
Aloe emodin inhibits the cytotoxic action of tumor necrosis factor
12
Aloe emodin inhibits the cytotoxic action of tumor necrosis factor Ljubica Harhaji a , Sanja Mijatovic a , Danijela Maksimovic-Ivanic a , Dusan Popadic b , Aleksandra Isakovic c , Biljana Todorovic-Markovic d , Vladimir Trajkovic b, ⁎ a Institute for Biological Research, Department of Immunology, Belgrade, Serbia and Montenegro b Institute of Microbiology and Immunology, School of Medicine, University of Belgrade, Belgrade, Serbia and Montenegro c Institute of Biochemistry, School of Medicine, University of Belgrade, Belgrade, Serbia and Montenegro d Vinca Institute of Nuclear Sciences, Belgrade, Serbia and Montenegro Received 24 November 2006; received in revised form 5 April 2007; accepted 12 April 2007 Available online 4 May 2007 Abstract We demonstrate the capacity of an herbal anthraquinone aloe emodin to reduce the cytotoxicity of the proinflammatory cytokine tumor necrosis factor (TNF) towards L929 mouse fibrosarcoma and U251 human glioma cell lines. Aloe emodin inhibited both TNF-induced cell necrosis and apoptosis, but it did not reduce cell death induced by UV radiation or hydrogen peroxide. Aloe emodin inhibited both basal and TNF-triggered activation of extracellular signal-regulated kinase (ERK), and a selective blockade of ERK activation mimicked the cytoprotective action of the drug. On the other hand, aloe emodin did not affect TNF-induced activation of p38 mitogen-activated protein kinase or generation of reactive oxygen species. The combination of aloe emodin and TNF caused an intracellular appearance of acidified autophagic vesicles, and the inhibition of autophagy with bafilomycin or 3-methyladenine efficiently blocked the cytoprotective action of aloe emodin. These data indicate that aloe emodin could prevent TNF-triggered cell death through mechanisms involving induction of autophagy and blockade of ERK activation. © 2007 Elsevier B.V. All rights reserved. Keywords: Aloe emodin; Tumor necrosis factor; Cytotoxicity; Apoptosis; Necrosis; Autophagy; Extracellular signal-regulated kinase 1. Introduction Tumor necrosis factor (TNF, previously known as TNF-α) is a pleiotropic cytokine produced by activated macrophages, as well as by several other cell types, including lymphocytes, fibroblasts, and hepatocytes (Vassalli, 1992). The effects of TNF are mediated by two distinct cell surface receptors, p55TNF-R1 and p75TNF-R2, which are expressed simultaneously on almost all cell types (Vandenabeele et al., 1995). The hallmark of this important cytokine is its fairly selective cytotoxic activity on tumor cells (Beyaert and Fiers, 1994), making it a plausible candidate for anticancer treatment, especially against surgically unresectable and chemotherapy- or radiation therapy-resistant tumors (Nakamoto et al., 2000). However, the clinical use of TNF has so far been hampered by the resistance of many tumors to TNF-mediated death (Shepard and Lewis, 1988), as well as because of the severe side-effects that included induction of hypotension and hepatotoxicity as the most prominent con- sequences of systemic TNF administration (Spriggs et al., 1988; Lucas et al., 2005). Production of large amounts of TNF has also been implicated in the liver damage during sepsis, viral hepatitis, alcoholic hepatitis, ischemia-reperfusion liver injury and fulminant hepatic failure (Ghavami et al., 2005), as well as in chemical-induced hepatotoxicity (Luster et al., 2000). Accord- ingly, TNF displayed a direct TNF-R1-dependent toxicity to- wards hepatocytes, both in vitro and in vivo (Leist et al., 1994). Therefore, investigating the mechanisms for both the enhance- ment of TNF-mediated tumor cell killing and decreasing its toxicity against normal tissues is important for developing TNF- related anticancer or anti-inflammatory strategies. Induction of tumor cell growth arrest and death by bioactive phytochemicals present in medical herbs and dietary plants is another attractive approach in cancer chemotherapy. Herbal European Journal of Pharmacology 568 (2007) 248 – 259 www.elsevier.com/locate/ejphar ⁎ Corresponding author. Institute of Microbiology and Immunology, School of Medicine, Dr. Subotica 1, 11000 Belgrade, Serbia and Montenegro. Tel./fax: +381 11 265 7258. E-mail address: [email protected] (V. Trajkovic). 0014-2999/$ - see front matter © 2007 Elsevier B.V. All rights reserved. doi:10.1016/j.ejphar.2007.04.029
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Aloe emodin inhibits the cytotoxic action of tumor necrosis factor

gy 568 (2007) 248–259www.elsevier.com/locate/ejphar
European Journal of Pharmacolo
Aloe emodin inhibits the cytotoxic action of tumor necrosis factor
Ljubica Harhaji a, Sanja Mijatovic a, Danijela Maksimovic-Ivanic a, Dusan Popadic b,Aleksandra Isakovic c, Biljana Todorovic-Markovic d, Vladimir Trajkovic b,⁎
a Institute for Biological Research, Department of Immunology, Belgrade, Serbia and Montenegrob Institute of Microbiology and Immunology, School of Medicine, University of Belgrade, Belgrade, Serbia and Montenegro
c Institute of Biochemistry, School of Medicine, University of Belgrade, Belgrade, Serbia and Montenegrod Vinca Institute of Nuclear Sciences, Belgrade, Serbia and Montenegro
Received 24 November 2006; received in revised form 5 April 2007; accepted 12 April 2007Available online 4 May 2007
Abstract
We demonstrate the capacity of an herbal anthraquinone aloe emodin to reduce the cytotoxicity of the proinflammatory cytokine tumornecrosis factor (TNF) towards L929 mouse fibrosarcoma and U251 human glioma cell lines. Aloe emodin inhibited both TNF-induced cellnecrosis and apoptosis, but it did not reduce cell death induced by UV radiation or hydrogen peroxide. Aloe emodin inhibited both basal andTNF-triggered activation of extracellular signal-regulated kinase (ERK), and a selective blockade of ERK activation mimicked thecytoprotective action of the drug. On the other hand, aloe emodin did not affect TNF-induced activation of p38 mitogen-activated protein kinaseor generation of reactive oxygen species. The combination of aloe emodin and TNF caused an intracellular appearance of acidified autophagicvesicles, and the inhibition of autophagy with bafilomycin or 3-methyladenine efficiently blocked the cytoprotective action of aloe emodin.These data indicate that aloe emodin could prevent TNF-triggered cell death through mechanisms involving induction of autophagy andblockade of ERK activation.© 2007 Elsevier B.V. All rights reserved.
Keywords: Aloe emodin; Tumor necrosis factor; Cytotoxicity; Apoptosis; Necrosis; Autophagy; Extracellular signal-regulated kinase
1. Introduction
Tumor necrosis factor (TNF, previously known as TNF-α) isa pleiotropic cytokine produced by activated macrophages, aswell as by several other cell types, including lymphocytes,fibroblasts, and hepatocytes (Vassalli, 1992). The effects of TNFare mediated by two distinct cell surface receptors, p55TNF-R1and p75TNF-R2, which are expressed simultaneously on almostall cell types (Vandenabeele et al., 1995). The hallmark of thisimportant cytokine is its fairly selective cytotoxic activity ontumor cells (Beyaert and Fiers, 1994), making it a plausiblecandidate for anticancer treatment, especially against surgicallyunresectable and chemotherapy- or radiation therapy-resistanttumors (Nakamoto et al., 2000). However, the clinical use of
⁎ Corresponding author. Institute of Microbiology and Immunology, School ofMedicine, Dr. Subotica 1, 11000 Belgrade, Serbia and Montenegro. Tel./fax:+381 11 265 7258.
E-mail address: [email protected] (V. Trajkovic).
0014-2999/$ - see front matter © 2007 Elsevier B.V. All rights reserved.doi:10.1016/j.ejphar.2007.04.029
TNF has so far been hampered by the resistance of many tumorsto TNF-mediated death (Shepard and Lewis, 1988), as well asbecause of the severe side-effects that included induction ofhypotension and hepatotoxicity as the most prominent con-sequences of systemic TNF administration (Spriggs et al., 1988;Lucas et al., 2005). Production of large amounts of TNF has alsobeen implicated in the liver damage during sepsis, viral hepatitis,alcoholic hepatitis, ischemia-reperfusion liver injury andfulminant hepatic failure (Ghavami et al., 2005), as well as inchemical-induced hepatotoxicity (Luster et al., 2000). Accord-ingly, TNF displayed a direct TNF-R1-dependent toxicity to-wards hepatocytes, both in vitro and in vivo (Leist et al., 1994).Therefore, investigating the mechanisms for both the enhance-ment of TNF-mediated tumor cell killing and decreasing itstoxicity against normal tissues is important for developing TNF-related anticancer or anti-inflammatory strategies.
Induction of tumor cell growth arrest and death by bioactivephytochemicals present in medical herbs and dietary plants isanother attractive approach in cancer chemotherapy. Herbal

249L. Harhaji et al. / European Journal of Pharmacology 568 (2007) 248–259
anthraquinone derivatives emodin (3-methyl-1,6,8-trihydrox-yanthraquninone) and aloe emodin (1,8-Dihydroxy-3-hydro-xymethyl-anthraquinone) have been shown efficient in limitingproliferation of various tumor cell lines in vitro (Zhang et al.,1995; Zhang and Hung, 1996; Lee et al., 2001; Kuo et al., 2002;Yeh et al., 2003; Pecere et al., 2003; Mijatovic et al., 2005a;Acevedo-Duncan et al., 2004) as well as in restricting tumorgrowth in vivo (Zhang et al., 1999; Pecere et al., 2000; Chaet al., 2005). The mechanisms underlying the observed anti-cancer actions of emodin and aloe emodin mainly involved theinduction of apoptotic death of tumor cells (Lee et al., 2001;Kuo et al., 2002; Yeh et al., 2003; Pecere et al., 2003; Mijatovicet al., 2005a; Acevedo-Duncan et al., 2004), their differentiationtoward more mature phenotype with reduced growth capacity(Zhang et al., 1995; Mijatovic et al., 2005a), or inhibition oftumor cell adhesion and migration (Huang et al., 2005, 2006).In addition, we have recently reported that the exposure ofglioma cells to aloe emodin triggers autophagy (Mijatovic et al.,2005a), in which the engulfment of cytoplasm and/or cyto-plasmic organelles by multiple-membrane cytoplasmic vesiclesleads to their destruction by the lysosomal system of the samecell (Codogno and Meijer, 2005). In contrast to its pro-apoptoticaction, emodin and/or aloe emodin displayed hepatoprotectiveeffect in carbon tetrachloride-or d-galactosamine-induced liverdamage in rats (Lin et al., 1996; Arosio et al., 2000), andprotected rat pancreas from sodium taurocholate toxicity (Zhanget al., 2002) or rat myocardium from ischemia-reperfusion injury(Yim et al., 1998). Chang et al. (1999) have demonstrated theability of emodin to increase the repair of UV- and cisplatin-induced DNA damage in normal human fibroblasts, throughmechanisms presumably involving increase in calcium influxand subsequent expression of nucleotide excision repaircomplex subunit ERCC1. Moreover, we have reported thataloe emodin can efficiently protect L929 fibrosarcoma and ratC6 glioma cells from cisplatin-induced (Mijatovic et al., 2005b)or cytokine-triggered death (Mijatovic et al., 2004) by blockingthe intracellular activation of extracellular signal-regulatedkinase (ERK) or the production of cytotoxic free radical nitricoxide, respectively. Therefore, it appears that emodin and struc-turally similar aloe emodin can act in a context-dependentmanner to either impede or promote cell survival.
Only few studies so far have addressed the biological inter-action between TNF and emodin. Emodin reduced proin-flammatory cytokine (interleukin-1 and interleukin-6)-mediatedproduction of TNF in human mesangial cells (Kuo et al., 2001),and inhibited TNF-induced expression of glutathione S-transfer-ase P1-1 gene in human leukemia cell lines (Duvoix et al., 2004),as well as the expression of adhesion molecules and secretion ofplasminogen activator inhibitor type 1 in human vascular endo-thelial cells (Hamaguchi et al., 2003). While emodin and aloeemodin also inhibited carbon tetrachloride-or sodium taurocho-late-induced TNF production in vivo (Arosio et al., 2000; Zhanget al., 2002), the influence of emodin or aloe emodin on thecytotoxicity of TNF, to our knowledge, has not been explored thusfar.
Having in mind both cytotoxic and cytoprotective potentialof aloe emodin, we have investigated its effect on the survival of
TNF-treated L929 mouse fibrosarcoma cell line, widely used asa model system for exploring TNF-induced cell death. Theresults of the present study for the first time demonstratethe ability of aloe emodin to antagonize the cytotoxic action ofTNF.
2. Materials and methods
2.1. Cells and cell culture
The mouse fibrosarcoma cell line L929 was obtained from theEuropean Collection of Animal Cell Cultures (Salisbury, UK),while a TNF-sensitive subclone of the human glioma cell lineU251 was kindly donated by Dr. Pedro Tranque (Universidad deCastilla-La Mancha, Albacete, Spain). Cells were maintained at37 °C in a humidified atmosphere with 5% CO2, in a HEPES-buffered RPMI 1640 cell culture medium supplemented with fetalcalf serum (5%), l-glutamine, 2-mercaptoethanol, pyruvate andantibiotics (all from Sigma, St. Louis, MO). The cells wereprepared for experiments using the conventional trypsinizationprocedure with trypsin/EDTA and incubated in 6-well plates forthe flow cytometric analysis (3 × 105 cells/well) or in 96-well flat-bottom plates (2 × 104 cells/well) for the crystal violet assay, cell-based enzyme-linked immunosorbent assay (ELISA) and super-oxide determination. Cells were rested until they reached nearconfluence (approx. 24 h). We have chosen the high-density cellcultures because TNF cytotoxicity has been reported to increasewith increasing L929 cell density (Kirstein et al., 1986). In someexperiments, however, the cells were treated 3 h after beingseeded, while they were still non-confluent. After resting period,the cell culture medium was replaced and the cells were treatedwith mouse recombinant TNF and/or aloe emodin, in the absenceor presence of the antioxidant agent N-acetylcysteine, mitogen-activated protein kinase (MAPK) antagonists 2′-Amino-3′-methoxyflavone (PD98059) and 4-[5-(4-fluorophenyl)-2-[4-(methylsulfonyl)phenyl]-1H-imidazol-4-yl]pyridine (SB203580),or an autophagy inhibitor bafilomycin A1 (all from Sigma). Sincealoe emodin, MAPK inhibitors and bafilomycin were initiallydissolved in dimethylsulfoxide (DMSO, Sigma), control cellcultures received the maximal amount of DMSO (always b 0.1%volume) used in the particular experiment. There was no signif-icant difference in any of the parameters tested between untreatedcell cultures and DMSO-containing controls (data not shown). Insome experiments, cell death was induced by addition of hydrogenperoxide (Sigma) to cell culture medium, or by a 5 min pulse ofUVB light (280–315 nm), delivered by a UV lamp at a distance of5 cm.
2.2. Determination of cell number
The cell number after treatment was determined by a crystalviolet staining exactly as previously described (Mijatovic et al.,2004). The absorbance of dissolved crystal violet, directly pro-portional to the number of adherent (viable) cells, was measuredby an automated microplate reader at 570 nm. The results werepresented as % of the control value (untreated cells), which wasarbitrarily set to 100%.

250 L. Harhaji et al. / European Journal of Pharmacology 568 (2007) 248–259
2.3. Detection of apoptosis and necrosis
Apoptotic and necrotic cell death were analyzed by doublestaining with annexin V-FITC and propidium iodide (PI), inwhich annexin V bound to the apoptotic cells with exposedphosphatidylserine, while PI labeled the necrotic cells with mem-brane damage. Staining was performed according to the instruc-tions by the manufacturer (BD Pharmingen, San Diego, CA), andflow cytometry was conducted using a FACSCalibur flow cytom-eter (BD). The percentage of apoptotic (annexin+/PI−) andnecrotic (annexin+/PI+) cells was determined using CellQuest Prosoftware.
2.4. Cell-based ELISA for MAPK activation
To measure MAPK activation, near confluent cells werestarved for 24 h in a cell culture medium with 0.1% fetal calfserum. Cells were then washed and incubated in fresh cell culturemedium (0.1% fetal calf serum) containing TNF and/or aloeemodin. The intracellular concentration of activated, phospho-p38MAPK and phospho-p42/p44MAPK (ERK)was determinedat various time points by cell-based ELISA as previouslydescribed (Mijatovic et al., 2005a). Mouse anti-phospho-ERKand anti-phospho-p38 MAPK (Santa Cruz Biotechnology, SantaCruz, CA) were used as primary antibodies, while horseradish
Fig. 1. Aloe emodin antagonizes the cytotoxic action of TNF. (A, C) Near confluconcentrations of TNF and/or aloe emodin (AE) (A, C), or with TNF and/or 20 μM alincubation. (D) The Chou–Talalay combination index was calculated based on theindicated in (C). The data are mean±S.D. values (each SD value in C was b10% of(B, C) experiments (⁎Pb0.05 refers to cells treated with TNF alone), or mean±Ssignificantly different from 1).
peroxidase-conjugated goat anti-mouse Ig (US Biochemical,Cleveland, OH)was used as a detection antibody. The absorbancevalues (492 nm) were normalized for the cell number by crystalviolet assay, and the results were presented as relative to thecontrol value (untreated cells), arbitrarily set to 1.
2.5. Measurement of reactive oxygen species
A slightlymodified version of a previously described assay forthe intracellular conversion of nitro blue tetrazolium (NBT) toformazan by superoxide anionwas used tomeasure the generationof reactive oxygen species (Vrablic et al., 2001). Briefly, 50 μMNBT (Sigma) was added to the media at the end of the treatmentperiod and cells were incubated for an additional 15 min at 37 °C.After removing the NBT solution, cells were allowed to air dry.The formazan content of the cells was solubilized with DMSO(Sigma), and the absorbance at 570 nm was measured using amicroplate reader. The results were presented as fold increaserelative to the control value (untreated cells).
2.6. Autophagy visualization
The acidic autophagic vesicles were visualized by supravitalacridine orange staining as previously described (Paglin et al.,2001). Briefly, cells (3 × 104) cultivated on glass chamber-slides
ent or (B) low density (2×104/well) L929 cells were incubated with differentoe emodin (B). The cell number was assessed by crystal violet assay after 24 h ofcytotoxicity of TNF, aloe emodin and their combinations at the concentrationsthe corresponding mean) of triplicates from a representative of four (A) or three.D. values from three independent experiments (D; ⁎Pb0.05 denotes values

Fig. 2. Aloe emodin prevents TNF-induced apoptosis and necrosis. (A, B) L929cells were incubated alone or with TNF (2 ng/ml), in the absence or presence ofaloe emodin (AE; 20 μM). After 24 h, cell morphology was analyzed usinginverted microscopy (A), while the number of apoptotic and necrotic cells wasdetermined by flow cytometric analysis of the cells stained with annexin V-FITCand propidium iodide (B). The representative dot plots are presented, while thedata are mean±S.D. values of three separate experiments (⁎Pb0.05 refers totreatment with TNF alone).
251L. Harhaji et al. / European Journal of Pharmacology 568 (2007) 248–259
were washed with PBS after the 6 h treatment and stained withacridine orange (1 μg/ml; Labo-Moderna, France) for 15 min at37 °C. Subsequently, cells were washed and analyzed usingfluorescence microscopy. Depending on their acidity, autophagiclysosomes appeared as yellow–orange to bright-red fluorescentcytoplasmic vesicles, while nuclei were stained green. Autophagywas blocked with bafilomycin (Sigma), an inhibitor of thevacuolar proton ATPase that blocks acidification of autophago-somes (Yamamoto et al., 1998), or 3-methyladenine, whichinhibits formation of autophagosomes, probably by interferingwith phosphatidylinositol 3-kinase activity (Blommaart et al.,1997).
2.7. Mathematical analysis of aloe emodin/TNF antagonism
To confirm the antagonistic interaction of aloe emodin andTNF in inducing tumor cell death, cells were treated withdifferent concentration of each agent alone and in combination.A combination index for mutually exclusive interactions wascalculated according to the method designed by Chou andTalalay (1984). The values of combination index N 1 indicateantagonistic interaction, while the values b 1 or not significantlydifferent from 1 specify synergistic or additive interaction,respectively.
2.8. Statistical analysis
The statistical significance of the differences betweentreatments was analyzed using t-test or ANOVA followed byStudent–Newman–Keuls test. The value of P b 0.05 was con-sidered to be significant.
3. Results
3.1. Aloe emodin protects L929 cells from TNF-induced death
Crystal violet test revealed that exposure to TNF lead to a dose-dependent reduction in cell number in cultures of TNF-sensitiveL929 cells (Fig. 1A). On the other hand, only the highest dose ofaloe emodin (80μM)displayed a slight antiproliferative/cytotoxicactivity, while lower concentrations (40 μM and less) did notsignificantly affect cell numbers in near confluent cell cultures(Fig. 1A). Moreover, treatment with aloe emodin partly, butsignificantly protected L929 cells from TNF toxicity (Fig. 1A).The effect was dose-dependent, showing a bell-shaped relationbetween concentration of aloe emodin and the observed protec-tion, with 20 μM being most efficient (Fig. 1A) and thereforechosen for further experiments. In contrast to results obtainedwithnear confluent cells, aloe emodin at 20 μMwas significantly toxicto low density cell cultures, but its protective effect against TNF-induced death was still readily observed (Fig. 1B). To obtain datafor the mathematical analysis of the interaction between TNF andaloe emodin in killing L929 cells, the cells were treated withvarious concentrations of each agent alone and in combination(Fig. 1C). Similarly to data presented in Fig. 1A and B, aloeemodin improved the survival of TNF-treated cells in this experi-mental setting (Fig. 1C). The combination index calculated using
the Chou-Talalay approach was significantly higher than 1throughout 0.1–0.99 efficiency range (Fig. 1D), thus confirmingthe ability of aloe emodin to antagonize the cytotoxic action ofTNF.
3.2. Aloe emodin inhibits TNF-induced necrosis and apoptosisof L929 cells
Treatment of L929 cells with TNF caused morphologicalchanges consistent with cell death, as most of the cells lost theirprocesses and became smaller, round, and detached from the

252 L. Harhaji et al. / European Journal of Pharmacology 568 (2007) 248–259
culture well surface (Fig. 2A). While aloe emodin alone did nothave any discernible effect on cellular morphology, its additionto TNF-treated cell cultures partly preserved the normal cuboidshape of confluent L929 cells (Fig. 2A). The TNF-induced celldeath apparently proceeded through both apoptotic and necroticpathways, as revealed by a marked increase in number of bothannexin+PI− (apoptotic) and annexin+PI+ (necrotic) cells fol-lowing TNF treatment (Fig. 2B). In accordance with data ob-tained in viability tests (Fig. 1), aloe emodin significantly
Fig. 3. The role of ERK, p38 MAPK and reactive oxygen species in aloe emodin-medor presence of different concentrations of the antioxidant N-acetyl cysteine (NAC) (AThe cell number was assessed by crystal violet assay after 24 h. (D–F) L929 cells wer20 μM). The production of superoxide was analyzed after 24 h using the NBTassay (Dactivation of ERK (E) and p38MAPK (F). (A–F) The data are mean±S.D. values of trefers to cells treated with TNF alone).
reduced the numbers of apoptotic and necrotic cells in TNF-treated cultures of L929 cells (Fig. 2B).
3.3. The role of ERK, p38 MAPK and reactive oxygen speciesin aloe emodin-mediated protection from TNF cytotoxicity
We next sought to explore the intracellular mechanisms re-sponsible for aloe emodin-mediated protection from TNFtoxicity. The antioxidant agent N-acetylcysteine and PD98059,
iated cytoprotection. (A–C) L929 cells were incubated with TNF, in the absence), ERK activation blocker PD98059 (B), or p38 MAPK inhibitor SB203580 (C).e incubated with TNF (2 ng/ml), in the absence or presence of aloe emodin (AE;), or a cell-based ELISAwas performed at the indicated time points to assess theriplicates from a representative of four (A) or three (B–F) experiments (⁎Pb0.05

253L. Harhaji et al. / European Journal of Pharmacology 568 (2007) 248–259
a selective inhibitor of the ERK upstream activator MEK, eachmarkedly reduced the cytotoxic effect of TNF (Fig. 3A, B). Onthe other hand, SB203580, a selective antagonist of p38 MAPK,significantly enhanced TNF-induced death of L929 cells(Fig. 3C). These data indicated that reactive oxygen species/ERK and p38 MAPK might play damaging and protective role,respectively, in TNF-induced cytotoxicity. We therefore assessedthe effect of aloe emodin on TNF-induced superoxide production,as well as on activation of ERK and p38 MAPK in L929 cells.Expectedly, the NBT assay revealed a significant increase insuperoxide production upon TNF treatment of L929 cells(Fig. 3D). However, aloe emodin itself displayed a similar
Fig. 4. Cytoprotective action of aloe emodin is stimulus-dependent, but not cell-speTNF and/or aloe emodin (AE) (A), 2 ng/ml TNF and/or 20 μM aloe emodin (B, C), ortreated with H2O2 (1 mM) and/or aloe emodin, or aloe emodin was added immediatelcrystal violet test (A, D–F) and the proportion of apoptotic and necrotic cells was detwhile ERK activation was determined after 15 min by cell-based ELISA (C). The da(C–F) experiments, or mean±S.D. values from three independent experiments (B) (
superoxide-inducing capacity, and superoxide generation inducedby combination of TNF and aloe emodin did not exceed the levelsobserved after treatment with each agent alone (Fig. 3D). TheTNF-induced activation of both p38 MAPK and ERK in L929cells wasmaximal after 15min of stimulation and then declined toa near-basal level during next 45min.While aloe emodin reducedboth basal and TNF-triggered activation of ERK (Fig. 3E), it didnot affect the phosphorylation of p38 MAPK in L929 cells(Fig. 3F). These data suggest that aloe emodin-mediatedinhibition of ERK activation, but not modulation of TNF-inducedsuperoxide generation or p38MAPK activation, might contributeto the observed protection from TNF cytotoxicity.
cific. (A–D) U251 glioma cells were incubated with different concentrations of2 ng/ml TNF and/or 20 μMPD98059 (D). (E, F) L929 (E) or U251 cells (F) werey after 5 min exposure to UVB light. After 24 h, the cell number was assessed byermined by flow cytometric analysis following annexin V-FITC/PI staining (B),ta are mean±S.D. values of triplicates from a representative of four (A) or three⁎Pb0.05 refers to cells treated with TNF alone).
254 L. Harhaji et al. / European Journal of Pharmacology 568 (2007) 248–259
3.4. Cytoprotective action of aloe emodin is stimulus-dependent,but not cell-specific
To investigate whether aloe emodin-mediated protectionfrom TNF cytotoxicity was restricted to L929 cells, we used aTNF-sensitive subclone of the human glioma cell line U251.
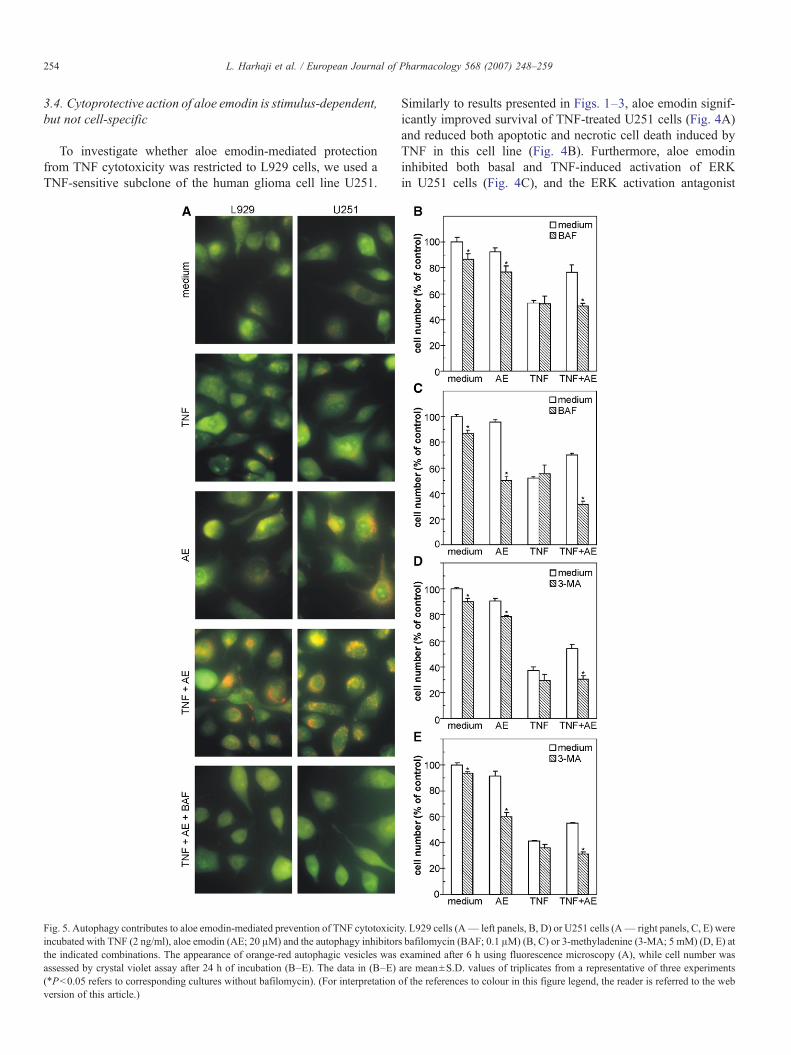
Fig. 5. Autophagy contributes to aloe emodin-mediated prevention of TNF cytotoxicitincubated with TNF (2 ng/ml), aloe emodin (AE; 20 μM) and the autophagy inhibitorthe indicated combinations. The appearance of orange-red autophagic vesicles wasassessed by crystal violet assay after 24 h of incubation (B–E). The data in (B–E)(⁎Pb0.05 refers to corresponding cultures without bafilomycin). (For interpretationversion of this article.)
Similarly to results presented in Figs. 1–3, aloe emodin signif-icantly improved survival of TNF-treated U251 cells (Fig. 4A)and reduced both apoptotic and necrotic cell death induced byTNF in this cell line (Fig. 4B). Furthermore, aloe emodininhibited both basal and TNF-induced activation of ERKin U251 cells (Fig. 4C), and the ERK activation antagonist
y. L929 cells (A— left panels, B, D) or U251 cells (A— right panels, C, E) weres bafilomycin (BAF; 0.1 μM) (B, C) or 3-methyladenine (3-MA; 5 mM) (D, E) atexamined after 6 h using fluorescence microscopy (A), while cell number wasare mean±S.D. values of triplicates from a representative of three experimentsof the references to colour in this figure legend, the reader is referred to the web

255L. Harhaji et al. / European Journal of Pharmacology 568 (2007) 248–259
PD98059 mimicked the observed protection of U251 cells fromTNF-triggered death (Fig. 4D). On the other hand, aloe emodincompletely failed to protect L929 and U251 cells from H2O2- orUV-induced death (Fig. 4E and F). While aloe emodin wasadded to cells immediately after UV irradiation in order to avoidits possible phototoxicity (Wamer et al., 2003), we did notobserve any improvement in cell survival even if the drug wasadministered before UV treatment (data not shown). These dataindicate that cytoprotective action of aloe emodin, while notcell-specific, could depend on cytotoxic stimulus.
3.5. Autophagy is involved in aloe emodin-mediated suppres-sion of TNF cytotoxicity
As aloe emodin and TNF have been reported to induceautophagy in glioma cells and leukaemic cells, respectively(Mijatovic et al., 2005a; Jia et al., 1997), we investigated therole of autophagy in aloe emodin-mediated protection fromTNF-induced cell death. Acridine orange staining of both L929and U251 cells incubated for 6 h with TNF revealed onlysporadic presence of a small number of red acidophylic auto-phagic vesicles (Fig. 5A). Similarly, a low level of autophagy
Fig. 6. The effect of aloe emodin pretreatment on sensitivity to TNF cytotoxicaction. (A) U251 or (B) L929 cells (1×106) were incubated without or with aloeemodin (AE; 20 μM) in 25 cm2 flasks. After 48 h, cells were trypsinized, washedand re-seeded in 96-well plates as described in Materials and methods. Cellswere treated with different doses of TNF and the cell number was assessed bycrystal violet assay after 24 h of incubation. The data are mean±S.D. values oftriplicates from a representative of three experiments (⁎Pb0.05 refers to cellsthat were not pretreated with aloe emodin).
was observed in L929 cells treated with aloe emodin only. Onthe other hand, a massive intracytoplasmic appearance ofautophagic vacuoles was clearly evident in U251 cells treatedwith aloe emodin alone, or both in L929 and U251 cellsexposed to combination of aloe emodin and TNF (Fig. 5A).Expectedly, the presence of acidophylic autophagolysosomes inaloe emodin + TNF-treated cells was abolished in the presenceof autophagy inhibitor bafilomycin (Fig. 5A). Bafilomycin and3-methyladenine, another autophagy inhibitor, were slightlytoxic to each cell line (Fig. 5B–E), and in combination with aloeemodin displayed an additive or even synergistic cytotoxicitytowards L929 (Fig. 5B and D) or U251 cells (Fig. 5C and E),respectively. This indicates that aloe emodin-mediated antican-cer action in the latter cell line might have been blocked by aconcomitant induction of autophagy. Moreover, bafilomycinand 3-methyladenine diminished the cytoprotective effect ofaloe emodin in either L929 or U251 cells treated with TNF(Fig. 5B–E), which suggests that autophagy was also involvedin the observed blockade of TNF cytotoxicity in both cell lines.
3.6. Pretreatment with aloe emodin reduces sensitivity to TNFcytotoxic action
Finally, we investigated whether aloe emodin-mediatedprotection requires simultaneous application of the drug withTNF, or the target cells could be in advance “programmed” bythe drug to cope with TNF toxicity. To that effect, cells werepretreated with aloe emodin for 48 h and the drug was removedbefore subsequent exposure to TNF. The dose-dependencycurves of TNF cytotoxicity clearly show that pretreatment withaloe emodin protected U251 glioma cells with the efficiencycomparable to that observed in the experiments with simulta-neous TNF/aloe emodin addition (Fig. 6). On the other hand, theprotection was considerably less pronounced in L929 fibrosar-coma cells (Fig. 6). Therefore, the observed decrease in sen-sitivity to TNF upon aloe emodin pretreatment appears to becell-type specific to a certain degree.
4. Discussion
The present report for the first time demonstrates the ability ofan herbal anthraquinone aloe emodin to antagonize TNFcytotoxicity. These data could have important implications forthe potential use of aloe emodin as an anticancer and/or antin-flammatory agent. It should be emphasized, however, that thestudy was performed solely on tumor cell lines. Therefore, acertain degree of caution seems required for interpretation ofthese results with regard to a possible role of aloe emodin inprotection of normal tissues from TNF-dependent inflammatorydamage.
In light of the known anticancer action of aloe emodin, itsability to improve survival of TNF-treated cells might seemsomewhat surprising. However, it is not unusual that certaincytotoxins, including aloe emodin and free radical nitric oxide,to name only the few, are able to interfere with cell deathinduced by other cytotoxic agents (Mijatovic et al., 2005b;Brune, 2003). It should be noted that cell culture conditions

256 L. Harhaji et al. / European Journal of Pharmacology 568 (2007) 248–259
employed in the present study (near confluent cells) favored theprotective over cytotoxic action of aloe emodin, since confluenttumor cells were less sensitive to its pro-apoptotic/antiproli-ferative activity (Mijatovic et al., 2004 and the present study).Nevertheless, the aloe emodin-mediated antagonistic effect onTNF toxicity was apparently independent of cell density, as thecytoprotective effect of the drug was also observed in non-confluent L929 cells. In addition, the protective effect of aloeemodin was not related to its ability to suppress the productionof nitric oxide (Mijatovic et al., 2004), as TNF-stimulated L929cells did not produce detectable levels of this cytotoxic freeradical (our unpublished observation).
The studies on the mode of TNF-induced death in L929 cellshave yielded conflicting results, as different groups reportedeither necrosis (Vercammen et al., 1998; Liu et al., 1999; Loset al., 2002) or apoptosis (Trent et al., 1996; Piret et al., 2004;Bulfone-Paus et al., 1999) to be responsible for TNF-mediatedL929 cell killing. While exploration of this discrepancy isbeyond the scope of the present report, we note that, at least insome studies, both apoptotic (internucleosomal DNA fragmen-tation and/or phosphatydilserine exposure) and necrotic changes(cell membrane damage) were observed in the same cell culturefollowing the TNF treatment (Humphreys and Wilson, 1999;Wang et al., 2004; Fady et al., 1995). Accordingly, the termssuch as “atypical apoptosis” and “necrapoptosis” have beencoined by some researchers (Strelow et al., 2000; Lemasters,1999). This is consistent with our data showing two distinct cellpopulations in TNF-treated L929 cells- one with phosphaty-dilserine exposed on the outer side of the intact cell membrane(apoptotic-like), and the other in which the phosphatydil serineexposure was associated with the loss of cell membrane integrity(necrotic-like). The appearance of the latter cells was probablynot due to a secondary necrosis, since the apoptotic/necrotic cellratio was similar at early (6 h) and late (18 h) time points (ourunpublished results). The addition of aloe emodin significantlyreduced TNF-mediated induction of both apoptosis andnecrosis, indicating that the drug could affect some intracellularevent that is shared by both cell death pathways. Indeed,mitochondrial permeability transition that precedes mitochon-drial depolarization might be a common cellular reaction tovarious death signals, including TNF, but that culminates inapoptosis, necrosis, or the mixture of both, depending on cellularATP level and other modifying factors (Lemasters, 1999;Denecker et al., 2001). However, the cytoprotective action ofaloe emodin was fairly selective for TNF-induced apoptotic/necrotic cell death, as it completely failed to reduce the cyto-toxicity of UV radiation or high-dose hydrogen peroxide(1 mM), which primarily induce apoptosis (Kitagawa et al.,2002) or necrosis (Arany et al., 2004), respectively.
The present study confirms the previous findings that reactiveoxygen species and ERK are the executors of TNF-mediated celldeath (Vercammen et al., 1998; Liu et al., 1999; Wang et al.,2004; Goossens et al., 1999; Chang, 1997), while p38 MAPKmight play a protective role in this process (Luschen et al., 2004).Therefore, the ability of aloe emodin to partly reduce TNF-triggered ERK activation in our experiments might contribute tothe observed cytoprotective effect. Accordingly, we have previ-
ously shown that inhibition of ERK activation was responsiblefor aloe emodin-mediated prevention of ERK-dependent cyto-toxicity of cisplatin (Mijatovic et al., 2005b), while emodin hasalso been reported to inhibit ERK phosphorylation in variouscancer cell lines (Huang et al., 2004). On the other hand, aloeemodin alone induced production of reactive oxygen species anddid not interfere with TNF-triggered oxidative stress or p38MAPK activation in L929 cells. These data are consistent withthe findings that emodin and aloe emodin in certain conditionsexerted pro-oxidant activity (Su et al., 2005; Yi et al., 2004; Vathet al., 2002) and failed to affect p38 MAPK activation in varioustumor cell lines (Mijatovic et al., 2004; Huang et al., 2004).Thus, it appears that aloe emodin-mediated protection from TNFcytotoxicity involved the inhibition of ERK activation, but notmodulation of TNF-induced production of reactive oxygenspecies or p38 MAPK activation in the target cells. Such ahypothesis is apparently consistent with the observed inability ofaloe emodin to block the toxicity of UV and H2O2, having inmind that ERK, at least in some experimental systems, did notseem to mediate UV- or H2O2- induced death and could evenplay a protective role in these two cell death models (Kitagawaet al., 2002; Arany et al., 2004). Even so, a relatively weakinhibitory effect of aloe emodin on ERK activation in our ex-periments indicates that the interference with this MAP kinasepathway was not a major mechanism in conferring protectionfrom TNF.
Autophagy is a process of self-cannibalization responsiblefor the removal of long-lived proteins and damaged organellesthrough a lysosomal degradation pathway, but also allowingcells to survive in the absence of nutrients and/or growth factors(Codogno and Meijer, 2005). However, recent data describingtumor cell death as a consequence of excessive autophagytriggered by some chemotherapeutic agents (Kanzawa et al.,2003; Bursch et al., 2000) indicate a possible therapeuticpotential of autophagy induction in anticancer treatment. On theother hand, inhibition of autophagy enhanced the susceptibilityof cancer cells to ionizing irradiation-or drug-triggered apopto-sis (Paglin et al., 2001; Kanzawa et al., 2004; Bauvy et al.,2001), suggesting that tumor cells in some conditions mightemploy autophagy as a mechanism to evade therapy-induceddeath. While we have previously reported the ability of aloeemodin to induce the appearance of autophagic vesicles inglioma cells (Mijatovic et al., 2005a), the role of autophagy inits anticancer action has not been investigated. In the presentstudy we demonstrate that inhibition of autophagy enhances thecytotoxic activity of aloe emodin against U251 glioma, but notL929 fibrosarcoma cells. This is consistent with different levelsof autophagy triggered by aloe emodin in these cell lines (highin U251 cells and almost negligible in L929 cells), and indicatesa protective role of autophagy in aloe emodin-induced death ofglioma cells. Importantly, autophagy inhibition diminished thealoe emodin-mediated protection from TNF-induced death inboth cell lines. These data suggest that autophagy, in addition toERK inhibition, might be involved in blockade of TNF cyto-toxicity by aloe emodin, thus reinforcing the concept of auto-phagy as a survival strategy against noxious stimuli. Having inmind the involvement of reactive oxygen in TNF cytotoxicity,

257L. Harhaji et al. / European Journal of Pharmacology 568 (2007) 248–259
such an assumption is consistent with the recently proposed roleof autophagy in cell protection from oxidative stress (Kiffinet al., 2006). However, the exact mechanisms underlying aloeemodin-mediated autophagy and its protective effect againstTNF toxicity are still to be investigated.
Finally, it should be noted that the protective effect of aloeemodin pretreatment excludes the chemical interaction of thedrug with TNF as a cause of reduced toxicity. Interestingly, theprotection exerted by pretreatment with aloe emodin, unlike theone observed upon simultaneous treatment, was markedly morepronounced in U251 glioma cells. This indicates that the cell-protective mechanisms of aloe emodin pretreatment might differfrom those responsible for the protection during simultaneousadministration with TNF. As the sensitivity to TNF cytotoxicaction could depend on the differentiation status of the cell(Kovarikova et al., 2004), the increased resistance to TNF mightbe a consequence of aloe emodin-induced differentiation ofU251 glioma to more mature glial cells with an astrocyte-likephenotype (Mijatovic et al., 2005a). On the other hand, it ispossible that L929 cells have not undergone differentiation uponthe drug treatment, or that differentiation-dependent decrease insensitivity to TNF could be a cell-type-specific feature. Thesepossibilities, as well as the relations between cell differentiationand other protective mechanisms employed by aloe emodin(autophagy and ERK activation), are currently being explored.
In conclusion, although the observed ability to antagonizeTNF cytotoxicity makes aloe emodin somewhat unlikelycandidate for enhancing the TNF-mediated tumor cell killing,our data suggest that simultaneous blockade of autophagy mightbe a plausible strategy to circumvent this problem. On the otherhand, the capacity of aloe emodin to directly interfere with thecytotoxic action of TNF might partly explain its hepatoprotec-tive effect in the TNF-dependent inflammatory liver damage.However, such an assumption remains to be confirmed byexamining the influence of aloe emodin on TNF cytotoxicityagainst normal cells, such as hepatocytes. Our data suggest thata potential value of the anthraquinone derivatives aloe emodinand emodin in a TNF-based anticancer or anti-inflammatory/cytoprotective therapy is worthy of further investigation.
Acknowledgements
This work was supported by the funds of The Ministry ofScience and Environmental Protection of the Republic of Serbia(Grant no. 143029B, 145066B). The authors are grateful toProf. Zorica Ramic (Institute of Microbiology and Immunology,School of Medicine, Belgrade) for the helpful discussion.
References
Acevedo-Duncan, M., Russell, C., Patel, S., Patel, R., 2004. Aloe emodinmodulates PKC isozymes, inhibits proliferation, and induces apoptosis in U-373MG glioma cells. Int. Immunopharmacol. 4, 1775–1784.
Arany, I., Megyesi, J.K., Kaneto, H., Tanaka, S., Safirstein, R.L., 2004.Activation of ERK or inhibition of JNK ameliorates H2O2 cytotoxicity inmouse renal proximal tubule cells. Kidney Int. 65, 1231–1239.
Arosio, B., Gagliano, N., Fusaro, L.M., Parmeggiani, L., Tagliabue, J., Galetti,P., De Castri, D., Moscheni, C., Annoni, G., 2000. Aloe emodin quinone
pretreatment reduces acute liver injury induced by carbon tetrachloride.Pharmacol. Toxicol. 87, 229–233.
Bauvy, C., Gane, P., Arico, S., Codogno, P., Ogier-Denis, E., 2001. Autophagydelays sulindac sulfide-induced apoptosis in the human intestinal coloncancer cell line HT-29. Exp. Cell Res. 268, 139–149.
Beyaert, R., Fiers, W., 1994. Molecular mechanisms of tumor necrosis factor-induced cytotoxicity. What we do understand and what we do not. FEBSLett. 340, 9–16.
Blommaart, E.F.C., Krause, U., Schellens, J.P.M., Vreeling-Sindelarova, H.,Meijer, A.J., 1997. The phosphatidylinositol 3-kinase inhibitors wortmanninand LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem.243, 240–246.
Brune, B., 2003. Nitric oxide: NO apoptosis or turning it ON? Cell Death Differ.10, 864–869.
Bulfone-Paus, S., Bulanova, E., Pohl, T., Budagian, V., Durkop, H., Ruckert, R.,Kunzendorf, U., Paus, R., Krause, H., 1999. Death deflected: IL-15 inhibitsTNF-α-mediated apoptosis in fibroblasts byTRAF2 recruitment to the IL-15Rαchain. FASEB J. 13, 1575–1585.
Bursch, W., Hochegger, K., Torok, L., Marian, B., Ellinger, A., Hermann, R.S.,2000. Autophagic and apoptotic types of programmed cell death exhibitdifferent fates of cytoskeletal filaments. J. Cell Sci. 113, 1189–1198.
Cha, T.L., Qiu, L., Chen, C.T., Wen, Y., Hung, M.C., 2005. Emodin down-regulates androgen receptor and inhibits prostate cancer cell growth. CancerRes. 65, 2287–2295.
Chang, N.S., 1997. Hyaluronidase enhancement of TNF-mediated cell death isreversed by TGF-β1. Am. J. Physiol. 273, C1987–C1994.
Chang, L.C., Sheu, H.M., Huang, Y.S., Tsai, T.R., Kuo, K.W., 1999. A novelfunction of emodin: enhancement of the nucleotide excision repair of UV-and cisplatin-induced DNA damage in human cells. Biochem. Pharmacol.58, 49–57.
Chou, T.C., Talalay, P., 1984. Quantitative analysis of dose–effect relationships:the combined effects of multiple drugs or enzyme inhibitors. Adv. EnzymeRegul. 22, 27–55.
Codogno, P., Meijer, A.J., 2005. Autophagy and signaling: their role in cellsurvival and cell death. Cell Death Differ. 12 (Suppl 2), 1509–1518.
Denecker, G., Vercammen, D., Declercq, W., Vandenabeele, P., 2001. Apoptoticand necrotic cell death induced by death domain receptors. Cell. Mol. LifeSci. 58, 356–370.
Duvoix, A., Delhalle, S., Blasius, R., Schnekenburger, M., Morceau, F.,Fougere, M., Henry, E., Galteau, M.M., Dicato, M., Diederich, M., 2004.Effect of chemopreventive agents on glutathione S-transferase P1-1 geneexpression mechanisms via activating protein 1 and nuclear factor-κBinhibition. Biochem. Pharmacol. 68, 1101–1111.
Fady, C., Gardner, A., Jacoby, F., Briskin, K., Tu, Y., Schmid, I., Lichtenstein,A., 1995. Atypical apoptotic cell death induced in L929 targets by exposureto tumor necrosis factor. J. Interferon Cytokine Res. 15, 71–80.
Ghavami, S., Hashemi, M., Kadkhoda, K., Alavian, S.M., Bay, G.H., Los, M.,2005. Apoptosis in liver diseases — detection and therapeutic applications.Med. Sci. Monit. 11, RA337–RA3345.
Goossens, V., De Vos, K., Vercammen, D., Steemans, M., Vancompernolle, K.,Fiers, W., Vandenabeele, P., Grooten, J., 1999. Redox regulation of TNFsignaling. Biofactors 10, 145–156.
Hamaguchi, E., Takamura, T., Shimizu, A., Nagai, Y., 2003. Tumor necrosisfactor-α and troglitazone regulate plasminogen activator inhibitor type 1production through extracellular signal-regulated kinase-and nuclear factor-κB-dependent pathways in cultured human umbilical vein endothelial cells.J. Pharmacol. Exp. Ther. 307, 987–994.
Huang, Q., Shen, H.M., Ong, C.N., 2004. Inhibitory effect of emodin on tumorinvasion through suppression of activator protein-1 and nuclear factor-κB.Biochem. Pharmacol. 68, 361–371.
Huang, Q., Shen, H.M., Ong, C.N., 2005. Emodin inhibits tumor cell migrationthrough suppression of the phosphatidylinositol 3-kinase-Cdc42/Rac1pathway. Cell. Mol. Life Sci. 62, 1167–1175.
Huang, Q., Shen, H.M., Shui, G., Wenk, M.R., Ong, C.N., 2006. Emodininhibits tumor cell adhesion through disruption of the membrane lipid Raft-associated integrin signaling pathway. Cancer Res. 66, 5807–5815.
Humphreys, D.T., Wilson, M.R., 1999. Modes of L929 cell death induced byTNF-α and other cytotoxic agents. Cytokine 11, 773–782.

258 L. Harhaji et al. / European Journal of Pharmacology 568 (2007) 248–259
Jia, L., Dourmashkin, R.R., Allen, P.D., Gray, A.B., Newland, A.C., Kelsey, S.M.,1997. Inhibition of autophagy abrogates tumour necrosis factor alpha inducedapoptosis in human T-lymphoblastic leukaemic cells. Br. J. Haematol. 98,673–685.
Kanzawa, T., Kondo, Y., Ito, H., Kondo, S., Germano, I., 2003. Induction ofautophagic cell death in malignant glioma cells by arsenic trioxide. CancerRes. 63, 2103–2108.
Kanzawa, T., Germano, I.M., Komata, T., Ito, H., Kondo, Y., Kondo, S., 2004.Role of autophagy in temozolomide-induced cytotoxicity for malignantglioma cells. Cell Death Differ. 11, 448–457.
Kiffin, R., Bandyopadhyay, U., Cuervo, A.M., 2006. Oxidative stress andautophagy. Antioxid. Redox Signal. 8, 152–162.
Kirstein, M., Fiers, W., Baglioni, C., 1986. Growth inhibition and cytotoxicity oftumor necrosis factor in L929 cells is enhanced by high cell density andinhibition of mRNA synthesis. J. Immunol. 137, 2277–2280.
Kitagawa,D., Tanemura, S., Ohata, S., Shimizu, N., Seo, J.,Nishitai, G.,Watanabe,T., Nakagawa,K., Kishimoto,H.,Wada, T., Tezuka, T., Yamamoto, T.,Nishina,H., Katada, T., 2002. Activation of extracellular signal-regulated kinase byultraviolet is mediated through Src-dependent epidermal growth factor receptorphosphorylation. Its implication in an anti-apoptotic function. J. Biol. Chem.277, 366–371.
Kovarikova, M., Hofmanova, J., Soucek, K., Kozubik, A., 2004. The effects ofTNF-alpha and inhibitors of arachidonic acid metabolism on human colonHT-29 cells depend on differentiation status. Differentiation 72, 23–31.
Kuo, Y.C., Tsai, W.J., Meng, H.C., Chen, W.P., Yang, L.Y., Lin, C.Y., 2001.Immune reponses in human mesangial cells regulated by emodin fromPolygonum hypoleucum Ohwi. Life Sci. 68, 1271–1286.
Kuo, P.L., Lin, T.C., Lin, C.C., 2002. The antiproliferative activity of aloeemodin is through p53-dependent and p21-dependent apoptotic pathway inhuman hepatoma cell lines. Life Sci. 71, 1879–1892.
Lee, H.Z., Hsu, S.L., Liu, M.C., Wu, C.H., 2001. Effects and mechanisms ofaloe emodin on cell death in human lung squamous cell carcinoma. Eur. J.Pharmacol. 431, 287–295.
Leist, M., Gantner, F., Bohlinger, I., Germann, P.G., Tiegs, G., Wendel, A.,1994. Murine hepatocyte apoptosis induced in vitro and in vivo by TNF-αrequires transcriptional arrest. J. Immunol. 153, 1778–1788.
Lemasters, J.J., 1999. Necrapoptosis and the mitochondrial permeability transi-tion: shared pathways to necrosis and apoptosis. Am. J. Physiol. 276, G1–G6.
Lin, C.C., Chang, C.H., Yang, J.J., Namba, T., Hattori, M., 1996. Hepatopro-tective effects of emodin from Ventilago leiocarpa. J. Ethnopharmacol. 52,107–111.
Liu, Y., Tergaonkar, V., Krishna, S., Androphy, E.J., 1999. Humanpapillomavirus type 16 E6-enhanced susceptibility of L929 cells to tumornecrosis factor-α correlates with increased accumulation of reactive oxygenspecies. J. Biol. Chem. 274, 24819–24827.
Los, M., Mozoluk, M., Ferrari, D., Stepczynska, A., Stroh, C., Renz, A., Herceg,Z., Wang, Z.Q., Schulze-Osthoff, K., 2002. Activation and caspase-mediatedinhibition of PARP: a molecular switch between fibroblast necrosis andapoptosis in death receptor signaling. Mol. Biol. Cell 13, 978–988.
Lucas, R., Kresse, M., Latta, M., Wendel, A., 2005. Tumor necrosis factor: how tomake a killermolecule tumor-specific? Curr. Cancer DrugTargets. 5, 381–392.
Luschen, S., Scherer, G., Ussat, S., Ungefroren, H., Adam-Klages, S., 2004.Inhibition of p38 mitogen-activated protein kinase reduces TNF-inducedactivation of NF-κB, elicits caspase activity, and enhances cytotoxicity. Exp.Cell Res. 293, 196–206.
Luster, M.I., Simeonova, P.P., Gallucci, R.M., Bruccoleri, A., Blazka, M.E.,Yucesoy, B., Matheson, J.M., 2000. The role of tumor necrosis factor alphain chemical-induced hepatotoxicity. Ann. N. Y. Acad. Sci. 919, 214–220.
Mijatovic, S., Maksimovic-Ivanic, D., Radovic, J., Popadic, D., Momcilovic,M., Harhaji, L., Miljkovic, D., Trajkovic, V., 2004. Aloe emodin preventscytokine-induced tumor cell death: the inhibition of auto-toxic nitric oxiderelease as a potential mechanism. Cell. Mol. Life Sci. 61, 1805–1815.
Mijatovic, S., Maksimovic-Ivanic, D., Radovic, J., Miljkovic, D., Harhaji, L.,Vuckovic, O., Stosic-Grujicic, S., Mostarica Stojkovic, M., Trajkovic, V.,2005a. Anti-glioma action of aloe emodin: the role of ERK inhibition. Cell.Mol. Life Sci. 62, 589–598.
Mijatovic, S., Maksimovic-Ivanic, D., Radovic, J., Miljkovic, D., Kaludjerovic,G.N., Sabo, T.J., Trajkovic, V., 2005b. Aloe emodin decreases the ERK-
dependent anticancer activity of cisplatin. Cell. Mol. Life Sci. 62,1275–1282.
Nakamoto, T., Inagawa, H., Takagi, K., Soma, G., 2000. A new method ofantitumor therapy with a high dose of TNF perfusion for unresectable livertumors. Anticancer Res. 20, 4087–4096.
Paglin, S., Hollister, T., Delohery, T., Hackett, N., McMahill, M., Sphicas, E.,Domingo, D., Yahalom, J., 2001. A novel response of cancer cells toradiation involves autophagy and formation of acidic vesicles. Cancer Res.61, 439–444.
Pecere, T., Gazzola, M.V., Mucignat, C., Parolin, C., Vecchia, F.D., Cavaggioni,A., Basso, G., Diaspro, A., Salvato, B., Carli, M., Palu, G., 2000. Aloeemodin is a new type of anticancer agent with selective activity againstneuroectodermal tumors. Cancer Res. 60, 2800–2804.
Pecere, T., Sarinella, F., Salata, C., Gatto, B., Bet, A., Dalla Vecchia, F., Diaspro,A., Carli, M., Palumbo, M., Palu, G., 2003. Involvement of p53 in specificanti-neuroectodermal tumor activity of aloe emodin. Int. J. Cancer 106,836–847.
Piret, J.P., Arnould, T., Fuks, B., Chatelain, P., Remacle, J., Michiels, C., 2004.Caspase activation precedes PTP opening in TNF-α-induced apoptosis inL929 cells. Mitochondrion 3, 261–278.
Shepard, H.M., Lewis, G.D., 1988. Resistance of tumor cells to tumor necrosisfactor. J. Clin. Immunol. 8, 333–341.
Spriggs, D.R., Sherman, M.L., Michie, H., Arthur, K.A., Imamura, K., Wilmore,D., Frei III, E., Kufe, D.W., 1988. Recombinant human tumor necrosis factoradministered as a 24-hour intravenous infusion. A phase I and pharmaco-logic study. J. Natl. Cancer Inst. 80, 1039–1044.
Strelow, A., Bernardo, K., Adam-Klages, S., Linke, T., Sandhoff, K., Kronke,M., Adam, D., 2000. Overexpression of acid ceramidase protects from tumornecrosis factor-induced cell death. J. Exp. Med. 192, 601–612.
Su, Y.T., Chang, H.L., Shyue, S.K., Hsu, S.L., 2005. Emodin induces apoptosis inhuman lung adenocarcinoma cells through a reactive oxygen species-dependentmitochondrial signaling pathway. Biochem. Pharmacol. 70, 229–241.
Trent, J.C., McConkey, D.J., Loughlin, S.M., Harbison, M.T., Fernandez, A.,Ananthaswamy, H.N., 1996. Ras signaling in tumor necrosis factor-inducedapoptosis. EMBO J. 15, 4497–4505.
Vandenabeele, P., Declerq, W., Beyaert, R., Fiers, W., 1995. Two tumor necrosisfactor receptors: structure and function. Trends Cell Biol. 5, 392–399.
Vassalli, P., 1992. The pathophysiology of tumor necrosis factor. Annu. Rev.Immunol. 10, 411–452.
Vath, P., Wamer, W.G., Falvey, D.E., 2002. Photochemistry and phototoxicity ofaloe emodin. Photochem. Photobiol. 75, 346–352.
Vercammen, D., Brouckaert, G., Denecker, G., Van de Craen, M., Declercq, W.,Fiers, W., Vandenabeele, P., 1998. Dual signaling of the Fas receptor:initiation of both apoptotic and necrotic cell death pathways. J. Exp. Med.188, 919–930.
Vrablic, A.S., Albright, C.D., Craciunescu, C.N., Salganik, R.I., Zeisel, S.H.,2001. Altered mitochondrial function and overgeneration of reactive oxygenspecies precede the induction of apoptosis by 1-O-octadecyl-2-methyl-rac-glycero-3-phosphocholine in p53-defective hepatocytes. FASEB J. 15,1739–1744.
Wamer, W.G., Vath, P., Falvey, D.E., 2003. In vitro studies on the photo-biological properties of aloe emodin and aloin A. Free Radic. Biol. Med. 34,233–242.
Wang, X., Li, N., Liu, B., Sun, H., Chen, T., Li, H., Qiu, J., Zhang, L., Wan, T.,Cao, X., 2004. A novel human phosphatidylethanolamine-binding proteinresists tumor necrosis factor-α-induced apoptosis by inhibiting mitogen-activated protein kinase pathway activation and phosphatidylethanolamineexternalization. J. Biol. Chem. 279, 45855–45864.
Yamamoto, A., Tagawa, Y., Yoshimori, T., Moriyama, Y., Masaki, R., Tashiro,Y., 1998. Bafilomycin A1 prevents maturation of autophagic vacuoles byinhibiting fusion between autophagosomes and lysosomes in rat hepatomacell line, H-4-II-E cells. Cell Struct. Funct. 23, 33–42.
Yeh, F.T., Wu, C.H., Lee, H.Z., 2003. Signalling pathway for aloe emodin-induced apoptosis in human H460 lung nonsmall carcinoma cell. Int. J.Cancer 106, 26–33.
Yi, J., Yang, J., He, R., Gao, F., Sang, H., Tang, X., Ye, R.D., 2004. Emodinenhances arsenic trioxide-induced apoptosis via generation of reactive oxygenspecies and inhibition of survival signaling. Cancer Res. 64, 108–116.

259L. Harhaji et al. / European Journal of Pharmacology 568 (2007) 248–259
Yim, T.K., Wu, W.K., Mak, D.H., Ko, K.M., 1998. Myocardial protective effectof an anthraquinone-containing extract of Polygonum multiflorum ex vivo.Planta Med. 64, 607–611.
Zhang, L., Hung, M.C., 1996. Sensitization of HER-2/neu-overexpressing non-small cell lung cancer cells to chemotherapeutic drugs by tyrosine kinaseinhibitor emodin. Oncogene 12, 571–576.
Zhang, L., Chang, C.J., Bacus, S.S., Hung, M.C., 1995. Suppressedtransformation and induced differentiation of HER-2/neu-overexpressingbreast cancer cells by emodin. Cancer Res. 55, 3890–3896.
Zhang, L., Lau, Y.K., Xia, W., Hortobagyi, G.N., Hung, M.C., 1999. Tyrosinekinase inhibitor emodin suppresses growth of HER-2/neu-overexpressingbreast cancer cells in athymic mice and sensitizes these cells to the inhibitoryeffect of paclitaxel. Clin. Cancer Res. 5, 343–353.
Zhang, X.P., Li, Z.F., Liu, X.G., Wu, Y.T., Wang, J.X., Wang, K.M., Zhou, Y.F.,2002. Effects of emodin and baicalein on rats with severe acute pancreatitis.World J. Gastroenterol. 11, 2095–2100.




















