
Bioconjugate Strategies for Antisense Therapeutic Delivery to ...
182
Bioconjugate Strategies for Antisense Therapeutic Delivery to Glioblastoma Stem Cells by Amy Elizabeth Arnold A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Department of Chemistry University of Toronto © Copyright by Amy Elizabeth Arnold 2020
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of Bioconjugate Strategies for Antisense Therapeutic Delivery to ...

Bioconjugate Strategies for Antisense Therapeutic Delivery to Glioblastoma Stem Cells
by
Amy Elizabeth Arnold
A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy
Department of Chemistry University of Toronto
© Copyright by Amy Elizabeth Arnold 2020

ii
Bioconjugate Strategies for Antisense Therapeutic Delivery to
Glioblastoma Stem Cells
Amy Elizabeth Arnold
Doctor of Philosophy
Department of Chemistry University of Toronto
2020
Abstract
Antisense therapeutics, including antisense oligonucleotides (AONs) and small interfering
ribonucleic acids (siRNAs), are powerful tools for regulating genes, making them a promising
therapy for diseases such as cancer where oncogenic genes are over-expressed. The delivery of
antisense therapeutics to target cells presents a significant challenge due to the many barriers a
nucleic acid must face in order to reach the cytoplasm where it exerts its effects. In this thesis, I
explored multiple strategies for delivery of AONs and siRNAs, focusing on targeting the desired
cell population, inducing endocytosis, and facilitating endosomal escape. This was done within
the context of glioblastoma (GBM), and specifically the glioblastoma stem cells (GSCs), an
aggressive subpopulation of GBM cells that are involved in resistance, migration, and
recurrence. Antisense oligonucleotides against a relevant GBM gene were conjugated to an
antibody engineered to target CD44, a cell surface receptor which is highly expressed on GSCs.
Using this system, we demonstrated functional targeting, endocytosis, and gene knockdown in
the GSCs, leading to a morphological change in the cells. This represented the first time an
antibody-oligonucleotide conjugate was used to target the GSC population. We were challenged
with a lack of endosomal escape when using the antibody delivery platform, so we next looked at
using a protein with a native endosomal escape mechanism to facilitate oligonucleotide delivery.
For the second strategy, I conjugated attenuated diphtheria toxin (aDT), a protein which escapes
the endolysosomal pathway, to siRNAs against relevant gene targets involved in GSC
proliferation and invasion. Using this aDT-siRNA conjugate, we could downregulate genes of
interest in the glioblastoma stem cells, leading to significant changes in cell viability and the

iii
invasive capacity of these cells. This is the first diphtheria toxin-based siRNA delivery vehicle
and represents a platform technology for siRNA- and AON-based therapies.

iv
Acknowledgments I would first and foremost like to thank my supervisor, Prof. Molly Shoichet, for five years’
worth of mentorship, support, and advice, and for all the doors that have been opened because of
my time in the Shoichet lab. I learned more than I ever expected, not just about science, but also
about how to communicate my science in a clear way, and how to effectively collaborate with
(many) colleagues. Thank you for being a true champion for women in science and for all the
time you devote to helping your students succeed – I am truly grateful.
Thank you to all of my collaborators, especially to Prof. Masad Damha, Prof. Roman Melnyk,
and Dr. Greg Beilhartz. Prof. Damha – you were instrumental in helping me succeed during my
PhD; although it finished at a different place than it started, you were there with advice at every
turn. Prof. Melnyk and Greg – you brought fresh perspectives and a new direction to my work; I
really appreciate it.
I would like to thank my committee who have continually encouraged me to learn new skills and
think creatively about my work. Prof. Jason Moffat – thank you for asking the tough questions
and inspiring me to see problems from a biologist’s perspective. Prof. Mitch Winnik – thanks for
encouraging me to see the exciting aspects of my work whenever I encountered roadblocks.
Thank you to all of the members of the Shoichet lab that have helped me along the way,
including my earliest mentors Jenn and Chris, as well as Vianney, Marian, Ahil, Ana, and Alex –
you are all fantastic mentors and great friends. Sonja, Laura B, Erics, and Carter – you guys
made up the best lab family ever. Above all, I am grateful to the other member of team
glioblastoma, Laura S. Thank you helping me through late nights and tough times and also
celebrating the good stuff from birthdays and weddings to afternoon trips to Jimmy’s.
I would like to acknowledge all of my family and friends whose endless encouragement and
support was invaluable throughout my PhD. Thank you especially to all of my aunts and uncles
who offered a home-away-from-home while I studied in Toronto. Mom and Dad – even though I
didn’t become a “real” doctor, you guys have always been there for me, and I truly believe that
every success I have had is because of you.

v
Finally, and most importantly, I would like to thank my husband, Jarret. From near or from far,
you never stopped believing in me. This thesis is dedicated to you.

vi
Table of Contents Acknowledgments .......................................................................................................................... iv
Table of Contents ........................................................................................................................... vi
List of Tables ................................................................................................................................. xi
List of Figures ............................................................................................................................... xii
Introduction .................................................................................................................................1
1.1 Rationale ..............................................................................................................................1
1.1.1 Hypothesis and objectives ........................................................................................2
1.2 Glioblastoma stem cells .......................................................................................................3
1.2.1 Glioblastoma ............................................................................................................3
1.2.2 Cancer stem cells .....................................................................................................4
1.2.3 Glioblastoma stem cells ...........................................................................................6
1.2.4 Therapeutic targets associated with GSC proliferation and invasion ......................8
1.3 Antisense therapeutics .......................................................................................................10
1.3.1 Antisense oligonucleotide mechanism ...................................................................11
1.3.2 Small interfering ribonucleic acid mechanism ......................................................12
1.3.3 Stability in the extracellular environment ..............................................................13
1.4 Nanoparticles as delivery vehicles .....................................................................................15
1.4.1 Polymeric formulations ..........................................................................................15
1.4.2 Lipid-based formulations .......................................................................................17
1.4.3 Virus-inspired formulations: cell penetrating and membranelytic peptides ..........19
1.5 Protein conjugates as delivery vehicles .............................................................................20
1.5.1 Antibodies for drug and oligonucleotide delivery .................................................21
1.5.2 Engineered toxins for biomolecule delivery ..........................................................27
Antibody-antisense oligonucleotide conjugate downregulates a key gene in glioblastoma stem cells ...................................................................................................................................32

vii
2.1 Abstract ..............................................................................................................................32
2.2 Introduction ........................................................................................................................33
2.3 Results ................................................................................................................................35
2.3.1 Antisense oligonucleotide activity .........................................................................35
2.3.2 Co-expression in GSCs of DRR and antigens for CD44, EphA2, and EGFR antibodies ...............................................................................................................37
2.3.3 Internalization of CD44 and EphA2 mAbs ............................................................37
2.3.4 Synthesis of mAb-dsDRR and mAb-dsScrambled conjugates ..............................39
2.3.5 DRR knockdown and cellular uptake of mAb-dsDRR conjugates ........................40
2.3.6 Cellular morphology following DRR knockdown .................................................44
2.4 Discussion and conclusions ...............................................................................................46
2.5 Materials and methods .......................................................................................................47
2.5.1 Cell lines ................................................................................................................47
2.5.2 Antibodies ..............................................................................................................48
2.5.3 AON synthesis .......................................................................................................48
2.5.4 AON duplex formation ..........................................................................................49
2.5.5 Preparation of mAb-dsDRR and mAb-dsScrambled conjugates ...........................49
2.5.6 DRR knockdown assays - lipofectamine2000 transfection protocol .....................49
2.5.7 DRR knockdown assays - treatment with mAb conjugate ....................................49
2.5.8 mAb internalization assay ......................................................................................50
2.5.9 Immunocytochemistry (CD44, EphA2, and DRR) ................................................50
2.5.10 Cellular uptake .......................................................................................................51
2.5.11 Lysosomal accumulation .......................................................................................51
2.5.12 Cellular morphology following DRR knockdown .................................................51
2.6 Acknowledgments ..............................................................................................................51
Attenuated diphtheria toxin mediates siRNA delivery .............................................................52

viii
3.1 Abstract ..............................................................................................................................52
3.2 Introduction ........................................................................................................................53
3.3 Results ................................................................................................................................55
3.3.1 Conjugation of siRNA to aDT ...............................................................................55
3.3.2 Glioblastoma stem cells (GSCs) express heparin-binding epidermal growth factor (HBEGF) .....................................................................................................57
3.3.3 aDT-siRNA conjugate downregulates ITGB1 and reduces cellular invasion .......58
3.3.4 aDT-siRNA conjugate downregulates eIF-3b and reduces cell viability ..............60
3.4 Discussion and conclusions ...............................................................................................62
3.5 Materials and methods .......................................................................................................64
3.5.1 Cell lines ................................................................................................................64
3.5.2 Attenuated diphtheria toxin ....................................................................................64
3.5.3 siRNAs ...................................................................................................................64
3.5.4 PCR primers ...........................................................................................................65
3.5.5 Immunocytochemistry (HBEGF) ...........................................................................65
3.5.6 Preparation of aDT-siRNA conjugate ....................................................................65
3.5.7 Gene knockdown assays - treatment with aDT-siRNA conjugate .........................65
3.5.8 Gene knockdown assays - positive control treatment with Lipofectamine RNAiMAX .............................................................................................................66
3.5.9 Invasion assay ........................................................................................................66
3.5.10 Adhesion assay .......................................................................................................66
3.5.11 Cell viability assays following treatment with aDT-eIF-3b ..................................66
3.6 Acknowledgements ............................................................................................................67
Thesis discussion .......................................................................................................................67
4.1 The interplay of stability and potency in antisense therapeutics .......................................68
4.2 The importance of endosomal escape in oligonucleotide delivery ....................................70
4.3 Measuring functional effects of oligonucleotide delivery .................................................71

ix
4.3.1 Functional effects of DRR knockdown ..................................................................72
4.3.2 Functional effects of eIF-3b knockdown ...............................................................72
4.3.3 Functional effects of ITGB1 knockdown ..............................................................73
4.4 Targeting the glioblastoma stem cell population ...............................................................74
4.4.1 Targeting cell surface receptors .............................................................................74
4.4.2 Targeting key genetic alterations for desirable phenotypes ...................................76
4.4.3 Optimizing the delivery of protein conjugates in vivo ..........................................77
Conclusions ...............................................................................................................................78
5.1 Completion of objectives ...................................................................................................79
Recommendations for future work ...........................................................................................80
6.1 Investigation into aDT-siRNA trafficking mechanism ......................................................80
6.1.1 Inhibition of translocation ......................................................................................81
6.1.2 IC50 shifts of modified DT .....................................................................................82
6.1.3 Split-reporter assays ...............................................................................................82
6.2 Optimization of specificity, potency, and endosomal escape ............................................83
6.2.1 Re-targeting the receptor binding domain .............................................................83
6.2.2 Stabilization of the siRNA .....................................................................................84
6.2.3 Exploring RNA analogues: morpholinos and peptide nucleic acids .....................85
6.2.4 Phenotypic assays as a first-line screening tool for oligonucleotide sequences ....86
6.3 Simultaneous delivery of siRNA and other therapeutics ...................................................86
6.3.1 Combination with native toxin domains ................................................................86
6.3.2 Combination with therapeutic proteins ..................................................................87
6.3.3 Combination with chemotherapeutic drugs ...........................................................88
Abbreviations .................................................................................................................................89
Appendix A: Supporting information for “Antibody-antisense oligonucleotide conjugate downregulates a key gene in glioblastoma stem cells” .............................................................93

x
Appendix B: Supporting information for “Attenuated diphtheria toxin mediates siRNA delivery” ....................................................................................................................................98
Appendix C: Effect of sugar 2’,4’-modifications on gene silencing activity of small interfering RNA duplexes .......................................................................................................102
Appendix D: Additional characterization of AONs and siRNAs against DRR ...........................117
Appendix E: In vivo biodistribution of a CD44 antibody conjugated to a fluorescent dye .........123
Appendix F: Polymeric nanomicelles for targeted therapeutic delivery to glioblastoma ............128
References ....................................................................................................................................134

xi
List of Tables Table 1. Common cell penetrating sequences. .............................................................................. 19
Table 2. Rates of selected bioorthogonal click chemistry reactions.183 ........................................ 25

xii
List of Figures Figure 1.1. Cancer stem cells resist treatment and promote recurrence. The bulk of the tumor is
susceptible to treatment including radiotherapy and chemotherapy, but some CSCs survive,
which can repopulate the tumor even in small numbers. ................................................................ 4
Figure 1.2. AON mechanism of action. The AON hybridizes to complementary strands of
mRNA and induces the activity of RNAseH, which recognizes the DNA/RNA hybrid and
degrades the RNA into its components (nucleoside monophosphates). ....................................... 11
Figure 1.3. siRNA mechanism of action. Dicer cleaves long, double-stranded RNA into short
fragments of siRNA. siRNA is then loaded into the RISC complex, binds to the complementary
mRNA, and RISC degrades the mRNA. ....................................................................................... 13
Figure 1.4. siRNA carriers protect it from nuclease degradation. (A) Free siRNA (blue double
helix) is rapidly degraded by nucleases (orange semi-circle) and (B) cleared by lymphatic
drainage (pale blue ovals). (C) Nanoparticle or protein carriers may protect siRNA from
nucleases and (D) reduce clearance. ............................................................................................. 14
Figure 1.5 Common modifications to oligonucleotides include modifications to both: (A) the
phosphodiester linkage and (B) the 2’ sugar. ................................................................................ 15
Figure 1.6. Reactions of native amino acids for site-specific protein conjugation. A) Thiols can
be modified through reactions with maleimides. B) Tyrosine can be reacted using diazonium
salts. C) Tryptophan can be modified through metallopeptide-catalyzed reactions. .................... 23
Figure 1.7. Cellular entry mechanisms of three “AB” toxins: diphtheria toxin, anthrax toxin, and
Pseudomonas exotoxin A. The R domain of DT binds to heparin-binding epidermal growth
factor (HBEGF) and induces internalization to the early endosomes where the connection
between the T and A domain is cleaved by furin, and the A domain translocates out of the early
endosome through a flexible α-helical pore. The protective antigen (T) domain of AT is cleaved
by extracellular furin, and oligomerizes on the cell surface, binding to the tumor endothelial
marker-8 (TEM-8) or capillary morphogenesis gene-2 (CMG-2) surface receptors. This leads to
association with the A domain and subsequent internalization into the early endosomes where A

xiii
domain translocations through a rigid β-barrel pore. Finally, the R domain of PE binds to the cell
surface receptor Low density lipoprotein receptor-related protein 1 (LRP1), and the connection
between the T and A domain is cleaved by furin in the early endosomes. PE is then trafficked to
the endoplasmic reticulum where it undergoes retrograde translocation. ..................................... 29
Figure 2.1. Downregulated in renal cell carcinoma (DRR) expression is reduced following
transfection of DRR+ U-251 MG cells with DRR antisense oligonucleotides. (A) Oligonucleotide
sequences used: all antisense strands comprise a phosphorothioate backbone while all sense
strands are synthesized with a phosphodiester backbone. (B) DRR expression following
treatment with single stranded DRR AON sequences normalized to untreated control. Data were
analyzed using one-way ANOVA followed by Dunnett’s post-hoc test compared to Scrambled
group (data is shown as mean+SD, n=3, *p<0.05, **p<0.01). (C) DRR expression following
treatment with double stranded anti-DRR oligonucleotide sequences normalized to untreated
control. Data were analyzed using unpaired t-test with Welch’s correction (data is shown as
mean+SD, n≥4, ***p<0.001). (D) Representative western blot showing single stranded and
double stranded antisense oligonucleotide DRR knockdown. ...................................................... 36
Figure 2.2. Patient-derived GSCs strongly co-express antigens CD44 and EphA2 with DRR and
weakly co-express EGFR with DRR. Representative confocal images are shown. Antigens
CD44, EphA2, and EGFR (green); DRR (red); cell nucleus (Hoechst, blue). All scale bars are 50
µM. ................................................................................................................................................ 37
Figure 2.3. EphA2 mAbs and CD44 mAbs are internalized upon binding to cells. (A-C) Flow
cytometry analysis of cell surface receptor internalization following 45 min incubation at 37 °C
with antibodies (A) CD44 mAb and (B) EphA2 mAb compared to (C) non-specific CTL mAb.
Cells without antibodies added (cells only) and cells incubated with antibodies, but held at 4 °C
(no internalization) curves are shown as a comparison to those cells that were allowed to
internalize the mAb for 45 min at 37 °C (45 min internalization). (D) Quantification of
internalized receptor following 45 min incubation period for CD44 mAb and EphA2 mAb
antibodies compared to CTL. Data were analyzed using one-way-ANOVA followed by
Dunnett’s post hoc test compared to CTL group (data is shown as mean+SD, n=3, *p<0.05,
***p<0.001). ................................................................................................................................. 38

xiv
Figure 2.4. CD44 mAb and EphA2 mAb can be efficiently conjugated to dsDRR using click
chemistry. (A) Scheme of antibody modification with dsDRR. (B) 10% PAGE analysis of CD44
mAb-dsDRR conjugation: Lane 1: dsDRR only; Lane 2: CD44 mAb conjugated to dsDRR via
NHS-PEG4-N3 crosslinker; Lane 3: dsDRR and CD44 mAb combined without crosslinker
present. (C) 10% PAGE analysis of EphA2 mAb-dsDRR conjugation: Lane 1: dsDRR only;
Lane 2: EphA2 mAb conjugated to dsDRR via NHS-PEG4-N3 crosslinker; Lane 3: dsDRR and
EphA2 mAb combined without crosslinker present. .................................................................... 40
Figure 2.5. DRR expression of patient-derived GSCs after treatment with CD44 mAb-dsDRR or
EphA2 mAb-dsRR (150 nM) normalized to untreated control. (A) Quantification of DRR
expression following CD44 mAb-dsDRR treatment. Data were analyzed using one-way-
ANOVA followed by Dunnett’s post hoc test compared to CD44 mAb-dsScrambled (data is
shown as mean+SD, n≥4, *p<0.05). (B) Representative western blot showing DRR knockdown
following treatment with CD44 mAb-dsDRR. (C) Quantification of DRR expression following
EphA2 mAb-dsDRR treatment. Data were analyzed using one-way-ANOVA followed by
Dunnett’s post hoc test compared to EphA2 mAb-dsScrambled (data is shown as mean+SD,
n≥3). (D) Representative western blot showing DRR expression following treatment with EphA2
mAb-dsDRR. ................................................................................................................................ 41
Figure 2.6. Antisense oligonucleotides conjugated to CD44 mAb are taken up by GSCs and
trafficked into the endolysosomal pathway. (A) Uptake of CD44 mAb-dsDRR (white arrows)
compared to (B) EphA2 mAb-dsDRR and (C) CTL-dsDRR after 3 h incubation at 37 °C. Control
(CTL) is a non-specific human IgG. (D) Colocalization (white arrows) of CD44 mAb-dsDRR
with the lysosomal compartments following a 2 h pulse and 1 h chase. Cell membrane (wheat
germ agglutinin (WGA), magenta); cell nucleus (Hoechst, blue); AON (Cy3, green); lysosome
(Dextran647, red). Representative z-stack images shown. All scale bars are 50 µM. .................. 43
Figure 2.7. GSCs treated with CD44 mAb-dsDRR have a rounder shape, fewer projections, and
centralized focal adhesions relative to the spindle-shaped cells of the control treatments. (A)
Cells treated with CD44 mAb-dsScrambled. (B) Cells treated with CD44 mAb-dsDRR. (C) Cells
treated with CD44 mAb alone. (D) Cells treated with dsDRR alone. Representative z-stack
images shown. All scale bars are 50 µM. Cell nucleus (Hoechst, blue); Actin (Phalloidin Alexa
Fluor 488, green). (B) Change in cellular morphology is quantified as actin area per cell

xv
normalized to a no treatment control. Data were analyzed using one-way-ANOVA compared to
CD44 mAb-dsScrambled with Dunnett’s post hoc correction (data is shown as mean+SD, n≥3,
***p<0.001). ................................................................................................................................. 45
Figure 3.1. Using attenuated diphtheria toxin for siRNA delivery. A) Attenuated AB toxins, such
as attenuated diphtheria toxin (aDT), consist of three main components: a receptor binding
domain (R) that binds to a receptor on the cell surface; a translocation domain (T) that allows for
endosomal escape; and a mutated active domain (a) in order for the protein to retain its
trafficking functions but is no longer toxic to cells. Cargo, such as siRNA, can be attached to this
“a” domain. B) siRNA delivered using attenuated DT occurs in five main steps: 1) binding to the
HB-EGF precursor cell surface receptor; 2) endocytosis of the aDT-siRNA cargo; 3)
translocation through the endosomal membrane, inserting the “a” domain and cargo into the
cytoplasm; 4) cleavage of the “a” domain from the rest of the protein; and 5) release of the
siRNA into the cytoplasm where it downregulates the relevant gene. ......................................... 54
Figure 3.2. siRNAs can be conjugated to attenuated diphtheria toxin. A) Attenuated diphtheria
toxin was engineered to contain a free cysteine as a functional handle (aDT-SH), protected by a
SUMO tag and purified using a histidine (His) tag. B) Attenuated diphtheria toxin was reacted
with a PEG crosslinker containing both maleimide and DBCO functional groups to obtain
DBCO-modified attenuated diphtheria toxin (aDT-DBCO). C) The presence of the DBCO
modification on attenuated diphtheria toxin was confirmed by reading the absorbance at 280 nm
(aDT) and 309 nm (DBCO). Curves shown are aDT before modification (blue) and after DBCO
modification (red). D) Azide-modified siRNA was reacted with the DBCO-functionalized
attenuated diphtheria toxin to obtain the aDT-siRNA conjugate. E) Modification of the aDT with
the siRNA was confirmed via PAGE stained with Coomassie blue to localize the diphtheria toxin
protein. Lane 1 shows the aDT-DBCO starting material (MW ~ 72 kDa) and lane 2 shows the
aDT-siRNA conjugate (MW~90 kDa) alone with some unreacted starting material. F)
Purification of the excess siRNA was confirmed via PAGE stained with GelRed to localize the
siRNA. Lane 3 shows the aDT-siRNA conjugate along with unreacted siRNA; lane 4 shows the
aDT-siRNA conjugate after nickel column purification, with only a small amount of excess
siRNA left over. ............................................................................................................................ 57

xvi
Figure 3.3. GSCs express HB-EGF, the native receptor for diphtheria toxin. Representative
confocal images are shown for HB-EGF (anti-HBEGF antibody, green) and nucleus (Hoechst,
blue). The secondary antibody only control confirms lack of non-specific binding. All scale bars
are 50 µm. ..................................................................................................................................... 58
Figure 3.4. aDT-siRNA downregulates ITGB1 expression in GSCs and reduces cellular invasion.
A) aDT-ITGB1 (light red bars) downregulates ITGB1 mRNA expression compared to negative
controls: aDT conjugated to a non-targeting siRNA (aDT-NT, black bars) and ITGB1 siRNA
only without lipofectamine (blue bars) at 24 h post treatment. Positive control is transfected
ITGB1-siRNA with lipofectamine (dark red bars). Data is shown as n=3, mean±SD, normalized
to an untreated control. Data was analyzed using one-way-ANOVA followed by Tukey’s
correction on the logarithmic data (* p<0.05, ** p<0.01). B) Cells were plated in a 3D hydrogel
assay on the surface of pre-formed hydrogels and treated with aDT-ITGB1 conjugates at the
beginning of the experiment. Invasion depth was measured after 5 days. C) aDT-ITGB1 reduces
invasion compared to controls (no treatment and aDT-NT) in a 3D hydrogel model.
Representative images shown. 15 µm red beads label the top of the hydrogel; blue cell nuclei are
labeled using Hoechst. All scale bars are 150 µm. D) Invasion depth was quantified as a
percentage of the untreated control. Data was analyzed using one-way-ANOVA followed by
Tukey’s correction (* p<0.05, ** p<0.01). E) aDT-ITGB1 did not reduce number of adhered
cells in a 3D hydrogel model. Representative images are shown. All scale bars are 150 µm. F)
Number of adherent cells was quantified by counting number of cell nuclei; no significant
difference was observed, demonstrating that differences in invasion were due to ITGB1
downregulation. Data was analyzed using one-way-ANOVA followed by Tukey’s correction. . 59
Figure 3.5. aDT-siRNA downregulates eIF-3b expression in GSCs and reduces cell viability. A)
aDT-eIF-3b (light red bars) downregulates eIF-3b mRNA expression compared to negative
controls: aDT conjugated to a non-targeting siRNA (aDT-NT, black bars) and eIF-3b siRNA
only without lipofectamine (blue bars) at 24 h post treatment. Positive control is transfected
siRNA with lipofectamine, dark red bars. Data is shown as n=3, mean±SD, normalized to an
untreated control. Data was analyzed using one-way-ANOVA followed by Tukey’s correction on
the logarithmic data (* p<0.05). B) aDT-eIF-3b (red bars) reduces cell viability of GSCs at 48 h
post treatment compared to aDT-NT (black bars) at 100 nM. Data is shown as n=3, mean+SD,

xvii
normalized to an untreated control. Data was analyzed using one-way-ANOVA followed by
Tukey’s correction (* p<0.05). ..................................................................................................... 62
Figure 6.1. Structures of nucleic acid analogues including morpholinos and peptide nucleic acids.
....................................................................................................................................................... 85

1
Introduction
Portions of this chapter are derived from the following manuscript:
Arnold, A. E.; Czupiel, P.; Shoichet, M.S. Engineered Polymeric Nanoparticles to Guide the
Cellular Internalization and Trafficking of Small Interfering Ribonucleic Acids. J. Control.
Release 2017, 259, 3–15.
A.E.A. and P.C. contributed equally to the research and writing of this article. M.S.S. edited the
manuscript.
1.1 Rationale
Glioblastoma (GBM) is a highly aggressive and invasive cancer, with the current standard of
care only offering marginal improvements in an already limited survival time. In particular, the
glioblastoma stem cell (GSC) population is hypothesized to be responsible for the resistance of
the cancer to chemotherapy, and these cells also invade deeply into the brain tissue, avoiding
surgical resection and promoting recurrence of the cancer. The basis of many cancers is gene
misregulation: due to underlying mutations, many genes become either under- or over-expressed,
leading to aberrant behaviour of the cells, malignancy, and metastasis.
Antisense technologies, which comprise oligonucleotides such as small interfering ribonucleic
acids (siRNAs) and antisense oligonucleotides (AONs), are powerful tools for the regulation of
gene expression, and offer an ideal therapeutic strategy for diseases caused by genetic mutations,
such as cancer. Cancer cells overexpress oncogenic genes, providing potential targets for gene
knockdown.1-2 siRNAs are short strands of ribonucleic acid (RNA) typically composed of 21-30
base pairs with overhanging 3’ ends that can induce sequence-specific gene silencing at low
(picomolar) concentrations when transfected into cells.3 AONs are short strands of
deoxyribonucleic acid (DNA) with sequence homology to the messenger RNA (mRNA) of
interest that, while typically less potent than siRNAs, can still effect potent gene silencing when
transfected into cells in a similar manner.4
Delivery materials for antisense therapeutics have traditionally been large, positively charged
nanomaterials. However, these materials often are challenged with poor biodistribution, with

2
most ending up in the clearance organs (liver, kidney, or spleen) or the lungs. For this reason,
many of these formulations target cancers or other diseases originating in these organs. In order
to engineer platform technologies that could be used for GSC therapeutics, we wanted to develop
small protein bioconjugates that would target specific receptors on the GSC cells and evade
clearance due to their small size and lack of positive charge. In this thesis, two modalities of
antisense therapeutic delivery are explored: the first using antibodies, which are targeting
proteins from the adaptive immune system that are highly stable in circulation; and the second
using attenuated toxins, which have the additional advantage of sophisticated trafficking
mechanisms once they penetrate cells. Proof-of-concept in vitro studies were carried out to
demonstrate the potential applications of these bioconjugates and their promise as platform
technologies for antisense therapeutic delivery to GSCs and potentially many other diseases.
1.1.1 Hypothesis and objectives
Hypothesis: Protein-conjugated oligonucleotides will selectively and effectively knockdown key
genes in brain cancer cells, including patient-derived models of GBM, and thereby reduce either
cellular proliferation or invasion in vitro.
1. Knockdown an essential GSC gene in vitro using a targeted delivery platform.
i. Validate knockdown using AONs or small interfering ribonucleic acids.
ii. Conjugate AONs to antibodies specific to GSCs.
iii. Evaluate uptake, gene knockdown, and changes in cellular morphology in vitro.
2. Achieve gene knockdown with enhanced endosomal escape.
i. Evaluate suitability of diphtheria toxin as a delivery vehicle for GSC treatment.
ii. Conjugate siRNAs to attenuated diphtheria toxin (aDT).
iii. Evaluate reduction of target mRNA following aDT-siRNA treatment in vitro.
iv. Evaluate functional effects of target knockdown including reduction in cell viability
reduction of cellular invasion in a 3D model.

3
1.2 Glioblastoma stem cells
This section will give an overview of GBM, the cancer stem cell concept, and ultimately focus
on GSCs as a targetable and therapeutically relevant cell population.
1.2.1 Glioblastoma
GBM is one of the deadliest brain cancers, with a median survival time of just over a year with
the current standard of care, which includes aggressive chemotherapy, radiation, and surgical
resection.5 The origin of GBM is unclear, although in at least some cases it develops from low-
grade astrocytomas. Recent studies have shown that the cells of origin could be astrocytes,
oligodendrocyte precursor cells, or neural stem/progenitor cells.6 Most GBMs demonstrate a
diffuse infiltrative growth pattern, meaning the cancer cells interweave with the existing healthy
brain tissue, with single cells or groups of cells diffusing outward into the brain tissue from the
main tumor mass.7 Cells in the invasive front of the GBM tend to take advantage of existing
‘supply lines’ for oxygen and nutrients rather than building their own.7
As many as 50% of GBMs have significant mutations in the Phosphoinositide 3-kinase/Protein
kinase B/Phosphatase and tensin homolog (PI3K/Akt/PTEN) pathway, which plays a significant
role in the cell cycle and cell motility and can have downstream effects including increasing
proliferation and invasion and avoiding apoptosis.8 However, many strategies to combat GBM
by inhibiting this pathway have led to disappointing results in clinical trials.9-11 Other important
oncogenic changes that are frequently observed in GBMs are mutations of isocitrate
dehydrogenase (IDH), which plays a key role in the production of NADPH, and methylation of
O6-methylguanine-DNA methyl-transferase (MGMT), a ubiquitously expressed enzyme
involved in DNA repair. These changes are important as both prognostic indicators and potential
therapeutic targets.12-14
The standard-of-care chemotherapy drug for GBM treatment is temozolomide (TMZ), which is
used because it crosses the blood brain barrier. Unfortunately, TMZ is only a modestly effective
chemotherapeutic and its clinical use is mainly palliative. The average increase in survival time
for patients treated with resection and radiotherapy concurrently with TMZ compared to patients
who received resection and radiotherapy is only 2.5 months.15 Treatment with TMZ also has
significant, dose-limiting toxic side effects which leads to adverse events in 15-20% of patients,

4
including thrombocytopenia, lymphopenia, neutropenia, and myelodysplastic syndrome (a
disturbance of the bone marrow).16 For these reasons, there is a significant unmet need to
develop new therapies for GBMs, and especially the cancer stem cell population, which may be
responsible for many of the aggressive characteristics of GBM (vide infra).
1.2.2 Cancer stem cells
Stem cells are immature cells within the body that have the capacity for self-renewal, meaning
they can divide to generate new stem cells, as well as multi- or pluri- potency, which means they
have the capacity to differentiate into multiple adult cell types.17 Although there are many genes
involved in the regulation of stemness, the transcription factors Oct4, Sox2, and Nanog are
essential transcription factors that maintain the self-renewal and pluripotency of pluripotent stem
cells. An increasing body of evidence shows that there is a sub-population of cells within many
cancers that exhibit characteristics of both cancer cells and stem cells, which means they express
markers of self-renewal and pluripotency while also containing oncogenic mutations. The cancer
stem cell (CSC) hypothesis suggests that because these cells have characteristics including self-
renewal, pluripotency, and tumorgenicity, they have the capacity to evade radio- and chemo-
therapy, initiate metastasis, and promote recurrence of the cancer (Figure 1.1).
Figure 1.1. Cancer stem cells resist treatment and promote recurrence. The bulk of the
tumor is susceptible to treatment including radiotherapy and chemotherapy, but some
CSCs survive, which can repopulate the tumor even in small numbers.
The first identification of a tumor-initiating subpopulation of cells was in 1994, when researchers
implanted an enriched fraction of CD34+/CD38- acute myeloid Leukemia cells into an immune-
compromised mouse and observed that the cells were more proliferative and tumorigenic than
the parent population.18 In 1997, Dick et al. studied multiple subtypes of AML and identified
tumor-initiating cells that could differentiate in vitro in all samples regardless of subtype.19
Discoveries of CSC populations in breast cancer20 and brain cancer, specifically

5
medulloblastoma and GBM,21 followed in 2003. Strikingly, as few as 100 cells were required to
initiate the formation of a solid tumor in mouse models in both breast and brain cancers. CSCs
have since been identified in a variety of cancers including colon, pancreas, lung, prostate, and
melanoma.22 There are conflicting theories about the origin of these cells: they may arise from
normal stem cells that gain oncogenic mutations, or from somatic cells that acquire stem-like
characteristics and develop malignant behavior.22 Notably, CSC markers are not ubiquitous - in
each different tissue type, or even subtype, the cells are characterized by expression of diverse
markers,23 which means their identification and isolation presents a significant challenge.24
CSCs play important roles in both resistance to chemo- and radio- therapy25 and cancer invasion
and metastasis. Meirelles et al. found that ovarian CSC populations are resistant to cisplatin and,
even more surprisingly, stimulated by doxorubicin (both common chemotherapy drugs).26 Li et
al. showed that the proportion of CSCs in human breast cancer biopsies increased from 4.7% to
13.6% after a common chemotherapy regimen while the percentage of epithelial cells remained
constant, indicating a higher resistance to chemotherapy in the CSC population.27 Interestingly,
Zielske et al. studied two different patient-derived breast cancer xenografts, and observed that
one CSC population was enhanced following radiotherapy, agreeing with earlier reports, while
the other CSC population was depleted by radiotherapy treatment, perhaps hinting towards a
high degree of variance in CSCs between different patients.28 CSCs are also important in cancer
migration processes. Kaplan et al. demonstrated that bone-marrow derived haematopoietic
progenitor cells hone to pre-metastatic sites and provide a permissive niche prior to the arrival of
tumor cells.29 Pandit et al. discovered that highly metastatic cancer cell lines express higher
proportions of CSC markers than non-metastatic cancer cell lines,30 and Wakamatsu et al. found
a higher proportion of CSC markers in diffuse lymph node metastases than in the primary tumor
site, suggesting that the metastases were initiated by the CSCs.31
Several approaches have been taken to try to specifically target the CSC population. One
approach is to target specific receptors on the CSC surface; since many markers are shared
between CSCs and normal stem cells, there are only a few targetable receptors that also
differentiate between the two populations. CD44 and CD133 are two common CSC markers that
have been used to target the CSC population. Jin et al. eradicated leukemia stem cells using an
anti-CD44 antibody and observed reduced leukemic repopulation.32 Perez et al. have developed
an anti-CD44 antibody that selectively targets the CSC population in head and neck squamous

6
cell carcinoma33 and two clinical trials using this strategy have been completed (NCT01358903
and NCT01641250). Zhao et al. inhibited tumor growth by developing a bispecific antibody for
both T-cells and CD133+ cells, encouraging the T cells to attack the CSC population.34 Another
approach is to target specific pathways that are active in CSCs. Several small molecule mTOR
inhibitors, including Everolimus and Metformin, have been explored to specifically target the
breast CSC population, which leads to significantly slower tumor growth.35-36 In GBM,
JAK/STAT3 inhibitors were used to target the GSC population resulting in prolonged survival in
a mouse model of GBM.37 Taken together, these studies demonstrate that specifically targeting
the CSC population can lead to significant benefit in tumor growth and patient survival, and thus
is a promising approach that is widely applicable to many cancers.
1.2.3 Glioblastoma stem cells
Brain tumor initiating cells were first identified by Singh et al. in 2003, primarily in samples of
aggressive medulloblastomas.21 In 2004 the same group discovered that GBM stem cells, which
comprised 20-30% of the total patient tumor samples, could be transplanted into mouse brains at
low cell numbers and recapitulate diversity of the original patient tumor, including neuron,
oligodendrocyte, and astrocyte lineages.38 These cells have characteristics including self-
renewal, pluripotency, neurosphere formation, proliferation, invasion, and high motility.39 The
major challenges that GSCs present are their highly infiltrative nature and their extreme
resistance to conventional treatments, which make them an important target for treatment of
GBM.39
GSCs are described by many terms, including cancer/tumor/glioma/brain tumor stem cell, stem-
like tumor cell, cancer-/tumor-/glioma-brain tumor-initiating cell, and glioma neural stem cell.40
While these terms are often coined in an attempt to delineate small variations in cell markers and
functional characteristics, for the purposes of this thesis they will all fall under the umbrella of
‘glioblastoma stem cells/GSCs’ that have been identified by one key functional assay:
propagation of a tumor upon transplantation in a rodent brain followed by differentiation into
multiple cell types, recapitulating the diversity of the parental tumor.
For specific targeting of the GSC population, it is important to pick markers that will be
expressed on the GSC cell surface. GSC samples are often thought of as a hierarchy, with a very
small fraction of the cells that are actually self-renewing, nearly quiescent stem cells maintaining

7
a larger population of progenitor cells and perhaps even differentiated progeny.41 This feature of
GSC populations makes them difficult to target, since many different markers will be expressed
at different stages of the cell cycle and even among different stem/progenitor cells within the
same population; so there is not one ubiquitous ‘GSC marker’. However, some common surface
markers have been identified which are discussed below.
1.2.3.1 Surface markers of GSCs
As previously mentioned, CD133 and CD44 are cell surface markers that are shared among
many CSC populations in different tissues and are two of the most common markers for GSCs.
Some additional surface markers that have been explored for the isolation, characterization, and
targeting of the GSC population are EphA2/EphA3,42-43 EGFR,44 CD15/SSEA1, A2B5,
L1CAM/CD171, ALDH1A3.39
CD133 (cluster of differentiation 133) was the first cell surface marker used to isolate GSCs.21, 38
Also known as prominin-1, CD133 is an antigen that is located on projections of the plasma
membrane and is typically expressed on hematopoietic stem and progenitor cells.45 Increased
CD133+ cells in in brain tumors correlates with a more aggressive tumor46 and a poorer
prognosis,47 and recurrent tumors often have higher proportions of CD133 cells,48 implying an
important role for CD133 in cancer recurrence and invasion. Singh et al. found that CD133+ cells
could initiate GBMs whereas CD133- cells could not.38 However, subsequent studies have shown
that CD133- cells can also give rise to GBMs with robust CD133 expression,49-50 implying that
not all GSCs express CD133.
CD44 (cluster of differentiation 44) is a cell membrane glycoprotein that serves as a hyaluronic
acid receptor.51 CD44 is involved in key pathways associated with GSCs including invasion and
proliferation.52 In neural stem cells, CD44 is often co-expressed with the stem cell markers nestin
and Sox2.53 CD44+ cells can generate new tumors that recapitulate the parental tumor in mouse
models, but CD44- cells cannot do the same.54 Importantly, inhibition of CD44 can also slow the
progression of GBM,55 indicating its key role in tumorgenesis and its potential as a therapeutic
target for GBM. GSCs often express a splice variant of CD44, CD44v6, which has been shown
to drive proliferation of GSCs.56

8
Some less common, but still important, proposed markers for GSC populations are
EphA2/EphA3, EGFR, CD15/SSEA1, and L1CAM/CD171. Binda et al. identified EphA2 as a
driver of self-renewal and tumorgenicity in GSCs,42 and Qazi et al. demonstrated that co-
targeting of EphA2 and EphA3 could be a promising approach for GSC targeting and GBM
treatment.43 EGFR is expressed in over half of newly diagnosed GBMs and the variant EGFRvIII
is amplified in GSCs in vivo; however, this amplification is often lost in in vitro culture.44 CD15,
also known as stage-specific embryonic antigen 1 (SSEA1) is a marker for central nervous
system stem cells and is also an enrichment marker for GSCs.57-58 L1CAM is a cell adhesion
molecule expressed in neurons and is involved in growth and migration during nervous system
development, and has been shown to be essential for the survival of CD133+ GSCs.59
1.2.4 Therapeutic targets associated with GSC proliferation and invasion
Many signaling pathways are activated to promote GSC self-renewal, proliferation, and
progression, including receptor tyrosine kinases, Akt, MAPK, Wnt, Notch, Hedgehog, and
JAK/STAT pathways.60 These complex, often inter-connected signaling pathways offer a
multitude of potential therapeutics for gene regulation in the GSC population. For example,
Esposito et al. demonstrated that knockdown of STAT3 reduced cell proliferation and migration
in vitro, leading to a decrease in tumor growth and angiogenesis in vivo.61 Berezovsky et al.
found that knockdown of Sox2, which can be downstream of both Akt and JAK/STAT signaling
pathways, reduced self-renewal and sphere forming properties in GSCs.62 This thesis will focus
on three selected genes for downregulation in GSCs, which are involved with GBM
proliferation, invasion, or both: downregulated in renal cell carcinoma (DRR), Eukaryotic
Translation Initiation Factor 3b (eIF-3b), and integrin b1 (ITGB1).
1.2.4.1 DRR
DRR, also known as Family with Sequence Similarity 107A (FAM107A), was first associated
with cancer as a tumor suppressor gene in renal cell carcinoma.63 In 2010, Petrecca et al.
identified DRR as a driver of GBM invasion that acts as a crosslinker between the actin and
microtubule cytoskeletons.64 They proposed the following theory of cancer progression relating
to DRR: low grade, infiltrative gliomas express high levels of DRR, and display an extremely
invasive phenotype. Over time, additional mutations in some cells lead to the loss of DRR and

9
invasion is reduced while proliferation is dramatically increased, as observed in higher grade
gliomas. This is supported by earlier work that shows invasion and proliferation as temporally
separate events in malignant gliomas.65
In 2014, Petrecca et al. expanded on their earlier work and demonstrated that DRR induces Akt
activation by recruiting kinases to focal adhesions.66 They also discovered a relationship between
the GSC population, and by knocking down DRR in the GSCs, they observed a corresponding
reduction in activated Akt levels and a reduction in invasion in vitro and in vivo. Therefore, DRR
is a promising target for reducing the invasion of GSCs.
1.2.4.2 eIF-3b
eIF-3b is a subunit of the 13-unit eukaryotic translation initiation factor 3 complex (eIF 3) which
is essential for assembling the machinery for protein synthesis within the cell.67 The composition
of eIF 3 is conserved within most eukaryotes.68 One of the first studies that proved eIF-3b played
a role in oncogenesis related eIF-3b expression to tumor grade, stage, and patient survival in
bladder and prostate tumor tissue samples.69 The same study demonstrated that robust eIF-3b
expression is required for proliferation of the tumor and colonization of the cancer cells in a
secondary site such as the lungs.69 Zang et al. studied the role of eIF-3b in clear cell renal cell
carcinoma (ccRCC) and observed that high levels of eIF-3b in patient tumor samples correlated
to a more aggressive tumor and reduced patient survival.70 Knockdown of eIF-3b in ccRCC
impaired the activation of the Akt pathway, inhibiting cell proliferation and inducing apoptosis.
Furthermore, the migration of the cells via epithelial-to-mesenchymal transition was reduced
following eIF-3b knockdown.70 Liang et al. discovered that eIF-3b also plays a key role in the
proliferation of GBM, and by knocking down eIF-3b the proliferation of the GBM cells was
significantly reduced,71 indicating the potential of eIF-3b as a therapeutic target in GBM.
1.2.4.3 ITGB1
Integrins are a large family of cell-adhesion molecules that are displayed on the surface of the
cell as heterodimers. In mammalian cells, there are 18 a integrins and 8 b integrins, with 24
identified dimers of a and b subunits.72 ITGB1 can form 12 different heterodimers to interact
with a variety of extracellular matrix materials, including fibronectin and collagen.73 Binding of
extracellular ligands to ITGB1 can initiate a signal cascade involving signaling partners such as

10
focal adhesion kinase (FAK) and Akt that initiates actin assembly or disassembly, an essential
step in the formation and dissolution of focal adhesions during cell migration.73 ITGB1 has been
shown to enhance cancer cell invasion and migration in breast cancer, pancreatic cancer, and
GBM.74-76 Interestingly, in GBM, knockdown of ITGB1 increased the sensitivity of cells to anti-
angiogenic therapy in addition to reducing the invasiveness of the cancer cells.76 While studies
connecting ITGB1 and GSCs have been limited, ITGB1 is overexpressed in at least some
subtypes of GSCs,77 and ITGB1 is involved in many invasion processes, so it is an interesting
target for preventing the invasion of the GSCs.
1.3 Antisense therapeutics
Antisense therapeutics are powerful tools for controlling gene expression. They were first
identified more than forty years ago: in 1978, the first example of an AON was reported as an
inhibitor of viral replication. Gene silencing by double-stranded siRNAs was first discovered in
C. Elegans in 1998,78 and naturally occurring siRNAs were reported in 1999 in plants.79
Synthetic siRNAs were used to effect gene knockdown in mammalian cells two years later.80
“Naked” AON and siRNA therapeutics have been successful in clinical trials: siRNAs for ocular
diseases when locally delivered at high concentrations, despite limitations including
inflammation and increased ocular pressure;81 and AONs for applications in muscular
dystrophy82 and spinal muscular atrophy.83 Because AONs and siRNAs can target and
downregulate specific genes within diseased cells, they are promising treatments for diseases
caused by genetic mutations such as cancer.84-85 However, efficient and targeted delivery of
nucleic acid therapeutics presents a significant clinical challenge.
Following intravenous injection in order to reach diseased (ie. cancerous) tissue, oligonucleotide
formulations must (1) evade the immune system, (2) avoid interactions with non-target cells, (3)
avoid premature renal clearance, and (4) reach target tissues. There are further issues once they
reach their site of action: the fragile nucleic acid material must avoid degradation by extracellular
nucleases, and overcome poor cell uptake and trafficking into the lysosomal compartments
where the RNA or DNA strands are quickly degraded.86-87 These challenges often require that
antisense therapeutics are combined with specialized delivery materials in order to be effective.
Some of the carriers that are currently used to carry these oligonucleotides into cells will be
presented in sections 1.4 and 1.5. This section will focus on the mechanism and stability of

11
AONs and siRNAs.
1.3.1 Antisense oligonucleotide mechanism
First generation AONs were strands of DNA, 8-50 nucleotides in length, which can bind to
complementary strands of mRNA and induce the activity of RNAseH, an enzyme that recognizes
the DNA/RNA hybrid and degrades the mRNA. (Figure 1.2).88 More recent studies have focused
mostly on chemically modified AONs, with modifications such as the ones shown in Figure 1.5,
which enhance the stability of the AON while reducing off-target effects.89 The AONs also
commonly include a phosphorothioate backbone or are completely replaced with other
backbones, such as morpholine rings, which further increase their resistance to nuclease and
protease degradation.4 The current clinically approved AONs are examples of these second-
generation motifs, including eteplirsen, an AON against Duchenne muscular dystrophy which is
administered as a “naked” AON intravenously,82 and nusinersen, an AON for treatment of spinal
muscular atrophy that is injected directly into the cerebral spinal fluid of the spinal column.83
Figure 1.2. AON mechanism of action. The AON hybridizes to complementary strands of
mRNA and induces the activity of RNAseH, which recognizes the DNA/RNA hybrid and
degrades the RNA into its components (nucleoside monophosphates).

12
1.3.2 Small interfering ribonucleic acid mechanism
siRNAs are 20-30 nucleotide long, double stranded, noncoding RNA that can degrade mRNA in
a sequence-specific manner.90 They are typically more potent, but somewhat less stable, than
their AON counterparts.90 Unlike AONs, siRNAs cannot be delivered without a carrier to
mediate delivery.91 In 2018, the FDA approved the first siRNA-based drug, patisiran, for the
treatment of transthyretin-mediated amyloidosis.92 While this drug uses a lipid nanoparticle
system for delivery, next-generation delivery systems that use smaller siRNA-biomolecule
conjugates are already in the clinical pipeline.92
As shown in Figure 1.3, one of the first steps in the siRNA mechanism is cleavage by an enzyme
called Dicer, which cuts long, double-stranded RNA into short siRNA fragments. The RNA-
induced silencing complex (RISC), which is a multi-protein assembly, then forms and
incorporates the strand of the siRNA with the more stable 5’-end. The complementary mRNA is
identified by sequence alignment to the siRNA and the catalytic protein of the RISC complex,
Argonaute 2, cleaves the complementary mRNA.90

13
Figure 1.3. siRNA mechanism of action. Dicer cleaves long, double-stranded RNA into
short fragments of siRNA. siRNA is then loaded into the RISC complex, binds to the
complementary mRNA, and RISC degrades the mRNA.
1.3.3 Stability in the extracellular environment
In order to increase the delivery efficiency of oligonucleotide payloads, they are often conjugated
or complexed to carriers that protect them from nucleases and rapid clearance (Figure 1.4).
While AONs are relatively stable to nuclease degradation,93 they are less potent than their siRNA
counterpart, and stability is a major challenge to siRNA delivery. Within 15 minutes of injection
in mice, more than 90% of standard 21-mer siRNAs are degraded by serum nucleases or lost via
renal or lymphatic clearance,94 underlining the importance of the delivery vehicle. Polymeric
nanoparticles can increase the stability of siRNAs against degradation: Raja et al. demonstrated
that crosslinked chitosan nanoparticles increased the stability of siRNAs against serum during a
15 day storage at 4°C95 while Zhu et al. increased the half-life of siRNA in the blood to
approximately 8 hours by encapsulating it within a PLGA-based delivery vehicle, resulting in
better tumor accumulation.96 Conjugation to a globular protein, such as an antibody, can also
protect nucleic acid materials from degradation: Bäumer et al. conjugated an siRNA to an anti-
EGFR antibody and saw a significant improvement in stability against serum.97

14
Figure 1.4. siRNA carriers protect it from nuclease degradation. (A) Free siRNA (blue
double helix) is rapidly degraded by nucleases (orange semi-circle) and (B) cleared by
lymphatic drainage (pale blue ovals). (C) Nanoparticle or protein carriers may protect
siRNA from nucleases and (D) reduce clearance.
Oligonucleotides can also be chemically modified in order to increase their stability against
nucleases. These modifications include any change to the native DNA or RNA structure,
typically employed on the phosphodiester bond or sugar ring (Figure 1.5). These modifications
enhance stability and potency, provide longer knockdown duration, reduced off-target effects,
and lower immunostimulatory effects.89, 98-100 Modified oligonucleotides are now commonly
used in research.101-103 As shown in Figure 1.5, some of the most common modifications of
oligonucleotides include modifications to the backbone or nucleosides. For example, backbone
modifications include phosphorothioate104 and boranophosphonate105 linkages, which increase
nuclease resistance, while nucleoside modifications include 2’-O-methyl,106-107 2’-deoxy-2’-
fluoro,108 and locked nucleic acids,109 which increase stability and target binding affinity.
Chemical modification of oligonucleotides and the effect on potency have been extensively
reviewed by Deleavey et al.89 Figure 1.5 is showing the RNA sugar but many of the same
modifications can be used with DNA for AON applications.
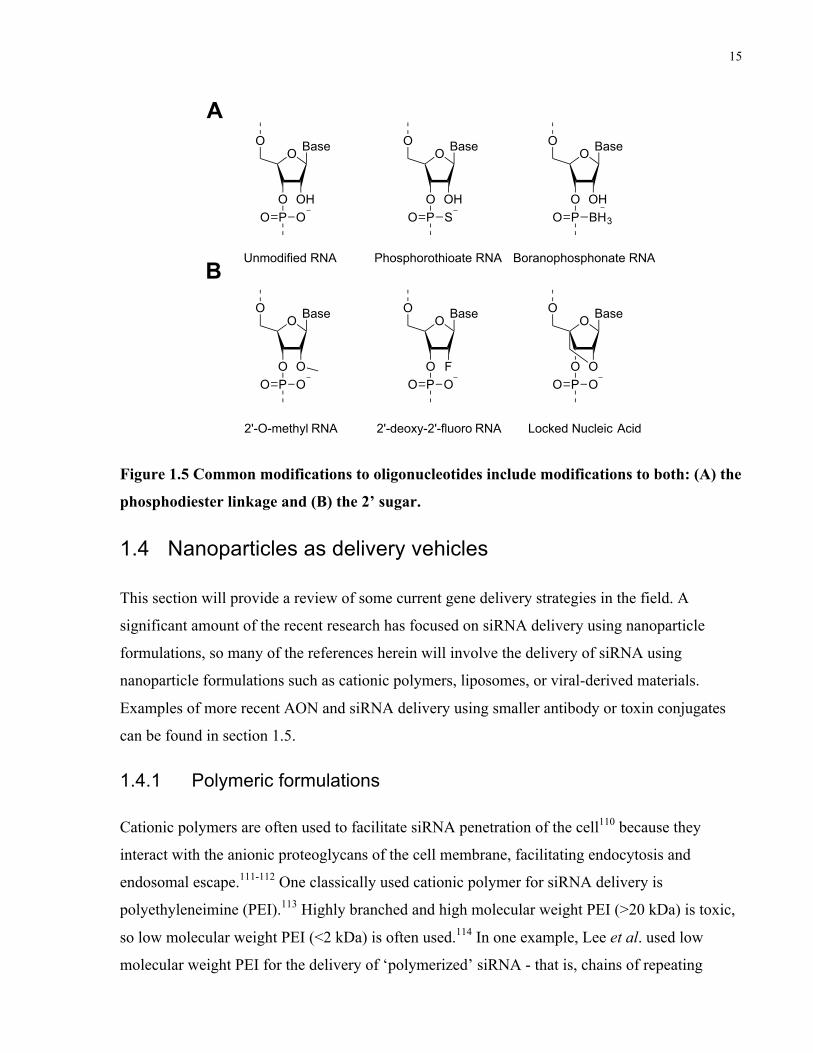
15
Figure 1.5 Common modifications to oligonucleotides include modifications to both: (A) the
phosphodiester linkage and (B) the 2’ sugar.
1.4 Nanoparticles as delivery vehicles
This section will provide a review of some current gene delivery strategies in the field. A
significant amount of the recent research has focused on siRNA delivery using nanoparticle
formulations, so many of the references herein will involve the delivery of siRNA using
nanoparticle formulations such as cationic polymers, liposomes, or viral-derived materials.
Examples of more recent AON and siRNA delivery using smaller antibody or toxin conjugates
can be found in section 1.5.
1.4.1 Polymeric formulations
Cationic polymers are often used to facilitate siRNA penetration of the cell110 because they
interact with the anionic proteoglycans of the cell membrane, facilitating endocytosis and
endosomal escape.111-112 One classically used cationic polymer for siRNA delivery is
polyethyleneimine (PEI).113 Highly branched and high molecular weight PEI (>20 kDa) is toxic,
so low molecular weight PEI (<2 kDa) is often used.114 In one example, Lee et al. used low
molecular weight PEI for the delivery of ‘polymerized’ siRNA - that is, chains of repeating
O
O OHPO O
BaseOO
O OHPO S
BaseOO
O OHPO BH3
BaseO
O
O OPO O
BaseOO
O FPO O
BaseOO
O OPO O
BaseO
A
BUnmodified RNA Phosphorothioate RNA Boranophosphonate RNA
2'-O-methyl RNA 2'-deoxy-2'-fluoro RNA Locked Nucleic Acid

16
siRNA segments connected by disulphide bonds. By using this PEI delivery system, they were
able to achieve significant knockdown in vitro of red fluorescent protein (RFP) in RFP+
melanoma cells.115 Despite efficient transfection, any cell will non-specifically take up PEI and
other positively charged polymers. Since most nano-scale formulations naturally accumulate in
the liver,116-117 many strategies deliver therapeutics against diseases of the liver.117-118 In order to
target other tissues, the positively charged polymer must be shielded until it reaches the tumor
site.
To temporarily shield their positive surface charge, cationic nanoparticles are modified with
sheddable poly(ethylene glycol) (PEG) coronas using various stimulus-responsive coupling
strategies. For example, Li et al. developed a polymeric nanoparticle that is responsive to matrix
metalloproteinase 7 (MMP-7), an enzyme that is overexpressed by breast cancer cells and found
at high concentrations in the tumor microenvironment.119 The nanoparticle corona is composed
of a PEG block linked by an MMP-7 cleavable peptide to a cationic block. When the
nanoparticle reaches the tumor microenvironment, extracellular MMPs cleave the peptide,
shedding the PEG layer and exposing the cationic layer, raising the zeta-potential of the
nanoparticle from +5.8 to +14.4 mV and increasing cellular internalization 2.5-fold.
Nanoparticles pre-treated with MMP-7 resulted in significant knockdown in vitro; however, this
system was not studied in vivo, so it is still unclear whether this strategy will result in improved
biodistribution.119 Despite the shielded cationic charge, significant toxicity was observed at high
nanoparticle:siRNA ratios, underlining the importance of nanoparticle safety to their utility.
Targeting ligands can be attached to polymeric delivery vehicles to increase the specificity of
cellular uptake. These specifically bind receptors overexpressed on cancer cell membranes,
facilitating receptor-mediated endocytosis of the nanoparticle.120 Interestingly, the MMP-7
responsive nanoparticle, previously discussed,119 was conjugated to folate ligands.121 In this case,
PEG cleavage was triggered by MMP-7 at the tumor site, exposing folate-conjugated
nanoparticles for receptor-mediated endocytosis. In vitro experiments, including MMP-7 pre-
treatment and folate ligand competition assays, revealed that knockdown was dependent on both
MMP-7 activity and folate receptor binding. Under optimal conditions, the formulation achieved
significant luciferase protein knockdown with no detectible cytotoxicity in a luciferase positive
breast cancer cell line.121

17
Antibodies can also be conjugated to nanoparticle formulations for targeted siRNA delivery,
triggering internalization via a receptor-mediated endocytosis pathway.122 Palanca-Wessels et al.
synthesized a nanoparticle in which siRNA was encapsulated and to which anti-human epidermal
growth factor receptor 2 (HER2) antibodies were conjugated for cellular internalization.123
Delivery of siRNAs against a variety of chemotherapy resistance-associated mRNAs resulted in
significant target gene knockdown in vitro and in vivo in a mouse model of ovarian cancer.
However, the authors noted a slight immune response in some of the streptavidin-containing
control groups.123-124
1.4.2 Lipid-based formulations
One of the most common delivery methods for siRNAs and AONs is lipid-based formulations,
which incorporate bulky alkyl chain ‘tail’ groups with small, hydrophilic ‘head’ groups that often
incorporate at least some cationic components in order to enhance cellular uptake and endosomal
escape.125 Additional components that are often included in lipid-based formulations are
cholesterol, which can associate with lipid bilayers;126-127 PEGylated lipids, which prevent
aggregation and off-target uptake;128 and targeting ligands to enhance uptake in the target
tissue.129
Lipid-based materials were one of the first materials investigated for siRNA delivery, and
include formulations such as liposomes, micelles, microemulsions, and solid lipid
nanoparticles.84 One of the most well-known examples of a lipid nanoparticle formulation of
siRNA is Patisiran, a liposomal formulation of siRNA to treat hereditary transthyretin
amyloidosis that marks the first siRNA drug to achieve FDA approval.130
To optimize delivery of nucleic acids, the pKa of an ionizable lipid should be low enough that it
is relatively neutral during circulation (to avoid off-target uptake and minimize toxicity), but
high enough that it is protonated in the early or late endosomes.131 Endosomal escape is induced
by the protonation of the lipid head group and subsequent association with the negatively
charged plasma membrane.132 Jayaraman et al. conducted a study of 53 different ionizable lipids
and their effects on siRNA delivery and gene silencing in vivo. They determined that the
silencing efficacy reached an optimized point around an average pKa of 6.44.133

18
The optimum amount of cholesterol incorporated in the lipid formulation depends on the system,
but some studies have shown that the rate of drug release depends on having at least a minimum
amount of cholesterol incorporated into the lipid bilayers.134 Zhang et al. achieved optimal
siRNA delivery using a cholesterol:lipid ratio of 1:1 or 1:2 (30-50% cholesterol) in a liposomal
formulation.135
Interestingly, Zhang et al. also found that PEGylation of liposomes decreases transfection
efficiency, perhaps due to steric hinderance of the bulky PEG preventing the lipids from
interacting with cell membranes.135 Bao et al. further investigated the effect of PEGylation on
lipid nanoparticle systems and discovered that an optimal level of siRNA-mediated gene
silencing was obtained at approximately 0.5 mol% PEG, which was enough to limit toxic side
effects of the lipid nanoparticle while maintaining a high level of gene silencing.128
Targeting ligands may also be added to lipid formulations to enhance their on-target effects. For
example, Akinc et al. investigated the use of the lipoprotein ApoE, which targets liver cells, for
delivery of siRNAs to hepatocytes in vivo.136 They confirmed the effect of their formulation by
delivering the ApoE-targeted formulation in ApoE-knockout mice and observing a strong degree
of gene silencing, as opposed to wild type mice where they observed less than 20% silencing at
the same dose.136 Other targeting moieties that have been investigated for lipid formulations are
Vitamin A, which is shuttled to hepatic cells,137 and N-acetylgalactosamine (GalNAc), a small
molecule that has been investigated for delivery to the liver because of its ability to bind to the
asialoglycoprotein receptor displayed on the surface of hepatocytes.136 Importantly, most
targeted liposomal formulations have involved binding receptors in the liver, which is probably
because most lipid-based formulations naturally accumulate in the liver and it is a major
challenge to achieve biodistribution of these formulations to other organs.117
Although most of these examples involve siRNA delivery, liposomal formulations have also
been investigated for the delivery of AONs. Wyrozumska et al. used a cationic lipid formulation
incorporating the cationic lipid 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) as well as
cholesterol and PEGylated lipids to deliver AONs against a gene BCL-2 that is overexpressed in
leukemia cells, and observed improved biodistribution of the PEGylated carriers and a prolonged
survival in the mouse model of leukemia.138

19
1.4.3 Virus-inspired formulations: cell penetrating and membranelytic
peptides
Cell penetrating peptides (CPPs) have been exploited to bring oligonucleotide cargo into cells.
The CPPs are typically < 40 amino acids, cationic, and viral-derived.139 There have been many
reviews focused on the characteristics and mechanisms of CPPs139-141 and while there are
numerous CPP sequences (Table 1), they usually lack specificity as they will cross any cell
membrane. While the internalization pathways of most CPPs are not well-defined, internalization
is initialized via interactions of the cationic CPPs with the phospholipids of the cell
membrane.142
Table 1. Common cell penetrating sequences.
Name Sequence Origin Chargec
TAT (48-60)143-
144
GRKKRRQRRRPPQ Derived from HIV type 1 +8
Penetratin145 RQIKIWFQNRRMKWKK Antennapedia homeodomain +7
TP10 146-147 GWTLNS/AGYLLGKINL
KALAALAKKILa
Neuropeptide galanin-
mastoparan fusion
+4
VP22148 NAKTRRHERRRKLAIER Herpes simplex virus +7
Polyarginine149 Rna, n=8-9 Engineered for positive
charge
+8 or +9
Pep-1150 KETWWETWWTEWSQP
KKKRKVb
Fusion of NLS from simian
Virus 40 and reverse
transcriptase of HIV-1
+3
CADY151 GLWRALWRLLRSLWRL
LWRAb
Derived from PPTG1 peptide,
addition of W and charged
amino acids
+5

20
a C-terminal amide. b C-terminal cysteamide. c pH 7.4.
To achieve greater specificity of CPPs, one of three strategies is typically employed: (1)
triggering CPP deprotection at the tumor site; (2) local delivery of the CPP to the tumor site; or
(3) conjugation to cell-targeting ligands. Using the first strategy, Sun et al. synthesized a
polyarginine CPP, used for siRNA complexation and siRNA release, sandwiched between a
hydrophobic poly(caprolactone) (PCL) block and a hydrophilic PEG block.152 The PEG corona
was conjugated to the CPP through 2-propionic-3-methylmaleic anhydride linkers, which are
cleavable under the acidic environment of the tumor site, deshielding the CPP and facilitating
cellular uptake in vitro and in vivo. For the second strategy of local CPP delivery, Kanazawa et
al. used an intranasal delivery route to carry siRNA directly to the brain in a mouse model of
brain cancer using a CPP-nanoparticle formulation.153 PCL nanoparticles were conjugated to a
TAT CPP and hydrophilic PEG. The authors were able to use the TAT peptide for siRNA
complexation and delivery. For the third strategy, conjugating CPPs to cell targeting peptides can
increase their specificity. Fang et al. conjugated the TAT CPP to A1, a peptide with high affinity
for vascular endothelial growth factor receptor-1 (VEGFR1) and demonstrated selective delivery
to tumor cells overexpressing VEGFR.154 Similarly, R9 can be fused to a cyclic arginine-glycine-
aspartic acid (cRGD) peptide for targeting.155
Another use for viral-derived CPPs is endosomal escape of oligonucleotide formulations.
Endosomolytic peptides destabilize membranes when a critical concentration of the peptide is
located near a membrane, followed by interaction with the negatively charged phospholipid
bilayer and pore formation. The resultant pore then destabilizes and disrupts the membrane.156
For example, Cheng et al. synthesized virus-inspired polymers for endosomal release by grafting
‘caged’ melittin, a membranolytic peptide, through a disulfide bridge to a polymer containing a
hydrophilic cationic block for therapeutic loading and a pH-sensitive block for the triggered
display of the melittin peptide.157 The authors demonstrated greater transfection efficiency in
vitro and in vivo relative to commercially available reagents.
1.5 Protein conjugates as delivery vehicles
Recently, there have been several examples of smaller biomolecules conjugated to siRNAs or
AONs for targeted delivery to tissues. One of the most clinically advanced examples is siRNA

21
directly conjugated to GalNAc (a sugar derivative that is taken up by liver cells), a product from
Alnylam that is close to FDA approval.158 These small conjugates have advantages over large,
cationic nanoparticles including reduced immune response, specificity of targeting, and
monodispersity of particle size.84 It has also been demonstrated that the stability of nucleic acid
material against nucleases can be enhanced by conjugation to biomolecules such as globular
proteins.97 This section will provide a review of current delivery strategies for nucleic acids
using proteins including antibodies and toxins, as well as some background on antibody-drug
conjugates (ADCs) and engineered toxins for other therapeutic applications.
1.5.1 Antibodies for drug and oligonucleotide delivery
One of the most common ways of targeting therapeutics to specific cell types is by using
antibodies or antibody fragments (Fabs).159 Antibodies are proteins that are produced by the
immune system to be highly specific for antigens that are displayed on the surface of cells,
making them ideal candidates for targeted drug delivery with limited off-target binding.160
Antibodies can be engineered to bind to specific antigens, expressed using mammalian protein
expression systems, and functionalized via expression as fusion proteins or chemical
conjugation.161 Most of the work exploring antibodies for drug delivery has been as conjugates
between antibodies and small molecule drugs. This section will include a brief overview of
bioconjugation strategies that are applicable to antibodies as well as other types of proteins; some
key examples of antibody-drug conjugates; and some recent work with antibody-oligonucleotide
conjugates.
1.5.1.1 Bioconjugation strategies
There are abundant options available for conjugating a protein including (but not limited to): (1)
lysine modification via NHS chemistry; (2) reacting a native or engineered cysteine, tyrosine,
tryptophan, or N-terminal residue; (3) introduction of a non-native amino acid; or (4) use of a
peptide-tagging sequence.162 The selection of the appropriate conjugation chemistry requires
consideration of site-specificity, native amino acid availability, and stability of an engineered
protein.
(1) Lysine or N-terminal amine modification

22
One of the most widely-used bioconjugation strategies is modification of lysine residues. Most
proteins contain many available lysine residues - for example, an antibody usually contains at
least 20 free lysines for modification.163 Typically this is done using reactive N-
hydroxysuccinimide (NHS) esters, although there are numerous other amine-reactive functional
groups that can be chosen.163 NHS esters are relatively unstable in aqueous solutions so must be
quickly used, especially at basic pH,164 but the adduct that forms after reaction is a stable amide
bond with a half-life of at least 7 years in water.165 There are some strategies that can specifically
target the N-terminal amine for modification,166 but in most cases this modification is not site-
specific and several modifications will be introduced in a single protein, so care must be taken to
avoid labeling the active site of the protein of interest.
(2) Reacting cysteine, tyrosine, tryptophan
If site specificity is required, one of the options to achieve limited and specific functionalization
is targeting an amino acid that is less abundant on the native protein, or engineering a protein
with a unique residue for functionalization, such as a cysteine, tyrosine, or tryptophan. Cysteines
are a common option for site-specific modification because they are readily reactive but their
abundance in protein structures is low.167 Available cysteines can be modified using reactive
groups such as maleimides or activated thiols (
Figure 1.6A).168 Proteins, such as antibodies, can be reduced to yield free thiols for
modification;169 however, caution must be taken to avoid destabilizing the protein, inducing
HSN
O
O
+R R N
O
O
S
Protein
HONO2
N2
+R O
NH
R
HNRO2C
N2
Ph+
Dirhodium Peptide Cat.HN
RO2C
Ph
A
B
C
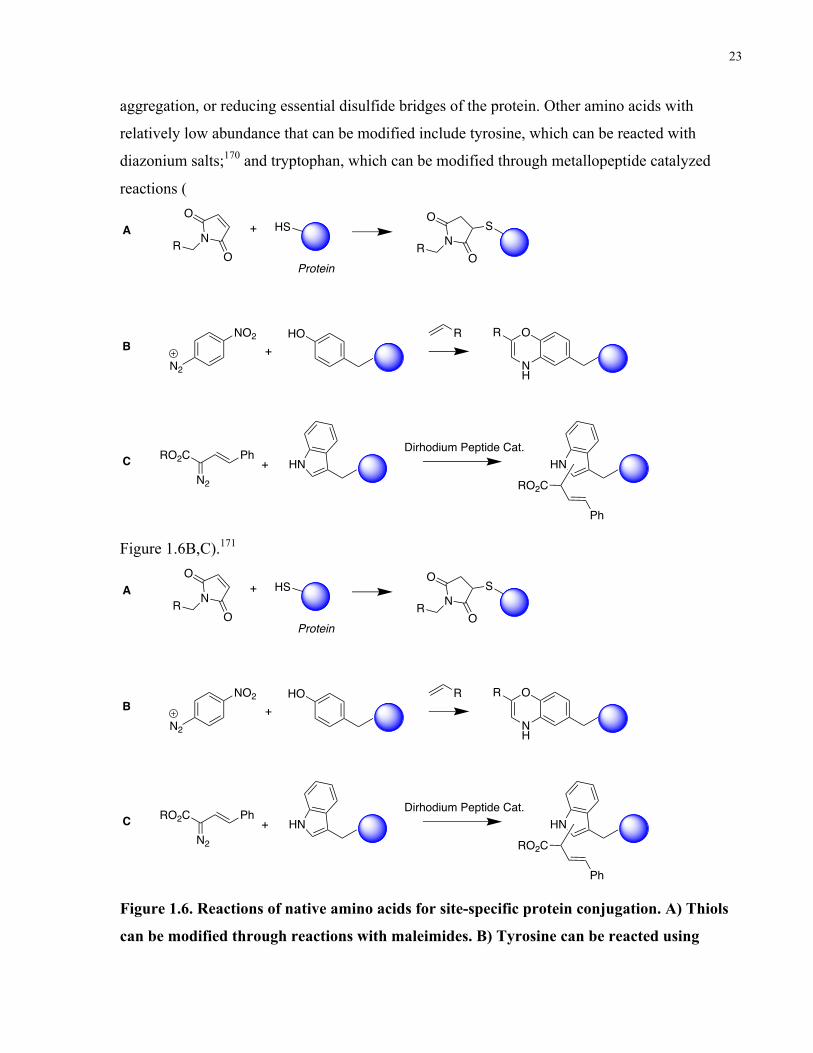
23
aggregation, or reducing essential disulfide bridges of the protein. Other amino acids with
relatively low abundance that can be modified include tyrosine, which can be reacted with
diazonium salts;170 and tryptophan, which can be modified through metallopeptide catalyzed
reactions (
Figure 1.6B,C).171
Figure 1.6. Reactions of native amino acids for site-specific protein conjugation. A) Thiols
can be modified through reactions with maleimides. B) Tyrosine can be reacted using
HSN
O
O
+R R N
O
O
S
Protein
HONO2
N2
+R O
NH
R
HNRO2C
N2
Ph+
Dirhodium Peptide Cat.HN
RO2C
Ph
A
B
C
HSN
O
O
+R R N
O
O
S
Protein
HONO2
N2
+R O
NH
R
HNRO2C
N2
Ph+
Dirhodium Peptide Cat.HN
RO2C
Ph
A
B
C

24
diazonium salts. C) Tryptophan can be modified through metallopeptide-catalyzed
reactions.
(3) Introduction of non-native amino acid
When engineering proteins, it is possible to introduce a non-native amino acid through the use of
a unique codon and introduction of the corresponding transfer RNA/synthetase pair.172 This
strategy can be used to introduce site-specific functional handles for bioorthogonal conjugation.
For example, moieties such as ketones,173 azides,174 alkynes,175 and anilines176 have been added
to proteins in this manner, and used for applications including labeling of live cell surfaces177 and
photocrosslinking.178 This strategy can yield highly specific conjugations for diverse
applications, but engineering the expression of a protein is a non-trivial process and care must be
taken to avoid destabilizing or deactivating the protein of interest.
(4) Use of a peptide-tagging sequence
Another site-specific method for introducing modifications to proteins is through the use of a
peptide-tagging sequence that an enzyme will recognize and label with the desired functional
group following protein expression. This method is particularly effective if the protein of interest
is present in a complex mixture but specific labeling is required.162 Some options for enzymatic
tagging via a peptide sequence include: biotin ligase, an enzyme that recognizes a peptide
sequence to biotinylate a specific site;179 transglutaminase, an enzyme which recognizes a
substrate sequence, and catalyzes a transamination reaction to introduce the desired
functionalization;180 lipoic acid ligase, which has been used to introduce non-native functionality
such as alkyl azides at the site of an acceptor peptide;181 and sortase, which recognizes one
peptide sequence on the protein of interest and another on the group to be conjugated and
mediates ligation.182
In addition to considerations of the site of protein conjugation, there is also the selection of linker
chemistry. One option that yields highly efficient reactions, amenable to aqueous conditions,
with limited or no by-products is conjugation via click chemistry.183 The relative rates of several
bioorthogonal click chemistry reactions are shown below. The selection of the chemistry requires
considerations including hydrophobicity of the reactive groups, available functional sites, and the
required reaction rate.

25
Table 2. Rates of selected bioorthogonal click chemistry reactions.183
Reaction Rate (Approx) (M-1s-1)
Azide-strained alkyne 10-1 – 10-2 (0.067)
Copper-mediated azide-alkyne 10-200
Maleimide-furan 10-5
Tetrazine-norbornene 106
(Oxime or hydrazide)-(aldehyde or ketone) 2-20
1.5.1.2 Antibody-drug conjugates
ADCs are conjugates between antibodies that bind to cell surface antigens and chemotherapy
drugs. An antibody could be an ideal delivery vehicle for drugs due to its favorable
pharmacokinetic profile, with a typical elimination half-life of 18-21 days for IgGs.184 The
promise of an ADC is a highly specific and selective delivery combined with the potency of the
conjugated drug,185 but ADCs have encountered some limitations in clinical trials due to the
relatively low potency of chemotherapeutics.186 Four antibody-drug conjugates have reached
FDA approval: three for hematologic cancers and one to treat breast cancer.187 The first ADC to
reach clinical approval was gemtuzumab ozogamicin (GO), a CD33-antibody delivering
calicheamicin, a DNA-damaging drug.188 Post-approval, GO encountered some roadblocks due
to off-target toxicity, but was later re-introduced to the clinic following optimization of
pharmacokinetic and safety profiles.188 More recently, a HER2-antibody (trastuzumab) carrying
emtansine, a cytotoxic chemotherapeutic, obtained FDA approval for breast cancer treatment;189
one of the most significant findings from this ADC was that significant efficacy was only
observed if the patients were carefully selected for high HER2-expression in the tumor tissue.189
In pre-clinical and clinical studies, antibodies-drug conjugates have been explored for numerous
other solid tumors including GBM.190 The majority of antibody-drug conjugates for GBM
treatment have been using an antibody against either EGFR, a receptor tyrosine kinase which is

26
expressed in the majority of primary GBMs, or the variant EGFRvIII, which is expressed in 25-
50% of GBM patients.191-193 Interestingly, there is some evidence that EGFRvIII may be
expressed in the GSC population.194 Many initial studies that focused on targeting GBM with
unmodified EGFR or EGFRvIII antibodies have yielded disappointing results.190, 195 Recently,
ABT-414, comprising an EGFR monoclonal antibody conjugated to a cytotoxic payload
monomethyl auristatin F (MMAF) underwent a three-arm clinical trial and resulted in a
progression-free survival (PFS) of 30% at six months, which could be a significant improvement
over the standard of care which generally yields a PSF of 10-20%.196 A clinical trial to compare
ABT-414 directly to ABT-414 combined with TMZ or TMZ alone is currently underway for
EGFR amplified GBM patients (NCT01800695).
1.5.1.3 Antibody-oligonucleotide conjugates
Since ADC efficacy has encountered limitations due to the low potency of many
chemotherapeutic drugs, as well as off-target cytotoxicity, a potential way forward is to
conjugate antibodies to highly specific and potent antisense therapeutics such as AONs or
siRNAs.
One of the first examples of an antibody-siRNA conjugate was via avidin-biotin conjugation:
Xia et al. conjugated streptavidin with an antibody against the human insulin receptor (HIR) and
combined this system with a biotinylated siRNA against luciferase, observing gene silencing at
low nanomolar concentrations.197 In vivo, they were able to observe luciferase silencing in
multiple organs including the brain.198 Following a similar biotin-avidin approach, Shankland et
al. used an antibody against podocytes in the kidney and conjugated the antibody to a positively
charged protamine molecule via neutravidin-biotin conjugation. siRNA against two relevant
targets for kidney disease, nephrin and transient receptor potential channel 6 (TRPC6), was
complexed to the positively charged protamine for delivery and significant gene silencing was
observed in vivo.199 Bäumer et al. conjugated the same protamine molecule directly to an anti-
EGFR antibody for complexation with an anti-KRAS siRNA and demonstrated that the therapy
induced apoptosis and slowed tumor growth in an in vivo model of colon cancer.200
Some potential limitations of antibody-siRNA conjugates were explored by Siebel et al., who
have developed a platform technology for the production of antibody-siRNA conjugates via
cysteine conjugation and generated a variety of these bioconjugates with different siRNA and

27
antibody targets.201 This led to several key findings about antibody siRNA conjugates: first, they
used both cleavable and non-cleavable linkers and observed no difference between the silencing
activity between the two types of linkers, indicating that release from the protein is not essential
for siRNA activity following delivery. Second, they discovered that only some combinations of
antibodies and cell lines could lead to efficient siRNA delivery and gene silencing, so the
antibody-siRNA platform was not universally applicable. Third, they observed a significant
amount of overlap between the delivered antibody-siRNA conjugate and the endolysosomal
pathway, and hypothesized that an enhanced endosomal escape mechanism could lead to a
higher degree of gene silencing. They used this information to develop an ‘optimized’ antibody-
siRNA conjugate for delivery of a lethal siRNA to prostate cancer xenografts and observed a
significant reduction in tumor growth.201
Despite some limitations of antibody-oligonucleotide conjugates, antibodies continue to be
explored for delivery of both AONs and siRNAs. Sugo et al. developed an antibody-siRNA
conjugate targeted to cardiac and skeletal muscles and were able to downregulate myostatin in
vivo, which translated into a recovery of movement ability.202 Satake et al. developed an
antibody-AON conjugate where the antibody was targeted to a leukemia target, CD22, for the
AON-mediated knockdown of a novel target MAX dimerization protein 3 (MXD3) and observed
a significant improvement in survival time in a mouse model of leukemia.203 In one of the most
recent examples, Wang et al. developed an antibody-siRNA conjugate for the treatment of
malignant melanoma, targeting the oncogene livin, and observed significant silencing in vivo.204
These diverse examples of antibody-siRNA and -AON conjugates and their efficacy in different
tissues exemplify the promise of antibody conjugates as truly targeted therapies where delivery is
not limited to the clearance organs. I explored this strategy for AON delivery as described in
more detail in Chapter 2.
1.5.2 Engineered toxins for biomolecule delivery
“AB” toxins, including toxins such as diphtheria toxin (DT), anthrax toxin (AT), and
Pseudomonas exotoxin A (PE), comprise two main subunits: The “A” domain, which is the
active toxin domain that is highly lethal to cells, and the “B” domain, made up of a receptor
binding component (R) and a translocation component (T).205 These toxins are efficient at
binding to the target cells, inducing endocytosis, and inserting the toxin domain into the

28
cytoplasm.206 These characteristics, combined with the recent expansion of protein engineering
techniques, make the AB toxin class an intriguing candidate as both targeted cytotoxic agents
and as vehicles for delivery of other drugs.
DT, AT, and PE all have slightly different cellular entry mechanisms (Figure 1.7) but they all
bind to an extracellular receptor and are eventually translocated into the cytosol. All three toxins
bind to a target receptor, induce endocytosis, and undergo cleavage by an enzyme called furin in
the endosomes; however, the protein domains of DT and PE remain linked by an essential
disulfide bond.207-208 PE induces trafficking to the endoplasmic reticulum where the disulfide
linkage is broken by protein disulfide isomerase (PDI) before it undergoes retrograde
translocation into the cytosol.209 Translocation of DT and AT occurs at the reduced pH of the
early endosome, and while the DT creates a flexible α-helical pore that is highly permissive to
different types of cargo,210 AT creates a rigid β-barrel pore that may require the complete
unfolding and refolding of the cargo.211 The disulfide bond of DT is cleaved after translocation
by intracellular enzymes.206 Each of these toxins has been exploited and re-engineered for the
targeted killing of diseased cells, or as attenuated delivery vehicles for diverse cargo, usually of
other therapeutic proteins. This section will provide some key examples of drug delivery using
these AB toxins.
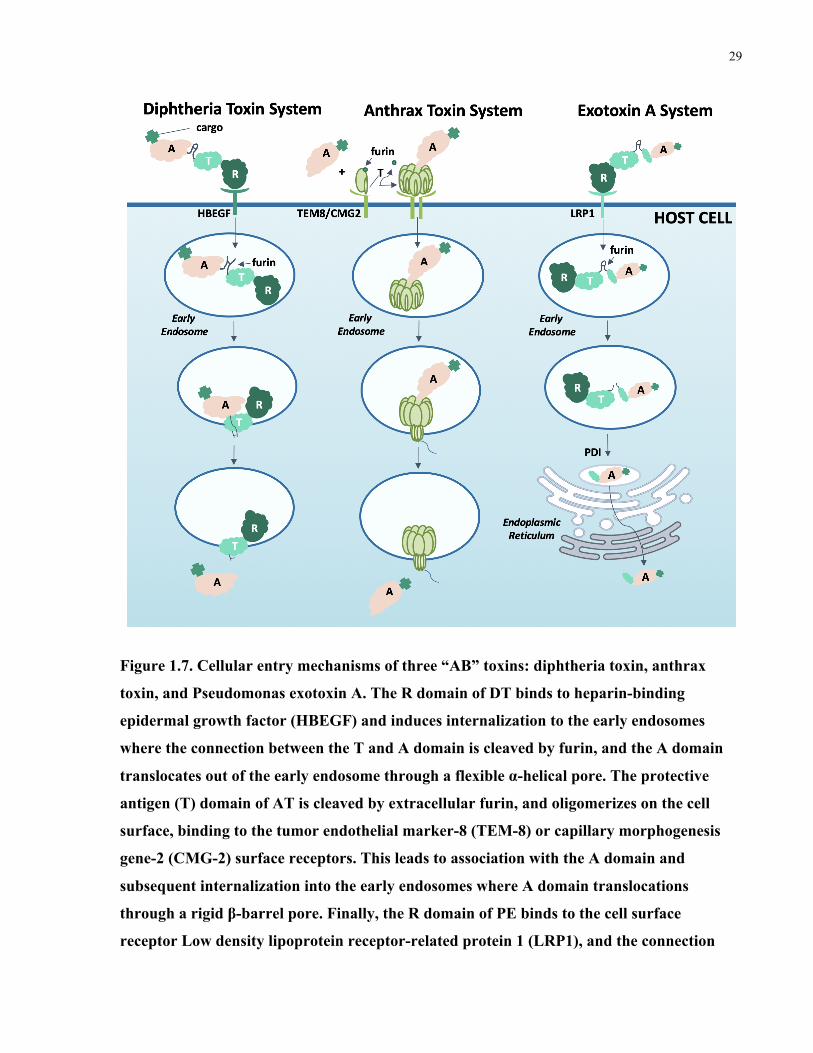
29
Figure 1.7. Cellular entry mechanisms of three “AB” toxins: diphtheria toxin, anthrax
toxin, and Pseudomonas exotoxin A. The R domain of DT binds to heparin-binding
epidermal growth factor (HBEGF) and induces internalization to the early endosomes
where the connection between the T and A domain is cleaved by furin, and the A domain
translocates out of the early endosome through a flexible α-helical pore. The protective
antigen (T) domain of AT is cleaved by extracellular furin, and oligomerizes on the cell
surface, binding to the tumor endothelial marker-8 (TEM-8) or capillary morphogenesis
gene-2 (CMG-2) surface receptors. This leads to association with the A domain and
subsequent internalization into the early endosomes where A domain translocations
through a rigid β-barrel pore. Finally, the R domain of PE binds to the cell surface
receptor Low density lipoprotein receptor-related protein 1 (LRP1), and the connection

30
between the T and A domain is cleaved by furin in the early endosomes. PE is then
trafficked to the endoplasmic reticulum where it undergoes retrograde translocation.
1.5.2.1 Targeted toxins as cytotoxic agents
One therapeutic strategy that utilizes toxins is by targeting the native toxic domain to a specific
cell-surface receptor. The modification of AT presents a significant challenge as alteration or
removal of the R domain can destabilize the protein.212 However, DT and PE are both amenable
to alteration or removal of the R domain and re-engineering of the protein structure to target
different cell surface receptors.
There are several examples of PE-based immunotoxins in the clinic, including moxetumomab
pasudotox, a PE fragment fused to an antibody fragment against CD22 for treatment of
lymphoma, which recently demonstrated positive results in a phase I clinical trial.213 Similarly, a
PE immunotoxin targeting CD25 for lymphoma and leukemia treatment recently underwent an
initial clinical trial and achieved complete remission in at least one leukemia patient.214 There is
at least one clinically approved therapeutic based on the delivery of the DT toxic domain:
denileukin difitox (Ontak) which is a DT variant targeted to the IL-2 receptor that has shown
significant benefit to cutaneous T-cell lymphoma patients.215 In clinical studies, Resimmune,
which combines the A and T domains of the DT with antibody fragments against CD3, has
shown promising effects in a clinical trial for lymphoma patients, including four complete
remissions (16% of patients).216 Interestingly, DT variants have also been investigated for GBM
treatment. This is an exciting strategy because DT has the potential to naturally cross the blood
brain barrier.217 The DT A and T domains were fused with an antibody fragment targeting EGFR
and EGFRvIII for targeting GBM and a robust anti-tumor efficacy was observed in an orthotopic
in vivo model of malignant glioma, prompting FDA approval of a phase I/II clinical study that is
currently underway.218 Taken together, these examples highlight the capacity of DT and PE for
targeted therapeutic delivery tailored to virtually any desired cell surface receptor.
1.5.2.2 Attenuated toxins for therapeutic delivery

31
More recently, some researchers have shifted to taking advantage of the receptor binding and
translocation components of AB toxins to deliver desired cargo into cells, while the toxic domain
is attenuated or completely removed.210, 219 This has most commonly been done with AT, and
one of the first examples was from Arora et al. where they removed the native AT toxic domain
and replaced it with the toxic domains of shiga and diphtheria toxin to show that the modified
protein still had cytotoxic effects on cells.220 Delivery of more diverse cargo soon followed, with
Ballard et al. using attenuated AT to delivery cytotoxic T-cell epitopes into mammalian cells,
indicating that the disarmed toxin fragment could deliver cargo other than AB toxin domains.221
More recently, non-canonical cargo including antibody mimics, polypeptides, and
chemotherapeutic drugs have been delivered into cells using the attenuated AT as a delivery
vehicle. Liao et al. conjugated small antibody mimics including monobodies and affibodies to
the attenuated AT for the perturbation of intracellular protein-protein interactions.222 Using
functional readouts including intracellular protein disruption and apoptosis, they confirmed that
delivery was more effective than a comparable vehicle such as a cell penetrating peptide.222
Shortly thereafter, Rabideau et al. demonstrated that AT could deliver linear peptides and small
molecule drugs including doxorubicin and MMAF to the cytosol of cells and that these remained
intact and functional following translocation.223 However, AT was not able to deliver cyclic
peptides or larger, polycyclic drugs such as docetaxel, perhaps due to the structural rigidity of the
AT translocation pore.223 Surprisingly, despite this limitation Dyer et al. were able to deliver
AONs and siRNAs to cells using disarmed AT and observed an impressive knockdown
efficiency of a proof-of-concept target, Syntaxin5; however, they have yet to evaluate the
functional effects of AT-mediated knockdown in vitro or in vivo.224
Some evidence indicates that DT might have a more permissive translocation pore. Auger et al.
demonstrated that DT could deliver intact, folded proteins directly into the cytosol by
conjugating DT to a fluorescent protein mCherry, which has high conformational stability and is
not likely to unfold.210, 225 In comparison, AT is not able to deliver mCherry into cells, likely
because it does not unfold prior to translocation.226 Auger et al. also optimized the linker length
between the cargo and the DT and determined that the appended cargo was invisible to the
translocation machinery with the correctly engineered construct.210 This strategy has been used
by Park et al. to deliver a therapeutically relevant protein, human purine nucleoside
phosphorylase (PNP), to rescue function in PNP-depleted cells.227 This is a novel treatment

32
modality for patients with inherited PNP mutations and could be extended for the delivery of
other essential enzymes involved in disease. Another potential advantage of DT as a delivery
vehicle is its reported ability to cross the blood-brain barrier (BBB) via HBEGF-mediated
transcytosis.228 As proof-of-principle, Gaillard et al. demonstrated that an attenuated DT
conjugated to horseradish peroxidase (HRP) could reach the brain tissue following intravenous
injection, whereas free HRP could not.217 Together, these studies imply that DT has a relatively
permissive translocation pore and can specifically target and cross the blood brain barrier, so it
has great potential as a drug delivery vehicle to brain disease including GBM. I explored
attenuated diphtheria toxin (aDT) for delivery of siRNA as described in Chapter 3.
Antibody-antisense oligonucleotide conjugate downregulates a key gene in glioblastoma stem cells
This chapter was published in Molecular Therapy - Nucleic Acids:
Arnold, A. E.; Malek-Adamian, E.; Le, P. U.; Meng, A.; Martínez-Montero, S.; Petrecca, K.;
Damha, M. J.; Shoichet, M. S. Antibody-Antisense Oligonucleotide Conjugate Downregulates a
Key Gene in Glioblastoma Stem Cells. Mol Ther Nucleic Acids 2018, 11, 518–527.
A.E.A. conceived and performed experiments and wrote the first and subsequent drafts of the
manuscript. E.M.-A. synthesized and characterized all oligonucleotide materials and provided
major revisions of the first draft. P.U.L. provided cell lines, advice, and feedback. A.M.
performed flow cytometry experiments. S.M.-M. conceptualized fundamental work on the
project and provided feedback on the manuscript. M.J.D., K.P., and M.S.S. secured funding,
supervised and guided the research, and participated in writing the manuscript.
2.1 Abstract Glioblastoma stem cells (GSCs) are invasive, treatment-resistant brain cancer cells that express
downregulated in renal cell carcinoma (DRR), also called FAM107A, a genetic driver of GSC
invasion. We developed antibody-antisense oligonucleotide conjugates to target and reduce
DRR/FAM107A expression. Specifically, we used antibodies against antigens expressed on the
glioblastoma stem cells, such as CD44 and EphA2, conjugated to chemically modified antisense
oligonucleotides (AONs) against DRR/FAM107A, which were designed as chimeras of DNA
and 2ʹ-deoxy-2ʹ-fluoro-beta-D-arabinonucleic acid (FANA) for increased nuclease stability and

33
mRNA affinity. We demonstrate that these therapeutic conjugates successfully internalize,
accumulate, and reduce DRR/FAM107A expression in patient-derived GSCs. This is the first
example of an antibody-antisense strategy against cancer stem cells.
2.2 Introduction Glioblastoma cancer stem cells (GSCs) are hypothesized to account, at least partially, for
treatment failures in aggressive glioblastoma multiformes.229-233 The GSCs are highly
invasive,234-235 resistant to radiation and conventional chemotherapy,236-237 and have the capacity
to initiate new tumor growth.232, 238
Downregulated in renal cell carcinoma (DRR/FAM107A)63 is an established genetic driver of
GSC invasion. It acts by regulating focal adhesion dynamics at the leading edge of migrating
cells64 and by activating Akt signaling, making it an ideal anti-invasion target.64, 66, 239
Antisense oligonucleotides (AONs), which are short (15-21 nucleotide) strands of DNA, are
potent regulators of gene expression.93 There are several clinically approved antisense
oligonucleotide therapeutics, including Eteplirsen, a treatment for muscular dystrophy,240 and
Nusinersen, which is used in the treatment of spinal muscular atrophy.83
Herein, we engineered antisense oligonucleotides (AONs) for DRR/FAM107A knockdown in
patient-derived GSCs. The AON is stabilized against nuclease degradation by substituting some
of the DNA residues for 2ʹ-deoxy-2ʹ-fluoro-beta-D-arabinonucleic acid (FANA) residues,
thereby increasing treatment longevity while maintaining potency.89, 241 We designed a “gapmer”
AON with FANA modifications flanking the DNA core because FANA gapmers bind target
mRNA with high affinity and elicit mRNA degradation while protecting the 3ʹ-end from
exonuclease degradation.242 This construct also includes a phosphorothioate (PS) backbone in
lieu of the naturally-occurring phosphodiester (PO) backbone for added nuclease resistance and
effective RNaseH-mediated cleavage of mRNA.243-244 Given that PS-DNA chemistry may
increase immunostimulation, we incorporated a 2ʹ-deoxy-5-methyl-cytidine at a CpG motif
within the AON sequence to obviate this issue in future studies.245-246
Antibody targeting is a powerful tool to guide therapeutic delivery to specific cell types, both as
pendant groups on larger nanoparticle systems247-250 and as direct antibody-drug conjugates
(ADCs).251-255 In glioblastomas, administration of an anti-EGFR antibody-monomethyl auristatin

34
F ADC has resulted in a survival benefit for EGFR-amplified glioblastoma patients256 whereas
administration of an anti-EGFR antibody alone failed.257 To mediate AON delivery, we chose to
investigate three monoclonal antibodies (mAbs) against three GSC surface markers: CD44, a
neural stem cell marker and a marker of GSCs within the glioblastoma tumor;258-260 EphA2, a
key component of cell-cell signaling that is overexpressed in many cancers and also in GSCs;42
and EGFR, which is amplified in 40-60% of all glioblastomas.261
We developed an antibody-conjugated double-stranded antisense oligonucleotide (dsAON)
therapeutic using click chemistry between an azide-modified antibody and an alkyne-modified
dsAON with a unique architecture: the sense strand is modified at the 5ʹ and 3ʹ ends with
functionalities for click chemistry (dibenzylcyclooctyne, DBCO) and for imaging (Cyanine 3
fluorophore, Cy3), respectively. While the sense strand is covalently conjugated to the antibody,
the antisense strand is conjugated only via hybridization to this sense strand to facilitate its
release once inside the cells. Direct conjugation of large molecules such as antibodies to the
antisense strand can interfere with RNase H recognition of the corresponding AON:mRNA
duplex.262 This approach is an improvement over the few antibody-antisense therapeutic
conjugates reported, with the majority of these being non-covalent, cationic complexation97 or
disulfide linkages directly to the antisense strand, which are unstable and prone to
degradation.263-264 Effective delivery and gene knockdown using antisense oligonucleotides has
been demonstrated with dsDNA systems where the DNA sense strand is left unmodified and as
such susceptible to nuclease-mediated digestion by endogenous cellular enzymes.265 This
degradation is the necessary driving force to release the antisense strand, which is then available
for hybridization with the target mRNA.265
We chose to use antibodies engineered by phage-display in order to target the glioblastoma stem
cells.266 Based on immunocytochemistry data, we identified CD44 and EphA2 as the best
candidates for AON delivery. We then verified that these antibodies were internalized upon
binding prior to conjugating them to the dsAONs via click chemistry. We found that the CD44
mAb-dsAON conjugate significantly reduced DRR expression, which correlated with a change
in cellular morphology. Thus, we can reduce expression of a key GSC target, DRR/FAM107A,
using antibody-dsAON conjugates and this provides a framework for antibody-AON conjugate
testing against GSCs in vivo.

35
2.3 Results
2.3.1 Antisense oligonucleotide activity
We tested four modification patterns of the DRR/FAM107A AON and compared these to a
scrambled sequence control (Figure 2.1) in order to maximize efficacy and reduce
immunogenicity: (i) Gapmer motif, consisting of flanking FANA modifications with a DNA
core; (ii) Altimer motif, where FANA substitutions were alternated every 3 nucleotides with
DNA;267 (iii) Gapmer-MeC with a 5-methylcytidine modification at the CpG motif (5MeCG) in
order to reduce immunogenicity; and (iv) Altimer-MeC with a similar 5MeCG modification
(Figure 2.1A). We observed significant DRR knockdown following transfection into DRR-
overexpressing (DRR+) U-251 MG glioblastoma cells with all single stranded antisense
oligonucleotides by western blot compared to the scrambled control (Figure 2.1B). Although not
statistically significant, we observed a trend towards greater knockdown when using Gapmer
MeC compared to Altimer MeC. Therefore, all future studies were carried out using the Gapmer
MeC strand.
We hybridized the therapeutically active Gapmer MeC antisense strand to a carrier sense strand
modified with a 3ʹ Cy3 for imaging studies and a 5ʹ DBCO for click chemistry (see melting
curves in Figure A.1). The Cy3 and DBCO functionalities were incorporated into the sense
strand during solid-phase oligonucleotide synthesis. We transfected this double stranded
antisense oligonucleotide into DRR+ GBM cells and demonstrated significant reduction of DRR
expression compared to a scrambled control Figure 2.1C). Representative western blots are
shown (Figure 2.1D).

36
Figure 2.1. Downregulated in renal cell carcinoma (DRR) expression is reduced following
transfection of DRR+ U-251 MG cells with DRR antisense oligonucleotides. (A)
Oligonucleotide sequences used: all antisense strands comprise a phosphorothioate
backbone while all sense strands are synthesized with a phosphodiester backbone. (B) DRR
expression following treatment with single stranded DRR AON sequences normalized to
untreated control. Data were analyzed using one-way ANOVA followed by Dunnett’s post-
hoc test compared to Scrambled group (data is shown as mean+SD, n=3, *p<0.05,
**p<0.01). (C) DRR expression following treatment with double stranded anti-DRR
oligonucleotide sequences normalized to untreated control. Data were analyzed using
unpaired t-test with Welch’s correction (data is shown as mean+SD, n≥4, ***p<0.001). (D)
Representative western blot showing single stranded and double stranded antisense
oligonucleotide DRR knockdown.

37
2.3.2 Co-expression in GSCs of DRR and antigens for CD44, EphA2, and EGFR antibodies
We screened the patient-derived GSCs for co-expression of DRR with potential antigens for
targeted delivery of the antisense oligonucleotides: CD44, EphA2 or EGFR. All of the GSCs
used in this study strongly co-expressed DRR with CD44 and EphA2 in all fields of view, yet
only weakly expressed EGFR (Figure 2.2). Thus, CD44 mAb and EphA2 mAb were pursued for
further study.
Figure 2.2. Patient-derived GSCs strongly co-express antigens CD44 and EphA2 with DRR
and weakly co-express EGFR with DRR. Representative confocal images are shown.
Antigens CD44, EphA2, and EGFR (green); DRR (red); cell nucleus (Hoechst, blue). All
scale bars are 50 µM.
2.3.3 Internalization of CD44 and EphA2 mAbs
We quantified the internalization of the CD44 and EphA2 mAbs at 15, 45, and 90 min using a
flow cytometry-based assay with DRR+ U-251 MG cells. We first incubated the cells with the
CD44 (Figure 2.3A), EphA2 (Figure 2.3B), or non-specific CTL mAb (Figure 2.3C) at 4 °C,
incubated this sample for the given time period at 37 °C in order for internalization to occur, and
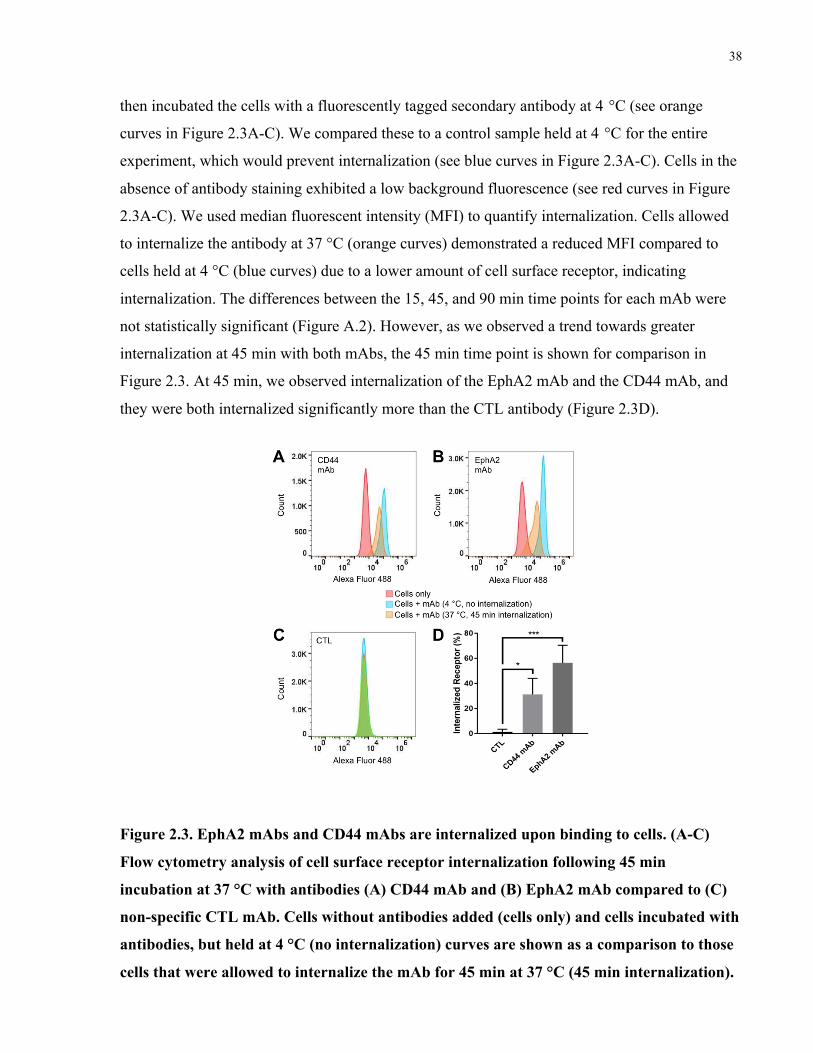
38
then incubated the cells with a fluorescently tagged secondary antibody at 4 °C (see orange
curves in Figure 2.3A-C). We compared these to a control sample held at 4 °C for the entire
experiment, which would prevent internalization (see blue curves in Figure 2.3A-C). Cells in the
absence of antibody staining exhibited a low background fluorescence (see red curves in Figure
2.3A-C). We used median fluorescent intensity (MFI) to quantify internalization. Cells allowed
to internalize the antibody at 37 °C (orange curves) demonstrated a reduced MFI compared to
cells held at 4 °C (blue curves) due to a lower amount of cell surface receptor, indicating
internalization. The differences between the 15, 45, and 90 min time points for each mAb were
not statistically significant (Figure A.2). However, as we observed a trend towards greater
internalization at 45 min with both mAbs, the 45 min time point is shown for comparison in
Figure 2.3. At 45 min, we observed internalization of the EphA2 mAb and the CD44 mAb, and
they were both internalized significantly more than the CTL antibody (Figure 2.3D).
Figure 2.3. EphA2 mAbs and CD44 mAbs are internalized upon binding to cells. (A-C)
Flow cytometry analysis of cell surface receptor internalization following 45 min
incubation at 37 °C with antibodies (A) CD44 mAb and (B) EphA2 mAb compared to (C)
non-specific CTL mAb. Cells without antibodies added (cells only) and cells incubated with
antibodies, but held at 4 °C (no internalization) curves are shown as a comparison to those
cells that were allowed to internalize the mAb for 45 min at 37 °C (45 min internalization).

39
(D) Quantification of internalized receptor following 45 min incubation period for CD44
mAb and EphA2 mAb antibodies compared to CTL. Data were analyzed using one-way-
ANOVA followed by Dunnett’s post hoc test compared to CTL group (data is shown as
mean+SD, n=3, *p<0.05, ***p<0.001).
2.3.4 Synthesis of mAb-dsDRR and mAb-dsScrambled conjugates
For antibody-mediated delivery of DRR, we first modified each of CD44 and EphA2 antibodies
with an azide functional group by covalently bonding NHS-PEG4-N3 to one of the lysines
therein. The azide modification enabled click conjugation of the DBCO-modified sense
oligonucleotide to the antibody following hybridization with either the Gapmer MeC (dsDRR) or
a scrambled sequence (dsScrambled) (Figure 2.4A). We determined conjugation efficiency by
gel electrophoresis followed by ImageJ quantification to be 60% for dsDRR conjugation to both
CD44 mAb (Figure 2.4B) and EphA2 mAb (Figure 2.4C). We observed the appearance of two
bands for both the CD44 mAb-dsDRR and the EphA2 mAb-dsDRR conjugates (lane 2 of Figure
2.4B,C), which we expect is due to the conjugation of either one or two antisense oligonucleotide
strands per antibody. To confirm the absence of non-specific aggregation or adsorption, the
dsDRR was mixed together with either the CD44 or EphA2 mAb (without prior azide
modification of the antibodies) and no evidence of conjugation was detected (lane 3 of Figure
2.4B,C).
To further verify the conjugation of the antisense oligonucleotides to the antibodies, we reacted
azide-modified mAb with the DRR sense strand and this conjugate was analyzed by MALDI-
TOF mass spectrometry (Figure A.3). Experimental results closely matched the expected
conjugate mass (Mobt = 160.0 kDa vs. Mth = 160.2 kDa).

40
Figure 2.4. CD44 mAb and EphA2 mAb can be efficiently conjugated to dsDRR using click
chemistry. (A) Scheme of antibody modification with dsDRR. (B) 10% PAGE analysis of
CD44 mAb-dsDRR conjugation: Lane 1: dsDRR only; Lane 2: CD44 mAb conjugated to
dsDRR via NHS-PEG4-N3 crosslinker; Lane 3: dsDRR and CD44 mAb combined without
crosslinker present. (C) 10% PAGE analysis of EphA2 mAb-dsDRR conjugation: Lane 1:
dsDRR only; Lane 2: EphA2 mAb conjugated to dsDRR via NHS-PEG4-N3 crosslinker;
Lane 3: dsDRR and EphA2 mAb combined without crosslinker present.
2.3.5 DRR knockdown and cellular uptake of mAb-dsDRR conjugates
To determine the knockdown efficiency of the EphA2 mAb-dsDRR and CD44 mAb-dsDRR
conjugates, we incubated patient-derived GSCs with each formulation for 72 h at 150 nM and
compared to mAb-dsScrambled, dsDRR alone, and antibody alone controls. We quantified DRR
protein expression using western blot analysis normalized first to α-tubulin and then to a no
treatment control. The CD44 mAb-dsDRR conjugate significantly reduced DRR expression in

41
patient-derived GSCs compared to the CD44 mAb-dsScrambled control (Figure 2.5A,B). We
observed no knockdown with the negative controls (dsDRR or CD44 mAb alone).
Unexpectedly, treatment with EphA2 mAb-dsDRR conjugate did not reduce DRR expression
relative to any of the EphA2 mAb-dsScrambled, dsDRR, or EphA2 mAb controls (Figure 2.5C,
D).
Figure 2.5. DRR expression of patient-derived GSCs after treatment with CD44 mAb-
dsDRR or EphA2 mAb-dsRR (150 nM) normalized to untreated control. (A)
Quantification of DRR expression following CD44 mAb-dsDRR treatment. Data were
analyzed using one-way-ANOVA followed by Dunnett’s post hoc test compared to CD44
mAb-dsScrambled (data is shown as mean+SD, n≥4, *p<0.05). (B) Representative western
blot showing DRR knockdown following treatment with CD44 mAb-dsDRR. (C)
Quantification of DRR expression following EphA2 mAb-dsDRR treatment. Data were
analyzed using one-way-ANOVA followed by Dunnett’s post hoc test compared to EphA2

42
mAb-dsScrambled (data is shown as mean+SD, n≥3). (D) Representative western blot
showing DRR expression following treatment with EphA2 mAb-dsDRR.
To better understand this discrepancy between the knockdown observed for CD44 mAb-dsDRR
and EphA2 mAb-dsDRR, we examined cellular uptake with patient-derived GSCs. We analyzed
uptake of the conjugates by confocal microscopy of Cy3-labeled dsDRR after a 3h incubation
with the mAb-dsDRR conjugates at 75 nM and 150 nM (Figure A.4). At 150 nM, we observed
considerable uptake of CD44 mAb-dsDRR (Figure 2.6A) whereas we observed minimal uptake
of both EphA2 mAb-dsDRR (Figure 2.6B) and control, IgG-dsDRR (Figure 2.6C). This suggests
that the lack of knockdown observed for EphA2 mAb-dsDRR is correlated to a reduced cell
uptake of the EphA2 mAb-dsDRR conjugate.
To gain greater insight into the fate of the CD44 mAb-dsDRR conjugate, we incubated the
patient-derived GSCs with the CD44 mAb-dsDRR conjugate and a lysosomal marker for 2 h,
and then incubated the GSCs cells with fresh media for an additional 1 h to allow for complete
internalization. The dsDRR Cy3 signal almost completely co-localized with the lysosomal
marker (Figure 2.6D), suggesting that the majority of the conjugate was trafficked into the
endolysosomal pathway, likely with a small percentage trafficked to the cytoplasm to account for
the gene knockdown observed.

43
Figure 2.6. Antisense oligonucleotides conjugated to CD44 mAb are taken up by GSCs and
trafficked into the endolysosomal pathway. (A) Uptake of CD44 mAb-dsDRR (white
arrows) compared to (B) EphA2 mAb-dsDRR and (C) CTL-dsDRR after 3 h incubation at
37 °C. Control (CTL) is a non-specific human IgG. (D) Colocalization (white arrows) of
CD44 mAb-dsDRR with the lysosomal compartments following a 2 h pulse and 1 h chase.
Cell membrane (wheat germ agglutinin (WGA), magenta); cell nucleus (Hoechst, blue);

44
AON (Cy3, green); lysosome (Dextran647, red). Representative z-stack images shown. All
scale bars are 50 µM.
2.3.6 Cellular morphology following DRR knockdown
DRR knockdown often results in a change in cell phenotype from spindle- to rounder-shaped
cells, reflecting a less invasive morphology.64 While untreated GSCs have a spindle-shaped
morphology, we found that GSCs treated with CD44 mAb-dsDRR exhibit a rounder cell
morphology with fewer, shorter projections and centralized focal adhesions, suggesting reduced
invasive capacity (Figure 2.7A). We quantified this effect by measuring cell area per nucleus,
with actin staining delineating the cell membrane and normalized to an untreated control.
Although the cell area was variable due to the complex cell bundles, we observed a significant
decrease in cell area per nucleus with the CD44 mAb-dsDRR treatment relative to controls
including the CD44 mAb alone, dsDRR alone, and CD44 mAb-dsScrambled (Figure 2.7B).

45
Figure 2.7. GSCs treated with CD44 mAb-dsDRR have a rounder shape, fewer projections,
and centralized focal adhesions relative to the spindle-shaped cells of the control
treatments. (A) Cells treated with CD44 mAb-dsScrambled. (B) Cells treated with CD44
mAb-dsDRR. (C) Cells treated with CD44 mAb alone. (D) Cells treated with dsDRR alone.
Representative z-stack images shown. All scale bars are 50 µM. Cell nucleus (Hoechst,
blue); Actin (Phalloidin Alexa Fluor 488, green). (B) Change in cellular morphology is
quantified as actin area per cell normalized to a no treatment control. Data were analyzed
using one-way-ANOVA compared to CD44 mAb-dsScrambled with Dunnett’s post hoc
correction (data is shown as mean+SD, n≥3, ***p<0.001).

46
2.4 Discussion and conclusions Here we report, for the first time, an antisense oligonucleotide-antibody conjugate constructed
via covalent click conjugation through a non-therapeutic, sense carrier strand and show how each
component of the system is optimized for maximum knockdown of DRR.
The antisense therapeutic strand was optimized for effective knockdown first through fluoro
modification of specific oligonucleotides and then by 5-methylation of the CpG containing
oligonucleotide. FANA modification has been shown to stabilize the especially sensitive
flanking regions of the ASO while an unmodified gap consisting of 9 DNA nucleotides allows
for efficient endonucleolytic ribonuclease H activity.268 We found a trend towards more potent
DRR knockdown with the Gapmer motif compared to the alternating oligonucleotide FANA-
modified Altimer. CpG motifs trigger an innate immune response;245, 269 yet, methylation of the
cytosine residue in strands containing this motif can decrease this immune response, thereby
decreasing off-target effects.246, 270 While the immune response was not examined herein, with an
ultimate view of testing this strategy in vivo, we confirmed the activity of the methylated CpG
therapeutic strand and used it in all of the in vitro studies.
We chose a targeted antibody-mediated approach for antisense oligonucleotide delivery because,
like many polyanions, AONs do not readily cross the cell membrane, especially at low
concentrations.108 To determine the optimal antibody for targeted delivery, first we screened the
GSCs for antigen expression and then for antibody internalization. While CD44, EphA2 and
EGFR are all expressed in many patient-derived glioblastoma stem cells,42, 258, 261, 271 only CD44
and EphA2 were strongly co-expressed with DRR. Notably, EGFR amplification is often lost in
vitro, which correlates with our observations.272 By flow cytometry, it appeared that both CD44
and EphA2 mAbs were internalized by the GSCs; however, the extent of internalization did not
correlate with silencing efficacy. CD44 mAb-dsDRR more effectively knocked down DRR
expression than EphA2 mAb-dsDRR, which was corroborated by greater accumulation of
dsDRR in the GSCs when delivered with CD44 mAb. This may be due to slight differences in
internalization pathway or route of endosomal escape, which are difficult to observe using
established techniques. Our results highlight that the pathway or percentage of receptor
internalization does not necessarily correlate to silencing efficacy, which is consistent with

47
earlier reports;273 however, the increased uptake observed by fluorescence microscopy did
correlate with better knockdown.
To gain insight into the trafficking of the CD44 mAb-dsDRR, we used a pulse-chase experiment
and found that the internalized CD44 mAb-dsDRR colocalized with the lysosomes. While it is
recognized that antibody-oligonucleotide conjugates are typically trafficked into the
endolysosomal pathway,264, 273-276 the DRR knockdown observed demonstrates that some dsDRR
is trafficked into the cytoplasm. While the Cy3 signal associated with the duplex sense strand is
colocalized with the lysosomes, some of the antisense strand may have dissociated from the
sense strand and diffused into the cytoplasm. To further increase the potency of knockdown,
incorporation of moieties to prevent lysosomal accumulation and facilitate early endosomal
escape in order to optimize knockdown are being actively pursued.275
We show the first example of an antibody-antisense therapeutic conjugate used to modulate the
genetic expression of cancer stem cells. With sufficient dsDRR uptake, we observe both gene
knockdown and a change in cell morphology, consistent with a less invasive phenotype: we
observe a clear distinction from spindle-shaped cells without treatment to a more rounded
morphology with CD44 mAb-dsDRR treatment. This lays the foundation for future studies
where the goal will be to knockdown the DRR gene in GSCs in vivo and thereby reduce tissue
invasion and cell metastasis.
2.5 Materials and methods
2.5.1 Cell lines
GBM DRR+ cells (U-251 MG glioblastoma cells with stable transfection of DsredDRR fusion
protein) were cultured as previously described.64 Patient-derived GSCs were provided by the
Petrecca lab at McGill University following consent from the patients and approval by the
hospital ethics committee. GSCs were expanded as neurosphere in complete neurocult-
proliferation media (Neurocult NS-A Proliferation kit (Stem Cell 05751), 20ng/ml recombinant
EGF, 20ng/ml recombinant bFGF, and 2µg/ml heparin).

48
2.5.2 Antibodies
CD44 and EphA2 mAbs were provided by the Toronto Recombinant Antibody Centre
(TRAC).266 EGFR mAb (cetuximab) is a clinically available formulation (Erbitux 2 mg/mL, Eli
Lilly). CTL antibody (IgG from human serum) was purchased from Sigma and used as received
(Cat. No. I4506).
2.5.3 AON synthesis
DNA amidites and gene machine compatible reagents were purchased from ChemGenes and
used as received, and all DBCO-TEG (Cat No 10-1941) and Cyanine 3 CPG (Cat No 20-5913-
41) were purchased from GlenResearch. All antisense oligonucleotides were synthesized on an
Applied Biosystems (ABI) 3400 DNA synthesizer at 1 µmol scale using Uny-linker CPG as solid
support, except for the sense delivery oligonucleotide which utilized Cyanine 3 CPG as the solid
support. The synthesis cycle conditions were as previously described239 with the exception that
0.1 M I2 in 1:2:10 pyridine/water/THF was used for oxidation of antisense oligonucleotides, and
0.02 M I2 in 1:2:10 pyridine/water/THF was used for the Cy3/DBCO modified sense delivery
oligonucleotides. When a phosphorothioate backbone was needed, a 0.10 M solution of
((Dimethylamino-methylidene)amino)-3H-1,2,4-dithiozaoline-3-thione (DDTT) in Py:MeCN
(9:1) was used for the oxidation step instead of the aqueous I2 solution. Deprotection and
cleavage from the solid support was achieved in pure aqueous NH4OH for 48 h at room
temperature. Purifications were performed by HPLC, using a Protein Pak DEAE 5PW analytical
anion-exchange column. A stationary phase of Milli-Q water and a mobile phase of 1 M LiClO4
in water was used for analysis and purification using a gradient of 0−50% over 46 min.
Following purification, excess LiClO4 salts were removed using NAP-25 sephadex size-
exclusion columns. The oligonucleotides were analyzed by LC-MS using a Dionex Ultimate
3000 coupled to a Bruker Maxis Impact QTOF in negative ESI mode. Samples were run through
an Acclaim RSLC 120 C18 column (2.2 uM 120A 2.1 x 50 mm) using a gradient of 98% mobile
phase A (100 mM HFIP and 5 mM TEA in H2O) and 2 % mobile phase B (MeOH) to 40 %
mobile phase A and 60% mobile phase B in 8 minutes. The data was processed and
deconvoluted using the Bruker DataAnalysis software version 4.1 (Table S1).

49
2.5.4 AON duplex formation
Sense and antisense oligonucleotide strands were annealed in annealing buffer (10 mM Tris, pH
7.5–8.0, 50 mM NaCl, 1 mM EDTA) by heating at 95 °C for 2 minutes followed by slow cooling
to room temperature over 1 h. The annealed oligonucleotides were stored at 4 °C until use.
Duplex formation was assessed by native PAGE in Tris/Borate/EDTA buffer followed by
imaging on a Typhoon FLA 9500 biomolecular imager.
2.5.5 Preparation of mAb-dsDRR and mAb-dsScrambled conjugates
mAbs were modified with NHS-PEG4-N3 (Thermo 26130) according to the provided protocol
from Thermo Fisher Scientific. Briefly, a 100 mM stock solution of NHS-PEG4-N3 was prepared
in dimethyl sulfoxide (DMSO, Sigma 472301). 10 eq. of this solution was added to the mAb in
PBS (Sigma D8537) and this reaction was shaken for 1 h at RT. After purification by dialysis for
24 h against PBS, 2 eq. of dsDRR or dsScrambled was added to the N3-modified mAb and this
reaction was allowed to proceed for 3 h at 37 oC. The resulting product was stored at -80 oC until
use or at 4 oC for up to 24 h. Conjugation efficiency was analyzed by 10% PAGE.
2.5.6 DRR knockdown assays - lipofectamine2000 transfection protocol
This protocol was adapted from Anzahaee et al.239 Briefly, DRR+ GBM cells were seeded at
120,000 cells/well in a 6-well plate and allowed to adhere overnight. Antisense oligonucleotides
(AONs) were complexed to lipofectamine2000 (Thermo 11668027, at 3 µL/well) for 20 minutes
in Opti-MEM media (Thermo 31985062) according to the lipofectamine2000 reagent protocol
and added to the cells in DMEM media (11995605) for a final DMEM:Opti-MEM ratio of
1.5:0.5 and AON concentration of 25 nM. At 24 h, an additional 1 mL DMEM media was added.
After a total of 72 hours of incubation at 37 oC, cells were collected and lysed using 0.1% NP-40
(Fluka 74385). Protein expression was assessed via western blot analysis of DRR with beta-
tubulin as a loading control.
2.5.7 DRR knockdown assays - treatment with mAb conjugate
GSCs were seeded at 250,000 cells/well in poly(l-ornithine), PLO (Sigma P4957) and laminin
(Thermo CB-40232) coated 6-well plates and allowed to adhere overnight in Neurocult NS-A

50
proliferation media (Stem Cell 05751). mAb conjugates (or AONs or mAb alone) were added to
500 uL Opti-MEM media and added to the cells for a final Opti-MEM:Neurocult ratio of 1.5:0.5
v/v and mAb-AON concentration of 150 nM. 24 hours later, an additional 1 mL Neurocult media
was added. After a total of 72 hours of incubation at 37 °C, cells were collected and lysed using
0.1% NP-40 (add company info). Protein expression was assessed via western blot analysis of
DRR with alpha-tubulin as a loading control.
2.5.8 mAb internalization assay
This protocol was adapted from Mumper et al.277 Briefly, the internalization of the CD44 and
EphA2 mAbs were determined after measuring surface levels of mAb after various incubation
periods at 37 °C. CD44 mAb at 4 µg/mL in 2% FBS (Sigma F1051) in PBS (FACS buffer) was
added to an equivalent volume of 1x106 cells in FACS buffer for one hour on ice at 4 °C. The
cells were then washed 3X in ice cold FACS buffer and suspended in 200 µL at 37 °C for the
indicated time. A control sample was held at 4 °C throughout the experiment. At the indicated
time point, cells were removed from 37 °C, quenched with 1 mL ice cold FACS buffer, and then
spun to a pellet and resuspended in 2 µg/mL goat anti-human IgG, Alexa Fluor 488 (Thermo A-
11013) in FACS buffer. Cells were washed 2X with ice cold FACS buffer, 1X with ice cold
PBS, and then fixed with 2% PFA (Bioshop PAR070) in PBS and stored at 4 °C until analysis on
a BD Accuri C6 flow cytometer. Internalized receptor was quantified using the median
fluorescence intensity calculated from FlowJo 10 software.
2.5.9 Immunocytochemistry (CD44, EphA2, and DRR)
GSCs were seeded at 20,000 cells/well in a PLO/laminin coated 8-well coverglass plate and
allowed to adhere overnight at 37 °C. The cells were then fixed using 4% PFA. Cells were
permeabilized using 1% Triton X-100 (Sigma T-9284) and then stained with either Epha2 mAb
or CD44 mAb together with an DRR antibody provided by the Petrecca lab at McGill University.
Secondary antibodies were anti-human IgG-Alexa Fluor 488 and goat anti-rabbit IgG-Alexa
Fluor 647 (Thermo A-21245). The cells were then counter-stained with Hoechst 33342 and
images were captured on an Olympus FV1000 confocal microscope.

51
2.5.10 Cellular uptake
GSCs were seeded at 20,000 cells/well in a PLO/laminin coated 8-well coverglass plate and
allowed to adhere overnight at 37 °C. The cells were then treated with the indicated formulation
for 3 h at 37 °C, washed with PBS, and fixed using 4% PFA. The cells were stained with WGA-
Alexa Fluor 488 or 647 (Thermo W11261 or W32466), counterstained with Hoechst (Invitrogen
H1399), and imaged on an Olympus FV1000 confocal microscope.
2.5.11 Lysosomal accumulation
GSCs were seeded at 20,000 cells/well in a PLO/laminin coated 8-well coverglass plate and
allowed to adhere overnight at 37 °C. The cells were then treated with the CD44 mAb-dsDRR at
150 nM and the lysosomal marker Dextran Alexa Fluor 647 (Thermo D22914) at 25 µg/mL for 2
h at 37 °C (pulse). The media was then replaced and the cells were incubated for an additional 1
h at 37 °C (chase), then washed with PBS and fixed using 4% PFA. The cells were then stained
with WGA Alexa Fluor 488, counterstained with Hoechst, and images were captured on an
Olympus FV1000 confocal microscope.
2.5.12 Cellular morphology following DRR knockdown
GSCs were seeded at 15,000 cells/well in a PLO/laminin coated 8-well coverglass plate and
allowed to adhere overnight at 37 °C. The cells were then treated with the indicated formulation
for 72 h at 37 °C, washed with PBS, and fixed using 4% PFA. The cells were then stained with
mouse anti-vinculin antibody (Sigma V9131) and anti-mouse Alexa Fluor 647 (Thermo A-
21236) antibodies, followed by Alexa Fluor 488 Phalloidin (Thermo A-12379) to label actin
filaments. The cells were counter-stained with Hoechst and images were captured on an
Olympus FV1000 confocal microscope.
2.6 Acknowledgments
We are grateful to the Canadian Institute for Health Research (CHRP to MSS, MJD, KP) and the
Natural Sciences & Engineering Council of Canada (CHRP to MSS, MJD, KP and a CGSD to
AEA). We thank Professors Dev Sidhu and Jason Moffat and Dr. Jarrett Adams of the TRAC
facility for thoughtful discussions and providing us with CD44 mAbs and EphA2 mAbs. We

52
thank members of the Shoichet, Damha, and Petrecca labs for thoughtful review of this
manuscript.
Attenuated diphtheria toxin mediates siRNA delivery
This chapter was submitted for publication in Science Advances:
Arnold, A. E.; Smith, L. J.; Beilhartz, G.; Bahlmann, L. C.; Jameson, E.; Melnyk, R.; Shoichet,
M. S. Attenuated diphtheria toxin mediates siRNA delivery. Sci Adv (in submission).
A.E.A. devised and performed experiments and drafted the initial manuscript. L.J.S. synthesized
materials, performed experiments, and revised the manuscript. G.B. and R.M. provided
materials, assisted in conceptualization and experimental design, and revised the manuscript.
L.C.B. contributed to conceptualization and experimental design and revised the manuscript. E.J.
assisted in performing experiments. M.S.S. provided guidance and supervision in
conceptualization and experimental design and assisted in writing the manuscript.
3.1 Abstract
Toxins can act as sophisticated delivery vehicles to cells by binding to cell surface ligands,
inducing endocytosis, and quickly escaping the endolysosomal pathway to release their cargo
directly into the cytoplasm. To take advantage of this mechanism without the inherent toxicity of
toxins, we engineered conjugates between an attenuated diphtheria toxin and siRNAs to achieve
gene downregulation in glioblastoma cells. Since glioblastoma is highly invasive, we delivered
siRNA against integrin-ß1 (ITGB1) - a gene that promotes invasion and metastasis - and siRNA
against eIF-3b - a survival gene. We demonstrated mRNA downregulation of both genes in
patient-derived glioblastoma cells and the relevant associated functional outcomes: knockdown
of ITGB1 led to a significant inhibition in the invasive behavior of the glioblastoma cells using
an innovative 3D hydrogel model; and knockdown of eIF-3b resulted in significant cell death.
This is the first example of diphtheria toxin being used to deliver siRNAs, and the first time a
toxin-based siRNA delivery strategy has been shown to induce relevant genotypic and
phenotypic effects in cancer cells.

53
3.2 Introduction
Pathogens produced by bacteria have evolved over millions of years to achieve highly
sophisticated mechanisms of penetrating cells, responding to intracellular cues to guide their
trafficking, and to deliver their payload to a desired site of action.278 In particular, “AB” type
toxins, such as anthrax toxin (AT) and diphtheria toxin (DT), bind to the cell surface, are
endocytosed, and then escape the endosomal pathway, and translocate into the cytosol.279 These
toxins are made up of three domains: an active (toxic) domain, “A,” that can be mutated to
attenuate its toxicity for delivery applications (ie. “a”); a translocation domain, “T,” that
facilitates escape from the endosomes; and a receptor binding domain, “R,” that binds a receptor
on the cell surface for targeting and inducing endocytosis (Figure 3.1A).205-206 Taking advantage
of the cell entry mechanism of AB toxins through cargo-toxin fusion constructs has been
explored for delivery of diverse protein cargos including peptides, effector proteins, and
antibody-like proteins.210, 219, 222, 227 There has been only one example of oligonucleotide delivery
with attenuated anthrax toxin reported; however, this preliminary study did not demonstrate any
functional effects of gene knockdown.224
Oligonucleotides, such as small interfering ribonucleic acids (siRNAs), are powerful tools to
regulate gene expression in diseased cells, with the potential to return them to a normal
phenotype.280-281 However, therapeutic siRNA delivery represents a significant clinical challenge
due to the many physiological barriers that must be overcome for efficacy,282 as well as the
toxicity and immunogenicity of many currently used delivery vehicles.111, 283 One of the biggest
barriers to siRNA delivery is entrapment in the endolysosomal pathway after endocytosis,
quickly leading to degradation of the fragile, nuclease-prone siRNA.284-285 Strategies that
enhance the endosomal escape of siRNA have recently attracted much attention.286-288 Many of
these strategies utilize liposomal formulations for the delivery of siRNA; however, the
biodistribution of these nanoparticles is largely limited to clearance organs such as the liver and
kidney.289 To overcome biodistribution challenges, targeting proteins such as antibodies have
been explored to deliver siRNAs to specific tissues with some success;97, 264, 276 however, only a
very small percentage of antibody-siRNA conjugates escape the endosomal pathway, limiting
their efficacy.273 AB toxins, such as DT, have the capacity for both endosomal escape and
specific targeting,290 and are thus attractive candidates for siRNA delivery into the cytoplasm
(Figure 3.1B).

54
Figure 3.1. Using attenuated diphtheria toxin for siRNA delivery. A) Attenuated AB toxins,
such as attenuated diphtheria toxin (aDT), consist of three main components: a receptor
binding domain (R) that binds to a receptor on the cell surface; a translocation domain (T)
that allows for endosomal escape; and a mutated active domain (a) in order for the protein
to retain its trafficking functions but is no longer toxic to cells. Cargo, such as siRNA, can
be attached to this “a” domain. B) siRNA delivered using attenuated DT occurs in five
main steps: 1) binding to the HB-EGF precursor cell surface receptor; 2) endocytosis of the
aDT-siRNA cargo; 3) translocation through the endosomal membrane, inserting the “a”
domain and cargo into the cytoplasm; 4) cleavage of the “a” domain from the rest of the
protein; and 5) release of the siRNA into the cytoplasm where it downregulates the relevant
gene.

55
We explored whether attenuated DT could be used as a delivery vehicle for siRNAs against two
cancer gene targets: (1) eIF-3b, a transcription factor that is overexpressed in many cancers and
inhibition of which slows growth and initiates apoptosis;291 and (2) ITGB1, which is involved in
the formation of focal adhesions and activation of actin rearrangement, the inhibition of which
reduces cellular invasion and metastasis.77, 292 We chose to focus on the delivery of siRNA to
glioblastoma (GBM) cells, and specifically glioblastoma stem cells (GSCs), because: (i) DT has
been reported to cross the blood brain barrier, making it an attractive vehicle against intractable
diseases like GBM;293-294 (ii) the diphtheria toxin receptor, heparin-binding epidermal growth
factor (HBEGF), is widely expressed in the central nervous system295 and is overexpressed in at
least a subset of malignant gliomas;296 (iii) there is a growing body of evidence that the
aggressiveness of glioblastoma is, at least in part, caused by the GSC subpopulation;232, 297 and
(iv), the GSCs are rapidly dividing and highly invasive,258, 298 allowing functional readouts for
both cell viability and invasiveness.
We conjugated an engineered, attenuated DT (aDT) to Dicer-substrate siRNAs against eIF-3b
and ITGB1 using azide-alkyne click chemistry. There are many conjugation strategies available
to conjugate cargo to proteins; however, the essential disulfide bond linking the A and T domains
of diphtheria toxin precluded the use of reducing agents during synthesis of the aDT-siRNA
conjugate.299 The click chemistry approach that we used was efficient, bio-orthogonal, produced
no byproducts,183 and maintained the integrity of the rest of the protein by avoiding the use of
reducing agents. We examined the biological effects of these conjugates by measuring mRNA
expression levels and assessed cell viability and invasiveness, and thus demonstrate this platform
strategy in vitro with both gene knockdown and functional assays in primary human
glioblastoma cells.
3.3 Results
3.3.1 Conjugation of siRNA to aDT
For aDT mediated siRNA delivery, we used an engineered, attenuated diphtheria toxin (aDT)
containing a cysteine for chemical modification, a SUMO tag for increased stability and a His
tag for purification (Figure 3.2A). We modified this aDT-cysteine variant with a maleimide-
modified crosslinker containing a dibenzocyclooctyne (DBCO) as a handle for further chemical
modification (Figure 3.2B). Successful synthesis of aDT-DBCO was demonstrated by
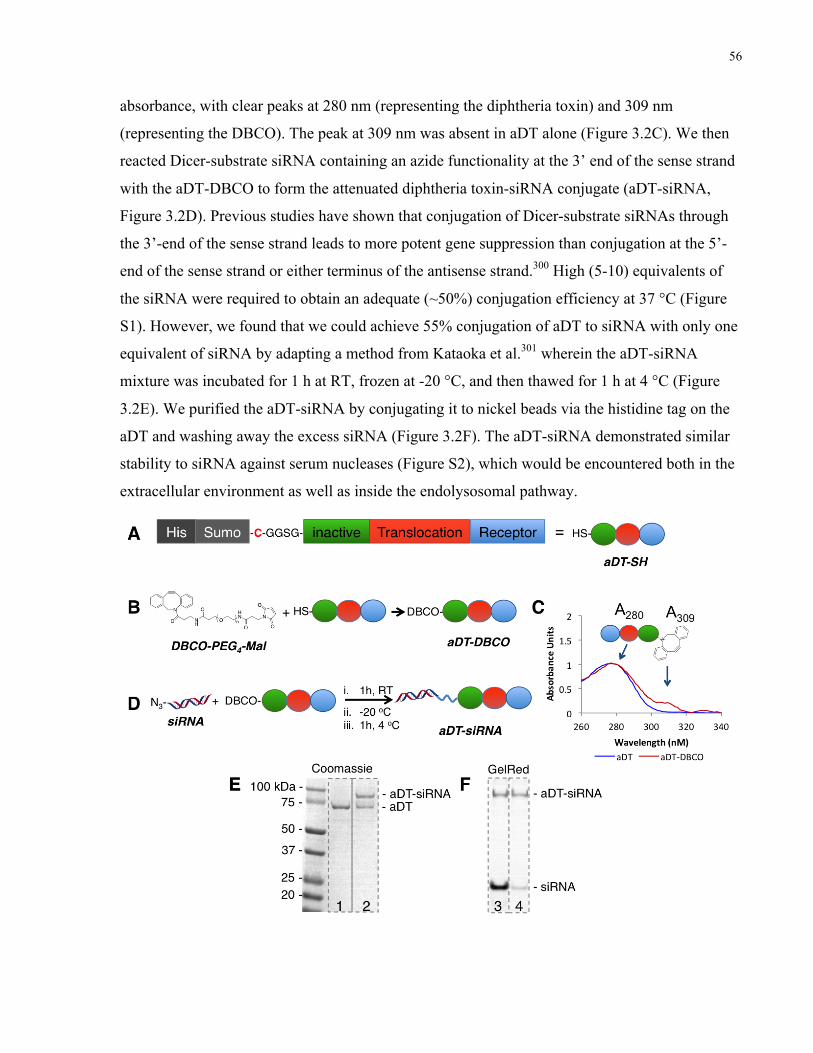
56
absorbance, with clear peaks at 280 nm (representing the diphtheria toxin) and 309 nm
(representing the DBCO). The peak at 309 nm was absent in aDT alone (Figure 3.2C). We then
reacted Dicer-substrate siRNA containing an azide functionality at the 3’ end of the sense strand
with the aDT-DBCO to form the attenuated diphtheria toxin-siRNA conjugate (aDT-siRNA,
Figure 3.2D). Previous studies have shown that conjugation of Dicer-substrate siRNAs through
the 3’-end of the sense strand leads to more potent gene suppression than conjugation at the 5’-
end of the sense strand or either terminus of the antisense strand.300 High (5-10) equivalents of
the siRNA were required to obtain an adequate (~50%) conjugation efficiency at 37 °C (Figure
S1). However, we found that we could achieve 55% conjugation of aDT to siRNA with only one
equivalent of siRNA by adapting a method from Kataoka et al.301 wherein the aDT-siRNA
mixture was incubated for 1 h at RT, frozen at -20 °C, and then thawed for 1 h at 4 °C (Figure
3.2E). We purified the aDT-siRNA by conjugating it to nickel beads via the histidine tag on the
aDT and washing away the excess siRNA (Figure 3.2F). The aDT-siRNA demonstrated similar
stability to siRNA against serum nucleases (Figure S2), which would be encountered both in the
extracellular environment as well as inside the endolysosomal pathway.

57
Figure 3.2. siRNAs can be conjugated to attenuated diphtheria toxin. A) Attenuated
diphtheria toxin was engineered to contain a free cysteine as a functional handle (aDT-SH),
protected by a SUMO tag and purified using a histidine (His) tag. B) Attenuated diphtheria
toxin was reacted with a PEG crosslinker containing both maleimide and DBCO functional
groups to obtain DBCO-modified attenuated diphtheria toxin (aDT-DBCO). C) The
presence of the DBCO modification on attenuated diphtheria toxin was confirmed by
reading the absorbance at 280 nm (aDT) and 309 nm (DBCO). Curves shown are aDT
before modification (blue) and after DBCO modification (red). D) Azide-modified siRNA
was reacted with the DBCO-functionalized attenuated diphtheria toxin to obtain the aDT-
siRNA conjugate. E) Modification of the aDT with the siRNA was confirmed via PAGE
stained with Coomassie blue to localize the diphtheria toxin protein. Lane 1 shows the aDT-
DBCO starting material (MW ~ 72 kDa) and lane 2 shows the aDT-siRNA conjugate
(MW~90 kDa) alone with some unreacted starting material. F) Purification of the excess
siRNA was confirmed via PAGE stained with GelRed to localize the siRNA. Lane 3 shows
the aDT-siRNA conjugate along with unreacted siRNA; lane 4 shows the aDT-siRNA
conjugate after nickel column purification, with only a small amount of excess siRNA left
over.
3.3.2 Glioblastoma stem cells (GSCs) express heparin-binding epidermal growth factor (HBEGF)
The GSCs used in this study were patient-derived glioblastoma cells grown in stem-cell
conditions, as previously described.302 We verified that aDT was a good candidate for targeting
and internalization in GSCs by both staining the cells for the diphtheria toxin receptor, HBEGF,
which was abundantly evident (green staining in Figure 3.3), and measuring the IC50 of the
native (toxic) diphtheria toxin against these cells to ensure the correct mechanism of action. The
IC50 of the native diphtheria toxin in the GSCs was 6.0 ± 1.0 pM (Figure S3), indicating high
sensitivity to the toxin and a conserved internalization and translocation mechanism therein. All
further experiments were conducted with a non-native, attenuated diphtheria toxin with an
insignificant level of toxicity as previously reported.210

58
Figure 3.3. GSCs express HB-EGF, the native receptor for diphtheria toxin. Representative
confocal images are shown for HB-EGF (anti-HBEGF antibody, green) and nucleus
(Hoechst, blue). The secondary antibody only control confirms lack of non-specific binding.
All scale bars are 50 µm.
3.3.3 aDT-siRNA conjugate downregulates ITGB1 and reduces cellular
invasion
We conjugated attenuated aDT to Dicer-substrate siRNA against two relevant gene targets:
ITGB1 (to make aDT-ITGB1) and eIF-3b (to make aDT-eIF-3b). We treated the GSCs with the
aDT-ITGB1 conjugate and observed a significant reduction in the target mRNA compared to
negative controls of siRNA only (without lipofectamine) and aDT conjugated to a non-targeting
siRNA (aDT-NT) at 50 nM (Figure 3.4A). To ascertain whether the reduction in ITGB1
expression would correspond to a phenotype of either reduced invasion or adhesion, we seeded
the cells on top of a crosslinked hyaluronic-acid based 3D hydrogel into which the cells normally
invade (Figure 3.4B).303 Impressively, we observed a striking decrease in invasiveness in aDT-
ITGB1-treated cells compared to untreated or aDT-NT controls (Figure 3.4C, D). To determine
whether cell adhesion influenced these results, we pre-treated the cells cultured in 2D tissue
culture polystyrene flasks with both aDT-ITGB1 and aDT-NT prior to plating them on the 3D
hydrogels. After several wash steps, we observed no significant difference in the number of
adhered cells between any of the treatment groups, demonstrating that cell adhesion did not
impact the reduced cell invasion observed with aDT-ITGB1 treatment (Figure 3.4E, F).

59
Figure 3.4. aDT-siRNA downregulates ITGB1 expression in GSCs and reduces cellular
invasion. A) aDT-ITGB1 (light red bars) downregulates ITGB1 mRNA expression
compared to negative controls: aDT conjugated to a non-targeting siRNA (aDT-NT, black

60
bars) and ITGB1 siRNA only without lipofectamine (blue bars) at 24 h post treatment.
Positive control is transfected ITGB1-siRNA with lipofectamine (dark red bars). Data is
shown as n=3, mean±SD, normalized to an untreated control. Data was analyzed using one-
way-ANOVA followed by Tukey’s correction on the logarithmic data (* p<0.05, ** p<0.01).
B) Cells were plated in a 3D hydrogel assay on the surface of pre-formed hydrogels and
treated with aDT-ITGB1 conjugates at the beginning of the experiment. Invasion depth
was measured after 5 days. C) aDT-ITGB1 reduces invasion compared to controls (no
treatment and aDT-NT) in a 3D hydrogel model. Representative images shown. 15 µm red
beads label the top of the hydrogel; blue cell nuclei are labeled using Hoechst. All scale bars
are 150 µm. D) Invasion depth was quantified as a percentage of the untreated control.
Data was analyzed using one-way-ANOVA followed by Tukey’s correction (* p<0.05, **
p<0.01). E) aDT-ITGB1 did not reduce number of adhered cells in a 3D hydrogel model.
Representative images are shown. All scale bars are 150 µm. F) Number of adherent cells
was quantified by counting number of cell nuclei; no significant difference was observed,
demonstrating that differences in invasion were due to ITGB1 downregulation. Data was
analyzed using one-way-ANOVA followed by Tukey’s correction.
3.3.4 aDT-siRNA conjugate downregulates eIF-3b and reduces cell viability
aDT-eIF-3b treated glioblastoma cells exhibited significant downregulation in the target mRNA
compared to the relevant negative controls of siRNA only controls (without lipofectamine) and
aDT-NT at 50 nM (Figure 3.5A). We confirmed that the eIF-3b siRNA could reduce cell
viability in the GSCs by complexing eIF-3b siRNA and NT siRNA with a commercially
available transfection reagent and observing a significant difference in cell viability with eIF-3b
siRNA-mediated knockdown (Figure S4). Furthermore, we observed a significant (albeit modest)
reduction of cell viability with an aDT-eIF-3b concentration of 100 nM (Figure 3.5B).
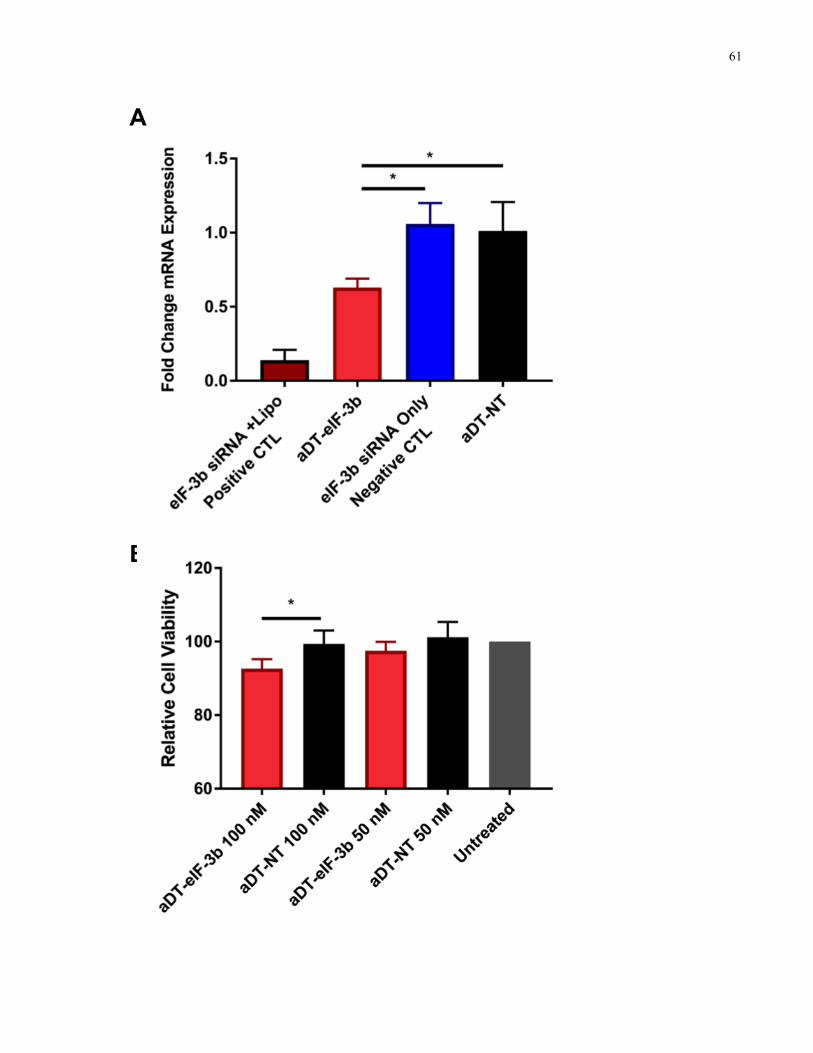
61

62
Figure 3.5. aDT-siRNA downregulates eIF-3b expression in GSCs and reduces cell
viability. A) aDT-eIF-3b (light red bars) downregulates eIF-3b mRNA expression
compared to negative controls: aDT conjugated to a non-targeting siRNA (aDT-NT, black
bars) and eIF-3b siRNA only without lipofectamine (blue bars) at 24 h post treatment.
Positive control is transfected siRNA with lipofectamine, dark red bars. Data is shown as
n=3, mean±SD, normalized to an untreated control. Data was analyzed using one-way-
ANOVA followed by Tukey’s correction on the logarithmic data (* p<0.05). B) aDT-eIF-3b
(red bars) reduces cell viability of GSCs at 48 h post treatment compared to aDT-NT
(black bars) at 100 nM. Data is shown as n=3, mean+SD, normalized to an untreated
control. Data was analyzed using one-way-ANOVA followed by Tukey’s correction (*
p<0.05).
3.4 Discussion and conclusions
We show the first example of siRNA delivery with an attenuated diphtheria toxin. Exploiting the
sophisticated trafficking mechanism of proteins, such as that of attenuated diphtheria toxin, has
the potential to overcome many of the barriers to siRNA delivery. Using our aDT-siRNA
conjugate, we were able to downregulate two distinct genetic targets and observe significant
changes in cell proliferation and invasion, supporting the robust nature of aDT-mediated drug
delivery and its potential as a novel treatment strategy for aggressive cancers such as
glioblastoma. Attenuated diphtheria toxin is an advantageous siRNA delivery vehicle for
glioblastoma treatment for two main reasons: (1) targeting via the receptor binding domain and
(2) endosomal escape via the translocation domain. HBEGF proved to be a good target for
siRNA delivery as it is expressed in the GSCs that are used in this study and widely expressed in
brain tissue.295 It is essential to develop strategies that can target the GSCs in addition to the bulk
tumor as GSCs may be responsible for tumor recurrence.304
Endosomal escape is important for the delivery of siRNAs in order to avoid trafficking into the
late endosomes/lysosomes and subsequent degradation by nucleases.305-306 Attenuated diphtheria
toxin has been reported to translocate out of the endolysosomal pathway at a relatively early
endosomal stage (pH ~6.5), protecting its cargo from the harsh degradation environment of the
lysosomes.307-308 The attenuated diphtheria toxin and its cargo translocate through a flexible α-
helical pore,210 making endosomal escape difficult to assay directly because most current
methods measure widespread membrane leakage.309-310 However, the gene knockdown and

63
subsequent phenotypic effects of siRNA delivery that we observed confirmed successful aDT-
mediated translocation, as previous work has shown that without translocation and endosomal
escape the aDT cannot deliver cargo.210, 311 We confirmed the robust nature of the aDT-siRNA
system by downregulating two genes of interest in the target cells. In comparison, in systems that
do not have inherent endosomal escape capabilities, such as antibody-siRNA conjugates,
delivery has been shown to be highly target dependent.201 While an anthrax toxin was used for
siRNA delivery previously, knockdown of only a proof-of-concept target was shown without
downstream functional effects.224 Using our aDT conjugate system, we observed phenotypic
changes in both cell invasion and growth.
Diffuse tumor cell infiltration is a hallmark of GBM, and recent findings have suggested GSCs
play important roles in migration and invasion.298, 312 It has been reported that inhibiting GSC
invasion is essential to slowing the progression of GBM.313 ITGB1 is involved in cellular
binding to many extracellular matrix components, including fibronectin,72 and ITGB1
knockdown has been shown to reduce invasion in cancer cells.314 Thus, we hypothesized that
ITGB1 knockdown would reduce the invasive behavior of GSCs. We demonstrated the
effectiveness of the aDT-ITGB1 conjugate in reducing of cell invasion using a previously
validated 3D hydrogel platform,303 confirming a functional effect of siRNA-mediated
knockdown. Moreover, as GSCs have also been shown to drive tumor growth,41 we also were
interested in using our treatment strategy to reduce cellular proliferation. We employed an
siRNA targeted against eIF-3b, which is known to reduce protein synthesis leading to a reduction
in cell growth and viability.315 Using the aDT-eIF-3b conjugate, we successfully reduced eIF-3b
expression and observed a corresponding change in cell viability. Thus, we demonstrated
functional effects of gene knockdown in two different pathways, indicating that aDT-siRNA can
be considered a platform strategy.
For the first time, we demonstrated a robust platform for siRNA delivery to glioblastoma stem
cells using an attenuated diphtheria toxin with an inherent endosomal escape mechanism. In
future work, this conjugate will be used for siRNA delivery in vivo to orthotopic models of
glioblastoma. Further extensions of this work could include incorporation of chemically
stabilized siRNAs89 and combination with chemotherapeutics in order to develop novel
treatments for a broad range of cancers and other diseases.

64
3.5 Materials and methods
3.5.1 Cell lines Patient-derived glioma neural stem cells (GNS 411) were a gift from the lab of Dr. Peter Dirks,
with Research Ethics Board approval at the Hospital for Sick Children, Toronto, and the
University of Toronto. These cells are referred to as glioblastoma stem cells (GSCs) throughout
the manuscript. Cells were maintained in an incubator (37 °C, 5% CO2, 95% humidity) grown in
Corning Primaria flasks (Corning 353808) coated with poly-L-ornithine (PLO, Sigma-Aldrich
P4957) and laminin (Sigma-Aldrich 11243217001). GSC growth media contained serum-free
NeuroCult NS-A Basal Media (StemCell Technologies 05750) supplemented with N2, B27,
EGF, FGF, and heparin as previously described for neural stem cells.316
3.5.2 Attenuated diphtheria toxin Attenuated diphtheria toxin (aDT) was expressed as previously described.210 Briefly, aDT was
expressed in E. coli BL21(DE3) cells, induced with 1 mM isopropyl-β-d-1-thiogalactopyranoside
(IPTG) for 4 h at 37 °C using the Champion pet-SUMO expression system (Invitrogen). Cells
were harvested by centrifugation, resuspended in lysis buffer (20 mM Tris-HCl pH 8.0, 0.5 M
NaCl, 20 mM imidazole, benzonase, lysozyme, and protease inhibitor cocktail) and lysed by an
EmulsiFlex C3 microfluidizer (Avestin) at 15,000 psi. The lysates were centrifuged at 18,000
× g for 20 min. The histidine-tagged DT was purified by Nickel affinity chromatography using a
His-Trap FF column (GE-Healthcare).
3.5.3 siRNAs Dicer-substrate siRNAs were purchased as annealed duplexes from Integrated DNA
Technologies. All siRNAs were suspended at a concentration of ~50 nM using nuclease free
duplex buffer (Dharmacon B-002000-UB-100). Concentrations were verified by measuring the
absorbance at 260 nm. Sequences of Dicer-substrate siRNAs used were as follows: (eIF-3b
sense) 5’-rGrG rArUrA rCrGrC rUrUrA rGrCrA rUrCrU rArUrG rArArA CT-Azide-3’; (eIF-3b
antisense) 5’-rArG rUrUrU rCrArU rArGrA rUrGrC rUrArA rGrCrG rUrArU rCrCrA rG-3’;
(ITGB1 sense) 5’-rGrA rCrUrG rUrUrC rUrUrU rGrGrA rUrArC rUrArG rUrArC TT-Azide-3’;
(ITGB1 antisense) rArA rGrUrA rCrUrA rGrUrA rUrCrC rArArA rGrArA rCrArG rUrCrA rC-
3’.

65
3.5.4 PCR primers Primers used were purchased from ACGT Corp. for eIF-3b317 (forward: 5’-
TGTGAAAGGTACCTGGTGAC-3’ and reverse: 5’-AATAGGCCAATGGGCTGAG-3’) and
for ITGB1318 (forward: 5’- GAAAACAGCGCATATCTGGAAATT-3’ and reverse: 5’-
CAGCCAATCAGTGATCCACAA-3’).
3.5.5 Immunocytochemistry (HBEGF) GSCs were plated to reach 60-80% confluency on chambered coverglass slides coated with poly-
L-ornithine (PLO) and laminin. Cells were then fixed with 4% PFA and stained using an
antibody specific for HBEGF (Abcam, ab66792) followed by a fluorescently labeled secondary
antibody (Thermo Fisher A-11001). The cells were then counter-stained with Hoechst 33342 and
images were captured on an Olympus FV1000 confocal microscope.
3.5.6 Preparation of aDT-siRNA conjugate Attenuated diphtheria toxin was modified with a dibenzocyclooctyne-PEG4-maleimide
crosslinker (DBCO-PEG4-Mal, Sigma-Aldrich 760676) by adding 4 equivalents of a 10 mM
solution in DMSO of the DBCO-PEG4-Mal crosslinker to a ~10 µM solution of the protein in 20
mM Tris, 150 mM NaCl, 5% glycerol, pH 7.5 and incubating at RT for 30 min. The aDT-DBCO
conjugate was then purified by dialysis against Tris pH 7.5, 1% glycerol for 48 h to remove any
excess crosslinker. The aDT-DBCO conjugate was characterized by absorbance to confirm a
presence of a peak at 309 nm representing the incorporation of the DBCO moiety (75% yield).
The aDT-DBCO conjugate was then mixed with an azide-containing siRNA at a 1:1 ratio,
followed by a 1 h incubation at RT, overnight incubation at -20 °C, and 1 h incubation at 4 °C.
The conjugate was then bound to nickel beads for 1 h at 4 °C, eluted from the beads in 500 mM
imidazole, and buffer exchanged into Tris pH 7.5, 1% glycerol. Successful conjugation was
confirmed by PAGE analysis and conjugation efficiency was quantified using imageJ software
(45% total yield; 80% recovery of DT, 55% siRNA conjugation efficiency).
3.5.7 Gene knockdown assays - treatment with aDT-siRNA conjugate aDT-siRNA was mixed with Opti-MEM for a final concentration of 50 nM and added to 24-well
Primaria plates coated with PLO/laminin. Cells were then added to the plates with the conjugate
at a density of 50,000 cells per well. 24 hours following treatment, cells were collected and

66
lysed. RNA was purified from the cells and gene expression was analyzed via quantitative RT-
PCR (qPCR).
3.5.8 Gene knockdown assays - positive control treatment with Lipofectamine RNAiMAX
Transfection complexes with Lipofectamine RNAiMAX (Thermo Fisher 13778075) were
prepared according to the manufacturer’s protocol in a reverse transfection procedure. Briefly,
siRNA and Lipofectamine were mixed in Opti-MEM serum reduced media (Thermo Fisher
31985062) and added to 24-well Primaria plates (Corning 353847) coated with PLO/laminin.
Cells were then added to the plates with the treatment at a density of 150,000 cells per well. 24
hours following transfection cells were collected and lysed. RNA was purified from the cells and
gene expression was analyzed via qPCR.
3.5.9 Invasion assay Hydrogels were synthesized as previously described by Tam et al.7 Cells were plated on
hydrogels at a density of 3500 cells/hydrogel and allowed to adhere for 24 h. Cells were then
treated with aDT-ITGB1 conjugates at a concentration of approximately 50 nM. 48 h after
treatment, fresh media was added to each well and cells were fixed with 4% PFA 4-5 days after
treatment. Cells were stained with Hoechst to identify cell nuclei and 15 µm fluorescent beads
were added to each well to label the surface of the hydrogel. Hydrogels were imaged using
confocal microscopy and depth of invasion was analyzed using a custom MATLAB script.
3.5.10 Adhesion assay aDT-siRNA was mixed with Opti-MEM for a final concentration of 50 nM and added to 24-well
Primaria plates coated with PLO/laminin. Cells were then added to the plates with the conjugate
at a density of 50,000 cells per well. 24 h after treatment, cells were lifted off the 2D plates using
accutase and re-plated on hydrogels at a density of 15,000 cells per hydrogel. 1 h after re-plating
on the hydrogel, cells were washed 5 times with PBS and adhered cells were fixed with 4% PFA,
stained with Hoechst and imaged using confocal microscopy. The number of adhered cells was
quantified using custom computational software.
3.5.11 Cell viability assays following treatment with aDT-eIF-3b aDT-eIF-3b was mixed with Opti-MEM for the desired final concentration and then added to 96-
well plates coated with PLO/laminin. Cells were then added to the plates with the conjugate at a

67
density of 8,000 cells per well. Fresh media was added to each well 24 h following treatment. At
48 h, cell viability was measured via PrestoBlue viability assay and quantified as a percentage of
untreated controls.
3.6 Acknowledgements
We thank the lab of Dr. Peter Dirks (Hospital for Sick Children, Toronto) for providing the
patient-derived glioblastoma stem cells. We thank Professor Masad J. Damha (McGill
University) for thoughtful advice on siRNA chemistry. We thank members of the Shoichet and
Melnyk labs for thoughtful review of this manuscript. We are grateful to the Canadian Institute
for Health Research (CHRP and Foundation to MSS) and the Natural Sciences & Engineering
Council of Canada (CHRP and Discovery to MSS, CGSD to AEA and LCB, PGSD to LJS).
MSS also acknowledges salary support from the Canada Research Chairs program.
Thesis discussion
The promise of antisense therapeutics is the reversal of disease symptoms through controlling
cellular behavior a genetic level. This could include phenotypes including reduced cellular
invasion, increased apoptosis, or lower resistance to chemotherapeutic treatment. However,
despite recent advances in siRNA/AON delivery, including several clinical approvals, many
formulations do not target specific tissues and struggle to escape degradation in the
endolysosomal pathway, limiting their efficacy.
In addition, it is essential to target the right cell population: there is growing recognition in the
field that the successful treatment of cancer requires the obliteration of not just the bulk tumor
but also the CSCs, which are more resistant to conventional treatment methods. In particular,
GBM is a highly recurrent and aggressive tumor even after resection of tumor tissue,
chemotherapy, and radiotherapy, emphasizing the need for a therapy that targets the GSC
population.
This thesis attempts to address two main challenges in the field of cancer treatment: the delivery
of antisense therapeutics and the targeting of GSCs. First, we explored targeted delivery of
AONs to GSCs by using antibodies against specific cell surface receptors of the GSCs, including
CD44 and EphA2. For this work, we selected a relevant target for knockdown, DRR, which has

68
implications in the survival, proliferation, and invasion of GSCs. Appendix F describes an initial
attempt to achieve targeted knockdown of DRR by functionalizing a nanoparticle with antibody
and oligonucleotide pendant groups; however, we quickly realized this approach would not be
successful, and so pivoted to utilizing simpler antibody-oligonucleotide conjugates. Using
antibody bioconjugates as delivery vehicles, we were able to deliver AONs to cells, observe
uptake and knockdown, and see a corresponding change in the morphology of the cells. This
represented the first time that an antibody-oligonucleotide conjugate had been used for delivery
to GSCs. We were limited by a high amount of uptake of the antibody-AON formulation into the
endolysosomal pathway, and wanted to use a delivery vehicle with an inherent endosomal escape
mechanism, which led us to next explore using DT as a delivery vehicle for oligonucleotides. We
conjugated DT to highly potent siRNAs to modulate GSC phenotypes, including siRNAs against
eIF-3b, a target for cell proliferation, and against ITGB1, a target for cell invasion. Using the DT
as a delivery vehicle, we observed efficient knockdown and corresponding changes in
proliferation and invasion phenotypes, demonstrating the potential of DT as a platform
technology for oligonucleotide delivery. In the following discussion we will explore the
importance of these findings to the field of drug delivery.
4.1 The interplay of stability and potency in antisense
therapeutics
Throughout the work presented in this thesis, a variety of AONs and siRNAs were explored for
gene knockdown. Modifications to increase oligonucleotide potency have been extensively
explored,89 and while many of these modifications have been shown to increase potency,239, 319-
320 there is also the possibility that modifications will interfere with RISC loading and mRNA
cleavage by making the duplex too stable or forcing the nucleic acid to adopt a suboptimal
geometry.321-322 In addition, it is widely accepted that AONs are more stable but less potent than
siRNAs,323 but AONs still can be useful for many applications, and continue to progress in the
clinical pipeline.324 Clearly, both stability and potency play important roles in the selection and
overall efficacy of modified oligonucleotides.
In the variety of modification patterns for AONs and siRNAs explored in this thesis, particularly
as described in Chapter 2, Appendix C, and Appendix D, we observed that higher stability of the
oligonucleotides often correlated to lower potency. These differences are most striking in

69
comparing the AONs and siRNAs against DRR. DRR siRNAs are extremely potent for
knockdown, especially the unmodified control siRNA, but this unmodified siRNA is the least
stable strand against nuclease-mediated degradation. The siRNAs against eIF-3b and ITGB1 are
also very potent for knockdown of the target genes, but degrade quickly in the presence of
nucleases. In contrast, the AONs against DRR are the most stable out of the oligonucleotides
tested, but less potent than all of the other siRNA sequences against DRR. As described in
Appendix C and Appendix D, we also observed this trend with other modification patterns of the
DRR siRNA: siRNAs with higher degrees of modification were more stable to nuclease
degradation but less potent. This could be due to sub-optimal design of modification chemistries
and patterns, but the data demonstrates the importance of measuring and optimizing both
stability and potency while considering the impact on the desired functional effect.
The correlation between stability and potency that we observed throughout different aspects of
this thesis should be considered for any future work. The trade-off between stability and potency
for the oligonucleotides used is a major limitation; a possible improvement would be to search
for a pattern of chemical modifications that would stabilize the oligonucleotide while also
enhancing potency. For example, this has been observed with alternating 2’-methoxy and 2’-
fluoro modifications within siRNA sequences.325 Interestingly, Patisiran, the first FDA-approved
formulation of siRNA, employs siRNA that contains a combination of 2’-methoxy-modified and
unmodified ribonucleosides; unfortunately, the exact pattern of modifications is protected.326 In
contrast, the siRNAs used as described in Appendix C and D contained mostly 2’-fluoro and
some 4’-methoxy modifications, which seem to have limited the observed potency. It is known
that small differences in chemical modifications of siRNA can have large effects on the loading
of the siRNA into the RISC complex and therefore negatively impact gene silencing;327
therefore, a larger screen should be performed on ITGB1 or eIF-3b siRNAs to identify
sequences, potentially a combination of 2’-fluoro and 2’-methoxy modifications, that maintain or
increase the potency of silencing. In this way, nuclease stability could be improved without
sacrificing, and perhaps even increasing, potency of gene knockdown. Such a strategy could
dramatically increase the efficacy of the delivery systems discussed in this work and will be
discussed further in section 6.2.2.

70
4.2 The importance of endosomal escape in oligonucleotide
delivery
Endosomal escape is a key challenge in the field of oligonucleotide delivery. The fragile nucleic
acid material must escape the degradation pathway of the endolysosomes before the acidic pH
and the activity of nucleases and other enzymes render the AONs or siRNAs inactive.120
Numerous strategies have been explored to escape the endosomal pathway, many of them
involving incorporation of cationic components.127, 285, 328 While enhancing endosomal escape
can improve transfection efficiency,329-331 most of these strategies lead to increased cytotoxicity
and lack of targeting capacity, limiting biodistribution to a few clearance organs.332 For delivery
methods that rely on “spontaneous” endosomal escape, such as antibody-siRNA conjugates, it is
estimated that only a fraction of one percent of conjugates are released into the cytoplasm.122
We also observed this challenge in our own work: as described in Chapter 2, when we delivered
a fluorescently labeled Ab-AON conjugate to GSCs, a significant amount of material was
determined to be overlapping with the lysosomes, indicating a high degree of degradation was
likely occurring. This is a significant limitation of the system, and agrees with other recent work
in the field of antibody-siRNA conjugates.122 The knockdown we observed at 150 nM of the Ab-
AON conjugate was significant but did not reach the levels of knockdown observed with 25 nM
of transfected AON using a commercially available lipofectamine system. One can assume that if
a larger fraction of the AON or siRNA was escaping the endosomal pathway, it would lead to a
higher degree of knockdown efficiency.
After observing these limitations with the Ab-AON system, we started exploring aDT as a
delivery system for antisense therapeutics as described in Chapter 3, and the toxin has the
advantage of an inherent endosomal escape mechanism.205 We also moved to using siRNAs
instead of AONs as we wanted to deliver a more potent therapeutic to give a large therapeutic
window for observing significant knockdown. Previous literature has shown that without
translocation and endosomal escape of the aDT, its delivery efficiency is decreased at least 1000-
fold,210 so we knew that if we observed any knockdown by the siRNA, it would indicate that the
translocation of the siRNA had occurred. We were excited to explore the aDT as an
oligonucleotide delivery vehicle because only one other AB toxin, anthrax, has been explored for
AON and siRNA delivery, and while the strategy appeared to be successful, they did not explore

71
the downstream functional effects of siRNA delivery.224 Thus, we were the first to explore a
toxin for siRNA delivery and functional gene knockdown.
When we conjugated siRNAs against eIF-3b and ITGB1 to aDT, we could observe
downregulation of both targets, indicating successful delivery and endosomal escape of the aDT-
siRNA conjugate. We were also encouraged that we could see potency of the aDT-siRNA at 50
nM, whereas we did not see any effect of the Ab-AON until the concentration of the conjugate
reached 150 nM. This supports our theory that aDT is a more efficient delivery vehicle than the
antibodies for antisense therapeutic delivery, at least in this example. The potency that we
observed with the aDT-siRNA conjugate did not reach the levels of the transfected siRNA, and
some possible future improvements to the system will be discussed in section 6.2. Nevertheless,
we were excited to see effective gene knockdown using aDT as a delivery vehicle, and the
protein function is dependent on the translocation through the endosomal membrane, indicating
that endosomal escape plays a key role in the diphtheria-toxin mediated siRNA delivery. As
discussed above, endosomal escape is a major challenge in the field of siRNA delivery,120, 122, 305
and we even observed this challenge with the Ab-AON system, where a significant portion of the
Ab-conjugate material colocalized with the lysosomal compartments. The improved delivery that
we observed with the aDT-mediated delivery compared to the Ab-mediated delivery supports our
hypothesis that improved endosomal escape can enhance siRNA delivery, and using aDT as a
delivery vehicle is a significant first step in overcoming this challenge.
A limitation of this work is that we were not able to directly assay the translocation and cytosolic
accumulation of siRNA, which would further support our hypothesis that endosomal escape is
enhancing siRNA delivery. Possible future strategies for examining the trafficking of aDT-
siRNA within the cells will be discussed in section 6.1.
4.3 Measuring functional effects of oligonucleotide delivery
The translation of oligonucleotide therapeutics has been met with many practical challenges, and
efficacy in the clinic remains elusive: with a few exceptions, most trials have concluded at the
Phase I and early Phase II stages due to a lack of efficacy.333 There is a significant number of
recent publications that report only on the gene silencing at the mRNA/protein level, stopping
short of a functional result, or only demonstrate knockdown of a non-endogenous, proof-of-

72
concept target such as luciferase or GFP.224, 334-336 An in vivo study to look at outcomes such as
tumor growth and animal survival would be of significant value, but these studies are lengthy
and not cost-effective. In order to determine whether the delivery of AONs and siRNAs that we
achieved using antibodies and aDT as delivery vehicles was sufficient to observe real, functional
effects, we evaluated the phenotype following knockdown using several assay including
assessment of cell shape, measuring cell viability, and monitoring cell invasion following
treatment. The results of these assays increase our confidence that these delivery strategies will
be robust strategies for in vivo gene knockdown and GBM treatment.
4.3.1 Functional effects of DRR knockdown
DRR is a cytoskeletal crosslinker that organizes microtubules and actin at focal adhesions,64 and
also recruits Akt to focal adhesions, activating several downstream pathways.66 Cells with lower
levels of DRR have been shown to lead to functional effects including decreased invasion in a
collagen matrix, a more rounded phenotype, and less invasion following direct injection of the
cells into brain tissue.64, 66
As described in Chapter 2, we observed significant DRR knockdown with a CD44-targeted
antibody as a delivery vehicle for the DRR AON. A simple phenotypic assay to measure whether
the DRR knockdown was having a significant functional effect involved measuring the area of
the cell to determine whether it had a rounded morphology, as has been observed when DRR
expression is reduced, or the more spread-out, spindle-like morphology of the native cell
population. We were able to observe a significant difference in the morphology of the cells using
this assay, indicating that the knockdown measured at the protein level was sufficient to lead to a
phenotypic change. As demonstrated by Petrecca et al., a similar change in the cellular
morphology has been shown to lead to reduced invasion following in vivo implantation of
treated cells.64, 66 Thus, the difference in phenotype that we observed is a promising indicator of a
reduced capacity for invasion in future in vivo studies, which is significant as inhibition of GSC
invasion can lead to a significantly reduction in tumor growth, at least in preclinical studies.313
4.3.2 Functional effects of eIF-3b knockdown
The silencing of the translation initiation factor eIF-3b has been shown to reduce protein
synthesis and impair the activity of the Akt pathway, leading to downstream effects including

73
apoptosis and cell death.70 This impact on cell viability has been demonstrated in colon cancer,337
esophageal squamous carcinoma,338 and bladder cancer,69 and interestingly has also been
observed in GBM,71 which is one of the reasons why eIF-3b was chosen as a genetic target for
this work.
As described in Chapter 3, we delivered siRNA against eIF-3b to GSCs using the aDT-siRNA
conjugate. We assessed the functional effects of eIF-3b knockdown in GSCs by measuring the
cell viability after treatment. Importantly, we determined there was a significant effect on cell
viability following aDT-mediated eIF-3b knockdown. Although the effect we observed is
significant, it was modest and other studies have demonstrated more dramatic inhibition
following eIF-3b knockdown; for example, the proliferation of some bladder cancer cell lines is
almost halted following eIF-3b knockdown.69 Although eIF-3b activation has been demonstrated
in GBM,71 it has not been studied in the context of GSCs and it is possible that this cell
population is more resistant to apoptosis initiated by eIF-3b knockdown. To clarify the
relationship between the GSCs and eIF-3b expression, future work could include comparing
expression of eIF-3b in GSC cells compared to bulk tumor cells, as well as measuring more
proximal effects of eIF-3b knockdown, such as the rate of total protein synthesis or the increase
in apoptotic markers.
4.3.3 Functional effects of ITGB1 knockdown
Integrin β1 (ITGB1) is essential to invasion in several cancers, often via dimerization with α5
integrins and association with fibronectin or fibronectin-mimetic peptides.74, 339-340 The
association with integrins to extracellular ligands initiates a signaling cascade that leads to
reorganization of actin fibres,341 and ITGB1 knockdown has been shown to decrease cellular
invasion and/or adhesion in multiple models.314, 342
As described in Chapter 3, we measured cellular invasion and adhesion as functional readouts for
ITGB1 knockdown. We first delivered aDT-ITGB1 to GSCs and determined that there was a
decrease in ITGB1 expression. To examine the effect on invasion, we used a previously
developed 3D hydrogel that has been validated as a drug screening tool for both proliferation and
invasion.303 This hydrogel contained fibronectin-mimetic peptides, which the cells bind to via a
dimer of ITGB1 and integrin alpha 5,343 so we hypothesized that ITGB1 knockdown would
inhibit GSC invasion in the hydrogel. Previous studies have demonstrated reduced ITGB1

74
expression can lead to a reduction of invasion in head and neck squamous cell carcinoma cells in
a Transwell assay of invasion, where cells migrate through a permeable membrane from one
chamber to another.314 When we treated the cells with aDT-ITGB1 in the 3D hydrogel model, we
observed a significant decrease in the depth of invasion of the GSCs. We also measured adhesion
to determine whether the effect was due to reduced adhesion or invasion and observed no
significant difference in the initial adhesion of the cells, indicating the observed decrease in cell
depth was due to an effect on cell invasion. There are no existing examples in the literature of a
synthetic hydrogel used to measure the effects of ITGB1 knockdown in 3D, and this represents
the first time the 3D hydrogel model developed in our lab has been used as a screening tool for a
novel therapeutic. Thus, we were excited to see that we could use it to measure phenotypic
effects of siRNA delivery against invasion targets such as ITGB1.
4.4 Targeting the glioblastoma stem cell population
GSCs contribute to the aggressive behavior of GBM, including the invasion, metastasis,
proliferation, and recurrence of the tumor.39-40, 312 Recent evidence has demonstrated that if a
therapeutic can target the GSCs, then in vivo tumor formation arising from GSC implantation is
significantly inhibited.344 In this thesis, we explored several methods of targeting GSCs,
including targeting cell surface receptors that have been identified as markers for GSCs,
targeting genes responsible for the aggressive phenotypes of GSCs, and exploring the optimal
delivery strategy for GSC therapeutics in vivo.
4.4.1 Targeting cell surface receptors
One method of targeting GSCs is to specifically target cell surface receptors that will be
expressed on the GSCs. This strategy requires careful consideration of the off-target effects of
the delivered drug, since many receptors that are expressed on GSCs are also expressed in the
bulk tumor and in neural stem cells.345
As described in Chapter 2, we used antibodies against specific antigens in order to deliver
antisense therapeutics to the GSCs. One of the cell surface receptors that we targeted in this work
was CD44, a potential GSC marker that is also expressed by neural stem cells.346-347 We were
able to successfully deliver AONs against DRR to GSCs and observe significant knockdown of
DRR at the protein level using the CD44-targeted strategy. We confirmed specificity of binding

75
by also conjugating the AON to a non-specific IgG and did not observe any internalization of the
AON. We also explored another antibody against EphA2, which is a cell surface marker shown
to play a role in the tumorgenicity of GSCs and is overexpressed in at least some GBM cells with
stem-like characteristics.42-43 Using this antibody for AON delivery we did not detect any
downregulation of the DRR following delivery.
One potential explanation for the difference in delivery efficacy is the internalization pathways
of the different antibodies. There is evidence in the literature that in some cases the CD44
receptor is recycled following internalization into the early endosomes,348-349 which could save
the AON from degradation in the lysosomes, with some Ab-AON conjugate spontaneously
escaping from the early endosomes prior to recycling. In contrast, the EphA2 receptor is likely
shuttled to the lysosomes for degradation following internalization,350 which could lead to the
degradation of any cargo that it is carrying into the cells. We saw some evidence of this
difference in internalization pathways by conducting a flow cytometry experiment, as described
in Appendix A; at every timepoint, the EphA2 antibody was internalized to a greater extent than
the CD44 antibody, suggesting that the EphA2 antibody is internalized and is not recycled back
to the cell membrane.
Despite the success of our CD44-targeted strategy, recent evidence has suggested that CD44 is
not an ideal marker for the GSCs because some cells that are low in CD44 expression have the
most GSC-like traits including sphere formation and stem cell marker expression.351 Another
marker that we could have chosen to target the GSCs is CD133, one of the first markers that was
used to identify the GSC population.38, 347 However, it has also been demonstrated that CD133-
negative cells have GSC characteristics including initiation of tumor formation.49 A successful
strategy for GSC therapeutics will likely need to target several different “GSC markers,” or
deliver a targeted drug to a broader cell population, in order to overcome these challenges.
As described in Chapter 3, we used aDT as a delivery vehicle for siRNA. aDT in its native form
targets the cell surface receptor HBEGF,352 which is widely expressed in many tissues and
especially the brain.295 HBEGF itself may play a role in GBM initiation,296 although it has not
been shown to be overexpressed specifically in the GSC population. We confirmed that HBEGF
was expressed in the GSCs studied as described in Chapter 3 by immunocytochemistry and also
by measuring the IC50 of the native toxin to confirm the correct binding and trafficking of the

76
toxin. We did not confirm whether aDT-siRNA preferentially targets the GSC population in
comparison to bulk tumor or other neural cells, which may be a limitation of this targeting
strategy. However, since the delivered therapeutic is siRNA that targets expression of a specific
gene, HBEGF targeting could still be a good strategy for GSC therapy with the selection of the
correct gene for downregulation in order to limit negative off-target effects.
4.4.2 Targeting key genetic alterations for desirable phenotypes
Another way that we targeted the GSC population is by selecting genes for downregulation that
had been shown to be over-expressed in the GSCs, or were associated with specific phenotypes
that we wanted to modulate in the GSCs. Some previous work has targeted genes involved in
stemness, including Sox2 or Nanog,39, 62 but we wanted to avoid targeting these genes as they are
also essential for neural stem cells. We also tried to select genes for downregulation that would
have obvious phenotypic effects, such as reduction in proliferation or invasion, if the
downregulation was effective.
As previously discussed, DRR has been shown to be involved in the migration and invasion of
GSCs by acting as a cytoskeletal crosslinker.64, 66 Petrecca et al. discovered that the knockdown
of DRR using AONs decreased the invasion of patient-derived GSCs in an in vivo model.66 As
described in Chapter 2, we delivered AONs against DRR to GSCs using CD44-targeted
antibodies and observed a reduction of DRR expression which correlated to a significant change
in the morphology of the cells, suggesting a less invasive phenotype. However, as described in
Appendix D, we delivered DRR AONs to DRR-overexpressing cells and, while there was a trend
towards a reduced capacity for invasion, the cell number significantly increased indicating a
greater rate of cell proliferation. This dichotomy that we observed between DRR-mediated
proliferation and invasion, where cells with reduced DRR expression leading to reduced invasion
also have increased proliferation, was originally suggested by Giese et al.65 and also observed by
Petrecca et al. where they observed increased proliferation with reduced DRR expression.64
Although this complicates DRR as an anti-cancer target, since proliferation of cancer cells is not
desirable, there is some evidence that increased proliferation of cancer cells could also sensitize
them to chemotherapeutic treatment,353 and thus this still could be a good strategy for
specifically targeting the GSC population.

77
To separate the proliferation and invasion phenotypes, as described in Chapter 3, we explored the
delivery of two different siRNAs, one associated with proliferation and one associated with
invasion, using the aDT as a delivery vehicle. Although there is likely a small population of
nearly-quiescent ‘stem’ cells within the GSC population,40 they give rise to a larger population of
progenitor-like cells that are quickly dividing,347 so proliferation is a common phenotype in GSC
populations.41 EIF-3b has been shown to promote proliferation in GBM and knockdown of eIF-
3b can reduce proliferation.71 Using aDT to deliver eIF-3b siRNA, we observed a significant
reduction in the proliferation of the GSCs. GSCs have also been shown to be highly invasive and
contribute to migration and metastasis of GBM,37, 77, 298, 312 and ITGB1 is involved in many
invasion mechanisms,74-75, 339-341 so we wanted to try to reduce ITGB1 expression and determine
whether this had a significant effect on the invasion of the cells. We conjugated an siRNA
against ITGB1 to aDT and the invasion of the cells treated with this conjugate was significantly
reduced in our 3D hydrogel model. Reduction of invasion in GBM, particularly when combined
with other therapeutic strategies, is a promising strategy for reducing tumor growth and
recurrence rates.354-356 These proof-of-concept experiments have shown that we can use aDT to
modulate specific phenotypes in the GSCs and future work will include expanding this strategy
for the knockdown of other genetic targets, for example genes involved in GSC fitness or TMZ
resistance that have been identified in CRISPR screens.302
4.4.3 Optimizing the delivery of protein conjugates in vivo
As described in Appendix E, we examined the behavior of an Ab-fluorophore conjugate
following injection in vivo. This study incorporated multiple injection paradigms, including
direct injection into the brain tissue, intraventricular injections, and intravenous injections, all in
an attempt to maximize the delivery of the Ab material to the tumor cells. We observed that the
best way to target the GSCs was to inject the Ab-fluorophore directly into the tumor space, and
with this strategy multiple areas of colocalization were observed between the GSCs and the
fluorophore-labeled antibody. We expect that this direct injection strategy would be the best way
to deliver the maximal amount of bioconjugate, whether Ab-AON or aDT-siRNA, to the GSCs
for preliminary in vivo studies and potentially for future clinical applications as well. This agrees
with current literature that suggests local administration of therapeutics to glioblastomas
overcomes several significant barriers, including rapid clearance in circulation and penetrance of
the blood-brain-barrier.357

78
Conclusions
In this thesis, I developed two strategies for the delivery of antisense therapeutics to GSCs. First,
AONs against a gene of interest, DRR, were conjugated to antibodies that were specific for
GSCs including CD44 or EphA2. Primary validation steps included the optimization of the
modification pattern of the AON for the best DRR knockdown efficacy, as well as an
internalization assay performed with the antibodies of interest to confirm that they would be
endocytosed following receptor binding. The best DRR AON had a “Gapmer”-motif with
modifications on the flanking ends of the oligonucleotide strand, and we found that both
antibodies were internalized by the GSCs. Next, we conjugated the AONs to the antibodies and
treated the GSCs with the conjugates in vitro. We found that the CD44-conjugated DRR AON
could significantly reduce DRR expression in GSCs, whereas the EphA2-conjugated DRR AON
did not. This satisfied the first objective, to “knockdown DRR/FAM107A in vitro using a
targeted delivery platform.” Additionally, we used the fluorophore-labelled AON to examine
conjugate uptake and colocalization with the endolysosomal pathway, and saw that although the
CD44 Ab-AON conjugate was taken up more than the EphA2 Ab-AON conjugate, supporting
our knockdown results, there was still a high degree of colocalization between the conjugate and
the lysosomes, suggesting a significant amount of endolysosomal trafficking and degradation.
Importantly, this study demonstrated that we could knock out a gene of interest in the GSCs
using a targeted antibody delivery platform.
Next, we explored an alternate delivery platform: aDT, which binds to cell surface receptors but
almost immediately escapes the endolysosomal pathway following endocytosis. We validated
that the GSCs were a suitable cell type for aDT-mediated delivery by staining the stem cells for
the aDT receptor, HBEGF, and by measuring the IC50 of the native toxin to confirm the correct
trafficking mechanism. The patient-derived GSCs used for these studies displayed a high amount
of HBEGF staining and a low IC50, confirming the suitability of the aDT for delivery in these
cells. aDT was then conjugated to two different siRNAs, one against a target for cell
proliferation, eIF-3b, and one against a target for invasion, ITGB1. We successfully reduced
expression of both eIF-3b and ITGB1 using the aDT-siRNA delivery platform and confirmed
functional effects including reduced cell proliferation and invasion. These studies confirmed that
aDT could successfully deliver siRNA to GSCs, representing the first time that aDT had been

79
used for siRNA delivery and highlighting the promise of attenuated toxins as a platform delivery
technology for antisense therapeutics to diseases including cancer.
5.1 Completion of objectives
This research was motivated by the following hypothesis:
Protein-conjugated oligonucleotides will selectively and effectively knockdown key genes in
brain cancer cells, including patient-derived models of GBM, and thereby reduce cellular
proliferation or invasion in vitro.
Herein we described the delivery of AONs and siRNAs to GSCs using either antibodies or toxins
as delivery vehicles. Achievements of the objectives originally outlined in Chapter 1 are outlined
below.
1. Knockdown of an essential GSC gene in vitro using a targeted delivery platform.
• Screened several modification patterns of DRR AONs to determine the best
oligonucleotide sequence for DRR knockdown.
• Confirmed cell-surface staining of CD44 and EphA2 receptors, as well as efficient
internalization of CD44 and EphA2 antibodies in GSCs to support their suitability as
AON delivery vehicles to GSCs.
• Conjugated AONs to antibodies and confirmed successful conjugation via PAGE and
mass spectrometry.
• Delivered Ab-AON conjugates to GSCs and induced significant DRR downregulation
using CD44 antibody-mediated delivery.
• Visualized significant internalization of CD44 Ab-AON into GSCs and colocalization
with the endolysosomal pathway.
• Measured a significant difference in cell morphology following DRR knockdown,
potentially indicating a less invasive phenotype.

80
These data were presented in Chapter 2 and published in Molecular Therapy-Nucleic Acids.
2. Achieve gene knockdown with enhanced endosomal escape.
• Confirmed HBEGF staining and low IC50 of native DT in GSCs, indicating the
suitability of the DT as a delivery vehicle for GSCs.
• Conjugated siRNAs to aDT using a freeze-thaw method and confirmed successful
conjugation using PAGE.
• Reduced eIF-3b expression in GSCs using aDT-eIF-3b and measured a corresponding
significant difference in cell viability following knockdown.
• Reduced ITGB1 expression in GSCs using aDT-ITGB1 and measured a corresponding
significant difference in cellular invasion in a 3D hydrogel model following knockdown.
These data were published in Chapter 3 and submitted for publication to Science Advances.
Recommendations for future work
In this thesis, novel delivery strategies for antisense therapeutics were developed and used to
target GSCs. Three areas of future work stem from the research conducted in this thesis:
investigation into the aDT-siRNA trafficking mechanism; optimization of specificity, potency,
and endosomal escape; and combination therapies with native toxins, therapeutic proteins, or
chemotherapeutic drugs.
6.1 Investigation into aDT-siRNA trafficking mechanism
Recently, Lacroix et al. have shown that it is difficult to draw any reliable conclusions about
trafficking from fluorescently labeled DNA nanostructures, because nuclease degradation can
separate the fluorophore from the nucleic acid material and give misleading information about its
intracellular location.358 While fluorescent labeling of the AON gave us some qualitative
information about trafficking as described in Chapter 2, we hesitated to fluorescently label the
siRNA bound to the aDT as described in Chapter 3 because it would be much more quickly
degraded than the AON and likely to give inaccurate trafficking information. However, there are

81
still several methods that could allow insights into the trafficking mechanism of the aDT-siRNA
conjugate.
6.1.1 Inhibition of translocation
One assay that could prove translocation of the aDT is essential for siRNA delivery is inhibiting
the translocation of the toxin and proving that this eliminates the knockdown efficiency observed
for targets such as eIF-3b and ITGB1. One possibility would be to use a small molecule inhibitor
of endosomal acidification, which would inhibit the translocation of the aDT. Bafilomycin A1 is
a small molecule that has been used for this purpose,210 as it inhibits the ATP-mediated transport
of protons across the endosomal membrane.359 For example, it has been shown to reduce the
efficiency of gene delivery with poly(L-lysine) carriers, which rely on the acidification of the
endosome to escape the endolysosomal pathway.360 However, many of these small molecule
inhibitors of endosomal acidification, including Bafilomyin A1, are highly toxic because
endosomal trafficking is essential for the survival of cells,361 which limits the possibility for
downstream assays such as quantitative PCR or western blot.
A better option would be to express a mutant version of the aDT containing a point mutation in
the translocation domain that prevents the mechanism of translocation, and prove that the siRNA
delivery is no longer effective with the mutant toxin. The point mutation of a glutamic acid
residue to a lysine residue at amino acid position 349 (E349K) has been shown to strongly inhibit
cytotoxicity and translocation compared to native DT.311 A crystallographic model of DT shows
that Glu349 sits in a short loop connecting two long alpha-helixes of the translocation domain,
and the E349K mutation introduces a cationic amino acid at this site that interferes with the
mechanism of translocation.311 This method has been used to explore mechanisms of DT-
mediated delivery in the past; for example, Shaw et al. used the E349K mutant to demonstrate
that the delivery of T-cell antigens fused to DT was no longer effective with the mutant DT. This
strategy would require expressing the DT with the same structure as described in Chapter 2,
where there is an available cysteine for modification and pendant SUMO and His tags, and also
containing the E349K point mutation.

82
6.1.2 IC50 shifts of modified DT
Another method to probe the delivery efficiency is to measure the shift in IC50 following
modification, if the native toxic domain is conserved during the modification process (ie. DT
instead of aDT).210 A higher IC50 would indicate a less efficient delivery, most likely from a
reduced capacity for either receptor binding or translocation. If the IC50 was conserved, it would
indicate that the cargo (ie. siRNA) is not interfering with the DT trafficking mechanism.
However, we were not able to use this strategy because a significant amount of unlabeled DT
(without siRNA) was present after synthesis and purification, and this unmodified DT would
interfere with any IC50 assays because it would retain the native IC50. In order to use this strategy
a labeling efficiency of at least 90% would be ideal.
We could have tried modifying the siRNA with the crosslinker first, instead of modifying the DT
with the crosslinker first to see if this different process of labeling could improve the reaction
efficiency and reduce the amount of unlabeled DT. The labeling of the siRNA with the
crosslinker, forming siRNA-maleimide, might be more efficient than labeling the DT with the
crosslinker, which formed DT-DBCO. However, even if this led to a more efficient conjugation
it is unlikely that we would reach >90% labeling of the DT with this method.
Another way to purify the DT-siRNA conjugate would be incorporate a tag for purification on
the distal end of the siRNA, for example a small peptide such as a Strep-tag that can be
recognized and purified using a streptavidin column.362 In this way, we could first use the Nickel
column to purify the DT from the siRNA, and then use a streptavidin column to purify the DT-
siRNA from the unlabeled DT. This would allow for downstream assays including
measurements of IC50 with the labeled DT, but could lead to a low recovery of the DT-siRNA
conjugate.
6.1.3 Split-reporter assays
Another way to localize the aDT fragment once it reaches the cytoplasm and confirm effective
delivery would be a split-reporter assay, where an enzyme or fluorescent reporter is split into two
parts. One part is included in the conjugate to be delivered and the other part is expressed in the
cytoplasm by the target cell population. Once the two parts rejoin, which requires successful
delivery of the conjugate into the cytoplasm, a signal such as fluorescence or luminescence can

83
be detected. This has been used for reporters such as GFP, which fluoresces once it recombines
in the cytoplasm,363 or for luciferase, which can turn over its substrate following recombination
to give a bright luminescent signal.364 The first step would be to express a fusion toxin with the
split fragment and confirm effective delivery; the fusion protein could then be modified with a
cargo such as siRNA. This would be a very useful strategy if testing different variations of an
aDT-siRNA conjugate, since different levels of fluorescence/luminescence could be used to
compare efficacy of delivery.
6.2 Optimization of specificity, potency, and endosomal escape
Although the work in this thesis involved testing two distinct delivery vehicles, an antibody and
a bacterial toxin, there are many similarities between the two: they are both proteins that bind to
extracellular receptors, induce endocytosis, and can deliver oligonucleotides into the cytoplasm.
There are several ways that we could combine concepts learned using these two different
delivery vehicles to synthesize a targeted delivery vehicle that would escape the endosome and
deliver a highly potent oligonucleotide. In this section, four main ideas are proposed: re-targeting
the receptor binding domain of the aDT to target more specific cell populations; stabilization of
the siRNA to yield stable oligonucleotides that are still very potent; exploring nucleic acid
analogues (morpholinos or peptide nucleic acids) that could have the same action as
oligonucleotides but with different backbone character and better stability; and finally using
phenotypic assays as a first-line selection tool for the optimal oligonucleotide.
6.2.1 Re-targeting the receptor binding domain
As previously discussed, the native cell surface receptor of DT, HBEGF, is a promiscuous
marker and not specific to one cell population. To combine the specific targeting capacity of an
antibody with the trafficking and/or toxic properties of AB toxins, it is possible to replace the
receptor binding domain of the toxin with an antibody mimic called a single-chain variable
fragment (scFv). These scFvs can be engineered to bind to desired cell surface receptors in a
similar way to engineered antibodies.365 For example, this strategy has been used with DT
variants for the in vivo targeting of T cell lymphoma cells366 and even in the clinic to target B-
cell malignancies.367 A version of pseudomonas exotoxin A has been expressed with a CD133
scFv to specifically target CSCs in head and neck cancer;368 a similar strategy could be used to

84
fuse a CD133 or CD44 scFv to DT to more specifically target the GSCs. However, transcytosis
of the DT across the blood brain barrier may be dependent on targeting HBEGF,217 so this trade-
off would have to be considered for future in vivo applications. However, a receptor mutant
could be propelled across the blood brain barrier by recent methods such as focused
ultrasound,369 or this re-targeting strategy could be used to target cancers in tissues other than the
brain.
6.2.2 Stabilization of the siRNA
As described in Chapter 3, the siRNAs that were tested against the gene targets eIF-3b and
ITGB1 were extremely potent for knockdown, reducing the mRNA expression by 80-90%.
However, these siRNAs were not very stable: we showed that almost all of the siRNA is
degraded by 24h when exposed to serum nucleases. These siRNAs were Dicer-substrate siRNAs,
meaning they were 27-mer RNA duplexes that must be recognized by an enzyme called Dicer to
be effective in the cells. Different modification patterns of these siRNAs should be explored to
determine structure-functional relationships between modifications and siRNA properties,
thereby optimizing the stability of the siRNAs while maintaining or increasing their potency.
A significant amount of work has already been done to optimize modification patterns for Dicer-
substrate siRNAs. Chan et al. used siRNAs with 2’-fluoro-modifications and observed similar
potency to unmodified siRNAs.370 Allerson et al. demonstrated that a combination of 2’-fluoro
and 2’-methoxy modifications to the siRNA was more effective than unmodified siRNA.325
Collingwood et al. showed that siRNAs with 2’-methoxy-based modifications were better at
evading immune activation than 2’-fluoro modifications and siRNAs containing 2’-methoxy
modifications were generally more potent than siRNAs containing 2’-fluoro modifications,
although these effects appeared to be target- and sequence-specific.371 Importantly, Patisiran, the
first FDA-approved siRNA therapeutic, incorporates siRNA with some combination of
unmodified and 2’-methoxy modified ribonucleosides.326 Thus, the most potent modification
pattern probably incorporates primarily 2’-methoxy modifications, although there is a whole
library of additional modifications that could be explored,89 and these may have to be re-
optimized for each target/sequence tested.

85
6.2.3 Exploring RNA analogues: morpholinos and peptide nucleic acids
A major concern when conjugating oligonucleotides to the aDT was that the negative nature of
the phosphodiester linkages within the RNA or DNA would interfere with the translocation of
the cargo. While the aDT was able to deliver siRNA, a backbone with a more neutral charge
might be more easily shuttled through the translocation pore. There are several options for
neutrally charged backbones with the same base-pair capacity as nucleic acids, including
morpholinos, where the 5-membered ring of RNA or DNA is replaced with a 6-membered
morpholino ring and the phosphodiester linkages are replaced by phosphorodiamidate
linkages;372 or peptide nucleic acids, where the DNA/RNA backbone is completely replaced by a
peptide backbone, but with the same pendant bases as a nucleic acid.373 The structures of these
analogues are shown in Figure 6.1. These structures have key advantages aside from their neutral
backbone: they are highly stable to nuclease degradation because they are not recognized in the
same way as DNA/RNA;374-375 and they can still be potent for gene knockdown in a sequence-
specific manner with a mechanism similar to AONs.374, 376 Conjugation of the morpholinos or
peptide nucleic acids would be done in a similar manner as the siRNA conjugation presented in
this thesis, and it could be an exciting way forward to achieve gene knockdown with a much
more stable biomolecule.
Figure 6.1. Structures of nucleic acid analogues including morpholinos and peptide nucleic
acids.
O Base
OPO OO
O Base
O
O
5’
OH
OH
3’
RNA
O
N
O Base
OPO OO
O Base
O
O
5’
3’
DNA
O
5’
PO NO
O
NO
3’
Base
Base
Morpholino
N
NH
N
O
NHO
N
N-terminus (5’)
C-terminus (3’)
O
Base
O
Base
O
Base
Peptide Nucleic Acid

86
6.2.4 Phenotypic assays as a first-line screening tool for oligonucleotide
sequences
As discussed in section 4.1, there was a negative correlation between stability and potency for
the oligonucleotides tested in this work. This means that either the stability or potency was a
constant limitation. For most of this work, we used potency as a screening tool to pick the best
oligonucleotide; however, stability and duration of silencing also play significant roles in
achieving the desired effect when delivered to cells.
Screening antisense therapeutics on the basis of a phenotypic effect, such as cell death or
invasion as were used in this work, or any other desired phenotypes, would be a more rational
selection strategy wherever possible. For example, several modification patterns of the ITGB1
siRNA could be tested in the 3D hydrogel platform and the most efficient oligonucleotide would
give the strongest reduction in invasion. Once the best phenotypic outcome is observed, it would
be possible to work backwards and determine the best combination of stability and potency in
order to achieve a desired effect. In addition, screening in this way would provide greater
confidence in downstream phenotypic assays when delivering the oligonucleotides using novel
platform technologies.
6.3 Simultaneous delivery of siRNA and other therapeutics
An extension of this work would be to use the aDT delivery platform developed in this thesis and
combine it with the delivery of other therapeutics. Most of the literature concerning AB toxins
for delivery has focused on delivering protein- or peptide-based therapeutics, so these types of
cargo could be combined with the siRNA delivery for therapy of many diseases, not only cancer.
Small-molecule drugs, such as chemotherapeutics, could also be combined with the siRNA
delivery to increase the therapeutic effect. This section will suggest several combination
therapies that draw on both the ideas developed in this thesis as well as previous work that has
been done to explore aDT as a delivery vehicle.
6.3.1 Combination with native toxin domains
Fusion toxins combining the toxic active domain of DT with re-directed receptor binding regions
have already been explored for the treatment of GBM. For example, Weaver et al. fused

87
transferrin, which is highly expressed in GBM, to the translocation and toxin domains of DT for
the treatment of malignant brain tumors and observed tumor response without significant off-
target toxicity in at least a third of patients in a phase I/II clinical trial.377 The work with DT
presented in this thesis was with an attenuated DT, where the active domain was mutated to
eliminate its cytotoxicity; this was important so that we could isolate the effects of siRNA
delivery. However, a future strategy could involve keeping the native toxic domain while also
conjugating an oligonucleotide for gene knockdown; with careful selection of the gene target,
this could result in synergistic effects between the toxin and the siRNA/AON for improved
toxicity towards cancer cells.
6.3.2 Combination with therapeutic proteins
Another delivery strategy that has been heavily pursued is the delivery of therapeutic proteins
that are depleted in diseased cells using attenuated toxins. An attenuated toxin variant could be
engineered that could bind to a desired receptor and maintain its translocation capacity, but it
would also contain a cysteine group for siRNA conjugation and also be expressed as a fusion
protein with a therapeutic protein domain of interest. For example, in cancer treatment, the toxin
could be used to carry both a therapeutic siRNA as well as a protein that would act as a tumor
suppressor such as caveolin-1 which represses apoptosis inhibitors.378
This combination delivery of therapeutic proteins and siRNA could also be a sophisticated
approach for complex diseases other than cancer, such as Alzheimer’s disease (AD). There are
many mis-regulated genes at work in AD, including downregulation of the MAPK/ERK pathway
and upregulation of clathrin proteins, which lead to increased internalization of amyloid
precursor proteins (APPs) and increased intracellular levels of amyloid beta plaques.379 Recent
studies have suggested that targeted RNA interference may be able to limit the progression of
AD, such as the study by Rodriguez-Lebron et al. which showed that short-hairpin RNA against
APP could reduce AD phenotypes in a mouse model.380 At the proteome level, brain-derived
neurotrophic factor (BDNF) plays an important role in synaptic plasticity and memory formation
and abnormalities in its expression have been reported in AD patients.381 Shin et al. delivered
Neuropep-1, a peptide that increases intracellular levels of BDNF, to a mouse model of AD and
observed increased BDNF expression and dramatically reduced amyloid beta plaque formation, a
promising sign of an effective AD treatment strategy.382 aDT could be engineered to carry

88
siRNA against APP in combination with Neuropep-1 or BDNF for a dual-factor treatment
strategy against Alzheimer’s disease. Of course, this is just one example of a dual delivery
strategy using the aDT platform; it could be used for virtually any disease where it is important
to downregulate expression of some genes and also introduce therapeutic proteins into cells.
6.3.3 Combination with chemotherapeutic drugs
aDT could be used to deliver other chemotherapeutic drugs alone or in combination with siRNA.
There is abundant literature where antibodies are used as carriers for small-molecule
chemotherapeutics; in a similar manner, aDT could be conjugated to chemotherapeutic drugs and
siRNA for co-delivery. This could result in synergistic effects if the siRNA target was chosen to
modulate the response of the cells to the chemotherapeutic drug. Chemotherapeutic resistance is
a significant challenge in cancer treatment and often arises from underlying genetic mutations.383
Triple-negative breast cancer often acquires chemotherapeutic resistance through over-
expression of the multidrug resistance protein 1 (MDR1), and siRNA-mediated knockdown of
MDR1 can lead to enhanced susceptibility of the tumor cells to common chemotherapeutics such
as doxorubicin; aDT could simultaneously deliver MDR1 siRNA and doxorubicin to cancer
cells, overcoming their chemotherapeutic resistance while simultaneously delivering a potent
chemotherapy drug. In GBM, resistance of DNA damage by TMZ is mediated by poly(ADP-
ribose) polymerase-1 and -2 (PARP-1 and -2), and inhibition of PARP can increase the
sensitivity of GBM cells to TMZ.384 Although TMZ is not the most potent chemotherapeutic,
aDT could be used to deliver PARP siRNA and TMZ synergistically and this would align with
the current standard of care of TMZ chemotherapy.

89
Abbreviations
AD Alzheimer's disease
ADC Antibody-drug conjugate
aDT Attenuated diphtheria toxin
Akt Protein kinase B
ALDH1A3 Aldehyde dehydrogenase 1A3
AON Antisense oligonucleotide
ApoE Apolipoprotein E
APP Amyloid precursor proteins
AT Anthrax Toxin
BBB Blood-brain-barrier
BCL-2 B-cell lymphoma 2
BDNF Brain-derived neurotrophic factor
ccRCC Clear cell renal cell carcinoma
CD133 Cluster of differentiation 133
CD15/SSEA1 Cluster of differentiation 15/stage-specific embryonic antigen 1
CD171/L1CAM Cluster of differentiation 171/L1 cell adhesion molecule
CD22 Cluster of differentiation 22
CD25 Cluster of differentiation 25
CD3 Cluster of differentiation 3
CD33 Cluster of differentiation 33
CD34 Cluster of differentiation 34
CD38 Cluster of differentiation 38

90
CD44 Cluster of differentiation 44
CPP Cell penetrating peptide
CSC Cancer stem cell
DNA Deoxyribonucleic acid
DRR Downregulated in renal cell carcinoma
DT Diphtheria toxin
EGFR Epidermal growth factor receptor
eIF-3b Eukaryotic translation initiation factor 3 subunit B
EphA2 Ephrin receptor A2
EphA3 Ephrin receptor A3
FAK Focal adhesion kinase
FAM107A Family with sequence homology 107A
FANA 2ʹ-deoxy-2ʹ-fluoro-beta-D-arabinonucleic acid
FDA Food and drug administration
GalNAc N-acetylgalactosamine
GBM Glioblastoma
GO Gemtuzumab ozogamicin
GSC Glioblastoma stem cell
HBEGF Heparin-binding epidermal growth factor receptor
HER2 Human epidermal growth factor receptor 2
HIR Human insulin receptor
HIV Human immunodeficiency virus
HRP Horseradish Peroxidase
IDH Isocitrate dehydrogenase

91
IgG Immunoglobulin G
ITGB1 Integrin beta 1
JAK Janus kinase
KRAS Kirsten rat sarcoma viral oncogene homolog
mAb Monoclonal antibody
MALDI-TOF Matrix-Assisted Laser Desorption/Ionization-Time Of Flight
MAPK Mitogen-activated protein kinase
MDR1 Multi-drug resistance protein 1
MFI Median fluorescent intensity
MGMT O6-methylguanine-DNA methyl-transferase
MMAF Monomethyl auristatin F
MMP Matrix metalloprotease
mRNA Messenger RNA
mTOR Mammalian target of rapamycin
MXD3 MAX dimerization protein 3
NADPH Nicotinamide adenine dinucleotide phosphate hydrogen
NHS N-hydroxysuccinimide
NLS Nuclear localization signal
PAGE Polyacrylamide gel electrophoresis
PARP Poly(ADP-ribose) polymerase
PCL Poly(caprolactone)
PCR Polymerase chain reaction
PDI Protein disulfide isomerase
PE Pseudomonas exotoxin A

92
PEG Poly(ethylene glycol)
PEI Polyethyleneimine
PFS Progression-free survival
PI3K Phosphoinositide 3-kinase
PLGA Poly(lactic-co-glycolic) acid
PLO Poly(L-ornithine)
PNP Human purine nucleoside phosphorylase
PO-DNA Phosphodiester backbone DNA
PS-DNA Phosphorothioate backbone DNA
RFP Red fluorescent protein
RISC RNA-induced silencing complex
RNA Ribonucleic acid
RNAseH Ribonuclease H
siRNA Small interfering ribonucleic acid
STAT Signal transducer and activator of transcription proteins
TAT Transactivator of transcription
TMZ Temozolomide
TP10 Transportan 10
TRPC6 Transient receptor potential channel 6
VEGFR1 Vascular endothelial growth factor receptor-1
Wnt Wingless-related integration site

93
Appendix A: Supporting information for “Antibody-antisense
oligonucleotide conjugate downregulates a key gene in
glioblastoma stem cells”
A.1 Melting Point Determination
Equimolar amounts (1.5 nmol) of complementary sequences were combined and dried. To the
resulting pellet was added 10 mM sodium phosphate buffer (pH 7.2) containing 100 mM NaCl
and 0.1 mM EDTA (1 mL). They were then transferred into cuvettes in a Varian UV
spectrophotometer. The samples were heated to 90 °C and then cooled to 5 °C. The change in
absorbance at 260 nm was then monitored upon heating from 5 to 90 °C at a rate of 0.5 oC/min.
The dissociation temperatures were calculated as the midpoint of the transition (T1/2) using the
first derivative of the melting curve.
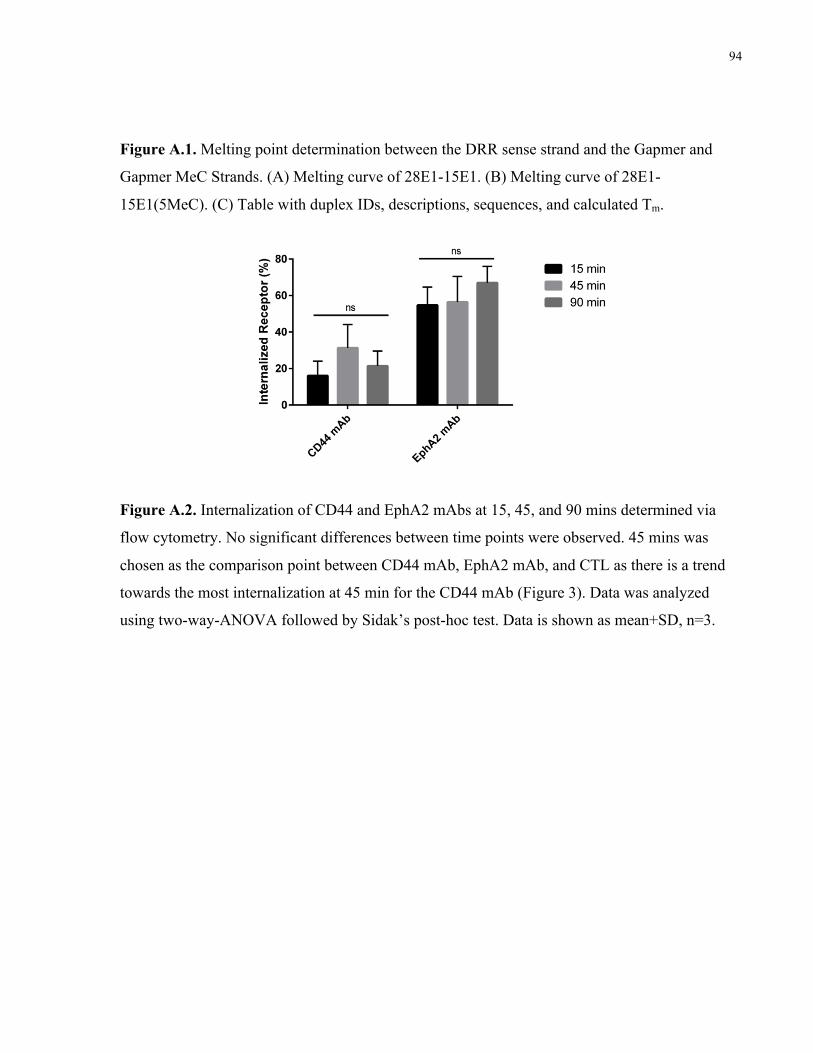
94
Figure A.1. Melting point determination between the DRR sense strand and the Gapmer and
Gapmer MeC Strands. (A) Melting curve of 28E1-15E1. (B) Melting curve of 28E1-
15E1(5MeC). (C) Table with duplex IDs, descriptions, sequences, and calculated Tm.
Figure A.2. Internalization of CD44 and EphA2 mAbs at 15, 45, and 90 mins determined via
flow cytometry. No significant differences between time points were observed. 45 mins was
chosen as the comparison point between CD44 mAb, EphA2 mAb, and CTL as there is a trend
towards the most internalization at 45 min for the CD44 mAb (Figure 3). Data was analyzed
using two-way-ANOVA followed by Sidak’s post-hoc test. Data is shown as mean+SD, n=3.
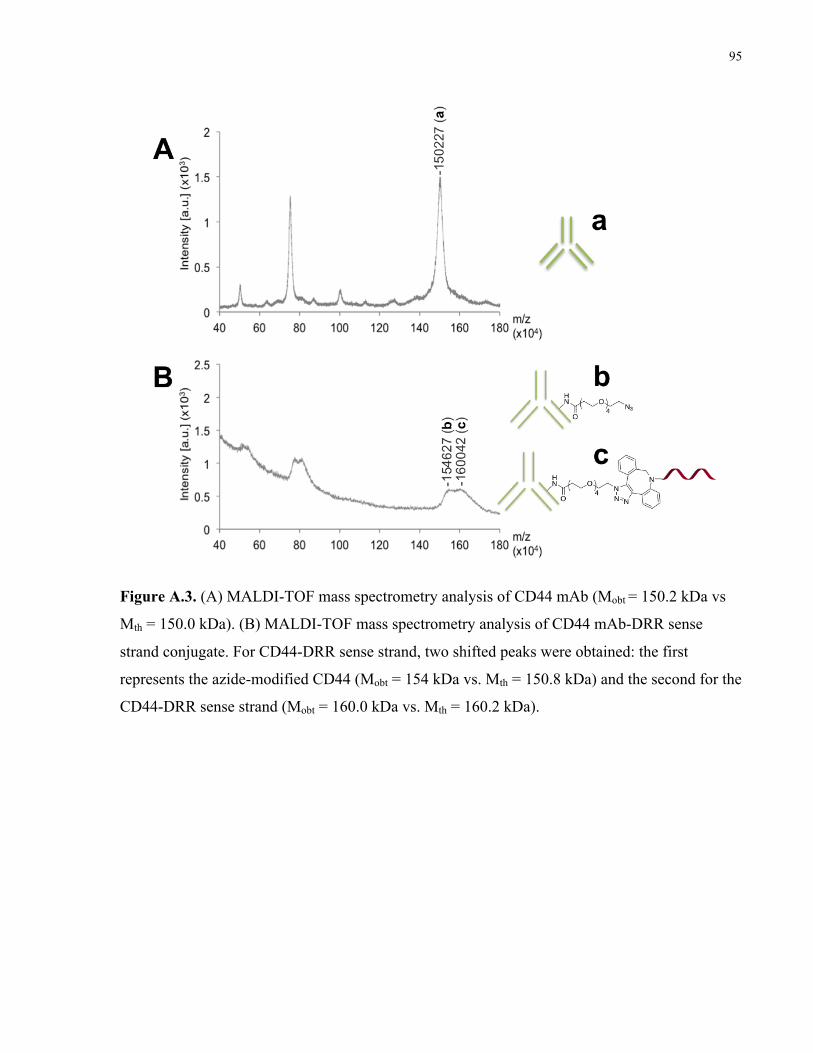
95
Figure A.3. (A) MALDI-TOF mass spectrometry analysis of CD44 mAb (Mobt = 150.2 kDa vs
Mth = 150.0 kDa). (B) MALDI-TOF mass spectrometry analysis of CD44 mAb-DRR sense
strand conjugate. For CD44-DRR sense strand, two shifted peaks were obtained: the first
represents the azide-modified CD44 (Mobt = 154 kDa vs. Mth = 150.8 kDa) and the second for the
CD44-DRR sense strand (Mobt = 160.0 kDa vs. Mth = 160.2 kDa).

96
Figure A.5. Comparison of uptake at 75 nM vs. 150 nM for EphA2 mAb-dsDRR and CD44
mAb-dsDRR. DsDRR only is shown as a negative control. A significant increase in uptake was
observed for both EphA2 mAb-dsDRR and CD44 mAb-dsDRR at 150 nM. Cell nucleus
(Hoechst, blue); AON (Cy3, green); lysosome (Dextran647, red). Representative z-stack images
shown. All scale bars are 50 µM.
Table A.1. Mass analysis of the oligonucleotide strands used in this study. High-resolution mass
spectrometry (HRMS) acquisition was performed in negative ion mode using electrospray
ionization (ESI).
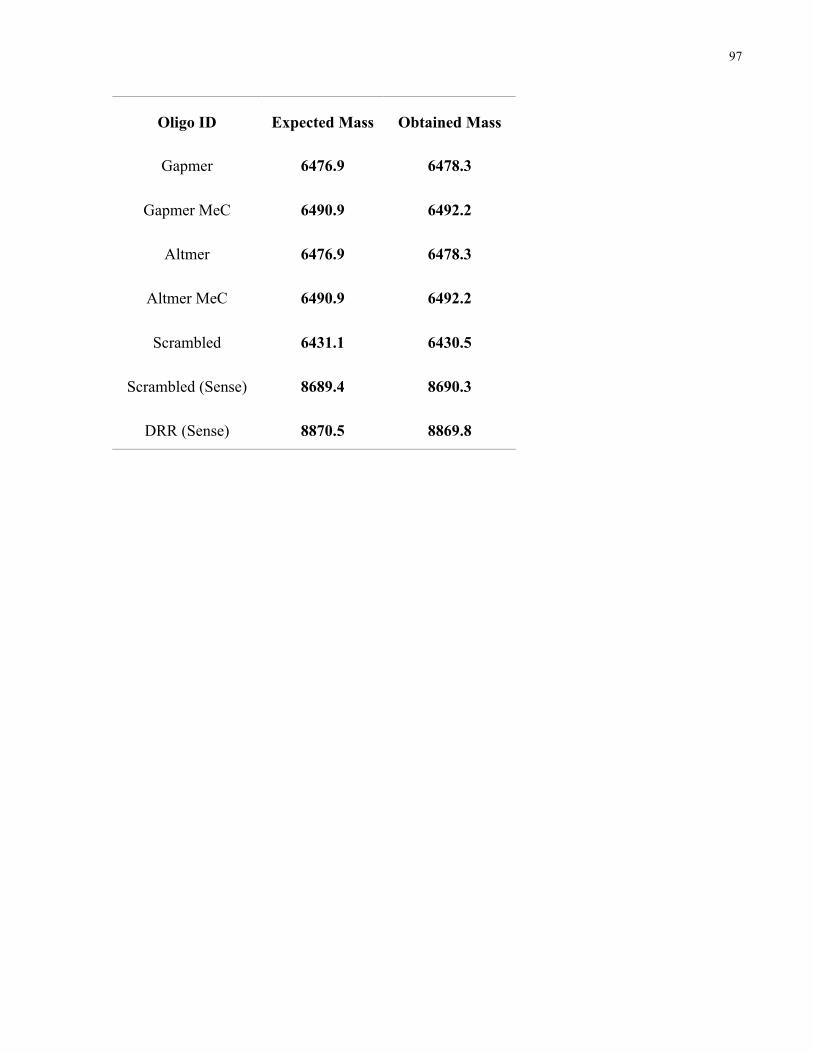
97
Oligo ID Expected Mass Obtained Mass
Gapmer 6476.9 6478.3
Gapmer MeC 6490.9 6492.2
Altmer 6476.9 6478.3
Altmer MeC 6490.9 6492.2
Scrambled 6431.1 6430.5
Scrambled (Sense) 8689.4 8690.3
DRR (Sense) 8870.5 8869.8

98
Appendix B: Supporting information for “Attenuated diphtheria
toxin mediates siRNA delivery”
Supplemental Methods
Preparation of aDT-siRNA conjugate (incubation at 37 °C)
The synthesis was performed as described in the main methods, with the exception of the
incubation at -20 °C. Instead, the mixture was held at 6h or 24 h at 37 °C with between 1 and 10
equivalents of oligonucleotide to 1 equivalent of diphtheria toxin.
Nuclease stability assay
siRNA or aDT-siRNA was incubated with a solution of 10% FBS in 20 mM Tris, 150 mM NaCl,
1% glycerol, pH 7.5 for 1, 4, 8, or 24 h. The entire contents of the sample were analyzed using
acrylamide electrophoresis to separate nucleic acid material based on size. The remaining full
length siRNA was quantified by imageJ and the data was reported as a percentage of intact
siRNA.
IC50 of native diphtheria toxin
Cells were plated to reach 60-80% confluency on 96-well Primaria plates (Corning 353872).
Native diphtheria toxin was added at varying concentrations to cells in full media and the cells
were incubated with the treatment for 48 h. At 48 h, PrestoBlue Cell Viability reagent (Thermo
Fisher A13262) was added for a final concentration of 10% in media and the viability was
measured by reading the fluorescent signal in each well. The IC50 was estimated using an online
calculator (aatbio.com/tools/ic50-calculator).
Cell viability assays following transfection with eIF-3b siRNA

99
siRNA and Lipofectamine RNAiMAX were incubated together with Opti-MEM for the desired
final concentration. The transfection complex was incubated at RT for at least five minutes then
added to 96-well plates coated with PLO/laminin. Cells were then added to the plates with the
transfection solution at a density of 8,000 cells per well. Fresh media was added to each well 24h
following treatment. At 72 hours, cell viability was measured via PrestoBlue viability assay and
quantified as a percentage of untreated controls.
Figure S1. Incubation at 37 °C leads to inefficient aDT-siRNA conjugation. A) Coomassie-stained SDS-PAGE demonstrating conjugation of DT with different equivalents of oligonucleotides conjugated for 6 or 24h at 37 °C. B) Quantification of % conjugation of aDT to

100
siRNA at different equivalents and different incubation times. Green star represents approximate conjugation efficiency using the freeze-thaw method presented in the main methods (~50-60%).
Figure S2. aDT conjugation does not change siRNA susceptibility to nuclease degradation. A) Coomassie-stained SDS PAGE gel shows that siRNAs are degraded in serum-containing solutions over a period of 24 hours. The lanes show the 0, 1, 4, and 8 h timepoints (left-right). No band was detected at 24 h. B) Quantification of remaining intact siRNA at 1, 4, and 8 hours demonstrates no significant difference between the degradation of siRNA and aDT-siRNA. Data is shown as n=3, mean±SD, and was analyzed using 2-way-ANOVA.
Figure S3. Native diphtheria toxin mechanism is conserved in GSCs. The IC50 of native diphtheria toxin in HB-EGF was determined; data is shown as cell viability normalized to an untreated control, n=3, mean+SD.
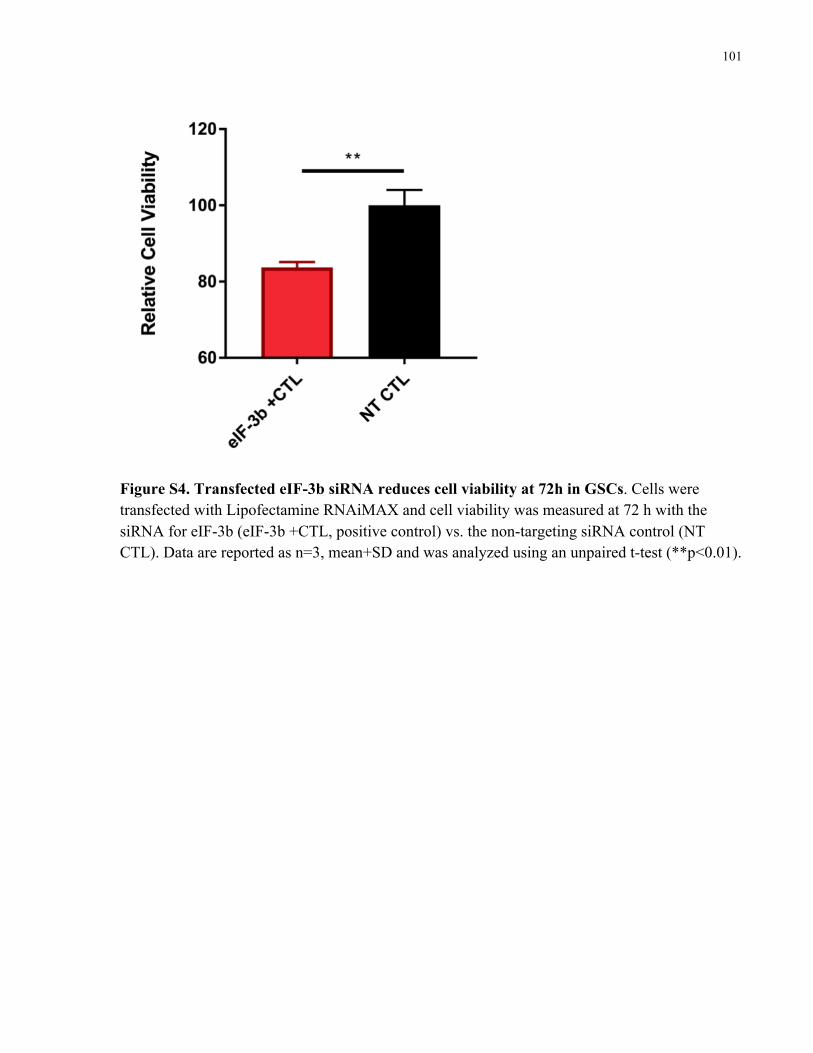
101
Figure S4. Transfected eIF-3b siRNA reduces cell viability at 72h in GSCs. Cells were transfected with Lipofectamine RNAiMAX and cell viability was measured at 72 h with the siRNA for eIF-3b (eIF-3b +CTL, positive control) vs. the non-targeting siRNA control (NT CTL). Data are reported as n=3, mean+SD and was analyzed using an unpaired t-test (**p<0.01).

102
Appendix C: Effect of sugar 2’,4’-modifications on gene silencing
activity of small interfering RNA duplexes
This Appendix was published in Nucleic Acid Therapeutics:
Malek-Adamian, E.; Fakhoury, J.; Arnold, A. E.; Martínez-Montero, S.; Shoichet, M. S.; Damha,
M. J. Effect of Sugar 2’,4’-Modifications on Gene Silencing Activity of Small Interfering RNA
Duplexes. Nucleic Acid Ther 2019.
E.M.-A. synthesized and characterized all oligonucleotide materials and wrote the manuscript.
A.E.A. and J.F. performed cell experiments and edited the manuscript. S.M.-M. conceptualized
fundamental work on the project and provided feedback on the manuscript. M.J.D. and M.S.S.
secured funding, supervised and guided the research, and participated in writing the manuscript.
ABSTRACT
In this study, we explore the effect of a library of 2’-, 4’-, and 2’,4’-modified uridine
nucleosides and their impact on silencing firefly luciferase and on Downregulated in Renal Cell
Carcinoma (DRR) gene targets. The modifications studied were 2’-F-ribose, 2’-F-arabinose, 2’-
OMe-ribose, 2’-F,4’-OMe-ribose, 2’-F,4’-OMe-arabinose, and 2’-OMe,4’-F-ribose. We found
that 2’,4’-modifications are well tolerated within A-form RNA duplexes, leading to virtually no
change in melting temperature as assessed by UV thermal melting. The impact of the dual
(2’,4’) modification was assessed by comparing gene silencing ability to 2’- or 4’- (singly)
modified siRNA counterparts. siRNAs with (2’,4’)-modified overhangs generally outperformed
the native siRNA as well as siRNAs with a 2’- or 4’-modified overhang, suggesting that 2’,4’-
modified nucleotides interact favorably with Argonaute protein’s PAZ domain. Among the most
active siRNAs were those with 2’-F,4’-OMe-ribose or 2’-F,4’-OMe-arabinose at the overhangs.
When modifications were placed at both overhangs and internal positions, a duplex with the 2’-F
(internal) and 2’-F,4’-OMe (overhang) combination was found to be the most potent, followed
by the duplex with 2’-OMe (internal) and 2’,4’-diOMe (overhang) modifications. Given the
nuclease resistance exhibited by 2’,4’-modified siRNAs, particularly when the modification is

103
placed at or near the overhangs, these findings may allow the creation of superior siRNAs for
therapy.
INTRODUCTION
Exploiting the therapeutic potential of RNAi has been fueled with the recent approval of
Patisiran, the first siRNA-based drug approved for the treatment of hereditary transthyretin
amyloidosis.326 This siRNA is comprised of unmodified and 2’-OMe pyrimidine nucleotides
that require encapsulation in a lipid-based nanoparticle for efficient delivery to the target organ
[326,2]. The pursuit of novel chemical modifications that improve the biochemical (nuclease
resistance, potency) and biophysical properties of oligonucleotides continues to be an important
endeavour towards the development of more effective siRNA drugs that do not require a lipid-
complex formulation. In this regard, although the 2’-F modification is widely introduced into
siRNAs, it provides minimal stabilization towards nuclease degradation. Modifications that
retain some of the properties of 2’-F nucleotides while increasing nuclease resistance are desired.
In a collaborative study, we reported recently on the synthesis, conformational analysis and gene
silencing activity of 2’-F,4’-OMe modified siRNAs in cultured cells, and showed that they
promote efficient gene silencing [7]. More recently, our laboratory reported on the synthesis and
hybridization properties of duplexes containing a variety of other 2’,4’-modified uridine
nucleosides, e.g., 2’,4’-di-OMe, 2’-OMe,4’-F, and ara-2’-F, 4’-OMe uridines[3]. As previously
found with 2’-F, 4’-OMe uridine, this new library of uridines exhibited primarily a C3’-endo
(“North”) sugar conformation[4] and their incorporation into siRNAs led to virtually no change
in duplex melting temperature [3,5]. This property, together with the observation that C2’/C4’
modifications impart much desirable resistance towards endo/exo nuclease degradation [6-8],
prompted us to evaluate whether these analogues are compatible with RISC and able to regulate
gene expression via the RNAi pathway. Herein we report studies which assess the gene
silencing efficiency of C2’/C4’ modified siRNA duplexes (Figure C.1). Modifications were first
introduced at either internal (positions 6, 13 and 14 of the guide strand) or terminal positions
(positions 20 and 21 of the guide strand); the most potent modifications were then combined to
produce siRNAs with modifications at both overhangs and internal positions. Of the various

104
modifications tested, duplexes with a C2’-F/OMe and C4’-OMe modified ribose sugar at the 3’-
overhangs were among the most potent.
Figure C.1. A) Sequence of siRNA targeting luciferase mRNA. Modified nucleotides were
introduced at positions 6, 13, and 14 and 3’-overhang of the antisense (guide) strand; B) 2’,4’-
modified nucleosides in this study.
MATERIALS AND METHODS
Oligonucleotide Synthesis. All 2’,4’-modified phosphoramidites and oligonucleotides were
synthesized as previously reported [3]. The 4’-OMe-dT phosphoramidite was synthesized from a
C4’-C5’ alkene dT precursor according to Liboska et al. [9] (Section S3, Supplementary
Information). Oligonucleotides were purified by ion exchange HPLC (1 M lithium perchlorate
buffer) or by PAGE (22% acrylamide), and desalted as previously described [7]. All
oligonucleotides were characterized by ESI or QTOF mass spectrometry (Table S1,
Supplementary Information).
Thermal Denaturation Studies. Complementary sequences were combined in equimolar
amounts (1.5 nmol), dried, and diluted in a buffer containing 10 mM sodium phosphate (pH 7.2)
with 100 mM NaCl and 0.1 mM EDTA (1 mL). Each solution was then transferred into cuvettes
in a Varian UV spectrophotometer. Each sample was heated to 93 °C and the cooled to 15 °C at
a rate of 0.5 oC/min. The change in absorbance at 260 nm was then monitored upon heating from
15 to 93 °C. The melting temperatures were calculated from the first derivative of the melting
curves (local maxima at dy/dt=0).

105
Luciferase Assays. HeLa cells stably expressing Luciferase were counted and seeded at a
density of 7500 cells/well in a 96-well plate. Cells were then left to recover for 24 hours at 37 °C
with 5% CO2. Subsequently, cells were washed once with serum-free DMEM media and then 80
µl of serum-free DMEM media was added. siRNA and control nucleic acid preparations were
prepared by heating at 93 oC in a heat block in 1x siRNA buffer (instructions for preparation by
Dharmacon) and were allowed to anneal slowly followed by overnight cooling at 5 oC in a
fridge. siRNA duplexes were then diluted up to 20 µl with serum-free media and transfection
reagent (Oligofectamine, Invitrogen) and added to the appropriate well (for a total of 100 uL) at
increasing concentrations (0.16, 0.8, 4, 20, and 100 nM). Cells were incubated overnight (for a
total of 24 hours post-siRNA addition). Then 50 µL of ONE-Glo luciferin reagent (Promega,
USA) was added to each well and luminescence was measured and normalized to protein levels
using a Biotek Synergy HT plate reader. Data was acquired with the Gen5 software suite and
data was manipulated and plotted using Graphpad Prism software suite.
DsRed DRR Cell Transfection. This protocol was adapted from Anzahaee et al. [10]. Briefly,
DsredDRR cells were seeded in a 6-well or 24-well plate such that they would reach 60-80%
confluency at the time of transfection. siRNAs were complexed to Lipofectamine RNAiMax
(Thermo 13778075) for five minutes in Opti-MEM media (Thermo 31985062) according to the
manufacturer’s reagent protocol and added to the cells in DMEM media (Gibco 11995605) for a
final DMEM:Opti-MEM ratio of 1.5:0.5 and desired siRNA concentration. At 24 h, additional
serum-containing DMEM media was added. After a total of 72 hours of incubation at 37 oC, cells
were collected and lysed using 0.1% NP-40 (Fluka 74385). Protein expression was assessed via
western blot analysis of DRR with alpha-tubulin as a loading control.
MTS Assay. Cytotoxicity of tested nucleosides was assayed by measuring cell viability using the
Cell Titer Blue Assay (Promega). Briefly, HeLa cells were seeded at 5,000 cells/well in a 96-
well plate. The following day, samples were prepared at stock concentrations starting at 1 M in
autoclaved H2O, and were serially diluted 10-fold down to 1 µM. 10 µL of each stock solution
was then added per well at the desired concentration to be incubated with HeLa cells for 24
hours. Incubations were performed in triplicate for each concentration. After overnight
incubation, 20 µL of Cell-Titer Blue reagent was added per well and sample fluorescence was
measured using a Biotek Synergy Plate Reader (Excitation = 560 nm and Emission = 590 nm)

106
after 1 hour following addition. Analysis was performed using Graphpad Prism software and
samples were normalized to the buffer control.
RESULTS AND DISCUSSION
siRNAs with internal 2ʹ/4ʹ-modifications in the guide strand
Melting temperature of internally modified duplexes were studied first using a model siRNA
duplex targeting firefly luciferase as previously studied [11]. This model siRNA was chosen due
to its modification pattern which allocates a modified nucleotide in the sensitive seed region
(position 6) and allows exploration of the impact of two centrally located and consecutive
modified nucleotides (positions 13 and 14). Each duplex was analyzed by UV thermal melting in
order to assess how these modifications affected duplex formation. In all cases, no distortions in
duplex formation were noted during the melting-annealing cycle (Section S1, Supplementary
Information). Relative to the native siRNA, Tm values of duplexes increased upon incorporation
of modified ribouridines and 4’-OMe-dT (+1-2 oC/nt), whereas a slight decrease (ca. -0.3-0.7 oC/nt) was observed for the arabinose modifications (Table 1; Figure S1-S3, Supplementary
Information). As previously observed, duplexes with 2’,4’ modifications were more stable than
the unmodified duplexes (+0.8-1.4 oC/nt), although they were not as stable as duplexes with 2’-
modifications at the same positons [3,7,11].
Table C.1. Melting Temperature of Luciferase siRNA Duplexes with Internal Modifications at Positions 6, 13, and 14 in the Guide Strand

107
Duplexes were transfected into HeLa cells stably expressing firefly luciferase, and the
reduction of firefly luciferase protein levels was evaluated in a dose-response manner (Figure
C.2). The best activity was granted by the 2’-OMe-rU modification, performing similarly to rU.
As previously observed with 2’,4’-diF-rU [11], siRNAs with 2’-F,4’-OMe-rU and 2’-OMe,4’-
F-rU modifications also conferred excellent activity. By contrast, decreased activity was
observed with 2’,4’-diOMe-rU and 2’-F,4’-OMe-araU modifications, in line with observed
results when incorporating LNA at these positions [11]. The negative impact of 2’,4’-diOMe-rU
on gene silencing was not expected given the excellent activities of siRNAs with 2’-OMe-rU or
4’-OMe-dT at the same positions. This may reflect possible disruption of hAGO2-RNA
interactions caused by the bulkier 2’,4’-diOMe-rU residues [12-14].
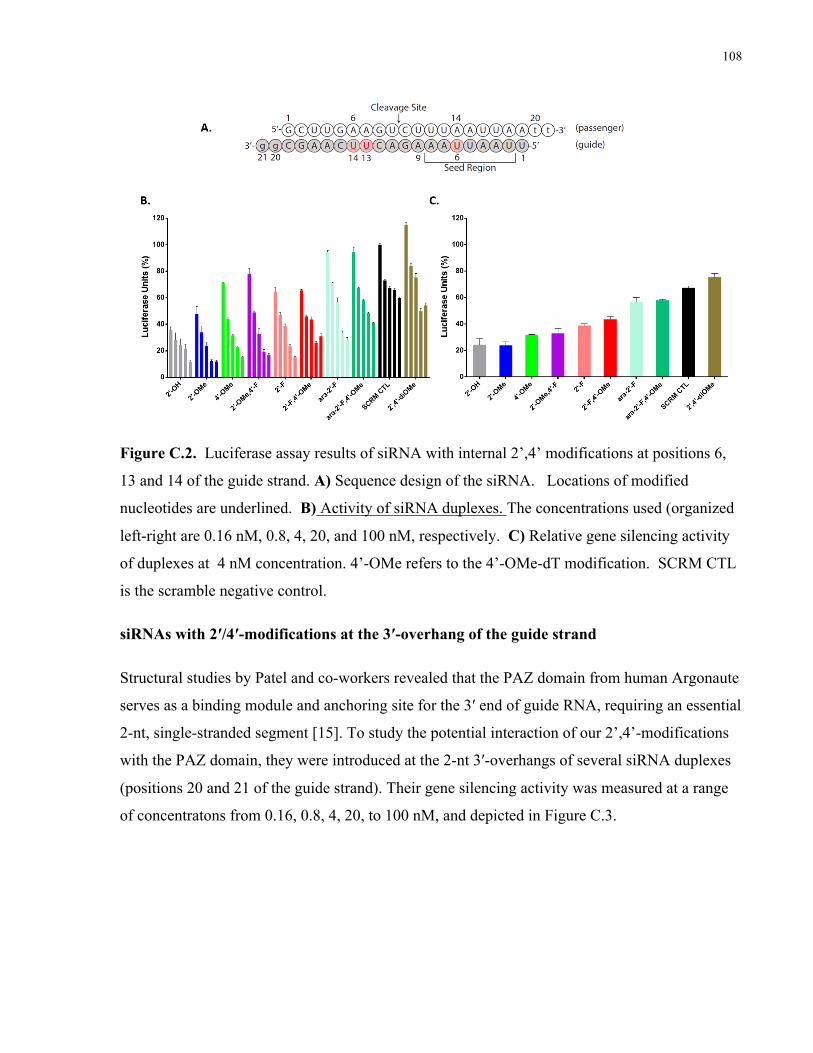
108
Figure C.2. Luciferase assay results of siRNA with internal 2’,4’ modifications at positions 6,
13 and 14 of the guide strand. A) Sequence design of the siRNA. Locations of modified
nucleotides are underlined. B) Activity of siRNA duplexes. The concentrations used (organized
left-right are 0.16 nM, 0.8, 4, 20, and 100 nM, respectively. C) Relative gene silencing activity
of duplexes at 4 nM concentration. 4’-OMe refers to the 4’-OMe-dT modification. SCRM CTL
is the scramble negative control.
siRNAs with 2ʹ/4ʹ-modifications at the 3′-overhang of the guide strand
Structural studies by Patel and co-workers revealed that the PAZ domain from human Argonaute
serves as a binding module and anchoring site for the 3′ end of guide RNA, requiring an essential
2-nt, single-stranded segment [15]. To study the potential interaction of our 2’,4’-modifications
with the PAZ domain, they were introduced at the 2-nt 3′-overhangs of several siRNA duplexes
(positions 20 and 21 of the guide strand). Their gene silencing activity was measured at a range
of concentratons from 0.16, 0.8, 4, 20, to 100 nM, and depicted in Figure C.3.
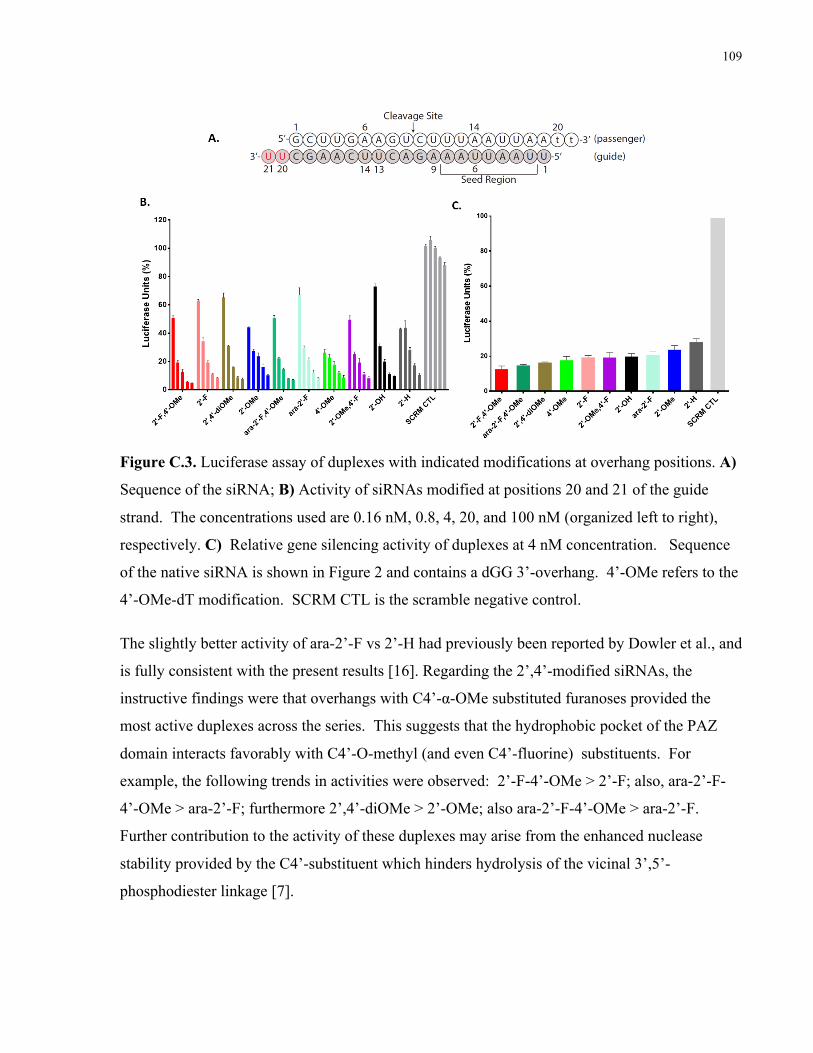
109
Figure C.3. Luciferase assay of duplexes with indicated modifications at overhang positions. A)
Sequence of the siRNA; B) Activity of siRNAs modified at positions 20 and 21 of the guide
strand. The concentrations used are 0.16 nM, 0.8, 4, 20, and 100 nM (organized left to right),
respectively. C) Relative gene silencing activity of duplexes at 4 nM concentration. Sequence
of the native siRNA is shown in Figure 2 and contains a dGG 3’-overhang. 4’-OMe refers to the
4’-OMe-dT modification. SCRM CTL is the scramble negative control.
The slightly better activity of ara-2’-F vs 2’-H had previously been reported by Dowler et al., and
is fully consistent with the present results [16]. Regarding the 2’,4’-modified siRNAs, the
instructive findings were that overhangs with C4’-α-OMe substituted furanoses provided the
most active duplexes across the series. This suggests that the hydrophobic pocket of the PAZ
domain interacts favorably with C4’-O-methyl (and even C4’-fluorine) substituents. For
example, the following trends in activities were observed: 2’-F-4’-OMe > 2’-F; also, ara-2’-F-
4’-OMe > ara-2’-F; furthermore 2’,4’-diOMe > 2’-OMe; also ara-2’-F-4’-OMe > ara-2’-F.
Further contribution to the activity of these duplexes may arise from the enhanced nuclease
stability provided by the C4’-substituent which hinders hydrolysis of the vicinal 3’,5’-
phosphodiester linkage [7].

110
siRNAs combining internal and overhang 2’/4’-modifications
Next, we sought to combine some of the best performing internal modifications (modifications at
positions 6, 13 and 14) with the best performing overhang modifications (modifications at
positions 20 and 21) with the aim of maximizing chemical modification without compromising
activity (Table 2). The sequence and melting temperatures of the duplexes prepared are provided
in Table 2, and their activity summarized in Figure C.4. For comparison, the Tm and gene
silencing activity of these duplexes were compared to siRNAs containing modifications at
overhangs or internal positions.
Table C.2. Melting Temperature of Luciferase siRNA Duplexes with a Modified Guide Strand.

111
Figure C.4. Luciferase assay of duplexes modified at at positions 6, 13, 14, 20 and 21. A)
Sequence of the siRNA; B) Activity of modified duplexes. The concentrations used are 0.16 nM,
0.8, 4, 20, and 100 nM (left to right), respectively; C) Relative gene silencing activity of
duplexes at 4 nM concentration. Sequence of the native siRNA is shown in Figure 2 and
contains a dGG 3’-overhang. 4’-OMe refers to the 4’-OMe-dT modification. SCRM CTL is the
scramble negative control.
UV experiments were performed in order to verify if the additional modifications affected the
melting temperature of the duplexes (Table 2, Figures S1-S3). Interestingly, the Tm of duplexes
with combined modifications at positions 6, 13, 14, 20 and 21 displayed similar or slightly higher
values compared to duplexes with internal modification at positions 6, 13, and 14 (DTm = + 0.3-
3.0 oC). It is also interesting to note that the modified rPy overhangs tested at positions 20 and
21, namely 2’-F-4’-OMe-rU, 2’,4’-diOMe-rU and 2’-F-4’-OMe-araU all provided significant
increases in Tm (+2.2 oC /nt) relative to the duplex with a native rPu (dGG) overhang. While the
origin for this difference cannot be reconciled with the data available, the larger stabilizing effect
of the modified rPy ‘dangling’ ends may be attributed to an overall more cross-stacking of the
modified residues which better conform to A-form structure of the siRNA duplex [17]. In other
words, the propensity of the unstacked dangling nucleotide to a stacked geometry should be

112
easier for a nucleotide pre-organized in the A-like C3’-endo conformation (such as 2’,4’-
modified rU) relative to a more flexible deoxynucleotide.
Regarding gene silencing activity, the combinations of modifications afforded siRNAs with
potencies similar to those seen for siRNAs with a solely overhang or a solely internally modified
guide strand. This implies that a desired level of activity can be achieved while maintaining a
high level of modification necessary for nuclease protection and immune system evasion.
Activity of the combination of best internal and overhang motifs against DRR gene
To validate the generality of our observations, we designed a new set of siRNAs targeting the
Downregulated in Renal (DRR) Cell Carcinoma gene as previously reported [18,19]. The DRR
protein contributes to the aggressive and invasive nature of glioblastoma stem cells by cross
linking microtubules and actin at focal adhesions and activating downstream pathways of Protein
Kinase B [18]. Knockdown of the DRR gene is a promising therapeutic strategy for decreasing
the invasion of brain cancer stem cells [19,20]. One duplex contained 2’-F-4’-OMe-rU residues
at the 2-nt 3’-overhang of the guide strand; the other contained three additional 2’-F -rU residues
at internal positions. Their sequences are shown in Table 3, along with that of the unmodified
siRNA control. DsedDRR cells were transfected with modified and control siRNA duplexes.
All siRNA duplexes targeting the DRR gene provided significant knockdown compared to the
scrambled control; the DRR positive CTRL duplex was the most active, followed by the siRNA
duplex with modified overhangs (Figure C.5). The least active was the most modified (112-2),
however it still exhibited excellent activity (IC50 ca. 4 nM) under the conditions tested.
Table 3. Sequences of Down-Regulated in Renal Cell Carcinoma (DRR) siRNAs.
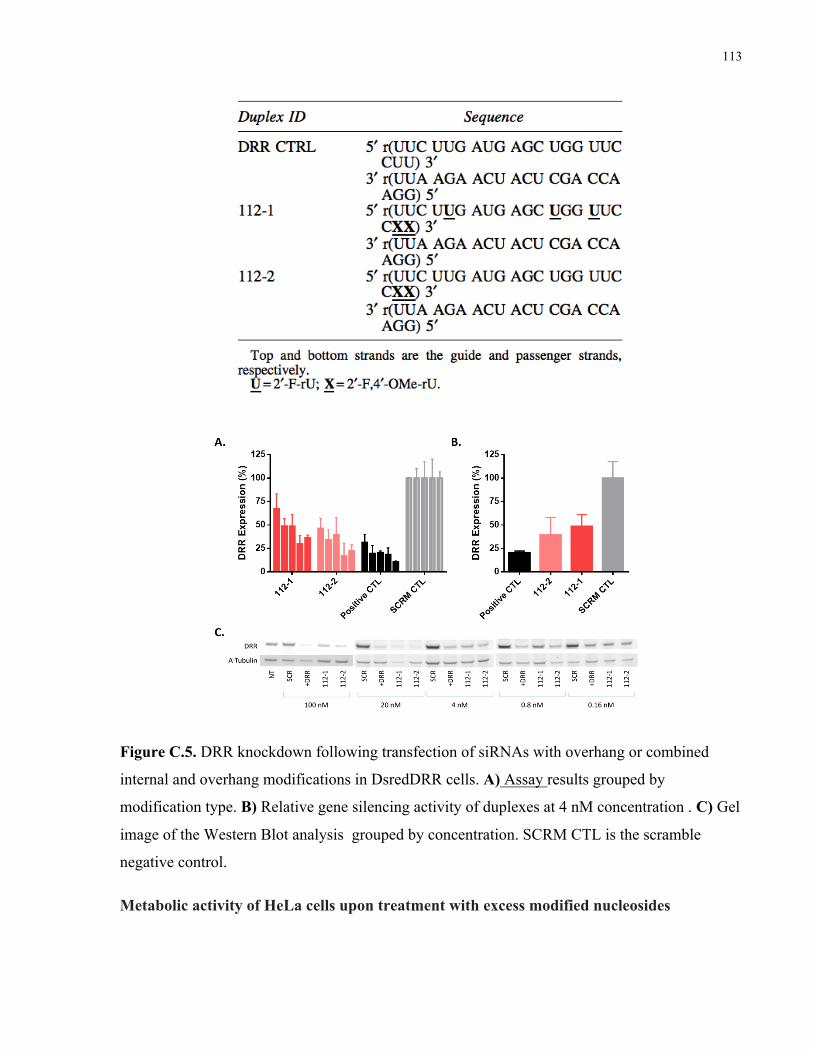
113
Figure C.5. DRR knockdown following transfection of siRNAs with overhang or combined
internal and overhang modifications in DsredDRR cells. A) Assay results grouped by
modification type. B) Relative gene silencing activity of duplexes at 4 nM concentration . C) Gel
image of the Western Blot analysis grouped by concentration. SCRM CTL is the scramble
negative control.
Metabolic activity of HeLa cells upon treatment with excess modified nucleosides
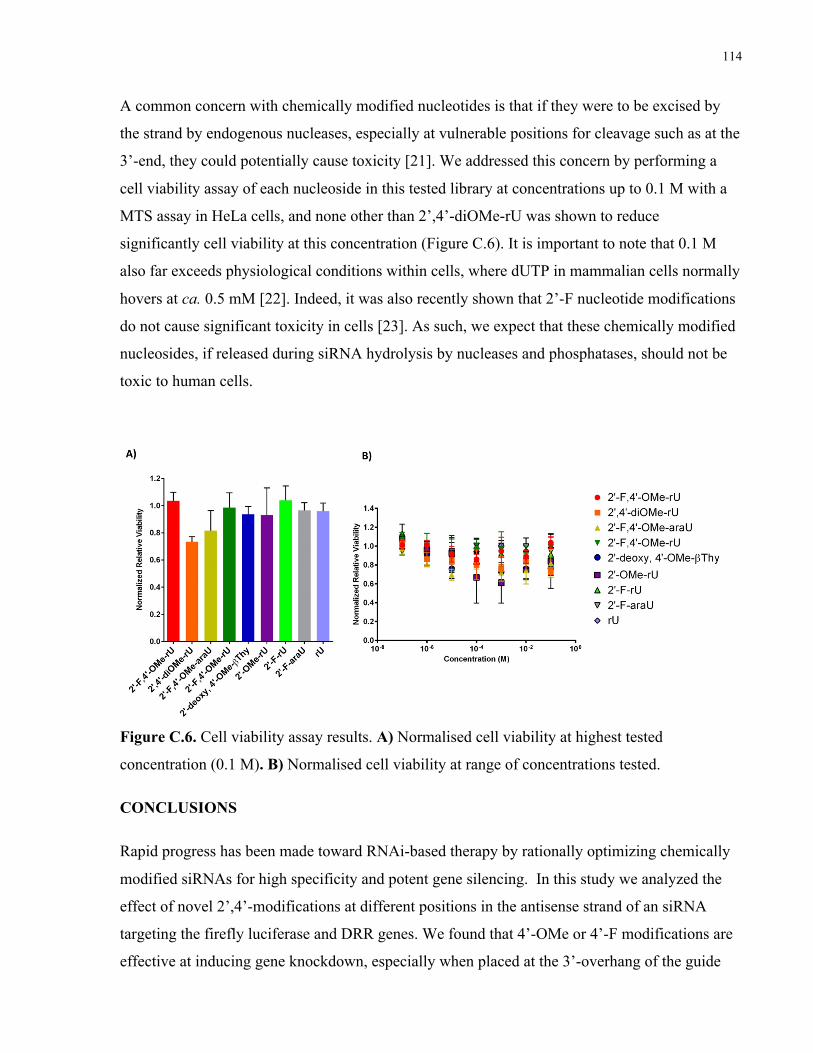
114
A common concern with chemically modified nucleotides is that if they were to be excised by
the strand by endogenous nucleases, especially at vulnerable positions for cleavage such as at the
3’-end, they could potentially cause toxicity [21]. We addressed this concern by performing a
cell viability assay of each nucleoside in this tested library at concentrations up to 0.1 M with a
MTS assay in HeLa cells, and none other than 2’,4’-diOMe-rU was shown to reduce
significantly cell viability at this concentration (Figure C.6). It is important to note that 0.1 M
also far exceeds physiological conditions within cells, where dUTP in mammalian cells normally
hovers at ca. 0.5 mM [22]. Indeed, it was also recently shown that 2’-F nucleotide modifications
do not cause significant toxicity in cells [23]. As such, we expect that these chemically modified
nucleosides, if released during siRNA hydrolysis by nucleases and phosphatases, should not be
toxic to human cells.
Figure C.6. Cell viability assay results. A) Normalised cell viability at highest tested
concentration (0.1 M). B) Normalised cell viability at range of concentrations tested.
CONCLUSIONS
Rapid progress has been made toward RNAi-based therapy by rationally optimizing chemically
modified siRNAs for high specificity and potent gene silencing. In this study we analyzed the
effect of novel 2’,4’-modifications at different positions in the antisense strand of an siRNA
targeting the firefly luciferase and DRR genes. We found that 4’-OMe or 4’-F modifications are
effective at inducing gene knockdown, especially when placed at the 3’-overhang of the guide

115
strand. We hypothesize that favourable interaction with the PAZ domain of hAGO2 and
enhanced nuclease stability provided by these modifications both contribute to the improved
activity of these modified siRNAs. This study expands the toolbox of desirable chemical
modifications for gene silencing applications and opens the avenue for further studies of these
chemical modifications against other gene targets.
ACKNOWLEDGEMENTS
We would like to thank Dr. Hanadi Sleiman for her generous contribution of resources to this
project. Financial support was provided by the Natural Sciences and Engineering Research
Council of Canada (Discovery grant to M.J.D.; CGSD to A.E.A), and the Canadian Institute for
Health Research (CHRP to M.J.D., M.S.S.).
Conflict of Interest Statement: No competing financial interests exist.
REFERENCES
1. Adams D, A Gonzalez-Duarte, WD O’Riordan, CC Yang, M Ueda, AV Kristen, I Tournev, HH Schmidt, T Coelho, JL Berk, K.-P. Lin, G Vita, S Attarian, V Planté-Bordeneuve, MM Mezei, JM Campistol, J Buades, TH Brannagan, BJ Kim, J Oh, Y Parman, Y Sekijima, PN Hawkins, SD Solomon, M Polydefkis, PJ Dyck, PJ Gandhi, S Goyal, J Chen, AL Strahs, SV Nochur, MT Sweetser, PP Garg, AK Vaishnaw, JA Gollob and OB Suhr. (2018). Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 379:11-21.
2. Kulkarni JA, PR Cullis and R van der Meel. (2018). Lipid nanoparticles enabling gene therapies: from concepts to clinical utility. Nucleic Acid Ther 28: 146-157.
3. Malek-Adamian E, MB Patrascu, SK Jana, S Martínez-Montero, N Moitessier and MJ Damha. (2018). Adjusting the structure of 2’-modified nucleosides and oligonucleotides via C4’-α-F or C4’-α-OMe. substitution: synthesis and conformational analysis. J Org Chem 83: 9839-9849.
4. Damha MJ and KK Ogilvie. (2018). Conformational properties of branched RNA fragments in aqueous solution. Biochemistry 27: 6403-6416.
5. Harp J, DC Guenther, A Bisbe, L Perkins, S Matsuda, GR Bommineni, I Zlatev, DJ Foster, N Taneja and K Charisse. (2018). Structural basis for the synergy of 4'- and 2'-modifications on siRNA nuclease resistance, thermal stability and RNAi activity. Nucleic Acids Res 46: 8090-8104.
6. Petrová M, O Páv, M Buděšínský, E Zborníková, P Novák, Š. Rosenbergová, O Pačes, R Liboska, I Dvořáková, O Šimák and I Rosenberg. (2015). Straightforward synthesis of purine 4′-alkoxy-2′-deoxynucleosides: first report of mixed purine–pyrimidine 4′-alkoxyoligodeoxynucleotides as new RNA mimics. Org Letts 17: 3426-3429.
7. Malek-Adamian E., DC Guenther, S Matsuda, S Martínez-Montero, I Zlatev, J Harp, M Burai Patrascu, DJ Foster, J Fakhoury, L Perkins, N Moitessier, RM Manoharan, N Taneja, A Bisbe, K Charisse, M Maier, KG Rajeev, M Egli, M Manoharan and MJ Damha. (2017).

116
4′-C-Methoxy-2′-deoxy-2′-fluoro Modified Ribonucleotides Improve Metabolic Stability and Elicit Efficient RNAi-Mediated Gene Silencing. J Am Chem Soc 139: 14542-14555.
8. He XY, J Wang, DD Lu and SQ Wang. (2018). Synthesis and antisense properties of 2′β-F-arabinouridine modified oligonucleotides with 4′-C-OMe substituent. Molecules 23: 2374.
9. Liboska R, J Snasel, I Barvik, M Budesinsky, R Pohl, Z Tocik, O Pav, D Rejman, P Novak and I Rosenberg. (2011). 4′-Alkoxy oligodeoxynucleotides: a novel class of RNA mimics. Org Biomol Chem 9: 8261-8267.
10. Anzahaee MY, GF Deleavey, PU Le, J Fakhoury, K Petrecca and MJ Damha. (2014). Arabinonucleic acids: 2'-stereoisomeric modulators of siRNA activity. Nucleic Acid Ther 24, 336-343.
11. Martínez-Montero S, GF Deleavey, N Martín-Pintado, JF Fakhoury, C González and MJ Damha. (2015). Locked 2′-deoxy-2′,4′-difluororibo modified nucleic acids: thermal stability, structural studies, and siRNA activity. ACS Chem Biol 10: 2016-2023.
12. Shukla S, CS Sumaria and PI Pradeepkumar. (2010). Exploring chemical modifications for siRNA therapeutics: a structural and functional outlook. ChemMedChem 5: 328-349.
13. Deleavey GF and MJ Damha. (2012). Designing chemically modified oligonucleotides for targeted gene silencing. Chem Biol 19, 937-954.
14. Wang Y, S Juranek, H Li, G Sheng, GS Wardle, T Tuschl and DJ Patel. (2009). Nucleation, propagation and cleavage of target RNAs in Ago silencing complexes. Nature 461: 754-761.
15. Ma JB, K Ye and DJ Patel. (2004). Structural basis for overhang-specific small interfering RNA recognition by the PAZ domain. Nature 429: 318-322.
16. Dowler T, D Bergeron, AL Tedeschi, L Paquet, N Ferrari and MJ Damha. (2006). Improvements in siRNA properties mediated by 2'-deoxy-2'-fluoro-beta-D-arabinonucleic acid (FANA). Nucleic Acids Res 34: 1669-1675.
17. Kara M and M Zacharias. (2014). Stabilization of duplex DNA and RNA by dangling ends studied by free energy simulations. Biopolymers 101: 418-427.
18. Le PU, A Angers-Loustau, RMW de Oliveira, A Ajlan, CL Brassard, A Dudley,H Brent, V Siu, G Trinh, G Molenkamp, J Wang, M Seyed Sadr, B Bedell, RF Del Maestro and K Petrecca. (2010). DRR drives brain cancer invasion by regulating cytoskeletal-focal adhesion dynamics. Oncogene 29: 4636-4647.
19. Dudley A, M Sater, PU Le, G Trinh, MS Sadr, J Bergeron, GF Deleavey, B Bedell, MJ Damha and K Petrecca. (2014). DRR regulates AKT activation to drive brain cancer invasion. Oncogene 33: 4952-4960.
20. Arnold AE, E Malek-Adamian, PU Le, A Meng, S Martínez-Montero, K Petrecca, MJ Damha and MS Shoichet. (2018). Antibody-antisense oligonucleotide conjugate downregulates a key gene in glioblastoma stem cells. Mol Ther Nucleic Acids 11: 518-527.
21. Andersson S, M Antonsson, M Elebring, R Jansson-Löfmark and L Weidolf, L. (2018). Drug metabolism and pharmacokinetic strategies for oligonucleotide- and mRNA-based drug development. Drug Discov Today 23: 1733-1745.
22, Traut TW. (1994). Physiological concentrations of purines and pyrimidines. Mol Cell Biochem 140: 1-22.
23. Janas MM, I Zlatev, J Liu, Y Jiang, SA Barros, JE Sutherland, WP Davis, J Liu, CR Brown, X Liu, MK Schlegel, L Blair, X Zhang, B Das, C Tran, K Aluri, J Li, S Agarwal, R Indrakanti, K Charisse, J Nair, S Matsuda, KG Rajeev, T Zimmermann, L Sepp-Lorenzino, Y Xu, A Akinc, K Fitzgerald, AK Vaishnaw, PF Smith, M Manoharan, V Jadhav, JT Wu and MA Maier. (2019). Safety evaluation of 2′-deoxy-2′-fluoro nucleotides in GalNAc-siRNA conjugates. Nucleic Acids Res. https://doi.org/10.1093/nar/gkz140.

117
Appendix D: Additional characterization of AONs and siRNAs
against DRR
Purpose
Many of the AONs and siRNAs used in during the course of this thesis were characterized in
terms of their stability and functional effects; not all of this data was published, but it is
important for the understanding of the stability and potency of the different AONs and siRNAs
against DRR used in this thesis.
Methods
Nuclease stability assays
AON samples (dsDRR and mAb-dsDRR) or DRR siRNAs were incubated with a solution of
10% FBS in PBS, pH 7.4 for up to 24h. At each timepoint, the entire contents of the sample were
analyzed using acrylamide electrophoresis to separate nucleic acid material based on size. The
remaining full length siRNA or AON was quantified by imageJ and the data was reported as a
percentage of intact AON or siRNA.
Invasion assay
Hydrogels were synthesized as previously described by Tam et al.7 Cells were plated on
hydrogels at a density of 3500 cells/hydrogel and allowed to adhere for 24h. Cells were then
treated with transfection complexes at a concentration of approximately 25 nM. 48h after
treatment fresh media was added to each well and cells were fixed with 4% PFA 4-5 days after
treatment. Cells were stained with Hoechst to identify cell nuclei and 15 µM beads were added to
each well to label the surface of the hydrogel. Hydrogels were imaged using confocal
microscopy and depth of invasion was analyzed using custom computational software.
Figures
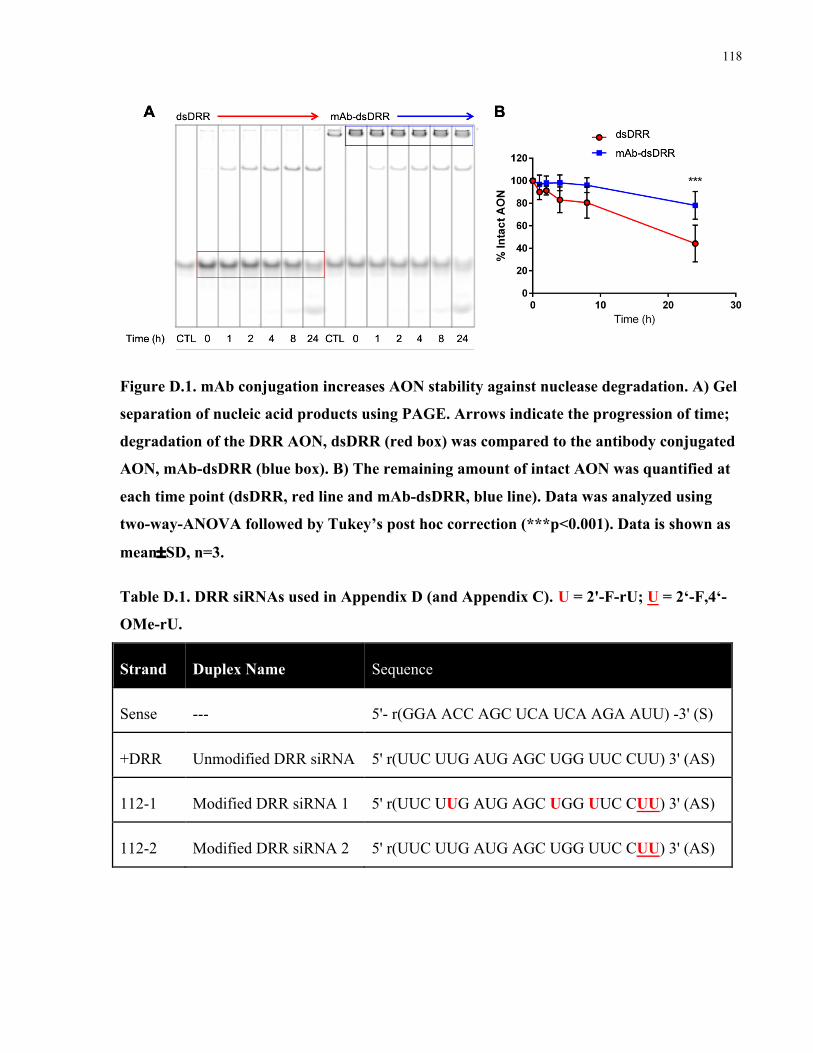
118
Figure D.1. mAb conjugation increases AON stability against nuclease degradation. A) Gel
separation of nucleic acid products using PAGE. Arrows indicate the progression of time;
degradation of the DRR AON, dsDRR (red box) was compared to the antibody conjugated
AON, mAb-dsDRR (blue box). B) The remaining amount of intact AON was quantified at
each time point (dsDRR, red line and mAb-dsDRR, blue line). Data was analyzed using
two-way-ANOVA followed by Tukey’s post hoc correction (***p<0.001). Data is shown as
mean±SD, n=3.
Table D.1. DRR siRNAs used in Appendix D (and Appendix C).U = 2'-F-rU; U = 2‘-F,4‘-
OMe-rU.
Strand Duplex Name Sequence
Sense --- 5'- r(GGA ACC AGC UCA UCA AGA AUU) -3' (S)
+DRR Unmodified DRR siRNA 5' r(UUC UUG AUG AGC UGG UUC CUU) 3' (AS)
112-1 Modified DRR siRNA 1 5' r(UUC UUG AUG AGC UGG UUC CUU) 3' (AS)
112-2 Modified DRR siRNA 2 5' r(UUC UUG AUG AGC UGG UUC CUU) 3' (AS)

119
Figure D.2. Chemical modifications increase siRNA stability against nuclease degradation.
At each time point, the amount of intact siRNA was quantified. Data was analyzed using
two-way-ANOVA followed by Tukey’s post hoc correction (***p<0.001). Data is shown as
mean±SD, n=3.
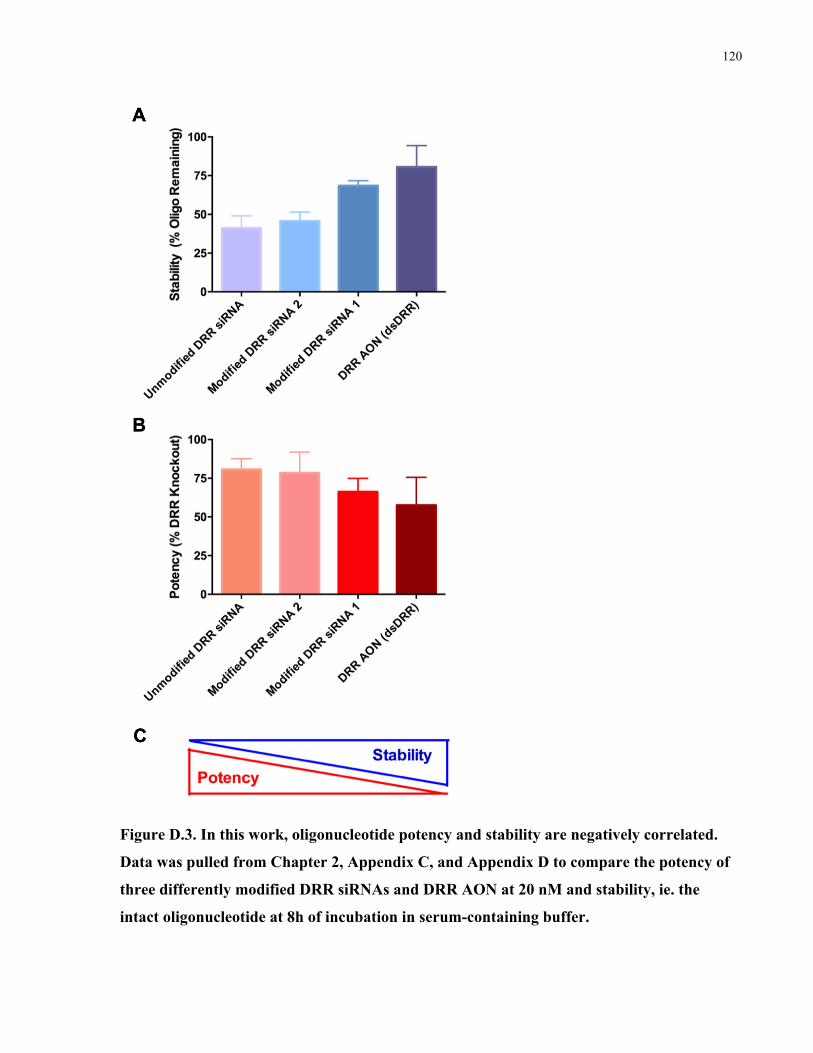
120
Figure D.3. In this work, oligonucleotide potency and stability are negatively correlated.
Data was pulled from Chapter 2, Appendix C, and Appendix D to compare the potency of
three differently modified DRR siRNAs and DRR AON at 20 nM and stability, ie. the
intact oligonucleotide at 8h of incubation in serum-containing buffer.
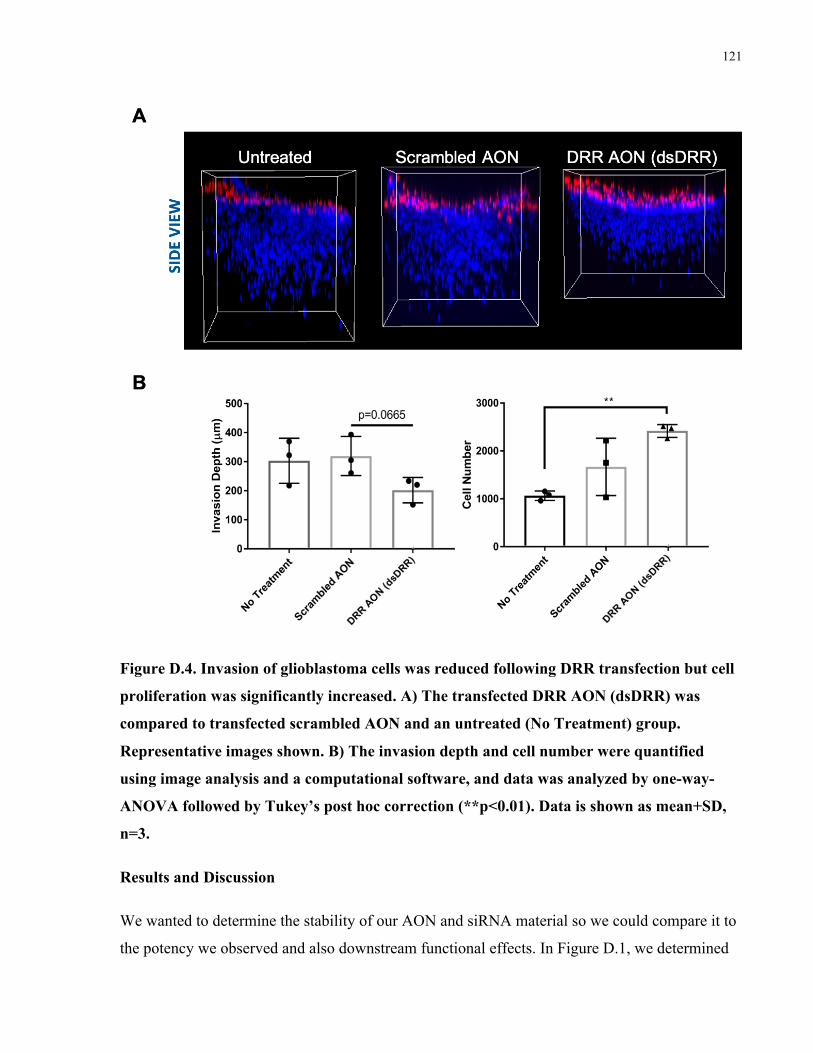
121
Figure D.4. Invasion of glioblastoma cells was reduced following DRR transfection but cell
proliferation was significantly increased. A) The transfected DRR AON (dsDRR) was
compared to transfected scrambled AON and an untreated (No Treatment) group.
Representative images shown. B) The invasion depth and cell number were quantified
using image analysis and a computational software, and data was analyzed by one-way-
ANOVA followed by Tukey’s post hoc correction (**p<0.01). Data is shown as mean+SD,
n=3.
Results and Discussion
We wanted to determine the stability of our AON and siRNA material so we could compare it to
the potency we observed and also downstream functional effects. In Figure D.1, we determined

122
that the AON was relatively stable (~50% remaining at 24h) and that conjugation to a protein,
such as an antibody, could further significantly increase its stability.
Several siRNAs against DRR were described in Appendix C; in Figure C.5, these are labelled
Positive control, 112-1, and 112-2; they are also known as Unmodified DRR siRNA, Modified
DRR siRNA 1, and Modified DRR 2, respectively. These siRNA strands are shown in Table D.1
and they contain different modification patterns. The stability of these siRNAs is shown in
Figure D.2, and the Modified DRR siRNA 1 is significantly more stable than the other strands at
8h.
In Figure D.3, data was pulled from Chapter 2, Appendix C, and Appendix D to correlate the
potency of these AONs and siRNAs at 25 nM and the stability at 8h of nuclease incubation. We
observed a negative correlation between stability in potency; that is, the more intact nucleic acid
material persisted, the less the oligonucleotides worked to reduce expression of DRR. This is
discussed further in the thesis discussion chapter.
Finally, in Figure D.4, we wanted to test the effect of transfected DRR antisense oligonucleotide
(dsDRR) on invasion of glioblastoma cells in a 3D hydrogel model. We saw that the invasion
was reduced, but it was not a statistically significant reduction in invasion. Interestingly, we also
observed that the proliferation of the cells was significantly increased following transfection of
the DRR antisense oligonucleotide in the 3D hydrogel model.

123
Appendix E: In vivo biodistribution of a CD44 antibody conjugated
to a fluorescent dye
Purpose
In order to determine the best injection route for an antibody conjugate to reach glioblastoma
stem cells, we carried out a biodistribution study using a Cy5-labelled antibody (ADC(CD44-
Cy5)). The control was a non-specific IgG (ADC(CTL-Cy5)). We injected the antibodies in five
different ways, including intravenous, intraventricular, and intratumoral injections into mice with
pre-established tumors from GSC injections, as well as co-injection with GSCs into naïve mice.
Methods
Conjugation of CD44 Ab to Cy5 Fluorophore
CD44 Ab or IgG control was reacted with Sulfo-Cy5-NHS Ester (ab146459) according to the
manufacturer’s protocols. Briefly, 10 eq. of the Cy5-NHS Ester was mixed with the antibody for
30 min at RT. The excess Cy5 was then purified by dialysis and successful labeling of the
antibody was confirmed by absorbance at 650 nM for the Cy5 fluorophore.
In Vivo Injections of CD44-Cy5 Conjugate
ADC(CD44-Cy5) or ADC(CTL-Cy5) were dialyzed into PBS and sterile filtered prior to
injection. All animal procedures were approved by the Institution’s Animal Care Committee and
performed according to the guidelines of the Canadian Council of Animal Care. Female NSG
mice (Charles River, Canada) were anesthetized at 6 weeks of age using intraperitoneal injection
containing Ketamine, Xylazine and Acepromazine. For all injections, the mice were placed on a
stereotaxic apparatus and a midline scalp incision was made. A burr-hole (3–5 mm) was created
2.2 mm lateral to the bregma using a high-powered drill. Tumor cells were GFP-labeled GSCs
provided by Dr. Kevin Petrecca, McGill University. The conjugate was injected at 15 ug/mouse
for all groups, except for the IV injection high dose, which was injected at 100 ug/mouse. IV
injections, intraventricular injections, and intratumoral injections were carried out in mice that
had pre-established tumors by stereotactically injecting GSCs 2 weeks before the start of the
study. IV injections were via tail vein; intraventricular injections were into the left ventricle; and
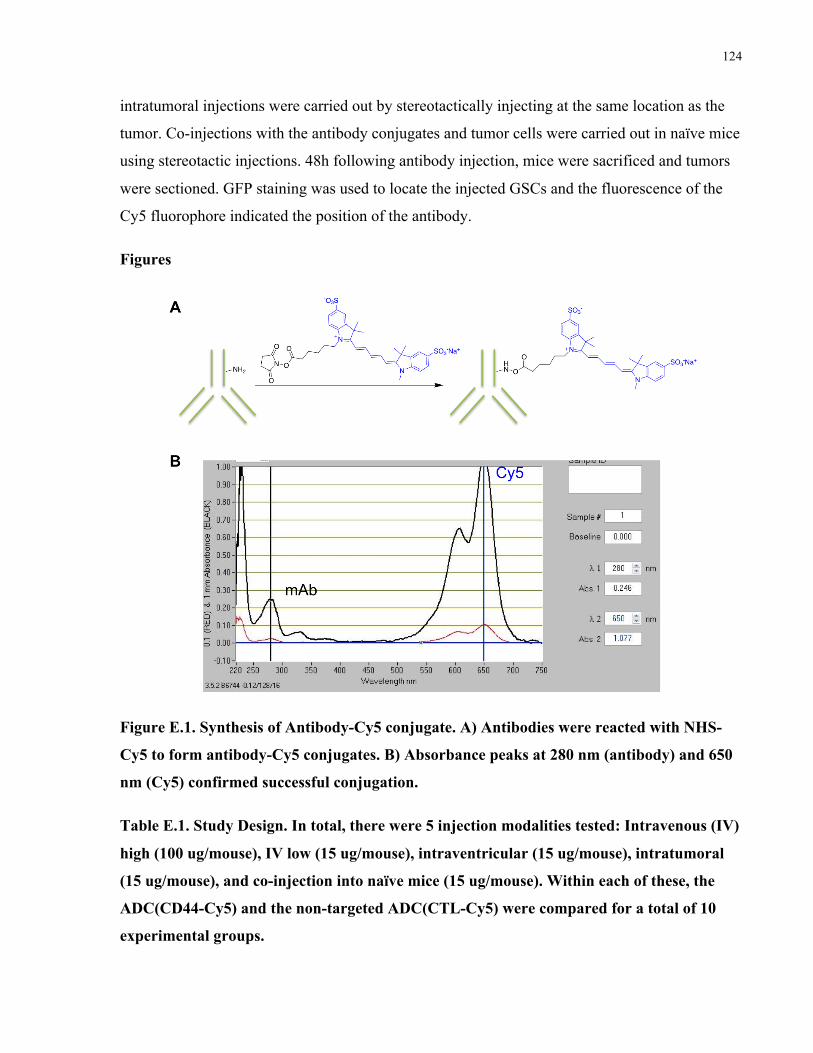
124
intratumoral injections were carried out by stereotactically injecting at the same location as the
tumor. Co-injections with the antibody conjugates and tumor cells were carried out in naïve mice
using stereotactic injections. 48h following antibody injection, mice were sacrificed and tumors
were sectioned. GFP staining was used to locate the injected GSCs and the fluorescence of the
Cy5 fluorophore indicated the position of the antibody.
Figures
Figure E.1. Synthesis of Antibody-Cy5 conjugate. A) Antibodies were reacted with NHS-
Cy5 to form antibody-Cy5 conjugates. B) Absorbance peaks at 280 nm (antibody) and 650
nm (Cy5) confirmed successful conjugation.
Table E.1. Study Design. In total, there were 5 injection modalities tested: Intravenous (IV)
high (100 ug/mouse), IV low (15 ug/mouse), intraventricular (15 ug/mouse), intratumoral
(15 ug/mouse), and co-injection into naïve mice (15 ug/mouse). Within each of these, the
ADC(CD44-Cy5) and the non-targeted ADC(CTL-Cy5) were compared for a total of 10
experimental groups.
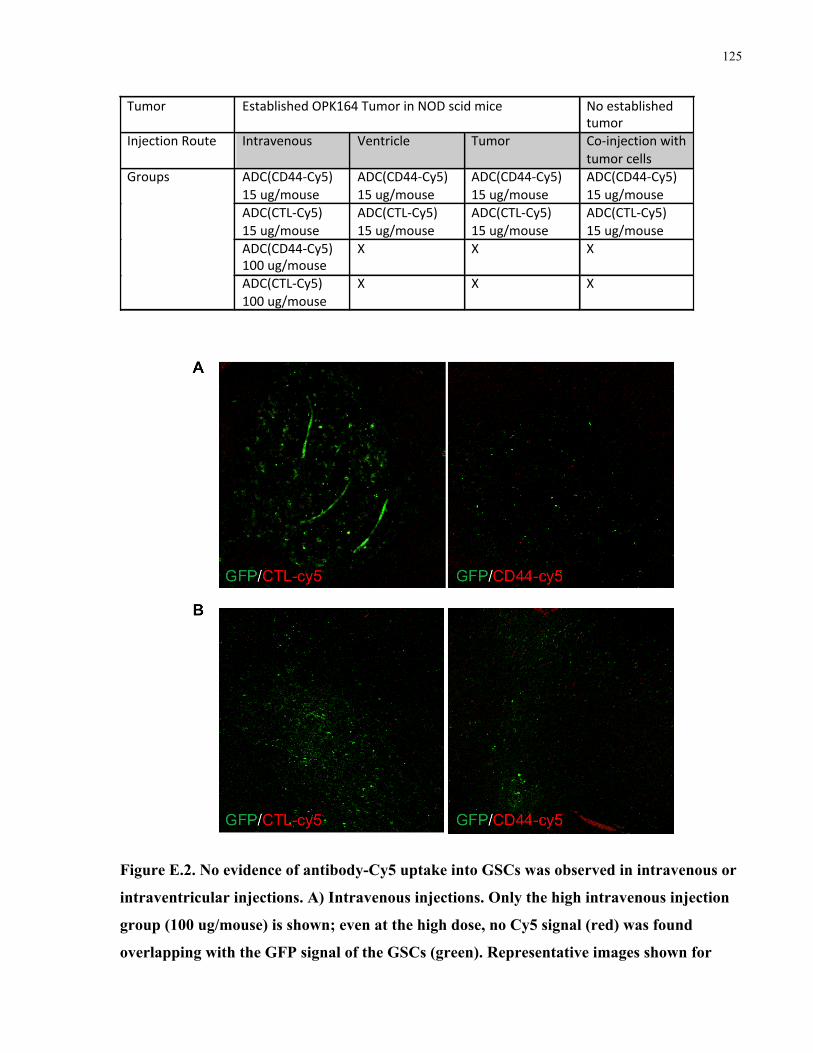
125
Figure E.2. No evidence of antibody-Cy5 uptake into GSCs was observed in intravenous or
intraventricular injections. A) Intravenous injections. Only the high intravenous injection
group (100 ug/mouse) is shown; even at the high dose, no Cy5 signal (red) was found
overlapping with the GFP signal of the GSCs (green). Representative images shown for
Tumor EstablishedOPK164TumorinNODscidmice Noestablishedtumor
InjectionRoute Intravenous Ventricle Tumor Co-injectionwithtumorcells
Groups ADC(CD44-Cy5)15ug/mouse
ADC(CD44-Cy5)15ug/mouse
ADC(CD44-Cy5)15ug/mouse
ADC(CD44-Cy5)15ug/mouse
ADC(CTL-Cy5)15ug/mouse
ADC(CTL-Cy5)15ug/mouse
ADC(CTL-Cy5)15ug/mouse
ADC(CTL-Cy5)15ug/mouse
ADC(CD44-Cy5)100ug/mouse
X X X
ADC(CTL-Cy5)100ug/mouse
X X X

126
ADC(CTL-Cy5) and ADC (CD44-Cy5). B) Intraventricular injections. No evidence of Cy5
signal (red) was found to be colocalized with the GFP (green) following injection into the
left ventricle.
Figure E.3. Intratumoral injections. Some colocalization with the antibody (red) and the
GSCs (green) was observed following direct intratumoral injection, and potentially a
higher amount of overlap between the ADC(CD44-Cy5), as indicated with yellow arrows,
compared to the ADC(CTL-Cy5). Representative images shown.
Figure E.4 Co-injections. Following co-injection of antibody conjugates (red) and GSCs
(green) into naïve mice, a significant amount of colocalization (yellow) was observed
between the ADC(CD44-Cy5) and the GSCs. Little to no overlap was observed between the
ADC(CTL-Cy5) and the GSCs. Representative images shown.

127
Results and Discussion
In order to track the biodistribution of an antibody in vivo, we modified the antibody with a
fluorophore, Cy5 (Figure E.1). We then designed a study that would compare multiple injection
routes and doses to find the best overlap between the delivered fluorescent antibody and the
GSCs in vivo (Table E.1). No uptake into the GSCs was observed when the antibody conjugates
were injected into the tail vein (Figure E.2A) or into the ventricle (Figure E.2B), indicating that
these delivery methods would not have a high chance of success for delivering an antibody-
conjugate to cells within the brain tissue.
With a future in vivo study in mind, we also explored injection directly into pre-established
tumors or co-injection that involved pre-mixing of the antibody and the GSCs immediately
followed by injection of the cocktail into naïve mice. When we injected the antibody-Cy5
conjugates into pre-established tumors, we observed some overlap between the antibody and the
GSCs (Figure E.3) and perhaps more areas of overlap with the targeted CD44 antibody
compared to the non-targeted CTL antibody, suggesting that this could be a possible route for
delivering antibody conjugates in vivo. Finally, we also co-injected GSCs and the antibody
conjugates, and observed a significant amount of overlap between the targeted CD44 antibody
and the GSCs but not for the non-targeting CTL antibody (Figure E.4). Although this would not
be practical as a treatment strategy, it could be a good way to carry out a proof-of-concept in
vivo study and ensure plenty of uptake of the antibody conjugate into the GSCs. This study had
quite a few limitations: for most groups we only tested one dose of the antibody conjugates; we
only analyzed images from one mouse for each injection strategy; and we did not quantify the
extent of colocalization between the antibody and the GSCs. Nevertheless, this data qualitatively
suggests that the best injection route for achieving colocalization between antibodies and tumor
cells is co-injection of tumor cells and the antibody treatment.

128
Appendix F: Polymeric nanomicelles for targeted therapeutic
delivery to glioblastoma
Rationale
Glioblastoma multiformes (GBMs), a type of brain cancer, is one of the deadliest cancers and
represents a significant clinical challenge. It is hypothesized that GBM cancer stem cells (GSCs)
are the major cell type responsible for glioblastoma invasion, resistance, and recurrence.21 Small
interfering ribonucleic acids (siRNAs) are potent therapeutics that can be engineered to target
specific proteins responsible for cancer cell proliferation and invasion.81 For this work, DRR
(downregulated in renal cell carcinoma), a gene associated with the GSC invasion, resistance,
and stemness, was chosen as the genetic target in a series of primary brain cancer cell lines.64, 66
Polymeric nanomicelles are promising vehicles for the delivery of genetic therapeutics to
cancerous tissue, stabilizing the nucleic acids with respect to cleavage and enabling targeted
delivery strategies. We have synthesized poly(D,L-lactide-co-2-methyl-2-carboxytrimethylene
carbonate)-graft-poly(ethylene glycol)-azide (P(LA-co-TMCC)-g-PEG-N3), a polymeric
nanomicellular platform with azide reactive sites for ligand conjugation. The azide functional
groups at the terminal ends of the poly(ethylene glycol) polymers are capable of undergoing
Huisgen [3+1] click-type conjugations. This delivery platform is being investigated for the
simultaneous conjugation of both targeting antibodies and oligonucleotides specific to GSCs.
This appendix describes the synthesis and application of polymeric nanomicelles functionalized
with anti-EphA2 Fabs and anti-DRR oligonucleotides.
Methods
P(LA-co-TMCC)-g-PEG-N3 Synthesis
P(LA-co-TMCC)-g-PEG-N3 micelles were synthesized and characterized according to
previously reported methods.385-386 Briefly, The P(LA-co-TMCC) backbone is synthesized by a
ring-opening polymerization of the monomers D,L-lactide and 5-methyl-5-benzyloxycarbonyl-
1,3- trimethylene carbonate (TMCC-Bn) initiated by 1-pyrenebutanol and catalyzed by an
organic thiourea compound. The TMCC-Bn is deprotected by palladium-catalyzed
hydrogenolysis. A graft-to approach is used bifunctional azide-poly(ethylene glycol)-amine

129
(NH2−PEG-N3) onto the P(LA-co-TMCC) backbone using diisopropylcarbodiimide (DIC) and
hydroxybenzotriazole (HOBt) coupling chemistry in DMF. The resulting polymer is 90 mol %
LA and 10 mol % TMCC, with an average of 2-3 PEG chains per backbone. Finally, micelles are
formed by a dialysis self-assembly procedure against phosphate buffered saline (1×, PBS).
P(LA-co-TMCC)-g-PEG-N3 Functionalization
P(LA-co-TMCC)-g-PEG-N3 micelles were mixed with DBCO-modified anti-DRR
oligonucleotides and DBCO-modified streptavidin at a 1:1:1 ratio for 1h at RT. The micelles
were then mixed with biotinylated Fab at a 1:1 ratio for 1h at RT. Conjugates were analyzed by
PAGE.
Cellular uptake
Functionalized nanomicelles were added to DsredDRR GBM cells or to GSCs at a desired
concentration (50-150 nM) and incubated for 3h at 37 °C. Cells were then fixed with 4% PFA,
counterstained with Hoechst, images were captured on an Olympus FV1000 confocal
microscope.
Gene knockdown
Functionalized nanomicelles were added to DsredDRR GBM cells or to GSCs at a desired
concentration (50-100 nM) and incubated for 72h at 37 °C in a 6-well format. Cells were then
lysed and protein expression was analyzed by western blot.
Figures and Results
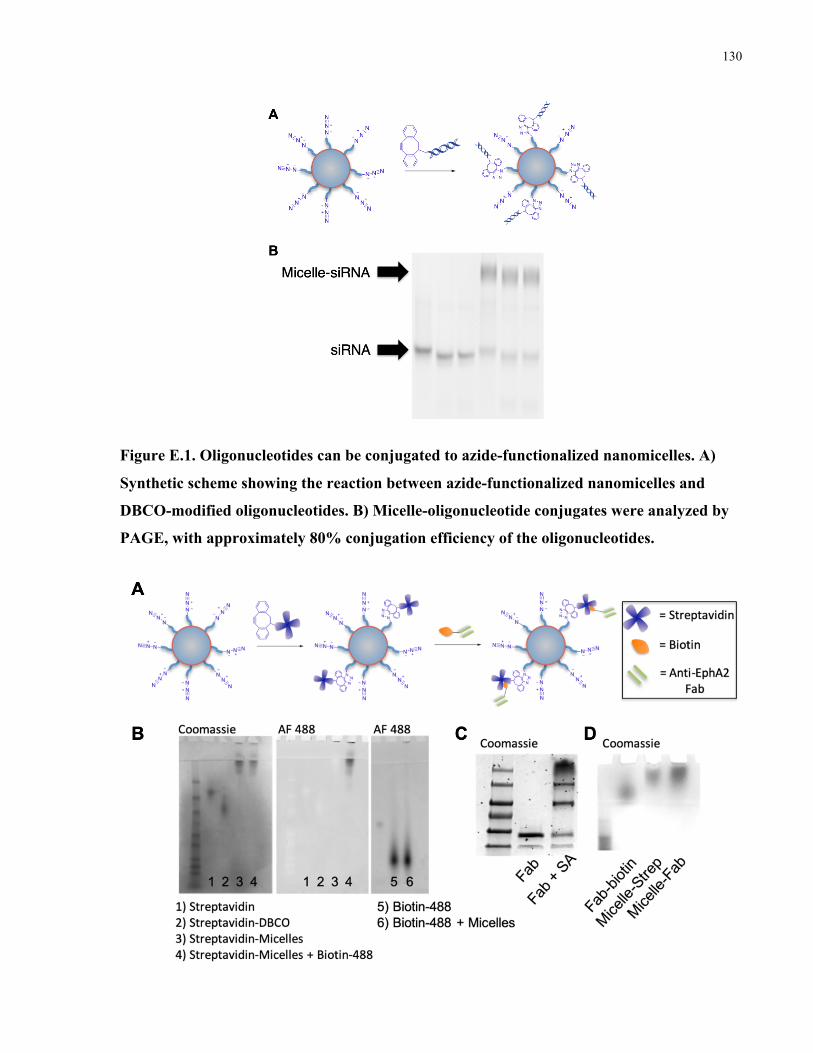
130
Figure E.1. Oligonucleotides can be conjugated to azide-functionalized nanomicelles. A)
Synthetic scheme showing the reaction between azide-functionalized nanomicelles and
DBCO-modified oligonucleotides. B) Micelle-oligonucleotide conjugates were analyzed by
PAGE, with approximately 80% conjugation efficiency of the oligonucleotides.
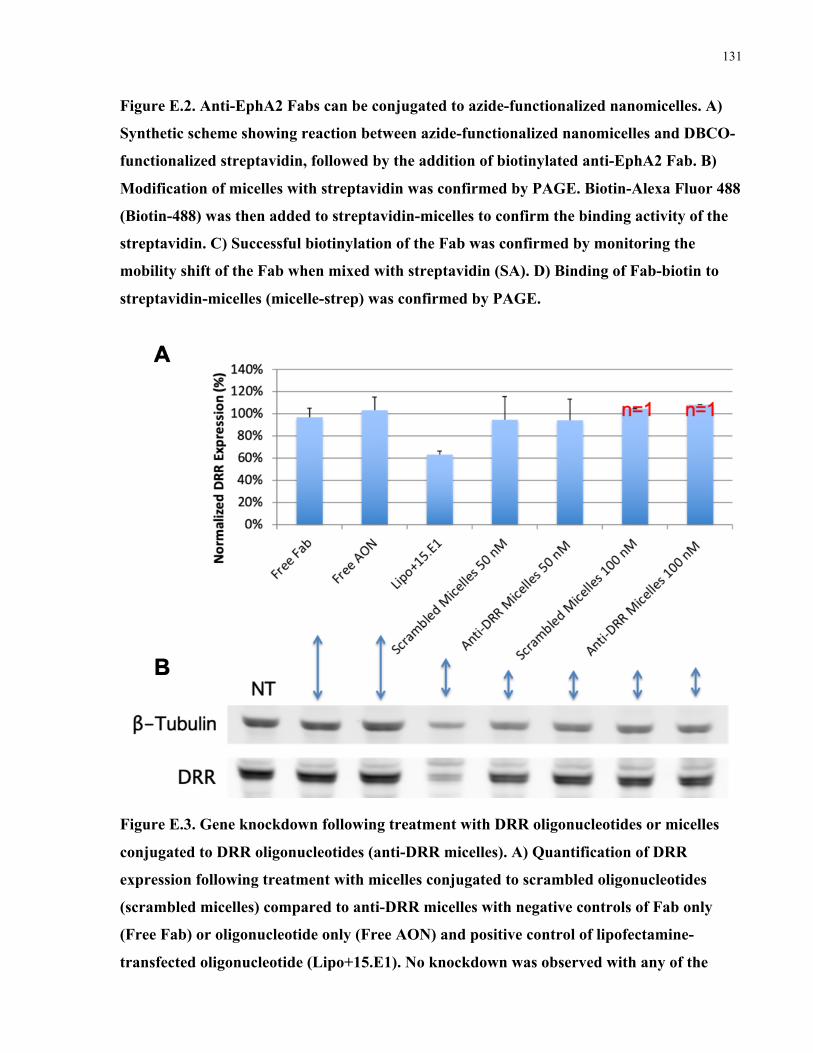
131
Figure E.2. Anti-EphA2 Fabs can be conjugated to azide-functionalized nanomicelles. A)
Synthetic scheme showing reaction between azide-functionalized nanomicelles and DBCO-
functionalized streptavidin, followed by the addition of biotinylated anti-EphA2 Fab. B)
Modification of micelles with streptavidin was confirmed by PAGE. Biotin-Alexa Fluor 488
(Biotin-488) was then added to streptavidin-micelles to confirm the binding activity of the
streptavidin. C) Successful biotinylation of the Fab was confirmed by monitoring the
mobility shift of the Fab when mixed with streptavidin (SA). D) Binding of Fab-biotin to
streptavidin-micelles (micelle-strep) was confirmed by PAGE.
Figure E.3. Gene knockdown following treatment with DRR oligonucleotides or micelles
conjugated to DRR oligonucleotides (anti-DRR micelles). A) Quantification of DRR
expression following treatment with micelles conjugated to scrambled oligonucleotides
(scrambled micelles) compared to anti-DRR micelles with negative controls of Fab only
(Free Fab) or oligonucleotide only (Free AON) and positive control of lipofectamine-
transfected oligonucleotide (Lipo+15.E1). No knockdown was observed with any of the

132
micelle-AON treatment conditions. Data is shown as n=3, mean+SD. B) Representative
western blot is shown.
Figure E.4. Uptake of micelle-oligonucleotide (micelle-AON) into GBM cells (DsredDRR)
and GBM stem cells (OPK126) compared to free AON control and no treatment controls.
No uptake was observed of micelles into glioblastoma stem cells (OPK126). A significant
amount of background uptake of free AON was observed in DsredDRR cells, and did not
appear to increase with the micelle formulation.
Discussion
Conjugation of the azide-functionalized nanomicelles to oligonucleotides and anti-EphA2 Fabs
was successful, with approximately 80% conjugation of the oligonucleotides (Figure E.1) and
nearly 100% conjugation of the anti-EphA2 Fabs (Figure E.2). Unfortunately, although the
positive control transfected by lipofectamine effectively knocked down DRR expression, no
DRR knockdown was observed in any of the anti-DRR-micelle treatment conditions (Figure
E.3). Finally, we examined uptake of the micelle formulations into GBM (DsredDRR) and GSC
(OPK126) cells (Figure E.4); although significant background uptake of the free AON was
observed in the DsredDRR cells, this did not translate to DRR knockdown as described in Figure
E.3. No uptake of the AON was observed with any treatment conditions for the GSCs (OPK126).

133
Following these results, we decided to pursue a direct conjugate between targeting antibodies
and antisense oligonucleotides instead of continuing with the functionalized nanomicelle system
due to a lack of observed efficacy. One hypothesis for the challenges faced with this approach is
the inefficient PEG conjugation achieved when using the graft-to approach for conjugating the
PEG chains to the backbone PLA-co-TMCC polymer; however, this hypothesis was not robustly
explored.

134
References 1. Kobayashi, K.; Nishioka, M.; Kohno, T.; Nakamoto, M.; Maeshima, A.; Aoyagi, K.; Sasaki, H.; Takenoshita, S.; Sugimura, H.; Yokota, J., Identification of genes whose expression is upregulated in lung adenocarcinoma cells in comparison with type II alveolar cells and bronchiolar epithelial cells in vivo. Oncogene 2004, 23 (17), 3089-3096.
2. van den Boom, J.; Wolter, M.; Blaschke, B.; Knobbe, C. B.; Reifenberger, G., Identification of novel genes associated with astrocytoma progression using suppression subtractive hybridization and real-time reverse transcription-polymerase chain reaction. Int. J. Cancer 2006, 119 (10), 2330-2338.
3. Bumcrot, D.; Manoharan, M.; Koteliansky, V.; Sah, D. W. Y., RNAi therapeutics: a potential new class of pharmaceutical drugs. Nat. Chem. Biol. 2006, 2 (12), 711-719.
4. Rinaldi, C.; Wood, M. J. A., Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2017, 14, 9.
5. Johnson, D. R.; O’Neill, B. P., Glioblastoma survival in the United States before and during the temozolomide era. J. Neuro-Oncol. 2012, 107 (2), 359-364.
6. Zong, H.; Parada, L. F.; Baker, S. J., Cell of origin for malignant gliomas and its implication in therapeutic development. Cold Spring Harb. Perspect. Biol. 7 (5), a020610.
7. Claes, A.; Idema, A. J.; Wesseling, P., Diffuse glioma growth: a guerilla war. Acta Neuropathol. 2007, 114 (5), 443-458.
8. Bleeker, F. E.; Lamba, S.; Zanon, C.; Molenaar, R. J.; Hulsebos, T. J. M.; Troost, D.; van Tilborg, A. A.; Vandertop, W. P.; Leenstra, S.; van Noorden, C. J. F.; Bardelli, A., Mutational profiling of kinases in glioblastoma. BMC Cancer 2014, 14, 718-718.
9. Grzmil, M.; Hemmings, B. A., Overcoming resistance to rapalogs in gliomas by combinatory therapies. Biochim. Biophys. Acta 2013, 1834 (7), 1371-1380.
10. Sami, A.; Karsy, M., Targeting the PI3K/AKT/mTOR signaling pathway in glioblastoma: novel therapeutic agents and advances in understanding. Tumor Biol. 2013, 34 (4), 1991-2002.
11. Wick, W.; Puduvalli, V. K.; Chamberlain, M. C.; van den Bent, M. J.; Carpentier, A. F.; Cher, L. M.; Mason, W.; Weller, M.; Hong, S.; Musib, L.; Liepa, A. M.; Thornton, D. E.; Fine, H. A., Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J. Clin. Oncol. 2010, 28 (7), 1168-1174.
12. Molenaar, R. J.; Verbaan, D.; Lamba, S.; Zanon, C.; Jeuken, J. W. M.; Boots-Sprenger, S. H. E.; Wesseling, P.; Hulsebos, T. J. M.; Troost, D.; van Tilborg, A. A.; Leenstra, S.; Vandertop, W. P.; Bardelli, A.; van Noorden, C. J. F.; Bleeker, F. E., The combination of IDH1 mutations and MGMT methylation status predicts survival in glioblastoma better than either IDH1 or MGMT alone. Neuro Oncol. 2014, 16 (9), 1263-1273.

135
13. Cohen, A. L.; Holmen, S. L.; Colman, H., IDH1 and IDH2 mutations in gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13 (5), 345-345.
14. Shah, N.; Schroeder, B.; Cobbs, C., MGMT methylation in glioblastoma: tale of the tail. Neuro Oncol. 2015, 17 (1), 167-168.
15. Stupp, R.; Mason, W. P.; van den Bent, M. J.; Weller, M.; Fisher, B.; Taphoorn, M. J. B.; Belanger, K.; Brandes, A. A.; Marosi, C.; Bogdahn, U.; Curschmann, J.; Janzer, R. C.; Ludwin, S. K.; Gorlia, T.; Allgeier, A.; Lacombe, D.; Cairncross, J. G.; Eisenhauer, E.; Mirimanoff, R. O., Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. New Engl. J. Med. 2005, 352 (10), 987-996.
16. Chamberlain, M. C., Temozolomide: therapeutic limitations in the treatment of adult high-grade gliomas. Expert Rev. Neurother. 2010, 10 (10), 1537-1544.
17. Zhang, H.; Wang, Z. Z., Mechanisms that mediate stem cell self-renewal and differentiation. J. Cell. Biochem. 2008, 103 (3), 709-718.
18. Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M. A.; Dick, J. E., A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367 (6464), 645-648.
19. Bonnet, D.; Dick, J. E., Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3 (7), 730-737.
20. Al-Hajj, M.; Wicha, M. S.; Benito-Hernandez, A.; Morrison, S. J.; Clarke, M. F., Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U. S. A. 2003, 100 (7), 3983-3988.
21. Singh, S. K.; Clarke, I. D.; Terasaki, M.; Bonn, V. E.; Hawkins, C.; Squire, J.; Dirks, P. B., Identification of a Cancer Stem Cell in Human Brain Tumors. Cancer Res. 2003, 63 (18), 5821.
22. Yu, Z.; Pestell, T. G.; Lisanti, M. P.; Pestell, R. G., Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44 (12), 2144-2151.
23. Klonisch, T.; Wiechec, E.; Hombach-Klonisch, S.; Ande, S. R.; Wesselborg, S.; Schulze-Osthoff, K.; Los, M., Cancer stem cell markers in common cancers – therapeutic implications. Trends Mol. Med. 2008, 14 (10), 450-460.
24. Brien, C. A.; Kreso, A.; Jamieson, C. H. M., Cancer Stem Cells and Self-renewal. Clin. Cancer. Res. 2010, 16 (12), 3113.
25. Bao, S.; Wu, Q.; McLendon, R. E.; Hao, Y.; Shi, Q.; Hjelmeland, A. B.; Dewhirst, M. W.; Bigner, D. D.; Rich, J. N., Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756.
26. Meirelles, K.; Benedict, L. A.; Dombkowski, D.; Pepin, D.; Preffer, F. I.; Teixeira, J.; Tanwar, P. S.; Young, R. H.; MacLaughlin, D. T.; Donahoe, P. K.; Wei, X., Human ovarian

136
cancer stem/progenitor cells are stimulated by doxorubicin but inhibited by Mullerian inhibiting substance. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (7), 2358-2363.
27. Li, X.; Lewis, M. T.; Huang, J.; Gutierrez, C.; Osborne, C. K.; Wu, M.-F.; Hilsenbeck, S. G.; Pavlick, A.; Zhang, X.; Chamness, G. C.; Wong, H.; Rosen, J.; Chang, J. C., Intrinsic Resistance of Tumorigenic Breast Cancer Cells to Chemotherapy. J. Natl. Cancer Inst. 2008, 100 (9), 672-679.
28. Zielske, S. P.; Spalding, A. C.; Wicha, M. S.; Lawrence, T. S., Ablation of breast cancer stem cells with radiation. Transl. Oncol. 2011, 4 (4), 227-233.
29. Kaplan, R. N.; Riba, R. D.; Zacharoulis, S.; Bramley, A. H.; Vincent, L.; Costa, C.; MacDonald, D. D.; Jin, D. K.; Shido, K.; Kerns, S. A.; Zhu, Z.; Hicklin, D.; Wu, Y.; Port, J. L.; Altorki, N.; Port, E. R.; Ruggero, D.; Shmelkov, S. V.; Jensen, K. K.; Rafii, S.; Lyden, D., VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438 (7069), 820-827.
30. Pandit, T. S.; Kennette, W.; Mackenzie, L.; Zhang, G.; Al-Katib, W.; Andrews, J.; Vantyghem, S. A.; Ormond, D. G.; Allan, A. L.; Rodenhiser, D. I.; Chambers, A. F.; Tuck, A. B., Lymphatic metastasis of breast cancer cells is associated with differential gene expression profiles that predict cancer stem cell-like properties and the ability to survive, establish and grow in a foreign environment. Int. J. Oncol. 2009, 35 (2), 297-308.
31. Wakamatsu, Y.; Sakamoto, N.; Oo, H. Z.; Naito, Y.; Uraoka, N.; Anami, K.; Sentani, K.; Oue, N.; Yasui, W., Expression of cancer stem cell markers ALDH1, CD44 and CD133 in primary tumor and lymph node metastasis of gastric cancer. Pathol. Int. 2012, 62 (2), 112-119.
32. Jin, L.; Hope, K. J.; Zhai, Q.; Smadja-Joffe, F.; Dick, J. E., Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat. Med. 2006, 12 (10), 1167-1174.
33. Perez, A.; Neskey, D. M.; Wen, J.; Goodwin, J. W.; Slingerland, J.; Pereira, L.; Weigand, S.; Franzmann, E. J., Abstract 2521: Targeting CD44 in head and neck squamous cell carcinoma (HNSCC) with a new humanized antibody RO5429083. Cancer Res. 2012, 72 (8 Supplement), 2521.
34. Zhao, L.; Yang, Y.; Zhou, P.; Ma, H.; Zhao, X.; He, X.; Wang, T.; Zhang, J.; Liu, Y.; Zhang, T., Targeting CD133high Colorectal Cancer Cells In Vitro and In Vivo With an Asymmetric Bispecific Antibody. J. Immunother. 2015, 38 (6), 217-228.
35. Liu, Y.; Zhang, X.; Liu, J.; Hou, G.; Zhang, S.; Zhang, J., Everolimus in combination with letrozole inhibit human breast cancer MCF-7/Aro stem cells via PI3K/mTOR pathway: an experimental study. Tumor Biol. 2014, 35 (2), 1275-1286.
36. Kaniyampadi, S. S.; Prathesha, P.; Aleena, M. C.; Shantikumar, V. N.; Vinoth-Kumar, L., Anti-diabetic Drug Metformin: Challenges and Perspectives for Cancer Therapy. Curr. Cancer Drug Targets 2014, 14 (8), 727-736.
37. Stechishin, O. D.; Luchman, H. A.; Ruan, Y.; Blough, M. D.; Nguyen, S. A.; Kelly, J. J.; Cairncross, J. G.; Weiss, S., On-target JAK2/STAT3 inhibition slows disease progression in

137
orthotopic xenografts of human glioblastoma brain tumor stem cells. Neuro Oncol. 2013, 15 (2), 198-207.
38. Singh, S. K.; Hawkins, C.; Clarke, I. D.; Squire, J. A.; Bayani, J.; Hide, T.; Henkelman, R. M.; Cusimano, M. D.; Dirks, P. B., Identification of human brain tumour initiating cells. Nature 2004, 432 (7015), 396-401.
39. Kalkan, R., Glioblastoma Stem Cells as a New Therapeutic Target for Glioblastoma. Clin. Med. Insights Oncol. 2015, 9, 95-103.
40. Lathia, J. D.; Mack, S. C.; Mulkearns-Hubert, E. E.; Valentim, C. L. L.; Rich, J. N., Cancer stem cells in glioblastoma. Genes Dev. 2015, 29 (12), 1203-1217.
41. Bradshaw, A.; Wickremsekera, A.; Tan, S. T.; Peng, L.; Davis, P. F.; Itinteang, T., Cancer Stem Cell Hierarchy in Glioblastoma Multiforme. Front. Surg. 2016, 3, 21-21.
42. Binda, E.; Visioli, A.; Giani, F.; Lamorte, G.; Copetti, M.; Pitter, K. L.; Huse, J. T.; Cajola, L.; Zanetti, N.; DiMeco, F.; De Filippis, L.; Mangiola, A.; Maira, G.; Anile, C.; De Bonis, P.; Reynolds, B. A.; Pasquale, E. B.; Vescovi, A. L., The EphA2 receptor drives self-renewal and tumorigenicity in stem-like tumor-propagating cells from human glioblastomas. Cancer Cell 2012, 22 (6), 765-780.
43. Qazi, M. A.; Vora, P.; Venugopal, C.; Adams, J.; Singh, M.; Hu, A.; Gorelik, M.; Subapanditha, M. K.; Savage, N.; Yang, J.; Chokshi, C.; London, M.; Gont, A.; Bobrowski, D.; Grinshtein, N.; Brown, K. R.; Murty, N. K.; Nilvebrant, J.; Kaplan, D.; Moffat, J.; Sidhu, S.; Singh, S. K., Cotargeting Ephrin Receptor Tyrosine Kinases A2 and A3 in Cancer Stem Cells Reduces Growth of Recurrent Glioblastoma. Cancer Res. 2018, 78 (17), 5023.
44. Liffers, K.; Lamszus, K.; Schulte, A., EGFR Amplification and Glioblastoma Stem-Like Cells. Stem Cells Int. 2015, 2015, 427518-427518.
45. Fargeas, C. A.; Corbeil, D.; Huttner, W. B., AC133 Antigen, CD133, Prominin-1, Prominin-2, Etc.: Prominin Family Gene Products in Need of a Rational Nomenclature. Stem Cells 2003, 21 (4), 506-508.
46. Yan, X.; Ma, L.; Yi, D.; Yoon, J.-g.; Diercks, A.; Foltz, G.; Price, N. D.; Hood, L. E.; Tian, Q., A CD133-related gene expression signature identifies an aggressive glioblastoma subtype with excessive mutations. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (4), 1591-1596.
47. Zeppernick, F.; Ahmadi, R.; Campos, B.; Dictus, C.; Helmke, B. M.; Becker, N.; Lichter, P.; Unterberg, A.; Radlwimmer, B.; Herold-Mende, C. C., Stem Cell Marker CD133 Affects Clinical Outcome in Glioma Patients. Clin. Cancer. Res. 2008, 14 (1), 123.
48. Kaoru, T.; Masaru, A.; Noboru, A.; Takahiro, O.; Hiroaki, W.; Masaaki, Y.; Kikuo, O., Expansion of CD133-positive glioma cells in recurrent de novo glioblastomas after radiotherapy and chemotherapy. J. Neurosurg. 2013, 119 (5), 1145-1155.
49. Shmelkov, S. V.; Butler, J. M.; Hooper, A. T.; Hormigo, A.; Kushner, J.; Milde, T.; St Clair, R.; Baljevic, M.; White, I.; Jin, D. K.; Chadburn, A.; Murphy, A. J.; Valenzuela, D. M.;

138
Gale, N. W.; Thurston, G.; Yancopoulos, G. D.; D'Angelica, M.; Kemeny, N.; Lyden, D.; Rafii, S., CD133 expression is not restricted to stem cells, and both CD133+ and CD133- metastatic colon cancer cells initiate tumors. J. Clin. Invest. 2008, 118 (6), 2111-2120.
50. Wang, J.; Sakariassen, P. Ø.; Tsinkalovsky, O.; Immervoll, H.; Bøe, S. O.; Svendsen, A.; Prestegarden, L.; Røsland, G.; Thorsen, F.; Stuhr, L.; Molven, A.; Bjerkvig, R.; Enger, P. Ø., CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int. J. Cancer 2008, 122 (4), 761-768.
51. Misra, S.; Hascall, V. C.; Markwald, R. R.; Ghatak, S., Interactions between Hyaluronan and Its Receptors (CD44, RHAMM) Regulate the Activities of Inflammation and Cancer. Front. Immunol. 2015, 6, 201-201.
52. Mooney, K. L.; Choy, W.; Sidhu, S.; Pelargos, P.; Bui, T. T.; Voth, B.; Barnette, N.; Yang, I., The role of CD44 in glioblastoma multiforme. J. Clin. Neurosci. 2016, 34, 1-5.
53. Naruse, M.; Shibasaki, K.; Yokoyama, S.; Kurachi, M.; Ishizaki, Y., Dynamic Changes of CD44 Expression from Progenitors to Subpopulations of Astrocytes and Neurons in Developing Cerebellum. PloS One 2013, 8 (1), e53109.
54. Prince, M. E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G. T.; Kaplan, M. J.; Dalerba, P.; Weissman, I. L.; Clarke, M. F.; Ailles, L. E., Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (3), 973-978.
55. Roger, B.; Sami, H.; Dorel, L. R.; Klaus-Martin, P.; Sven, G.; Hartmut, H.; Madjid, S.; Gerhard, F. W.; Alexandru, C. S., Disruption of intracerebral progression of rat C6 glioblastoma by in vivo treatment with anti-CD44 monoclonal antibody. J. Neurosurg. 2000, 92 (1), 140-149.
56. Jijiwa, M.; Demir, H.; Gupta, S.; Leung, C.; Joshi, K.; Orozco, N.; Huang, T.; Yildiz, V. O.; Shibahara, I.; de Jesus, J. A.; Yong, W. H.; Mischel, P. S.; Fernandez, S.; Kornblum, H. I.; Nakano, I., CD44v6 regulates growth of brain tumor stem cells partially through the AKT-mediated pathway. PloS One 2011, 6 (9), e24217-e24217.
57. Reifenberger, G.; Sieth, P.; Niederhaus, M.; Wechsler, W., Expression of CD15 in tumours of the nervous system. J. Histochem. 1992, 24 (11), 890-901.
58. Son, M. J.; Woolard, K.; Nam, D.-H.; Lee, J.; Fine, H. A., SSEA-1 Is an Enrichment Marker for Tumor-Initiating Cells in Human Glioblastoma. Cell Stem Cell 2009, 4 (5), 440-452.
59. Brescia, P.; Richichi, C.; Pelicci, G., Current Strategies for Identification of Glioma Stem Cells: Adequate or Unsatisfactory? 2012; Vol. 2012, p 376894.
60. Cheng, L.; Bao, S.; Rich, J. N., Potential therapeutic implications of cancer stem cells in glioblastoma. Biochem. Pharmacol. 2010, 80 (5), 654-665.
61. Esposito, C. L.; Nuzzo, S.; Catuogno, S.; Romano, S.; de Nigris, F.; de Franciscis, V., STAT3 Gene Silencing by Aptamer-siRNA Chimera as Selective Therapeutic for Glioblastoma. Mol. Ther. Nucleic Acids 2018, 10, 398-411.

139
62. Berezovsky, A. D.; Poisson, L. M.; Cherba, D.; Webb, C. P.; Transou, A. D.; Lemke, N. W.; Hong, X.; Hasselbach, L. A.; Irtenkauf, S. M.; Mikkelsen, T.; deCarvalho, A. C., Sox2 promotes malignancy in glioblastoma by regulating plasticity and astrocytic differentiation. Neoplasia 2014, 16 (3), 193-206.e2.06E25.
63. Wang, L.; Darling, J.; Zhang, J.-S.; Liu, W.; Qian, J.; Bostwick, D.; Hartmann, L.; Jenkins, R.; Bardenhauer, W.; Schutte, J.; Opalka, B.; Smith, D. I., Loss of expression of the DRR 1 gene at chromosomal segment 3p21.1 in renal cell carcinoma. Genes Chromosomes Cancer 2000, 27 (1), 1-10.
64. Le, P. U.; Angers-Loustau, A.; de Oliveira, R. M. W.; Ajlan, A.; Brassard, C. L.; Dudley, A.; Brent, H.; Siu, V.; Trinh, G.; Mölenkamp, G.; Wang, J.; Seyed Sadr, M.; Bedell, B.; Del Maestro, R. F.; Petrecca, K., DRR drives brain cancer invasion by regulating cytoskeletal-focal adhesion dynamics. Oncogene 2010, 29, 4636.
65. Giese, A.; Loo, M. A.; Tran, N.; Haskett, D.; Coons, S. W.; Berens, M. E., Dichotomy of astrocytoma migration and proliferation. Int. J. Cancer 1996, 67 (2), 275-282.
66. Dudley, A.; Sater, M.; Le, P. U.; Trinh, G.; Sadr, M. S.; Bergeron, J.; Deleavey, G. F.; Bedell, B.; Damha, M. J.; Petrecca, K., DRR regulates AKT activation to drive brain cancer invasion. Oncogene 2013, 33, 4952.
67. Browning, K. S.; Gallie, D. R.; Hershey, J. W. B.; Hinnebusch, A. G.; Maitra, U.; Merrick, W. C.; Norbury, C., Unified nomenclature for the subunits of eukaryotic initiation factor 3. Trends Biochem. Sci. 2001, 26 (5), 284.
68. Hinnebusch, A. G., The Scanning Mechanism of Eukaryotic Translation Initiation. Annu. Rev. Biochem. 2014, 83 (1), 779-812.
69. Wang, H.; Ru, Y.; Sanchez-Carbayo, M.; Wang, X.; Kieft, J. S.; Theodorescu, D., Translation Initiation Factor eIF3b Expression in Human Cancer and Its Role in Tumor Growth and Lung Colonization. Clin. Cancer. Res. 2013, 19 (11), 2850.
70. Zang, Y.; Zhang, X.; Yan, L.; Gu, G.; Li, D.; Zhang, Y.; Fang, L.; Fu, S.; Ren, J.; Xu, Z., Eukaryotic Translation Initiation Factor 3b is both a Promising Prognostic Biomarker and a Potential Therapeutic Target for Patients with Clear Cell Renal Cell Carcinoma. J. Cancer 2017, 8 (15), 3049-3061.
71. Liang, H.; Ding, X.; Zhou, C.; Zhang, Y.; Xu, M.; Zhang, C.; Xu, L., Knockdown of eukaryotic translation initiation factors 3B (EIF3B) inhibits proliferation and promotes apoptosis in glioblastoma cells. Neurol. Sci. 2012, 33 (5), 1057-1062.
72. Humphries, J. D.; Byron, A.; Humphries, M. J., Integrin ligands at a glance. J. Cell Sci. 2006, 119 (19), 3901.
73. Howe, G. A.; Addison, C. L., β1 integrin: an emerging player in the modulation of tumorigenesis and response to therapy. Cell Adhes. Migr. 2012, 6 (2), 71-77.

140
74. Mierke, C. T.; Frey, B.; Fellner, M.; Herrmann, M.; Fabry, B., Integrin α5β1 facilitates cancer cell invasion through enhanced contractile forces. J. Cell Sci. 2011, 124 (3), 369.
75. Arao, S.; Masumoto, A.; Otsuki, M., β1 Integrins Play an Essential Role in Adhesion and Invasion of Pancreatic Carcinoma Cells. Pancreas 2000, 20 (2).
76. Carbonell, W. S.; DeLay, M.; Jahangiri, A.; Park, C. C.; Aghi, M. K., β1 Integrin Targeting Potentiates Antiangiogenic Therapy and Inhibits the Growth of Bevacizumab-Resistant Glioblastoma. Cancer Res. 2013, 73 (10), 3145.
77. Malric, L.; Monferran, S.; Gilhodes, J.; Boyrie, S.; Dahan, P.; Skuli, N.; Sesen, J.; Filleron, T.; Kowalski-Chauvel, A.; Cohen-Jonathan Moyal, E.; Toulas, C.; Lemarié, A., Interest of integrins targeting in glioblastoma according to tumor heterogeneity and cancer stem cell paradigm: an update. Oncotarget 2017, 8 (49), 86947-86968.
78. Fire, A.; Xu, S.; Montgomery, M. K.; Kostas, S. A.; Driver, S. E.; Mello, C. C., Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391 (6669), 806-811.
79. Hamilton, A. J.; Baulcombe, D. C., A Species of Small Antisense RNA in Posttranscriptional Gene Silencing in Plants. Science 1999, 286 (5441), 950-952.
80. Elbashir, S. M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T., Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411 (6836), 494-498.
81. Ozcan, G.; Ozpolat, B.; Coleman, R. L.; Sood, A. K.; Lopez-Berestein, G., Preclinical and clinical development of siRNA-based therapeutics. Adv. Drug Del. Rev. 2015, 87, 108-119.
82. Kesselheim, A. S.; Avorn, J., Approving a Problematic Muscular Dystrophy Drug: Implications for FDA PolicyImplications of the FDA Approval of Eteplirsen for Muscular DystrophyImplications of the FDA Approval of Eteplirsen for Muscular Dystrophy. JAMA 2016, 316 (22), 2357-2358.
83. Stein, C. A.; Castanotto, D., FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther. 2017, 25 (5), 1069-1075.
84. Oh, Y.-K.; Park, T. G., siRNA delivery systems for cancer treatment. Adv. Drug Del. Rev. 2009, 61 (10), 850-862.
85. Castanotto, D.; Stein, C. A., Antisense oligonucleotides in cancer. Curr. Opin. Oncol. 2014, 26 (6).
86. Hickerson, R. P.; Vlassov, A. V.; Wang, Q.; Leake, D.; Ilves, H.; Gonzalez-Gonzalez, E.; Contag, C. H.; Johnston, B. H.; Kaspar, R. L., Stability Study of Unmodified siRNA and Relevance to Clinical Use. Oligonucleotides 2008, 18 (4), 345-354.
87. Whitehead, K. A.; Langer, R.; Anderson, D. G., Knocking down barriers: advances in siRNA delivery. Nature reviews. Drug discovery 2009, 8 (2), 129-138.

141
88. Opalinska, J. B.; Gewirtz, A. M., Nucleic-acid therapeutics: basic principles and recent applications. Nat. Rev. Drug Discov. 2002, 1 (7), 503-514.
89. Deleavey, Glen F.; Damha, Masad J., Designing Chemically Modified Oligonucleotides for Targeted Gene Silencing. Chem. Biol. 2012, 19 (8), 937-954.
90. Dana, H.; Chalbatani, G. M.; Mahmoodzadeh, H.; Karimloo, R.; Rezaiean, O.; Moradzadeh, A.; Mehmandoost, N.; Moazzen, F.; Mazraeh, A.; Marmari, V.; Ebrahimi, M.; Rashno, M. M.; Abadi, S. J.; Gharagouzlo, E., Molecular Mechanisms and Biological Functions of siRNA. Int. J. Biomed. Sci. 2017, 13 (2), 48-57.
91. Guo, P.; Coban, O.; Snead, N. M.; Trebley, J.; Hoeprich, S.; Guo, S.; Shu, Y., Engineering RNA for targeted siRNA delivery and medical application. Adv. Drug Del. Rev. 2010, 62 (6), 650-666.
92. Mullard, A., FDA approves landmark RNAi drug. Nat. Rev. Drug Discov. 2018, 17, 613.
93. Dias, N.; Stein, C. A., Antisense Oligonucleotides: Basic Concepts and Mechanisms. Mol. Cancer Ther. 2002, 1 (5), 347-355.
94. Iversen, F.; Yang, C. X.; Dagnaes-Hansen, F.; Schaffert, D. H.; Kjems, J.; Gao, S., Optimized siRNA-PEG Conjugates for Extended Blood Circulation and Reduced Urine Excretion in Mice. Theranostics 2013, 3 (3), 201-209.
95. Abdul Ghafoor Raja, M.; Katas, H.; Jing Wen, T., Stability, Intracellular Delivery, and Release of siRNA from Chitosan Nanoparticles Using Different Cross-Linkers. PLoS ONE 2015, 10 (6), e0128963.
96. Zhu, X.; Xu, Y.; Solis, L. M.; Tao, W.; Wang, L.; Behrens, C.; Xu, X.; Zhao, L.; Liu, D.; Wu, J.; Zhang, N.; Wistuba, I. I.; Farokhzad, O. C.; Zetter, B. R.; Shi, J., Long-circulating siRNA nanoparticles for validating Prohibitin1-targeted non-small cell lung cancer treatment. Proc. Natl. Acad. Sci. U.S.A. 2015, 112 (25), 7779-7784.
97. Bäumer, S.; Bäumer, N.; Appel, N.; Terheyden, L.; Fremerey, J.; Schelhaas, S.; Wardelmann, E.; Buchholz, F.; Berdel, W. E.; Müller-Tidow, C., Antibody-Mediated Delivery of Anti-KRAS-siRNA In Vivo Overcomes Therapy Resistance in Colon Cancer. Clin. Cancer. Res. 2015, 21 (6), 1383-1394.
98. Watts, J. K.; Katolik, A.; Viladoms, J.; Damha, M. J., Studies on the hydrolytic stability of 2 '-fluoroarabinonucleic acid (2 ' F-ANA). Org. Biomol. Chem. 2009, 7 (9), 1904-1910.
99. Kenski, D. M.; Butora, G.; Willingham, A. T.; Cooper, A. J.; Fu, W. L.; Qi, N.; Soriano, F.; Davies, I. W.; Flanagan, W. M., siRNA-optimized Modifications for Enhanced In Vivo Activity. Mol. Ther. Nucleic Acids 2012, 1, e5.
100. Rettig, G.; Behlke, M., Progress Toward In Vivo Use of siRNAs-II. Mol. Ther. 2012, 20, 483-512.

142
101. Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Geick, A.; Hadwiger, P.; Harborth, J.; John, M.; Kesavan, V.; Lavine, G.; Pandey, R. K.; Racie, T.; Rajeev, K. G.; Rohl, I.; Toudjarska, I.; Wang, G.; Wuschko, S.; Bumcrot, D.; Koteliansky, V.; Limmer, S.; Manoharan, M.; Vornlocher, H.-P., Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432 (7014), 173-178.
102. Takabatake, Y.; Isaka, Y.; Mizui, M.; Kawachi, H.; Takahara, S.; Imai, E., Chemically modified siRNA prolonged RNA interference in renal disease. Biochem. Biophys. Res. Commun. 2007, 363 (2), 432-437.
103. Zhang, W.; Müller, K.; Kessel, E.; Reinhard, S.; He, D.; Klein, P. M.; Höhn, M.; Rödl, W.; Kempter, S.; Wagner, E., Targeted siRNA Delivery Using a Lipo-Oligoaminoamide Nanocore with an Influenza Peptide and Transferrin Shell. Adv. Healthc. Mater. 2016, 5 (12), 1493-1504.
104. Eckstein, F., Phosphorothioate Oligodeoxynucleotides: What Is Their Origin and What Is Unique About Them? Antisense Nucleic Acid Drug Dev. 2000, 10 (2), 117-121.
105. Hall, A. H. S.; Wan, J.; Shaughnessy, E. E.; Ramsay Shaw, B.; Alexander, K. A., RNA interference using boranophosphate siRNAs: structure–activity relationships. Nucleic Acids Res. 2004, 32 (20), 5991-6000.
106. Rettig, G. R.; Behlke, M. A., Progress Toward In Vivo Use of siRNAs-II. Mol. Ther. 2012, 20 (3), 483-512.
107. Whitehead, K. A.; Dahlman, J. E.; Langer, R. S.; Anderson, D. G., Silencing or Stimulation? siRNA Delivery and the Immune System. Annu. Rev. Chem. Biomol. Eng. 2011, 2 (1), 77-96.
108. Bennett, C. F.; Swayze, E. E., RNA Targeting Therapeutics: Molecular Mechanisms of Antisense Oligonucleotides as a Therapeutic Platform. Annu. Rev. Pharmacool. Toxicol. 2010, 50 (1), 259-293.
109. Elmén, J.; Thonberg, H.; Ljungberg, K.; Frieden, M.; Westergaard, M.; Xu, Y.; Wahren, B.; Liang, Z.; Ørum, H.; Koch, T.; Wahlestedt, C., Locked nucleic acid (LNA) mediated improvements in siRNA stability and functionality. Nucleic Acids Res. 2005, 33 (1), 439-447.
110. Zhang, S.; Zhao, B.; Jiang, H.; Wang, B.; Ma, B., Cationic lipids and polymers mediated vectors for delivery of siRNA. J. Control. Release 2007, 123 (1), 1-10.
111. Wang, J.; Lu, Z.; Wientjes, M. G.; Au, J. L. S., Delivery of siRNA Therapeutics: Barriers and Carriers. AAPS J. 2010, 12 (4), 492-503.
112. Poon, G. M. K.; Gariépy, J., Cell-surface proteoglycans as molecular portals for cationic peptide and polymer entry into cells. Biochem. Soc. Trans. 2007, 35 (4), 788-793.
113. Lungwitz, U.; Breunig, M.; Blunk, T.; Göpferich, A., Polyethylenimine-based non-viral gene delivery systems. Eur. J. Pharm. Biopharm. 2005, 60 (2), 247-266.

143
114. Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J., Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 2006, 114 (1), 100-109.
115. Lee, S.-Y.; Huh, M. S.; Lee, S.; Lee, S. J.; Chung, H.; Park, J. H.; Oh, Y.-K.; Choi, K.; Kim, K.; Kwon, I. C., Stability and cellular uptake of polymerized siRNA (poly-siRNA)/polyethylenimine (PEI) complexes for efficient gene silencing. J. Control. Release 2010, 141 (3), 339-346.
116. Blanco, E.; Shen, H.; Ferrari, M., Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotech 2015, 33 (9), 941-951.
117. Lorenzer, C.; Dirin, M.; Winkler, A.-M.; Baumann, V.; Winkler, J., Going beyond the liver: Progress and challenges of targeted delivery of siRNA therapeutics. J. Control. Release 2015, 203, 1-15.
118. Wittrup, A.; Lieberman, J., Knocking down disease: a progress report on siRNA therapeutics. Nat. Rev. Genet. 2015, 16 (9), 543-552.
119. Li, H.; Yu, S. S.; Miteva, M.; Nelson, C. E.; Werfel, T.; Giorgio, T. D.; Duvall, C. L., Matrix Metalloproteinase Responsive, Proximity-activated Polymeric Nanoparticles for siRNA Delivery. Adv. Funct. Mater. 2013, 23 (24), 3040-3052.
120. Bareford, L. M.; Swaan, P. W., Endocytic mechanisms for targeted drug delivery. Adv. Drug Del. Rev. 2007, 59 (8), 748-758.
121. Li, H.; Miteva, M.; Kirkbride, K. C.; Cheng, M. J.; Nelson, C. E.; Simpson, E. M.; Gupta, M. K.; Duvall, C. L.; Giorgio, T. D., Dual MMP7-proximity-activated and folate receptor-targeted nanoparticles for siRNA delivery. Biomacromolecules 2015, 16 (1), 192-201.
122. Cuellar, T. L.; Barnes, D.; Nelson, C.; Tanguay, J.; Yu, S.-F.; Wen, X.; Scales, S. J.; Gesch, J.; Davis, D.; van Brabant Smith, A.; Leake, D.; Vandlen, R.; Siebel, C. W., Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of THIOMAB-siRNA conjugates. Nucleic Acids Res. 2015, 43 (2), 1189-1203.
123. Palanca-Wessels, M. C.; Booth, G. C.; Convertine, A. J.; Lundy, B. B.; Berguig, G. Y.; Press, M. F.; Stayton, P. S.; Press, O. W., Antibody targeting facilitates effective intratumoral siRNA nanoparticle delivery to HER2-overexpressing cancer cells. Oncotarget 2016, 7 (8), 9561-9575.
124. Yumura, K.; Ui, M.; Doi, H.; Hamakubo, T.; Kodama, T.; Tsumoto, K.; Sugiyama, A., Mutations for decreasing the immunogenicity and maintaining the function of core streptavidin. Protein Sci. 2013, 22 (2), 213-221.
125. Tseng, Y.-C.; Mozumdar, S.; Huang, L., Lipid-based systemic delivery of siRNA. Adv. Drug Del. Rev. 2009, 61 (9), 721-731.
126. Virtanen, J.; Ruonala, M.; Vauhkonen, M.; Somerharju, P., Lateral Organization of Liquid-Crystalline Cholesterol-Dimyristoylphosphatidylcholine Bilayers. Evidence for Domains

144
with Hexagonal and Centered Rectangular Cholesterol Superlattices. Biochemistry 1995, 34 (36), 11568-11581.
127. Lechanteur, A.; Sanna, V.; Duchemin, A.; Evrard, B.; Mottet, D.; Piel, G., Cationic Liposomes Carrying siRNA: Impact of Lipid Composition on Physicochemical Properties, Cytotoxicity and Endosomal Escape. Nanomaterials 2018, 8 (5), 270.
128. Bao, Y.; Jin, Y.; Chivukula, P.; Zhang, J.; Liu, Y.; Liu, J.; Clamme, J.-P.; Mahato, R. I.; Ng, D.; Ying, W.; Wang, Y.; Yu, L., Effect of PEGylation on Biodistribution and Gene Silencing of siRNA/Lipid Nanoparticle Complexes. Pharm. Res. 2013, 30 (2), 342-351.
129. Noble, G. T.; Stefanick, J. F.; Ashley, J. D.; Kiziltepe, T.; Bilgicer, B., Ligand-targeted liposome design: challenges and fundamental considerations. Trends Biotechnol. 2014, 32 (1), 32-45.
130. Hoy, S. M., Patisiran: First Global Approval. Drugs 2018, 78 (15), 1625-1631.
131. Kanasty, R.; Dorkin, J. R.; Vegas, A.; Anderson, D., Delivery materials for siRNA therapeutics. Nat. Mater. 2013, 12 (11), 967-977.
132. Zhang, J.; Fan, H.; Levorse, D. A.; Crocker, L. S., Ionization Behavior of Amino Lipids for siRNA Delivery: Determination of Ionization Constants, SAR, and the Impact of Lipid pKa on Cationic Lipid−Biomembrane Interactions. Langmuir 2011, 27 (5), 1907-1914.
133. Jayaraman, M.; Ansell, S. M.; Mui, B. L.; Tam, Y. K.; Chen, J.; Du, X.; Butler, D.; Eltepu, L.; Matsuda, S.; Narayanannair, J. K.; Rajeev, K. G.; Hafez, I. M.; Akinc, A.; Maier, M. A.; Tracy, M. A.; Cullis, P. R.; Madden, T. D.; Manoharan, M.; Hope, M. J., Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. 2012, 51 (34), 8529-8533.
134. Zhigaltsev, I. V.; Maurer, N.; Wong, K. F.; Cullis, P. R., Triggered release of doxorubicin following mixing of cationic and anionic liposomes. Biochim. Biophys. Acta 2002, 1565 (1), 129-135.
135. Zhang, Y.; Li, H.; Sun, J.; Gao, J.; Liu, W.; Li, B.; Guo, Y.; Chen, J., DC-Chol/DOPE cationic liposomes: A comparative study of the influence factors on plasmid pDNA and siRNA gene delivery. Int. J. Pharm. 2010, 390 (2), 198-207.
136. Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K. N.; Jayaraman, M.; Rajeev, K. G.; Cantley, W. L.; Dorkin, J. R.; Butler, J. S.; Qin, L.; Racie, T.; Sprague, A.; Fava, E.; Zeigerer, A.; Hope, M. J.; Zerial, M.; Sah, D. W. Y.; Fitzgerald, K.; Tracy, M. A.; Manoharan, M.; Koteliansky, V.; Fougerolles, A. d.; Maier, M. A., Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010, 18 (7), 1357-1364.
137. Sato, Y.; Murase, K.; Kato, J.; Kobune, M.; Sato, T.; Kawano, Y.; Takimoto, R.; Takada, K.; Miyanishi, K.; Matsunaga, T.; Takayama, T.; Niitsu, Y., Resolution of liver cirrhosis using vitamin A–coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat. Biotechnol. 2008, 26, 431.

145
138. Wyrozumska, P.; Meissner, J.; Toporkiewicz, M.; Szarawarska, M.; Kuliczkowski, K.; Ugorski, M.; Walasek, M. A.; Sikorski, A. F., Liposome-coated lipoplex-based carrier for antisense oligonucleotides. Cancer Biol. Ther. 2014, 16 (1), 66-76.
139. Copolovici, D. M.; Langel, K.; Eriste, E.; Langel, Ü., Cell-penetrating peptides: design, synthesis, and applications. ACS Nano 2014, 8 (3), 1972-1994.
140. Bechara, C.; Sagan, S., Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013, 587 (12), 1693-1702.
141. Margus, H.; Padari, K.; Pooga, M., Cell-penetrating Peptides as Versatile Vehicles for Oligonucleotide Delivery. Mol. Ther. 2012, 20 (3), 525-533.
142. Di Pisa, M.; Chassaing, G.; Swiecicki, J.-M., Translocation Mechanism(s) of Cell-Penetrating Peptides: Biophysical Studies Using Artificial Membrane Bilayers. Biochemistry 2015, 54 (2), 194-207.
143. Kaplan, I. M.; Wadia, J. S.; Dowdy, S. F., Cationic TAT peptide transduction domain enters cells by macropinocytosis. J. Control. Release 2005, 102 (1), 247-253.
144. Brooks, H.; Lebleu, B.; Vivès, E., Tat peptide-mediated cellular delivery: back to basics. Adv. Drug Del. Rev. 2005, 57 (4), 559-577.
145. Derossi, D.; Joliot, A. H.; Chassaing, G.; Prochiantz, A., The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269 (14), 10444-50.
146. Pooga, M.; Hällbrink, M.; Zorko, M.; Langel, U.; lo, Cell penetration by transportan. The FASEB Journal 1998, 12 (1), 67-77.
147. Soomets, U.; Lindgren, M.; Gallet, X.; Hällbrink, M.; Elmquist, A.; Balaspiri, L.; Zorko, M.; Pooga, M.; Brasseur, R.; Langel, Ü., Deletion analogues of transportan. Biochim. Biophys. Acta 2000, 1467 (1), 165-176.
148. Elliott, G.; O'Hare, P., Intercellular Trafficking and Protein Delivery by a Herpesvirus Structural Protein. Cell 1997, 88 (2), 223-233.
149. Futaki, S.; Suzuki, T.; Ohashi, W.; Yagami, T.; Tanaka, S.; Ueda, K.; Sugiura, Y., Arginine-rich Peptides: An Abundant Source of Membrane-Permeable Peptides Having Potential as Carriers for Intracellular Protein Delivery. J. Biol. Chem. 2001, 276 (8), 5836-5840.
150. Chaloin, L.; Vidal, P.; Lory, P.; Méry, J.; Lautredou, N.; Divita, G.; Heitz, F., Design of Carrier Peptide-Oligonucleotide Conjugates with Rapid Membrane Translocation and Nuclear Localization Properties. Biochem. Biophys. Res. Commun. 1998, 243 (2), 601-608.
151. Crombez, L.; Aldrian-Herrada, G.; Konate, K.; Nguyen, Q. N.; McMaster, G. K.; Brasseur, R.; Heitz, F.; Divita, G., A New Potent Secondary Amphipathic Cell-penetrating Peptide for siRNA Delivery Into Mammalian Cells. Mol. Ther. 2008, 17 (1), 95-103.

146
152. Sun, C.-Y.; Shen, S.; Xu, C.-F.; Li, H.-J.; Liu, Y.; Cao, Z.-T.; Yang, X.-Z.; Xia, J.-X.; Wang, J., Tumor Acidity-Sensitive Polymeric Vector for Active Targeted siRNA Delivery. J. Am. Chem. Soc. 2015, 137 (48), 15217-15224.
153. Kanazawa, T.; Morisaki, K.; Suzuki, S.; Takashima, Y., Prolongation of life in rats with malignant glioma by intranasal siRNA/drug codelivery to the brain with cell-penetrating peptide-modified micelles. Mol. Pharm. 2014, 11 (5), 1471-1478.
154. Fang, B.; Jiang, L.; Zhang, M.; Ren, F. Z., A novel cell-penetrating peptide TAT-A1 delivers siRNA into tumor cells selectively. Biochimie 2013, 95 (2), 251-257.
155. Liu, Y.; Ran, R.; Chen, J.; Kuang, Q.; Tang, J.; Mei, L.; Zhang, Q.; Gao, H.; Zhang, Z.; He, Q., Paclitaxel loaded liposomes decorated with a multifunctional tandem peptide for glioma targeting. Biomaterials 2014, 35 (17), 4835-4847.
156. Hou, K. K.; Pan, H.; Schlesinger, P. H.; Wickline, S. A., A role for peptides in overcoming endosomal entrapment in siRNA delivery - A focus on melittin. Biotechnol. Adv. 2015, 33 (6), 931-940.
157. Cheng, Y.; Yumul, R.; Pun, S., Virus-Inspired Polymer for Efficient In Vitro and In Vivo Gene Delivery. Angew. Chem. 2016, 55, 12013-12017.
158. Janas, M. M.; Schlegel, M. K.; Harbison, C. E.; Yilmaz, V. O.; Jiang, Y.; Parmar, R.; Zlatev, I.; Castoreno, A.; Xu, H.; Shulga-Morskaya, S.; Rajeev, K. G.; Manoharan, M.; Keirstead, N. D.; Maier, M. A.; Jadhav, V., Selection of GalNAc-conjugated siRNAs with limited off-target-driven rat hepatotoxicity. Nat. Commun. 2018, 9 (1), 723.
159. Kumar, A.; White, J.; James Christie, R.; Dimasi, N.; Gao, C., Chapter Twelve - Antibody-Drug Conjugates. In Annu. Rep. Med. Chem., Goodnow, R. A., Ed. Academic Press: 2017; Vol. 50, pp 441-480.
160. Dowd, S. E.; Halonen, M. J.; Maier, R. M., Chapter 12 - Immunological Methods. In Environmental Microbiology (Second Edition), Maier, R. M.; Pepper, I. L.; Gerba, C. P., Eds. Academic Press: San Diego, 2009; pp 225-241.
161. Jain, M.; Kamal, N.; Batra, S. K., Engineering antibodies for clinical applications. Trends Biotechnol. 2007, 25 (7), 307-316.
162. Stephanopoulos, N.; Francis, M. B., Choosing an effective protein bioconjugation strategy. Nat. Chem. Biol. 2011, 7, 876.
163. Koniev, O.; Wagner, A., Developments and recent advancements in the field of endogenous amino acid selective bond forming reactions for bioconjugation. Chem. Soc. Rev. 2015, 44 (15), 5495-5551.
164. Lomant, A. J.; Fairbanks, G., Chemical probes of extended biological structures: Synthesis and properties of the cleavable protein cross-linking reagent [35S]dithiobis(succinimidyl propionate). J. Mol. Biol. 1976, 104 (1), 243-261.

147
165. Kahne, D.; Still, W. C., Hydrolysis of a peptide bond in neutral water. J. Am. Chem. Soc. 1988, 110 (22), 7529-7534.
166. Kalkhof, S.; Sinz, A., Chances and Pitfalls of Chemical Cross-Linking with Amine-Reactive N-Hydroxy Succinimide Esters. 2008; Vol. 392, p 305-12.
167. F. Doolittle, R., Redundancies in Protein Sequences. 1989; pp 599-623.
168. Hermanson, G. T., Chapter 2 - Functional Targets for Bioconjugation. In Bioconjugate Techniques (Third Edition), Hermanson, G. T., Ed. Academic Press: Boston, 2013; pp 127-228.
169. Robotham, A. C.; Kelly, J. F., Detection and quantification of free sulfhydryls in monoclonal antibodies using maleimide labeling and mass spectrometry. mAbs 2019, 11 (4), 757-766.
170. Hooker, J. M.; Kovacs, E. W.; Francis, M. B., Interior Surface Modification of Bacteriophage MS2. J. Am. Chem. Soc. 2004, 126 (12), 3718-3719.
171. Popp, B. V.; Ball, Z. T., Structure-Selective Modification of Aromatic Side Chains with Dirhodium Metallopeptide Catalysts. J. Am. Chem. Soc. 2010, 132 (19), 6660-6662.
172. Xie, J.; Schultz, P. G., A chemical toolkit for proteins — an expanded genetic code. Nat. Rev. Mol. Cell Biol. 2006, 7 (10), 775-782.
173. Cornish, V. W.; Hahn, K. M.; Schultz, P. G., Site-Specific Protein Modification Using a Ketone Handle. J. Am. Chem. Soc. 1996, 118 (34), 8150-8151.
174. Strable, E.; Prasuhn, D. E.; Udit, A. K.; Brown, S.; Link, A. J.; Ngo, J. T.; Lander, G.; Quispe, J.; Potter, C. S.; Carragher, B.; Tirrell, D. A.; Finn, M. G., Unnatural Amino Acid Incorporation into Virus-Like Particles. Bioconjugate Chem. 2008, 19 (4), 866-875.
175. van Hest, J. C. M.; Kiick, K. L.; Tirrell, D. A., Efficient Incorporation of Unsaturated Methionine Analogues into Proteins in Vivo. J. Am. Chem. Soc. 2000, 122 (7), 1282-1288.
176. Carrico, Z. M.; Romanini, D. W.; Mehl, R. A.; Francis, M. B., Oxidative coupling of peptides to a virus capsid containing unnatural amino acids. Chem. Commun. 2008, (10), 1205-1207.
177. Beatty, K. E.; Fisk, J. D.; Smart, B. P.; Lu, Y. Y.; Szychowski, J.; Hangauer, M. J.; Baskin, J. M.; Bertozzi, C. R.; Tirrell, D. A., Live-cell imaging of cellular proteins by a strain-promoted azide-alkyne cycloaddition. ChemBioChem 2010, 11 (15), 2092-2095.
178. Chin, J. W.; Martin, A. B.; King, D. S.; Wang, L.; Schultz, P. G., Addition of a photocrosslinking amino acid to the genetic code of Escherichiacoli. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (17), 11020-11024.
179. Chen, I.; Howarth, M.; Lin, W.; Ting, A. Y., Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat. Methods 2005, 2 (2), 99-104.

148
180. Sato, H.; Ikeda, M.; Suzuki, K.; Hirayama, K., Site-Specific Modification of Interleukin-2 by the Combined Use of Genetic Engineering Techniques and Transglutaminase. Biochemistry 1996, 35 (40), 13072-13080.
181. Fernández-Suárez, M.; Baruah, H.; Martínez-Hernández, L.; Xie, K. T.; Baskin, J. M.; Bertozzi, C. R.; Ting, A. Y., Redirecting lipoic acid ligase for cell surface protein labeling with small-molecule probes. Nat. Biotechnol. 2007, 25 (12), 1483-1487.
182. Popp, M. W.; Antos, J. M.; Grotenbreg, G. M.; Spooner, E.; Ploegh, H. L., Sortagging: a versatile method for protein labeling. Nat. Chem. Biol. 2007, 3, 707.
183. McKay, C. S.; Finn, M. G., Click chemistry in complex mixtures: bioorthogonal bioconjugation. Chem. Biol. 2014, 21 (9), 1075-1101.
184. Ryman, J. T.; Meibohm, B., Pharmacokinetics of Monoclonal Antibodies. CPT Pharmacometrics Syst. Pharmacol. 2017, 6 (9), 576-588.
185. Hughes, B., Antibody–drug conjugates for cancer: poised to deliver? Nat. Rev. Drug Discov. 2010, 9, 665.
186. Diamantis, N.; Banerji, U., Antibody-drug conjugates--an emerging class of cancer treatment. Br. J. Cancer 2016, 114 (4), 362-367.
187. Coats, S.; Williams, M.; Kebble, B.; Dixit, R.; Tseng, L.; Yao, N.-S.; Tice, D. A.; Soria, J.-C., Antibody Drug Conjugates: Future Directions in Clinical and Translational Strategies to Improve the Therapeutic Index. Clin. Cancer. Res. 2019, clincanres.0272.2019.
188. Godwin, C. D.; Gale, R. P.; Walter, R. B., Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia 2017, 31, 1855.
189. von Minckwitz, G.; Huang, C.-S.; Mano, M. S.; Loibl, S.; Mamounas, E. P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; Fischer, H. H.; Jacot, W.; Conlin, A. K.; Arce-Salinas, C.; Wapnir, I. L.; Jackisch, C.; DiGiovanna, M. P.; Fasching, P. A.; Crown, J. P.; Wülfing, P.; Shao, Z.; Rota Caremoli, E.; Wu, H.; Lam, L. H.; Tesarowski, D.; Smitt, M.; Douthwaite, H.; Singel, S. M.; Geyer, C. E., Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. New Engl. J. Med. 2018, 380 (7), 617-628.
190. Gan, H. K.; van den Bent, M.; Lassman, A. B.; Reardon, D. A.; Scott, A. M., Antibody–drug conjugates in glioblastoma therapy: the right drugs to the right cells. Nat. Rev. Clin. Oncol. 2017, 14, 695.
191. Reardon, D. A.; Lassman, A. B.; van den Bent, M.; Kumthekar, P.; Merrell, R.; Scott, A. M.; Fichtel, L.; Sulman, E. P.; Gomez, E.; Fischer, J.; Lee, H.-J.; Munasinghe, W.; Xiong, H.; Mandich, H.; Roberts-Rapp, L.; Ansell, P.; Holen, K. D.; Gan, H. K., Efficacy and safety results of ABT-414 in combination with radiation and temozolomide in newly diagnosed glioblastoma. Neuro Oncol. 2017, 19 (7), 965-975.
192. Hamblett, K. J.; Kozlosky, C. J.; Siu, S.; Chang, W. S.; Liu, H.; Foltz, I. N.; Trueblood, E. S.; Meininger, D.; Arora, T.; Twomey, B.; Vonderfecht, S. L.; Chen, Q.; Hill, J. S.; Fanslow,

149
W. C., AMG 595, an Anti-EGFRvIII Antibody–Drug Conjugate, Induces Potent Antitumor Activity against EGFRvIII-Expressing Glioblastoma. Mol. Cancer Ther. 2015, 14 (7), 1614.
193. Linna, L.; Tony, S. Q.; Ed, J. G.; Ji, H. K.; Jacqueline, G. E.; Theodore, E. Y.; Joseph, M. J.; Steven, C. C.; Perry, B.; Luther, W. B., A Phase II study of anti–epidermal growth factor receptor radioimmunotherapy in the treatment of glioblastoma multiforme. J. Neurosurg. 2010, 113 (2), 192-198.
194. Emlet, D. R.; Gupta, P.; Holgado-Madruga, M.; Del Vecchio, C. A.; Mitra, S. S.; Han, S.-Y.; Li, G.; Jensen, K. C.; Vogel, H.; Xu, L. W.; Skirboll, S. S.; Wong, A. J., Targeting a glioblastoma cancer stem-cell population defined by EGF receptor variant III. Cancer Res. 2014, 74 (4), 1238-1249.
195. Westphal, M.; Heese, O.; Steinbach, J. P.; Schnell, O.; Schackert, G.; Mehdorn, M.; Schulz, D.; Simon, M.; Schlegel, U.; Senft, C.; Geletneky, K.; Braun, C.; Hartung, J. G.; Reuter, D.; Metz, M. W.; Bach, F.; Pietsch, T., A randomised, open label phase III trial with nimotuzumab, an anti-epidermal growth factor receptor monoclonal antibody in the treatment of newly diagnosed adult glioblastoma. Eur. J. Cancer 2015, 51 (4), 522-532.
196. Van Den Bent, M. J.; Gan, H. K.; Lassman, A. B.; Kumthekar, P.; Merrell, R.; Butowski, N. A.; Lwin, Z.; Mikkelsen, T.; Nabors, L. B.; Papadopoulos, K. P.; Penas-Prado, M.; Simes, J.; Wheeler, H.; Gomez, E. J.; Lee, H.-J.; Roberts-Rapp, L.; Xiong, H.; Bain, E. E.; Holen, K. D.; Reardon, D. A., Efficacy of a novel antibody-drug conjugate (ADC), ABT-414, as monotherapy in epidermal growth factor receptor (EGFR) amplified, recurrent glioblastoma (GBM). J. Clin. Oncol. 2016, 34 (15_suppl), 2542-2542.
197. Xia, C.-F.; Boado, R. J.; Pardridge, W. M., Antibody-Mediated Targeting of siRNA via the Human Insulin Receptor Using Avidin−Biotin Technology. Mol. Pharm. 2009, 6 (3), 747-751.
198. Xia, C.-F.; Zhang, Y.; Zhang, Y.; Boado, R. J.; Pardridge, W. M., Intravenous siRNA of Brain Cancer with Receptor Targeting and Avidin–Biotin Technology. Pharm. Res. 2007, 24 (12), 2309-2316.
199. Hauser, P. V.; Pippin, J. W.; Kaiser, C.; Krofft, R. D.; Brinkkoetter, P. T.; Hudkins, K. L.; Kerjaschki, D.; Reiser, J.; Alpers, C. E.; Shankland, S. J., Novel siRNA Delivery System to Target Podocytes In Vivo. PloS One 2010, 5 (3), e9463.
200. Bäumer, S.; Bäumer, N.; Appel, N.; Terheyden, L.; Fremerey, J.; Schelhaas, S.; Wardelmann, E.; Buchholz, F.; Berdel, W. E.; Müller-Tidow, C., Antibody-Mediated Delivery of Anti-KRAS-siRNA Overcomes Therapy Resistance in Colon Cancer. Clin. Cancer. Res. 2015, 21 (6), 1383.
201. Cuellar, T. L.; Barnes, D.; Nelson, C.; Tanguay, J.; Yu, S.-F.; Wen, X.; Scales, S. J.; Gesch, J.; Davis, D.; van Brabant Smith, A.; Leake, D.; Vandlen, R.; Siebel, C. W., Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of THIOMAB-siRNA conjugates. Nucleic Acids Res. 2015, 43 (2), 1189-1203.

150
202. Sugo, T.; Terada, M.; Oikawa, T.; Miyata, K.; Nishimura, S.; Kenjo, E.; Ogasawara-Shimizu, M.; Makita, Y.; Imaichi, S.; Murata, S.; Otake, K.; Kikuchi, K.; Teratani, M.; Masuda, Y.; Kamei, T.; Takagahara, S.; Ikeda, S.; Ohtaki, T.; Matsumoto, H., Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control. Release 2016, 237, 1-13.
203. Satake, N.; Duong, C.; Yoshida, S.; Oestergaard, M.; Chen, C.; Peralta, R.; Guo, S.; Seth, P. P.; Li, Y.; Beckett, L.; Chung, J.; Nolta, J.; Nitin, N.; Tuscano, J. M., Novel Targeted Therapy for Precursor B Cell Acute Lymphoblastic Leukemia: anti-CD22 Antibody-MXD3 Antisense Oligonucleotide Conjugate. Mol. Med. 2016, 22, 632-642.
204. Wang, H.; Yang, Y.; Wang, W.; Guan, B.; Xun, M.; Zhang, H.; Wang, Z.; Zhao, Y., Single-chain antibody–delivered Livin siRNA inhibits human malignant melanoma growth in vitro and in vivo. Tumor Biol. 2017, 39 (5), 1010428317701645.
205. Odumosu, O.; Nicholas, D.; Yano, H.; Langridge, W., AB toxins: a paradigm switch from deadly to desirable. Toxins (Basel) 2010, 2 (7), 1612-1645.
206. Beilhartz, G. L.; Sugiman-Marangos, S. N.; Melnyk, R. A., Repurposing bacterial toxins for intracellular delivery of therapeutic proteins. Biochem. Pharmacol. 2017, 142, 13-20.
207. D’Astolfo, Diego S.; Pagliero, Romina J.; Pras, A.; Karthaus, Wouter R.; Clevers, H.; Prasad, V.; Lebbink, Robert J.; Rehmann, H.; Geijsen, N., Efficient Intracellular Delivery of Native Proteins. Cell 2015, 161 (3), 674-690.
208. Naglich, J. G.; Metherall, J. E.; Russell, D. W.; Eidels, L., Expression cloning of a diphtheria toxin receptor: Identity with a heparin-binding EGF-like growth factor precursor. Cell 1992, 69 (6), 1051-1061.
209. Michalska, M.; Wolf, P., Pseudomonas Exotoxin A: optimized by evolution for effective killing. Front. Microbiol. 2015, 6, 963-963.
210. Auger, A.; Park, M.; Nitschke, F.; Minassian, L. M.; Beilhartz, G. L.; Minassian, B. A.; Melnyk, R. A., Efficient Delivery of Structurally Diverse Protein Cargo into Mammalian Cells by a Bacterial Toxin. Mol. Pharm. 2015, 12 (8), 2962-2971.
211. Feld, G. K.; Thoren, K. L.; Kintzer, A. F.; Sterling, H. J.; Tang, I. I.; Greenberg, S. G.; Williams, E. R.; Krantz, B. A., Structural basis for the unfolding of anthrax lethal factor by protective antigen oligomers. Nat. Struct. Mol. Biol. 2010, 17 (11), 1383-1390.
212. Milne, J. C.; Blanket, S. R.; Hanna, P. C.; Collier, R. J., Protective antigen-binding domain of anthrax lethal factor mediates translocation of a heterologous protein fused to its amino- or carboxy-terminus. Mol. Microbiol. 1995, 15 (4), 661-666.
213. Kreitman, R. J.; Pastan, I., Antibody fusion proteins: anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin. Cancer. Res. 2011, 17 (20), 6398-6405.
214. Kreitman, R. J.; Wilson, W. H.; White, J. D.; Stetler-Stevenson, M.; Jaffe, E. S.; Giardina, S.; Waldmann, T. A.; Pastan, I., Phase I Trial of Recombinant Immunotoxin Anti-

151
Tac(Fv)-PE38 (LMB-2) in Patients With Hematologic Malignancies. J. Clin. Oncol. 2000, 18 (8), 1622-1636.
215. Kaminetzky, D.; Hymes, K. B., Denileukin diftitox for the treatment of cutaneous T-cell lymphoma. Biologics 2008, 2 (4), 717-724.
216. Frankel, A. E.; Woo, J. H.; Ahn, C.; Foss, F. M.; Duvic, M.; Neville, P. H.; Neville, D. M., Resimmune, an anti-CD3ε recombinant immunotoxin, induces durable remissions in patients with cutaneous T-cell lymphoma. Haematologica 2015, 100 (6), 794-800.
217. Gaillard, P. J.; Brink, A.; de Boer, A. G., Diphtheria toxin receptor-targeted brain drug delivery. Int. Congr. Ser. 2005, 1277, 185-198.
218. Bao, X.; Pastan, I.; Bigner, D. D.; Chandramohan, V., EGFR/EGFRvIII-targeted immunotoxin therapy for the treatment of glioblastomas via convection-enhanced delivery. Receptors Clin. Investig. 2016, 3 (4), e1430.
219. Rabideau, A. E.; Pentelute, B. L., Delivery of Non-Native Cargo into Mammalian Cells Using Anthrax Lethal Toxin. ACS Chem. Biol. 2016, 11 (6), 1490-1501.
220. Arora, N.; Leppla, S. H., Fusions of anthrax toxin lethal factor with shiga toxin and diphtheria toxin enzymatic domains are toxic to mammalian cells. Infect. Immun. 1994, 62 (11), 4955-4961.
221. Ballard, J. D.; Collier, R. J.; Starnbach, M. N., Anthrax toxin-mediated delivery of a cytotoxic T-cell epitope in vivo. Proc. Natl. Acad. Sci. U. S. A. 1996, 93 (22), 12531-12534.
222. Liao, X.; Rabideau, A. E.; Pentelute, B. L., Delivery of antibody mimics into mammalian cells via anthrax toxin protective antigen. ChemBioChem 2014, 15 (16), 2458-2466.
223. Rabideau, A. E.; Liao, X.; Akçay, G.; Pentelute, B. L., Translocation of Non-Canonical Polypeptides into Cells Using Protective Antigen. Sci. Rep. 2015, 5, 11944.
224. Dyer, P. D. R.; Shepherd, T. R.; Gollings, A. S.; Shorter, S. A.; Gorringe-Pattrick, M. A. M.; Tang, C.-K.; Cattoz, B. N.; Baillie, L.; Griffiths, P. C.; Richardson, S. C. W., Disarmed anthrax toxin delivers antisense oligonucleotides and siRNA with high efficiency and low toxicity. J. Controlled Release 2015, 220, 316-328.
225. Stepanenko, O. V.; Verkhusha, V. V.; Kazakov, V. I.; Shavlovsky, M. M.; Kuznetsova, I. M.; Uversky, V. N.; Turoverov, K. K., Comparative Studies on the Structure and Stability of Fluorescent Proteins EGFP, zFP506, mRFP1, “dimer2”, and DsRed1. Biochemistry 2004, 43 (47), 14913-14923.
226. Zornetta, I.; Brandi, L.; Janowiak, B.; Dal Molin, F.; Tonello, F.; Collier, R. J.; Montecucco, C., Imaging the cell entry of the anthrax oedema and lethal toxins with fluorescent protein chimeras. Cell. Microbiol. 2010, 12 (10), 1435-1445.
227. Park, M.; Xu, X.; Min, W.; Sugiman-Marangos, S. N.; Beilhartz, G. L.; Adams, J. J.; Sidhu, S. S.; Grunebaum, E.; Melnyk, R. A., Intracellular Delivery of Human Purine Nucleoside

152
Phosphorylase by Engineered Diphtheria Toxin Rescues Function in Target Cells. Mol. Pharm. 2018, 15 (11), 5217-5226.
228. Wang, P.; Xue, Y.; Shang, X.; Liu, Y., Diphtheria Toxin Mutant CRM197-Mediated Transcytosis across Blood–Brain Barrier In Vitro. Cell. Mol. Neurobiol. 2010, 30 (5), 717-725.
229. Michel Lacroix; Dima Abi-Said; Daryl R. Fourney; Ziya L. Gokaslan; Weiming Shi; Franco DeMonte; Frederick F. Lang; Ian E. McCutcheon; Samuel J. Hassenbusch; Eric Holland; Kenneth Hess; Christopher Michael; Daniel Miller; Raymond Sawaya, A multivariate analysis of 416 patients with glioblastoma multiforme: prognosis, extent of resection, and survival. J. Neurosurg. 2001, 95 (2), 190-198.
230. Nieder, C.; Grosu, A. L.; Astner, S.; Molls, M., Treatment of Unresectable Glioblastoma Multiforme. Anticancer Res. 2005, 25 (6C), 4605-4610.
231. Chen, J.; McKay, Renée M.; Parada, Luis F., Malignant Glioma: Lessons from Genomics, Mouse Models, and Stem Cells. Cell 2012, 149 (1), 36-47.
232. Singh, S. K.; Hawkins, C.; Clarke, I. D.; Squire, J. A.; Bayani, J.; Hide, T.; Henkelman, R. M.; Cusimano, M. D.; Dirks, P. B., Identification of human brain tumour initiating cells. Nature 2004, 432, 396-401.
233. Gilbertson, R. J.; Rich, J. N., Making a tumour's bed: glioblastoma stem cells and the vascular niche. Nature Reviews Cancer 2007, 7, 733-736.
234. Molina, J. R.; Hayashi, Y.; Stephens, C.; Georgescu, M.-M., Invasive Glioblastoma Cells Acquire Stemness and Increased Akt Activation. Neoplasia 2010, 12 (6), 453-463.
235. Paw, I.; Carpenter, R. C.; Watabe, K.; Debinski, W.; Lo, H.-W., Mechanisms regulating glioma invasion. Cancer Lett. 2015, 362 (1), 1-7.
236. Suvà, Mario L.; Rheinbay, E.; Gillespie, Shawn M.; Patel, Anoop P.; Wakimoto, H.; Rabkin, Samuel D.; Riggi, N.; Chi, Andrew S.; Cahill, Daniel P.; Nahed, Brian V.; Curry, William T.; Martuza, Robert L.; Rivera, Miguel N.; Rossetti, N.; Kasif, S.; Beik, S.; Kadri, S.; Tirosh, I.; Wortman, I.; Shalek, A. K.; Rozenblatt-Rosen, O.; Regev, A.; Louis, David N.; Bernstein, Bradley E., Reconstructing and Reprogramming the Tumor-Propagating Potential of Glioblastoma Stem-like Cells. Cell 2014, 157 (3), 580-594.
237. Lathia, J. D.; Hitomi, M.; Gallagher, J.; Gadani, S. P.; Adkins, J.; Vasanji, A.; Liu, L.; Eyler, C. E.; Heddleston, J. M.; Wu, Q.; Minhas, S.; Soeda, A.; Hoeppner, D. J.; Ravin, R.; McKay, R. D. G.; McLendon, R. E.; Corbeil, D.; Chenn, A.; Hjelmeland, A. B.; Park, D. M.; Rich, J. N., Distribution of CD133 reveals glioma stem cells self-renew through symmetric and asymmetric cell divisions. Cell Death Dis. 2011, 2 (9), e200.
238. Bradshaw, A.; Wickremsekera, A.; Tan, S. T.; Peng, L.; Davis, P. F.; Itinteang, T., Cancer Stem Cell Hierarchy in Glioblastoma Multiforme. Front. Surg. 2016, 3, 21.

153
239. Anzahaee, M. Y.; Deleavey, G. F.; Le, P. U.; Fakhoury, J.; Petrecca, K.; Damha, M. J., Arabinonucleic Acids: 2′-Stereoisomeric Modulators of siRNA Activity. Nucleic Acid Ther. 2014, 24 (5), 336-343.
240. Aartsma-Rus, A.; Krieg, A. M., FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2016, 27 (1), 1-3.
241. Ferrari, N.; Bergeron, D.; Tedeschi, A.-L.; Mangos, M. M.; Paquet, L. U. C.; Renzi, P. M.; Damha, M. J., Characterization of Antisense Oligonucleotides Comprising 2′-Deoxy-2′-Fluoro-β-d-Arabinonucleic Acid (FANA). Ann. N. Y. Acad. Sci. 2006, 1082 (1), 91-102.
242. Lok, C.-N.; Viazovkina, E.; Min, K.-L.; Nagy, E.; Wilds, C. J.; Damha, M. J.; Parniak, M. A., Potent Gene-Specific Inhibitory Properties of Mixed-Backbone Antisense Oligonucleotides Comprised of 2‘-Deoxy-2‘-fluoro-d-arabinose and 2‘-Deoxyribose Nucleotides. Biochemistry 2002, 41 (10), 3457-3467.
243. Agrawal, S.; Kandimalla, E. R., Antisense therapeutics: is it as simple as complementary base recognition? Mol. Med. Today 2000, 6 (2), 72-81.
244. Eckstein, F., Phosphorothioates, Essential Components of Therapeutic Oligonucleotides. Nucleic Acid Ther. 2014, 24 (6), 374-387.
245. Krieg, A. M.; Yi, A.-K.; Matson, S.; Waldschmidt, T. J.; Bishop, G. A.; Teasdale, R.; Koretzky, G. A.; Klinman, D. M., CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 1995, 374, 546-549.
246. Zhao, Q.; Temsamani, J.; Iadarola, P. L.; Jiang, Z.; Agrawal, S., Effect of different chemically modified oligodeoxynucleotides on immune stimulation. Biochem. Pharmacol. 1996, 51 (2), 173-182.
247. Owen, S. C.; Patel, N.; Logie, J.; Pan, G.; Persson, H.; Moffat, J.; Sidhu, S. S.; Shoichet, M. S., Targeting HER2+ breast cancer cells: Lysosomal accumulation of anti-HER2 antibodies is influenced by antibody binding site and conjugation to polymeric nanoparticles. J. Controlled Release 2013, 172 (2), 395-404.
248. Chen, F.; Hong, H.; Zhang, Y.; Valdovinos, H. F.; Shi, S.; Kwon, G. S.; Theuer, C. P.; Barnhart, T. E.; Cai, W., In Vivo Tumor Targeting and Image-Guided Drug Delivery with Antibody-Conjugated, Radiolabeled Mesoporous Silica Nanoparticles. ACS Nano 2013, 7 (10), 9027-9039.
249. Riley, R. S.; Day, E. S., Frizzled7 Antibody-Functionalized Nanoshells Enable Multivalent Binding for Wnt Signaling Inhibition in Triple Negative Breast Cancer Cells. Small 2017, 13 (26), 1700544.
250. Sivaram, A. J.; Wardiana, A.; Howard, C. B.; Mahler, S. M.; Thurecht, K. J., Recent Advances in the Generation of Antibody–Nanomaterial Conjugates. Adv. Healthc. Mater., 1700607.

154
251. D Senter, P.; L Sievers, E., The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631-637.
252. Lambert, J. M.; Chari, R. V. J., Ado-trastuzumab Emtansine (T-DM1): An Antibody–Drug Conjugate (ADC) for HER2-Positive Breast Cancer. J. Med. Chem. 2014, 57 (16), 6949-6964.
253. Lyon, R. P.; Setter, J. R.; Bovee, T. D.; Doronina, S. O.; Hunter, J. H.; Anderson, M. E.; Balasubramanian, C. L.; Duniho, S. M.; Leiske, C. I.; Li, F.; Senter, P. D., Self-hydrolyzing maleimides improve the stability and pharmacological properties of antibody-drug conjugates. Nat. Biotechnol. 2014, 32, 1059-1062.
254. Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N., Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315-337.
255. McCombs, J. R.; Owen, S. C., Antibody Drug Conjugates: Design and Selection of Linker, Payload and Conjugation Chemistry. The AAPS Journal 2015, 17 (2), 339-351.
256. van den Bent, M.; Gan, H. K.; Lassman, A. B.; Kumthekar, P.; Merrell, R.; Butowski, N.; Lwin, Z.; Mikkelsen, T.; Nabors, L. B.; Papadopoulos, K. P.; Penas-Prado, M.; Simes, J.; Wheeler, H.; Walbert, T.; Scott, A. M.; Gomez, E.; Lee, H.-J.; Roberts-Rapp, L.; Xiong, H.; Bain, E.; Ansell, P. J.; Holen, K. D.; Maag, D.; Reardon, D. A., Efficacy of depatuxizumab mafodotin (ABT-414) monotherapy in patients with EGFR-amplified, recurrent glioblastoma: results from a multi-center, international study. Cancer Chemother. Pharmacol. 2017, 80 (6), 1209-1217.
257. Neyns, B.; Sadones, J.; Joosens, E.; Bouttens, F.; Verbeke, L.; Baurain, J. F.; D'Hondt, L.; Strauven, T.; Chaskis, C.; In't Veld, P.; Michotte, A.; De Greve, J., Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Ann. Oncol. 2009, 20 (9), 1596-1603.
258. Brown, D. V.; Filiz, G.; Daniel, P. M.; Hollande, F.; Dworkin, S.; Amiridis, S.; Kountouri, N.; Ng, W.; Morokoff, A. P.; Mantamadiotis, T., Expression of CD133 and CD44 in glioblastoma stem cells correlates with cell proliferation, phenotype stability and intra-tumor heterogeneity. PLOS ONE 2017, 12 (2), e0172791.
259. Brown, D. V.; Daniel, P. M.; D'Abaco, G. M.; Gogos, A.; Ng, W.; Morokoff, A. P.; Mantamadiotis, T., Coexpression analysis of CD133 and CD44 identifies Proneural and Mesenchymal subtypes of glioblastoma multiforme. Oncotarget 2015, 6 (8), 6267-6280.
260. Anido, J.; Sáez-Borderías, A.; Gonzàlez-Juncà, A.; Rodón, L.; Folch, G.; Carmona, M. A.; Prieto-Sánchez, R. M.; Barba, I.; Martínez-Sáez, E.; Prudkin, L.; Cuartas, I.; Raventós, C.; Martínez-Ricarte, F.; Poca, M. A.; García-Dorado, D.; Lahn, M. M.; Yingling, J. M.; Rodón, J.; Sahuquillo, J.; Baselga, J.; Seoane, J., TGF-β Receptor Inhibitors Target the CD44high/Id1high Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell 2010, 18 (6), 655-668.
261. Liffers, K.; Lamszus, K.; Schulte, A., EGFR Amplification and Glioblastoma Stem-Like Cells. Stem Cells International 2015, 2015, 11.

155
262. Lima, W. F.; Nichols, J. G.; Wu, H.; Prakash, T. P.; Migawa, M. T.; Wyrzykiewicz, T. K.; Bhat, B.; Crooke, S. T., Structural Requirements at the Catalytic Site of the Heteroduplex Substrate for Human RNase H1 Catalysis. J. Biol. Chem. 2004, 279 (35), 36317-36326.
263. Uckun, F. M.; Qazi, S.; Dibirdik, I.; Myers, D. E., Rational design of an immunoconjugate for selective knock-down of leukemia-specific E2A-PBX1 fusion gene expression in human Pre-B leukemia. Integr. Biol. 2013, 5 (1), 122-132.
264. Sugo, T.; Terada, M.; Oikawa, T.; Miyata, K.; Nishimura, S.; Kenjo, E.; Ogasawara-Shimizu, M.; Makita, Y.; Imaichi, S.; Murata, S.; Otake, K.; Kikuchi, K.; Teratani, M.; Masuda, Y.; Kamei, T.; Takagahara, S.; Ikeda, S.; Ohtaki, T.; Matsumoto, H., Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Controlled Release 2016, 237 (Supplement C), 1-13.
265. Astriab-Fisher, A.; Fisher, M. H.; Juliano, R.; Herdewijn, P., Increased uptake of antisense oligonucleotides by delivery as double stranded complexes. Biochem. Pharmacol. 2004, 68 (3), 403-407.
266. Hornsby, M.; Paduch, M.; Miersch, S.; Sääf, A.; Matsuguchi, T.; Lee, B.; Wypisniak, K.; Doak, A.; King, D.; Usatyuk, S.; Perry, K.; Lu, V.; Thomas, W.; Luke, J.; Goodman, J.; Hoey, R. J.; Lai, D.; Griffin, C.; Li, Z.; Vizeacoumar, F. J.; Dong, D.; Campbell, E.; Anderson, S.; Zhong, N.; Gräslund, S.; Koide, S.; Moffat, J.; Sidhu, S.; Kossiakoff, A.; Wells, J., A High Through-put Platform for Recombinant Antibodies to Folded Proteins. Mol. Cell. Proteomics 2015, 14 (10), 2833-2847.
267. Min, K.-L.; Viazovkina, E.; Galarneau, A.; Parniak, M. A.; Damha, M. J., Oligonucleotides comprised of alternating 2′-Deoxy-2′-fluoro-β-d-arabinonucleosides and d-2′-deoxyribonucleosides (2′F-ANA/DNA ‘Altimers’) induce efficient RNA cleavage mediated by RNase H. Bioorg. Med. Chem. Lett. 2002, 12 (18), 2651-2654.
268. Champoux, J. J.; Schultz, S. J., Ribonuclease H: properties, substrate specificity and roles in retroviral reverse transcription. FEBS J. 2009, 276 (6), 1506-1516.
269. Klinman, D. M.; Yamshchikov, G.; Ishigatsubo, Y., Contribution of CpG motifs to the immunogenicity of DNA vaccines. J. Immunol. 1997, 158 (8), 3635-3639.
270. Henry, S.; Stecker, K.; Brooks, D.; Monteith, D.; Conklin, B.; Bennett, C. F., Chemically Modified Oligonucleotides Exhibit Decreased Immune Stimulation in Mice. J. Pharmacol. Exp. Ther. 2000, 292 (2), 468-479.
271. Persano, L.; Rampazzo, E.; Basso, G.; Viola, G., Glioblastoma cancer stem cells: Role of the microenvironment and therapeutic targeting. Biochem. Pharmacol. 2013, 85 (5), 612-622.
272. Pandita, A.; Aldape, K. D.; Zadeh, G.; Guha, A.; James, C. D., Contrasting in vivo and in vitro fates of glioblastoma cell subpopulations with amplified EGFR. Genes Chromosomes Cancer 2004, 39 (1), 29-36.
273. Cuellar, T. L.; Barnes, D.; Nelson, C.; Tanguay, J.; Yu, S.-F.; Wen, X.; Scales, S. J.; Gesch, J.; Davis, D.; van Brabant Smith, A.; Leake, D.; Vandlen, R.; Siebel, C. W., Systematic

156
evaluation of antibody-mediated siRNA delivery using an industrial platform of THIOMAB–siRNA conjugates. Nucleic Acids Res. 2015, 43 (2), 1189-1203.
274. Liu, B., Exploring cell type-specific internalizing antibodies for targeted delivery of siRNA. Brief. Funct. Genomics 2007, 6 (2), 112-119.
275. Varkouhi, A. K.; Scholte, M.; Storm, G.; Haisma, H. J., Endosomal escape pathways for delivery of biologicals. J. Controlled Release 2011, 151 (3), 220-228.
276. Bäumer, N.; Berdel, W. E.; Bäumer, S., Immunoprotein-Mediated siRNA Delivery. Mol. Pharm. 2017, 14 (5), 1339-1351.
277. Glatt, D. M.; Beckford Vera, D. R.; Parrott, M. C.; Luft, J. C.; Benhabbour, S. R.; Mumper, R. J., The Interplay of Antigen Affinity, Internalization, and Pharmacokinetics on CD44-Positive Tumor Targeting of Monoclonal Antibodies. Mol. Pharm. 2016, 13 (6), 1894-1903.
278. Rüter, C.; Hardwidge, P. R., ‘Drugs from Bugs’: bacterial effector proteins as promising biological (immune-) therapeutics. FEMS Microbiol. Lett. 2014, 351 (2), 126-132.
279. Henkel, J. S.; Baldwin, M. R.; Barbieri, J. T., Toxins from bacteria. EXS 2010, 100, 1-29.
280. Yao, Y.-d.; Sun, T.-m.; Huang, S.-y.; Dou, S.; Lin, L.; Chen, J.-n.; Ruan, J.-b.; Mao, C.-q.; Yu, F.-y.; Zeng, M.-s.; Zang, J.-y.; Liu, Q.; Su, F.-x.; Zhang, P.; Lieberman, J.; Wang, J.; Song, E., Targeted Delivery of PLK1-siRNA by ScFv Suppresses Her2<sup>+</sup> Breast Cancer Growth and Metastasis. Science Translational Medicine 2012, 4 (130), 130ra48.
281. Zheng, M.; Tao, W.; Zou, Y.; Farokhzad, O. C.; Shi, B., Nanotechnology-Based Strategies for siRNA Brain Delivery for Disease Therapy. Trends Biotechnol. 2018, 36 (5), 562-575.
282. Whitehead, K. A.; Langer, R.; Anderson, D. G., Knocking down barriers: advances in siRNA delivery. Nature Reviews Drug Discovery 2009, 8, 129.
283. Kanasty, R. L.; Whitehead, K. A.; Vegas, A. J.; Anderson, D. G., Action and reaction: the biological response to siRNA and its delivery vehicles. Molecular therapy : the journal of the American Society of Gene Therapy 2012, 20 (3), 513-524.
284. Lönn, P.; Kacsinta, A. D.; Cui, X.-S.; Hamil, A. S.; Kaulich, M.; Gogoi, K.; Dowdy, S. F., Enhancing Endosomal Escape for Intracellular Delivery of Macromolecular Biologic Therapeutics. Sci. Rep. 2016, 6, 32301.
285. Dominska, M.; Dykxhoorn, D. M., Breaking down the barriers: siRNA delivery and endosome escape. J. Cell Sci. 2010, 123 (8), 1183.
286. Zhu, J.; Qiao, M.; Wang, Q.; Ye, Y.; Ba, S.; Ma, J.; Hu, H.; Zhao, X.; Chen, D., Dual-responsive polyplexes with enhanced disassembly and endosomal escape for efficient delivery of siRNA. Biomaterials 2018, 162, 47-59.

157
287. Zhang, T.; Huang, Y.; Ma, X.; Gong, N.; Liu, X.; Liu, L.; Ye, X.; Hu, B.; Li, C.; Tian, J.-H.; Magrini, A.; Zhang, J.; Guo, W.; Xing, J.-F.; Bottini, M.; Liang, X.-J., Fluorinated Oligoethylenimine Nanoassemblies for Efficient siRNA-Mediated Gene Silencing in Serum-Containing Media by Effective Endosomal Escape. Nano Lett. 2018, 18 (10), 6301-6311.
288. Dong, Y.; Siegwart, D. J.; Anderson, D. G., Strategies, design, and chemistry in siRNA delivery systems. Adv. Drug Del. Rev. 2019.
289. Bartlett, D. W.; Su, H.; Hildebrandt, I. J.; Weber, W. A.; Davis, M. E., Impact of tumor-specific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging. Proc Natl Acad Sci U S A 2007, 104 (39), 15549-15554.
290. Li, Y. M.; Vallera, D. A.; Hall, W. A., Diphtheria toxin-based targeted toxin therapy for brain tumors. Journal of Neuro-Oncology 2013, 114 (2), 155-164.
291. Liang, H.; Ding, X.; Zhou, C.; Zhang, Y.; Xu, M.; Zhang, C.; Xu, L., Knockdown of eukaryotic translation initiation factors 3B (EIF3B) inhibits proliferation and promotes apoptosis in glioblastoma cells. 2012; Vol. 33, p 1057-62.
292. Sun, Q.; Zhou, C.; Ma, R.; Guo, Q.; Huang, H.; Hao, J.; Liu, H.; Shi, R.; Liu, B., Prognostic value of increased integrin-beta 1 expression in solid cancers: a meta-analysis. Onco Targets Ther 2018, 11, 1787-1799.
293. Pereira, M. M. A.; Mahú, I.; Seixas, E.; Martinéz-Sánchez, N.; Kubasova, N.; Pirzgalska, R. M.; Cohen, P.; Dietrich, M. O.; López, M.; Bernardes, G. J. L.; Domingos, A. I., A brain-sparing diphtheria toxin for chemical genetic ablation of peripheral cell lineages. Nature Communications 2017, 8, 14967.
294. Harder, B. G.; Blomquist, M. R.; Wang, J.; Kim, A. J.; Woodworth, G. F.; Winkles, J. A.; Loftus, J. C.; Tran, N. L., Developments in Blood-Brain Barrier Penetrance and Drug Repurposing for Improved Treatment of Glioblastoma. Front. Oncol. 2018, 8, 462-462.
295. Oyagi, A.; Hara, H., Essential roles of heparin-binding epidermal growth factor-like growth factor in the brain. CNS Neurosci. Ther. 2012, 18 (10), 803-810.
296. Shin, C. H.; Robinson, J. P.; Sonnen, J. A.; Welker, A. E.; Yu, D. X.; VanBrocklin, M. W.; Holmen, S. L., HBEGF promotes gliomagenesis in the context of Ink4a/Arf and Pten loss. Oncogene 2017, 36 (32), 4610-4618.
297. Lan, X.; Jörg, D. J.; Cavalli, F. M. G.; Richards, L. M.; Nguyen, L. V.; Vanner, R. J.; Guilhamon, P.; Lee, L.; Kushida, M. M.; Pellacani, D.; Park, N. I.; Coutinho, F. J.; Whetstone, H.; Selvadurai, H. J.; Che, C.; Luu, B.; Carles, A.; Moksa, M.; Rastegar, N.; Head, R.; Dolma, S.; Prinos, P.; Cusimano, M. D.; Das, S.; Bernstein, M.; Arrowsmith, C. H.; Mungall, A. J.; Moore, R. A.; Ma, Y.; Gallo, M.; Lupien, M.; Pugh, T. J.; Taylor, M. D.; Hirst, M.; Eaves, C. J.; Simons, B. D.; Dirks, P. B., Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 2017, 549 (7671), 227-232.

158
298. Cheng, L.; Wu, Q.; Guryanova, O. A.; Huang, Z.; Huang, Q.; Rich, J. N.; Bao, S., Elevated invasive potential of glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2011, 406 (4), 643-648.
299. Pappenheimer, A. M., Diphtheria Toxin. Annu. Rev. Biochem 1977, 46 (1), 69-94.
300. Kubo, T.; Takei, Y.; Mihara, K.; Yanagihara, K.; Seyama, T., Amino-Modified and Lipid-Conjugated Dicer-Substrate siRNA Enhances RNAi Efficacy. Bioconjugate Chem. 2012, 23, 164-73.
301. Takemoto, H.; Miyata, K.; Ishii, T.; Hattori, S.; Osawa, S.; Nishiyama, N.; Kataoka, K., Accelerated Polymer–Polymer Click Conjugation by Freeze–Thaw Treatment. Bioconjugate Chem. 2012, 23 (8), 1503-1506.
302. MacLeod, G.; Bozek, D. A.; Rajakulendran, N.; Monteiro, V.; Ahmadi, M.; Steinhart, Z.; Kushida, M. M.; Yu, H.; Coutinho, F. J.; Cavalli, F. M. G.; Restall, I.; Hao, X.; Hart, T.; Luchman, H. A.; Weiss, S.; Dirks, P. B.; Angers, S., Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep. 2019, 27 (3), 971-986.e9.
303. Tam, R. Y.; Yockell-Lelièvre, J.; Smith, L. J.; Julian, L. M.; Baker, A. E. G.; Choey, C.; Hasim, M. S.; Dimitroulakos, J.; Stanford, W. L.; Shoichet, M. S., Rationally Designed 3D Hydrogels Model Invasive Lung Diseases Enabling High-Content Drug Screening. Adv. Mater. 2019, 31 (7), 1806214.
304. Jackson, M.; Hassiotou, F.; Nowak, A., Glioblastoma stem-like cells: at the root of tumor recurrence and a therapeutic target. Carcinogenesis 2014, 36 (2), 177-185.
305. Dominska, M.; Dykxhoorn, D. M., Breaking down the barriers: siRNA delivery and endosome escape. J. Cell Sci. 2010, 123 (Pt 8), 1183-1189.
306. Johannes, L.; Lucchino, M., Current Challenges in Delivery and Cytosolic Translocation of Therapeutic RNAs. Nucleic Acid Ther. 2018, 28 (3), 178-193.
307. Ladokhin, A. S.; Vargas-Uribe, M.; Rodnin, M. V.; Ghatak, C.; Sharma, O., Cellular Entry of the Diphtheria Toxin Does Not Require the Formation of the Open-Channel State by Its Translocation Domain. Toxins (Basel) 2017, 9 (10), 299.
308. Leka, O.; Vallese, F.; Pirazzini, M.; Berto, P.; Montecucco, C.; Zanotti, G., Diphtheria toxin conformational switching at acidic pH. FEBS J. 2014, 281 (9), 2115-2122.
309. Brock, D. J.; Kustigian, L.; Jiang, M.; Graham, K.; Wang, T.-Y.; Erazo-Oliveras, A.; Najjar, K.; Zhang, J.; Rye, H.; Pellois, J.-P., Efficient cell delivery mediated by lipid-specific endosomal escape of supercharged branched peptides. Traffic 2018, 19 (6), 421-435.
310. Najjar, K.; Erazo-Oliveras, A.; Mosior, J. W.; Whitlock, M. J.; Rostane, I.; Cinclair, J. M.; Pellois, J.-P., Unlocking Endosomal Entrapment with Supercharged Arginine-Rich Peptides. Bioconjugate Chem. 2017, 28 (12), 2932-2941.

159
311. O'Keefe, D. O.; Cabiaux, V.; Choe, S.; Eisenberg, D.; Collier, R. J., pH-dependent insertion of proteins into membranes: B-chain mutation of diphtheria toxin that inhibits membrane translocation, Glu-349----Lys. Proc. Natl. Acad. Sci. U. S. A. 1992, 89 (13), 6202-6206.
312. Sattiraju, A.; Sai, K. K. S.; Mintz, A., Glioblastoma Stem Cells and Their Microenvironment. In Stem Cell Microenvironments and Beyond, Birbrair, A., Ed. Springer International Publishing: Cham, 2017; pp 119-140.
313. Marín-Ramos, N. I.; Thein, T. Z.; Cho, H.-Y.; Swenson, S. D.; Wang, W.; Schönthal, A. H.; Chen, T. C.; Hofman, F. M., NEO212 Inhibits Migration and Invasion of Glioma Stem Cells. Mol. Cancer Ther. 2018, 17 (3), 625.
314. Koshizuka, K.; Kikkawa, N.; Hanazawa, T.; Yamada, Y.; Okato, A.; Arai, T.; Katada, K.; Okamoto, Y.; Seki, N., Inhibition of integrin β1-mediated oncogenic signalling by the antitumor microRNA-29 family in head and neck squamous cell carcinoma. Oncotarget 2017, 9 (3), 3663-3676.
315. Wang, H.; Ru, Y.; Sanchez-Carbayo, M.; Wang, X.; Kieft, J. S.; Theodorescu, D., Translation initiation factor eIF3b expression in human cancer and its role in tumor growth and lung colonization. Clin. Cancer. Res. 2013, 19 (11), 2850-2860.
316. Pollard, S. M.; Yoshikawa, K.; Clarke, I. D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; Squire, J. A.; Smith, A.; Dirks, P., Glioma Stem Cell Lines Expanded in Adherent Culture Have Tumor-Specific Phenotypes and Are Suitable for Chemical and Genetic Screens. Cell Stem Cell 2009, 4 (6), 568-580.
317. Wagner, S.; Herrmannová, A.; Šikrová, D.; Valášek, L. S., Human eIF3b and eIF3a serve as the nucleation core for the assembly of eIF3 into two interconnected modules: the yeast-like core and the octamer. Nucleic Acids Res. 2016, 44 (22), 10772-10788.
318. Kopp, S.; Krüger, M.; Feldmann, S.; Oltmann, H.; Schütte, A.; Schmitz, B.; Bauer, J.; Schulz, H.; Saar, K.; Huebner, N.; Wehland, M.; Nassef, M. Z.; Melnik, D.; Meltendorf, S.; Infanger, M.; Grimm, D., Thyroid cancer cells in space during the TEXUS-53 sounding rocket mission - The THYROID Project. Sci. Rep. 2018, 8 (1), 10355-10355.
319. Deleavey, G. F.; Watts, J. K.; Alain, T.; Robert, F.; Kalota, A.; Aishwarya, V.; Pelletier, J.; Gewirtz, A. M.; Sonenberg, N.; Damha, M. J., Synergistic effects between analogs of DNA and RNA improve the potency of siRNA-mediated gene silencing. Nucleic Acids Res. 2010, 38 (13), 4547-4557.
320. Ge, Q.; Dallas, A.; Ilves, H.; Shorenstein, J.; Behlke, M. A.; Johnston, B. H., Effects of chemical modification on the potency, serum stability, and immunostimulatory properties of short shRNAs. RNA 2010, 16 (1), 118-130.
321. Khvorova, A.; Watts, J. K., The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35 (3), 238-248.

160
322. Matranga, C.; Tomari, Y.; Shin, C.; Bartel, D. P.; Zamore, P. D., Passenger-Strand Cleavage Facilitates Assembly of siRNA into Ago2-Containing RNAi Enzyme Complexes. Cell 2005, 123 (4), 607-620.
323. Watts, J. K.; Corey, D. R., Silencing disease genes in the laboratory and the clinic. J. Pathol. 2012, 226 (2), 365-379.
324. McClorey, G.; Wood, M. J., An overview of the clinical application of antisense oligonucleotides for RNA-targeting therapies. Curr. Opin. Pharmacol. 2015, 24, 52-58.
325. Allerson, C. R.; Sioufi, N.; Jarres, R.; Prakash, T. P.; Naik, N.; Berdeja, A.; Wanders, L.; Griffey, R. H.; Swayze, E. E.; Bhat, B., Fully 2‘-Modified Oligonucleotide Duplexes with Improved in Vitro Potency and Stability Compared to Unmodified Small Interfering RNA. J. Med. Chem. 2005, 48 (4), 901-904.
326. Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W. D.; Yang, C.-C.; Ueda, M.; Kristen, A. V.; Tournev, I.; Schmidt, H. H.; Coelho, T.; Berk, J. L.; Lin, K.-P.; Vita, G.; Attarian, S.; Planté-Bordeneuve, V.; Mezei, M. M.; Campistol, J. M.; Buades, J.; Brannagan, T. H.; Kim, B. J.; Oh, J.; Parman, Y.; Sekijima, Y.; Hawkins, P. N.; Solomon, S. D.; Polydefkis, M.; Dyck, P. J.; Gandhi, P. J.; Goyal, S.; Chen, J.; Strahs, A. L.; Nochur, S. V.; Sweetser, M. T.; Garg, P. P.; Vaishnaw, A. K.; Gollob, J. A.; Suhr, O. B., Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. New England Journal of Medicine 2018, 379 (1), 11-21.
327. Hassler, M. R.; Turanov, A. A.; Alterman, J. F.; Haraszti, R. A.; Coles, A. H.; Osborn, M. F.; Echeverria, D.; Nikan, M.; Salomon, W. E.; Roux, L.; Godinho, B. M. D. C.; Davis, S. M.; Morrissey, D. V.; Zamore, P. D.; Karumanchi, S. A.; Moore, M. J.; Aronin, N.; Khvorova, A., Comparison of partially and fully chemically-modified siRNA in conjugate-mediated delivery in vivo. Nucleic Acids Res. 2018, 46 (5), 2185-2196.
328. Beloor, J.; Zeller, S.; Choi, C. S.; Lee, S.-K.; Kumar, P., Cationic cell-penetrating peptides as vehicles for siRNA delivery. Ther. Deliv. 2015, 6 (4), 491-507.
329. Wang, X.-L.; Xu, R.; Wu, X.; Gillespie, D.; Jensen, R.; Lu, Z.-R., Targeted Systemic Delivery of a Therapeutic siRNA with a Multifunctional Carrier Controls Tumor Proliferation in Mice. Mol. Pharm. 2009, 6 (3), 738-746.
330. Patil, M. L.; Zhang, M.; Taratula, O.; Garbuzenko, O. B.; He, H.; Minko, T., Internally Cationic Polyamidoamine PAMAM-OH Dendrimers for siRNA Delivery: Effect of the Degree of Quaternization and Cancer Targeting. Biomacromolecules 2009, 10 (2), 258-266.
331. Kwon, E. J.; Bergen, J. M.; Pun, S. H., Application of an HIV gp41-Derived Peptide for Enhanced Intracellular Trafficking of Synthetic Gene and siRNA Delivery Vehicles. Bioconjugate Chem. 2008, 19 (4), 920-927.
332. Park, J.; Park, J.; Pei, Y.; Xu, J.; Yeo, Y., Pharmacokinetics and biodistribution of recently-developed siRNA nanomedicines. Adv. Drug Del. Rev. 2016, 104, 93-109.
333. Xue, H. Y.; Liu, S.; Wong, H. L., Nanotoxicity: a key obstacle to clinical translation of siRNA-based nanomedicine. Nanomedicine 2014, 9 (2), 295-312.

161
334. Hegde, V.; Hickerson, R. P.; Nainamalai, S.; Campbell, P. A.; Smith, F. J. D.; McLean, W. H. I.; Leslie Pedrioli, D. M., In vivo gene silencing following non-invasive siRNA delivery into the skin using a novel topical formulation. J. Control. Release 2014, 196, 355-362.
335. Shrivats, A. R.; Mishina, Y.; Averick, S.; Matyjaszewski, K.; Hollinger, J. O., In Vivo GFP Knockdown by Cationic Nanogel-siRNA Polyplexes. Bioengineering 2015, 2 (3), 160-175.
336. Lee, S. H.; Mok, H.; Lee, Y.; Park, T. G., Self-assembled siRNA–PLGA conjugate micelles for gene silencing. J. Control. Release 2011, 152 (1), 152-158.
337. Wang, Z.; Chen, J.; Sun, J.; Cui, Z.; Wu, H., RNA interference-mediated silencing of eukaryotic translation initiation factor 3, subunit B (EIF3B) gene expression inhibits proliferation of colon cancer cells. World J. Surg. Oncol. 2012, 10, 119-119.
338. Xu, F.; Xu, C.-Z.; Gu, J.; Liu, X.; Liu, R.; Huang, E.; Yuan, Y.; Zhao, G.; Jiang, J.; Xu, C.; Chu, Y.; Lu, C.; Ge, D., Eukaryotic translation initiation factor 3B accelerates the progression of esophageal squamous cell carcinoma by activating β-catenin signaling pathway. Oncotarget 2016, 7 (28), 43401-43411.
339. Zeng, Z.-Z.; Yao, H.; Staszewski, E. D.; Rockwood, K. F.; Markwart, S. M.; Fay, K. S.; Spalding, A. C.; Livant, D. L., α5β1 Integrin Ligand PHSRN Induces Invasion and α5 mRNA in Endothelial Cells to Stimulate Angiogenesis. Transl. Oncol. 2009, 2 (1), 8-20.
340. Brockbank, E. C.; Bridges, J.; Marshall, C. J.; Sahai, E., Integrin β1 is required for the invasive behaviour but not proliferation of squamous cell carcinoma cells in vivo. Br. J. Cancer 2005, 92 (1), 102-112.
341. Calderwood, D. A., Integrin activation. J. Cell Sci. 2004, 117 (5), 657.
342. Zhang, L.; Zou, W., Inhibition of integrin β1 decreases the malignancy of ovarian cancer cells and potentiates anticancer therapy via the FAK/STAT1 signaling pathway. Mol. Med. Report. 2015, 12 (6), 7869-7876.
343. Ruoslahti, E., FIBRONECTIN AND ITS RECEPTORS. Annu. Rev. Biochem. 1988, 57 (1), 375-413.
344. Lucki, N. C.; Villa, G. R.; Vergani, N.; Bollong, M. J.; Beyer, B. A.; Lee, J. W.; Anglin, J. L.; Spangenberg, S. H.; Chin, E. N.; Sharma, A.; Johnson, K.; Sander, P. N.; Gordon, P.; Skirboll, S. L.; Wurdak, H.; Schultz, P. G.; Mischel, P. S.; Lairson, L. L., A cell type-selective apoptosis-inducing small molecule for the treatment of brain cancer. Proc. Natl. Acad. Sci. U.S.A. 2019, 116 (13), 6435.
345. Imad Saeed, K.; Moneeb, E., Targeting glioblastoma cancer stem cells: the next great hope? Neurosurg. Focus 2014, 37 (6), E7.
346. Xu, Y.; Stamenkovic, I.; Yu, Q., CD44 Attenuates Activation of the Hippo Signaling Pathway and Is a Prime Therapeutic Target for Glioblastoma. Cancer Res. 2010, 70 (6), 2455.

162
347. Brown, D. V.; Filiz, G.; Daniel, P. M.; Hollande, F.; Dworkin, S.; Amiridis, S.; Kountouri, N.; Ng, W.; Morokoff, A. P.; Mantamadiotis, T., Expression of CD133 and CD44 in glioblastoma stem cells correlates with cell proliferation, phenotype stability and intra-tumor heterogeneity. PloS One 2017, 12 (2), e0172791-e0172791.
348. Tammi, R.; Rilla, K.; Pienimäki, J.-P.; MacCallum, D. K.; Hogg, M.; Luukkonen, M.; Hascall, V. C.; Tammi, M., Hyaluronan Enters Keratinocytes by a Novel Endocytic Route for Catabolism. J. Biol. Chem. 2001, 276 (37), 35111-35122.
349. López-Ortega, O.; Santos-Argumedo, L., Myosin 1g Contributes to CD44 Adhesion Protein and Lipid Rafts Recycling and Controls CD44 Capping and Cell Migration in B Lymphocytes. Front. Immunol. 2017, 8, 1731-1731.
350. Boissier, P.; Chen, J.; Huynh-Do, U., EphA2 signaling following endocytosis: role of Tiam1. Traffic 2013, 14 (12), 1255-1271.
351. Wang, H.-H.; Liao, C.-C.; Chow, N.-H.; Huang, L. L.-H.; Chuang, J.-I.; Wei, K.-C.; Shin, J.-W., Whether CD44 is an applicable marker for glioma stem cells. Am. J. Transl. Res. 2017, 9 (11), 4785-4806.
352. Mitamura, T.; Higashiyama, S.; Taniguchi, N.; Klagsbrun, M.; Mekada, E., Diphtheria Toxin Binds to the Epidermal Growth Factor (EGF)-like Domain of Human Heparin-binding EGF-like Growth Factor/Diphtheria Toxin Receptor and Inhibits Specifically Its Mitogenic Activity. J. Biol. Chem. 1995, 270 (3), 1015-1019.
353. Yan, M.; Yang, X.; Shen, R.; Wu, C.; Wang, H.; Ye, Q.; Yang, P.; Zhang, L.; Chen, M.; Wan, B.; Zhang, Q.; Xia, S.; Lu, X.; Shao, G.; Zhou, X.; Yu, J.; Shao, Q., miR-146b promotes cell proliferation and increases chemosensitivity, but attenuates cell migration and invasion via FBXL10 in ovarian cancer. Cell Death Dis. 2018, 9 (11), 1123.
354. Zhang, I.; Beus, M.; Stochaj, U.; Le, P. U.; Zorc, B.; Rajić, Z.; Petrecca, K.; Maysinger, D., Inhibition of glioblastoma cell proliferation, invasion, and mechanism of action of a novel hydroxamic acid hybrid molecule. Cell Death Discov. 2018, 4 (1), 41.
355. Hatoum, A.; Mohammed, R.; Zakieh, O., The unique invasiveness of glioblastoma and possible drug targets on extracellular matrix. Cancer Manag. Res. 2019, 11, 1843-1855.
356. Al Hassan, M.; Fakhoury, I.; El Masri, Z.; Ghazale, N.; Dennaoui, R.; El Atat, O.; Kanaan, A.; El-Sibai, M., Metformin Treatment Inhibits Motility and Invasion of Glioblastoma Cancer Cells. Anal. Cell. Pathol. 2018, 2018, 9.
357. Nam, L.; Coll, C.; Erthal, L. C. S.; de la Torre, C.; Serrano, D.; Martínez-Máñez, R.; Santos-Martínez, M. J.; Ruiz-Hernández, E., Drug Delivery Nanosystems for the Localized Treatment of Glioblastoma Multiforme. Materials 2018, 11 (5), 779.
358. Lacroix, A.; Vengut-Climent, E.; de Rochambeau, D.; Sleiman, H. F., Uptake and Fate of Fluorescently Labeled DNA Nanostructures in Cellular Environments: A Cautionary Tale. ACS Cent. Sci. 2019, 5 (5), 882-891.

163
359. Johnson, L. S.; Dunn, K. W.; Pytowski, B.; McGraw, T. E., Endosome acidification and receptor trafficking: bafilomycin A1 slows receptor externalization by a mechanism involving the receptor's internalization motif. Mol. Biol. Cell 1993, 4 (12), 1251-1266.
360. Cervia, L. D.; Chang, C.-C.; Wang, L.; Yuan, F., Distinct effects of endosomal escape and inhibition of endosomal trafficking on gene delivery via electrotransfection. PloS One 2017, 12 (2), e0171699-e0171699.
361. DeVorkin, L.; Lum, J. J., Chapter 8 - Strategies to Block Autophagy in Tumor Cells. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging, Hayat, M. A., Ed. Academic Press: Amsterdam, 2014; pp 121-130.
362. Schmidt, T. G. M.; Skerra, A., The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2007, 2, 1528.
363. Cabantous, S.; Terwilliger, T. C.; Waldo, G. S., Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 2005, 23 (1), 102-107.
364. Kim, S. J.; Hatch, S. T.; Dixon, A. S.; Owen, S. C., Split-enzyme fragment as a single affinity tag that enables protein expression, purification, and functional assays. Biotechnol. Bioeng. 2019, 116 (7), 1575-1583.
365. Ahmad, Z. A.; Yeap, S. K.; Ali, A. M.; Ho, W. Y.; Alitheen, N. B. M.; Hamid, M., scFv Antibody: Principles and Clinical Application. Clin. Dev. Immunol. 2012, 2012, 15.
366. Wang, Z.; Wei, M.; Zhang, H.; Chen, H.; Germana, S.; Huang, C. A.; Madsen, J. C.; Sachs, D. H.; Wang, Z., Diphtheria-toxin based anti-human CCR4 immunotoxin for targeting human CCR4+ cells in vivo. Mol. Oncol. 2015, 9 (7), 1458-1470.
367. Bachanova, V.; Cao, Q.; Weisdorf, D. J.; Curtsinger, J. M.; Cooley, S. A.; Miller, J.; Vallera, D., Bispecific ligand-directed toxin targeting CD22 and CD19 (DT2219) for refractory B-cell malignancies: Results of phase I-II trial. J. Clin. Oncol. 2019, 37 (15_suppl), e19066-e19066.
368. Waldron, N. N.; Kaufman, D. S.; Oh, S.; Inde, Z.; Hexum, M. K.; Ohlfest, J. R.; Vallera, D. A., Targeting tumor-initiating cancer cells with dCD133KDEL shows impressive tumor reductions in a xenotransplant model of human head and neck cancer. Mol. Cancer Ther. 2011, 10 (10), 1829-1838.
369. Lipsman, N.; Meng, Y.; Bethune, A. J.; Huang, Y.; Lam, B.; Masellis, M.; Herrmann, N.; Heyn, C.; Aubert, I.; Boutet, A.; Smith, G. S.; Hynynen, K.; Black, S. E., Blood–brain barrier opening in Alzheimer’s disease using MR-guided focused ultrasound. Nat. Commun. 2018, 9 (1), 2336.
370. Chan, D. P. Y.; Deleavey, G. F.; Owen, S. C.; Damha, M. J.; Shoichet, M. S., Click conjugated polymeric immuno-nanoparticles for targeted siRNA and antisense oligonucleotide delivery. Biomaterials 2013, 34 (33), 8408-8415.

164
371. Collingwood, M. A.; Rose, S. D.; Huang, L.; Hillier, C.; Amarzguioui, M.; Wiiger, M. T.; Soifer, H. S.; Rossi, J. J.; Behlke, M. A., Chemical modification patterns compatible with high potency dicer-substrate small interfering RNAs. Oligonucleotides 2008, 18 (2), 187-200.
372. Corey, D. R.; Abrams, J. M., Morpholino antisense oligonucleotides: tools for investigating vertebrate development. Genome Biol. 2001, 2 (5), REVIEWS1015-REVIEWS1015.
373. Pellestor, F.; Paulasova, P., The peptide nucleic acids (PNAs), powerful tools for molecular genetics and cytogenetics. Europ. J. Hum. Genet. 2004, 12 (9), 694-700.
374. Moulton, J. D.; Yan, Y.-L., Using Morpholinos to Control Gene Expression. Curr. Protoc. Mol. Biol. 2008, 83 (1), 26.8.1-26.8.29.
375. Corey, D. R., Peptide nucleic acids: Cellular delivery and recognition of DNA and RNA targets. Lett. Pept. Sci. 2003, 10 (3), 347-352.
376. Kolevzon, N.; Nasereddin, A.; Naik, S.; Yavin, E.; Dzikowski, R., Use of peptide nucleic acids to manipulate gene expression in the malaria parasite Plasmodium falciparum. PloS One 2014, 9 (1), e86802-e86802.
377. Weaver, M.; Laske, D. W., Transferrin Receptor Ligand-Targeted Toxin Conjugate (Tf-CRM107) for Therapy of Malignant Gliomas. J. Neuro-Oncol. 2003, 65 (1), 3-14.
378. Torres, V. A.; Tapia, J. C.; Rodríguez, D. A.; Párraga, M.; Lisboa, P.; Montoya, M.; Leyton, L.; Quest, A. F. G., Caveolin-1 controls cell proliferation and cell death by suppressing expression of the inhibitor of apoptosis protein survivin. J. Cell Sci. 2006, 119 (9), 1812.
379. Hallock, P.; Thomas, M. A., Integrating the Alzheimer's disease proteome and transcriptome: a comprehensive network model of a complex disease. OMICS 2012, 16 (1-2), 37-49.
380. Rodríguez-Lebrón, E.; Gouvion, C. M.; Moore, S. A.; Davidson, B. L.; Paulson, H. L., Allele-specific RNAi Mitigates Phenotypic Progression in a Transgenic Model of Alzheimer's Disease. Mol. Ther. 2009, 17 (9), 1563-1573.
381. Nagahara, A. H.; Merrill, D. A.; Coppola, G.; Tsukada, S.; Schroeder, B. E.; Shaked, G. M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J. M.; Rockenstein, E.; Chao, M. V.; Koo, E. H.; Geschwind, D.; Masliah, E.; Chiba, A. A.; Tuszynski, M. H., Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat. Med. 2009, 15 (3), 331-337.
382. Shin, M.-K.; Kim, H.-G.; Baek, S.-H.; Jung, W.-R.; Park, D.-I.; Park, J.-S.; Jo, D.-G.; Kim, K.-L., Neuropep-1 ameliorates learning and memory deficits in an Alzheimer's disease mouse model, increases brain-derived neurotrophic factor expression in the brain, and causes reduction of amyloid beta plaques. Neurobiol. Aging 2014, 35 (5), 990-1001.
383. Alfarouk, K. O.; Stock, C.-M.; Taylor, S.; Walsh, M.; Muddathir, A. K.; Verduzco, D.; Bashir, A. H. H.; Mohammed, O. Y.; Elhassan, G. O.; Harguindey, S.; Reshkin, S. J.; Ibrahim,

165
M. E.; Rauch, C., Resistance to cancer chemotherapy: failure in drug response from ADME to P-gp. Cancer Cell Int. 2015, 15 (1), 71.
384. Yuan, A. L.; Ricks, C. B.; Bohm, A. K.; Lun, X.; Maxwell, L.; Safdar, S.; Bukhari, S.; Gerber, A.; Sayeed, W.; Bering, E. A.; Pedersen, H.; Chan, J. A.; Shen, Y.; Marra, M.; Kaplan, D. R.; Mason, W.; Goodman, L. D.; Ezhilarasan, R.; Kaufmann, A. B.; Cabral, M.; Robbins, S. M.; Senger, D. L.; Cahill, D. P.; Sulman, E. P.; Cairncross, J. G.; Blough, M. D., ABT-888 restores sensitivity in temozolomide resistant glioma cells and xenografts. PloS One 2018, 13 (8), e0202860.
385. Lu, J.; Shoichet, M., Self-Assembled Polymeric Nanoparticles of Organocatalytic Copolymerizated D, L-Lactide and 2-Methyl 2-Carboxytrimethylene Carbonate. Macromolecules 2010, 43.
386. Logie, J.; Owen, S.; McLaughlin, C.; Shoichet, M., PEG-Graft Density Controls Polymeric Nanoparticle Micelle Stability. Chem. Mater. 2014, 26, 2847–2855.




















