
Aspects of Antisense and Antigene Chemistry of ... - DiVA Portal
68
!!! " # $% #&#'#"#(") !!*#)+'#!#(!,#+*# ((!,!-..-
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of Aspects of Antisense and Antigene Chemistry of ... - DiVA Portal


Aspects of Antisense and Antigene Chemistry of Oligonucleotides Tethered
to Intercalators
BY
DIMITRI OSSIPOV

Dissertation for the Degree of Doctor of Philosophy in Bioorganic Chemistry presented at Uppsala University in 2002 Abstract Ossipov, D., 2002. Aspects of Antisense and Antigene Chemistry of Oligonucleotides Tethered to Intercalators. Acta Univ. Ups., Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 735. 67 pp. Uppsala. ISBN 91-554-5365-1. Synthetic and physicochemical studies on appropriately functionalized ODN-conjugates have been performed to evaluate their abilities to act as antisense agents against RNA or as intramolecular DNA cross-linking agents. Intercalating aromatic systems [phenazine (Pnz), dipyridophenazine (DPPZ)] and metallointercalators such as Ru2+(phen)2(DPPZ) and Ru2+(tpy)(DPPZ)L [where L = chemically or photochemically labile ligand, phen = phenanthroline, tpy = terpyridine], which are covalently tethered to the oligodeoxynucleotides (ODNs), have been chosen for this purpose. The ODN-conjugates were typically prepared by automated solid phase synthesis using phosphoramidite building blocks, or on solid supports, both functionalized with the chromophore groups. The photosensitive metal complex, Ru2+(tpy)(DPPZ)(CH3CN), has been incorporated by post-synthetic coupling to the amino-linker modified ODNs via an amide bond. The intercalating ability of the tethered chromophores gave enhanced stability of the duplexes and triplexes formed with ODN-conjugates and their complementary targets: DNA, RNA, or double-stranded DNA. The conjugation of DPPZ chromophore to ODN (at 3', 5' or at the middle) led us to incorporate Ru2+(phen)2(DPPZ) through the DPPZ ligand, for the first time. The corresponding (Ru2+-ODN)•DNA duplexes showed dramatic stabilization (∆Tm = 19.4 – 22.0ºC). The CD and DNase I footprinting experiments suggest that the stabilization is owing to metallointercalation by threading of the Ru2+(phen)2 moiety through the ODN•DNA duplex core, thus “stapling” the two helical strands from the minor to major groove. On the other hand, Ru2+(tpy)(DPPZ)(CH3CN)-ODN conjugates represent a new class of oligonucleotides containing the photoactivatible Ru2+ complexes, which can successfully crosslink to the complementary strand. The mechanism of cross-linking upon photoirradiation of [Ru2+(tpy)(DPPZ)(CH3CN)-ODN]•DNA involves in situ conversion to the reactive [Ru2+(tpy)(DPPZ)(H2O)-ODN]•DNA which are subsequently cross-linked through the G residue of the complementary DNA strand. All starting materials and products have been purified by HPLC and/or by PAGE and subsequently charcterized by MALDI-TOF as well as ESI mass spectroscopy. Terminal conjugation of the planar Pnz and DPPZ groups through the flexible linkers were also shown to improve thermal stability of the ODN•RNA hybrid duplexes without alteration of the initial AB-type global helical structure as revealed from CD experiments. As a result, RNase H mediated cleavage of the RNA strand in the intercalator-tethered ODN•RNA duplexes was more efficient compared to the natural counterpart. The RNase H cleavage pattern was also found to be dependent on the chemical nature of the chromophore. It appeared that introduction of a tether at the 3'-end of the ODN can be most easily tolerated by the enzyme regardless of the nature of the appending chromophore. The tethered DPPZ group has also been shown to chelate Cu2+ and Fe3+, like phenanthroline group, followed by the formation of redox-active metal complex which cleaves the complementary DNA strand in a sequence-specific manner. This shows that the choice of appropriate ligand is useful to (i) attain improved intercalation giving Tm enhancement, and (ii) sequence-specifically inactivate target RNA or DNA molecules using multiple modes of chemistry (RNase H mediated cleavage, free-radical, oxidative pathways or photocross-linkage).
Dimitri Ossipov, Department of Bioorganic Chemistry, Biomedical Centre, Box 581, Uppsala University, SE-751 23 Uppsala, Sweden © Dimitri Ossipov 2002 ISSN 1104-232X ISBN 91-554-5365-1 Printed in Sweden by Tryck & Medier, Uppsala 2002

To memory of my father
who should have lived
to see this thesis

THE ORIGINAL PUBLICATIONS
This thesis is based on the following original publications, which are referred to by the Roman numerals. I. Ossipov, D.; Chattopadhyaya, J. Synthesis of 1'-Phenazine-Tethered Psicofuranosyl
Oligonucleotides: The Thermal Stability and Fluorescence Properties of Their Duplexes and Triplexes.
Tetrahedron 1998, 54, 5667-5682. II. Ossipov, D.; Zamaratski, E.; Chattopadhyaya, J. Dipyrido[3,2-a:2',3'-c]phenazine-Tethered Oligo-
DNA: Synthesis and Thermal Stability of Their DNA-DNA and DNA-RNA Duplexes and Triplexes.
Helvetica Chimica Acta 1999, 82, 2186-2200. III. Zamaratski, E.; Ossipov, D.; Pradeepkumar, P.I.; Amirkhanov, N.; Chattopadhyaya, J. The 3'-
Modified Antisense Oligos Promote Faster Hydrolysis of the Target RNA by RNase H than the Natural Counterpart.
Tetrahedron 2001, 57, 593-606. IV. Ossipov, D.; Pradeepkumar, P.I.; Chattopadhyaya, J. Synthesis of [Ru(phen)2dppz]2+-Tethered
Oligo-DNA and Studies on the Metallointercalation Mode into the DNA Duplex. Journal of the American Chemical Society 2001, 123, 3551-3562. V. Ossipov, D.; Gohil, S.; Chattopadhyaya, J. First Synthesis and Reactivity of the DNA-
[Ru(tpy)(dppz)(CH3CN)]2+ Conjugates. Journal of the American Chemical Society 2002 (submitted).
Reprints were made with permission from the publishers.

Table of Contents Abbreviations
1. Introduction 8
1.1 Formation of duplex and triplex structures by oligonucleotide-intercalator conjugates 9 1.2 Antisense oligodeoxynucleotides activating RNase H mediated cleavage of mRNA 10 1.3 Use of metal ions in the antisense and antigene strategies 11
2. Present Work 2.1 Synthesis of the ODNs tethered to phenazine, dipyridophenazine, and Ru2+- dipyridophenazine chromophores 14
2.1.1 Synthesis of Pnz labeled ODNs (Paper I) 14 2.1.2 Synthesis of amino-linkers of different lengths tethered to the glycerol moiety (Paper II) 16 2.1.3 Derivatization of DPPZ containing chromophores for conjugation (Papers II, IV & V) 20 2.1.4 Synthesis of DPPZ and Ru2+(phen)2(DPPZ) labeled ODNs (Papers II & IV) 21 2.1.5 Synthesis of Ru2+(tpy)(DPPZ)(CH3CN) labeled ODNs (Paper V) 23 2.1.6 Characterization of Ru2+(tpy)(DPPZ)(CH3CN) labeled ODNs (Paper V) 26 2.2 Thermal stability of duplexes and triplexes formed by phenazine, dipyridophenazine, and Ru2+-dipyridophenazine conjugated ODNs 27 2.2.1 Thermal stability of conjugated ODN•DNA duplexes (Papers I, II, IV & V) 27 2.2.2 Thermal stability of conjugated ODN•RNA duplexes (Papers II & IV) 30 2.2.3 Thermal stability of conjugated ODN×DNA•DNA triplexes (Papers I, II & IV) 31 2.2.4 Circular dichroism (CD) and DNase I footprinting studies on [Ru(phen)2(DPPZ)]2+- labeled ODN•DNA duplexes performed for the elucidation of the metallointercalation binding mode in the DNA double helixes (Paper IV) 34 2.3 RNase H degradation studies on the AON•RNA duplexes conjugated with various intercalators at AON strand (Paper III) 39 2.3.1 Physicochemical properties of RNA targets and their duplexes formed with AON conjugates 39 2.3.2 RNase H mediated cleavage studies on the conjugated AON•RNA duplexes 42 2.3.3 Nuclease resistance of the 3'-Pnz and 3'-DPPZ-conjugated AONs 46 2.4 Specific chemical modifications of the complementary DNA strand by DPPZ and Ru2+- DPPZ tethered ODNs in ODN•DNA duplexes 46 2.4.1 Sequence-specific cleavage of target DNA by metal-binding DPPZ-group linked to the internucleotide position of the complementary ODN strand (unpublished data) 46 2.4.3 Photochemical generation of Ru2+(tpy)(DPPZ)(H2O)-ODN (Paper V) 48 2.4.4 Photochemical cross-linking of Ru2+(tpy)(DPPZ)(CH3CN)-ODN conjugates to the complementary DNA strand in ODN•DNA duplexes (Paper V) 49 2.4.5 The extension of the idea of the Ru2+ complex activation via photoaquation (Paper V) 55 3. Acknowledgements 58 4. References 59

Abbreviations
AON antisense oligodeoxynucleotide
biq 2,2'-biquinoline
bp base pair
bpy 2,2'-bypyridine
CPG control pore glass
dA 2'-deoxyadenosine
dC 2'-deoxycytidine
dG 2'-deoxyguanosine
dmbpy 6,6'-dimethyl-2,2'-bypyridine
DMF dimethylformamide
DMT 4,4'-dimethoxytrityl
dpp 4,7-diphenyl-1,10-phenanthroline
DPPZ dipyrido[3,2-a;2',3'-c]phenazine
DTT dithiothreitol
dsDNA double-stranded DNA
EDTA ethylenediaminetetraacetic acid
ESI electrospray ionisation
Fmoc 9-fluorenylmethoxycarbonyl
HPLC high perfomance liquid chromatography
MALDI-TOF matrix-assisted laser desorption ionization – time-of-flight
MPE methidiumpropyl-EDTA
mRNA messenger RNA
MS mass spectroscopy
NHS N-hydroxysuccinimidyl
ODN oligodeoxynucleotide
ON oligonucleotide
PAGE polyacrylamide gel electrophoresis
phen 1,10-phenanthroline
phi 9,10-phenanthrenequinone diimine
Pnz phenazine
PO phosphodiester

PS phosphorothioate
pypz 1-(2'-pyridyl)-3,5-dimethylpyrazole
ssDNA single-stranded DNA
T thymidine
tap 1,4,5,8-tatraazaphenanthrene
TBAF n-tetrabutylammonium fluoride
THF tetrahydrofurane
Tm melting temperature
TPDS 1,1,3,3-tetraisopropyldisiloxan-1,3-diyl
tpy 2,2':6',2''-terpyridine
TSU N,N,N',N'-tetramethyl(succinimidyl)uronium tetrafluoroborate
U uridine
UV-vis ultraviolet-visible
• Watson-Crick hydrogen bond
× Hoogsteen hydrogen bond

8
1. Introduction.
The idea to regulate gene expression by the use of synthetic oligonucleotides (ONs), which can
specifically recognize particular base sequence of nucleic acids, was first proposed in 1978 by
Zamecnik and Stephenson.1,2 Later, two main strategies have been considered: in the antisense
strategy,3-7 the ON is targeted to a specific mRNA via Watson-Crick8 base paring (ON•RNA duplex
formation), thereby inhibiting its translation into the corresponding protein;9,10 in the antigene
strategy,11-13 a dsDNA sequence is the target of the ON which is then expected to hybridize with
dsDNA involving Hoogsteen hydrogen bonding14 (ON×DNA•DNA triplex formation) and block
transcription of the specific gene where the target is located (symbols × and • denote here Hoogsteen
and Watson-Crick hydrogen bonding, respectively). Although short ONs are able to bind selectively to
target sequences under physiological conditions, their potential therapeutic use as artificial gene control
agents is possible if they comply with the following requirements:15 (1) ONs must be able to penetrate
the cell membrane to reach its site of action. Unlike low molecular weight agents, natural ONs are
polyanionic and cannot passively diffuse across cell membranes. (2) ONs must be resistant to
intracellular enzymes (endo- and exonucleases) that degrade nucleic acids. This precludes the use of
phosphodiester (PO) linked natural ONs as therapeutics because of their instability against nucleases.
(3) ONs must be able to interact with their cellular targets (such as mRNA or genomic dsDNA) i.e.
target sequences should be accessible for hybridization. As targets are invariably protein bound or
structurally folded, many sites are probably not accessible for interaction with ONs of defined
sequences. (4) ONs should not interact in a non-specific manner with other macromolecules as well as
their interaction within target nucleic acid should be sequence specific. (5) The complex formed
between the ON and its complementary target sequence must be sufficiently stable under physiological
conditions. Especially it concerns the triplex forming oligonucleotides (TFOs) bound to an oligopurine
target sequence in dsDNA, since the formation of dC+×dG•dC triplet is pH dependent with an optimum
stability at non-physiological pH 5.6-6.0, and the T×dA•T triplet is only stable under conditions of high
ionic strength.16,17 (6) Antisense oligodeoxynucleotides (AONs) have been found to inhibit translation
by steric blocking of the ribosomal machinery18-21, as well as by recruitment of an endogenous nuclease
RNase H which degrades the RNA strand in the AON•RNA duplex.22,23 Thus, the ability of an
AON•RNA duplex to be a substrate for the cellular RNase H enzyme is essential because of irreversible
degradation of mRNA.
Whereas the requirement for specificity is satisfactorily met by the natural ONs, sufficient passage
through membranes, adequate stability to nucleases, and improved binding affinity to the target
molecule can be achieved only by modification of the ON. Moreover, modification of natural ON by

9
tethering of chemically reactive groups can be used to induce irreversible sequence specific
modifications and/or cleavage of targeted nucleic acids24 thus permitting permanent inactivation of
unwanted genetic information. Among different chemical modifications of ONs the conjugation is
considered as a result of the coupling of ON to one or more molecules with distinct properties. In this
way the affinity of ON residue to complementary sequence is combined with specific characteristics of
the appending group(s) in the resulting ON conjugate.25 We report here the synthesis of ODN
conjugates, which enhance the binding affinity of the parent natural ODNs toward various targets
(single-stranded DNA or RNA molecules, and DNA duplexes), as well as show improvement of their
properties to elicit RNase H activity for complementary RNA cleavage in the corresponding
heteroduplex. Finally, we show when an appropriate functional group is tethered to a synthetic ODN, it
can specifically modify the target DNA strand in the ODN•DNA duplex.
1.1 Formation of duplex and triplex structures by oligonucleotide-intercalator conjugates.
In order to increase the stability of duplexes and triplexes formed by natural ONs, intercalating agents
have been attached to oligonucleotides without loss of specificity. It has generally been accepted that
planar aromatic chromophores can increase the π-stacking interactions if they are inserted between
adjacent base pairs of a duplex thus stabilizing the helical structure.26,27 Such polyaromatic agents as
phenazinium,28,29 acridine,30-34 ethidium,35 anthraquinone,36-38 fagaronine,39 pyrine,40-42 daunomycin,43
anthracene,44,45 and fluorescein42 were conjugated to a number of ONs at the 3'-, 5'-terminals,
internucleotide position, or through the base residue. Most of these ON conjugates were shown to
significantly increase the thermodinamic stability (expressed with melting temperature Tm) by 10-30°C.
The stabilizing effect of a chromophore was found to be dependent on (i) the type of the tether, (ii) the
linker length, and (iii) its site of attachment within the duplex. Generally, 5'-conjugation had more
noticeable effect on Tm than conjugation at the 3'-terminal of ODN sequence, whereas incorporation of a
potential intercalator at the central position of oligonucleotide chain mostly led to the destabilizing of a
resulting duplex.
Conjugation of intercalating agents was also applied to the triplex forming oligonucleotides
(TFOs) with the expectation of stabilization of resulting triplex provided by insertion of an intercalator
between base pairs at the triplex-duplex junction. This was indeed observed upon triplex formation by
TFOs derivatized with acridine at 5'-46-48 or 3'-ends49. Similar results were obtained with psoralen,50
phenanthroline,48 daunorubicin,51 and naphthalene diimade derivative52 attached to the 5'-end of an
oligonucleotide.53 Triple helix-specific intercalators, benzopyridoindoles and benzopyridoquinoxalines,
were covalently linked54,55 to the 5'-end as well as to the interior of a TFO for the remarkable
improvement of triplex stability under physiological conditions as compared with unmodified and

10
acridine modified counterparts. Despite the attractive idea to efficiently block dsDNA instead of many
copies of mRNA which are continuously produced from one equivalent of DNA, the major limitation of
antigene strategy is: (i) the low accessibility of the DNA strands which are protected with bound
proteins forming complex chromatin structure, and (ii) the requirement of the polypurine sequence of a
dsDNA target recognized by oligopyrimidine TFO. Therefore, several studies have been aimed at
circumventing this limitation. The extension of recognition sequences has been realized by
simultaneous binding of the homopyrimidine TFO to adjacent purine tracts on alternate strands of the
DNA duplex.56-58 The reverse orientation of the pyrimidine oligo-α-nucleotide TFO strand with respect
to that of the corresponding β-oligomer59 was also used to construct another set of molecules which are
able to bind to adjacent homopurine sequences on alternate DNA strands. Varios strategies have been
developed to accommodate a single pyrimidine mismatch in the homopurine tract. The incorporation of
an abasic site into a TFO at a site opposite a single interruption resulted in considerable destabilization
of the triplex.60 The reduction of stability was also observed upon incorporation of base triads
characterized by single hydrogen bond between the third strand base and the pyrimidine interruption
(like dG×T•dA).61 The recognition of base pair inversions within polypurine•polypyrimidine tract was
also achieved by TFO carrying a non-natural nucleoside, N4-(6-amino-2-pyridinyl)-deoxycytidine,62
which simultaneously form two hydrogen bonds with both bases of T•dA interruption. The
incorporation of acridine at an internal site of TFO allowed to stabilize the mismatched triplexes,63,64
thus extending the range of DNA sequences available for triplex formation. The stabilizing effect
brought by acridine intercalation at the base pair inversion site compensated the destabilization
introduced by mismatched base triplet. However, this intercalation did not provide much specificity.
1.2 Antisense oligodeoxynucleotides activating RNase H mediated cleavage of mRNA.
It has been demonstrated that the heteroduplex between natural phosphodiester (PO) AONs and the
target RNA is recognized by an intracellular nuclease RNase H that cleaves only the RNA strand of this
duplex. The RNase H is highly sensitive to structural alterations of the AON, and therefore only limited
number of certain modifications have been shown to support the RNase H-mediated cleavage of RNA
target of an AON•RNA duplex.65,66 On the other hand, it has been found that the inhibition of
translation does not necessarily occur with many antisense oligonucleotides, which do not recruit RNase
H despite their high binding affinity to the target RNA.18,20,67,68 Contrary, naturally occurring AONs
with lower Tm were very effective in inhibition of gene expression through RNase H mechanism. It
appears that irreversible degradation of mRNA by RNase H can amplify the efficacy of AONs
compared to those which are only able to arrest translation by steric blockage of mRNA via the
formation of AON•RNA duplex (RNase H-independent mechanism).69 For instance, 2'-O-alkyl,70 2'-

11
aminoalkyl,71,72 2'-fluoro-modified73 AONs, as well as locked nucleic acids (LNA)74 hybridizes to RNA
resulting in significantly stabilized but not RNase H cleavable AON•RNA duplexes. The same negative
result has been observed for AON phosphodiester backbone modifications as in
methylphosphonates,9,75 phosphoro-N-morpholidates,76 phosphoro-N-butylamidates,76
methylenemethyl-imines,77 and N3'→P5' phosphoramidates.78
To date, only few examples of modified AONs have been shown to support RNase H hydrolysis,
among of which are backbone modified phosphorothioates (PS)15,67 and boranophosphates79 and sugar
modified 2'-deoxy-2'-fluoro-arabinonucleic acids (2'F-ANA).80-82 Unfortunately, such modifications
were accompanied by lowering of AON•RNA duplex stability and, as a consequence, led to the less
RNase H cleavage efficiency compared to the native counterpart.80 To improve AON•RNA duplex
stability without loss of ability to activate RNase H, chimeric oligonucleotides consisting of the middle
part, which supports RNase H hydrolysis (PO or PS-backbone) and flanked with modified sequences to
provide high binding affinity to RNA (2'-O-methyl-ribonucleic acid, locked nucleic acid (LNA), were
synthesized.83-89 The introduction of one or more oxetane modified thymidine blocks into
oligonucleotide allowed to modulate the cleavage pattern of complementary RNA strand by changing
the location of modification.90-92 Recent work has shown that in the gapmer constructs with 2'-O-methyl
nucleosides, one requires at least a minimum stretch of four or five deoxyresidues89. In oxetane92 and
LNA89 modified AONs, one requires at least a stretch of six deoxy nucleotide residues from the
modified site (cleavage site at the sixth phosphate from the 3'-end). Another approach is based on the
terminal conjugation of intercalating agent which can significantly stabilize AON•RNA duplex without
alteration of conformation inherent in the native DNA•RNA hybrid.93 However, only few reports have
been devoted to the investigation of RNase H activity induced by such conjugates. Acridine,94
psoralen,95 and phenanthroline-derivatized95 AONs have been shown to promote specific RNase H
cleavage when bound to their RNA targets.
1.3 Use of metal ions in the antisense and antigene strategies.
Another attractive mechanism by which the potency of ONs might be increased is to synthesize
derivatives that inactivate their nucleic acid targets directly by either irreversible cross-linkage to the
target or by secondary reaction leading ultimately to the disruption of the target molecule. Photocross-
linking96 and cross-linking by an alkylating electrophile97,98 are among the most widely applied
approaches. Cleavage of target strands either hydrolytically or through redox chemistry was mainly
achieved by metal complexes attached to ONs. In the case of cleavage reactions through backbone
hydrolysis, it is relatively easy to hydrolyze RNA target while the estimated half-life of a
phosphodiester bond in DNA is 200 million years at pH 7 and 25°C – unless metal ions come into play

12
when this process can be accelerated enormously.99 A number of cleaving agents,100 Cu2+-terpyridine,101
Lu3+-,Th3+-,Eu3+-iminodiacetate,102 Eu3+-,103 Dy3+-texaphyrin,104,105 Cu2+-N-(2-
mercaptopropionyl)glycine106 complexes, have been covalently attached to ONs and shown to hydrolyze
RNA target. Among the metal ions which cleave DNA at an unprecedented rate are ions of the
lanthanides and combinations thereof, e.g. Ce4+ and Pr3+.107 The introduction of these ions into ONs
represents a major challenge to preparative chemistry. In the case of oxidative cleavage reactions, the
most widely applied pathway is based on the initial generation of hydroxyl radical from molecular
oxygen by its reduction with transition metal ion and subsequent HO•-radical attack on heterocyclic
bases or ribose residues of nucleic acid (Fenton-type chemistry). Fe2+-EDTA-ON conjugates, pioneered
by Dervan, cleaved ssDNA (or RNA)108,109 as well as dsDNA (after triplex formation)110-112 under mild
conditions. The nuclease activity of Cu+-phenanthroline (Cu+(phen)2), introduced by Sigman, was
aimed at ssDNA,113-115 RNA,116 and dsDNA115,117 by attachment of phen ligand at different positions of
the ON. Once delivered to the target strand(s), some metal complexes covalently linked to ONs were
oxidized to the reactive higher oxidation states with co-reactants, such as H2O2 or another peroxo
compounds. The resulting oxo complexes are able to initiate oxidative cleavage of target molecule as it
was shown for metalloporphyrine-11,118,119 and Fe2+-bleomycin-linked120,121 oligonucleotides.
Polypyridyl oxo complexes of ruthenium, electrochemically or chemically obtained from their
aquaruthenium(II) precursors, cleave dsDNA and RNA122-124 through the guanine base oxidation and
H1' abstraction from the ribose moiety, but no attempts have been undertaken to covalently link this
metal species to ODNs because of their high reactivity. Finally, the cleavage of target nucleic acid has
been observed upon photoactivation of some metal complexes tethered to oligonucleotides125-128:
Excited Rh3+(phi)2(bpy)-ODN conjugates underwent the formation of radical cation centered on phi-
ligand which can, depending on the excitation wavelength, either decompose125 or initiate electron
transfer from the remote GG site of the target sequence. High energy activation (313 nm) led to the
direct DNA cleavage near the complex binding site while low energy activation (365 nm) promoted
long-range electron transfer mediated oxidation of distal GG doublet.126,128 Several Ru2+-complexes
with reduction potential in their excited state above 1.1 V (vs. SCE) have been found to promote DNA
strand breaks via an electron transfer.129-132 However, ODN labeled with one of that complexes
(Ru2+(tap)2(dpp)) surprisingly revealed that the dominant process is not a strand break, but the
formation of a cross-linked ODN•DNA duplex.133 The energy transfer from the photoexcited metal
complex to the molecular oxygen has been also proposed to occur thus generating reactive singlet
oxygen (1O2) species which damage the DNA.134
Much less success has been attained in sequence specific cross-linking of nucleic acids by metal
complexes tethered to oligonucleotides. Numerous functional metal complexes, cisplatin and its various

13
platinum analogs,135 as well as RuIICl2(DMSO)4,136 RuII(bpy)2Cl2,137,138 RuIII(tpy)Cl3,137,139
[RuII(NH3)5Cl]Cl,140,141 showed antitumor activity which was generally accepted as a result of their
covalent binding to dsDNA. The profound spatial disturbance and destabilization of the double-helix
caused by metal complex coordination lead to the inhibition of dsDNA replication. Structural studies
performed on model compounds revealed that N7 of a guanine base is the most preferable coordination
site for metal complex binding.135,139,142-145 Unfortunately, these compounds act on dsDNA with low
selectivity, and the problem of toxicity thus becomes an important factor in the use of such agents. The
only reactive metal complexes covalently linked to oligonucleotides to give sequence specific
interstrand cross-linked products were platinum derivatives, currently used in cancer chemotherapy. In
the early studies, the bifunctional platinum complex was directly introduced to ODN containing
exclusively the pyrimidine bases T and dC or one dG residue.146-149 Apart from non-selective
platination at N3 of different cytosine residues, all platinated species formed were sufficiently reactive
to form intrastrand cross-links or could be inactivated by the reactions with other nucleophiles present
in the reaction medium. Thus, the undesirable toxicity cannot be excluded for such ODN conjugates as
a consequence of the presence of the reactive group. Keeping a monofunctionally bound platinum entity
in a reactive state for cross-linking is still a major challenge. Automated insertion of platinated building
blocks during the ODN solid phase synthesis150,151 solved the problem of selective incorporation of Pt2+
to the ODN sequence but not the problem of side reactions. Ideally, functional metal species have to be
protected and back activated after binding to the target molecule. Recently, some efforts have been
made in this direction when [Pt(NH3)2(X)Cl]+-ODN conjugate (X is a 5'-aminolinker of ODN) was
obtained by acidic substitution of cyclohexylmethylthyminate group to Cl on the last stage of ODN
preparation.152,153 However, the possible occurrence of depurination154 in the acid-mediated (HCl, pH
2.3) exchange process limits this scheme to homopyrimidine ODNs only. The activation of protected
Pt2+-ODN conjugates which is exclusively triggered by the duplex formation with target DNA, has been
observed, but the activation was also accompanied by the complete release of the platinum
complex.155,156

14
2. Present Work.
2.1 Synthesis of the ODNs tethered to phenazine, dipyridophenazine, and Ru2+-
dipyridophenazine chromophores.
In the present work, we have covalently linked two sets of chromophore groups to the terminal or
internucleotide positions of ODNs listed in Table 1 and 2. One set consists of simple planar aromatic
labels phenazine (Pnz) and dipyrido[3,2-a;2',3'-c]phenazine (DPPZ), while in the second set the DPPZ
ligand is coordinated to the Ru2+ ion together with other ancillary polypyridyl ligands in octahedral
geometry. Generally, the conjugation sites have been designed in all cases as follows:
ChromophoreLinker Nucleoside residue or non-nucleosidic analog
R2
R2 = 3'-ODN part or H
R1 = 5'-ODN part or H
R1
Two different strategies have been exploited for the coupling of conjugate groups to ODNs: (i) coupling
of chromophore through a linker to a nucleoside residue or non-nucleosidic analog and subsequent
incorporation of the resulting building block into ODN during the solid phase synthesis, and (ii) post-
synthetic modification of amino-linker functionalized ODN with N-hydroxysuccinimidyl (NHS) ester of
the appropriate carboxyl-containing chromophore.
2.1.1 Synthesis of Pnz labeled ODNs (Paper I)
To label ODNs with Pnz, 2-(N-methylamino)ethanol has been chosen as a linker and coupled first to
Pnz at C2 position by nucleophilic substitution in 9-methyl phenazinium methylsulphate 69 (Scheme 1),
which gave us 2-(N-(2-hydroxyethyl)-N-methyl)aminophenazine 70.53,157 Compound 70 was
converted in the usual way158 to the corresponding phosphoramidite 71 and then coupled via tetrazole
activation159 to the 4',6'-TPDS-protected 1-(3'-deoxy-β-D-psicofuranosyl)uracil nucleoside 73 prepared
in six steps from D-fructose.160 Nucleosides 72 and 73 mimics 2'-deoxyribonucleosides but has an
additional hydroxymethyl functionality at 1'-position which can be used for the attachment of conjugate
and linker groups. The coupling product 74, in which Pnz is connected to the nucleoside unit through
the methyl phosphotriester, was deprotected by treatment with TBAF in THF and then 6'-O-DMT-
blocked (74→75→76, Scheme 1). After 75 was phosphitylated or anchored to the

15
Table 1. Oligonucleotides synthesized for ODN•DNA and ODN•RNA duplex study
Sequence Oligo N
5'-TCCAAACAT-3'
5'-TCCATACAT-3'
1
2
5'-PnzTCCAAACAT-3'
5'-PnzUCCAAACAT-3'
5'-TCCAAACAPnzU-3'
5'-TCCAPnzUACAT-3'
5'-PnzUCCAAACAPnzU-3'
5'-TCCAAACAUPnz-3'
3
4
5
6
7
8
DPPZ-Ln-5'-pTCCAAACAT-3'
5'-TCCAAACATp-3'-Ln-DPPZ
5'-TCCAp-Ln(DPPZ)-pAACAT-3'
5'-TCCAp-Ln(DPPZ)-pACAT-3'
9,10,11 (n=1,2,3)
12,13,14 (n=1,2,3)
15,16,17 (n=1,2,3)
18,19,20 (n=1,2,3)
Phos
phod
iest
er
back
bone
(phen)2Ru2+DPPZ-L2-5'-pTCCAAACAT-3'
5'-TCCAAACATp-3'-L2-DPPZ Ru2+(phen)2
5'-TCCAp-Ln[DPPZ Ru2+(phen)2]-pAACAT-3'
21
22
23
Sequ
ence
I
Phos
phor
othi
oate
bac k
bone
PS-5'-TCCAAACAT-3'
PS-5'-PnzTCCAAACAT-3'
PS-5'-TCCAAACAUPnz-3'
PS-DPPZ-L2-5'-pTCCAAACAT-3'
PS-5'-TCCAAACATp-3'-L3-DPPZ
24
25
26
27
28
DNA targets
for sequence I
5'-CATGTTTGGAC-3'
5'-CATGTATGGAC-3'
29
30
RNA targets
for sequence I
5'-r(CAUGUUUGGAC)-3'
5'-r(ACUCAUGUUUGGACUCU)-3'
5'-r(UAACAUGUUUGGACUCU)-3'
31
32
33
5'-CTTACCAATC-3' 34
L2-5'-pCTTACCAATC-3'
5'-CTTACCAATCp-3'-L2
5'-CTTACp-L2-pCAATC-3'
L2-5'-pCTTACCAATCp-3'-L2
35
36
37
38
Sequ
ence
II
Phos
phod
iest
er
back
bone
MeCN(tpy)Ru2+DPPZ-L2-5'-pCTTACCAATC-3'
5'-CTTACCAATCp-3'-L2-DPPZ Ru2+(tpy)NCMe
5'-CTTACp-L2[DPPZ Ru2+(tpy)NCMe]-pCAATC-3'
X-5'-pCTTACCAATCp-3'-X, X=L2-DPPZ Ru2+(tpy)NCMe
39
40
41
42
DNA target
for sequence II 5'-TGATTGGTAAG-3' 43

16
Table 2. Oligonucleotides synthesized for ODN×DNA•DNA triplex study
Sequence Oligo N
5'-TTCT6CT6CT-3'
44
5'-PnzTTCT6CT6CT-3'
5'-PnzUTCT6CT6CT-3'
5'-TTCT6CT6CPnzU-3'
5'-TTCT5PnzUCT6CT-3'
5'-PnzUTCT6CT6CPnzU-3'
45
46
47
48
49
DPPZ-Ln-5'-pTTCT6CT6CT-3'
5'-TTCT6CT6CTp-3'-Ln-DPPZ
5'-TTCT5Tp-Ln(DPPZ)-pCT6CT-3'
5'-TTCT5Tp-Ln(DPPZ)-pT6CT-3'
50,51,52 (n=1,2,3)
53,54,55 (n=1,2,3)
56,57,58 (n=1,2,3)
59,60,61 (n=1,2,3)
Triplex forming
oligos
(TFOs)
(phen)2Ru2+DPPZ-L2-5'-pTTCT6CT6CT-3'
5'-TTCT6CT6CTp-3'-L2-DPPZ Ru2+(phen)2
5'-TTCT5Tp-L2[DPPZ Ru2+(phen)2]-pCT6CT-3'
62
63
64
Duplex targets
for triplex
formation
5'-GCCAAGA6GA6GACGC-3'
3'-CGGTTCT6CT6CTGCG-5'
5'-GCCAAAAAAGA6GA6GACGC-3'
3'-CGGTTTTTTTCT6CT6CTGCG-5'
65
66
67
68
3-aminopropyl-CPG support,161 the resulting 4'-O-phosphoramidite 77 and Pnz-modified support 78
were used as building blocks in solid phase synthesis, permitting Pnz incorporation to the 5'-, 3'- or
internucleotide positions of the oligo chain (ODNs 4 –7, 46 – 49, Tables 1 and 2). Phosphoramidite 71
(R = CH2CH2CN) was also directly used for 5'-Pnz-conjugation as in ODNs 3, 25, 45.
2.1.2 Synthesis of amino-linkers of different lengths tethered to the glycerol moiety (Paper II)
The DPPZ chromophore including its Ru2+ complexes have been attached to the non-nucleosidic
glycerol moiety through the ethylene glycol linkers of different length. The shortest 3-aminopropyl
linker has been connected to the glycerol moiety by the addition of readily available solketal 79 to
acrylonitrile with subsequent reduction of the adduct 80 with NaBH4 in the presence of CoCl2 to give
finally 3-(alkyloxy)propanamine 81 (Scheme 2).162,163 Longer tri- and penta(ethylene glycol) arms 82

17
Abbreviations used in Table 1 and Table 2
N
N
N
Ru
N
N
N
N
N
CH3
O2+
NH
O
ON
OOPO
O-
O
ON
N N
NH
O
ON
OO
P O
-O
OO
N
NNO
CH3
CH3
NH
O
ON
OO
ON
N
O
PO-O
OHN
N
N
N
NO
NN
NN
Ru
2+
N
N
N
NO
HN
OO
O
O
n
HNO
O
O5'
3'
5'
5'
3'
3'
5'
3'
3'
PnzT =
PnzU =
UPnz =
L1 = L2 (n=2)L3 (n=4) =
DPPZ =
(phen)2Ru2+DPPZ =
MeCN(tpy)Ru2+DPPZ =

18
N
NOH
N
N N
CH3
O
N
N N
CH3
PN(iPr)2
OR
NH
O
ON
OHO
OHOH
NH
O
ON
OO
OOH
Si
SiO
NH
O
ON
OO
P O
O
OCH3O
N
NNO
CH3
Si
SiO
NH
O
ON
OR1O
P O
O
OCH3OR2
N
NNO
CH375: R1 = H, R2 = H76: R1 = DMT, R2 = H
Pnz 70 71: R = CH3 or CH2CH2CN
73
7274
NH
O
ON
ODMTO
P O
O
OCH3O
N
NNO
CH377
78
POCH2CH2CN(iPr)2N
NH
O
ON
ODMTO
P O
O
OCH3O
N
NNO
CH3
HN
O
O
S
N
N
69CH3CH3OSO3
-
Scheme 1. Synthesis of building blocks for Pnz incorporation into ODNs 3 – 7, 25, 45 – 49 (Tables 1 and 2).
and 83 were first mono-tosylated and the tosyloxy groups in the resulting compounds 84 and 85 were
then substituted with phthalimido function to afford the protected precursors 86 and 87. They were
treated with allyl bromide in the presence of NaH affording olefins 88 and 89. Subsequently, double
bond oxidation with OsO4 or KMnO4 gave diols 90 and 91, in order to build the sn-glycerol moiety

19
OHO
O
OO
O
CNO
O
O
NH2
O
DMTO
O
DMTO
POCH2CH2CN(iPr)2N
HN
O
O
S
OHO OR
n
OHO
nN
O
O
OO
nN
O
O
OO
N
O
OR2O
R1O
n
OO
NHR2R1O
DMTO
n
79 8180
82: n = 2, R = H83: n = 4, R = H
84: n = 2, R = Ts85: n = 4, R = Ts
86: n = 2
90: n = 2, R1 = R2 = H91: n = 4, R1 = R2 = H
92: n = 2, R1 = DMT, R2 = H93: n = 4, R1 = DMT, R2 = H
94: n = 2, R1 = R2 = H95: n = 4, R1 = R2 = H
96: n = 2, R1 = H, R2 = Fmoc
OO
NHFmoc
2
OO
NHFmoc
297 98
87: n = 488: n = 289: n = 4
Scheme 2. Preparation of linkers for conjugation with dipyridophenazine (DPPZ) containing chromophores (including building blocks 97 and 98 which were directly used for the synthesis of ODNs 35 – 38, Table 1). connected to the linker arms.164 DMT-protection of primary hydroxyl in these diols with subsequent
removal of phthalimido group by refluxing with hydrazine165 gave finally sn-glycerol conjugated
aminolinkers 94 and 95. The amino functionalized compounds 81, 94, and 95 were then used for
condensation with carboxyl derivatives of DPPZ and its Ru2+ complexes. Moreover, 94 was also
utilized for the preparation of amino-linkertethered ODNs 35 – 38 (Table 1) which were subsequently
subjected for the post-synthetic labeling reactions. For this purpose, amino group in 94 was Fmoc-
protected to give compound 96, which was converted into O-(2-cyanoethyl)(N,N-
diisopropyl)phosphoramidite block 97.158 The remaining part of 96 was succinylated and the
corresponding succinate was attached166 to the 3-aminopropyl-CPG support to provide 49 µmol of
bound 96 per gram CPG (98).

20
N
N
N
NO
NN
NN
RuOH
2+
N
N
N
NO
OH
N
N
N
N O
O
99 (phen) 100 101 (DPPZ-COOH)
RuCl3 Ru(phen)2Cl2
102
99(i) 100
2 PF6-
103
H2N
H2NO
OH
N
N
N
Ru
N
N
N
N
COOH
Cl
+
N
N
N
Ru
N
N
N
N
COOH
N
2+
CH3
O
O
N
N
NN
NN
Ru
2+ 2 PF6-
2 Cl-Cl-Ru(tpy)Cl3
terp
yrid
ine
101
104 105
(ii) NH4PF6
Scheme 3. Preparation of dipyridophenazine (DPPZ) containing chromophores for conjugation.
2.1.3 Derivatization of DPPZ containing chromophores for conjugation (Papers II, IV & V)
To be able to attach DPPZ and its Ru2+ complexes to the amino-linker containing blocks 81, 94, 95
(Scheme 2) or to the aminolinker modified ODNs 35 – 38 (Table 1), we prepared dipyrido[3,2- a;2',3'-
c]phenazine-11-carboxylic acid 101 in two steps from 1,10-phenanthroline 99 by its oxidation to 1,10-
phenanthroline-5,6-dione 100 and the following condensation with 3,4-diaminobenzoic acid in ethanol
(Scheme 3).167 Next, we coordinated DPPZ-COOH ligand 101 around Ru2+ center to obtain
tris(polypyridyl) ruthenium complex [Ru(phen)2(dppz-COOH)]2+ 103 as well as bis(polypyridyl)
ruthenium complexes, such as [Ru(tpy)(DPPZ-COOH)Cl]+ 104 and [Ru(tpy)(DPPZ-
COOH)(CH3CN)]2+ 105 containing monodentate ligands (Cl, CH3CN). Common synthetic approaches
to the preparation of complexes of general formulas [Ru(NN)2(NN)']2+ and [Ru(NNN)(NN)X]n+ (NNN

21
and NN denote tri- and bidentate polypyridyl ligands respectively, X is Cl or CH3CN) have been
applied in the synthesis of 103 - 105. Ru(phen)2Cl2167 was coordinated with ligand 100 in refluxing
ethanol-water and the product 102 was condensed with 3,4-diaminobenzoic acid to give finally complex
103.168 In a similar manner, complex 104 was formed starting from Ru(tpy)Cl3169 by coordination with
DPPZ-COOH (101) in refluxing ethanol-water in the presence of LiCl and triethylamine.170 In this
reaction triethylamine functions as a reducing agent to help the dissociation of Cl from Ru(tpy)Cl3,
whereas LiCl is used to prevent any dissociation of Cl from the product. The Cl ligand was then
readily replaced in 104 by CH3CN ligand upon heating under reflux in acetonitrile-water mixture171,172
to afford complex 105.
2.1.4 Synthesis of DPPZ and Ru2+(phen)2DPPZ labeled ODNs (Papers II & IV)
Carboxyl-derivatized DPPZ chromophore 101 (Scheme 3) was subjected for the condensation with the
amines 81, 94, and 95 (Scheme 2) using N,N'-carbonyldiimidazole173 as a condensing agent in pyridine
under heating (80ºC) which resulted in the formation of corresponding amides 106, 111, and 112
(Scheme 4). These compounds were highly contaminated with side products arising from heating of the
reaction mixture but poor solubility of 101 in any organic solvent prevented utilization of other more
soft amide-forming strategies. The DPPZ derivative 106 was then treated with 1M HCl / THF (1:1, v/v)
to remove isopropylidene protection, and the crude product 107 was tritylated to give 108. Finally,
compounds 108, 111, and 112 were converted in the usual manner158 to the corresponding
phosphoramidites 109, 113, and 114 as well as immobilized161 onto 3-aminopropyl-CPG support
(110, 115, 116). The building blocks thus prepared enabled incorporation of the DPPZ chromophore
through the linkers of different lengths at any desired position of an oligo sequence as in ODNs 9 –
20, 27 – 28, and 50 – 61 (Tables 1 and 2) by solid phase synthesis. The present ODN conjugates are the
first examples of oligonucleotides tethered with DPPZ chromophore.
Analogously, hexafluorophosphate salt of Ru2+ complex 103 (Scheme 3) was treated with N,N'-
carbonyldiimidazole174 in pyridine at room temperature and the intermediate acylimidazole was
quenched with amine 94 (Scheme 2) in one pot giving compound 117 (Scheme 4). Next, 2-
cyanoethylchloro-(N,N-diisopropyl)phosphoramidite was reacted with 117 in dry CH3CN175 to afford
metallo-phosphoramidite 118. After drying, 118 was used in the solid phase synthesis of 5'-Ru2+-ODN
conjugates 21, 62, and internucleotide conjugated Ru2+-ODNs 23, 64 (Tables 1 and 2). Compound 117
was also attached161 to the 3-aminopropyl-CPG to assemble ODNs modified with [Ru(phen)2DPPZ]2+-
moiety at the 3'-terminal as in 22 and 63 (Tables 1 and 2). Thus, the novel type of attachment through
the DPPZ moiety of [Ru(phen)2DPPZ]2+ complex has been implemented for its ODN conjugation.

22
OO
OHN DPPZ
O
OHO
ROHN DPPZ
O
OO
DMTOHN DPPZ
O
OO
DMTOHN DPPZ
OPOCH2CH2CN(iPr)2N
HN
O
O
S
N
N
N
NO
OH
101 (DPPZ-COOH)
81
106 107: R = H108: R = DMT
OO
HN
HO
DMTO
n
DPPZ
O
94
95or
110
109
111: n = 2112: n = 4
OO
HN
O
DMTO
n
DPPZ
O113: n = 2114: n = 4
POCH2CH2CN(iPr)2N
OO
HN
O
DMTO
n
DPPZ
O115: n = 2116: n = 4
HN
O
O
SN
N
N
NO
NN
NN
Ru OH
2+ 2 PF6-
103
OO
HN
HO
DMTO
2
DPPZ Ru2+(phen)2
O
94117
OO
HN
O
DMTO
2
DPPZ Ru2+(phen)2
O118
OO
HN
O
DMTO
2
DPPZ Ru2+(phen)2
O119
POCH2CH2CN(iPr)2N
HN
O
O
S
Scheme 4. Synthesis of building blocks for DPPZ and (phen)2Ru2+DPPZ incorporation into ODNs 9 – 23, 27 - 28, and 50 – 64 (Tables 1 and 2).

23
2.1.5 Synthesis of Ru2+(tpy)(DPPZ)(CH3CN) labeled ODNs (Paper V)
At present time, incorporation of metal complexes into ODNs is generally restricted to the linking of
only substitution-inert polypyridyl complexes of Ru2+ 133,134,174-190 and Rh3+.126,127,191,192 One of the
biggest advantages of utilizing such complexes is the fact that their chelation chemistry is less
demanding than that of the functional metal species, and can be compatible both with the synthesis of
ODN building blocks and post-synthetic labeling strategy. Contrary, conjugation of functional metal
complexes requires the use of protection ligand which should be chemically and thermally stable on one
hand, and on the other hand, it should be readily substituted onto a labile ligand under specific
conditions without altering of the ODN part. In the course of our attempts to synthesize
[Ru(tpy)(DPPZ)(H2O)]2+-ODN conjugates, we used [Ru(tpy)(DPPZ)(X)]n+-precursors (X = Cl,
CH3CN) in which X can be thermally (X = Cl)171 or photolytically (X = CH3CN)172,193,194 replaced by
aqua ligand. Due to the thermal dissociation of Cl ligand in [Ru(tpy)(DPPZ-COOH)Cl]+ complex 104
(Scheme 3) occurring in aqueous solutions, its conjugation is not compatible with the conditions
normally applied for the post-synthetic deprotection of the synthesized ODN (conc. aq. NH3, 55ºC) and
subsequent HPLC purification. Replacement of Cl¯ by NH3 during deprotection step as well as by
CH3CN or other coordinating ligands present in HPLC buffers leads to the mixture of conjugates in
which different monodentate ligands are bonded to the Ru2+ center. On the other hand, Meyer at
al.195,196 reported on the incorporation of [Ru(tpy)(bpy')Cl]+ (bpy' = 4-carboxy-4'-methyl-2,2'-
bypyridine) into polymers and peptides indicating that Cl remains unaltered under conditions of
condensation in anhydrous solutions, although no clear spectroscopic evidence was given. This
prompted us to prepare anchored to the solid support building block 123 (Scheme 5A) and use it in
automated solid phase ODN synthesis with subsequent substitution of Cl ligand in ODN-conjugated
Ru2+ complex by thermally stable CH3CN ligand after mild deprotection with conc. aq. NH3 at room
temperature. According to Scheme 5A, the key compound 122 was prepared through either amide bond
formation (as in 104→124→122) or through the coordination of compounds 111 or 120 to Ru(tpy)Cl3
(as in 111→122 or 120→121→122). The amide bond formation has been accomplished by
conversion of a chloride salt of 104 to its NHS-ester 124 upon treatment with TSU in dry DMF.133 The
activated complex 124 was then reacted with amine 94 (Scheme 2) in dry DMF and chloride salt of the
product was metathesized to the hexafluorophosphate salt 122 by treatment with excess ethanolic
NH4PF6. Alternatively, complex 121 was formed by coordination of Ru(tpy)Cl3 with the functionalized
DPPZ ligand 120 in boiling ethanol-water (120 was obtained by deprotecton of 111 with aqueous 80%
acetic acid). The presence of reducing agent triethylamine in the reaction between 120 and Ru(tpy)Cl3

24
N
N
N
Ru
N
N
N
N
COOH
Cl
+
N
N
N
Ru
N
N
N
N
COOH
N
2+
CH3
2 Cl-
Cl-
Ru(tpy)Cl3
104
105
OO
HN
HO
RO
2
DPPZ
O120: R = H111: R = DMT
OO
HN
HO
RO
2
DPPZ Ru2+(tpy)Cl
O
121: R = H122: R = DMT
PF6-
N
N
N
Ru
N
N
N
N
Cl
+ Cl-
124
OO
NO
O
OO
HN
O
DMTO
2
DPPZ Ru2+(tpy)Cl
OPF6
-
HN
O
O
S
(i) 94
123
N
N
N
Ru
N
N
N
N
125
OO
NO
O
N
CH3
2+ 2 Cl-
Oligonucleotides 39 - 42
OO
NH2R2O
R1O
235: R1 = H, R2 = pCTTACCAATC-3'36: R1 = 5'-CTTACCAATCp, R2 = H37: R1 = 5'-CTTACp, R2 = pCAATC-3'38: R1 = H, R2 = pCTTACCAATCp-3'-CH2CH(OH)((OCH2CH2)3NH2)
A
B
(ii) NH4PF6
Scheme 5. (A) Synthesis of solid support for the 3'-incorporation of MeCN(tpy)Ru2+DPPZ moiety (ODN 40, Table 1). (B) Post-synthetic labeling of ODNs 35 – 38 for MeCN(tpy)Ru2+DPPZ incorporation at different terminals (ODNs 39 – 42, Table 1).

25
enabled us to similarly modify DMT-protected analog 111 and prepare compound 122.
Correspondingly, both 121 and 122 were isolated as PF6 salts to ensure their solubility in organic
solvents. Complex 121 was also shown to produce 122 upon treatment with DMT-Cl in dry pyridine at
room temperature. Attachment of 122 to 3-aminopropyl-CPG allowed to prepare [Ru(tpy)(dppz)Cl]+-
modified solid support and use it for automated assembly of 3'-[Ru(tpy)(dppz)Cl]+-ODN conjugate. The
fast deprotecting 5'-O-DMT-N4-isobutyryl-2'-deoxycytidine-3'-O-(β-cyanoethyl-N,N-diisopropyl)- and
5'-O-DMT-N6-phenoxyacetyl-2'-deoxyadenosine-3'-O-(β-cyanoethyl-N,N-diisopropyl)-phosphor-
amidites197,198 utilized in this ODN synthesis enabled to apply mild ammonia deprotection at room
temperature which does not effect the Cl displacement. This was proven by us on a model
[Ru(tpy)(dppz)Cl](PF6) complex. After removal from solid support and lyophilization, the crude
material was incubated in CH3CN-H2O (1:1, v/v) at 55ºC for 17 h to substitute Cl to the
thermally stable CH3CN at the metal center to give 3'-[Ru(tpy)(dppz)(CH3CN)]2+-ODN conjugate 40
(Table 1). Unfortunately, our attempts to prepare metallo-phosphoramidite from compound 122 and use
it for 5'- or internucleotide incorporation failed. This can be explained by the possible displacement of
Cl¯ during interaction between the Ru2+ and phosphite center which accompanies the phosphitilation of
OH group.199,200
Contrary to the [Ru(tpy)(dppz)Cl]+, its Ru2+-CH3CN analog undergoes base hydrolysis201 of the
coordinated CH3CN ligand to acetamide which is released very rapidly from the coordination sphere of
Ru2+, since amides are poor π-acceptor ligands.202 Such decomposition excludes the use of Ru2+-
CH3CN species in the solid phase synthesis, requiring removal of the ODN from solid support and
deprotection with conc. aq. NH3. Hence, the [Ru(tpy)(DPPZ-COOH)(CH3CN)]2+ complex 105 (Scheme
3) was introduced to ODN by amide bond formation between NHS-ester 125 and aminolinker-
functionalized ODNs 35 – 38 (Scheme 5B). For this purpose, complex 105 was activated analogously
as in 104→124 step (Scheme 5A) to give 125 which was incubated with appropriate ODN in 33%
CH3CN / 10 mM sodium tetraborate (pH 8.5) buffer and the product was separated from the excess of
Ru2+ complex by cation exchange column chromatography. This post-synthetic modification has been
proven to be valuable for incorporation of metallating species containing sensitive functionality, as in
ODNs 39 – 42 (Table 1). The methodology described here shows for the first time that mono-
acetonitrile polypyridyl Ru2+ complex tethered to a oligodeoxynucleotide (as in ODNs 39 – 42) can be
constructed by post-synthetic labeling of amino-linker modified ODN, thus opening the way to design
new class of metal-ODN conjugates carrying photochemically reactive metal entity.

26
2.1.6 Characterization of Ru2+(tpy)(DPPZ)(CH3CN) labeled ODNs (Paper V)
The main structural feature of the newly synthesized ODNs 39 – 42 (Table 1) tethered with
monofunctional Ru2+(tpy)(DPPZ)(CH3CN) complex is that they carry photolabile CH3CN ligand in
contrast to other Ru2+- 133,134,174-190 or Rh3+-conjugated126,127,191,192 ODNs prepared till present and
characterized by inertness towards the ligand substitution reactions in the metal coordination sphere. To
show the presence of the sensitive functionality, ODNs 39 – 42 have been carefully characterized by
enzymatic digestion nucleobase analysis, PAGE, as well as by UV-vis and MS-spectroscopy. All
metallated ODNs exhibited the low-energy metal-to-ligand charge transfer (MLCT Ru2+→CH3CN)
bands at 453 nm and DPPZ intraligand transition at 376 nm identically with those for the not tethered
monomer block [Ru(tpy)(DPPZ-CO-L2)(CH3CN)]2+ (L2 is a linker, see page 17).172,194,201 The
substitution of CH3CN by other monodentate ligands leads to the shift of the MLCT band as has been
shown for [Ru(tpy)(DPPZ-CO-L2)X]2+ (X = Cl, py) complexes. The A260/A453 absorption ratio for the
ODN conjugates was found to be higher (9.1) then the corresponding ratio for the not tethered Ru2+-
CH3CN complex (3.5) reflecting the contribution of the DNA bases at 260 nm in the ruthenated oligos.
PAGE analysis revealed that the metal complex-ODN conjugates have less electrophoretic mobility
compared to those of the amino-linker L2 modified counterparts (ODNs 35 – 38, Table 1). This is
owing to the increase of molecular weight (685 dalton per [Ru(tpy)(DPPZ-CO-)(CH3CN)]2+ residue)
and the decrease of the overall negative charge by a factor of 2 in the Ru2+-ODNs. The base
composition as well as the presence of tethered Ru2+ complex was further confirmed by the degradation
of ODN conjugates to nucleosides upon treatment with a mixture of snake venom phosphodiesterase
(SVDP) and alkaline phosphotase (AP).203 The identity of nucleoside peaks in the digest mixtures was
proven by comparison of their retention times with those of the authentic mixture of dC, dT, and dA
nucleosides. The ratio of normalized areas under nucleoside peaks in the digest of mono-Ru2+-labeled
ODNs was found 4.1 : 3 : 2.9 which is close to the nucleoside composition in the native ODN 34 (Table
1). On the other hand, the appearance of two strongly retarded peaks in the HPLC profile of the digest
mixtures was assigned to the Ru2+ species. We suspect that they are the products of the hydrolysis of
coordinated CH3CN ligand under digestion conditions (pH 9.0)201 and subsequent acetamide
substitution onto other entering groups existing in the enzymatic coctail.
The final proof of the identity of [Ru(tpy)(DPPZ)(CH3CN)]2+-ODN conjugates has been obtained
by MALDI-TOF and ESI-mass spectrometry. The development of these soft ionization techniques
enabled us to characterize both natural and synthetic oligonucleotides at the structural level.204-208 The
negative ion mode ESI MS of the mono-metallated ODNs 39 – 41 (calculated MW 3916 in the fully
phosphodiester protonated and 2+ charged state) and bis-metallated ODN 42 (calculated MW 4885 for
the 4+ charged state) showed a number of multiply charged ions, which after deconvolution, revealed

27
the expected ODN anions in the range of mass/charge (m/z) = 3912.5 – 3913.8 for the ODNs 39 – 41
and 4880.9 for the ODN 42 corresponding respectively to [M-3H+] and [M-5H+] anions. From this
data one can conclude that all conjugates have the CH3CN ligand intact in the coordination sphere of
their Ru2+-label. On the other hand, when the ODNs were ionized in the negative mode by laser pulse
(MALDI-TOF MS), the observed mass/charge (m/z) ratio were in the range of 3871.9 – 3872.9 for the
single-modified ODNs 39 – 41 in the 1- charge state. The difference between the m/z values obtained
from MALDI and ESI ionizations for the single Ru2+ modified ODNs 39 – 41 is approximately 41 Da
and expected from the photolabilization of CH3CN ligand occurring during the laser irradiation.
Similarly, laser ionization of the 5',3'-bis-Ru2+- modified ODN 42 led to the loss of two acetonitrile
ligands affording the 1- charged ions with m/z of 4798.4: M[(C186H208N51O74P11102Ru2)4+]-M[2CH3CN
+ 5H+]. Thus, MS-analysis of the Ru2+-ODN conjugates established the presence of the covalently
linked [Ru(tpy)(DPPZ)(CH3CN)]2+ monofunctional complex in the ODN chain. Comparison of the MS
data obtained on the bases of use of the different ionization techniques clearly showed that synthesized
ODN conjugates are photochemically active and undergo the expected CH3CN ligand decomposition
under light irradiation.
2.2 Thermal stability of duplexes and triplexes formed by phenazine, dipyridophenazine,
and Ru2+-dipyridophenazine conjugated ODNs.
2.2.1 Thermal stability of conjugated ODN•DNA duplexes (Papers I, II, IV & V)
Different chromophore groups tethered to natural ODNs are expected to influence the hybridization
properties of the resulting ODN conjugates with the complementary DNA and RNA molecules. The
analysis of melting temperatures of differently conjugated ODN•DNA duplexes shows (Table 3) that
Pnz chromophore tethered at the 1'-position of uridine moiety (PnzU) through the relatively short linker
has the least ability to stabilize a duplex. The stabilization was only achieved when the Pnz-tethered PnzU was attached at the 5'-terminal (∆Tm = 6.1°C) while 3'-conjugation gave slight destabilization (∆Tm
= -0.9°C) and middle-conjugation led to almost complete loss of stability (∆Tm = -12.9°C) of the
duplex. Double Pnz-modification at both 5'- and 3'-termini gave also positive ∆Tm value (2.5°C), albeit
lower than for single 5'-Pnz-conjugation, probably as a result of negative influence of 3'-Pnz-tether.
Interestingly, the same type of Pnz-tethering at 5'-hydroxyl of a 5'-terminal T (PnzT as in the duplex
3•29) allowed to strengthen the stabilization effect (∆Tm = 10.0°C) reflecting better positioning of a
stacker when it is delivered from the 5'-hydroxy terminal of the duplex. Contrary, DPPZ conjugation
allowed us to stabilize ODN•DNA duplexes more effectively. As expected, the stabilization effect of
the DPPZ chromophore depended on the linker length as well as on the position of the linker
attachment to the ODN. As a whole, 5'- and 3'-DPPZ-conjugation lead to comparably equal
28
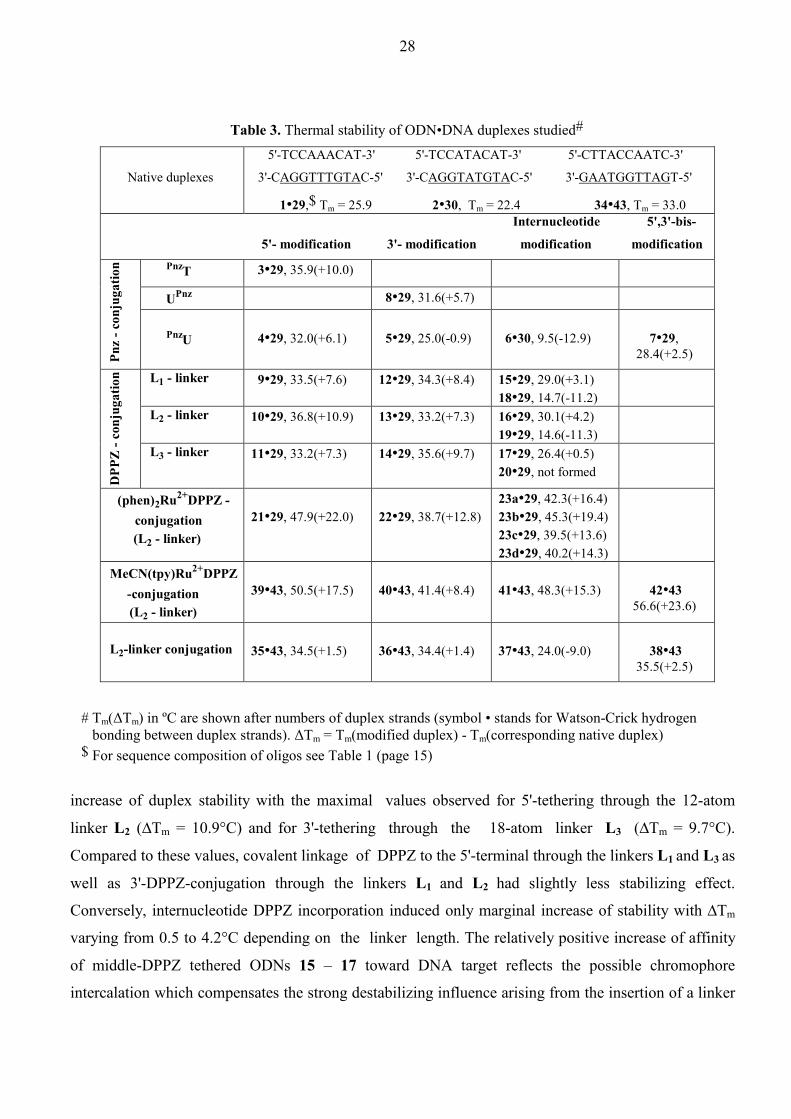
Table 3. Thermal stability of ODN•DNA duplexes studied#
Native duplexes
5'-TCCAAACAT-3' 5'-TCCATACAT-3' 5'-CTTACCAATC-3'
3'-CAGGTTTGTAC-5' 3'-CAGGTATGTAC-5' 3'-GAATGGTTAGT-5'
1•29,$ Tm = 25.9
2•30,
Tm = 22.4
34•43, Tm = 33.0
Internucleotide 5',3'-bis-
5'- modification
3'- modification modification
modification
PnzT 3•29, 35.9(+10.0)
UPnz 8•29, 31.6(+5.7)
Pnz
- con
juga
tion
PnzU
4•29, 32.0(+6.1)
5•29, 25.0(-0.9)
6•30, 9.5(-12.9)
7•29, 28.4(+2.5)
L1 - linker 9•29, 33.5(+7.6)
12•29, 34.3(+8.4) 15•29, 29.0(+3.1) 18•29, 14.7(-11.2)
L2 - linker 10•29, 36.8(+10.9)
13•29, 33.2(+7.3) 16•29, 30.1(+4.2) 19•29, 14.6(-11.3)
DPP
Z - c
onju
gatio
n
L3 - linker 11•29, 33.2(+7.3)
14•29, 35.6(+9.7) 17•29, 26.4(+0.5) 20•29, not formed
(phen)2Ru2+DPPZ - conjugation
(L2 - linker)
21•29, 47.9(+22.0)
22•29, 38.7(+12.8)
23a•29, 42.3(+16.4) 23b•29, 45.3(+19.4) 23c•29, 39.5(+13.6) 23d•29, 40.2(+14.3)
MeCN(tpy)Ru2+DPPZ -conjugation
(L2 - linker)
39•43, 50.5(+17.5)
40•43, 41.4(+8.4)
41•43, 48.3(+15.3)
42•43 56.6(+23.6)
L2-linker conjugation
35•43, 34.5(+1.5)
36•43, 34.4(+1.4)
37•43, 24.0(-9.0)
38•43 35.5(+2.5)
# Tm(∆Tm) in ºC are shown after numbers of duplex strands (symbol • stands for Watson-Crick hydrogen bonding between duplex strands). ∆Tm = Tm(modified duplex) - Tm(corresponding native duplex) $ For sequence composition of oligos see Table 1 (page 15)
increase of duplex stability with the maximal values observed for 5'-tethering through the 12-atom
linker L2 (∆Tm = 10.9°C) and for 3'-tethering through the 18-atom linker L3 (∆Tm = 9.7°C).
Compared to these values, covalent linkage of DPPZ to the 5'-terminal through the linkers L1 and L3 as
well as 3'-DPPZ-conjugation through the linkers L1 and L2 had slightly less stabilizing effect.
Conversely, internucleotide DPPZ incorporation induced only marginal increase of stability with ∆Tm
varying from 0.5 to 4.2°C depending on the linker length. The relatively positive increase of affinity
of middle-DPPZ tethered ODNs 15 – 17 toward DNA target reflects the possible chromophore
intercalation which compensates the strong destabilizing influence arising from the insertion of a linker

29
between internal dA residues of natural ODN 1. Such linker insertion structurally increases the distance
between the neighboring dAs which should lead to bulge formation in the middle-modified duplexes to
provide Watson-Crick paring of given dAs with complementary nucleobases.34 This strongly disrupts
the helix as is clearly seen from the introduction of a 12-atom linker L2 into the interior position of a
duplex as in 37•43 (∆Tm = -9.0°C). Note that linker L2 externally tethered at 5'-, 3'- or both terminals
does not have a great influence on the hybridization affinity of ODNs 35, 36, 38 as is seen from the
thermal stability of the corresponding duplexes (∆Tm = 1.4 – 2.5°C). In order to exclude the bulge
formation effected by internal insertion of DPPZ unit, we prepared duplexes 18•29, 19•29, and 20•29,
in which the DPPZ-linker units were introduced instead of the central dA nucleotide of the parent
sequence 1, with the expectation that the DPPZ chromophore would place the opposite T base of the
complementary target outside of the helix.34 Experimental results showed that duplexes so formed are
very unstable indicating appearance of a mismatch pair DPPZ-T. In general, more efficient
enhancement of hybridization affinity of DPPZ-ODN conjugates towards DNA compared to the Pnz-
tethered analogs can be explained on the basis of the following considerations: (1) DPPZ itself has more
ability for intercalation because of its more extended planar surface. (2) The use of flexible linkers
attaching the chromophore to the non-nucleosidic glecerol residue of ODN sequence provides the
delivery and optimal orientation for the stacker more effectively.
When coordinated to Ru2+, as in Ru2+(DPPZ)-labeled ODNs, the intercalating ability of DPPZ is
modulated by other ancillary ligands. It was shown earlier that positively charged polypyridyl Ru2+
complexes can bind to dsDNA electrostatically209,210 as well as through hydrophobic interactions in
DNA grooves211 or intercalation by one of the extended ligands.209,212,213 Because in our Ru2+(DPPZ)-
ODN conjugates the DPPZ ligand is sterically hampered by covalent attachment of a linker arm L2 (see
abbreviations on page 17) we first evaluated the ability of complex [Ru(phen)2(DPPZ-CO-L2)]2+ to
intercalate into the native duplex 1•29 by comparison with binding affinity of typical metallointercalator
[Ru(phen)2DPPZ]2+ and groove-binder [Ru(phen)3]2+.214-217 The results of titration of duplex 1•29 with
Ru2+ complexes clearly showed that our linker-tethered Ru2+ complex and [Ru(phen)2DPPZ]2+ behave
similarly, sharply increasing duplex stability upon increasing Ru2+ concentration up to 3 – 4 equiv. per
duplex after which Tm increase becomes slow. This trend is general for intercalation of metal complexes
taking place until all intercalation sites are saturated. After this the stabilization is due to electrostatic
binding, and Tm increases less steeply. On the contrary, growing amount of [Ru(phen)3]2+ does not
practically result in the duplex stabilization.129 Thus, the [Ru(phen)2(DPPZ-CO-L2)]2+ binding should
proceed via intercalation of DPPZ ligand despite its derivatization with a long linker arm.
In conformity with the above data, tethering of the [Ru(phen)2DPPZ]2+ and
[Ru(tpy)(DPPZ)(CH3CN)]2+ complexes to ODN through the linker L2 dramatically enhanced the

30
hybridization affinity of the corresponding conjugates towards target DNA (Table 3). The magnitude of
duplex stabilization for various site-specific [Ru(tpy)(DPPZ)(CH3CN)]2+ incorporations increases as
follows: 3'-Ru2+(∆Tm = 8.4°C) < middle Ru2+(∆Tm = 15.3°C) < 5'-Ru2+(∆Tm = 17.5°C) < 5',3'-bis-
Ru2+(∆Tm = 23.6°C). The same trend was also found for the [Ru(phen)2DPPZ]2+-ODNs with ∆Tm
varying from 12.8°C to 22.0°C. The absolute ligand configuration at the Ru2+ center of the separated
middle-[Ru(phen)2DPPZ]2+-ODN conjugates plays an essential role in the duplex formation. It has been
observed that Λ-Ru2+ tethered complexes (as in duplexes 23a•29 and 23b•29) gave higher stabilization
(∆Tm = 16.4°C and 19.4°C respectively) while ∆-Ru2+ tethered stereoisomers (as in duplexes 23c•29
and 23d•29) yielded relatively poorer stabilization (∆Tm = 13.6°C and 14.3°C respectively). The
stabilization effect gained thus by attachment of metal complexes considerably exceeds those observed
with DPPZ- and Pnz-ODN conjugates irrespective of the attachment site. It is also noteworthy that the
best stabilization achieved before this work was 3°C134 and 8°C181 for 5'-tethered [Ru(phen)2DPPZ]2+
when the phen ligand was used for covalent attachment, while for other types of Ru2+-ODN
conjugates133,182,184,185 no improvement in the stabilization has been observed.
2.2.2 Thermal stability of conjugated ODN•RNA duplexes (Papers II & IV)
The DPPZ- and [Ru(phen)2DPPZ]2+-labeled ODNs, prepared in this work, were also tested (Table 4)
for their ability to act as antisense ONs to form stable duplexes with complementary RNA target.
Generally, all ODN•RNA duplexes, including the native counterpart, were less stable than the
corresponding ODN•DNA analogs as well as the stabilizing effect of intercalator was less pronounced
on ODN•RNA hybrids compared to the ODN•DNA counterparts. Analogously to (DPPZ-ODN)•DNA
duplexes (Table 3), 12-atom linker L2 provided maximal stability for 5'-DPPZ-tethered ODN•RNA
hybrids (∆Tm = 5.4°C for 10•31 in Table 4) while 3'-DPPZ-ODN conjugates showed greater affinity
towards RNA in case of chromophore linking through the 18-atom linker L3 (∆Tm = 7.4°C for 14•31).
The thermal stability of ODN•RNA duplexes internally modified with DPPZ was comparable with that
of the native duplex 1•31, indicating that destabilization induced by the bulge formation is compensated
by the stabilizing impact of DPPZ moiety within ODN•RNA helical structure as it was found for the
middle-DPPZ-modified ODN•DNA duplexes.
The advantage of [Ru(phen)2DPPZ]2+-conjugation for the aim of ODN•RNA hybrid stabilization
was not so remarkable as for ODN•DNA duplexes. Interesting to note that the effect of metal
conjugation at the 3'-terminal of both types of duplexes was almost equal [∆Tm = 7.9°C (Table 4) and
12.8°C (Table 3) for ODN•RNA 22•31 and ODN•DNA 22•29 duplexes respectively]. For the 5'-

31
Table 4. Thermal stability of DNA•RNA duplexes studied#
Native duplex
5'-TCCAAACAT-3'
3'-r(CAGGUUUGUAC)-5'
1•31,$ Tm = 20.6
Internucleotide
5'- modification
3'- modification modification
L1 - linker 9•31, 25.2(+4.6)
12•31, 27.1(+6.5) 15•31, 19.2(-1.5) 18•31, not formed
L2 - linker 10•31, 26.1(+5.4)
13•31, 25.2(+4.5) 16•31, 21.5(+0.9) 19•31, not formed
DPP
Z - c
onju
gatio
n
L3 - linker 11•31, 25.5(+4.9)
14•31, 28.0(+7.4) 17•31, 21.1(+0.4) 20•31, not formed
(phen)2Ru2+DPPZ - conjugation
(L2 - linker)
21•31, 29.7(+9.1)
22•31, 28.5(+7.9)
23a•31, 20.6(+0.0) 23b•31, 21.6(+1.0) 23c•31, 20.1(-0.5) 23d•31, 20.1(-0.5)
# Tm(∆Tm) in ºC are shown after numbers of duplex strands (symbol • stands for Watson-Crick hydrogen bonding between duplex strands). ∆Tm = Tm(modified duplex) - Tm(corresponding native duplex) $ For sequence composition of oligos see Table 1 (page 15)
terminal conjugation the difference in stabilization effect was strikingly higher [∆Tm = 9.1°C (Table 4)
and 22.0°C (Table 3) for ODN•RNA 21•31 and ODN•DNA 21•29 duplexes respectively], much more
than the corresponding difference between (DPPZ-ODN)•RNA and (DPPZ-ODN)•DNA duplexes
(compare ∆Tms for 9•31 - 17•31 with those for 9•29 - 17•29). Finally, the ODN•RNA duplexes carrying
metal complex at the internucleotide position (as in stereoisomeric 23a-d•31, Table 4) were as stable as
the native counterpart 1•31 as opposed to very stabilized (middle-[Ru(phen)2DPPZ]2+-ODN)•DNA
duplexes 23a-d•29 (Table 3). This indicates that interactions of the bulky Ru2+ complex with duplex
nucleobases are sensitive to the duplex conformation, while the planar DPPZ moiety can be more easily
tolerated by both DNA•DNA and DNA•RNA helical structures.
2.2.3 Thermal stability of conjugated ODN×DNA•DNA triplexes (Papers I, II & IV)
All triplex forming ODN 18mer conjugates (TFOs) consisted of pyrimidines T and dC with the
expectation of Hoogsteen base pair formation in a parallel orientation to the complementary purine-rich
strand of a duplex target.11,218 It can be seen (Table 5) that conjugation of the different chromophores to
the native ODN 44 had no or much less stabilization effect compared to the influence of chromophore
conjugation on the duplex formation. Pnz-modified uridine (PnzU) nucleotide when placed at the 5'-end
32
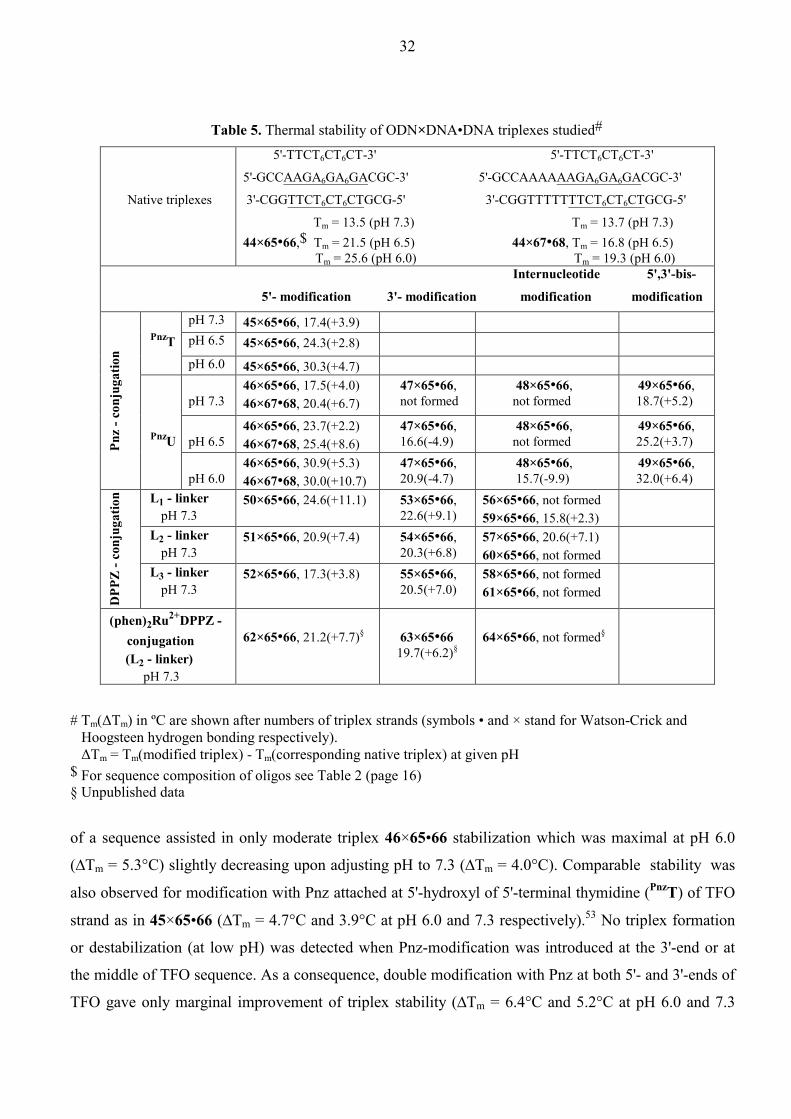
Table 5. Thermal stability of ODN×DNA•DNA triplexes studied#
Native triplexes
5'-TTCT6CT6CT-3' 5'-TTCT6CT6CT-3'
5'-GCCAAGA6GA6GACGC-3' 5'-GCCAAAAAAGA6GA6GACGC-3'
3'-CGGTTCT6CT6CTGCG-5' 3'-CGGTTTTTTTCT6CT6CTGCG-5'
Tm = 13.5 (pH 7.3) Tm = 13.7 (pH 7.3) 44×65•66,$
Tm = 21.5 (pH 6.5) 44×67•68, Tm = 16.8 (pH 6.5)
Tm = 25.6 (pH 6.0)
Tm = 19.3 (pH 6.0) Internucleotide 5',3'-bis-
5'- modification
3'- modification modification
modification
pH 7.3 45×65•66, 17.4(+3.9) pH 6.5 45×65•66, 24.3(+2.8)
PnzT pH 6.0 45×65•66, 30.3(+4.7)
pH 7.3
46×65•66, 17.5(+4.0) 46×67•68, 20.4(+6.7)
47×65•66, not formed
48×65•66, not formed
49×65•66, 18.7(+5.2)
pH 6.5
46×65•66, 23.7(+2.2) 46×67•68, 25.4(+8.6)
47×65•66, 16.6(-4.9)
48×65•66, not formed
49×65•66, 25.2(+3.7)
Pnz
- con
juga
tion
PnzU
pH 6.0 46×65•66, 30.9(+5.3) 46×67•68, 30.0(+10.7)
47×65•66, 20.9(-4.7)
48×65•66, 15.7(-9.9)
49×65•66, 32.0(+6.4)
L1 - linker pH 7.3
50×65•66, 24.6(+11.1)
53×65•66, 22.6(+9.1)
56×65•66, not formed 59×65•66, 15.8(+2.3)
L2 - linker pH 7.3
51×65•66, 20.9(+7.4)
54×65•66, 20.3(+6.8)
57×65•66, 20.6(+7.1) 60×65•66, not formed
DPP
Z - c
onju
gatio
n
L3 - linker pH 7.3
52×65•66, 17.3(+3.8)
55×65•66, 20.5(+7.0)
58×65•66, not formed 61×65•66, not formed
(phen)2Ru2+DPPZ - conjugation (L2 - linker) pH 7.3
62×65•66, 21.2(+7.7)§
63×65•66 19.7(+6.2)§
64×65•66, not formed§
# Tm(∆Tm) in ºC are shown after numbers of triplex strands (symbols • and × stand for Watson-Crick and Hoogsteen hydrogen bonding respectively). ∆Tm = Tm(modified triplex) - Tm(corresponding native triplex) at given pH $ For sequence composition of oligos see Table 2 (page 16) § Unpublished data
of a sequence assisted in only moderate triplex 46×65•66 stabilization which was maximal at pH 6.0
(∆Tm = 5.3°C) slightly decreasing upon adjusting pH to 7.3 (∆Tm = 4.0°C). Comparable stability was
also observed for modification with Pnz attached at 5'-hydroxyl of 5'-terminal thymidine (PnzT) of TFO
strand as in 45×65•66 (∆Tm = 4.7°C and 3.9°C at pH 6.0 and 7.3 respectively).53 No triplex formation
or destabilization (at low pH) was detected when Pnz-modification was introduced at the 3'-end or at
the middle of TFO sequence. As a consequence, double modification with Pnz at both 5'- and 3'-ends of
TFO gave only marginal improvement of triplex stability (∆Tm = 6.4°C and 5.2°C at pH 6.0 and 7.3

33
respectively) with respect to single Pnz-modification at the 5'-terminal. Insertion of the five extra A•T
base paires into the duplex target 65•66 at the triplex-duplex junction, as in triplexes 44×67•68 and
46×67•68, allowed to enhance the stabilization effect of 5'-tethered Pnz group in comparison with its
affinity to the parent duplex target 65•66. This testifies that Pnz binds more strongly at T×A•T - A•T
triplex-duplex junction then at T×A•T - C•G site. As expected, the stability of Pnz-modified triplexes as
well as their native counterparts was found to increase upon lowering the pH.
It has been shown that DPPZ-conjugated ODNs have more affinity towards ssDNA target
(ODN•DNA duplex formation) than Pnz-modified analogs. The same trend was also observed with the
triplex formation, yet there are significant variations depending on the ODN attachment site (5'-, 3'-, or
internucleotide position) and the length of the linker arm used. The most essential stabilization was seen
with DPPZ conjugation at the 5'-terminal through the shortest linker L1 (∆Tm = 11.1°C) whereas the
increase of the linker length led to a sharp decrease of stabilization (up to ∆Tm = 3.8°C for the longest
linker L3). This tendency was also found for 3'-terminal DPPZ conjugation, albeit more gradually (∆Tms
were changed from 9.1°C to 6.8°C). Such effect can be attributed to the entropy decrease in the course
of triplex formation which should be higher upon employment of more extended flexible linkers. On the
other hand, longer linkers may interfere with the triplex strands thus destabilizing triple helical
structure. Triplex formation by 18mers carrying DPPZ at the central position was especially
sensitive to the linker length: only 12-atom linker L2 was found to provide optimal DPPZ configuration
to stabilize the triplex (57×67•68, ∆Tm = 7.1°C) while with DPPZ tethered through the shorter or longer
linkers no triplex formation was detected. Triplex 57×67•68 was, in fact, the only one among other
middle-modified analogs, studied in this work, which showed improved stability.
Despite the extensive investigation of polypyridyl Ru2+ complex interaction with dsDNA, only
few reports were devoted to the elucidation of the binding affinity of those complexes towards triple
helical DNA.219,220 It has been shown that [Ru(phen)2DPPZ]2+ intermolecularly bound to poly(T×dA•T)
or poly(dC+×dG•dC) significantly increases the triplex stability. Another issue to be addressed is the
metal complex binding mode. Because the major groove in triplexes is blocked by the third strand, the
major groove interacting drugs should not bind to the triple-stranded DNA or their binding mode must
be changed. Since the spectral characteristics of [Ru(phen)2DPPZ]2+ bound to poly(T×dA•T) triplex
were similar to those bound to poly(dA•T) duplex, it was concluded that the DPPZ ligand of the metal
complex is intercalated with the two phen ligands located in the minor groove, thereby stabilizing the
third strand by expansion of the stacking interaction. From this point of view, the tethering of potential
metallointercalator to the different sites of a TFO strand could shed light on a still contraversial
question: from which direction (major or minor groove) are the metal complexes intercalated? To date,
the available literature data on triplex formation assisted with metal complex conjugated TFO strand are

34
limited by two works in which [Ru(phen)2DPPZ]2+ 189 or [Ru(bpy)3]2+ 183 complexes were tethered
eithert at the 3'-terminal or at the middle of TFO respectively.
In our [Ru(phen)2DPPZ]2+-ODNs the metal complex was uniformly tethered through the DPPZ
ligand to different positions of 18mer ODN strand (not published data). The metal complex conjugation
at the 5'- or 3'-terminals allowed us to enhance the stability of the resulting triplexes (∆Tm = 7.7°C and
6.2°C for 62×65•66 and 63×65•66 respectively, Table 5). The insertion of metal complex at the interior
of a TFO strand led to the complete loss of ability to form a triplex. Following comparisons are
noteworthy: (1) Ligation of Ru2+(phen)2 moiety to the DPPZ ligand did not improve the TFOs binding
affinity with respect to DPPZ-tethered TFOs, if the modification is located at terminals of TFO
(compare stability of 62×65•66 and 51×65•66 as well as 63×65•66 and 54×65•66 in Table 5), which is
in contrast with the trend observed for the duplexes. (2) Middle-modification of an ODN strand with the
metal complex allowed to stabilize ODN•DNA duplexes but, it does not make the ODN able to bind to
dsDNA and form a triplex. These observations only strengthen the fact that the binding modes of the
tethered metal complex in the metal conjugated duplexes and triplexes are different. These binding
modes are directly dictated by structural compatibility of the metal complex and various types of DNA
hybrids. Most probably the bulky Ru2+ complex can not intercalate into the triplex from the minor
groove as well, when it is conjugated to the TFO strand because of the high steric interference of the
linker which connects the metal complex and the third strand.
2.2.4 Circular dichroism (CD) and DNase I footprinting studies on [Ru(phen)2DPPZ]2+-
labeled ODN•DNA duplexes performed for the elucidation of the metallointercalation
binding mode in the DNA double helixes (Paper IV, in collaboration with Mr. P. I.
Pradeepkumar)
CD spectroscopy221-223 was employed to determine the difference in the global structure of the
ODN•DNA duplexes site-specifically tethered with the metal complex at various positions. The CD
spectra of the racemic Ru2+-ODN conjugates 21, 22, and 23 hybridized with the DNA target 29 were
recorded to exclude the contribution of the CD signals originated from each of the pure Ru2+complex
diastereomers in order to observe the global helical conformation of the DNA duplex. The spectral
characteristics for 5'- and middle-modified duplexes 21•29 and 23•29 were significantly different from
those of the native counterpart. Positive (267.5 nm) and negative (240.0 nm) bands of the native duplex
were shifted correspondingly to 260 nm and 232.5 nm of modified analogs, while CD spectra of 3'-
conjugated duplex 22•29 exhibited some features which are characteristic for natural duplex: i.e. the
same negative band wavelength (240 nm) and a shoulder at 267.5 nm which coincides with the native
duplex positive band. At the same time, crossover point (246.3 nm) and the wavelength shift of main

35
positive band for 3'-modified duplex 22•29 were identical with those of the 5'- and middle-modified
analogs. Generally, it appears that the metal conjugation to ODN applied in the present work allowed to
bring the metal complex in tight contact with nucleobases which leads to the essential global helical
structure alteration from the native B-type helix. This alteration was more pronounced in the case of 5'-
and middle-conjugation. The different structural changes in the enantiomerically pure Ru2+-ODNs 23a-
d upon its hybridization with DNA target 29 further proved that the metal complex strongly interact
within double helix: distinct CD changes upon hybridization of ∆-isomers 23c-d compared with those
of Λ-isomers 23a-b implies their different metal complex binding geometry in the duplex. This
situation is expected when ancillary phen ligands are rigidly located within helical grooves but not
when the metal complex is externally bound.224
DNase I provides simple and quick method for identifying or confirming the preferred dsDNA
binding sites for several ligands.225-227 We exploited this technique to evaluate the extent of alterations
from the B-type DNA duplex structure caused by tethered metal complex as well as to study the DNase I
cleavage pattern as a reflection of structural accessibility of phosphodiester groups which can be
protected from the enzyme digestion by the appending Ru2+ complex. The extent of the cleavage by
DNase I (Figure 1) decreases as follows: native duplex 1•29 (97%) ~ 3'-Ru2+-modified 22•29 (98%) >
middle-Ru2+-modified 23•29 (48%) > 5'-Ru2+-modified 21•29 (38%) signifying that in case of 5'- and
middle-modification the duplex structure is deeply altered from the canonical B-form which is ideal
for DNase I recognition.228 Nevertheless, conformational reorganization in the 5'- and middle-modified
duplexes do not completely preclude the recognition by the enzyme. DNase I is known to bind at least 4
bp to the 5'-end and 6 bp to the 3'-end from the scissile phosphodiester bond of the substrate duplex.228
Thus, the full length of studied 9mer + 11mer duplex should be covered by DNase I. Cleavage patterns of
5'- and 3'- ruthenated duplexes are not distinguishable from the native counterpart, while middle
modified analog has completely different digestion pattern with more or less one cleavage site only
(between 8G and 9T, Figure 1). Moreover, it was found that the cleavage sites of the middle-DPPZ-
modified duplex 16•29 were stretched from 5T to 8G (Figure 2). This shows that the bulky
[Ru(phen)2(dppz)]2+ moiety, tethered at the middle of ODN sequence, acts as a protection against the
enzyme cleaving site (E75-H131-H2O228), blocking those phosphodiester groups which are normally
cleaved when the metal entity is distal from the center of a duplex (as in case of 5'- and 3'-metal complex
conjugation) or when simple planar DPPZ group is used for the internucleotide conjugation. The
cleavage patterns of the diastereomerically pure middle-modified (Ru2+-ODN)•DNA duplexes were also
found (Figure 3) to be distinct from each other confirming the CD spectral data that binding geometries
of ∆- isomers 23c-d and Λ-isomers 23a-b are indeed different. Altogether, the CD and DNase I
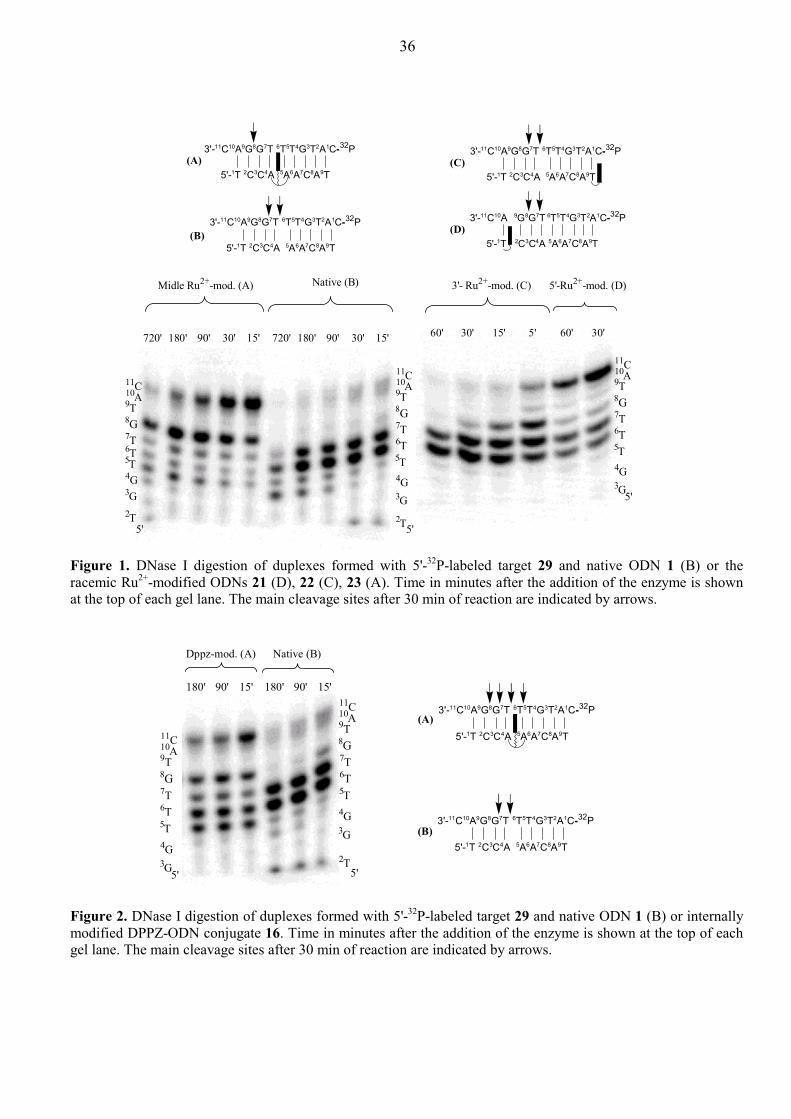
36
Figure 1. DNase I digestion of duplexes formed with 5'-32P-labeled target 29 and native ODN 1 (B) or the racemic Ru2+-modified ODNs 21 (D), 22 (C), 23 (A). Time in minutes after the addition of the enzyme is shown at the top of each gel lane. The main cleavage sites after 30 min of reaction are indicated by arrows.
Figure 2. DNase I digestion of duplexes formed with 5'-32P-labeled target 29 and native ODN 1 (B) or internally modified DPPZ-ODN conjugate 16. Time in minutes after the addition of the enzyme is shown at the top of each gel lane. The main cleavage sites after 30 min of reaction are indicated by arrows.
5'-1T 2C3C4A 5A6A7C8A9T
3'-11C10A9G8G7T 6T5T4G3T2A1C-32P(B)
5'-1T 2C3C4A 5A6A7C8A9T
3'-11C10A9G8G7T 6T5T4G3T2A1C-32P(A)
5'-1T 2C3C4A 5A6A7C8A9T
3'-11C10A9G8G7T 6T5T4G3T2A1C-32P(C)
5'-1T 2C3C4A 5A6A7C8A9T
3'-11C10A 9G8G7T 6T5T4G3T2A1C-32P(D)
720' 180' 90' 30' 15' 720' 180' 90' 30' 15'
Midle Ru2+-mod. (A) Native (B) 3'- Ru2+-mod. (C) 5'-Ru2+-mod. (D)
60' 30' 15' 5' 60' 30'
11C10A9T8G7T6T5T4G3G5'
11C10A9T8G7T6T5T4G3G
5'2T
11C10A9T8G7T6T5T4G3G
5'2T
5'-1T 2C3C4A 5A6A7C8A9T
3'-11C10A9G8G7T 6T5T4G3T2A1C-32P(A)
5'-1T 2C3C4A 5A6A7C8A9T
3'-11C10A9G8G7T 6T5T4G3T2A1C-32P(B)
11C10A9T8G7T6T5T4G3G
2T5'
180' 90' 15' 180' 90' 15'
Native (B)Dppz-mod. (A)
11C10A9T8G7T6T5T4G3G5'

37
footprinting data performed on ruthenated duplexes point to some special binding mode which allow
the metal complex strongly interact with nucleobases which leads to (1) the high increase of duplex
stability, (2) conformational reorganization of DNA double helix, and (3) protecting phosphodiester
groups in helical grooves.
Figure 3. DNase I digestion of duplexes formed with 5'-32P-labeled target 29 and native ODN 1 or different diastereomers of Ru2+ middle-modified ODNs 23a-d. Time in minutes after addition of the enzyme is shown at the top of each gel lane. The main cleavage sites after 30 min of reaction are indicated by arrows.
The results of CD, DNase I footprinting, and thermal denaturation studies on ODN•DNA
duplexes tethered with [Ru(phen)2DPPZ]2+ complex and their comparison with the thermal stabilities of
analogous ODN•RNA duplexes and ODN×DNA•DNA triplexes, allowed us to suggest the most
probable mode of complex interaction within the DNA double helix. As it was shown, the DPPZ
moiety of [Ru(phen)2(DPPZ-CO-L2)]2+ complex intercalates into the native duplex 1•29. We proposed
a similar intercalation mode of DPPZ subunit in the [Ru2+(DPPZ)-ODN]•DNA duplexes. Tethering of
[Ru(phen)2DPPZ]2+ or [Ru(tpy)(DPPZ)(CH3CN)]2+ to an ODN through the DPPZ ligand leaves the
only possibility for DPPZ to intercalate in a duplex: via its threading across the double helix core thus
delivering Ru2+(phen)2 or Ru2+(tpy)(CH3CN) moieties to the final position in the opposite groove
(Figure 4). This type of binding229 can be envisaged to give a very rigid structure in which covalently
attached complex acts as a staple, holding the bases firmly stacked together near the intercalation site.
On the other hand, upon such “stapling” dsDNA is likely to undergo a much larger conformational
reorganization then in case of classical intercalation of a planar chromophore. With the above scenario,
the resulting ODN•DNA duplexes should be very stable, their global structure should be altered from
the parent B-type, and the position of the Ru2+(phen)2 moiety is restricted in the helical groove
protecting some nearest backbone phosphodiester groups. The protecting ability is also expected to be
5'-1T 2C3C4A 5A6A7C8A9T
3'-11C10A9G8G7T 6T5T4G3T2A1C-32P23a , 23b
5'-1T 2C3C4A 5A6A7C8A9T
3'-11C10A9G8G7T 6T5T4G3T2A1C-32P23c
5'-1T 2C3C4A 5A6A7C8A9T
3'-11C10A9G8G7T 6T5T4G3T2A1C-32P23d
90' 180' 90' 180' 90' 180' 90' 180' 90' 180'
23a(Λ) 23b(Λ) 23c(∆) 23d(∆) Native
11C10A9T8G7T6T5T
4G3G
2T5'
11C10A9T8G7T6T
5T4G3G
2T5'

38
dependent on the ∆- or Λ-configuration of the two ancillary phen ligands around Ru2+ center. These
expectations were in agreement with the results obtained for our 5'- and middle-modified duplexes
21•29 and 23•29. The net free-energy gain which can be reached by metal complex threading is
Figure 4. Proposed metallointercalation mode shown for the ∆-[Ru(phen)2DPPZ]2+ complex tethered to the internal position of the duplex 23•29 through the linker L2. The linker-complex unit –L2-DPPZ Ru2+(phen)2 covalently linked to the glycerol moiety of the ODN 23 is depicted in bold.
determined by balancing between the positive deposit of additional π-π stacking interactions and
negative impact of structural reorganization of the DNA helix near the metallointercalation site. The
last factor is very sensitive to the initial helical conformation undergoing structural changes upon the
threading of metal complex. This explains the fact that Ru2+ middle-modification gave us highly
stabilized ODN•DNA duplex while analogous ODN•RNA duplex had the same stability as the native
counterpart. Finally, the presence of the third strand bound to the dsDNA in the major groove
completely inhibited triplex formation consisting of Ru2+ middle-modified TFO strand. Interesting is to
note also that comparison of Tms of [Ru(tpy)(DPPZ)(CH3CN)]2+-ODN•DNA duplexes 39•43 - 42•43
with Tms of structurally related amino-linker modified duplexes 35•43 - 38•43 allows to estimate the
stabilization effect inherent in [Ru(tpy)(DPPZ)(CH3CN)]2+ itself. Such effect is more pronounced in
case of internucleotide conjugation (∆∆Tm = 24.3°C for 41•43 and 37•43) which is more than for 5'- and
3'-metalated duplexes (in 1.5 and 3.5 times respectively). This observation can be interpreted by the
structural feature of the applied internucleotide modification: insertion of a bulged abasic site magnifies
the distance between adjacent base pairs providing more space for accommodation of the bulky metal

39
complex (Figure 5A and 5D). In the 5'-Ru2+-modified ODN•DNA duplexes the internucleotide
distance expected for metallointercalation is not structurally increased giving the less positive net
Ru
Ru
Ru
5'-1T2C3C4A 5A6A7C8A9T
3'-11C10A9G8G7T 6T5T4G3T2A1C
5'-1T 2C3C4A5A6A7C8A9T
3'-11C10A 9G8G7T6T5T4G3T2A1C
5'-1T2C3C4A5A6A7C8A9T
3'-11C10A9G8G7T6T5T4G3T2A 1C
(E)
(A)
(B)
(C)
(D)
(F)
5'-1C2T3T4A5C 6C7A8A9T10C
3'-11G10A9A8T7G 6G5T4T3A2G1TRu
Ru
Ru
5'-1C 2T3T4A5C6C7A8A9T10C
3'-11G 10A9A8T7G6G5T4T3A2G1T
5'-1C2T3T4A5C6C7A8A9T10C
3'-11G10A9A8T7G6G5T4T3A2G 1T
Figure 5. Schematic illustration of the positioning of the metal complex in the various ODN•DNA duplexes site-specifically labeled with [Ru(phen)2(DPPZ)]2+ (A – C) or [Ru(tpy)(DPPZ)(CH3CN)]2+ (D – F) metal complexes.
stabilization effect (Figure 5B and 5E). The relatively low stabilization of 3'-Ru2+-modified duplexes,
which is comparable with those of (DPPZ-ODN)•DNA duplexes 11•29 and 14•29 (Table 3), can be a
result of the less effective π-π stacking interactions between the metal complex and the terminal base
pair instead of intercalation between internal base pairs (Figure 5C and 5F).
2.3 RNase H degradation studies on the AON•RNA hybrid duplexes conjugated with various
intercalators at the ODN strand (Paper III, in collaboration with Dr. E. Zamaratski and Mr. P.
I. Pradeepkumar).
2.3.1 Physicochemical properties of RNA targets and their duplexes formed with AON conjugates.
To evaluate the potency of ODNs (abbreviated in this chapter as AONs) conjugated with intercalating
agents to form stable ODN•RNA duplexes, three RNA targets 31 – 33 (Table 1) have been chosen
consisting of the same central 9mer tract which is complementary to the AON 1 sequence and flanked
with different patches of deoxynucleotides with the expectation to give different self-folding capacities
of the resulting RNA molecules. The self-aggregation of these targets was examined by CD and UV-
spectroscopy. Thermal melting of the 11mer RNA 31 showed no sigmoidal change in UV absorbance
while clear concentration dependent transition was observed for the 17mer RNA 33 which allowed us
to determine Tm (29.6°C and 31.2°C at 1 µM and 3 µM concentration respectively) and the
thermodynamics of its self-aggregation: ∆H° = -342 kJ/mol, -T∆S° = 303.9 kJ/mol, and ∆G°298 = -38.3

40
kJ/mol. Melting experiments with another 17mer RNA 32 revealed only slight hyperchromic effect. The
temperature-dependent CD was found to be more sensitive tool230 to monitor structural transitions in
given RNA targets. The CD spectra recorded at three different temperatures (6°C, 20°C, and 50°C)
were almost identical for 11mer 31 whereas ellipticity of 17mer 33 strongly decreased upon heating,
suggesting a high degree of self-organization. Although 17mer 32 did not show any clear transition in
the UV melting experiments, it exhibited a definite (but less pronounced compared to RNA 33)
temperature dependency of its CD spectra. The ellipticity measured at 265 nm in the range of
temperatures from 5°C to 60°C was found to have the sigmoidal dependence from temperature for both
RNAs 32 and 33, giving the Tms values for RNA aggregation (24.7°C for target 32 and 28.8°C for
target 33 at 1 µM concentration). The concentration dependence of Tm found from the CD experiments
for both targets allowed us to tentatively propose the structures of self-aggregated RNAs 32 and 33:
Thus, the above RNA targets with different tertiary structures231,232 gave the possibility to study the
effect of intercalator tethered to the AON to influence on the two competing and reversible
hybridizations leading correspondingly to RNA•RNA and AON•RNA structures.
Native AON 1 and its conjugates tethered with Pnz and DPPZ chromophores at 5'- or 3'-terminals
as in 3, 8, 10, and 14 (Table 1) have been chosen to study the effect of covalently linked intercalator on
the antisense activity of the resulting ODN conjugates. The phosphorothioate (PS) analogs of the above
AONs have also been synthesized (PS-AONs 24 – 28) to compare the effect of chromophore
conjugation with their phosphodiester (PO) counterparts. All chromophore-conjugated (both PO and
PS) AON•RNA duplexes formed with non-aggregated RNA target 31 demonstrated higher stability
compared to the non-conjugated analogs (Table 6). The additional π-π stacking interactions between
the aromatic rings of the chromophore and the adjacent base pairs provide a significant gain in enthalpy
(∆H°) upon hybridization of conjugated AONs to the RNA target 31. On the other hand, the competing
decrease of entropy (∆S°) is much higher when conformationally flexible tethers participate in the
duplex formation which restrict their flexibility due to the positioning of a chromophore near the paired
nucleobases. The net change of free energy (∆G°298) was yet found to be more negative for the
hybridization of conjugated AONs to the non-aggregated target 31 compared to the native counterparts,
which correlates with the enhanced thermal stability of chromophore-tethered AON•RNA duplexes. As
expected, PS-AON•RNA duplexes revealed poorer stability then their PO-analogs (0.5 - 1°C Tm
decrease for each PS-backbone modification)71 as well as the stabilizing effect of a chromophore in the
case of PS-AON•RNA duplexes was less then for the corresponding PO-hybrids.
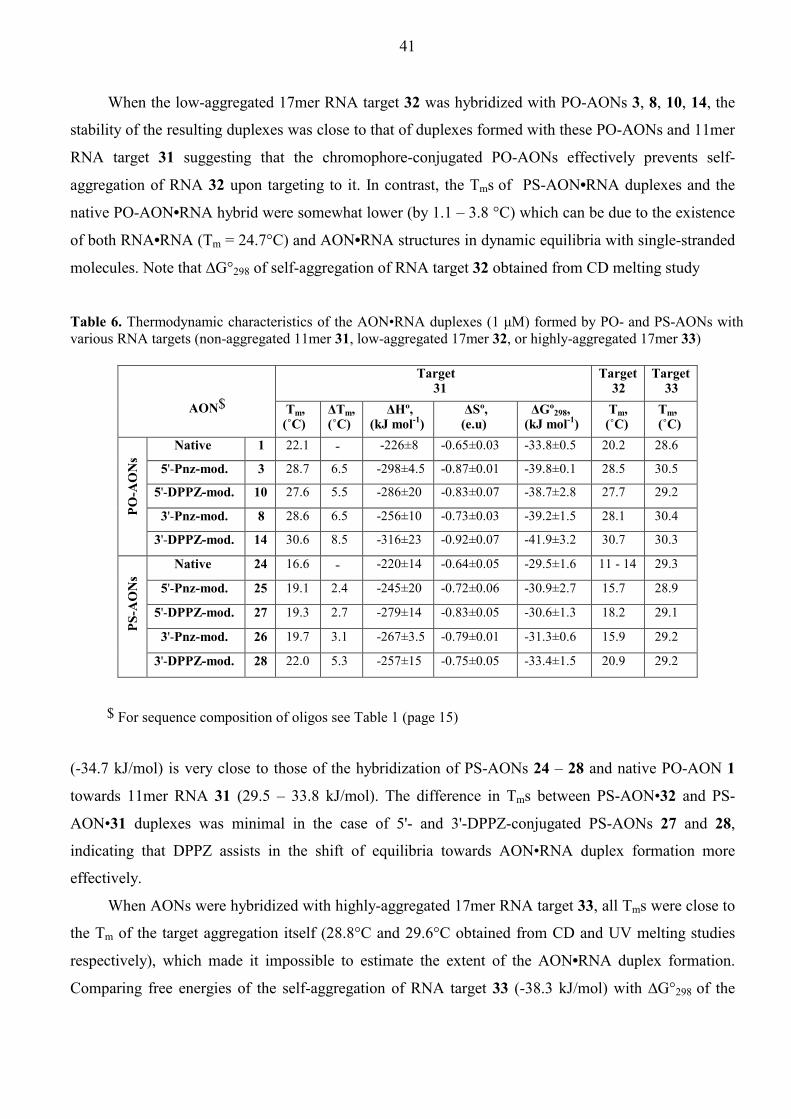
41
When the low-aggregated 17mer RNA target 32 was hybridized with PO-AONs 3, 8, 10, 14, the
stability of the resulting duplexes was close to that of duplexes formed with these PO-AONs and 11mer
RNA target 31 suggesting that the chromophore-conjugated PO-AONs effectively prevents self-
aggregation of RNA 32 upon targeting to it. In contrast, the Tms of PS-AON•RNA duplexes and the
native PO-AON•RNA hybrid were somewhat lower (by 1.1 – 3.8 °C) which can be due to the existence
of both RNA•RNA (Tm = 24.7°C) and AON•RNA structures in dynamic equilibria with single-stranded
molecules. Note that ∆G°298 of self-aggregation of RNA target 32 obtained from CD melting study
Table 6. Thermodynamic characteristics of the AON•RNA duplexes (1 µM) formed by PO- and PS-AONs with various RNA targets (non-aggregated 11mer 31, low-aggregated 17mer 32, or highly-aggregated 17mer 33)
Target 31
Target 32
Target 33
AON$ Tm,
(˚C) ∆Tm, (˚C)
∆Hº, (kJ mol-1)
∆Sº, (e.u)
∆Gº298, (kJ mol-1)
Tm, (˚C)
Tm, (˚C)
Native 1 22.1 - -226±8 -0.65±0.03 -33.8±0.5 20.2 28.6
5'-Pnz-mod. 3 28.7 6.5 -298±4.5 -0.87±0.01 -39.8±0.1 28.5 30.5
5'-DPPZ-mod. 10 27.6 5.5 -286±20 -0.83±0.07 -38.7±2.8 27.7 29.2
3'-Pnz-mod. 8 28.6 6.5 -256±10 -0.73±0.03 -39.2±1.5 28.1 30.4
PO-A
ON
s
3'-DPPZ-mod. 14 30.6 8.5 -316±23 -0.92±0.07 -41.9±3.2 30.7 30.3
Native 24 16.6 - -220±14 -0.64±0.05 -29.5±1.6 11 - 14 29.3
5'-Pnz-mod. 25 19.1 2.4 -245±20 -0.72±0.06 -30.9±2.7 15.7 28.9
5'-DPPZ-mod. 27 19.3 2.7 -279±14 -0.83±0.05 -30.6±1.3 18.2 29.1
3'-Pnz-mod. 26 19.7 3.1 -267±3.5 -0.79±0.01 -31.3±0.6 15.9 29.2
PS-A
ON
s
3'-DPPZ-mod. 28 22.0 5.3 -257±15 -0.75±0.05 -33.4±1.5 20.9 29.2
$ For sequence composition of oligos see Table 1 (page 15)
(-34.7 kJ/mol) is very close to those of the hybridization of PS-AONs 24 – 28 and native PO-AON 1
towards 11mer RNA 31 (29.5 – 33.8 kJ/mol). The difference in Tms between PS-AON•32 and PS-
AON•31 duplexes was minimal in the case of 5'- and 3'-DPPZ-conjugated PS-AONs 27 and 28,
indicating that DPPZ assists in the shift of equilibria towards AON•RNA duplex formation more
effectively.
When AONs were hybridized with highly-aggregated 17mer RNA target 33, all Tms were close to
the Tm of the target aggregation itself (28.8°C and 29.6°C obtained from CD and UV melting studies
respectively), which made it impossible to estimate the extent of the AON•RNA duplex formation.
Comparing free energies of the self-aggregation of RNA target 33 (-38.3 kJ/mol) with ∆G°298 of the

42
AON•31 duplex formation, we assumed that only a fraction of AON strands was able to form the
desired duplex with highly aggregated RNA 33. Generally, these data show that the thermodynamic
accessibility of RNA tertiary structure for AON•RNA duplex formation can be increased by tethering of
the intercalating agents to an AON strand.
Another important aspect of successful application of conjugated AONs in the RNase H mediated
degradation of target RNA molecules is the deviation of the global AON•RNA heteroduplex structure
from the native DNA•RNA conformation, in which RNA strand is of the A-type and the DNA strand
has a conformation close to the B-type.233,234 This type of the heteroduplex conformation was proven to
be essential for the substrate recognition by RNase H.233-236 CD spectra of our chromophore-conjugated
duplexes (both PO and PS) formed with 11mer RNA target 31 were very similar to the native duplexes
1•31 and 24•31, and rather different from those of the highly aggregated 17mer RNA 33 and natural
AON•DNA duplex 1•29 representing the canonical A- and B-type structures respectively. The shape of
all heteroduplex spectra supports that the global structure of AON•RNA duplexes is inremediate
between A- and B-type. Thus, terminal conjugation of the planar chromophores through the flexible
linkers does not alter the helical structure inherent in the native DNA•RNA duplex.
2.3.2 RNase H mediated cleavage studies on the conjugated AON•RNA duplexes
5'-32P-labeled RNA targets were hybridized with complementary AON strands in equimolar ratio (0.8
µM) and incubated with Escherichia coli RNase H at 21 °C and aliquots were taken after 15 and 120 min
intervals. All modified AONs (both PO and PS) promoted a higher degree of RNA hydrolysis compared
to the natural counterparts 1 and 24 (Table 7) irrespective of the nature of the RNA target (31, 32, or 33).
The following comparisons are noteworthy: (1) DPPZ conjugated PO-AONs and PS-AONs promoted
exceptionally higher extent of the cleavage of all RNA targets compared to the Pnz-modified analogs.
(2) Generally, 3'-conjugation of a chromophore assisted in slightly better hydrolytic activity of RNase H
relative to the corresponding 5'-conjugates (especially in the case of DPPZ-modification). (3) The extent
of hydrolysis for the PS-AON•RNA substrates was somewhat lower in comparison with the PO-
counterparts as expected from the lower thermal stability of these duplexes. (4) When PO- and PS-
AONs were hybridized with the low- or highly-aggregated 17mer RNAs 32 and 33, the hydrolysis yields
remained approximately the same as those with the non-aggregated 11mer RNA 31 despite different
accessibility of the single strand region in these RNA targets. This suggets that the rate of conversion of
the folded RNA structures to the single-stranded form is much faster than the rate limiting RNase H
promoted cleavage and the kinetic accessibility of the single strand region in all three targets becomes
equal. (5) No clear correlation between the thermal stability of AON•RNA duplexes and the extent of
their cleavage by RNase H can be drawn on the basis of present data. According to the Michaelis-Menten
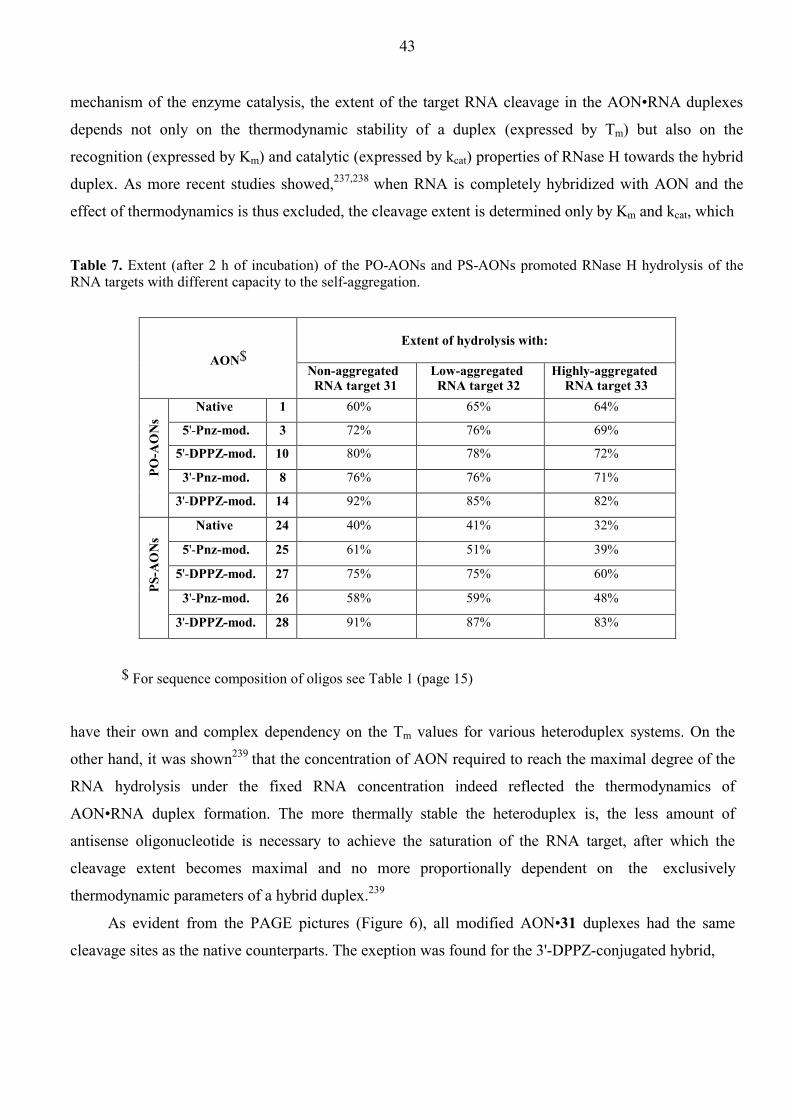
43
mechanism of the enzyme catalysis, the extent of the target RNA cleavage in the AON•RNA duplexes
depends not only on the thermodynamic stability of a duplex (expressed by Tm) but also on the
recognition (expressed by Km) and catalytic (expressed by kcat) properties of RNase H towards the hybrid
duplex. As more recent studies showed,237,238 when RNA is completely hybridized with AON and the
effect of thermodynamics is thus excluded, the cleavage extent is determined only by Km and kcat, which
Table 7. Extent (after 2 h of incubation) of the PO-AONs and PS-AONs promoted RNase H hydrolysis of the RNA targets with different capacity to the self-aggregation.
Extent of hydrolysis with:
AON$
Non-aggregated RNA target 31
Low-aggregated RNA target 32
Highly-aggregated RNA target 33
Native 1 60% 65% 64%
5'-Pnz-mod. 3 72% 76% 69%
5'-DPPZ-mod. 10 80% 78% 72%
3'-Pnz-mod. 8 76% 76% 71%
PO-A
ON
s
3'-DPPZ-mod. 14 92% 85% 82%
Native 24 40% 41% 32%
5'-Pnz-mod. 25 61% 51% 39%
5'-DPPZ-mod. 27 75% 75% 60%
3'-Pnz-mod. 26 58% 59% 48%
PS-A
ON
s
3'-DPPZ-mod. 28 91% 87% 83%
$ For sequence composition of oligos see Table 1 (page 15)
have their own and complex dependency on the Tm values for various heteroduplex systems. On the
other hand, it was shown239 that the concentration of AON required to reach the maximal degree of the
RNA hydrolysis under the fixed RNA concentration indeed reflected the thermodynamics of
AON•RNA duplex formation. The more thermally stable the heteroduplex is, the less amount of
antisense oligonucleotide is necessary to achieve the saturation of the RNA target, after which the
cleavage extent becomes maximal and no more proportionally dependent on the exclusively
thermodynamic parameters of a hybrid duplex.239
As evident from the PAGE pictures (Figure 6), all modified AON•31 duplexes had the same
cleavage sites as the native counterparts. The exeption was found for the 3'-DPPZ-conjugated hybrid,
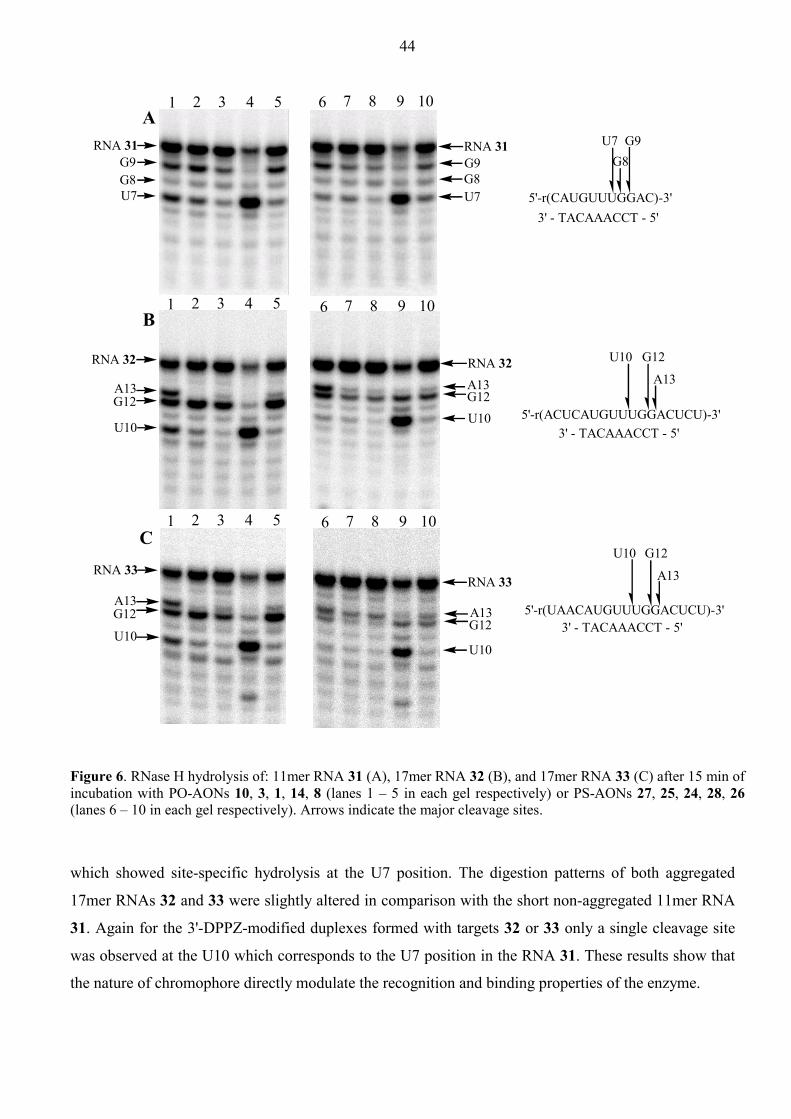
44
RNA 31RNA 31
RNA 32RNA 32
RNA 33RNA 33
G8G9
U7
G9G8U7
A13G12U10
A13G12
U10
A13
U10
G12
A13G12
U10
A
B
C
5'-r(CAUGUUUGGAC)-3'3' - TACAAACCT - 5'
5'-r(ACUCAUGUUUGGACUCU)-3'3' - TACAAACCT - 5'
5'-r(UAACAUGUUUGGACUCU)-3'3' - TACAAACCT - 5'
G9G8
U7
A13
A13
G12
G12
U10
U10
1 2 3 4 5 6 7 8 9 10
1 2 3 4 5
1 2 3 4 5
6 7 8 9 10
6 7 8 9 10
Figure 6. RNase H hydrolysis of: 11mer RNA 31 (A), 17mer RNA 32 (B), and 17mer RNA 33 (C) after 15 min of incubation with PO-AONs 10, 3, 1, 14, 8 (lanes 1 – 5 in each gel respectively) or PS-AONs 27, 25, 24, 28, 26 (lanes 6 – 10 in each gel respectively). Arrows indicate the major cleavage sites.
which showed site-specific hydrolysis at the U7 position. The digestion patterns of both aggregated
17mer RNAs 32 and 33 were slightly altered in comparison with the short non-aggregated 11mer RNA
31. Again for the 3'-DPPZ-modified duplexes formed with targets 32 or 33 only a single cleavage site
was observed at the U10 which corresponds to the U7 position in the RNA 31. These results show that
the nature of chromophore directly modulate the recognition and binding properties of the enzyme.
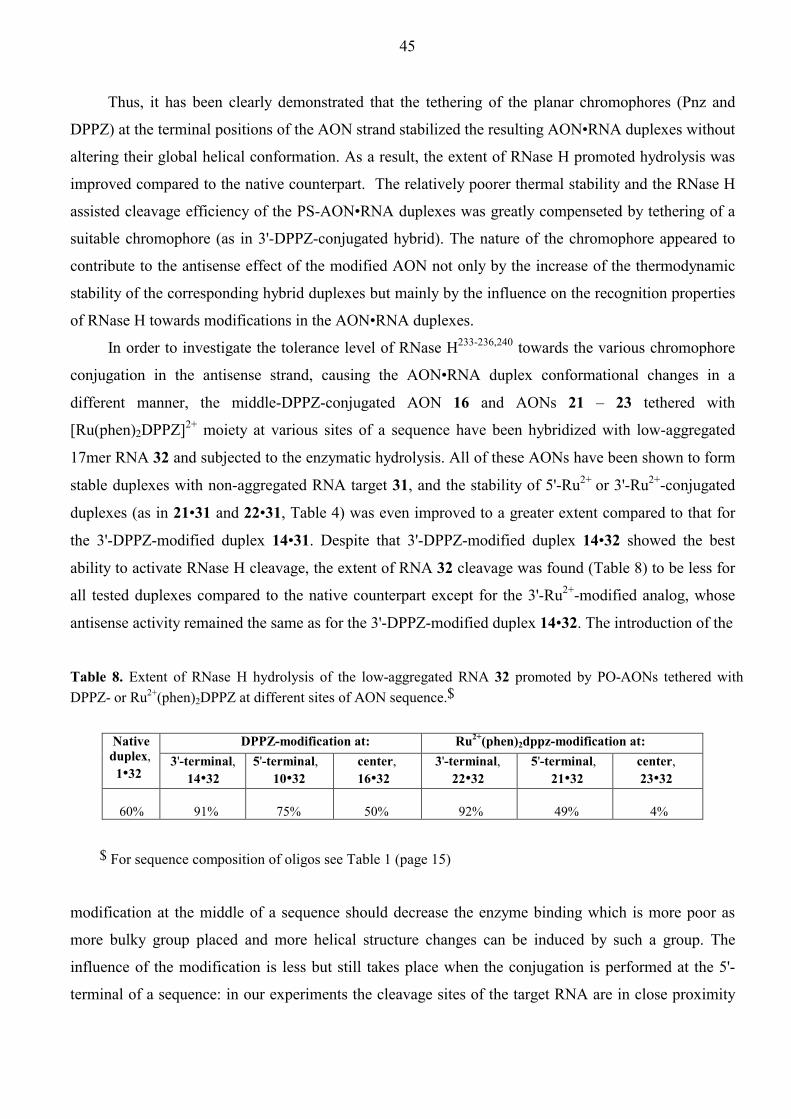
45
Thus, it has been clearly demonstrated that the tethering of the planar chromophores (Pnz and
DPPZ) at the terminal positions of the AON strand stabilized the resulting AON•RNA duplexes without
altering their global helical conformation. As a result, the extent of RNase H promoted hydrolysis was
improved compared to the native counterpart. The relatively poorer thermal stability and the RNase H
assisted cleavage efficiency of the PS-AON•RNA duplexes was greatly compenseted by tethering of a
suitable chromophore (as in 3'-DPPZ-conjugated hybrid). The nature of the chromophore appeared to
contribute to the antisense effect of the modified AON not only by the increase of the thermodynamic
stability of the corresponding hybrid duplexes but mainly by the influence on the recognition properties
of RNase H towards modifications in the AON•RNA duplexes.
In order to investigate the tolerance level of RNase H233-236,240 towards the various chromophore
conjugation in the antisense strand, causing the AON•RNA duplex conformational changes in a
different manner, the middle-DPPZ-conjugated AON 16 and AONs 21 – 23 tethered with
[Ru(phen)2DPPZ]2+ moiety at various sites of a sequence have been hybridized with low-aggregated
17mer RNA 32 and subjected to the enzymatic hydrolysis. All of these AONs have been shown to form
stable duplexes with non-aggregated RNA target 31, and the stability of 5'-Ru2+ or 3'-Ru2+-conjugated
duplexes (as in 21•31 and 22•31, Table 4) was even improved to a greater extent compared to that for
the 3'-DPPZ-modified duplex 14•31. Despite that 3'-DPPZ-modified duplex 14•32 showed the best
ability to activate RNase H cleavage, the extent of RNA 32 cleavage was found (Table 8) to be less for
all tested duplexes compared to the native counterpart except for the 3'-Ru2+-modified analog, whose
antisense activity remained the same as for the 3'-DPPZ-modified duplex 14•32. The introduction of the
Table 8. Extent of RNase H hydrolysis of the low-aggregated RNA 32 promoted by PO-AONs tethered with DPPZ- or Ru2+(phen)2DPPZ at different sites of AON sequence.$
DPPZ-modification at: Ru2+(phen)2dppz-modification at: Native duplex, 1•32
3'-terminal, 14•32
5'-terminal, 10•32
center, 16•32
3'-terminal, 22•32
5'-terminal, 21•32
center, 23•32
60%
91%
75%
50%
92%
49%
4%
$ For sequence composition of oligos see Table 1 (page 15)
modification at the middle of a sequence should decrease the enzyme binding which is more poor as
more bulky group placed and more helical structure changes can be induced by such a group. The
influence of the modification is less but still takes place when the conjugation is performed at the 5'-
terminal of a sequence: in our experiments the cleavage sites of the target RNA are in close proximity

46
to the 5'-end of the AON sequence and the presence of any tether at this position might interfere with
the enzyme active sites. Finally, the 3'-end of the AON seems to be the most suitable for the
introduction of different chemical functions as bulky as [Ru(phen)2DPPZ]2+ complex. The marked
difference in the activation of RNase H cleavage for the middle or 5'-metal Ru2+ complex tethered
heteroduplexes and their corresponding DPPZ-tethered analogs might be a consequence of the
specificity of metallointercalation which, as was shown on ODN•DNA duplexes, strongly alters the
conformation of a duplex.
2.3.3 Nuclease resistance of the 3'-Pnz and 3'-DPPZ-conjugated AONs
To be suitable for pharmaceutical application, the AONs are required to be resistant to the nucleic acid
degrading enzymes.240,241 The 3'-exonuclease activity was found to be responsible for most of the AON
degradation,240-242 therefore, modification of AONs at the 3'-end has been used to minimize the extent
of degradation.243,244 Under incubation of our AONs with snake venom phosphodiesterase, the non-
modified PO-AON 1 had a half-life time of 4 min, however, 3'-Pnz and 3'-DPPZ-modified PO-AONs 8
and 14 did not show any sign of degradation even after 2 h of incubation. Similar enhancement of
stability was observed with the phosphorothioate analogs. Most probably the steric hindrance caused by
the tethered polyaromatic system as well as the flexible linker prevents the enzyme binding to the
modified AON.
2.4 Specific chemical modifications of the complementary DNA strand by DPPZ and Ru2+-
DPPZ tethered ODNs in ODN•DNA duplexes.
2.4.1 Sequence-specific cleavage of target DNA by metal-binding DPPZ-group linked to an
internucleotide position of the complementary ODN strand (unpublished data)
The intercalating ability of some chromophores can be endowed with chemical properties such that they
can be used to induce irreversible modifications in their target sequence. These chromophore-ODN
conjugates appear particularly attractive for those ODNs which do not allow cleavage of mRNA by
RNase H and, for this reason, often have little or no antisense activity.9,68,70,75 Moreover, the reactivity
of interactive groups can be directed to specific sites in both single-stranded mRNA and double-
stranded genomic DNA allowing to improve the potency of synthetic ODNs as antisense and antigene
agents.11,245
The DPPZ ligand can be considered as the extended structural analog of the phenanthroline, with
two chelating nitrogens which are able to coordinate transition metal ions. Metal complexes formed
with ODN-tethered ligands have been shown to act as sequence-specific cleavers of RNA101-106 and
DNA targets.110-115,117-121 Such polypyridyl ligands as terpyridine (tpy),101,246-249 phenanthroline

47
(phen),249,250 and bypyridine (bpy)251 have been linked to ODNs or monomeric 2'-deoxynucleosides to
give artificial ribonucleases which successfully hydrolzed RNA in ODN•RNA duplex. On the other
hand, nuclease activity of Cu+(phen)2 and the related complexes towards dsDNA has been
comprehensively studied252-263 and found to proceed through the generation of hydroxyl radicals which
probably abstract H atom from the deoxyribose of DNA with subsequent break of the phosphodiester
backbone:
M(n-1)+ + O2 → Mn+ + O2• (HO2
•)
O2• (HO2
•) + H2A → HO2 (H2O2) + HA•
M(n-1)+ + H2O2 → Mn+ + HO• + OH
Mn+ + H2A or HA• → M(n-1)+ + HA• or A + H+
HO• + H-DNA → H2O + DNA•; DNA• → scission products,
where M(n-1)+/ Mn+ corresponds to the Cu+/Cu2+ or Fe2+/Fe3+ redox active pairs and H2A is the auxiliary
reducing agent.264 Correspondingly, phen ligand was covalently tethered to sequence-specific DNA
binding protein,265,266 minor groove binder agents,266 short DNA113-117,267 or RNA molecules268,269 for
directing Cu+(phen)2 nuclease activity in a sequence specific manner towards RNA,266,116 ssDNA,113-
115,267 and dsDNA.115,117,265,266,268 From this point of view, the ODN conjugates covalently linked to the
metal chelating DPPZ group are expected to sequence specifically hydrolyze RNA (artificial
ribonucleases and ribozymes) or oxidatively cleave dsDNA when hybridized to their targets. In this
sense linking of DPPZ group to ODNs would allow to prepare new antisense or antigene agents which
act through the chemical inactivation of genetic information.
To test the ability of the DPPZ moiety to participate in the formation of redox-active complex
with the subsequent cleavage of target DNA in the ODN•DNA duplex, the middle-modified DPPZ-
ODN conjugates 15 – 17 (Table 1) were hybridized with 5'-32P-labeled DNA target 29 and then
incubated with ferrous ammonium sulfate and DTT as a reducing agent. The presence of DPPZ-ODN
conjugates led to the highly specific formation of scission products at the positions of the expected
ligand intercalation (T6 and T7, Figure 7) while the native ODN 1 did not induce any target degradation
after 5 h of incubation. The extent of cleavage was greater for the internucleotide DPPZ-conjugation
through the linker L2 (28% of the target 29 was cleaved with 66% of the cleavage occurring at T6 and
T7). The cleavage was less for DPPZ-L1- and DPPZ-L3-ODN conjugates which correlated with the
lower stability of their duplexes. It should be noted that the extent of cleavage with our DPPZ-ODN
conjugate is of the same order of magnitude compared to that, which was detected with phen-tethered
ODN under the same conditions (38%).267 These experiments proved that our DPPZ-modified ODNs
have the potential to act as sequence-specific redox-based artificial nucleases, almost as good as phen-
ODN conjugates.267

48
Figure 7. Scission of the duplexes formed with 5'-32P-labeled 11mer DNA 29 and ODNs covalently linked to the DPPZ chromophore at the middle position of a sequence. (A) Target DNA 29 (1 µM) was hybridized with complementary ODN (5 µM) and then incubated for 5 h at r.t. with 10 µM Fe2+ and 4 mM DTT in a buffer containing 40 mM Tris•HCl and 0.2 M NaCl. Lanes 2 – 5 correspond to the experiments with native ODN 1 and DPPZ-ODN conjugates 15, 16, 17, respectively. Lane 1: footprinting of the native duplex 1•29 (10 µM) with Fe2+-MPE (50 µM) in the presence of 4 mM DTT in a buffer containing 10 mM Tris•HCl and 50 mM NaCl. (B) Sites of primary cleavage.
2.4.2 Photochemical generation of Ru2+(tpy)(DPPZ)(H2O)-ODN (Paper V)
[Ru(tpy)(DPPZ)(CH3CN)]2+-ODN conjugates represent another type of ODNs attached to reactive
groups inducing chemical modifications in its target nucleic acids in a sequence-specific manner. Basing
on the photochemical properties of Ru2+-CH3CN complexes172,193,194 one can expect the dissociation of
the CH3CN ligand in the [Ru(tpy)(DPPZ)(CH3CN)]2+-ODNs upon light exposure. This has been indeed
observed by MALDI-TOF MS analysis (see chapter 2.1.6). The photochemical behavior of
[Ru(tpy)(DPPZ)(CH3CN)]2+-ODN conjugates has also been examined by UV-vis spectroscopy in
aqueous solutions not containing any potential nucleophiles. Under light irradiation (1 h, λ > 300 nm) the
MLCT absorption bands initially centered at 454 nm shifted to longer wavelength (482 nm) exhibiting
three isobestic points in the 300 – 600 nm region (309, 387, and 465 nm respectively). The same change
in the UV-vis characteristics was also observed in the course of light irradiation of the not tethered
[Ru(tpy)(DPPZ-CO-L2)(CH3CN)]2+ complex. The photosubstitution of CH3CN ligand by solvent
molecule has been established for [Ru(tpy)(bpy)(CH3CN)]2+ or related complexes193-195 which allowed us
to assume that [Ru(tpy)(DPPZ-CO-L2)(CH3CN)]2+ complex should undergo photoaquation affording
[Ru(tpy)(DPPZ-CO-L2)(H2O)]2+ if no any potential nucleophiles are present in the mixture. To confirm
this assumption, [Ru(tpy)(DPPZ-CO-L2)(H2O)]2+ has been chemically synthesized from its
[Ru(tpy)(DPPZ-CO-L2)Cl]+ precursor by treatment with a silver p-toluenesulphonate270,271 and the
product was found by UV-vis and 1H-NMR spectroscopy to be identical to that which was photolytically
obtained from the corresponding Ru2+-CH3CN analog. The similarity of the spectroscopic changes
DNA 29
T7
T6
G8
G9
A10
T5
G4
T7
T6
A B
5'-CATGTT TGGAC-3'
3'-TACAA ACCT-5'
T7T61 2 3 4 5

49
occurring upon photoirradiation of the non-tethered complex [Ru(tpy)(DPPZ-CO-L2)(CH3CN)]2+ and
[Ru(tpy)(DPPZ)(CH3CN)]2+-ODN conjugates enable us to conclude that the photosubstitution of CH3CN
ligand by H2O molecule takes place also on an oligonucleotide level affording [Ru(tpy)(DPPZ)(H2O)]2+-
ODN conjugates. The metallated ODNs were analyzed by PAGE before and after photolysis to show that
no ODN decomposition occurs upon the irradiation conditions applied. The above observations based
on MS and UV-vis spectroscopic data thus clearly point that our [Ru(tpy)(DPPZ)(CH3CN)]2+-ODN
conjugates 39 – 42 (Table 1) are indeed thermally stable, and can be activated by light through the
dissociation of the CH3CN ligand. Acetonitrile thus serves as a protective group for the ODN tethered
rective monofunctional [Ru(tpy)(DPPZ)(H2O)]2+ complex. This also suggests that CH3CN is expected to
be replaced by other potential coordinating species if they exist in the reaction mixture.
2.4.3 Photochemical cross-linking of Ru2+(tpy)(DPPZ)(CH3CN)-ODN conjugates to the
complementary DNA strand in ODN•DNA duplexes (Paper V)
Since the initial reports by Rosenberg at al. on the biological affects of some platinum complexes, in
particular cisplatin,272,273 there has been a great deal of interest in the development of transition metal
complexes for use in medicine.274 Most research on potentially chemotherapeutic transition metal
complexes has involved thermal aquation.135-145 Particularly, it has been established that mono- and
diaqua polypyridyl Ru2+ complexes covalently binds to DNA (presumably at N7 of guanine residue)
with different efficiency depending on the steric effects caused by polypyridyl ligands in the octahedral
geometry.275,276 A striking increase of dsDNA thermal stability was detected upon going from mono-
aqua complex binding to the binding of diaqua complexes pointing out that the difunctional metal
complexes are able to cross-link the two DNA strands.276 On the other hand, all of the complexes
requiring thermal activation are toxic because of the facile ligand substitution. Photochemotherapy has
the potential to minimize such toxicity due to that drug activation can be highly controlled in time and
space. Most work in this area has focused on the development of photosensitizing drugs which produce
DNA-damaging oxygen species.277 Recently, some Rh3+ and Cr3+ complexes have been shown to
undergo aquation and form covalent adducts with calf thymus DNA upon irradiation with UV light.278-
285 However, the photoreactivity of such complexes is still not sequence specific and it is desirable to
design ODN conjugates covalently linked to the photoactivatable metal complexes to precisely direct
the drug reactivity towards the sequence of interest.
From this point of view, [Ru(tpy)(DPPZ)(CH3CN)]2+-ODN conjugates can be considered as the
molecules which are activated by light owing to the photoaquation of their Ru2+-label and site
specifically cross-link to the dG residue in the complementary DNA strand after ODN•DNA duplex
formation. To prove this proposal, first, the reactivity of the non-tethered [Ru(tpy)(DPPZ)(H2O)]2+ and its

50
derivatives towards nucleic acid components and natural dsDNA has been investigated. The
monofunctional complex [Ru(tpy)(DPPZ-CONHEt)(H2O)]2+ has been photolytically prepared from its
Ru2+-CH3CN precursor and showed no reaction with thymidine, 2'-deoxycytidine, and 2'-deoxyadenosine
5'-monophosphates (TMP, dCMP, and dAMP respectively). Mixing of equimolar amounts of the
complex with 2'-deoxyguanosine 5'-monophosphate (dGMP) exhibited the formation of a product, which
is characterized by upfield shifts of 1H NMR resonances from 8.2 to 6.8 ppm for H8 and 6.2 to 6.0 for
H1'. The composition of this mixture was identified by positive mode MALDI-TOF MS (Figure 8A) as a
mixture of the starting complex, which is ionized with the loss of H2O molecule [Ru2+(tpy)(DPPZ-
CONHEt)-H+] and the product. The isotopic pattern observed for the product corresponds to a singly
charged species containing Ru2+(tpy)(DPPZ-CONHEt) (688 Da) plus the mass of the singly protonated
dGMP ([C10H13N5O7P], 346 Da). Thus, the product composition fits the expected adduct of the formula
(DPPZ-CONHEt)(tpy)Ru2+-dGMP. The photolysis of [Ru(tpy)(DPPZ)(CH3CN)]2+ in the presence of the
native duplex 34•43 revealed that the photoactivated monofunctional Ru2+ complexes bind to the
dsDNA. From the MALDI-TOF MS analysis of the photolyzed mixture (Figure 8B) it is seen that the
10mer 34 (containing only dA, dC, and T nucleotides) remained completely unaltered (calculated mass is
2945.5, the observed mass is 2944.9), while the target 11mer 43 was modified giving rise to new
m/z2500 3000 3500 4000 4500 5000 55000
3000
6000
9000
12000
15000
3500 4000 4500 50000
1000
(2944.9)34
43 (3409.2)
43+Ru2+
(4024.0)
43+2Ru2+
(4642.6)43+3Ru2+
(5263.4)
43+Ru2+
43+2Ru2+ 43
m/z600 700 800 900 1000 11000
2000
4000
6000
8000
1010 1020 1030 10400
400
800
1200
1600
Figure 8. (A) MALDI-TOF MS (positive mode) of the 1:1 reaction between the 5'-dGMP and complex [Ru(tpy)(DPPZ-CONHEt)(H2O)]2+ in the acetone-H2O mixture (5.4 ×10-3 mol l-1). (B) Negative ion MALDI-TOF MS spectra of the native duplex 34•43 (10-5 mol l-1, reaction volume - 1600 µl) photolyzed with 5 equivalent excess of [Ru(tpy)(dppz)(CH3CN)]2+ for 2 h in 20 mM PO4
3-, 0.1M NaClO4, pH 6.6 buffer and passed after photolysis through Sephadex G-25 column to remove salts. The peaks corresponding to the target ODN 43 bound with Ru2+(tpy)(dppz) residues are depicted as 43+nRu2+ (n = 1,2,3).
687.08 1034.04
Intensity IntensityA B

51
products with the observed m/z values corresponding to the molecular weight of the [43 +
Ru2+(tpy)(DPPZ)] (m/z 4024.0) and [43 + 2×Ru2+(tpy)(DPPZ)] (m/z 4642.6) adducts. It is of interest that
only target ODN 43 containing dG nucleotides is the substrate for the covalent binding of the mono-aqua
Ru2+ complexes in accordance with our observation that dAMP, dCMP, and TMP do not react with such
complexes. Thus, these data clearly esiablish that [Ru(tpy)(DPPZ)(CH3CN)]2+ and its derivatives, upon
light activation, can bind to the guanine containing compounds and to the dG nucleotides of the double
stranded DNA.
From the above experiments, it is clear that when a monofunctional Ru2+ complex is tethered to an
ODN strand of an ODN•DNA duplex it should form a cross-linkage, upon photoactivation, to the
complementary strand containing dG residue in the direct proximity to the appended Ru2+ center. Figure
9 shows the autoradiogram from the photolysis experiments immediately analyzed after irradiation (2 h)
by PAGE in denaturing conditions. All of the Ru2+-ODN conjugates 39 – 42 produced a higher molecular
1 2 3 4 5 6 7 8 9 Figure 9. Autoradiogram of a 20% denaturing polyacrylamide gel showing the formation of interstrand photo cross-linked product (P) upon irradiation of duplexes (10-5 mol l-1) formed with 5'-32P-labeled 11mer 43 and 10mer 34 or different Ru2+ labeled ODNs 39 – 42 in 20 mM PO4
3-, 0.1M NaClO4, pH 6.6 buffer. Lane 1: 42•43 without irradiation. Duplexes irradiated for 2 h: 42•43 (lane 2), 41•43 (lane 3), 39a•43 (lane 4), 39b•43 (lane 5), 40a•43 (lane 6), 40b•43 (lane 7), 34•43 (lane 8). Lane 9: 5'-32P-labeled 11mer 43. 39a and 39b stand for two fractions obtained in the course of HPLC purification of ODN 39. Corresponding two fractions of ODN 40 are designated as 40a and 40b.
weight band under light exposure, indicating the formation of a cross-linked product. As expected, the
highest yield (34%) was obtained with the ODN 42 carrying reactive centers both at the 5'- and 3'-
terminals. For the ODNs carrying only one Ru2+ complex the cross-coupling efficiency is decreased in
the order of 3'-(22%) > 5'-(9%) > middle-(7%) modified duplex which is reversible to the duplex
stabilization effect caused by the tethered metal complex (see Table 3). This trend can be explained in
P→
32P-labeled ODN 43 →

52
terms of structural rigidity of the metallointercalation site in the duplex. The more rigid structure arising
from the intercalation of Ru2+ complex by its threading through the duplex core (see chapter 2.2.4), the
higher stabilization effect and less the metal complex flexibility which dictates the accessibility of the
target dG moiety. As a consequence, the probability of the favorable structural geometry resulting in
cross-coupling reaction is less for the duplexes in which Ru2+ center is more rigidly packed within the
double helix. It is noteworthy that the electrophoretic mobility of the target strand is not effected by the
irradiation of the native duplex (lane 8, Figure 9) as well as without irradiation of Ru2+ labeled duplexes
(lane 1, Figure 9), indicating that indeed the [Ru(tpy)(DPPZ)(CH3CN)]2+ moiety exposed to the light
source is responsible for the cross-coupling effect. MALDI-TOF MS analysis of the irradiated 3'-Ru2+
modified duplex 40•43 confirmed the formation of the cross-linked product generating 1- charged ions at
m/z 7282.1 which corresponds to the sum of the m/z values observed for the non-reacted target strand 43
(m/z 3410.3) and singly Ru2+ modified ODN 40 with decoordinated CH3CN ligand (m/z 3872.8).
The time course of photoreaction examined for the 5',3'-bis-Ru2+ modified ODN 42 (Figure 10A,
lanes 1 - 6) showed that the yield of the adduct is almost steady after 1 h of irradiation achieving the
maximal value of ~33% (Figure 10B). The reason that the cross-coupling reaction proceeds till
saturation at such low yield can be two-fold: (1) the reversibility of the cross-coupling because of the low
product stability, (2) the cross-coupling is accompanied by side reactions deactivating the intermediate
aquaruthenium-ODN species. Note that both undesired processes could take place in the present system.
To explore the stability of the cross-coupling product, the reaction mixture obtained after irradiation for
5 h was kept at room temperature or 37°C and analyzed by PAGE (Figure 10A, lanes 7 – 12). One can
see that the high molecular weight band continuously disappears faster as the incubation temperature is
higher indicating that the cross-coupling product is not stable. When the 32P-labeled slow-migrating band
corresponding to the cross-coupled duplex (as in lane 5, Figure 10A) was excised and subsequently
extracted from the gel with sodium acetate (0.3 M), the analytical PAGE of the resulting extract revealed
a partial regeneration of the non-modified 32P-labeled target ODN 43 (Figure 10C).
The reversibility of the coss-coupling was also proven by MS analysis of the material obtained in
the course of isolation of the PAGE low-migrated band. For this purpose, two photo-cross-linking
reactions were performed on a 17 nmol scale for 3'-Ru2+ modified duplex 40•43 and 5',3'-bis-Ru2+
modified duplex 42•43. The photolyzed duplexes were separated by denaturing PAGE (Figure 11) and
the low-migrated bands were isolated by excision and extraction from gel analogously as it has been
done in the analytical photolysis experiments with 32P-labeled target ODN 43. Optimal separation of the
photoproduct was achived in the case of 3'-Ru2+-labeled duplex (lane 1 in Figure 11), while in the case of
5',3'-bis-Ru2+ modified duplex the electrophoretic mobility of the cross-coupled duplex was very similar
to that of the ODN 42 (lane 2 in Figure 11). UV-Shadowing PAGE at 366 nm clearly exhibits
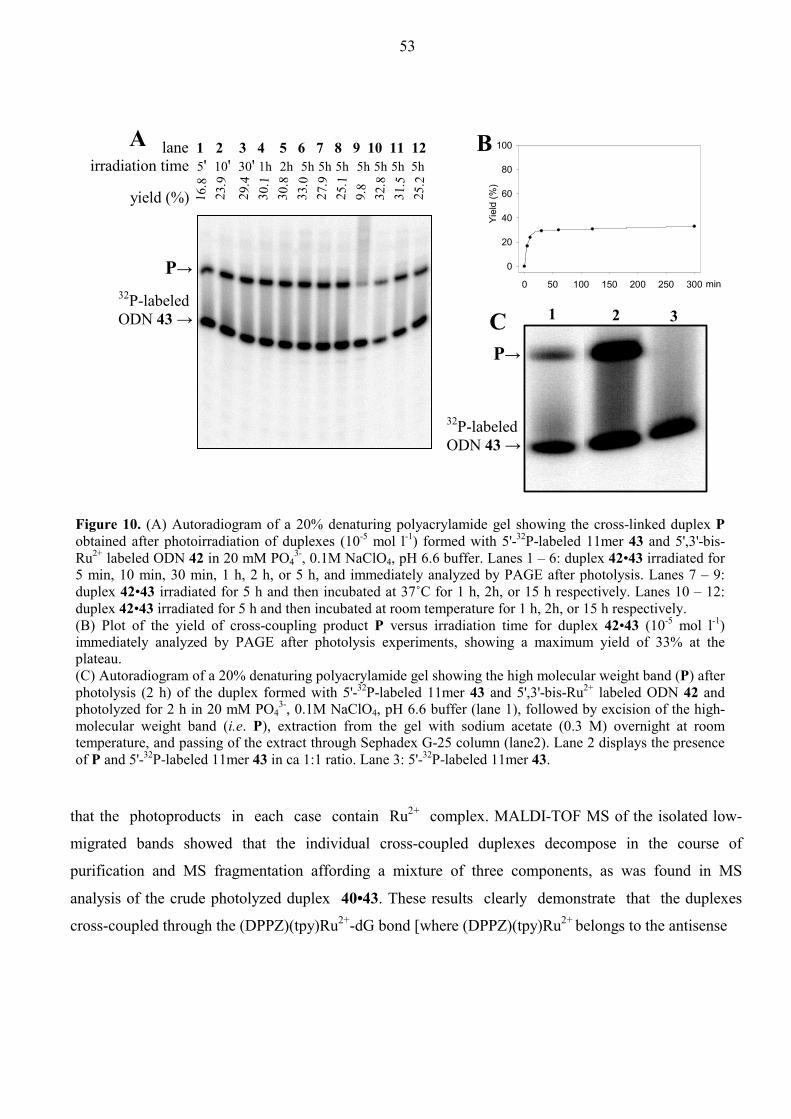
53
lane 1 2 3 4 5 6 7 8 9 10 11 12 irradiation time 5' 10' 30' 1h 2h 5h 5h 5h 5h 5h 5h 5h
yield (%) 16.8
23.9
29.4
30.1
30.8
33.0
27.9
25.1
9.8
32.8
31.5
25.2
that the photoproducts in each case contain Ru2+ complex. MALDI-TOF MS of the isolated low-
migrated bands showed that the individual cross-coupled duplexes decompose in the course of
purification and MS fragmentation affording a mixture of three components, as was found in MS
analysis of the crude photolyzed duplex 40•43. These results clearly demonstrate that the duplexes
cross-coupled through the (DPPZ)(tpy)Ru2+-dG bond [where (DPPZ)(tpy)Ru2+ belongs to the antisense
min0 50 100 150 200 250 300
Yiel
d (%
)
0
20
40
60
80
100BA
C 1 2 3
Figure 10. (A) Autoradiogram of a 20% denaturing polyacrylamide gel showing the cross-linked duplex Pobtained after photoirradiation of duplexes (10-5 mol l-1) formed with 5'-32P-labeled 11mer 43 and 5',3'-bis-Ru2+ labeled ODN 42 in 20 mM PO4
3-, 0.1M NaClO4, pH 6.6 buffer. Lanes 1 – 6: duplex 42•43 irradiated for 5 min, 10 min, 30 min, 1 h, 2 h, or 5 h, and immediately analyzed by PAGE after photolysis. Lanes 7 – 9: duplex 42•43 irradiated for 5 h and then incubated at 37˚C for 1 h, 2h, or 15 h respectively. Lanes 10 – 12: duplex 42•43 irradiated for 5 h and then incubated at room temperature for 1 h, 2h, or 15 h respectively. (B) Plot of the yield of cross-coupling product P versus irradiation time for duplex 42•43 (10-5 mol l-1) immediately analyzed by PAGE after photolysis experiments, showing a maximum yield of 33% at the plateau. (C) Autoradiogram of a 20% denaturing polyacrylamide gel showing the high molecular weight band (P) after photolysis (2 h) of the duplex formed with 5'-32P-labeled 11mer 43 and 5',3'-bis-Ru2+ labeled ODN 42 and photolyzed for 2 h in 20 mM PO4
3-, 0.1M NaClO4, pH 6.6 buffer (lane 1), followed by excision of the high-molecular weight band (i.e. P), extraction from the gel with sodium acetate (0.3 M) overnight at room temperature, and passing of the extract through Sephadex G-25 column (lane2). Lane 2 displays the presence of P and 5'-32P-labeled 11mer 43 in ca 1:1 ratio. Lane 3: 5'-32P-labeled 11mer 43.
P→ 32P-labeled ODN 43 →
P→
32P-labeled ODN 43 →

54
Oligo T
Oligo VI
Photoproduct
1 2 21
254 nm 366 nm
Photoproduct + Oligo VIII
Figure 11. UV-Shadowing 20% denaturing polyacrylamide gel (λ = 254 nm and 366 nm) of the reaction mixtures obtained after photolysis of 3'-Ru2+ labeled duplex 40•43 (lane 1) and 5',3'-bis-Ru2+ labeled duplex 42•43 (lane 2) in 20 mM PO4
3-, 0.1M NaClO4, pH 6.6 buffer (duplex concentration in each experiment was 10-5 mol l-1, reaction volume: 1600 µl) and passed through Sephadex G-25 column to remove salts. Comparison of the bands visualized at 254 nm and 366 nm shows that the slow-migrating bands (i.e.high molecular weight photoproduct) indeed show fluorescence at 366 nm, which indicates that the photoproduct consists of Ru2+ cross-linkage. The slow-migrating photoproduct bands in each lane were excised and then extracted from the gel with sodium acetate (0.3 M). The extracts were passed through Sephadex G-25 column and analyzed by MALDI-TOF MS.
strand and dG belongs to the target strand] are not stable and can reversibly decomposed liberating the
target strand without any secondary modifications.
To understand the reason of instability of cross-coupling products formed with the target
DNA strand 43 and photochemically generated [Ru(tpy)(DPPZ)(H2O)]2+-ODN conjugates, the time
dependence of the 1:1 reaction between [Ru(tpy)(dppz-CONHEt)(H2O)]2+ and 5'-dGMP in
acetone-d6/D2O mixture was monitored by 1H NMR at various temperatures. It was found that this
reaction never goes to completion, and in fact reaches an equilibrium depending upon the reaction
condition (Figure 12A). Thus, the experimental data pointed that coordination of dGMP to the mono-
aqua Ru2+ complex belongs to reversible reaction of type, A + B ←→ X + Y, where X is [Ru(tpy)(dppz-
CONHEt)(dGMP)] and A and B are starting complex [Ru(tpy)(dppz-CONHEt)(H2O)]2+ and dGMP
respectively. The equilibrium was only slightly shifted to the product side with the decrease of
temperature, which indicates that the reaction is poorly exothermic. Fitting of the observed equilibrium
constants K to the van’t-Hoff equation (Figure 12B) gave 39.3±1.7 kJ/mol for ∆Hº and –0.038±0.006
kJ/(mol K) for ∆Sº. Our experimental results show that the product-complex, [Ru(tpy)(dppz-
CONHEt)(dGMP)], thus formed suffers reverse aquation and exists in an equilibrium with
[Ru(tpy)(dppz-CONHEt)(H2O)]2+. Next, we added an excess of CH3CN to the reaction mixture when it
has already reached the equilibrium, and incubated in the dark at room temperature. After 17 h of

55
incubation with CH3CN, 1H NMR spectra indicated the presence of starting dGMP and [Ru(tpy)(dppz-
CONHEt)(CH3CN)](PF6)2 only, which confirms the reversibility of the reaction. The poor stability of the
adduct could be a consequence of high distortions of the octahedral geometry which arise to minimize
the steric constraints between the bulky guanine and polypyridyl ligands (tpy and dppz). The present data
Figure 12. (A) Kinetic study of the 1:1 reaction between complex [Ru(tpy)(dppz-CONHEt)(H2O)]2+ and 5'-dGMP in CD3COCD3-D2O (1:1, v/v) solution at 20˚C (●), 33˚C (○), 44˚C (▼), and 56˚C (∇ ), which shows that the yield of the photo adduct varies from 80% to 60% in the temperature range of 20-56˚C. (B) Plot of RlnK vs 1/T obtained from the corresponding kinetic studies, giving the enthalpy ∆Hº, and entropy ∆Sº from the slope and intercept respectively: RlnK = ∆Sº - ∆Hº(1/T).
are in accordance with the low efficiency of the cross-coupling of the two strands in Ru2+ labeled
duplexes 39•43 - 42•43, and correlate with the thermal instability of cross-coupled duplexes. The present
study also explains the very low binding efficiency of the polypyridyl mono-aquaruthenium(II)
complexes towards the double-stranded DNA observed by Thorp.275
2.4.4 The extension of the idea of the Ru2+ complex activation via photoaquation (unpublished data)
The idea to activate the thermally stable polypyridyl Ru2+ complexes by quantative exclusion of the
labile ligands with subsequent aquation of the metal complexes under light irradiation can also be
applied for the bifunctional Ru2+ analogs. It is expected that increasing the number of sites available in
the Ru2+ coordination sphere for photoaquation and subsequent binding to nucleobases, one can
influence on tethered complex reactivity and strikingly improve the stability of the cross-linked product.
We utilized that the mixed Ru2+ complexes containing sterically congested bidentate ligands such as
dmbpy, pypz, or biq undergo clean and selective ligand photosubstitution by solvent molecules.286-294
1/T (K-1)
0.0030 0.0032 0.0034R
ln(K
)80
85
90
95
100
time (h)
Con
cent
ratio
n of
5'-d
GM
P (m
ol/l)
0.000
0.001
0.002
0.003
0.004
0.005
0.0000.0010.0020.0030.0040.005
0 6 12 18 24
0 20 40 60 80 100 120 140 160 180
BA
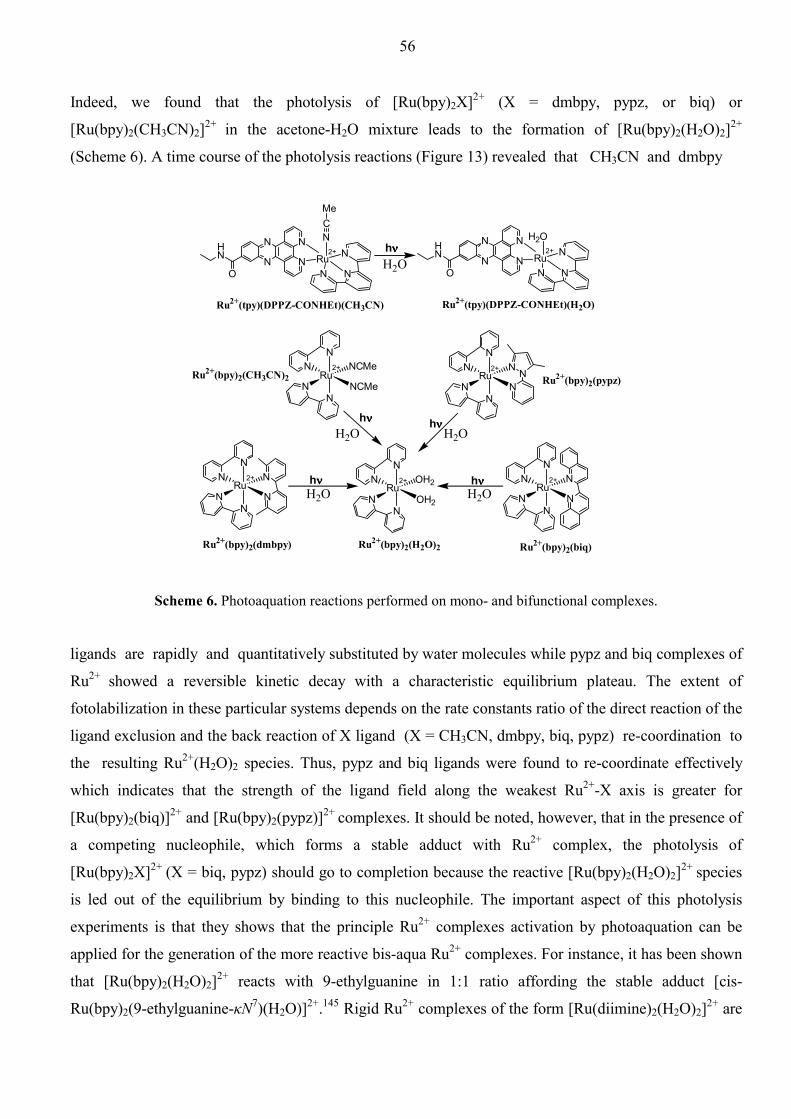
56
Indeed, we found that the photolysis of [Ru(bpy)2X]2+ (X = dmbpy, pypz, or biq) or
[Ru(bpy)2(CH3CN)2]2+ in the acetone-H2O mixture leads to the formation of [Ru(bpy)2(H2O)2]2+
(Scheme 6). A time course of the photolysis reactions (Figure 13) revealed that CH3CN and dmbpy
HN
NN
NRu
N
N
N
N
NCMe
O
RuN
N NCMe
NCMe
N
N
RuN
N OH2
OH2
N
N
HN
NN
NRu
N
N
N
N
H2O
O
RuN
N N
N
N
N
N
RuN
N N
N
N
N
RuN
N N
N
N
N
H2O
H2O H2O
H2O H2O
2+
2+
2+
2+
2+
2+ 2+
hνννν
hνννν hνννν
hνννν hνννν
Ru2+(bpy)2(dmbpy) Ru2+(bpy)2(H2O)2 Ru2+(bpy)2(biq)
Ru2+(bpy)2(pypz)Ru2+(bpy)2(CH3CN)2
Ru2+(tpy)(DPPZ-CONHEt)(CH3CN) Ru2+(tpy)(DPPZ-CONHEt)(H2O)
Scheme 6. Photoaquation reactions performed on mono- and bifunctional complexes.
ligands are rapidly and quantitatively substituted by water molecules while pypz and biq complexes of
Ru2+ showed a reversible kinetic decay with a characteristic equilibrium plateau. The extent of
fotolabilization in these particular systems depends on the rate constants ratio of the direct reaction of the
ligand exclusion and the back reaction of X ligand (X = CH3CN, dmbpy, biq, pypz) re-coordination to
the resulting Ru2+(H2O)2 species. Thus, pypz and biq ligands were found to re-coordinate effectively
which indicates that the strength of the ligand field along the weakest Ru2+-X axis is greater for
[Ru(bpy)2(biq)]2+ and [Ru(bpy)2(pypz)]2+ complexes. It should be noted, however, that in the presence of
a competing nucleophile, which forms a stable adduct with Ru2+ complex, the photolysis of
[Ru(bpy)2X]2+ (X = biq, pypz) should go to completion because the reactive [Ru(bpy)2(H2O)2]2+ species
is led out of the equilibrium by binding to this nucleophile. The important aspect of this photolysis
experiments is that they shows that the principle Ru2+ complexes activation by photoaquation can be
applied for the generation of the more reactive bis-aqua Ru2+ complexes. For instance, it has been shown
that [Ru(bpy)2(H2O)2]2+ reacts with 9-ethylguanine in 1:1 ratio affording the stable adduct [cis-
Ru(bpy)2(9-ethylguanine-κN7)(H2O)]2+.145 Rigid Ru2+ complexes of the form [Ru(diimine)2(H2O)2]2+ are
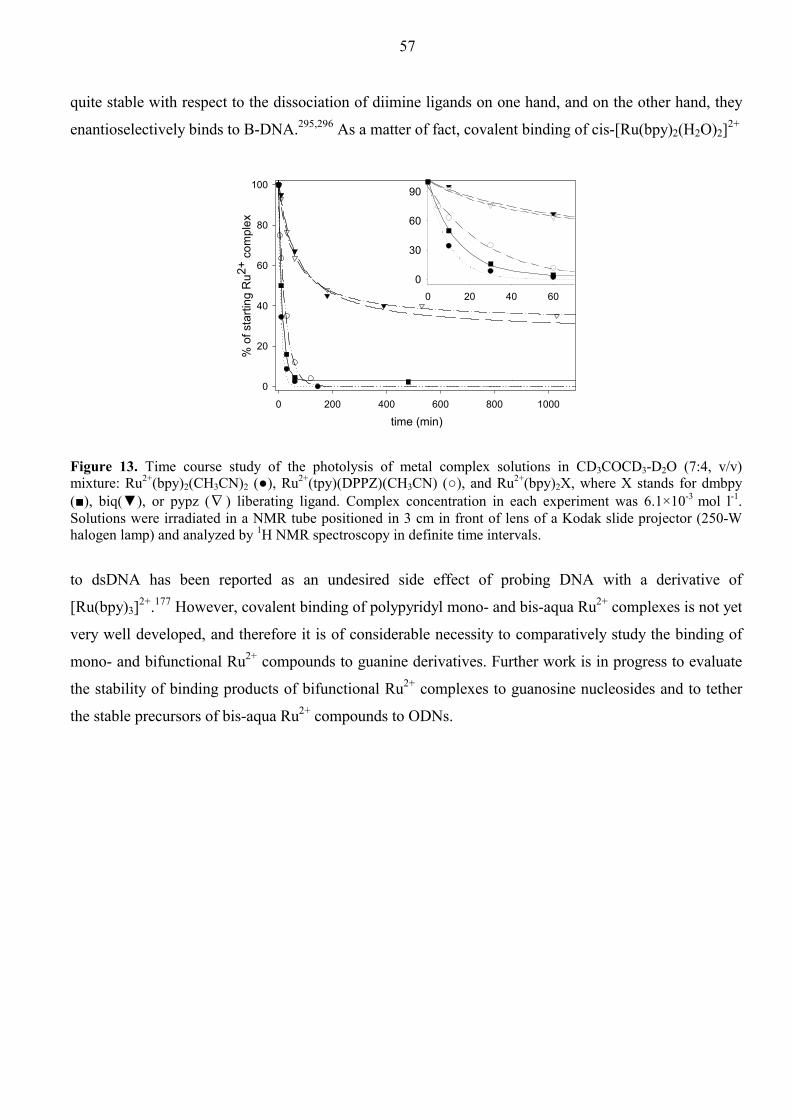
57
quite stable with respect to the dissociation of diimine ligands on one hand, and on the other hand, they
enantioselectively binds to B-DNA.295,296 As a matter of fact, covalent binding of cis-[Ru(bpy)2(H2O)2]2+
time (min)0 200 400 600 800 1000
% o
f sta
rting
Ru2+
com
plex
0
20
40
60
80
100
0 20 40 600
30
60
90
Figure 13. Time course study of the photolysis of metal complex solutions in CD3COCD3-D2O (7:4, v/v) mixture: Ru2+(bpy)2(CH3CN)2 (●), Ru2+(tpy)(DPPZ)(CH3CN) (○), and Ru2+(bpy)2X, where X stands for dmbpy (■), biq(▼), or pypz (∇ ) liberating ligand. Complex concentration in each experiment was 6.1×10-3 mol l-1. Solutions were irradiated in a NMR tube positioned in 3 cm in front of lens of a Kodak slide projector (250-W halogen lamp) and analyzed by 1H NMR spectroscopy in definite time intervals.
to dsDNA has been reported as an undesired side effect of probing DNA with a derivative of
[Ru(bpy)3]2+.177 However, covalent binding of polypyridyl mono- and bis-aqua Ru2+ complexes is not yet
very well developed, and therefore it is of considerable necessity to comparatively study the binding of
mono- and bifunctional Ru2+ compounds to guanine derivatives. Further work is in progress to evaluate
the stability of binding products of bifunctional Ru2+ complexes to guanosine nucleosides and to tether
the stable precursors of bis-aqua Ru2+ compounds to ODNs.

58
3. Acknowledgements This work was carried out at the Department of Bioorganic Chemistry, Biomedical Center, Uppsala University, Uppsala, during the years 1996-2002. I would like to express my appreciation to my supervisor Professor Jyoti Chattopadhyaya for giving me this opportunity to work and learn in his well-equipped laboratory, for introducing me to the field of nucleic acids chemistry and its biological applications, for stimulating and constructive criticism, thoughtful guidance throughout the years and valuable discussions. I wish to express my deep gratitude to all the former and present colleagues at the Department of Bioorganic Chemistry: Parag Acharya, Sandipta Acharya, Peter Agback, Jharna Barman, Somer Bekiroglu, Alexei Denisov, Krishna Kumar Dubey, Melker Holmer, Johan Isaksson, Anil Kumar, Mrinal Kanti Kundu, Brian Lawrence, Xiaoguang Luo, Ingrid Luyten, Tatiana Maltseva, Jan Milecki, Frans Peder Nilson, Dimitry Ovcharenko, Catherine Petit, Janez Plavec, Nitin Puri, Anders Sandström, Christian Sund, Christophe Thibaudeau, Anna Trifonova, Andrei Vasiliev, Irina Velikian, Shun-ichi Yamakage, Peter Weiler, Pei Wen and Edouard Zamaratski for their contribution in creation of exiting scientific atmosphere on the Department. I am specially grateful to: Pradeep Kumar for his splendid human character and readiness to help András Földesi who helped me a lot with synthesis, instruments and in chemicals search Nariman Amirkhanov for the fruitful scientific discussions which allowed me to understand many not trivial aspects of my work Oles Plashkevich for solution of computer problems Gopal Datta for his brotherhood and the same excitement by magic of mountains Kristiina Komminaho, Pernilla Brohage, Rabab Elkarib and Jaana Öst for their kind assitance, availability and competent secretarial work. Alexander Papchikhin for importing me to Sweden Suresh Gohil (Department of Chemistry, Swedish University of Agricultural Sciences) for the rare ability to share difficulties, which I had a lot with my last project, and for the qualified MS analysis. I am deeply thankful to Konstantin Kovalev, the only person I’ve ever met abroad, with whom I felt myself most easy and comfortable – for his attractively ironic point of view on things. My warmest thanks I express to Alex and Olga Shebanov for polemic discussions throughout where we sat and drank, drank, and drank. My work in Uppsala would never be somehow possible without that warm and re-animating atmosphere created by my friends, Yuri Khotyaintsev and Tania Leonova, Maxim and Larisa Smirnov, Oleg Travnikov, Svjatoslav Korepov and Lena Vlasova, Konstantin Matveev, Tatyana Tentler, Mikis and Mette Larsen and many others. I am very grateful to Line Strand and Joel Sohlberg for taking care of my son and exporting of Swedish traditions to my family. I would like to thank my wife Elena for love, patience, creating the home comfort which served as a buffer against aggressive “chemical medium” present in a chemist life, and for my son Daniil, who is specially acknowledged for his love and that endless energy reservoir which I have now in his person on my back. Last but not least, I thank my mother and father who gave me life, love, and everything to be the person who I am. I thank my sister Sasha, mother-in-law, and all relatives for their love, support, and waiting, waiting, and waiting for me all these years.

59
4. References
1 Zamecnik, P. C.; Stephenson, M. L. Proc. Natl. Acad. Sci. USA 1978, 75, 280 2 Stephenson, M. L.; Zamecnik, P. C. Proc. Natl. Acad. Sci. USA 1978, 75, 285 3 Uhlmann, E.; Peyman, A. Chem. Rev. 1990, 90, 543 4 De Mesmaeker, A.; Häner, R.; Martin, P.; Moser, H. E. Acc. Chem. Res. 1995, 28, 366 5 Agrawal, S.; Kandimalla, E. Mol. Med. Today 2000, 6, 72 6 Toulme, J.-J. Nat. Biotechnol. 2001, 19, 17 7 Braasch, D. A.; Corey, D. R. Biochemistry 2002, 41, 4503 8 Watson, J. D.; Crick, F. H. C. Nature 1953, 171, 737 9 Oligodeoxynucleotides. Antisense Inhibitors of Gene Expression, Ed. Cohen, J. S.; Topics in Molecular and Structural Biology, MacMillan, 1989 10 Hélène, C.; Saison-Behmoaras, T. Medicine/Science, 1994, 10, 257 11 Thuong, N. T., Hélène, C. Angew. Chem. Int. Ed. Engl. 1993, 32, 666 12 Sun, J.-S.; Garestier, T., Hélène, C. Curr. Opin. Struct. Biol. 1996, 6, 327 13 Vasquez, K. M.; Wilson, J. Trends Biochem. Sci. 1998, 23, 4 14 Hoogsteen, K. Acta Crystallogr. 1959, 12, 822 15 Stain, C. A.; Cheng, Y.-C. Science, 1993, 261, 1004 16 Moser, H. E.; Dervan, P. B. Science, 1987, 238, 645 17 Povsic, T. J.; Dervan, P. B. J. Am. Chem. Soc. 1990, 112, 9428 18 Bertrand, J.-R.; Imbach, J.-L.; Paoletti, C. Biochem. Biophys. Res. Commun. 1989, 164, 311 19 Blacke, K. R.; Murakami, A.; Spitz, S. A. Biochemistry 1985, 24, 6139 20 Boiziau, C.; Kurfurst, R.; Cazenave, C. Nucleic Acids Res. 1991, 19, 1113 21 Johansson, H. E.; Belsham, G. J.; Sproat, B. S. Nucleic Acids Res. 1994, 22, 4591 22 Haeuptle, M. T.; Frank, R.; Dobberstain, B. Nucleic Acids Res. 1986, 14, 1427 23 Minshull, J.; Hunt, T. Nucleic Acids Res. 1986, 14, 6433 24 Knorre, D. G.; Vlassov, V. V. Progr. Nucleic Acids Res. Mol. Biol. 1985, 32, 291 25 Goodchild, J. Bioconjugate Chem. 1990, 1, 165 26 Lerman, L. S. J. Mol. Biol. 1961, 3, 18 27 Asseline, U.; Thuong, N. T.; Hélène, C. New J. Chem. 1997, 21, 5 28 Lokhov, S.; Podyminogin, M. A.; Sregeev, D. S.; Silnikov, V. N.; Kutyavin, I. V.; Sishkin, G. V.; Zarytova, V. P. Bioconjugate Chem. 1992, 3, 414 29 Balbi, A.; Sottofattori, E.; Grandi, T.; Mazzei, M. Tetrahedron 1994, 50, 4009 30 Asseline, U.; Delarue, M.; Lancelot, G.; Toulme, F.; Thuong, N. T.; Montenay-Garestier, T.; Hélène, C. Proc. Natl. Acad. Sci. USA 1984, 81, 3297 31 Asseline, U.; Bonfils, E.; Dupret, D.; Thuong, N. T. Bioconjugate Chem. 1996, 7, 369 32 Fukui, K.; Morimoto, M.; Segawa, H.; Tanaka, K.; Shimidzu, T. Bioconjugate Chem. 1996, 7, 349 33 Fukui, K.; Iwane, K.; Shimidzu, T.; Tanaka, K. Tetrahedron Lett. 1996, 37, 4983 34 Fukui, K.; Tanaka, K.; Nucleic Acids Res. 1996, 24, 3962 35 Asseline, U.; Thuong, N. T. Nucleosides & Nucleotides 1991, 10, 359 36 Lin, K.-Y.; Matteuchi, M. Nucleic Acids Res. 1991, 19, 3111 37 Ono, A.; Dan, A.; Matsuda, A. Bioconjugate Chem. 1993, 4, 499 38 Dan, A.; Yoshimura, Y.; Ono, A.; Matsuda, A. Bioorg. Med. Chem. Lett. 1993, 3, 615 39 Chen, J. K.; Carlson, D. V.; Weith, H. L.; O’Brien, J. A.; Goldman, M. E.; Cushman, M. Tetrahedron Lett. 1992, 33, 2275 40 Mann, J. S.; Shibata, Y.; Meehan, T. Bioconjugate Chem. 1992, 3, 554 41 Yamana, K.; Ohashi, Y.; Nunota, K.; Kitamura, M.; Nakana, H.; Sangen, O.; Shimidzu,

60
T. Tetrahedron Lett. 1991, 32, 6347 42 Telser, J.; Cruickshank, K. A.; Morrison, L. E.; Netzel, T. L.; J. Am. Chem. Soc. 1989, 111, 9428 43 Dikalov, S. I.; Rumyantseva, G. V.; Weiner, L. M.; Sergejev, D. S.; Frolova, E. I.; Godovikova, T. S.; Zarytova, V. F. Chem. Biol. Interactions 1991, 77, 325 44 Kasai, H.; Goto, M.; Ikeda, K.; Zama, M.; Takemura, S.; Matsuura, S.; Sugimoto, T.; Goto, T. Biochemistry 1976, 15, 898 45 Yamana, K.; Aota, R.; Nakano, H. Tetrahedron Lett. 1995, 46, 8427 46 Sun, J. S.; François, J.-C.; Montenay-Garestier, T.; Saison-Behmoaras, T.; Roig, V.; Thuong, N. T.; Hélène, C. Proc. Natl. Acad. Sci. USA, 1989, 86, 9198 47 Collier, D. A.; Thuong, N. T.; Hélène, C. J. Am. Chem. Soc. 1991, 113, 1457 48 Collier, D. A.; Mergny, J.-L.; Thuong, N. T.; Hélène, C. Nucleic Acids Res. 1991, 19, 4219 49 Montenay-Garestier, T.; Sun, J. S.; Chomilier, J.; Mergny, J. L.; Takasugi, M.; Asseline, U.; Thuong, N. T.; Rougée, M.; Hélène, C. In Molecular Basis of Specificity in Nucleic Acid-Drug-Interactions, Pullman, B.; Lortner, J. Eds.; Kluwer, Dordrecht, 1990, 275 50 Takasugi, M.; Guendouz, A.; Chassignol, M.; Decout, J. L.; Lhomme, J.; Thuong, N. T.; Hélène, C. Proc. Natl. Acad. Sci. USA, 1991, 88, 5602 51 Garbesi, A.; Bonazzi, S.; Zanella, S.; Capobianco, M. L.; Giannini, G.; Arcamone, F. Nucleic Acids Res. 1997, 25, 2121 52 Gianolio, D. A.; McLaughlin, L. W. J. Am. Chem. Soc. 1999, 121, 6334 53 Puri, N.; Zamaratski, E.; Sund, C.; Chattopadhyaya, J. Tetrahedron 1997, 53, 10409 54 Silver, G. C.; Nguyen, C. H.; Boutorine, A. S.; Bisagni, E.; Garestier, T.; Hélène, C. Bioconjugate Chem. 1997, 8, 15 55 Silver, G. C.; Sun, J.-S.; Nguyen, C. H.; Boutorine, A. S.; Bisagni, E.; Hélène, C. J. Am. Chem. Soc. 1997, 119, 263 56 Horne, D. A.; Dervan, P. B. J. Am. Chem. Soc. 1990, 112, 2435 57 Ono, A.; Chen, L. S.; Kan, L. S. Biochemistry 1991, 30, 9914 58 Froehler, B. C.; Terhorst, T.; Shaw, J. P.; McCurdy, S. N. Biochemistry 1992, 31, 1603 59 Sun, J. S.; Giovannangeli, C.; François, J. C.; Kurfurst, R.; Montenay-Garestier, T.; Asseline, U.; Saison-Behmoaras, T.; Thuong, N. T.; Hélène, C. Proc. Natl. Acad. Sci.
USA, 1991, 88, 6023 60 Horne, D. A.; Dervan, P. B. Nucleic Acids Res. 1991, 19, 4963 61 Rahakrishnan, I.; Patel, D. J. J. Mol. Biol. 1994, 241, 600 62 Huang, C.-Y.; Bi, G.; Miller, P. S. Nucleic Acids Res. 1996, 24, 2606 63 Zhou, B. W.; Puga, E.; Sun, J. S.; Garestier, T.; Hélène, C. J. Am. Chem. Soc. 1995, 117, 10425 64 Kukreti, S.; Sun, J. S.; Garestier, T.; Hélène, C. Nucleic Acids Res. 1997, 25, 4264 65 Milligan, J. F.; Matteucci, M. D.; Martin, J. C. J. Med. Chem. 1993, 36, 1923 66 Zamaratski, E.; Pradeepkumar, P. I.; Chattopadhyaya, J. J. Biochem. Biophys. Methods 2001, 48, 189 67 Cazenave, C.; Stain, C. A.; Loreau, N.; et al. Nucleic Acids Res. 1989, 17, 4255 68 Gagnor, C.; Bertrand, J.-R.; Thenet, S.; Lemaitre, M.; Morvan, F.; Rayner, B.; Malvy, C.; Lebleu, B.; Imbach, J.-L.; Paoletti, C. Nucleic Acids Res. 1987, 15, 10419 69 Crooke, S. T. Biochimica et Biophysica Acta 1999, 1489, 31 70 Lesnik, E. A.; Guinosso, C. J.; Kawasaki, A. M.; Sasmor, H.; Zounes, M.; Cummins, L. L.; Ecker, D. J.; Cook, P. D.; Freier, S. M. Biochemistry 1993, 32, 7832 71 Monoharan, M. Biochimica et Biophysica Acta 1999, 1489, 117 72 Martin, P. Helv. Chim. Acta 1995, 78, 486 73 Kawasaki, A. M.; Casper, M. D.; Freier, S. M.; Lesnik, E. A.; Zounes, M. C.;

61
Cummins, L. L.; Gonzalez, C.; Cook, P. D. J. Med. Chem. 1993, 36, 831 74 Wengel, J. Acc. Chem. Res. 1999, 32, 301 75 Furdon, P. J.; Dominski, Z.; Kole, R. Nucleic Acids Res. 1989, 17, 9193 76 Agrawal, S.; Mayrand, S. H.; Zamecnik, P. C.; Pederson, T. Proc. Natl. Acad. Sci. USA 1990, 87, 1401 77 Morvan, F.; Sanghvi, Y. S.; Perbost, M.; Vasseur, J.-J.; Bellon, L. J. Am. Chem. Soc. 1996, 118, 255 78 Gryaznov, S. M. Biochimica et Biophysica Acta 1999, 1489, 131 79 Rait, V. K.; Shaw, B. R. Antisense Nucleic Acid Drug Dev. 1999, 9, 53 80 Damha, M. J.; Wilds, C. J.; Noronha, A.; Brukner, I.; Borkow, G.; Arion, D.; Parniak, M. A. J. Am. Chem. Soc. 1998, 120, 12976 81 Noronha, A. M.; Wilds, C. J.; Lok, C.-N.; Viazovkina, K.; Arion, D.; Parniak, M. A.; Damha, M. J. Biochemistry 2000, 39, 7050 82 Manisov, G.; Teplova, M.; Nielsen, P.; Wengel, J.; Egli, M. Biochemistry 2000, 39, 3525 83 Giles, R. V.; Tidd, D. M. Nucleic Acids Res. 1992, 20, 763 84 Agrawal, S.; Jiang, Z.; Zhao, Q.; Shaw, D.; Cai, Q.; Roskey, A.; Channavajjala, L.; Saxinger, C.; Zhang, R. Proc. Natl. Acad. Sci. USA 1997, 94, 2620 85 Argawal, S. Biochimica et Biophysica Acta 1999, 1489, 53 86 Altmann, K.-H.; Fabbro, D.; Dean, N. M.; Geiger, T.; Monia, B. P.; Nicklin, P. Biochem. Soc. Trans. 1996, 24, 630 87 Lok, C.-N.; Viazovkina, E.; Min, K.-L.; Nagy, E.; Wilds, C. J.; Damha, M. J.; Parniak, M. A. Biochemistry 2002, 41, 3457 88 Wahlestedt, C.; Salmi, P.; Good, L.; Kela, J.; Johnsson, T.; Hökfelt,T.; Broberger, C.;
Porreca, F.; Lai, J.; Ren, K.; Ossipov, M.; Koshkin, A.; Jakobsen, N.; Skouv, J.; Oerum, H.; Jacobsen, M. H.; Wengel, J. Proc. Natl. Acad. Sci. USA 2000, 97, 5633 89 Kurreck, J.; Wyszko, E.; Gillen, C.; Erdmann, V. A. Nucleic Acids Res. 2002, 30, 1911 90 Pradeepkumar, P. I.; Zamaratski, E.; Földesi, A.; Chattopadhyaya, J. Tetrahedron Lett. 2000, 41, 8601 91 Pradeepkumar, P. I.; Zamaratski, E.; Földesi, A.; Chattopadhyaya, J. J. Chem. Soc., Perkin Trans. 2, 2001, 402 92 Pradeepkumar, P. I.; Chattopadhyaya, J. J. Chem. Soc., Perkin Trans. 2, 2001, 2074 93 Puri, N.; Chattopadhyaya, J. Nucleosides & Nucleotides, 1999, 18, 2785 94 Cazenave, C.; Loreau, N.; Thoung, N. T.; Toulme, J.-J.; Hélène, C. Nucleic Acids Res. 1987, 15, 4717 95 Godard, G.; François, J.-C.; Duroux, I.; Asseline, U.; Chassignol, M.; Thuong, N.; Hélène, C.; Saison-Behmoaras, T. Nucleic Acids Res. 1994, 22, 4789 96 Boutorine, A. S.; Brault, D.; Takasugi, M.; Delgado, O.; Hélène, C. J. Am. Chem. Soc. 1996, 118, 9469 97 Kutyavin, I. V.; Gamper, H. B.; Gall, A. A.; Meyer, R. B.; J. Am. Chem. Soc. 1993, 115, 9303 98 Maruenda, H.; Tomasz, M. Bioconjugate Chem. 1996, 7, 541 99 Tsubouchi, A.; Bruice, T. C. J. Am. Chem. Soc. 1995, 117, 7399 100 Trawick, B. N.; Daniher, A. T.; Bashkin, J. K. Chem. Rev. 1998, 98, 939 101 Bashkin, J. K.; Frolova, E. I.; Sampath, U. J. Am. Chem. Soc. 1994, 116, 5981 102 Matsumura, K.; Endo, M.; Komiyama, M. J. Chem. Soc., Chem. Commun. 1994, 2019 103 Magda, D.; Miller, R. A.; Sessler, J. L.; Iverson, B. L. J. Am. Chem. Soc. 1994, 116, 7439 104 Magda, D.; Crofts, S.; Lin, A.; Miles, D.; Wright, M.; Sessler, J. L. J. Am. Chem. Soc. 1997, 119, 2293

62
105 Magda, D.; Wright, M.; Crofts, S.; Lin, A.; Sessler, J. L. J. Am. Chem. Soc. 1997, 119, 6947 106 Baker, B. F.; Ramasamy, K.; Keily, J. Bioorg. Med. Chem. Lett. 1996, 6, 1647 107 Takeda, N.; Imai, T.; Irisawa, M.; Sumaoka, J.; Yashiro, M.; Shigekawa, H.; Komiyama, M. Chem. Lett. 1996, 599 108 Chu, B. C.; Orgel, L. E. Proc. Natl. Acad. Sci. USA 1985, 82, 963 109 Lin, S.-B.; Blake, K. R.; Miller, P. S.; Ts’o, P. O. Biochemistry 1989, 28, 1054 110 Moser, H. E.; Dervan, P. B. Science 1987, 238, 645 111 Strobel, S. A.; Dervan, P. B. Science 1990, 249, 73 112 Han, H.; Dervan, P. B. Proc. Natl. Acad. Sci. USA 1993, 90, 3806 113 François, J.-C.; Saison-Behmoaras, T.; Chassignol, M.; Thuong, N. T.; Sun, J.-S.; Hélène, C. Biochemistry 1988, 27, 2272 114 Chen, C. B.; Sigman, D. C. Proc. Natl. Acad. Sci. USA 1986, 83, 7147 115 François, J.-C.; Saison-Behmoaras, T.; Chassignol, M.; Thuong, N. T.; Hélène, C.; J. Biol. Chem. 1989, 264, 5891 116 Chen, C. B.; Sigman, D. C. J. Am. Chem. Soc. 1988, 110, 6570 117 François, J.-C.; Saison-Behmoaras, T.; Barbier, C.; Chassignol, M.; Thuong, N. T.; Hélène, C.; Proc. Natl. Acad. Sci. USA 1989, 86, 9702 118 Le Doan, T.; Perrouault, L.; Chassignol, M.; Thuong, N. T.; Hélène, C. Nucleic Acids Res. 1987, 15, 8643 119 Frolova, E. I.; Fedorova, O. S.; Knorre, D. G. Biochimie, 1993, 75, 5 120 Sergeev, D. S.; Zarytova, V. F.; Mamaev, S. V.; Godovikova, T. S.; Vlassov, V. V. Antisense Res. Dev. 1992, 2, 235 121 Sergeev, D. S.; Vorobjev, P. E.; Zarytova, V. F. Nucleosides & Nucleotides 1997, 16,
1575 122 Carter, P. J.; Cheng, C.-C.; Thorp, H. H. J. Am. Chem. Soc. 1998, 120, 632 123 Carter, P. J.; Cheng, C.-C.; Thorp, H. H. Inorg. Chem. 1996, 35, 3348 124 Cheng, C.-C.; Goll, J. G.; Neyhart, G. A.; Welch, T. W.; Singh, P.; Thorp, H. H. J. Am. Chem. Soc. 1995, 117, 2970 125 Pyle, A. M.; Chiang, M. Y.; Barton, J. K. Inorg. Chem. 1990, 29, 4487 126 Murphy, C. J.; Arkin, M. R.; Jenkins, Y.; Ghatlia, N. D.; Bossmann, S. H.; Turro, N. J.;
Barton, J. K. Science 1993, 262, 1025 127 Hall, D. B.; Holmlin, R. E.; Barton, J. K. Nature 1996, 382, 731 128 Daniel, B. H.; Barton, J. K. J. Am. Chem. Soc. 1997, 119, 5045 129 Tossi, A. B.; Kelly, J. M. Photochem. Photobiol. 1989, 49, 545 130 Sentagne, C.; Chambron, J.-C.; Sauvage, J.-P.; Paillous, N. J. Photochem. Photobiol. B 1994,
26, 165 131 Moucheron, C.; Kirsch-De Mesmaeker, A.; Kelly, J. M. J. Photochem. Photobiol. B 1997, 40,
91 132 Kirsch-De Mesmaeker, A.; Moucheron, C.; Boutonnet, N. J. Phys. Org. Chem. 1998, 11, 566 133 Ortmans, I.; Content, S.; Boutonnet, N.; Kirsch-De Mesmaeker, A.; Bannwarth, W.; Constant,
J.-F.; Defrancq, E.; Lhomme, J. Chem. Eur. J. 1999, 5, 2712 134 Arkin, M. R.; Stemp, E. D. A.; Pulver, S. C.; Barton, J. K. Chem. Biol. 1997, 4, 389 135 Gelasco, A.; Lippard, S. J. Anticancer Activity of Cisplatin and Related Complexes; Clarke, M.
J.; Sadler, P. J., Ed.; “Metallopharmaceuticals I : DNA Interactions”, Springer-Verlag Berlin Heidelberg: Berlin, 1999, p117
136 Mestroni, G.; Alessio, E.; Calligaris, M.; Attia, W. M.; Quadrifoglio, F.; Cauci, S.; Sava, G.; Zorzet, S.; Pacor, S.; Monti-Bragadin, C.; Tamaro, M.; Dolzani, L. In Ruthenium and Other Non-Platinum Metal Complexes in Cancer Chemotherapy (Clarke, M. J. ed.), Springer-Verlag, Heidelberg, 1989, p74

63
137 Nováková, O.; Kašpárková, J.; Vrána, O.; van Vliet, P. M.; Reedijk, J.; Brabec, V. Biochemistry 1995, 34, 12369
138 Cheng, C.-C.; Lee, W.-L.; Su, J.-G.; Liu, C.-L. J. Chin. Chem. Soc. 2000, 47, 213 139 van Vliet, P. M.; Toekimin, S. M. S.; Haasnoot, J. G.; Reedijk, J.; Nováková, O.; Vrána, O.;
Brabec, V. Inorg. Chim. Acta 1995, 231, 57 140 Armor, J. personal communication 1970 141 Bottomley, F. Can. J. Chem. 1977, 55, 2788 142 Cauci, S.; Viglino, P.; Esposito, G.; Quadrifoglio, F. J. Inorg. Biochem. 1991, 43, 739 143 Esposito, G.; Cauci, S.; Fogolari, F.; Alessio, E.; Scocchi, M.; Quadrifoglio, F.; Viglino, P.
Biochemistry 1992, 31, 7094 144 Anagnostopoulou, A.; Moldrheim, E.; Katsaros, N.; Sletten, E. J. Biol. Inorg. Chem. 1999, 4,
199 145 van Vliet, P. M.; Haasnoot, J. G.; Reedijk, J. Inorg. Chem. 1994, 33, 1934 146 Vlassov, V. V.; Gorn, V. V.; Ivanova, E. M.; Kazakov, S. A.; Mamaev. S. V.; FEBS Lett.
1983, 162, 286 147 Gruff, E. S.; Orgel, L. E. Nucleic Acids Res. 1991, 19, 6849 148 Colombier, C.; Lippert, B.; Leng, M. Nucleic Acids Res. 1996, 24, 4519 149 Bernal-Méndez, E.; Sun, J.-S.; Francisco, G.-V.; Leng, M. New J. Chem. 1998, 1479 150 Manchanda, R.; Dunham, S. U.; Lippard, S. J. J. Am. Chem. Soc. 1996, 118, 5144 151 Schliepe, J.; Berghoff, U.; Lippert, B.; Cech, D. Angew. Chem. Int. Ed. Engl. 1996, 35, 646 152 Berghoff, U.; Schmidt, K.; Janik, M.; Schröder, G.; Lippert, B. Inorg. Chim. Acta 1998, 269,
135 153 Schmidt, K. S.; Filippov, D. V.; Meeuwenoord, N. J.; van der Marel, G. A.; van Boom, J. H.;
Lippert, B.; Reedijk, J. Angew. Chem. Int. Ed. Engl. 2000, 39, 375 154 Zoltewicz, J. A.; Clark, D. F.; Sharpless, T. W.; Grahe, G. J. Am. Chem. Soc. 1970, 92, 1741 155 Gaucheron, F.; Malinge, J.-M.; Blacker, A. J.; Lehn, J.-M.; Leng, M. Proc. Natl. Acad. Sci.
USA 1991, 88, 3516 156 Payet, D.; Gaucheron, F.; Sip, M.; Leng, M. Nucleic Acids Res. 1993, 21, 5846 157 Kehrmann, F.; Havas, E. Ber. 1913, 46, 341 158 Sinha, N. D.; Biernat, J.; McManus, J.; Köster, H. Nucleic Acids Res. 1984, 12, 4539 159 McBride, L.; Caruthers, M. Tetrahedron Lett. 1983, 24, 245 160 Holy, A. Nucleic Acids Res. 1974, 1, 289 161 Oligonucleotide Synthesis. A Practical Approach; Gait, M. J., Ed.; IRL Press: Oxford, 1984, p 45 162 Misiura, K.; Durrant, I.; Evans, M. R.; Gait, M. J. Nucleic Acids Res. 1990, 18, 4345 163 Theisen, P.; McCollum, C.; Upadhya, K.; Jacobson, K.; Vu, H.; Andrus, A. Tetrahedron Lett. 1992, 33, 5033 164 Willis, M. C.; Collins, B.; Zhang, T.; Green, L. S.; Sebesta, D. P.; Bell, C.; Kellogg, E.; Gill, S. C.; Magallanez, A.; Knauer, S.; Bendele, R. A.; Gill, P. S.; Janjić, N.
Bioconjugate Chem. 1998, 9, 573 165 Ing; Manske J. Chem. Soc. 1926, 2348 166 Pon, R. T.; Yu, S. Tetrahedron Lett. 1997, 38, 3331 167 Sullivan, B. P.; Salmon, D. J.; Meyer, T. J. Inorg. Chem. 1978, 17, 3334 168 Hartshorn, R. M.; Barton, J. K. J. Am. Chem. Soc. 1992, 114, 5919 169 Sullivan, B. P.; Calvert, J. M.; Meyer, T. J. Inorg. Chem. 1980, 19, 4104 170 Takeuchi, K. J.; Thompson, M. S.; Pipes, D. W.; Meyer, T. J. Inorg. Chem. 1984, 23, 1845 171 Pramanik, N. C. ; Pramanik, K.; Ghosh, P.; Bhattacharya, S. Polyhedron 1998, 17, 1525 172 Laemmel, A.-C.; Collin, J.-P.; Sauvage, J.-P.; C. R. Acad. Sci. Paris 2000, 3, 43

64
173 Paul, R.; Anderson, G. W. J. Am. Chem. Soc. 1960, 82, 4596 174 Hu, X.; Smith, G. D.; Sykora, M.; Lee, S. J.; Gristaff, M. W. Inorg. Chem. 2000, 39, 2500 175 Khan, S. I.; Beilstein, A. E.; Sykora, M.; Smith, G. D.; Hu, X.; Gristaff, M. W. Inorg. Chem. 1999, 38, 3922 176 Kelly, J. M.; Tossi, A. B.; McConnel, D. J.; OhUigin, C.; Hélène, C.; Le Doan, T. In Free Radicals, Metal Ions and Biopolymers (Beaumont, P. C.; Deedle, D. J.; Parsons, B. J.; Rice-Evans, C., eds.), Richelieu Press, London, 1989, p 143 177 Telser, J.; Cruickshank, K. A.; Schanze, K. S.; Netzel, T. N. J. Am. Chem. Soc. 1989, 111, 7221 178 Bannwarth, W.; Schmidt, D.; Stallard, R. L.; Hornung, C.; Knorr, R.; Müller, F. Helv. Chim. Acta 1988, 71, 2085 179 Bannwarth, W.; Schmidt, D. Tetrahedron Lett. 1989, 30, 1513 180 Jenkins, Y.; Barton, J. K.; J. Am. Chem. Soc. 1992, 114, 8736 181 Meggers, E.; Kursch, D.; Giese, B. Helv. Chim. Acta 1997, 80, 640 182 Hurley, D. J.; Tor, Y. J. Am. Chem. Soc. 1998, 120, 2194 183 Wiederholt, K.; McLaughlin, L. W. Nucleic Acids Res. 1999, 27, 2487 184 Khan, S. I.; Beilstain, A. E.; Gristaff, M. W. Inorg. Chem. 1999, 38, 418 185 Khan, S. I.; Beilstain, A. E.; Sykora, M.; Smith, G. D.; Hu, X.; Gristaff, M. W. Inorg. Chem. 1999, 38, 3922 186 Khan, S. I.; Beilstain, A. E.; Tierney, M. T.; Sykora, M.; Gristaff, M. W. Inorg. Chem. 1999, 38, 5999 187 Lewis, F. D.; Helvoigt, S. A.; Letsinger, R. L. Chem. Commun. 1999, 327 188 Rack, J. J.; Krider, E. S.; Meade, T. J. J. Am. Chem. Soc. 2000, 122, 6287 189 Grimm, G. N.; Boutorine, A. S.; Lincoln, P.; Hélène, C. Nucleosides & Nucleotides 2001, 20, 909 190 Vargas-Baca, I.; Mitra, D.; Zulyniak, H. J.; Banerjee, J.; Sleiman, H. F. Angew. Chem. Int. Ed. 2001, 40, 4629 191 Dandliker, P. J.; Holmlin, R. E.; Barton, J. K. Science, 1997, 275, 1465 192 Holmlin, R. E.; Dandliker, P. J.; Barton, J. K. Bioconjugate Chem. 1999, 10, 1122 193 Suen, H.-F.; Wilson, S. W.; Pomerantz, M.; Walsh, J. L. Inorg. Chem. 1989, 28, 786 194 Hecker, C. R.; Fanwick, P. E.; McMillin, D. R. Inorg. Chem. 1991, 30, 659 195 Hartshorn, S. M.; Maxwell, K. A.; White, P. S.; DeSimone, J. M.; Meyer, T. J. Inorg.
Chem. 2001, 40, 601 196 Serron, S. A.; Aldridge III, W. S.; Danell, R. M.; Meyer, T. J. Tetrahedron Lett. 2001, 41, 4039 197 Sinha, N. D.; Biernat, J.; McManus, J.; Koster, H. Nucleic Acids Res. 1984, 12, 4539. 198 Sinha, N. D.; Biernat, J.; Koster, H. Tetrahedron Lett. 1983, 24, 5843. 199 Ren, S.; Cai, L.; Segal, B. M. J. Chem. Soc., Dalton Trans. 1999, 1413 200 Cai, L.; Lim, K.; Ren, S.; Cadena, R. S.; Beck, W. T. J. Med. Chem. 2001, 44, 2959 201 Fagalde, F.; Lis de Katz, N. D.; Katz, N. E. Polyhedron 1997, 16, 1921 202 Naal, Z.; Tfouni, E.; Benedetti, A. V. Polyhedron 1994, 13, 133 203 Weizman, H.; Tor, Y. J. Am. Chem. Soc. 2001, 123, 3375 204 Potier, N.; Van Dorsselaer, A.; Cordier, Y.; Roch, O.; Bischoff, R. Nucleic Acids Res. 1994, 22, 3895 205 Apffel, A.; Chakel, J. A.; Fischer, S.; Lichtenwalter, K.; Hancock, W. Anal. Chem. 1997, 69, 1320 206 Huber, C. G.; Buchmeiser, M. R. Anal. Chem. 1998, 70, 5288 207 De Bellis, G.; Salani, G.; Battaglia, C.; Pietta, P.; Rosti, E.; Mauri, P. Rapid Commun. Mass Spectrom. 2000, 14, 243

65
208 Koomen, J. M.; Russell, W. K.; Tichy, S. E.; Russell, D. H. J. Mass. Spectrom. 2002, 37, 357 209 Hiort, C.; Norden, B.; Rodger, A. J. Am. Chem. Soc. 1990, 112, 1971 210 Kelly, J. M.; Tossi, A. B.; McConnel, D. J.; OhUigin, C. Nucleic Acids Res. 1985, 13, 6017 211 Rehmann, J.; Barton, J. K. Biochemistry 1990, 29, 1701 212 Barton, J. K. J. Biol. Struct. Dyn. 1983, 1, 621 213 Long, E. C.; Barton, J. K. Acc. Chem. Res. 1990, 13, 271 214 Hiort, C.; Linkoln, P.; Norden, B. J. Am. Chem. Soc. 1993, 115, 3448 215 Friedman, A. E.; Chambron, J. C.; Sauvage, J.-P.; Turro, N. J.; Barton, J. K. J. Am. Chem. Soc. 1990, 112, 4960 216 Dupureur, C. M.; Barton, J. K. J. Am. Chem. Soc. 1994, 116, 10286 217 Jenkins, Y.; Friedman, A. E.; Turro, N. J.; Barton, J. K. Biochemistry 1992, 31, 10809 218 Barawkar, D.; Rajeev.; Kumar, V.; Ganesh, K. Nucleic Acids Res. 1996, 24, 1229 219 Choi, S.-D.; Kim, M.-S.; Linkoln, P.; Tuite, E.; Norden, B. Biochemistry 1997, 36, 214 220 Cho, C.-B.; Cho, T.-S.; Kim, S. K.; Kim, B.-J.; Han, S. W.; Jung, M. J. Bull. Korean Chem. Soc. 2000, 21, 995 221 Tinoco, I.; Sauer, K.; Wang, J. C. In Physical Chemistry: Principles and Applications in Biological Scinces (Young, D.; Cavanaugh, D., Eds.), Prentice Hall, Upper Saddle River, NJ, 1995, p 559 222 Allen, F. S.; Gray, D. M.; Roberts, G. P.; Tinoco, I. Biopolymers 1972, 11, 853 223 Fasman, G. Handbook of Biochemistry and Molecular Biology, CRC Press: Ohio, 1975, Vol. 1, p 589 224 Eriksson, M.; Leijon, M.; Hiort, C.; Norden, B.; Gräslund, A. Biochemistry 1994, 33, 5031 225 Lomonossoff, G. P.; Butler, P. J. G.; Klug, A. J. Mol. Biol. 1981, 149, 745 226 Fox, K. R.; Waring, M. J. Nucleic Acids Res. 1984, 12, 9271 227 Fox, K. R. In Methods in Molecular Biology (Fox, K. R., Ed.), Humana Press Inc., Totowa, NJ, Vol. 90, p 1 228 Suck, D.; Oefner, C. Nature 1986, 321, 620 229 Önfelt, B.; Linkoln, P.; Norden, B. J. Am. Chem. Soc. 1999, 121, 10846 230 Davis, T. M.; McFail-Isom, L.; Keane, E.; Williams, L. D. Biochemistry 1998, 37, 6975 231 Li, J.; Wartell, R. M. Biochemistry 1998, 37, 5154 232 Lima, W. F.; Mohan, V.; Crooke, S. T. J. Biol. Chem. 1997, 29, 18191 233 Salazar, M.; Fedoroff, O. Y.; Miller, J. M.; Riberio, N. S.; Reid, B. R. Biochemistry 1993, 32, 4207 234 Fedoroff, O. Y.; Salazar, M.; Reid, B. R. J. Mol. Biol. 1993, 233, 509 235 Uchiyama, Y.; Iwai, S.; Ueno, Y.; Ikehara, M.; Ohtsuka, E. J. Biochem. 1994, 116, 1322 236 Uchiyama, Y.; Miura, Y.; Inoue, H.; Ohtsuka, E.; Ueno, Y.; Ikehara, M.; Iwai, S. J. Mol. Biol. 1994, 4, 782 237 Amirkhanov, N. V.; Chattopadhyaya, J. J. Chem. Soc., Perkin Trans. 2 2002, 271 238 Amirkhanov, N. V.; Pradeepkumar, P. I.; Chattopadhyaya, J. J. Chem. Soc., Perkin Trans. 2 2002, in press 239 Amirkhanov, N. V.; Zamaratski, E.; Chattopadhyaya, J. Tetrahedron Lett. 2001, 42, 489 240 Temsamani, J.; Tang, J.-Y.; Padmapriya, A.; Kubert, M.; Argawal, S. Antisense Res. Dev. 1993, 3, 277 241 Nicklin, P. L.; Ambler, J.; Mitchelson, A.; Bayley, D.; Phillips, J. A.; Craig, S. J.;

66
Monia, B. P. Proc. Natl. Acad. Sci. USA 1997, 16, 1145 242 Shaw, J.-P.; Kent, K.; Fishback, J.; Froehler, B. Nucleic Acids Res. 1991, 19, 747 243 Boutorin, A. S.; Guskova, L. V.; Ivanova, E. M.; Kobetz, N. D.; Zarytova, V. F.; Ryte, A. S.; Yurchenko, L. V.; Vlassov, V. V. FEBS Lett. 1989, 254, 129 244 Ryte, A. S.; Karamyshev, V. N.; Nechaeva, M. N.; Guskova, L. V.; Ivanova, E. M.; Zarytova, V. F.; Vlassov, V. V. FEBS Lett. 1992, 299, 124 245 Hélène, C. Current Opinion of Biotechnology 1993, 4, 29 246 Hall, J.; Hüsken, D.; Häner, R. Nucleic Acids Res. 1996, 24, 3522 247 Daniher, A. T.; Bashkin, J. K. J. Chem. Soc., Chem. Commun. 1998, 1077 248 Trawick, B. N.; Osiek, T. A.; Bashkin, J. K. Bioconjugate Chem. 2001, 12, 900 249 Putnam, W. C.; Bashkin, J. K. J. Chem. Soc., Chem. Commun. 2000, 767 250 Åström, H.; Strömberg, R. Nucleosides, Nucleotides & Nucleic Acids, 2001, 20, 1385 251 Kodak, A. S.; Gard, J. K.; Merriman, M. C.; Winkeler, K. A.; Bashkin, J. K.; Stern, M. K. J.
Am. Chem. Soc. 1991, 113, 283 252 Spassky, A.; Sigman, D. S. Biochemistry 1985, 24, 8050 253 Hélène, C.; Thuong, N. T.; Saison-Behmoaras, T.; François, J. C. Trends Biotechnol. 1989, 7,
310 254 Sigman, D. S. Biochemistry 1990, 29, 9097 255 Sigman, D. S.; Mazumder, A.; Perrin, D. M. Chem. Rev. 1993, 93, 2295 256 Sigman, D. S.; Bruice, T. W.; Mazumder, A.; Sutton, C. L. Acc. Chem. Res. 1993, 26, 98 257 Patviel, G.; Bernadou, J.; Meunier, B. Angew. Chem. Int. Ed. Engl. 1995, 34, 746 258 Cowan, J. A. Curr. Opin. Chem. Biol. 2001, 5, 634 259 Baker, A. D.; Morgan, R. J.; Strekas, T. C. J. Chem. Soc., Chem. Commun. 1992, 1099 260 Gallagher, J.; Chen, C. B.; Pan, C. Q.; Perrin, D. M.; Cho, Y.-M.; Sigman, D. S. Bioconjugate Chem. 1996, 7, 413 261 Meijler, M. M.; Zelenko, O.; Sigman, D. S. J. Am. Chem. Soc. 1997, 119, 1135 262 Baudoin, O.; Teulade-Fichou, M.-P.; Vigneron, J.-P.; Lehn, J.-M. Chem. Commun. 1998, 2349 263 Pitie, M.; Meunier, B. Bioconjugate Chem. 1998, 9, 604 264 Knorre, D. G.; Vlassov, V. V.; Zarytova, V. F.; Lebedev, A. V.; Fedorova, O. S. Design and targeted reactions of oligonucleotide derivatives; CRC Press: 1994 265 Ebright, R. H.; Ebright, Y. W.; Pendergrast, P. S.; Gunasekera, A. Proc. Natl. Acad. Sci. USA 1990, 87, 2882 266 Chen, C. B.; Mazumder, A.; Constant, J.-F.; Sigman, D. S. Bioconjugate Chem. 1993, 4,
69 267 Bergstrom, D. E.; Gerry, N. P. J. Am. Chem. Soc. 1994, 116, 12067 268 Chen, C. B.; Gorin, M. B.; Sigman, D. S. Proc. Natl. Acad. Sci. USA 1993, 90, 4206 269 Chen, C. B.; Milne, L.; Landgraf, R.; Perrin, D. M.; Sigman, D. S. ChemBioChem 2001, 2, 735 270 Gupta, N.; Grover, N.; Neyhart, G. A.; Liang, W.; Singh, P.; Thorp, H. H. Angew. Chem. Int. Ed. Engl. 1992, 31, 1048 271 Ho, C.; Che, C.-M.; Lau, T.-C. J. Chem. Soc., Perkin Trans. 1990, 967 272 Rosenberg, B.; Vancamp, L.; Krigas, T. Nature 1965, 205, 689 273 Rosenberg, B.; Vancamp, L.; Trosko, J. E.; Mansour, V. H. Nature 1969, 222, 385 274 Clarke, M. J.; Stubbs, M. Interactions of Metallopharmaceuticals with DNA; Sigel, A.; Sigel, H., Ed.; “Metal Ions in Biological Systems”, Marcel Dekker Inc.: New York, 1996, vol. 32, p 727 275 Grover, N.; Gupta, N.; Thorp, H. H. J. Am. Chem. Soc. 1992, 114, 3390 276 Grover, N.; Welch, T. W.; Fairlay, T. A.; Cory, M.; Thorp, H. H. Inorg. Chem. 1994, 33, 3544

67
277 Oseroff, A. R.; Ara, G.; Ohuoha, D.; Aprille, J.; Bommer, J. C.; Yarmush, M. L.; Foley, J.; Cincotta, L. Photochem. Photobiol. 1987, 46, 83 278 Billadeau, M. A.; Morrison, H. Photolytic Covalent Binding of Metal Complexes to DNA; Sigel, A.; Sigel, H., Ed.; “Metal Ions in Biological Systems”, Marcel Dekker Inc.: New York, 1996, vol. 33, p 269 279 Mahnken, R. E.; Bina, M.; Deibel, R. M.; Luebke, K.; Morrison, H. Photochem. Photobiol. 1989, 49, 519 280 Mahnken, R. E.; Billadeau, M. A.; Niconowicz, E. P.; Morrison, H. J. Am. Chem. Soc. 1992, 114, 9253 281 Harmon, H. L.; Morrison, H. Inorg. Chem. 1995, 34, 4937 282 Billadeau, M. A.; Wood, K. V.; Morrison, H. Inorg. Chem. 1994, 33, 5780 283 Mohammad, T.; Tessman, I.; Morrison, H.; Kennedy, M. A.; Simmonds, S. W. Photochem. Photobiol. 1994, 59, 189 284 Mohammad, T.; Chen, C.; Guo, P.; Morrison, H. Bioorg & Med. Chem. Lett. 1999, 9, 1703 285 Billadeau, M. A.; Morrison, H. J. Inorg. Biochem. 1995, 57, 249 286 Steel, P. J.; Lahousse, F.; Lerner, D.; Marzin, C. Inorg. Chem. 1983, 22, 1488 287 von Zelewski, A.; Gremaud, G. Helv. Chim. Acta 1988, 71, 1108 288 Laemmel, A.-C.; Collin, J.-P.; Sauvage, J.-P. Eur. J. Inorg. Chem. 1999, 383 289 Collin, J.-P.; Laemmel, A.-C.; Sauvage, J.-P. New J. Chem. 2001, 25, 22 290 Baranoff, E.; Collin, J.-P.; Furusho, Y.; Laemmel, A.-C.; Sauvage, J.-P. Chem. Commun. 2000, 1935 291 Wang, R.; Vos, J. G.; Schmehl, R. H.; Hage, R. J. Am. Chem. Soc. 1992, 114, 1964 292 Buchanan, B. E.; Degn, P.; Pavon Velasco, J. M.; Hughes, H.; Creaven, B. S.; Long, C.; Vos, J. G.; Howie, R. A.; Hage, R.; van Diemen, J. H.; Haasnoot, J. G.; Reedijk, J. J. Chem. Soc., Dalton Trans. 1992, 1177 293 Buchanan, B. E.; Hughes, H.; van Diemen, J. H.; Hage, R.; Haasnoot, J. G.; Reedijk, J.; Vos, J. G. Chem. Commun. 1991, 300 294 Baranoff, E.; Collin, J.-P.; Furusho, J.; Furusho, Y.; Laemmel, A.-C.; Sauvage, J.-P. Inorg. Chem. 2002, 41, 1215 295 Barton, J. K.; Lolis, E. J. Am. Chem. Soc. 1985, 107, 708 296 Danishefsky, A. Ph.D. Thesis, Columbia University, 1987




















