
Alteration of Golgi structure in senescent cells and its regulation by a G protein γ subunit
9
Alteration of Golgi structure in senescent cells and its regulation by a G protein γ subunit Joon-Ho Cho a,1 , Deepak Kumar Saini a,1 , W.K. Ajith Karunarathne a , Vani Kalyanaraman a , N. Gautam a,b, ⁎ a Department of Anesthesiology, Washington University School of Medicine, St. Louis, MO 63110, USA b Department of Genetics, Washington University School of Medicine, St. Louis, MO 63110, USA abstract article info Article history: Received 3 November 2010 Received in revised form 21 December 2010 Accepted 5 January 2011 Available online 14 January 2011 Keywords: G protein Betagamma subunits Cell senescence Translocation Cellular senescence is a process wherein proliferating cells undergo permanent cell cycle arrest while remaining viable. Senescence results in enhanced secretion of proteins that promote cancer and inflammation. We report here that the structure of the Golgi complex which regulates secretion is altered in senescent cells. In cells where senescence is achieved by replicative exhaustion or in cells wherein senescence has been induced with BrdU treatment dependent stress, the Golgi complex is dispersed. The expression of a G protein γ subunit, γ11, capable of translocation from the plasma membrane to the Golgi complex on receptor activation increases with senescence. Knockdown of γ11 or overexpression of a dominant negative γ3 subunit inhibits Golgi dispersal induced by senescence. Overall these results suggest that in cellular senescence an upregulated G protein gamma subunit mediates alterations in the structure of the Golgi. © 2011 Elsevier Inc. All rights reserved. 1. Introduction Replicatively senescent cells or cells that have undergone genotoxic stress exhibit increased secretion of a number of factors including cytokines, growth factors, metalloproteinases and extracel- lular matrix proteins [1]. The enhanced secretion of these factors is known to induce inflammation and has been demonstrated to facilitate epithelial mesenchymal transition which promotes tumor- igenesis [1]. Since cellular secretion is mediated by the Golgi complex, we examined the status of the Golgi in senescent cells resulting from stress or replicative exhaustion. Based on previous reports, 5-bromo 2-deoxyuridine (BrdU) exposure to cells was used as a stress induced model for senescence [2–4] which mimics the properties of replicative senescence. It has been shown that BrdU treatment induces cellular senescence likely by inducing the DNA-damage response [5]. DNA damage has been shown to trigger senescence [6]. It has been shown that it induces senescence in stem cells and inhibits proliferation of cancer cells [15,16]. We also confirmed a previous finding that a heterotrimeric G protein subunit, γ11 (GNG11) is upregulated in senescent cells [7]. The γ11 subunit is capable of translocation from the plasma membrane to the Golgi on receptor activation as a βγ complex [8,9] and regulates the structure of the Golgi [10]. We therefore examined the possibility that the G protein γ11 subunit plays a role in the regulation of the Golgi structure in senescence. 2. Materials and methods 2.1. Constructs, cell lines and chemicals The tagged and untagged G protein constructs, various Golgi markers and PH-mCh used in this study have been previously described [9–12]. Mammalian expression vector containing cDNA encoding γ11 shRNA and control scrambled shRNAs were from the TRC library of Broad Institute (Sigma) and CFP-tubulin was from E. Bertrand (CNRS, Montpellier, France). The HeLa cell line was from ATCC; the WI38 and IMR90 cell lines were from NIA Aging Cell Repository at Coriell Institute for Medical Research (Camden, NJ). Antibodies to the Golgi network marker, TGN46 were obtained from Sigma; antibodies to the cis Golgi marker, GM130 were from A. Lindstedt (Carnegie Mellon University, Pittsburgh, PA) and were used at a dilution of 1:100. TRITC-conjugated goat anti-rabbit secondary antibody was from Sigma and was used at a 1:1000 dilution. 5-bromo deoxyuridine was procured from Sigma and was dissolved in DMSO to prepare a 200 μM solution. The solution was prepared just before use. 2.2. Cell culture, transfections and lentiviral transduction HeLa cells were cultured in DMEM (Cellgro, Manassas, VA) containing 10% dialyzed FBS (Atlanta Biologicals), while WI38 and IMR90 cells were grown in MEM containing 10% non-dialyzed FBS, at 37 °C, and 5% CO 2 . Cells were transfected with Lipofectamine 2000 (Invitrogen) as per the manufacturer's protocol. To obtain stable knock down HeLa cell lines, lentiviral particles containing specific shRNAs were used as per the protocol provided by Sigma. The cells Cellular Signalling 23 (2011) 785–793 ⁎ Corresponding author. Box 8054, Washington University School of Medicine, St. Louis, MO 63110, USA. Tel.: +1 314 362 8568; fax: +1 314 362 8571. E-mail address: [email protected] (N. Gautam). 1 These authors contributed equally to this work. 0898-6568/$ – see front matter © 2011 Elsevier Inc. All rights reserved. doi:10.1016/j.cellsig.2011.01.001 Contents lists available at ScienceDirect Cellular Signalling journal homepage: www.elsevier.com/locate/cellsig
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Alteration of Golgi structure in senescent cells and its regulation by a G protein γ subunit

Cellular Signalling 23 (2011) 785–793
Contents lists available at ScienceDirect
Cellular Signalling
j ourna l homepage: www.e lsev ie r.com/ locate /ce l l s ig
Alteration of Golgi structure in senescent cells and its regulation by a G proteinγ subunit
Joon-Ho Cho a,1, Deepak Kumar Saini a,1, W.K. Ajith Karunarathne a, Vani Kalyanaraman a, N. Gautam a,b,⁎a Department of Anesthesiology, Washington University School of Medicine, St. Louis, MO 63110, USAb Department of Genetics, Washington University School of Medicine, St. Louis, MO 63110, USA
⁎ Corresponding author. Box 8054, Washington USt. Louis, MO 63110, USA. Tel.: +1 314 362 8568; fax: +
E-mail address: [email protected] (N. Gautam).1 These authors contributed equally to this work.
0898-6568/$ – see front matter © 2011 Elsevier Inc. Aldoi:10.1016/j.cellsig.2011.01.001
a b s t r a c t
a r t i c l e i n f oArticle history:Received 3 November 2010Received in revised form 21 December 2010Accepted 5 January 2011Available online 14 January 2011
Keywords:G proteinBetagamma subunitsCell senescenceTranslocation
Cellular senescence is a process wherein proliferating cells undergo permanent cell cycle arrest whileremaining viable. Senescence results in enhanced secretion of proteins that promote cancer andinflammation. We report here that the structure of the Golgi complex which regulates secretion is alteredin senescent cells. In cells where senescence is achieved by replicative exhaustion or in cells whereinsenescence has been induced with BrdU treatment dependent stress, the Golgi complex is dispersed. Theexpression of a G protein γ subunit, γ11, capable of translocation from the plasma membrane to the Golgicomplex on receptor activation increases with senescence. Knockdown of γ11 or overexpression of adominant negative γ3 subunit inhibits Golgi dispersal induced by senescence. Overall these results suggestthat in cellular senescence an upregulated G protein gamma subunit mediates alterations in the structure ofthe Golgi.
niversity School of Medicine,1 314 362 8571.
l rights reserved.
© 2011 Elsevier Inc. All rights reserved.
1. Introduction
Replicatively senescent cells or cells that have undergonegenotoxic stress exhibit increased secretion of a number of factorsincluding cytokines, growth factors, metalloproteinases and extracel-lular matrix proteins [1]. The enhanced secretion of these factors isknown to induce inflammation and has been demonstrated tofacilitate epithelial mesenchymal transition which promotes tumor-igenesis [1]. Since cellular secretion is mediated by the Golgi complex,we examined the status of the Golgi in senescent cells resulting fromstress or replicative exhaustion. Based on previous reports, 5-bromo2-deoxyuridine (BrdU) exposure to cells was used as a stress inducedmodel for senescence [2–4] whichmimics the properties of replicativesenescence. It has been shown that BrdU treatment induces cellularsenescence likely by inducing the DNA-damage response [5]. DNAdamage has been shown to trigger senescence [6]. It has been shownthat it induces senescence in stem cells and inhibits proliferation ofcancer cells [15,16]. We also confirmed a previous finding that aheterotrimeric G protein subunit, γ11 (GNG11) is upregulated insenescent cells [7]. The γ11 subunit is capable of translocation fromthe plasma membrane to the Golgi on receptor activation as a βγcomplex [8,9] and regulates the structure of the Golgi [10]. Wetherefore examined the possibility that the G protein γ11 subunitplays a role in the regulation of the Golgi structure in senescence.
2. Materials and methods
2.1. Constructs, cell lines and chemicals
The tagged and untagged G protein constructs, various Golgimarkers and PH-mCh used in this study have been previouslydescribed [9–12]. Mammalian expression vector containing cDNAencoding γ11 shRNA and control scrambled shRNAs were from theTRC library of Broad Institute (Sigma) and CFP-tubulin was from E.Bertrand (CNRS, Montpellier, France). The HeLa cell line was fromATCC; the WI38 and IMR90 cell lines were from NIA Aging CellRepository at Coriell Institute for Medical Research (Camden, NJ).Antibodies to the Golgi network marker, TGN46 were obtained fromSigma; antibodies to the cis Golgi marker, GM130 were from A.Lindstedt (Carnegie Mellon University, Pittsburgh, PA) and were usedat a dilution of 1:100. TRITC-conjugated goat anti-rabbit secondaryantibody was from Sigma and was used at a 1:1000 dilution. 5-bromodeoxyuridine was procured from Sigma andwas dissolved in DMSO toprepare a 200 μM solution. The solution was prepared just before use.
2.2. Cell culture, transfections and lentiviral transduction
HeLa cells were cultured in DMEM (Cellgro, Manassas, VA)containing 10% dialyzed FBS (Atlanta Biologicals), while WI38 andIMR90 cells were grown in MEM containing 10% non-dialyzed FBS, at37 °C, and 5% CO2. Cells were transfected with Lipofectamine 2000(Invitrogen) as per the manufacturer's protocol. To obtain stableknock down HeLa cell lines, lentiviral particles containing specificshRNAs were used as per the protocol provided by Sigma. The cells

786 J.-H. Cho et al. / Cellular Signalling 23 (2011) 785–793
were transduced in a 96-well plate and after 48 h, 2 μg/ml of pu-romycin was added to screen for cells expressing shRNAs. The cellswere cultured for several generations and the reduction in theexpression of γ11wasmonitored to evaluate the stability of the knockdown cell line. For all experiments, the cell line was evaluated forknock down before use by real time PCR.
2.3. Quantitative real time-PCR
Total cellular RNA was isolated from various cell lines using theRNeasy Plus Mini Kit (QIAGEN). Reverse transcription of RNA wasperformed using Themoscript RT-PCR system (Invitrogen, Carlsbad,CA) as per manufacturer's instructions and as previously described[10]. Quantitative real time PCR was performed using SYBR Green PCRmaster mix (Applied Biosystems) in 20 μl reaction volume as permanufacturer's instructions. Melting curve analyses were performedon all reactions to check for specificity of the amplicons. Expressionlevels of β-actin were used to normalize the data. The followingprimer pairs were used for quantitative RT-PCR analysis:
Fibronectin — 5′GGTGGCTGTCAGTCAAAGC3′ and 5′CGCATTGC-CTAGGTAGGTC3′p21 — 5′GGAGCAGGCTGAAGGGTC3′ and 5′CCGGCGTT-TGGAGTGGTAG3′γ10 — 5′TGCCTTCAAGCACAAAGTGA3′ and 5′TATAGG-ACCAGGCCACAGGA3′γ11 — 5′GTGCCCTTCACATCGAAGAT3′ and 5′CACTTGTT-GTCTCTGCAACTTCA3′β-actin — 5′CCAACCGCGAGAAGATGAC3′ and 5′CAGAGGC-GTACAGGGATAGC3′.
Fig. 1. Characterization of BrdU stress induced cellular senescence in immortal HeLa cells andinduce cellular senescence. Nonsenescent PDL17 and close to senescence PDL46WI38 cells wcells (0.2 million/well) were seeded in triplicate in six well plates out of which three wells wnumber of cells at 72 h to the initial number at seeding was calculated. This ratio for BrdUmorphology of cells exposed to BrdU. DIC images of HeLa cells before and after exposure tomode using 1.4 N.A. 60× plan apochromat objective. C. Changes in gene expression in BrdUlevels for p21, fibronectin, G protein γ10 and γ11 subunits as described in Materials and mnormalized using β-actin as an internal control, and relative expression was calculated usingreplicatively senescent cells. Early PDL and late PDL WI38 cells were used for expression anadescribed in Materials and methods. Error bars represent SEM (n=3).
2.4. IL-8 secretion
HeLa cells were seeded in 6-well plates andwere grown overnight.The next day the cells were treated with 200 μM BrdU and after 24 hthe growth media was replaced with serum free media (DMEM-F121:1mix with Peprotech serum free mix 1) containing 200 μMBrdU foranother 48 h. The media and cells were collected and IL-8 levels wereestimated in the media using an ELISA kit (R&D systems). The cellswere lysed with M-Per protein extraction reagent (Pierce) and totalprotein was estimated using BCA protein estimation kit (Pierce). TheIL-8 levels were then calculated per μg total protein.
2.5. Immunofluorescence microscopy
Immunofluorescence microscopy was performed as describedpreviously [10]. Briefly, cells grown on cover slips were subjected toBrdU treatment and fixed with 4% freshly prepared paraformaldehydefor 10 min, followed by permeabilization with 0.5% Triton X-100 for10 min. The permeabilized cellswere blockedwith 2% BSA in PBS for 1 hfollowed by treatment with primary antibodies at recommendeddilution for 2 h followed by incubation with goat anti-rabbit-TRITCconjugate Ab (Sigma, MO; 1:1000) for 1 h. The coverslips were washedand mounted on a slide with antifade solution (Calbiochem, CA) andimaged using a Nikon TE2000 widefield fluorescence microscope usinga 60× Plan-Apochromat objective (1.4 NA) as described previously [10].
2.6. Live cell imaging
Live cell imaging was performed essentially as described[8,9,11,13]. An inverted wide field epifluorescence microscope with
replicative senescence in mortal WI38 cells. HeLa cells were exposed to 200 μMBrdU toere used. A. Inhibition of cell proliferation on treatment with BrdU. An equal number ofere exposed to 200 μM of BrdU for 72 h. Cell numbers were counted and the ratio of thetreated cells is distinctly different from untreated cells (pb0.01) (n=3). B. Change inBrdU are shown. Cells were imaged in glass bottom dishes using Leica DMI6000 in DICtreated senescent cells. Quantitative real time PCR was performed for measuring mRNAethods. Cells were treated with 200 μM BrdU for 24 h and 48 h. Levels of RNAs werethe 2−ΔΔCt method. Error bars represent SEM (n=3). D. Changes in gene expression inlysis as above. E. Induction of IL-8 secretion from senescent cells. IL-8 was measured as

787J.-H. Cho et al. / Cellular Signalling 23 (2011) 785–793
1.4 NA 60× Plan Apochromat objective (Nikon TE2000) was used. Themicroscope was equipped with an Exfo X-Cite light source containinga 103 W Hg-Arc lamp for illumination, Sutter lambda 10-2 opticalfilter wheel controller for wavelength selection (Sutter InstrumentCompany), specific wavelength filters (Chroma), Orca-ER CCD camera(Hamamatsu Corp.) for image acquisition, 8-channel automated fluiddelivery system (Automate Scientific) and Metamorph 6.3.7 software(Molecular Devices) for controlling all the devices. Imaging wasperformed at room temperature at variable exposure times withvariable binning. Filters used to acquire specific fluorescence were asfollows: for CFP, 436/10 excitation and D465/30 emission; YFP, D492/18 excitation and D535/30 emission; DsRed, D580/20 excitation andD630/60 emission. Endomembrane regions for intensity measure-ments were selected using MetaMorph. The images were enhanced
Fig. 2. Cotranslocation of YFP-β1 in HeLa cells. HeLa cells expressing M3, αq, YFP-β1 andmounted on a 25 μl volume perfusion chamber (RC30, Warner Instruments) and imaged inreceptor agonist, 100 μM carbachol followed by antagonist, 100 μM atropine. The images shcell. Representative images are shown (nN4). A. YFP-β1 translocation is not detectable in btranslocation of YFP-β1 on receptor activation (upper panel) are shown. Lower panel shows tβ1 translocation is detectable in BrdU treated HeLa cells. Images were taken at 5 s interval. Reacquired at 10 s (basal) and at 2 min 20 s (agonist treated). The intracellular region used for ito the endomembranes. The intensity measurement from the endomembranes is plotted aswere added. D. Plot showing translocation of PH-mCh to the endomembranes (as reported pplotted as a function of time (seconds). Arrows indicate the point at which agonist and anreceptor is present in the cells and is active.
for brightness and/or contrast uniformly post acquisition either usingMetamorph, ImageJ or Microsoft Word image editing tool.
3. Results
3.1. BrdU (5-bromo deoxyuridine) induces cellular senescence
We used BrdU treated HeLa cells as a rapidly assayable model forcellular senescence. It has been observed that BrdU induces cellularsenescence by activating the DNA damage response [5,14]. Consistentwith previous reports [2,4,5,14,15,17] we found that BrdU inducescellular senescence. HeLa cells treated with 200 μM BrdU for 24–48 hshowed several characteristics similar to replicatively senescentIMR90 cells. BrdU treated cells slowed down in growth, changed
PH-mCh were used. The cells were treated with 200 μM BrdU for 48 h. The cells werea continuous flow of HBSS at a rate of 0.5 ml/ml. The cells were stimulated with M3
ow distribution of YFP-β1 and PH-mCh before and after agonist treatment in the sameasal state HeLa cells. Images from basal HeLa cells (not exposed to BrdU) depicting noranslocation of PH-mCh in the same cells to confirm the presence of the receptor. B. YFP-presentative images of a cell before and after agonist treatment. The images shownwerentensity measurements is shown (white circle). C. Plot showing translocation of YFP-β1a function of time (seconds). Arrows indicate the point at which agonist and antagonistreviously) [11]. The intensity measurement from the endomembranes (white circle) istagonist were added. The translocation of PH domain confirmed that the introduced

788 J.-H. Cho et al. / Cellular Signalling 23 (2011) 785–793
shape (Fig. 1A and B) and showed enhanced transcription of aninhibitor of cyclin dependent kinase 4, p21 (Fig. 1C) [18]. BrdU treatedHeLa cells and replicatively senescent IMR90 and WI38 cells alsoexpressed higher levels of fibronectin [19] and a G protein γ11subunit type (Fig. 1C and D) consistent with previous reports [7]. Inour experiments, a G protein γ10 subunit served as an internal controlthat did not show any change in expression when exposed to BrdU(Fig. 1C and D). To further confirm that BrdU treatment mimicsreplicative senescence, we monitored the secreted levels of the pro-inflammatory cytokine IL-8 which has been shown to be an integralpart of the senescence associated secretory phenotype [1,20]. Asexpected we observed increased secretion of IL-8 levels from BrdUtreated cells (Fig. 1E). These observations together confirmed thatBrdU treatment mimics cellular senescence induced by replicativeexhaustion.
3.2. The G protein βγ complex translocates from the plasma membraneto the Golgi complex on receptor activation
Recent results show that the γ11 subunit type is capable oftranslocating from the plasma membrane to the Golgi complex onreceptor activation [8,9], regulating the structure of the Golgi [10].Here, we examined the potential role of the G protein γ11 subunit inregulating the structure of the Golgi in senescent cells since itstranscript levels are upregulated in these cells (Fig. 1C and D). First,we examined whether the upregulation of the γ11 transcript wasreflected in its translocation properties in senescent cells. The β1subunit taggedwith a fluorescent protein (FP) was cotransfected withthe M3 muscarinic receptor and αq into HeLa cells. We utilized this
Fig. 3. Dispersal of trans Golgi and the trans Golgi network (TGN) in senescent cells. A. Disperwere transfected with GalT-DsRed using lipofectamine 2000 (Invitrogen, Carlsbad, CA). TheGolgi network labeling, HeLa cells were grown on coverslips, treated for 24 h with BrdU toMO). Top panels, basal cells, bottom panel, senescent cells (nN3). B. Replicative senescence iwere fixed and stained for TGN46 distribution. Images were acquired using widefieldmicroscthe number of cells with compact or dispersed TGN in late and early PDL IMR90 andWI38 cewas b10%.
strategy based on our previous observations wherein we havedetected cotranslocation of tagged β1 in cells which endogenouslycontain a translocation proficient γ subunit [10]. Receptor mediatedtranslocation was examined before and after BrdU treatment. Inuntreated cells, agonist stimulation (100 μM carbachol) did not evokesignificantly detectable translocation of the YFP-β1 subunit to theinside of the cell (Fig. 2A upper panel). In contrast, when cells weretreated with BrdU, significant YFP-β1 translocation to the intracellularregion was detected (Fig. 2B). The translocation was reversible andrapid (Fig. 2C) as reported previously [8–10]. To confirm that theobserved effect was due to activation of the M3 muscarinic receptor,we also monitored the activation of PLCβ by monitoring thegeneration of IP3 inside the same cell using the PH-mCh sensor aspreviously described [11] (Fig. 2A lower panels and 2D).
3.3. The Golgi complex is dispersed in senescent cells
To observe the status of the Golgi complex in HeLa cells, a markerfor the Golgi region, Galactosyl transferase tagged with red fluores-cent protein, DsRed (GalT-DsRed) [21] was transiently expressed inthe cells. Senescence was induced post transfection by treating thecells with BrdU. While untreated cells contained compact trans Golgi,BrdU treated senescent cells contained dispersed Golgi (Fig. 3A).Similarly, when replicatively senescent IMR90 and WI38 cells wereexamined by immunofluorescence microscopy using antibodiesspecific to the endogenous trans Golgi network marker, TGN46 [21],early PDL cells contained predominantly compact TGN and late PDLcells contained TGN thatwas dispersed (Fig. 3B). The cells were scoredfor compact versus dispersed Golgi. For both IMR90 and WI38 cells,
sal of Golgi in stress induced senescence in HeLa cells. For trans Golgi labeling, HeLa cellslocalization of GalT in live cells was monitored by epifluorescence microscopy. For transinduce senescence, fixed and stained using the anti-TGN46 antibody (Sigma, St. Louis,n IMR90 andWI38 cells. Early (16–17 PDL) and late PDL (46 PDL) IMR90 andWI38 cellsopy. Top panels, early PDL cells, and bottom panels, late PDL cells. C. Bar graph depictinglls. Average plot of two experiments. The percentage variability across the experiments

Fig. 4. Redistribution of cis and medial Golgi in senescent cells. A. Control and BrdUtreated senescent HeLa cells. B. Early and late PDLs of WI38 and IMR90 cells. In A and Bcis Golgi structure was observed using anti-GM130 antibodies. Representative cellswith compact or dispersed cis Golgi (n=2). C. Redistribution ofαmannosidase, medialGolgi marker, in senescent HeLa cells. HeLa cells were transfected with CFP-αmannosidase. The distribution of α mannosidase was monitored by fluorescencemicroscopy (n=2).
Fig. 5. Tubulin independent redistribution of trans Golgi in stress induced senescent HeLa ceBrdU to induce senescence. The cells were then visualized for GalT and tubulin distributioncells. C. Nocodazole treated HeLa cells, which lead to both depolymerization of microtubulesnuclear region due to depolymerization) (n=2).
789J.-H. Cho et al. / Cellular Signalling 23 (2011) 785–793
~70% of cells exhibited compact Golgi in the pre-senescent (early PDL)stage and the profile was reversed in senescent cells (late PDL) wheremore than 70% of cells had dispersed Golgi (Fig. 3C). These resultssuggested that (i) the Golgi structure is altered in senescent cells and(ii) the changes in the Golgi structure alternations were not peculiarto BrdU treatment but were common to cellular senescence.
3.4. The cis and medial Golgi are also altered in senescent cells
We examined whether the alterations in the Golgi structure wererestricted to the trans Golgi and trans Golgi network. A specificantibody for GM130 which localizes to the cis Golgi [22] was used toexamine the cis Golgi structure in control and BrdU treated Hela cellsas well as early and late PDL IMR90 and WI38 cells. Similarly, thestructure of the medial Golgi was examined by expressing CFP-mannosidase II which is known to be localized specifically to thisregion of the Golgi complex [23]. The results showed that both the cisand medial Golgi were dispersed in senescent cells (Fig. 4).
3.5. Golgi alterations in senescent cells are not due to microtubuledisassembly
Microtubule disassembly is known todisrupt the structure of theGolgi[24].We examinedwhether the senescence associated changes are due tomicrotubule disassemblyduring senescence. Tubulin taggedwithCFPwascoexpressed with GalT-DsRed and their distribution was examined inHeLa cells before and after treatment with BrdU. Cells treated with BrdUshowed GalT-DsRed scattered over the cells suggesting disruption of thetrans Golgi. But the microtubule distribution in these cells was unalteredand similar to the control untreated cells (Fig. 5A and B, right panels).WhenHeLa cellswere treatedwithnocodazole, an agent known to lead tothe depolymerization of microtubules [24,25], the microtubule polymerswere no longer detectable (Fig. 5C). In these cells, GalT-DsRed wasdistributed in dispersed structures indicating that the trans Golgi wasbroken down as expected from previous reports [24]. The pattern of transGolgi distribution was distinctly different in nocodazole treated cells
lls. HeLa cells transfected with GalT-DsRed and CFP-tubulin were treated with 200 μMusing epifluorescence microscopy. A. Basal HeLa cells. B. BrdU treated senescent HeLaas well as Golgi redistribution (note that tubulin distribution is distinctly absent in the

790 J.-H. Cho et al. / Cellular Signalling 23 (2011) 785–793
compared to BrdU treated cells (left panels of Fig. 5B and C) suggestingthat the Golgi dispersal which occurs during senescence induction isdistinct from the Golgi fragmentation due to microtubule depolymeriza-
Fig. 6. Effect of G protein γ11 subunit knockdown on Golgi structure in HeLa cells. A. Specificicells stably transfected with γ11-shRNA and control scrambled shRNAs were used. RNA waanalyzed by real time PCR. The expression levels were normalized to β-actin expression leveHeLa cells expressing M3, αq, YFP-β1, PH-mCh and γ11-shRNA were treated with 200 μM Brshown. Upper panel shows images of YFP-β1 indicating absence of translocation on receptorpresence of the receptor in the same cell. C. Knock down of γ11 in HeLa cells reduces senescepercentage of cells with compact or dispersed trans Golgi detected by TGN46 staining. Stablespecific shRNA particles (Sigma) and Golgi structure was analyzed using TGN46 staining ascells expressing control shRNA and γ11-shRNA are shown. An averaged plot of all the experimfrom basal cells and similarly BrdU treated γ11-shRNA was significantly different from contrexpressing various shRNAs immunostained with TGN46 antibodies are shown. D. Effect of inwas introduced transiently into cells expressing γ11 shRNA or scrambled shRNA. Only cells trof two experiments. Percentage variability across experiments was b30%. Representative imshown.
tion. Overall, these results suggested that the structural changes observedin the Golgi in senescent cells are not likely to be a result of microtubuledepolymerization.
ty of γ11-shRNA on the expression of G protein γ10 and γ11 subunits in HeLa cells. HeLas isolated from stable cell lines and mRNA expression of the γ10 and γ11 subunits wasls (n=3). B. YFP-β1 translocation is not detectable in HeLa cells expressing γ11-shRNA.dU for 24 h. Representative images of cells at basal level and after agonist treatment areactivation. Lower panel shows translocation of PH-mCh in the same cells to confirm thence induced Golgi dispersal (n=4). Bar graphs from representative experiments depictknock down of the γ11 subunit was generated using lentiviral transduction containingmentioned above. A scrambled shRNA was used as a control. Representative images ofents is shown. Golgi structure in wild type BrdU treated cells was significantly different
ol-shRNA expressing cells (t test) (pb0.01). Representative images of BrdU treated cellstroducing the mouse YFP-γ11 subunit into γ11 knock down HeLa cells. Mouse YFP-γ11ansfected with YFPwere counted for compact or dispersed Golgi (n=2). Plots aremeanages of basal and BrdU treated cells expressing γ11-shRNA and mouse γ11-cDNA are
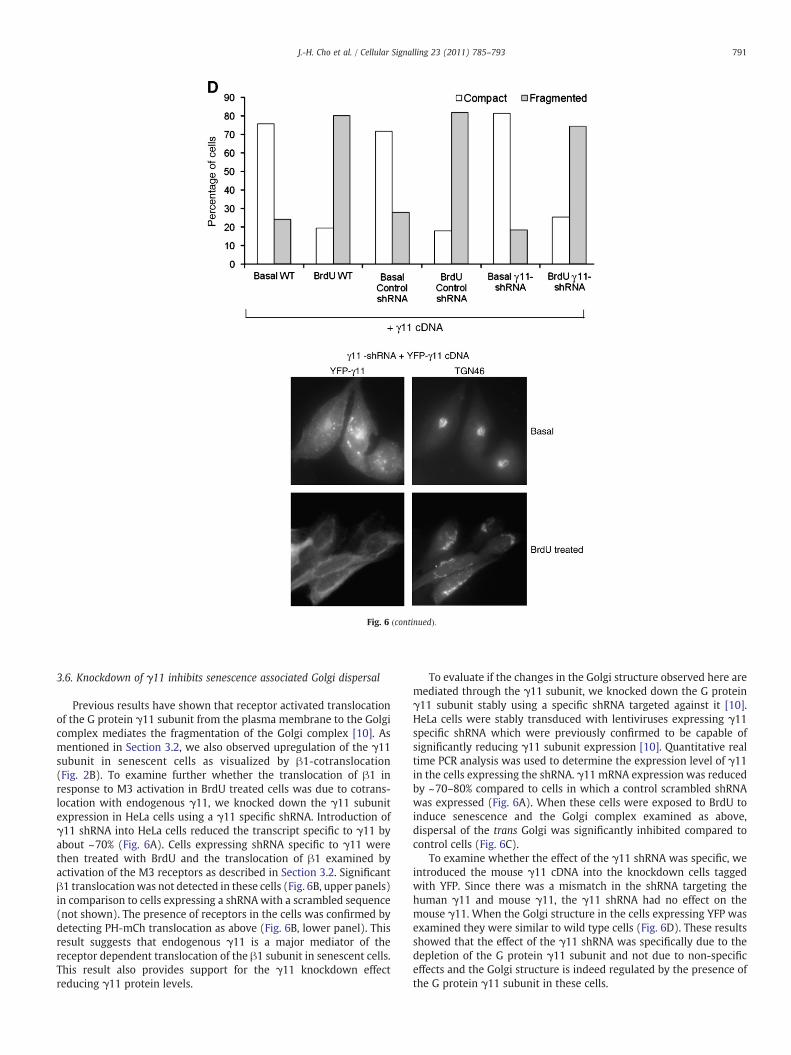
Fig. 6 (continued).
791J.-H. Cho et al. / Cellular Signalling 23 (2011) 785–793
3.6. Knockdown of γ11 inhibits senescence associated Golgi dispersal
Previous results have shown that receptor activated translocationof the G protein γ11 subunit from the plasma membrane to the Golgicomplex mediates the fragmentation of the Golgi complex [10]. Asmentioned in Section 3.2, we also observed upregulation of the γ11subunit in senescent cells as visualized by β1-cotranslocation(Fig. 2B). To examine further whether the translocation of β1 inresponse to M3 activation in BrdU treated cells was due to cotrans-location with endogenous γ11, we knocked down the γ11 subunitexpression in HeLa cells using a γ11 specific shRNA. Introduction ofγ11 shRNA into HeLa cells reduced the transcript specific to γ11 byabout ~70% (Fig. 6A). Cells expressing shRNA specific to γ11 werethen treated with BrdU and the translocation of β1 examined byactivation of the M3 receptors as described in Section 3.2. Significantβ1 translocationwas not detected in these cells (Fig. 6B, upper panels)in comparison to cells expressing a shRNAwith a scrambled sequence(not shown). The presence of receptors in the cells was confirmed bydetecting PH-mCh translocation as above (Fig. 6B, lower panel). Thisresult suggests that endogenous γ11 is a major mediator of thereceptor dependent translocation of the β1 subunit in senescent cells.This result also provides support for the γ11 knockdown effectreducing γ11 protein levels.
To evaluate if the changes in the Golgi structure observed here aremediated through the γ11 subunit, we knocked down the G proteinγ11 subunit stably using a specific shRNA targeted against it [10].HeLa cells were stably transduced with lentiviruses expressing γ11specific shRNA which were previously confirmed to be capable ofsignificantly reducing γ11 subunit expression [10]. Quantitative realtime PCR analysis was used to determine the expression level of γ11in the cells expressing the shRNA. γ11 mRNA expression was reducedby ~70–80% compared to cells in which a control scrambled shRNAwas expressed (Fig. 6A). When these cells were exposed to BrdU toinduce senescence and the Golgi complex examined as above,dispersal of the trans Golgi was significantly inhibited compared tocontrol cells (Fig. 6C).
To examine whether the effect of the γ11 shRNA was specific, weintroduced the mouse γ11 cDNA into the knockdown cells taggedwith YFP. Since there was a mismatch in the shRNA targeting thehuman γ11 and mouse γ11, the γ11 shRNA had no effect on themouse γ11. When the Golgi structure in the cells expressing YFP wasexamined they were similar to wild type cells (Fig. 6D). These resultsshowed that the effect of the γ11 shRNA was specifically due to thedepletion of the G protein γ11 subunit and not due to non-specificeffects and the Golgi structure is indeed regulated by the presence ofthe G protein γ11 subunit in these cells.

792 J.-H. Cho et al. / Cellular Signalling 23 (2011) 785–793
3.7. Translocation of the G protein βγ complex is likely required tomediate the Golgi structural changes associated with senescence
Receptor stimulated translocation of the G protein βγ complex fromthe plasma membrane to the Golgi complex has been shown to lead tostructural changes in the Golgi complex [10]. We examined if amechanism similar to that seen here was at the basis of the dispersalof Golgi elements in cellular senescence. We have previously reportedthat activation of the muscarinic receptor leads to the translocation ofsixmembers of theG proteinγ subunit familywhile translocation of theremaining sixmemberswas not detected under the conditions used [9].We have also shown that one of these γ subunits, γ3 whosetranslocation could not be detected, is dominant negative and reducesthe translocation of an endogenous γ subunit so that it is not detectableon receptor activation and inhibits the breakdown of the Golgi complexmediated by the translocation [10]. Using a similar strategy, weexpressed YFP-γ3 in HeLa cells and induced senescence with BrdU.The structure of the Golgi complex in these cells in comparison tocontrol cells inwhich YFP alonewas expressedwas then observed usingTGN46 as a marker for the trans Golgi network (Fig. 7). A significantdecrease in the number of cells containing dispersed Golgi was noted incells expressing the dominant negative γ3 subunit. These observationssuggest that the translocation ability of the γ11 subunit is a likelyrequirement for Golgi dispersal during senescence.
4. Discussion
The structure of the Golgi complex in senescent cells has not beenexamined before to our knowledge. Results here show that the Golgicomplex is compact in non-senescent or pre-senescent cells but isdispersed in senescent cells. The consistently altered appearance ofthe Golgi in both stress induced senescent cells and replicatively
Fig. 7. Inhibition of Golgi dispersal in BrdU treated senescent HeLa cells by the γ3subunit. Trans Golgi dispersal induced by incubation of HeLa cells with 200 μM BrdU for48 h is inhibited in HeLa cells transiently transfected with a G protein γ3 subunit. TheGolgi structure was observed by immunofluorescence using an antibody specific to thetrans Golgi network, TGN46. Bar diagram representing the percentage of cells (±SEM)that contained either compact or dispersed Golgi. Total cells analyzed the range from100–250 (n=3). Representative images of cells expressing YFP-γ3 are shown. Upperpanel, control untreated cells and lower panel, BrdU treated cells.
senescent cells suggests that the Golgi structure could serve as auseful marker for cellular senescence. Senescent cells show signifi-cantly increased secretion of cytokines such as IL-6 and IL-8; growthfactors such as VEGF and extracellular matrix proteins such asfibronectin [1,19,27]. One potential reason for the alteration in thestructure of the Golgi complex could be that it is a result of theincreased secretion of various proteins which traffic from the Golgi tothe plasma membrane.
It has been shown that the G protein γ11 subunit is upregulated instress induced and replicatively senescent cells [7] and we haveconfirmed this finding here. We show here that the upregulated Gprotein γ11 subunit in senescent cells mediates the translocation of theβγ11 complex from the plasma membrane to the Golgi in response toreceptor activation. The translocation is mediated by the upregulatedγ11 subunit because in the presence of the γ11 shRNA, translocation isnot seen. Recent evidence shows that receptor mediated translocationof the βγ complex breaks down the Golgi complex [10]. The changesseen in the structure of theGolgi complex in senescent cells are similarlymediated by the βγ11 complex because the γ11 shRNA inhibits thedispersal of trans Golgi and the trans Golgi network. Furthermore, theability of a known γ subunit whose translocation could not be detected,γ3, to inhibit senescence associated Golgi structure alterations suggestsstrongly that it is mediated by the translocation of the βγ complex fromthe plasma membrane to the Golgi.
The results here suggest that G protein γ11 upregulation plays arole in senescence and are consistent with an earlier report thatoverexpression of γ11 shortens the number of PDLs required to reachreplicative senescence [7]. Cellular senescence is thought to be cancerprotective since cell division is arrested. Consistent with such a roleγ11 transcription levels have been reported to be reduced inmedullary thyroid carcinoma [28] and splenic marginal zonelymphoma [29] indicating that its downregulation is accompaniedby cell proliferation.
It is expected that secretory changes in a cell will be primarilyreflected at the level of the trans Golgi network and trans Golgiregions. However, the structurally altered cis and medial Golgi seenhere in senescent cells may be because of the increased secretion of alarge number of proteins from senescent cells [1]. There is previousevidence that the entire Golgi can be broken downwhen regulators ofsecretory vesicle formation such as the translocating βγ complexesand PKD are overactive [10,26]. An alternative possibility is that theGolgi structure changes seen in senescent cells are due to the celldivision arrest which is a consequence of senescence induction. It isknown that during cell division and specifically mitosis, the Golgicomplex breaks down completely and is then reassembled indaughter cells [30]. It is possible that during senescence the initialchanges in the Golgi that occur inmitosis are observed but the processdoes not continue further than the dispersal of the Golgi elements.
Is it possible that Golgi structural changes accompany senescencebut do not directly play a role in regulating senescence? Previousresults show that overexpression of the G protein γ11 protein inducessenescent growth arrest at an earlier PDL compared to control mortalcells [7]. This result suggests that the translocation of the γ11 subunitfrom the plasma membrane to the Golgi likely plays a direct role inregulating senescence. The results here also suggest that a G proteincoupled receptor (GPCR) regulates senescence through the translo-cation of the βγ11 complex. GPCRs are the single most importanttarget for therapeutic drugs because they are accessible to extracel-lular ligands and modulate an enormous variety of physiologicalprocesses. The results here suggest that it may be possible to target aspecific GPCR to regulate senescence.
Acknowledgements
This work was supported by NIH grants (GM69027 andGM080558) to N.G. and an AHA postdoctoral fellowship (D.K.S.).

793J.-H. Cho et al. / Cellular Signalling 23 (2011) 785–793
References
[1] J.P. Coppe, P.Y. Desprez, A. Krtolica, J. Campisi, Annu. Rev. Pathol. 5 (2010) 99.[2] E. Michishita, K. Nakabayashi, T. Suzuki, S.C. Kaul, H. Ogino, M. Fujii, Y. Mitsui, D.
Ayusawa, J. Biochem. 126 (6) (1999) 1052.[3] T. Suzuki, S. Minagawa, E. Michishita, H. Ogino, M. Fujii, Y. Mitsui, D. Ayusawa, Exp.
Gerontol. 36 (3) (2001) 465.[4] S. Minagawa, K. Nakabayashi, M. Fujii, S.W. Scherer, D. Ayusawa, Exp. Cell Res. 304
(2) (2005) 552.[5] J.C. Masterson, S. O'Dea, Anticancer Drugs 18 (9) (2007) 1053.[6] F. Rodier, J.P. Coppe, C.K. Patil, W.A. Hoeijmakers, D.P. Munoz, S.R. Raza, A. Freund,
E. Campeau, A.R. Davalos, J. Campisi, Nat. Cell Biol. 11 (8) (2009) 973.[7] M.N. Hossain, R. Sakemura, M. Fujii, D. Ayusawa, Biochem. Biophys. Res. Commun.
351 (3) (2006) 645.[8] M. Akgoz, V. Kalyanaraman, N. Gautam, J. Biol. Chem. 279 (49) (2004) 51541.[9] D.K. Saini, V. Kalyanaraman, M. Chisari, N. Gautam, J. Biol. Chem. 282 (33) (2007)
24099.[10] D.K. Saini, W.K. Karunarathne, N. Angaswamy, D. Saini, J.H. Cho, V. Kalyanaraman,
N. Gautam, Proc. Natl Acad. Sci. USA 107 (25) (2010) 11417.[11] M. Chisari, D.K. Saini, J. Cho, V. Kalyanaraman, N. Gautam, PLoS ONE 4 (11) (2009)
e7797.[12] M. Chisari, D.K. Saini, V. Kalyanaraman, N. Gautam, J. Biol. Chem. 282 (33) (2007)
24092.[13] D.K. Saini, N. Gautam, Meth. Mol. Biol. 617 (2010) 191.[14] S. Hubackova, Z. Novakova, K. Krejcikova, M. Kosar, J. Dobrovolna, P. Duskova, H.
Hanzlikova, M. Vancurova, P. Barath, J. Bartek, Z. Hodny, Cell Cycle 9 (15) (2010)3085.
[15] L.H. Levkoff, G.P. Marshall 2nd, H.H. Ross, M. Caldeira, B.A. Reynolds, M. Cakiroglu,C.L. Mariani, W.J. Streit, E.D. Laywell, Neoplasia 10 (8) (2008) 804.
[16] H.H. Ross, L.H. Levkoff, G.P. Marshall 2nd, M. Caldeira, D.A. Steindler, B.A.Reynolds, E.D. Laywell, Stem Cells 26 (12) (2008) 3218.
[17] Z. Novakova, S. Hubackova, M. Kosar, L. Janderova-Rossmeislova, J. Dobrovolna, P.Vasicova, M. Vancurova, Z. Horejsi, P. Hozak, J. Bartek, Z. Hodny, Oncogene 29 (2)(2010) 273.
[18] I.B. Roninson, Cancer Lett. 179 (1) (2002) 1.[19] T. Kumazaki, R.S. Robetorye, S.C. Robetorye, J.R. Smith, Exp. Cell Res. 195 (1)
(1991) 13.[20] J.C. Acosta, A. O'Loghlen, A. Banito, M.V. Guijarro, A. Augert, S. Raguz, M. Fumagalli,
M. Da Costa, C. Brown, N. Popov, Y. Takatsu, J. Melamed, F. d'Adda di Fagagna, D.Bernard, E. Hernando, J. Gil, Cell 133 (6) (2008) 1006.
[21] A.R. Prescott, J.M. Lucocq, J. James, J.M. Lister, S. Ponnambalam, Eur. J. Cell Biol. 72(3) (1997) 238.
[22] N. Nakamura, C. Rabouille, R. Watson, T. Nilsson, N. Hui, P. Slusarewicz, T.E. Kreis,G. Warren, J. Cell Biol. 131 (6 Pt 2) (1995) 1715.
[23] B.S.Wilson, C. Nuoffer, J.L. Meinkoth, M.McCaffery, J.R. Feramisco,W.E. Balch, M.G.Farquhar, J. Cell Biol. 125 (3) (1994) 557.
[24] B. Storrie, W. Yang, Biochim. Biophys. Acta 1404 (1–2) (1998) 127.[25] J. Hoebeke, G. Van Nijen, M. De Brabander, Biochem. Biophys. Res. Commun. 69
(2) (1976) 319.[26] C. Bossard, D. Bresson, R.S. Polishchuk, V. Malhotra, J. Cell Biol. 179 (6) (2007)
1123.[27] A.R. Davalos, J.P. Coppe, J. Campisi, P.Y. Desprez, Cancer Metastasis Rev. 29 (2)
(2010) 273.[28] T.J. Musholt, J. Hanack, C. Brehm, R. von Wasielewski, P.B. Musholt, World J. Surg.
29 (4) (2005) 472.[29] E. Ruiz-Ballesteros, M. Mollejo, A. Rodriguez, F.I. Camacho, P. Algara, N. Martinez,
M. Pollan, A. Sanchez-Aguilera, J.Menarguez, E. Campo, P.Martinez,M.Mateo,M.A.Piris, Blood 106 (5) (2005) 1831.
[30] S. Puri, H. Telfer, M. Velliste, R.F. Murphy, A.D. Linstedt, J. Cell Sci. 117 (Pt 3) (2004)451.




















