
and N, O- Containing Heterocycles by - OhioLINK ETD Center
234
A Dissertation entitled Halogen Mediated Alkene Difunctionalization for Synthesis of N, S- and N, O- Containing Heterocycles by Nur-E Alom Submitted to the Graduate Faculty as partial fulfillment of the requirements for the Doctor of Philosophy Degree in Chemistry ___________________________________________ Dr. Wei Li, Committee Chair ___________________________________________ Dr. Steven J. Sucheck, Committee Member ___________________________________________ Dr. Jianglong Zhu, Committee Member ___________________________________________ Dr. Ana C. Alba-Rubio, Committee Member ___________________________________________ Dr. Amanda C. Bryant-Friedrich, Dean College of Graduate Studies The University of Toledo August 2020
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of and N, O- Containing Heterocycles by - OhioLINK ETD Center

A Dissertation
entitled
Halogen Mediated Alkene Difunctionalization for Synthesis of N, S- and N, O-
Containing Heterocycles
by
Nur-E Alom
Submitted to the Graduate Faculty as partial fulfillment of the requirements for the
Doctor of Philosophy Degree in
Chemistry
___________________________________________
Dr. Wei Li, Committee Chair
___________________________________________
Dr. Steven J. Sucheck, Committee Member
___________________________________________
Dr. Jianglong Zhu, Committee Member
___________________________________________
Dr. Ana C. Alba-Rubio, Committee Member
___________________________________________
Dr. Amanda C. Bryant-Friedrich, Dean
College of Graduate Studies
The University of Toledo
August 2020

© 2020 Nur-E Alom
This document is copyrighted material. Under copyright law, no parts of this document
may be reproduced without the expressed permission of the author.

iii
An Abstract of
Halogen Mediated Alkene Difunctionalization for Synthesis of N, S- and N, O-
Containing Heterocycles
by
Nur-E Alom
Submitted to the Graduate Faculty as partial fulfillment of the requirements for the
Doctor of Philosophy Degree in
Chemistry
The University of Toledo
August 2020
N, S- and N, O-containing heterocycles are ubiquitous structural motifs in
pharmaceuticals, agrochemicals, and bioactive natural products. These types of compounds
exhibit a broad range of bioactivities such as anti-cancer, anti-HIV, antibiotics, and
antidepressants. To gain access to those heterocycles, alkene sulfenoamination and alkene
oxyamination have been considered as powerful strategies because of its ability to rapidly
increase the molecular complexity and functional diversity. Moreover, the wide availability
of alkenes in natural products and commercial sources, in addition to the ease of synthetic
accesses make these alkene functionalization strategies more appealing in the synthetic
community for the modular synthesis of those heterocycles. Our group is interested in the
utilization of classic halonium ion as a regioselective template for the functionalization of
alkenes. The research efforts presented in this dissertation focus on the development of
simple and efficient methods for alkene sulfenoamination and alkene oxyamination by
demonstrating a series of alkene sulfenoamination protocols for accessing interesting N,
S–containing heterocycles.

iv
In Chapter 2, a simple and convenient method for the synthesis of thiazoline from
readily available chemical feedstocks such as alkenes and thioamides is described. The
reaction goes through the in-situ generation of 1,2-dibromoalkane from the bromination of
alkene, followed by the nucleophilic attack of thioamide on the 1,2-dibromoalkane
intermediate leading to thiazoline formation. The synthetic application of this method was
further demonstrated by hydrolysis of thiazoline to 1,2-amino thiol and oxidation to
thiazole, a common scaffold in drugs.
In Chapter 3, the regio- and stereoselective sulfenoamination of alkene with
thioimidazoles for the synthesis of N, S-containing heterocycle is reported. In this reaction,
Selectfluor was used as a halogen source to convert the nucleophilic sulfur reagent into an
electrophilic sulfur source through the formation of a sulfur-fluorine bond. Nucleophilic
attack of an alkene on the sulfur electrophile led to the formation of a thiiranium ion
intermediate, then intramolecular cyclization on thiiranium ion intermediate or open
carbocation resulted in the product formation. The opposite regioisomer of the product
could also be achieved by using bromine as a halogen source. This method exhibited good
functional group tolerance and a broad range of substrate scope in a highly regio- and
stereoselective manner.
In Chapter 4, an efficient approach for the synthesis of 1,4-benzothiazine via alkene
sulfenoamination is presented. This method is an improvement over our previous two
strategies, in which we can use a catalytic amount of an iodide salt, that offers an
environmentally benign and more economic strategy for alkene sulfenoamination.
Moreover, the use of unprotected aminothiophenol as a coupling partner represents another
significant advance in accessing 1,4-benzothiazine. The reaction proceeds through

v
inversion of the thiol polarity with the formation of a sulfur-iodine bond. The investigation
of the mechanism suggests that both polar thiiranium and radical pathways are plausible in
this reaction.
In Chapter 5, the development of a method for the alkylation of arene using alcohol
via carbon-carbon bond formation is disclosed. The highlighting feature of this reaction is
the utilization of an alkene as a catalyst for iodonium formation that can activate an alcohol,
leading to the generation of the product with an all-carbon quaternary centers. The method
shows a good functional group tolerance with a wide range of arene and alcohol substrate
scope. This strategy can be extended to the formation of sterically congested carbon-
heteroatom bonds. In addition, this method is chemoselective for tertiary alcohols in
preference to the primary and secondary alcohols.
In Chapter 6, an iodide-catalyzed alkene oxyamination reaction for the synthesis of
oxazolidinone is discovered. The reaction utilizes unfunctionalized carbamate as a
nucleophilic coupling partner, and Selectfluor as an oxidant to oxidize iodide to iodine.
The reaction proceeds through the generation of catalytic iodonium intermediate, followed
by nucleophilic attack of carbamate, leading to the formation of oxazolidinone product.
The complementary regioisomeric product can also be achieved utilizing NBS instead as a
halogen source.

vi
This work is dedicated to my parents Harun Moral and Basiron.
Thanks for your support and love.

vii
Acknowledgements
At first, I would like to express my sincere gratitude to my supervisor, Dr. Wei Li
for his continuous support and guidance during my PhD study. His trust and confidence in
my learning capacity as a newly minted graduate student in 2015 fostered my growth,
confidence in my organic chemistry skills, and curiosities as a scientist. I am profoundly
grateful to him for the training, encouragement and freedom that I received to explore
halogen mediated chemistry and will be a great asset for my future research endeavors.
I wish to thank my research committee members Dr. Steve Sucheck, Dr. Jianglong
Zhu, Dr. Ana C. Alba-Rubio, and Dr. Michael Young for their insightful comments and
valuable advice to my research as well as the time they have dedicated to me.
Next, I would like to thank my labmates Fan, Jeewani and Navdeep for all the fun
we had, discussions, feedbacks of the project, proofreading of my papers and thesis as well
as their collaboration for finishing my projects. I also wish to thank all the graduate students
that I have known and worked with, and Bangladeshi Communities in Toledo for always
bringing me joys. I would like to thank the Department of Chemistry and Biochemistry,
the University of Toledo for providing me facilities to conduct research.
I am greatly indebted to my parents for their love, support and sacrifice for me. My
family members and friends from home have always been there for me and have always
helped me remember my roots.
Finally, I am thankful to my wife for her relentless care, love and emotional support.

viii
Table of Contents
Abstract .............................................................................................................................. iii
Acknowledgements ........................................................................................................... vii
Table of Contents ............................................................................................................. viii
List of Tables .................................................................................................................... xii
List of Figures .................................................................................................................. xiii
List of Schemes ................................................................................................................ xiv
List of Abbreviations .........................................................................................................xv
Preface............................................................................................................................. xvii
1 Halogen Mediated Alkene Difunctionalization for the Synthesis of N, S- and N,
O-Containing Heterocycles ......................................................................................1
1.1 Introduction of Alkene Difuctionalization ...................................................1
1.1.1 Abundance and Reactivity of Alkene ..............................................1
1.1.2 Difunctionalization of Alkene..........................................................2
1.2 Significance of N, S- and N, O-Containing Heterocycles ...........................4
1.3 Our Proposed Halogen Mediated Difunctionalization of Alkene for the
Synthesis of N-Containing Heterocycles .....................................................6
2 One-Pot Strategy for Thiazoline Synthesis from Alkenes and Thioamides ..........11
2.1 Introduction ................................................................................................11
2.2 Results and Discussions for Thiazoline Synthesis .....................................14

ix
2.2.1 Reaction Design and Optimization ................................................14
2.2.2 Reaction Scope...............................................................................16
2.3 Proposed Reaction Mechanism ..................................................................19
2.4 Synthetic Application.................................................................................20
2.5 Conclusion .................................................................................................20
2.6 Experimental ..............................................................................................21
3 Intramolecular Regio- and Stereoselective Alkene Sulfenoamination With
Thioimidazoles .......................................................................................................52
3.1 Introduction ................................................................................................52
3.2 Results and Discussions for Sulfenoamination ..........................................55
3.2.1 Reaction Design and Optimization ................................................55
3.2.2 Reaction Scope...............................................................................56
3.2.3 Regiodivergent Sulfenoamination .................................................60
3.3 Reaction Mechanism ..................................................................................61
3.3.1 Solvent Effect on Reaction ............................................................61
3.3.2 Temperature-Dependent Diastereoselectivity ................................62
3.2.3 Proposed Mechanism for Intermolecular Sulfenoamination ........63
3.4 Conclusion .................................................................................................64
3.5 Experimental ..............................................................................................64
4 Catalytic Regio- and Stereoselective Alkene Sulfenoamination and Arene
Sulfenylation for 1,4-Benzothiazine Synthesis ......................................................95
4.1 Introduction ................................................................................................95
4.2 Results and Discussions for 1,4-Benzothiazine Synthesis .........................99

x
4.2.1 Reaction Design and Optimization ................................................99
4.2.2 Substrate Scope ............................................................................101
4.2.3 Substrate Scope for Arene Sulfenylation .....................................104
4.3 Reaction Mechanism ................................................................................106
4.4 Conclusion ...............................................................................................109
4.5 Experimental ............................................................................................109
5 Alkene Catalyzed Construction of All-Carbon Quaternary Center Via C-H
Alkylation of Arene .............................................................................................139
5.1 Introduction ..............................................................................................139
5.2 Results and Discussions for C-H Alkylation of Arene ............................144
5.2.1 Reaction Design and Optimization ..............................................144
5.2.2 Substrate Scope ............................................................................144
5.2.3 Chemoselectivity of Alcohols ......................................................148
5.3 Synthetic Exploration of Our Strategy .....................................................149
5.4 Reaction Mechanism ................................................................................150
5.5 Conclusion ...............................................................................................151
5.6 Experimental ............................................................................................151
6 Iodide-Catalyzed Oxyamination of Olefins for Synthesis of Oxazolidinone from
Unfunctionalized Carbamate ...............................................................................182
6.1 Introduction ..............................................................................................182
6.2 Results and Discussions for Alkene Oxyamination .................................186
6.2.1 Reaction Design and Optimization ..............................................186
6.2.2 Substrate Scope ............................................................................186

xi
6.3 Regiodivergent Alkene Oxyamination ....................................................188
6.4 Reaction Mechanism ................................................................................189
6.5 Conclusion ...............................................................................................191
6.6 Experimental ............................................................................................191
References ........................................................................................................................201

xii
List of Tables
2.1 Optimization of Reaction for Thiazoline Synthesis ...............................................15
2.2 Alkene Substrate Scope for Thiazoline Synthesis .................................................17
2.3 Thioamide Substrate Scope for Thiazoline Synthesis ...........................................18
3.1 Optimization of Reaction for Sulfenoamination ....................................................56
3.2 Alkene Substrate Scope for Sulfenoamination ......................................................58
3.3 Thioimidazole Substrate Scope for Sulfenoamination ..........................................59
4.1 Optimization for 1,4-Benzothiazine Synthesis ....................................................100
4.2 Alkene Substrate Scope for 1,4-Benzothiazine Synthesis ...................................102
4.3 Thioamine Substrate Scope for 1,4-Benzothiazine Synthesis .............................103
4.4 Substrate Scope for C-H Sulfenylation of Arene .................................................105
5.1 Optimization of Reaction Condition ...................................................................143
5.2 Arene Substrate Scope ........................................................................................145
5.3 Alcohol Substrate Scope .....................................................................................146
6.1 Optimization of Reaction Condition ...................................................................185
6.2 Substrate Scope for Alkene Oxyamination .........................................................187

xiii
List of Figures
1 – 1 Significance of Heterocycle .....................................................................................5
2 – 1 Biologically Active Compounds of Thiazoline .....................................................12
2 – 2 Validation of Hypothesis .......................................................................................14
3 – 1 Temperature-Dependent Diastereoselective Study of Cis-β-Methylstyrene .........62
3 – 2 Proposed Reaction Mechanism for Alkene Sulfenoamination ..............................63
4 – 1 Importance of 1,4-Benzothiazine Structural Motifs ..............................................96
4 – 2 Unexpected CSP2‒H Sulfenylation .......................................................................104
4 – 3 Synthesis of Thiomorpholine ...............................................................................106
5 – 1 Proposed Reaction Mechanism for Alkylation of Arene .....................................150

xiv
List of Schemes
1 – 1 Examples of Alkene Difunctionalization .................................................................3
1 – 2 Challenges of Alkene and Nucleophile Coupling ....................................................8
1 – 3 Our Proposed Halogen Mediated Coupling of Alkenes and Dinucleophile ............9
2 – 1 Literature Background of Thiazoline Synthesis ....................................................13
2 – 2 Mechanism of Thiazoline Formation .....................................................................19
2 – 3 Synthetic Exploration of Thiazoline ......................................................................20
3 – 1 Background on Sulfenoamination of Alkene .........................................................53
3 – 2 Regiodivergent of Alkene Sulfenoamination .........................................................60
3 – 3 Participation of DMF in the Sulfenoamination Reaction ......................................62
4 – 1 Alkene Sulfenoamination Background for 1,4-Benzothiazine Synthesis ..............97
4 – 2 Mechanistic Experiments for 1,4-Benzothiazine Synthesis.................................107
4 – 3 Proposed Reaction Mechanism ............................................................................108
5 – 1 Literature Background of Alcohol Activation .....................................................140
5 – 2 Chemoselectivity of Different Alcohols ..............................................................148
5 – 3 Synthetic Utilities of Our Method........................................................................149
6 – 1 Literature Background of Alkene Oxyamination ................................................183
6 – 2 Origin of Regiodivergent for Alkene Oxyamination ...........................................189
6 – 3 Proposed Reaction Mechanism for Alkene Oxyamination ..................................190

xv
List of Abbreviation
Ac ...............................acetyl
acac ............................acetylacetone
AcOH .........................acetic acid
ALS ............................amyotrophic lateral sclerosis
aq ................................aqueous
Bn ...............................benzyl
br ................................broad
CH3CN .......................acetonitrile
COD ...........................1,5-cyclooctadiene
cy-Pr ...........................cyclopropyl
δ ..................................chemical shift
d..................................doublet
DCM ..........................dichloromethane
DCE............................1,2-dichloroethane
dd................................doublet of doublet
DDQ ...........................2,3-dichloro-5,6-dicyano-1,4-benzoquinone
DMF ...........................dimethylformamide
DMSO ........................dimethylsulfoxide
dr ................................diastereomeric ratio
equiv ...........................equivalent
EtOAc ........................ethylacetate
FTIR ...........................fourier transform infrared spectroscopy
g..................................gram
h..................................hour
HFIP ...........................hexafluoroisopropanol
HIV ............................human immunodeficiency virus
HMPA ........................hexamethylphosphoramide
HRMS ........................high resolution mass spectrometry
μL ...............................microliter
m ................................multiplet

xvi
M ................................molarity
MHz ...........................megahertz
mmol ..........................millimole
NIS .............................N-iodosuccinimide
NMR ..........................nuclear magnetic resonance
Ph ...............................phenyl
Piv ..............................pivaloyl
ppm ............................parts per million
PTH ............................phenothiazine
rr .................................regioisomeric ratio
rt .................................room temperature
s ..................................singlet
SAR ............................structure-activity relationship
t ..................................triplet
TBS ............................tert-butyldimethylsilyl
TEMPO ......................(2,2,6,6-tetramethylpiperidin-1-yl)oxyl
TFA ............................trifluoroacetic acid
Ts................................tosyl
TsOH ..........................tosylic acid
UHP............................urea hydrogen peroxide

xvii
Preface
This thesis has been adapted from the following published articles co-written by author
1. Alom, N.-E.; Wu, F.; Li, W. “One-Pot Strategy for Thiazoline Synthesis from
Alkenes and Thioamides” Org. Lett. 2017, 19, 930-933.
2. Alom, N.-E.; Rina, Y. A.; Li, W. “Intermolecular Regio- and Stereoselective
Sulfenoamination of Alkenes with Thioimidazoles” Org. Lett. 2017, 19, 6204-
6207.
3. Alom, N.-E.; Kaur, N.; Wu, F.; S. J. Saluga, S. J.; Li, W. “Catalytic Regio- and
Stereoselective Alkene Sulfenoamination for 1,4-Benzothiazine Synthesis” Chem.
Eur. J. 2019, 25, 6902-6906.

1
Chapter 1
Halogen Mediated Alkene Difunctionalization for the
Synthesis of N, S- and N, O-Containing Heterocycles
1.1 Introduction of Alkene Difunctionalization
1.1.1 Abundance and Reactivity of Alkene
Alkenes are widely used chemical feedstocks in organic transformations due to
their reactivities in synthetic processes involving acid, base, mildly oxidative, and
reductive conditions. Alkenes are regarded as attractive synthetic building blocks because
of their versatility, diversity, and abundance from petrochemicals and natural product
extracts.1 Moreover, many alkenes are commercially available, cheap, and easily
accessible. The wide availability of alkenes renders them highly appealing as reagents in
bioactive molecule syntheses in terms of structural diversity and complexity. There is a
plethora of simple and reliable procedures to synthesize alkenes, further augmenting the
synthetic utility of alkenes. Particularly, several efficient methods have been developed to
synthesize alkenes in a stereoselective manner. For example, the Wittig and the Horner-
Wadsworth-Emmons reactions provide either Z- or E-olefins under specific conditions
while the Julia olefination offers E-alkenes.2-3 The transition metal-catalyzed cross-
coupling reactions such as Suzuki, Stille, and Heck are also common methods to access
aryl olefins.4-6 The olefin metathesis, catalyzed by a variety of transition-metal complexes,

2
has been extensively used in industries.7-8 Due to the easy accessibility of alkenes, they
have been used in a broad range of classic transformations. For example, syn-selective
dihydroxylation of alkene has been achieved using OsO4 or KMnO4.
9 Moreover, alcohols
can also be synthesized in a regioselective manner from alkenes by using acid-catalyzed
hydration or hydroboration-oxidation sequence.10-11 The halogenation of alkene is another
fundamentally important reaction in organic synthesis to access dihalocompounds, which
are common synthons for a wide variety of useful compounds. The syn-selective
cyclopropanation of alkenes can be done with the Simmons-Smith reaction.12 The
Markovnikov selective ether formation can be achieved by oxymercuration and subsequent
reduction with sodium borohydride.13 More importantly, olefins play a significant role in
the petrochemical industry. The polymerization of terminal olefins, catalyzed by the
Ziegler-Natta catalyst, is an essential catalytic process in polymer synthesis.14-15
Asymmetric hydrogenation, catalyzed by the chiral ruthenium and rhodium catalysts,
developed by Noyori and Knowles, has been employed in countless pharmaceutical
syntheses.16-18
1.1.2 Difunctionalization of Alkene
The difunctionalization of alkene has received significant attention among the
synthetic communities for the rapid assembly of molecular complexity via simultaneous
construction of multiple carbon-carbon, carbon-heteroatom bonds. In this regard, metal-
catalyzed difunctionalization of alkene is one of the most powerful and widely used
strategies for the modular synthesis of complex structures from simple chemical

3
feedstocks. Significant efforts have been devoted to the direct installation of nitrogen and
oxygen functionalities for the oxyamination of alkenes. In 1996, Sharpless developed
Scheme 1-1: Examples of Alkene Difunctionalization
osmium-catalyzed asymmetric hydroxyamination of alkenes for 1,2-amino alcohol
synthesis (Scheme 1-1a).19-20 The toxicity of the catalyst and poor regioselectivity have

4
encouraged several other groups to develop alternative oxyamination protocols. The Stahl
group reported the palladium-catalyzed aminoacetoxyllation of allylic and homoallylic
ether and ester (Scheme 1-1b).21 The Yoon group discovered a series of elegant strategies
utilizing copper and iron catalyst for regiodivergent oxyamination of terminal alkenes with
oxaziridines (Scheme 1-1c).22-24 Along with the continuous success of oxyamination,
transition metal catalysis reaction has emerged as an efficient tool in many other
difunctionalization reactions. For example, the Brown, Engle, and Giri groups have
independently reported nickel-catalyzed diarylation of olefins along with other C-C bond
formation reactions from alkene (Scheme 1-1d) 25-27, and the Wolfe group reported Pd-
catalyzed intramolecular and asymmetric carboamination reaction for the synthesis of
carbocycle (Scheme 1-1e).28 Despite the advancement of above mentioned metal-catalyzed
alkene difunctionalization reactions, alkene sulfenoamination strategies for the synthesis
of N, S-containing heterocycles remains a formidable challenge due to the thiophilic nature
of the metal catalyst, which in turn can significantly alter their desired reactivity. In this
context, the development of a metal-free alkene sulfenoamination method to swiftly
construct a custom compound library of N, S-containing heterocycles is highly desirable.
1.2 Significance of N, S- and N, O-Containing Heterocycles
Sulfur is a common chemical element utilized in many important biological
processes. For example, gram-negative bacteria often rely on glutathione, whereas gram-
positive bacteria can synthesize mycothiol to neutralize oxidative stress.29 On the other
hand, nitrogen is an essential element included in many therapeutic agents. Besides, N-
containing heterocycles are common structural motifs in many biologically active

5
compounds, pharmaceuticals, and value-added products.30-31 N- and S-containing
heterocycles are also found in many natural products. For example, fully unsaturated
thiazole, is often found in natural products such as leinamycin, barakacin, and epothilone.32
Figure 1-1: Significance of Heterocycles. Reprinted with permission from reference 40
(Copyright 2014, American Chemical Society) and Reprinted by permission from Springer
Nature, Top Curr Chem (Z), Kevin A. Scott, 376:5, Copyright 2018.
Moreover, these heterocycle often exhibit a wide range of bioactivities such as anticancer,
antibiotic, anti-HIV, and neurological activities.33-34 For example, Penicillin, a class of
antibiotics containing N- and S-based thiazolidine ring, , discovered by Alexander
Flemming in 1928, have saved millions of lives from bacterial infections (Figure1-1).35

6
Pramipexole encoded with a thiazole, is a dopamine agonist, that has been used for the
treatment of Parkinson’s disorder.36 Firefly luciferin, a molecule that is responsible for
bioluminescence of fireflies, also consists of multiple S- and N-heterocycles.37 Nizatidine
is used for acid-related disorders.38 In addition, N, S-containing heterocycles can function
as photocatalysts in organic transformation. The photocatalytic activity of phenothiazine
has been shown by the Jui group39 for the reduction of aryl halide by excited photocatalyst.
Those heterocycles can also be used as a ligand in transition metal-catalyzed reactions.40
Moreover, N-containing heterocycle act can as a chiral auxiliary for the asymmetric
synthesis of active pharmaceuticals.41 The importance of those heterocycles (Figure 1-1) is
further highlighted by a recent database analysis of 1086 U.S. FDA approved small-
molecule drugs through 2012, which reveals that approximately 84% of these drugs contain
at least one nitrogen atom and 60% incorporating the nitrogen atom in a heterocycle.
Among them, there are 250 and 379 FDA approved drugs that contain five-membered and
six-membered nitrogen-containing heterocycles respectively.42 Development of new
therapeutic agents is an important solution of the global health concern on fighting against
existing and emerging diseases. Design and development of simple and reliable methods
to facilitate the synthesis of those heterocycles from simple alkenes, can improve and
expedite the discovery of new small-molecule therapeutics.40
1.3 Our Proposed Halogen Mediated Difunctionalization of
Alkene for the Synthesis of N-Containing Heterocycles
The research program proposed herein is directed towards the development of
chemical methods for the synthesis of interesting heterocycles from simple alkene

7
feedstocks and dinucleophiles. Alkenes are considered as nucleophiles whereas
heteroatoms such as nitrogen (N), oxygen (O), and sulfur (S) in any nucleophiles are also
nucleophilic atoms. The polarity mismatch between these reagents often makes these
alkene addition reactions difficult (Scheme 1-2a). Moreover, the control of regioselectivity
associated with alkene is often regarded as a prominent challenge. For example, the
addition of two heteroatoms across a simple terminal alkene can result in two regioisomeric
products (Scheme 1-2b). Chemical methods that can predictably install nitrogen, sulfur and
oxygen atoms across an alkene are of high importance and value in generating bioactive
chemical libraries. To overcome these difficulties, we will focus on enabling the concept
of halogenation for either alkene activation or thiol activation, for the synthesis of highly
functionalized N, and S- containing heterocycles. The halogenation of alkene is a viable
approach because it can reverse the polarity of an alkene to generate a dielectrophile.
However, the exact nature of the dielectrophilic intermediate can be crucial in defining the
scope and variability of both the alkenes and nucleophiles. One possibility of the
dielectrophile is dihaloalkane from halogenation of alkenes, while the second potential
dielectrophile involves the classic halonium ion.
Our group is interested in the utilization of cyclic halonium ion for the
regioselective synthesis of N, S- and N, O- containing heterocycles. The three-membered
cyclic halonium ion was first proposed by Kimball and Olah in 1937, which holds an
interest of organic chemists to account for the trans addition of halogens across alkenes.43-
45 Structural characterization of these intermediate by Nugent,46 Brown,47-49 and Kochi50
set the foundation for wide acceptance and recognition of their importance in organic
synthesis (Scheme 1-2d). Since then, halonium ion has emerged as a fundamental

8
intermediate for many organic transformations. The traditional routes to prepare cyclic
halonium ion or vicinal dihalo compounds are the addition of molecular halogens to
Scheme 1-2: Challenges of Alkene and Nucleophile Coupling.
alkenes (Scheme 1-3a). However, molecular halogens are toxic and corrosive reagents that
causes significant health concerns. To circumvent this problem, several environmentally
benign reagents have been developed. In these contexts, N-bromosuccinimide and N-
bromoacetamide are considered as safer reagents in comparison with Br2, but the cost and
low atom economy of these reagents make it less attractive to chemists.51-52 Oxidative

9
halogenation using inorganic halides has gained significant attention among the synthetic
community to preclude the use of detrimental halogens. A broad range of protocols has
been developed to achieve the bromination of alkene using bromide salts with different
oxidants such as H2O221-23, TBHP53, Oxone54-55, NaIO4, and hypervalent iodine.56-58
Another potential way to accomplish the goal of heterocycle synthesis is the
activation of sulfur-based nucleophiles into electrophilic sulfur reagents by halogenation
of thiol via the formation of a sulfur-halogen bond. Upon nucleophilic attack of an alkene
on sulfur electrophile generates thiiranium ion intermediate which can be in equilibrium
with open carbocation. Then, intramolecular attack from tethered nucleophile on the
thiiranium ion or open carbocation leads to a cyclized product (Scheme 1-3b). In addition,
a sulfur-halogen bond can undergo homolysis to generate thiyl radical, which can combine
with an alkene to form carbon-centered radical. Alternatively, the oxidation of radical into
carbocation followed by cyclization can lead to polar pathway for product formation. On
the other hand, the reversal of the alkene polarity with a simple halogenation process can
directly engage in two consecutive nucleophilic displacements to form the heterocyclic
Scheme 1-3: Our Proposed Halogen Mediated Coupling of Alkene and Dinucleophile.

10
compounds (Scheme 1-3a). A particular issue that needs to be addressed is that 1,2-
dihaloalkanes are often regarded as poor electrophiles that can undergo reversion to alkenes
or elimination in the presence of bases or hard nucleophiles. In this case, one can argue that
the careful selection of nucleophiles and fine-tuning of halogenation can be a suitable
approach to get access to various potential heterocyclic compounds. If successful,
significant diversity in structure and functionality can then be achieved with olefin
difunctionalizations.
With these nucleophile activation strategies, we hope to develop alkene
sulfenoamination and alkene oxyamination protocols for accessing valuable heterocycles
that would (i) provide regioselectivity based on the electronic and steric bias from alkene
activation and (ii) promote opposite regioisomer from thiol activation.
Aiming at achieving the aforementioned goals, the results presented herein focus
on 1) the development of a one-pot strategy for thiazoline synthesis from alkenes and
thioamides (Chapter 2) as a proof of concept on halogen activation of alkene; 2) the
demonstration of intermolecular regio- and stereoselective sulfenoamination of alkenes
with thioimidazoles (Chapter 3) as evidence of halogen activation of thiol for accessing
opposite regioisomer; (3) the discovery of catalytic regio- and stereoselective alkene
sulfenoamination for 1,4-benzothiazine synthesis (Chapter 4) as a demonstration of
catalytic utilization of iodide salt; (4) the illustration of alkene-catalyzed generation of all-
carbon quaternary centers via alkylation of arene (Chapter 5) as evidence of iodonium
activation of alcohol; (5) the development of an iodide-catalyzed alkene oxyamination for
the synthesis of oxazolidinone (Chapter 6) as an example of catalytic iodonium ion as a
regiocontrol template.

11
Chapter 2
One-Pot Strategy for Thiazoline Synthesis from Alkenes
and Thioamides
2.1 Introduction
Nitrogen- and sulfur-containing heterocycles are important structural motifs and
highly attractive synthetic targets due to their interesting biochemical properties.
Particularly, thiazoline, an important N, S-containing heterocycle, is a ubiquitous structural
unit in pharmaceuticals, bioactive compounds, and value-added molecules (Figure 2-1).
Moreover, these types of compounds show a broad range of bioactivities such as
anticancer, antibiotic, anti-HIV, and neurological activities.33-34 For example, pramipexole
encoded with a thiazole is a dopamine agonist that has been used for the treatment of
Parkinson’s disorder,20 and Ritonavir, an antiviral drug is used to treat HIV, also contains
two thiazole units. In addition, thiazoline is present in many other biologically active
natural products such as largazole, curacin A, tantazole B and pyochelin.59-62 On the other
hand, fully unsaturated thiazole structural unit is found in drugs such as abafungin,
sulfathiazole and natural product such as leinamycin, barakacin, and epothilone.32 Firefly
luciferin embedded with thiazoline and benzothiazole, is responsible for the
bioluminescence of fireflies (Figure 2-1).37 Riluzole is another important drug molecule
that is used for the treatment of anxiety disorder63 while Nizatidine is used for acid-related

12
Figure 2-1: Biologically Active Compounds of Thiazoline
disorders.42 Thiazoline rings can also act as ligands and directing groups for transition
metal-catalyzed reactions.40 Due to a broad range of bioactivities of the thiazoline scaffold
in pharmaceuticals, the synthesis of this structural motif has received significant attention
from the synthetic communities. To gain access for this heterocycle, β-aminothiols are the
most useful and convenient synthetic building blocks.64 The condensation of cysteamine
with acids, esters, and amides is the traditional way to synthesize thiazolines (Scheme 2-
1a).65-67 In this context, the coupling of aryl nitrile with cysteine provides thiazoline bearing
carboxylic acid (Scheme 2-1b).68 However, the scarcity of naturally occurring β-aminothiol
often hinders potential structure-activity relationship (SAR) studies with this interesting
bioactive scaffold. To overcome the availability problem of β-aminothiol, methods
involving the condensation of β-amino alcohols with carbonyl precursors via thionation of
amide intermediate have also been developed (Scheme 2-1c).69 Finally, intramolecular
strategies that require multistep syntheses can also be utilized to gain access to
thiazolines.69-71 In this regard, the Pierce group disclosed an intramolecular protocol for the
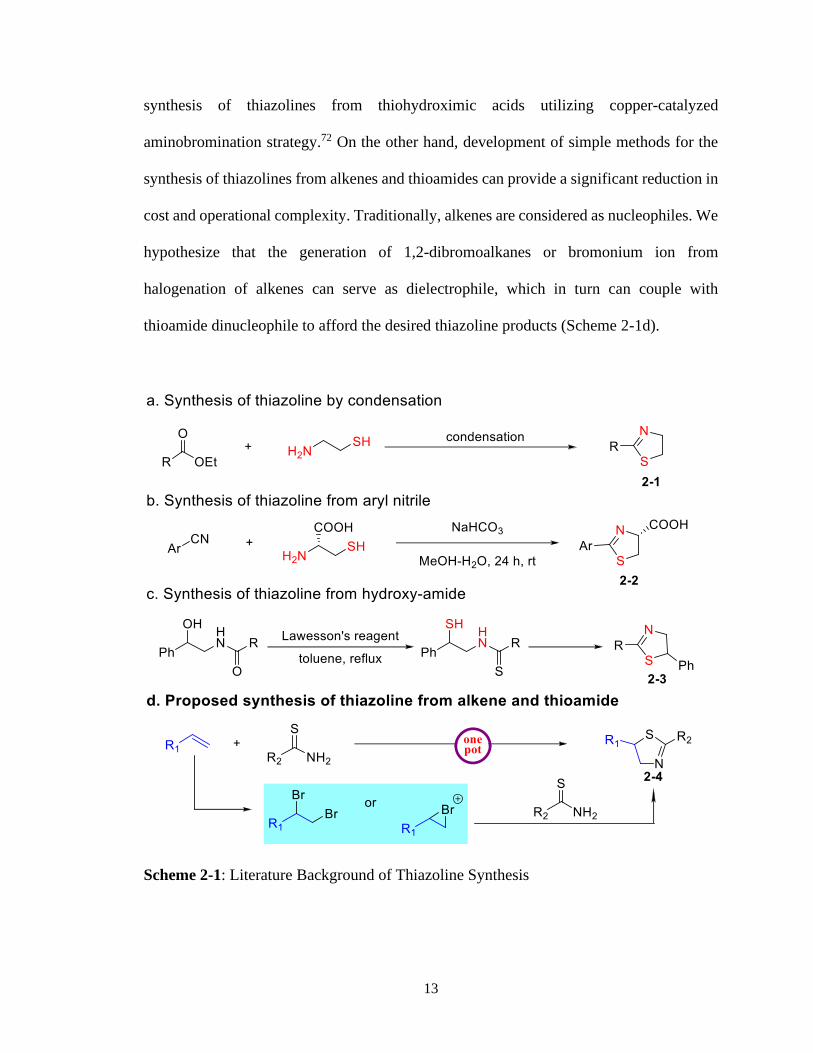
13
synthesis of thiazolines from thiohydroximic acids utilizing copper-catalyzed
aminobromination strategy.72 On the other hand, development of simple methods for the
synthesis of thiazolines from alkenes and thioamides can provide a significant reduction in
cost and operational complexity. Traditionally, alkenes are considered as nucleophiles. We
hypothesize that the generation of 1,2-dibromoalkanes or bromonium ion from
halogenation of alkenes can serve as dielectrophile, which in turn can couple with
thioamide dinucleophile to afford the desired thiazoline products (Scheme 2-1d).
Scheme 2-1: Literature Background of Thiazoline Synthesis

14
2.2 Results and Discussions for Thiazoline Synthesis
2.2.1 Reaction Design and Optimization
To validate our hypothesis, 1,2-dibromoethylbenzene (Figure 2-2, product 2-5) was
synthesized from the bromination of styrene in 94% yield in acetonitrile. The treatment of
1,2-dibromoethylbenzene with thiobenzamide provided thiazoline product in 63% yield in
acetonitrile at 80 ºC. This result explicitly revealed that a common solvent acetonitrile
could be used for both the 1,2-dibromoalkane formation and the subsequent nucleophilic
displacements. This experimental data suggested that we could proceed with a one-pot
strategy directly from a simple alkene.
Figure 2-2: Validation of Hypothesis
To our delight, when a one-pot reaction was carried out by adding the thioamide to the
reaction mixture after 1 h, the desired thiazoline was obtained in 39% yield (Table 2.1,
entry 1). To avoid the use of corrosive molecular bromine, oxidation of bromide salt was
carried out for in-situ generation of bromine. The combination of lithium bromide (LiBr)
and aqueous hydrogen peroxide provided the desired thiazoline in 7% yield (Table 2.1,
entry 2). The addition of trifluoroacetic acid (TFA) was realized to facilitate the oxidation
of bromide salt, increased the yield to 50% (Table 2.1, entry 3). The screening of halides,

15
Table 2.1: Optimization of Reaction for Thiazoline Synthesis
oxidants, solvents, and acids revealed the initial optimal condition to provide 56% of the
thiazoline product (Table 2.1, entries 4-13). Further testing of concentration,
stoichiometrey, and temperature didn’t provide a higher yield. We reasoned that acid

16
produced from SN2 reaction could be detrimental to subsequent nucleophilic displacement
reactions. To validate our hypothesis, LiOAc was added along with the thioamide
nucleophile, which indeed improved the yield to 60% (Table 2.1, entry 14). Further
refinement of the reaction conditions by screening different inorganic bases afforded 69%
yield with 3 equivalents of NaHCO3 (Table 2.1, entries 15-19). Finally, increasing the
oxidant to 1.5 equiv and halide source to 3.0 equiv provided the highest yield of 73% for
this reaction (Table 2.1, entry 20). However, increasing the concentration or decreasing the
temperature led to lower yields (Table 2.1, entries 21 and 22).
2.2.2 Reaction Scope
With the optimized conditions in hand, the alkene substrate scope was tested. A range of
styrene derivatives provided moderate to good yields of the desired thiazolines (Table 2.2).
For example, substituents at the para position of the benzene ring with electron-donating
or -neutral properties worked well, providing acceptable yields (Table 2.2, products 2-8 to
2-10, 2-13, 2-19). On the other hand, electron-withdrawing group at the para position
afforded the product in moderate yield (Table 2.2, product 2-11). Moreover, halogen
substituents at either the para or ortho position delivered good yields of thiazoline products
(Table 2.2, products 2-12, 2-14 to 2-16). Aliphatic alkenes comprised of alkyl, alcohol,
ether, ester, and imides were also compatible with the reaction conditions (Table 2.2,
products 2-17, 2-18, 2-20 to 2-25). In the case of regioselectivity for aliphatic alkenes, the
initial nucleophilic attack from the sulfur atom was reversed compared to styrene
derivatives because the primary C-Br bond was the more electrophilic site due to steric
hinderance whereas secondary benzylic C-Br bond for styrene derivatives is more
electrophilic due to electronic reason.
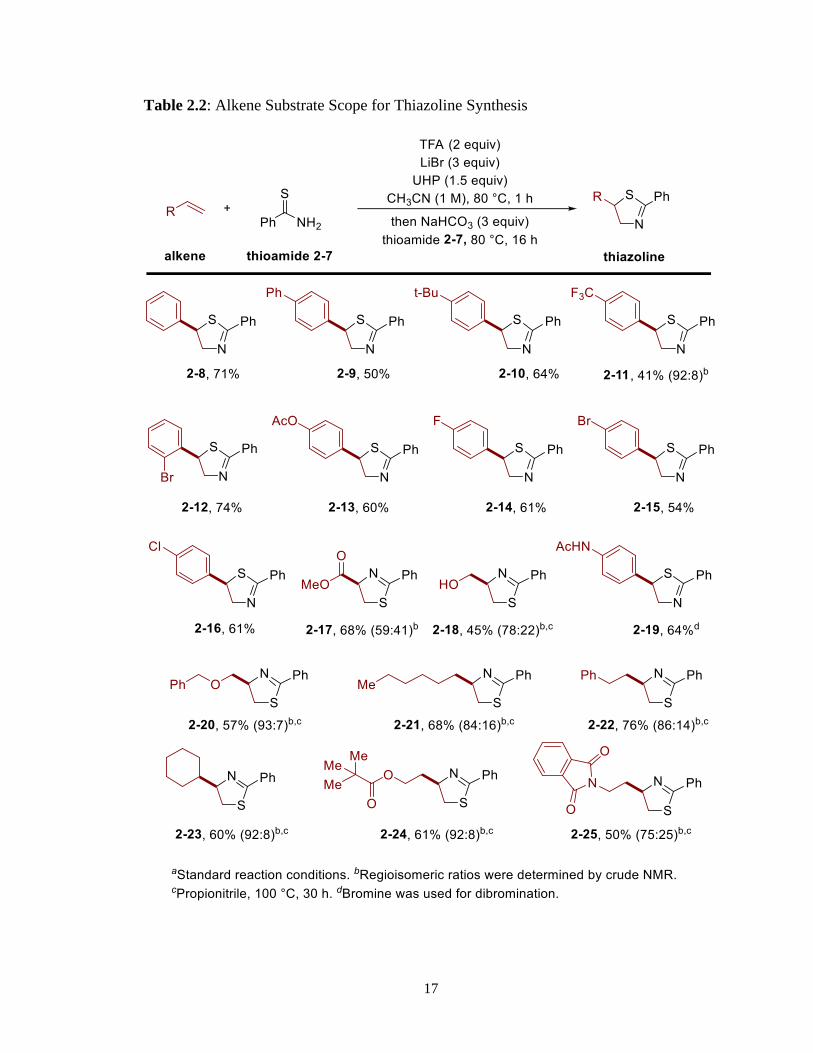
17
Table 2.2: Alkene Substrate Scope for Thiazoline Synthesis

18
Table 2.3: Thioamide Substrate Scope for Thiazoline Synthesis
For the thioamide substrate scope, the methyl group at either the para or ortho position
provided the products in good yields (Table 2.3, products 2-26 and 2-27). For electron-
donating substituents such as alcohol, ether, and amine, the thiazolines were obtained in
moderate

19
yields (Table 2.3, products 2-28, 2-37 to 2-39). The different halogen-substituted
thioamides proceeded to generate the products efficiently (Table 2.3, products 2-29 to 2-
35). Alkylthioamide could also accomplish the corresponding thiazoline product (Table
2.3, products 2-36). Moreover, thioamides containing electron-deficient aromatics such as
pyridines and pyrimidine could also furnish the desired thiazoline products with high
regioselectivities albeit in lower yields (Table 2.3, products 2-40 to 2-43). The thiazoline
from 2-pyridinethioamide could function as potential ligand in metal-catalyzed reactions.
(Table 2.3, product 2-40). The ability of our strategy to incorporate these heteroaromatic
structures further highlights the versatility and utility of the method.
2.3 Proposed Reaction Mechanism
At first, bromine is generated from the oxidation of LiBr by urea hydrogen peroxide
(UHP). The bromination of alkene leads to the formation of 1,2-dibromoalkanene A which
can act as a dielectrophile. Then, the reaction goes through a double nucleophilic attack
from thioamide onto the 1,2-dibromoalkane or bromonium ion intermediate, leading to the
formation of thiazoline.
Scheme 2-2: Mechanism of Thiazoline Formation

20
2.4 Synthetic Application
After demonstrating a broad substrate scope, the synthetic utility of thiazoline was
explored. The gram-scale synthesis of thiazoline 2-8 was achieved in 61% yield using the
standard reaction conditions. Under oxidation of thiazoline 2-8 by DDQ in
dichloromethane afforded thiazole in 95% yield, one of the most attractive structural units
in sulfur-containing FDA approved drugs (Scheme 2-3, product 2-44). On the other hand,
hydrolysis of thiazoline by using 5 M HCl provided a single β-aminothiol in 92% yield
(Scheme 2-3, product 2-45) which could overcome the β-aminothiol availability problem.
Scheme 2-3: Synthetic Exploration of Thiazoline.
2.5 Conclusion
We have developed a simple and reliable method for the synthesis of thiazoline
from the coupling of alkenes with thioamides. The method offers a wide variety of
thiazolines with structural diversities and good functional group compatibility. The
derivatization of the thiazoline to thiazole and β-aminothiol via oxidation and hydrolysis
respectively further demonstrates the synthetic utility of our method. We have also

21
compiled a small library of thiazoline compounds suitable for biochemical evaluations. We
hope that this method will be beneficial for synthetic communities in the synthesis of
potentially bioactive compounds bearing thiazoline structural motifs.
2.6 Experimental
General Information. Commercial reagents and solvents were purchased from Sigma
Aldrich, Oakwood Chemicals, Alfa Aesar, Matrix Scientific, Acros Organic, and were used
as received. The substrates for the products 2-20,73 2-24,74 2-25,75 were synthesized
according to the reported procedure. Organic solutions were concentrated under reduced
pressure on a Büchi rotary evaporator using an acetone-dry ice bath. Chromatographic
purification of products was accomplished using flash chromatography on 230-400 mesh
silica gel. Thin-layer chromatography (TLC) was performed on Analtech 250 mm silica
gel HLF UV-254 plates. Visualization of the developed plates was performed by
fluorescence quenching, potassium permanganate and iodine-silica gel system. 1H and 13C
NMR spectra were recorded on a Bruker 600 instrument (600 and 150 MHz) or INOVA
600 (600 and 150 MHz) and are internally referenced to residual protio solvent signals (for
CDCl3, 7.26 and 77.0 ppm, respectively). Data for 1H NMR are reported as follows:
chemical shift ( ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, h =
heptet, m = multiplet, br = broad), integration, coupling constant (Hz). 13C spectra were
reported as chemical shifts in ppm and multiplicity where appropriate. IR spectra were
recorded on a Perkin Elmer FT-IR spectrophotometer and are reported in terms of
wavenumber of absorption (cm-1). High resolution mass spectra were obtained on Waters
Synapt High Definition Mass Spectrometer (HDMS) by electrospray ionization at

22
University of Toledo, OH, USA.
General Procedure A: Urea hydrogen peroxide (71 mg, 0.75 mmol) was added to a
mixture of LiBr (130 mg, 1.5 mmol), TFA (77 L, 1.0 mmol) and alkene (0.5 mmol) in
acetonitrile (0.5 mL, 1.0 M). Then, the reaction mixture was stirred at 80 °C for 1 h. After
the reaction mixture was cooled to room temperature, NaHCO3 (126 mg, 1.5 mmol) and
thioamide (1.5 mmol) were added sequentially to the reaction mixture and stirred at 80 °C
for 15 h. The reaction mixture was cooled to room temperature, followed by dilution with
water (3 mL), extraction with EtOAc (3 x 4 mL). The combined organic layer was
concentrated in vacuo and purified by flash chromatography on SiO2 (5-50% EtOAc in
hexanes) to provide the desired product. The regioisomeric ratio was determined by crude
NMR.
General Procedure B: Urea hydrogen peroxide (71 mg, 0.75 mmol) was added to a
mixture of LiBr (130 mg, 1.5 mmol), TFA (77 L, 1.0 mmol) and alkene (0.5 mmol) in
propionitrile (0.5 mL, 1.0 M). Then, the reaction mixture was stirred at 100 °C for 1 h.
After the reaction mixture was cooled to room temperature, NaHCO3 (126 mg, 1.5 mmol)
and thioamide (1.5 mmol) were added sequentially to the reaction mixture and stirred at
100 °C for 15 h. The reaction mixture was cooled to room temperature, followed by dilution
with water (3 mL), extraction with EtOAc (3 x 4 mL). The combined organic layer was
concentrated in vacuo and purified by flash chromatography on SiO2 (5-50% EtOAc in
hexanes) to provide the desired product. The regioisomeric ratio was determined by crude
NMR.
Spectral Characterization of the Products

23
2,5-diphenyl-4,5-dihydrothiazole (2-8): This compound was prepared according to the
General Procedure A using styrene (52 mg, 0.5 mmol), thiobenzamide (206 mg, 1.5 mmol).
After purification by column chromatography on SiO2 (5% EtOAc in hexanes, Rf = 0.3),
the title compound was isolated as a light yellow oil (85 mg, 71% yield, >95:5).
1H NMR (600 MHz, CDCl3): = 7.90 (d, J = 7.6 Hz, 2 H), 7.54-7.40 (m, 3 H), 7.40-
7.24 (m, 5 H), 5.09 (dd, J = 5.6, 9.0 Hz, 1 H), 4.80 (dd, J = 9.0, 16.1 Hz, 1 H), 4.64 (dd, J
= 5.6, 16.1 Hz, 1 H);
13C NMR (150MHz, CDCl3): = 168.1, 141.9, 132.8, 131.3, 128.8, 128.5, 128.4, 127.8,
127.0, 73.0, 54.4;
IR (neat): 3059, 3027, 2846, 1648, 1600, 1489, 1447, 1311, 1236, 1007, 944, 763 cm-1;
HRMS (ESI) m/z calcd for C15H14NS [(M+H)+] 240.0847, found 240.0850.
5-(1,1'-biphenyl]-4-yl)-2-phenyl-4,5-dihydrothiazole (2-9): This compound was
prepared according to the General Procedure A using 4-vinylbiphenyl (90 mg, 0.5 mmol),
thiobenzamide (206 mg, 1.5 mmol). After purification by column chromatography on SiO2
(5% EtOAc in hexanes, Rf = 0.2), the title compound was isolated as a light yellow solid
(79 mg, 50% yield, >95:5).

24
1H NMR (600 MHz, CDCl3): = 7.90 (d, J = 7.3 Hz, 2 H), 7.56 (d, J = 8.3 Hz, 2 H),
7.58 (d, J = 7.8 Hz, 2 H), 7.52-7.48 (m, 1 H), 7.48-7.40 (m, 6 H), 7.39-7.32 (m, 1 H),
5.13 (dd, J = 5.5, 8.8 Hz, 1 H), 4.82 (dd, J = 8.8, 16.1 Hz, 1 H), 4.69 (dd, J = 5.5, 16.1
Hz, 1 H);
13C NMR (150MHz, CDCl3): = 167.6, 141.2, 140.7, 140.5, 133.1, 131.3, 128.8, 128.5,
128.4, 127.6, 127.5, 127.4, 127.0, 73.3, 54.2;
IR (neat): 2921, 1610, 1484, 1308, 1006, 823, 761, 601, 558 cm-1;
HRMS (ESI) m/z calcd for C21H18NS [(M+H)+] 316.1160, found 316.1168.
5-(4-(tert-butyl)phenyl)-2-phenyl-4,5-dihydrothiazole (2-10): This compound was
prepared according to the General Procedure A using 4-tert-butylstyrene (86 mg, 93%
purity, 0.5 mmol), thiobenzamide (206 mg, 1.5 mmol). After purification by column
chromatography on SiO2 (5% EtOAc in hexanes, Rf = 0.3), the title compound was isolated
as a light yellow solid (95 mg, 64% yield, >95:5);
1H NMR (600 MHz, CDCl3): = 7.91 (d, J = 7.3 Hz, 2 H), 7.53-7.42 (m, 3 H), 7.40-
7.34 (m, 2 H), 7.34-7.28 (m, 2 H), 5.09 (dd, J = 5.9, 8.8 Hz, 1 H), 4.78 (dd, J = 8.8, 16.1
Hz, 1 H), 4.64 (dd, J = 5.9, 16.1 Hz, 1 H), 1.33 (s, 9 H).
13C NMR (150MHz, CDCl3): = 167.7, 150.7, 138.8, 133.1, 131.1, 128.4, 128.3, 126.7,
125.7, 73.1, 54.3, 34.4, 31.2;
IR (neat): 3057, 2959, 1660, 1602, 1490, 1445, 1310, 1225cm–1;

25
HRMS (ESI) m/z calcd for C19H22NS [(M+H)+] 296.1473, found 296.1475.
2-phenyl-5-(4-(trifluoromethyl)phenyl)-4,5-dihydrothiazole (2-11): This compound
was prepared according to the General Procedure A using 4-(trifluoromethyl)styrene (86
mg, 0.5 mmol), thiobenzamide (206 mg, 1.5 mmol). After purification by column
chromatography on SiO2 (5% EtOAc in hexanes, Rf = 0.2), the title compound was isolated
as a light yellow solid (63 mg, 41% yield, 92:8).
1H NMR (600 MHz, CDCl3): = 7.87 (d, J = 7.6 Hz, 2 H), 7.58 (d, J = 8.1 Hz, 2 H), 7.54-
7.39 (m, 5 H), 5.09 (dd, J = 4.9, 8.8 Hz, 1 H), 4.81 (dd, J = 8.8, 16.2 Hz, 1 H), 4.63 (dd, J
= 4.9, 16.2 Hz, 1 H);
13C NMR (150MHz, CDCl3): = 167.4, 146.3, 132.8, 131.5, 128.6, 128.4, 127.4, 125.9,
125.8, 77.2, 76.8, 73.1, 53.7;
IR (neat): 2983, 1607, 1324, 1158, 1068, 953, 767, 666, 525 cm-1;
HRMS (ESI) m/z calcd for C16H13F3NS [(M+H)+] 308.0721, found 308.0720.
5-(2-bromophenyl)-2-phenyl-4,5-dihydrothiazole (2-12): This compound was prepared
according to the General Procedure A using 2-bromostyrene (92 mg, 0.5 mmol),
thiobenzamide (206 mg, 1.5 mmol). After purification by column chromatography on SiO2

26
(5% EtOAc in hexanes, Rf = 0.3), the title compound was isolated as a colorless liquid (118
mg, 74% yield, >95:5).
1H NMR (600 MHz, CDCl3): = 7.86 (d, J = 7.3 Hz, 2 H), 7.56 (d, J = 8.1 Hz, 1 H), 7.51-
7.44 (m, 2 H), 7.44-7.38 (m, 2 H), 7.29-7.23 (m, 1 H), 7.14-7.07 (m, 1 H), 5.46 (dd, J =
3.8, 8.7 Hz, 1 H), 4.74 (dd, J = 8.7, 16.2 Hz, 1 H), 4.66 (dd, J = 3.8, 16.2 Hz, 1 H);
13C NMR (150 MHz, CDCl3): = 167.5, 141.3, 133.0, 132.9, 132.9, 132.7, 131.5,
131.2, 129.3, 128.9, 128.7, 128.4, 128.2, 128.0, 127.9, 123.2, 71.4, 71.3, 71.2, 53.1, 53.0;
IR (neat): 2897, 1595,1575, 1483, 1445, 1236, 1005, 943, 771, 676, 503 cm-1;
HRMS (ESI) m/z calcd for C15H13BrNS [(M+H)+] 317.9952, found 317.9963.
4-(2-phenyl-4,5-dihydrothiazol-5-yl)phenyl acetate (2-13): This compound was
prepared according to the General Procedure A using 4-acetoxystyrene (81 mg, 0.5 mmol),
thiobenzamide (206 mg, 1.5 mmol). After purification by column chromatography on SiO2
(10% to 20% EtOAc in hexanes, Rf = 0.3), the title compound was isolated as a light yellow
solid (89 mg, 60% yield, >95:5).
1H NMR (600 MHz, CDCl3): = 7.87 (d, J = 7.6 Hz, 2 H), 7.51-7.46 (m, 1 H), 7.46-
7.41 (m, 2 H), 7.36 (d, J = 8.4 Hz, 2 H), 7.04 (d, J = 8.4 Hz, 2 H), 5.06 (dd, J = 5.4, 8.8
Hz, 1 H), 4.74 (dd, J = 8.8, 16.1 Hz, 1 H), 4.61 (dd, J = 5.4, 16.1 Hz, 1 H), 2.29 (s, 3 H);
13C NMR (150MHz, CDCl3): = 169.4, 167.5, 149.9, 139.6, 132.9, 131.3, 128.5, 128.3,
128.1, 121.9, 73.2, 53.8, 21.0;

27
IR (neat): 3024, 2929, 1753, 1604, 1504, 1490, 1369, 1308 cm–1;
HRMS (ESI) m/z calcd for C17H16NO2S [(M+H)+] 298.0902, found 298.0900.
5-(4-fluorophenyl)-2-phenyl-4,5-dihydrothiazole (2-14): This compound was prepared
according to the General Procedure A using 4-fluorostyrene (61 mg, 0.5 mmol),
thiobenzamide (206 mg, 1.5 mmol). After purification by column chromatography on SiO2
(5% EtOAc in hexanes, Rf = 0.3), the title compound was isolated as a gummy liquid (78
mg, 61% yield, >95:5).
1H NMR (600 MHz, CDCl3): = 7.87 (d, J = 7.3 Hz, 2 H), 7.48 (d, J = 7.3 Hz, 1 H), 7.46-
7.40 (m, 2 H), 7.35-7.28 (m, 2 H), 7.04-6.96 (m, 2 H), 5.05 (dd, J = 5.2, 8.9 Hz, 1 H), 4.77
(dd, J = 8.9, 16.1 Hz, 1 H), 4.59 (dd, J = 5.2, 16.1 Hz, 1 H);
13C NMR (151MHz, CDCl3): = 167.5, 162.9, 161.3, 138.0, 138.0, 132.9, 131.3, 128.7,
128.6, 128.5, 128.4, 115.7, 115.6, 73.3, 53.7;
IR (neat): 3060, 2846, 1601, 1577, 1505, 1311, 1223, 1006, 763, 604, 525 cm-1;
HRMS (ESI) m/z calcd for C15H13FN S[(M+H)+] 258.0753, found 258.0755.
5-(4-bromophenyl)-2-phenyl-4,5-dihydrothiazole (2-15): This compound was prepared

28
according to the General Procedure A using 4-bromostyrene (92 mg, 0.5 mmol),
thiobenzamide (206 mg, 1.5 mmol). After purification by column chromatography on SiO2
(5% EtOAc in hexanes, Rf = 0.3), the title compound was isolated as a yellow solid (86 mg,
54% yield, >95:5).
1H NMR (600 MHz, CDCl3): = 7.87 (d, J = 7.8 Hz, 2 H), 7.52-7.46 (m, 1 H), 7.46-7.40
(m, 4 H), 7.22 (d, J = 8.3 Hz, 2 H), 5.01 (dd, J = 5.1, 9.0 Hz, 1 H), 4.77 (dd, J = 9.0, 16.1
Hz, 1 H), 4.59 (dd, J = 5.1, 16.1 Hz, 1 H);
13C NMR (150MHz, CDCl3): = 167.5, 141.3, 132.8, 131.9, 131.4, 128.7, 128.6, 128.4,
121.6, 73.1, 53.7;
IR (neat): 2897, 1595, 1575, 1483, 1445, 1236, 1005, 943, 771, 676, 503 cm-1;
HRMS (ESI) m/z calcd for C15H13BrNS [(M+H)+] 317.9952, found 317.9962.
5-(4-chlorophenyl)-2-phenyl-4,5-dihydrothiazole (2-16): This compound was prepared
according to the General Procedure A using 4-chlorostyrene (69 mg, 0.5 mmol),
thiobenzamide (206 mg, 1.5 mmol). After purification by column chromatography on SiO2
(5% EtOAc in hexanes, Rf = 0.3), the title compound was isolated as a yellow solid (84 mg,
61% yield, >95:5).
1H NMR (600 MHz, CDCl3): = 7.90-7.83 (m, 2 H), 7.52-7.46 (m, 1 H), 7.46-7.40 (m, 2
H), 7.31-7.24 (m, 4 H), 5.02 (dd, J = 5.2, 8.9 Hz, 1 H), 4.77 (dd, J = 8.9, 16.1 Hz, 1 H),
4.59 (dd, J = 5.2, 16.1 Hz, 1 H);
13C NMR (150MHz, CDCl3): = 167.5, 140.7, 133.5, 132.8, 131.4, 128.9, 128.6, 128.4,

29
128.4, 77.2, 76.8, 73.2, 53.7;
IR (neat): 2897, 1595, 1574, 1486, 1398, 1235, 1086, 944, 771, 603, 577, 506 cm-1;
HRMS (ESI) m/z calcd for C15H13ClNS [(M+H)+] 274.0457, found 274.0467.
Methyl-2-phenyl-4,5-dihydrothiazole-4-carboxylate (2-17): This compound was
prepared according to the General Procedure A using methyl acrylate (43 mg, 0.5 mmol),
thiobenzamide (206 mg, 1.5 mmol). After purification by column chromatography on SiO2
(20% EtOAc in hexanes, Rf = 0.4), the inseparable mixture of both regioisomers were
isolated as a colorless liquid (75 mg, 68% yield, 59:41).
1H NMR (600 MHz, CDCl3): = 7.87 (d, J = 7.8 Hz, 2 H), 7.80 (d, J = 7.8 Hz, 1 H), 7.48
(q, J = 7.6 Hz, 2 H), 7.44-7.37 (m, 3 H), 5.29 (t, J = 9.2 Hz, 1 H), 4.94 (d, J = 11.5 Hz, 1
H), 4.61-4.53 (m, 1 H), 3.84 (s, 3 H), 3.77 (s, 2 H), 3.72 (dd, J = 9.2, 10.9 Hz, 1 H), 3.64
(t, J = 10.3 Hz, 1 H);
13C NMR (150MHz, CDCl3): = 171.7, 171.2, 170.9, 165.7, 132.5, 132.3, 131.6, 131.3,
128.5, 128.5, 128.4, 128.3, 78.4, 67.6, 52.9, 52.7, 50.1, 35.3;
IR (neat): 2951, 1733, 1596, 1434, 1315, 1197, 991, 764, 603 cm-1;
HRMS (ESI) m/z calcd for C11H12NO2S [(M+H)+] 222.0589, found 222.0583.
2-phenyl-4,5-dihydrothiazol-4-yl)methanol (2-18): This compound was prepared

30
according to the General Procedure B using allyl alcohol (29 mg, 0.5 mmol),
thiobenzamide (206 mg, 1.5 mmol). After purification by column chromatography on SiO2
(50% EtOAc in hexanes, Rf = 0.4), the inseparable mixture of both regioisomers were
isolated as a white solid (43 mg, 45% yield, 78:22).
1H NMR (600 MHz, CDCl3): = 7.84-7.75 (m, 3 H), 7.49-7.43 (m, 1 H), 7.43-7.37 (m, 3
H), 4.83-4.74 (m, 1 H), 4.10-3.98 (m, 1 H), 3.78 (dd, J = 5.6, 11.2 Hz, 1 H), 3.46-3.39 (m,
1 H), 3.34-3.26 (m, 1 H), 2.75 (br. s., 1 H);
13C NMR (150MHz, CDCl3): = 169.7, 132.8, 131.4, 131.3, 128.5, 128.4, 128.3, 128.3,
79.3, 77.2, 76.8, 66.9, 64.9, 64.4, 52.4, 34.3;
IR (neat): 3239, 3064, 2924, 1603, 1576, 1440, 1324, 1228, 1069, 947, 766, 687 cm-1;
HRMS (ESI) m/z calcd for C10H12NOS [(M+H)+] 194.0640, found 194.0638.
N-(4-(2-phenyl-4,5-dihydrothiazol-5-yl)phenyl)acetamide (2-19): This compound was
prepared according to the General Procedure A using 2-vinylacetanilide (81 mg, 0.5
mmol), thiobenzamide (206 mg, 1.5 mmol) and Br2 (79 mg, 0.5 mmol) instead of urea
hydrogen peroxide, LiBr, and TFA. After purification by column chromatography on SiO2
(50% EtOAc in hexanes, Rf = 0.2), the title compound was isolated as a yellow solid (95
mg, 64% yield, >95:5).
1H NMR (600 MHz, CDCl3): = 7.92-7.83 (m, 3 H), 7.50-7.38 (m, 5 H), 7.25 (d, J = 8.5
Hz, 1 H), 5.02 (dd, J = 5.4, 8.8 Hz, 1 H), 4.73 (dd, J = 8.8, 16.1 Hz, 1 H), 4.56 (dd, J = 5.4,
16.1 Hz, 1 H), 2.13 (s, 3 H);

31
13C NMR (150MHz, CDCl3): = 168.6, 167.9, 137.8, 137.5, 133.0, 131.3, 128.5, 128.3,
127.6, 120.2, 73.1, 54.0, 24.4;
IR (neat): 3322, 2926, 1660, 1596, 1365, 1010, 763, 647, 524 cm-1;
HRMS (ESI) m/z calcd for C17H17N2OS [(M+H)+] 297.1062, found 297.1068.
4-((benzyloxy)methyl)-2-phenyl-4,5-dihydrothiazole (2-20): This compound was
prepared according to the General Procedure B using ((allyloxy)methyl)benzene (74 mg,
0.5 mmol), thiobenzamide (206 mg, 1.5 mmol). After purification by column
chromatography on SiO2 (10% EtOAc in hexanes, Rf = 0.4), the title compound (major
regioisomer) was isolated as a yellow liquid (75 mg, 53% yield).
1H NMR (600 MHz, CDCl3): = 7.84 (d, J = 7.8 Hz, 2 H), 7.49-7.44 (m, 1 H), 7.44-7.34
(m, 6 H), 7.34-7.28 (m, 1 H), 4.92 (dd, J = 4.4, 7.8 Hz, 1 H), 4.63 (s, 2 H), 3.87 (dd, J =
4.4, 9.0 Hz, 1 H), 3.68-3.58 (m, 1 H), 3.55-3.47 (m, 1 H), 3.44-3.36 (m, 1 H);
13C NMR (150MHz, CDCl3): = 168.8, 138.0, 133.1, 131.2, 128.4, 128.3, 127.7, 127.7,
77.2, 73.3, 70.8, 35.8;
IR (neat): 3028, 2855, 1601, 1576, 1447, 1360,1251, 1093, 940, 687, 607 cm-1;
HRMS (ESI) m/z calcd for C17H18NOS [(M+H)+] 284.1109, found 284.1108.
4-hexyl-2-phenyl-4,5-dihydrothiazole (2-21): This compound was prepared according to

32
the General Procedure B using 1-octene (56 mg, 0.5 mmol), thiobenzamide (206 mg, 1.5
mmol). After purification by column chromatography on SiO2 (2% to 5% EtOAc in
hexanes, Rf = 0.4), the title compound (major regioisomer) was isolated as a colorless liquid
(71 mg, 57% yield).
1H NMR (600 MHz, CDCl3): = 7.83 (d, J = 7.6 Hz, 2 H), 7.48-7.36 (m, 3 H), 4.65-
4.60 (m, 1 H), 3.48 (dd, J = 8.4, 10.5 Hz, 1 H), 3.08 (dd, J = 8.4, 10.5 Hz, 1 H), 1.96-1.84
(m, 1 H), 1.71-1.62 (m, 1 H), 1.61-1.42 (m, 2 H), 1.42-1.22 (m, 6 H), 0.89 (t, J = 6.6 Hz,
3 H);
13C NMR (150MHz, CDCl3): = 166.2, 133.4, 131.0, 128.4, 128.3, 77.9, 38.1, 35.1,
31.8, 29.3, 26.7, 22.6, 14.1;
IR (neat): 2924, 2854, 1598, 1490, 1447, 1252, 764 cm–1;
HRMS (ESI) m/zcalcd for C15H22NS [(M+H)+] 248.1473, found 248.1475.
5-hexyl-2-phenyl-4,5-dihydrothiazole (2-21’): This compound was prepared according
to the General Procedure B using 1-octene (56 mg, 0.5 mmol), thiobenzamide (206 mg, 1.5
mmol). After purification by column chromatography on SiO2 (2% to 5% EtOAc in
hexanes, Rf = 0.2), the title compound (minor regioisomer) was isolated as a colorless liquid
(13 mg, 11% yield).
1H NMR (600 MHz, CDCl3): = 7.83 (d, J = 7.3 Hz, 2 H), 7.48-7.37 (m, 3 H), 4.39 (dd,
J = 8.2, 15.7 Hz, 1 H), 4.24 (dd, J = 4.8, 15.7 Hz, 1 H), 3.98-3.89 (m, 1 H), 1.71-1.63 (m,

33
2 H), 1.44-1.35 (m, 2 H), 1.35-1.19 (m, 6 H), 0.88 (t, J = 6.7 Hz, 3 H).
13C NMR (150MHz, CDCl3): = 168.1, 133.7, 131.2, 128.7, 128.4, 70.6, 52.1, 36.7, 31.9,
29.2, 28.3, 22.8, 14.3;
IR (neat): 2924, 2853, 1598, 1446, 1252, 688 cm–1;
HRMS (ESI) m/zcalcd for C15H22NS [(M+H)+] 248.1473, found 248.1477.
4-phenethyl-2-phenyl-4,5-dihydrothiazole (2-22): This compound was prepared
according to the General Procedure B using 4-phenyl-1-butene (66 mg, 0.5 mmol),
thiobenzamide (206 mg, 1.5 mmol). After purification by column chromatography on SiO2
(5% EtOAc in hexanes, Rf = 0.4), the title compound (major regioisomer) was isolated as
a yellow liquid (87 mg, 65% yield).
1H NMR (600 MHz, CDCl3): = 7.85 (d, J = 7.6 Hz, 2 H), 7.49-7.38 (m, 3 H), 7.35-7.24
(m, 4 H), 7.24-7.17 (m, 1 H), 4.65 (t, J = 7.4 Hz, 1 H), 3.51 (t, J = 9.5 Hz, 1 H), 3.17-3.07
(m, 1 H), 2.97-2.82 (m, 2 H), 2.29-2.16 (m, 1 H), 2.06-1.94 (m, 1 H);
13C NMR (150MHz, CDCl3): = 166.5, 141.7, 133.4, 131.1, 128.5, 128.4, 128.4, 128.4,
125.9, 77.1, 38.1, 36.8, 33.1;
IR (neat): 3025, 2921, 1602, 1576, 1490, 1447, 1312, 938, 764 cm-1;
HRMS (ESI) m/z calcd for C17H18NS [(M+H)+] 268.1160, found 268.1159.
5-phenethyl-2-phenyl-4,5-dihydrothiazole (2-22’): This compound was prepared

34
according to the General Procedure B using 4-phenyl-1-butene (66 mg, 0.5 mmol),
thiobenzamide (206 mg, 1.5 mmol). After purification by column chromatography on SiO2
(5% EtOAc in hexanes, Rf = 0.2), the title compound (minor regioisomer) was isolated as
a yellow liquid (15 mg, 11% yield).
1H NMR (600 MHz, CDCl3): = 7.85 (d, J = 7.3 Hz, 2 H), 7.49-7.39 (m, 3 H), 7.32-7.27
(m, 2 H), 7.23-7.16 (m, 3 H), 4.39 (dd, J = 8.3, 15.9 Hz, 1 H), 4.30 (dd, J = 4.4, 15.9 Hz, 1
H), 3.95-3.88 (m, 1 H), 2.82-2.66 (m, 2 H), 2.03-1.93 (m, 2 H);
13C NMR (150MHz, CDCl3): = 167.9, 141.1, 133.6, 131.3, 128.7, 128.7, 128.5, 126.3,
70.5, 51.1, 38.4, 34.3 167.6, 140.9, 133.3, 131.1, 128.5, 128.4, 128.2, 126.1, 70.3, 50.8,
38.1, 34.0;
IR (neat): 3026, 2923, 1603, 1578, 1491, 1447, 1313, 944, 765, 690 cm-1;
HRMS (ESI) m/z calcd for C17H18NS [(M+H)+] 268.1160, found 268.1157.
4-cyclohexyl-2-phenyl-4,5-dihydrothiazole (2-23): This compound was prepared
according to the General Procedure B using Vinylcyclohexane (55 mg, 0.5 mmol),
thiobenzamide (206 mg, 1.5 mmol). After purification by column chromatography on SiO2
(5% EtOAc in hexanes, Rf = 0.4), the title compound (major regioisomer) was isolated as
a white solid (67 mg, 55% yield).
1H NMR (600 MHz, CDCl3): = 7.84 (d, J = 7.1 Hz, 2 H), 7.48-7.35 (m, 3 H), 4.48-4.37
(m, 1 H), 3.40 (dd, J = 8.7, 10.6 Hz, 1 H), 3.17 (t, J = 10.6 Hz, 1 H), 2.07 (d, J = 12.7 Hz,
1 H), 1.84-1.64 (m, 5 H), 1.34-1.14 (m, 5H);

35
13C NMR (150MHz, CDCl3): = 165.9, 133.5, 130.9, 128.3, 128.3, 83.2, 43.1, 35.7,
30.5, 29.5, 26.5, 26.2;
IR (neat): 2921, 2850, 1602, 1575, 1490, 1446, 1308, 1253, 933, 763, 682 cm-1;
HRMS (ESI) m/z calcd for C15H20NS [(M+H)+] 246.1316, found 246.1315.
5-cyclohexyl-2-phenyl-4,5-dihydrothiazole (2-23’): This compound was prepared
according to the General Procedure B using Vinylcyclohexane (55 mg, 0.5 mmol),
thiobenzamide (206 mg, 1.5 mmol). After purification by column chromatography on SiO2
(5% EtOAc in hexanes, Rf = 0.4), the title compound (minor regioisomer) was isolated as
a white solid (6 mg, 5% yield.
1H NMR (600 MHz, CDCl3): = 7.86-7.80 (m, 2 H), 7.48-7.37 (m, 3 H), 4.38 (dd, J =
8.5, 15.9 Hz, 1 H), 4.30 (dd, J = 5.6, 15.9 Hz, 1 H), 3.88-3.82 (m, 1 H), 1.83-1.69 (m, 5 H),
1.66 (d, J = 12.7 Hz, 1 H), 1.54-1.45 (m, 1 H), 1.29-1.18 (m, 3 H), 1.05-0.96 (m, 1 H);
13C NMR (150MHz, CDCl3): = 168.1, 133.4, 131.0, 128.4, 128.2, 67.9, 58.1, 43.1,
30.8, 30.6, 26.2, 26.0;
IR (neat): 2922, 2850, 1605, 1578, 1447, 1310, 1013, 765, 607 cm-1;
HRMS (ESI) m/z calcd for C15H20NS [(M+H)+] 246.1316, found 246.1317.
2-(2-phenyl-4,5-dihydrothiazol-4-yl)ethyl pivalate (2-24): This compound was prepared

36
according to the General Procedure B using but-3-en-1-yl pivalate (78 mg, 0.5 mmol),
thiobenzamide (206 mg, 1.5 mmol). After purification by column chromatography on SiO2
(5% to 10% EtOAc in hexanes, Rf = 0.3), the title compound (major regioisomer) was
isolated as a light yellow oil (87 mg, 59% yield).
1H NMR (600 MHz, CDCl3): = 7.82 (d, J = 7.3 Hz, 2 H), 7.44 (t, J = 7.3 Hz, 1 H), 7.39
(t, J = 7.3 Hz, 2 H), 4.72 (t, J = 7.6 Hz, 1 H), 4.34-4.28 (m, 2 H), 3.52 (dd, J = 8.3, 10.7
Hz, 1 H), 3.13 (dd, J = 8.1, 10.7 Hz, 1 H), 2.23-2.17 (m, 1 H), 2.03-1.98 (m, 1 H), 1.21 (s,
9 H);
13C NMR (150MHz, CDCl3): = 178.5, 167.1, 133.1, 131.1, 128.4, 128.3, 74.5, 62.1,
38.7, 38.1, 34.0, 27.2;
IR (neat): 3061, 2969, 1723, 1603, 1479, 1447, 1147, 765 cm–1;
HRMS (ESI) m/z calcd for C16H22NO2S [(M+H)+] 292.1371, found 292.1375.
2-(2-phenyl-4,5-dihydrothiazol-5-yl)ethyl pivalate (2-24’): This compound was
prepared according to the General Procedure B using but-3-en-1-yl pivalate (78 mg, 0.5
mmol), thiobenzamide (206 mg, 1.5 mmol). After purification by column chromatography
on SiO2 (10% to 20% EtOAc in hexanes, Rf = 0.2), the title compound (minor regioisomer)
was isolated as a light yellow oil (3 mg, 2% yield).
1H NMR (600 MHz, CDCl3): = 7.83 (d, J = 7.3 Hz, 2 H), 7.50-7.44 (m, 1 H), 7.44-7.37
(m, 2 H), 4.43 (dd, J = 8.1,15.6 Hz, 1 H), 4.33 (dd, J = 4.3, 15.7 Hz, 1 H), 4.23-4.12 (m, 2
H), 4.05 - 3.97 (m, 1 H), 2.11-2.02 (m, 1 H), 2.00-1.91 (m, 1 H), 1.22 (s, 9 H);

37
13C NMR (125 MHz, CDCl3): = 178.7, 167.9, 133.4, 131.5, 128.7, 128.5, 70.6, 62.6,
48.4, 39.0, 35.5, 27.4;
IR (neat): 2970, 1727, 1479, 1284, 1156, 691 cm–1;
HRMS (ESI) m/z calcd for C16H22NO2S [(M+H)+] 292.1371, found 292.1379.
2-(2-(2-phenyl-4,5-dihydrothiazol-4-yl)ethyl)isoindoline-1,3-dione (2-25): This
compound was prepared according to the General Procedure B using N-(3-buten-1-yl)
phthalimide (101 mg, 0.5 mmol), thiobenzamide (206 mg, 1.5 mmol). After purification
by column chromatography on SiO2 (30% EtOAc in hexanes, Rf = 0.4), the title compound
(major regioisomer) was isolated as a yellow liquid (84 mg, 50% yield, 75:25). The minor
regioisomer was not isolable.
1H NMR (600 MHz, CDCl3): = 7.89-7.81 (m, 2 H), 7.76-7.69 (m, 2 H), 7.65 (d, J = 8.3
Hz, 2 H), 7.43-7.37 (m, 1 H), 7.33-7.27 (m, 2 H), 4.71-4.61 (m, 1 H), 4.08-3.99 (m, 1 H),
3.98-3.89 (m, 1 H), 3.58 (dd, J = 8.4, 10.9 Hz, 1 H), 3.13 (dd, J = 8.4, 10.9 Hz, 1 H), 2.31-
2.21 (m, 1 H), 2.06 (m, 1 H);
13C NMR (150MHz, CDCl3): = 168.4, 167.0, 133.9, 133.1, 132.2, 131.0, 128.3, 123.2,
75.6, 38.2, 35.9, 33.7;
IR (neat): 3059, 2935, 1769, 1704, 1597, 1576, 1490, 1395, 1368, 1026, 766, 718 cm-1;
HRMS (ESI) m/z calcd for C19H17N2O2S [(M+H)+] 337.1011, found 337.1012.

38
5-phenyl-2-(p-tolyl)-4,5-dihydrothiazole (2-26): This compound was prepared according
to the General Procedure A using styrene (52 mg, 0.5 mmol), 4-methylthiobenzamide (227
mg, 1.5 mmol). After purification by column chromatography on SiO2 (5% EtOAc in
hexanes, Rf = 0.3), the title compound was isolated as a light yellow solid (67 mg, 53%
yield, >95:5).
1H NMR (600 MHz, CDCl3): = 7.78 (d, J = 8.1 Hz, 2 H), 7.40-7.30 (m, 4 H), 7.30-7.18
(m, 3 H), 5.06 (dd, J = 5.4, 8.8 Hz, 1 H), 4.78 (dd, J = 8.8, 16.1 Hz, 1 H), 4.61 (dd, J = 5.4,
16.1 Hz, 1 H), 2.41 (s, 3 H);
13C NMR (150MHz, CDCl3): = 167.5, 142.1, 141.6, 130.4, 129.2, 128.8, 128.3, 127.7,
127.0, 77.2, 76.8, 73.2, 54.4, 21.5;
IR (neat): 2920, 1601, 1452, 1180, 1077, 959, 823, 759, 704, 602 cm-1;
HRMS (ESI) m/z calcd for C16H16NS [(M+H)+] 254.1003, found 254.0995.
5-phenyl-2-(o-tolyl)-4,5-dihydrothiazole (2-27): This compound was prepared according
to the General Procedure A using styrene (52 mg, 0.5 mmol), 2-methylthiobenzamide (227
mg, 1.5 mmol.). After purification by column chromatography on SiO2 (5% EtOAc in
hexanes, Rf = 0.2), the title compound was isolated as a colorless liquid (83 mg, 65% yield,
>95:5).

39
1H NMR (600 MHz, CDCl3): = 7.61 (d, J = 7.6 Hz, 1 H), 7.42-7.22 (m, 8 H), 5.07 (dd,
J = 5.4, 9.0 Hz, 1 H), 4.83 (dd, J = 9.0, 16.0 Hz, 1 H), 4.70 (dd, J = 5.4, 16.0 Hz, 1 H), 2.59
(s, 3 H).
13C NMR (150MHz, CDCl3): = 167.6, 142.3, 137.0, 132.8, 131.1, 129.9, 129.6, 128.8,
127.6, 126.9, 125.6, 73.8, 54.6, 21.0;
IR (neat): 3025, 1610, 1598, 1489, 1453, 1224, 933, 757 cm–1;
HRMS (ESI) m/zcalcd for C16H16NS [(M+H)+] 254.1003, found 254.1013.
4-(5-phenyl-4,5-dihydrothiazol-2-yl)phenol (2-28): This compound was prepared
according to the General Procedure A using styrene (52 mg, 0.5 mmol), 4-
hydroxythiobenzamide (230 mg, 1.5 mmol). After purification by column chromatography
on SiO2 (30% EtOAc in hexanes, Rf = 0.2), the title compound was isolated as a light yellow
solid (78 mg, 61% yield, >95:5).
1H NMR (600 MHz, CDCl3): = 7.71 (d, J = 8.3 Hz, 2 H), 7.37-7.30 (m, 4 H), 7.30-7.24
(m, 1 H), 6.78 (d, J = 8.3 Hz, 2 H), 5.06 (dd, J = 5.9, 8.8 Hz, 1 H), 4.74 (dd, J = 8.8, 15.9
Hz, 1 H), 4.57 (dd, J = 5.9, 15.9 Hz, 1 H);
13C NMR (150MHz, CDCl3): = 159.1, 141.8, 130.4, 128.9, 127.8, 127.0, 125.1, 115.5,
72.5, 54.3
IR (neat): 2921, 2849, 1598, 1507, 1289, 1243, 1018, 834, 697, 608 cm-1;
HRMS (ESI) m/z calcd for C15H14NOS [(M+H)+] 256.0796, found 256.0795.

40
5-phenyl-2-(4-(trifluoromethyl)phenyl)-4,5-dihydrothiazole (2-29): This compound
was prepared according to the General Procedure A using styrene (52 mg, 0.5 mmol), 4-
(trifrluoromethyl)thiobenzamide (308 mg, 1.5 mmol). After purification by column
chromatography on SiO2 (5% EtOAc in hexanes, Rf = 0.2), the title compound was isolated
as a white solid (93 mg, 60% yield, 94:6).
1H NMR (600 MHz, CDCl3): = 7.99 (d, J = 8.1 Hz, 2 H), 7.70 (d, J = 8.1 Hz, 2 H), 7.39-
7.27 (m, 5 H), 5.13 (dd, J = 5.6, 8.9 Hz, 1 H), 4.82 (dd, J = 8.9, 16.5 Hz, 1 H), 4.66 (dd, J
= 5.6, 16.5 Hz, 1 H);
13C NMR (150MHz, CDCl3): = 166.4, 141.7, 136.2, 132.9, 132.6, 128.9, 128.7, 127.9,
127.0, 125.5, 73.4, 55.0;
IR (neat): 3032, 2915, 1596, 1574, 1408, 1322, 1224, 1109, 845 cm–1;
HRMS (ESI) m/z calcd for C16H13F3NS [(M+H)+] 308.0721, found 308.0719.
2-(4-fluorophenyl)-5-phenyl-4,5-dihydrothiazole (2-30): This compound was prepared
according to the General Procedure A using styrene (52 mg, 0.5 mmol), 4-
fluorothiobenzamide (233 mg, 1.5 mmol). After purification by column chromatography
on SiO2 (5% EtOAc in hexanes, Rf = 0.2), the title compound was isolated as a light yellow

41
oil (80 mg, 62% yield, >95:5).
1H NMR (600 MHz, CDCl3): = 7.91-7.84 (m, 2 H), 7.39-7.31 (m, 4 H), 7.31-7.25 (m, 1
H), 7.12 (t, J = 8.7 Hz, 2 H), 5.09 (dd, J = 5.5, 9.0 Hz, 1 H), 4.77 (dd, J = 9.0, 16.1 Hz, 1
H), 4.61 (dd, J = 5.5, 16.1 Hz, 1 H);
13C NMR (150MHz, CDCl3): = 166.5, 165.3, 163.7, 141.8, 130.5, 130.4, 129.3, 129.3,
128.9, 127.8, 127.0, 115.7, 115.5, 73.2, 54.9;
IR (neat): 3028, 2912, 1599, 1503, 1401, 1231, 1154, 999, 842, 698, 617 cm-1;
HRMS (ESI) m/z calcd for C15H13FNS [(M+H)+] 258.0753, found 258.0765.
2-(4-bromophenyl)-5-phenyl-4,5-dihydrothiazole (2-31): This compound was prepared
according to the General Procedure A using styrene (52 mg, 0.5 mmol), 4-
bromothiobenzamide (324 mg, 1.5 mmol). After purification by column chromatography
on SiO2 (5% EtOAc in hexanes, Rf = 0.4), the title compound was isolated as a white solid
(102 mg, 64% yield, >95:5).
1H NMR (600 MHz, CDCl3): = 7.74 (d, J = 8.5 Hz, 2 H), 7.57 (d, J = 8.5 Hz, 2 H), 7.37-
7.30 (m, 4 H), 7.30-7.24 (m, 1 H), 5.10 (dd, J = 5.6, 8.8 Hz, 1 H), 4.77 (dd, J = 8.8, 16.1
Hz, 1 H), 4.60 (dd, J = 5.6, 16.1 Hz, 1 H);
13C NMR (150MHz, CDCl3): = 166.6, 141.8, 132.0, 131.7, 129.8, 128.9, 127.9, 127.0,
125.8, 73.3, 54.9;
IR (neat): 2971, 1598, 1585, 1482, 1394, 1074, 926, 825, 754, 699, 594 cm-1;

42
HRMS (ESI) m/z calcd for C15H13BrNS [(M+H)+] 317.9952, found 317.9957.
2-(4-chlorophenyl)-5-phenyl-4,5-dihydrothiazole (2-32): This compound was prepared
according to the General Procedure A using styrene (52 mg, 0.5 mmol), 4-
chlorothiobenzamide (258 mg, 1.5 mmol). After purification by column chromatography
on SiO2 (5% EtOAc in hexanes, Rf = 0.3), the title compound was isolated as a yellow solid
(85 mg, 62% yield, >95:5).
1H NMR (600 MHz, CDCl3): = 7.81 (d, J = 8.5 Hz, 2 H), 7.41 (d, J = 8.5 Hz, 2 H), 7.37-
7.31 (m, 4 H), 7.31-7.25 (m, 1 H), 5.10 (dd, J = 5.6, 8.8 Hz, 1 H), 4.78 (dd, J = 8.8, 16.1
Hz, 1 H), 4.62 (dd, J = 5.6, 16.1 Hz, 1 H);
13C NMR (150MHz, CDCl3): = 166.5, 141.8, 137.3, 131.5, 129.6, 128.9, 128.7, 127.8,
127.0, 73.2, 54.9;
IR (neat): 3063, 2918, 1599, 1486, 1398, 1090, 830, 754, 693, 596 cm-1;
HRMS (ESI) m/z calcd for C15H13ClNS [(M+H)+] 274.0457, found 274.0456.
2-(2,4-difluorophenyl)-5-phenyl-4,5-dihydrothiazole (2-33): This compound was

43
prepared according to the General Procedure A using styrene (52 mg, 0.5 mmol), 2,4-
difluorothiobenzamide (260 mg, 1.5 mmol). After purification by column chromatography
on SiO2 (5% EtOAc in hexanes, Rf = 0.3), the title compound was isolated as a white solid
(80 mg, 58% yield, >95:5).
1H NMR (600 MHz, CDCl3): = 7.96-7.89 (m, 1 H), 7.37-7.31 (m, 4 H), 7.30-7.24 (m, 1
H), 6.98-6.87 (m, 2 H), 5.06 (dd, J = 5.7, 9.2 Hz, 1 H), 4.74 (dd, J = 9.2, 16.4 Hz, 1 H),
4.58 (dd, J = 5.7, 16.4 Hz, 1 H);
13C NMR (151MHz, CDCl3): = 165.2, 165.2, 163.6, 163.5, 161.8, 141.7, 132.1, 132.0,
132.0, 132.0, 128.9, 127.8, 127.0, 111.9, 111.8, 111.7, 111.7, 104.9, 104.7, 104.6, 72.3,
54.4, 54.4;
IR (neat): 2916, 2851, 1614, 160 0, 1494, 1314, 1250, 1139, 1097, 971, 851, 763, 670,
542 cm-1;
HRMS (ESI) m/z calcd for C15H12F2NS [(M+H)+] 276.0659, found 276.0663.
2-(3-bromophenyl)-5-phenyl-4,5-dihydrothiazole (2-34): This compound was prepared
according to the General Procedure A using styrene (52 mg, 0.5 mmol), 3-
bromothiobenzamide (324 mg, 1.5 mmol). After purification by column chromatography
on SiO2 (5% EtOAc in hexanes, Rf = 0.2), the title compound was isolated as a light yellow
oil (55 mg, 35% yield, 94:6).
1H NMR (600 MHz, CDCl3): = 8.07 (s, 1 H), 7.77 (d, J = 7.9 Hz, 1 H), 7.61 (d, J = 7.9

44
Hz, 1 H), 7.37-7.24 (m, 7 H), 5.10 (dd, J = 5.6, 9.0 Hz, 1 H), 4.78 (dd, J = 9.0, 16.2 Hz, 1
H), 4.63 (dd, J = 5.6, 16.2 Hz, 1 H);
13C NMR (150MHz, CDCl3): = 166.2, 141.8, 134.9, 134.1, 131.1, 130.0, 128.9, 127.9,
127.0, 127.0, 122.6, 73.2, 54.8;
IR (neat): 3026, 2938, 1602, 1560, 1419, 1226, 1008, 696 cm–1;
HRMS (ESI) m/z calcd for C15H13BrNS [(M+H)+] 317.9952, found 317.9960.
2-(2-bromophenyl)-5-phenyl-4,5-dihydrothiazole (2-35): This compound was prepared
according to the General Procedure A using styrene (52 mg, 0.5 mmol), 2-
bromothiobenzamide (324 mg, 1.5 mmol). After purification by column chromatography
on SiO2 (5% EtOAc in hexanes, Rf = 0.2), the title compound was isolated as a yellow
liquid (100 mg, 63% yield, 93:7).
1H NMR (600 MHz, CDCl3): = 7.65 (d, J = 8.1 Hz, 1 H), 7.61-7.55 (m, 1 H), 7.41 (d, J
= 7.1 Hz, 2 H), 7.39-7.32 (m, 3 H), 7.32-7.24 (m, 2 H), 5.13 (dd, J = 5.4, 9.0 Hz, 1 H), 4.81
(dd, J = 9.0, 16.2 Hz, 1 H), 4.66 (dd, J = 5.4, 16.2 Hz, 1 H);
13C NMR (150MHz, CDCl3): = 166.4, 142.0, 134.8, 133.6, 131.1, 130.5, 128.8, 127.8,
127.2, 127.0, 121.2, 73.2, 55.8;
IR (neat): 3026, 2937, 2845, 1620, 1466, 1224, 995, 752, 643 cm-1;
HRMS (ESI) m/z calcd for C15H13BrNS [(M+H)+] 317.9952, found 317.9965.

45
2-isopropyl-5-phenyl-4,5-dihydrothiazole (2-36): This compound was prepared
according to the General Procedure A using styrene (52 mg, 0.5 mmol), thioisobutyramide
(155 mg, 1.5 mmol). After purification by column chromatography on SiO2 (10% EtOAc
in hexanes, Rf = 0.3), the title compound was isolated as a yellow solid (31mg, 30% yield,
>95:5);
1H NMR (600 MHz, CDCl3): = 7.41-7.21 (m, 5 H), 4.91 (dd, J = 5.5, 9.0 Hz, 1 H), 4.54
(dd, J = 9.0, 15.6 Hz, 1 H), 4.36 (dd, J = 5.5, 15.6 Hz, 1 H), 2.92-2.77 (m, 1 H), 1.28 (s, 6
H);
13C NMR (150MHz, CDCl3): = 176.7, 142.5, 128.8, 127.6, 126.9, 72.6, 54.1, 34.0, 21.1,
21.1;
IR (neat): 2965, 1625, 1454,1324,984,760,696 cm-1;
HRMS (ESI) m/z calcd for C12H16NS [(M+H)+] 206.1003, found 206.0997.
2-(4-methoxyphenyl)-5-phenyl-4,5-dihydrothiazole (2-37)66: This compound was
prepared according to the General Procedure A using styrene (52 mg, 0.5 mmol), 4-
methoxythiobenzamide (251 mg, 1.5 mmol). After purification by column chromatography
on SiO2 (10% EtOAc in hexanes, Rf = 0.3), the title compound was isolated as a yellow
solid (70 mg, 52% yield, >95:5).
1H NMR (600 MHz, CDCl3): = 7.82 (d, J = 8.8 Hz, 2 H), 7.38-7.30 (m, 4 H), 7.29-7.24

46
(m, 1 H), 6.93 (d, J = 8.8 Hz, 2 H), 5.05 (dd, J = 5.4, 8.8 Hz, 1 H), 4.75 (dd, J = 8.8, 16.0
Hz, 1 H), 4.58 (dd, J = 5.4, 16.0 Hz, 1 H), 3.86 (s, 3 H).
2-(3,4-dimethoxyphenyl)-5-phenyl-4,5-dihydrothiazole (2-38): This compound was
prepared according to the General Procedure A using styrene (52 mg, 0.5 mmol), 3,4-
dimethoxythiobenzamide (296 mg, 1.5 mmol). After purification by column
chromatography on SiO2 (10% to 20% EtOAc in hexanes, Rf = 0.3), the title compound
was isolated as a white solid (72 mg, 48% yield, >95:5).
1H NMR (600 MHz, CDCl3): = 7.49 (s, 1 H), 7.40-7.21 (m, 6 H), 6.86 (d, J = 8.3 Hz, 1
H), 5.04 (dd, J = 5.3, 8.5 Hz, 1 H), 4.74 (dd, J = 8.5, 16.0Hz, 1 H), 4.58 (dd, J = 5.3, 16.0
Hz, 1 H), 3.91 (s, 3 H), 3.93 (s, 3 H);
13C NMR (150MHz, CDCl3): = 167.1, 151.6, 148.7, 142.0, 128.8, 127.7, 127.0, 125.9,
122.5, 110.2, 109.9, 73.0, 55.9, 55.9, 54.5;
IR (neat): 3051, 2925, 1605, 1581, 1507, 1412, 1262 cm–1;
HRMS (ESI) m/zcalcd for C17H18NO2S [(M+H)+] 300.1058, found 300.1060.
4-(5-phenyl-4,5-dihydrothiazol-2-yl)aniline (2-39): This compound was prepared

47
according to the General Procedure A using styrene (52 mg, 0.5 mmol), 4-
aminothiobenzamide (254 mg, 1.5 mmol). After purification by column chromatography
on SiO2 (40% EtOAc in hexanes, Rf = 0.4), the title compound was isolated as a yellow
solid (41 mg, 32% yield, >95:5).
1H NMR (600 MHz, CDCl3): = 7.69 (d, J = 8.5 Hz, 2 H), 7.39-7.22 (m, 5 H), 6.66 (d, J
= 8.5 Hz, 2 H), 5.01 (dd, J = 5.4, 8.8 Hz, 1 H), 4.72 (dd, J = 8.8, 15.9 Hz, 1 H), 4.56 (dd,
J= 5.4, 15.9 Hz, 1 H), 3.98 (br. s., 2 H);
13C NMR (150MHz, CDCl3): = 167.1, 149.3, 142.3, 130.1, 128.7, 127.6, 127.0, 123.3,
114.2, 73.0, 54.3;
IR (neat): 3446, 3184, 2852, 1634, 1592, 1513, 1304, 1171, 1009, 930, 930, 763 cm-1;
HRMS (ESI) m/z calcd for C15H15N2S [(M+H)+] 255.0956, found 255.0956.
5-phenyl-2-(pyridin-2-yl)-4,5-dihydrothiazole (2-40): This compound was prepared
according to the General Procedure A using styrene (52 mg, 0.5 mmol), pyridine-2-
carbothioic acid amide (207 mg, 1.5 mmol). After purification by column chromatography
on SiO2 (DCM to 40% EtOAc in DCM, Rf = 0.3), the title compound was isolated as a
white solid (64 mg, 53% yield, >95:5).
1H NMR (600 MHz, CDCl3): = 8.66 (d, J = 4.2 Hz, 1 H), 8.12 (d, J = 8.1 Hz, 1 H), 7.78
(t, J = 7.7 Hz, 1 H), 7.40-7.33 (m, 3 H), 7.31 (t, J = 7.2 Hz, 2 H), 7.28-7.22 (m, 1 H), 5.05
(dd, J = 5.8, 9.1 Hz, 1 H), 4.88 (dd, J = 9.1, 16.6 Hz, 1 H), 4.67 (dd, J = 5.8, 16.6 Hz, 1 H).

48
13C NMR (150MHz, CDCl3: = 170.0, 151.0, 149.2, 142.1, 136.5, 128.7, 127.5, 127.0,
125.3, 121.4, 73.7, 53.2;
IR (neat): 3054, 2903, 1599, 1466, 1434, 1321, 961, 700 cm–1;
HRMS (ESI) m/zcalcd for C14H13N2S [(M+H)+] 241.0799, found 241.0798.
5-phenyl-2-(pyridin-3-yl)-4,5-dihydrothiazole (2-41)66: This compound was prepared
according to the General Procedure A using styrene (52 mg, 0.5 mmol), pyridine-3-
carbothioic acid amide (207 mg, 1.5 mmol). After purification by column chromatography
on SiO2 (50% EtOAc in hexanes, Rf = 0.3), the title compound was isolated as a yellow
liquid (40 mg, 33% yield, >95:5).
1H NMR (600 MHz, CDCl3): = 9.10-9.05 (m, 1 H), 8.71 (d, J= 4.6 Hz, 1 H), 8.14 (d, J
= 8.1 Hz, 1 H), 7.41-7.23 (m, 6 H), 5.13 (dd, J = 5.6, 9.0 Hz, 1 H), 4.80 (dd, J = 9.0, 16.4
Hz, 1 H), 4.64 (dd, J = 5.6, 16.4 Hz, 1 H).
5-phenyl-2-(pyridin-4-yl)-4,5-dihydrothiazole (2-42): This compound was prepared
according to the General Procedure A using styrene (52 mg, 0.5 mmol), pyridine-4-
carbothioic acid amide (207 mg, 1.5 mmol). After purification by column chromatography
on SiO2 (50% EtOAc in hexanes, Rf = 0.3), the title compound was isolated as a yellow
solid (21 mg, 18% yield, >95:5).

49
1H NMR (600 MHz, CDCl3): = 8.72 (d, J = 5.4 Hz, 2 H), 7.70 (d, J = 5.4 Hz, 2 H), 7.37-
7.24 (m, 5 H), 5.14 (dd, J = 5.6, 9.0 Hz, 1 H), 4.81 (dd, J = 9.0, 16.7 Hz, 1 H), 4.66 (dd, J
= 5.6, 16.7 Hz, 1 H);
13C NMR (150MHz, CDCl3): = 166.1, 150.4, 141.4, 139.9, 129.0, 128.0, 127.0, 122.1,
73.4, 54.9;
IR (neat): 3027, 2920, 2847,1591, 1550, 1406, 1321, 1243, 1006, 822,696.54, 611 cm-1;
HRMS (ESI) m/z calcd for C14H13N2S [(M+H)+] 241.0799, found 241.0797.
5-phenyl-2-(pyrimidin-2-yl)-4,5-dihydrothiazole (2-43): This compound was prepared
according to the General Procedure A using styrene (52 mg, 0.5 mmol), Pyrimidine-2-
carbothioamide (209 mg, 1.5 mmol). After purification by column chromatography on
SiO2 (5% MeOH in DCM, Rf = 0.3), the title compound was isolated as a light yellow oil
(38 mg, 31% yield, >95:5).
1H NMR (600 MHz, CDCl3): = 8.87 (d, J = 4.9 Hz, 2 H), 7.39-7.34 (m, 3 H), 7.31 (t, J
= 7.4 Hz, 2 H), 7.28-7.22 (m, 1 H), 5.12 (dd, J = 5.9, 9.3 Hz, 1 H), 4.95 (dd, J = 9.3, 17.1
Hz, 1 H), 4.76 (dd, J = 5.9, 17.1 Hz, 1 H);
13C NMR (150MHz, CDCl3): = 168.2, 159.1, 157.5, 141.8, 128.8, 127.7, 127.1, 121.7,
73.9, 54.1;
IR (neat): 3029, 2928, 1611, 1559, 1416, 1007, 698 cm–1;
HRMS (ESI) m/z calcd for C13H12N3S [(M+H)+] 242.0752, found 242.0756.

50
Procedure for oxidation of thiazoline76:
2,5-diphenylthiazole (2-44): The molecular sieve (4Å MS, 400 mg/ mmol, 100 mg) was
added to solution of thiazoline 2-8 (60 mg, 0.25 mmol) in CH2Cl2 (1.5 mL, 0.17 M); After
10 min, DDQ (86 mg, 0.38 mmol) was added to the reaction mixture and stirred at 50 °C.
After 16 h, the reaction mixture was cooled to room temperature and quenched with 10%
NaOH (4 mL) and extracted with CH2Cl2 (3 x 2 mL). The combined organic layer was
concentrated in vacuo. Purification by flash chromatography on SiO2 (5% EtOAc in
hexanes, Rf = 0.4) provided the title compound as a white solid (56 mg, 95% yield)77.
1H NMR (600 MHz, CDCl3): = 8.03 (s, 1 H), 8.01-7.95 (m, 2 H), 7.61 (d, J = 7.8 Hz, 2
H), 7.51-7.39 (m, 5 H), 7.38-7.32 (m, 1 H).
Procedure for hydrolysis of thiazoline71:
2-amino-1-phenylethane-1-thiol hydrochloride (2-45): The thiazoline 2-8 (120 mg, 0.5
mmol) was added to the solution of HCl 5N (2.5 mL) and heated under reflux for 16 h.
After 16 h, the reaction mixture was cooled to room temperature and extracted with EtOAc
(2 x 2 mL), aqueous layer was concentrated in vacuo to provide the desired product as a
white solid (87 mg, 92% yield).
1H NMR (600MHz, D2O): = 7.46-7.38 (m, 4 H), 7.36 (d, J = 5.9 Hz, 1 H), 4.22 (t, J =
7.9 Hz, 1 H), 3.52-3.38 (m, 2 H);

51
13C NMR (150 MHz, D2O): =139.6, 129.6, 128.8, 127.2, 46.2, 40.5;
IR (neat): 3313.62, 3029, 2142, 1451, 1159, 692 cm-1;
HRMS (ESI) m/z calcd for C12H16NS [(M+H)+] 154.0690, found 154.0701.

52
Chapter 3
Intermolecular Regio- and Stereoselective Alkene
Sulfenoamination With Thioimidazoles
3.1 Introduction
Nitrogen and sulfur-containing heterocycles are common structural motifs in
natural products, bioactive compounds, and agrochemicals.42 Expedient incorporation of
nitrogen and sulfur heteroatoms into simple chemical feedstocks like alkenes, represents
an important and attractive strategy for the direct synthesis of pharmaceuticals. However,
the absolute control of regio- and stereoselectivity associated with alkene is often
considered as a prominent challenge. Therefore, chemical methods that can selectively
install nitrogen and sulfur atoms at the desired carbon position of the alkenes are of high
importance and value for accessing biomedically-relevant chemical libraries. In this
context, electrophilic activation has been representing a powerful tool for alkene
functionalization reactions.78 Typically, the electrophilic activating group converts alkene
into -onium intermediate, followed by nucleophilic displacement to give the desired
product. The utilization of such a strategy for the development of vicinal sulfenoamination
of the alkene in a regioselective manner has drawn significant attention from the synthetic
and medicinal communities due to the frequent appearances of nitrogen (N) and sulfur (S)
atoms in pharmaceuticals and natural products. In addition, the abundant, versatile, and

53
Scheme 3-1: Background on Sulfenoamination of Alkene
diverse nature of the alkene substrate make the method even more attractive to gain access
to valuable heterocycles with diverse functionalities. In this regard, several methods have
been well documented for intramolecular alkene sulfenoamination utilizing electrophilic
sulfur precursors.79-80 The pioneering work from Denmark group has demonstrated the
enantioselective sulfenoamination of N-alkenyl sulfonamide with phenylthiophthalimide
(PhthSPh) for the synthesis of N-containing heterocycles in intramolecular settings

54
(Scheme 3-1a).79, 81-82 Similarly, Shi and coworkers also reported intramolecular
thioamination using benzenesulfenate (MeOSPh) (Scheme 3-1a).83 The origin and extent
of regioselectivity, in these cases, are dependent on the competing endo vs. exo trapping
of the thiiranium intermediate (compound 3-1) by a pendant nucleophile. Later, Wirth
group developed hypervalent iodine-catalyzed thioamination of alkenes with sodium
thiophenolate via activation of the alkene by iodine (III) reagents.80 However, in contrast
to intramolecular sulfenoamination, the development of intermolecular thioamination
which is thermodynamically and kinetically disfavored, is more challenging and highly
desirable.84 In a demonstration of this strategy, sulfenoamination for the synthesis of β-
acetamido sulfide have been reported using nitrile as solvent and nitrogen source, and thiol
as a sulfur source via Ritter type reaction (Scheme 3-1b).85-87 However, this approach is
only limited to nitrile while intermolecular protocols with a broad range of alkenes and
thioamine structures can provide a modular approach for increasing structural complexity
and functional group diversity of the molecules.88 Direct intermolecular sulfenoamination
of alkene using unadorned thioamine as a nucleophile has not been realized. In addition,
the utilization of stable sulfur precursor instead of pre-generated electrophilic sulfur
reagents can further enhance the practicality of the alkene sulfenoamination strategy.89-91
As part of our interest in heterocycles synthesis, we aim to develop an
intermolecular alkene sulfenoamination strategy for accessing structurally diverse N- and
S-containing heterocycles. As a proof of concept for our halogen activation, we have
previously developed an intermolecular and regioselective sulfenoamination of terminal
olefins for the synthesis of thiazoline, utilizing thioamide as a stable sulfur and nitrogen
precursor, in a one-pot process (Scheme 3-1c).92 The regioselectivity for thiazoline

55
synthesis originates from the initial nucleophilic displacement of a 1,2-dibromoalkane,
generated from the halogenation of alkenes, with which the more nucleophilic sulfur atom
preferentially attacks the more electrophilic carbon of the 1,2-dibromoalkane.
In contrast to this previous halogenation approach, we hypothesize that
halogenation of thiol can lead to an in-situ generation of electrophilic sulfur reagent instead
of formation of 1,2-dihaloalkane.93-95 Attack of the resulting thiiranium ion (compound 3-
6) by a pendant nitrogen nucleophile, can then afford the desired N- and S-containing
heterocycle.96-100 With this hypothesis in mind, we report herein an intermolecular regio-
and stereoselective alkene sulfenoamination reaction with thioimidazoles (Scheme 3-1d).
3.2 Results and Discussions for Sulfenoamination
3.2.1 Reaction Design and Optimization
We commenced our study with the evaluation of a range of sulfur- and nitrogen-
containing structures with different halogen source for exploration of the compatibility of
the nucleophile with halogen. To our delight, our screenings revealed that Selectfluor was
a suitable halogen source that could enable 2-mercaptobenzimidazole to readily couple
with styrene. The reaction provided desired heterocyclic product 3-10 with high
regioselectivity albeit in low yield (7%) using acetonitrile as solvent (Table 3.1, entry 1).101
The validation of the hypothesis inspired us to perform further optimization, and testing of
different solvents revealed that N, N-dimethylformamide (DMF) was a superior solvent,
affording the product in 53% yield (Table 3.1, entries 2 and 3). The screening of the
concentration and stoichiometry of Selectfluor improved the yield of the product to 77%
(Table 3.1, entries 4-7). Additional time studies confirmed that 16 he was needed for the

56
Table 3.1: Optimization of Reaction for Sulfenoamination
reaction to proceed to completion (Table 3.1, entries 8 and 9). Further evaluation of the
stoichiometry of alkene and thiobenzimidazole demonstrated that the optimal condition
was achieved by increasing the stoichiometry of styrene to 1.5 equiv, providing the
expected product in 87% yield in a single regioisomer (Table 3.1, entries 10-12).
3.2.2 Reaction Scope
With the optimized condition in hand, we were excited to explore the scope of the

57
alkene substrate for our sulfenoamination strategy. We were pleased to find that a wide
range of alkenes with different functionalities was tolerable in our reaction condition,
providing good to excellent yields of the products. The styrene derivatives with different
halogen substituents at para - position provided the products with good yields and high
regioselectivities (Table 3.2, products 3-11 to 3-13). Moreover, the styrene derivatives with
electron-donating substituents such as methoxy, acetoxy, and electron-withdrawing groups
such as ester and trifluoromethyl groups were compatible with the reaction conditions,
affording the products in excellent regioselectivities with high efficiencies (Table 3.2,
products 3-14 to 3-17, 3-22, 3-23). The regioisomers observed in these reactions are shown
in Table 3.2 with a sulfur atom attached to the terminal position and nitrogen atom to the
benzylic position. To our delight, sterically hindered 1,1-disubstituted styrene derivatives
provided the desired products with the formation of highly steric congested quaternary
carbon centers (Table 3.2, products 3-18 to 3-21). The regioselectivities, in these cases,
were also consistent with the standard substrate. In addition, 1,2-disubstituted alkenes such
as trans-β-methyl styrene, Indene, and trans-4-octene were participated in the reaction,
affording the products in good yields with excellent diastereoselectivities (Table 3.2,
products 3-24 to 3-26). Interestingly, aliphatic disubstituted alkenes are also viable
substrates, providing exotic tetracyclic structures 3-27 and 3-28 further highlighting the
synthetic utility of this sulfenoamination strategy (Table 3.2, products 3-27, 3-28). In
general, the regioselectivity of the aliphatic alkenes is predominately governed by steric
factors. In our strategy, the regioselectivity with sulfur being in terminal carbon, was
decreased with increasing steric bulkiness in alkene and completely reversing the normally
observed regioselectivity for product 3-32 (Table 3.2, products 3-29 to 3-32).

58
Table 3.2: Alkene Substrate Scope for Sulfenoamination
Excited by the comprehensive alkene substrate scope, we focused on extending our
protocol to the incorporation of thioimidazole structure in this reaction. We were pleased
to find that our strategy could be extended to a wide range of functionalities. A number of

59
Table 3.3: Thioimidazole Substrate Scope for Sulfenoamination
useful functional groups including electron-donating and -withdrawing groups such as
alkyl, halogen, ether, nitro, and difluoromethyl ether provided the thioaminated products
efficiently (Table 3.3, Products 3-33 to 3-39). In each of these cases, regioselectivity shown
in Table 3.3, were obtained from the aspect that both nitrogen atoms in the benzimidazole
ring were capable of nucleophilic additions. However, regioselectivity with respect to N-
and S- additions to the alkene structure, remained intact with only nitrogen addition to the
benzylic position and sulfur to the terminal carbon. Moreover, 2-thioimidazoles were also
viable substrates providing the corresponding products in moderate yields with excellent
regioselectivities (Table 3.3, products 3-40 and 3-41).

60
3.2.3 Regiodivergent Sulfenoamination
In our previous sulfenoamination study for thiazoline synthesis with thioamides,
we observed the opposite regioselectivity for styrene derivatives, in which sulfur was added
to the benzylic positions and nitrogen was attached to the terminal carbons. We wondered
if we could develop potential regiodivergent sulfenoamination processes on both the
halogenation and electrophilic sulfur activation strategies. In this regard, utilization of
Scheme 3-2: Regiodivergent of Alkene Sulfenoamination
Selectfluor as a halogen source resulted in the formation of 3-10, 3-14, and 3-22 as the only
regioisomeric products with sulfur being in terminal position and nitrogen is attached to
benzylic carbon. On the other hand, the opposite regioisomers 3-42 to 3-44 were obtained
when bromine was used as the halogen source via 1,2-dibromoethylarene, from alkene and
thiobenzimidazole coupling (Scheme 3.2).102 Moreover, the electronic nature of the styrene
substrates did not have any impact on the regioisomeric outcome of the product,
highlighting the robustness of regiocontrol in these reactions.

61
3.3 Reaction Mechanism
To get insight into the mechanism of the reaction, we considered several
experiments for intermolecular sulfenoamination reaction. The formation of product 3-21
proceeds with cyclopropane ring remaining intact. This data suggests that pathways leading
to the formation of radical at the benzylic position are less likely, as such pathways
generally will lead to cyclopropane ring opening.103 In addition, the regioselectivity of the
product 3-32, obtained from 3,3-dimethylbutene is not characteristic of thiyl radical
addition. In radical addition, we would expect a product with sulfur being attached to the
terminal carbon of alkene. Instead, the ability of steric factors to influence regioselectivity,
suggested the thiiranium ion pathway. Furthermore, NMR studies by mixing
thiobenzimidazole and Selectfluor in deuterated DMF revealed a new set of fluorine peaks
showing up at 38 ppm, in agreement with literature data of a sulfur-fluorine bond.
3.3.1 Solvent Effect on Reaction
To further understand the mechanism, we also considered the effect of DMF, only
solvent that works efficiently for the reaction. DMF can act as a nucleophile and can
participate in opening up the thiiraniun ion intermediate A to form intermediate B which
can undergo nucleophile attack from the nitrogen of thiobenzimidazole, resulting in
cyclized product 3-10 (Scheme 3-3). To test the effect of the solvent, the reaction was
carried out in acetonitrile (CH3CN). The increasing the equivalent of DMF increased the
yield to 32% for 5 equiv of DMF. That yield is not comparable to the yield in standard
reaction conditions, suggesting that DMF can act as a Lewis base, with having little effect
in sulfenoamination reaction.
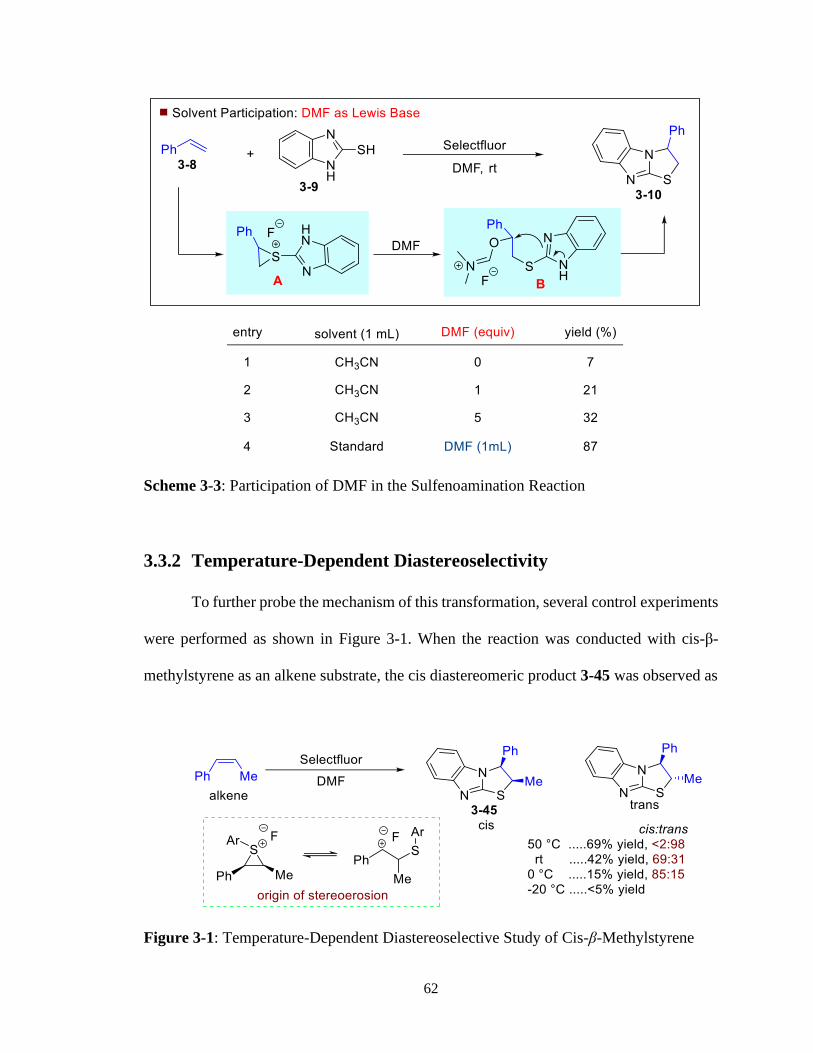
62
Scheme 3-3: Participation of DMF in the Sulfenoamination Reaction
3.3.2 Temperature-Dependent Diastereoselectivity
To further probe the mechanism of this transformation, several control experiments
were performed as shown in Figure 3-1. When the reaction was conducted with cis-β-
methylstyrene as an alkene substrate, the cis diastereomeric product 3-45 was observed as
Figure 3-1: Temperature-Dependent Diastereoselective Study of Cis-β-Methylstyrene

63
the major product in 15% yield and 85:15 dr at 0 ˚C. On the other hand, increasing
temperatures favoring the formation of trans product and at 50 ˚C, the trans isomer is
obtained in >98:2 ratio. This temperature-dependent behavior is consistent with previous
studies on the configurational stability of the thiiranium ions conducted by Smit and
Denmark.104-106
3.3.3 Proposed Mechanism for Intermolecular Sulfenoamination
After considering the above-mentioned experiments, our proposed mechanism for
this reaction is depicted in Figure 3-2. Fluorination of thioimidazole 3-46 by Selectfluor
results in the formation of the active sulfur electrophile 3-47. Alkene attack on this
Figure 3-2: Proposed Reaction Mechanism for Alkene Sulfenoamination
sulfur electrophile leads to the formation of thiiranium ion 3-48. Generally, the thiiranium
ion is unstable, equilibration to the open carbocation 3-49 can occur, and subsequent
nucleophile trapping results in the product formation 3-50. Alternatively, DMF can act as
a nucleophile and participate in thiiranium ring opening. The ensuing intramolecular

64
cyclization then preserves the stereochemistry in the product, as evidence in the cis-β-
methyl styrene case at low temperature.
3.4 Conclusion
We have developed a highly regio- and stereoselective sulfenoamination reaction
of alkenes. The method exhibits good functional group tolerance and broad substrate scope.
The mechanistic studies suggest the reaction pathway involving an in-situ generation of
the sulfur electrophile. Moreover, we have observed an interesting temperature-dependent
equilibrium between the thiiranium ion and the open carbocation species. In addition, we
have also developed a highly regiodivergent protocol to access both regioisomeric products
utilizing two different halogen sources. Finally, this modular and practical synthetic
approach will enable the generation of exotic heterocyclic motifs for potentially useful
biological studies.
3.5 Experimental
General Information. Commercial reagents and solvents were purchased from Sigma
Aldrich, Oakwood Chemicals, Alfa Aesar, Matrix Scientific, Acros Organic, and were used
as were used as received. The alkene substrates for products 3-21,107 3-23,108 3-25,75 were
synthesized according to reported procedure. Organic solutions were concentrated under
reduced pressure on a Büchi rotary evaporator using an acetone-dry ice bath.
Chromatographic purification of products was accomplished using flash chromatography
on 230-400 mesh silica gel. Thin-layer chromatography (TLC) was performed on Analtech

65
250 mm silica gel HLF UV-254 plates. Visualization of the developed plates was
performed by fluorescence quenching, potassium permanganate and iodine-silica gel
system. 1H and 13C NMR spectra were recorded on a Bruker 600 instrument (600 and 150
MHz) or INOVA 600 (600 and 150 MHz) and are internally referenced to residual protio
solvent signals (for CDCl3, 7.26 and 77.0 ppm, respectively). Data for 1H NMR are
reported as follows: chemical shift ( ppm), multiplicity (s = singlet, d = doublet, t = triplet,
q = quartet, h = heptet, m = multiplet, br = broad), integration, coupling constant (Hz). 13C
spectra were reported as chemical shifts in ppm and multiplicity where appropriate. IR
spectra were recorded on a PerkinElmer FT-IR spectrophotometer and are reported in terms
of wavenumber of absorption (cm-1). High resolution mass spectra were obtained on
Waters Synapt High Definition Mass Spectrometer (HDMS) by electrospray ionization at
University of Toledo, OH, USA.
Experimental Procedures
General Procedure A: Alkene (0.75 mmol) was added to a solution of 2-
mercaptoimidazole (0.5 mmol) in DMF (1 mL, 0.5 M). After adding Selectfluor (213 mg,
0.6 mmol), the reaction mixture was stirred at room temperature for 16 h. After completion,
the reaction mixture was quenched with 1 M NaOH (2 mL). The aqueous layer was
extracted with DCM (2 x 4 mL) and combined organic layers were washed with water (3
mL) three times. The organic solution was dried with anhydrous Na2SO4, filtered,
concentrated in vacuo by rotavapor and purified by flash chromatography on SiO2 (30-70%
EtOAc in hexanes) to provide the desired product. The regioisomeric ratio was determined
by crude NMR.

66
General Procedure B: Alkene (0.75 mmol) was added to a solution of 2-
mercaptoimidazole (0.5 mmol) in DMF (1 mL, 0.5 M). After adding Selectfluor (213 mg,
0.6 mmol), the reaction mixture was stirred at 50 ºC for 16 h. After completion, the reaction
mixture was cooled to room temperature and quenched with 1 M NaOH (2 mL). The
aqueous layer was extracted with DCM (2 x 4 mL) and combined organic layers were
washed with water (3 mL) three times. The organic solution was dried with anhydrous
Na2SO4, filtered, concentrated in vacuo by rotavapor and purified by flash chromatography
on SiO2 (30-70% EtOAc in hexanes) to provide the desired product. The regioisomeric
ratio was determined by crude NMR.
General Procedure C: Alkene (0.75 mmol) was added to a solution of bromine (0.5
mmol) in DMF (1 mL, 0.5 M) and stirred at room temperature for 1 h. After adding 2-
mercaptobenzimidazole (113 mg, 0.75 mmol), the reaction mixture was stirred at 50 ºC for
16 h. Then, the reaction mixture was cooled to room temperature and quenched with 1 M
NaOH (2 mL) and 20% aq Na2S2O3 (1 mL). The aqueous layer was extracted with DCM
(2 x 4 mL) and combined organic layers were washed with water (3 mL) three times. The
organic solution was dried with anhydrous Na2SO4, filtered, concentrated in vacuo by
rotavapor and purified by flash chromatography on SiO2 (30-50% EtOAc in hexanes) to
provide the desired product. The regioisomeric ratio was determined by crude NMR.
3. Spectral Characterization of the Products

67
3-phenyl-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-10): This compound was
prepared according to the General Procedure A using styrene (78 mg, 0.75 mmol), 2-
mercaptobenzimidazole (75 mg, 0.5 mmol). After purification by column chromatography
on SiO2 (30-50% EtOAc in hexanes), the title compound was isolated as a white solid (110
mg, 87% yield, >98:2 rr).
1H NMR (600 MHz, CDCl3): = 7.62 (d, J = 8.1 Hz, 1 H), 7.42-7.35 (m, 3 H), 7.29-7.22
(m, 2 H), 7.13 (t, J = 7.7 Hz, 1 H), 6.94 (t, J = 7.7 Hz, 1 H), 6.57 (d, J = 8.1 Hz, 1 H), 5.49
(t, J = 7.6 Hz, 1 H), 4.14 (dd, J = 7.6, 11.2 Hz, 1 H), 3.76 (dd, J = 7.6, 11.2 Hz, 1 H);
13C NMR (150 MHz, CDCl3): = 158.5, 149.5, 136.8, 133.4, 129.2, 129.1, 126.5,
121.8, 121.5, 118.6, 109.2, 77.2, 76.8, 61.0, 43.2;
IR (neat): 3027, 1612, 1469, 1448, 1248, 733 cm-1;
HRMS (ESI) m/z calcd for C15H13N2S [(M+H)+] 253.0799, found 253.0804.
3-(4-fluorophenyl)-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-11): This
compound was prepared according to the General Procedure A using 4-fluorostyrene (92
mg, 0.75 mmol), 2-mercaptobenzimidazole (75 mg, 0.5 mmol). After purification by
column chromatography on SiO2 (30-50% EtOAc in hexanes), the title compound was
isolated as a white solid (106 mg, 78% yield, >98:2 rr).
1H NMR (600 MHz, CDCl3): = 7.61 (d, J = 8.1 Hz, 1 H), 7.25 (dd, J = 5.2, 8.1 Hz, 2

68
H), 7.14 (t, J = 7.7 Hz, 1 H), 7.08 (t, J = 8.1 Hz, 2 H), 6.97 (t, J = 7.7 Hz, 1 H), 6.57 (d, J
= 8.1 Hz, 1 H), 5.49 (t, J = 7.3 Hz, 1 H), 4.15 (dd, J = 7.3, 11.2 Hz, 1 H), 3.72 (dd, J = 7.3,
11.2 Hz, 1 H);
13C NMR (150 MHz, CDCl3): = 163.7, 162.1, 158.4, 149.5, 133.3, 132.7, 132.6,
128.4, 128.3, 122.0, 121.6, 118.7, 116.3, 116.2, 109.1, 60.3, 43.2;
IR (neat): 3070, 1677, 1603, 1582, 1469, 1275, 928 cm-1;
HRMS (ESI) m/z calcd for C15H12FN2S [(M+H)+] 271.0705, found 271.0711.
3-(4-chlorophenyl)-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-12): This
compound was prepared according to the General Procedure A using 4-chlorostyrene (104
mg, 0.75 mmol), 2-mercaptobenzimidazole (75 mg, 0.5 mmol). After purification by
column chromatography on SiO2 (30-50% EtOAc in hexanes), the title compound was
isolated as a white solid (113 mg, 79% yield, >98:2 rr).
1H NMR (600 MHz, CDCl3): = 7.62 (d, J = 8.1 Hz, 1 H), 7.36 (d, J = 8.1 Hz, 2 H), 7.20
(d, J = 8.1 Hz, 2 H), 7.15 (t, J = 7.7 Hz, 1 H), 6.98 (t, J = 7.7 Hz, 1 H), 6.60 (d, J = 8.1 Hz,
1 H), 5.49 (t, J = 7.3 Hz, 1 H), 4.17 (dd, J = 7.3, 11.4 Hz, 1 H), 3.71 (dd, J = 7.3, 11.4 Hz,
1 H);
13C NMR (150 MHz, CDCl3): = 158.4, 149.5, 135.4, 135.0, 133.2, 129.4, 127.8,
122.0, 121.7, 118.7, 109.1, 60.2, 43.1;
IR (neat): 3054, 1609, 1472, 1219, 926, 737 cm-1;

69
HRMS (ESI) m/z calcd for C15H12ClN2S [(M+H)+] 287.0410, found 287.0412.
3-(4-bromophenyl)-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-13): This
compound was prepared according to the General Procedure A using 4-bromostyrene (137
mg, 0.75 mmol), 2-mercaptobenzimidazole (75 mg, 0.5 mmol). After purification by
column chromatography on SiO2 (30-50% EtOAc in hexanes), the title compound was
isolated as a white solid (145 mg, 88% yield, >98:2 rr).
1H NMR (600 MHz, CDCl3): = 7.61 (d, J = 8.5 Hz, 1 H), 7.51 (d, J = 8.5 Hz, 2 H), 7.18
-7.10 (m, 3 H), 6.98 (t, J = 7.6 Hz, 1 H), 6.60 (d, J = 7.6 Hz, 1 H), 5.48 (t, J = 7.4 Hz, 1 H),
4.16 (dd, J = 7.4, 11.2 Hz, 1 H), 3.71 (dd, J = 7.4, 11.2 Hz, 1 H);
13C NMR (150 MHz, CDCl3): = 158.4, 149.4, 135.9, 133.2, 132.4, 128.1, 123.1, 122.0,
121.7, 118.7, 109.1, 60.2, 43.1;
IR (neat): 3051, 1595, 1472, 1219, 820, 737 cm-1;
HRMS (ESI) m/z calcd for C15H12BrN2S [(M+H)+] 330.9904, found 330.9906.
3-(4-(tert-butyl)phenyl)-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-14): This

70
compound was prepared according to the General Procedure A using 4-tert-butylstyrene
(120 mg, 0.75 mmol), 2-mercaptobenzimidazole (75 mg, 0.5 mmol). After purification by
column chromatography on SiO2 (30-50% EtOAc in hexanes), the title compound was
isolated as a white solid (130 mg, 84% yield, >98:2 rr).
1H NMR (600 MHz, CDCl3): = 7.62 (d, J = 8.1 Hz, 1 H), 7.40 (d, J = 8.1 Hz, 2 H),
7.19 (d, J = 8.1 Hz, 2 H), 7.15 (t, J = 7.6 Hz, 1 H), 6.96 (t, J = 7.6 Hz, 1 H), 6.62 (d, J =
8.1 Hz, 1 H), 5.48 (t, J = 7.4 Hz, 1 H), 4.13 (dd, J = 7.4, 11.1 Hz, 1 H), 3.75 (dd, J = 7.4,
11.1 Hz, 1 H), 1.32 (s, 9 H);
13C NMR (150 MHz, CDCl3): = 158.6, 152.2, 149.5, 133.7, 133.5, 126.2, 126.1, 121.7,
121.4, 118.5, 109.3, 60.7, 43.3, 34.6, 31.2;
IR (neat): 3051, 1596, 1472, 1276, 737 cm-1;
HRMS (ESI) m/z calcd for C19H21N2S [(M+H)+] 309.1425, found 309.1418.
4-(2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazol-3-yl)phenyl acetate (3-15): This
compound was prepared according to the General Procedure A using 4-acetoxystyrene
(122 mg, 0.75 mmol), 2-mercaptobenzimidazole (75 mg, 0.5 mmol). After purification by
column chromatography on SiO2 (30-70% EtOAc in hexanes), the title compound was
isolated as a white gummy solid (114 mg, 73% yield, >98:2 rr).
1H NMR (600 MHz, CDCl3): = 7.60 (d, J = 8.1 Hz, 1 H), 7.26 (d, J = 8.3 Hz, 2 H), 7.16-
7.08 (m, 3 H), 6.95 (t, J = 7.6 Hz, 1 H), 6.60 (d, J = 8.1 Hz, 1 H), 5.50 (t, J = 7.3 Hz, 1 H),

71
4.14 (dd, J = 7.3, 11.2 Hz, 1 H), 3.72 (dd, J = 7.3, 11.2 Hz, 1 H), 2.29 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 169.1, 158.4, 151.0, 149.4, 134.3, 133.3, 127.6, 122.4,
121.9, 121.6, 118.6, 109.2, 60.3, 43.2, 21.0;
IR (neat): 3053, 1757, 1608, 1472, 1187, 738 cm-1;
HRMS (ESI) m/z calcd for C17H15N2O2S [(M+H)+] 311.0854, found 311.0859.
3-(4-methoxyphenyl)-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-16): This
compound was prepared according to the General Procedure A using 4-methoxystyrene
(101 mg, 0.75 mmol), 2-mercaptobenzimidazole (75 mg, 0.5 mmol). After purification by
column chromatography on SiO2 (30-70% EtOAc in hexanes), the title compound was
isolated as a white solid (103 mg, 73% yield, >98:2 rr).
1H NMR (600 MHz, CDCl3): = 7.60 (d, J = 8.1 Hz, 1 H), 7.21 (d, J = 8.5 Hz, 2 H), 7.13
(t, J = 7.7 Hz, 1 H), 6.98-6.87 (m, 3 H), 6.57 (d, J = 8.1 Hz, 1 H), 5.46 (t, J = 7.7 Hz, 1 H),
4.09 (dd, J = 7.7, 11.2 Hz, 1 H), 3.81 (s, 3 H), 3.76 (dd, J = 7.7, 11.2 Hz, 1 H);
13C NMR (150 MHz, CDCl3): = 160.0, 158.3, 149.3, 133.4, 128.5, 127.8, 121.7,
121.4, 118.4, 114.4, 109.2, 60.7, 55.2, 43.2;
IR (neat): 3012, 1607, 1474, 1226, 768 cm-1;
HRMS (ESI) m/z calcd for C16H15N2OS [(M+H)+] 283.0905, found 283.0911.

72
3-(2-methoxyphenyl)-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-17): This
compound was prepared according to the General Procedure A using 2-methoxystyrene
(101 mg, 0.75 mmol), 2-mercaptobenzimidazole (75 mg, 0.5 mmol). After purification by
column chromatography on SiO2 (30-70% EtOAc in hexanes), the title compound was
isolated as a white gummy solid (121mg, 86% yield, >98:2 rr).
1H NMR (600 MHz, CDCl3): = 7.64 (d, J = 8.1 Hz, 1 H), 7.35-7.29 (m, 1 H), 7.15 (t, J
= 7.7 Hz, 1 H), 7.04-6.93 (m, 2 H), 6.86-6.77 (m, 2 H), 6.68 (d, J = 6.8 Hz, 1 H), 5.99 (dd,
J = 5.6, 7.6 Hz, 1 H), 4.28 (dd, J = 7.6, 11.0 Hz, 1 H), 3.90 (s, 3 H), 3.74- 3.67 (m, 1 H);
13C NMR (150 MHz, CDCl3): = 159.1, 156.2, 149.4, 133.4, 129.8, 126.5, 124.7, 121.7,
121.5, 120.7, 118.5, 110.6, 109.3, 55.4, 54.9, 42.1;
IR (neat): 3053, 1601, 1449, 1242, 737 cm-1;
HRMS (ESI) m/z calcd for C16H15N2OS [(M+H)+] 283.0905, found 283.0899.
3-methyl-3-phenyl-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-18): This
compound was prepared according to the General Procedure A using α-methylstyrene (89
mg, 0.75 mmol), 2-mercaptobenzimidazole (75 mg, 0.5 mmol). After purification by
column chromatography on SiO2 (30-50% EtOAc in hexanes), the title compound was

73
isolated as a white gummy solid (90 mg, 68% yield, >98:2 rr).
1H NMR (600 MHz, CDCl3): =7.64 (d, J = 8.1 Hz, 1 H), 7.42-7.35 (m, 3 H), 7.28 (d, J
= 8.1 Hz, 2 H), 7.16 (t, J = 7.7 Hz, 1 H), 6.98 (t, J = 7.7 Hz, 1 H), 6.69 (d, J = 8.1 Hz, 1 H),
3.98 (d, J = 11.2 Hz, 1 H), 3.84 (d, J = 11.2 Hz, 1 H), 2.05 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 158.0, 149.7, 140.9, 132.8, 129.1, 128.5, 125.6, 121.9,
121.5, 118.8, 109.3, 66.4, 50.3, 23.1;
IR (neat): 3056, 1610, 1443, 1219, 738 cm-1;
HRMS (ESI) m/z calcd for C16H15N2S [(M+H)+] 267.0956, found 267.0948.
3-isopropyl-3-phenyl-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-19): This
compound was prepared according to the General Procedure A using α-isopropylstyrene
(110 mg, 0.75 mmol), 2-mercaptobenzimidazole (75 mg, 0.5 mmol). After purification by
column chromatography on SiO2 (30-50% EtOAc in hexanes), the title compound was
isolated as a white solid (93 mg, 63% yield, >98:2 rr).
1H NMR (600 MHz, CDCl3): = 7.60 (d, J = 7.8 Hz, 1 H), 7.44 (d, J = 7.8 Hz, 2 H), 7.36-
7.25 (m, 3 H), 7.18-7.11 (m, 2 H), 7.06 (t, J = 7.7 Hz, 1 H), 4.10 (d, J = 11.7 Hz, 1 H), 3.99
(d, J = 11.7 Hz, 1 H), 3.24-3.15 (m, 1 H), 1.17-1.03 (m, 6 H);
13C NMR (150 MHz, CDCl3): = 158.3, 149.4, 140.9, 133.8, 128.8, 128.1, 126.0,
121.7, 121.3, 118.7, 110.5, 73.3, 43.1, 35.8, 19.1, 17.7;
IR (neat): 2959, 1607, 1473, 1221, 738 cm-1;

74
HRMS (ESI) m/z calcd for C18H19N2S [(M+H)+] 295.1269, found 295.1256.
3,3-diphenyl-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-20): This compound was
prepared according to the General Procedure A using α-phenylstyrene (135 mg, 0.75
mmol), 2-mercaptobenzimidazole (75 mg, 0.5 mmol). After purification by column
chromatography on SiO2 (30-50% EtOAc in hexanes), the title compound was isolated as
a white solid (110 mg, 67% yield, >98:2 rr).
1H NMR (600 MHz, CDCl3): = 7.65 (d, J = 8.1 Hz, 1 H), 7.41-7.32 (m, 6 H), 7.27-7.20
(m, 4 H), 7.14 (s, 1 H), 6.90 (s, 1 H), 6.33 (d, J = 8.1 Hz, 1 H), 4.43 (s, 2 H);
13C NMR (150 MHz, CDCl3): = 157.5, 149.0, 138.9, 133.9, 128.7, 128.6, 127.8, 121.8,
121.7, 118.9, 109.8, 73.1, 51.5;
IR (neat): 3028, 1611, 1475, 1227, 733 cm-1;
HRMS (ESI) m/z calcd for C21H17N2S [(M+H)+] 329.1112, found 329.1107.
3-cyclopropyl-3-phenyl-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-21): This
compound was prepared according to the General Procedure A using α-cyclopropylstyrene
(108 mg, 0.75 mmol), 2-mercaptobenzimidazole (75 mg, 0.5 mmol). After purification by

75
column chromatography on SiO2 (30-50% EtOAc in hexanes), the title compound was
isolated as a white solid (107 mg, 73% yield, >98:2 rr).
1H NMR (600 MHz, CDCl3): = 7.62 (d, J = 8.1 Hz, 1 H), 7.41-7.33 (m, 5 H), 7.14 (t, J
= 7.7 Hz, 1 H), 6.96 (t, J = 7.7 Hz, 1 H), 6.76 (d, J = 8.1 Hz, 1 H), 3.89 (d, J = 11.2 Hz, 1
H), 3.75 (d, J = 11.2 Hz, 1 H), 1.87-1.80 (m, 1 H), 0.96-0.88 (m, 1 H), 0.68-0.61 (m, 1 H),
0.57 (dd, J = 5.7, 9.6 Hz, 1 H), 0.45 (dd, J = 5.5, 9.6 Hz, 1 H);
13C NMR (150 MHz, CDCl3): = 158.1, 149.2, 141.2, 133.4, 128.8, 128.4, 126.4,
121.7, 121.5, 118.8, 109.8, 69.3, 47.0, 18.2, 3.6, 1.2;
IR (neat): 3016, 1610, 1473, 1216, 744 cm-1;
HRMS (ESI) m/z calcd for C18H17N2S [(M+H)+] 293.1121, found 293.1112.
3-(4-(trifluoromethyl)phenyl)-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-22):
This compound was prepared according to the General Procedure A using 4-
(trifluoromethyl)styrene (129 mg, 0.75 mmol), 2-mercaptobenzimidazole (75 mg, 0.5
mmol). After purification by column chromatography on SiO2 (30-50% EtOAc in
hexanes), the title compound was isolated as a white solid (79 mg, 49% yield, >98:2 rr).
1H NMR (600 MHz, CDCl3): = 7.70-7.62 (m, 3 H), 7.38 (d, J = 8.1 Hz, 2 H), 7.18 (t, J
= 7.8 Hz, 1 H), 7.01 (t, J = 7.7 Hz, 1 H), 6.63 (d, J = 8.1 Hz, 1 H), 5.64 (t, J = 7.1 Hz, 1 H),
4.28 (dd, J = 7.6, 11.2 Hz, 1 H), 3.75 (dd, J = 6.7, 11.2 Hz, 1 H);
13C NMR (150 MHz, CDCl3): = 158.5, 149.6, 141.0, 133.2, 131.6, 131.3, 126.9,

76
126.3, 122.3, 122.0, 118.9, 109.1, 77.2, 76.8, 60.2, 43.2;
IR (neat): 2924, 1611, 1470, 1320, 741 cm-1;
HRMS (ESI) m/z calcd for C16H12F3N2S [(M+H)+] 321.0670, found 321.0674.
methyl 4-(2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazol-3-yl)benzoate (3-23): This
compound was prepared according to the General Procedure A using methyl 4-
vinylbenzoate (122 mg, 0.75 mmol), 2-mercaptobenzimidazole (75 mg, 0.5 mmol). After
purification by column chromatography on SiO2 (30-70% EtOAc in hexanes), the title
compound was isolated as a white solid (65 mg, 42% yield, >98:2 rr).
1H NMR (600 MHz, CDCl3): = 8.06 (d, J = 8.1 Hz, 2 H), 7.62 (d, J = 8.1 Hz, 1 H), 7.35
(d, J = 8.1 Hz, 2 H), 7.15 (t, J = 7.6 Hz, 1 H), 6.97 (t, J = 7.6 Hz, 1 H), 6.58 (d, J = 8.1 Hz,
1 H), 5.60 (t, J = 7.3 Hz, 1 H), 4.23 (dd, J = 7.3, 11.2 Hz, 1 H), 3.92 (s, 3 H), 3.77 (dd, J =
7.3, 11.2 Hz, 1 H);
13C NMR (150 MHz, CDCl3): = 166.2, 158.5, 149.5, 141.8, 133.3, 131.0, 130.5,
126.6, 122.1, 121.8, 118.8, 109.1, 60.6, 52.3, 43.1;
IR (neat): 2947, 1725, 1477, 1276, 737 cm-1;
HRMS (ESI) m/z calcd for C17H15N2O2S [(M+H)+] 311.0844, found 311.0854.

77
6a,11b-dihydro-7H-benzo[4,5]imidazo[2,1-b]indeno[1,2-d]thiazole (3-24): This
compound was prepared according to the General Procedure A using indene (87 mg, 0.75
mmol), 2-mercaptobenzimidazole (75 mg, 0.5 mmol). After purification by column
chromatography on SiO2 (30-50% EtOAc in hexanes), the title compound was isolated as
a yellow solid (108 mg, 82% yield, >98:2 rr, >98:2 dr).
1H NMR (600 MHz, CDCl3): =7.60 (d, J = 8.1 Hz, 1 H), 7.51 (d, J = 7.8 Hz, 2 H), 7.34-
7.27 (m, 2 H), 7.27-7.17 (m, 3 H), 5.95 (d, J = 7.1 Hz, 1 H), 5.21 (dt, J = 2.9, 7.1 Hz, 1 H),
3.58 (dd, J = 7.1, 17.0 Hz, 1 H), 3.35 (dd, J = 2.9, 17.0 Hz, 1 H);
13C NMR (150 MHz, CDCl3): = 157.7, 149.5, 140.7, 137.9, 133.7, 129.8, 128.0, 125.7,
125.7, 122.2, 122.1, 119.1, 109.3, 65.8, 55.5, 39.2;
IR (neat): 3025, 1613, 1467, 1230, 727 cm-1;
HRMS (ESI) m/z calcd for C16H13N2S [(M+H)+] 265.0799, found 265.0793.
2-methyl-3-phenyl-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-25): This
compound was prepared according to the General Procedure A using trans-β-
methylstyrene (89 mg, 0.75 mmol), 2-mercaptobenzimidazole (75 mg, 0.5 mmol). After
purification by column chromatography on SiO2 (30-50% EtOAc in hexanes), the title

78
compound was isolated as a white solid (107 mg, 80% yield, >98:2 rr, >98:2 dr).
1H NMR (600 MHz, CDCl3): = 7.61 (d, J = 8.1 Hz, 1 H), 7.43 (d, J = 4.9 Hz, 3 H), 7.34-
7.28 (m, 2 H), 7.12 (t, J = 7.7 Hz, 1 H), 6.91 (t, J = 7.7 Hz, 1 H), 6.43 (d, J = 8.1 Hz, 1 H),
4.98 (d, J = 8.5 Hz, 1 H), 4.37-4.28 (m, 1 H), 1.61 (d, J = 6.8 Hz, 3 H);
13C NMR (150 MHz, CDCl3): = 158.0, 149.2, 135.9, 133.8, 129.7, 129.5, 127.5, 122.1,
121.7, 118.9, 109.6, 69.3, 56.3, 18.6;
IR (neat): 2914, 1612, 1466, 1249, 735 cm-1;
HRMS (ESI) m/z calcd for C16H15N2S [(M+H)+] 267.0956, found 267.0945.
2,3-dipropyl-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-26): This compound was
prepared according to the General Procedure B using trans-4-octene (84 mg, 0.75 mmol),
2-mercaptobenzimidazole (75 mg, 0.5 mmol). After purification by column
chromatography on SiO2 (30-50% EtOAc in hexanes), the title compound was isolated as
a colorless oil (94 mg, 72% yield, >98:2 dr).
1H NMR (600 MHz, CDCl3): =7.59 (d, J = 8.3 Hz, 1 H), 7.20 (d, J = 7.8 Hz, 1 H), 4.33-4.27 (m,
1 H), 4.00-3.92 (m, 1 H), 1.88-1.76 (m, 4 H), 1.54-1.36 (m, 4 H), 0.99-0.89 (m, 6 H);
13C NMR (150 MHz, CDCl3): = 157.6, 149.5, 133.6, 121.8, 121.7, 118.9, 109.0, 62.4,
58.2, 39.5, 35.1, 20.5, 19.0, 14.2, 13.8;
IR (neat): 2957, 1613, 1447, 1252, 737 cm-1;
HRMS (ESI) m/z calcd for C15H21N2S [(M+H)+] 261.1425, found 261.1417.

79
2,3,3a,10a-tetrahydrobenzo[4,5]imidazo[2,1-b]furo[2,3-d]thiazole (3-27): This
compound was prepared according to the General Procedure A using 2,3-dihydrofuran (53
mg, 0.75 mmol), 2-mercaptobenzimidazole (75 mg, 0.5 mmol). After purification by
column chromatography on SiO2 (30-70% EtOAc in hexanes), the title compound was
isolated as a white solid (74 mg, 68% yield, >98:2 rr, >98:2 dr).
1H NMR (600 MHz, CDCl3): = 7.58 (d, J = 8.1 Hz, 1 H), 7.37 (d, J = 7.3 Hz, 1 H), 7.24-
7.14 (m, 2 H), 6.35 (d, J = 6.3 Hz, 1 H), 4.94 (t, J = 7.0 Hz, 1 H), 4.15 (t, J = 8.2 Hz, 1 H),
3.89 (ddd, J = 4.8, 9.2, 11.2 Hz, 1 H), 2.49-2.37 (m, 1 H), 2.27 (dd, J = 4.8, 13.4 Hz, 1 H);
13C NMR (150 MHz, CDCl3): = 157.1, 150.1, 133.1, 122.7, 122.4, 118.8, 109.4, 88.7,
67.1, 54.3, 35.6;
IR (neat): 2965, 1613,1468, 1251, 753 cm-1;
HRMS (ESI) m/z calcd for C11H11N2OS [(M+H)+] 219.0592, found 219.0583.
2H-spiro[benzo[4,5]imidazo[2,1-b]thiazole-3,1'-cyclopentane] (3-28): This compound
was prepared according to the General Procedure A using methylenecyclopentane (62mg,
0.75 mmol), 2-mercaptobenzimidazole (75 mg, 0.5 mmol). After purification by column
chromatography on SiO2 (30-50% EtOAc in hexanes), the title compound was isolated as
a white solid (54 mg, 47% yield, >98:2 rr).
1H NMR (600 MHz, CDCl3): = 7.60 (d, J = 8.1 Hz, 1 H), 7.28 (d, J = 7.6 Hz, 1 H), 7.20-

80
7.10 (m, 2 H), 3.70 (s, 2 H), 2.40-2.30 (m, 2 H), 2.10-1.97 (m, 4 H), 1.87-1.76 (m, 2 H);
13C NMR (150 MHz, CDCl3): = 158.1, 150.1, 132.5, 121.9, 121.6, 119.2, 108.8, 71.4,
48.1, 36.1, 25.1;
IR (neat): 3047, 1610, 1475, 1229, 743 cm-1;
HRMS (ESI) m/z calcd for C13H15N2S [(M+H)+] 231.0956, found 231.0950.
3-hexyl-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-29): This compound was
prepared according to the General Procedure B using 1-octene (84 mg, 0.75 mmol), 2-
mercaptobenzimidazole (75 mg, 0.5 mmol). After purification by column chromatography
on SiO2 (30-50% EtOAc in hexanes), the inseparable mixture of both regioisomers was
isolated as a colorless oil (103 mg, 79% yield, 75:25 rr).
1H NMR (600 MHz, CDCl3): = 7.58 (d, J = 7.8 Hz, 1 Hmajor + 1 Hminor), 7.23-7.08 (m, 3
Hmajor + 3 Hminor), 4.58 (tt, J = 3.5, 7.6 Hz, 1 Hmajor), 4.41 (d, J = 7.3 Hz, 1 Hminor), 4.31 (dd,
J = 7.3, 9.8 Hz, 1 Hminor), 3.99 (dd, J = 7.6, 11.1 Hz, 1 Hmajor), 3.86 (dd, J = 7.3, 9.8 Hz, 1
Hminor), 3.53 (dd, J = 3.5, 11.1 Hz, 1 Hmajor), 1.99 (ddd, J = 4.5, 9.5, 13.8 Hz, 1 Hmajor), 1.95-
1.78 (m, 1 Hmajor + 2 Hminor), 1.44-1.39 (m, 1 Hmajor), 1.39-1.23 (m, 7 Hmajor + 8 Hminor),
0.92-0.79 (m, 3 Hmajor + 3 Hminor);
13C NMR (150 MHz, CDCl3): = 158.2, 149.7, 133.6, 121.9, 121.9, 121.8, 118.9, 118.8,
109.1, 108.9, 57.3, 53.9, 49.7, 39.8, 35.5, 32.7, 31.8, 31.8, 29.3, 29.1, 28.1, 25.6, 22.8, 14.3;
IR (neat): 2925, 1614, 1448, 1248, 736 cm-1;
HRMS (ESI) m/z calcd for C15H21N2S [(M+H)+] 261.1425, found 261.1419.

81
2-(2-(2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazol-3-yl)ethyl)isoindoline-1,3-dione (3-
30): This compound was prepared according to the General Procedure B using N-(3-buten-
1-yl)phthalimide (151 mg, 0.75 mmol), 2-mercaptobenzimidazole (75 mg, 0.5 mmol).
After purification by column chromatography on SiO2 (30-70% EtOAc in hexanes), the
inseparable mixture of both regioisomers was isolated as a white solid (149 mg, 85% yield,
76:24 rr).
1H NMR (600 MHz, CDCl3): = 7.87-7.78 (m, 2 Hmajor + 2 Hminor), 7.75-7.68 (m, 2 Hmajor
+ 2 Hminor), 7.57-7.54 (m, 1 Hminor), 7.51 (d, J = 7.6 Hz, 1 Hmajor), 7.19 (d, J = 7.6 Hz, 1
Hmajor), 7.17-7.04 (m, 2 Hmajor + 3 Hminor ), 4.73-4.58 (m, 1 Hmajor), 4.42-4.31(m, 2 Hminor),
4.13 (dd, J = 7.4, 11.4 Hz, 1 Hmajor), 4.02 (dd, J = 4.9, 9.8 Hz, 1 Hminor), 3.92-3.78 (m, 2
Hmajor + 2 Hminor), 3.74 (dd, J = 4.2, 11.4 Hz, 1 Hmajor), 2.49 - 2.39 (m, 1 Hmajor), 2.25 (d, J =
6.8 Hz, 2 Hminor), 2.31-2.15 (m, 1 Hmajor);
13C NMR (150 MHz, CDCl3): = 168.3, 158.1, 149.7, 134.5, 133.2, 132.0, 131.9,
123.7, 122.2, 122.1, 122.0, 119.0, 118.9, 109.0, 109.0, 55.0, 50.5, 49.4, 39.4, 35.8, 34.9,
34.4, 31.1;
IR (neat): 2937, 1768, 1701, 1248, 715 cm-1;
HRMS (ESI) m/z calcd for C19H16N3O2S [(M+H)+] 350.0963, found 350.0951.

82
3-cyclohexyl-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-31): This compound was
prepared according to the General Procedure B using vinylcyclohexane (83 mg, 0.75
mmol), 2-mercaptobenzimidazole (75 mg, 0.5 mmol). After purification by column
chromatography on SiO2 (30-50% EtOAc in hexanes), the inseparable mixture of both
regioisomers were isolated as a white solid (80 mg, 62% yield, 50:50 rr).
1H NMR (600 MHz, CDCl3): = 7.59 (d, J = 7.6 Hz, 1 Hmajor + 1 Hminor), 7.23 (d, J = 7.6
Hz, 1 Hmajor), 7.19-7.10 (m, 2 Hmajor + 3 Hminor), 4.53-4.47 (m, 1 Hmajor), 4.36-4.27 (m, 2
Hminor), 3.97-3.90 (m, 1 Hmajor + 1 Hminor), 3.68 (dd, J = 3.7, 11.2 Hz, 1 Hmajor), 2.25-2.15
(m, 1 Hmajor), 1.84-1.64 (m, 4 Hmajor + 5 Hminor), 1.49 (d, J = 11.7 Hz, 1 Hmajor), 1.34-1.10
(m, 5 Hmajor + 6 Hminor);
13C NMR (150 MHz, CDCl3): = 158.7, 158.5, 149.5, 148.9, 134.0, 133.8, 122.2, 122.0,
121.9, 121.8, 118.9, 118.8, 109.5, 108.9, 61.7, 60.0, 47.8, 42.8, 40.9, 36.7, 31.4, 31.1, 29.9,
27.3, 26.3, 26.3, 26.2, 26.0, 25.9;
IR (neat): 2922, 1614, 1471, 1245, 738 cm-1;
HRMS (ESI) m/z calcd for C15H19N2S [(M+H)+] 259.1276, found 259.1269.
2-(tert-butyl)-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-32): This compound was
prepared according to the General Procedure B using 3,3-dimethylbut-1-ene (63 mg, 0.75
mmol), 2-mercaptobenzimidazole (75 mg, 0.5 mmol). After purification by column
chromatography on SiO2 (30-50% EtOAc in hexanes), the title compound was isolated as
a white solid (94 mg, 81% yield, >98:2 rr).

83
1H NMR (600 MHz, CDCl3): = 7.58 (d, J = 7.6 Hz, 1 H), 7.20-7.11 (m, 3 H), 4.40 (t, J
= 8.1 Hz, 1 H), 4.21 (dd, J = 8.1, 10.3 Hz, 1 H), 4.02 (dd, J = 8.1, 10.3 Hz, 1 H), 1.07 (s, 9
H);
13C NMR (150 MHz, CDCl3): = 158.5, 149.0, 133.9, 122.0, 121.8, 118.7, 108.8, 65.0,
45.3, 34.5, 27.4;
IR (neat): 2949, 1614, 1445, 1250, 738 cm-1;
HRMS (ESI) m/z calcd for C13H17N2S [(M+H)+] 233.1112, found 233.1118.
7-methyl-3-phenyl-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-33): This
compound was prepared according to the General Procedure A using styrene (78 mg, 0.75
mmol), 2-mercapto-5-methylbenzimidazole (82 mg, 0.5 mmol). After purification by
column chromatography on SiO2 (30-50% EtOAc in hexanes), the inseparable mixture of
both nitrogen addition products was isolated as a yellow solid (117 mg, 88% yield, >98:2
rr, 54:46 N1:N2).
1H NMR (600 MHz, CDCl3): = 7.49 (d, J = 8.1 Hz, 1 Hminor), 7.42-7.36 (m, 4 Hmajor + 3
Hminor), 7.29-7.23 (m, 2 Hmajor + 2 Hminor), 6.96 (d, J = 8.1 Hz, 1 Hminor), 6.77 (d, J = 8.3 Hz,
1 Hmajor), 6.46 (d, J = 8.3 Hz, 1 Hmajor), 6.40 (s, 1 Hminor), 5.49 (t, J = 7.4 Hz, 1 Hmajor + 1
Hminor), 4.15 (ddd, J = 7.4, 11.4, 13.2 Hz, 1 Hmajor + 1 Hminor), 3.79-3.70 (m, 1 Hmajor + 1
Hminor), 2.39 (s, 3 Hmajor), 2.24 (s, 3 Hminor);
13C NMR (150 MHz, CDCl3): = 158.4, 157.8, 149.8, 147.5, 137.0, 136.9, 133.6, 131.5,
131.5, 129.2, 129.2, 129.1, 129.0, 126.5, 126.4, 123.2, 122.7, 118.6, 118.1, 109.3, 108.7,

84
61.0, 60.7, 43.3, 43.3, 21.5, 21.4;
IR (neat): 2917, 1615, 1472, 1112, 746 cm-1;
HRMS (ESI) m/z calcd for C16H15N2S [(M+H)+] 267.0956, found 267.0946.
7-methoxy-3-phenyl-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-34): This
compound was prepared according to the General Procedure A using styrene (78 mg, 0.75
mmol), 2-mercapto-5-methoxybenzimidazole (90 mg, 0.5 mmol). After purification by
column chromatography on SiO2 (30-70% EtOAc in hexanes), the inseparable mixture of
both nitrogen addition products was isolated as a white solid (100 mg, 71% yield, >98:2 rr,
60:40 N1:N2).
1H NMR (600 MHz, CDCl3): = 7.49 (d, J = 8.8 Hz, 1 Hminor), 7.44 - 7.36 (m, 3 Hmajor +
3 Hminor), 7.31-7.25 (m, 2 Hmajor + 2 Hminor), 7.14 (d, J = 2.4 Hz, 1 Hmajor), 6.76 (dd, J = 2.4,
8.8 Hz, 1 Hminor), 6.59 (dd, J = 2.4, 8.8 Hz, 1 Hmajor), 6.45 (d, J = 8.8 Hz, 1 Hmajor), 6.08 (d,
J = 2.4 Hz, 1 Hminor), 5.53-5.45 (m, 1 Hmajor + 1 Hminor), 4.20-4.09 (m, 1 Hmajor + 1 Hminor),
3.84-3.73 (m, 4 Hmajor + 1 Hminor), 3.61 (s, 3 Hminor);
13C NMR (150 MHz, CDCl3): = 158.7, 157.1, 155.8, 155.4, 150.4, 143.9, 136.9, 136.7,
134.0, 129.2, 129.2, 128.1, 126.6, 126.6, 118.9, 110.5, 109.6, 109.4, 102.0, 94.3, 77.2, 76.8,
61.4, 60.9, 55.7, 55.6, 43.3, 43.3;
IR (neat): 2993, 1619, 1437, 1225, 695 cm-1;
HRMS (ESI) m/z calcd for C16H15N2OS [(M+H)+] 283.0905, found 283.0894.

85
7-nitro-3-phenyl-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-35): This compound
was prepared according to the General Procedure A using styrene (78 mg, 0.75 mmol), 2-
mercapto-5-nitrobenzimidazole (98 mg, 0.5 mmol). After purification by column
chromatography on SiO2 (30-70% EtOAc in hexanes), the inseparable mixture of both
nitrogen addition products was isolated as a yellow gummy solid (95 mg, 64% yield, >98:2
rr, 55:45 N1:N2).
1H NMR (600 MHz, CDCl3): = 8.45-8.41 (m, 1 Hminor), 8.05 (dd, J = 2.0, 9.0 Hz, 1
Hmajor), 7.87 (dd, J = 1.7, 8.8 Hz, 1 Hminor), 7.60 (d, J = 9.0 Hz, 1 Hmajor), 7.50-7.47 (m, 1
Hmajor), 7.47-7.39 (m, 3 Hmajor + 3 Hminor), 7.33-7.26 (m, 2 Hmajor + 2 Hminor), 6.57 (d, J = 9.0
Hz, 1 Hminor), 5.72-5.62 (m, 1 Hmajor + 1 Hminor), 4.34-4.22 (m, 1 Hmajor + 1 Hminor), 3.88 (td,
J = 6.7, 11.4 Hz, 1 Hmajor + 1 Hminor);
13C NMR (150 MHz, CDCl3): = 164.4, 162.5, 154.0, 148.8, 143.2, 142.3, 137.4, 135.7,
132.5, 129.8, 129.7, 129.6, 129.6, 129.4, 126.5, 126.3, 118.2, 118.2, 117.7, 114.7, 108.7,
105.5, 61.3, 61.3, 43.4, 43.4;
IR (neat): 2922, 1616, 1438, 1315, 1054, 696 cm-1;
HRMS (ESI) m/z calcd for C15H12N3O2S [(M+H)+] 298.0653, found 298.0650.
7-chloro-3-phenyl-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-36): This
compound was prepared according to the General Procedure A using styrene (78 mg, 0.75

86
mmol), 5-chloro-2-mercaptobenzimidazole (92 mg, 0.5 mmol). After purification by
column chromatography on SiO2 (30-50% EtOAc in hexanes), the inseparable mixture of
both nitrogen addition products was isolated as a white solid (109 mg, 76% yield, >98:2 rr,
55:45 N1:N2).
1H NMR (600 MHz, CDCl3): = 7.59-7.56 (m, 1 Hminor), 7.49 (d, J = 8.5 Hz, 1 Hmajor),
7.45-7.37 (m, 3 Hmajor + 3 Hminor), 7.27 (d, J = 5.1 Hz, 2 Hmajor + 2 Hminor), 7.09 (dd, J = 1.7,
8.5 Hz, 1 Hmajor), 6.90 (dd, J = 1.7, 8.3 Hz, 1 Hminor), 6.57-6.53 (m, 1 Hmajor), 6.45 (d, J =
8.3 Hz, 1 Hminor), 5.51 (q, J = 7.6 Hz, 1 Hmajor + 1 Hminor), 4.17 (ddd, J = 4.5, 7.3, 11.4 Hz, 1
Hmajor + 1 Hminor), 3.83-3.75 (m, 1 Hmajor + 1 Hminor);
13C NMR (150 MHz, CDCl3): = 160.1, 159.5, 150.2, 148.1, 136.4, 136.3, 133.9, 132.0,
129.5, 129.4, 129.4, 129.4, 127.6, 127.2, 126.5, 126.5, 122.5, 121.8, 119.3, 118.5, 109.8,
109.4, 61.3, 61.1, 43.3, 43.3;
IR (neat): 2919, 1618, 1437, 1027, 696 cm-1;
HRMS (ESI) m/z calcd for C15H12ClN2S [(M+H)+] 287.0410, found 287.0396.
7-(difluoromethoxy)-3-phenyl-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-37):
This compound was prepared according to the General Procedure A using styrene (78 mg,
0.75 mmol), 5-(difluoromethoxy)-2-mercaptobenzimidazole (108 mg, 0.5 mmol). After
purification by column chromatography on SiO2 (30-70% EtOAc in hexanes), the
inseparable mixture of both nitrogen addition products was isolated as a colorless oil (107
mg, 67% yield, >98:2 rr, 53:47 N1:N2).

87
1H NMR (600 MHz, CDCl3): = 7.53 (d, J = 8.8 Hz, 1 Hmajor), 7.43-7.39 (m, 3 Hmajor + 3
Hminor), 7.38-7.36 (m, 1 Hminor), 7.31-7.23 (m, 2 Hmajor + 2 Hminor), 6.92 (dd, J = 2.1, 8.7 Hz,
1 Hmajor), 6.75 (dd, J = 1.8, 8.7 Hz, 1 Hminor), 6.49 (d, J = 8.8 Hz, 1 Hminor), 6.44 (t, J = 72
Hz, 1 Hminor), 6.33 (d, J = 2.0 Hz, 1 Hmajor), 6.32-6.30 (m, 1 Hmajor), 5.52 (dt, J = 2.9, 7.6
Hz, 1 Hmajor + 1 Hminor), 4.17 (ddd, J = 3.5, 7.5, 11.2 Hz, 1 Hmajor + 1 Hminor), 3.83-3.76 (m,
1 Hmajor + 1 Hminor);
13C NMR (150 MHz, CDCl3): = 160.3, 159.5, 149.9, 147.1, 146.5, 145.9, 136.4, 136.2,
133.4, 131.2, 129.4, 129.4, 129.3, 126.5, 126.5, 119.0, 118.1, 117.9, 116.4, 116.2, 114.6,
114.4, 114.4, 114.4, 110.1, 109.4, 101.5, 61.3, 61.1, 43.3, 43.3;
IR (neat): 2988, 1621, 1470, 1111, 697 cm-1;
HRMS (ESI) m/z calcd for C16H13F2N2OS [(M+H)+] 319.0716, found 319.0717.
6-chloro-7-fluoro-3-phenyl-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-38): This
compound was prepared according to the General Procedure A using styrene (78 mg, 0.75
mmol), 6-chloro-5-fluoro-2-mercaptobenzimidazole (101 mg, 0.5 mmol). After
purification by column chromatography on SiO2 (30-50% EtOAc in hexanes), the
inseparable mixture of both nitrogen addition products was isolated as a white solid (101
mg, 66% yield, >98:2 rr, 51:49 N1:N2).
1H NMR (600 MHz, CDCl3): = 7.59 (d, J = 6.6 Hz, 1 Hminor), 7.47-7.40 (m, 3 Hmajor + 3
Hminor), 7.36 (d, J = 9.5 Hz, 1 Hmajor), 7.31-7.24 (m, 2 Hmajor + 2 Hminor), 6.55 (d, J = 6.6 Hz,
1 Hmajor), 6.32 (d, J = 8.8 Hz, 1 Hminor), 5.55-5.48 (m, 1 Hmajor + 1 Hminor), 4.19 (dt, J = 7.6,

88
10.5 Hz, 1 Hmajor + 1 Hminor), 3.86-3.78 (m, 1 Hmajor + 1 Hminor);
13C NMR (150 MHz, CDCl3): = 160.8, 160.1, 155.1, 154.4, 153.5, 152.8, 148.4,148.3,
145.7, 136.0, 135.9, 131.9, 131.8, 130.0, 129.7, 129.6, 129.5, 126.6, 126.5, 119.4, 115.4,
115.3, 114.9, 114.8, 109.9, 106.0, 105.8, 97.4, 97.2, 61.5, 61.4, 43.4, 43.3;
IR (neat): 2923, 1618, 1515, 1444, 1335, 696 cm-1;
HRMS (ESI) m/z calcd for C15H11ClFN2S [(M+H)+] 305.0320, found 305.0316.
6,7-dichloro-3-phenyl-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-39): This
compound was prepared according to the General Procedure A using styrene (78 mg, 0.75
mmol), 5,6-dichloro-2-mercaptobenzimidazole (110 mg, 0.5 mmol). After purification by
column chromatography on SiO2 (30-50% EtOAc in hexanes), the title compound was
isolated as a yellow solid (108 mg, 67% yield, >98:2 rr).
1H NMR (600 MHz, CDCl3): = 7.66 (s, 1 H), 7.43 (d, J = 4.9 Hz, 3 H), 7.27 (d, J = 4.9
Hz, 2 H), 6.63 (s, 1 H), 5.52 (t, J = 7.6 Hz, 1 H), 4.20 (dd, J = 7.6, 11.2 Hz, 1 H), 3.81 (dd,
J = 7.6, 11.2 Hz, 1 H);
13C NMR (150 MHz, CDCl3): = 161.0, 148.7, 135.9, 132.5, 129.6, 129.5, 126.4, 126.1,
125.4, 119.7, 110.4, 61.3, 43.4;
IR (neat): 3058, 1614, 1445, 1252, 740, 693 cm-1;
HRMS (ESI) m/z calcd for C15H11Cl2N2S [(M+H)+] 321.0013, found 321.0020.

89
3-phenyl-2,3-dihydroimidazo[2,1-b]thiazole (3-40): This compound was prepared
according to the General Procedure B using styrene (104 mg, 1.0 mmol), 2-
mercaptoimidazole (50 mg, 0.5 mmol), Selectfluor (266 mg, 0.75 mmol). After purification
by column chromatography on SiO2 (30-50% EtOAc in hexanes), the title compound was
isolated as a yellow oil (37 mg, 37% yield, >98:2 rr).
1H NMR (600 MHz, CDCl3): = 7.44-7.37 (m, 3 H), 7.25-7.20 (m, 2 H), 7.03 (s, 1 H),
6.72 (s, 1 H), 5.34 (t, J = 7.6 Hz, 1 H), 4.07 (dd, J = 7.2, 11.2 Hz, 1 H), 3.70 (dd, J = 8.1,
11.2 Hz, 1 H);
13C NMR (150 MHz, CDCl3): = 149.6, 137.7, 134.3, 129.2, 129.1, 126.5, 116.5, 62.1,
43.7;
IR (neat): 2935, 1465, 1234, 696 cm-1;
HRMS (ESI) m/z calcd for C11H11N2S [(M+H)+] 203.0648, found 203.0643.
3,5,6-triphenyl-2,3-dihydroimidazo[2,1-b]thiazole (3-41): This compound was prepared
according to the General Procedure B using styrene (78 mg, 0.75 mmol), 4,5-diphenyl-2-
imidazolethiol (126 mg, 0.5 mmol). After purification by column chromatography on SiO2
(30-50% EtOAc in hexanes), the title compound was isolated as a yellow solid (90 mg,
51% yield, >98:2 rr).

90
1H NMR (600 MHz, CDCl3): = 7.53-7.49 (m, 2 H), 7.28-7.12 (m, 9 H), 7.05-7.01 (m, 2
H), 7.00-6.95 (m, 2 H), 5.38 (dd, J = 2.1, 7.8 Hz, 1 H), 4.37 (dd, J = 7.8, 11.2 Hz, 1 H),
3.55 (dd, J = 2.1, 11.2 Hz, 1 H);
13C NMR (150 MHz, CDCl3): = 149.1, 143.0, 138.7, 134.4, 130.4, 129.7, 128.8,128.5,
128.5, 128.1, 128.1, 127.5, 126.6, 126.5, 125.8, 60.5, 43.6;
IR (neat): 3031, 1601, 1440, 1317, 1137, 695 cm-1;
HRMS (ESI) m/z calcd for C23H19N2S [(M+H)+] 355.1268, found 355.1269.
2-phenyl-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-42): This compound was
prepared according to the General Procedure C using styrene (78 mg, 0.75 mmol), 2-
mercaptobenzimidazole (113 mg, 0.75 mmol). After purification by column
chromatography on SiO2 (30-50% EtOAc in hexanes), the title compound was isolated as
a white solid (96 mg, 76% yield, 94:6 rr).
1H NMR (600 MHz, CDCl3): = 7.66 (d, J = 7.6 Hz, 1 H), 7.48 (d, J = 7.3 Hz, 2 H), 7.42-
7.34 (m, 3 H), 7.24-7.15 (m, 3 H), 5.58 (t, J = 7.9 Hz, 1 H), 4.61 (dd, J = 7.9, 10.3 Hz, 1
H), 4.26 (dd, J = 7.9, 10.3 Hz, 1 H);
13C NMR (150 MHz, CDCl3): = 158.0, 148.9, 137.5, 133.5, 129.1, 129.0, 127.7, 121.9,
121.9, 118.8, 108.8, 56.0, 51.7;
IR (neat): 2927, 1612, 1473, 1249, 742 cm-1;
HRMS (ESI) m/z calcd for C15H13N2S [(M+H)+] 253.0799, found 253.0798.

91
2-(4-(tert-butyl)phenyl)-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-43): This
compound was prepared according to the General Procedure C using 4-tert-butylstyrene
(120 mg, 0.75 mmol), 2-mercaptobenzimidazole (113 mg, 0.75 mmol). After purification
by column chromatography on SiO2 (30-50% EtOAc in hexanes), the title compound was
isolated as a white solid (101 mg, 65% yield, 95:5 rr).
1H NMR (600 MHz, CDCl3): = 7.66 (d, J = 7.8 Hz, 1 H), 7.43-7.37 (m, 4 H), 7.23-7.12
(m, 3 H), 5.53 (t, J = 7.9 Hz, 1 H), 4.53 (dd, J = 7.9, 10.3 Hz, 1 H), 4.22 (dd, J = 7.9, 10.3
Hz, 1 H), 1.33 (s, 9 H);
13C NMR (150 MHz, CDCl3): = 158.1, 152.1, 148.8, 134.2, 133.5, 127.3, 126.0, 121.8,
121.8, 118.6, 108.8, 55.7, 51.5, 34.6, 31.1;
IR (neat): 2957, 1612, 1465, 1229, 734 cm-1;
HRMS (ESI) m/z calcd for C19H21N2S [(M+H)+] 309.1425, found 309.1422.
2-(4-(trifluoromethyl)phenyl)-2,3-dihydrobenzo[4,5]imidazo[2,1-b]thiazole (3-44):
This compound was prepared according to the General Procedure C using 4-
(trifluoromethyl)styrene (129 mg, 0.75 mmol), 2-mercaptobenzimidazole (113 mg, 0.75
mmol). After purification by column chromatography on SiO2 (30-50% EtOAc in
hexanes), the title compound was isolated as a yellow solid (91 mg, 57% yield, >98:2).
1H NMR (600 MHz, CDCl3): = 7.65 (t, J = 7.9 Hz, 3 H), 7.58 (d, J = 8.1 Hz, 2 H), 7.28-

92
7.15 (m, 3 H), 5.63-5.55 (m, 1 H), 4.64 (dd, J = 7.7, 10.4 Hz, 1 H), 4.31-4.22 (m, 1 H);
13C NMR (150 MHz, CDCl3): = 157.4, 149.0, 141.9, 133.5, 131.2, 131.0, 128.0, 126.2
(q, JC-F = 3.5 Hz), 124.5, 122.2, 118.9, 108.9, 55.0, 51.4;
IR (neat): 2950, 1615, 1473, 1113, 746 cm-1;
HRMS (ESI) m/z calcd for C16H12F3N2S [(M+H)+] 321.0673, found 321.0665.
Temperature dependent diastereoselectivity:
The study of diastereoselctivity was carried out according to the General Procedure A using
cis-β-methylstyrene (89 mg, 0.75 mmol), 2-mercaptobenzimidazole (75 mg, 0.5 mmol) for
8 h. The temperature 50 ºC, rt, 0 ºC and -20 ºC provided 69% (cis:trans <2:98), 42%
(cis:trans 69:31), 15% (cis:trans 85:15), and <5% based on crude NMR yield, and ratio
respectively. After purification of the rt reaction by column chromatography on SiO2 (25%
EtOAc in hexanes), the inseparable mixture of two diastereomers was isolated as a white
solid (53 mg, 40% yield).
1H NMR (600 MHz, CDCl3): = 7.60 (d, J = 8.3 Hz, 1 Htrans), 7.51-7.47 (m, 2 Hcis), 7.47
-7.39 (m, 3 Htrans + 2 Hcis), 7.35-7.29 (m, 2 Htrans + 2 Hcis), 7.29-7.23 (m, 1 Hcis), 7.20-7.15
(m, 2 Hcis), 7.13 (t, J = 7.6 Hz, 1 Htrans), 6.94 (t, J = 7.6 Hz, 1 Htrans), 6.45 (d, J = 8.1 Hz, 1
Htrans), 4.99 (d, J = 8.5 Hz, 1 Htrans), 4.85 (d, J = 7.3 Hz, 1 Hcis), 4.34 (dd, J = 6.8, 8.5 Hz,
1 Htrans), 3.71 (t, J = 7.2 Hz, 1 Hcis), 1.60 (d, J = 6.8 Hz, 3 Htrans), 1.32 (d, J = 7.3 Hz, 3
Hcis);
13C NMR (150 MHz, CDCl3): = 157.7, 149.9, 148.4, 143.1, 135.4, 133.4, 129.4, 129.3,
128.2, 127.6, 127.2, 126.6, 122.2, 122.0, 121.6, 118.3, 109.4, 79.1, 69.1, 56.2, 50.8, 18.7,
18.2;

93
IR (neat): 2916, 1615, 1470, 1247, 737 cm-1;
HRMS (ESI) m/z calcd for C16H15N2S [(M+H)+] 267.0956, found 267.0950.
NMR Study for Sulfur-Fluorine Bond:
The Selectfluor (213 mg, 0.6 mmol) was added to the deuterated dimethylformamide in
NMR tube and 19F NMR was taken for Selectfluor. Then, 2-mercaptobenzimidazole (75
mg, 0.5 mmol) was added to the solution of Selectfluor in NMR tube and 19F NMR was
taken after 5 min, 30 min, 2 h, 6 h, 7 days. The intensity of new peak (38 ppm) was
increasing with time. The peak at 38 ppm indicates the S-F bond, and peak at -182 ppm is
responsible for BF4-.

94
5 mmol scale reaction:
The large-scale reaction was carried out according to the General Procedure A using
styrene (781 mg, 7.5 mmol), 2-mercaptobenzimidazole (750 mg, 5 mmol) and Selectfluor
(2125 mg, 6 mmol). After purification by column chromatography on SiO2 (30-50%
EtOAc in hexanes), the title compound was isolated as a white solid (828 mg, 66% yield,
>98:2 rr).

95
Chapter 4
Catalytic Regio- and Stereoselective Alkene
Sulfenoamination and Arene Sulfenylation for 1,4-
Benzothiazine Synthesis
4.1 Introduction
Alkene functionalization is an important strategy for the rapid construction of carbon-
carbon, carbon-heteroatom bond across the alkene, enabling the molecular complexity and
functional diversity of the molecule.25, 78, 109-113 In this regard, alkene difunctionalizations
such as oxyamination21, 24, diamination114, diarylation25, and carboamination28 have widely
been studied and well documented in the literature. On the other hand, alkene
sulfenoamination is a difficult process to achieve, hold significant potential for the
construction of N, S-containing molecule.42 A major challenge for this functionalization is
that the thiophilic nature of many metal catalysts often precludes the use of transition metal
for alkene sulfenoaminations.72, 115-116 Therefore, stable electrophilic sulfur reagents need
to be used for alkene activation and subsequent nucleophilic attack from another
heteroatom on the alkene can afford vicinal thioamine product.93-95 In this context,
Denmark and Shi groups have independently reported intramolecular and enantioselective
alkene thioamination for the synthesis of N-containing heterocycles.79, 81, 83, 117-118 In
addition, intermolecular sulfenoamination was achieved using nitriles as nucleophile.85-87
Despite their success on sulfenoamination of alkene, the construction of N, S-containing

96
Figure 4-1: Importance of 1,4-Benzothiazine Structural Motifs
heterocycles via alkene sulfenoamination from readily available feedstock chemicals like
alkene and unfunctionalized thiol-amine is still very rare, but highly desirable due to their
easy synthetic elaborations.
N- and S- containing heterocycles are ubiquitous structural motifs in natural
products and agrochemicals. Particularly, benzothiazine bearing N, S atoms, and its
derivatives are important scaffolds in commercially approved drugs (Figure 4-1).
Moreover, benzothiazine also plays a significant role in biological and pharmaceutical
processes.119-121 These compounds exhibit a wide variety of bioactivities such as
anticancer, antitumor, antimicrobial, antifungal, and neurodegenerative diseases, and KATP
channel openers.122 For example, Rufloxacin, a worldwide drug embedding benzothiazine
ring, has been used for bacterial infection.123 DHBT-NE-1 has been known to be relevant
in neurodegenerative diseases,124-125 and Promethazine, a first-generation antihistamine of

97
Scheme 4-1: Alkene Sulfenoamination Background for 1,4-Benzothiazine Synthesis
phenothiazine family, is used as a sedative and antiallergic medication.126 Additionally,
these structural units can also serve as photosensitizers for many organic transformations,
and ligands in metal-catalyzed reactions.127 Due to their significance in pharmaceuticals
and organic transformation, the syntheses of 1,4-benzothiazine have garnered enormous

98
attention among the medicinal and synthetic communities.128-131 Several methods have
been developed for the syntheses of benzothiazine.132-135 The traditional method involves
condensation of 2-aminothiophenol/its sodium salt with α-bromoacetophenone, followed
by reduction to get access 1,4-benzothiazine (Scheme 4-1a).134, 136-137 Alternatively, the o-
aminobenzenethiols can couple with various ketones in a radical pathway to afford the
unsaturated 1,4-benzothiazine.138 Moreover, Kumar group reported a two-step strategy
utilizing sulfenoacetoxylation of alkene, followed by TFA mediated cyclization to afford
1,4-benzothiazine (Scheme 4-1b).139 Jiang group developed an interesting strategy of
accessing benzothiazine, engaging Pd-catalyzed coupling of presynthesized diiodo
compound bearing tosyl protected amine with sodium thiosulfate as sulfurating agent.140
Gao group disclosed a one-pot protocol for the construction of benzothiazine from the
alkene, involving sufenoamination using N-sulfanylsuccinimides and sulfonamide,
followed by stoichiometric copper-mediated intramolecular cyclization (Scheme 4-1c).84
Nevertheless, most of the methods suffer from certain limitations, such as limited substrate
scope, Ts- or Ac-protected amine functionalities, strong acid, and stoichiometric transition
metal.141 The requirement of multistep syntheses and tedious procedure to get access
starting materials, make the methods less attractive to medicinal chemists for potential SAR
studies, and screening for various bioactivities.142-144 The development of an efficient and
convenient method utilizing unadorned 2-aminothiophenols and alkenes for accessing
structurally diverse 1,4-benzothiazine is in great demand.
As a part of our program to access bioactive heterocycles from the simple chemical
feedstock, we are interested in the utilization of alkene as building blocks for 1,4 -
benzothiazine synthesis. In our previous studies, we realized that halogen could activate a

99
thiol into an electrophilic sulfur reagent, enabling the alkene to thiiranium ion formation,
and then a subsequent nucleophilic attack of tethered amine on thiiranium intermediate or
carbocation would lead to cyclized product (Scheme 4-1d). We wondered whether in-situ
generated catalytic halogen source would be capable of thiol polarity inversion. Herein, we
report an iodide-catalyzed regioselective synthesis of 1,4-benzothiazine using simple
feedstock alkene and free thioamine (Scheme 4-1e). In addition, we also extend our
coupling protocol to achieve a highly versatile Csp2‒H sulfenylation of aniline derivatives,
to afford 1,4-benzothiazine structure that is complementary to the alkene coupling protocol.
4.2 Results and Discussions for 1,4-Benzothiazine Synthesis
4.2.1 Reaction Design and Optimization
With the aforementioned hypothesis in mind, our study was commenced with α-
methylstyrene as alkene partner, 2-aminothiophenol, sodium iodide (NaI), potassium
persulfate (K2S2O8) as an oxidant in 1,2-dichloroethane (DCE) at 80 ˚C. To our delight,
the reaction provided 1,4-benzothiazine in 10% yield with excellent regioselectivity (Table
4.1, entry 1). Further evaluation of reaction solvents revealed that acetonitrile (CH3CN)
was the superior solvent affording product 4-16 in 65% yield (Table 4.1, entries 2 and 3).
However, other oxidants were less effective to deliver the product (Table 4.1, entries 4 and
5). Interestingly, increasing stoichiometry of the alkene to 2.5 equiv provided the optimal
condition, affording the desired product in 98% yield (Table 4.1 entries 6 and 7). Screening
of different halides demonstrated that all halides could afford the product with comparable
yields (Table 4.1, entries 7-10). Further, increasing or decreasing the concentration resulted
in lower yields (Table 4.1, entries 11 and 12). Decreasing the halide loading or lowering

100
Table 4.1: Optimization for 1,4-Benzothiazine Synthesis
the temperature provided the product in diminished yields (Table 4.1, entries 13 and 14).
The control experiment demonstrated that only oxidant without halide could furnish the

101
product in a very low yield of 12%, and no product was observed in the absence of oxidant
in optimized condition (Table 4.1, entries 15 and16).
4.2.2 Substrate Scope
With optimized conditions in hand, we sought to explore the alkene substrate scope.
A range of 1,1-disubstituted styrene derivatives provided highly steric congested 1,4-
benzothiazines in excellent yields (Table 4.2, products 4-16 to 4-21). The regioselectivity
outcomes in these cases were identical to our previous sulfenoamination chemistry
involving thiiranium ion intermediate, in which sulfur attached to terminal carbon and
nitrogen added on benzylic carbon. Moreover, cross-coupling ready functional groups
such as bromo, fluoro, and methoxy were tolerated, affording the products in excellent
yields. The styrene with different functionalities and vinyl naphthalene proceeded
smoothly to afford desired products with high regioselectivities (Table 4.2, products 4-22
to 4-26). Both cis-, trans-1,2-disubstituted styrene participated in the reaction smoothly and
delivered the 1,4-benzothiazines with high diastereoselectivities (Table 4.2, products 4-27
to 4-29). Interestingly, trisubstituted alkene which is usually ineffective for most of the
alkene functionalization reactions, furnished products in good yields with excellent
diastereoselectivities (Table 4.2, products 4-30 and 4-31). The aliphatic monosubstituted
alkenes were also viable substrates to deliver the product with moderate regioselectivities
(Table 4.2, products 4-32 and 4-33). Finally, the complex steroid derived alkene resulted
in the product formation in good yield (Table 4.2, products 4-34).
Encouraged by these promising substrate scope of alkenes, we, then, turned our
attention to explore the thioamine substrate scope. A range of 2-aminothiophenol

102
Table 4.2: Alkene Substrate Scope for 1,4-Benzothiazine Synthesis

103
possessing different halogen substituents such as fluoro, chloro, and bromo performed well
and afforded the corresponding 1,4-benzothiazine products in nearly quantitative yields
(Table 4.3, products 4-35 to 4-38). In addition, pharmaceutically relevant trifluoromethyl
was tolerated and provided the product in excellent yield (Table 4.3, product 4-39). The 2-
aminothiophenol bearing electron-donating substituents were also viable substrates to
furnish the products in good yields (Table 4.3, products 4-40 and 4-41). Gratifyingly,
unprotected carboxylic acid substituent was tolerable in this reaction and furnished the 1,4-
benzothiazine in high yield with excellent regioselectivity (Table 4.3, product 4-42).
Furthermore, we could also synthesis 1,4-benzooxathiine when 2-hydroxythiophenol was
used as a substrate (Table 4.3, product 4-43). Finally, these substrates all uniformly
afforded a single regioisomeric product in good yields.
Table 4.3: Thioamine Substrate Scope for 1,4-Benzothiazine Synthesis

104
4.2.3 Substrate Scope for Arene Sulfenylation
While we were satisfied with the scope for alkene sulfenoamination reaction, we
were concerned by the modest regioselectivity of the aliphatic olefins in the reaction.
Fortuitously, during our studies toward thiomorpholine synthesis with the 2-
(phenylamino)ethane-1-thiol substrate shown in figure 4-2, we instead observed the 1,4-
benzothiazine product, in which the thiol cyclized intramolecularly via Csp2‒H
sulfenylation of the arene. Intrigued by this finding, we were delighted that this strategy
could provide complementary products comparable to the alkene sulfenoamination
protocol of aliphatic olefins. In the case of 4-44, the structure is equivalent to the product
derived from the coupling of ethylene with 2-aminothiophenol in the alkene
Figure 4-2: Unexpected Csp2‒H Sulfenylation

105
sulfenoamination. Excited by this result, we decided to examine the preliminary scope of
this method. Simply removing the olefin from the reaction and with a slight increase in
catalyst loading resulted in the formation of product 4-44 in high yield. With this modified
procedure, we screened a number of thioamine structures. This sulfenylation protocol
impressively tolerated highly substituted carbon centers and tertiary aniline structure
(Table 4.4, products 4-45 to 4-48). In addition, different functional groups with varying
electronic nature all proceeded smoothly (Table 4.4, products 4-49 to 4-52). Notably,
diastereoselectivity could be completely transferred from the thioamine starting material
to the 1,4-benzothiazine as demonstrated in the case of product 4-46. Interestingly, when
the ortho-positions of the benzene ring were blocked, the thioamine substrate 4-53 could
Table 4.4: Substrate Scope for C-H Sulfenylation of Arene

106
then participate in the alkene sulfenoamintion reaction (Figure 4-3). In this case, a
sterically encumbered product 4-54 was formed as a single regioisomer. Further studies
of this thiomorpholine synthesis protocol are currently underway in our laboratory.
Figure 4-3: Synthesis of Thiomorpholine
4.3 Reaction Mechanism
In order to gain insight into the reaction mechanism, first, we considered the
regiochemical outcome of the reaction. For the styrene derived substrates, nitrogen was
attached to the benzylic position whereas the sulfur was attached to the terminal carbon.
This regiochemical feature is distinct from the previously developed alkene
sulfenoamination based on halogenation of alkene in which more nucleophilic sulfur was
added to the benzylic position. This regioselectivity suggests that reaction can go through
either radical or thiiranium pathway. To further understand the mechanism, several control
experiments were performed as shown in scheme 4-2. When the standard reaction was
carried out with α-cycloproylstyrene, cyclopropane ring intact product 4-55 was obtained
in 12% suggesting the polar pathway was operative (Scheme 4-2a). We also observed a
complex mixture of ring-opening adducts from the product isolation. Moreover, when the

107
Scheme 4-2: Mechanistic Experiments for 1,4-Benzothiazine Synthesis
reaction was performed using disulfide as a thioamine structure 4-56, the desired product
4-16 was observed in a comparable 70% yield, which indicates that active catalytic halogen
source is capable of cleaving the S-S bond to form sulfur electrophile (Scheme 4-2b). Our
control experiments revealed that in the absence of the oxidant, the iodide salt could also
promote disulfide 4-56 for product formation to 12 % yield, and in the absence of NaI, less
than 2% product was observed. Moreover, when TEMPO was added to the standard
reaction as a free radical scavenger, the percentage of yield for product formation was
decreasing with the increasing amount of TEMPO (Scheme 4-2c). These results further
support the radical pathways for product formation. It is noteworthy that only oxidant could
afford benzothiazine in 12% (Table 4.1, entry 15) which again confirmed the radical

108
Scheme 4-3: Proposed Reaction Mechanism
process might be involved in the reaction. Hence, the radical trapping experiment could
not completely inhibit the formation of 1,4-benzothiazine, we cannot rule out the ionic
thiiranium ion intermediate for product formation.
Based on the experimental results and literature reports, two potential mechanisms are
proposed in the Scheme 4-3. Initially, sodium iodide is oxidized by potassium persulfate to
generate iodine which can react with thioamine to generate sulfur electrophile A. The
intermediate A then undergoes electrophilic attack on alkene to form the thiiranium ion B.
The thiiranium ion can be in equilibrium with the open carbocation C which can then be
trapped by the tethered nucleophile to provide the final heterocyclic product. Alternatively,
pathway B from intermediate A involves homolytic cleavage of sulfur iodine bond to thiyl

109
radical D. The radical intermediate D combines with the alkene to generate more stable
benzylic radical E which can undergo single electron transfer from oxidant to produce
carbocation C. Finally, the intramolecular cyclization on the carbocation leads to desired
product 4-22.
4.4 Conclusion
We have developed an iodide-catalyzed regioselective alkene sulfenoamination for
1,4-benzothiazine synthesis from free thioamine. A wide range of alkenes and thioamine
with a variety of functional groups are tolerable with the reaction conditions. We have also
demonstrated that disulfide is a viable substrate for this protocol. We have discovered an
arene ortho sulfenylation reaction to afford 1,4-benzothiazine. The feasibility of this
catalytic strategy towards thiomorpholine synthesis is also validated. Finally, the
mechanistic experiment demonstrates that both thiiraniun and radical pathways are
operative in our reaction protocol.
4.5 Experimental
General Information. Commercial reagents and solvents were purchased from Sigma
Aldrich, Oakwood Chemicals, Alfa Aesar, Matrix Scientific, Acros Organic, and were used
as received. The thioamine substrates for products 4-35 to 4-37, 4-40 to 4-42 were
synthesized according to reported procedure.145 The alkene substrates for products 4-30
and 4-31 were synthesized according to reported procedure.146 Organic solutions were
concentrated under reduced pressure on a Büchi rotary evaporator using an acetone-dry ice
bath. Chromatographic purification of products was accomplished using flash

110
chromatography on 230-400 mesh silica gel. Thin-layer chromatography (TLC) was
performed on Analtech 250 mm silica gel HLF UV-254 plates. Visualization of the
developed plates was performed by fluorescence quenching, potassium permanganate and
iodine-silica gel system. 1H and 13C NMR spectra were recorded on a Bruker 600
instrument (600 and 150 MHz) or INOVA 600 (600 and 150 MHz) and are internally
referenced to residual protio solvent signals (for CDCl3, 7.26 and 77.0 ppm, respectively).
Data for 1H NMR are reported as follows: chemical shift ( ppm), multiplicity (s = singlet,
d = doublet, t = triplet, q = quartet, h = heptet, m = multiplet, br = broad), integration,
coupling constant (Hz). 13C spectra were reported as chemical shifts in ppm and
multiplicity where appropriate. IR spectra were recorded on a PerkinElmer FT-IR
spectrophotometer and are reported in terms of wavenumber of absorption (cm-1). High
resolution mass spectra were obtained on Waters Synapt High Definition Mass
Spectrometer (HDMS) by electrospray ionization at The University of Toledo, OH, USA
and University of Cincinnati, OH, USA.
Experimental Procedures
General Procedure A: The alkene (0.625 mmol) and 2-aminothiophenol (0.375 mmol)
were added to the solution of NaI (10 mol%, 3.7 mg) in acetonitrile (4 mL). Then, the
K2S2O8 (0.25 mmol, 67.6 mg) was added to the reaction mixture and stirred at 80 ºC for 16
h. The reaction was quenched with saturated NaHCO3 (1 mL), 20% Na2S2O3 (1 mL) and
EtOAc (2 mL) was added. The organic layer was separated, and aqueous layer was
extracted with EtOAc (2 x 2 mL). The combined organic solution was dried with anhydrous
Na2SO4, filtered, concentrated in vacuo by rotavapor and purified by flash Chromatography

111
on SiO2 (5%-10% EtOAc in hexanes or 10%-30% DCM in hexanes) to provide the desired
product. The regioisomeric ratio was determined by crude NMR.
General Procedure B for Arene Sulfenylation: The thioamine (0.325 mmol) were added
to the solution of NaI (20 mol%, 7.5 mg) in acetonitrile (2.5 mL). Then, the K2S2O8 (0.25
mmol, 67.6 mg) was added to the reaction mixture and stirred at 80 ºC for 16 h. The reaction
was quenched with saturated NaHCO3 (1 mL), 20% Na2S2O3 (1 mL) and EtOAc (2 mL)
was added. The organic layer was separated, and aqueous layer was extracted with EtOAc
(2 x 2 mL). The combined organic solution was dried with anhydrous Na2SO4, filtered,
concentrated in vacuo by rotavapor and purified by flash chromatography on SiO2 (5%-
10% EtOAc in hexanes or 10%-30% DCM in hexanes) to provide the desired product. The
regioisomeric ratio was determined by crude NMR.
Procedure for synthesis of N-arylthioamine substrates:
The aniline (25 mmol) was added to the solution of sulfide (20 mmol) in ethanol (0.5 M,
40 mL) stirred at 35 ºC for 16 h. The ethanol was concentrated in vacuo by rotavapor and
product was purified by flash chromatography on SiO2 (5%-10% EtOAc in hexanes) to
provide the desired product. For cyclohexane sulfide, the reaction was run at 50 ºC for 36
h. The purified products were characterized by NMR techniques.

112
Spectral Characterization of the Products
3-methyl-3-phenyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-16): This compound was
prepared according to the General Procedure A using α-methylstyrene (74 mg, 0.625
mmol), 2-aminobenzenethiol (40 μL, 0.375 mmol). After purification by column
chromatography on SiO2 (10% DCM in hexanes), the title compound was isolated as a
white solid (58 mg, 96% yield).
1H NMR (600 MHz, CDCl3): = 7.45-7.40 (m, 2 H), 7.35 (t, J = 7.8 Hz, 2 H), 7.30-7.25
(m, 1 H), 7.03-6.94 (m, 2 H), 6.67-6.58 (m, 2 H), 4.19 (br. s., 1 H), 3.14 (d, J = 12.7 Hz, 1
H), 2.94 (d, J = 12.7 Hz, 1 H), 1.70 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 146.8, 141.1, 128.5, 127.6, 127.1, 125.7, 125.4, 117.7,
115.1, 114.6, 54.6, 37.8, 29.3;
IR (neat): IR (neat): 3413, 2930, 1588, 1442, 750 cm-1;
HRMS (ESI) m/z calcd for C15H16NS [(M+H)+] 242.1003, found 242.1007.
3-isopropyl-3-phenyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-17): This compound
was prepared according to the General Procedure A using α-isopropylstyrene (91 mg, 0.625
mmol), 2-aminobenzenethiol (40 μL, 0.375 mmol). After purification by column
chromatography on SiO2 (10% DCM in hexanes), the title compound was isolated as a

113
white solid (45 mg, 67% yield).
1H NMR (600 MHz, CDCl3): = 7.32-7.19 (m, 5 H), 6.95 (t, J = 7.6 Hz, 1 H), 6.90 (d, J
= 7.6 Hz, 1 H), 6.67 (d, J = 8.1 Hz, 1 H), 6.57 (t, J = 7.4 Hz, 1 H), 4.32 (br. s., 1 H), 3.30
(d, J = 12.7 Hz, 1 H), 3.08 (d, J = 12.7 Hz, 1 H), 2.31-2.21 (m, 1 H), 1.03 (d, J = 6.8 Hz, 3
H), 0.81 (d, J = 7.1 Hz, 3 H);
13C NMR (150 MHz, CDCl3): = 145.2, 141.3, 128.2, 127.6, 126.6, 126.2, 125.6, 117.5,
115.3, 115.0, 60.8, 38.4, 33.7, 17.6, 16.7;
IR (neat): 3407, 2947, 1590, 1473, 747 cm-1 ,
HRMS (ESI) m/z calcd for C17H20NS [(M+H)+] 270.1316, found 270.1319.
3,3-diphenyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-18): This compound was
prepared according to the General Procedure A using α-phenylstyrene (113 mg, 0.625
mmol), 2-aminobenzenethiol (40 μL, 0.375 mmol). After purification by column
chromatography on SiO2 (10% DCM in hexanes), the title compound was isolated as a
white solid (55 mg, 73% yield).
1H NMR (600 MHz, CDCl3): = 7.37-7.25 (m, 10 H), 6.99-6.92 (m, 2 H), 6.66-6.59 (m,
2 H), 4.58 (s, 1 H), 3.55 (s, 2 H);
13C NMR (150 MHz, CDCl3): = 145.4, 141.0, 128.4, 127.5, 127.4, 127.1, 125.5, 118.0,
115.3, 114.8, 61.3, 35.8;
IR (neat): 3402, 2937, 1589, 1474, 695 cm-1;
HRMS (ESI) m/z calcd for C20H18NS [(M+H)+] 304.1160, found 304.1165.

114
3-(4-fluorophenyl)-3-methyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-19): This
compound was prepared according to the General Procedure A using 4-fluoro-α-
methylstyrene (85 mg, 0.625 mmol), 2-aminobenzenethiol (40 μL, 0.375 mmol). After
purification by column chromatography on SiO2 (10% DCM in hexanes), the title
compound was isolated as a white solid (53 mg, 81% yield).
1H NMR (600 MHz, CDCl3): = 7.41-7.34 (m, 2 H), 7.04-6.94 (m, 4 H), 6.68-6.57 (m, 2
H), 4.16 (br. s., 1 H), 3.10 (d, J = 12.7 Hz, 1 H), 2.94 (d, J = 12.7 Hz, 1 H), 1.70 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 162.6, 160.9, 142.6, 140.9, 127.6, 127.2, 127.2, 125.8,
117.9, 115.3, 115.2, 115.1, 114.5, 54.4, 37.8, 29.6;
IR (neat): 3406, 2976,1589, 1475, 740 cm-1;
HRMS (ESI) m/z calcd for C15H15FNS [(M+H)+] 260.0909, found 260.0905.
3-(4-bromophenyl)-3-methyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-20): This
compound was prepared according to the General Procedure A using 4-bromo-α-
methylstyrene (123 mg, 0.625 mmol), 2-aminobenzenethiol (40 μL, 0.375 mmol). After
purification by column chromatography on SiO2 (10% DCM in hexanes), the title
compound was isolated as a white solid (57 mg, 71% yield).
1H NMR (600 MHz, CDCl3): = 7.43 (d, J = 8.5 Hz, 2 H), 7.26 (d, J = 8.5 Hz, 2 H), 7.01-
6.93 (m, 2 H), 6.67-6.57 (m, 2 H), 4.15 (br. s., 1 H), 3.07 (d, J = 12.7 Hz, 1 H), 2.93 (d, J

115
= 12.7 Hz, 1 H), 1.66 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 145.9, 140.7, 131.5, 127.6, 127.3, 125.8, 121.0, 117.9,
115.2, 114.4, 54.6, 37.4, 29.7;
IR (neat): 3400, 2912, 1588, 1473, 748 cm-1
HRMS (ESI) m/z calcd for C15H15BrNS [(M+H)+] 320.0109, found 320.0105.
3-(4-methoxyphenyl)-3-methyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-21): This
compound was prepared according to the General Procedure A using 4-methoxy-α-
methylstyrene (93 mg, 0.625 mmol), 2-aminobenzenethiol (40 μL, 0.375 mmol) and
benzoyl peroxide (75% pure, 81 mg, 0.25 mmol). After purification by column
chromatography on SiO2 (5% EtOAc in hexanes), the title compound was isolated as a
yellow gummy solid (44 mg, 65% yield).
1H NMR (600 MHz, CDCl3): = 7.37-7.31 (m, J = 8.8 Hz, 2 H), 7.02 (d, J = 7.6 Hz, 1
H), 6.99-6.94 (m, 1 H), 6.90-6.84 (m, J = 8.5 Hz, 2 H), 6.64 (t, J = 7.4 Hz, 1 H), 6.59 (d, J
= 8.1 Hz, 1 H), 4.16 (br. s., 1 H), 3.80 (s, 3 H), 3.11 (d, J = 12.7 Hz, 1 H), 2.91 (d, J = 12.7
Hz, 1 H), 1.69 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 158.5, 141.2, 138.9, 127.5, 126.6, 125.6, 117.7, 115.2,
114.5, 113.7, 55.2, 54.0, 38.0, 29.2;
IR (neat): 3373, 2929, 1588, 1509, 742 cm-1;
HRMS (ESI) m/z calcd for C16H18NOS [(M+H)+] 272.1109, found 272.1110.

116
3-phenyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-22): This compound was prepared
according to the General Procedure A using styrene (65 mg, 0.625 mmol), 2-
aminobenzenethiol (40 μL, 0.375 mmol), NaI (20 mol%, 7.5 mg) for 16 h. After
purification by column chromatography on SiO2 (10% DCM in hexanes), the title
compound was isolated as a colorless liquid (29 mg, 51% yield). The spectral data match
with literature134.
1H NMR (600 MHz, CDCl3): = 7.45-7.31 (m, 5 H), 7.07 (d, J = 7.6 Hz, 1 H), 6.98-6.90
(m, 1 H), 6.71-6.64 (m, 1 H), 6.57-6.50 (m, 1 H), 4.68 (dd, J = 2.2, 9.0 Hz, 1 H), 4.13 (br.
s., 1 H), 3.18 (dd, J = 9.0, 12.5 Hz, 1 H), 3.04-2.98 (m, 1 H);
13C NMR (150 MHz, CDCl3): = 142.8, 142.2, 128.9, 128.2, 127.4, 126.7, 125.6, 118.3,
115.4, 115.3, 56.1, 33.1;
IR (neat): 3384, 2921, 1590, 1476, 697 cm-1;
HRMS (ESI) m/z calcd for C14H14NS [(M+H)+] 228.0847, found 228.0844.
3-(4-fluorophenyl)-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-23): This compound was
prepared according to the General Procedure A using 4-fluorostyrene (76 mg, 0.625 mmol),
2-aminobenzenethiol (40 μL, 0.375 mmol), NaI (20 mol%, 7.5 mg) for 16 h. After
purification by column chromatography on SiO2 (10% DCM in hexanes), the title

117
compound was isolated as a yellow solid (29 mg, 47% yield). The spectral data match with
literature.136
1H NMR (600 MHz, CDCl3): = 7.34 (dd, J = 6.1, 7.3 Hz, 2 H), 7.10-7.04 (m, 3 H), 6.94
(t, J = 7.6 Hz, 1 H), 6.68 (t, J = 7.6 Hz, 1 H), 6.53 (d, J = 8.1 Hz, 1 H), 4.68 (d, J = 8.5 Hz,
1 H), 4.10 (br. s., 1 H), 3.17-3.08 (m, 1 H), 2.98 (d, J = 12.5 Hz, 1 H);
13C NMR (150 MHz, CDCl3): = 163.3, 161.7, 142.0, 138.6, 138.6, 128.3, 128.3, 127.5,
125.7, 118.5, 115.8, 115.7, 115.4, 115.3, 55.4, 33.1.
3-(4-methoxyphenyl)-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-24): This compound
was prepared according to the General Procedure A using 4-methoxystyrene (84 mg, 0.625
mmol), 2-aminobenzenethiol (40 μL, 0.375 mmol), benzoyl peroxide (75% pure, 81 mg,
0.25 mmol), NaI (20 mol%, 7.5 mg) for 16 h. After purification by column chromatography
on SiO2 (5% EtOAc in hexanes), the title compound was isolated as a yellow solid (32 mg,
50% yield). The spectral data match with literature.134
1H NMR (600 MHz, CDCl3): = 7.30 (d, J = 8.5 Hz, 2 H), 7.07 (d, J = 7.8 Hz, 1 H), 6.96
- 6.90 (m, 3 H), 6.67 (t, J = 7.6 Hz, 1 H), 6.52 (d, J = 8.1 Hz, 1 H), 4.62 (dd, J = 2.0, 9.3
Hz, 1 H), 4.08 (br. s., 1 H), 3.83 (s, 3 H), 3.15 (dd, J = 9.3, 12.5 Hz, 1 H), 3.01-2.93 (m, 1
H);
13C NMR (150 MHz, CDCl3): = 159.4, 142.3, 134.9, 127.8, 127.4, 125.5, 118.2, 115.3,
115.3, 114.2, 55.5, 55.3, 33.1.

118
4-(3,4-dihydro-2H-benzo[b][1,4]thiazin-3-yl)phenyl acetate (4-25): This compound
was prepared according to the General Procedure A using 4-acetoxystyrene (101 mg, 0.625
mmol), 2-aminobenzenethiol (40 μL, 0.375 mmol), NaI (20 mol%, 7.5 mg) for 16 h. After
purification by column chromatography on SiO2 (5% EtOAc in hexanes), the title
compound was isolated as a yellow solid (41 mg, 57% yield).
1H NMR (600 MHz, CDCl3): = 7.39 (d, J = 8.3 Hz, 2 H), 7.14 - 7.03 (m, 3 H), 6.94 (t,
J = 7.6 Hz, 1 H), 6.68 (t, J = 7.4 Hz, 1 H), 6.52 (d, J = 7.8 Hz, 1 H), 4.68 (d, J = 8.5 Hz, 1
H), 4.13 (br. s., 1 H), 3.15 (dd, J = 8.5, 12.3 Hz, 1 H), 3.00 (d, J = 12.3 Hz, 1 H), 2.32 (s,
3 H);
13C NMR (150 MHz, CDCl3): = 169.5, 150.4, 142.0, 140.3, 127.8, 127.4, 125.6, 122.0,
118.4, 115.3, 115.3, 55.5, 33.0, 21.1;
IR (neat): 3393, 2924, 1760, 1588, 1477, 743 cm-1;
HRMS (ESI) m/z calcd for C16H16NO2S [(M+H)+] 286.0902, found 286.0909.
3-(naphthalen-1-yl)-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-26): This compound was
prepared according to the General Procedure A using 2-vinylnaphthalene (96 mg, 0.625
mmol), 2-aminobenzenethiol (40 μL, 0.375 mmol), NaI (20 mol%, 7.5 mg) for 16 h. After
purification by column chromatography on SiO2 (10% DCM in hexanes), the title

119
compound was isolated as a yellow gummy solid (47 mg, 68% yield). The spectral data
match with literature.136
1H NMR (600 MHz, CDCl3): = 7.91-7.81 (m, 4 H), 7.55-7.45 (m, 3 H), 7.11 (d, J = 7.8
Hz, 1 H), 7.01-6.95 (m, 1 H), 6.71 (t, J = 7.6 Hz, 1 H), 6.58 (d, J = 8.1 Hz, 1 H), 4.85 (dd,
J = 2.0, 9.0 Hz, 1 H), 4.24 (s, 1 H), 3.27 (dd, J = 9.0, 12.5 Hz, 1 H), 3.07 (d, J = 12.5 Hz,
1 H);
13C NMR (150 MHz, CDCl3): = 142.2, 140.1, 133.3, 133.2, 128.7, 127.9, 127.7, 127.5,
126.4, 126.2, 125.7, 125.6, 124.5, 118.3, 115.4, 115.4, 56.2, 33.0.
Trans-2-methyl-3-phenyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-27): This
compound was prepared according to the General Procedure A using trans-β-
methylstyrene (74 mg, 0.625 mmol), 2-aminobenzenethiol (40 μL, 0.375 mmol), NaI (20
mol%, 7.5 mg) for 16 h. After purification by column chromatography on SiO2 (10% DCM
in hexanes), the title compound was isolated as a white solid (13 mg, 22% yield, >95:5 dr).
1H NMR (600 MHz, CDCl3): = 7.42-7.30 (m, 5 H), 7.06 (d, J = 7.8 Hz, 1 H), 6.92 (t, J
= 7.6 Hz, 1 H), 6.68 (t, J = 7.4 Hz, 1 H), 6.52 (d, J = 7.8 Hz, 1 H), 4.18 (d, J = 8.1 Hz, 1
H), 4.12 (br. s., 1 H), 3.33 (quin, J = 7.1 Hz, 1 H), 1.08 (d, J = 6.8 Hz, 3 H);
13C NMR (150 MHz, CDCl3): = 142.0, 141.4, 128.8, 128.4, 127.6, 126.9, 125.4, 118.3,
116.8, 114.7, 63.1, 39.4, 18.0;
IR (neat): 3371, 2923, 1590, 1480,739 cm-1;
HRMS (ESI) m/z calcd for C15H16NS [(M+H)+] 242.1003, found 242.0996.

120
Trans-(4-methoxyphenyl)-2-methyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-28):
This compound was prepared according to the General Procedure A using 4-methoxy-
trans-β-methylstyrene (93 mg, 0.625 mmol), 2-aminobenzenethiol (40 μL, 0.375 mmol),
NaI (20 mol%, 7.5 mg) for 16 h. After purification by column chromatography on SiO2
(5% EtOAc in hexanes), the title compound was isolated as a white solid (27 mg, 40%
yield, >95:5 dr).
1H NMR (600 MHz, CDCl3): = 7.22 (d, J = 8.5 Hz, 2 H), 7.03 (d, J = 7.6 Hz, 1 H), 6.93-
6.85 (m, 3 H), 6.64 (t, J = 7.4 Hz, 1 H), 6.48 (d, J = 7.8 Hz, 1 H), 4.10 (d, J = 8.3 Hz, 1 H),
4.05 (br. s., 1 H), 3.81 (s, 3 H), 3.30-3.23 (m, 1 H), 1.04 (d, J = 6.8 Hz, 3 H);
13C NMR (150 MHz, CDCl3): = 159.5, 142.1, 133.4, 128.6, 126.8, 125.3, 118.2, 116.9,
114.7, 114.1, 62.5, 55.3, 39.5, 17.9;
IR (neat): 3344, 2961, 1589, 1455, 1242, 743 cm-1;
HRMS (ESI) m/z calcd for C16H18NOS [(M+H)+] 272.1109, found 272.1100.
Cis-4b,5,10a,11-tetrahydrobenzo[b]indeno[1,2-e][1,4]thiazine (4-29): This compound
was prepared according to the General Procedure A using indene (73 mg, 0.625 mmol), 2-
aminobenzenethiol (40 μL, 0.375 mmol), NaI (20 mol%, 7.5 mg) for 16 h. After
purification by column chromatography on SiO2 (10% DCM in hexanes), the title

121
compound was isolated as a brown solid (30 mg, 50% yield, >95:5 dr).
1H NMR (600 MHz, CDCl3): = 7.30 (d, J = 4.2 Hz, 1 H), 7.27-7.20 (m, 3 H), 7.08 (dd,
J = 1.2, 7.6 Hz, 1 H), 6.98-6.91 (m, 1 H), 6.70-6.60 (m, 2 H), 4.92 (d, J = 5.1 Hz, 1 H),
4.38 (br. s., 1 H), 3.91-3.81 (m, 1 H), 3.34 (dd, J = 6.3, 16.0 Hz, 1 H), 3.02 (dd, J = 4.3,
16.0 Hz, 1 H);
13C NMR (150 MHz, CDCl3): = 144.2, 142.4, 140.6, 128.1, 128.1, 127.1, 126.0, 125.4,
123.8, 118.6, 117.8, 115.5, 60.8, 42.9, 38.3;
IR (neat): 3340, 2922, 1590, 1479, 744 cm-1;
HRMS (ESI) m/z calcd for C15H14NS [(M+H)+] 240.0847, found 240.0846.
Trans-2,3-dimethyl-3-phenyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-30) This
compound was prepared according to the General Procedure A using trans-but-2-en-2-
ylbenzene (83 mg, 0.625 mmol), 2-aminobenzenethiol (40 μL, 0.375 mmol). After
purification by column chromatography on SiO2 (10% DCM in hexanes), the title
compounds were isolated as white gummy solid (48 mg, 75% yield, >95:5 dr).
1H NMR (600 MHz, CDCl3): =7.47 (d, J = 7.8 Hz, 2 H), 7.37 (t, J = 7.6 Hz, 2 H), 7.32-
7.27 (m, 1 H), 7.02 (d, J = 7.6 Hz, 1 H), 6.98-6.92 (m, 1 H), 6.67 (t, J = 7.4 Hz, 1 H), 6.57
(d, J = 8.1 Hz, 1 H), 4.14 (br. s., 1 H), 3.43 (q, J = 6.8 Hz, 1 H), 1.61 (s, 3 H), 1.11 (d, J =
6.8 Hz, 3 H);
13C NMR (150 MHz, CDCl3): = 146.3, 141.0, 128.5, 127.3, 127.1, 126.0, 125.3, 117.8,
115.7, 115.1, 57.2, 43.1, 22.5, 16.5;

122
IR (neat): 3403, 2934, 1590, 1474, 739 cm-1;
HRMS (ESI) for m/z calcd for C16H18NS [(M+H)+] 256.1160, found 256.1152.
Trans-3-methyl-3-phenyl-2-propyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-31): This
compound was prepared according to the General Procedure A using trans-hex-2-en-2-
ylbenzene (100 mg, 0.625 mmol), 2-aminobenzenethiol (40 μL, 0.375 mmol). After
purification by column chromatography on SiO2 (10% DCM in hexanes), the title
compounds were isolated as white solid (47 mg, 66% yield, >95:5 dr).
1H NMR (600 MHz, CDCl3): = 7.49-7.44 (m, 2 H), 7.37 (t, J = 7.6 Hz, 2 H), 7.33-7.28
(m, 1 H), 7.04 (d, J = 7.8 Hz, 1 H), 6.98-6.91 (m, 1 H), 6.69-6.63 (m, 1 H), 6.55 (d, J = 8.1
Hz, 1 H), 4.12 (s, 1 H), 3.29 (dd, J = 2.6, 10.9 Hz, 1 H), 1.63 (s, 3 H), 1.60-1.51 (m, 1 H),
1.43-1.34 (m, 1 H), 1.34-1.16 (m, 2 H), 0.80 (t, J = 7.3 Hz, 3 H);
13C NMR (150 MHz, CDCl3): = 146.5, 141.1, 128.4, 127.3, 127.3, 126.1, 125.3, 117.7,
115.5, 115.0, 57.3, 49.0, 32.2, 23.0, 20.7, 13.7;
IR (neat): 3423, 2950, 1592, 1478, 968 cm-1;
HRMS (ESI) m/z calcd for C18H22NS [(M+H)+] 284.1473, found 284.1473.
2-hexyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-32): This compound was prepared
according to the General Procedure A using octene (70 mg, 0.625 mmol), 2-

123
aminobenzenethiol (40 μL, 0.375 mmol), NaI (20 mol%, 7.5 mg) for 36 h. After
purification by column chromatography on SiO2 (10% DCM in hexanes), the inseparable
mixture of two regioisomers was isolated as a red gummy solid (21 mg, 36% yield, 2.5:1
rr).
1H NMR (600 MHz, CDCl3): = 7.02-6.96 (m, 1 Hmajor + 1 Hminor ), 6.91-6.85 (m, 1 Hmajor
+ 1 Hminor), 6.64-6.58 (m, 1 Hmajor + 1 Hminor), 6.50-6.44 (m, 1 Hmajor + 1 Hminor), 3.95 (br.
s., 1 Hmajor + 1 Hminor), 3.66-3.57 (m, 1 Hmajor), 3.56-3.47 (m, 1 Hminor), 3.31-3.18 (m, 2
Hmajor), 2.95 (dd, J = 2.4, 12.5 Hz, 1 Hminor), 2.82 (dd, J = 8.1, 12.2 Hz, 1 Hminor), 1.69-1.55
(m, 2 Hmajor + 2 Hminor), 1.54-1.38 (m, 2 Hmajor + 2 Hminor), 1.38-1.28 (m, 5 Hmajor + 4 Hminor),
1.28-1.15 (m, 1 Hmajor + 2 Hminor), 0.95-0.78 (m, 3 Hmajor + 3 Hminor);
13C NMR (150 MHz, CDCl3): = 141.5, 127.5, 127.4, 125.5, 125.2, 118.2, 117.9, 116.5,
115.2, 115.0, 51.3, 47.7, 39.6, 36.4, 33.6, 31.7, 31.7, 30.7, 29.2, 29.1, 26.8, 25.5, 22.6, 14.1;
IR (neat): 3406, 2924, 1591, 1484, 740 cm-1;
HRMS (ESI) m/z calcd for C14H22NS [(M+H)+] 236.1473, found 236.1476.
2-phenethyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-33): This compound was
prepared according to the General Procedure A using 4-phenyl-1-butene (83 mg, 0.625
mmol), 2-aminobenzenethiol (40 μL, 0.375 mmol), NaI (20 mol%, 7.5 mg) for 36 h. After
purification by column chromatography on SiO2 (10% DCM in hexanes), the title
compound was isolated as a colorless liquid (28 mg, 44% yield, 2.5:1 rr).

124
1H NMR (600 MHz, CDCl3) for major: = 7.33-7.27 (m, 2 H), 7.25-7.18 (m, 3 H), 7.01
(d, J = 7.6 Hz, 1 H), 6.89 (t, J = 7.6 Hz, 1 H), 6.64 (t, J = 7.4 Hz, 1 H), 6.48 (d, J = 8.1 Hz,
1 H), 3.96 (br. s., 1 H), 3.63 (d, J = 11.7 Hz, 1 H), 3.31 (dd, J = 7.3, 11.7 Hz, 1 H), 3.25-
3.17 (m, 1 H), 2.87 (td, J = 7.1, 13.9 Hz, 1 H), 2.83-2.73 (m, 1 H), 2.04-1.92 (m, 2 H);
13C NMR (150 MHz, CDCl3) for major: = 141.4, 141.2, 128.5, 128.4, 127.6, 126.0,
125.3, 118.2, 116.0, 115.0, 47.6, 38.7, 35.3, 32.9;
IR (neat) for major: 3390, 2925, 1587, 1492, 742 cm-1;
HRMS (ESI) for major m/z calcd for C16H18NS [(M+H)+] 256.1160, found 256.1158.
1H NMR (600 MHz, CDCl3) for minor: = 7.33-7.28 (m, 2 H), 7.24-7.20 (m, 3 H), 6.99
(d, J = 7.6 Hz, 1 H), 6.88 (t, J = 7.6 Hz, 1 H), 6.61 (t, J = 7.4 Hz, 1 H), 6.41 (d, J = 8.1 Hz,
1 H), 3.87 (br. s., 1 H), 3.59 (d, J = 6.1 Hz, 1 H), 2.99 (dd, J = 2.6, 12.3 Hz, 1 H), 2.86 (dd,
J = 7.4, 12.3 Hz, 1 H), 2.82-2.71 (m, 2 H), 2.01-1.88 (m, 2 H);
13C NMR (150 MHz, CDCl3) for minor: = 141.4, 141.1, 128.6, 128.3, 127.4, 126.2,
125.5, 118.0, 115.7, 115.4, 50.7, 37.7, 32.0, 30.6;
IR (neat) for minor: 3400, 2921, 1589, 1498, 750 cm-1;
HRMS (ESI) for minor m/z calcd for C16H18NS [(M+H)+] 256.1160, found 256.1158.
(8R,9S,13S,14S,17S)-2-(3,4-dihydro-2H-benzo[b][1,4]thiazin-3-yl)-13-methyl-
7,8,9,11,12,13,14,15, 16,17-decahydro-6H-cyclopenta[a]phenanthren-17-yl pivalate
(4-34): This compound was prepared according to the General Procedure A using

125
(8R,9S,13S,14S,17S)-13-methyl-2-vinyl-7,8,9,11,12,13,14,15,16,17-decahydro-6H-
cyclopenta[a]phenanthren-17-yl pivalate (229 mg, 0.625 mmol), 2-aminobenzenethiol (40
μL, 0.375 mmol), NaI (20 mol%, 7.5 mg) for 16 h. After purification by column
chromatography on SiO2 (3% EtOAc in hexanes), the title compound was isolated as a
white solid (61 mg, 50% yield, 1:1 dr).
1H NMR (600 MHz, CDCl3): = 7.31 (d, J = 8.1 Hz, 1 H), 7.15 (d, J = 8.1 Hz, 1 H), 7.10
(s, 1 H), 7.06 (d, J = 7.6 Hz, 1 H), 6.95-6.89 (m, 1 H), 6.66 (t, J = 7.4 Hz, 1 H), 6.50 (d, J
= 8.1 Hz, 1 H), 4.67 (t, J = 8.4 Hz, 1 H), 4.60 (dd, J = 2.0, 9.0 Hz, 1 H), 4.08 (br. s., 1 H),
3.21-3.13 (m, 1 H), 2.98 (d, J = 12.5 Hz, 1 H), 2.92-2.85 (m, 2 H), 2.37-2.17 (m, 3 H),
1.96-1.84 (m, 2 H), 1.81-1.71 (m, 1 H), 1.56-1.45 (m, 3 H), 1.45-1.36 (m, 3 H), 1.35-1.27
(m, 1 H), 1.21 (s, 9 H), 0.88-0.81 (m, 3 H);
13C NMR (150 MHz, CDCl3): = 178.6, 142.3, 140.4, 140.0, 140.0, 137.3, 127.4, 127.2,
125.9, 125.5, 124.0, 123.9, 118.2, 118.2, 115.4, 115.3, 115.2, 82.2, 55.9, 55.8, 49.8, 44.2,
44.2, 43.0, 38.9, 38.3, 38.3, 36.9, 33.1, 29.6, 29.5, 27.5, 27.2, 27.1, 26.0, 26.0, 23.3, 12.1;
IR (neat): 3344, 2922, 1722, 1590, 1478, 743 cm-1;
HRMS (ESI) m/z calcd for C31H40NO2S [(M+H)+] 490.2780, found 490.2780.
7-fluoro-3-methyl-3-phenyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-35): This
compound was prepared according to the General Procedure A using α-methylstyrene (74
mg, 0.625 mmol), 2-amino-5-fluorobenzenethiol (54 mg, 0.375 mmol). After purification
by column chromatography on SiO2 (10% DCM in hexanes), the title compound was

126
isolated as a purple solid (64 mg, 99% yield).
1H NMR (600 MHz, CDCl3): = 7.45-7.40 (m, 2 H), 7.36 (t, J = 7.6 Hz, 2 H), 7.32-7.27
(m, 1 H), 6.76 (dd, J = 2.7, 8.8 Hz, 1 H), 6.70 (dt, J = 2.7, 8.4 Hz, 1 H), 6.55 (dd, J = 4.8,
8.8 Hz, 1 H), 4.11 (br. s., 1 H), 3.17 (d, J = 12.7 Hz, 1 H), 2.96 (d, J = 12.7 Hz, 1 H), 1.70
(s, 3 H);
13C NMR (150 MHz, CDCl3): = 156.1, 154.5, 146.5, 137.3, 128.5, 127.2, 125.3, 116.0,
115.8, 115.7, 113.6, 113.4, 112.6, 112.4, 54.3, 37.6, 29.2;
IR (neat): 3398, 2925, 1577,1476, 799, 698 cm-1;
HRMS (ESI) m/z calcd for C15H15FNS [(M+H)+] 260.0909, found 260.0901.
7-chloro-3-methyl-3-phenyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-36): This
compound was prepared according to the General Procedure A using α-methylstyrene (74
mg, 0.625 mmol), 2-amino-5-chlorobenzenethiol (60 mg, 0.375 mmol). After purification
by column chromatography on SiO2 (10% DCM in hexanes), the title compound was
isolated as a white solid (65 mg, 94% yield).
1H NMR (600 MHz, CDCl3): = 7.43-7.33 (m, 4 H), 7.32-7.27 (m, 1 H), 7.00 (d, J = 2.2
Hz, 1 H), 6.92 (dd, J = 2.2, 8.5 Hz, 1 H), 6.54 (d, J = 8.5 Hz, 1 H), 4.22 (s, 1 H), 3.14 (d, J
= 12.7 Hz, 1 H), 2.94 (d, J = 12.7 Hz, 1 H), 1.71 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 146.3, 139.7, 128.6, 127.2, 126.7, 125.5, 125.3, 122.0,
116.3, 116.0, 54.6, 37.5, 29.4;
IR (neat): 3407, 2928, 1580,1475,797, 698 cm-1;

127
HRMS (ESI) m/z calcd for C15H15ClNS [(M+H)+] 276.0614, found 276.0616.
7-bromo-3-methyl-3-phenyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-37): This
compound was prepared according to the General Procedure A using α-methylstyrene (74
mg, 0.625 mmol), 2-amino-5-bromobenzenethiol (77 mg, 0.375 mmol). After purification
by column chromatography on SiO2 (10% DCM in hexanes), the title compound was
isolated as a white solid (78 mg, 97% yield).
1H NMR (600 MHz, CDCl3): =7.42-7.33 (m, 4 H), 7.32-7.27 (m, 1 H), 7.13 (s, 1 H),
7.05 (dd, J = 2.0, 8.5 Hz, 1 H), 6.49 (d, J = 8.5 Hz, 1 H), 4.23 (br. s., 1 H), 3.13 (d, J = 12.7
Hz, 1 H), 2.93 (d, J = 12.7 Hz, 1 H), 1.71 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 146.3, 140.1, 129.5, 128.6, 128.3, 127.2, 125.2, 116.8,
116.4, 108.9, 54.6, 37.5, 29.4;
IR (neat): 3400, 2968, 1576, 1474, 799 cm-1;
HRMS (ESI) m/z calcd for C15H15BrNS [(M+H)+] 320.0109, found 320.0119.
6-chloro-3-methyl-3-phenyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-38): This
compound was prepared according to the General Procedure A using α-methylstyrene (74
mg, 0.625 mmol), 2-amino-4-chlorobenzenethiol (60 mg, 0.375 mmol). After purification

128
by column chromatography on SiO2 (10% DCM in hexanes), the title compound was
isolated as a white solid (66 mg, 96% yield).
1H NMR (600 MHz, CDCl3): = 7.43-7.34 (m, 4 H), 7.33-7.27 (m, 1 H), 6.94-6.89 (m, 1
H), 6.64-6.59 (m, 2 H), 4.27 (s, 1 H), 3.13 (d, J = 12.7 Hz, 1 H), 2.93 (d, J = 12.7 Hz, 1 H),
1.72 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 146.3, 142.0, 130.8, 128.6, 128.5, 127.2, 125.3, 117.6,
114.5, 113.0, 54.7, 37.6, 29.4;
IR (neat): 3402, 2920, 1577, 1474, 792, 698 cm-1;
HRMS (ESI) m/z calcd for C15H15ClNS [(M+H)+] 276.0614, found 276.0609.
3-methyl-3-phenyl-6-(trifluoromethyl)-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-39):
This compound was prepared according to the General Procedure A using α-methylstyrene
(74 mg, 0.625 mmol), 2-amino-4-(trifluoromethyl)benzenethiol hydrochloride (86 mg,
0.375 mmol). After purification by column chromatography on SiO2 (10% DCM in
hexanes), the title compound was isolated as a white solid (76 mg, 98% yield).
1H NMR (600 MHz, CDCl3): = 7.45-7.35 (m, 4 H), 7.34-7.28 (m, 1 H), 7.10 (d, J = 8.1
Hz, 1 H), 6.90-6.83 (m, 2 H), 4.40 (s, 1 H), 3.19 (d, J = 12.7 Hz, 1 H), 2.97 (d, J = 12.7 Hz,
1 H), 1.74 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 146.1, 141.0, 128.6, 127.9, 127.8, 127.7, 127.3, 125.2,
125.1, 123.3, 119.2, 114.0, 114.0, 113.9, 113.9, 111.4, 111.4, 111.4, 111.4, 54.7, 37.5, 29.4;
IR (neat): 3406, 2927, 1600, 1482, 1071, 864, 701 cm-1;

129
HRMS (ESI) m/z calcd for C16H15F3NS [(M+H)+] 310.0877, found 310.0875.
7-methoxy-3-methyl-3-phenyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-40): This
compound was prepared according to the General Procedure A using α-methylstyrene (74
mg, 0.625 mmol), 2-amino-5-methoxybenzenethiol (58 mg, 0.375 mmol). After
purification by column chromatography on SiO2 (5% EtOAc in hexanes), the title
compound was isolated as a yellow gummy solid (47 mg, 69% yield).
1H NMR (600 MHz, CDCl3): = 7.44 (d, J = 8.1 Hz, 2 H), 7.35 (t, J = 7.7 Hz, 2 H), 7.28
(d, J = 7.3 Hz, 1 H), 6.63 - 6.54 (m, 3 H), 3.96 (s, 1 H), 3.72 (s, 3 H), 3.18 (d, J = 12.7 Hz,
1 H), 2.96 (d, J = 12.7 Hz, 1 H), 1.68 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 151.9, 146.8, 135.1, 128.5, 127.0, 125.4, 116.4, 115.6,
112.8, 111.7, 55.7, 54.2, 37.9, 29.2;
IR (neat): 3375, 2924, 1600, 1491, 697 cm-1;
HRMS (ESI) m/z calcd for C16H18NOS [(M+H)+] 272.1109, found 272.1112.
3,6,7-trimethyl-3-phenyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-41): This
compound was prepared according to the General Procedure A using α-methylstyrene (74
mg, 0.625 mmol), 2-amino-4,5-dimethylbenzenethiol (57 mg, 0.375 mmol). After
purification by column chromatography on SiO2 (10% DCM in hexanes), the title

130
compound was isolated as a yellow gummy solid (65 mg, 97% yield).
1H NMR (600 MHz, CDCl3): = 7.45 (d, J = 7.8 Hz, 2 H), 7.37 (t, J = 7.6 Hz, 2 H), 7.30
(d, J = 7.3 Hz, 1 H), 6.81 (s, 1 H), 6.47 (s, 1 H), 4.05 (br. s., 1 H), 3.14 (d, J = 12.7 Hz, 1
H), 2.93 (d, J = 12.7 Hz, 1 H), 2.20 (s, 3 H), 2.15 (s, 3 H), 1.71 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 147.0, 138.9, 134.1, 128.4, 128.2, 127.0, 126.0, 125.4,
116.6, 111.2, 54.4, 38.0, 29.2, 19.4, 18.6;
IR (neat): 3281, 2920, 1600, 1481, 744 cm-1;
HRMS (ESI) m/z calcd for C17H20NS [(M+H)+] 270.1316, found 270.1310.
3-methyl-3-phenyl-3,4-dihydro-2H-benzo[b][1,4]thiazine-7-carboxylic acid (4-42):
This compound was prepared according to the General Procedure A using α-methylstyrene
(74 mg, 0.625 mmol), 4-amino-3-mercaptobenzoic (63 mg, 0.375 mmol). After
purification by column chromatography on SiO2 (25% DCM in hexanes), the title
compound was isolated as a yellow solid (60 mg, 84% yield).
1H NMR (600 MHz, CDCl3): = 7.82 (s, 1 H), 7.72 (d, J = 8.3 Hz, 1 H), 7.40-7.33 (m, 4
H), 7.32-7.27 (m, 1 H), 6.61 (d, J = 8.5 Hz, 1 H), 4.77 (br. s., 1 H), 3.12 (d, J = 12.7 Hz, 1
H), 2.95 (d, J = 12.7 Hz, 1 H), 1.79-1.72 (m, 3 H);
13C NMR (150 MHz, CDCl3): = 171.6, 146.0, 145.8, 130.6, 128.7, 128.5, 127.4, 125.2,
118.1, 114.1, 114.0, 55.6, 37.5, 29.5;
IR (neat): 3371, 2924, 1656, 1560, 1251, 697 cm-1;
HRMS (ESI) m/z calcd for C16H16NO2S [(M+H)+] 286.0902, found 286.0904.

131
2-methyl-6-nitro-2-phenyl-2,3-dihydrobenzo[b][1,4]oxathiine (4-43): This compound
was prepared according to the General Procedure A using α-methylstyrene (74 mg, 0.625
mmol), 2-mercapto-4-nitrophenol (64 mg, 0.375 mmol). After purification by column
chromatography on SiO2 (10% DCM in hexanes), the title compound was isolated as a
yellow gummy solid (57 mg, 79% yield).
1H NMR (600 MHz, CDCl3): = 7.98 (d, J = 2.4 Hz, 1 H), 7.94 (dd, J = 2.4, 9.0 Hz, 1
H), 7.37 (d, J = 4.2 Hz, 4 H), 7.32 (dd, J = 4.2, 8.3 Hz, 1 H), 7.08 (d, J = 9.0 Hz, 1 H), 3.33
(d, J = 13.7 Hz, 1 H), 3.20 (d, J = 13.7 Hz, 1 H), 1.79 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 155.9, 143.2, 141.5, 128.7, 127.8, 124.5, 123.5, 121.6,
119.2, 118.3, 77.7, 34.7, 28.5;
IR (neat): 2927, 1510, 1337, 1066, 698 cm-1;
HRMS (ESI) m/z calcd for C15H14NO3S [(M+H)+] 288.0694, found 288.0687.
3,4-dihydro-2H-benzo[b][1,4]thiazine (4-44): This compound was prepared according to
the General Procedure B using 2-(phenylamino)ethane-1-thiol (50 mg, 0.325 mmol). After
purification by column chromatography on SiO2 (20% DCM in hexanes), the title
compound was isolated as a yellow liquid (30 mg, 79% yield).
1H NMR (600 MHz, CDCl3): = 7.02-6.97 (m, 1 H), 6.92-6.86 (m, 1 H), 6.62 (t, J = 7.6
Hz, 1 H), 6.46 (d, J = 8.1 Hz, 1 H), 3.97 (br. s., 1 H), 3.67-3.58 (m, 2 H), 3.10-3.01 (m, 2

132
H);
13C NMR (150 MHz, CDCl3): = 141.7, 127.7, 125.4, 118.1, 115.9, 115.3, 42.2, 26.0;
IR (neat): 3400, 2926, 1587, 1483, 739 cm-1;
HRMS (ESI) m/z calcd for C8H10NS [(M+H)+] 152.0534, found 152.0538.
4-methyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-45): This compound was prepared
according to the General Procedure B using 2-(methyl(phenyl)amino)ethane-1-thiol (54
mg, 0.325 mmol). After purification by column chromatography on SiO2 (20% DCM in
hexanes), the title compound was isolated as a yellow liquid (39 mg, 94% yield).
1H NMR (600 MHz, CDCl3): = 7.09-7.00 (m, 2 H), 6.71-6.63 (m, 2 H), 3.59-3.52 (m, 2
H), 3.12-3.06 (m, 2 H), 2.96 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 144.6, 127.4, 125.9, 118.6, 117.6, 112.5, 51.6, 40.1,
26.3;
IR (neat): 2924, 1585, 1489, 738 cm-1;
HRMS (ESI) m/z calcd for C9H12NS [(M+H)+] 166.0690, found 166.0681
Trans-2,3,4,4a,10,10a-hexahydro-1H-phenothiazine (4-46): This compound was
prepared according to the General Procedure B using 2-(phenylamino)cyclohexane-1-thiol

133
(67 mg, 0.325 mmol). After purification by column chromatography on SiO2 (20% DCM
in hexanes), the title compound was isolated as a white solid (47 mg, 92% yield, >95:5 dr).
1H NMR (600 MHz, CDCl3): = 7.02-6.94 (m, 1 H), 6.91-6.83 (m, 1 H), 6.61 (t, J = 7.1
Hz, 1 H), 6.47 (d, J = 7.8 Hz, 1 H), 3.85-3.65 (m, 1 H), 3.19-3.07 (m, 1 H), 2.94 (ddd, J =
3.5, 8.8, 11.8 Hz, 1 H), 2.02 (d, J = 12.9 Hz, 1 H), 1.98-1.92 (m, 1 H), 1.88-1.79 (m, 2 H),
1.51-1.27 (m, 4 H);
13C NMR (150 MHz, CDCl3): = 141.4, 126.8, 125.2, 118.0, 117.0, 114.7, 56.1, 42.8,
33.4, 30.5, 25.6, 24.4;
IR (neat): 3353, 2922, 1588, 1482, 1305, 736 cm-1;
HRMS (ESI) m/z calcd for C12H16NS [(M+H)+] 206.1003, found 206.1004.
2-methyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-47): This compound was prepared
according to the General Procedure B using 1-(phenylamino)propane-2-thiol (54 mg, 0.325
mmol). After purification by column chromatography on SiO2 (20% DCM in hexanes), the
title compound was isolated as a yellow solid (35 mg, 85% yield).
1H NMR (600 MHz, CDCl3): = 6.98 (d, J = 7.6 Hz, 1 H), 6.89 (t, J = 7.6 Hz, 1 H), 6.63
(t, J = 7.6 Hz, 1 H), 6.49 (d, J = 8.1 Hz, 1 H), 4.01 (br. s., 1 H), 3.59 (dd, J = 2.2, 11.7 Hz,
1 H), 3.41-3.32 (m, 1 H), 3.22 (dd, J = 8.3, 11.7 Hz, 1 H), 1.36 (d, J = 6.8 Hz, 3 H);
13C NMR (150 MHz, CDCl3): = 141.1, 127.4, 125.2, 118.1, 116.5, 115.0, 49.2, 34.0,
19.1;
IR (neat): 3403, 2921, 1490, 1484, 739 cm-1;

134
HRMS (ESI) m/z calcd for C9H12NS [(M+H)+] 166.0690, found 166.0682.
2,2-dimethyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-48): This compound was
prepared according to the General Procedure B using 2-methyl-1-(phenylamino)propane-
2-thiol (59 mg, 0.325 mmol). After purification by column chromatography on SiO2 (20%
DCM in hexanes), the title compound was isolated as a colorless liquid (32 mg, 71% yield).
1H NMR (600 MHz, CDCl3): = 6.96 (d, J = 7.8 Hz, 1 H), 6.91 (t, J = 7.6 Hz, 1 H), 6.63
(t, J = 7.4 Hz, 1 H), 6.52 (d, J = 8.1 Hz, 1 H), 4.12 (br. s., 1 H), 3.25 (s, 2 H), 1.42 (s, 6 H);
13C NMR (150 MHz, CDCl3): = 140.1, 127.6, 125.2, 118.0, 116.4, 114.7, 54.5, 39.5,
27.9;
IR (neat): 3409, 2959, 1590, 1484, 739 cm-1;
HRMS (ESI) m/z calcd for C10H14NS [(M+H)+] 180.0847, found 180.0844.
5-fluoro-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-49): This compound was prepared
according to the General Procedure B using 2-((2-fluorophenyl)amino)ethane-1-thiol (56
mg, 0.325 mmol). After purification by column chromatography on SiO2 (20% DCM in
hexanes), the title compound was isolated as a yellow liquid (40 mg, 95% yield).
1H NMR (600 MHz, CDCl3): = 6.81-6.69 (m, 2 H), 6.52 (dt, J = 5.5, 8.0 Hz, 1 H), 4.24

135
(br. s., 1 H), 3.67 (td, J = 2.7, 7.3 Hz, 2 H), 3.11-3.03 (m, 2 H);
13C NMR (150 MHz, CDCl3): = 151.6, 150.1, 130.4, 130.3, 122.8, 122.8, 117.5, 117.5,
116.5, 116.4, 111.1, 110.9, 41.4, 25.7;
IR (neat): 3419, 2929, 1608, 1496, 757 cm-1;
HRMS (ESI) m/z calcd for C8H9FNS [(M+H)+] 170.0440, found 170.0433.
8-chloro-3,4-dihydro-2H-benzo[b][1,4]thiazine and 6-chloro-3,4-dihydro-2H-
benzo[b][1,4]thiazine (4-50): This compound was prepared according to the General
Procedure B using 2-((3-chlorophenyl)amino)ethane-1-thiol (61 mg, 0.325 mmol). After
purification by column chromatography on SiO2 (20% DCM in hexanes), the title
compounds were isolated as clear liquid (major) and white solid (minor) (44 mg, 95%
yield, 1.5:1 rr).
1H NMR (600 MHz, CDCl3) for major: = 6.81 (t, J = 7.9 Hz, 1 H), 6.72 (d, J = 7.9 Hz,
1 H), 6.37 (d, J = 8.1 Hz, 1 H), 4.11 (br. s., 1 H), 3.63-3.54 (m, 2 H), 3.12-3.04 (m, 2 H);
13C NMR (150 MHz, CDCl3) for major: = 143.1, 131.6, 125.2, 118.6, 115.6, 113.2,
41.5, 26.3;
IR (neat) for major: 3403, 2925, 1580, 1304, 750 cm-1
HRMS (ESI) for major m/z calcd for C8H9ClNS [(M+H)+] 186.0144, found 186.0145.
1H NMR (600 MHz, CDCl3) for minor: = 6.89 (d, J = 8.2 Hz, 1 H), 6.57 (dd, J = 1.8,
8.2 Hz, 1 H), 6.45 (d, J = 1.8 Hz, 1 H), 4.05 (br. s., 1 H), 3.66-3.58 (m, 2 H), 3.05-2.97 (m,

136
2 H);
13C NMR (150 MHz, CDCl3) for minor: = 142.4, 130.7, 128.6, 117.8, 114.5, 114.1,
42.1, 25.5;
IR (neat) for minor: 3408, 2928, 1581, 1307, 759 cm-1;
HRMS (ESI) for minor m/z calcd for C8H9ClNS [(M+H)+] 186.0144, found 186.0143.
5,7-difluoro-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-51): This compound was
prepared according to the General Procedure B using 2-((2,4-
difluorophenyl)amino)ethane-1-thiol (61 mg, 0.325 mmol). After purification by column
chromatography on SiO2 (20% DCM in hexanes), the title compound was isolated as a
clear liquid (45 mg, 96% yield).
1H NMR (600 MHz, CDCl3): = 6.60-6.49 (m, 2 H), 4.05 (br. s., 1 H), 3.62 (dt, J = 2.7,
4.8 Hz, 2 H), 3.12-3.06 (m, 2 H);
13C NMR (150 MHz, CDCl3): = 154.6, 154.5, 153.0, 152.9, 151.1, 151.0, 149.5, 149.4,
126.8, 126.8, 118.7, 118.7, 118.6, 118.6, 109.0, 108.9, 108.8, 108.8, 100.1, 100.0, 99.9,
99.8, 41.1, 26.2;
IR (neat): 3424, 1621, 1494, 1103, 994, 585 cm-1;
HRMS (ESI) m/z calcd for C8H8F2NS [(M+H)+] 188.0346, found 188.0338.

137
8-fluoro-5-methoxy-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-52): This compound was
prepared according to the General Procedure B using 2-((5-fluoro-2-
methoxyphenyl)amino)ethane-1-thiol (65 mg, 0.325 mmol). After purification by column
chromatography on SiO2 (30% DCM in hexanes), the title compound was isolated as a
yellow liquid (49 mg, 98% yield).
1H NMR (600 MHz, CDCl3): = 6.44 (dd, J = 4.9, 8.8 Hz, 1 H), 6.33 (t, J = 8.9 Hz, 1 H),
4.65 (br. s., 1 H), 3.80 (s, 3 H), 3.69-3.60 (m, 2 H), 3.06-2.99 (m, 2 H);
13C NMR (150 MHz, CDCl3): = 154.5, 153.0, 142.1, 142.1, 132.5, 132.4, 105.5, 105.4,
103.6, 103.5, 101.1, 100.9, 55.8, 41.1, 24.5;
IR (neat): 3418, 2931, 1609, 1498, 775 cm-1;
HRMS (ESI) m/z calcd for C9H11FNOS [(M+H)+] 200.0545, found 200.0538.
4-(2,6-dimethylphenyl)-3-phenylthiomorpholine (4-54): This compound was prepared
according to the General Procedure A using styrene (65 mg, 0.625 mmol), 2-((2,6-
dimethylphenyl)amino)ethane-1-thiol (68 mg, 0.375 mmol), NaI (20 mol%, 7.5 mg) for 16
h. After purification by column chromatography on SiO2 (10% DCM in hexanes), the title
compound was isolated as a white solid (22 mg, 31% yield).

138
1H NMR (600 MHz, CDCl3): = 7.18 (d, J = 7.8 Hz, 2 H), 7.11-7.02 (m, 3 H), 6.92 (d, J
= 7.3 Hz, 1 H), 6.77 (t, J = 7.3 Hz, 1 H), 6.64 (d, J = 7.3 Hz, 1 H), 4.63 (d, J = 10.0 Hz, 1
H), 3.58 (t, J = 12.2 Hz, 1 H), 3.30 (d, J = 12.9 Hz, 1 H), 3.18-3.09 (m, 1 H), 2.99-2.90 (m,
1 H), 2.58-2.50 (m, 2 H), 2.43 (s, 3 H), 2.31 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 147.8, 142.2, 137.0, 136.0, 129.4, 128.2, 127.8, 127.1,
126.9, 125.1, 66.9, 53.4, 37.5, 28.4, 20.3, 19.4;
IR (neat): 2897, 1583, 1450, 771, 518 cm-1;
HRMS (ESI) m/z calcd for C18H22NS [(M+H)+] 284.1473, found 284.1465.
3-cyclopropyl-3-phenyl-3,4-dihydro-2H-benzo[b][1,4]thiazine (4-55): This compound
was prepared according to the General Procedure A using α-cycloopropylstyrene (90 mg,
0.625 mmol), 2-aminobenzenethiol (40 μL, 0.375 mmol). After purification by column
chromatography on SiO2 (10% DCM in hexanes), the title compound was isolated as a
clear liquid (8 mg, 12% yield).
1H NMR (600 MHz, CDCl3): = 7.38 (d, J = 7.8 Hz, 2 H), 7.31 (t, J = 7.8 Hz, 2 H), 7.28-
7.21 (m, 1 H), 6.97-6.91 (m, 2 H), 6.64-6.56 (m, 2 H), 4.27 (br. s., 1 H), 3.27 (d, J = 12.7
Hz, 1 H), 3.12 (d, J = 12.7 Hz, 1 H), 1.38-1.30 (m, 1 H), 0.58-0.44 (m, 2 H), 0.34-0.21 (m,
2 H);
13C NMR (150 MHz, CDCl3): = 143.9, 141.1, 128.2, 127.7, 127.1, 126.8, 125.7, 117.6,
115.1, 114.9, 57.6, 36.1, 22.6, 1.5, 0.6;
IR (neat): 3328, 2956, 1587, 1478, 740 cm-1;
HRMS (ESI) m/z calcd for C17H20NS [(M+H)+] 268.1160, found 268.1159

139
Chapter 5
Alkene Catalyzed Construction of All-Carbon Quaternary
Center Via C-H Alkylation of Arene
5.1 Introduction
Alcohols are prevalent in many natural products and biologically active
compounds. Alcohols are considered as important chemical feedstocks for many organic
transformations because of easy accessibility, versatility, commercial availability, and
efficient synthetic procedures, and can be modified into other functional groups via
nucleophilic substitution.147-149 Direct nucleophilic substitution of alcohol is quite
challenging because of their thermodynamic and kinetic barrier for removal of hydroxide
anion, other chemical activators are required.150 Traditionally, protonation, using strong
acids, conversion of alcohol to sulfonate, phosphite ester are the widely used strategies to
achieve the goal of substitution of alcohol.151-152 Conventionally, the Mitsunobu reaction
is one of the most frequently used strategy, despite fact that it involves the use of hazardous
and mass intensive stoichiometric reagents.153-154 In addition, several catalytic strategies
using Lewis acids, transition metals have been developed for the conversion of alcohols to
the various useful compounds.155-156 The Shenvi group developed an elegant strategy for
Lewis acid-catalyzed stereoinversion of tertiary alcohols to tertiary alkyl isonitrile via
conversion of alcohol to their corresponding trifluoroacetate (Scheme 5-1a).157

140
Scheme 5-1: Literature Background of Alcohol Activation

141
Moreover, the Lambert group has made a significant contribution to the nucleophilic
substitution of alcohols into various valuable compounds by utilizing the Breslow-type
cyclopropenium cation (Scheme 5-1e).158-162 Similarly, the Nguyen group demonstrated
catalytic activity of the organic Lewis acid tropylium for the activation of hetero atom
containing compounds (Scheme 5-1e).163-167 Recently, the Denton group reported an
organic phosphine oxide catalyzed Mitsunobu reaction of alcohol for C-O, C-N, C-S bond
formations (Scheme 5-1b).168 Despite the significant advancement of primary and
secondary alcohol functionalizations, tertiary alcohol functionalization remains significant
challenges due to their competing elimination reaction.169 Moreover, the functionalization
of tertiary alcohol for the construction of all-carbon quaternary center is difficult due to
strong steric hindrance.150
Our group is interested in the utilization of halogen a widely used synthetic reagent
in many organic transformations that garnered enormous attention from synthetic and
medicinal chemists. A significant effort has been devoted to identifying the reactive
intermediates and their reaction mechanism. In 1937, Kimball first proposed the cyclic
three-membered halonium ion as an intermediate for the halogenation of ethylene.43 Later,
the syntheses of the retively more stable bromonium, chloronium, and iodonium of
adamantylideneadamantane and their characterization has been done by Olah,44-45
Nugent,46 Brown47 followed by the elucidation of X-ray structure of bridged 2,2’-
bis(adamant-2-ylidene) chloronium cation by Kochi lab (Scheme 5-1c).50 With such
success, synthetic utilities of those halonium intermediates have been demonstrated by
halocyclization of alkenols and alkenoic acids, which often proceeded via halonium
transfer from adamantylideneadamantane to the acceptor alkene.48-49 Later, the Denmark

142
lab studied the absolute configurational stability of bromonium and chloronium ions by
examining the retention of enantiospecificity of nucleophilic substituted products.170 In
some cases, bromonium ion transfer has been observed between substrate and alkene,
similar to the previously reported alkene to alkene transfer processes for thiiranium and
seleniranium (Scheme 5-1d).105 Recently, the Matsubara group further expanded this
promising strategy by demonstrating the sterically hindered alkene-catalyzed
bromolactonization of alkenoic acids by utilizing the alkene to alkene transfer phenomenon
of an in-situ generated bromonium ion.171 Besides, halogen or halonium ion has been
extensively used in the activation of alkene for halofunctionalization and
difunctionalization reactions. The Denmark lab has been a pioneer, that has made
significant contribution in the halofunctionalization of alkene.78 Muniz lab developed a
bromonium-catalyzed intramolecular diamination of the alkene.114 Moreover, our group
and a few others have been focusing on halogen activation of alkene for heterocycle
syntheses.172-174 Despite these advances, halonium activation of heteroatom has been
underdeveloped, represents significant potential in organic chemistry. To the best of our
knowledge, iodonium transfer from alkene to alcohol has not been realized so far.
As a part of our general program in halogen activation, we propose here an alkene
catalyzed C-H alkylation of arene for the construction of quaternary carbon center via
iodonium transfer from an alkene to an alcohol (Scheme 5-1f). We envision that
electrophilic iodinating reagent could generate iodonium with an alkene, which would
transfer to alcohol due to nucleophilicity of alcohol, resulting in carbocation formation with
the elimination of HOI. The newly generated carbocation could be attacked by arene,
leading to the formation of an alkylated quaternary carbon center.

143
Table 5.1: Optimization of Reaction Condition

144
5.2 Results and Discussions for C-H Alkylation of Arene
5.2.1 Reaction Design and Optimization
With this hypothesis in mind, the reaction was commenced with adamantanol, NIS as the
halogen source, 1-methylcyclohexene as the catalyst, and Ts-protected pyrrole as a
nucleophile in hexafluoroisopropanol (HFIP) at rt for 16 h. To our delight, the reaction
provided 47% yield of the expected product as a single regioisomer (Table 5.1, entry 1).
Surprisingly, other solvents were unable to afford any product (Table 5.1 entries 2 and 3).
After the screening of stoichiometry of nucleophile and concentration revealed that 1.5
equiv of arene and 1.5 mL of HFIP was the best combination, providing the product in 94%
yield (Table 5.1, entries 4-7). Other alkene catalysts did not improve the yield of the
product (Table 5.1, entries 8-11). As expected, reactions in absence of either the alkene or
NIS did not generate any product, demonstrating that both NIS and alkene catalyst were
required for alkylation of arene (Table 5.1, entries 12 and 13).
5.2.2 Substrate Scope
With optimized conditions in hand, we focused on the exploration of the arene
substrate scope for the alkylation of arenes. Different indole derivatives including electron-
withdrawing substituent afforded the products in very good yields with excellent
regioselectivities (Table 5.2, products 5-10 to 5-12). In these cases, the regioselectivity
favoring the alkylation at C-3 position of indoles and pyrrole. Surprisingly, the reaction
occurred when the C-3 position was blocked with ethyl group, delivering the product with
adamantyl group attached to C-2 and C-5 position of indole in almost equal
regioselectivities (Table 5.2, product 5-13). Moreover, 2-oxindole was a compatible
145
Table 5.2: Arene Substrate Scope

146
Table 5.3: Alcohol Substrate Scope
substrate, providing the product with excellent regioselectivity albeit in low yield (Table

147
5.2, product 5-14). The sulfur-containing heterocycles such as benzothiophene, thiophene
afforded products in good yields (Table 5.2, products 5-15 and 5-16). The aniline
derivatives delivered the products with excellent regioselectivities (Table 5.2, products 5-
17 to 5-19. Interestingly, the adamantyl group was added chemoselectively to 4-position
of aniline in the presence of a methoxy group in another aromatic ring (Table 5.2, product
5-19). Different nucleophilic functional groups such as sulfur, ether, and alcohol were well
tolerated and provided the products in good yields with high regioselectivities (Table 5.2,
products 5-20 to 5-24). To our surprise, electron neutral ring toluene furnished the product
in good yield (Table 5.2, product 5-25). A natural product such as estradiol provided
product with demonstrating the chemoselectivity for the aromatic ring instead of a
secondary alcohol (5.2, product 5-26). The late-stage functionalization of Tyrosine,
Gemifibrozil, Naproxen further highlights the synthetic utility of our method (Table 5.2,
product 5-27 to 5-29).
After satisfied with the arene substrate scope, we then turned our attention to
discover alcohol substrate scope. Different six-membered cyclic alcohols provided the
products in very good yields with exceptional regioselectivities (Table 5.3, products 5-30
to 5-34). The methyl, tert-butyl substituted alcohols furnished the products with good
diastereoselectivities (Table 5.3, products 5-33 and 5-34). Four-, five-, eight-, twelve-
membered cyclic alcohols were also excellent substrates in product formation with a good
regioselective manner (Table 5.3, products 5-35 to 5-38). O- and N-containing heterocyclic
alcohols were compatible and delivered the products without loss of efficiency (Table 5.3,
products 5-39 and 5-40). The benzylic alcohols with different substituents provided the
sterically encumbered products in a highly regioselective manner (Table 5.3, products 5-

148
41 to 5-45). Tert-butanol and tert-amyl alcohol also provided products with good
regioisomers (Table 5.3, products 5-46 and 5-47). The complex alcohol, derived from
lithocholic acid afforded the product with a quaternary carbon center, demonstrating
chemoselectivity for tertiary alcohol over secondary alcohol (Table 5.3, product 5-48).
5.2.3 Chemoselectivity of Alcohols
The synthetic applicability of our protocol was further demonstrated by the
chemoselectivity of different alcohols for C-C bond formation via alkylation of arene
(Scheme 5-2). Surprisingly, the aliphatic tertiary alcohol showed preference over benzylic
tertiary alcohol for the alkylation of pyrrole, providing the product in high yield (Scheme
5-2a). Moreover, pyrrole was added selectively to Adamantanol in the presence of primary
and secondary alcohol, highlighting the utility of our method (Scheme 5-2b,c).
Scheme 5-2: Chemoselectivity of Different Alcohols
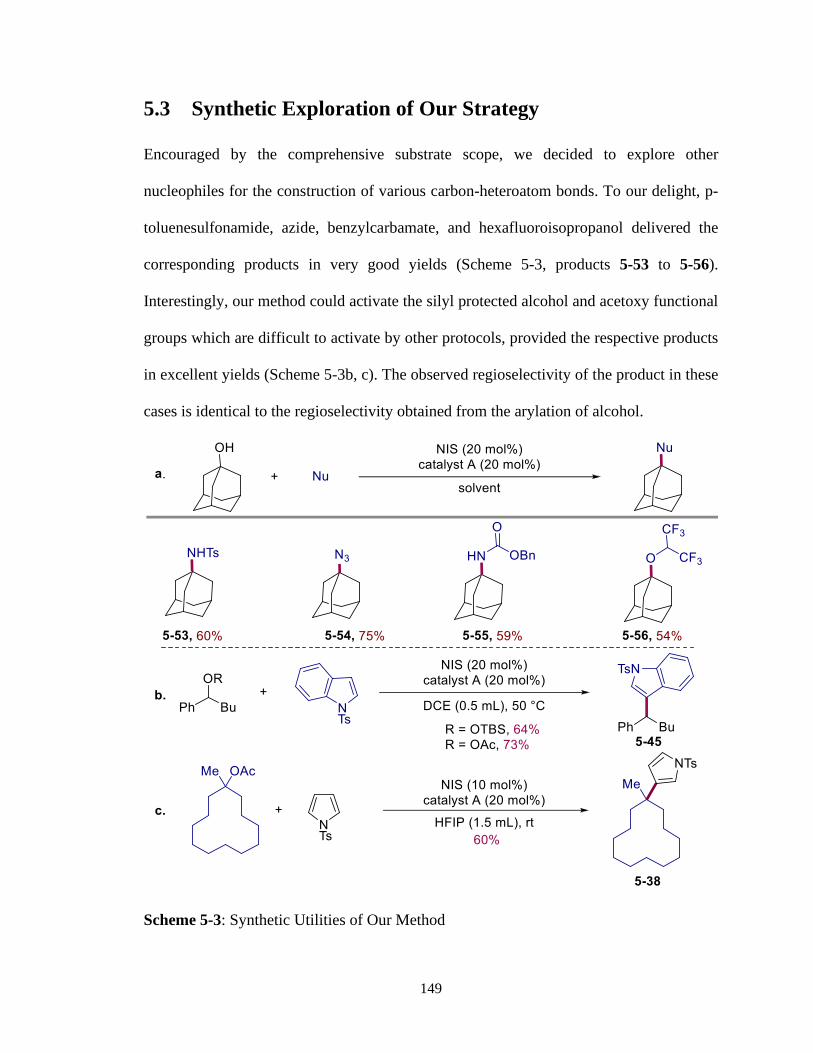
149
5.3 Synthetic Exploration of Our Strategy
Encouraged by the comprehensive substrate scope, we decided to explore other
nucleophiles for the construction of various carbon-heteroatom bonds. To our delight, p-
toluenesulfonamide, azide, benzylcarbamate, and hexafluoroisopropanol delivered the
corresponding products in very good yields (Scheme 5-3, products 5-53 to 5-56).
Interestingly, our method could activate the silyl protected alcohol and acetoxy functional
groups which are difficult to activate by other protocols, provided the respective products
in excellent yields (Scheme 5-3b, c). The observed regioselectivity of the product in these
cases is identical to the regioselectivity obtained from the arylation of alcohol.
Scheme 5-3: Synthetic Utilities of Our Method
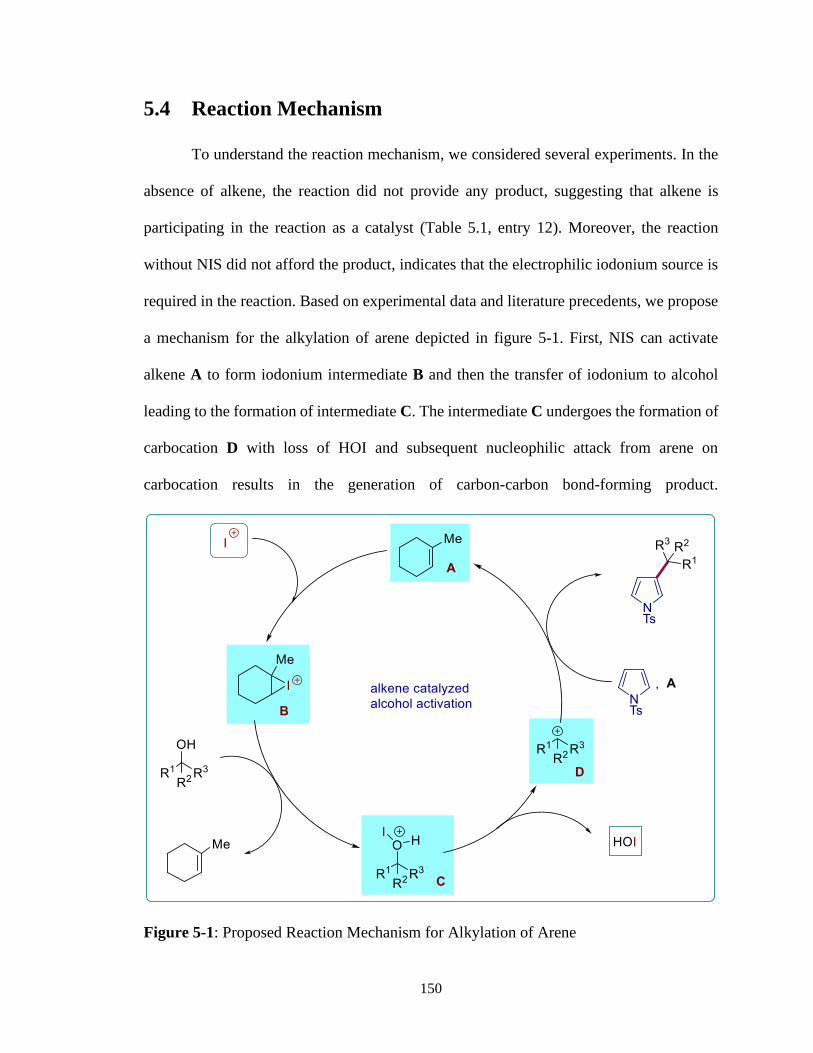
150
5.4 Reaction Mechanism
To understand the reaction mechanism, we considered several experiments. In the
absence of alkene, the reaction did not provide any product, suggesting that alkene is
participating in the reaction as a catalyst (Table 5.1, entry 12). Moreover, the reaction
without NIS did not afford the product, indicates that the electrophilic iodonium source is
required in the reaction. Based on experimental data and literature precedents, we propose
a mechanism for the alkylation of arene depicted in figure 5-1. First, NIS can activate
alkene A to form iodonium intermediate B and then the transfer of iodonium to alcohol
leading to the formation of intermediate C. The intermediate C undergoes the formation of
carbocation D with loss of HOI and subsequent nucleophilic attack from arene on
carbocation results in the generation of carbon-carbon bond-forming product.
Figure 5-1: Proposed Reaction Mechanism for Alkylation of Arene

151
5.5 Conclusion
We have developed a simple and highly efficient method for carbon-carbon bond
formation via alkylation of the arene. The reaction utilizes an alkene as a catalyst for the
in-situ generation of iodonium and subsequent activation of alcohol. The method shows
good functional group compatibility and a broad range of substrate scope. The reaction is
chemoselective for tertiary alcohol in preference to secondary or primary alcohols. We
have also demonstrated that various other nucleophiles could also couple with alcohol to
provide the products in a highly efficient manner. Moreover, we have shown that our
reaction protocol could apply to activate ether and ester to generate their corresponding
products which will enable the expansion of this chemistry to the related systems. We
expect that this reaction will lead to the development of other alkene catalyzed reactions
and further advances the carbocation chemistry.
5.6 Experimental
General Information. Commercial reagents and solvents were purchased from Sigma
Aldrich, Oakwood Chemicals, Alfa Aesar, Matrix Scientific, Acros Organic, and were
used as received. The alcohol substrates were synthesized according to the reported
procedure. Organic solutions were concentrated under reduced pressure on a Büchi rotary
evaporator using an acetone-dry ice bath. Chromatographic purification of products was
accomplished using flash chromatography on 230-400 mesh silica gel. Thin-layer
chromatography (TLC) was performed on Analtech 250 mm silica gel HLF UV-254 plates.
Visualization of the developed plates was performed by fluorescence quenching, potassium

152
permanganate, and iodine-silica gel system. 1H and 13C NMR spectra were recorded on a
Bruker 600 instrument (600 and 150 MHz) or INOVA 600 (600 and 150 MHz) and are
internally referenced to residual protio solvent signals (for CDCl3, 7.26 and 77.0 ppm,
MeOH-d4 3.31, 4.90 and 49.0 ppm, DMSO-d6 2.50 and 39.53 respectively). Data for 1H
NMR are reported as follows: chemical shift ( ppm), multiplicity (s = singlet, d = doublet,
t = triplet, q = quartet, h = heptet, m = multiplet, br = broad), integration, coupling constant
(Hz). 13C spectra were reported as chemical shifts in ppm and multiplicity where
appropriate.
Experimental Procedures
General Procedure A: The alcohol (0.25 mmol) and arene (0.375 mmol), 1-methyl-1-
cyclohexene (10 mol%, 3 µL), NIS (10 mol%, 6 mg) were added to 8 mL vial. Then, HFIP
(1.5 mL) was added to the reaction mixture and stirred at room temperature for 16 h. After
the reaction, HFIP was removed by rotavapor. The reaction was quenched with 20%
Na2S2O3 (1 mL) and EtOAc (2 mL) was added. The organic layer was separated, and
aqueous layer was extracted with EtOAc (2 x 2 mL). The combined organic solution was
dried with anhydrous Na2SO4, filtered, concentrated in vacuo by rotavapor and purified by
flash chromatography on SiO2 (2%-20% EtOAc in hexanes) to provide the desired product.
General Procedure B: The alcohol (0.25 mmol) and arene (0.375 mmol), 1-methyl-1-
cyclohexene (20 mol%, 6 µL), NIS (20 mol%, 11 mg) were added to 8 mL vial. Then, DCE
(0.5 mL) was added to the reaction mixture and stirred at 50 ºC for 16 h. The reaction was
quenched with 20% Na2S2O3 (1 mL) and EtOAc (2 mL) was added. The organic layer was
separated, and aqueous layer was extracted with EtOAc (2 x 2 mL). The combined organic

153
solution was dried with anhydrous Na2SO4, filtered, concentrated in vacuo by rotavapor
and purified by flash chromatography on SiO2 (2%-5% EtOAc in hexanes) to provide the
desired product.
3. Spectral Characterization of the Products
3-adamantan-1-yl-1-tosyl-1H-pyrrole (5-9): This compound was prepared according to
the General Procedure A using 1-adamantanol (38 mg, 0.25 mmol), 1-tosyl-1H-pyrrole (83
mg, 0.375 mmol). After purification by column chromatography on SiO2 (2% EtOAc in
hexanes), the title compound was isolated as a white solid (79 mg, 89% yield).
1H NMR (600 MHz, CDCl3): = 7.71 (d, J = 8.3 Hz, 2 H), 7.28 (d, J = 8.3 Hz, 2 H), 7.06
(t, J = 2.2 Hz, 1 H), 6.85 (s, 1 H), 6.27-6.22 (m, 1 H), 2.40 (s, 3 H), 2.00 (br. s., 3 H), 1.78-
1.67 (m, 12 H);
13C NMR (150 MHz, CDCl3): = 144.6, 140.9, 136.4, 129.9, 126.7, 120.6, 114.8, 111.7,
43.2, 36.7, 32.6, 28.6, 21.6.

154
3-(adamantan-1-yl)-1-tosyl-1H-indole (5-10): This compound was prepared according to
the General Procedure A using 1-adamantanol (38 mg, 0.25 mmol), 1-tosyl-1H-indole (102
mg, 0.375 mmol). After purification by column chromatography on SiO2 (2% EtOAc in
hexanes), the title compound was isolated as a white solid (98 mg, 97% yield).
1H NMR (600 MHz, CDCl3): = 7.97 (d, J = 8.3 Hz, 1 H), 7.78 (d, J = 7.8 Hz, 1 H),
7.73 (d, J = 8.3 Hz, 2 H), 7.27-7.22 (m, 2 H), 7.22-7.14 (m, 3 H), 2.32 (s, 3 H), 2.08 (br.
s., 3 H), 2.03 (br. s., 6 H), 1.85-1.76 (m, 6 H);
13C NMR (150 MHz, CDCl3): = 144.6, 135.9, 135.3, 132.8, 129.7, 129.3, 126.7,
123.9, 122.3, 122.0, 121.0, 113.9, 42.1, 36.9, 34.0, 28.5, 21.5.
3-(adamantan-1-yl)-5-bromo-1-tosyl-1H-indole (5-11): This compound was prepared
according to the General Procedure A using 1-adamantanol (38 mg, 0.25 mmol), 5-bromo-
1-tosyl-1H-indole (131 mg, 0.375 mmol). After purification by column chromatography
on SiO2 (2% EtOAc in hexanes), the title compound was isolated as a white solid (83 mg,
69% yield).
1H NMR (600 MHz, CDCl3): = 7.88 (s, 1 H), 7.85 (d, J = 8.8 Hz, 1 H), 7.71 (d, J = 8.1
Hz, 2 H), 7.37-7.33 (m, 1 H), 7.25-7.20 (m, 3 H), 2.35 (s, 3 H), 2.10 (br. s., 3 H), 2.00 (br.
s., 6 H), 1.86-1.74 (m, 6 H);
13C NMR (150 MHz, CDCl3): = 145.0, 135.0, 134.6, 132.3, 131.0, 129.9, 126.8, 126.7,

155
124.6, 122.3, 115.9, 115.2, 42.1, 36.8, 34.0, 28.4, 21.6.
3-(adamantan-1-yl)-5-nitro-1-tosyl-1H-indole (5-12): This compound was prepared
according to the General Procedure A using 1-adamantanol (38 mg, 0.25 mmol), 5-nitro-
1-tosyl-1H-indole (119 mg, 0.375 mmol) at 80 ˚C. After purification by column
chromatography on SiO2 (2% EtOAc in hexanes), the title compound was isolated as a
white solid (86 mg, 76% yield).
1H NMR (600 MHz, CDCl3): = 8.68 (s, 1 H), 8.16 (d, J = 9.0 Hz, 1 H), 8.06 (d, J = 9.0
Hz, 1 H), 7.82-7.74 (m, J = 8.1 Hz, 2 H), 7.41 (s, 1 H), 7.32-7.24 (m, J = 8.1 Hz, 2 H), 2.37
(s, 3 H), 2.12 (br. s., 3 H), 2.04 (br. s., 6 H), 1.83 (br. s., 6 H);
13C NMR (150 MHz, CDCl3): = 145.6, 143.2, 138.8, 134.7, 133.4, 130.1, 128.9, 126.8,
123.8, 119.2, 118.3, 113.8, 42.2, 36.7, 34.1, 28.4, 21.6.
5-(adamantan-1-yl)-3-ethyl-1-tosyl-1H-indole (5-13): This compound was prepared
according to the General Procedure A using 1-adamantanol (38 mg, 0.25 mmol), 3-ethyl-

156
1-tosyl-1H-indole (112 mg, 0.375 mmol) 50 ˚C. After purification by column
chromatography on SiO2 (2% EtOAc in hexanes), the inseparable mixture of two
regioisomers was isolated as a white solid (82 mg, 76% yield, 59:41 rr).
1H NMR (600 MHz, CDCl3): = 7.99 (s, 1 H major), 7.90 (d, J = 8.5 Hz, 1 H minor),
7.79- 7.72 (m, 2 H major + 2 H minor), 7.40 (d, J = 8.5 Hz, 1 H major + 1 H minor), 7.36
(d, J = 8.8 Hz, 1 H minor), 7.29-7.26 (m, 2 H major + 1 H minor), 7.19 (d, J = 8.3 Hz, 2 H
major + 2 H minor), 2.73-2.59 (m, 2 H major + 2 H minor), 2.32 (s, 3 H major + 3 H
minor), 2.14 (br. s., 3 H major), 2.10 (br. s., 3 H minor), 2.02-1.90 (m, 6 H major + 6 H
minor), 1.87-1.71 (m, 6 H major + 6 H minor), 1.37-1.23 (m, 3 H major + 3 H minor);
13C NMR (150 MHz, CDCl3): = 148.5, 146.3, 144.5, 144.4, 135.8, 135.5, 135.3, 133.4,
130.8, 129.7, 129.6, 128.8, 126.7, 126.7, 125.4, 125.1, 122.1, 121.8, 121.7, 120.3, 118.7,
115.1, 113.1, 110.1, 43.5, 36.7, 36.5, 36.1, 29.0, 29.0, 21.5, 18.2, 18.1, 13.3, 13.2.
5-(-adamantan-1-yl)indolin-2-one (5-14): This compound was prepared according to the
General Procedure A using 1-adamantanol (38 mg, 0.25 mmol), indolin-2-one (50 mg,
0.375 mmol) at 80 ºC. After purification by column chromatography on SiO2 (10% EtOAc
in hexanes), the title compound was isolated as a white solid (14 mg, 21% yield).
1H NMR (600MHz, DMSO-d6): = 10.21 (s, 1 H), 7.17 (s, 1 H), 7.08 (d, J = 8.1 Hz, 1
H), 6.69 (d, J = 8.1 Hz, 1 H), 3.39 (s, 2 H), 1.99 (br. s., 3 H), 1.78 (br. s., 6 H), 1.72-1.62

157
(m, 6 H);
13C NMR (150 MHz, DMSO-d6): = 176.5, 144.0, 141.3, 125.5, 123.4, 121.0, 108.5,
42.9, 36.2, 36.0, 35.4, 28.4.
3-(adamantan-1-yl)benzo[b]thiophene (5-15): This compound was prepared according
to the General Procedure A using 1-adamantanol (38 mg, 0.25 mmol), benzothiophene (50
mg, 0.375 mmol). After purification by column chromatography on SiO2 (hexanes), the
title compound was isolated as a white solid (53 mg, 79% yield).
1H NMR (600 MHz, CDCl3): = 8.18 (d, J = 8.1 Hz, 1 H), 7.86 (d, J = 7.8 Hz, 1 H), 7.34
(t, J = 7.3 Hz, 1 H), 7.29 (t, J = 7.3 Hz, 1 H), 7.07 (s, 1 H), 2.18 (s, 6 H), 2.14 (br. s., 3 H),
1.85 (br. s., 6 H);
13C NMR (150 MHz, CDCl3): = 146.1, 141.7, 137.5, 124.6, 123.4, 123.3, 123.0, 119.7,
42.0, 37.0, 36.9, 28.8.
2-(adamantan-1-yl)-3-iodothiophene (5-16): This compound was prepared according to
the General Procedure A using 1-adamantanol (38 mg, 0.25 mmol), 3-iodothiophene (79
mg, 0.375 mmol). After purification by column chromatography on SiO2 (hexanes), the
158
title compound was isolated as a colorless liquid (72 mg, 84% yield, 78:15:7).
1H NMR (600 MHz, CDCl3): = 7.22-7.19 (m, 1 H minor1), 7.05 (d, J = 5.1 Hz, 1 H major),
7.02 (d, J = 5.1 Hz, 1 H major), 6.80-6.77 (m, 1 H minor1), 6.72 (s, 1 H minor2), 2.28-2.24 (m, 6
H major), 2.24-2.21 (m, 6 H minor1 + 6 H minor2), 2.10 (br. s., 3 H major + 6 H minor2), 1.95-1.88
(m, 3 H minor1 + 6 H minor2), 1.82-1.68 (m, 6 H major + 6 H minor1 + 12 H minor2);
13C NMR (150 MHz, CDCl3): = 150.6, 138.2, 123.1, 71.3, 41.1, 36.5, 36.4, 28.7.
N-(4-adamantan-1-yl)phenyl)acetamide (5-17): This compound was prepared according
to the General Procedure A using 1-adamantanol (38 mg, 0.25 mmol), N-phenylacetamide
(51 mg, 0.375 mmol) at 80 ºC. After purification by column chromatography on SiO2 (10%
EtOAc in hexanes), the title compound was isolated as a white solid (53 mg, 79% yield).
1H NMR (600 MHz, CDCl3): = 7.42 (d, J = 8.5 Hz, 3 H), 7.29 (d, J = 8.5 Hz, 2 H), 2.15
(s, 3 H), 2.08 (br. s., 3 H), 1.88 (br. s., 6 H), 1.82-1.70 (m, 6 H);
13C NMR (150 MHz, CDCl3): = 168.3, 147.5, 135.2, 125.3, 119.8, 43.1, 36.7, 35.8, 28.9,
24.5.

159
N-(4-((1s,3s)-adamantan-1-yl)phenyl)benzamide (5-18): This compound was prepared
according to the General Procedure A using 1-adamantanol (38 mg, 0.25 mmol), N-
phenylbenzamide (74 mg, 0.375 mmol) at 50 ºC. After purification by column
chromatography on SiO2 (10% EtOAc in hexanes), the title compound was isolated as a
white solid (48 mg, 58% yield).
1H NMR (600 MHz, CDCl3): = 7.94 (br. s., 1 H), 7.85 (d, J = 7.6 Hz, 2 H), 7.62-7.56
(m, J = 8.3 Hz, 2 H), 7.53 (t, J = 7.4 Hz, 1 H), 7.45 (t, J = 7.6 Hz, 2 H), 7.39-7.31 (m, J =
8.8 Hz, 2 H), 2.10 (br. s., 3 H), 1.91 (br. s., 6 H), 1.84-1.71 (m, 6 H);
13C NMR (150 MHz, CDCl3): = 165.7, 147.8, 135.3, 135.0, 131.7, 128.7, 127.0, 125.4,
120.1, 43.2, 36.7, 35.9, 28.9.
N-(4-adamantan-1-yl)phenyl)-4-methoxybenzamide (5-19): This compound was
prepared according to the General Procedure A using 1-adamantanol (38 mg, 0.25 mmol),
4-methoxy-N-phenylbenzamide (85 mg, 0.375 mmol) at 50 ºC. After purification by
column chromatography on SiO2 (20% EtOAc in hexanes), the title compound was isolated
as a white solid (58 mg, 64% yield).
1H NMR (600 MHz, CDCl3): = 7.94 (s, 1 H), 7.82 (d, J = 8.8 Hz, 2 H), 7.60-7.53 (m, J
= 8.5 Hz, 2 H), 7.36-7.29 (m, J = 8.8 Hz, 2 H), 6.91 (d, J = 8.8 Hz, 2 H), 3.84 (s, 3 H), 2.10
(br. s., 3 H), 1.90 (br. s., 6 H), 1.83-1.69 (m, 6 H);

160
13C NMR (150 MHz, CDCl3): = 165.2, 162.2, 147.5, 135.5, 128.9, 127.2, 125.4, 120.1,
113.8, 55.4, 43.1, 36.7, 35.8, 28.9.
1-(4-methoxyphenyl)adamantane (5-20): This compound was prepared according to the
General Procedure A using 1-adamantanol (38 mg, 0.25 mmol), anisole (41 mg, 0.375
mmol). After purification by column chromatography on SiO2 (2% EtOAc in hexanes), the
title compound was isolated as a colorless liquid (60 mg, 99% yield).
1H NMR (600 MHz, CDCl3): = 7.33-7.28 (d, J = 8.5 Hz, 2 H), 6.91-6.85 (d, J = 8.5 Hz,
2 H), 3.81 (s, 3 H), 2.10 (br. s., 3 H), 1.91 (br. s., 6 H), 1.83-1.71 (m, 6 H);
13C NMR (150 MHz, CDCl3): = 157.3, 143.7, 125.8, 113.4, 55.2, 43.4, 36.8, 35.5, 29.0.
3-(3-adamantan-1-yl)-4-methoxyphenyl)propan-1-ol (5-21): This compound was
prepared according to the General Procedure A using 1-adamantanol (38 mg, 0.25 mmol),
3-(4-methoxyphenyl)propan-1-ol (62 mg, 0.375 mmol). After purification by column
chromatography on SiO2 (20% EtOAc in hexanes), the title compound was isolated as a
white solid (46 mg, 61% yield).

161
1H NMR (600 MHz, CDCl3): = 7.04 (s, 1 H), 7.00 (d, J = 8.1 Hz, 1 H), 6.80 (d, J = 8.1
Hz, 1 H), 3.81 (s, 3 H), 3.69 (t, J = 6.3 Hz, 2 H), 2.64 (t, J = 7.8 Hz, 2 H), 2.09 (br. s., 6
H), 2.06 (br. s., 3 H), 1.93-1.84 (m, 2 H), 1.77 (br. s., 6 H);
13C NMR (150 MHz, CDCl3): = 157.0, 138.4, 133.4, 126.7, 126.2, 111.7, 62.5, 55.1,
40.6, 37.1, 36.9, 34.6, 31.6, 29.1.
(4-adamantan-1-yl)phenyl)(phenyl)sulfane (5-22): This compound was prepared
according to the General Procedure A using 1-adamantanol (38 mg, 0.25 mmol),
diphenylsulfane (70 mg, 0.375 mmol). After purification by column chromatography on
SiO2 (1% EtOAc in hexanes), the title compound was isolated as a white solid (52 mg, 65%
yield).
1H NMR (600 MHz, CDCl3): = 7.39-7.25 (m, 8 H), 7.25-7.18 (m, 1 H), 2.10 (br. s., 3
H), 1.90 (br. s., 6 H), 1.83-1.71 (m, 6 H);
13C NMR (150 MHz, CDCl3): = 150.8, 136.7, 131.5, 131.5, 130.2, 129.0, 126.5, 125.9,
43.0, 36.7, 36.1, 28.9;

162
4-(adamantan-1-yl)-2-ethylphenol (5-23): This compound was prepared according to the
General Procedure A using 1-adamantanol (38 mg, 0.25 mmol), 2-ethylphenol (46 mg,
0.375 mmol). After purification by column chromatography on SiO2 (5% EtOAc in
hexanes), the title compound was isolated as a white solid (52 mg, 81% yield).
1H NMR (600 MHz, CDCl3): = 7.14 (br. s., 1 H), 7.08 (d, J = 8.3 Hz, 1 H), 6.72 (d, J =
8.3 Hz, 1 H), 4.56 (s, 1 H), 2.65 (q, J = 7.5 Hz, 2 H), 2.09 (br. s., 3 H), 1.90 (br. s., 6 H),
1.77 (q, J = 12.1 Hz, 6 H), 1.25 (t, J = 7.5 Hz, 3 H);
13C NMR (150 MHz, CDCl3): = 151.0, 144.0, 129.1, 125.9, 123.3, 114.7, 43.4, 36.8,
35.6, 29.0, 23.4, 14.2.
5-(adamantan-1-yl)-2-hydroxybenzoic acid (5-24): This compound was prepared
according to the General Procedure A using 1-adamantanol (38 mg, 0.25 mmol), 2-
acetoxybenzoic acid (68 mg, 0.375 mmol) 80 ºC. After purification by column
chromatography on SiO2 (20% EtOAc in hexanes), the title compound was isolated as a
white solid (38 mg, 51% yield).

163
1H NMR (600 MHz, Methanol-d4): = 7.81 (d, J = 2.4 Hz, 1 H), 7.52 (dd, J = 2.4, 8.5
Hz, 1 H), 6.86 (d, J = 8.5 Hz, 1 H), 2.09 (br. s., 3 H), 1.95-1.88 (m, 6 H), 1.86-1.76 (m, 6
H);
13C NMR (150 MHz, Methanol-d4): = 173.8, 161.0, 143.3, 133.6, 127.3, 117.8, 113.2,
44.4, 37.8, 36.6, 30.4.
1-(p-tolyl)adamantane (5-25): This compound was prepared according to the General
Procedure A using 1-adamantanol (38 mg, 0.25 mmol), toluene (35 mg, 0.375 mmol) at 80
ºC. After purification by column chromatography on SiO2 (hexanes), the title compound
was isolated as a colorless liquid (38 mg, 67% yield).
1H NMR (600 MHz, CDCl3): = 7.31-7.27 (m, J = 8.1 Hz, 2 H), 7.18-7.13 (m, J = 8.1
Hz, 2 H), 2.34 (s, 3 H), 2.11 (br. s., 3 H), 1.92 (br. s., 6 H), 1.78-1.72 (m, 6 H) ;
13C NMR (150 MHz, CDCl3): = 148.4, 134.9, 128.8, 124.7, 43.2, 36.8, 35.8, 29.0, 20.9.
(8R,9S,13S,14S,17S)-2-((1s,3R)-adamantan-1-yl)-13-methyl 7,8,9,11,12,13,14,15,16,
17-decahydro-6H-cyclopenta[a]phenanthrene-3,17-diol (5-26): This compound was

164
prepared according to the General Procedure A using 1-adamantanol (38 mg, 0.25 mmol),
estradiol (102 mg, 0.375 mmol). After purification by column chromatography on SiO2
(20% EtOAc-10% DCM in hexanes), the title compound was isolated as a white solid (81
mg, 80% yield).
1H NMR (600 MHz, CDCl3): = 7.16 (s, 1 H), 6.40 (s, 1 H), 4.87 (s, 1 H), 3.75 (t, J = 8.2
Hz, 1 H), 2.85-2.71 (m, 2 H), 2.42-2.33 (m, 1 H), 2.26-2.17 (m, 1 H), 2.15-2.10 (m, 6 H),
2.10-2.05 (m, 3 H), 2.00-1.92 (m, 1 H), 1.90-1.82 (m, 1 H), 1.78 (br. s., 6 H), 1.74-1.65 (m,
1 H), 1.56-1.41 (m, 4 H), 1.41-1.24 (m, 4 H), 1.24-1.16 (m, 1 H), 0.79 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 152.2, 135.1, 133.7, 131.9, 124.0, 116.7, 82.0, 50.0,
44.3, 43.2, 40.7, 38.9, 37.1, 36.7, 36.6, 30.6, 29.0, 28.9, 27.2, 26.4, 23.1, 11.1.
Methyl 3-(3-(adamantan-1-yl)-4-hydroxyphenyl)-2-(1,3-dioxoisoindolin-2-yl)
Propanoate (5-27): This compound was prepared according to the General Procedure A
using 1-adamantanol (38 mg, 0.25 mmol), methyl 2-(1,3-dioxoisoindolin-2-yl)-3-(4-
hydroxyphenyl)propanoate (122 mg, 0.375 mmol) 50 ºC. After purification by column
chromatography on SiO2 (10% EtOAc in hexanes), the title compound was isolated as a
white solid (47 mg, 41% yield).
1H NMR (600 MHz, CDCl3): = 7.84-7.78 (m, 2 H), 7.74-7.67 (m, 2 H), 6.90 (d, J = 8.1
Hz, 1 H), 6.70 (s, 1 H), 6.48 (d, J = 8.1 Hz, 1 H), 5.26 (s, 1 H), 5.01 (dd, J = 4.9, 11.5 Hz,
1 H), 3.79 (s, 3 H), 3.50-3.32 (m, 2 H), 1.89 (br. s., 3 H), 1.79-1.69 (m, 6 H), 1.64 (d, J =
165
12.5 Hz, 3 H), 1.55 (d, J = 11.7 Hz, 3 H);
13C NMR (150 MHz, CDCl3): = 169.4, 167.4, 153.4, 136.1, 134.1, 131.6, 128.1, 127.5,
127.0, 123.5, 117.0, 53.8, 52.8, 40.1, 36.9, 36.2, 34.0, 28.8.
N-(4-(adamantan-1-yl)phenyl)-5-(2,5-dimethylphenoxy)-2,2-dimethylpentanamide
(5-28): This compound was prepared according to the General Procedure A using 1-
adamantanol (38 mg, 0.25 mmol), 5-(2,5-dimethylphenoxy)-2,2-dimethyl-N-
phenylpentanamide (122 mg, 0.375 mmol) 80 ºC. After purification by column
chromatography on SiO2 (10% EtOAc in hexanes), the title compound was isolated as a
white solid (80 mg, 70% yield).
1H NMR (600 MHz, CDCl3): = 7.46 (d, J = 8.5 Hz, 2 H), 7.35-7.29 (m, 3 H), 7.01 (d, J
= 7.6 Hz, 1 H), 6.67 (d, J = 7.6 Hz, 1 H), 6.61 (s, 1 H), 3.94 (t, J = 5.1 Hz, 2 H), 2.30 (s, 3
H), 2.18 (s, 3 H), 2.09 (br. s., 3 H), 1.90 (br. s., 6 H), 1.84-1.72 (m, 10 H), 1.34 (s, 6 H);
13C NMR (150 MHz, CDCl3): = 175.5, 156.8, 147.5, 136.5, 135.3, 130.3, 125.3, 123.5,
120.7, 119.8, 112.0, 67.8, 43.2, 42.7, 37.7, 36.7, 35.8, 28.9, 25.6, 25.1, 21.4, 15.8.

166
(2S)-N-(4-(adamantan-1-yl)phenyl)-2-(6-methoxynaphthalen-2-yl)propenamide (5-
29): This compound was prepared according to the General Procedure A using 1-
adamantanol (38 mg, 0.25 mmol), (S)-2-(6-methoxynaphthalen-2-yl)-N-
phenylpropanamide (115 mg, 0.375 mmol) at 80 ˚C. After purification by column
chromatography on SiO2 (10% EtOAc in hexanes), the title compound was isolated as a
white solid (74 mg, 67% yield).
1H NMR (600 MHz, CDCl3): = 7.77-7.69 (m, 3 H), 7.46-7.40 (m, 1 H), 7.38-7.32 (m, J
= 8.8 Hz, 2 H), 7.25-7.21 (m, J = 8.8 Hz, 2 H), 7.20-7.15 (m, 2 H), 7.15-7.12 (m, 1 H),
3.93 (s, 3 H), 3.85 (d, J = 7.1 Hz, 1 H), 2.06 (br. s., 3 H), 1.84 (br. s., 6 H), 1.80-1.69 (m, 6
H), 1.66 (d, J = 7.1 Hz, 3 H);
13C NMR (150 MHz, CDCl3): = 172.3, 157.8, 147.4, 136.1, 135.2, 133.8, 129.2, 129.0,
127.8, 126.2, 126.2, 125.2, 119.4, 119.2, 105.6, 55.3, 47.9, 43.1, 36.7, 35.8, 28.8, 18.5.
3-(1-methylcyclohexyl)-1-tosyl-1H-pyrrole (5-30): This compound was prepared
according to the General Procedure A using 1-methylcyclohexan-1-ol (29 mg, 0.25 mmol),
1-tosyl-1H-pyrrole (83 mg, 0.375 mmol). After purification by column chromatography on
SiO2 (2% EtOAc in hexanes), the title compound was isolated as a colorless liquid (57 mg,
72% yield).
1H NMR (600 MHz, CDCl3): = 7.68 (d, J = 8.3 Hz, 2 H), 7.27-7.21 (m, 2 H), 7.03 (t, J
= 2.7 Hz, 1 H), 6.87 (s, 1 H), 6.19 (dd, J = 1.6, 3.1 Hz, 1 H), 2.37 (s, 3 H), 1.72-1.62 (m, 2

167
H), 1.47-1.38 (m, 4 H), 1.38-1.29 (m, 4 H), 1.06 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 144.6, 139.5, 136.4, 129.8, 126.6, 120.8, 116.1, 112.7,
38.1, 34.1, 29.7, 26.2, 22.4, 21.6.
3-(1-phenylcyclohexyl)-1-tosyl-1H-indole (5-31): This compound was prepared
according to the General Procedure B using 1-phenylcyclohexan-1-ol (44 mg, 0.25 mmol),
1-tosyl-1H-indole (102 mg, 0.375 mmol). After purification by column chromatography
on SiO2 (2% EtOAc in hexanes), the title compound was isolated as a white solid (97 mg,
90% yield).
1H NMR (600 MHz, CDCl3): = 7.94 (d, J = 8.3 Hz, 1 H), 7.73 (d, J = 8.3 Hz, 2 H), 7.52
(s, 1 H), 7.30-7.24 (m, 2 H), 7.24-7.15 (m, 5 H), 7.15-7.10 (m, 2 H), 6.97 (t, J = 7.6 Hz, 1
H), 2.39-2.31 (m, 5 H), 2.24-2.16 (m, 2 H), 1.66-1.45 (m, 6 H);
13C NMR (150 MHz, CDCl3): = 147.3, 144.7, 136.1, 135.0, 130.0, 129.8, 129.7, 128.1,
126.8, 126.7, 125.8, 124.2, 124.1, 122.7, 121.9, 113.9, 43.1, 36.8, 26.3, 22.9, 21.6.
1-tosyl-3-(1,3,3-trimethylcyclohexyl)-1H-pyrrole (5-32): This compound was prepared
according to the General Procedure A using 1,3,3-trimethylcyclohexan-1-ol (36 mg, 0.25

168
mmol), 1-tosyl-1H-pyrrole (83 mg, 0.375 mmol), NIS (20 mol%), catalyst (20 mol%).
After purification by column chromatography on SiO2 (2% EtOAc in hexanes), the title
compound was isolated as a white solid (53 mg, 61% yield).
1H NMR (600 MHz, CDCl3): = 7.68 (d, J = 8.1 Hz, 2 H), 7.25 (d, J = 8.1 Hz, 2 H), 7.04
(br. s., 1 H), 6.87 (s, 1 H), 6.23-6.18 (m, 1 H), 2.38 (s, 3 H), 1.92 (d, J = 13.4 Hz, 1 H),
1.65 (d, J = 13.9 Hz, 1 H), 1.56-1.45 (m, 2 H), 1.28-1.19 (m, 3 H), 1.15-1.07 (m, 1 H), 1.04
(s, 3 H), 0.84 (s, 3 H), 0.35 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 144.5, 139.3, 136.5, 129.7, 126.5, 120.8, 115.9, 113.6,
50.9, 39.6, 37.1, 34.4, 34.0, 33.1, 31.2, 27.6, 21.6, 19.6.
3-(1,2-dimethylcyclohexyl)-1-tosyl-1H-pyrrole (5-33): This compound was prepared
according to the General Procedure A using 1,2-dimethylcyclohexan-1-ol (32 mg, 0.25
mmol), 1-tosyl-1H-pyrrole (83 mg, 0.375 mmol), NIS (20 mol%), catalyst (20 mol%).
After purification by column chromatography on SiO2 (2% EtOAc in hexanes), the title
compound was isolated as a colorless liquid (41 mg, 49% yield).
1H NMR (600 MHz, CDCl3): = 7.69 (d, J = 8.3 Hz, 2 H), 7.27 (d, J = 8.3 Hz, 2 H), 7.08-
7.02 (m, 1 H), 6.93-6.87 (m, 1 H), 6.24 (dd, J = 1.5, 2.9 Hz, 1 H), 2.39 (s, 3 H), 1.70 (d, J
= 12.7 Hz, 1 H), 1.60 (ddd, J = 3.9, 6.8, 10.9 Hz, 1 H), 1.54-1.41 (m, 5 H), 1.33-1.17 (m,
2 H), 1.09-1.04 (m, 3 H), 0.56 (d, J = 6.8 Hz, 3 H);
13C NMR (150 MHz, CDCl3): = 144.5, 140.6, 136.4, 129.8, 126.6, 120.8, 116.5, 112.7,

169
41.3, 39.7, 37.4, 30.3, 26.4, 22.1, 21.6, 16.8, 16.4.
3-(4-(tert-butyl)-1-methylcyclohexyl)-1-tosyl-1H-pyrrole (5-34): This compound was
prepared according to the General Procedure A using 4-(tert-butyl)-1-methylcyclohexan-
1-ol (43 mg, 0.25 mmol), 1-tosyl-1H-pyrrole (83 mg, 0.375 mmol). After purification by
column chromatography on SiO2 (2% EtOAc in hexanes), the title compound was isolated
as a colorless liquid (69 mg, 63%, 4:1 dr yield).
1H NMR (600 MHz, CDCl3): = 7.71 (d, J = 8.3 Hz, 2 H major), 7.66 (d, J = 8.3 Hz, 2
H minor), 7.27 (d, J = 8.1 Hz, 2 H major), 7.23 (d, J = 8.1 Hz, 2 H minor), 7.08-7.04 (m, 1
H major + 1 H minor), 6.91-6.87 (m, 1 H major + 1 H minor), 6.25 (br. s., 1 H major), 6.18
(br. s., 1 H minor), 2.39 (s, 3 H major), 2.36 (s, 3 H minor), 1.99 (d, J = 13.4 Hz, 2 H
minor), 1.67 (s, 1 H major), 1.65 (s, 1 H major), 1.61 (s, 1 H major), 1.59 (s, 1 H major),
1.47 (dt, J = 3.2, 13.1 Hz, 2 H major + 2 H minor), 1.34-1.18 (m, 2 H major + 2 H minor),
1.13 (s, 3 H major), 1.04 (s, 3 H minor), 1.00 - 0.91 (m, 1 H major + 3 H minor), 0.90 -
0.79 (m, 9 H major), 0.68 (s, 9 H minor);
13C NMR (150 MHz, CDCl3) major: = 144.6, 142.2, 136.4, 129.9, 126.7, 120.6, 114.9,
112.2, 48.0, 38.6, 33.0, 32.4, 27.5, 23.3, 22.7, 21.6;
13C NMR (150 MHz, CDCl3) minor: = 144.5, 142.2, 136.9, 129.8, 126.4, 121.2, 117.4,
113.4, 48.0, 38.4, 34.4, 34.1, 32.2, 27.4, 23.5, 21.5.

170
1-methoxy-4-(1-phenylcyclobutyl)benzene (5-35): This compound was prepared
according to the General Procedure B using 1-phenylcyclobutan-1-ol (37 mg, 0.25 mmol),
anisole (41 mg, 0.375 mmol) in HFIP (0.5 mL). After purification by column
chromatography on SiO2 (2% EtOAc in hexanes), the title compound was isolated as a
colorless liquid (45 mg, 76% yield).
1H NMR (600 MHz, CDCl3): = 7.33-7.21 (m, 6 H), 7.18-7.10 (m, 1 H), 6.83 (d, J = 8.5
Hz, 2 H), 3.77 (s, 3 H), 2.76-2.69 (m, 4 H), 2.02-1.91 (m, 2 H);
13C NMR (150 MHz, CDCl3): = 157.3, 150.2, 141.8, 128.2, 127.2, 126.0, 125.3, 113.5,
55.2, 50.5, 35.1, 16.6.
3-(1-methyl-2,3-dihydro-1H-inden-1-yl)-1-tosyl-1H-indole (5-36): This compound was
prepared according to the General Procedure B using 1-methyl-2,3-dihydro-1H-inden-1-
ol (37 mg, 0.25 mmol), 1-tosyl-1H-indole (102 mg, 0.375 mmol). After purification by
column chromatography on SiO2 (2% EtOAc in hexanes), the title compound was isolated
as a white solid (79 mg, 79% yield).
1H NMR (600 MHz, CDCl3): = 7.94 (d, J = 8.3 Hz, 1 H), 7.73 (d, J = 8.3 Hz, 2 H), 7.30
(d, J = 7.6 Hz, 1 H), 7.25-7.19 (m, 5 H), 7.11 (t, J = 7.4 Hz, 1 H), 7.03 (t, J = 7.4 Hz, 1 H),

171
6.97 (d, J = 8.1 Hz, 1 H), 6.87 (d, J = 7.6 Hz, 1 H), 3.05 (td, J = 7.8, 15.8 Hz, 1 H), 2.98-
2.88 (m, 1 H), 2.58 (td, J = 8.2, 12.7 Hz, 1 H), 2.39-2.31 (m, 3 H), 2.10 (ddd, J = 4.5, 8.2,
12.7 Hz, 1 H), 1.67 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 149.6, 144.7, 142.7, 136.1, 135.2, 130.3, 129.8, 129.4,
126.9, 126.8, 126.7, 124.7, 124.1, 123.5, 123.0, 122.6, 121.5, 113.8, 47.4, 40.4, 30.4, 26.8,
21.6.
3-(1-methylcyclooctyl)-1-tosyl-1H-pyrrole (5-37): This compound was prepared
according to the General Procedure A using 1-methylcyclooctan-1-ol (36 mg, 0.25 mmol),
1-tosyl-1H-pyrrole (83 mg, 0.375 mmol). After purification by column chromatography on
SiO2 (2% EtOAc in hexanes), the title compound was isolated as a white solid (60 mg, 69%
yield).
1H NMR (600 MHz, CDCl3): = 7.69 (d, J = 8.3 Hz, 2 H), 7.27 (d, J = 8.3 Hz, 2 H), 7.05
(t, J = 2.3 Hz, 1 H), 6.89 (s, 1 H), 6.24-6.20 (m, 1 H), 2.39 (s, 3 H), 1.78-1.70 (m, 2 H),
1.66-1.58 (m, 2 H), 1.56-1.43 (m, 8 H), 1.42-1.34 (m, 2 H), 1.08 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 144.5, 139.9, 136.3, 129.8, 126.6, 120.8, 116.2, 113.1,
37.1, 35.6, 29.9, 28.6, 25.2, 23.1, 21.6.

172
3-(1-methylcyclododecyl)-1-tosyl-1H-pyrrole (5-38): This compound was prepared
according to the General Procedure A using 1-methylcyclododecan-1-ol (50 mg, 0.25
mmol), 1-tosyl-1H-pyrrole (83 mg, 0.375 mmol). After purification by column
chromatography on SiO2 (2% EtOAc in hexanes), the title compound was isolated as a
white solid (68 mg, 68% yield).
1H NMR (600 MHz, CDCl3): = 7.69 (d, J = 8.1 Hz, 2 H), 7.26 (d, J = 8.1 Hz, 2 H), 7.09-
7.02 (m, 1 H), 6.89-6.84 (m, 1 H), 6.22 (dd, J = 1.6, 3.1 Hz, 1 H), 2.42-2.37 (m, 3 H), 1.54-
1.43 (m, 4 H), 1.35-1.14 (m, 17 H), 1.08-1.00 (m, 4 H);
13C NMR (150 MHz, CDCl3): = 144.5, 139.5, 136.3, 129.8, 126.6, 120.7, 116.1, 113.0,
36.2, 34.2, 28.2, 26.6, 26.1, 22.6, 22.1, 21.6, 19.5.
3-(4-phenyltetrahydro-2H-pyran-4-yl)-1-tosyl-1H-indole (5-39): This compound was
prepared according to the General Procedure A using 4-phenyltetrahydro-2H-pyran-4-ol
(45 mg, 0.25 mmol), 1-tosyl-1H-indole (102 mg, 0.375 mmol). After purification by
column chromatography on SiO2 (20% EtOAc in hexanes), the title compound was isolated
as a white solid (89 mg, 82% yield).

173
1H NMR (600 MHz, CDCl3): = 7.97 (d, J = 8.3 Hz, 1 H), 7.76 (d, J = 8.1 Hz, 2 H), 7.55
(s, 1 H), 7.30-7.22 (m, 6 H), 7.20 (t, J = 7.8 Hz, 1 H), 7.18-7.13 (m, 1 H), 7.10 (d, J = 8.1
Hz, 1 H), 6.99 (t, J = 7.7 Hz, 1 H), 3.87-3.79 (m, 2 H), 3.76-3.66 (m, 2 H), 2.49-2.38 (m, 4
H), 2.36 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 145.9, 144.9, 136.1, 134.9, 129.8, 129.3, 128.4, 128.2,
126.7, 126.6, 126.3, 124.4, 124.1, 122.9, 121.6, 114.0, 64.6, 40.8, 36.3, 21.6.
3-(4-phenyl-1-tosylpiperidin-4-yl)-1-tosyl-1H-indole (5-40): This compound was
prepared according to the General Procedure A using 4-phenyl-1-tosylpiperidin-4-ol (76
mg, 0.23 mmol), 1-tosyl-1H-indole (102 mg, 0.375 mmol) at 50 ˚C. After purification by
column chromatography on SiO2 (20% EtOAc in hexanes), the title compound was isolated
as a white solid (79 mg, 59% yield).
1H NMR (600 MHz, CDCl3): = 7.88 (d, J = 8.3 Hz, 1 H), 7.65 (d, J = 8.1 Hz, 4 H), 7.37
(s, 1 H), 7.31 (d, J = 8.1 Hz, 2 H), 7.21-7.15 (m, 5 H), 7.15-7.11 (m, 3 H), 7.04 (d, J = 8.1
Hz, 1 H), 6.97 (t, J = 7.6 Hz, 1 H), 3.43-3.34 (m, 2 H), 3.03 (td, J = 6.0, 12.1 Hz, 2 H), 2.50
(t, J = 5.4 Hz, 4 H), 2.43 (s, 3 H), 2.35 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 44.9, 144.8, 143.5, 135.8, 134.7, 133.9, 129.8, 129.7,
128.9, 128.5, 127.5, 127.4, 126.6, 126.5, 124.5, 123.9, 122.9, 121.5, 113.8, 43.0, 41.1, 35.2,
21.5.

174
3-(2-phenylpropan-2-yl)-1-tosyl-1H-indole (5-41): This compound was prepared
according to the General Procedure B using 2-phenylpropan-2-ol (36 mg, 0.25 mmol), 1-
tosyl-1H-indole (102 mg, 0.375 mmol). After purification by column chromatography on
SiO2 (2% EtOAc in hexanes), the title compound was isolated as a white solid (72 mg, 74%
yield).
1H NMR (600 MHz, CDCl3): = 7.97 (d, J = 8.3 Hz, 1 H), 7.80 (d, J = 8.3 Hz, 2 H),
7.56 (s, 1 H), 7.29-7.14 (m, 8 H), 6.97 (t, J = 7.4 Hz, 1 H), 6.87 (d, J = 7.8 Hz, 1 H), 2.37
(s, 3 H), 1.73 (s, 6 H);
13C NMR (150 MHz, CDCl3): = 147.9, 144.7, 136.0, 135.2, 132.2, 129.8, 129.5,
128.2, 126.7, 126.0, 125.9, 124.1, 122.6, 122.3, 121.9, 113.7, 39.0, 30.0, 21.5.
3-(2-phenylbutan-2-yl)-1-tosyl-1H-indole (5-42): This compound was prepared
according to the General Procedure B using 2-phenylbutan-2-ol (38 mg, 0.25 mmol), 1-
tosyl-1H-indole (102 mg, 0.375 mmol). After purification by column chromatography on
SiO2 (2% EtOAc in hexanes), the title compound was isolated as a white solid (88 mg, 87%
yield).
1H NMR (600 MHz, CDCl3): = 7.94 (d, J = 8.3 Hz, 1 H), 7.76 (d, J = 8.3 Hz, 2 H), 7.51

175
(s, 1 H), 7.25-7.11 (m, 8 H), 6.93 (t, J = 7.6 Hz, 1 H), 6.79 (d, J = 8.1 Hz, 1 H), 2.36 (s, 3
H), 2.26-2.08 (m, 2 H), 1.62 (s, 3 H), 0.66 (t, J = 7.4 Hz, 3 H);
13C NMR (150 MHz, CDCl3): = 147.1, 144.7, 136.1, 135.1, 131.0, 129.7, 128.1, 126.7,
126.7, 125.9, 124.2, 123.4, 122.7, 121.9, 113.8, 42.7, 32.8, 26.4, 21.6, 8.8.
3-(2-(4-methoxyphenyl)butan-2-yl)-1-tosyl-1H-indole (5-43): This compound was
prepared according to the General Procedure B using 2-(4-methoxyphenyl)butan-2-ol (45
mg, 0.25 mmol), 1-tosyl-1H-indole (102 mg, 0.375 mmol). After purification by column
chromatography on SiO2 (5% EtOAc in hexanes), the title compound was isolated as a
white solid (76 mg, 70% yield).
1H NMR (600 MHz, CDCl3): = 7.96 (d, J = 8.3 Hz, 1 H), 7.80-7.73 (m, J = 8.1 Hz, 2
H), 7.50 (s, 1 H), 7.25-7.21 (m, J = 8.1 Hz, 2 H), 7.19 (t, J = 7.7 Hz, 1 H), 7.10-7.04 (m, J
= 8.5 Hz, 2 H), 6.95 (t, J = 7.6 Hz, 1 H), 6.83 (d, J = 8.1 Hz, 1 H), 6.78-6.72 (m, J = 8.5
Hz, 2 H), 3.76 (s, 3 H), 2.36 (s, 3 H), 2.23-2.15 (m, 1 H), 2.15-2.06 (m, 1 H), 1.60 (s, 3 H),
0.66 (t, J = 7.2 Hz, 3 H);
13C NMR (150 MHz, CDCl3): = 157.5, 144.7, 139.2, 136.0, 135.1, 131.2, 129.7, 129.7,
127.7, 126.7, 124.1, 123.2, 122.6, 122.0, 113.7, 113.3, 55.1, 42.0, 33.0, 26.5, 21.5, 8.8.

176
3-(3-methyl-2-phenylbutan-2-yl)-1-tosyl-1H-indole (5-44): This compound was
prepared according to the General Procedure B using 3-methyl-2-phenylbutan-2-ol (41 mg,
0.25 mmol), 1-tosyl-1H-indole (102 mg, 0.375 mmol). After purification by column
chromatography on SiO2 (2% EtOAc in hexanes), the title compound was isolated as a
white solid (85 mg, 81% yield).
1H NMR (600 MHz, CDCl3): = 7.94 (d, J = 8.3 Hz, 1 H), 7.76 (d, J = 8.3 Hz, 2 H), 7.55
(s, 1 H), 7.23 (d, J = 8.3 Hz, 2 H), 7.21-7.12 (m, 6 H), 6.92 (t, J = 7.6 Hz, 1 H), 6.82 (d, J
= 8.1 Hz, 1 H), 2.67-2.58 (m, 1 H), 2.36 (s, 3 H), 1.63 (s, 3 H), 0.94 (d, J = 6.8 Hz, 3 H),
0.83 (d, J = 6.6 Hz, 3 H);
13C NMR (150 MHz, CDCl3): = 145.2, 144.7, 135.9, 135.1, 130.8, 130.3, 129.7, 127.7,
126.7, 125.8, 124.0, 123.3, 122.6, 122.4, 113.7, 46.0, 34.6, 22.7, 21.5, 18.9, 18.7.
3-(1-phenylpentyl)-1-tosyl-1H-indole (5-45): This compound was prepared according to
the General Procedure B using -1-phenylpentan-1-ol (41 mg, 0.25 mmol), 1-tosyl-1H-
indole (102 mg, 0.375 mmol). After purification by column chromatography on SiO2 (2%
EtOAc in hexanes), the title compound was isolated as a white solid (74 mg, 71% yield).
1H NMR (600 MHz, CDCl3): = 7.93 (d, J = 8.3 Hz, 1 H), 7.73 (d, J = 8.3 Hz, 2 H), 7.44

177
(s, 1 H), 7.25-7.13 (m, 9 H), 7.08 (t, J = 7.6 Hz, 1 H), 3.99 (t, J = 7.4 Hz, 1 H), 2.34 (s, 3
H), 2.15-2.06 (m, 1 H), 2.00-1.92 (m, 1 H), 1.39-1.19 (m, 4 H), 0.87 (t, J = 7.1 Hz, 3 H);
13C NMR (150 MHz, CDCl3): = 144.7, 143.6, 135.6, 135.2, 130.7, 129.7, 128.4, 127.8,
127.0, 126.7, 126.3, 124.6, 123.0, 122.7, 120.1, 113.7, 42.6, 35.3, 30.0, 22.7, 21.5, 14.0.
3-(tert-butyl)-1-tosyl-1H-pyrrole (5-46): This compound was prepared according to the
General Procedure A using tert-butanol (19 mg, 0.25 mmol), 1-tosyl-1H-pyrrole (83 mg,
0.375 mmol) at 50 ˚C. After purification by column chromatography on SiO2 (2% EtOAc
in hexanes), the title compound was isolated as a colorless liquid (55 mg, 79%, 93:7 rr
yield).
1H NMR (600 MHz, CDCl3): = 7.74-7.70 (m, J = 8.1 Hz, 2 H), 7.31-7.27 (m, J = 8.1
Hz, 2 H), 7.06 (t, J = 2.6 Hz, 1 H), 6.88 (s, 1 H), 6.25-6.22 (m, 1 H), 2.40 (s, 3 H), 1.17 (s,
9 H);
13C NMR (150 MHz, CDCl3): = 144.6, 140.4, 136.3, 129.9, 126.7, 120.7, 114.9, 112.5,
30.9, 30.7, 21.6.
3-(tert-pentyl)-1-tosyl-1H-pyrrole (5-47): This compound was prepared according to the

178
General Procedure A 50 ˚C using 2-methylbutan-2-ol (22 mg, 0.25 mmol), 1-tosyl-1H-
pyrrole (83 mg, 0.375 mmol). After purification by column chromatography on SiO2 (2%
EtOAc in hexanes), the title compound was isolated as a colorless liquid (50 mg, 69%
yield).
1H NMR (600 MHz, CDCl3): = 7.70 (d, J = 8.1 Hz, 2 H), 7.27 (d, J = 8.1 Hz, 2 H), 7.06
(t, J = 2.6 Hz, 1 H), 6.86 (s, 1 H), 6.21-6.15 (m, 1 H), 2.40 (s, 3 H), 1.45 (q, J = 7.4 Hz, 2
H), 1.13 (s, 6 H), 0.64 (t, J = 7.4 Hz, 3 H);
13C NMR (150 MHz, CDCl3): = 144.6, 138.8, 136.4, 129.8, 126.6, 120.9, 116.2, 112.9,
36.1, 34.0, 27.9, 21.6, 9.0.
(3R,5R,8R,9S,10S,13R,14S)-10,13-dimethyl-17-((R)-5-methyl-5-(1-tosyl-1H-indol-3-
yl)hexan-2-yl)hexadecahydro-1H-cyclopenta[a]phenanthren-3-ol (5-48): This
compound was prepared according to the General Procedure A using
(3R,5R,8R,9S,10S,13R,14S)-17-((R)-5-hydroxy-5-methylhexan-2-yl)-10,13-dimethyl
hexadecahydro-1H-cyclopenta[a]phenanthren-3-ol (98 mg, 0.25 mmol), 1-tosyl-1H-indole
(102 mg, 0.375 mmol). After purification by column chromatography on SiO2 (2% EtOAc
in hexanes), the title compound was isolated as a white solid (53 mg, 33% yield).
1H NMR (600 MHz, CDCl3): = 7.99 (dd, J = 2.4, 8.3 Hz, 1 H major), 7.74-7.63 (m, 3 H
major + 3 H minor), 7.51 (d, J = 7.8 Hz, 1 H minor), 7.31-7.13 (m, , 5 H major + 5 H minor), 3.86

179
(br. s., 1 H minor), 3.62 (d, J = 4.2 Hz, 1 H major), 2.38-2.27 (m, 3 H major + 3 H minor), 2.10 (m,
2 H minor), 1.92-1.53 (m, 9 H major + 7 H minor), 1.52-1.41 (m, 3 H major + 3 H minor), 1.41-1.21
(m, 9 H major + 9 H minor), 1.14 -0.63 (m, 10 H major + 4 H minor), 0.55-0.48 (3 H major, dr1 + 6
H minor), 0.35 (d, J = 6.6 Hz, 3 H major, dr2);
13C NMR (150 MHz, CDCl3): = 144.5, 143.4, 136.9, 136.2, 136.1, 135.8, 135.2, 131.5,
131.4, 129.8, 129.7, 129.7, 129.6, 126.7, 126.6, 126.5, 124.5, 124.1, 124.0, 123.0, 122.6,
122.6, 122.5, 122.4, 122.3, 121.7, 119.8, 114.0, 113.9, 71.9, 71.9, 56.4, 55.6, 52.7, 48.9,
43.4, 42.5, 42.3, 42.0, 41.7, 41.7, 41.6, 40.4, 40.1, 39.3, 39.1, 38.7, 37.7, 37.6, 37.1, 36.9,
36.5, 36.4, 36.2, 36.1, 35.8, 35.8, 35.3, 35.2, 35.1, 34.9, 34.9, 34.6, 34.5, 34.5, 34.4, 31.6,
31.1, 30.6, 30.5, 30.5, 29.1, 28.4, 28.4, 28.2, 28.2, 28.2, 27.9, 27.6, 27.2, 27.1, 26.9, 26.6,
26.4, 25.5, 25.3, 25.3, 25.1, 25.1, 24.1, 23.4, 23.3, 23.1, 23.1, 22.6, 22.6, 22.3, 21.6, 21.5,
20.7, 20.7, 19.3, 18.7, 14.9, 14.1, 12.0, 11.9.
N-(adamantan-1-yl)-4-methylbenzenesulfonamide (5-53): This compound was
prepared according to the General Procedure A using 1-adamantanol (38 mg, 0.25 mmol),
p-toluene sulfonamide (65 mg, 0.375 mmol). After purification by column chromatography
on SiO2 (10% EtOAc in hexanes), the title compound was isolated as a white solid (46 mg,
60% yield). The spectral data match with literature.175
1H NMR (600 MHz, CDCl3): = 7.77 (d, J = 8.1 Hz, 2 H), 7.25 (d, J = 8.1 Hz, 2 H), 4.83
(s, 1 H), 2.40 (s, 3 H), 1.97 (br. s., 3 H), 1.76 (br. s., 6 H), 1.61-1.48 (m, 6 H);
13C NMR (150 MHz, CDCl3): = 142.6, 141.1, 129.4, 126.9, 55.0, 43.0, 35.8, 29.4, 21.5.

180
1-azidoadamantane (5-54): This compound was prepared according to the General
Procedure B using 1-adamantanol (38 mg, 0.25 mmol), trimethylsilyl azide (49 µL, 0.375
mmol) in MeNO2 (0.5 mL) at 80 ̊ C. After purification by column chromatography on SiO2
(5% EtOAc in hexanes), the title compound was isolated as a white solid (33 mg, 75%
yield). The spectral data match with literature.176
1H NMR (600 MHz, CDCl3): = 2.15 (br. s., 3 H), 1.80 (br. s., 6 H), 1.73-1.59 (m, 6 H);
13C NMR (150 MHz, CDCl3): = 59.0, 41.5, 35.9, 29.8.
benzyl (adamantan-1-yl)carbamate (5-55): This compound was prepared according to
the General Procedure B using 1-adamantanol (38 mg, 0.25 mmol), benzyl carbamate (57
mg, 0.375 mmol) in HFIP (0.5 mL) at 80 ̊ C. After purification by column chromatography
on SiO2 (5% EtOAc in hexanes), the title compound was isolated as a white solid (42 mg,
59% yield). The spectral data match with literature.177
1H NMR (600 MHz, CDCl3): = 7.39 - 7.33 (m, 4 H), 7.33 - 7.28 (m, 1 H), 5.03 (br. s.,
2 H), 4.63 (br. s., 1 H), 2.08 (br. s., 3 H), 1.94 (br. s., 6 H), 1.67 (br. s., 6 H);
13C NMR (150 MHz, CDCl3): = 154.2, 136.8, 128.5, 128.1, 128.0, 65.9, 50.7, 41.8, 36.3,
29.4.

181
1-((1,1,1,3,3,3-hexafluoropropan-2-yl)oxy)adamantane (5-56): This compound was
prepared according to the General Procedure A using 1-adamantanol (38 mg, 0.25 mmol).
After purification by column chromatography on SiO2 (1% EtOAc in hexanes), the title
compound was isolated as a colorless liquid (41 mg, 54% yield).
1H NMR (600 MHz, CDCl3): = 4.45-4.35 (m, 1 H), 2.21 (br. s., 3 H), 1.79 (br. s., 6 H),
1.57-1.69 (m, 6 H);
13C NMR (150 MHz, CDCl3): = 121.6 (J = 283.5 Hz), 78.5, 67.8 (J = 31.5 Hz), 41.6,
35.8, 30.8, 30.7.

182
Chapter 6
Iodide-Catalyzed Oxyamination of Olefins for Synthesis of
Oxazolidinone from Unfunctionalized Carbamate
6.1 Introduction
Alkene difunctionalization has emerged as a powerful tool to rapidly construct
molecular complexity via simultaneous construction of multiple carbon-carbon, carbon-
heteroatom bonds across alkenes.113, 178 In this regard, oxyamination of alkene is one of the
most attractive and widely used transformations for the modular synthesis of vicinal amino
alcohol which is a common motif in natural products, bioactive compounds, and ligand
frameworks.109 In this context, absolute control of regio- and stereoselectivity in alkene
oxyamination is highly appealing but difficult to achieve in the alkene difunctionalization
field. Sharpless osmium-catalyzed hydroxyamination reaction became prominent after its
discovery in 1996 and remains as a benchmark for the asymmetric synthesis of 1,2-amino
alcohol (Scheme 6-1a).19-20 The toxicity of the catalyst and poor regioselectivity have
encouraged several other groups to develop alternative oxyamination protocols. Significant
efforts have been devoted to broader substrate scope and address the regio- and
enantioselectivity issues via intramolecular oxyamination of alkenes, achieved by
Palladium,179-184 Copper,185-187 Gold,188 and non-metal catalysis.189-193 However, catalytic
and regioselective intermolecular oxyamination of alkene remains significant challenges.
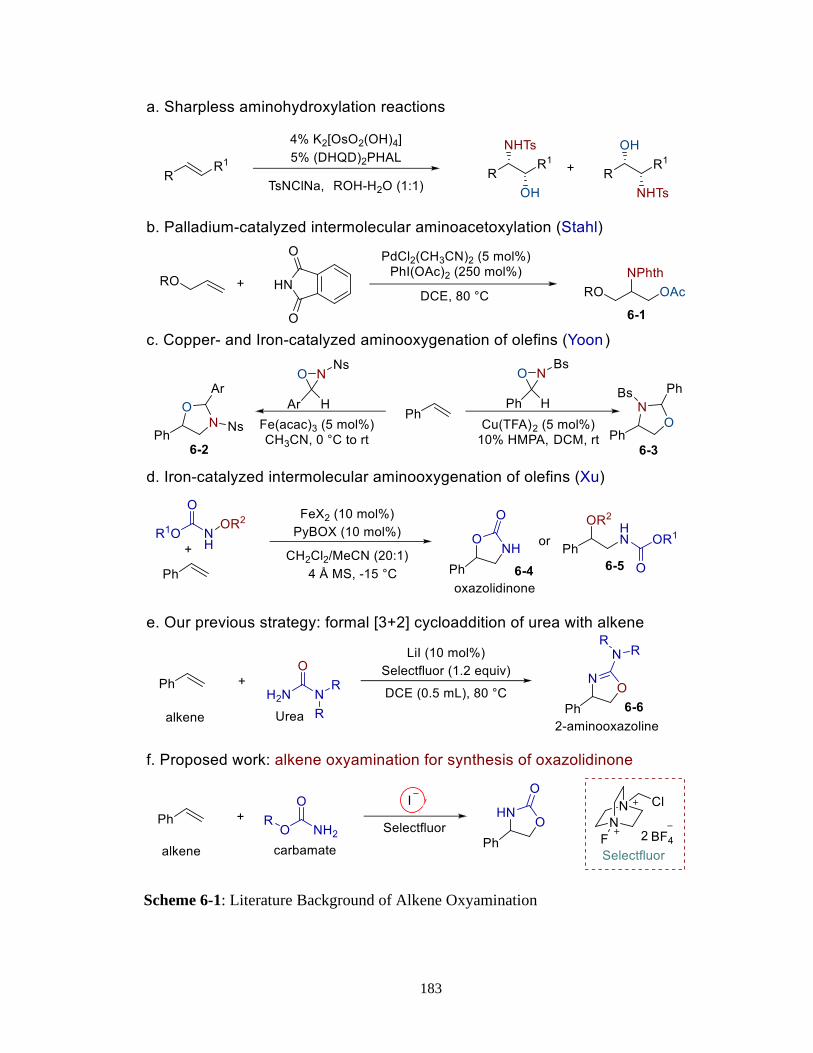
183
Scheme 6-1: Literature Background of Alkene Oxyamination

184
In this regard, Stahl group reported palladium-catalyzed aminoacetoxylation of allylic and
homoallylic ether and ester (Scheme 6-1b).21 Yoon group discovered elegant strategies
utilizing copper and iron catalyst for regiodivergent oxyamination of terminal alkenes with
oxaziridines (Scheme 6-1c).22-24 Despite the advancement of the above-mentioned
reactions, direct intermolecular oxyamination of alkene for the synthesis of oxazolidinone
is underdeveloped due to the poor nucleophilicity of carbamate substrate. To address this
issue, Xu group disclosed an iron-catalyzed strategy for oxazolidinone synthesis,
proceeded via iron-nitrenoid species generated by the cleavage of N-O bond of
functionalized hydroxylamine.194 In addition, the significance of oxazolidinones as
bioactive compounds and chiral auxiliary in asymmetric synthesis has greatly inspired
many synthetic chemists to develop different strategies to afford the oxazolidinones from
other useful precursors.30, 41 The common methods to access to this oxazolidinone
structures involves the coupling of carbon dioxide with aziridine, amino alcohol or
epoxide.195-198 Moreover, Beller group developed a ruthenium-catalyzed synthesis of
oxazolidinone from diol and urea via hydrogen atom transfer.199 However, most of the
methods are limited to presynthesized starting materials, functionalized nucleophile, and
often poor regioselectivity, make the methods unattractive to medicinal chemists.
Therefore, development of a simple intermolecular oxyamination method for swift
construction of a custom compound library of oxazolidinone heterocycles is highly
desirable.
Our group has previously discovered an iodide-catalyzed formal [3+2]
cycloaddition of ureas with alkenes.173 From mechanistic studies, it revealed that reaction
went through an in-situ generation of iodonium intermediate with alkenes, followed by the
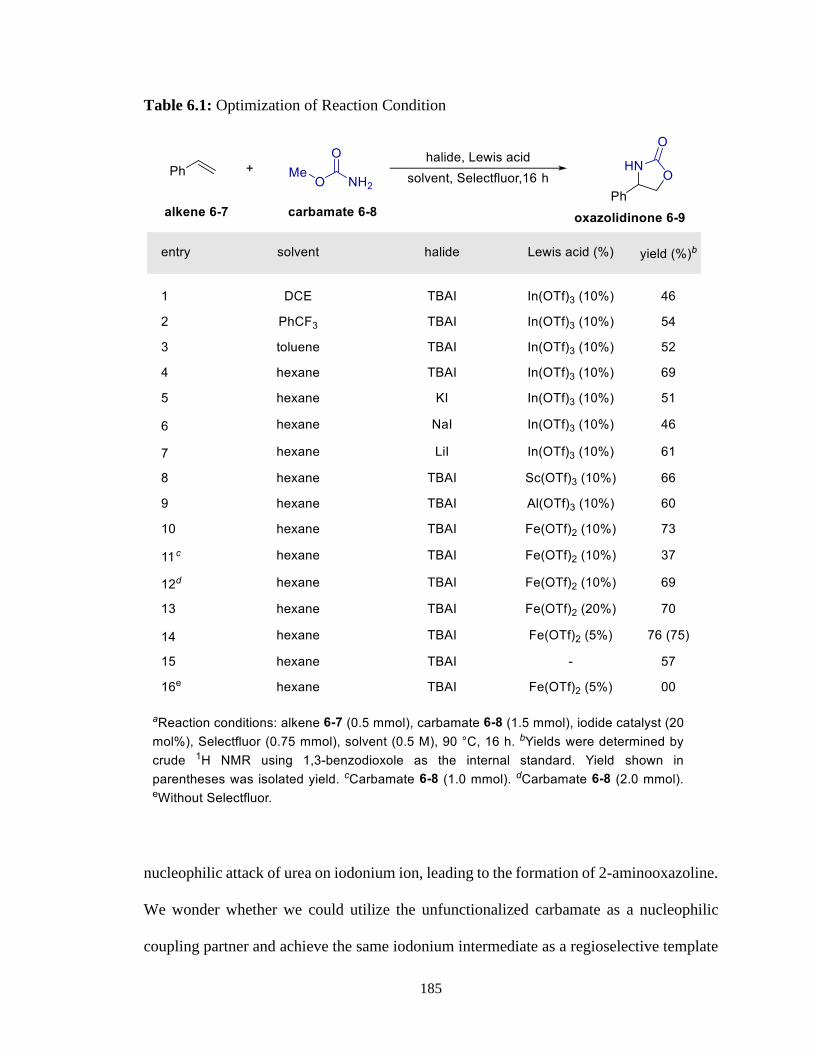
185
Table 6.1: Optimization of Reaction Condition
nucleophilic attack of urea on iodonium ion, leading to the formation of 2-aminooxazoline.
We wonder whether we could utilize the unfunctionalized carbamate as a nucleophilic
coupling partner and achieve the same iodonium intermediate as a regioselective template

186
for the synthesis of oxazolidinone. Herein, we disclose an iodide-catalyzed oxyamination
of alkenes for oxazolidinones synthesis from carbamates.
6.2 Results and Discussions for Alkene Oxyamination
6.2.1 Reaction Design and Optimization
To validate our hypothesis, we commenced our reaction with styrene, methylcarbamate,
TBAI, and Selectfluor as an oxidant in DCE at 90 ˚C for 16 h. To our delight, the reaction
provided 47% yield of our expected oxazolidinone product in a single regioisomer (Table
6.1, entry 1). After the screening of solvents revealed that hexane was the superior solvent
in this oxyamination reaction (Table 6.1, entries 2-4). The other iodide sources did not
improve the yield of products (Table 6.1, entries 5-7). The testing of Lewis acids further
demonstrated that Fe(OTf)2 was the best Lewis acid to provide the product in 73% yield
(Table 6.1, entries 8-10). The optimal yield of the product was achieved by the evaluation
of the stoichiometry of carbamate and Fe(OTf)2, affording oxazolidinone in 76% (Table
6.1, entries 11-14). In the absence of Selectfluor, the reaction did not furnish any product,
suggested that Selectfluor was essential in this reaction (Table 6.1, entry 16).
6.2.2 Substrate Scope
Based on the optimized conditions, we sought to explore the alkene substrate scope
for this oxyamination reaction. Different halogen-substituted styrenes provided the
products in high yields (Table 6.2, products 6-9 to 6-11). The electron-donating and
electron-withdrawing substituents were compatible, affording the products in good yields
with high regioselectivities (Table 6.2, products 6-12 to 6-14). The regioselectivity
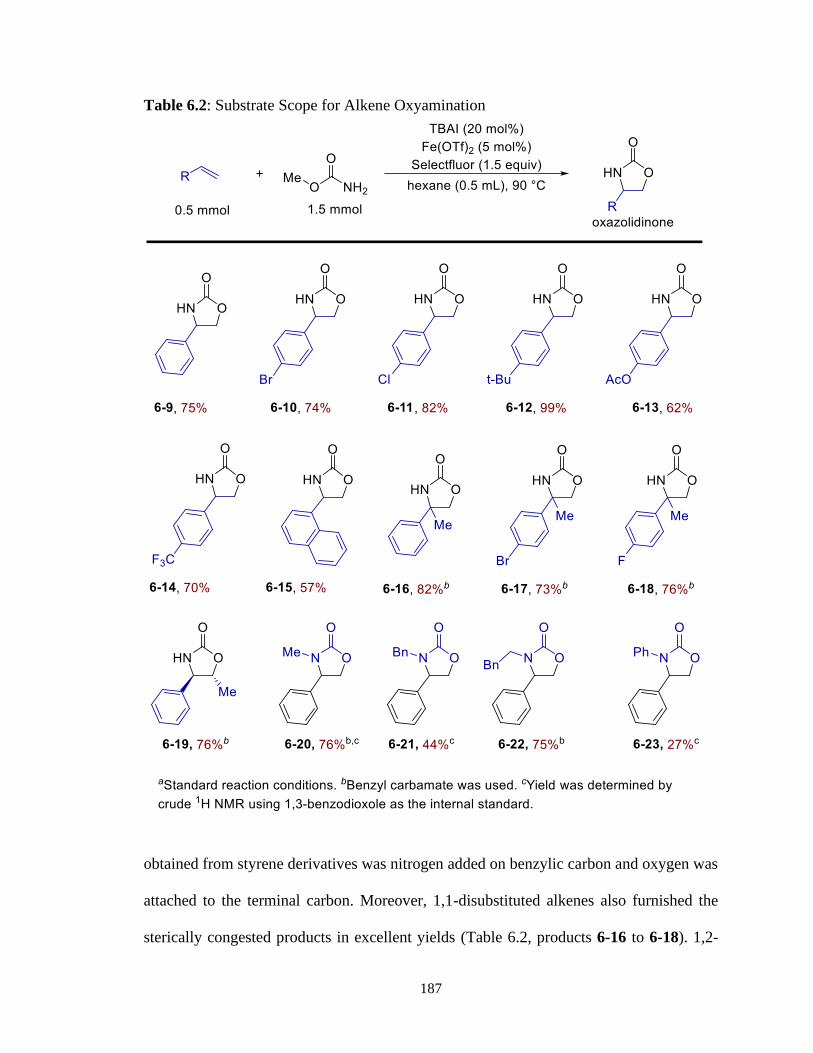
187
Table 6.2: Substrate Scope for Alkene Oxyamination
obtained from styrene derivatives was nitrogen added on benzylic carbon and oxygen was
attached to the terminal carbon. Moreover, 1,1-disubstituted alkenes also furnished the
sterically congested products in excellent yields (Table 6.2, products 6-16 to 6-18). 1,2-

188
Disubstituted alkene such as trans-β-methyl styrene delivered the product with high
diastereoselectivity (Table 6.2, product 6-19). With satisfied with the alkene substrate
scope, we then focused on the carbamate substrate scope. Different substituents on the
nitrogen of carbamate were capable of providing the oxazolidinones in moderate to high
yields (Table 6.2, products 6-20 to 6-23).
6.3 Regiodivergent Alkene Oxyamination
The actual regioisomer of oxazolidinone product was with nitrogen on benzylic
carbon and oxygen on the homobenzylic position. We wondered whether we could oppose
the regioselectivity by controlling the halonium intermediate, generated from the alkene.
To our delight, when we used N-bromo succinimide (NBS) instead of iodonium condition,
the reaction provided opposite regioisomer of oxazolidinone (Scheme 6-2, product 6-24).
The regioselectivity of these reactions is governed by the relative rate of nucleophilic attack
on the halonium ion vs equilibrium between two resonance forms of carbamate. If the
reaction generates more electrophilic halonium, more nucleophilic nitrogen of carbamate
attack preferentially on the benzylic position whereas nucleophilic attack is slower,
equilibrium predominates the negative charge on the oxygen of carbamate. For example,
Lewis acid condition, the reaction goes through the formation of more electrophilic
iodonium and nitrogen attacks on iodonium before carbamate equilibrates to its resonance
form. The resulted intermediate 6-27 undergo cyclization to afford product 6-9. On the
other hand, in NBS condition, generated bromonium ion is less electrophilic than iodonium
ion and carbamate can equilibrate faster to its resonance form 6-26 where oxygen is
negatively charged, and nucleophilic attack occur from the more nucleophilic oxygen of

189
Scheme 6-2: Origin of Regiodivergent for Alkene Oxyamination
carbamate on bromonium, leading to the generation of complementary regioisomer of
oxazolidinone 6-24.
6.4 Reaction Mechanism
In order to shed light on the reaction mechanism, several experiments were
conducted. The control experiments in the absence of either TBAI or Selectfluor did not
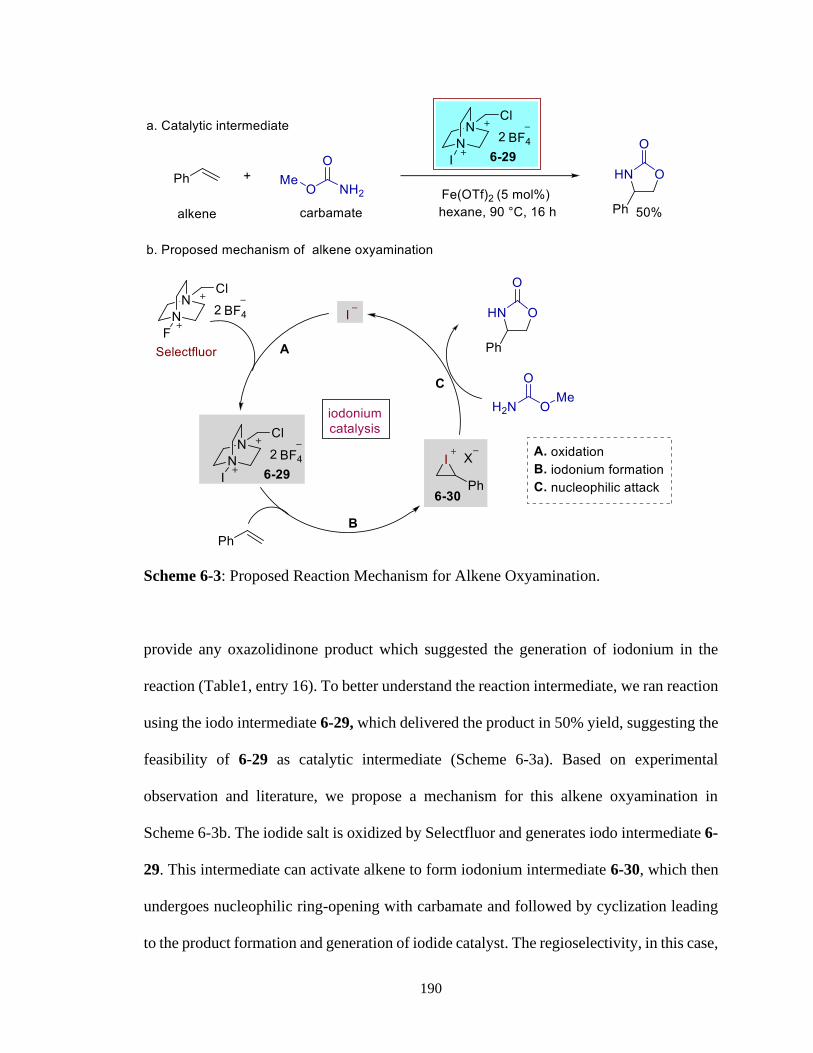
190
Scheme 6-3: Proposed Reaction Mechanism for Alkene Oxyamination.
provide any oxazolidinone product which suggested the generation of iodonium in the
reaction (Table1, entry 16). To better understand the reaction intermediate, we ran reaction
using the iodo intermediate 6-29, which delivered the product in 50% yield, suggesting the
feasibility of 6-29 as catalytic intermediate (Scheme 6-3a). Based on experimental
observation and literature, we propose a mechanism for this alkene oxyamination in
Scheme 6-3b. The iodide salt is oxidized by Selectfluor and generates iodo intermediate 6-
29. This intermediate can activate alkene to form iodonium intermediate 6-30, which then
undergoes nucleophilic ring-opening with carbamate and followed by cyclization leading
to the product formation and generation of iodide catalyst. The regioselectivity, in this case,

191
is achieved by the nucleophilic attack from the nitrogen of carbamate on more electrophilic
benzylic carbon of iodonium intermediate.
6.5 Conclusion
We have developed an iodide-catalyzed alkene oxyamination protocol for the
synthesis of oxazolidinone. A broad range of alkenes and thioamides with various
functionalities are capable of affording products. The halide exchange of Selectfluor with
TBAI is the viable pathway for the reaction to occur. We have also demonstrated the
regiodivergent synthesis of oxazolidinone by utilizing two different halogen sources. The
exploration of this protocol with expanding substrate scope and achieving different
heterocycles is underway in our laboratory.
6.6 Experimental
General Information. Commercial reagents and solvents were purchased from Sigma
Aldrich, Oakwood Chemicals, Alfa Aesar, Matrix Scientific, Acros Organic, and were
used as received. The carbamate substrates were synthesized according to the reported
procedure. Organic solutions were concentrated under reduced pressure on a Büchi rotary
evaporator using an acetone-dry ice bath. Chromatographic purification of products was
accomplished using flash chromatography on 230-400 mesh silica gel. Thin-layer
chromatography (TLC) was performed on Analtech 250 mm silica gel HLF UV-254 plates.
Visualization of the developed plates was performed by fluorescence quenching, potassium

192
permanganate, and iodine-silica gel system. 1H and 13C NMR spectra were recorded on a
Bruker 600 instrument (600 and 150 MHz) or INOVA 600 (600 and 150 MHz) and are
internally referenced to residual protio solvent signals (for CDCl3, 7.26 and 77.0 ppm,
MeOH-d4 3.31, 4.90 and 49.0 ppm, DMSO-d6 2.50 and 39.53 respectively). Data for 1H
NMR are reported as follows: chemical shift ( ppm), multiplicity (s = singlet, d = doublet,
t = triplet, q = quartet, h = heptet, m = multiplet, br = broad), integration, coupling constant
(Hz). 13C NMR spectra were reported as chemical shifts in ppm and multiplicity where
appropriate.
Experimental Procedures
General Procedure: To a 8 mL vial equipped with a stir bar was added Fe(OTf)2 (9 mg,
5 mol% ), TBAI (20 mol%, 37 mg), F-TEDA-BF4 (266 mg, 0.75 mmol) and carbamate
substrate (1.5 mmol). Then hexane (1 mL) was added via syringe, followed by alkene
substrate (0.5 mmol). The reaction mixture was then heated to 90 ˚C and stirred for 16 h.
After cooling to room temperature, the reaction mixture was diluted with EtOAc (3 mL)
and quenched with 20% aq. Na2S2O3 (1 mL) and saturated NaHCO3 solution (1 mL). The
organic layer was separated and the aqueous layer was extracted with EtOAc (2×2 mL).
The combined organic layer was dried over Na2SO4 and concentrated under reduced
pressure to give the crude product, which was purified by column chromatography on silica
gel to afford the pure product.

193
Spectral Characterization of the Product
4-phenyloxazolidin-2-one (6-9): This compound was prepared according to the General
Procedure using styrene (58 µL, 0.5 mmol), methyl carbamate (113 mg, 1.5 mmol). After
purification by column chromatography on SiO2 (40% EtOAc in hexanes), the title
compound was isolated as a white solid (61 mg, 75% yield).
1H NMR (600 MHz, CDCl3): = 7.43-7.29 (m, 5 H), 6.65 (br. s., 1 H), 4.94 (t, J = 7.8
Hz, 1 H), 4.74-4.67 (m, 1 H), 4.18-4.12 (m, 1 H);
13C NMR (150 MHz, CDCl3): = 160.1, 139.5, 129.0, 128.6, 125.9, 72.4, 56.3.
4-(4-bromophenyl)oxazolidin-2-one (6-10): This compound was prepared according to
the General Procedure using 4-bromostyrene (65 µL, 0.5 mmol), methyl carbamate (113
mg, 1.5 mmol). After purification by column chromatography on SiO2 (40% EtOAc in
hexanes), the title compound was isolated as a white solid (90 mg, 74% yield).
1H NMR (600 MHz, CDCl3): = 7.51 (d, J = 8.3 Hz, 2 H), 7.20 (d, J = 8.3 Hz, 2 H), 6.77
(br. s., 1 H), 4.91 (t, J = 7.8 Hz, 1 H), 4.69 (t, J = 8.7 Hz, 1 H), 4.10 (dd, J = 7.1, 8.3 Hz, 1
H);

194
13C NMR (150 MHz, CDCl3): = 160.0, 138.6, 132.2, 127.6, 122.6, 77.2, 76.8, 72.2,
55.7.
4-(4-chlorophenyl)oxazolidin-2-one (6-11): This compound was prepared according to
the General Procedure using 4-chlorostyrene (60 µL, 0.5 mmol), methyl carbamate (113
mg, 1.5 mmol). After purification by column chromatography on SiO2 (40% EtOAc in
hexanes), the title compound was isolated as a white solid (81 mg, 82% yield).
1H NMR (600 MHz, CDCl3): = 7.37 (d, J = 8.3 Hz, 2 H), 7.27 (d, J = 8.3 Hz, 2 H),
6.66 (br. s., 1 H), 4.94 (t, J = 7.9 Hz, 1 H), 4.71 (t, J = 8.8 Hz, 1 H), 4.12 (dd, J = 7.2, 8.2
Hz, 1 H);
13C NMR (150 MHz, CDCl3): = 159.9, 138.0, 134.5, 129.3, 127.4, 72.3, 55.7.
4-(4-(tert-butyl)phenyl)oxazolidin-2-one (6-12): This compound was prepared
according to the General Procedure using 4-tert-butylstyrene (98 µL, 0.5 mmol), methyl

195
carbamate (113 mg, 1.5 mmol). After purification by column chromatography on SiO2
(40% EtOAc in hexanes), the title compound was isolated as a white solid (109 mg, 99%
yield).
1H NMR (600 MHz, CDCl3): = 7.41 (d, J = 8.3 Hz, 2 H), 7.27 (d, J = 8.3 Hz, 2 H), 6.31
(br. s., 1 H), 4.93 (t, J = 7.8 Hz, 1 H), 4.70 (t, J = 8.7 Hz, 1 H), 4.17 (t, J = 7.8 Hz, 1 H),
1.35-1.28 (m, 9 H);
13C NMR (150 MHz, CDCl3): = 159.9, 151.8, 136.3, 126.0, 125.8, 72.5, 56.1, 34.6, 31.2.
4-(2-oxooxazolidin-4-yl)phenyl acetate (6-13): This compound was prepared according
to the General Procedure using 4-acetoxystyrene (77 µL, 0.5 mmol), methyl carbamate
(113 mg, 1.5 mmol) without Lewis acid. After purification by column chromatography on
SiO2 (50% EtOAc in hexanes), the title compound was isolated as a white solid (69 mg,
62% yield).
1H NMR (600 MHz, CDCl3): = 7.33 (d, J = 8.6 Hz, 2 H), 7.10 (d, J = 8.6 Hz, 2 H), 6.41
(s, 1 H), 4.93 (t, J = 7.8 Hz, 1 H), 4.68 (t, J = 8.6 Hz, 1 H), 4.13 (dd, J = 7.0, 8.6 Hz, 1 H),
2.29 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 169.4, 159.8, 150.7, 137.1, 127.2, 122.3, 72.3, 55.8,
21.0.

196
4-(4-(trifluoromethyl)phenyl)oxazolidin-2-one (6-14): This compound was prepared
according to the General Procedure using 1-(trifluoromethyl)-4-vinylbenzene (74 µL, 0.5
mmol), methyl carbamate (113 mg, 1.5 mmol). After purification by column
chromatography on SiO2 (40% EtOAc in hexanes), the title compound was isolated as a
white solid (81 mg, 70% yield).
1H NMR (600 MHz, CDCl3): = 7.65 (d, J = 8.1 Hz, 2 H), 7.45 (d, J = 8.1 Hz, 2 H),
7.02 (br. s., 1 H), 5.03 (t, J = 7.8 Hz, 1 H), 4.74 (t, J = 8.8 Hz, 1 H), 4.15-4.08 (m, 1 H);
13C NMR (150 MHz, CDCl3): = 160.1, 143.5, 130.9 (q, 2JC-F = 33 Hz), 126.4,126.1(q,
3JC-F = 4.5 Hz), 125.5, 123.8 (q, 1JC-F = 271.5 Hz), 72.1, 55.8.
4-(naphthalen-1-yl)oxazolidin-2-one (6-15): This compound was prepared according to
the General Procedure using 2-vinylnaphthalene (77 mg, 0.5 mmol), methyl carbamate
(113 mg, 1.5 mmol). After purification by column chromatography on SiO2 (40% EtOAc
in hexanes), the title compound was isolated as a white solid (61 mg, 57% yield).

197
1H NMR (600 MHz, CDCl3): = 7.96-7.80 (m, 3 H), 7.77 (s, 1 H), 7.59-7.47 (m, 2 H),
7.43 (dd, J = 1.8, 8.4 Hz, 1 H), 6.22 (br. s., 1 H), 5.09 (t, J = 7.9 Hz, 1 H), 4.77 (t, J = 8.8
Hz, 1 H), 4.25 (dd, J = 7.0, 8.4 Hz, 1 H);
13C NMR (150 MHz, CDCl3): = 159.8, 136.6, 133.3, 133.1, 129.4, 127.9, 127.7, 126.7,
126.6, 125.4, 123.2, 72.3, 56.4.
4-methyl-4-phenyloxazolidin-2-one (6-16): This compound was prepared according to
the General Procedure using α-methylstyrene (65 µL, 0.5 mmol), benzyl carbamate (227
mg, 1.5 mmol). After purification by column chromatography on SiO2 (40% EtOAc in
hexanes), the title compound was isolated as a white solid (65 mg, 73% yield).
1H NMR (600 MHz, CDCl3): = 7.43-7.34 (m, 4 H), 7.31 (t, J = 7.0 Hz, 1 H), 6.78 (br.
s., 1 H), 4.43-4.31 (m, 2 H), 1.75 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 159.4, 143.5, 128.9, 127.8, 124.5, 78.0, 60.5, 27.8.
4-(4-bromophenyl)-4-methyloxazolidin-2-one (6-17): This compound was prepared
according to the General Procedure using 4-bromo-α-methylstyrene (99 mg, 0.5 mmol),

198
benzyl carbamate (227 mg, 1.5 mmol). After purification by column chromatography on
SiO2 (40% EtOAc in hexanes), the title compound was isolated as a white solid (93 mg,
73% yield).
1H NMR (600 MHz, CDCl3): = 7.49 (d, J = 8.5 Hz, 2 H), 7.39 (s, 1 H), 7.23 (d, J = 8.5
Hz, 2 H), 4.36 (d, J = 8.5 Hz, 1 H), 4.30 (d, J = 8.5 Hz, 1 H), 1.70 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 159.6, 142.7, 132.0, 126.4, 121.8, 77.7, 60.3, 27.8.
4-(4-fluorophenyl)-4-methyloxazolidin-2-one (6-18): This compound was prepared
according to the General Procedure using 4-fluoro-α-methylstyrene (68 mg, 0.5 mmol),
benzyl carbamate (227 mg, 1.5 mmol). After purification by column chromatography on
SiO2 (40% EtOAc in hexanes), the title compound was isolated as a white solid (69 mg,
76% yield).
1H NMR (600 MHz, CDCl3): = 7.33 (dd, J = 5.1, 8.8 Hz, 2 H), 7.28 (br. s., 1 H), 7.05
(t, J = 8.7 Hz, 2 H), 4.37 (d, J = 8.3 Hz, 1 H), 4.31 (d, J = 8.3 Hz, 1 H), 1.72 (s, 3 H);
13C NMR (150 MHz, CDCl3): = 162.1 (d, J = 244.5 Hz), 159.6, 139.4 (d, J = 3 Hz),
126.4 (d, J = 7.5 Hz), 115.8, 115.7 (d, J = 21.0 Hz), 78.0, 60.2, 27.9.

199
5-methyl-4-phenyloxazolidin-2-one (6-19): This compound was prepared according to
the General Procedure using trans-β-methylstyrene (65 µL, 0.5 mmol), benzyl carbamate
(227 mg, 1.5 mmol). After purification by column chromatography on SiO2 (40% EtOAc
in hexanes), the title compound was isolated as a white solid (67 mg, 76% yield).
1H NMR (600 MHz, CDCl3): = 7.41-7.36 (m, 2 H), 7.36-7.30 (m, 3 H), 6.50 (br. s., 1
H), 4.45 (d, J = 7.3 Hz, 1 H), 4.42-4.36 (m, 1 H), 1.48 (d, J = 6.2 Hz, 3 H);
13C NMR (150 MHz, CDCl3): = 159.4, 138.7, 129.0, 128.7, 126.1, 81.6, 64.0, 19.2.
3-phenethyl-4-phenyloxazolidin-2-one (6-22): This compound was prepared according
to the General Procedure using styrene (58 µL, 0.5 mmol), benzyl phenethylcarbamate
(383 mg, 1.5 mmol). After purification by column chromatography on SiO2 (40% EtOAc
in hexanes), the title compound was isolated as a white solid (100 mg, 75% yield).
1H NMR (600 MHz, CDCl3): = 7.43-7.33 (m, 3 H), 7.28 (t, J = 7.3 Hz, 2 H), 7.23 (t, J
= 7.3 Hz, 1 H), 7.20-7.15 (m, 2 H), 7.12 (d, J = 7.1 Hz, 2 H), 4.55-4.45 (m, 2 H), 4.06
(dd, J = 6.3, 8.1 Hz, 1 H), 3.71 (ddd, J = 5.7, 8.4, 14.2 Hz, 1 H), 3.01-2.93 (m, 1 H), 2.92-
2.82 (m, 1 H), 2.75 (ddd, J = 5.9, 7.9, 13.6 Hz, 1 H);

200
13C NMR (150 MHz, CDCl3): = 158.0, 138.4, 137.6, 129.2, 129.0, 128.6, 128.5,
127.0, 126.5, 69.7, 60.2, 43.2, 33.5.

201
References
1. Rimando, A. M.; Kalt, W.; Magee, J. B.; Dewey, J.; Ballington, J. R. Resveratrol,
Pterostilbene, and Piceatannol in Vaccinium Berries. J. Agric. Food Chem. 2004,
52, 4713-4719.
2. Maryanoff, B. E.; Reitz, A. B. The Wittig Olefination Reaction and Modifications
Involving Phosphoryl-Stabilized Carbanions. Stereochemistry, Mechanism, and
Selected Synthetic Aspects. Chem. Rev.. 1989, 89, 863-927.
3. Blakemore, P. R. The Modified Julia Olefination: Alkene Synthesis Via the
Condensation of Metallated Heteroarylalkylsulfones with Carbonyl Compounds. J.
Chem. Soc., Perkin Trans. 1. 2002, 2563-2585.
4. Miyaura, N.; Suzuki, A. Stereoselective Synthesis of Arylated (E)-Alkenes by the
Reaction of Alk-1-Enylboranes with Aryl Halides in the Presence of Palladium
Catalyst. J. Chem. Soc., Chem. Commun. 1979, 866-867.
5. Scott, W. J.; Stille, J. K. Palladium-Catalyzed Coupling of Vinyl Triflates with
Organostannanes. Synthetic and Mechanistic Studies. J. Am. Chem. Soc. 1986, 108,
3033-3040.
6. Felpin, F.-X.; Nassar-Hardy, L.; Le Callonnec, F.; Fouquet, E. Recent Advances in
the Heck–Matsuda Reaction in Heterocyclic Chemistry. Tetrahedron. 2011, 67,
2815-2831.
7. Chatterjee, A. K.; Choi, T.-L.; Sanders, D. P.; Grubbs, R. H. A General Model for
Selectivity in Olefin Cross Metathesis. J. Am. Chem. Soc. 2003, 125, 11360-11370.
8. Peterson, D. J. Carbonyl Olefination Reaction Using Silyl-Substituted
Organometallic Compounds. J. Org. Chem. 1968, 33, 780-784.
9. Junttila, M. H.; Hormi, O. E. O. Sodium Chlorite as an Efficient Oxidant and
Hydroxy Ion Pump in Osmium-Catalyzed Asymmetric Dihydroxylation. J. Org.
Chem. 2004, 69, 4816-4820.
10. Kanth, J. V. B.; Brown, H. C. Hydroboration. 97. Synthesis of New Exceptional
Chloroborane−Lewis Base Adducts for Hydroboration.
Dioxane−Monochloroborane as a Superior Reagent for the Selective
Hydroboration of Terminal Alkenes. J. Org. Chem. 2001, 66, 5359-5365.

202
11. Brown, H.; Rao, B. C. Selective Conversion of Olefins into Organoboranes through
Competitive Hydroboration, Isomerization and Displacement Reactions. J. Org.
Chem. 1957, 22, 1137-1138.
12. Simmons, H. E.; Smith, R. D. A New Synthesis of Cyclopropanes from Olefins. J.
Am. Chem. Soc. 1958, 80, 5323-5324.
13. Pasto, D. J.; Gontarz, J. A. Mechanism of the Oxymercuration of Substituted
Cyclohexenes. J. Am. Chem. Soc. 1971, 93, 6902-6908.
14. Eisch, J. J. Fifty Years of Ziegler–Natta Polymerization: From Serendipity to
Science. A Personal Account. Organometallics. 2012, 31, 4917-4932.
15. Shamiri, A.; Chakrabarti, M. H.; Jahan, S.; Hussain, M. A.; Kaminsky, W.;
Aravind, P. V.; Yehye, W. A. The Influence of Ziegler-Natta and Metallocene
Catalysts on Polyolefin Structure, Properties, and Processing Ability. Materials.
2014, 7, 5069-5108.
16. Cui, X.; Burgess, K. Catalytic Homogeneous Asymmetric Hydrogenations of
Largely Unfunctionalized Alkenes. Chem. Rev. 2005, 105, 3272-3296.
17. Verendel, J. J.; Pamies, O.; Dieguez, M.; Andersson, P. G. Asymmetric
Hydrogenation of Olefins Using Chiral Crabtree-Type Catalysts: Scope and
Limitations. Chem. Rev. 2014, 114, 2130-2169.
18. Church, T. L.; Andersson, P. G. Iridium Catalysts for the Asymmetric
Hydrogenation of Olefins with Nontraditional Functional Substituents. Coord.
Chem. Rev. 2008, 252, 513-531.
19. O'Brien, P. Sharpless Asymmetric Aminohydroxylation: Scope, Limitations, and
Use in Synthesis. Angew. Chem., Int. Ed. 1999, 38, 326-329.
20. Bodkin, J. A.; McLeod, M. D. The Sharpless Asymmetric Aminohydroxylation. J.
Chem. Soc., Perkin Trans. 1. 2002, 2733-2746.
21. Liu, G.; Stahl, S. S. Highly Regioselective Pd-Catalyzed Intermolecular
Aminoacetoxylation of Alkenes and Evidence for Cis-Aminopalladation and SN2
C−O Bond Formation. J. Am. Chem. Soc. 2006, 128, 7179-7181.
22. Michaelis, D. J.; Ischay, M. A.; Yoon, T. P. Activation of N-Sulfonyl Oxaziridines
Using Copper (II) Catalysts: Aminohydroxylations of Styrenes and 1,3-Dienes. J.
Am. Chem. Soc. 2008, 130, 6610-6615.
23. Williamson, K. S.; Yoon, T. P. Iron-Catalyzed Aminohydroxylation of Olefins. J.
Am. Chem. Soc. 2010, 132, 4570-4571.

203
24. Michaelis, D. J.; Shaffer, C. J.; Yoon, T. P. Copper (II)-Catalyzed
Aminohydroxylation of Olefins. J. Am. Chem. Soc. 2007, 129, 1866-1867.
25. Derosa, J.; Kleinmans, R.; Tran, V. T.; Karunananda, M. K.; Wisniewski, S. R.;
Eastgate, M. D.; Engle, K. M. Nickel-Catalyzed 1,2-Diarylation of Simple Alkenyl
Amides. J. Am. Chem. Soc. 2018, 140, 17878-17883.
26. Basnet, P.; Kc, S.; Dhungana, R. K.; Shrestha, B.; Boyle, T. J.; Giri, R. Synergistic
Bimetallic Ni/Ag and Ni/Cu Catalysis for Regioselective λ,δ,-Diarylation of
Alkenyl Ketimines: Addressing β-H Elimination by in Situ Generation of Cationic
Ni(II) Catalysts. J. Am. Chem. Soc. 2018, 140, 15586-15590.
27. Gao, P.; Chen, L.-A.; Brown, M. K. Nickel-Catalyzed Stereoselective Diarylation
of Alkenylarenes. J. Am. Chem. Soc. 2018, 140, 10653-10657.
28. White, D. R.; Hutt, J. T.; Wolfe, J. P. Asymmetric Pd-Catalyzed Alkene
Carboamination Reactions for the Synthesis of 2-Aminoindane Derivatives. J. Am.
Chem. Soc. 2015, 137, 11246-11249.
29. Loi, V. V.; Rossius, M.; Antelmann, H. Redox Regulation by Reversible Protein S-
Thiolation in Bacteria. Frontiers in Microbiology. 2015, 6.
30. Mukhtar, T. A.; Wright, G. D. Streptogramins, Oxazolidinones, and Other
Inhibitors of Bacterial Protein Synthesis. Chem. Rev. 2005, 105, 529-542.
31. Zampieri, D.; Vio, L.; Fermeglia, M.; Pricl, S.; Wunsch, B.; Schepmann, D.;
Romano, M.; Mamolo, M. G.; Laurini, E. Computer-Assisted Design, Synthesis,
Binding and Cytotoxicity Assessments of New 1-(4-(Aryl(Methyl)Amino)Butyl)-
Heterocyclic Sigma 1 Ligands. Eur. J. Med. Chem. 2016, 121, 712-726.
32. Jin, Z. Muscarine, Imidaozle, Oxazole and Thiazole Alkaloids. Natural Product
Reports. 2013, 30, 869-915.
33. Abd-Elzaher, M. M.; Labib, A. A.; Mousa, H. A.; Moustafa, S. A.; Ali, M. M.; El-
Rashedy, A. A. Synthesis, Anticancer Activity and Molecular Docking Study of
Schiff Base Complexes Containing Thiazole Moiety. Beni-Suef Univ. J. Basic Appl.
Sci. 2016, 5, 85-96.
34. Jurkiewicz, E.; Jansen, R.; Kunze, B.; Trowitzsch-Kienast, W.; Forche, E.;
Reichenbach, H.; Höfle, G.; Hunsmann, G. Three New Potent HIV-1 Inhibitors
from Myxobacteria. Antiviral Chemistry and Chemotherapy. 1992, 3, 189-193.
35. Kardos, N.; Demain, A. L. Penicillin: The Medicine with the Greatest Impact on
Therapeutic Outcomes. Applied microbiology and biotechnology. 2011, 92, 677.

204
36. Bennett, J. P., Jr.; Piercey, M. F. Pramipexole - a New Dopamine Agonist for the
Treatment of Parkinson's Disease. J. Neurol. Sci. 1999, 163, 25-31.
37. McElroy, W. D. The Energy Source for Bioluminescence in an Isolated System.
Proc. Natl. Acad. Sci. U. S. A. 1947, 33, 342-345.
38. Morton, D. Pharmacology and Toxicology of Nizatidine. Scand. J. Gastroenterol.
1987, 22, 1-8.
39. Boyington, A. J.; Seath, C. P.; Zearfoss, A. M.; Xu, Z.; Jui, N. T. Catalytic Strategy
for Regioselective Arylethylamine Synthesis. J. Am. Chem. Soc. 2019, 141, 4147-
4153.
40. Mellah, M.; Voituriez, A.; Schulz, E. Chiral Sulfur Ligands for Asymmetric
Catalysis. Chem. Rev. 2007, 107, 5133-5209.
41. Farina, V.; Reeves, J. T.; Senanayake, C. H.; Song, J. J. Asymmetric Synthesis of
Active Pharmaceutical Ingredients. Chem. Rev. 2006, 106, 2734-2793.
42. Vitaku, E.; Smith, D. T.; Njardarson, J. T. Analysis of the Structural Diversity,
Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA
Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257-10274.
43. Roberts, I.; Kimball, G. E. The Halogenation of Ethylenes. J. Am. Chem. Soc. 1937,
59, 947-948.
44. Olah, G. A.; Schilling, P.; Westerman, P. W.; Lin, H. C. Electrophilic Reactions at
Multiple Bonds. II. Observation and Differentiation of Intermediate. Sigma. And.
Pi. Complexes in Electrophilic Additions to Ethene, 2, 3-Dimethyl-2-Butene, and
Adamantylideneadamantane. J. Am. Chem. Soc. 1974, 96, 3581-3589.
45. Olah, G. A.; Bollinger, J. M. Stable Carbonium Ions. XlVIII. Halonium Ion
Formation Via Neighboring Halogen Participation. Tetramethylethylene Halonium
Ions. J. Am. Chem. Soc. 1967, 89, 4744-4752.
46. Nugent, W. A. Unusual Reactions of Adamantylideneadamantane with Metal
Oxidants. Isolation of Stable Chloronium Salts. J. Org. Chem. 1980, 45, 4533-
4534.
47. Brown, R.; Nagorski, R.; Bennet, A.; McClung, R.; Aarts, G.; Klobukowski, M.;
McDonald, R.; Santarsiero, B. Stable Bromonium and Iodonium Ions of the
Hindered Olefins Adamantylideneadamantane and Bicyclo [3.3.1]
Nonylidenebicyclo [3.3.1] Nonane. X-Ray Structure, Transfer of Positive Halogens
to Acceptor Olefins, and Ab Initio Studies. J. Am. Chem. Soc. 1994, 116, 2448-
2456.

205
48. Bennet, A.; Brown, R.; McClung, R.; Klobukowski, M.; Aarts, G.; Santarsiero, B.;
Bellucci, G.; Bianchini, R. An Unprecedented Rapid and Direct Direct Br+
Transfer from the Bromonium Ion of Adamantylideneadamantane to Acceptor
Olefins. J. Am. Chem. Soc. 1991, 113, 8532-8534.
49. Neverov, A. A.; Muise, T. L.; Brown, R. X+ Transfer from the Halonium Ions of
Adamantylideneadamantane to Acceptor Olefins. The Possibility of Chiral
Induction in the Transfer Process. Can. J. Chem. 1997, 75, 1844-1850.
50. Mori, T.; Rathore, R. X-Ray Structure of Bridged 2, 2′-Bi (Adamant-2-Ylidene)
Chloronium Cation and Comparison of Its Reactivity with a Singly-Bonded
Chloroarenium Cation. Chem. Commun. 1998, 927-928.
51. Shao, L.-X.; Shi, M. N-Bromosuccinimide and Lithium Bromide: An Efficient
Combination for the Dibromination of Carbon-Carbon Unsaturated Bonds. Synlett.
2006, 2006, 1269-1271.
52. Zhu, M.; Lin, S.; Zhao, G.-L.; Sun, J.; Córdova, A. Organocatalytic
Diastereoselective Dibromination of Alkenes. Tetrahedron Lett. 2010, 51, 2708-
2712.
53. Barhate, N. B.; Gajare, A. S.; Wakharkar, R. D.; Bedekar, A. V. Simple and
Practical Halogenation of Arenes, Alkenes and Alkynes with Hydrohalic
Acid/H2O2 (or TBHP). Tetrahedron. 1999, 55, 11127-11142.
54. Kim, K.-M.; Park, I.-H. A Convenient Halogenation of Α, Β-Unsaturated Carbonyl
Compounds with Oxone® and Hydrohalic Acid (HBr, HCl). Synthesis. 2004, 2004,
2641-2644.
55. Macharla, A. K.; Chozhiyath Nappunni, R.; Nama, N. Regio- and Stereoselective
Hydroxybromination and Dibromination of Olefins Using Ammonium Bromide
and Oxone®. Tetrahedron Lett. 2012, 53, 1401-1405.
56. Dewkar, G. K.; Narina, S. V.; Sudalai, A. NaIO4-Mediated Selective Oxidative
Halogenation of Alkenes and Aromatics Using Alkali Metal Halides. Org. Lett.
2003, 5, 4501-4504.
57. Das, B.; Srinivas, Y.; Sudhakar, C.; Damodar, K.; Narender, R. Efficient
Bromination of Alkenes and Alkynes Using Potassium Bromide and Diacetoxy
Iodobenzene. Synth. Commun. 2008, 39, 220-227.
58. Karki, M.; Magolan, J. Bromination of Olefins with HBr and DMSO. J. Org. Chem.
2015, 80, 3701-3707.
59. Taori, K.; Paul, V. J.; Luesch, H. Structure and Activity of Largazole, a Potent
Antiproliferative Agent from the Floridian Marine Cyanobacterium Symploca Sp.

206
J. Am. Chem. Soc. 2008, 130, 1806-1807.
60. Gerwick, W. H.; Proteau, P. J.; Nagle, D. G.; Hamel, E.; Blokhin, A.; Slate, D. L.
Structure of Curacin A, a Novel Antimitotic, Antiproliferative and Brine Shrimp
Toxic Natural Product from the Marine Cyanobacterium Lyngbya Majuscula. J.
Org. Chem. 1994, 59, 1243-1245.
61. Carmeli, S.; Moore, R. E.; Patterson, G. M. L.; Corbett, T. H.; Valeriote, F. A.
Tantazoles, Unusual Cytotoxic Alkaloids from the Blue-Green Alga Scytonema
Mirabile. J. Am. Chem. Soc. 1990, 112, 8195-8197.
62. Schlegel, K.; Taraz, K.; Budzikiewicz, H. The Stereoisomers of Pyochelin, a
Siderophore of Pseudomonas Aeruginosa. Biometals. 2004, 17, 409-414.
63. Bensimon, G.; Lacomblez, L.; Meininger, V. f.; Group, A. R. S. A Controlled Trial
of Riluzole in Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 1994, 330, 585-591.
64. Fukuhara, T.; Hasegawa, C.; Hara, S. A Facile Synthesis of Oxazolines,
Thiazolines, and Imidazolines Using α,α-Difluoroalkylamines. Synthesis. 2007,
2007, 1528-1534.
65. Charette, A. B.; Chua, P. Mild Method for the Synthesis of Thiazolines from
Secondary and Tertiary Amides. J. Org. Chem.1998, 63, 908-909.
66. Seijas, J. A.; Vázquez-Tato, M. P.; Crecente-Campo, J. Straightforward
Microwave-Assisted Synthesis of 2-Thiazolines Using Lawesson's Reagent under
Solvent-Free Conditions. Tetrahedron. 2008, 64, 9280-9285.
67. Busacca, C. A.; Dong, Y.; Spinelli, E. M. A One Step Synthesis of Thiazolines
from Esters. Tetrahedron Lett. 1996, 37, 2935-2938.
68. Maltsev, O. V.; Walter, V.; Brandl, M. J.; Hintermann, L. Medium Buffer Effects
on the Condensation of L-Cysteine and Aryl Nitriles to (R)-2-Aryl-4,5-
Dihydrothiazole-4-Carboxylic Acids. Synthesis. 2013, 45, 2763-2767.
69. Nishio, T. Sulfur-Containing Heterocycles Derived by the Reaction of Hydroxy-
Amides and Lawesson's Reagent. Tetrahedron Lett. 1995, 36, 6113-6116.
70. Wipf, P.; Fritch, P. C. Synthesis of Peptide Thiazolines from β-Hydroxythioamides.
An Investigation of Racemization in Cyclodehydration Protocols. Tetrahedron
Lett. 1994, 35, 5397-5400.
71. Mercey, G.; Brégeon, D.; Gaumont, A.-C.; Levillain, J.; Gulea, M. Efficient
Synthesis of Primary 2-Aminothiols from 2-Aminoalcohols and
Methyldithioacetate. Tetrahedron Lett. 2008, 49, 6553-6555.

207
72. Lemercier, B. C.; Pierce, J. G. Synthesis of Thiazolines by Copper Catalyzed
Aminobromination of Thiohydroximic Acids. Org. Lett. 2014, 16, 2074-2076.
73. Pollex, A.; Hiersemann, M. Catalytic Asymmetric Claisen Rearrangement in
Natural Product Synthesis: Synthetic Studies toward (−)-Xeniolide F. Org. Lett.
2005, 7, 5705-5708.
74. Vettel, S.; Vaupel, A.; Knochel, P. Nickel-Catalyzed Preparations of
Functionalized Organozincs. J. Org. Chem. 1996, 61, 7473-7481.
75. Fujii, S.; Lehn, J. M. Structural and Functional Evolution of a Library of
Constitutional Dynamic Polymers Driven by Alkali Metal Ion Recognition. Angew.
Chem., Int. Ed. 2009, 48, 7635-7638.
76. Li, X.; Li, C.; Yin, B.; Li, C.; Liu, P.; Li, J.; Shi, Z. DDQ‐Induced Dehydrogenation
of Heterocycles for C‒C Double Bond Formation: Synthesis of 2‐Thiazoles and 2‐
Oxazoles. Chem. Asian J. 2013, 8, 1408-1411.
77. Miura, T.; Funakoshi, Y.; Fujimoto, Y.; Nakahashi, J.; Murakami, M. Facile
Synthesis of 2, 5-Disubstituted Thiazoles from Terminal Alkynes, Sulfonyl Azides,
and Thionoesters. Org. Lett. 2015, 17, 2454-2457.
78. Denmark, S. E.; Kuester, W. E.; Burk, M. T. Catalytic, Asymmetric
Halofunctionalization of Alkenes—a Critical Perspective. Angew. Chem., Int. Ed.
2012, 51, 10938-10953.
79. Denmark, S. E.; Chi, H. M. Catalytic, Enantioselective, Intramolecular
Sulfenoamination of Alkenes with Anilines. J. Org. Chem. 2017, 82, 3826-3843.
80. Mizar, P.; Niebuhr, R.; Hutchings, M.; Farooq, U.; Wirth, T. Thioamination of
Alkenes with Hypervalent Iodine Reagents. Chemistry. 2016, 22, 1614-7.
81. Denmark, S. E.; Chi, H. M. Lewis Base Catalyzed, Enantioselective, Intramolecular
Sulfenoamination of Olefins. J. Am. Chem. Soc. 2014, 136, 8915-8.
82. Denmark, S. E.; Hartmann, E.; Kornfilt, D. J.; Wang, H. Mechanistic,
Crystallographic, and Computational Studies on the Catalytic, Enantioselective
Sulfenofunctionalization of Alkenes. Nat. Chem. 2014, 6, 1056-64.
83. Li, L.; Li, Z.; Huang, D.; Wang, H.; Shi, Y. Chiral Phosphoric Acid Catalyzed
Enantioselective Sulfamination of Amino-Alkenes. RSC Adv. 2013, 3, 4523-4525.
84. Liu, T.; Tian, J.; Gao, W. C.; Chang, H. H.; Liu, Q.; Li, X.; Wei, W. L.
Intermolecular Sulfenoamination of Alkenes with Sulfonamides and N-
Sulfanylsuccinimides to Access Beta-Sulfonylamino Sulfides and
Dihydrobenzothiazines. Org. Biomol. Chem. 2017, 15, 5983-5992.

208
85. Wang, D.; Yan, Z.; Xie, Q.; Zhang, R.; Lin, S.; Wang, Y. Three-Component
Difunctionalization of Alkenes Leading to β-Acetamido Sulfides and β-Acetoxy
Sulfides. Org. Biomol. Chem. 2017, 15, 1998-2002.
86. Zheng, Y.; He, Y.; Rong, G.; Zhang, X.; Weng, Y.; Dong, K.; Xu, X.; Mao, J. NaI-
Mediated Acetamidosulphenylation of Alkenes with Nitriles as the Nucleophiles:
A Direct Access to Acetamidosulfides. Org. Lett. 2015, 17, 5444-7.
87. Cui, H.; Liu, X.; Wei, W.; Yang, D.; He, C.; Zhang, T.; Wang, H. Molecular Iodine-
Mediated Difunctionalization of Alkenes with Nitriles and Thiols Leading to Beta-
Acetamido Sulfides. J. Org. Chem. 2016, 81, 2252-60.
88. Borisov, A.; Goncharova, T.; Matsulevich, Z. V.; Borisova, G.; Osmanov, V.
Directions of Heterocyclization in Reactions of 4,6-Dimethyl-2-Pyrimidinesulfenyl
Chloride with 2-Allylphenol. Chem. Heterocycl. Compd. 2001, 6, 783-784.
89. Jarboe, S. G.; Terrazas, M. S.; Beak, P. The Endocyclic Restriction Test: The
Geometries of Nucleophilic Substitutions at Sulfur (VI) and Sulfur (II). J. Org.
Chem. 2008, 73, 9627-9632.
90. Iwasaki, M.; Fujii, T.; Yamamoto, A.; Nakajima, K.; Nishihara, Y. Palladium-
Catalyzed Regio- and Stereoselective Chlorothiolation of Terminal Alkynes with
Sulfenyl Chlorides. Chem. Asian J. 2014, 9, 58-62.
91. Li, Y.; Shi, Y.; Huang, Z.; Wu, X.; Xu, P.; Wang, J.; Zhang, Y. Catalytic Thia-
Sommelet− Hauser Rearrangement: Application to the Synthesis of Oxindoles.
Org. Lett. 2011, 13, 1210-1213.
92. Alom, N.-E.; Wu, F.; Li, W. One-Pot Strategy for Thiazoline Synthesis from
Alkenes and Thioamides. Organic Letters. 2017, 19, 930-933.
93. Wallbaum, J.; Garve, L. K.; Jones, P. G.; Werz, D. B. Ring-Opening 1,3-
Halochalcogenation of Cyclopropane Dicarboxylates. Org. Lett. 2017, 19, 98-101.
94. Iwasaki, M.; Fujii, T.; Nakajima, K.; Nishihara, Y. Iron-Induced Regio- and
Stereoselective Addition of Sulfenyl Chlorides to Alkynes by a Radical Pathway.
Angew. Chem., Int. Ed. 2014, 53, 13880-4.
95. Hostier, T.; Ferey, V.; Ricci, G.; Gomez Pardo, D.; Cossy, J. TFA-Promoted Direct
C-H Sulfenylation at the C2 Position of Non-Protected Indoles. Chem. Commun.
2015, 51, 13898-901.
96. Pettitt, D.; Helmkamp, G. Stable Oxonium Salts and Alkylation of Episulfides and
Disulfides. J. Org. Chem. 1963, 28, 2932-&.

209
97. Raynolds, P.; Zonnebelt, S.; Bakker, S.; Kellogg, R. M. Chemistry of Cis- and
Trans-2,3-Di-Tert-Butylthiiranes (Episulfides). Consequences of Steric
Overcrowding in Small Ring Compounds. J. Am. Chem. Soc. 1974, 96, 3146-3154.
98. Lucchini, V.; Modena, G.; Pasquato, L. Novel Type of Selectivity in Anionotropic
Rearrangements. J. Am. Chem. Soc. 1988, 110, 6900-6901.
99. Lucchini, V.; Modena, G.; Pasquato, L. Anionotropic Rearrangements of Tert-
Butyl-and Adamantylthiiranium Ions into Thietanium Ions. A Novel Case of
Selectivity. J. Am. Chem. Soc. 1991, 113, 6600-6607.
100. Fachini, M.; Lucchini, V.; Modena, G.; Pasi, M.; Pasquato, L. Nucleophilic
Reactions at the Sulfur of Thiiranium and Thiirenium Ions. New Insight in the
Electrophilic Additions to Alkenes and Alkynes. Evidence for an Episulfurane
Intermediate. J. Am. Chem. Soc. 1999, 121, 3944-3950.
101. Wagh, S. J.; Tawde, T. S.; Sapre, J. V.; Khose, V. N.; Karnik, A. V. A Convenient
Route to Benzimidazole Fused Chiral Heterocyclic Bases. 2016.
102. Krasovskii, A.; Klyuev, N.; Roman, A.; Kochergin, P.; Dank, E. K. Synthesis and
Physicochemical Properties of Thiazolino [3,2-a] Benzimidazoles. Chem.
Heterocycl. Compd. 1983, 19, 756-762.
103. Griller, D.; Ingold, K. U. Free-Radical Clocks. Acc. Chem. Res. 1980, 13, 317-323.
104. Smit, V.; Zefirov, N. S.; Bodrikov, I. V.; Krimer, M. Z. Episulfonium Ions: Myth
and Reality. Acc. Chem. Res. 1979, 12, 282-288.
105. Denmark, S. E.; Collins, W. R.; Cullen, M. D. Observation of Direct Sulfenium and
Selenenium Group Transfer from Thiiranium and Seleniranium Ions to Alkenes. J.
Am. Chem. Soc. 2009, 131, 3490-3492.
106. Denmark, S. E.; Vogler, T. Synthesis and Reactivity of Enantiomerically Enriched
Thiiranium Ions. Chemistry. 2009, 15, 11737-45.
107. Ke, M.; Song, Q. Copper/B2Pin2-Catalyzed C–H Difluoroacetylation–
Cycloamidation of Anilines Leading to the Formation of 3,3-Difluoro-2-Oxindoles.
Chem. Commun. 2017, 53, 2222-2225.
108. Movahhed, S.; Westphal, J.; Dindaroğlu, M.; Falk, A.; Schmalz, H. G. Low‐
Pressure Cobalt‐Catalyzed Enantioselective Hydrovinylation of Vinylarenes.
Chem.-Eur. J. 2016, 22, 7381-7384.
109. Donohoe, T. J.; Callens, C. K.; Flores, A.; Lacy, A. R.; Rathi, A. H. Recent
Developments in Methodology for the Direct Oxyamination of Olefins. Chem. Eur.
J. 2011, 17, 58-76.

210
110. Chemler, S. R.; Fuller, P. H. Heterocycle Synthesis by Copper Facilitated Addition
of Heteroatoms to Alkenes, Alkynes and Arenes. Chem. Soc. Rev. 2007, 36, 1153-
1160.
111. McDonald, R. I.; Liu, G.; Stahl, S. S. Palladium (II)-Catalyzed Alkene
Functionalization Via Nucleopalladation: Stereochemical Pathways and
Enantioselective Catalytic Applications. Chem. Rev. 2011, 111, 2981-3019.
112. Yin, G.; Mu, X.; Liu, G. Palladium(II)-Catalyzed Oxidative Difunctionalization of
Alkenes: Bond Forming at a High-Valent Palladium Center. Acc Chem Res. 2016,
49, 2413-2423.
113. Lan, X.-W.; Wang, N.-X.; Xing, Y. Recent Advances in Radical
Difunctionalization of Simple Alkenes. Eur. J. Org. Chem. 2017, 2017, 5821-5851.
114. Muniz, K.; Martinez, C. Development of Intramolecular Vicinal Diamination of
Alkenes: From Palladium to Bromine Catalysis. J. Org. Chem. 2013, 78, 2168-
2174.
115. Ni, Y.; Zuo, H.; Li, Y.; Wu, Y.; Zhong, F. Copper-Catalyzed Regioselective
Intramolecular Electrophilic Sulfenoamination Via Lewis Acid Activation of
Disulfides under Aerobic Conditions. Org. Lett. 2018, 20, 4350-4353.
116. Dickerson, T. J.; Reed, N. N.; LaClair, J. J.; Janda, K. D. A Precipitator for the
Detection of Thiophilic Metals in Aqua. J. Am. Chem. Soc. 2004, 126, 16582-
16586.
117. Denmark, S. E.; Hartmann, E.; Kornfilt, D. J.; Wang, H. Catalytic, Enantioselective
Sulfenofunctionalisation of Alkenes: Mechanistic, Crystallographic, and
Computational Studies. Nat. Chem. 2014, 6, 1056.
118. Denmark, S. E.; Kornfilt, D. J.; Vogler, T. Catalytic Asymmetric
Thiofunctionalization of Unactivated Alkenes. J. Am. Chem. Soc. 2011, 133,
15308-15311.
119. Schiaffella, F.; Macchiarulo, A.; Milanese, L.; Vecchiarelli, A.; Costantino, G.;
Pietrella, D.; Fringuelli, R. Design, Synthesis, and Microbiological Evaluation of
New Candida Albicans Cyp51 Inhibitors. J. Med. Chem. 2005, 48, 7658-7666.
120. Matsumoto, Y.; Tsuzuki, R.; Matsuhisa, A.; Yoden, T.; Yamagiwa, Y.;
Yanagisawa, I.; Shibanuma, T.; Nohira, H. Novel Potassium Channel Openers. Part
4: Transformation of the 1, 4-Benzoxazine Skeleton into 1, 4-Benzothiazine, 1,2,
3, 4-Tetrahydroquinoline, 1, 2, 3, 4-Tetrahydroquinoxaline, Indoline, and 1,5-
Benzoxazepine. Bioorg. Med. Chem. 2000, 8, 393-404.
121. Hasegawa, K.; Ito, S.; Inoue, S.; Wakamatsu, K.; Ozeki, H.; Ishiguro, I. Dihydro-

211
1,4-Benzothiazine-6,7-Dione, the Ultimate Toxic Metabolite of 4-S-
Cysteaminylphenol and 4-S-Cysteaminylcatechol. Biochem. Pharmacol. 1997, 53,
1435-1444.
122. Ajani, O. O. Functionalized 1, 4‐Benzothiazine: A Versatile Scaffold with Diverse
Biological Properties. Archiv der Pharmazie. 2012, 345, 841-851.
123. Cecchetti, V.; Fravolini, A.; Fringuelli, R.; Mascellani, G.; Pagella, P.; Palmioli,
M.; Segre, G.; Terni, P. Quinolonecarboxylic Acids. 2. Synthesis and Antibacterial
Evaluation of 7-Oxo-2,3-Dihydro-7h-Pyrido[1,2,3-De][1,4]Benzothiazine-6-
Carboxylic Acids. J. Med. Chem. 1987, 30, 465-473.
124. Shen, X.-M.; Dryhurst, G. Further Insights into the Influence of L-Cysteine on the
Oxidation Chemistry of Dopamine: Reaction Pathways of Potential Relevance to
Parkinson's Disease. Chem. Res. Toxicol. 1996, 9, 751-763.
125. Shen, X.-M.; Dryhurst, G. Oxidation Chemistry of (−)-Norepinephrine in the
Presence of L-Cysteine. J. Med. Chem. 1996, 39, 2018-2029.
126. Taurand, G. Phenothiazine and Derivatives. Ullmann's Encyclopedia of Industrial
Chemistry. 2000, 601-615.
127. Bicherov, A. V.; Kharisov, B. I.; Blanco, L. M.; Korshunov, O. Y.; Koroleva, E.
L.; Burlov, A. S.; Borodkin, G. S.; Kurbatov, V. P.; Uflyand, I. E.; Garnovskii, A.
D. Metal Chelates of New Ligands: 1,2-Benzothiazine-1,1-Dioxide Derivatives. J.
Coord. Chem. 2001, 54, 337-342.
128. Dabholkar, V. V.; Gavande, R. P. Synthesis and Antimicrobial Activities of Novel
1,4-Benzothiazine Derivatives. Arab. J. Chem. 2016, 9, S225-S229.
129. Fringuelli, R.; Schiaffella, F.; Utrilla Navarro, M. P.; Milanese, L.; Santini, C.;
Rapucci, M.; Marchetti, C.; Riccardi, C. 1,4-Benzothiazine Analogues and
Apoptosis. Bioorg. Med. Chem. 2003, 11, 3245-3254.
130. Sanicanin, Z.; Juric, A.; Tabakovic, I.; Trinajstic, N. Synthesis and Electrochemical
Study of Benzothiazine and Phenothiazine Derivatives. J. Org. Chem. 1987, 52,
4053-4057.
131. Sonawane, A. E.; Pawar, Y. A.; Nagle, P. S.; Mahulikar, P. P.; More, D. H.
Synthesis of 1, 4‐Benzothiazine Compounds Containing Isatin Hydrazone Moiety
as Antimicrobial Agent. Chin. J. Chem. 2009, 27, 2049-2054.
132. Saadouni, M.; Ghailane, T.; Boukhris, S.; Hassikou, A.; Habbadi, N.; Ghailane, R.;
Harcharras, M.; Souizi, A.; Amri, H. Regioselective Synthesis of New Variety of
1, 4-Benzothiazines. Organic Communications. 2014, 7, 77.

212
133. Ishikawa, Y.; Terao, Y.; Suzuki, K.; Shikano, N.; Sekiya, M. Cyclization of α- and
β-Alkylthio-Substituted Amines Possessing Positively Charged Carbon at the
Nitrogen. A New Synthetic Method for Thiazolidines, Thiomorpholines and
Dihydro-1,4-Benzothiazines. Chem. Pharm. Bull. 1984, 32, 438-446.
134. Qiao, X.; Bao, Z.; Xing, H.; Yang, Y.; Ren, Q.; Zhang, Z. Organocatalytic
Approach for Transfer Hydrogenation of Quinolines, Benzoxazines and
Benzothiazines. Catal. Lett. 2017, 147, 1673-1678.
135. Bhattacharya, S.; Ghosh, P.; Basu, B. Graphene Oxide (Go): An Efficient
Carbocatalyst for the Benign Synthesis of Functionalized 1,4-Benzothiazines.
Tetrahedron Lett. 2017, 58, 926-931.
136. Rueping, M.; Antonchick, A. P.; Theissmann, T. Remarkably Low Catalyst
Loading in Bronsted Acid Catalyzed Transfer Hydrogenations: Enantioselective
Reduction of Benzoxazines, Benzothiazines, and Benzoxazinones. Angew. Chem.,
Int. Ed. 2006, 45, 6751-6755.
137. Chao, H. J.; Tuerdi, H.; Kick, E. K.; Yang, W., Lxr Modulators. Google Patents:
2010.
138. Lin, Y.-M.; Lu, G.-P.; Wang, R.-K.; Yi, W.-B. Radical Route to 1, 4-Benzothiazine
Derivatives from 2-Aminobenzenethiols and Ketones under Transition-Metal-Free
Conditions. Org. Lett. 2016, 18, 6424-6427.
139. Prasad, C. D.; Verma, A.; Sattar, M.; Kumar, S. Silver-Mediated Thio-
Acetoxylation and TFA Triggered Cyclization of Amino Disulfides with
Unactivated Alkenes: Synthesis of 3-Aryl/Alkyl-1,4-Benzothiazines. RSC Adv.
2015, 5, 75881-75888.
140. Qiao, Z.; Liu, H.; Xiao, X.; Fu, Y.; Wei, J.; Li, Y.; Jiang, X. Efficient Access to 1,
4-Benzothiazine: Palladium-Catalyzed Double C–S Bond Formation Using
Na2S2O3 as Sulfurating Reagent. Org. Lett. 2013, 15, 2594-2597.
141. Babudri, F.; Di Nunno, L.; Florio, S. An Easy and Efficient Synthesis of 4H‐
Thiazolo [5, 4, 3‐ij] Quinolin‐4‐Ones and 5H‐1,4‐Thiazino [2, 3, 4‐i,j] Quinolin‐5‐
Ones. J. Heterocycl. Chem. 1981, 18, 1273-1274.
142. Olagbemiro, T.; Nyakutse, C.; Lajide, L.; Agho, M.; Chukwu, C. Synthesis and
Reactions of 3‐Phenyl‐3,4‐Dihydro‐1,4‐Quinoxalin‐2(1H)‐One and Its
Heterocyclic Analogues. Bulletin des Sociétés Chimiques Belges. 1987, 96, 473-
480.
143. Hori, M.; Kataoka, T.; Shimizu, H.; Ueda, N. Non-Stereospecific Ring Expansions
of Benzothiazoline Sulfoxides. Tetrahedron Lett. 1981, 22, 1701-1704.

213
144. Lin, Y. M.; Lu, G. P.; Wang, R. K.; Yi, W. B. Radical Route to 1,4-Benzothiazine
Derivatives from 2-Aminobenzenethiols and Ketones under Transition-Metal-Free
Conditions. Org. Lett. 2016, 18, 6424-6427.
145. Gao, X.; Yu, B.; Yang, Z.; Zhao, Y.; Zhang, H.; Hao, L.; Han, B.; Liu, Z. Ionic
Liquid-Catalyzed C–S Bond Construction Using CO2 as a C1 Building Block under
Mild Conditions: A Metal-Free Route to Synthesis of Benzothiazoles. ACS Catal.
2015, 5, 6648-6652.
146. Brown, M.; Kumar, R.; Rehbein, J.; Wirth, T. Enantioselective Oxidative
Rearrangements with Chiral Hypervalent Iodine Reagents. Chem.-Eur. J. 2016, 22,
4030-4035.
147. Nawrat, C. C.; Jamison, C. R.; Slutskyy, Y.; MacMillan, D. W.; Overman, L. E.
Oxalates as Activating Groups for Alcohols in Visible Light Photoredox Catalysis:
Formation of Quaternary Centers by Redox-Neutral Fragment Coupling. J. Am.
Chem. Soc. 2015, 137, 11270-11273.
148. Jin, J.; MacMillan, D. W. Alcohols as Alkylating Agents in Heteroarene C–H
Functionalization. Nature. 2015, 525, 87-90.
149. Huy, P. H.; Hauch, T.; Filbrich, I. Lewis Base Catalyzed Nucleophilic Substitutions
of Alcohols. Synlett. 2016, 27, 2631-2636.
150. An, J.; Denton, R. M.; Lambert, T. H.; Nacsa, E. D. The Development of Catalytic
Nucleophilic Substitution Reactions: Challenges, Progress and Future Directions.
Org. Biomol. Chem. 2014, 12, 2993-3003.
151. Motokura, K.; Nakagiri, N.; Mizugaki, T.; Ebitani, K.; Kaneda, K. Nucleophilic
Substitution Reactions of Alcohols with Use of Montmorillonite Catalysts as Solid
Brønsted Acids. J. Org. Chem. 2007, 72, 6006-6015.
152. Sanz, R.; Martínez, A.; Miguel, D.; Alvarez‐Gutierrez, J. M.; Rodriguez, F.
Brønsted Acid‐Catalyzed Nucleophilic Substitution of Alcohols. Adv. Synth. Catal.
2006, 348, 1841-1845.
153. Mitsunobu, O. The Use of Diethyl Azodicarboxylate and Triphenylphosphine in
Synthesis and Transformation of Natural Products. Synthesis. 1981, 1981, 1-28.
154. Mitsunobu, O.; Yamada, M. Preparation of Esters of Carboxylic and Phosphoric
Acid Via Quaternary Phosphonium Salts. Bull. Chem. Soc. Jpn. 1967, 40, 2380-
2382.
155. Dryzhakov, M.; Richmond, E.; Moran, J. Recent Advances in Direct Catalytic
Dehydrative Substitution of Alcohols. Synthesis. 2016, 48, 935-959.

214
156. Sundararaju, B.; Achard, M.; Bruneau, C. Transition Metal Catalyzed Nucleophilic
Allylic Substitution: Activation of Allylic Alcohols Via Π-Allylic Species. Chem.
Soc. Rev. 2012, 41, 4467-4483.
157. Pronin, S. V.; Reiher, C. A.; Shenvi, R. A. Stereoinversion of Tertiary Alcohols to
Tertiary-Alkyl Isonitriles and Amines. Nature. 2013, 501, 195-199.
158. Hardee, D. J.; Kovalchuke, L.; Lambert, T. H. Nucleophilic Acyl Substitution Via
Aromatic Cation Activation of Carboxylic Acids: Rapid Generation of Acid
Chlorides under Mild Conditions. J. Am. Chem. Soc. 2010, 132, 5002-5003.
159. Kelly, B. D.; Lambert, T. H. Cyclopropenium-Activated Cyclodehydration of
Diols. Org. Lett. 2011, 13, 740-743.
160. Kelly, B. D.; Lambert, T. H. Aromatic Cation Activation of Alcohols: Conversion
to Alkyl Chlorides Using Dichlorodiphenylcyclopropene. J. Am. Chem. Soc. 2009,
131, 13930-13931.
161. Nacsa, E. D.; Lambert, T. H. Cyclopropenone Catalyzed Substitution of Alcohols
with Mesylate Ion. Org. Lett. 2013, 15, 38-41.
162. Vanos, C. M.; Lambert, T. H. Development of a Catalytic Platform for Nucleophilic
Substitution: Cyclopropenone‐Catalyzed Chlorodehydration of Alcohols. Angew.
Chem., Int. Ed. 2011, 50, 12222-12226.
163. Nguyen, T. V.; Bekensir, A. Aromatic Cation Activation: Nucleophilic Substitution
of Alcohols and Carboxylic Acids. Org. Lett. 2014, 16, 1720-3.
164. Nguyen, T. V.; Lyons, D. J. A Novel Aromatic Carbocation-Based Coupling
Reagent for Esterification and Amidation Reactions. Chem. Commun. 2015, 51,
3131-4.
165. Oss, G.; Ho, J.; Nguyen, T. V. Tropylium Ion Catalyzes Hydration Reactions of
Alkynes. Eur. J. Org. Chem. 2018, 2018, 3974-3981.
166. Lyons, D. J. M.; Crocker, R. D.; Enders, D.; Nguyen, T. V. Tropylium Salts as
Efficient Organic Lewis Acid Catalysts for Acetalization and Transacetalization
Reactions in Batch and Flow. Green Chem. 2017, 19, 3993-3996.
167. Tran, U. P. N.; Oss, G.; Pace, D. P.; Ho, J.; Nguyen, T. V. Tropylium-Promoted
Carbonyl-Olefin Metathesis Reactions. Chem. Sci. 2018, 9, 5145-5151.
168. Beddoe, R. H.; Andrews, K. G.; Magné, V.; Cuthbertson, J. D.; Saska, J.; Shannon-
Little, A. L.; Shanahan, S. E.; Sneddon, H. F.; Denton, R. M. Redox-Neutral
Organocatalytic Mitsunobu Reactions. Science. 2019, 365, 910-914.

215
169. Chen, L.; Yin, X.-P.; Wang, C.-H.; Zhou, J. Catalytic Functionalization of Tertiary
Alcohols to Fully Substituted Carbon Centres. Org. Biomol. Chem. 2014, 12, 6033-
6048.
170. Denmark, S. E.; Burk, M. T.; Hoover, A. J. On the Absolute Configurational
Stability of Bromonium and Chloronium Ions. J. Am. Chem. Soc. 2010, 132, 1232-
1233.
171. Einaru, S.; Shitamichi, K.; Nagano, T.; Matsumoto, A.; Asano, K.; Matsubara, S.
Trans‐Cyclooctenes as Halolactonization Catalysts. Angew. Chem., Int. Ed. 2018,
57, 13863-13867.
172. Wu, F.; Ariyarathna, J. P.; Alom, N.-E.; Kaur, N.; Li, W. Oxyamination of
Unactivated Alkenes with Electron-Rich Amines and Acids Via a Catalytic
Triiodide Intermediate. Org. Lett. 2020, 22, 884-890.
173. Wu, F.; Alom, N. E.; Ariyarathna, J. P.; Naß, J.; Li, W. Regioselective Formal [3+
2] Cycloadditions of Urea Substrates with Activated and Unactivated Olefins for
Intermolecular Olefin Aminooxygenation. Angew. Chem., Int. Ed. 2019, 58, 11676-
11680.
174. Kaur, N.; Wu, F.; Alom, N.-E.; Ariyarathna, J. P.; Saluga, S. J.; Li, W.
Intermolecular Alkene Difunctionalizations for the Synthesis of Saturated
Heterocycles. Org. Biomol. Chem. 2019, 17, 1643-1654.
175. Nishikata, T.; Nagashima, H. N Alkylation of Tosylamides Using Esters as Primary
and Tertiary Alkyl Sources: Mediated by Hydrosilanes Activated by a Ruthenium
Catalyst. Angew. Chem., Int. Ed. 2012, 51, 5363-5366.
176. Dryzhakov, M.; Richmond, E.; Li, G.; Moran, J. Catalytic B(C6F5)3 H2O-Promoted
Defluorinative Functionalization of Tertiary Aliphatic Fluorides. J. Fluorine Chem.
2017, 193, 45-51.
177. Ohshima, T.; Ipposhi, J.; Nakahara, Y.; Shibuya, R.; Mashima, K. Aluminum
Triflate as a Powerful Catalyst for Direct Amination of Alcohols, Including
Electron‐Withdrawing Group‐Substituted Benzhydrols. Adv. Synth. Catal. 2012,
354, 2447-2452.
178. Costa, D. C. S. Additions to Non-Activated Alkenes: Recent Advances. Arab. J.
Chem. 2020, 13, 799-834.
179. Alexanian, E. J.; Lee, C.; Sorensen, E. J. Palladium-Catalyzed Ring-Forming
Aminoacetoxylation of Alkenes. J. Am. Chem. Soc. 2005, 127, 7690-7691.
180. Borsini, E.; Broggini, G.; Fasana, A.; Galli, S.; Khansaa, M.; Piarulli, U.;
Rigamonti, M. Intramolecular Palladium‐Catalyzed Aminocarboxylation of

216
Olefins as a Direct Route to Bicyclic Oxazolidinones. Adv. Synth. Catal. 2011, 353,
985-994.
181. Zhu, H.; Chen, P.; Liu, G. Palladium-Catalyzed Intramolecular
Aminoacetoxylation of Unactivated Alkenes with Hydrogen Peroxide as Oxidant.
Org. Lett. 2015, 17, 1485-1488.
182. Kou, X.; Li, Y.; Wu, L.; Zhang, X.; Yang, G.; Zhang, W. Palladium-Catalyzed
Aerobic Aminooxygenation of Alkenes for Preparation of Isoindolinones. Org.
Lett. 2015, 17, 5566-5569.
183. Li, J.; Grubbs, R. H.; Stoltz, B. M. Palladium-Catalyzed Aerobic Intramolecular
Aminoacetoxylation of Alkenes Enabled by Catalytic Nitrate. Org. Lett. 2016, 18,
5449-5451.
184. Desai, L. V.; Sanford, M. S. Construction of Tetrahydrofurans by PdII/PdIV‐
Catalyzed Aminooxygenation of Alkenes. Angew. Chem., Int. Ed. 2007, 46, 5737-
5740.
185. Noack, M.; Göttlich, R. Copper (I) Catalysed Cyclisation of Unsaturated N-
Benzoyloxyamines: An Aminohydroxylation Via Radicals. Chem. Commun. 2002,
536-537.
186. Fuller, P. H.; Kim, J.-W.; Chemler, S. R. Copper Catalyzed Enantioselective
Intramolecular Aminooxygenation of Alkenes. J. Am. Chem. Soc. 2008, 130,
17638-17639.
187. Karyakarte, S. D.; Smith, T. P.; Chemler, S. R. Stereoselective Isoxazolidine
Synthesis Via Copper-Catalyzed Alkene Aminooxygenation. J. Org. Chem. 2012,
77, 7755-7760.
188. de Haro, T.; Nevado, C. Flexible Gold‐Catalyzed Regioselective Oxidative
Difunctionalization of Unactivated Alkenes. Angew. Chem., Int. Ed. 2011, 50, 906-
910.
189. Mahoney, J. M.; Smith, C. R.; Johnston, J. N. Brønsted Acid-Promoted Olefin
Aziridination and Formal a Nti-Aminohydroxylation. J. Am. Chem. Soc. 2005, 127,
1354-1355.
190. Xu, H.-C.; Moeller, K. D. Intramolecular Anodic Olefin Coupling Reactions: The
Use of a Nitrogen Trapping Group. J. Am. Chem. Soc. 2008, 130, 13542-13543.
191. Schmidt, V. A.; Alexanian, E. J. Metal-Free Oxyaminations of Alkenes Using
Hydroxamic Acids. J. Am. Chem. Soc. 2011, 133, 11402-11405.
192. Farid, U.; Wirth, T. Highly Stereoselective Metal‐Free Oxyaminations Using
Chiral Hypervalent Iodine Reagents. Angew. Chem., Int. Ed. 2012, 51, 3462-3465.

217
193. Danneman, M. W.; Hong, K. B.; Johnston, J. N. Oxidative Inter-/Intermolecular
Alkene Diamination of Hydroxy Styrenes with Electron-Rich Amines. Org. Lett.
2015, 17, 2558-2561.
194. Lu, D.-F.; Zhu, C.-L.; Jia, Z.-X.; Xu, H. Iron (II)-Catalyzed Intermolecular Amino-
Oxygenation of Olefins through the N–O Bond Cleavage of Functionalized
Hydroxylamines. J. Am. Chem. Soc. 2014, 136, 13186-13189.
195. Du, Y.; Wu, Y.; Liu, A.-H.; He, L.-N. Quaternary Ammonium Bromide
Functionalized Polyethylene Glycol: A Highly Efficient and Recyclable Catalyst
for Selective Synthesis of 5-Aryl-2-Oxazolidinones from Carbon Dioxide and
Aziridines under Solvent-Free Conditions. J. Org. Chem. 2008, 73, 4709-4712.
196. Niemi, T.; Fernandez, I.; Steadman, B.; Mannisto, J. K.; Repo, T. Carbon Dioxide-
Based Facile Synthesis of Cyclic Carbamates from Amino Alcohols. Chem.
Commun. 2018, 54, 3166-3169.
197. Yang, Z.-Z.; He, L.-N.; Peng, S.-Y.; Liu, A.-H. Lewis Basic Ionic Liquids-
Catalyzed Synthesis of 5-Aryl-2-Oxazolidinones from Aziridines and CO2 under
Solvent-Free Conditions. Green Chem. 2010, 12.
198. Seo, U. R.; Chung, Y. K. Potassium Phosphate-Catalyzed One-Pot Synthesis of 3-
Aryl-2-Oxazolidinones from Epoxides, Amines, and Atmospheric Carbon Dioxide.
Green Chem. 2017, 19, 803-808.
199. Pena-Lopez, M.; Neumann, H.; Beller, M. (Enantio)Selective Hydrogen
Autotransfer: Ruthenium-Catalyzed Synthesis of Oxazolidin-2-Ones from Urea
and Diols. Angew. Chem., Int. Ed. 2016, 55, 7826-30.




















