[25] Wijffels MC, Kirchhof CJ, Dorland R, Power J, Allessie MA. Electrical remodelingdue to atrial fibrillation in chronically instrumented conscious goats: roles of

680 S.T. Mathew et al. / European Journal of Internal Medicine 20 (2009) 672–681
neurohumoral changes, ischemia, atrial stretch, and high rate of electricalactivation. Circulation 1997;96:3710–20.
[26] Sun H, Chartier D, Leblanc N, Nattel S. Intracellular calcium changes andtachycardia-induced contractile dysfunction in canine atrial myocytes. Cardio-vasc Res 2001;49:751–61.
[27] Nattel S, Opie LH. Controversies in atrial fibrillation. Lancet 2006;367:262–72.[28] Nattel S. New ideas about atrial fibrillation 50 years on. Nature 2002;415:219–26.[29] Yue L, Feng J, Gaspo R, Li GR, Wang Z, Nattel S. Ionic remodeling underlying
action potential changes in a canine model of atrial fibrillation. Circ Res 1997;81:512–25.
[30] Allessie M, Ausma J, Schotten U. Electrical, contractile and structural remodelingduring atrial fibrillation. Cardiovasc Res 2002;54:230–46.
[31] Falk RH. Atrial fibrillation. N Engl J Med 2001;344:1067–78.[32] Falk RH. Etiology and complications of atrial fibrillation: insights from pathology
studies. Am J Cardiol 1998;82:10N–7N.[33] Aviles RJ, Martin DO, Apperson-Hansen C, Houghtaling PL, Rautaharju P, Kronmal
RA, et al. Inflammation as a risk factor for atrial fibrillation. Circulation 2003;108:3006–10.
[34] Frustaci A, Chimenti C, Bellocci F, Morgante E, Russo MA, Maseri A. Histologicalsubstrate of atrial biopsies in patients with lone atrial fibrillation. Circulation1997;96:1180–4.
[35] Nakamura Y, Nakamura K, Fukushima-Kusano K, Ohta K, Matsubara H, Hamuro T,et al. Tissue factor expression in atrial endothelia associated with nonvalvularatrial fibrillation: possible involvement in intracardiac thrombogenesis. ThrombRes 2003;111:137–42.
[36] Kumagai K, Nakashima H, Saku K. The HMG-CoA reductase inhibitor atorvastatinprevents atrial fibrillation by inhibiting inflammation in a canine sterilepericarditis model. Cardiovasc Res 2004;62:105–11.
[37] Dernellis J, Panaretou M. C-reactive protein and paroxysmal atrial fibrillation:evidence of the implication of an inflammatory process in paroxysmal atrialfibrillation. Acta Cardiol 2001;56:375–80.
[38] ChungMK,MartinDO, SprecherD,WazniO,KanderianA, CarnesCA, et al. C-reactiveprotein elevation in patients with atrial arrhythmias: inflammatory mechanismsand persistence of atrial fibrillation. Circulation 2001;104:2886–91.
[39] Yared JP, Starr NJ, Torres FK, Bashour CA, Bourdakos G, Piedmonte M, et al. Effectsof single dose, postinduction dexamethasone on recovery after cardiac surgery.Ann Thorac Surg 2000;69:1420–4.
[40] Halonen J, Halonen P, Jarvinen O, Taskinen P, Auvinen T, Tarkka M, et al.Corticosteroids for the prevention of atrial fibrillation after cardiac surgery: arandomized controlled trial. JAMA 2007;297:1562–7.
[41] Engelmann MD, Svendsen JH. Inflammation in the genesis and perpetuation ofatrial fibrillation. Eur Heart J 2005;26:2083–92.
[42] Korantzopoulos P, Kolettis T, Siogas K, Goudevenos J. Atrial fibrillation andelectrical remodeling: the potential role of inflammation and oxidative stress.Med Sci Monit 2003;9 RA225-9.
[43] Benjamin EJ, Levy D, Vaziri SM, D'Agostino RB, Belanger AJ, Wolf PA. Independentrisk factors for atrial fibrillation in a population-based cohort. The FraminghamHeart Study. JAMA 1994;271:840–4.
[44] Kato T, Yamashita T, Sekiguchi A, Sagara K, Takamura M, Takata S, et al. What arearrhythmogenic substrates in diabetic rat atria? J Cardiovasc Electrophysiol2006;17:890–4.
[45] Patlolla V, Alsheikh-Ali AA, Al-Ahmad AM. The renin–angiotensin system: atherapeutic target in atrialfibrillation. PacingClin Electrophysiol 2006;29: 1006–12.
[46] Kumagai K, Nakashima H, Urata H, Gondo N, Arakawa K, Saku K. Effects ofangiotensin II type 1 receptor antagonist on electrical and structural remodelingin atrial fibrillation. J Am Coll Cardiol 2003;41:2197–204.
[47] Pedersen OD, Bagger H, Kober L, Torp-Pedersen C. Trandolapril reduces theincidence of atrial fibrillation after acute myocardial infarction in patients withleft ventricular dysfunction. Circulation 1999;100:376–80.
[48] Vermes E, Tardif JC, Bourassa MG, Racine N, Levesque S, White M, et al. Enalaprildecreases the incidence of atrial fibrillation in patients with left ventriculardysfunction: insight from the Studies Of Left Ventricular Dysfunction (SOLVD)trials. Circulation 2003;107:2926–31.
[49] Hobbs WJ, Van Gelder IC, Fitzpatrick AP, Crijns HJ, Garratt CJ. The role of atrialelectrical remodeling in the progression of focal atrial ectopy to persistent atrialfibrillation. J Cardiovasc Electrophysiol 1999;10:866–70.
[50] Wu TJ, Ong JJ, Chang CM, Doshi RN, Yashima M, Huang HL, et al. Pulmonary veinsand ligament of Marshall as sources of rapid activations in a canine model ofsustained atrial fibrillation. Circulation 2001;103:1157–63.
[51] Kumagai K, Yasuda T, Tojo H, Noguchi H, Matsumoto N, Nakashima H, et al. Roleof rapid focal activation in the maintenance of atrial fibrillation originating fromthe pulmonary veins. Pacing Clin Electrophysiol 2000;23:1823–7.
[52] Sueda T, Imai K, Ishii O, Orihashi K,Watari M, Okada K. Efficacy of pulmonary veinisolation for the elimination of chronic atrial fibrillation in cardiac valvularsurgery. Ann Thorac Surg 2001;71:1189–93.
[53] Nathan H, Gloobe H. Myocardial atrio-venous junctions and extensions (sleeves)over the pulmonary and caval veins. Anatomical observations in variousmammals.Thorax 1970;25:317–24.
[54] Saito T, Waki K, Becker AE. Left atrial myocardial extension onto pulmonary veinsin humans: anatomic observations relevant for atrial arrhythmias. J CardiovascElectrophysiol 2000;11:888–94.
[55] Perez-Lugones A, McMahon JT, Ratliff NB, Saliba WI, Schweikert RA, MarroucheNF, et al. Evidence of specialized conduction cells in human pulmonary veins ofpatients with atrial fibrillation. J Cardiovasc Electrophysiol 2003;14:803–9.
[56] Ho SY, Cabrera JA, Tran VH, Farre J, Anderson RH, Sanchez-Quintana D. Architecture ofthe pulmonary veins: relevance to radiofrequency ablation. Heart 2001;86:265–70.
[57] Jalife J. Rotors and spiral waves in atrial fibrillation. J Cardiovasc Electrophysiol2003;14:776–80.
[58] Wu RC, Berger R, Calkins H. Catheter ablation of atrial flutter and macroreentrantatrial tachycardia. Curr Opin Cardiol 2002;17:58–64.
[59] Randall WCAJ. Cardiac electrophysiology: from cell to bedside. In: Zipes DPJJ,editor. Nervous control of the heart: anatomy and pathophysiology. Philadel-phia: WB Saunders, Co; 1990. p. 291–9.
[60] Coumel P. Paroxysmal atrial fibrillation: a disorder of autonomic tone? Eur HeartJ 1994;15(Suppl A):9–16.
[61] Coumel P. Autonomic influences in atrial tachyarrhythmias. J CardiovascElectrophysiol 1996;7:999–1007.
[62] Coumel P, Attuel P, Lavallee J, Flammang D, Leclercq JF, Slama R. The atrialarrhythmia syndrome of vagal origin. Arch Mal Coeur Vaiss 1978;71:645–56.
[63] Chen YJ, Chen SA, Chang MS, Lin CI. Arrhythmogenic activity of cardiac muscle inpulmonary veins of the dog: implication for the genesis of atrial fibrillation.Cardiovasc Res 2000;48:265–73.
[64] Singh JP, Larson MG, Levy D, Evans JC, Tsuji H, Benjamin EJ. Is baseline autonomictone associated with new onset atrial fibrillation?: Insights from the Framinghamheart study. Ann Noninvasive Electrocardiol 2004;9:215–20.
[65] Tomita T, Takei M, Saikawa Y, Hanaoka T, Uchikawa S, Tsutsui H, et al. Role ofautonomic tone in the initiation and termination of paroxysmal atrial fibrillation inpatientswithout structural heart disease. J Cardiovasc Electrophysiol 2003;14:559–64.
[66] Nagasawa H, Fujiki A, Fujikura N, Matsuda T, Yamashita T, Inoue H. Effects of anovel class III antiarrhythmic agent, NIP-142, on canine atrial fibrillation andflutter. Circ J 2002;66:185–91.
[67] Coumel P. Atrial fibrillation: one more sporting inconvenience? Eur Heart J2002;23:431–3.
[68] Nattel S. Therapeutic implications of atrial fibrillation mechanisms: can mech-anistic insights be used to improve AF management? Cardiovasc Res 2002;54:347–60.
[69] Van Gelder IC, Hagens VE, Bosker HA, Kingma JH, Kamp O, Kingma T, et al. Acomparison of rate control and rhythm control in patients with recurrentpersistent atrial fibrillation. N Engl J Med 2002;347:1834–40.
[70] Wyse DG, Waldo AL, DiMarco JP, Domanski MJ, Rosenberg Y, Schron EB, et al. Acomparison of rate control and rhythm control in patients with atrial fibrillation.N Engl J Med 2002;347:1825–33.
[71] Hohnloser SH, Kuck KH, Lilienthal J. Rhythm or rate control in atrial fibrillation—Pharmacological Intervention in Atrial Fibrillation (PIAF): a randomised trial.Lancet 2000;356:1789–94.
[72] Carlsson J, Miketic S,Windeler J, Cuneo A, Haun S,Micus S, et al. Randomized trial ofrate-control versus rhythm control in persistent atrial fibrillation: the Strategies ofTreatment of Atrial Fibrillation (STAF) study. J Am Coll Cardiol 2003;41:1690–6.
[73] Opolski G, Torbicki A, Kosior DA, Szulc M,Wozakowska-Kaplon B, Kolodziej P, et al.Rate control vs rhythm control in patients with nonvalvular persistent atrialfibrillation: the results of the Polish How to Treat Chronic Atrial Fibrillation (HOTCAFE) Study. Chest 2004;126:476–86.
[74] Fuster V, Ryden LE, Cannom DS, Crijns HJ, Curtis AB, Ellenbogen KA, et al. ACC/AHA/ESC 2006 Guidelines for the Management of Patients with Atrial Fibrilla-tion: a report of the American College of Cardiology/American Heart AssociationTask Force on Practice Guidelines and the European Society of CardiologyCommittee for Practice Guidelines (Writing Committee to Revise the 2001Guidelines for the Management of Patients with Atrial Fibrillation): developed incollaboration with the European Heart Rhythm Association and the HeartRhythm Society. Circulation 2006;114:e257–354.
[75] Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation.Analysis of pooled data from five randomized controlled trials. Arch Intern Med1994;154:1449–1457.
[76] Medi C, Hankey GJ, Freedman SB. Atrial fibrillation. Med J Aust 2007;186:197–202.[77] Albers GW. Antithrombotic therapy for prevention and treatment of ischemic
stroke. J Thromb Thrombolysis 2001;12:19–22.[78] VermaA,Natale A. Should atrial fibrillation ablation be consideredfirst-line therapy
for some patients? Why atrial fibrillation ablation should be considered first-linetherapy for some patients. Circulation 2005;112:1214–22 discussion 1231.
[79] Corley SD, Epstein AE, DiMarco JP, Domanski MJ, Geller N, Greene HL, et al.Relationships between sinus rhythm, treatment, and survival in the AtrialFibrillation Follow-Up Investigation of Rhythm Management (AFFIRM) Study.Circulation 2004;109:1509–13.
[80] Falk RH. Management of atrial fibrillation—radical reform or modest modifica-tion? N Engl J Med 2002;347:1883–4.
[81] Chung MK. Atrial fibrillation: rate control is as good as rhythm control for some,but not all. Cleve Clin J Med 2003;70:567–73.
[82] Pappone C, Rosanio S, Augello G, Gallus G, Vicedomini G, Mazzone P, et al.Mortality, morbidity, and quality of life after circumferential pulmonary veinablation for atrial fibrillation: outcomes from a controlled nonrandomized long-term study. J Am Coll Cardiol 2003;42:185–97.
[83] Crijns HJ. Rate versus rhythm control in patients with atrial fibrillation: what thetrials really say. Drugs 2005;65:1651–67.
[84] Cooper HA, Bloomfield DA, Bush DE, Katcher MS, Rawlins M, Sacco JD, et al.Relation between achieved heart rate and outcomes in patients with atrialfibrillation (from the Atrial Fibrillation Follow-up Investigation of RhythmManagement [AFFIRM] Study). Am J Cardiol 2004;93:1247–53.
[85] Bjerregaard P, BaileyWB, Robinson SE. Rate control in patients with chronic atrialfibrillation. Am J Cardiol 2004;93:329–32.
[86] Kowey PR. You only get so many heartbeats. J Am Coll Cardiol 2004;43:1209–10.[87] Van Gelder IC, Rienstra M, Van den Berg MP, Van Veldhuisen DJ. Rate control in
atrial fibrillation. J Am Coll Cardiol 2004;44:2417–8 author reply 2418–9.

681S.T. Mathew et al. / European Journal of Internal Medicine 20 (2009) 672–681
[88] Wood MA, Brown-Mahoney C, Kay GN, Ellenbogen KA. Clinical outcomes afterablation and pacing therapy for atrial fibrillation: a meta-analysis. Circulation2000;101:1138–44.
[89] Dorian P, Connors SP. Pharmacological and nonpharmacological methods for ratecontrol. Can J Cardiol 2005;21(Suppl B):26B–30B.
[90] Levy T, Walker S, Mason M, Spurrell P, Rex S, Brant S, et al. Importance of ratecontrol or rate regulation for improving exercise capacity and quality of life inpatients with permanent atrial fibrillation and normal left ventricular function: arandomised controlled study. Heart 2001;85:171–8.
[91] Weerasooriya R, Davis M, Powell A, Szili-Torok T, Shah C, Whalley D, et al. TheAustralian Intervention Randomized Control of Rate in Atrial Fibrillation Trial(AIRCRAFT). J Am Coll Cardiol 2003;41:1697–702.
[92] Ahmad K, Dorian P. Rate control in atrial fibrillation: looking beyond the averageheart rate. Curr Opin Cardiol 2006;21:88–93.
[93] Bailin SJ, Adler S, Giudici M. Prevention of chronic atrial fibrillation by pacing inthe region of Bachmann's bundle: results of a multicenter randomized trial.J Cardiovasc Electrophysiol 2001;12:912–7.
[94] Padeletti L, Porciani MC, Michelucci A, Colella A, Costoli A, Ciapetti C, et al.Prevention of short term reversible chronic atrial fibrillation by permanentpacing at the triangle of Koch. J Interv Card Electrophysiol 2000;4:575–83.
[95] Hermida JS, Kubala M, Lescure FX, Delonca J, Clerc J, Otmani A, et al. Atrial septalpacing to prevent atrial fibrillation in patients with sinus node dysfunction:results of a randomized controlled study. Am Heart J 2004;148:312–7.
[96] Doshi RN, Daoud EG, Fellows C, Turk K, Duran A, Hamdan MH, et al. Leftventricular-based cardiac stimulation post AV nodal ablation evaluation (thePAVE study). J Cardiovasc Electrophysiol 2005;16:1160–5.
[97] Cox JL, Boineau JP, Schuessler RB, Jaquiss RD, Lappas DG. Modification of themazeprocedure for atrial flutter and atrial fibrillation. I. Rationale and surgical results.J Thorac Cardiovasc Surg 1995;110:473–84.
[98] Gillinov AM, McCarthy PM. Advances in the surgical treatment of atrialfibrillation. Cardiol Clin 2004;22:147–57.
[99] Damiano Jr RJ, Gaynor SL, Bailey M, Prasad S, Cox JL, Boineau JP, et al. The long-term outcome of patients with coronary disease and atrial fibrillation undergoingthe Cox maze procedure. J Thorac Cardiovasc Surg 2003;126:2016–21.
[100] Chen SA, Hsieh MH, Tai CT, Tsai CF, Prakash VS, Yu WC, et al. Initiation of atrialfibrillation by ectopic beats originating from the pulmonary veins: electro-physiological characteristics, pharmacological responses, and effects of radio-frequency ablation. Circulation 1999;100:1879–86.
[101] Haissaguerre M, Shah DC, Jais P, Hocini M, Yamane T, Deisenhofer I, et al.Electrophysiological breakthroughs from the left atrium to the pulmonary veins.Circulation 2000;102:2463–5.
[102] Haissaguerre M, Jais P, Shah DC, Garrigue S, Takahashi A, Lavergne T, et al.Electrophysiological end point for catheter ablation of atrial fibrillation initiatedfrom multiple pulmonary venous foci. Circulation 2000;101:1409–17.
[103] Pappone C, Rosanio S, Oreto G, Tocchi M, Gugliotta F, Vicedomini G, et al.Circumferential radiofrequency ablation of pulmonary vein ostia: a newanatomic approach for curing atrial fibrillation. Circulation 2000;102:2619–28.
[104] Gerstenfeld EP, Callans D, Dixit S, Lin D, Cooper J, Russo AM, et al. Characteristicsof patients undergoing atrial fibrillation ablation: trends over a seven-yearperiod 1999–2005. J Cardiovasc Electrophysiol 2007;18:23–8.
[105] Haissaguerre M, Shah DC, Jais P, Hocini M, Yamane T, Deisenhofer I, et al.Mapping-guided ablation of pulmonary veins to cure atrial fibrillation. Am JCardiol 2000;86:9K–19K.
[106] Keane D, Ruskin J. Pulmonary vein isolation for atrial fibrillation. Rev CardiovascMed 2002;3:167–75.
[107] Oral H, Knight BP, Tada H, Ozaydin M, Chugh A, Hassan S, et al. Pulmonary veinisolation for paroxysmal and persistent atrial fibrillation. Circulation 2002;105:1077–81.
[108] Morady F. Treatment of paroxysmal atrial fibrillation by pulmonary veinisolation. Circ J 2003;67:567–71.
[109] Pappone C, Oreto G, Rosanio S, Vicedomini G, Tocchi M, Gugliotta F, et al. Atrialelectroanatomic remodeling after circumferential radiofrequency pulmonaryvein ablation: efficacy of an anatomic approach in a large cohort of patients withatrial fibrillation. Circulation 2001;104:2539–44.
[110] Schwartzman D, Parizhskaya M, Devine WA. Linear ablation using an irrigatedelectrode electrophysiologic and histologic lesion evolution comparisonwith ablationutilizing a non-irrigated electrode. J Interv Card Electrophysiol 2001;5:17–26.
[111] Ho SY, Sanchez-Quintana D, Cabrera JA, Anderson RH. Anatomy of the leftatrium: implications for radiofrequency ablation of atrial fibrillation. J CardiovascElectrophysiol 1999;10:1525–33.
[112] Seidl K, SchwackeH, ZahnR, RamekenM,Drogemuller A, Senges J. Catheter ablationof chronic atrialfibrillationwith noncontactmapping: are continuous linear lesionsassociated with ablation success? Pacing Clin Electrophysiol 2003;26:534–43.
[113] Ernst S, Ouyang F, Goya M, Lober F, Schneider C, Hoffmann-Riem M, et al. Totalpulmonary vein occlusion as a consequence of catheter ablation for atrial fib-rillation mimicking primary lung disease. J Cardiovasc Electrophysiol 2003;14:366–70.
[114] Keane D, Mansour M, Singh J. Detection by intracardiac echocardiography ofearly formation of left atrial thrombus during pulmonary vein isolation. Europace2004;6:109–10.
[115] Schwab JO, Burkhardt D, Yang A, Schrickel J, Luderitz B, Lewalter T. ECG signsmimicking acute inferior wall myocardial infarction are associated with elevatedmyocardial enzymes during isolation of pulmonary vein for focal atrial fibrillation.Europace 2004;6:111–5.
[116] Packer DL, Asirvatham S, Munger TM. Progress in nonpharmacologic therapy ofatrial fibrillation. J Cardiovasc Electrophysiol 2003;14:S296–309.
[117] Patterson E, Po SS, Scherlag BJ, Lazzara R. Triggered firing in pulmonary veinsinitiated by in vitro autonomic nerve stimulation. Heart Rhythm 2005;2:624–31.
[118] Marron K, Wharton J, Sheppard MN, Fagan D, Royston D, Kuhn DM, et al.Distribution, morphology, and neurochemistry of endocardial and epicardialnerve terminal arborizations in the human heart. Circulation 1995;92:2343–51.
[119] Kaye DM, Vaddadi G, Gruskin SL, Du XJ, Esler MD. Reduced myocardial nervegrowth factor expression in human and experimental heart failure. Circ Res2000;86:E80–4.
[120] Ginty DD, Segal RA. Retrograde neurotrophin signaling: Trk-ing along the axon.Curr Opin Neurobiol 2002;12:268–74.
[121] Cao JM, Fishbein MC, Han JB, Lai WW, Lai AC, Wu TJ, et al. Relationship betweenregional cardiac hyperinnervation and ventricular arrhythmia. Circulation2000;101:1960–9.
[122] KimDT, LuthringerDJ, LaiAC, SuhG,Czer L, Chen LS, et al. Sympathetic nerve sproutingafter orthotopic heart transplantation. J Heart Lung Transplant 2004;23:1349–58.
[123] Cao JM, Chen LS, KenKnight BH, Ohara T, LeeMH, Tsai J, et al. Nerve sprouting andsudden cardiac death. Circ Res 2000;86:816–21.
[124] Swissa M, Zhou S, Gonzalez-Gomez I, Chang CM, Lai AC, Cates AW, et al. Long-term subthreshold electrical stimulation of the left stellate ganglion and a caninemodel of sudden cardiac death. J Am Coll Cardiol 2004;43:858–64.
[125] Lafuente-Lafuente C, Mouly S, Longas-Tejero MA, Mahe I, Bergmann JF.Antiarrhythmic drugs for maintaining sinus rhythm after cardioversion of atrialfibrillation: a systematic review of randomized controlled trials. Arch InternMed2006;166:719–28.
[126] Page RL. Medical management of atrial fibrillation: future directions. HeartRhythm 2007;4:S91–4.
[127] Olsson SB. Atrial fibrillation—new aspects on mechanism and treatment. J InternMed 1996;239:3–15.
Copyright © 2022 FDOKUMEN




















