
Analysis of the Quaternary Structure of the MutL C-terminal Domain
15
Analysis of the Quaternary Structure of the MutL C-terminal Domain Jan Kosinski 1,2 , Ina Steindorf 1 , Janusz M. Bujnicki 2 , Luis Giron-Monzon 1 and Peter Friedhoff 1 * 1 Institut fu ¨r Biochemie (FB 08) Justus-Liebig Universita ¨t Giessen D-35392, Germany 2 Laboratory of Bioinformatics and Protein Engineering International Institute of Molecular and Cell Biology 4 Ks. Trojdena, 02-109 Warsaw Poland The dimeric DNA mismatch repair protein MutL has a key function in communicating mismatch recognition by MutS to downstream repair processes. Dimerization of MutL is mediated by the C-terminal domain, while activity of the protein is modulated by the ATP-dependent dimerization of the highly conserved N-terminal domain. Recently, a crystal structure analysis of the Escherichia coli MutL C-terminal dimerization domain has been reported and a model for the biological dimer was proposed. In this model, dimerization is mediated by the internal (In) subdomain comprising residues 475–569. Here, we report a computational analysis of all protein interfaces observed in the crystal structure and suggest that the biological dimer interface is formed by a hydrophobic surface patch of the external (Ex) subdomain (residues 432–474 and 570–615). Moreover, sequence analysis revealed that this surface patch is conserved among the MutL proteins. To test this hypothesis, single and double-cysteine variants of MutL were generated and tested for their ability to be cross-linked with chemical cross-linkers of various size. Finally, deletion of the C-terminal residues 605–615 abolished homodimerization. The biochemical data are fully compatible with a revised model for the biological dimer, which has important implications for understanding the heterodimerization of eukaryotic MutL homologues, modeling the MutL holoenzyme and predicting protein–protein interaction sites. q 2005 Elsevier Ltd. All rights reserved. Keywords: mismatch repair; MutL; protein–protein interaction; homo- dimers; crystal packing *Corresponding author Introduction DNA repair systems are crucial for maintaining a stable genome. 1 One of these systems, DNA mismatch repair (MMR), is present in most organisms and enhances the fidelity of DNA replication by a factor of up to 1000 by correcting replication errors. 2,3 As mismatch repair proteins MutS and MutL are evolutionarily conserved among the three kingdoms of life, it is believed that the basic mechanisms of mismatch repair are similar. In the presence of ATP and a mismatch, MutS recruits MutL, and together they activate proteins involved in downstream processes of MMR, e.g. in the Escherichia coli system, strand discrimination by the MutH endonuclease and DNA unwinding by the UvrD helicase. 4,5 Bacterial MutL proteins are homodimers, while their eukaryotic homologs form heterodimers consisting of the MutL homolog MLH1 and either PMS1, PMS2 or MLH3. 6–8 Moreover, MutL is a member of the GHKL superfamily of ATPases, which includes gyrase, a type II topoisomerase, Hsp90, histidine kinase, and MutL. 9 Structurally, MutL can be dissected into a highly conserved N-terminal ATPase domain (NTD); which is able to dimerize upon ATP binding, a flexible and poorly conserved linker, and a less conserved C-terminal domain (CTD), which is involved in homo- and heterodimerization. 10,11 On the basis of the mutual (dis)similarity of the CTD, MutL homologs can be classified into at least three groups: MutL, PMS2 (HexB) and MLH1. 7,12 Recently, Yang and 0022-2836/$ - see front matter q 2005 Elsevier Ltd. All rights reserved. Abbreviations used: ASA, accessible surface area; PDB, Protein Data Bank; BMOE, bis-maleimidoethane; BM[PEO] 4 , 1,11-bis-maleimidotetraethyleneglycol; CTD, C-terminal domain; NTD, N-terminal domain. E-mail address of the corresponding author: [email protected] doi:10.1016/j.jmb.2005.06.044 J. Mol. Biol. (2005) 351, 895–909
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Analysis of the Quaternary Structure of the MutL C-terminal Domain

doi:10.1016/j.jmb.2005.06.044 J. Mol. Biol. (2005) 351, 895–909
Analysis of the Quaternary Structure of theMutL C-terminal Domain
Jan Kosinski1,2, Ina Steindorf1, Janusz M. Bujnicki2, Luis Giron-Monzon1
and Peter Friedhoff1*
1Institut fur Biochemie (FB 08)Justus-Liebig UniversitatGiessen D-35392, Germany
2Laboratory of Bioinformaticsand Protein EngineeringInternational Institute ofMolecular and Cell Biology4 Ks. Trojdena, 02-109 WarsawPoland
0022-2836/$ - see front matter q 2005 E
Abbreviations used: ASA, accessiProtein Data Bank; BMOE, bis-maleBM[PEO]4, 1,11-bis-maleimidotetraeC-terminal domain; NTD, N-termin
E-mail address of the [email protected]
The dimeric DNA mismatch repair protein MutL has a key function incommunicating mismatch recognition by MutS to downstream repairprocesses. Dimerization of MutL is mediated by the C-terminal domain,while activity of the protein is modulated by the ATP-dependentdimerization of the highly conserved N-terminal domain. Recently, acrystal structure analysis of the Escherichia coli MutL C-terminaldimerization domain has been reported and a model for the biologicaldimer was proposed. In this model, dimerization is mediated by theinternal (In) subdomain comprising residues 475–569. Here, we report acomputational analysis of all protein interfaces observed in the crystalstructure and suggest that the biological dimer interface is formed by ahydrophobic surface patch of the external (Ex) subdomain (residues432–474 and 570–615). Moreover, sequence analysis revealed that thissurface patch is conserved among the MutL proteins. To test thishypothesis, single and double-cysteine variants of MutL were generatedand tested for their ability to be cross-linked with chemical cross-linkers ofvarious size. Finally, deletion of the C-terminal residues 605–615 abolishedhomodimerization. The biochemical data are fully compatible with arevised model for the biological dimer, which has important implicationsfor understanding the heterodimerization of eukaryotic MutL homologues,modeling the MutL holoenzyme and predicting protein–protein interactionsites.
q 2005 Elsevier Ltd. All rights reserved.
Keywords: mismatch repair; MutL; protein–protein interaction; homo-dimers; crystal packing
*Corresponding authorIntroduction
DNA repair systems are crucial for maintaininga stable genome.1 One of these systems, DNAmismatch repair (MMR), is present in mostorganisms and enhances the fidelity of DNAreplication by a factor of up to 1000 by correctingreplication errors.2,3 As mismatch repair proteinsMutS and MutL are evolutionarily conservedamong the three kingdoms of life, it is believedthat the basic mechanisms of mismatch repair aresimilar. In the presence of ATP and a mismatch,MutS recruits MutL, and together they activate
lsevier Ltd. All rights reserve
ble surface area; PDB,imidoethane;thyleneglycol; CTD,al domain.ing author:
proteins involved in downstream processes ofMMR, e.g. in the Escherichia coli system, stranddiscrimination by the MutH endonuclease andDNA unwinding by the UvrD helicase.4,5
Bacterial MutL proteins are homodimers, whiletheir eukaryotic homologs form heterodimersconsisting of the MutL homolog MLH1 and eitherPMS1, PMS2 or MLH3.6–8 Moreover, MutL is amember of the GHKL superfamily of ATPases,which includes gyrase, a type II topoisomerase,Hsp90, histidine kinase, and MutL.9 Structurally,MutL can be dissected into a highly conservedN-terminal ATPase domain (NTD); which is able todimerize upon ATP binding, a flexible and poorlyconserved linker, and a less conserved C-terminaldomain (CTD), which is involved in homo- andheterodimerization.10,11 On the basis of the mutual(dis)similarity of the CTD, MutL homologs canbe classified into at least three groups: MutL,PMS2 (HexB) and MLH1.7,12 Recently, Yang and
d.
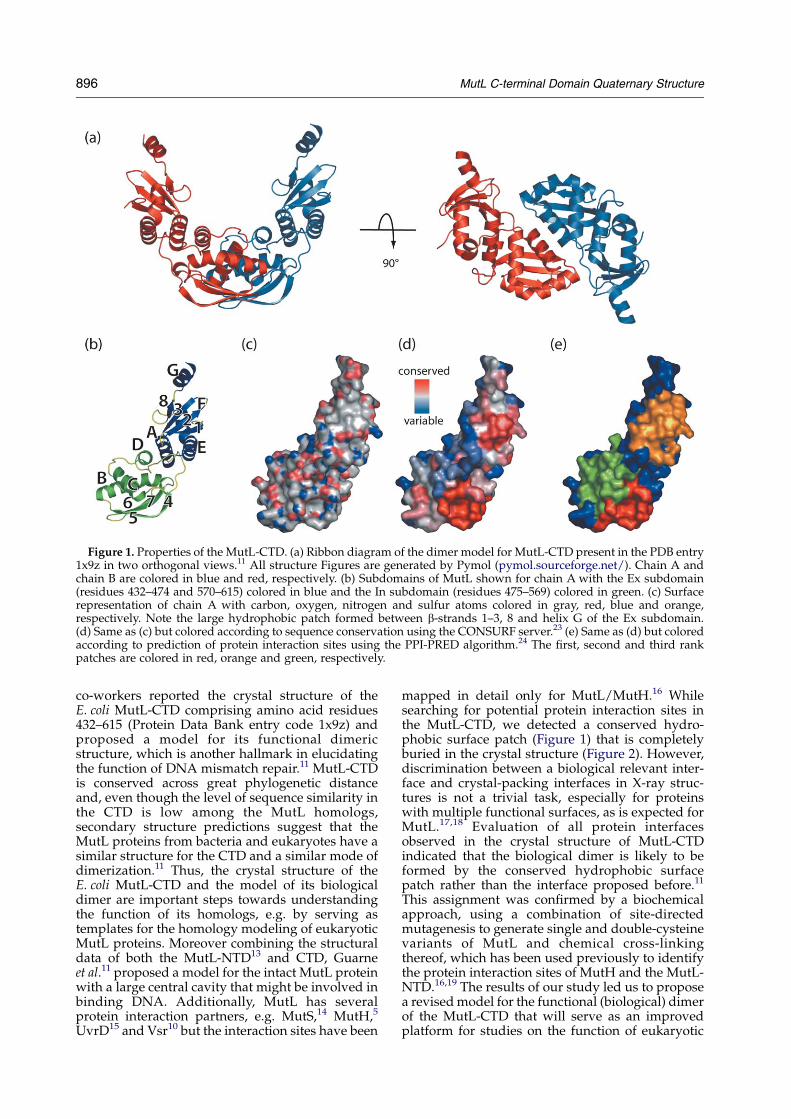
Figure 1. Properties of the MutL-CTD. (a) Ribbon diagram of the dimer model for MutL-CTD present in the PDB entry1x9z in two orthogonal views.11 All structure Figures are generated by Pymol (pymol.sourceforge.net/). Chain A andchain B are colored in blue and red, respectively. (b) Subdomains of MutL shown for chain A with the Ex subdomain(residues 432–474 and 570–615) colored in blue and the In subdomain (residues 475–569) colored in green. (c) Surfacerepresentation of chain A with carbon, oxygen, nitrogen and sulfur atoms colored in gray, red, blue and orange,respectively. Note the large hydrophobic patch formed between b-strands 1–3, 8 and helix G of the Ex subdomain.(d) Same as (c) but colored according to sequence conservation using the CONSURF server.23 (e) Same as (d) but coloredaccording to prediction of protein interaction sites using the PPI-PRED algorithm.24 The first, second and third rankpatches are colored in red, orange and green, respectively.
896 MutL C-terminal Domain Quaternary Structure
co-workers reported the crystal structure of theE. coli MutL-CTD comprising amino acid residues432–615 (Protein Data Bank entry code 1x9z) andproposed a model for its functional dimericstructure, which is another hallmark in elucidatingthe function of DNA mismatch repair.11 MutL-CTDis conserved across great phylogenetic distanceand, even though the level of sequence similarity inthe CTD is low among the MutL homologs,secondary structure predictions suggest that theMutL proteins from bacteria and eukaryotes have asimilar structure for the CTD and a similar mode ofdimerization.11 Thus, the crystal structure of theE. coli MutL-CTD and the model of its biologicaldimer are important steps towards understandingthe function of its homologs, e.g. by serving astemplates for the homology modeling of eukaryoticMutL proteins. Moreover combining the structuraldata of both the MutL-NTD13 and CTD, Guarneet al.11 proposed a model for the intact MutL proteinwith a large central cavity that might be involved inbinding DNA. Additionally, MutL has severalprotein interaction partners, e.g. MutS,14 MutH,5
UvrD15 and Vsr10 but the interaction sites have been
mapped in detail only for MutL/MutH.16 Whilesearching for potential protein interaction sites inthe MutL-CTD, we detected a conserved hydro-phobic surface patch (Figure 1) that is completelyburied in the crystal structure (Figure 2). However,discrimination between a biological relevant inter-face and crystal-packing interfaces in X-ray struc-tures is not a trivial task, especially for proteinswith multiple functional surfaces, as is expected forMutL.17,18 Evaluation of all protein interfacesobserved in the crystal structure of MutL-CTDindicated that the biological dimer is likely to beformed by the conserved hydrophobic surfacepatch rather than the interface proposed before.11
This assignment was confirmed by a biochemicalapproach, using a combination of site-directedmutagenesis to generate single and double-cysteinevariants of MutL and chemical cross-linkingthereof, which has been used previously to identifythe protein interaction sites of MutH and the MutL-NTD.16,19 The results of our study led us to proposea revised model for the functional (biological) dimerof the MutL-CTD that will serve as an improvedplatform for studies on the function of eukaryotic

Figure 2. Protein contacts observed in the crystal structure of MutL C-terminal domain. Stereo view with the sameorientation as in Figure 1(a) showing cartoons of chain A (blue) and chain B (red) of 1x9z. Crystallographic neighbors(symmetry mates) corresponding to chain A or chain B are shown as ribbons colored in light blue and red, respectively.Only those neighbors are shown that have at least ten residues within 6 A of either chain A or chain B. Four differenttypes of interfaces are observed: interface 1 (A/B-I); interface 2 (A/A-II and B/B-II); interface 3 (A/A-III and B/B-III);and interface 4 (A/B-IV and B/A-IV). See Tables 1 and 2 for a detailed analysis of the interfaces.
MutL C-terminal Domain Quaternary Structure 897
MutL homologs and the modeling of their quater-nary structure, and as the starting point for theprediction and identification of docking sites forUvrD and MutH proteins.
Results and Discussion
Protein interaction site prediction
Since the MutL-CTD has been shown to bephysically interacting with the UvrD helicase,11,15
we asked whether it is possible to predict theprotein interaction site using available bioinfor-matics tools such as surface hydrophobicity,interface residue propensity,20,21 residue conserva-tion22,23and support vector machine approach(Figure 1).18,24 The interface patch with the highestlevel of sequence conservation and highest rankedprediction score24 is formed by residues Pro483–Leu488 and Arg531–Gln536 located in the Insubdomain of MutL-CTD with an accessible surfacearea (ASA) of 968 A2. In the search for the potentialprotein interaction sites of the MutL-CTD it did notescape our notice that a conserved hydrophobicpatch (residues 439–459 of strand b1–b3 and597–612 of b8 and helix aG) on the surface ofMutL the external (Ex) subdomain (Figure 1) alsowas predicted to be a potential protein interactionsite. The analysis of Contacts of Structural Units(CSU)25 revealed that this hydrophobic patch wascompletely buried in the crystal structure (Figure 2).It had been noticed before that, although abiological partner may not be present in the crystal,the corresponding functional interface may providea non-natural crystal contact.17 To determinewhether the observed crystallographic contact ofthis surface patch is of biological relevance, wegenerated all protein dimers present in the crystalstructure of MutL-CTD and investigated in detailfour protein interfaces (numbered 1–4) that had at
least ten residues within 6 A of another subunit(Figure 2). Guarne et al.11 proposed that thebiological dimer is formed by interface 1 consistingof residues 489–496 (b5 and aB), 533–548 (aC) and563–570 (aD and loop LDE), while the above-mentioned conserved, hydrophobic patch is almostidentical with interface 2 consisting of residues437–459 (b1–3) and 598–612 (b8 and aG).
Analysis of interfaces observed in the crystalstructure of MutL-CTD
Next, we subjected the dimer models formed bythe four interfaces to an analysis using the protein–protein interaction server26 and by a proceduresimilar to that performed by Bahadur et al.(Table 1).27 One of the best discriminators betweena biological relevant interface and a crystal contactis the ASA buried upon the dimer formation.28
Based on the buried ASA (B/2) of 400 A2 as anarbitrary lower-limit criterion for defining anoligomeric contact,29 only interfaces 1–3 are suf-ficiently large to be classified as potential biologicalrelevant interfaces. Since the protein fragment ofMutL that had been crystallized was shown byanalytical ultracentrifugation to be a dimer insolution,11 this raises the question of which of thethree larger interfaces observed in the crystal isthe biological relevant dimer interface. However,discrimination of the biological dimer from crystalcontacts is far from being trivial.27,28
In this case, other parameters and combinationsthereof are needed to improve discrimination. Thegap volume (GV) indices30 relating the volume ofcavities to the interface area for interface 1 (GVZ5.4) and interface 3 (GVZ7.4) are high compared tothe one of interface 2 (GVZ2.2). Thus, interfaces 1and 3 resemble more a crystal dimer interface(mean GVZ4.0G0.9),20 while interface 2 resemblesa bone fide homodimer interface (mean GVZ2.2G0.9).26 Moreover, other parameters such as a fraction
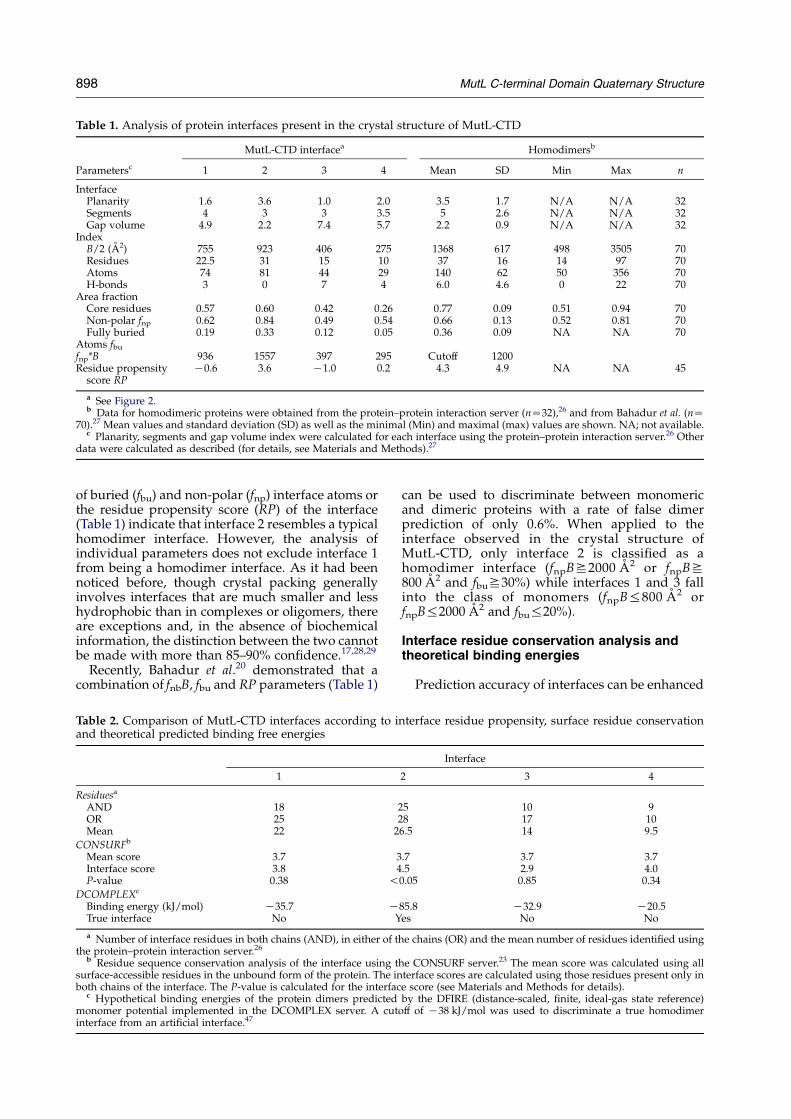
Table 1. Analysis of protein interfaces present in the crystal structure of MutL-CTD
MutL-CTD interfacea Homodimersb
Parametersc 1 2 3 4 Mean SD Min Max n
InterfacePlanarity 1.6 3.6 1.0 2.0 3.5 1.7 N/A N/A 32Segments 4 3 3 3.5 5 2.6 N/A N/A 32Gap volume
Index4.9 2.2 7.4 5.7 2.2 0.9 N/A N/A 32
B/2 (A2) 755 923 406 275 1368 617 498 3505 70Residues 22.5 31 15 10 37 16 14 97 70Atoms 74 81 44 29 140 62 50 356 70H-bonds 3 0 7 4 6.0 4.6 0 22 70
Area fractionCore residues 0.57 0.60 0.42 0.26 0.77 0.09 0.51 0.94 70Non-polar fnp 0.62 0.84 0.49 0.54 0.66 0.13 0.52 0.81 70Fully buried
Atoms fbu
0.19 0.33 0.12 0.05 0.36 0.09 NA NA 70
fnp*B 936 1557 397 295 Cutoff 1200Residue propensity
score RPK0.6 3.6 K1.0 0.2 4.3 4.9 NA NA 45
a See Figure 2.b Data for homodimeric proteins were obtained from the protein–protein interaction server (nZ32),26 and from Bahadur et al. (nZ
70).27 Mean values and standard deviation (SD) as well as the minimal (Min) and maximal (max) values are shown. NA; not available.c Planarity, segments and gap volume index were calculated for each interface using the protein–protein interaction server.26 Other
data were calculated as described (for details, see Materials and Methods).27
898 MutL C-terminal Domain Quaternary Structure
of buried (fbu) and non-polar (fnp) interface atoms orthe residue propensity score (RP) of the interface(Table 1) indicate that interface 2 resembles a typicalhomodimer interface. However, the analysis ofindividual parameters does not exclude interface 1from being a homodimer interface. As it had beennoticed before, though crystal packing generallyinvolves interfaces that are much smaller and lesshydrophobic than in complexes or oligomers, thereare exceptions and, in the absence of biochemicalinformation, the distinction between the two cannotbe made with more than 85–90% confidence.17,28,29
Recently, Bahadur et al.20 demonstrated that acombination of fnbB, fbu and RP parameters (Table 1)
Table 2. Comparison of MutL-CTD interfaces according to iand theoretical predicted binding free energies
1
Residuesa
AND 18OR 25Mean 22 2
CONSURFb
Mean score 3.7 3Interface score 3.8 4P-value 0.38 !
DCOMPLEXc
Binding energy (kJ/mol) K35.7 KTrue interface No Y
a Number of interface residues in both chains (AND), in either of tthe protein–protein interaction server.26
b Residue sequence conservation analysis of the interface using tsurface-accessible residues in the unbound form of the protein. The inboth chains of the interface. The P-value is calculated for the interfac
c Hypothetical binding energies of the protein dimers predictedmonomer potential implemented in the DCOMPLEX server. A cutinterface from an artificial interface.47
can be used to discriminate between monomericand dimeric proteins with a rate of false dimerprediction of only 0.6%. When applied to theinterface observed in the crystal structure ofMutL-CTD, only interface 2 is classified as ahomodimer interface (fnpBS2000 A2 or fnpBS800 A2 and fbuS30%) while interfaces 1 and 3 fallinto the class of monomers (fnpB%800 A2 orfnpB%2000 A2 and fbu%20%).
Interface residue conservation analysis andtheoretical binding energies
Prediction accuracy of interfaces can be enhanced
nterface residue propensity, surface residue conservation
Interface
2 3 4
25 10 928 17 106.5 14 9.5
.7 3.7 3.7
.5 2.9 4.00.05 0.85 0.34
85.8 K32.9 K20.5es No No
he chains (OR) and the mean number of residues identified using
he CONSURF server.23 The mean score was calculated using allterface scores are calculated using those residues present only ine score (see Materials and Methods for details).by the DFIRE (distance-scaled, finite, ideal-gas state reference)
off of K38 kJ/mol was used to discriminate a true homodimer
MutL C-terminal Domain Quaternary Structure 899
by including a residue conservation analysis.22,31–34
Consequently, we performed such an analysis usingan alignment of 30 MutL protein sequencesobtained from several bacterial genomes (completeand incomplete) that also harbor a gene for MutH.This alignment was used to calculate the residuesurface conservation using the CONSURF server23
(see Figure 1) and mapped onto the surface ofMutL-CTD (Figure 1(e)). A statistical analysisof residue conservation of the four different typesof interfaces revealed that only interface 2 had asignificantly high residue conservation score(P-value !0.05) compared to a random selected
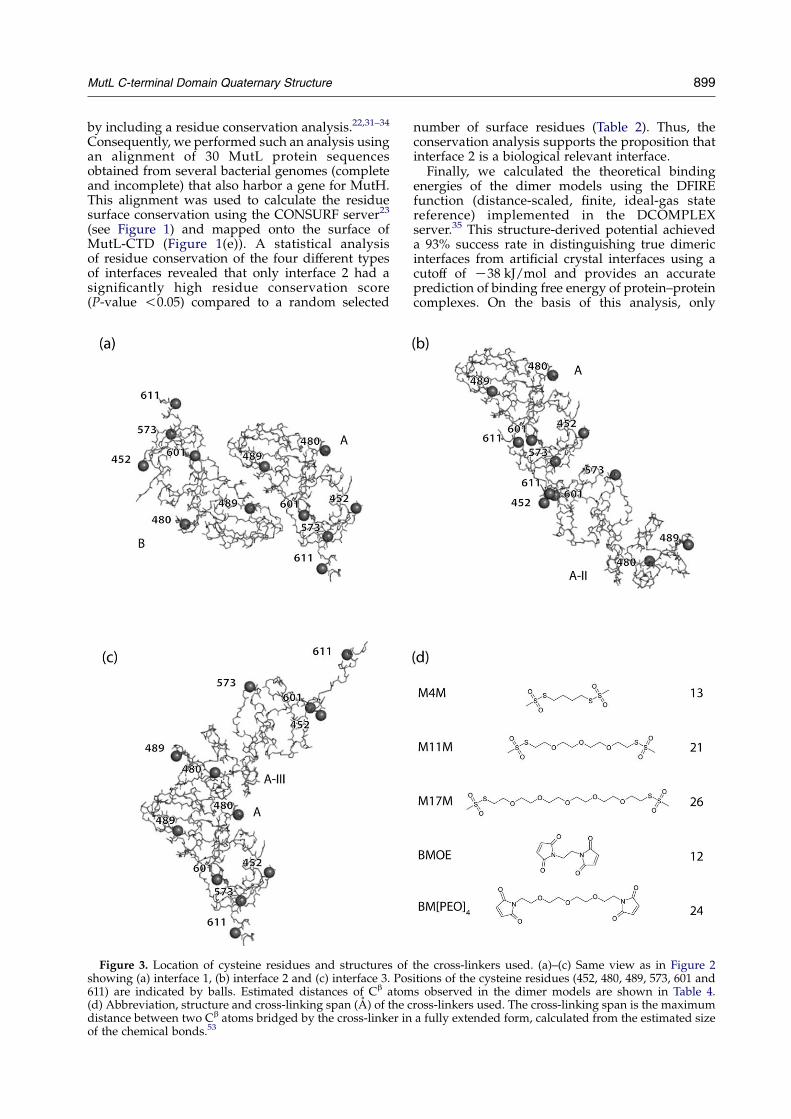
Figure 3. Location of cysteine residues and structures ofshowing (a) interface 1, (b) interface 2 and (c) interface 3. Pos611) are indicated by balls. Estimated distances of Cb atom(d) Abbreviation, structure and cross-linking span (A) of the cdistance between two Cb atoms bridged by the cross-linker inof the chemical bonds.53
number of surface residues (Table 2). Thus, theconservation analysis supports the proposition thatinterface 2 is a biological relevant interface.
Finally, we calculated the theoretical bindingenergies of the dimer models using the DFIREfunction (distance-scaled, finite, ideal-gas statereference) implemented in the DCOMPLEXserver.35 This structure-derived potential achieveda 93% success rate in distinguishing true dimericinterfaces from artificial crystal interfaces using acutoff of K38 kJ/mol and provides an accurateprediction of binding free energy of protein–proteincomplexes. On the basis of this analysis, only
the cross-linkers used. (a)–(c) Same view as in Figure 2itions of the cysteine residues (452, 480, 489, 573, 601 ands observed in the dimer models are shown in Table 4.ross-linkers used. The cross-linking span is the maximuma fully extended form, calculated from the estimated size
900 MutL C-terminal Domain Quaternary Structure
interface 2 was judged to be a “true dimer” interface(see Table 2) having a calculated binding energy ofabout K90 kJ/mol, which is almost three timeslower than that of any of the other dimer models.
In summary, on the basis of the different types ofanalyses described above, we conclude that inter-face 2 consisting of residues 437–459 (b1–b 3) and598–612 (b8 and aG) is the true biological interfaceto form the homodimer of the MutL-CTD.
Single cysteine variants of MutL
In order to substantiate our prediction, we carriedout biochemical experiments to distinguishbetween the alternative dimer models for theMutL-CTD. To this end, we generated a series ofsingle-cysteine variants of MutL starting from apreviously constructed cysteine-free variant ofMutL (LCF). Cross-linking of these variants withreagents of various length (see Figure 3) shouldallow discrimination between the different dimermodels. A prerequisite for the successful mappingof protein interactions via cross-linking is that themutation does not affect the function of the protein.We therefore assayed the MutL variants for theirin vivo function in mismatch repair. MutL variantswere tested for their ability to complement amutator phenotype in the mutL-deficient E. colistrain TX2652.36 All variants were able to comple-ment a mutL-mutator phenotype at least tenfoldbetter than the vector control (Table 3). Next, theMutL variants were purified by affinity chromato-graphy and assayed for their ability to dimerize viathe CTD as judged by size-exclusion chromato-graphy and to activate MutH in a mismatch-
Table 3. In vivo and in vitro analysis of single-cysteine MutL
In vivo
Variant Mediana Rangeb Repai
Wild-type (wt) 3 0–69 10Vector (mutL null) 1023 806–1891 0N-terminal domain 906 143–1219 1LCF 13 6–66 9LR74C 27 24–90 9LN131C 6 4–40 10LK135C 60 39–106 9LD452C 6 1–33 10L480C 10 3–34 10LR489C 18 4–49 10LS573C 9 4–50 10LQ601C 6 0–50 10LA611C 86 24–97 9LQ601C/A611C (SH)f 65 45–150 9(S-S)f NA NA NLD605–615 201 99–589 8
a For in vivo activity the rpo mutation assay was used (see Materialsclones that arise by spontaneous mutation.
b Minimum and maximum number of rifampicin-resistant clones.c Repair is scored by calculating the ratio: (mediannullKmedianvard MutL activity was measured by mismatch and MutS-dependent a
containing a single d(GATC) site as described in Materials and Methe Quaternary structure was determined by size-exclusion chromatf This variant was tested in vitro either in the reduced form or in t
dependent manner by MutS (Table 3). All MutLvariants retained the ability to dimerize and tomediate communication between MutS and MutH,in good agreement with our in vivo complementa-tion data. Finally, we tested the variants for theaccessibility of cysteine residues by modificationwith tetramethylrhodamine-5-maleimide. Allsingle-cysteine variants could be modified to asimilar extent (data not shown). Hence, we con-cluded that these variants are suitable to determinea low-resolution distance map of the selectedpositions.
Thiol-thiol cross-linking of single-cysteine MutLand MutH variants
The single-cysteine MutL variants were subjectedto cross-linking using the homobifunctionalcysteine-specific cross-linkers bis-maleimidoethane(BMOE) and 1,11-bis-maleimidotetraethyleneglycol(BM[PEO]4) (Figure 4). Under the experimentalconditions used, we never observed cross-linkformation using the cysteine-free MutL variantLCF, indicating that the reaction is specific forcysteine residues. We have used the same strategyto map the protein–protein interaction betweenMutH and the MutL-NTD.16 Since the structure ofthe dimeric MutL-NTD is known,13 distances (d)between Cb atoms of cysteine residues in the MutLvariants could be estimated and compared toexperimental results of cross-linking with reagentsof different length (Table 4). Variant LR489C wasexpected to form cross-links if interface 1 would bethe correct biological interface, while variants LS573C,LQ601Cand LA611C should produce cross-link
variants
In vitro
r (%)c % Activityd % SDQuaternarystructuree
0 100 8 DimerNA NA NA
1 !3 ND NA9 103 5 Dimer8 162 9 Dimer0 62 6 Dimer
4 56 5 Dimer1 61 3 Dimer0 79 4 Dimer0 45 4 Dimer0 83 5 Dimer1 90 11 Dimer
3 53 13 Dimer5 62 10 DimerA 95 7 Dimer2 !5 ND Monomer
and Methods for details). Median number of rifampicin-resistant
iant)/(mediannullKmedianwt)!100.ctivation of MutH to cleave a linear 484 bp heteroduplex substrateods. Errors shown are G one standard deviation.ography of the C-terminal fragment of thrombin-cleaved MutL.he oxidized form.

Figure 4. Chemical cross-linking of MutL single-cysteine variants. (a) Single-cysteine MutL variants(1–10 mM) were cross-linked for 30 seconds with a 50-fold molar excess of either BMOE or BM[PEO]4. Reactionswere stopped with 10 mM DTT and analyzed by SDS-PAGE followed by staining with Coomassie brilliant blue.(b) Quantitative evaluation of cross-linking yield fromtwo to four independent experiments.
MutL C-terminal Domain Quaternary Structure 901
products if interface 2 were correct. Variants L480C
and LD452C were not expected to produce anycross-link if either interface 1 or interface 2 werecorrect. In addition to the previously tested MutLvariants LN131C and LK135C,16 we included anothervariant, LR74C. Starting with the long-range cross-linker (BM[PEO]4), which was able to cross-linkLN131C (dZ28 A) but not LK135C (dZ50 A),16 we
Table 4. Comparison of dimer models with data from cross-l
Distance (A) in interface
Variant CB-CB 1 2 3 4
LR74C 74–74 9LN131C 131–131 28LK135C 135–135 50LD452C 452–452 67 28 52 26LC480 480–480 49 73 16 47LR489C 489–489 14 100 22 87LS573C 573–573 58 27 66 26LQ601C 601–601 38 20 65 39LA611C 611–611 68 21 97 48LQ601C/
A611C601–611 53 4 82 45
a Yield of cross-linking after subtraction of background of 5–10% (CCC, O 40%; ND, not determined. Information for the cross-linker
were able to generate cross-links for variants LR74C
(dZ9 A), LQ601C, LA611C and, to a smaller extent,LS573C (Table 4 and Figure 4). In contrast to this, theLD452C, LC480 and LR489C variants did not form cross-links to an extent greater than the background(Table 4). Similar results were obtained using theshorter cross-linker BMOE (Figure 4). Furthermore,we used a series of methanethiosulfonate cross-linkers with four, 11 and 17 spacer atoms (Figures 3and 5). The results obtained were similar to thosefor the bis-maleimide cross-linkers and are sum-marized in Table 4. Moreover, thrombin cleavage ofthe MutL variants splitting the protein into anN-terminal and a C-terminal half did not change theobserved cross-linking pattern (data not shown).11
In conclusion, the results of this cross-linkinganalysis are compatible only with Cb–Cb distancesobserved in the dimer model formed by interface 2but not with any of the other dimer modelsincluding that proposed by Guarne et al.11
Engineering a disulfide bond in the MutL-CTD
Next, we used the revised dimer model for theMutL-CTD formed by interface 2 to design a doublecysteine variant that can form an intermoleculardisulfide linkage, thereby forming a covalentlylinked protein dimer (Figure 6). To this end, wegenerated the double-cysteine variant LA601C/Q611C,which was tested for in vivo and in vitro activity asshown before for the single-cysteine variants(Table 3). Indeed, we observed that upon removalof DTT (by dialysis or gel-filtration), the doublevariant LQ601C/A611C readily forms a covalentlylinked dimer (O90%) as judged by SDS-PAGE(Figure 6), while the corresponding single-cysteinevariants (LA601C and LQ611C, respectively) had only alow tendency to form covalently linked proteindimers (!5%) in the absence of a reducing reagent.Finally, we tested whether the dimeric variant is stillable to activate the MutH endonuclease in amismatch and MutS-dependent manner. Interest-ingly, the double-cysteine variant LQ601C/A611C hadeven higher activity in the presence of a disulfide
inking analysis
Cross-link witha
S-S M4M M11M M17M BMOEBM[PE-
O]4
K CCC CCC CCC CC CCK C CC CC C CCK ND ND ND K KK K K K K KK K K K K KK K K K K KK K C CC K CK CCC CCC CCC CC CCK CCC CCC CCC CCC CCC
CCC ND ND ND ND ND
i.e. no cross-linker added): K, ! 10%; C, 10–20%; CC, 20–40%;s is given in Figure 3.
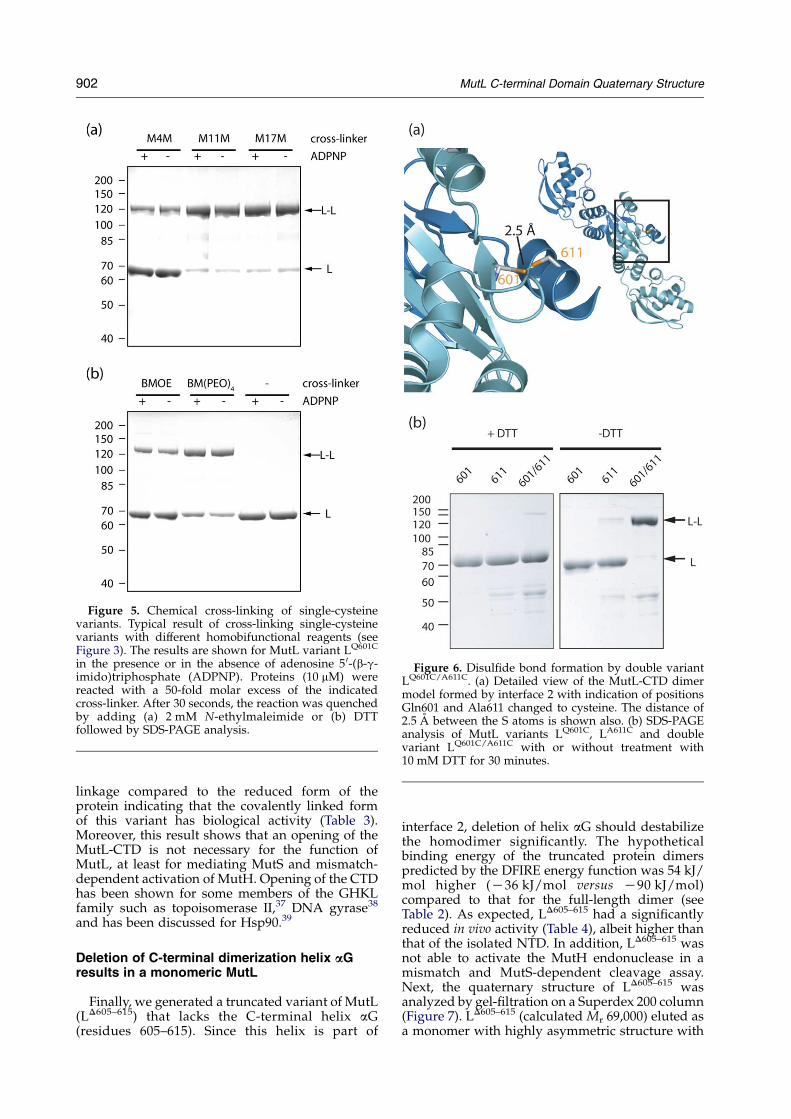
Figure 5. Chemical cross-linking of single-cysteinevariants. Typical result of cross-linking single-cysteinevariants with different homobifunctional reagents (seeFigure 3). The results are shown for MutL variant LQ601C
in the presence or in the absence of adenosine 5 0-(b-g-imido)triphosphate (ADPNP). Proteins (10 mM) werereacted with a 50-fold molar excess of the indicatedcross-linker. After 30 seconds, the reaction was quenchedby adding (a) 2 mM N-ethylmaleimide or (b) DTTfollowed by SDS-PAGE analysis.
Figure 6. Disulfide bond formation by double variantLQ601C/A611C. (a) Detailed view of the MutL-CTD dimermodel formed by interface 2 with indication of positionsGln601 and Ala611 changed to cysteine. The distance of2.5 A between the S atoms is shown also. (b) SDS-PAGEanalysis of MutL variants LQ601C, LA611C and doublevariant LQ601C/A611C with or without treatment with10 mM DTT for 30 minutes.
902 MutL C-terminal Domain Quaternary Structure
linkage compared to the reduced form of theprotein indicating that the covalently linked formof this variant has biological activity (Table 3).Moreover, this result shows that an opening of theMutL-CTD is not necessary for the function ofMutL, at least for mediating MutS and mismatch-dependent activation of MutH. Opening of the CTDhas been shown for some members of the GHKLfamily such as topoisomerase II,37 DNA gyrase38
and has been discussed for Hsp90.39
Deletion of C-terminal dimerization helix aGresults in a monomeric MutL
Finally, we generated a truncated variant of MutL(LD605–615) that lacks the C-terminal helix aG(residues 605–615). Since this helix is part of
interface 2, deletion of helix aG should destabilizethe homodimer significantly. The hypotheticalbinding energy of the truncated protein dimerspredicted by the DFIRE energy function was 54 kJ/mol higher (K36 kJ/mol versus K90 kJ/mol)compared to that for the full-length dimer (seeTable 2). As expected, LD605–615 had a significantlyreduced in vivo activity (Table 4), albeit higher thanthat of the isolated NTD. In addition, LD605–615 wasnot able to activate the MutH endonuclease in amismatch and MutS-dependent cleavage assay.Next, the quaternary structure of LD605–615 wasanalyzed by gel-filtration on a Superdex 200 column(Figure 7). LD605–615 (calculated Mr 69,000) eluted asa monomer with highly asymmetric structure with
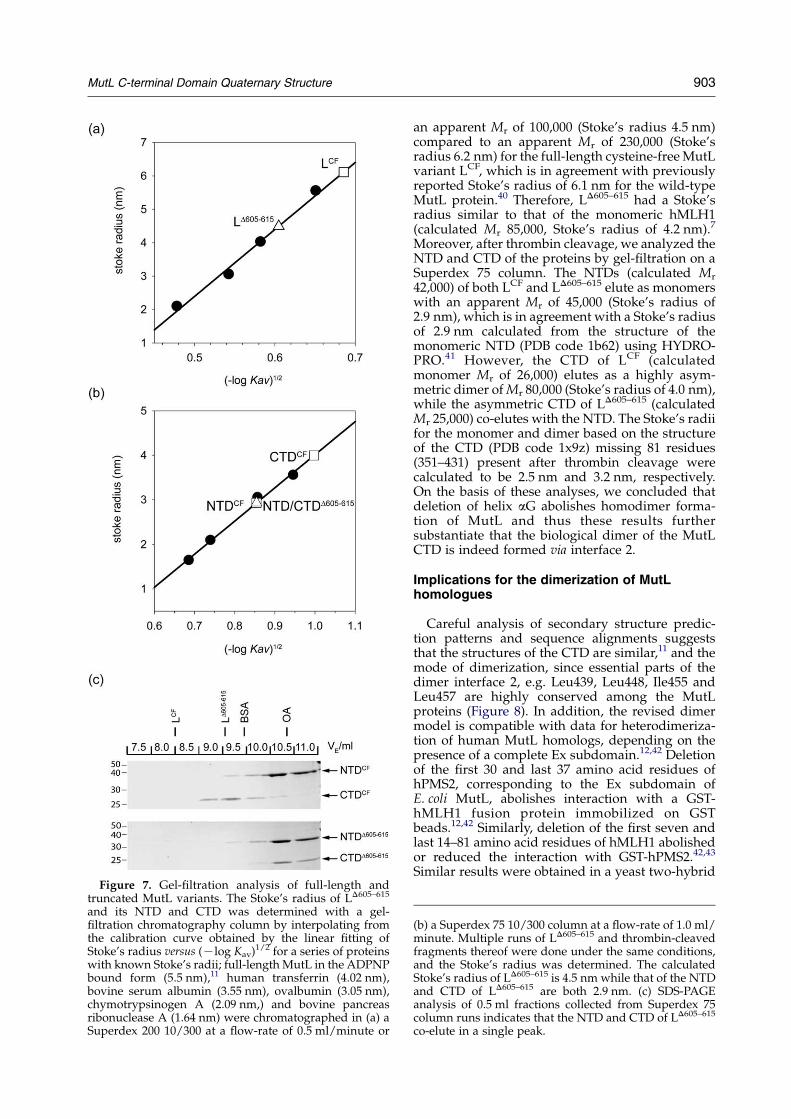
Figure 7. Gel-filtration analysis of full-length andtruncated MutL variants. The Stoke’s radius of LD605–615
and its NTD and CTD was determined with a gel-filtration chromatography column by interpolating fromthe calibration curve obtained by the linear fitting ofStoke’s radius versus (Klog Kav)1/2 for a series of proteinswith known Stoke’s radii; full-length MutL in the ADPNPbound form (5.5 nm),11 human transferrin (4.02 nm),bovine serum albumin (3.55 nm), ovalbumin (3.05 nm),chymotrypsinogen A (2.09 nm,) and bovine pancreasribonuclease A (1.64 nm) were chromatographed in (a) aSuperdex 200 10/300 at a flow-rate of 0.5 ml/minute or
MutL C-terminal Domain Quaternary Structure 903
an apparent Mr of 100,000 (Stoke’s radius 4.5 nm)compared to an apparent Mr of 230,000 (Stoke’sradius 6.2 nm) for the full-length cysteine-free MutLvariant LCF, which is in agreement with previouslyreported Stoke’s radius of 6.1 nm for the wild-typeMutL protein.40 Therefore, LD605–615 had a Stoke’sradius similar to that of the monomeric hMLH1(calculated Mr 85,000, Stoke’s radius of 4.2 nm).7
Moreover, after thrombin cleavage, we analyzed theNTD and CTD of the proteins by gel-filtration on aSuperdex 75 column. The NTDs (calculated Mr
42,000) of both LCF and LD605–615 elute as monomerswith an apparent Mr of 45,000 (Stoke’s radius of2.9 nm), which is in agreement with a Stoke’s radiusof 2.9 nm calculated from the structure of themonomeric NTD (PDB code 1b62) using HYDRO-PRO.41 However, the CTD of LCF (calculatedmonomer Mr of 26,000) elutes as a highly asym-metric dimer of Mr 80,000 (Stoke’s radius of 4.0 nm),while the asymmetric CTD of LD605–615 (calculatedMr 25,000) co-elutes with the NTD. The Stoke’s radiifor the monomer and dimer based on the structureof the CTD (PDB code 1x9z) missing 81 residues(351–431) present after thrombin cleavage werecalculated to be 2.5 nm and 3.2 nm, respectively.On the basis of these analyses, we concluded thatdeletion of helix aG abolishes homodimer forma-tion of MutL and thus these results furthersubstantiate that the biological dimer of the MutLCTD is indeed formed via interface 2.
Implications for the dimerization of MutLhomologues
Careful analysis of secondary structure predic-tion patterns and sequence alignments suggeststhat the structures of the CTD are similar,11 and themode of dimerization, since essential parts of thedimer interface 2, e.g. Leu439, Leu448, Ile455 andLeu457 are highly conserved among the MutLproteins (Figure 8). In addition, the revised dimermodel is compatible with data for heterodimeriza-tion of human MutL homologs, depending on thepresence of a complete Ex subdomain.12,42 Deletionof the first 30 and last 37 amino acid residues ofhPMS2, corresponding to the Ex subdomain ofE. coli MutL, abolishes interaction with a GST-hMLH1 fusion protein immobilized on GSTbeads.12,42 Similarly, deletion of the first seven andlast 14–81 amino acid residues of hMLH1 abolishedor reduced the interaction with GST-hPMS2.42,43
Similar results were obtained in a yeast two-hybrid
(b) a Superdex 75 10/300 column at a flow-rate of 1.0 ml/minute. Multiple runs of LD605–615 and thrombin-cleavedfragments thereof were done under the same conditions,and the Stoke’s radius was determined. The calculatedStoke’s radius of LD605–615 is 4.5 nm while that of the NTDand CTD of LD605–615 are both 2.9 nm. (c) SDS-PAGEanalysis of 0.5 ml fractions collected from Superdex 75column runs indicates that the NTD and CTD of LD605–615
co-elute in a single peak.

Figure 8. Sequence alignment and secondary structure (experimental and prediction) of MutL homologs. Core andrim residues of the dimer interface are indicated by red and green stars, respectively. Subdomain borders are indicatedfor the E. coli protein. Deletions within the CTD, which abolish in vitro heterodimerization of the human homologs andthe E. coli MutL homodimer (this study) are indicated by gray bars.
904 MutL C-terminal Domain Quaternary Structure
assay using the yeast MLH1p and PMS1p pro-teins.12 These observations are in agreement withour results, that deletion of the C-terminal residues605–615 abolishes homodimerization of E. coliMutL. However, since deletion of parts of a proteinmay affect its structural integrity, a detailed analysisincluding homology modeling of the bacterialand eukaryotic homologs and validation of thesemodels by statistical and biochemical analyses areneeded to substantiate this prediction but arebeyond the scope of this article.
Revised dimer model of the MutL-CTD: impli-cations for protein–protein interactions
We present a revised model for the biologicaldimer of the E. coli MutL-CTD that has implicationsfor the modes of heterodimerization by the eukary-otic MutL homologs and for the identification ofprotein–protein interaction sites. In the revisedmodel, the dimer interface is formed by interface 2consisting of residues 437–459 (b1–b 3) and 598–612(b8 and aG) located in the Ex subdomain withcontributions from both the N-terminal and theC-terminal halves of this subdomain in dimeri-zation (Figure 9). Following an approach similar tothat taken by Guarne,11 it is tempting to predict amodel for the MutL holoenzyme using the struc-tures of the NTD and CTD sharing a common dyadsymmetry axis. We decided to arrange the domainsin such a way that the conserved surface of the CTDis facing the C-terminal half of the NTD (Figure 9).However, even with this constraint, the NTD canadopt all possible orientations with respect torotation around the dyad axis relative to the CTDand a range of distances to the CTD along this axis.We have shown before that the binding site of MutH
in the NTD is located in a region close to residues169, 251, 314 and 327 (Figure 9).16 In addition, otherdata suggested that the CTD is also in directphysical contact to MutH and UvrD,5,11,44 probablyvia the conserved surface patch. The model shownin Figure 9 offers the attractive opportunity thatinteraction partners such as MutH and UvrD couldbind between the conserved patch of the CTD andthe highly conserved NTD.
Moreover, there are two molecules in theasymmetric unit cell of the crystal of the CTD.When superimposing the Ex subdomains of bothmolecules, the In-subdomain (residues 475–569)will rotate by 78 towards the dyad axis and thus inthe model shown in Figure 9 towards the NTD anda bound protein partner. Such a domain movementis further supported by a normal mode analysis45 ofthe CTD suggesting that a low-frequency normalmode would allow a rotation of the In subdomaincloser to the dyad symmetry axis (data not shown).This would offer an attractive, albeit speculative,model of how activation could be achieved usingboth the NTD and the CTD. The model would alsoallow for DNA binding in a tunnel made betweenthe NTD and CTD. However, the model of the MutLholoenzyme as shown in Figure 9 is not consistentwith some “DNA-binding” residues, i.e. Arg465and Arg468.11 The double mutant LR465E/R468E hadreduced in vivo activity as well as reduced ability tobind to DNA (five- to tenfold). However, inspectionof the positions of Arg465 and Arg468 indicates thatthese residues are close to the hinge region betweenthe In and the Ex subdomain, Arg468 forming a salt-bridge with Glu464. Hence, an alternative expla-nation for the reduced activities of observed effectsof LR465E/R468E could be a disturbed mobility of thehinge region.

Figure 9. Model for the biological dimer of MutL C-terminal domain. (a) Surface representation of the biological dimerof MutL-CTD in three views rotated around the x-axis (top, front and bottom) colored according to sequenceconservation using the CONSURF server.23 Note that the top surface contains highly conserved patches (red) while thebottom surface is apparently non-conserved (blue). (b) Front view in ribbon diagram of the homodimeric NTD and CTDaligned to a common dyad axis in an orientation that the conserved side of the CTD is facing the C-terminal half of theNTD. Subunits are colored in light blue and dark blue. The axial orientation around the dyad axis and distance of theNTD relative to the CTD are chosen arbitrarily. Residues of the NTD that have been shown to be close to bound MutH areindicated by orange spheres. The highly conserved surface residues of the CTD are indicated by light red and dark redspheres. (c) Superposition of one dimer formed by molecule A (blue) of the asymmetric unit and the correspondingdimer formed by molecule B (orange) using the Ex subdomains of molecule A (residues 433–474 and 570–613) as areference. The two subunits are colored in dark and light. Note that the In subdomain (residues 475–570) is rotated by 78towards the dyad axis. (d) Model of how the MutL holoenzyme might activate an interaction partner after ATP-binding,NTD dimerization and CTD subdomain movement.
MutL C-terminal Domain Quaternary Structure 905
Conclusions
On the basis of a series of bioinformatic analysesof the MutL-CTD crystal structure, we could showthat the functional dimer is formed by the Exsubdomain, leading to a revised model for theMutL-CTD dimer. This proposition has beenvalidated by comparison with distance restraintsderived from chemical cross-linking of single anddouble-cysteine variants of MutL fully compatiblewith the model of the biological dimer proposedhere. Moreover, the model is consistent with theobservation that deletion of the C-terminal helix ofMutL abolishes dimerization. This revised model ofthe functional dimer will have profound impact onthe analysis of the homologous MutL proteins,modeling of their structures and complexes withinteraction partners. Since some proteins e.g. UvrD
helicase and MutH, can interact with both the NTDand the CTD of MutL,5,11,13,44 prediction andidentification of protein interaction sites in theCTD provides a platform for studying possiblearrangements of the NTD and CTD of MutL.
Materials and Methods
Sequence alignment and calculation of evolutionaryrates
The amino acid sequence of E. coli MutL was used as aquery to search the non-redundant database (nr) usingPSI-BLAST.46 Sequences of MutL homologs from g-proteo-bacteria containing MutH homologs were collected.Additional databases of prokaryotic genome sequencingprojects were searched to increase the number of

906 MutL C-terminal Domain Quaternary Structure
sequences in the analysis. The initial multiple sequencealignment was generated using PCMA,47 and was refinedon the basis of the results of the fold-recognition analysisreported by the GeneSilico MetaServer.48 The phylo-genetic tree was calculated using the MaximumLikelihood method implemented in PHYML,49 based onthe alignment from which columns representing divergedregions were removed. The alignment and phylogenetictree were submitted to the Consurf 3.0 server to generatethe assignment of normalized evolutionary rates for eachposition (column) of the alignment (low rates ofdivergence correspond to high sequence conservation).23
Protein–protein interface analysis
Models for all possible dimers in the crystal structurewere obtained by applying symmetry operators for spacegroup P4322, in which the structure of the E. coli MutL-CTD (1x9z in the PDB) was determined.11 Selenomethio-nine residues were replaced by methionine residues priorto the following analysis. Protein dimer models wereanalyzed using the protein–protein zzz interactionserver†,26 and the DCOMPLEX server‡.35 Additionally,the interfaces were analyzed as described.27 To this end,the ASA was computed using the program GETAREAV1.150 with a probe sphere of radius 1.4 A. The subunitinterface area was estimated as B/2 where B wascalculated as:
B ZX
ASAmonomer1 CX
ASAmonomer2 KX
ASAdimer
where ASA is the accessible surface area for each atom inthe respective molecule. Briefly, atoms or amino acidresidues in the monomer that lose more than 0.1 A2 ASAin the dimer were counted as interface atoms or residues.Residues with one or more interface atoms completelyburied (zero ASA) in the dimer, were considered asbelonging to the core. Residues where all interface atomshave residual accessibility formed the rim of the inter-face.27 The area fraction of non-polar interface atoms (fnp)was calculated using the area contributed by carbonatoms. The area fraction of fully buried atoms (fbu) wascalculated using atoms with zero ASA after dimerformation. Finally, the residue propensity of the interfacewas calculated as RPZ
Pi niPi, where ni is the number of
interface residues of type i and Pi is the number-basedpropensity to be part of a homodimer interface citedbefore.20
Interface residue conservation analysis
Last, normalized evolutionary rates obtained fromConsurf were used to assess the significance of sequenceconservation at all interfaces. We used a similar approachto that applied by Valdar and Thornton for analysisof protein–protein interfaces in large datasets ofcomplexes.22 First, the mean of evolutionary rates ofresidues in the core of each interface was calculated.Second, the following null hypothesis was tested: themean evolutionary rate of the interface consisting of nresidues is not lower than the mean evolutionary rate ofthe random set of n residues drawn without replacementfrom the set of all surface residues. The test wasperformed by simulation, during which the random set
† http://www.biochem.ucl.ac.uk/bsm/PP/server/‡ http://phyyz4.med.buffalo.edu/czhang/complex.
html
of residues was drawn 107 times and the mean of whichrandom set was compared with the mean of the interfacebeing analyzed. The P-value expressing the support forthe null hypothesis was calculated as follows:
p Z tc=t
where tc is the number of times the mean evolutionaryrate of interface was lower than the mean of the randomset and t is a total number of iterations in the simulation(here 106). If the resulting P-value was lower than thearbitrary defined cut-off of 5%, the null hypothesis wasrejected and the residue interface was regarded assignificantly more conserved than expected from arandom process.
Strains, plasmids, enzymes and reagents
E. coli K12 strain TX2652 (CC106 mutLTU4 (BsaAI;Kanr) and the pET-15b (Novagen) derived plasmidspTX418 containing the mutL gene under control of thephage T7 promoter were kindly provided by Dr M.Winkler.36 For protein expression of MutL variants, theE. coli strain HMS174(lDE3) (Novagen) was used.
Site-directed mutagenesis
pTX418/Cys-free containing the gene for a cysteine-free MutL variant (C61S, C216L, C256F, C276F, C446S,C480S, and C588S; termed LCF) was used as a template tointroduce codons for single-cysteine residues and the stopcodon TAG at codon 605 using the Quikchange protocol(Stratagene) essentially as described.16,19 The substi-tutions R74C, D452C, S480C, R489C, S573C, Q611C,A611C and the deletion D605–615 are numbered withrespect to the E. coli MutL sequence. E. coli XL1-blueMRF’ was transformed with the full-length PCR product.Marker-positive clones were inoculated and grown over-night in LB containing ampicillin. Plasmid DNA wasisolated using the QIAprep Spin Miniprep (Qiagen) andthe entire mutL gene was sequenced. Protein variants arelabeled as LR74C, LD452C etc., where the superscriptindicates the position of the cysteine residues in theMutL protein and LD605–615 for the C-terminal deletionvariant lacking the last 11 amino acid residues.
Complementation mutator assay
Cells lacking a functional chromosomal mutL geneshow a mutator phenotype, which can be analyzed by thefrequency of occurrence of rifampicin-resistant clones.51
Single colonies of mutL-deficient TX2652 cells trans-formed with pET-15b vector alone or pET-15b containingthe wild-type or mutant MutL gene were grown over-night at 37 8C in 3 ml of LB containing 100 mg/ml ofampicillin. Aliquots of 50 ml from the undiluted culturewere plated on LB agar containing 25 mg/ml of ampicillinand 100 mg/ml of rifampicin. Colonies were counted afterincubation overnight at 37 8C.
Protein purification
Recombinant His6-tagged MutH, MutL and MutSproteins were purified by Ni-NTA chromatographyessentially as described.16 When necessary, proteinswere purified by gel-filtration on a Superdex 200 column(Pharmacia). MutH proteins were stored at K20 8C in10 mM Hepes-KOH (pH 7.9), 500 mM KCl, 1 mM EDTA,1 mM DTT, 50% (v/v) glycerol. MutL and MutS proteins

MutL C-terminal Domain Quaternary Structure 907
were snap-frozen in liquid nitrogen and stored at K70 8Cin 10 mM Hepes-KOH (pH 7.9), 200 mM KCl, 1 mMEDTA. Protein concentrations were determined usingtheoretical extinction coefficients.52
MutH endonuclease assay
MutH endonuclease was assayed on heteroduplexDNA substrate (484 bp) containing a G/T or A/Cmismatch at position 385 and a single unmethylatedd(GATC) site at position 210 as described.16 Briefly, theheteroduplex DNA (25 nM) was incubated at 37 8C with500 nM MutH, 2 mM MutL and 1 mM MutS in 10 mM Tris–HCl (pH 7.9), 5 mM MgCl2, 1 mM ATP, 125 mM KCl. Atsuitable time-points, 10 ml aliquots were removed and thereaction stopped by addition of 3 ml of 250 mM EDTA,25% (w/v) sucrose, 1.2% (w/v) SDS, 0.1% (w/v)bromophenol blue (pH 8.0) to 10 ml aliquots. Substrateand product were separated by electrophoresis on 2%(w/v) agarose gels. Initial rates were determined from thelinear portion of a plot of the time-course.
Chemical cross-linking
The homobifunctional cross-linkers BM[PEO]4 andBMOE were obtained from Pierce. M4M, 1,4-butanediylbismethanethiosulfonate; M11M, 3,6,9-trioxaoctane-1,11-diyl bismethanethiosulfonate and M17M, 3,6,9,12,15-pentaoxaheptadecane-1,17-diyl bismethanethiosulfonatewere from Toronto Research Chemicals (Figure 3). Stocksolutions at a concentration of 10 mM were made inwater, DMSO, or acetonitrile. The single-cysteine MutLvariants were preincubated on ice at 5 mM each for tenminutes in 10 mM Tris–HCl (pH 7.5), 10 mM MgCl2containing either 0.8 mM ADP or adenosine 5 0-(b-g-imido)triphosphate (ADPNP) followed by incubationwith a 100-fold molar excess of a cross-linker over thiolgroups. The reaction was quenched after 30 seconds witha fivefold molar excess of DTT or N-ethylmaleimide overthe cross-linker and samples were subjected to SDS-PAGEanalysis. All gels were analyzed with a video documen-tation system (Bio-Rad). The intensity of the stainedprotein bands was quantified using TINA v2.07dsoftware.
Gel-filtration analysis
Quaternary structures of MutL variants and thrombincleaved fragments (NTD and CTD) thereof11,13 wereanalyzed on Superdex 200 10/300 and Superdex 75 10/300 columns, respectively. Samples (100 ml) containing10 mM LCF and LD605–615 or thrombin-cleaved fragmentsthereof were injected onto the columns equilibrated with10 mM Hepes-KOH (pH 7.9), 500 mM KCl, 1 mM EDTA,at flow-rates of 0.5 ml/minute. Elution profiles weremonitored at 280 nm; 500 ml fractions were collected andsubjected to SDS-PAGE analysis.
Acknowledgements
We are grateful to Wei Yang for providingcoordinates of 1x9z prior to publication. This workwas supported by the Deutsche Forschungsge-meinschaft (Fr-1495/3-1) and the DAAD (Inter-national Quality Network “Biochemistry of Nucleic
Acids”). J.K. and J.M.B. were supported addition-ally by the Polish Ministry for Scientific Researchand Information Technology (grant PBZ-KBN-088/PO4/2003). J.K. was supported also by an ExchangeGrant from the European Science Foundation(ESF) “Integrated Approaches for FunctionalGenomics” program. J.M.B. is an EMBO/HHMIYoung Investigator.
References
1. Friedberg, E. C. (2003). DNA damage and repair.Nature, 421, 436–440.
2. Modrich, P. & Lahue, R. (1996). Mismatch repair inreplication fidelity, genetic recombination, and cancerbiology. Annu. Rev. Biochem. 65, 101–133.
3. Kunkel, T. A. (2004). DNA replication fidelity. J. Biol.Chem. 279, 16895–16898.
4. Mechanic, L. E., Frankel, B. A. & Matson, S. W. (2000).Escherichia coli MutL loads DNA helicase II ontoDNA. J. Biol. Chem. 275, 38337–38346.
5. Hall, M. C. & Matson, S. W. (1999). The Escherichia coliMutL protein physically interacts with MutH andstimulates the MutH-associated endonucleaseactivity. J. Biol. Chem. 274, 1306–1312.
6. Li, G. M. & Modrich, P. (1995). Restoration ofmismatch repair to nuclear extracts of H6 colorectaltumor cells by a heterodimer of human MutLhomologs. Proc. Natl Acad. Sci. USA, 92, 1950–1954.
7. Raschle, M., Marra, G., Nystrom-Lahti, M., Schar, P. &Jiricny, J. (1999). Identification of hMutLb, a hetero-dimer of hMLH1 and hPMS1. J. Biol. Chem. 274,32368–32375.
8. Lipkin, S. M., Wang, V., Jacoby, R., Banerjee-Basu, S.,Baxevanis, A. D., Lynch, H. T. et al. (2000). MLH3:a DNA mismatch repair gene associated withmammalian microsatellite instability. Nature Genet.24, 27–35.
9. Dutta, R. & Inouye, M. (2000). GHKL, an emergentATPase/kinase superfamily. Trends Biochem. Sci. 25,24–28.
10. Drotschmann, K., Aronshtam, A., Fritz, H. J. &Marinus, M. G. (1998). The Escherichia coli MutLprotein stimulates binding of Vsr and MutS toheteroduplex DNA. Nucl. Acids Res. 26, 948–953.
11. Guarne, A., Ramon-Maiques, S., Wolff, E. M.,Ghirlando, R., Hu, X., Miller, J. H. & Yang, W.(2004). Structure of the MutL C-terminal domain: amodel of intact MutL and its roles in mismatch repair.EMBO J. 23, 4134–4145.
12. Pang, Q., Prolla, T. A. & Liskay, R. M. (1997).Functional domains of the Saccharomyces cerevisiaeMlh1p and Pms1p DNA mismatch repair proteinsand their relevance to human hereditary non-polyposis colorectal cancer-associated mutations.Mol. Cell. Biol. 17, 4465–4473.
13. Ban, C., Junop, M. & Yang, W. (1999). Transformationof MutL by ATP binding and hydrolysis: a switch inDNA mismatch repair. Cell, 97, 85–97.
14. Allen, D. J., Makhov, A., Grilley, M., Taylor, J.,Thresher, R., Modrich, P. & Griffith, J. D. (1997).MutS mediates heteroduplex loop formation by atranslocation mechanism. EMBO J. 16, 4467–4476.
15. Hall, M. C., Jordan, J. R. & Matson, S. W. (1998).Evidence for a physical interaction between theEscherichia coli methyl- directed mismatch repairproteins MutL and UvrD. EMBO J. 17, 1535–1541.

908 MutL C-terminal Domain Quaternary Structure
16. Giron-Monzon, L., Manelyte, L., Ahrends, R., Kirsch,D., Spengler, B. & Friedhoff, P. (2004). Mappingprotein-protein interactions between MutL and MutHby cross-linking. J. Biol. Chem. 279, 49338–49345.
17. Valdar, W. S. & Thornton, J. M. (2001). Conservationhelps to identify biologically relevant crystal contacts.J. Mol. Biol. 313, 399–416.
18. Szilagyi, A., Grimm, V., Arakaki, A. K. & Skolnick, J.(2005). Prediction of physical protein–protein inter-actions. Phys. Biol 2, S1–S16.
19. Toedt, G., Krishnan, R. & Friedhoff, P. (2003). Site-specific protein modification to identify the MutLinterface of MutH. Nucl. Acids Res. 31, 819–825.
20. Bahadur, R. P., Chakrabarti, P., Rodier, F. & Janin, J.(2004). A dissection of specific and non-specificprotein-protein interfaces. J. Mol. Biol. 336, 943–955.
21. Chakrabarti, P. & Janin, J. (2002). Dissecting protein–protein recognition sites. Proteins: Struct. Funct. Genet.47, 334–343.
22. Valdar, W. S. & Thornton, J. M. (2001). Protein–proteininterfaces: analysis of amino acid conservation inhomodimers. Proteins: Struct. Funct. Genet. 42,108–124.
23. Glaser, F., Pupko, T., Paz, I., Bell, R. E., Bechor-Shental,D., Martz, E. & Ben-Tal, N. (2003). ConSurf: identifi-cation of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics,19, 163–164.
24. Bradford, J. R. & Westhead, D. R. (2004). Improvedprediction of protein–protein binding sites using asupport vector machines approach. Bioinformatics, 21,1487–1494.
25. Sobolev, V., Sorokine, A., Prilusky, J., Abola, E. E. &Edelman, M. (1999). Automated analysis of inter-atomic contacts in proteins. Bioinformatics, 15,327–332.
26. Jones, S. & Thornton, J. M. (1996). Principles ofprotein-protein interactions. Proc. Natl Acad. Sci.USA, 93, 13–20.
27. Bahadur, R. P., Chakrabarti, P., Rodier, F. & Janin, J.(2003). Dissecting subunit interfaces in homodimericproteins. Proteins: Struct. Funct. Genet. 53, 708–719.
28. Ponstingl, H., Henrick, K. & Thornton, J. M. (2000).Discriminating between homodimeric and mono-meric proteins in the crystalline state. Proteins: Struct.Funct. Genet. 41, 47–57.
29. Henrick, K. & Thornton, J. M. (1998). PQS: a proteinquaternary structure file server. Trends Biochem. Sci.23, 358–361.
30. Laskowski, R. A. (1995). Surfnet—a program forvisualizing molecular-surfaces, cavities, and inter-molecular interactions. J. Mol. Graph. Model. 13,323–330.
31. Halperin, I., Wolfson, H. & Nussinov, R. (2004).Protein–protein interactions; coupling of structu-rally conserved residues and of hot spots acrossinterfaces. Implications for docking. Structure, 12,1027–1038.
32. Ma, B. Y., Elkayam, T., Wolfson, H. & Nussinov, R.(2003). Protein–protein interactions: structurally con-served residues distinguish between binding sitesand exposed protein surfaces. Proc. Natl Acad. Sci.USA, 100, 5772–5777.
33. Elcock, A. H. & McCammon, J. A. (2001). Identifi-cation of protein oligomerization states by analysis ofinterface conservation. Proc. Natl Acad. Sci. USA, 98,2990–2994.
34. Keskin, O. & Nussinov, R. (2005). Favorable scaffolds:
proteins with different sequence, structure andfunction may associate in similar ways. Protein Eng.Des. Sel. 18, 11–24.
35. Liu, S., Zhang, C., Zhou, H. Y. & Zhou, Y. Q. (2004).A physical reference state unifies the structure-derived potential of mean force for protein foldingand binding. Proteins: Struct. Funct. Genet. 56,93–101.
36. Feng, G. & Winkler, M. E. (1995). Single-steppurifications of His6-MutH, His6-MutL and His6-MutS repair proteins of Escherichia coli K-12. Bio-techniques, 19, 956–965.
37. Roca, J. & Wang, J. C. (1994). DNA transport by a typeII DNA topoisomerase: evidence in favor of a two-gate mechanism. Cell, 77, 609–616.
38. Kampranis, S. C., Bates, A. D. & Maxwell, A. (1999). Amodel for the mechanism of strand passage by DNAgyrase. Proc. Natl Acad. Sci. USA, 96, 8414–8419.
39. Harris, S. F., Shiau, A. K. & Agard, D. A. (2004). Thecrystal structure of the carboxy-terminal dimerizationdomain of htpG, the Escherichia coli Hsp90, revealsa potential substrate binding site. Structure, 12,1087–1097.
40. Grilley, M., Welsh, K. M., Su, S. S. & Modrich, P.(1989). Isolation and characterization of the Escherichiacoli mutL gene product. J. Biol. Chem. 264, 1000–1004.
41. Garcia De La Torre, J., Huertas, M. L. & Carrasco, B.(2000). Calculation of hydrodynamic properties ofglobular proteins from their atomic-level structure.Biophys. J. 78, 719–730.
42. Guerrette, S., Acharya, S. & Fishel, R. (1999). Theinteraction of the human MutL homologues inhereditary nonpolyposis colon cancer. J. Biol. Chem.274, 6336–6341.
43. Kondo, E., Horii, A. & Fukushige, S. (2001). Theinteracting domains of three MutL heterodimers inman: hMLH1 interacts with 36 homologous aminoacid residues within hMLH3, hPMS1 and hPMS2.Nucl. Acids Res. 29, 1695–1702.
44. Jacquelin, D. K., Filiberti, A., Argarana, C. E. & Barra,J. L. (2005). The Pseudomonas aeruginosa MutL proteinfunctions in Escherichia coli. Biochem. J. 388, 879–887.
45. Suhre, K. & Sanejouand, Y. H. (2004). ElNemo: anormal mode web server for protein movementanalysis and the generation of templates for molecu-lar replacement. Nucl. Acids Res. 32, W610–W614.
46. Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J.,Zhang, Z., Miller, W. & Lipman, D. J. (1997). GappedBLAST and PSI-BLAST: a new generation of proteindatabase search programs. Nucl. Acids Res. 25,3389–3402.
47. Pei, J., Sadreyev, R. & Grishin, N. V. (2003). PCMA:fast and accurate multiple sequence alignment basedon profile consistency. Bioinformatics, 19, 427–428.
48. Kurowski, M. A. & Bujnicki, J. M. (2003). GeneSilicoprotein structure prediction meta-server. Nucl. AcidsRes. 31, 3305–3307.
49. Guindon, S. & Gascuel, O. (2003). A simple, fast, andaccurate algorithm to estimate large phylogenies bymaximum likelihood. System. Biol. 52, 696–704.
50. Fraczkiewicz, R. & Braun, W. (1998). Exact andefficient analytical calculation of the accessible surfaceareas and their gradients for macromolecules. J. Comput.Chem. 19, 319–333.
51. Loh, T., Murphy, K. C. & Marinus, M. G. (2001).Mutational analysis of the MutH protein fromEscherichia coli. J. Biol. Chem. 276, 12113–12119.
52. Pace, C. N., Vajdos, F., Fee, L., Grimsley, G. & Gray, T.

MutL C-terminal Domain Quaternary Structure 909
(1995). How to measure and predict the molarabsorption coefficient of a protein. Protein Sci. 4,2411–2423.
53. Green, N. S., Reisler, E. & Houk, K. N. (2001).
Quantitative evaluation of the lengths of homo-bifunctional protein cross-linking reagents used asmolecular rulers. Protein Sci. 10, 1293–1304.
Edited by R. Huber
(Received 24 March 2005; received in revised form 14 June 2005; accepted 17 June 2005)Available online 5 July 2005




















