































































Allen S Commercial Organic Analysis - Forgotten Books
531
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of Allen S Commercial Organic Analysis - Forgotten Books


ALLEN’
S
C OMMERC IAL ORGANIC ANALYS IS
FOURTH EDITION
IN many respects this edition '
of Allen will be a new work . Thefield of Commercial
‘
Organic Analysi s has been so enl arged and
special ised during the last few years that it was found necessary torewrite many parts and add much new matter . Obsolete methodsare omitted ; what little of the old text remains has been carefully revised and many new illustrations added .
To accomplish the obj ect in view,namely
,the furni sh ing of a
modern work of the greatest practical value to the analyst , i t wasdeemed advisable to secure the services of an English and an American editor and to organise a corps of writers particularly versed inthe subj ects discussed . Aside from those who have written for thisvolume , the following gentlemen have promised to write or revisearticles for the forthcoming ones J . A . Gardner, Chemical Department
,S t . George ’s Hospital
,London ; A . Marshall
,Chief Inspector
of E xplosives ; Indian Army ; A . H . Gill, Massachusetts Institute ofTechnology
,Boston ; F . C . Garrett
,Armstrong College
,Newcastle
ou—Tyne ; Samuel S . Sadtler, Ph iladelphia ; C . A . M i tchell , London ;Leonard Archbutt , Derby ; C . A . Klein
,London ; Will iam Robertson
,
London ; W. P . Dreaper , author of “ The Chemistry and Physics ofDyeing ” ; Cecil Revis , London ; Edward Horton ,
Chelsea ; Dr . M .
B . B lackler , Ilford ; E . W. Lewis,Laughton ; J . Merritt Matthews ,
formerly Professor of Chemistry and Dyeing,Philadelphia Textile
School ; Dr . T . M . Lowry,London ; E . J . Parry, London ; Charles H .
Lawall, Assistant in Theory and Practice of Pharmacy, PhiladelphiaCollege of Pharmacy ; J . T . Hewitt , Surbiton ; W. M . Gardner,Technical College
,Bradford ; Percy H . Walker, Washington ; A . F .
Seeker, New York .
The general arrangement of the volumes remains as before,only
such changes have been made as will bring the text into line with thelatest scientific classification . Great care has been exercised bythe editors and contributors in the choice of methods and only thoseof the highest degree of accuracy and rapidity selected . Effort hasbeen made to secure uniformity in weights and measures , nomenclature and abbreviations . References are to original sources , not totranslations or abstracts .The work will be issued in eight volumes
,numbered consecutively ,
and will be published as rapidly as possible . Volumes I and I I areready ; Volumes I I I and IV will be published very shortly ; the re
mainder will follow as quickly as is consistent with good work .
THE PUB LI SHERS .
S ee next page f or detai l announcement .

VOLUME 1 .
EDITED BY HENRY LEFFMANN AND W . A . DAVI S.Introduction ,
By W . A . DAVI S ; Alcoho ls , By G . C . JONES ; Malt and Malt
L iquors , By JULIAN L . BAKER ; Wines and Potable S pirits,By G . C . JONES ;Yeast, By EMIL SCHLICHTING ; Neutral Alcoho lic Derivatives
,By HENRY LEFFMANN ; Sugars , Starch and i ts I somers , By E . FRANKLAND ARMSTRONG ; Paper
and Paper-making Materials,By R . W . S INDALL ; Acid Derivatives of Alcoho l ,
By HENRY LEFFMANN ; Appendi x ; Index .
86 I llustrations . Octavo,x + 5 76 pages . Ready . C loth , net .
VOLUME II .
EDITED BY HENRY LEFFMANN AND W.
.
A . DAVIS.Fixed O i ls , Fats and Waxes
,By C . AINSWORTH M ITCHELL ; Specia l Char
acters and Modes of Examining Fats , O ils and Waxes,By LEONARD ‘
ARCHBUTT ;Butter Fat
,By CECIL REVI S and E . R . BOLTON ; Lard
,By C . AINSWORTH
M ITCHELL ; L inseed O i l, By C . A . KLE IN ; Hi gher Fatty Acids , By W . ROBERTSON;S oap
,By HENRY LEFFMANN ; G lycero l, By W . A . DAVIS ; Cho leste ro ls , By JOHN
ADDYMAN GARDNER ; Woo l-fat , C lo th O ils , By AUGUSTUS H. G ILL .
O ctavo,x + 5 20 pages . Ready . C loth ,
net .
VOLUME III .
EDITED BY W . A . DAVI S AND SAMUEL S . SADTLER .
Hydrocarbons , By F . C . GARRETT ; Naphthalene and i ts Deri vatives , ByW . A . DAVI S ; B itumens,By SAMUEL S . SADTLER ; Aromatic Ac ids , By EDWARD
HORTON ; Ga llic Acid and i ts Allies , By W..P . DREAPER ; Phthalic Acid and the
Phthale ins,By W. A . DAVI S ; Explosives , By A . MARSHALL .
Ready in May , 1 9 1 0 . C loth , $500.
VOLUME IV.
EDITED BY W . A . DAVI S AND SAMUEL S . SADTLER.
Resins,By M . B . BLACKLER ; Essential O i ls , By E . J . PARRY ; Hydrocarbons
and Ketones of Essential O ils , By T . M . LOWRY ; Caoutchouc and Guttapercha ,
By E . W . LEWI S ; Special Characters of Individual O i ls and T erpene less EssentialO ils
,By HENRY LEFFMANN and CHARLES H. LAWALL ; Tables of Essential O i ls ,
By the EDITORS. To be Publi shed August, 1 9 1 0 .
VOLUME V .
EDITED BY W . A . DAVI S AND SAMUEL S . SADTLER .
Tannins,Dyes and Co louring Matters
,By W . P . DREAPER ; Diph enylmethane
and Co louring Matters , By J . T . HEW ITT ; Co louring M atters of Natura l O ri gin ,
By W. M . GARDNER ; Ana lysis of Co louring Matters , By W . P . DREAPER ; Inks ,Carbon Papers, Typewriter Ribbons , e tc . ,
By PERCY H. WALKER ; Co louri ngMatters in Food
,By A . F . SEEKER .
In P reparati on .
VOLUME VI .
EDITED BY W . A . DAVI S AND SAMUEL S . SADTLER .
Am ines and Hydrazines ; Ani line and i ts Allies ; O ther Bases from Tar ;
Alkalo ids ; Vo latile Bases ; N icotine and Tobacco Products ; Aconite Bases and
Atropine ; Coca Alkalo ids ; Opium Alkaloids ; S trychno s Alka lo ids ; C inchonaAlkalo ids ; Barberine ; Caffe ine ; Cocoa and Choco late . In P reparati on .
VOLUME VI I .
ED ITED BY W . A. DAVI S AND SAMUEL S . SADTLER .
M iscellaneous Alkalo ids ; Non-Basic Vegetable B itter P rinciples ; Animal
Bases ; Animal Ac ids ; Cyanogen and i ts Derivatives .In P reparatwn .
VOLUME VI I I .EDITED BY W . A . DAVIS AND SAMUEL S . SADTLER .
P rote ids of P lants ; P rote ids of M i lk and M i lk Products ; M i lk ; Meat andMeat Products ; Prote ids of Diges tion ; Haemoglobin and i ts Allies ; P rote ids or
Albumenoids and G lue ; Ultra M icroscopy .In P reparatwn .


CONTRIBUTORS
TO VOLUME II
C . AINSWORTH M I TCHELL,B . A . F . I . C .
,London .
LEONARD ARCHBUTT, F . I . C .
,Derby
,England .
CECIL REVI S , A . c . G. London .
E . R . BOLTON, London .
C . A . KLE IN, London .
W. ROBERTSON ,A . R . c . S .
,London .
HENRY LEBEMANN, M. A .,M. D . , Philadelphia .
W. A . DAVIS , B . Sc .
,A . C . G. I .
,Bromley
,England .
JOHN ADDYMAN GARDNER , M. A .,F . I . C .
,London .
AUGUSTUS H . GI LL,PH. D .
,Boston
,U . S . A .

ALLEN’
S COMMERC IAL
ROAN C ANALYS SA TREATISE ON
PRO PERTIES , MODES OF ASSAYING , AND PROXIMATEANALYTICAL EXAMINATION OF THE VARIOUS
ORGANIC CHEMICALS AND PRODUCTSEMPLOYED IN THE ARTS , MANU
FACTURES , MEDIC INE, Etc .
WITH CONC IS E METHODS FOR
THE DETECTION AND ESTIMATION OF THEIR IMPURITIES,
ADULTERATIONS , AND PRODUCTS OF DECOMPOS ITION
VOLUME II
Fixed O ils , Fats and Waxes , S pecial Characters and Methods , ButterFat , Lard , Linseed O il , Higher Fatty Acids , S oap , Glycerol ,
C holesterols , Wool—fat , C loth O ils
BY THE EDITORS AND THE FOLLOWING CONTRIBUTORSC . AINSWORTH MITCHELL, LEONARD ARCHBUTT , C . REVIS ,
E. R. BOLTON, C . A . KLEIN, w. ROBERTSON,
JOHN ADDYMAN GARDNER, AUGUSTUS H. GILL
FOURTH EDITION. ENTIRELY REWRITTEN
EDITED BY
HENRY LEFFMANN, and W . A . DAVIS , B .Sc . ,
PROFE S S OR OF CHEM I S TRY AND TOX ICOLOGY FORMERLY LECTURER AND A S S I S TANT IN THEIN THE W
'
OMAN ’
S MED I CAL COLLEGE OF CHEM ICAL R ES EARCH LAB ORATORY . C ITYP ENN S YLV AN IA AND IN THE W AGNER AND GU ILDS COLLEG E , IMP ER IAL
FREE IN S T ITUTE OF S C I ENCE , COLLEG E OF S C I ENC E ANDPH ILADELP H IA TECHNOLOGY , LONDON
PH ILAD ELPH IAP . BLAKISTON
’
S S ON C O .
1 01 2 W ALNUT S TREET
1 9 1 0


REFAC E
of the articles .otherwise noted
,temperatures are centigrade .
me,the same methods have
o i the first volume of this edition . Thesubject matter was included in Vol . 2
,
of “ Commercial Organic Analysis ,” butexplosives
,partly included in that volume
same edition,Will appear in a later volume .
have been revised by those especiallytext has been completely rewritten
,and a
has been added .
for the care and atten


C O NTE NTS .
FIXED O ILS , FATS AND WAXES .
C . AINSWORTH MITCHELL.
General Properties and Analytical Methods , 1 ; Classification, 64 ;Analytical Data
,69 ; General Examination of Fats and O ils , 74 ;
Identification of Fats and O i ls , 84 .
SPECIAL CHARACTERS AND METHODS .
LEONARD ARCHBUTT .
Olive O il Group , 9 1 ; Rape O il Group , 1 20 ; Cottonseed Oil Group ,1 3 1 ; Linseed O il Group , 1 48 ; Castor O il Group , 1 59 ; CacaoButter Group
,1 76 ; Coconut Oil Group , 1 87 ; Lard O il Group ,
1 97 ; Tallow and Butter Group,
204 ; Whale O il Group , 2 1 3 ;
Sperm O il Group , 23 2 ; B eeswax Group , 242 .
BUTTER FAT .
CECIL REVI S AND E . R . B oLTON .
Butter Fat, 2 79 ; Butter , 302 ; Butter Substitutes , 3 1 3 .
LARD .
C . A INSWORTH M ITCHELL .
Lard, 3 1 7 .
L INSEED O IL .
C . A . KLE IN .
Linseed O il, 3 23 ; B lown O ils , 36 1 .
H IGHER FATTY AC IDS .
W. RoBERTsoN .
Synopsis of Higher Fatty Acids , 3 7 2 ; Palmitic S tearicAcid
, 398 ; Oleic Acid , 402

CONTENTS .
SOAP .
HENRY LEBEMANN.
Commercial Soaps , 4 1 7 ; Assay of S oap , 42 1 .
GLYCEROL .
W. A . DAVI S .
Properties, 447 ; Estimation, 45 7 ; Commercial Glycerol (G1)
466 .
CHOLESTEROLS .
JOHN ADDYMAN GARDNER.
Cholesterols , 479 ; Vegetable Cholesterols , 484 ; Phytosterol , 4WOOL-FAT
, CLOTH O ILS .
AUGUSTUS H . GILL.
Wool-fat , 495 ; Lanolin , 502 ; French Degras , 502 ; Sod oil ,504 ; Cloth O ils, 5 1 1 .
INDEX

ILS , FATS , ANDWAXES .
ion .
are the general properties characterising the true fats
pure,most of them are colourless or pale yellow . Impure
rcial oils vary in colour from light yellow to red,and even
nd black . Many vegetable oils have a distinct shade ofthe presence of chlorophyll
,and Show absorption spectra
,
h oils of animal origin .
taste are often peculiar, and are characteristic of
se characters become less perceptible the morepurified
,they may be due to the presence of as
ers not readily removed,rather than to the con
s tituents of the o il .3 . If dropped in a liquid condition on paper they leave a permanent grease-Spot
,unless they are crystalline and hard enough to be
4 . They are not fluorescent and,as a rule
,have but little rotatory
action on a ray of polarised light . Castor and croton oils,however
,are
MITCHELL,B . A . (Oxom,
F . I . c .
IES AND ANALYTICAL METHODS .
ils,fatty oils
,fats
,and waxes are classed
animal and vegetable structures .is generally used for such members of
quid at ordinary temperatures . Those havinga relatively large proportion of olein or otherp .
,but beyond this there is no absolu te dis
oils and fatswell-defined physical characters , and differ infrom the true fats . They are
,however
,in
related to them,and are conveniently descr ibed

FIXED OI LS,FATS , AND WAXE S .
5 . The Sp . gr . i s less than that of water,ranging between the limits
of and but if certain anomalous oils from marine animal sbe excluded , the lowest density is about at a temperature of I 5° C .
In the fluid state,at the temperature of boiling water
,the sp . grs .
range from to about The waxes and all ied substancesare still lighter in the melted condition their sp . gr . ranging fromto6 . The fusing or melting points range within Wide l imits , and areliable to modification in an obscure manner by special treatment .7 . They are practically insoluble in water
,but dissolve to some
extent in absolute alcohol or strong spirit,especially when hot
,and are
readily soluble in ether,chloroform
,carbon tetrachloride
,carbon
disulphide , benzene , petroleum Spirit,turpentine
,and other volat ile
solvents . They are readily m iscible with one another .8 . The fixed oils and fats are composed of carbon
,hydrogen
,and
oxygen,the nitrogen
,sulphur
,phosphorus
,and iron present in many of
them being due to foreign matters,which often cannot be completely
removed .
9 . They do not emit inflammable vapours at the ordinary temperature
,but may be burnt by means of a wick . They are not capable of
being d i stilled -at the ordinary atmospheric pressure without decomposition . When heated alone they darken and evolve acrid off ensivevapours ; and when further heated to about 3 1 5° carbon dioxide isevolved
,together with the peculiarly irritating vapours of acrolein ,
C3H
4O
,various volatile organ ic acids
,and gaseous
,l iquid
,and solid
hydrocarbons . The temperature at which this decomposition occurshas been improperly called the “ boiling point ” of the oil , thephenomenon of apparent ebullition being really due to the escape of thegases formed by the decomposition .
1 0 . On distillation with superheated steam ,they undergo a simpler
decomposition,with formation of glycerol and fatty acids . This
change may also be effected by acting on them with sulphuric acidor a strong base . The action is known as “
saponification,
” orhydrolysis and its analytical application is discussed in anothersection .
1 1 . If air is excluded,the fixed oils may be preserved unchanged
for a lengthened period,but
,on exposure to air , many of them thicken
owing to absorption of oxygen,and are ultimately converted ( if ex
posed in sufficiently th in layers) into a yellowish transparent Skin or

EXTRACTION AND PURIFICATION . 3
g .,linseed
,walnut
,hempseed
,and poppy-seed
oi ls behave in a different manner on exposuree rancid; that is , lose the ir colour (and toand acqu ire an acrid , disagreeable taste ,
This alteration is primarily anhe action of air and l ight , and i sfatty acids and other bodies . I t
of foreign matters,such as the
plant from which the oil was exnourishment for bacteria
,which
11 once the decomposition processating such rancid oil with hot water
,and subse
th a cold and dilute solution of sodium carbonate,
mposition may often be removed and the fat reoriginal state .
CTION AND PURIFICATION OF FIXED OILS ANDFATS.
ethod of extraction and subsequent treatment have coninfluence upon the analytical characteristics of the product .
f oils and fats from animal ti ssues it is often suffi cient(e . g .
,cod l iver) to become somewhat putrid ,
when some of the oil drains from i t,or may be obtained by slight
pressure . A further quantity can be extracted by warming or boilingthe tissue with water
,as is done with blubber . In the case of lard and
tallow , it is merely necessary to heat the substance alone , and strainthe melted fat away from the membranous matter . From compacttissue , such as bone , the whole of the fat can be extracted by a solvent
The extraction of the fat or oil from vegetable tissue may be eff ectedby boiling the crushed substance with water or by subjecting it topowerful pressure
,either at the ordinary temperature or between plates
heated to slightly above the m . p . of the fat The product obtained inthe last manner will usually contain more stearin ” or solid fat thanthe “ cold—drawn ” oil . In either case a certain quantity of the fat is
0
1 Under certa in condit ions , a s wh en co t ton -wast e , sh oddy , o r hemp i s moistened withand ex o sed to th e a ir , th e oxidat ion of th e o i l become s so energe t i c a s t o lead t o con
sxderable e evat ion of t empera ture , and even ac tua l inflammat ion (see p . 3

FIXED OI LS,FATS
,AND WAXE S .
mechanically retained by the tissues,and hence a larger yield can be
obtained by the use of carbon disulphide or petroleum spirit,which
,
on being distilled off,leaves the fat behind .
The proportion of oil or fat yielded by any particular material depends on many conditions .Tables of the yields usually obtained from different seeds
,nuts
,etc .
are given in Schaedler ’s Untersuchungen derFette , Oele und Wacli sarten,
1 89 2 , p . 2 5 , and in Wright and M itchell ’sOi ls
,Fats and Waxes
,1 903 , 297 .
O ils obtained by the use of solvents aremore likely to contain impurities than thoseobtained by pressure .Est imat i on of Oi ls and Fa ts .
—_
In thelaboratory
,the estimation of the oil in
solid animal and vegetable matters iseffected by treating the finely divided and
previously dried substance I with a suitablesolvent under such conditions as to ensurecomplete extraction . Carbon disulphide orpetroleum Spiri t may be employed for thepurpose
,but ether or carbon tetrachloride
is,as a rule
,preferable .
The exhausti on of seeds , bones , shoddy,oil-cakes
,milk residues
,etc .
,by simply
digesting the substance with the solvent atthe ordinary temperature
,with frequent
agitation ,in a closed flask
,i s unsatisfactory
,as it requires a consider
able quantity of the solvent,of which a notable proportion is likely to
be lost . The apparatus devised by Szombathy (see Vol . 1,p . 7 7)
obviates these drawbacks . The substance to be exhausted of oil isenclosed in a plaited filter or cylinder of filter-paper ; or if i t becoarse
,i t is suffi cient to place i t loose in a large test—tube having an
aperture at the bottom closed by a plug of glass-wool .A very Simple and convenient form of exhauster , adapted either forextraction or re-percolation
,has been described by Dunstan and Short
(Pharm . J .
,1 882
,I 3 ,
A form of exhau ster (Fig . I ) , suitable for the extraction of very
F IG . 1 .
1 In th e case of lin se ed and o th er substance s con tain ing dry ing o ils , th e de sicca t ion mu st
e ith e r b e om it t ed or conduc t ed in an a tmo sph ere of h y drogen or I llum i nat i ng gas .

EXTRACTION AND PURIFICATION . 5
ties of material,was devised by West-Knights (Analyst,
I t has the advantage of being readily constructed in theA percolator is made by cutting ofi‘ the bottom from asuitable Size
,and blowing a hole or two (A A) in the Side
about an inch from the top . A disc of filter-paper or fineis tied over the lower end of the tube . The substance tois placed in the tube
,and kept in
some glass-wool or a perforatedand the tube with its contents
the lower end of the tube(C) . This is fitted by a
larger cork (D) to the neck of an ordinary flask«containing the volatile solvent . On heatingthe flask the vaporised solvent passes throughthe holes in the Side of the test—tube up into thetube of the condenser
,where it is liquefied .
The condensed liquid drops back into the testtube
,percolates through the substance to be
extracted,and falls to the bottom of the flask
,
to be again volatilised . As the percolator isinside the flask
,its contents are kept constantly
at the b . p . of the solvent,and
,the action being
continuous and automatic,very rapid exhaus
tion may be effected .
Other forms of exhauster have been con
trived by Church,Drechsel
,Angell
,Thorns
,
Thresh (P /i arm.
‘
J .,
1 884 , 1 5 ,
Fr i'
i hling (Zei t. angew. Cli ent,
1 889 ,
(See also Vol . FIG ' 2 ‘
To recover the oil from its solution in the ether or other liqu id em
ployed,the solvent Should be distilled off at a steam-heat
,and the last
traces of it removed by placing the flask on its side and heating it in thewater-oven until constant in weight . In some cases the complete removal of the solvent is best effected by blowing a gentle stream of air ,previously filtered through cotton-wool
,through the flask while it is
maintained at a temperature ofLarge quantities of niaterial may be readily extracted in the ap
paratus (Fig . which is constructed on the principle of the Szombathy extractor .

FIXED OILS , FATS , AND WAXES .
In the case of liquids containing oil in the form of emulsion, a separation may often be effected by agitation with ether . For the extractionof unsaponifiable matter FOrster has devised an apparatus which isfigured and described in Vol . 1 , page 82 .
Pur ifi cat ion Of Oi ls .-The methods used in the refining and puri
fication of crude oils have often considerable influence upon the analytical characteristics of the final products .Acti on of Heat—S imple application of heat may effect coagulationof protein impurities in an oil .Mechani cal Attracti on and Fi ltrati on—Substances such as Spanishclay
,full er ’s earth
,and the like are used as mechanical precipitants of
the suspended matter in oils . The clarified Oil,which is not chemi
cally altered by this treatment,is subsequently decanted or passed
through a filter.Treatment wi th Aci ds .
—Rape,l inseed
,and some fish o ils are fre
quently refined by treatment with a small proportion of sulphuric acid,
which chars the impurities and causes them to subside w ithoutmaterially attacking the oil i tself . The objection to the process is thattraces of free mineral acid may remain
,even after the subsequent
washing with water,and
,if the oil is used as -a lubricant, may lead to
corrosion of bearings,etc .
,or to charring of the wick in the case of
lamp oils . Treatment with sulphuric or hydrochloric acid is also employed in the removal of the lime which is present in bone fat .Treatment wi th Alkali es .
—Certain oils , notably cottonseed , olive ,and sperm oils
,are frequently purified by treatment with a solution of
caustic soda,the quantity of which depends upon the amount of free
fatty acids and impurities to be removed . Cottonseed oil contains anotable proportion of a resin-l ike substance which gives a blue coloration with the alkali . Ammonia
,sodium carbonate
,magnesium car
bonate,milk of lime
,and sodium peroxide are al so employed in certain
refining processes . O ils , which have been treated with alkali usuallycontain a much smaller amount of free fatty acids than even thefreshly—expressed crude oils
,and cottonseed oil used for cooking pur
poses is often practically neutral .Treatment wi th Oxidi sing Agents .
—Fish oils are purified , and tosome extent deodourised
,by treatment with a current of steam followed
by a current of hot air . Excessive treatment of this kind will alterthe character of the oil i tself
,so that it becomes heavier and more
viscous, and acquires other characteristics of “ oxidised ” or “ blown ”

FIXED OILS,FATS
,AND WAXE S .
C3HS occurs in glycerol , i t i s generally called glyceryl or glycyl , and
the esters are usually called glycerides .
The fatty acidsmost commonl y forming estersWith the glyceryl radiclein natural fats and oils are those belonging to the series with the generalformulae , Cn
H, nO 2 (acetic or stearic acid series) ; Cn
H (Oleicacid series) ; Cn
H2n (linolic acid series) ; Cn
H2n
' (l inolen i cacid series), and Cn
H (ricinoleic or hydroxyacrylic acid series).Glyceryl stearate , C
3H5 (C 1 8H3 SO , ) 3 C 5 7HH OO é , is known as
tristearin,or stearin ; i t is probably the chief constituent of beef and
mutton tallow . In l ike manner olein is probably the principal component of almond
,Olive
,and lard oils
,and palmitin
,
of palm oil .Esters of linolic acid are main constituents of cottonseed and maizeoils
,While the esters of linolenic and isolinolenic acid form an impor
tant part of linseed oil,and that of ric inoleic acid of castor oil .
Olein ,l inolein
, and l inolenin , being liquid , predominate in oils , whilestearin and palmitin are more abundant in solid fats .The view formerly held that the natural esters rarely contain morethan one acid radicle requires modification
,Since it has been shown
that mixed glycerides , in which the acid radicles are not all of the samekind
,are present in numerous fats . Thus Heise (Arbei t a . d. Kai serl .
Gesundhei tsamt, 1 896 , 540) and subsequently Henriques andKimne (Ben ,
1 899 , 3 2 , 387) isolated oleo-distearin from the fat of theseeds of the East African tallow tree (S tearodendron S tuhllmanni ); andthe bromides of mixed glycerides were separated by Hebner andM itchell(Analyst, 1 898 , 23 , 3 1 7) from l inseed oil , walnut oil , and marine animalOils . The separation and behaviour of these bromides is a valuabletest for distinguishing between diff erent classes of oils
,as is Shown in a
subsequent section .
The waxes proper contain the esters of higher alcohols of the methylseries. Thus spermaceti consists chiefly of cetyl palmitate , C1 6H3 3
C 1 6H3 IO Z,whilst Chinese wax
,beeswax
,and carna iiba wax contain
still h igher radicles . Sperm oil and bottlenose oil are chieflycomposed of substances having a constitution similar to that ofthe waxes .In addition to the esters which constitute the essential portions ,
most natural fats,oils
, and waxes contain more or less of free fattyacids
,and small proportions of colouring
,odorous , resinous , and other
matters,to which the characteristic colours , smells , and tastes are
mostly due . Small proportions of cholesterols or phytosterols are also

ACID VALUE .
n affords a means of distinguishing betweenand of vegetable origin .
ac ids in natural fats and oils are usually products ofaccelerated by the presence of mucilaginous or proOrdinary butter , which contains casein ,
readily turnshen contains free butyric acid ; but if all casein and waterby melting and filtering the butter
,the butter-fat may
anged for a much longer time . Over-treatment with sulprocess of refining oils often resultS ' in the formation
fatty acids . Commerc ial oils which have been refined by thisare apt to retain traces of free mineral acid .
Value .—The proportion of free fatty ac ids is best ascer
by shaking a weighed quantity of the fat with warm alcoholand t itrating the solution with a standard alkali solution
,with phenol
p hthalein as indicator .An accurately weighed quantity of the sample
,ranging from 5
O
grm .
of fatty acid to 50 grm . o f an ordinary oil,is introduced into a flask or
bottle furnished with a glass stopper,and from 50 to 1 00 c .c . of pure
neutralised al cohol containing a little phenolphthalein in solution isadded and raised to the boiling—point by immersing the bottle in hotwater . ‘ The contents are thoroughly agitated to efi ect as complete asolution of the fatty acids as possible . I f the sample of oil is whollyfree from acid
,the pink colour of the alcohol will remain unchanged
,but
otherwise i t will disappear . In the latter case,a N/2 solution of
sodium hydroxide is added in small amounts to the warm contents ofthe flask
,which is shaken thoroughly after each addition until the
pink colouration persists . The reaction is as well defined and
the neutralisation point as easy to perceive as in the titration ofmineral acids ; but owing to the very high combining weights of thefatty acids
,great care is necessary . Thus 1 c .c . of N 2 alkali used
corresponds to of palmi ti c,
of steari c,or grm . of
olei c aci d. For determining small proportions of free acid,i t is de
sirable to employ decinormal alkali,while in the case of samples con
taining much free acid the quantity taken for the assay Should be correSpondingly reduced . The result is usually expressed in terms of thenumber of mg . of potassium hydroxide neutralised by 1 grm . of thefat , and i s termed the Acid Value .
If the mean equ ivalent weight of the free fatty acids be known,their
percentage may readily be calculated from the acid value . For this

I O FIXED OILS,FATS
,AND WAXES .
purpose it is often assumed that the free fatty acids in oils consist solelyof oleic acid , and Since 282 parts of oleic acid are equivalent toparts of potassium hydroxide
,the percentage of free fatty acids
(expressed as oleic acid) i s Obtained by multiplying the acid value bythe factorThe amount of free fatty acids in commercial oils is Often very con
siderable . Thus in palm oil the free acid,calculated as palmitic acid
,
usually ranges from 1 2 to nearly In 89 samples of olive oil intended for lubricating use
,Archbutt (Analyst, 1 884, 9 , 1 7 1 ) found
from to of free (oleic) acid , the mean being In
the superior grades of olive oil the proportion‘
Of free acid is muchsmaller. In rape oil
0the percentage of free acid is generally fromto but cottonseed oil
,which is refined by means of alkali
,i s
generally free from any trace of acid .
The influence of free acid in an oil upon its tendency to act uponmetals is considered in the section on “Lubricating O ils .”In the case of fats of a dark colour Sharper readings may be obtainedby the use of the indicator
,known as Alkali blue 6 B (red with alkalies)
in place of phenolphthalein . About 2 c .c . of a 2% al coholic solutionare added .
In determining the acid value of artificially coloured fats the dyestuffmust
,if possible
,be removed before the titration by treatment with a
suitable solven t,such as 80% alcohol or petroleum spirit , which in some
cases dissolves the fat and leaves the dyestuff (e . g .,nigrosine in
leather fats). Sometimes the dyestuff may be removed by shaking anethereal solution of the fat with dilute hydrochlori c acid
,and washing
the residual fat solution with water . Or the petroleum spirit solutionof the fat may be thoroughly shaken with a measured quantity of N 1 0
alcoholic sodium hydroxide solution,and the aqueous layer subse
quently titrated with standard hydr ochloric acid until colourless tophenolphthalein .
Sapon ifi cation Of F ixed Oi ls .—Fatty oi ls heated with water under
a pressure of 8 to 1 2 atmospheres or distilled with superheated steamare hydrolysed into fatty acids and glycerol . This method of decomposing fats is employed in the industrial production of fatty acids andglycerol .Many natural oils and fats are partially hydrolysed into fatty acidsand glycerol probably by the action of air and l ight and possiblybacterial action in presence of traces of albuminous or other foreign

SAPONIFICATION OF FIXED OILS .
The free fatty acids often present in commercial palm oil,
and tallow are due to this cause .sent in castor and other oil seeds are
of fats in the presence of diluteshown by Connste in, Hoyer, and Wartenberg
3989)curs when a fatty oil is heated to with about 8%sulphuric acid . On washing the product with hoturic acid and glycerol are removed
,and the fatty acids
ly layer .An analogous action takes place when a fat or oil is treated with
“basic oxides or hydroxides . The change occurs more readily withsome O ils than with others
,and is promoted by heat and by using
alcohol or glycerol as a solvent for the alkali . A sal t (soap) of the fattyacid is produced
,glycerol being likewise formed . The soaps pro
duced by potassium,sodium
,or ammonium hydroxide are soluble in
water,but most other soaps are i nsoluble .
Waxes yield soaps and a monatomic alcohol,instead of glycerol .
The decomposition i s usually difficul t .When an ester is split up into an acid and an alcohol , the change isusually called “ saponification,” no matter whether the agent effectingthe change is water
,an acid
,or a base . The term is even extended to
the decomposition of esters that do not yield fatty acids . I t is evident ,therefore , that the saponification of fixed oils is a definite chemicalaction , precisely analogous to the decomposition of the ordinary salts .The table on page 1 2 gives the molecular weights and proportion offatty acids and glycerol theoretically obtainable from pure triglyceridesand other esters of common occurrence .Hence it appears that the majori ty of fats and oils yield , on saponification
,from 95 to 96% of fatty acids , and about 1 0% of glycerol .
The esters of butyric,valeric
,or lauric acid contained in butter-fat
,por
poise , and coconut oils , respectively, yield a larger proportion ofglycerol
,while rape oil
,containing an ester of erucic acid
,yields a
smaller proportion .
The waxes yield much smaller proportions of fatty acids,and , in
stead of glycerol,give large proportions of alcohols of the C
nH
2n+ ,
series , as solid bodies insoluble in water. The nature and proportionof the products of saponification Sharply distinguish sperm and bottlenose oils from all other fixed oils of commercial interest .
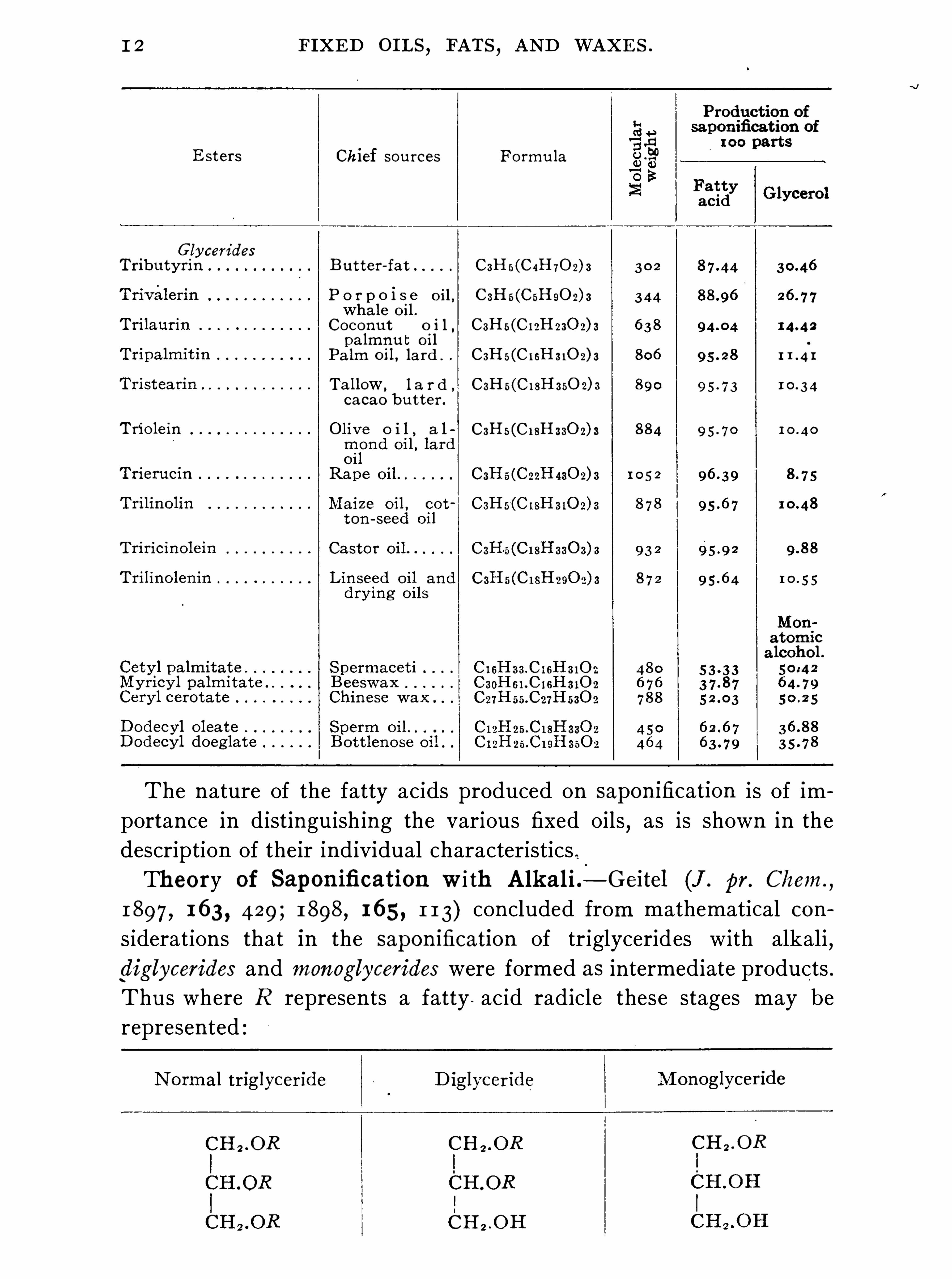
1 2 FIXED OI LS,FATS
,AND WAXE S .
E st ers
Gly ceri desTribut y ri n
0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0
O O O O O O O O O O
0 0 0 0 0 0 0 0
Chief sou rces Formu la
B ut ter-fa t C3H6 (C 4H7O2) 3
P o r p o i s e o i l , C sH5 (C5H902)3wha le o i l .
Coconu t O i I , C3H5 (C 12H2302) 3pa lmnu t o i l
Pa lm o i l , lard . C3H5 (C 1 6H3 102)3
Ta llow ,l a r d , C3H5 (C 1 8H3 502) 3
cacao bu t t er .
C3H5 (C 18H3302) 3
0 0 0 0 0 0
1 0-3 4
1 0 5 2
The nature of the fatty acids produced on saponification i s of importance in distinguish ing the various fixed oils
,as is shown in the
description of their individual characteristics .Th eory of Sapon ifi cat ion w i th Alk ali f—Ge itel (J . pr . Chem.
,
1 897 , 1 63 , 429 ; 1 898, 1 65 , 1 1 3) concluded from mathematical considerations that in the saponification of triglycerides with alkali ,diglycerides and monoglycerides were formed as intermediate products .Thu s where R represents a fatty . acid radicle these stages may berepresented .
CH2 .OR
CH..OH
CH2 .OR

ALCOHOLYS I S OF FATS . 1 3
was opposed by Henriques (Zei t. angei . Chem,1 898 ,
quently Lewkowitsch (B er .,
1 900, 3 2 , 89 ; 1 906 , 3 9 ,
Chem. Ind ,1 903 , 2 2
, 596) has brought experimentalwhilst the Opposite View is main5 , 9 1 9 ; 1 907 , 28 , 3 83 ; Annalm ,
(Ber. ,1 906 , 3 9 , The question
unsettled .
lcoholic alkali the ethyl esters of the diff erentare formed as intermediate products
,and their
tion affords a means of distinguish ing between
A lcoh o lys is Of Fats .—When glycerides are subjected to the action
ofan alcohol containing a small quantity of an acid they are decomposed in a manner analogous to the hydrolysis effected by water in thepresence of acid . A useful method of estimating the composition‘of fats has been based on this reaction by Haller (Compt. rend ,
1 906 ,‘
1 43 , 65 7) who describes the process as “ alcoholysis .”Abou t 1 00 grm . of the dried fat are heated on the water-bath with
200 grm . of,e . g .
,methyl alcohol
,to which has been added 1 or 2% of
dry hydrochloric acid,fresh additions of acidified methyl alcohol be
ing made , i f required , until the mixture appears homogeneous . I t isthen treated with a large volume of water or salt solution
,which retains
the excess of methyl alcohol and the glycerol from the fat ,While themethyl esters o f the fatty acids rise to the surface . These may then beseparated by fractional distillation and the fatty ac ids in the distillatesseparated and identified . In the case of the methyl esters of bu tyric ,capro i c
,and caprylic acids the distillation may be carried out at the
ordinary temperature but from 1 94°
(the b . p . of methyl caprylate)upward reduced pressure is necessary . The method gives good resultsup to lauric acid
,but the separated esters of myristic
,palmitic
,and
stearic acids always retain some methyl oleate . ‘The latter may beseparated by chilling the fractions with ice and draining the crystals ona porous tile with the aid of a pump .
By this method Haller and Youssoufian 1 906 , 1 7 3 ,
803 ) found coconut oil to contain caproic , caprylic , lauric , myristic ,palmitic , stearic , -and oleic ac ids ; whilst Meyer (Chem. Zei t. , 1 907 , 3 1 ,
793) found cottonseed o il to consist chiefly (up to of palmi tin ,
with the glycerides of oleic,l inolic
,and probably stearic and arachidic
acids .

1 4 FIXED OI LS,FATS , AND WAXES .
Sapon ifi cat ion in Ana lys is .-The most convenient method of
saponifying oils,etc .
,for the further examination of their constituents
i s by treatment with an alcoholic solution of potassiumhydroxide andsubsequent evaporation of the alcohol :Analcoholic solution of alkali is prepared by dissolving 80 grm . ofpotassium hydroxide in 1 000 c . c . of strong alcohol
,which has been
previously redistilled with a little alkali . I t i s desirable to dehydratethe Spirit by keeping it over a large excess of dry potassium carbonate .About 5 grm . of the clarified fat or oil are weighed in a 4-oz i widenecked flask
,treated with 2 5 to 30 c . c . of the solution of alkali in
spirit,and the flask closed with a cork fitted with a long tube . The
flask is heated over boiling water,and as soon as the Spirit boils the con
tents are mixed by circular agitation . In most cases the whole of theoil will rapidly disappear
,form ing a clear solution of soap
,which may
be further heated for a Short time with occasional agitation to ensurecomplete saponification Of the fat . The cork is then removed andthe alcohol evaporated . In the presence of unsaponifiable oil thecontents of the flask Should be allowed to boil until nearly dry
,and the
residue treated with 25 c .c . of Spirit , and again boiled down . Whenthere is no danger of loss of hydrocarbon oils or esters of lowerfatty acids by incautious treatment
,the saponification and subsequent
evaporation may be satisfactorily conducted in a hemispherical porcelain basin
,placed over a small naked flame . The mixture is well
stirred with a glass rod,and kept gently boiling until the alcohol is
nearly driven off and the residual l iquid froths strongly . By this timethe whole of the oil Should have disappeared
,but
,if incomplete saponi
fication is suspected,1 0 c .c . of alcohol may be added , and the evapora
tion repeated .
To ensure the saponification of butter fat , codliver oil , the waxes ,and other substances difficult to decompose
,i t is better to place the
sample and alcoholic solution in a strong 200 c .c . bottle , closed by anIndia-rubber stopper firmly fastened by wire . The bottle is then keptat and frequently agitated during half an hour
,or until no glob
ules of oil can be seen,after which it is opened
,and the contents
rinsed into a basin and evaporated over boiling water till the alcohol isexpelled . Special precautions for ensuring the saponification of waxesare described in the section on “
B eeswax .
Saponifi cati on Va lues of Oi ls . Koettstorfer’s P rocess—The
saponification of fatty Oils being a perfectly definite reaction , not onl y

1 6 FIXED OI LS,FATS
,AND WAXE S .
The difference between the volumes of standard acid used in the 2
estimations gives the number of c .c . corresponding to the alkalineu tralised in saponifying the oil . Each c .c . of N 2 c .c . hydrochlori cacid 5 grm . HCl per 1 000 c . c .) thus employed represents
of KOH,whence the number of mg . of potassium hydroxide
required to saponify 1 grm . of the oil can readily be ascertained .
The sapo nificati on equivalent of the oil i s found by dividing theweight of the sample employed
,expressed in mg .
,by the number of
c .c . of N 1 (not N 2) acid corresponding to the alkali neutralised bythe oil . If the percentage of potassium hydroxide required is known
,
the saponification equivalent can be found by dividing this percentageinto 56 1 0 .
I t i s essent ial that the alcoholic alkali should be as free as possiblefrom any colour, since any brown or yellow tint aff ects the sensitivenessof the acid reaction with phenolphthalein . The saponification andtitration Should be conducted with as little access of air as possible
,
since the action is influenced by the presence of carbon i c ac i d .
I t i s absolutely necessary to ascertain the strength of the alcoholicalkali from day to day
,as such solu tions rapidly alter
,and the mere
heating is l iable to cau se a Slight change in the neutralising power .S tandard sulphuric ac id cannot be conveniently substituted for thehydrochloric acid recommended for the titration
,as its employment
causes a precipitation of sulphate , which masks the end—point .In the case of waxes the nature and amount of unsaponifiable matterrenders saponification more difficult , and i t i s necessary to boil thesubstance for at least an hour over a flame protected by wire-gauzewith an excess of 2N . alcoholic alkali prepared with alcohol of 96 to98% strength . To prevent dissociation i t is advisable to add . 20 c .c . ofneutral alcohol to the liqu id before t itration .
Cold S aponificati on .
-The method of cold saponification devisedby Henriques (Zei t. angew . Chem.
,1 89 1 , 7 2 1 ) may sometimes be found
of use for oils and fats,though it is not satisfactory in the case of waxes .
From 3 to 4 grm . of the fat are dissolved in 25 c .c . of light petroleumand treated with 2 5 c .c . of N / 1 alcoholic alkali solution , a blankestimation being Simultaneously made . Both flasks are closed
,
Shaken,and allowed to stand for 1 2 hours at the ordinary temperature
,
after which the excess of alkali is titrated with standard hydrochloricacid .
The following table gives the saponification values and saponification

SAPONIFICATION VALUE S .
as constituents of the natural
saponification—equ ivalents of the monatomic
their molecular weights,while those of the
rd of their molecular weights .
S aponifica
equ ivalent
B utte r-fa tPo rpo ise
,do lph in
,
and whale O i ls .
Coconut and palmnut o i ls
Palm o i l ; lard
T allow ; lard ; cacaobutterArach is o i l .
I O live , a lmond,and
I lard O i lsRape O i l
Cottonseed,maize Oi ls .
L inse ed O i l
Castor o i l
S permacetiB ee swaxCh ine se waxS perm o i l.
B o ttlenose o i l
S met with in practice do no t consist of a Singleapproximate purity
,the saponification values of
and fats are the resultants of the values of their constituents,
e Show less pronounced diff erences than do the pure esters .ess
,the peculiarity of constitution of many of the natural
is indicated by the results of this test . Thu s rape oil andontaining erucin have low saponification values , whilst , on
fat,containing butyrin and other glycerides of
high values .fication values Of oil and fats of commercial im
VOI. I I .
—2

1 8 FIXED OILS,FATS , AND WAXE S .
portance will be foun d in the tables on pp . 69—73 . From the
figures there given i t will be seen that glyceridic oils and fats may beroughly classified into 3 groups in accordance with their saponificationvalues 1 . Those with low values ( 1 69 to 1 8 1 , usually about suchas castor oil and members of the rape-oil group . 2 . Those withmedium values ( 1 83 to such as the majority of fats and oils ;and 3 . Those with high values due to the presence of lower fattyacids
,such as members of the coconut-oil group
,butter-fat
,and cer
tain marine-animal oils (group X). The waxes (Group X I I) andSperm oil have exceptionally low saponification values indicative oftheir peculiar composition .
S ince hydrocarbon oils do not interact with alkali the proportion ofsuch oils in admixture with fatty oils may be deduced from the saponification value of the mixture . Thus if a sample of so-called linseed oilhas a saponification value of only instead of about 1 90, i t may beassumed to contain approximately 95% of hydrocarbon oil .S eparati on of the P roducts of S aponifiw ti on—The solution ofsoap
,freed in the foregoing manner from alcohol
,Should then be
diluted with warm water till i t measures 70 to 80 c .c . A perfectlyclear solution will usually be obtained if a pure oil has been used andthe process has been successfully conducted
,but waxes and mixtures
containing hydrocarbons and other foreign matters will give a solutioncontaining solid matter or oily globules in suspension . These ad
mixtures may usually be removed and estimated by agitating the soapsolution in a glass separator
,with an immiscible solvent
,ether being the
most generally suitable for the purpose .
I The ethereal layer is thenseparated
,evaporated
,and the residue weighed . The best method
of manipulation is described later . Cholesterol and other unsaponifiable substances are present in small proportion
,even in the purest
fatty oils .2If ether has been employed
,i t Should be removed by keeping the
soap solution at a gentle heat for some time . On then treating the1Owing to th e lim it ed so lubility ofmy ric l a lcoh ol in most so lvent s , th e me thod d escribed
in th e t ext i s at tended with pract ica l d i cul t ie s in th e ca se o f bee swax and carnafi ba wax ,
th ough i t is adm irably adapt ed for th e analy s is of S permace t i . If th e remova l of th e sepa ’
rated high er alcoho l by an imm iscible solven t b e found im pract icable , th e so lu t i on of th e soapsh ou ld be trea t ed wi th ace t ic acid in quant ity ju st su ffi c ient t o dest roy th e pi nk co lorat ionp i oduced by ph enolphth a le in , and th e so lu t ion t rea t ed with lead acetat e . Th e p reci p ita t e sh oul d be washed , dried , m ixed with sand , and th e ,
wax-a lcoh ol d i sso lved In b orhngpet ro leum sp irit .
0
2In rigidly accurate experiment s i t i s de sirable to t reat th e un sa p on i fi ed re s i due i n thesame manner a s th e original o i l , as t races of fat are liable t o escape sa p on i fi cat i on b
ya
S ingle t reatmen t . If th e re sidu e left on evap orat in th e e th erea l so lu t ion b e t reat ed w i t a
lit t le h ot a lcoh ol , th e so lu t ion fil t ered h ot , and th e trat e coo led , and , i f n ecessary , a llowedt o evaporat e spontaneous ly , cry stall ine pla t e s of ch o lest e ro l will oft en b e d epo si t ed .

SAPONI FICATION VALUE S
with an acid,dilute sulphuric acid being generally preferable
,
precipitate is produced,which
,on warming the liquid
,will
and form an oily layer on the surface . This layertty acids produced from the oil . These acids diff eresters in being soluble in alcohol
,the solution having
and decomposing the carbonates of the alkali metals,
dioxide and forming soaps .ty acids are almost wholly insoluble in water and notat but from butter-fat
,coconut oil
, palmnut
and some others a notable amount of the lower fattyand hence the acids from these sources are partiallyand capable of distill ation with water at
For obtaining these soluble or volati le acids from oils , the soap solution is acidified with sulphuric acid in the manner already described
,
the aqueous liquid separated from the layer of fatty acids,and the
latter boiled several times with a considerable quantity of water ina flask furnished with a long tube or inverted condenser . The liquidsresulting from these operations are separated from the insoluble fattyacids
,which it is desirable to boil again with a moderate quantity of
water , whilst driving a current of steam through the flask in whichthey are contained
,collecting the distillate
,and treating it like the
washings . 1 The acidified aqueous liquid first separated from thelayer of fatty acids is then distilled to small bulk and the distillateexactly neutralised with a standard solution of sodium or bariumhydroxide , using phenolphthalein as an indicator . The first washingsfrom the insoluble fatty acids are then added to the contents of theretort , and the liquid again distilled to a low bulk , the process beingrepeated with the sti cceeding washings . The diff erent distillates obtained Should be titrated separately with N/ I O standard alkali andphenolphthalein
,as
,in this manner
,with but little extra trouble,
the progress and completion of the washing,etc . , can be followed ,
and useful information obtained as to the probable nature and
relative proportions of the lower fatty acids present .The several neutralised distillates may now be united and evapo
rated gently to dryness,the residue being dried at 1 00° till constant in
1When coconu t or palm nu t o i l 1 5 t rea t ed in this manne r , th e d ist illate will b e found toconta in lauric acid , wh ich , th ough a lmo st inso luble i n wat er , i s volat ile i n a curren t of s t eam .
It may b e separat ed from th e mo re so luble vola t ile fat ty acids by fi lte ring th e d ist illa t e .

20 FIXED OI LS,FATS
,AND WAXE S .
weight . I t consists of the sodium or barium salts of the acids whichpassed over in the preceding distillation . I f the total volume (in c .c .)of N/ 1 sodium hydroxide solution employed for the neutralisation bemultiplied by or the volume of N 1 barium hydroxide solutionby and the number so obtained be subtracted from the grossweight (in grm .) of the dry residue , the difference will be the weight ofthe volati le fatty acids ; Their mean combining equivalent will befound by dividing their weight by the volume (in c .c .) of normalalkali required for their neutralisation .
A further examination of the volatile fatty acids can be made bydistilling the barium or sodium salts with phosphoric or diluted sulphuric acid , and exam ining the distillate as indicated in Vol . 1 , p . 23 5 .
In Reichert ’s method (see below) an aliquot portion of the acidifiedsolution of the saponified fat is distilled , and the distillate titrated withstandard alkali .Kehne t Va lue .
—In cases in which the oil under examinationis known not to contain any appreciable quantity of esters of thelower acids
,the treatment for their isolation may be wholly omitted
,
and the insolublefatty acids are then practically identical wi th the totalfatty acids liberated on adding a dilute m ineral acid to the aqueoussolution of the soap . The oily layer thus obtained should be Shakenseveral times with warm water
,or until
,after separation
,the aqueous
liquid is no longer acid to litmus . The subsequent treatment of theinsoluble fatty acids will depend on the nature and extent of the information required . In some cases i t will
,be suffi cient to add alcohol
and ti trate with standard alkali with phenolphthalein as indicator . 0
I f the fatty acids are to be weighed,the best mode of operating is to
run them from the separator into a small paper filter previously wettedwith hot water . The funnel con taining the filter is placed in themouth of a small dry beaker
,and the whole heated inthe water-oven .
AS the filter dries,the greater part of the fatty acids will pass through
the paper into the beaker . When no more drops through , the funnelis removed to a small dry flask
,and the acids adhering to the separator
or other vessels removed by means of ether , carbon tetrachloride , orpetroleum Spiri t . The solution thus obtained is poured into thefilter and caught in the flask below . A fresh quanti ty of the solvent isu sed to effect complete solution and removal of the fatty acids fromthe filter
,these washings also being allowed to run into the flask .
The solvent i s then distilled off by immersing the flask in hot water ,

HEHNER VALUE .
atty acids further dried by blowing a current of airtill they begin to lose weight
,or till all odour of the
peared . The weight of fatty acids thus estimatedat of the main quanti ty contained in the beaker
,and the
insolublefatty acids in the amount of fat employed for the
pressed in percentage of the fat is, commonly termed theI t usually ranges from about to 96% in the caseonly minute quantities of soluble fatty acids .the estimation of the total insoluble fatty ac ids isdesired
,a further proximate analysis may be made by
cat ed in the section on“Higher Fatty Acids .”
ified aqueous liqu id remaining after the isolation oftty acids
,and the removal of any volatile fatty acids by
contains glycerol , which may be isolated by exactly neuith potassium hydroxide
,evaporating the solu
dryness on the water-bath,and exhausting the residue with
On fil tering and evaporating the alcoholic solution,the
is obtained as a sweet syrupy liquid,which may be further
by treatment with a m ixture of alcohol and ether and evaporahe filtered solution . Although glycerol resulting from the sa
may be readily isolated in this manner , the resultsobtained are only very roughly quantitative , owing to loss duringthe evaporations . The estimation of the glycerol produced by sa
ponification is most accurately effected by the methods described inthe s ection on “ Glycerol .”The following table Shows in a c ondensed form the general process ,just described
,for the separation of the products of saponification of
genuine fixed oils . The method of estimating foreign addi ti ons to1
fixed oils is described in a separate section .

2 2 FIXED OILS , FATS,AND WAXE S .
Sapon ify th e oi l , evaporate off th e a lcoh o l , d issolve th e re s idua ll soap in wat er , and agita teth e so lu t ion with e ther.
Ethereal Solu tion Aqueous Layer . Ac idify with d ilu t e su lphuric acid , and washconta ins c h o l e s li bera t ed fat t y acids W i th bo iling h ot water .
terat, phy tosterol ,hydrocarbons , u n
saponified oi l , and Oily Layer con sist s o f i n Aqueous Liqu id on d ist illa t ion giveshi gher a l c o h o l s s oluble fatty acids , wh ich( f r o m w a x e s , may b e convert ed int osperm o i l , lead salt s and part ia lly I n D i s t i l l a t e In Retort , an aq u eous
separat ed by t rea tment lower fa tty aci ds . liqu id , wh ich , wh enwith e th er . such as bu ty ri c, va neu t ralised . carefu llyleri c. caproi c, lau evaporat ed to dryn ess ,ri c , etc. ; e st imat ed a n d t h e r e S i d u e
More S o luble More Inso lu by t itrat ion with t reat ed with e therln Eth er . ble in Eth er . standard a l k a l i a lcoh o l , gi ve s a so luLead c o m Lead c o m and furth er e xam t ion of gly cerol , leftp o u n d s o f p o u n d s o f ined by frac t ional a s a sweet syru pyolei c, ri ci n my ri s ti c. pal d ist illa t ion , e t c . liqu id on evaporat ingoli c, li noli c, mi ti c , s teari c , th e so lven t , but wh ichli noleni c, h arach i di c , i s more accu ra t e lypogei c aci s , ceroti c aci ds . es tima ted in a separat eetc. etc. port ion by oxida t ion .
Re ich ert Value .—This term is applied to the number of c .c . of
N 1 0 alkali solution required to neutralise the distillate obtained fromthe acidified solution of a fat saponified under definite empirical conditions . I t was devised by Reichert (Zei t. anal. Chem.
,1 879 , 1 8,
and,though practically superseded by later modifications
,is still used
in some laboratories,and is the method by which the earlier recorded
values were obtained .
AS the process is an arbitrary one,only about 4/5 of the entire
volatile fatty acids obtainable from butter being found in the distill ateunder the conditions of Operation
,i t is necessary to adhere to the
following directions : Saponify grm . of the fat with 2 5 c .c . of approximately N/ 2 alcoholic potassium hydroxide , by heating it in aclosed bottle or flask fitted with a long tube . Transfer the productto a porcelain basin
,and evaporate off the alcohol completely at a steam
heat . D issolve the residual soap in water,add dilute sulphuric acid
in slight excess,dilute the liquid with water to 7 5 c .c . , add some frag
ments of pumice coiled round with platinum wire,and distil gently till
50 c .c . have passed over, Filter the distillate,i f not quite free from
white flakes or oily globules,wash the filter with a little hot water ,
and titrate the clear solution with N 1 0 alkali , using phenolphthaleinas an indicator .The following table gives typical Reichert values thus obtained

24 FIXED OILS,FATS
,AND WAXE S .
Acid—So lution o f sulphuric ac id conta ining 25 c .c . o f strongest sulphuric ac idin c .c . of wate r.
Barium Hydroxide .
—An accurately standard is ed,approximate ly N 1 0 So lution
of barium hydroxide .
Indi cator .
-1 grm . of phenolphthale in in 1 00 c .c . of alcohol.
S aponificati on flasks , of from 250 to 300 c .c . capacity, of hard , we ll-annealedglass
,capable of resisting the tens ion of alcoho l vapo r at
P ipette graduated to deliver 40 c .c .
D i sti lling Apparatus
Burette .
—An accurate ly calibrated burette , read ing to tenths of a c .c .
Estimati on—Weighing the f at—The butter o r fat to be exam ined should be
me lted and kept in a dry, warm place , at about 60° for 2 or 3 hours , until thewater and curd have entirely settled out . T he clear
,supernatant fat is poured o ff
and filtered through a dry fi lter-paper in a jacketed funne l conta in ing bo iling water.
Should the filtered fat,in a fused state
,no t be perfectly clear
,it must be filtered
a second time .
The saponification flasks are prepared by tho roughly wash ing with water, alcoho l,and ether
,wiping perfectly dry on the outs ide
,and heating for 1 hour at the
temperature of bo iling water. The flasks Should then be placed in a tray by thes ide of the balance and covered with a S ilk handkerch ief until they are perfectlycoo l. They must no t be wiped with a S ilk handkerch ief with in 1 5 o r 20 m inute s ofthe time they are we ighed . The we ight of the flasks having been accurately determ ined
,they are charged with the melted fat in the fo llowing way
The pipette with a long stem,marked to de liver c .c .
,is warmed to a tem
perature of about The fat,having been poured back and forth once o r twice
into a dry beaker in o rder to mix it thorough ly , is taken up in the pipette , and 5
c .c . of fat allowed to flow into the flask . After the flasks have been charged in th isway they should be re-covered with the s ilk handkerch ief and allowed to stand 1 5 or
20 m inutes,when they are aga in we ighed .
S aponificati on .
—1 0 c .c . of 95% alcohol are added to the fat in the flask, and then2 c .c . of sod ium hydroxide solution . A soft cork stopper is now
,
inserted in the flaskand tied down with a piece of twine . The saponification is then completed byplacing the flask upon the water or steam—bath . During the saponification , wh ichShould last 1 hour, the flask Should be gently rotated from time to time
,care be ing
taken not to project the soap for any d istance up its sides . At the end of an
hour the flask,after having been coo led to about the temperature of the room
,is
opened .
Removal of the Alcohol. -The stopper having been la id loosely in the mouth of
the flask , the alcohol is removed by d ipping the flask into a steam-bath . T he
steam Should cover the who le of the flask except the neck. After the alcoho l is
nearly removed,froth ing may be not iced in the soap
,and
,to avo id any loss from
th is cause or creeping of the soap up the s ides of the glass, the flask Should be removedfrom the bath and shaken to and fro until the froth ing d isappears . The lasttraces of alcoho l vapor may be removed from the flask by waving it bri skly, mouthdown
, to and fro .
D i ssolvi ng the S oap—Af ter the removal of the alcoho l the soap Should be
d isso lved by add ing 1 00 c .c . of recently-bo iled d istilled water, and warm ing

RE ICHERT-MEI S SL VALUE .
the steam—bath with occas ional Shaking unt il so lut ion of the so ap is
of the Fatty Acids .
—When the soap so lut ion has coo led to about 600add ing 40 c .c . o f the d ilute sulphuric ac id
F atty-Aci d Emuls ions
—The flask shou ld now be stoppe red as in the
and the fatty-ac id emuls ion me lted by replac ing the flask on the ,
The time requ ired for the fus ion may vary from a few m inutes toacco rd ing to the nature of the fat exam ined .
ac ids are completely me lted,form ing a transparent
surface of the water, the flask is cooled to the temperature of the
pieces of pum ice-stone added . The pum ice-stone is prepared byat a whi te heat, into d istilled water, and keeping it under water untilflask is now connected with a glass condenser, S lowly heated wi th a
until ebullit ion begins , and then the d ist illat ion continued by regulatingsuch a way as to co llect 1 1 0 c .c . of the d istillate in,
as nearly as poss ible ,The d istillate should
kbe rece ived in a flask accurate ly marked at
of the Volati le Acids .
—The 1 1 0 c .c . of d istillate , after thorough m ix1 00 c .c . of the filtrate pouredthe pheno lphthalei n so lutionred co lour is produced . The
returned to the measuring flask to remove any
in,poured aga in into the beaker, and the titration continued
co lour produced rema ins apparently, unchanged fo r 2 o r 3 m inutes .
of c .c . of N 1 0 barium hydroxide requ ired Should be increased
Leffmann and B eam ’s modification (Analyst, 1 89 1 , 1 6,
1 53 ;
1 896, 2 1 , 25 1 ) in which a solu tion of sodium hydroxide in glycerolis used for the saponification i s the offi c ial German method forthe examination of fats and cheese
,the estimation being made as
follows : 5 grm . of the fat are cautiously heated with constant Shaking over a small flame in a 300 c .c . Erlenmeyer flask with 20 c .c .of glycerol of sp . gr . and 2 c .c . of sodium hydr oxide solution(prepared by dissolving. 1 00 grm . of sodium hydroxide in 1 00 c .c . ofwater). After evaporation of the water , which usually takes from5 to 8 minutes , the liquid becomes clear , and is then completely saponified . I t is now allowed to cool to about and treated with 90 c .c .
of water at 80° to This solution is acidified with 50 c .c . of dilutesulphuric acid (50 c .c . of strong acid in c . c . of water) and thevolatile fatty acids distilled and titrated as in the Reichert-Meisslprocess .
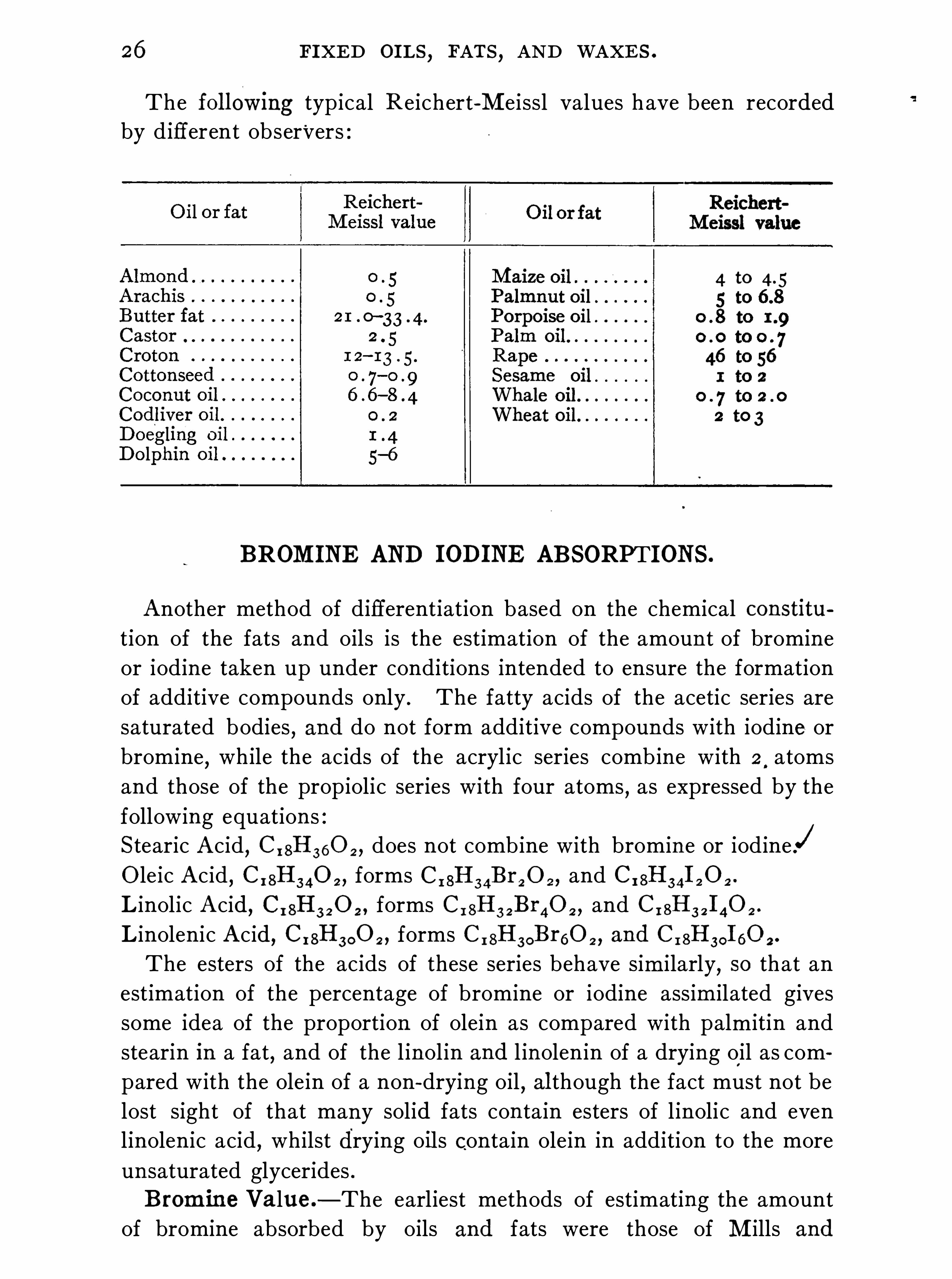
26 FIXED OILS,FATS , AND WAXES .
The followmg typical R eichert-Meissl values have been recordedby different obser 'vers
4 to
0 0 0 0 0 0
O O O O O O O O O O O O
0 0 0 0 0 0 0
o o o o o oo o o o o o o o o o o o o o o
0 0 0 0 0 0 0 0
BROMINE AND IODINE ABSORPTIONS .
Another method of diff erentiation based on the chemical constitution of the fats and oils is the estimation of the amount of bromineor iodine taken up under conditions intended to ensure the formationof additive compounds only . The fatty acids of the acetic series aresaturated bodies
,and do not form additive compounds with iodine or
bromine,while the acids of the acrylic series combine with 2
.
atomsand those of the propiolic series with four atoms
,as expressed by the
following equationsS tearic Acid
,C , 8H3 602 ,
does not combine with bromine or iodine/Ole ic Acid
,C 1 8H3 4O 2 ,
forms C 1 3H3 4Br 20 and C 1 3H3 4
I ZO , .
Linolic Acid, C 1 8H3 ,
O , , forms C 1 8H3 2Br
4O z , and C 1 8H3 2
14O z .
Linolenic Acid, C 1 3H3 o
O z , forms C l gHsoBr6O z ,
and C 1 8H301 602 .
The esters of the acids of these series behave Similarly, so that anestimation of the percentage of bromine or iodine assimilated givessome idea of the proportion of olein as compared with palm itin andstearin in a fat
,and of the l inolin and l inolenin of a drying oil as com
pared with the olein of a non-drying oil,al though the fact must not be
lost Sight of that many solid fats contain esters of linolic and evenlinolenic acid
,whilst drying oils contain olein in addition to the more
unsaturated glycerides .B romine Va lue .
—The earliest methods of estimating the amountof bromine absorbed by oils and fats were those of M ills and

BROMINE AND IODINE AB S ORPTIONS . 2 7
Chem. Ind. ,1 883 , 2 , 43 5) and M ills and Ak itt
but for most purposes these and Similar methodstically superseded by Hiibl’s iodine method and
t of bromine enters into combination by way ofaddition , andMcIlhiney (J. Amer. Chem. S oc.
1 084) has based a useful test for the detectionin drying oils upon a determination of the
hed quantity of the oil in 1 0 c .c . of carbon3. stoppered bottle with 20 c .c .of N.
bromine in the same solvent . Af ter the lapse of 2 or 3to ~
30 c .c . of a 1 0% solution of potassium iodide are introbottle thoroughly shaken
,and the liberated iodine titrated
ard th iosulphate solution , and calculated into the corbromine addition value . An addi tion of 5 c .c . of a neutralpotassium iodate is then made
, and the liberated iodine ,to the hydrobromic acid formed in the substitution
,
substitution val ue .acid is prevented by fixing athe neck of the bottle
,so as to
well into which the potassium iodide solution is poured . Thes then cooled in ice-water to create a partial vacuum beforewithdrawing the stopper .bromine
'
substitution value of ordinary fats and oils usuallyto whilst rosin and turpentine Show values
Logan (J . Amer. Chem. S ea,
1 90 1 , 23 , 1 56) madeestimations of the bromine value by this method and
of the iodine value by Hubl ’s method,and showed that the ratio
between the iodine value as estimated and as calculated from thebromine addition value might afford useful indications in the detectiono f marine animal oils in linseed oil
,etc . They found rosin to have a
bromine substitution value ofComparative results obtained by Wijs’ iodine chloride
'
method and
McI-lhiney’s bromine method are also given by Williams (J . S oc.
Chem. Ind .,1 900, 1 9 , A gravimetric bromine method was
devised by Hehner (Analyst, 1 895 , 20, 49 , J . S oc. Chem. Ind.,1 897 ,
1 6, and was discussed by Lewkowi tsch (J . S oc. Chem. Ind
,

28 FIXED OILS,FATS
,AND WAXE S .
1 896 , 1 5 , Williams (Analyst, 1 895 , 20,
and Jenkins(J . S oc. Chem. Ind
,1 89 7 , 1 6 ,
The main advantages of this method,where applicable
,are its
S implicity and speed,but both are possessed in greater
-
degree by thebromine thermal process (q . v.)Inso luble Brom ide Test —Hebner and M itchell found (Analyst,
1 898, 2 3 , 3 1 5) that on treating an ethereal solution of certain oils witha Slight excess of bromine an insoluble precipitate was obtained
,the
amount of which could frequently give valuable indications as to thepurity of an oil .These precipitates appear to be the bromides of mixed glyceridescontaining one radicle of linolenic acid or (in the case of marineanimal oils) the i someri c j ecoric acid . The bromide from linseed oilmelts at '
1 43 .5 to 1 44° and contains about 56% of bromine . The
Similar bromides from marine animal oils decompose before melting,
and this aff ords a means of detecting even a small amount of such oilsin linseed and other drying oils .The precipitate may be collected either in a Soxhlet tube , if thequantity taken is small
,or on a counterpoised filter
,but the method
employed for the estimation of stearic acid in mixtures of fatty acids(see page is the most satisfactory
,the best filtering material
in this case being thin,flexible Chamois leather tied over the end of the
small thistle funnel,from which any adhering precipitate can after
wards readily be removed by washing .
From 1 to 2 grm . of the sample are dissolved in 40c .c . of ether , towhich a few c .c . of glacial acetic acid are added
,the precipitate formed
being more granular from such a mixture than when ether aloneis employed . The solution is cooled in an ice-chest and bromineadded
,the flask being preferably left all night in the ice . This , how
ever,is not essential for ordinary working . The liquid is filtered off
by the suction funnel attached to a pump,the flask washed out with
four successive portions of ether at and the residue dried in theflask to constant weight . Even when ether at ordinary temperaturesis used
,no considerable error is introduced .
Various samples of pure linseed oil were examined by th is method ,with the following results

IODINE VALUE .
walnu t oil gave,in two determinations
,and
oppy oil gave no deposit,nor did Brazil nut oil , maize
oil,olive oil
,Japanese wood oil
,or almond oil . Mix
and other oils gave percentages of bromide in procentage of linseed oil
,as will be seen from the
0 0 0 0 0 0 0
s were Obtained in this way by Walker and
1 902 , 28,
Linseed oil,
nut oil,
Japan fish oil,
cOdl iver oil , cod Oil
shark-liver oil,
seal oil,
54 ; and Sperm oil, (and after 48 hours
standing,
A S a rule linseed oil yields abou t 2 5% of insoluble bromide , butMitchell has met with a Specimen yielding over 30% and Lewk owi tsch
with one giving O ther insoluble bromide values for marineanimal oils are given by Procter (J . S oc . Chem. 2 5 ,
Iod ine Va lue .—Free iodine is SO slowly absorbed by oils and
fats that i t has not been found possible to base a satisfactory methodupon i ts use
,and i t has long been discarded in favour of the Hubl proc
"ess (D ingler ’s polyt. J .
,1 884 , 2 5 3 , 28 1 ) in which the reagent i s an
alcoholic solution of iodine in conjunction with mercuric chloride in the

30 FIXED OILS,FATS , AND WAXE S .
proportion of at least 1 molecule (I , ) of the former to at least 1 (HgC12)of the latter .Hi tbl ’s Method—The reagent is prepared by d issolving 25 gr in . ofiodine in 500 c .c . of nearly absolute alcohol (free from fusel oil), and30 grm . of mercuric chloride in an equalmeasure of the same solvent .The latter solution is filtered
,if necessary
,and then added to the
tincture of iodine . The mixed solution should be allowed to stand for1 2 hours before being used
,as
,owing to the presence of impurities in
the alcohol employed,i t i s liable to undergo considerable reduction in
strength,and must in all cases be re—standardised immediately before
or after use . The strength is ascertained by titration with decinormalsolution of sodium thiosulphate
,which in its turn is standardised by a
solution of resublimed iodine in the usual way . The mercurial iodinesolution acts readily at ordinary temperatures on either free unsaturated fatty acids or their esters to form chloro- iodo-addition products
,
the total proportion of halogen assimilated being estimated in terms ofiodine .To estimate the iodine-absorption
,from to grm . of drying
oil,0 .3 to of non-drying oil
,or from to grm . of fat , is weighed
accurately,and dissolved in 1 0 c .c . of chloroform . The Solution is
mixed in a stoppered flask with 20 c .c . of the standard solution of iodomercuric chloride
,and if the liquid is not quite clear after agitation a
further addition of chloroform is made . If the mixture becomes decolorised
,or nearly so
,after standing a Short time
,a further addition
of 5 or 1 0 c .c . of iodine solution must be made . T0 ensure accurateresults
,the excess of iodine must be considerable
,and hence the
l iquid ought still to be quite brown after standing for 2 hours .
I Afterthat time
,from 1 0 to 1 5 c .c . of a aqueous solution of potassium
iodide Should be added,and the whole diluted with about 1 50 c .c . of
water . The free iodine,part of which exists in the aqueous and
part in the chloroform solution,i s then estimated by titration with
thiosulphate,the contents of the flask being frequently agitated , and
starch solution being added just before the end of the reaction . A
blank experiment with the same quantities of chloroform , iodinesolution
,etc .
,is made Side by Side with the actual test , so as to obtain a
1 Hfi b l found tha t wi th free fat t y ac ids th e act ion i s comple te with only a sma ll excessof iod ine , bu t with fa t s or o ils a larger exce ss mu st be em ploy ed , or th e re sult s will be t oolow . In pre sence o f a su ffi cient exce ss of iodine , variat ion s in th e concent rat ion of thefat ty solu t ion and in th e amount of m ercuric ch loride present do not aff ect th e re su lt sTh e react ion shou ld b e allowed t o cont inu e for at least 2 hours (or , accord ing t o Archbu t t , 6 hou rs) .
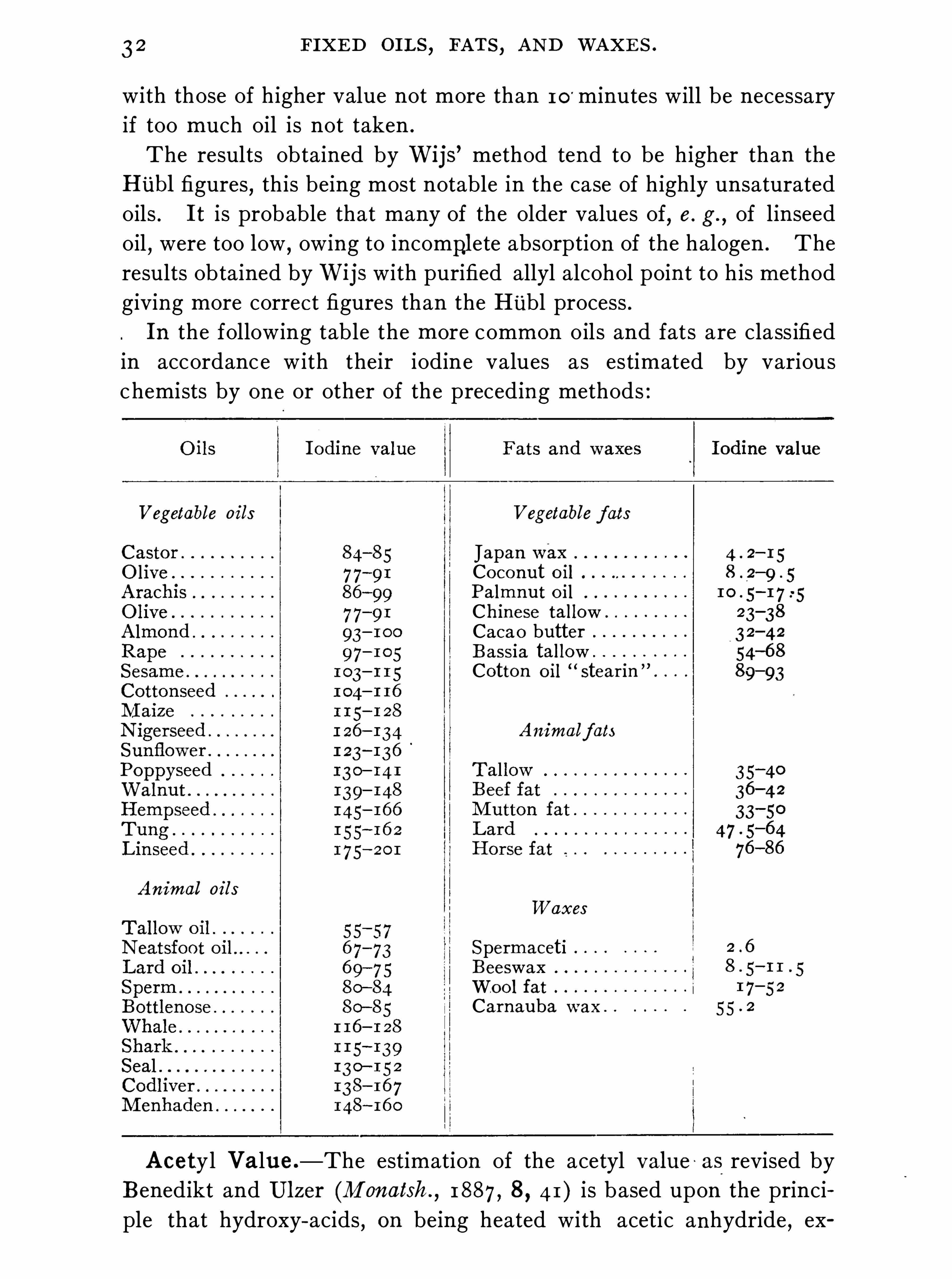
3 2 FIXED OILS , FATS , AND WAXE S .
with those of higher value not more than 1 0' minutes will be necessary
if too much oil i s not taken .
The results obtained by Wijs’ method tend to be higher than theHi
’
i bl figures , this being most notable in the case of highly unsaturatedoils . I t i s probable that many of the older values of
,e . g .
,of linseed
oil,were too low
,owing to incomplete absorption of the halogen . The
results obtained by Wijs with purified allyl alcohol point to h is methodgiving more correct figures than the Hubl process .In the following table the more common oils and fats are class ifiedin accordanc e with their iodine values as estimated by variousc hemists by one or other of the preceding methods :
Vegetable oi ls Vegetable f ats
Japan waxCoconut o i lPalmnut o i l
Ch ine se tallowCacao butterB ass ia tallowCotton o il
“stearin
Animalf ats
0 0 0 0 0 0 0 0
Animal oi lsWaxes
SpermacetiBeeswaxWoo l fat
Carnauba wax .
Acetyl Va lue .—The estimation of the acetyl value as revised by
B enedikt and Ulzer (Monatsh .
,1 887 , 8 , 4 1 ) i s based upon the princi
ple that hydroxy-acids,on being heated with acetic anhydride , ex

ACETYL VALUE . 33
atom of their hydroxyl group or groups for theThe operation is carried out by heating the
6 , 7 2 , 9 1 ) drew attention tosubsequently (J. S oc. Chem.
ng method , which is now inboiled for 2 hours with an
d flask beneath amixture then transferred to a large beaker
,
undred c .c . of water , bumping being meang a slow current of carbon dioxide through ato the bottom of the beaker.to separate into 2 layers
,the water is siphoned
ain boiled out in the same manner until theacid is removed . This is ascertained by testingThe acetylated product is freed from water andgh fil ter paper in a drying oven .
be carried out quantitatively, and in that caseon a 'weighed filter . An increase of weight
imilation of acetyl groups had taken place .seful to ascertain preliminarily whether ated acids is present in the sample under
acetylated substance are saponified by means ofhydroxide solution as in the estimation of the
ion value . If the fil tration process be used,the alcoholic
t be measured exactly , and this is also advisable with thedistillation process
,so as to obtain the saponification value of the
a cetylated fat . The alcohol is next evaporated and the soap dissolved
From this stage the determination is carried out either by the (a)distillation process or (b) “ filtration process .”(a) D i sti llati on P rocess .
—Add dilute sulphuric acid in morethan suffi cient quantity to saturate the alkali , and distil as usual inReichert ’s distillation process . S ince several portions of 1 00 c .c . eachmust be distilled off, either a current of steam is blown through theSuspended fatty acids or water is run into the distilling flask
,from
time to time,through a stoppered funnel fixed in the cork
,or any
other convenient device is adopted . It will be found quite sufl‘i cientVo l. I I .
—3

34 FIXED OILS,FATS , AND WAXE S .
to distil over 500 to 700 c .c . , as the last 1 00 c .c . contain practically noacid . Fi lter the distillates to remove any insoluble acids carried overby the steam , and titrate the filtrates with N 1 0 potassium hydroxidesolution , phenolphthalein being used as indicator . Multiply the number of c .c . by and divide the product by the weight of substancetaken . This gives the acetyl value .(b) Fi ltrati on P rocess —Add to the soap solution a quantity ofstandardised sulphuric acid exactly corresponding to the amount ofalcoholic potassium hydroxide solution employed and warm gently,when the fatty acids will readily collect on the top as an oily layer .(If the saponification value has been estimated , i t i s , of course ,necessary to take into account the volume of acid used for titratingback the excess of potassium hydroxide .) Filter off the liberatedfatty acids
,wash them with boiling water until the Washings are no
longer acid,and titrate the filtrate with N/ 1 0 potassium hydroxide
solution,using phenolphthalei n as indicator . The acetyl value is
calculated in the manner Shown above .B oth methods give identical results ; the latter will be found Shorter .The acetyl value indicates the number of mg . of potassium hy
droxide required for the neutralisation of the acetic acid obtained onsaponifying 1 grm . of the acetylated fat or wax .
In the case of those oils and fats which have a high Reichert value,
the apparent acetyl value will be too high,owing to the presence of the
volatile acids . This influence will aff ect the distillation process to agreater extent than the filtration process . To eliminate th is error ,the volatile acids of the original oil or fat should be estimated inprecisely the same manner
,and the value thus obtained Should be
deducted from the apparent acetyl value .I t Should be noted that in the case of a fat containing free alcohols(phytosterol , cholesterol), or , in .the case of waxes , the acetyl value willbe a measure of both the hydroxy-acids and the free alcohols . If
present,acetic acid radicles are also absorbed by them . If the free
alcohol is isolated its acetyl value may be determined as well . Thediff erence between the acetyl value of the fat or wax and the acetylvalue corresponding to the amount of free alcohol present will be thetrue measure of the hydroxy-acids .If a free alcohol is acetylated , no complicationthrough formation ofanhydrides can arise
,and in that case Simply the saponification value
of the acetylated product—the acetic ester of the alcohol—is deter

DRYING PROPERTIE S .
value is also the acetyl value of the alcohol (thevalue of the original alcohol being ni l) .
acetyl value,Lewkowitsch (Analyst, 1 899 ,
that i t may indicate : 1 . hydroxy-acids ; 2 . freefatty acids ; 4 . acids of unk nown composition ;cerides
,and 6 . rancidity . Hence
,until i t is
tent these several factors contributebe regarded as a constant .
results obes
free ac ids)
OXIDATION OF OILS—DRYING PROPERTIES .
(See also under L inseed and other o ils .)
Many of the fixed oils thicken on exposure to air,and
,under favour
able c ircumstances,gradually dry up into yellowish , transparent var
nishes or resin-like substances,to which in the case of linseed oil the
name linoxyn has been given . The nature of the oxidation changes

36 FIXED OILS , FATS , AND WAXE S .
that take place in the drying process is still very obscure,though the ‘
oils which possess th is property in the most marked degree appear(except in the case of tung oil) to be characterised by a high proportion of linolenic and isolinolenic acids .S trictly Speaking
,no hard and fast line can be drawn between dif
ferent classes of oils as regards their drying properties,though for
convenience of classification it is usual to group vegetable oils intodrying , semi
-drying , and non—drying oils .An experimental investigation of the process of drying of linseed oilhas been made by Genthe (Zei t. angew . Chem.
,1 896 , 1 9 , who
Shows that the presence of peroxides plays an important part in theprocess , and that polymerisation and formation of volatile acids accompany the oxidation . For other investigations of -the theory ofdrying of oils see L ivache (Compt. rend ,
1 895 , 1 20, and Fahrion(Zei t. angew . Chem. ,
1 89 1 , 540 ; 1 892 , 1 7 1 , and Chem. Zei t., 1 894,
For testing drying properties , a definite number of drops of thesample may be placed in a watch-glass or flat porcelain capsule
,and
exposed to a temperature of about 1 00° for 1 2 or 24 hours , side by sidewith samples of oil of known purity . Ol ive oil will be scarcely affectedby such treatment
,and rape oil will only become slightly th icker .
Cottonseed oil will be considerably affected , while good linseed oil willform a hard skin or varnish
,which can only with difi culty be ruptured
by pressure with the finger . In some respects,a preferable plan is to
flood a Slip of glass with the oil to be tested,in the manner in which a
glass-plate is covered with collodion . The glass with the adheringfilm of oil is then kept at and the progress of the drying followedby touching
,at intervals
,successive parts of the plate with the finger .
Another useful method is to soak a' definite measure of
o
thick fil terpaper in the sample of oil
,and then expose i t to 1 00 or 1 30° for some
hours,Side by side
,with samples of oil of known purity .
Livach e’s Meth od.—L ivache has shown (Compt. rend ,
1 886,1 02 ,
1 1 67) that the absorption of oxygen is accelerated by the addition offinely
-divided lead,and on this fact has based the following test ,
which enables numerical values to be obtained : About I grm . oflead I i s accurately weighed and spread in a thin layer on a watch-glass ,and to grm . of the oil allowed to drop from a pipette upon
1 Prepared by pre cipita t ing a lead salt with zinc , washing th e precipitat e rapidly insucce ssion wi th wat er , a lcoh o l , and e ther , and finally dry i ng i n a vacuum .

DRYING PROPERTIE S .
of the lead,care being taken that they do not run into
The watch-glass is then weighed and allowed to standlight at the ordinary temperature .
found to have absorbed the maximum quantityurs
,or in some cases after 3 days , whereas nont until the fourth or fifth day .
with the notable exception of cottonseed-oiloils
,i . e .
,their increase in weight corresponds
of the corresponding neutral oils . L ivache’s
Gain in we ight of 1 00 parts
Of o i l after
To obtain a correct estimate as to the drying properti es of anust be had not onl y to the increment in weight
,but also
of time required . Thus,of the two oils in the following
table,No . 1 must be considered the better
,although both finally reach
the same absorption of oxygen :

FIXED OI LS,FATS , AND WAXE S .
B ish op’s meth od (J . Pharm. Chim.,1 896 , 5 , 5 5) in which the
oil is ‘mixed with precipitated Silica and manganese resinate (as anoxygen carrier) gives the results more rapidly, but has not yet displaced L ivache ’
s method as a practical test .Oxygen Absorption —The tendency of fixed oils to absorb oxygenis in direct proportion to their capacity of absorbing bromine or iodine ,and to the rise of temperature produced on treating them withsulphuric acid . This is Shown by the fact that it i s possible to obtainozone values of oil s corresponding to the iodine values as was shownby Fenaroli (Gazzetta , 1 906 , 3 6 , When dry ozone is allowed tobubble through an oil at a temperature not exceeding the increasein weight of the oil exactly corresponds to an addition of 1 mol . ofozone for each double-bond in the mol . of the fat .Spontaneous Combustion.
—Gellatly has pointed Out the close relationsh ip which exists between the drying properties of oils and theirtendency to inflame spontaneously when exposed to the air in a finelydivided condition . Useful forms of apparatus for testing the liabilityof oils to Spontaneous combustion have been devised by Allbright andClark (J. S oc. Chem. Ind .
,1 892 , 1 1 , 547) and by Mackey (i bid.
,1 895 ,
1 4 , In the latter, 7 n n . of cotton-waste previously soaked
in 1 4 grm . of the oil,are placed in a roll of wire gauze 5 inches square
(24 meshes to the inch), and the whole placed in a water-oven in whichthe water is boiling . A thermometer is passed through the Opening ofthe oven
,so that its bulb reaches to the centre of the wool in the wire
roll,and the temperature is taken at regular intervals . All oils that
take fire or attain a temperature of over 200° in less than 2 hours in thistest must be regarded as dangerous . (See p .
Ox id ised Oi l . B lown Oi l . Base Oi l . -The commercial productssold under these names are produced by blowing a stream of airthrough a fatty oil—rape
,cottonseed
,or linseed oil being usually
chosen for the purpose . A certain initial temperature i s necessary tostart the action
,but afterwards the heat produced by the oxidation is
suffi cient to maintain the temperature required . By proper regulation ,
products can be obtained which closely Simulate castor oil , and equalthat body both in sp . gr . and viscosity . Methods of distinguish ingblown oils from castor oil are given in the section treating of the latterproduct.

40 FIXED OILS,FATS , AND WAXES .
A hard mass is yielded by olive o il,almond o il , arachis oil , lard
o il,Sperm oil , and sometimes neatsfoot oil .A product of the consi stence of bu tter is given by neatsfoot, bottlenosemustard
,and sometimes by arach is
,sperm
,and rape o ils .
A pasty or buttery mass whi ch separatesfrom afluid porti on i s yieldedby rape (mustard), sesame , cottonseed , sunflower , nigerseed, codliver
,seal
,whale
,and porpoise oils .
Li quid products are yielded by linseed , hempseed , walnut , and otherdrying o il s .In practice
,the elaidin test receives its most important application
in the examination of olive o il,wit h which i t gives a very characteri stic
result . The subject is further discussed in the sections treating ofolive and rape oils .Investigations of the elaidin reaction and attempts to apply itquantitatively have been made by Farnsteiner (Zei t. Unters . N ahr .
Genussm. , 1 899 , 2 , I ) ; by Edmed (Proc. Chem. S oc.,1 899 , 1 5 ,
and by Lidow (Pharm. Zei t. Russland. , 1 895 , 34, 1 05 ; Analyst,
1 895 , 20,
Interaction of Oi ls w ith Su lphur Ch loride .—The vegetable drying
oils are converted,on treatment with sulphur chloride (S 2C12), into
gelatinous or elastic masses,which are employed as substitutes for
india-rubber . Bruce Warren investigated the reaction with a Vi ew toits employment in the analysis of oils (Chem. N ews , 1 888
, 5 7 ,
COLOUR TESTS OF OILS .
Many fatty o ils give coloured products when treated with chemicalreagents
,and in some cases these afford valuable means of detecting
even small quantities of special oils in admixture with other oil s .The most characteristic of these tests for Special o ils are Becchi ’s and
Halphen’s tests for cottonseed oil and the Baudouin test for sesame
oil ; these are described under the special sections deal ing with thoseoils .Little value can be placed on the results of most of the older coloursts described by Calvert and Chateau , s ince the particular colorations were often due to accidental impurities in the o ils . Thetests with sulphuric and nitric acid
,however, have some value ,
especially,
when applied simultaneously to specimens of oil of knownpuri ty .

COLOUR TE S TS . 4 1
on of 1 or 2 drops Ofuces colorations which
,
s character
atuoils
fluorescence .more unsaturated oils
e effect of the heat producedof the oil and acid .
has a greater value in the examination ofanimal o ils
,S ince the presence of choles
ese oils cause s the producti on of characterred indistinct
,however
,
g action exerted by the reagent . This may be avoided1 drop of the oil in 20 drops of carbon disulphide ,he solution with a drop of strong sulphuric acid . Whaletreated
,gives a fine violet coloration , quickly changing
alone a red or reddish-brown
ur Test .—This is useful as a test for the presenceolive and other non-drying oils . 5 c .c . of the oi lare Shaken with an equal quanti ty of nitri c acidthe mixture left for 24 hours . In the presence of
“even a small percentage of cottonseed oil there should be a more orless pronounced brown coloration .
PHYSICAL PROPERTIES.
The general characteristics of the fixed oils have al ready beendescribed . Some of ' their physical properties are of importance fortheir recognition and estimation
,this being especially true of their
sp . gr ., melting and solidifying points , absorption-spectras refractiveindices, viscosity, and behaviour with solvents . The methods ofascertaining these characteristics are described in deta il in the following
Coh esion-figures of Oi ls .-The surface-tension of oils may in
certain cases be capable of useful application,though its value has been
much exaggerated . When a drop of oil is allowed to fall gently on to

42 FIXED OILS,FATS
,AND WAXE S .
the surface of water,i t often behaves in a characteristic fashion
,first
Spreading out and then contracting,forming figures which differ with
the nature of the oil . Descriptions and illustrations of typical cohesionfigures are given in Alder Wrigh t and M itche ll ’s Oi ls , Fats and Waxes ,
1 905 , p . 50 .
Absorpt ion-spectra of Oi ls .—The absorption-spectra of the fixed
oils occasionally afford valuable indications of their purity . For ob
serving them a micro-spectroscope may be used , but in many cases thel ight must be caused to pass through several cm . of the oil to be examined . Al though some vegetable o ils give exceedingly striking ah
sorp tion-bands,these are due not to the o ils themselves but to the
chlorophyl l and impurities contained in them . Hence the purificationor clarification of an oil tends to reduce the characteristic nature of theabsorption-bands
,which
,indeed
,may disappear altogether if the oil
be long exposed to sunlight . In one particular,however
,the absorp
tion-Spectrum furn ishes important inf ormation . Thus,no oils of
animal origin give definite absorption-bands,the Spectrum being
merely obscured at the more refrangible end,wh ilst in many vegetable
oils the absorption-bands of chlorophyll are exceedingly well marked ,especially a band having about the same refrangib ility as the linetermed B . In this way i t i s easy to detect the presence of rape ,olive
,or l inseed oil in Sperm
,cod
,or lard oil . Castor and almond oil ,
on the other hand,give no well-defined bands
,and the band at B in the
case of sesame oil i s faint,though there is strongly marked absorption
of the whole of the red portion nearly up to that point .Patterson (J . S oc. Chem. Ind .
,1 890 , 9 , 36) devised a Special absorp
tion Spectrum colorimeter for the Spectroscopic examination of oils .(See also Introduction to Vol .Refract ive Power.
—Valuable indica tions as to the purity of fatsand o ils
,especially butter-fat
,may be gained from the observation
of the refractive index . The instrumen t in general use for th is purpose is the bu tyrorefractometer, which is described in the section dealing with BUTTER . In certain r cases , however , such as tung oil androsin oils
,the indices are outside the Scale of the butyrorefractometer ,
and recourse must be had to the earl ier instrument of Abb e’ .
Abbé’
s Refractometer .
—The following method of using th is instrument is prescribed by the A . O . A . CA piece of fine t issue paper, 3 cm . in length by 1 .5 cm . in width , is p laced on
the lower of the two glass prisms of the apparatus. Two or three drops of the

REFRACTIVE POWER .
through an angle of
the instrument aga inis the true read ing .
ace,the tempe rature
For reduc ing the results to a standard temperafor every degree above that po int , s ince , as the tempe rafalls . The instrument used shou ld be set with d istilledrefractive index of water at that temperature be ing
ometer.
—The instrument devised by Amagat and1 889 , 1 09 , 6 1 6) enables a very rapid compari
F1G . 3 .
—Jean ’
s O leorefractometer . (Ba ird and Tatlocle.)
son to be made between the refractive power of a given oil and thatof a genuine oil taken as a standard . The oil to be observed is i htroduced into a hollow prism
,which is immersed in a vessel with

FIXED OILS,FATS
,AND WAXE S .
parallel sides filled with a standard oil . If the refractive power of thesample is the same as that of the standard
,no deviation of the ray of
light traversing the apparatus will take place ; but otherwise deviationwill occur, and can be measured on a micrometer-scale placed on theeye-piece . The angle of the prism
,the neutral or standard oil
,and the
divis ion of the scale are all arbitrary . The standard oil sold with theinstrument i s Sheepsfoot o il .In the figure , t t represent circular metal vessel with 2 opposite lenses ,
l l in front of the glass Sides . From these extend the 2 tubes C and
L , the former terminating in a coll imator, V,and the latter in a tele
scope , Oc. The glass prism-cell i s represented by Cy, whilst Ec is anarbitrary photographic scale . The instrument is illuminated by meansof a gas-j et , and the luminous field may be divided by means of a slideinto a light and dark portion .
In using the oleorefractometer,the outer vessel is charged with the
standard oil (which gives a zero reading), the oil to be tested placed inthe inner vessel
,and an outer trough (not Shown) filled with water .
The temperature i s then brought to 22° by means of the lamp , L P ,
and the deviation read upon the scale .The following resul ts were obtained by Pearmain (Analyst, 1 895 ,
20,1 35) with this instrument
Oi l or fat
Temperature
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
O O O O O O O O O O O O O O O O O O O O
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
CastorCo ttonseed (crude)Cottonseed (refined)Hempseed .
Lard o i l
L inseed (crude)L inseed (refined)
Peach kernelP ilchardPoppy seed
Ravison

GENERAL PHYS ICAL PROPERTIE S .
O il or fat
Temperature
Ole ic acidTemperature
Parafli n (soft)
Action on Po larised Light—Most of the vegetable oils in common use are either neutral or Slightly laevorotatory toin a 200 mm . tube in the polarimeter). Hence , rosin oil , which isstrongly dextrorotatory
,may often be detected by means of th is test
in drying o ils . Olive oil and sesame o il have a'
slight dextrorotation ,wh ilst castor o il and croton oil (q . v.) are strongly dextrorotatory .For the values obtained with various oil s see Al der Wright andMitchell ’s Oi ls
,Fats and Waxes
,1 903 , p . 5 2 , and Peter, Bull . S oc .
Chem. , 1 887 , 48 , 483 .
Electrical Conductivi ty .-The measurement of the specific resis t
ance off ered by solutions of the potassium soaps obtained understandard conditions from diff erent oils and fats affords a means ofdist inguishing between them . A method was devised by Herlant
(Bull . de l’Ass . belge Chim.
,1 896 , 1 0 ,
Heat of Combus t ion—Measurement of the heat of combustion ofdiff erent oils and fats may be used in their analytical diff erentiationand in obtaining data as to their food value . A table of the valuesobtained with the diff erent fatty acids is given by S tohmann
,Kleber,
Langbein and Offenhaur (J. pr. Chem.,1 894 , 49 , while results
obtained with various oils and fats and their adulterants are givenby Schw’einitz and Emery (J. Amer. Chem. S oc. , 1 896 , 1 8 , 1 74) and
Sherman and Snell (i bi d .,
1 901 , 23 , For the detection of

46 FIXED OI LS,FATS
,AND WAXE S .
adulteration the heat of combustion offers no advantage over Simplermethods of analysis .Viscos i ty .
—A useful physical test for o ils is based on their relativebody ” or viscosity
,a property which may be regarded as the converse
of fluidity . The viscos ity i s usually compared with that of rape o il,
but it may also be referred to water or glycerol as a standard . The
subj ect is fully discussed in the section on the “Examination of
Lubricating O ils .Specific Gravity .
—The sp . gr . of the fixed oils and fats is a property largely dependent ‘
on their constitution,and hence is more or
less characteristic of each particular oil . AS a rule,the Sp . gr. of
different samples of the same kind of oil varies within very narrowlimits , but i t i s l iable to be affected by the treatment to which the oilmay have been subjected in the process of refining , the presence of freefatty acids
,the age of the oil
,and the amount of oxidation it has under
gone,and by other circumstances .
The Sp . gr . of fixed oils may be ascertained by the usual methods,
but great care is necessary . Owing to the high coefli cient of expansion of oils , the temperature at which the observation is madeshould be carefully noted
,and in accurate observations the ther
mometer employed Should be an instrument the indications of whichhave been verified .
When a sufficient quanti ty of the sample i s available , and results ofextreme accuracy are not required
,sp . gr . can be ascertained readily
and satisfactorily by means of an accurate and delicate hydrometer .In any observations
,save those of the roughest kind , the oil Should
be brought accurately to the standard temperature by immersing thehydrometer-glass in water, cooled , if necessary, to 1 5 .5
° by dissolving in i t sodium thiosulphate or ammonium nitrate . The hydrometershould be immersed in the oil for 5 or 1 0 minutes , and the temperature again observed before taking a reading of the sp . gr. , as the use o fa warm hydrometer may cause an increase of several degrees in thetemperature of the oil . Of course , in taking sp . gr . by a hydrometer
,
the accuracy of the instrument employed is presupposed , but many ofthe instruments sold are inaccurate to the extent of several degrees .The sp . gr . bottle and Sprengel-tube (see Vol . 1 ) may also be employed for ascertaining Sp . gr . of oils , and allow of more accurateestimations than can be made with a hydrometer . The weight ofdistilled water which the bottle or tube holds at a temperature of

48 FIXED OILS , FATS,AND WAXE S .
Hager has devised an ingenious method of ascertaining the sp . gr .of solid fats at the ordinary temperature . The fat is melted anddrawn up into a pipette
,from which it is allowed to drop Slowly from
the height of 2 .5 cm . into cold alcohol contained in a flat-bottomed dish ,care being taken that each drop of fat falls in a different place . An
alternative plan is to melt the fat in a small—l ipped capsule and allowdrops of i t to fal l plate glass which has been previ ously wiped
with a wet cloth . On placing the glassin cold water
,the drops usually be
come detached on the sl ightest touch,
but if necessary can be removed witha knife after half an hour. The fatglobules obtained by one of the abovemethods are removed to a beakercontain ing dilute alcohol . The sp .
gr . of the liquid i s then adjusted byaddition of alcohol or water
,till
,after
careful stirring,the fat-globules re
main in equilibrium in any part ofthe liquid at a temperature of 1 5 .
Ammonia may be substituted for -thespirit if preferred . The sp . gr. ofthe liquid i s then taken
,and the result
obtained recorded as the Sp . gr. of thesuspended fat . The great obj ection
to this method is that fats and waxes which have undergone suddencooling have abnormal sp . gr. On this account it is far preferableto employ for the experiment fragments which have been cut off amass cooled under normal conditions .A good method of preparing beeswax and other waxes for this testis to pour the melted substance into a cylinder about inch inlength cut from narrow glass tub ing . The lower end of this is keptpressed against a glass plate until the wax solidifies
,after which the
outside of the glass is gently warmed,and thecore of wax pushed out ,
and allowed to stand for some hours at the ordinary temperature . I t
i s then brushed over with dilute alcohol,to prevent the subsequent
occurrence of air-bubbles, and i s immersed in Spirit which is diluted or
strengthened until of the same Sp . gr . as the wax .The following table Shows the sp . gr . (observed by Allen) at diff er
FIG. 4.

tances which are solidcase the examinations wereby the plummet method . A
in Sp . gr . corresponding to a
at the higher temperature were not corrected forass plummet of the apparatus
,and hence many
about to
I S . 5°
foregoing table represen t merely the sp . gr .samples of different oils . The limits of variavalue of it as a means of recognising and estithe various fixed oils in mixtures are discussed
ans ion .—I t is always desirable to determine
be standard temperature,but in many cases in
done a suitable correction may be made . The
oil may be found by estimating the sp . gr . at two

FIXED OILS,FATS
,AND WAXE S .
given temperatures as far apart as possible .
I The rates of expansionof the solid fats
,etc .
,are given in the table on page 5 1 , while from the
figures recorded on page 49 Allen calculated the rates of expansion ofvarious oils fluid at ordinary temperatures . The following table Showsthe values so obtained , together with certain data published by otherobservers
0 0 0 0 0 0 0
0 0 0 0 0 0
I t i s evident that the coefiici ent of expansi on of an oil may be deducedby dividing the temperature-correction by the sp . gr . Thus thecoeffi cient of expansion of olive oil will be
9 1 6for each de
gree centigrade .From the foregoing data Wright (J . S oc. Chem. Ind.
,1 907 , 2 6 , 5 1 3)
has calculated the numerical value of the approximate modulus of ex
pansi on,m. If S
O ,S ,, and S T represent the sp . gr . of the same fat at
the respective temperatures O°
,t°and T
°
(water at thenS,= S
O ( I—mt),
and ST= S
O ( 1 —mT ),The mean value of the m in the case of the 30 oils , etc . , examined byAllen is approximately whence is obtained the equation
St 1
ST 1 -0 .000 7T’
or i f5
—0 .000 7T
1 Thu s a sample o f rape o i l wa s found t o have a sp . gr . o f a t and
a t th e d iff erence be ing Divi d ing th is by th e d ifference be tween th e t em
p e rat u re s at wh ich th e observat ions were made ( 9 8 0 th e fi gu re i s
Obta ined as th e correc t ion t o b e made for a variat ion o f 1° from th e standard t empera ture .

this factor for each degree from
5° may be rap i dly corrected to the latter
Po ints .-The observation of the solidify
considerable importance,especially in the
hich too high a m . p . is a decided disaduitability of the solid fats for many of the
are applied is greatly dependen t on their 111 . p .
f solidifying or melting—point in the case ofis not to be expected
,as the natural fats consist
and solid substances,the proportions of which
erably in different samples of What is nominally theMoreover
,the melting-points
,l ike the Sp . gr . of the
fats,are l iable to obscure alterations on keeping
,and
dified by the presence of varying amounts of free acid .
een observed that many of the fats Sol id at the ordinaryave at least two distinct m . p . Thus the ordinaryof commerce
,if previously melted at a temperature con
ts m . p .
,Shows a m . p . of 3 5° to 36° If it be care
that temperature,cooled
,and the m . p . again taken ,
sometimes be found nearly 1 2° above the former

5 2 FIXED OILS,FATS
,AND WAXE S .
This phenomenon of double m. p.,which is a striking characteristic
of the mixed glycerides isolated from various fats,has been shown by
Griin and Schach t (Ber. , 1 907 , 40, 1 7 78) to be due to the existence oftwo isomeric modifications of such glycerides
,the one of lower m . p .
being the more unstable,and undergoing gradual transformation into
the modification of higher m . p .
Melting P oint.—The substance is melted at a temperatureS lightly above its fusing point
,and drawn up into a very narrow
glass tube (made by drawing out oneend of a piece of ordinary quill tubing),where it is allowed to solidify spontaneously. After an interval of not less thanI hour and preferably 1 2 hours the tube ,open at both ends
,is attached by a cork
or india-rubber ring to the stem of athermometer in such a manner that thepart of the tube containing the substanceis at the same level as
,and in close
proximity to,the bulb . The thermom
eter,with its tube
,i s then immersed in
water, which is gradually heated at a ratenot exceeding 0 .5
° per minute untilfusion of the contents of the cap illarytube takes place
,when the temperature
i s recorded . The flame is then removed,
and the temperature at which the fat resolidifies also observed . In
cases in which the melting and solidifying points are not notablyd ifferent
,i t is usual to record the mean of the two as the true m . p .
of the substance . It i s desirable to immerse the beaker of water containing the thermometer in an outer vessel also filled with water
, and
to apply the source of heat to the latter . A 500 c .c . flask , from whichthe neck has been cut off
,filled to the brim (Fig. 5) furnishes a very
convenient water-bath,and allows of a very regular and gradual ;
heating of the water contained in the beaker placed in its mouth .
The foregoing method is applicable only to bodies melting above thefreezing point of water
,but by substituting strong brine for the water
,
much lower temperatures may be observed .
By using a tube with its capillary end sealed up , and placing the fatin the upper part of the tube
,the softening point, which is frequently
FIG. 5 .

MELTING POINT . 53
appreciably lower than the m . p . (the temperature at which the fatfalls to the bottom of the tube), may be ascertained , while the point ofresolidification is also more accurately observed than is possible in anopen tube .The method of ascertaining the softening point devised byBevan and
Cross (J. Chem. S oc.,1 882
, 4 1 , 1 1 1 ) gives very accurate results , and isSpecially suitable for cases in which the fat does not melt readily (seeFig .
A 'very thin piece of Sheet- iron (ferrotype plate) is cut into an ellipti
cal form , about 1 in . long by in . wide . At one of the foci (A) asmall depression is made
,and at the
other a hole (B) is cut , of such sizeas to allow the plate to be fixed on tothe elongated bulb of a thermome
ter (C), so as to project therefromat right angles . A glass float (D) ismade by blowing a small bulb atthe end of a capillary glass tubeabout 3 in . long
,a small looped or
hoe-Shaped piece of platinum wire(E) being sealed into the bulb at theend opposite the stem of the float .To make an observation
,a very
small quanti ty of the sample ismelted in the indentation of theiron—plate ; and , while still liquid ,the Ioop or hoe of the platinumwire of the float is immersed in i t and allowed to become fixed bythe spontaneous solidification of the substance
,the stem of the
float being supported in a vertical position . A thermometer isthen cautiously introduced into the hole in the plate , and with i tsupported in a small beaker
,which is then filled with mercury or
other liquid . This is then gradually heated till the substance underobservation melts
,when the float is released and instantly rises to the
surface of the liquid . The results are very concordant , and free fromcertain sources of error to which observations made by the capill arytubemethod are liable .A very rapid method of ascertaining both the temperature of incipien tfusion and that of complete liquefaction is to place fragments of the
FIG. 6 .

54 FIXED OILS,FATS
,AND WAXE S .
material upon the surface of clean mercury (previously chilled ifnecessary) in a small basin , which is placed on a water-bath . Thetemperature is very gradually raised
,and the point at which the sub
stance changes its form is ascertained by means of a sensi tive thermome ter .
Modifications of th is method , in which the substance , on fusing , completes an electric circuitand rings a bell
,have been
devised by Loewe (D ingler
’s polyt. J .
,1 87 1 , 20 1
,
250) and Christomanos
‘
(Ber .,1 890, 23 , and
an arrangement of thekind is in use in the ParisMunicipa l Laboratory .
A . O . A . C . Method.
The melted and fil teredfat is allowed to fall froma dropping-tube from aheight of from 1 5 to 20
cm . on a smooth piece ofi ce floating in distilledwater that has been re
cently boiledfi The discsthus formed are from 1 to1 . 5 cm . in diameter , and
weigh abo ut 200 mg . By
pressing the ice under thewater the discs are madeto float on the surface
,
whence they are easilyremoved with a steelSpatula
,which Should be
Fm , 7 ,cooled in the ice-waterbefore using .
The apparatus (see Fig . 7) consists of a test-tube 30 cm . long andcm . in diameter , supported in a boiling tube 3 5 cm . long and 1 0 cm .
in diameter . The test-tube contains a m ixture of alcohol and waterprepared by separately boil ing distilled water and 95% alcohol for 1 0

56 FIXED OILS,FATS
,AND WAXE S .
in commercial work in England and France under the name of the“ti ter test.
”A test—tube
,about 5 in . in length by 2 /3 in . in diameter
,
is fitted with a ring or collar of cork or india-rubber,by which i t is
fixed in the mouth of an empty bottl e or flask . The melted substancei s then poured into the (warmed) tube t ill i t i s about 2 /3 fil led
,and a
delicate thermometer,previously warmed
,is suspended freely in the
liquid,so that the bulb may be wholly immersed . When the fat com
menees to solidify at the bottom of the tube,i t is slowly stirred by
giving the thermometer a circular movement fir st 3 times to the rightand then 3 times to the left . At first the mercury falls
,but subse
quently rises to a point at which i t remains stationary for about 2minutes . This temperature is taken as the solidification point of thesubstance
,and the results are very constant provided the same ap
paratus and method of working be employedModificati on of Dali can
’
s Method (Offi cial in United S tates). —Thefoll owing is the A . O . A. C . method : 2 5 grm . of the fat are saponified in a metal dish with 60 c .c . of 30% sodium hydroxide (36° B .)and 75 c . c . of 95% al cohol or 1 20 c .c . of water
,and the mass evapo
rated to dryness . The dry soap is dissolved in c .c . of boilingwater
,and the solution boiled for 40 m inutes to expel all alcohol
,
and then treated with 1 00 c .c . of 30% sulphuric acid (25° B .)and heated until the fatty acids are clear . The fatty acids are thenthoroughly washed
,separated from the water
,fil tered through a dry
fil ter and dried for 20 minutes atThe dried fatty acids are allowed to cool to 1 5
° to 20° above the
expected m . p .,and poured into a test-tube 1 00 mm . in length by
25 mm . in diameter,which is fitted by means of a cork into a flask 1 50
mm . in height by 70 mm . in diameter. The thermometer graduatedfrom 1 0
° to 60° in tenths of a degree is passed through an opening in thecork of the tube
,so that its bulb reaches to about the middl e of the
material . The 1 0° mark on the scale Should be 3 to 4 cm . above thebulb and the entire length of the thermometer Should be about 36 cm .
The estimation Should be made with the outside temperature about1 0
° lower than the solidification temperature . The readings Should bemade at Short
,preferably equal
,intervals , and the maximum point
reached in the short r i se after the mercury ceases to fall i s the solidification point or “ ti ter .” Duplicate observations Should agreewith inF inkener
’s Method (Ofl
‘i cial for Customs Examination in Germany) .

SOLIDIFYING POINT. 5 7
—This method is used in Germany for the technical differentiationof lard
,tallow
,and candle-fats . If the solidification point is below
30° i t belongs to the first class ; between 30° and to the second(tallows) and above to the third . Pressed tallow
,however
,may
still pass as tall ow even with a solidification point above providedit be declared as such and does not contain more than 50% of free fattyacids . In Finkener
’s method (Chem. Zeit .
,1 896 , 20, 1 3 2) the fatty
acids are dried as in Dal ican’s method and a larger quantity ( 1 50
grm .) introduced with a flask of prescribed dimensions up to adefinite mark
,the flask being then closed by means of a delicate ther
mometer on which is an expansionground to fit the neck of the flask .
The flask is then placed in a wooden box,in the top of which is an
opening for the stem of the thermometer. The whole is allowed tostand and the readings taken at intervals of 2 minutes after the temperature has fal len to about The solidification point thus es
timated is Slightly higher than that given by the original Dal icanmethod . This method offers no advantages over the ordinarymethod .
Wolf bauer’
s Method (used in Austria). -A test-tube 1 5 cm . longand 3 .5 cm . in diameter i s nearly fil led with the melted fatty acids
,
and closed by means of a cork through which passes the thermometer .The .tube is then fitted through an open ing in the cork of a widemouthed bottle . The mass i s stirred with the thermometer until i tbecomes turbid
,and the readings then taken without further stirring
(Mi tt. d. k . k . techn. Gew . Mus . in Wi en .,1 894,
S huhofi’
s Method (used in Russia). —The melted fatty acids areintroduced into a tube which is fitted into a bottle
,as in Wolfbauer
’s
method,though the dimensions are difi erent . As soon as the tem
perature has fallen to within 5° above the expected solidification
point,the apparatus is vigorously Shaken from the top to the bottom,
until the contents of the tube become turbid,the readings being then
taken . Results thus obtained agree closely with those given byWolfbauer
’s method. A vacuum-j acketed tube closed by means of
a rubber cork was also employed by Shukoff, but his later apparatusmentioned above is more commonly used (Chem. Rev. Fett. u . Harz.
Ind . , 1 899 , 6 , 1 1 ; Chem. Zei t. , 1 901 , 2 5 ,
In comparing results obtained by the foregoing methods it has beenfound that the method and duration of saponification have no materia linfluence
,provided that the alcohol used is subsequently completely

58 FIXED OILS , FATS , AND WAXE S .
expelled , and that the fatty acids are thoroughly dried . Thus thesolidification point of properly dried acids may be above that ofthe same acids in which a trace of moisture has been left . The formof the apparatus has an influence when the quantity of fatty acids usedis small
,and cooling from outside proceeds too rapidly . Until an in
ternational method has been fixed,i t i s advisable in giving results to
state the method by which they were obtained ;Tempera ture Tes ts .
—The ri se of temperature which ensues ontreating a fixed oil with concentrated sulphuric or nitric acid
,or
bromine,i s a measure of the extent and intensi ty of the chemical
action which ensues . The use of'
sulphuric acid was originally proposed by Maumené (Compt. rend.
,1 852 , 3 5 ,
Maumené Test—The following method of applying this test isrecommended by Archbutt
,and i s in common use : 50 grm . of the oil
are weighed into a 200 c .c . beaker,and the latter immersed in a
capacious vessel of water,together with the bottle of strong sulphuric
acid,until they are both at the same temperature
,which should not
be far from The beaker contain ing the oil i s then wiped,and
placed in a cotton-wool nest previously made for i t in a cardboarddrum
,or a wider beaker . The immersed thermometer i s then oh
served,and the temperature recorded . 1 0 c .c . of . the concentrated
sulphuric acid should then be withdrawn from the bottle with apipette
,and allowed to run into the oil . During the addition of the
acid,which Should occupy about 1 minute
,the mixture must be con
stantly stirred wi th the thermometer , and‘ the agitation continued
till no further rise of temperature ensues . This point i s readily ohserved
,as the indication remains constant for a minute or two , and
the temperature then begins to fall .The results obtained from a particular oil are remarkably constant ,when the acid i s of a uniform strength and a defined method of manipulation i s rigidly adhered to , but apparently insignificant diff erencesin the mode of operation result in serious discrepancies in the results .Thus
,Archbutt observed a rise of 78 . when the oil was stirred until
all the acid was added and the thermometer then held stationary inthe m iddle of the oil
,but when the stirring was continued until no
further ri se of temperature was observed,the increase was only 73 .
When the temperature exceeds 60° i t i s impossible to obtain concordant results . Maumené
,therefore
,advocated diluting highly unsatu
rated oil s,such as linseed or marine animal oil
,with olive oil , so as

of mineral oil for the purpose .preferable to either of these sub
6, 1 69) has
with tha t substance,the ri se of
in most cases,proportional to the
This indicates that,under such
4 1—43
1 04—1 24 1 04
—1 1 1
4 1—44
1 02—1 03 1 1 6
Skate-liver o i l 1 02
Menhaden o i l
0 0 0 0 0 0 0 0 0 0 0 0 0
O le ic ac id .

60 FIXED OI LS , FATS , AND WAXE S .
Owing to the notable difference in the rise of temperature caused bycomparatively slight variations in the mode of operating
,many of the
recorded figures obtained by Maumené’s test have little value . Hence
it i s desirable to compare a sample with one or more oils of knownpurity under exactly Similar conditions . The figures in the tableShow the kind of resul t to be expected from various oils , but they mustnot be relied on too rigidly .
From these figures i t will be seen that with some mixtures,for in
stance olive with cottonseed oil and rape with linseed oil,the rise of
temperature with sulphuric acid may afford a means of forming anapproximate estimate of the proportion of ingredients .In order to obviate the effects of the use of different strengths of acid,Thomson and B allantyne (J . S oc . Chem. Ind.
,1 89 1 , 1 0, 233) ascer
tain the ri se of temperature obtained on mixing 50 grm . of water wi th1 0 c .c . of strong sulphuric acid and under preci sely the same conditionsas those used for testing the oil . The specific temperature-reacti on ofthe oil i s obtained by multiplying the ri se of temperature of the oilacid mixture by 1 00
,and dividing by the ri se of temperature of the
water-acid mixture .I t i s probable that the Maumené test will
,ere long
,be entirely dis
placed by more accurate methods,though it is still frequently employed
as a rapid means of obtaining preliminary inf ormation .
The bromine thermal method devised by Hehner and M i tchell(Analyst, 1 895 , 20, 1 46) is based upon the fact that the heat evolved onthe addition of bromine to unsaturated fatty acids or glycerides is , as arul e
,proportional to the degree of u nsaturation . Thus , when once
a relationship has been established between the iodine value of anordinary unoxidised fat and i ts bromine thermal value obtained understandard conditions
,the degree of unsaturation (i . e .
,the iodine value)
of Similar fats may be rapidl y ascertained .
I grm . of, e. g .
,lard
,the iodine value of which has been accurately
determined by Hi ’i bl or Wi jS ’ method, i s dissolved in 1 0 c .c . ofchloroform or carbon tetrachloride in a small D ewar ’s vacuum-jacketed
tube , and the temperature of the solution taken by means of a standardthermometer graduated in tenths of a degree . 1 c .c . of bromine is thenintroduced by means of Hehner and M i tchell ’s bromine pipette , I the
1 This. consist s of a 1 c .c . pipet te , connected at th e top with a tube bent twice at
r igh t angles and conta in ing cau st ic lime kep t in po sit ion by means of a sbe sto s plugs .
2111
apply ing suct ion to th e end of this tube , a ll brom ine vapour i s reta ined by
t e ime .

S OLUBILITIE S .
mixture rapidly stirred with the thermometer, and the rise in temperature recorded .
The ratio between the values gives a factor (e . g .,
which,
when multiplied by the ri se of temperature observed under identicalconditions wi th a Similar fat or oil
,gives a result in close agreement
wi th the iodine value of the latter .An apparatus thus standardised for lard gives good results withother animal body fats
,with butter
,and wi th most unoxidised vege
table oils and fats . It does not give concordant values , however , withJapanese wool (tung) oil , blown rapeseed oil , blown cottonseed oil andboiled linseed oil
,evidently owing to substitution of the bromine
taking place in these cas es .The value and limitations of the bromine thermal process are discussed by Jenkins (J . S oc. Chem. Ind .
,1 897 , 1 6 , 1 94) and by Archbutt
(i bi d.,
and modifications have been proposed by Wiley (J.
Amer. Chem. S oc. ,1 896, 1 8 , 3 78) and by Gill and Hatch (ibid .
,1 899 ;
2 1 , These modifications off er no advantage over the originalmethod .
So lubi li ti es of Fats and Fixed Oils .
Fats and oils are,wi thout exception
,insoluble in water and aqueous
li quids generally .In cold alcohol the fixed oils are
,as a rule
,but little soluble , and the
solid fats and waxes still less so . In boiling alcohol,however, some of
the fluid oils dissolve to a considerable extent,especially if the solvent
is anhydrous . In many cases,statements as to the solubili ty can only
be regarded as giving a rough indication of the amount of free fattyacids in the sample examined . Speaking generally
,i t may be stated :
That oils containing the esters of lower fatty acids (e . g ., porpoise
oil , cocoanut oil , butter-fat) are exceptionally soluble in alcohol .That oils containing the glycerides of linolenic and isolinolenic acidsare fairly soluble . That castor and croton oil are readily soluble inalcohol
,and are sharply differentiated from most other oils by thi s
characteri stic .Ether
,chloroform,
carbon tetrachloride, benzene , and oi l of turpentinedissolve fixed oils readily
,and are in many cases miscible with them
in all proportions .P etroleum spi ri t is also an excellent solvent for most oils and fats ,though castor oil (q . v.) forms a striking exception, being practicallyinsoluble in that liquid .

FIXED OILS,FATS
,AND WAXE S .
Va len ta Tes t —A method of distinguishing between different fatsand oils was based by Valenta (Dingler’s polyt. J 1 884, 2 5 2 ,
296) on the differences in their solubility in glacial acetic acid . A hotsolution of the oil i s gradually chilled and the temperature at whichturbidity occurs recorded .
The incomplete solubili ty of rape oil and other oils from the Crucif erw,
even at the b . p . of acetic acid,i s noteworthy
,as are the low
figures found for linseed oil,nigerseed o il
,and menhaden oil
,as com
pared with those for the non-drying oils .The test i s open to the obj ection that the slightest vari ations in thestrength of acetic acid
,and in the method of stirring and observing the
turbidity point have the greatest influence upon the results .To obtain comparable results
,i t i s essenti al to follow invariably th e
same details of working,to use acids of exactly the same strength
,and to
see that the fat i s free from water . The most accurate method of ascertaining the strength of glacial acetic acid is to ascertain i ts sol idifying point
,and to compare the results with the figures given by Rudorff
(Pharm. J .,1 87 1
—2,
2, 24 1 )
Glacial acetic ac id containing So lidifying po int ;°
The modification of the method introduced by Chattaway , Pearmainand Moor (Analyst, 1 894 , 1 9 , 1 47) embodies the various precautionsnecessary for obtai ni ng concordant results ; but , at best , the methodShould only be regarded as a rapid preliminary sorting test , or as aconfirmation of results obtained by other methodsThe m ixture of grms
of the fat and 3 c .c . of glacial acetic ac id(99 . 5% strength) i s heated in a stoppered tube about 1 0 cm . long bycm . in diameter , which is immersed in hot water and Shaken until
a clear solution i s obtained . I t i s then left in warm water with a thermometer attached to i t until the contents become turbid . In the caseof oils that have been excessively heated
,no reliance can be placed upon
the test . The following results were thus obtained

64 FIXED OILS , FATS , AND WAXE S .
the cri tical temperature of solution . In this way the following resul tswere obtained :
W i th 90% alcoholB u t t er—fa t .MargarineArach is o i l .Cot ton seed Oi lS e same o i l .O live Oi l.Almond Oi l.Rape O i l (crudeRa pe Oi l (refined) .Hempseed o i l .Nu t Oi lCastor Oi lLin seed Oi l (oxid ised) .
Old and rancid butter-fat Shows a lower value than fresh butter-fat ,but after removal of the free fatty acids by treatment with sodiumcarbonate and washing the fat with hot water
,normal values are ob
tained .
Crismer’s figures for butter and margarine were in the main con
firmed by Asboth (Chem. Zei t. , 1 896, 20,
CLASSIFICATION OF FATS , OILS , AND WAXES .
In studying the characters of fixed oils , and identifying oils of unknown nature
,valuable assistance is obtained from a suitable arrange
ment of the oil s in classes or groups . The classification here adoptedis based on a joint consideration of the origin
, p hysical characters , andchemical constitution of the oils . An attempt i s likewise made toclassify the oils so that each group contains some important commercial oil which is typical of the other members of the group . Thus ,the oils included respectively in the rape-oil
,olive-oil
,and cocoanut-oil
groups present a more or less close resemblance to rape-oil , olive-oil ,and cocoanut-oil
,respectively .
I . Ol ive-O i l Group—Vegetable Oleins .
—The oils of this group havea sp . gr . ranging from to and hence are , as a rule , lighterthan the oils of Groups I I I
,IV
,and V. Their vi scosity i s notably
greater than that of the drying oils,but inferior to that of rape oil , and
they do not lose their power of producing a greasy stain on paper ,however long they may be exposed to the air. They yield very solid
Wi th alcohol of sp . gr.
Min eral Oils (variou s)Va lve O il.An ima l o i lS h eep sfoot o i lLard O i l .
Neat sfoot Oi lCo lza o i l . .
apan fi sh Oi l.u t t er-fat ( 1 0 sample s)

CLA S S I FI CATION OF FATS,OILS
,AND WAXE S .
products in the elaidin test,and are also characterised by their
relatively low iodine values and medium saponification values . Theyc ontain olein as a main constituent , with smaller quanti ties of theglycerides of saturated fatty acids (palmitin , stearin , arachidin ,
etc .)and in some cases
,at all events, of glycerides of more unsaturated
fatty acids such as linolic acid . They yield no insoluble bromides ontreatment with bromine
,and their fatty acids yield
,at most , only
traces of linolic tetrabromide .I I . Rape-O i l Group—The oils in this group are derived from the
Cruciferae. They are classed as non-drying oils,though this character
istic i s less pronounced than in the case of the oils in Group I , from whichthey may be distinguished by their low saponification values , and by forming a paste-like product in the elaidin reaction . Some of these oils resemble linseed oil in yielding an insoluble product on treatment withbromi ne , possibly the bromide of a mixed glyceride containing linolenicacid . Their low saponification values are probaby due to the presence of glycerides of erucic acid
,C 2 2H4 ,
O , ,whilst ralpic acid (i someric
with oleic acid) i s present in rape oil .I II . Co t tonseed-o i l Group —In sp . gr. these range from to
the values of the crude oils being somewhat higher than thoseof the refined products . They are usually classified as semi -dryingoi ls from the fact that they come between the non-drying oils and thedrying oils, both in this respect and in their chemical composition .
They have fairly high iodine values,and their fatty acids yield con
siderable amounts of linolic tetrabromide on treatment with bromine .
In the elaidin test they yield soft solid masses,intermediate between
the hard products given by the oils of Group I and the fluid productsfrom the drying oils . They consist largely of linolin and olein
,with
smaller quanti ties of glycerides of solid fatty acids and traces oflinolenin .
IV. Linseed-o i l Group—Drying Oi ls .
—They range in sp . gr .from to and are thus distinctly heavier than the oils ofthe preceding groups . They are not solidified by treatment with nitrousacid, evolve great heat in the Maumené test , and have high iodinevalues . Linseed oil (and to a less extent some of the other oils) yieldsa large amount of an insoluble bromide on treatment with bromine .On exposure to the air in thin l ayers they absorb oxygen and formvarni shes which are at first sticky
,but subsequently become plastic or
brittle . The viscosity of the drying oils i s less than that of the precedVo l . I I .
—5

66 FIXED OILS,FATS
,AND WAXE S .
ing groups . In composition they differ from the semi -drying oils incontaining a greater proportion of the glycerides of the highly unsat
urated acids (linolenic and isolinolenic ac ids). The composition andproperties of tung oil differ greatly from those of other members of thisgroup .
V. Cas tor-O i l Group —The oils in this group have little in common
,though some are characterised by their great viscosity and high
sp . gr . Castor,curcas and croton oils have also the characteristic
of ready solubili ty in alcohol and glacial acetic acid,and of marked
purgative properties,but curcas oil i s less soluble in alcohol than the
others . In castor oil and grapeseed oil glycerides of a hydroxy-acidsuch as ricinoleic acid predominate
,and are indi cated by the high
acetyl value of the oil . Croton oil has a high Reichert-Meissl value,
due to the presence of glycerides of volatile fatty acids .VI . Cacao-bu t ter Group —Vegetable Fats .
—This group includessolid fats
,consisting mainly of glycerides of higher fatty acids such as
myri stic,palmi tic
,stearic and oleic acids . They contain only small
amounts of glycerides of volatile acids as i s Shown by the low ReichertMei ssl values . The fairly high iodine values point to the presence ofa considerable proportion of glycerides of oleic and , probably , linolicacids .VI I . Coconu t-o i l Group — Vegetable Fats
—The members of thi sgroup are fats of high sp . gr . and with low saponification values . Theyalso include the two commercial vegetable “ stearins ” obtained fromcoconut and palmnut oils . The typical fats of the group (c oconutand palmnu t oils) contain a considerable amount of the glycerides ofthe lower fatty ac ids
,whence their high Reichert-Meissl values . They
are also distinguished from the fats of the preceding group by theirhigh saponification values ( indicating glycerides of lower fatty acids)and low iodine values ( indicating the small proportion of unsaturatedglycerides) .
VI I I . Lard-o i l Group .—Animal Oleins .
—In thi s group are included the oils
,fluid at ordinary temperatures , which are obtained
from terrestrial animals . They have lower iodine values than the corresponding vegetable non-drying oils (Group I), though they alsoyield more or less solid products in the elaidin test . They consis tmainly of olein with smaller quantities of palmitin , stearin , and probably linolin .
IX . Ta l lowGroup —Animal Fats .
—The tallow group comprises the

CLAS S I FICATION OF FATS , OILS,AND WAXE S . 67
which are solid or semi- solid at thedy fats consist of stearin ,
palmitin,and
n and other glycerides whilst bu ttermembers of the group by its high sp .
high Reichert-Meissl value,due to
considerable amount of the glycerides of butyric andother lower fatty acids . Animal fats may be distingui shed fromvegetable ~ fats by the phytosteryl acetate test (q .
X . Wha le-O i l Group —Marine Animal Oi ls —This group comprises the maj ority of the fluid oils obtained from fish and marinemammals . They are distinguished as aclass by their off ensive fishyodour
,which becomes more perceptible on warming ; by the reddish
brown colour they assume when subjec ted to the action of chlorine ; andby the reddish or reddish-brown colour produced on boiling them witha solution of caustic alkali . Wi th concentrated sulphuric acid theygive considerable rise of temperature and colorations
,varying from
light red to purple and brown . Most members of the group drymore or less on exposure to the air
,and yield but little solid elaidin on
treatment with ni trous acid .
i
In these respects they resemble the y egetable oils of the cottonseed group
,and have similar sp . gr . The oils
from the sperm and bottlenose whales are peculiar,as regards physical
characters and chemical constitution,and form a separate class (Group
X I) .
“ Train oil ” includes the oil from the blubber of any marinemammal .On treatment with bromine
,many of the oils of this group yield an
insoluble bromide,which may be distinguished from the similar
product given by linseed and other drying oils by turning black whenheated .
Porpoise oil is characteri sed by its high saponification value and
high Reichert-Meissl value due to the presence of glycerides of valericacid . The other oils in the group consist largely of glycerides of veryunsaturated acids
,some of which are i someric with linolenic acid , and
others still more unsaturated . Some of them ,such as codliver and
other liver oils,also contain a considerable amount of cholesterol and
allied biliary products .X I . Sperm-o i l Group.
—Liquid Waxes—The ,members of this
group differ from all the fatty oils of previous classes in consisting essentially of esters of the-ethyl series . In thi s respect they resemblethe true waxes
,but are fluid at the ordinary temperature . They are

FIXED OILS , FATS,AND W E S .
of less sp . gr . than the true oils at the ordinary temperature and at theb . p . of water ; and on saponification yield considerable proportions ofsolid higher homologues of ethyl alcohol . They do not dry or thickennotably on exposure to air and yield sol id elaidins on treatment wi thni trous acid .
X II . Spermaceti Group .-Waxes .
—The members of this groupare solid at ordinary temperatures
,and more or less resemble beesv ax ,
the prototype of the class . They consist essenti ally of esters of thehigher radicles of the ethyl series
,with in some cases an admixture of
higher monatomic alcohols and higher fatty acids in the free state .Carnauba wax seems also to contain diatomic alcohol radicles . Spermand bottlenose oils (Table X I) resemble the waxes in consti tution , butare liquid at ordinary temperatures . The substances known as Japanwax and myrtle wax (Table VI I) are fats , not true waxes . Parafli n
wax and mineral wax are hydrocarbons,and hence quite different in
chemical constitution from the true waxes of animal and vegetableorigin .
The following tables give the values likely to be obtained inthe examination of the chief oils
,fats
,and waxes of commercial
importance :
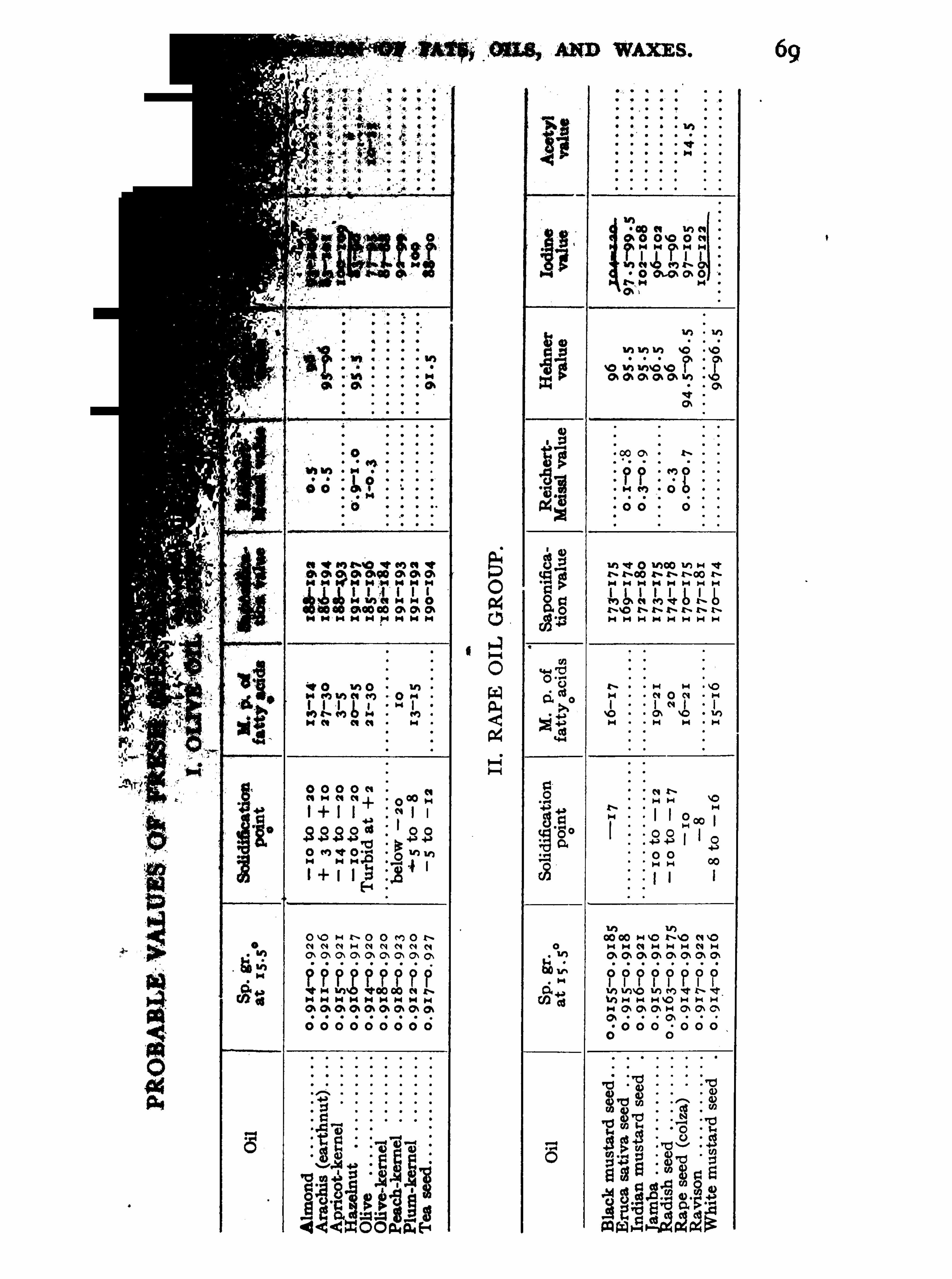
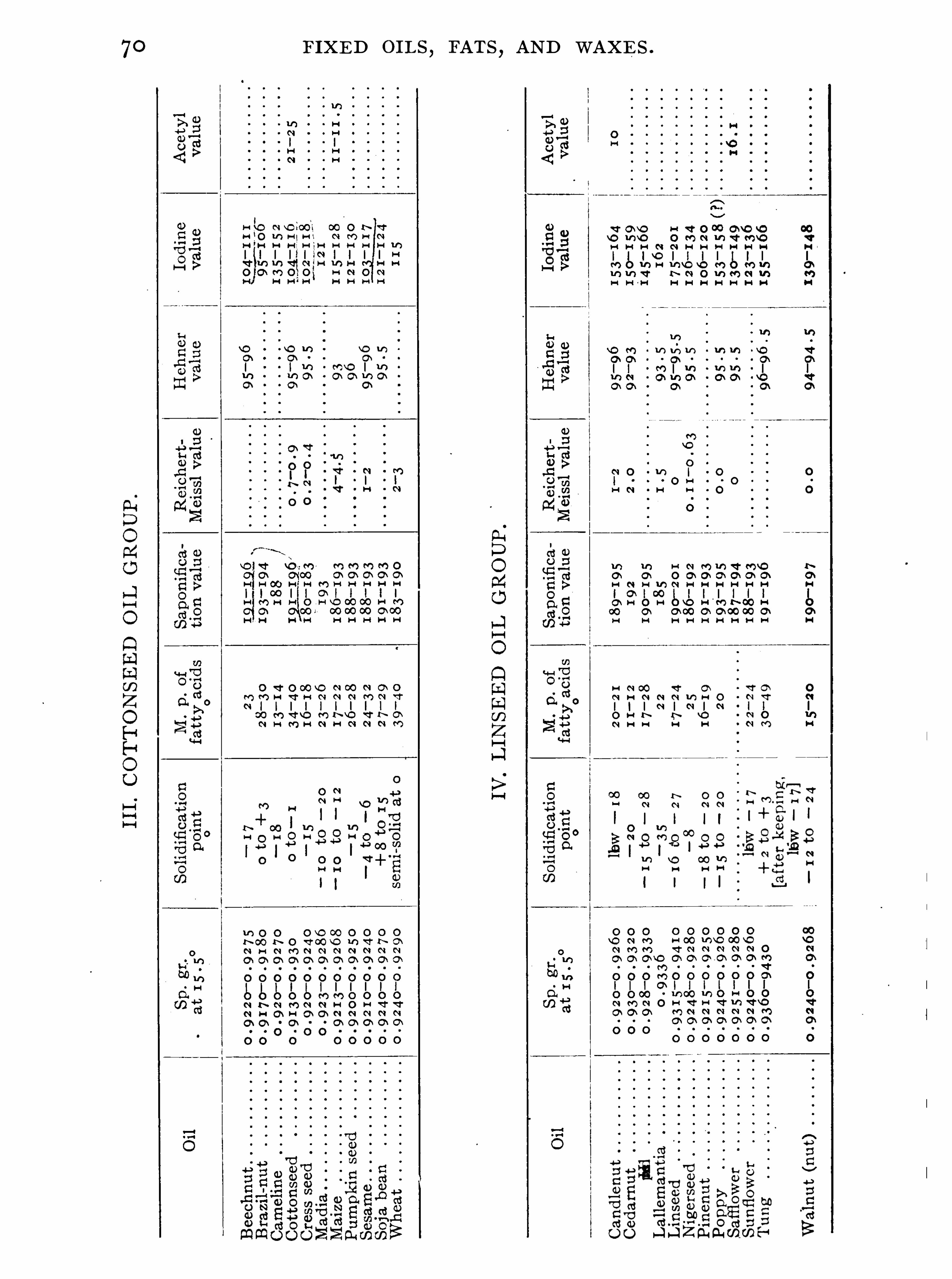
FIXED OILS,FATS
,AND WAXE S .
10 m mV )D‘
)
a
v
m
vmlmu
ou
OH
IO“
mm
vu
lva
a
n
wmlh
u
«H
IH
H
H
N
ION
va
l
ou
«H
I
IA
"3200.
.
ucm
aoo
x
c
o
te
“
m
+
ou
w
ar
SI
3200.
on
I
Op
mH
I
ON
I
On
mu
I
w
]
“m
l
00.
ca
l
mm
"
I
0»ma
l
ON
"
I
320
2
u
se
was“.
ao
aouczw
L
o
go
gam
haao
m
a
zco
cm
m
voomuo
wm
z
pcowa
s
mfice
ao
ze
d
“0
00
380
3
s
h
ampo
o
a
sso
zo
ca
o
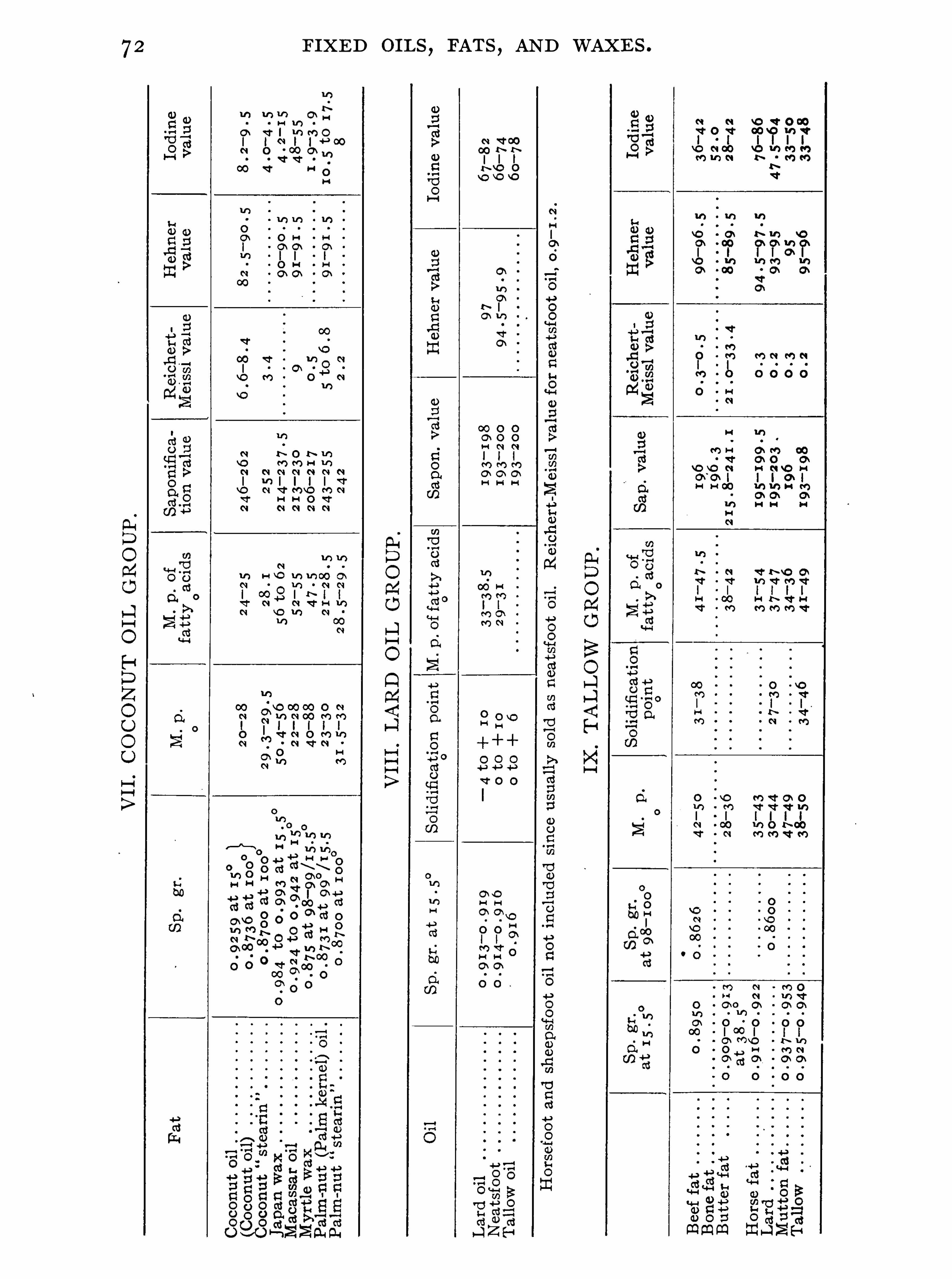
FIXED OILS,FATS , AND WAXES .
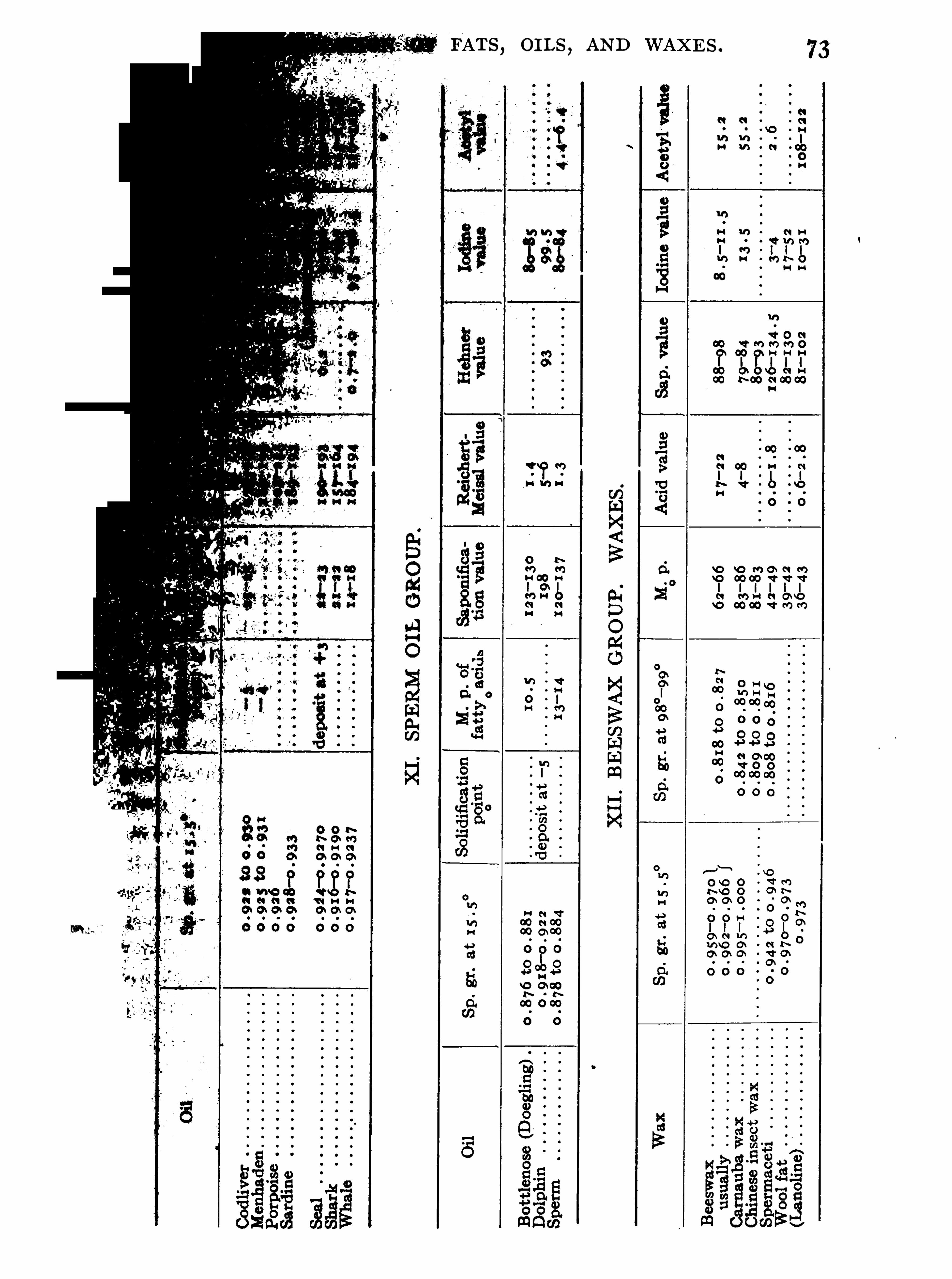
FATS,OI LS
,AND WAXE S .

FIXED OILS,FATS , AND WAXE S .
EXAMINATION OF FATS AND CRUDE OILS FOR FOREIGNMATTERS .
The term foreign matters used in this connection indicates substances added to the oils
,such as rosin
,soaps
,hydrocarbon
,water and
mineral matter as well as excess of free fatty acids,but does not apply
to substances whi ch are natural constituents of an oil such as colouringmatters
,cholesterol
,albuminous substances or chlorophyll .
T he estimation of water,curd
,and salt in fats such as butter and
margarine is described in the special section dealing with butter . An
oil,if clear
,may be regarded as free from such extraneous matters , and
their presence in a fat may usually be detected by melting the sample .If an opaque or opalescent oil result
,or one containing visible particles
of suspended matter or globules of water,i t Should be p ii rified from
these by filtration through dry paper before proceeding to search forrosin
,fatty ac ids
,soap
,or hydrocarbons .
Soap i s sometimes directly added to an oil , but i ts presence i s morefrequently due to the use of alkali employed to increase Sp . gr . andviscosi ty . Soap i s readily detected by dissolving the oil in about 3times its volume of ether or carbon tetrachloride
,adding a little water ,
and agitating the whole thoroughly in a separating funnel . The soapwill dissolve in the water
,while the other foreign matters will dissolve
with the oil,in the ether or carbon tetrachloride
,and may be recovered
therefrom by distillation . The soap may be estimated by evaporatingthe aqueous liquid and weighing the residue after drying atThe proportion of soap may also be inferred from the amount of carbonate left after igniting the oil .Inso luble soaps are not infrequently present in oils , waste greases,
and pharmaceutical preparations Though insoluble inwater
,many of them are soluble in ether or petroleum spiri t . They
may be decomposed by agitating the mixture with dilute sulphuricacid
,when the acid l iquid will contain the metal of the soap , and a
corresponding quanti ty of fatty acid will dissolve in the oily layer .When i t i s desired to ascertain the proportion of free fatty acidsoriginally present in the oil
,a titration with alkali Should be made both
before and after shaking with dilute acid . The difference betweenthe two estimations represents the fatty acid l iberated by thetreatment .Free Ac id in Oi ls .
—Commercial oils and fats very frequently con

EXAMINATION OF FATS AND CRUDE OI LS . 7 5
tain notable proportions of free acid,which may either be mineral
acid, as a result of incomplete separation after refining , or free fatty
acid resulting from unskilful refining or from the natural decomposition
Minera l ac i ds are only accidentally present in fixed oils,and
usually in very small proportions . Even minute quantities arehighly obj ectionable in oil intended for lubricating
,bu t are harmless
when th e article i s to be used for soap-making . M ineral acids may bereadily recognis ed by agitating the oil with warm water , separatingthe aqueous liquid
,and testing it with a solution of methyl-orange ,
which will give an orange or red colouration if any mineral acid bepresent . The nature of the mineral acid
,which is most commonly
sulphuric,can then be ascertained by testing the aqueous liquid with
barium chloride,silver nitrate
,and other appropriate reagents . O il s
which,from over-treatment with acid during refining
,contain a sul
phonated fatty acid,must be boiled with water for some time , in order
to decompose the compound .Free fa tty aci ds are often normally present
,and in some oils
(e . g ., olive and palm) may exist in very large proportion . Free oleic
acid is largely used as a lubricant in wool-spinning,and free palmitic
and - stearic acids are employed for making candles and night-lights .All are used for soap-making . Their proportion is estimated as described in the section (page 9) dealing with the Acid Value .
Rosin aci ds present in the sample will be estimated by the aboveprocess as fatty acids . Their separation from the latter is describedbelow . JI/I ineral aci ds will aff ect the accuracy of the results unless anallowance i s made for them or they are previously separated by re
peatedly agitating tli e ‘
oil with water . S oap and hydrocarbons do no tinterfere with the estimation .
The estimation of the acid value may be supplemented by agravimetric estimation . The resultant alcoholic liquid is separatedfrom the oil
,the alcohol evaporated
,and water added . This solution
i s agitated with a little petroleum spiri t (not ether) to dissolve suspendedoil , the aqueous liquid separated , and the fatty acid liberated from thesoap solution by adding dilute sulphuric acid . On agitating withether , separating the ethereal solution , and evaporating it to dryness ,the fatty acids can be weighed . This method should be used whenrosin acids may be present . In their absence , the estimationShould be fairly concordant with the resul t of the titration . S oap

76 FIXED OILS , FATS,AND WAXE S .
should be previously separated . Mineral aci ds and hydrocarbons donot interfere with the results .Ros in .
—Common rosin or colophony , which is described in a specialsection
,i s added to oils to impart certain properties
,but its employment
often r enders them wholly unsuitable for their intended purposes .One of the methods of detecting rosin is by the brown colour it imparts to sodium hydroxide . The original sample i s saponified , thealcohol boiled off
,and the liquid treated with sufli cient sodium hydrox
ide solution to cause precipitation of the soap . The solution,separated
from the soap by decantation or fil tration through glass-wool,will be
dark brown if rosin is present . The same method serves for the recogni tion of rosin in soap , previous saponification being unnecessary .
The method may also be applied to the mixture of fatty and resin acidsseparated in the manner described in the table on page 22 . Thedissolved rosin may be recovered by acidifying the alkaline liquidwith hydrochloric acid
,When a precipitate of resinous odour will be
formed . The rosin may be isolated by agitating the li quid with etherand evaporating the ethereal layer to dryness
,and may be identified by
its physical and other tcharacteristics .
In the absence of free fatty acids,rosin may be isolated from fixed
oils by agitating the sample with moderately strong alcohol,separating
the solution and evaporating i t to dryness . I t may also be '
i solated,
and approximately estimated,by ti trating the alcoholic solution of the
sample with alkali and phenolphthalei n as described elsewhere . AS
the several acids which ordinary colophony contains are not presentin constant proportion
,the neutralising power of rosin i s variable ,
ranging from to grm . of colophony for 1 c .c . of N/ Ialkali . The rosin subsequently extracted from the acidified aqueousliquid , and left on evaporating the ethereal solution to dryness , i sreadily recognisable by the ‘taste and smell on heating , and oftenshows the physical characteri stics of rosin .
In the last method of operating,the rosin is obtained in admixture
with any free fatty acids the sample may have contained . Thesemodify the physical properties of the extracted rosin very materi ally ,and render the method useless for quantitative purpo ses . In suchcases
,if there i s sufficient material for the purpose , a good indication
of the relative proportions of fatty and rosin acids in the mixture maybe obtained by observing the sp . gr . at the temperature of boilingwater
, as described on page 48 . As,however
,rosin varies consid

EXAMINATI ON OF FATS AND CRUDE OILS . 7 7
erably in sp . gr . and the fatty acids from various oils exhibit Similarvariations
,the method furni shes but very rough results unless the
source of the fatty acids be defini tely known .
Es t ima t ion of Ros in Aci ds —The most satisfactory method ofseparating rosin acids from fatty acids is that of Twitchell (J . S oc .
Chem. Ind,1 89 1 , 1 0, 804) which yields much more accurate results
than other methods . I t i s based upon the fact that aliphatic acids areconverted into ethyl esters when acted upon by hydrochloric acid gasin their alcoholic solution ; whereas colophony undergoes little or nochange under the treatment
,abietic acid separating from the solution .
The rosin gives an acid indication in alcoholic solution with phenolphthale‘i’n
,and interacts readily with potassium hydroxide to form a
soluble soap . All that i s necessary,therefore
,i s to make the fatty
acids interact with alcohol and to titrate the rosin acids with standardalkali ; or they may be treated with potassium hydroxide and theresulting rosin soap separated from the saponified fatty esters bymeans of petroleum spirit .(a). Gravimetric Method—From
'
z to 3 grm . of the mixture of fattyacids and rosin are dissolved in I O times their volume of absolutealcohol in a flask
,and dry hydrochlori c acid introduced in a moderate
stream . The flask is set in a vessel with water to keep it cool . Theacid is rapidly absorbed
,and
,after about 45 minutes , the esters sepa
rate,floating in the solution
,and no more hydrochloric acid is ab
sorbed . The current of gas i s now stopped,and the flask allowed to
stand for half an hour to complete the reaction . The liquid is dilutedwith about 5 times i ts volume of water and boiled until the acid solution i s clear
,the esters
,with resin in solution
,floating on the top . To
this i s added some,petroleum spiri t
,and the whole transferred to a
separating funnel,the flask being washed out with petroleum Spiri t .
The acid solution is then run off,and the petroleum-spirit solution
(which ought to measure about 50 c .c .) washed once with water andthen shaken in the funnel with a solution of grm . potassiumhydroxide and 5 c .c . of alcohol in 50 c .c . of water . The rosin i s immediately saponified and the two layers completely separated . Thesolution of rosin soap can then be run off
,treated with acid , the rosin
collected in any manner desired,dried
,and weighed . A second
washing of the soap with petroleum spiri t i s hardly necessary , as veryli ttle remains after the first extraction .
(b). Volumetri c Method.
—The first stages of the volumetric method

78 FIXED OILS,FATS , AND WAXE S .
are similar to the gravimetric,with the exception that the contents of
the flask are washed into the separating funnel with ether instead ofpetroleum spiri t
,and the ethereal solution in the funnel i s then thor
oughly washed with water until the wash-water i s no longer acid ; 50 c .c .of alcohol
,previously neutralised
,are then added and the -solution
titrated with standard sodium hydroxide solution with phenolphthale’i'n as indicator . If the combining equivalent of rosin is known
,i ts
percentage may be calculated,or some of the original mixture may be
also ti trated,when the diff erence in sodium hydroxide required will
correspond to the fatty acids converted into esters .The average combining equivalent of the samples of rosin examinedby Twitchell was 346 , and a closely Similar value was found by Lewkowi tsch (J . S oc . Chem. Ind
,1 893 , 1 2
,Hence an approxi
mately correct result for the amount of rosin may be Obtained bymultiplying the number of c .c . of N/ I alkali used in the titration by
Certain varieties of commercial rosin ,however
,have combin
ing equivalents differing very considerably from this average value .
The results of test experiments have shown that in practice the volumetric figures though usually too high
,are more accurate than the
gravimetric figures,which are usually too low . A critical examination
of this method was made by Lewkowi tsch (loc. ci t.)Hydrocarbons .
The hydrocarbons most commonly added to fatty oils are1 . Those produced from petroleum and by the distillation of bi tumi
nous shale .
2 . Those produced by the distillation oi common rosin,having the
nature and properties detailed in the section on “Rosin O il .”
3 . Neutral coal oi l; being the portion of the products of the distillation of coal-tar boiling above and freed from phenolic bodiesby treatment with soda .
4 . Solid paraffine , employed for the adulteration of beeswax andspermaceti
,and used in . admixture with stearic acid for making
candles .Detection of Hydrocarbons .
—The presence of hydrocarbons infats and fatty oils i s detected by the altered sp . gr . of the sample ,which i s decreased by members of the first class
,and increased by
rosin and coal-tar products ; by the lowering of the flashing and b . p .
by the fluorescence of members of the first two classes ; and by the ln

80 FIXED OI LS,FATS
,AND WAXE S .
The aqueous liqui d is drawn off through the tap into a beaker .About 1 0 c .c . of water and a few drops of caustic alkali solution areadded to the ether which remains in the separator
,and the whole
agitated . The washings are then run off in their turn,and after re
peating the treatment with water , which i s removed by the tap as before , the ethereal solution is poured off through the mouth into aweighed flask . The aqueous li quid and washings are then returnedto the separator
,and agi tated with a fresh quantity of ether , which i s
washed and poured into the flask as before .The agitation of the soap solution i s repeated once more
,to complete
the extraction of the hydrocarbon oil . The ethefeal solution willusually b e strongly fluorescent . The flask containing i t i s attached toa condensing arrangement
,and the greater part of the ether distilled
off by immersing the flask in boiling water . When distillation hasceased
,the condenser i s detached and the flask placed on the top of
the water-oven to eliminate the rest of the ether . Sometimes thehydrocarbon will contain globules of water ; in this case the flaskShould be held horizontally
,and rotated rapidly
,so as to spread the
oil over the Sides in a very thin layer,and facil i tate the evaporation of
the water . When no more water is vi sible,and the smell of ether i s
scarcely perceptible,the flask is placed on i ts Side in the water-oven
for 1 0 or 1 5 minutes and weighed, I when the increase of weight overthe original tare gives the amount of hydrocarbon oil extracted .
Prolonged heating Should be avoided,as many hydrocarbons are
appreciably volatile at This i s notably the case with coal-taroil
,and hence
,in analysing m ixtures containing it
,the heating in the
water-oven Should be wholly dispensed with . Wi th rosin oil, parafli n
wax,and the denser mineral oils there i s but little danger of loss by
volatili sation atThe results obtained are correct to within about 1 % in all ordinarycases .2 Where extreme accuracy i s desired
,i t i s necessary to re
member that most,if not all
,animal and vegetable oils contain traces
of matter wholly unacted on by alkalies . In certain cases,e . g .
,butter
1 S omet imes i t i s very d iffi cu lt t o obta in a constant we ight by th e means ind ica t ed inth e t ext . In such case s , inst ead Of heat ing th e fla sk on th e wat er-oven , i t Sh oul d b e ke pton th e bath o f bo iling wa te r and a m odera t e current of a ir , fi lt ered by pass ing i t througha tube conta ining co t t on-wood , sh ou ld b e blown th rough i t by a second tube passing throughth e cork. Th e fi t t ings are th en detach ed , and th e fla sk h ea t ed for a Sh ort t im e in th e wat erOven .
2 Traces of fat ty o ils which had e scaped sa p on ifi ca t ion and traces of soap are ap t to passinto th e e th ereal so lu t ion , and h ence th e proport ion o f unsapon ifi ab le mat t er found i s
oft en S ligh t ly reduced on t rea t ing th e e ther-residu e w ith a lcoh o lic po ta ssium h y droxideso lu t ion , and aga in ext ract ing th e so lu t ion of th e soap with e ith er .

HYDROCARBONS .
fat and cod-liver oil,thi s consists largely of cholesterol
, C QGHM O ,
which may be obtained in characteri stic crystalline tablets by warming the ethereal extract with alcohol , and allowing the solution tocool . The proportion of unsaponifiable matter soluble in ether whichi s naturally present in fixed oils and fats
,rarely exceeds and is
usually much less . Sperm and bottle-nose whale oils,however
,con
stitute an exception,yielding about 38 to 40% of matter soluble in ether .
Spermaceti and the other waxes yield after saponification large percentages of matter to ether
,and hence the p rocess i s not available for
the estimation of parafli n wax in admixture with these bodies , thoughit gives accurate results with the mixtures of paraffin and stearicacid so largely employed for making candles .The following table indicates the behaviour of the constituents ofcomplex mixtures of fats
,oils
,and waxes
,when the aqueous solution
of the saponified substance i s Shaken with ether :
Disso lved by the e the r Rema ining in the aqueous liqu id
Hydrocarbon o ils ; including Fatty acids .
Shale and petro leum o ils,Rosin o i l hydrocarbons , Rosin ac ids .
Coal-tar o i l,Paraffi n wax and ozokerite
,Pheno l,Vase line . Cre so ls
,Neutral re sins . and other pheno ls .
Unsaponified fat or o i l.
Unsaponifiable matter,as cho lestero l
from liver o ils,e tc . G lycerol (glycerine).Dodecyl alcoho l, from sperm and
bottlenose o ils .
Cetyl alcoho l, from spermaceti . Excess of potassium hydroxide .
Myricyl alcoho l, from beeswax .
Co louring matters,as from palm o i l.
The hydrocarbon having been isolated by saponifying the sampleand agitating with ether
,its nature may be ascertained by observing
its sp . gr .,taste
,and smell
,and behaviour with acids and bromine .
If the proportion be small,i t may be necessary to operate on a larger
quanti ty than 5 grm . of the sample . A good approximation of theSp . gr . of the extracted hydrocarbons may be made on Hager ’s principle ,by adding a drop of the oil to very dilute alcohol
,or ammonia , and ad
j usting the strength of the liqui d so that i t may be identical with that ofthe drop of oil (see p . The Sp . gr . of the dilute alcohol i sthen ascertained in the usual way . The fluorescence of hydrocar
Vo l. I I .
-6

82 FIXED OILS , FATS , AND WAXE S .
bons is best observed in the manner described on page 79 . It oftenbecomes intensified by treating the extracted hydrocarbon with anequal volume of strong sulphuric acid .
The odour and taste of the hydrocarbons are often highly characteristic of their origin . The smell of coal-tar oil i s readily observed ;and the taste
,especially the after-taste
,of rosin oil i s : not to be mis
taken . The smell produced on strongly heating a drop of the oil in aplatinum capsule i s also highly characteristic . Further details re
specting the tests for hydrocarbons are given in the section on “Min
eral Lubricating O ils .The higher alcohols from sperm and bottlenose oil may be separatedfrom hydrocarbons by treating the ether-residue with rectified spirit
,
which dissolves the alcohols without materially affecting the hydrocarbons .If the aqueous liquid separated from the ethereal layer be treatedwith dilute sulphuric acid
,the fatty acids are liberated
,and may be
weighed,ti trated with standard alkali
,or otherwise examined .
When i t i s merely desired to ascertain approximately the proportionof hydrocarbon oil in a mixture
,and not to i solate i t and examine i t
further,there i s no occasion to extract the solution of the saponified
oil with ether . Instead,the aqueous liquid may be at once acidified
with dilute sulphuric acid,a little ether added to promote the separa
tion of the mixed hydrocarbon oils and fatty acids,the aqueous
li quid drawn off,and the oily layer repeatedly agitated with water till
the washings are no longer acid to li tmus . Rectified spiri t and a fewdrops of phenolphthalei n solution are then added
,and the liquid
titrated with N/ 1 0 alkali .The amount of acid
,calculated as oleic acid
,multiplied by
gives the amount of saponifiable substances , and the difference may beregarded as unsaponifiable matter .The latter represents the hydrocarbons
,and the former the fat or
fixed oil of the mixture,provided that waxes
,including sperm and
bottlenose oils,are absent .
When the nature of the fat or oil i s known,and i t i s merely desired
to estimate the proportion of hydrocarbon present,and not to ascertain
i ts exact character,a very fair approximation to the truth can be
obtained by ascertaining the saponification-equivalent of the sample .The table on page 82 gives an outline of the processes described inthe foregoing section .
FORE IGN ADMIXTURE S .
.
0000
000
:
“200
300
00
000
500000
0
40000
0
-00
0
0
0
40000
0
$20
00
$30
0
0
$0000
50
00
00
-000
0
00
0
00
00
0
00
0
300
0
00
000
0
0
0
3000
000
0
0
000
00
0
030
0
0
5:
0
0000
00
N
.
00
0
0
000
300
0
0
200
0
00.0
0
000
0
000
00
0
00
000
00m00
m~00
o
n
00
00
00
0
00
300
M0
EO
0
000
3
0000
30
0
0
00
0
0
0
00
80
0000
.
0
0
00
30
0
00
0
3000
0
5
.
00
0
0
000
30
80
0
000
00
000
000000
0
00
00:
0
000
0
0
00
000
00
000
00
00
00
90.
.
00
00
0
000
3.
000
0
200
000
000
000.
000
0
3
800
300
0000
0
200
-00
0
400000
0
00
0
0
0
m.
EO
M
0
30
00
0
0000
000
300
00000
00000
00
0
00
00
00
0
03
@
40
0
.
000
0
3m000
00
000
30
00
00
30
00
000
00
0000
00
0
300
00
0
0000
.
v
OmmM00
00
0
0
00
20
00
0
0
00
:
Owa
m00
010
00
000
£0
3
80
0
000
00
50\
00
00
00
00
00
000
00
000
00
0
0
50
va
m00
000
00
00
000
00000
00
300
0
200
4
00
5:
0
000
00
4
00
00
0
0000
00
0
00
0
00
00
0
000
00
0
30
00
000
000.
00
00
0
000
3000
0
00
0
30
0000
20
000
3
30
20
0000
00
00
000
0
000
000
000
:0
0
00
00
000
040
500
0
0
00
00
20000
000
0
00
00
00
-0000
00
09
.
0
00
00
000
0
00
000000
0
0
00
0000
0
00
0
000
0
500
00
0000
000
00000
0
0
30
00
00
300
0
0000
00
.
0
00
0
30
0
00
0000000
00
000
0
300
0
00
0000
00
$00
000
300
20000
5000
00
08
.
000
0
200
0
80
000
00
0
000
0
0
00
000
0
300
0
00
<
.
00
00
0
00
:O
0
0
0000
30w
0
0
4000a
0
0
000
0
200
0
000
00000
0
3
00
00
0
0
0
.
0000
00
0000
-000
00
000
0.
0
0
0
0
000
00
.
0
0000
0
0
0
000
000
0
h0
0
0
0
-300
0
00
000
00
0
0
0
0
0
0
0
0
00
00
00
000
e
s
3
30
Boa.
00000
:0
00
0000
00m000
0
0
000
0:0
000
000
0
00
00
0
00
0000
000v
OmN
E00
000
000
300
000
040
.
ee
e
soe
no
00
000000
-0
0
0
00
00
300
00
00
88800
20
0
00
00
000
000
000
0
00
000
090
0
0
0
0
o
o
.
00
5
000
3
000
0000
0
0
3
0000
80.
.
eoo
no
o
00
00
80
898
00
00
00
000
00
-0
0
0
30000
0
00
0
00
000
.
o
000
0
0
0
0
0
0
30
0
00
000
0000030
00
0H
00
00
0000
zo
ne
30
S
0
0000
0
00
00
000
0
05:
0
000
00
4
.
om
00m
-0
0
300
000
0
0
0
0
0
0
:
0
300
00
0
00
000
00.
00
00
0
00
.
0m
0m0
0
0
0
m0
0
3
0
0
0
an
000m0000
0
t
mm
00
00
“
00
0
00
0
3
000
.
000
0
0
.
00
000
000
3
0
0
00
0
3
0
0
3000
0H
.
02000
000
00000
00
:
0
3
000
000
00
(00
0
8
500
00
00
00
0
0
0
0
0
30
80
000
0
0
“
.
0000
0w0
000
000
000
2
3
-00
0
00
0
30
000
0
2
00
00
00
00
0
3300
0
300
00
00
2.
0
00000
0
300
000
0
000
00
00
00
0
00
0w0
0
3
000
00
00
00000
0
00
00
000
000
000
0
80
00
0
300
000
0
0
0
80
0
8
300
380
3

84 FIX ED OILS , FATS , AND WAXES .
IDENTIFICATION OF FATS AND FIXED OILS .
The recognition ofan unmixed fat or fixed oil may usually be effectedby a careful application of the methods of examination already described . Systematic schemes for the purpose have been devised
,but
cannot be implicitly relied on,owing to the variable nature of the
substances themselves . Most of the colour tests are of li ttle value,
unl ess confirmed by other indications .In examining fats and oils for the detection of adulteration
,the
relative commercial value of the different kinds should be kept inview . In addition to the adulteration of the more valuable substanceswith the cheaper
,the use of hydrocarbons derived from the distillation
of petroleum,shale
,coal
,and rosin
,i s also extensively practi sed .
In practice i t i s often of less importance to know the origin of asample than whether i t may be used as a substitute for the genuineoil . This may be ascertained with tolerable certainty
,and
,in some
cases,the nature of the adulterants may be definitely detected .
By the following systematic method identification may generally beeffected
, and much inf ormation gained that will suggest the specialtests
,for the substances suspected to be present :
1 . Pl ace a drop of the oil on the back of the tongue by means of aglass rod and taste i t carefully
,avoiding too hasty a decision . In thi s
manner,marine animal oils
,l inseed
,croton
,mineral
,rosin
,and some
other oil s may often be detected . Rosin oil is remarkable for thenauseous after-taste produced by it . Rancidity may also be recognised by taste .
2 . Heat a portion of the sample in a porcelain or platinum capsuleto about 1 40 or and observe the odour ' careful ly . When suffi
ciently cool , pour a little into one hand , rub with the other , and smellagain . A li ttle practice will allow of vegetable oils being readily distingu i shed from animal oils , and the products of fish and marine mammals from those of terrestri al animals . The odour on heating will alsofrequently permit the recognition of mineral and rosin oils
,and , if the
remainder of the sample be strongly heated till i t igni tes and the flamethen blown out
,the vapours will often have a characteristic odour .
3 . Ascertain the sp . gr . of the sample at 1 5 . if fluid at that temperature ; but at the b . p . of water (page if solid at the ordinarytemperature . This test i s valuable
,but if the sample be very old , or a
mixture of several substances,or if much free acid be present , the
indications are less reli able . An unmixed substance may , as a

IDENTI FICATION .
ru le,be placed in one of the groups on pages 86—7 , though thi s
classification must only be regarded as giving a rough preliminaryindication . Many of the fats and oils might be classified in morethan one of the groups . More defini te figures are given in the tableson pages 69—73 .
Sperm and bottlenose oils are readily distinguished from shale andpetroleum products of similar density by the elaidin test
,their saponifi
cation values and the quanti tative results of their saponification .
Their estimation,when mixed with hydrocarbon oils
,may be effected
as described under “Sperm O il .
”Oleic acid is distinguished from
hydrocarbons by i ts solubili ty in an aqueous solution of sodium hy
droxide . M ixtures of oleic acid and hydrocarbons may be analysedby titration with standard alkali . If fixed oils are present
,the methods
given on page 84 should be used .
Di fferent ia t ion of An ima l andNou -dry ing Vegetable Oi ls —Thenon-drying vegetable oils may be distinguished from the similar o ils ofanimal origin by their taste and odour on heating . Their iodinevalues and the m . p
.of their fatty acids are higher . Many of the
vegetable oils show absorption spectra,which is never the case with the
animal oils . The phytosteryl acetate test (q . and the oleorefractometer reading are also valuable means of differentiation .
The vegetable non-drying oils may be distinguished from each otherby various tests . Rape and mustard oils are distinguished fromothers by relative insolubility in glacial acetic acid by low saponifi
cation values,and by yielding small amounts of an insoluble bromide
on treatment with bromine . B one oil usually gives an orange or reddish-yellow elaidin of a pasty consistence
,while lard oil and tallow
oil yield a firm product of a pale or lemon-yellow colour . Theproduct from neatsfoot oil i s vari able .Coconut olein i s distinguished from other vegetable oils by i ts high
saponification value , low iodine value, and the very moderate heatingproduced by sulphuric acid .
Differentiat ion of Sem i-drying and Non-dry ing Oi ls .—The
semi-drying oils,of which cottonseed and maize oils are typical , have
higher iodine values and sp . gr . than the non-drying oils . They mayalso be distinguished by the large amount of linolic tetrabromide(m . p . 1 1 3 to I which they yield on adding bromine to a solution oftheir insoluble fatty acids in petroleum Spiri t or carbon tetrachloride .The nature of the product formed in the elaidin test i s also instructive .
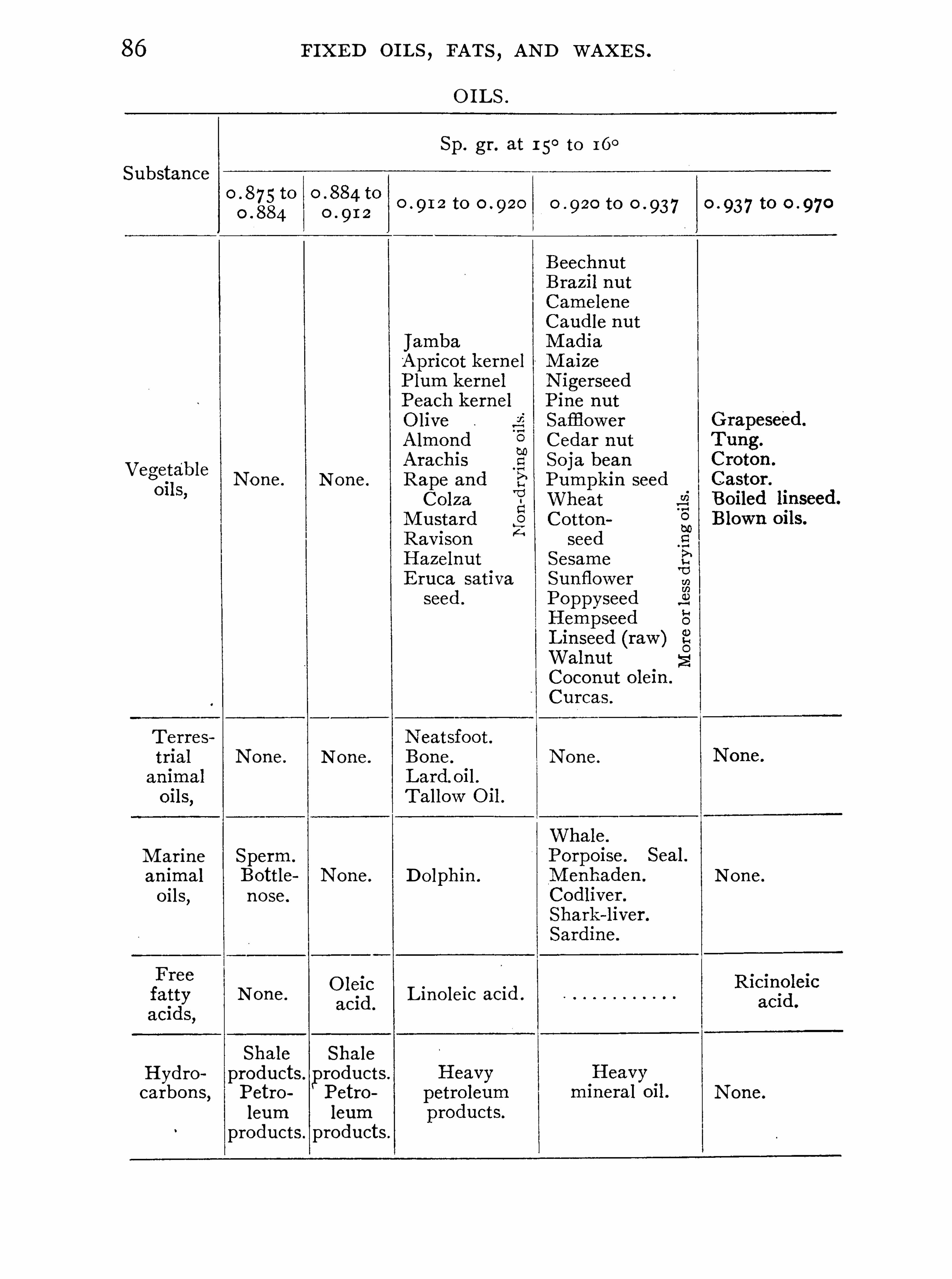
86 FIX ED OI LS , FATS , AND WAXE S .
O ILS .
Sp . gr. at 1 5° to 1 6°
Substance
No ne . None .
None . None . None .
M arine Sperm .
animal Bottle None . Dolph in . None .
o ils,
nose .
None . L ino le ic acid .
Shale Shale
Hydro products . products .
carbons, Petro Petro None .
leum leum
products . products .

88 FIXED OILS,FATS , AND WAXE S .
D iff eren t ia t ion of Drying and Sem i-dry ing Oi ls —The dryingoils differ from the semi-drying oils in having a higher iodi ne value
,
and in yielding a solid film in a short time,when exposed to the air
in a thin layer . Some of them give insoluble bromides on treatmentwith bromine
,and an insoluble deposi t of linolenic hexabromide
(m . p . 1 80 to on brominating a solution of their fatty acids .Different iat ion of Dry ing andMar ine An ima l Oi ls .
—These oils,
which often have similarly high iodine values,are best distinguished
by the behaviour of the deposit formed in the insoluble bromi de test(9 . i f .) when heated , and the nature of the unsaponifiable matter .The sulphuric acid colour test and L ivach e
’s drying test will also
afford valuable information in the case of m ixtures . On saponifi
cation,marine animal oils give a much darker soap solution than lin
seed or other drying oils .They may be distinguished from one another by their analyticalvalues (see tables). Porpoise oil and some varieties of whale oil contain a notable proportion of esters of lower acids
,and give characteristic
Reichert-Meissl values .Diff erent iat ion of Various Oi ls .
—O ils of sp . gr . above arefew and easily distingui shed . Croton and castor oil are purgativeand readily soluble in alcohol
,but have little further resemblance .
B oiled linseed oil and Japanese wood (tung) oil have sp . gr . between and dry rapidly on exposure
,and give a firm
brown or black clot with sulphuri c acid . B lown oils closely resemblecastor oil
,but may be distingui shed as described in the section treat
ing of that oil . Rosin oil has a sp . gr . exceeding and is notsaponified to any considerable extent by alkalies . I t i s readily identified by its strong after-taste , and the odour of turpentine developed ,when the sample i s heated till i t catches fire and the flame then blownout . M ixtures of rosin oil with fatty oils may be analysed as describedon page 84 .
Hydrocarbons and Waxes .-The solid hydrocarbons having a
density below at the b . p . of water are described under “ParaffineWax .
The distinctions between the various waxes are fully indicated inthe table on page 73 , and in the special sections on “ Spermaceti
,
”
“B eeswax
,
”and
“ Carnauba Wax .
”Free acids are at once distin
guished from the waxes by their solubility in alcohol , behaviour withalkalies
,and their saponification values ; from each other by their

IDENTIFICATI ON .
m . p . and combining weights . Vaseline and similar hydrocarbons aresharply distinguished from the waxes and fatty ac ids by being incapable of saponification .
D iff eren t ia t ion of An ima l and Vege table Fats —These are bestdifferen ti ated by the phytosteryl acetate test (9 . TA), and distinguishedfrom one another by a comparison of the various analytical values .Coconut and palmnut oils are soft
,melt readily
,and have high sa
ponification values , fairly high Reichert-Mei ssl values and low iodinevalues . Japan and myrtle wax are hard , wax-like bodies of comparatively high m . p . (See
“ Japan Wax”
) Palmnut oil i s distinguishedfrom coconut oil and coconut stearin ” by its taste and smell .Butter- fat i s the only common fat of animal origin that has a highReichert-Meissl value .The nature of the sample having been indicated
,further confirma
tion may be obtained by means of the tables commencing on page 69 .
The principal fats,oils
,and waxes are described at greater length in
the following sections .In the case of a sample consisting of a mixture of wholly unknown sub
stances , identification of the constituents is often a diffi cult problem,
but when the leading component i s known or can be recogni sed,the
detection of the others becomes more feasible . In most cases oils cannot be recogni sed by distinct and specific tests
,such as exist for the
different elements ; and , in arriving at a conclusion as to the compositiou of any sample of mixed oils , the analyst must be content to beguided in a great measure by c ircumstanti al evidence and a carefulconsideration of probabilities . The foregoing methods of examinationare of course employed
,and
,in addition
,such special tests as will be
found described under the various heads . The sub-articles descriptiveof the more important substances contain a li st of the admixturesmost commonly found in each and special tests suitable for theirdetect ion .

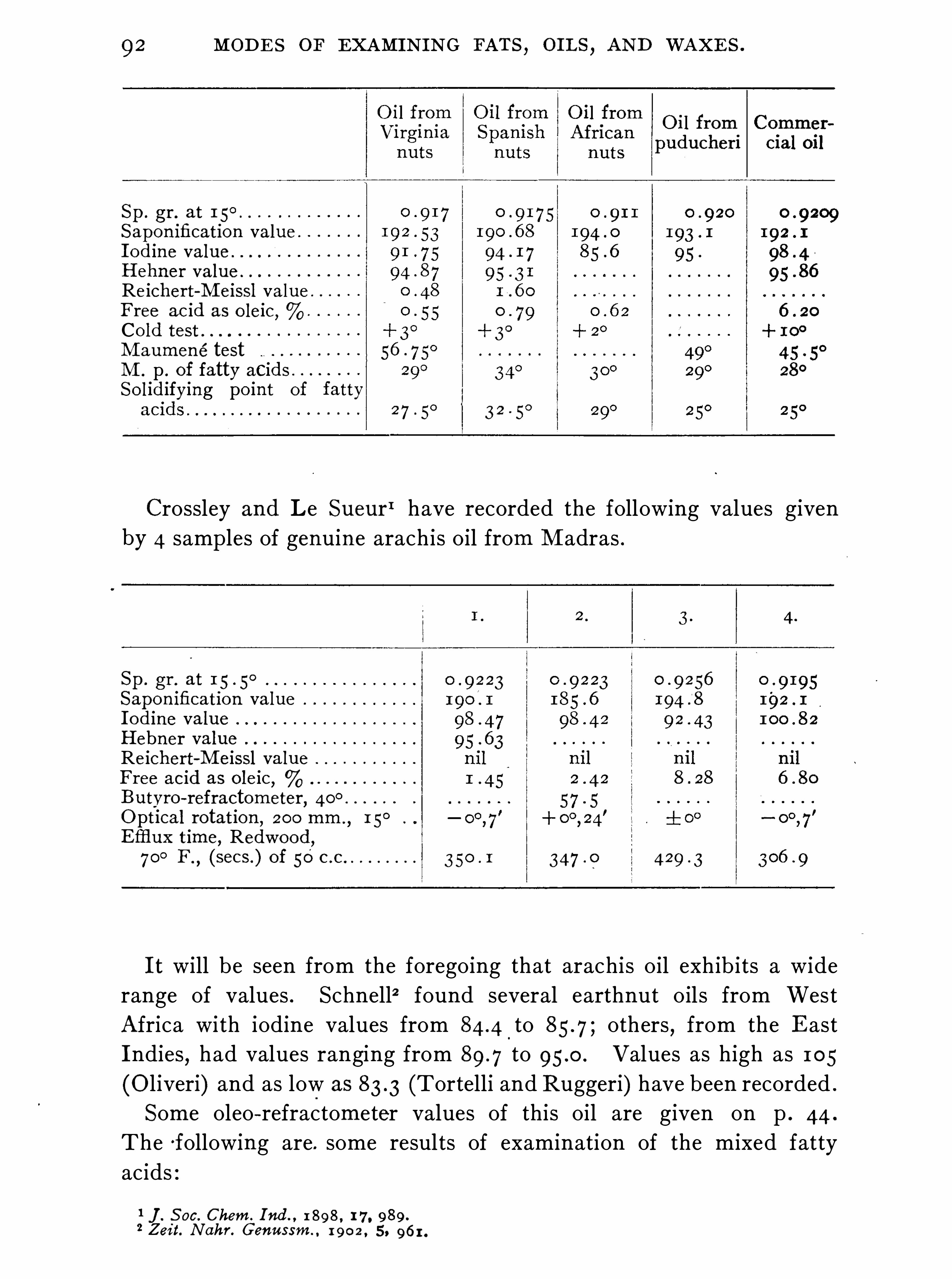
92 MODE S OF EXAMINING FATS,OI LS
,AND WAXE S .
Sp . gr. at
S aponification valueIod ine value .
Hehner value .
Re ichert-M e issl value .
Free ac id as o le ic,Co ld test
Maumené testM . p . of fatty acids.
So lidifying po int of
2 7 60
3 2 0 50
Crossley and Le Sueur I have recorded the following values givenby 4 samples of genuine arachis oil from Madras .
Sp . gr . at
Saponification valueIod ine valueHebner valueRe ichert—M e issl valueFree acid as ole ic
,
B utyro-refractometer
, 40°
Opt ical rotat ion,200 mm .
,1 5
°
Efllux t ime , Redwood ,70
° F ., (secs ) of 50c .c
I t will be seen from the foregoing that arachis oil exhibits a widerange of values . Schnell2 found several earthnut oils from Wes tAfrica with iodine values from 84 .4
_
to others,from the East
Indies,had values ranging from to Values as high as 1 05
(Oliveri) and as low as (Tortelli and Ruggeri) have been recorded .
Some oleo-refractometer values of this oil are given on p . 44 .
The f ollowing are. some results of examination of the mixed fattyacids1 S oc. Chem. Ind . , 1 89 8 , 1 7 . 9 89 .
2 Zei t. Nahr. Genussm . , 1 9 02 , 5 , 9 6 1 .

ARACHI S OI L .
Sp . gr. at
Sp . gr. at
T iter test .Refractive index at 60°Iod ine value of m ixed fatty ac idsIod ine value of liqu id fatty ac ids
Arachis oil contains from to of arachidic and lignoceric acids ,which
,owing to their sparing solubili ty in cold alcohol
,can be isolated
without much difli culty . O live and most other oils , except those ofrape and mustard seed
,contain not more than traces of these acids .
Upon this difference in composition,Renard I based the following
process for the detection and estimation of arachis oil,which is
described with some modifications in detail introduced by Archbutt :21 0 grrn . of the oil are saponified in a deep porcelain basin with 8 c .c .
of sodium hydroxide solution (containing approximately 50 grm .
sodium hydroxide in 1 00 c .c .) and 70 c .c . of alcohol , boiled downgently to about 20 c .c .
,rinsed with hot water into a separating funnel ,
decomposed with hydrochloric acid in excess,and Shaken with ether to
extract the fatty acids . After distilling off the ether in an 8—oz . widenecked flask
,the fatty acids are dried by heating the flask on a steam
bath and sucking out the vapour, and are then dissolved by pouring
50 c .c . of rectified alcohol (sp . gr . into the hot flask .
To the solution,which should not be hotter than and must not be
allowed to cool below lest crystals of fatty acids should separate ,5 c .c . of a 20% aqueous solution of lead acetate are added , which wil lprecipitate the whole of the arachidic and lignoceric acids as leadsalts
,together with some lead stearate and oleate .3 After cooling
to about 1 5° and allowing to stand for half an hour , the alcoholicliquid is decanted through a filter
,and the lead soaps are washed on
the filter once with ether . They are then rinsed back into the flaskand digested with ether
,again filtered and again rinsed back and
digested with ether . After doing this about 4 times , using the samefilter each time
,all the soluble lead salts will have been dissolved out .
The extraction with ether should be continued until the washings g ive1 Compt. rend . , 1 8 7 1 , 7 3 , 1 3 3 0 .
S oc. Chem . I nd . , 1 89 8 , 1 7 1 1 24 .
3 This quant ity o f lead i s suffi cient for 1 0 g rm . of arach is o i l . If more i s added , a largerprec i pitat e i s produced , conta in ing m ore lead o leat e . wh ich take s more wash ing ou t W i the th er , bu t no more arach id ic and lignoceri c acids are obta ined

94 MODE S 0F
‘
EXAMINING FATS , OILS , AND WAXE S .
no colour, or only a pale brown colour , when Shaken with aqueoushydrogen sulphide .The filter paper containing the lead arachidate
,etc .
,i s opened in a
l arge plain funnel placed in the neck of a separating funnel,and
,before
the soaps have had time to dry,they are rinsed into the separator with
a j et of ether from a washing bottle . The soaps which adhere to thepaper and flask may be decomposed and transferred by rinsing withwarm dilute hydrochloric acid
,followed with ether . About 20 c .c .
more hydrochloric acid sp . gr .) are poured into the separator ,which is stoppered and shaken until all the lead salts are decomposed .
The aqueous liquid is then run off,and the ethereal solution of the
fatty acids washed with small quantities of water until the leadchloride i s removed . The ether i s distilled off in an 8-oz . flask
,and
the residual fatty acids are heated in the water-oven until ‘dry . Theyare then dissolved by warming with 50 c .c . of 90% ethyl alcohol (sp .
gr . and the solution is cooled to I when arachidic and lignoceric acids
,if present
,will crystalli se out
,either at once or after stand
, ing a short time . The flask should be closed by a cork carrying athermometer .According to Tortelli and Ruggeri , I a rough estimate of the amountof earthnut oil present may be made at this stage by observing the temperature at which the crystals commence to form . For thi s purposethe liquid in the flask must be heated until the crystals have redissolved ,and then allowed to cool slowly .
commence to form ; Earthnut 011;
In order to estimate the proportion of earthnut oil more accurately,
the liquid is allowed to stand from 1 to 3 hours , with occasional agitation
,at 1 5
° or or at some intermediate temperature which i snearest to that of the laboratory ; the crystals are then collected on a
1 Chem . Zei t 1 89 8 , 2 2 , 600 .
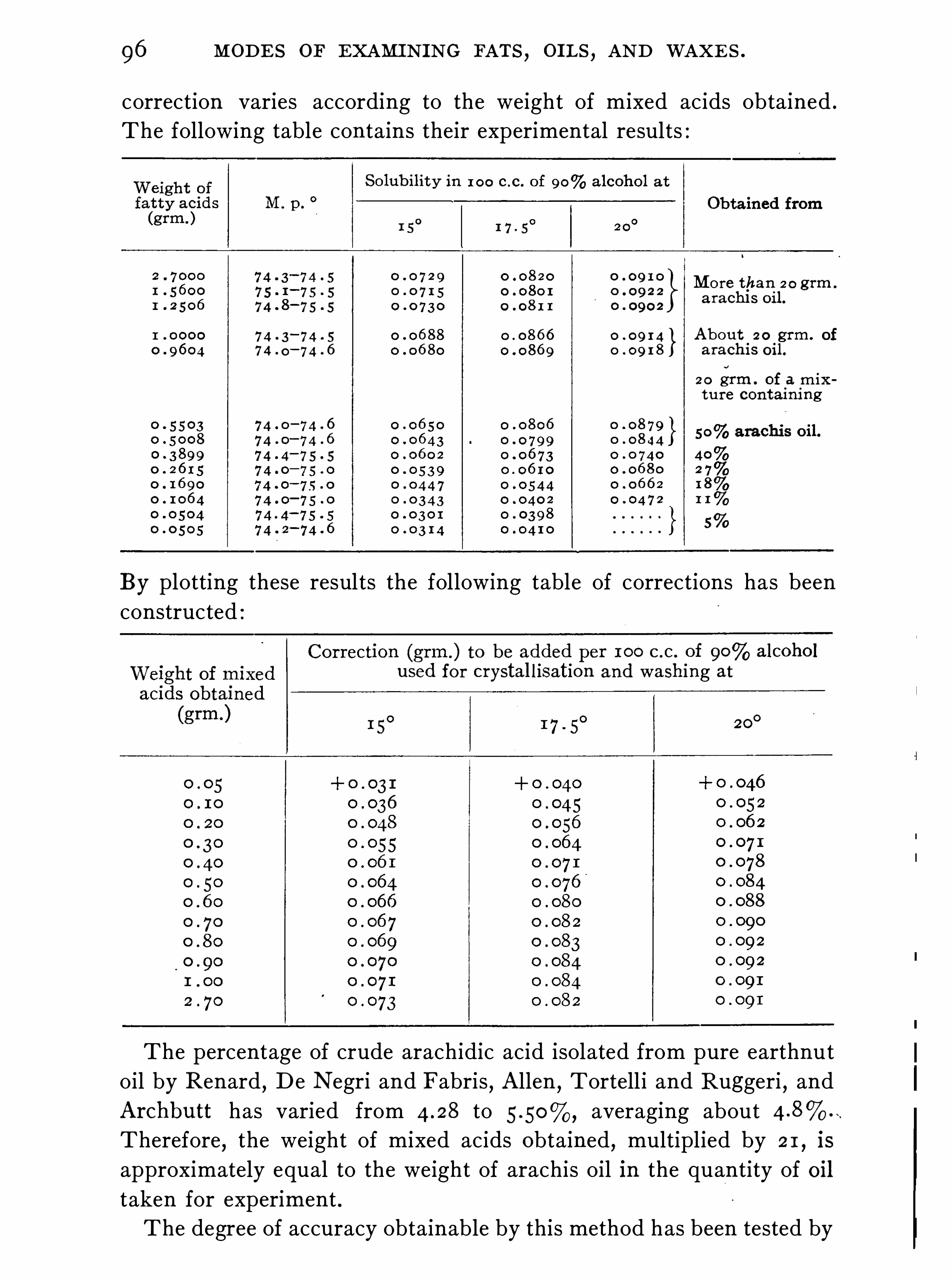
96 MODE S OF EXAMINING FATS , OILS , AND WAXES .
correction varies according to the weight of mixed acids obtained .
The following table contains their experimental results :
Mo re than 20 g rrn .
arach is o il .Abou t
.
20.grm .
7 4 0—7 4 6 o 09 1 8 arach i s 011.
d
20 grrn . of'
a. m ixture containi ng
By plotting these results the following table of corrections has beenconstructed
used for crystallisation and wash ing at
The percentage of crude arachidic acid isolated from pure earthnutoil by Renard
,D e Negri and Fabris , Allen , Tortelli and Ruggeri , and
Archbutt has varied from to averaging aboutTherefore
,the weight of mixed acids Obtained
,multiplied by 2 1
,i s
approximately equal to the weight of arachis oil in the quantity of Oiltaken for experiment .The degree of accuracy obtainable by this method has been tested by

his oils , and recrystalli s ing the
h4. p
7 4 -5
7 5 -I
7 4 -1
0 23 3 7 5
1 Lewk ow i t sch , Oils , Fat s and Waxes , 2 , 2 5 3 .
Vo l . II .
—7
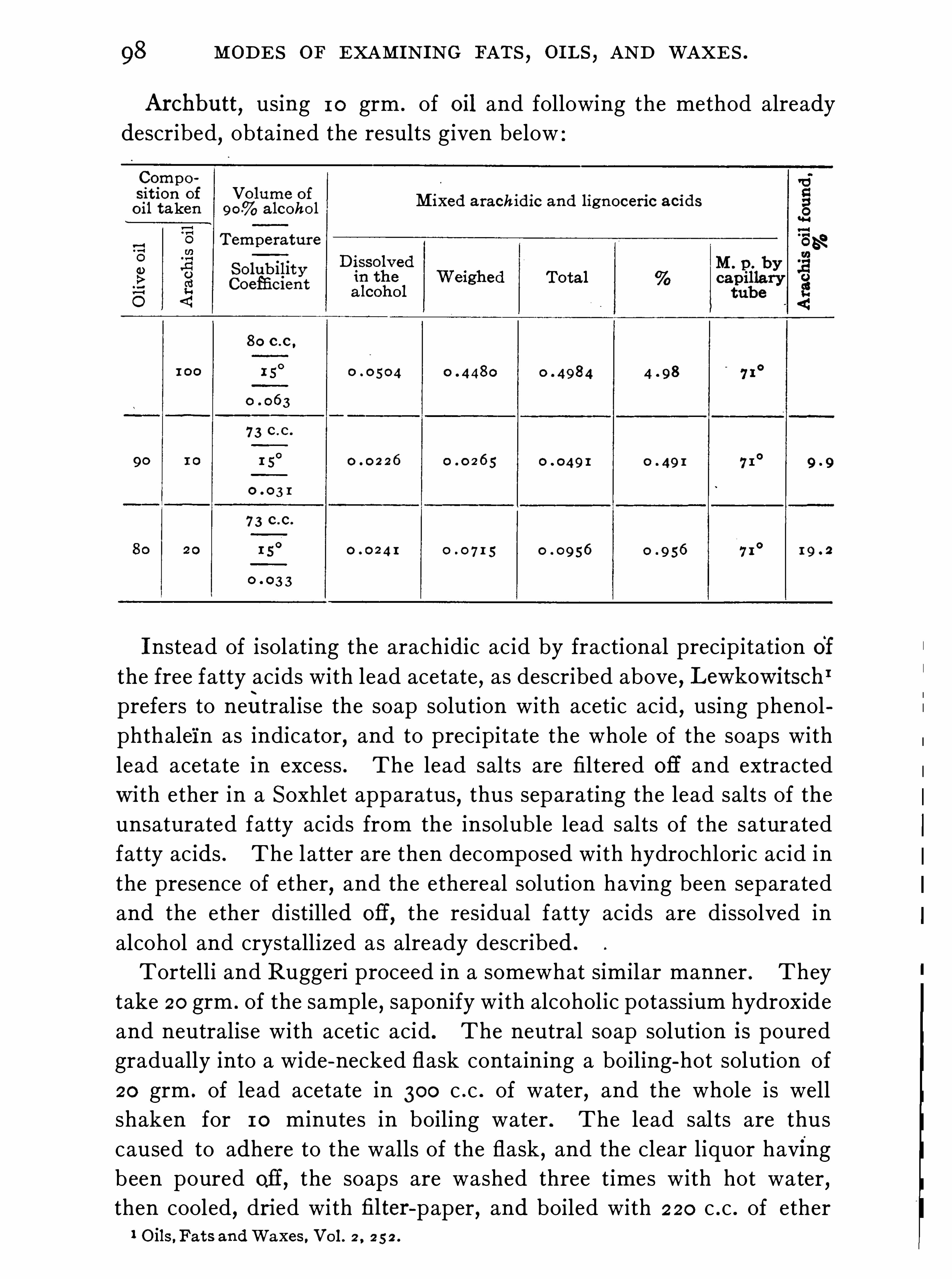
98 MODE S OF EXAMINING FATS , OILS , AND WAXE S .
Archbutt, using 1 0 grm . of Oil and following the method already
described, obtained the results given belowComposit ion Of Vo lume o f011 taken 9 0% a lcoho l Mi xed arach i d i c and li gnoceri c aci ds
TemperatureS olubilit yCoeffi c ient
80 c .c ,
1 00
7 3
7 3
Instead of isolating the arachidic acid by fractional precipitation Ofthe free fatty acids with lead acetate
,as described above , Lewkowitsch I
prefers to neutralise the soap solution with acetic acid , using phenolphthalei‘n as indicator
,and to precipitate the whole of the soaps with
lead acetate in excess . The lead salts are filtered Off and extractedwi th ether in a Soxhlet apparatus , thus separating the lead salts of theunsaturated fatty acids from the insoluble lead salts of the saturatedfatty acids . The latter are then decomposed with hydrochloric acid inthe presence of ether
,and the ethereal solution having been separated
and the ether distilled Off,the residual fatty acids are dissolved in
alcohol and crystallized as already described .
Tortelli and Ruggeri proceed in a somewhat similar manner . Theytake 20 grm . of the sample , saponify with alcoholic potassium hydroxideand neutralise with acetic acid. The neutral soap solution is pouredgradually into a wide-necked flask containing a boiling-hot solution of20 grm . of lead acetate in 300 c .c . of water , and the whole i s wellshaken for 1 0 minutes in boiling water . The lead salts are thuscaused to adhere to the walls of the flask
,and the clear liquor having
been poured off, the soaps are washed three times with hot water ,then cooled
,dried with filter-paper , and boiled with 220 c .c . of ether
1 Oils , Fat s andWaxes , Vo l . 2 , 2 5 2 .

I OO MODE S OF EXAMINING FATS,OI LS
,AND WAXE S .
acid diluted with water to 1 50 c .c . i s,approximately
,of the right
strength].Weigh I grm . of the sample into a dry boiling tube
,add 5 c .c . Of the
alkali solution and boil gently over a small flame,holding the tube in
the hand,until saponification i s complete , which will take rather more
than 2 minutes,avoiding evaporation as far as possible . '
Add 1 .5 c .c .of the acetic acid
,or just suffi cient to neutralise the 5 c .c . of alkali
solution,mix well
,rapidly cool by placing in water at 1 7° to 1 9
° andleave in the water for about 30 minutes (not less), shaking occasionally .
Then add 50 c .c . 70% alcohol containing 1 % by volume of hydrochloric acid shake well
,and again place in the water for 1 hour .
If no arachis Oil be present,a clear or opalescent liquid is formed ; i f
more than 1 0% of arachis Oil be present , a flocculent , crystalline precipitate remains ; even with 5% of arachis oil a distinct precipitate remains and separates on standing .
Industrial neutral olive Oils , known in commerce as'
Saponified
O ils and prepared from the olive residuum Oils and oils of the th irdpressing , which frequently contain as much as 3% or even more ofunsaponifiable matter , apparently derived from the Shell of th e Olive ‘
kernel,may give a floccul ent precipitate in Bellier’s test, though free
from arachis oil .For the quanti tative estimation of arachis oil , B ellier takes 5 grm .
,
saponifies with 25 c .c . of the alcoholic alkali solution ,exactly
neutralises with acetic acid,and places in running water for 1 hour .
The precipitate i s collected on a filter,and washed with 70% alcohol
containing 1 % of hydrochloric acid at 1 5° until the
filtrate does not become perceptibly turbid on the addition of water .The residue i s dissolved Off the filter with 25 to 50 c .c . of boilingrectified alcohol
,which is then mixed with sufficient water to reduce
the strength to and kept at 20° for 1 hour . The crude arachidicacid i s filtered
,washed with 70% alcohol free from hydrochlori c acid ,
and weighed . The 111 . p . should be about By thi s process , B ellierobtained of crude arachidic acid from B ordeaux earthnut Oiland from a sample of Marseilles oil . A large number Of
European and African olive oils which were examined yieldedifromnil to of fatty acid
,the latter amount , corresponding to
of earthnut oil,being Obtained from an Oil from Tunis . Known
mixtur es of olive Oil and arachis Oil gave correct results when analysedby this process . Samples of cottonseed oil and sesame oil also gave .

ARACHI S OIL .
small quanti ties of insoluble acids corresponding to andrespectively
,of earthnut oil . This process i s much shorter than
Renard ’s,but needs further investigation . The reviser has Obtained
good results by the quali tative,but low results by the quantitative
method .
In the examination of samples of arachis Oil for adulterants,an
estimation of the crude arachidic acid Should be made,as it i s the
most characteristic test for this Oil . Tortelli and Ruggeri found thefollowing percentages in arachis oil from different sources
Buenos Ayre s , expressed at 45°to
50°
Bueno s Ayres , extracted with ethe rRuffi sque , extra ,
1 st press ing .
Rufli sque , fine , 2d press ing .
Gambia,extra
,1 st pre ss ing
French (commerc ial O i l)S panish (commerc ial O i l)
Sesam e O i l should always be looked for,as i t‘ i s frequently present
in large quantity . Soltsein 1 found it by the B audouin test in allsamples of commerc ial arachis Oil examined by him
,and states that i t
i s customary to add sesame oil to the finest grades Of arachis oil withthe Object of lowering the cold test and improving the misc ibili ty of theoil for salads . As arachis Oil i s frequently offered as a lubricating oilin place of olive oil , the absence of sesame Oil i s important , as evengenuine arachis oil has more strongly marked drying properties thanolive o il
, and any addition of sesame oil increases the tendency tooxidise . Sesame Oil may be detected by ,
the furfural test . I t willraise the sp . gr .
,also the iodine
,Maumené
,and oleo-refractometer
values . Sesame oil contains more linolic acid than arachis Oil .Poppy o i l would also raise the sp . gr .
,iodine value
,Maumené value
and oleo-refractometer value of arachis Oil,and would lower the
solidifying point of the oil and of its mixed fatty ac ids , as well as increasing in a marked degree the tendency to oxidise.
Co t tonseed o i l would be indicated by Halphen’s colour test ,
1 Chem. Rev. F ett-Harz-Ind . , 1 9 0 1 , 8 , 20 2 .

1 02 MODE S OF EXAMINING FATS , OI LS, AND WAXE S .
and by the much higher iodine value of i ts liquid fatty acids and muchlarger yield of tetrabromides .
Rape o i l would lower the saponification value and increase theviscosity in a marked degree .
ALMOND OIL .
(See also p . Almond oil i s a fixed Oil expressed from eithersweet or bitter almonds
,the kernels of Prunus amygdalus . The oil of
commerce i s mostly obtained from bitter almonds (P . amygdalus
amara), the marc of which i s then distilled with water to obtain theessential oil . Fixed oil of almonds must not be confounded with theessential oil of bitter almonds . I t i s largely employed in the preparation of ointments and emulsions
,for which it i s better adapted than
Olive Oil .Almond oil i s nearly Odourless
,of a straw-yellow colour and bland
taste . I t does not solidify till cooled to about some samples onlybecoming turbid at that temperature . According to the GermanPharmacopoeia , almond oil should remain clear when exposed to atemperature of The sp . gr . ranges from to O .92O .
I I t i ssoluble in 24 parts of cold alcohol or in 6 parts at the b . p . It consistschiefly of olein and a small quantity of linolin .
2 Only small quantities of solid glycerides are present
,and no stearin .
3 I t i s not a dryingoil and
,according to Lewkowitsch
,does not easily turn rancid .
The chief physical and chemical constants of this Oil are given onp . 69 and the oleo-refractometer value on p . 44 . 7 samples Of Oilfrom sweet and bitter almonds tested by Lewkowi tsch4 in the butyrorefractometer at 40° gave numbers ranging from to Therefractive indices of 3 5 samples determined by Harvey5 in the Abbe refractometer ranged from to atThe mixed fatty acids have an exceptionally low m . p . (see p .
According to the German Pharmacopoeia,they should remain per
manently fluid at should give a clear solution with an equal volumeOf alcohol at and this solution should remain clear on adding twicethe volume Of alcohol .1 Mo st Observers gi ve a smaller range t o at2Farnst e iner . Zei t. Nahr . Genu ssm . , 1 89 9 , 2 , 1 .
3 Hehner and Mitch ell . A naly s t , , 1 89 6 2 1 , 3 1 6 .
4 Ana ly s t , 1 9 04 , 29 , 1 0 5 .
S oc. Chem . Ind . , 1 9 0 5 ,24 , 7 1 7 .

1 04 MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
almond Oil,but the difference is not great enough and the results
given by almond Oils from different sources are too vari able,for any
definite conclusion to be based upon this test in the case Of mixtures .Further investigation of the liquid fatty acids m ight lead to a test basedupon a difference in the yield of tetrabromides
,as Lewkowitsch I has
suggested,and an observation by D ieterich2 that the critical tempera
ture of solution determined by Crismer’
s method (p . 63 ) of almondoil i s much lower than that of apricot-kernel oil or peach-kernel Oil i salso worth following up . In the present state of our knowledge
,re
course must be had to colour tests,of which the following are
available :B ieber’s Test —5 volumes of the sample are shaken with 1 volumeof a cold mixture of strong sulphuric acid
,water
,and fuming nitric
acid in equal parts by weight . Pure almond Oil gives “a white oryellowish-white liniment
,apricot-kernel oil a deep salmon—red or peach
blossom colour,changing to dark orange . Lewkowitsch recommends
this test in preference to others . The reagent should be freshly prepared . M ixtures of almond Oil and apricot-kernel Oil containing 1 /3of the latter are distinctly coloured
,but with 25% the colour i s slight .
Peach-kernel oil gives the same test much more faintly and onlyafter standing for some time ; this oil will , therefore , be still moredifl
‘i cult to detect in mixtures .Kre is’ Ph lo rogluc ino l Tes t .3—A few c .c . Of the sample of Oil arepoured upon an equal volume at nitric acid of sp . gr . ; a similar quantity Of a solution of phloroglucinol in ether i s then added and thewhole well Shaken together . Peach-kernel and apricot-kernel Oilsgive an intense raspberry-red colour
,inclining to
.
violet . Chwolles ,
who recommended this test,found that genuine almond Oil gave no
colour or only a faint rose-red colouration and that 1 0% of peachkernel oil could be detected in admixture
,but Lewkowitsch found that
several genuine almond oils gave the coloration more or less stronglyand recommends great caution in the use of this test . I t should benoted that the raspberry-red colour is also Obtained with arachis , sesame
,cottonseed
,walnut
,and castor oils
,but not with olive or lard Oils
(Kreis).Ni tr ic Ac id Tes t .—Almond oil , if shaken with nitric acid ofSp . gr . becomes pale yellow ; apricot-kernel and peach—kernel Oils be
1 A na ly s t , 1 9 04 , 2
9,1 0 7 .
2 S ee Wri h t and i tch e ll , Oils , Fat s , Waxes , e tc . , p . 403 .
3 Chem . ei t , 1 9 02 , 26 , 8 9 7 .

HAZELNUT OIL .
come orange-coloured (Micko) . According to the B ritish Pharmaco ~
poeia, if 2 c .c . of almond oil “be well shaken with 1 c .c . of fumingnitric acid and 1 c .c . of water
,a whitish
,not brownish—red
,mixture
Should be formed , which , after standing for 6 hours at about shouldseparate into a solid white mass and a nearly colourless liquid (absenceof peach -kernel and other fixed The United S tates Pharmacopoeia and the Swiss Pharmacopoei a also
,g ive thi s test for the detectionof peach -kernel Oil
,but Umney I found the test incapable of detecting
peach—kernel Oil,though useful for detecting apricot—kernel Oil .
F . B . Power,in a paper read before the B ritish Pharmaceutical Con
ference in July,1 900 ,
suggests as an explanation of this apparent ’
dis
crepancy the statement of Hirsch that “PfirschkernOl ,” for which theSwiss Pharmacopoei a gives the test with fuming nitric acid as specific ,i s not the Oil from the kernels of the common peach
,but from a small
sort of the bitter almond,Amydgalus communi s . This oil , Power
states,shows the behaviour described in the Pharmacopoeia .
APRICOT-KERNEL OIL .
2 PEACH-KERNEL OIL .
2
PLUM-KERNEL OIL .
(See also p . These three Oils,Obtained
,respectively
,from
the kernels of the apricot,P runus armeniaca
,the peach
,Amygda
lus persi ca,and the plum
,P runus domesti ca
,closely resemble almond
Oil, for which they are largely used as adulterants and substitutes
(see“Almond Apricot and peach-kernel oils are known com
mercially as Ol-Amygdalce P ersi c. (Squire) .
HAZELNUT OIL .
(See also p . This is a golden-yellow coloured,non-drying
Oll Obtained from the seeds or nuts Of Corylus avellana , the commonhazel . The nuts contain from 50 to 60% Of the oil . I t i s used inperfumery , in pharmacy , and also as a lubricant for clocks .Hanus3 states the composition of the fatty acids Of this o il to be ,oleic ac i d 85 , palmitic and stearic acids 1 0 . About 1 % Of stearicacid was found
,but no arachidic acid
,and no linolic or linolenic acid .
The Oil forms a green-coloured,solid elaidin . I t contains about
of phytosterol . Hanus Obtained the following values1 Pharm .]ou r Ju ly , 1 89 9 , p . 1 06 , and Jan 1 9 00 ,
p . 8 .
2 Se e Lew kowrt sch , A na ly s t , 1 904 , 29 , 1 05 .
3 Zei t. Nahr. Genussm . , 1 89 9 , 2 , 6 1 7 .

1 06 MODES OF EX AMINING FATS,OILS
,AND WAXE S .
Sp . gr. at
Maumené test .S aponification valueIod ineHebner value .
Re ichert-M e issl valueAcetyl value . .
Tortelli and Ruggeri found the iodine value of the cold-pressed Oil
to be and that Of the liquid fatty acids which points to thepresence of fatty acid more unsaturated than oleic .
OLIVE OIL .
(See also p . O l ive oil i s expressed from the fruit of the olive ,Olea europcea , and oil of inf erior quality i s extracted from the residualmarc by carbon disulphide sulphocarbon oil ”) or petroleum ether.Peano I states that in determining the oil in Olives
,carbon disulphide
should be used and not ordinary ether,as the latter dissolves another
substance .Of the commerci al varieties
,Provence and Tuscan oils are among the
most esteemed . The finest grade in the market i s “ finest cream sublime Oil
,
” which i s imported from Leghorn . O ils of other origin areGallipoli
, S icili an , Spanish , Portuguese , Levant , and Mogador . Thatsold in the so-called “
Florence flasks,
” i s u sually of inferior quality .
Lucca and Gallipoli oils are well-known brands,and much excellent
oil i s expressed in Spain,and exported from Malaga and S eville .
Olive Oil i s now largely prepared in California,Tunis
,Algeria , and
Morocco . Much African oil goes to Nice and i s there blended with theoil of the district and sold as pure Nice Olive Oil .2The oil which exudes from the ripe olives under moderate pressurein the cold is sold as “ virgin
,
” “ sublime,
” or “ first expressed ” oil ; i t i sthe best edible oil . Ordinary oil
,from a second pressing with the aid of
hot water,has a less agreeable flavour than the first and is more li able
to become acid,but the two sorts are often mixed
,forming several va
rieties .
“Pyrene ’” Oil
,
“ bagasses ” oil,
“ huile tournante,
” “ huiled ’enfer
,etc .
,are very impure acid oils
,recovered from residues which
S oc Chem. Ind . , 1 9 0 22 , 3 5 .
2 Chemi s t and Drugg i s t , ug . 1 9 , 1 9 0 5 .

1 08 MODE S OF EXAMINING FATS,OILS , AND WAXE S .
’
as mixed esters containing 1 molecule of saturated fatty . acid and 2
molecules Of oleic acid .
I Hehner and M itchell found no stearic acidin olive Oil . M inute traces of arachidic acid have been i solated
,but not
sufli cient even in Tunisian oils to lead to erroneous conclusions beingdrawn from the results Of Renard ’s and B ellier ’s tests for arachis oil .2O live Oil i s the type Of a non-drying vegetable oil . I t does notthicken materially
,even on prolonged exposure to air , but gradually
becomes rancid,a change which appears to be mainly due to oxidation .
3
In very thin films it dries slowly . The following results obtained byArchbutt 4 Show how it compares with some other well-known oils inthis respect :
Kind of Oi l
surface 7 cm . square)
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
If free from acid it is only slightly soluble in alcohol,but dissolves in
about 1 . 5 times i ts weight Of ether , and is miscible in all proportionswith carbon disulphide
,chloroform
,and hydrocarbons .
When heated to about I Olive Oil becomes lighter in colour , and at2 20
° nearly colourless and at the same time rancid . At 3 1 5° i t suffers
decomposition,emitting a disagreeable Odour of acrolein .
The following are some observed analytical data of the mixed fattyacids of olive oil not given on p . 69 :
Authority
Sp gr at 5°
S p . gr. at
S o lid ify ing-p0int (titer testRefract ive index atIod ine value of m ixed fatty ac idsIod ine value Of liqu id fatty ac ids
1 B ern,2 40 2 ; 1 9 0 2 , 5 , 403 6 ; 1 24 7 .
2 Arc:libu t t , ]“ Soc Chem. 2 6 , 4 5 3 and 1 1 8 5 .
3 R y an andMarsha ll . Amer . f ou r. P harm . , 1 9 0 7 ,
S oc. Chem. Ind . , 1 89 9 ,

oil Often contains awhich increases by
Thomson and Dunlop3 found in 20
very varied sources free (Oleic) acida sample from the Levant to in a
found in 1 8 I talian oils from tos from towere observed by N . J . Lane in the Uni ted
Number of samples
Most of this free ac i d i s caused by allowing the fruit or the pulp toferment before the o il i s expressed from it
,or by storing the oil in a
crude state .

I I O MODE S OF EXAMINING FATS,OILS , AND WAXES .
I t has been shown I that if the Oil be fil tered immediately after expression , to remove the insoluble impurities , the acidity does not increase by storage
,or at any rate only very slowly .
Oil intended for table use,for lubricating
,and for burning in lamps
,
should be as free as possible from acid,a maximum of 4% being the
desirable limit . In lubricating oils,the free acid corrodes the bearings
,
forming metallic soaps which dissolve in and thicken the lubricant . 2In lamp oils excess of acid causes charring of the wick . For soapmaking
,free acid is no detriment
,and for Turkey-red dyeing a very
acid Oil (“ tournante oil ”) i s preferred , as i t readily emulsifies with a
solution of sodium carbonate . Lewkowitsch states that 25% of freefatty acids should be present in Turkey-red oil . For woolcombers ’
use free acid is not necessarily Objectionable,providing the oil
does not form sticky and varnish-like films on the wool . (See below .)The proportion Of free acid in olive Oil can be ascertained with easeand accuracy by titration in presence of alcohol with standard causticalkali and phenolphthalei n
,in the manner described on p . 9 .
B urstyn (D ingl . polyt. J ., 1 875 , 2 1 7 , 3 1 4 ; J . Chem. S oc . , 1 876 , 29 ,
769) has described the following method for estimating'
the free acidin olive Oil . The process appears well suited for rapid technical investigations , though the volumetric method described elsewhere willb epreferred by chemists . The oil i s shaken with an equal measure Ofrectified spirit of to sp . gr .
,the exact figure being accurately
determined . After the liquids have separated,the sp . gr . of the spiri t
i s determined . Burstyn finds that an oil,1 00 c .c . of which contains
free acid in quantity sufli cient to neutralise 1 c .c . of normal alkaliof oleic acid), will raise the gravi ty of the alcohol from
to and that each additional 1 c .c . of alkali neutralisedcorresponds to an increase of in the sp . gr . of the spiri t . Hence ,the increase due to the solution of a trace of neutral fat i s andthat each increase of in sp . gr . beyond this number represents
grm . of free acid per 1 00 c .c . Burstyn states that the actionof Olive oil on brass i s regularly and directly proportional to the percentage of free acid present .In examining Oil intended for cooking or table use , the flavour and
Odour should be carefully observed , as many apparently genuine specimens which are fairly free from acid are unsatisfactory in this respect .1 Millian , B erta inchaud andMale t . M oni t. S cient. , 1 9 00 , 5 6, 5 08 .
2 Archbu t t and Dee ley , Lubricat ion and Lubricant s , p . 2 1 3 .

1 1 2 MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
in America,i s a not unlikely adulterant
,and has been sold as a sub
sti tute for olive Oil (see below) . Lard oil,expressed at a low tempera
ture and specially refined,i s largely used when the price permits of it
,
“Superfine Lucca Oil ” being stated to contain sometimes as much as
60 to 70% of i t . Fish Oils are occasionally employed,menhaden oil
being said to be used frequently . Hydrocarbon oils are also used .
In the United S tates,cottonseed Oil i s largely sold under the name of
olive oil . In fact , until the adoption of the recent food laws , especiallythe Federal act
,the label “Huile d ’
Olive vierge , E . Loubon ,Nice
,
”
was generally understood in the grocery trade to indicate cottonseedOil . B ulletin No . 7 7 ( 1 905) of the United S tates Department ofAgriculture contains illustrations of several spurious labels . A bottlelabelled “
Freres 81 du Peaux,B ordeaux
,France . Huile D O l ive ”
contained cottonseed oil . Another containing mixed Olive ‘
and cottonseed oil bore the label Tisserand 81 Fils . Huile d ’
Olive extrasur
fine,B ordeaux , France . Falcon brand .
”A mixture of olive and
peanut oil was described as “Huile D ’Olive
,extra surfine
,Jules
Chambon C ie .,B ordeaux
,France . This label also bore the
signature of the alleged importer . On enquiry at Bordeaux,no trace
could be found of the above-named firms,and most likely the labels
as well as the oil were of American manufacture . A label of anotherkind was found on a bottle of maize oil . This
,at first glance
,appears
to read “Superior O live O il . Dove Pure O il but
,on closer in
spection ,i t i s seen to bear the words “ Superior in quali ty , purity and
flavour to any olive Oil in the market .In examining Olive oil
,the most important indications are sp . gr .
iodine value, saponification value , ri se of temperature on treatment
with sulphuric acid or bromine,amount and nature of the unsa
ponifiable matter , some form of oxidation test , and some colour indications . Some sophistications require the application of Specialmethods for their detection .
The sp. gr . Of Olive oil usually ranges from (rarely toat but genuine Tunisian and Californian and even some
I talian oil s may have as high a sp . gr . as Even has beenrecorded for Tunisian oil and for an Olive oil from the Punj ab .
1
On the other hand,a sp . gr . as low as has been observed in an
Oil containing 3 1 % of free (oleic) acid ? High gravi ty Oil s are usually
1 Cro ssley and Le Su eur , S oc. Chem. Ind . , 1 89 8 , I 7 , 9 9 8 .
2 B ull . NO. 7 7 ( 1 9 05 ) U . Dept . o f Agri cu lture , p . 1 5 .

OLIVE OIL . 1 1 3
dark in colour,and may contain o il from the kernel and endocarp . All
samples over in sp . gr. Should be submitted to a very criticalexamination for adulterants . An admixture of rape , lard , or arachisoil would not be indicated by the sp . gr . Cot tonseed , poppyseed , orsesame oil would tend to raise i t
,but the sp . gr . could be adjusted by a
judicious mixture of these with sperm or mineral oil which would ,however
,be readily detected by other tests .
The i odine value i s a most useful test , but for its correct interpretationa knowledge of the source of the oil i s needed . Genuine samplesusually absorb from to of iodine
,but lower and higher
values may be met with and the oil still be genuine . The results ofnumerous Observers for oils from various countries tabulated by Lewkowi tsch I range from to I talian and Spani sh O ils rarelyabsorb more than 86% of iodine ; the highest iodine values are to belooked for in the oils from California
,Tunis
,Algiers
,Morocco
,and
Dalmatia . .The following values have been recorded for single
samples
Observer Source of Oi l
Colby2 Califo rnia 93 . 5Crossley and Le Sueur3 . Punjab . 93 . 6 7Ahrens and Hett4 Mo rocco black o lives,hand pre ssed 9 1 . 5
Guozdenovics Dalmatia .
Thomson and Dunlop6 M ogado r . 94 . 3Archbutt? Mornag (Tunis). Olive (var.)
Che tu i .
B ize rte (Tunis). O live (var .)Chetu i .
M edjez-Amar
A few varieties Of olives grown in certain districts appear to give theseexceptionally high results
,Oils from other varieties and districts giving
normal or more nearly normal figures . In ordinary cases , an iodinevalue of over 87 would indicate adulteration . Goldberg ’s observationthat the solid and liquid portions of chilled olive oil absorb practicallythe same amount of iodine shows that the iodine value of demargari
mated Oil i s no t likely to be appreciably higher than that of the entireOil from the fru i t .
1 Ch em . Tech 2 , 2 7 5 .
2 Ca li forn i a A gr . Exp t. S ta . Rept 1 89 7—8 , 1 68 .
S oc . Chem . Ind . , 1 89 8 , 1 7 , 9 89 .
4 Zet ts . Oefientl . Chem . , 1 9 03 , 9 , 2 84 .
5 Lek W It sch , Chem . Tech. , 2 , 2 7 5 (footno t e ) .0A na ly s t , 1 9 06 , 3 1 , 2 8 1 .
7 J. S oc. Chem . I nd . , 1 9 0 7 , 26, 4 5 3 and 1 1 8 5 .
Vo l. I I .
—8
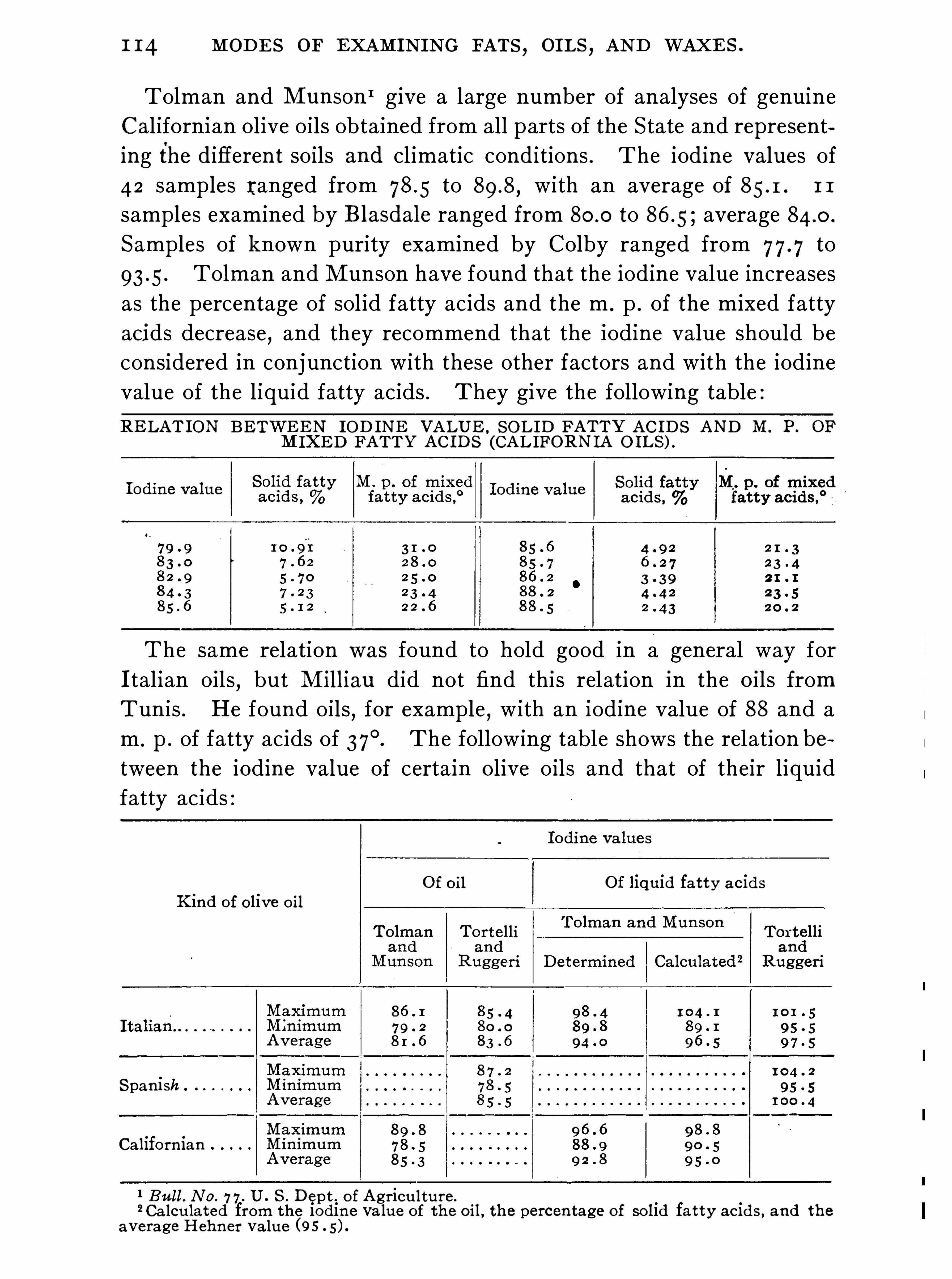
1 1 4 MODE S OF EXAMINING FATS , OILS,AND WAXE S .
Tolman and MunsonI give a large number of analyses of genuine
Californian olive oils obtained from all parts of the S tate and representing the diff erent soils and climatic conditions . The iodine values of42 samples ranged from to with an average of 1 1
samples examined by B lasdale ranged from to averageSamples of known purity examined by Colby ranged from to
Tolman and Munson have found that the iodine value increasesas the percentage of solid fatty acids and the m . p . of the mixed fattyaci ds decrease , and they recommend that the iodine value should beconsidered in conj unction with these other factors and with the iodinevalue of the liquid fatty acids . They give the following table :RELATION BETWEEN IOD INE VALUE , SOLID FATTY ACIDS AND M. P . OFMIXED FATTY ACIDS (CAL IFORNIA O ILS ) .
The same relation was found to hold good in a general way forI talian oils
,but M illiau did not find this relation in the O ils from
Tunis . He found oils,for example
,with an iodine value of 88 and a
m . p . of fatty acids of The following table shows the relation between the iodine value of certain Olive oils and that of their liquidfatty acids
Of o i l Of liqu id fat ty ac idsKind o f olive o i l
S pan ish
Californian
1 B u ll . No . 7 7 . U . S . Dept . O f Agricu lture .
1’ Ca lcu lat ed from th e iod ine va lue of th e o i l , th e percentage of so lid fat t y ac id s , and the
average Hehner value ( 9 5

1 1 6 MODE S OF EXAMINING FATS,OILS , AND WAXE S .
cocoanut Olein and tallow and lard oils,produce more heat than olive
oil,so that a rise of temperature of more than 45° with sulphuric acid
( 1 0 c .c . of acid containing 9 7% H, SO 4and 50 grm . of oil) may at
once be considered as indicating probable adulteration,and In some
cases i t allows of an approximate estimation of the extent of thesophistication .
Archbutt I has determined the heat of brominati on of 1 0 samples ofolive oil and Obtained results ranging from to using 1 grm .
of Oil and 1 c .c . of bromine . The thermal values when multiplied byagreed very nearly with the iodine values .The elaidin test i s of li ttle use unless carried out under standard con
ditions ? The reagent Should be prepared by dissolving 6 grm . ofmercury in c .c . of nitric acid in a 50 c .c . stoppered cylinderimmersed in cold water
,i t should be mixed with the oil in the propor
tion of 1 part of reagent to 1 2 of oil by weight and the mixture kept ata fixed temperature and Shaken every 1 0 minutes . Under these conditions
,at genuine olive oil i s converted into a solid pale yellow
coloured mass Of elaidin in about 1 hour,arachis oil rather more
slowly,but rape
,cottonseed
,sesame
,and other more strongly drying
oils remain partially or wholly liquid and are coloured orange or red .
To increase the delicacy of the test,i t should be made at Ol ive
oil will,then
,take from 200 to 400 minutes to solidify , and 1 0% of more
strongly drying Oils if present will delay the solidification , darken thecolour
,and soften the consistency of the elaidin formed . I t i s said that
as little as 5 of poppy oil can be detected by this test at which requ i res confirmation
,but most adulterants are more readily detected by
other tests . O l ive Oil which has become bleached by exposure to sunlight no longer forms a solid elaidin .
Useful information as regards the ox idi sing properti es of an Ol ive Oilmay be obtained by exposing 0 . 5 grm . on a watch-glass to the air in awater-oven at 1 00° for about 1 6 hours side by side with an equalweight of a standard sample on a watch-glass of the same curvature .
Livache’s or B ishop ’s tests (pp . 36 and 3 8) may also be used .
Reference to the tables on pp . 44—45 will Show that the oleo-refractam
eter of Amagat and Jean i s a valuable instrument for the rapid testingof Olive oil
,the recorded deviation of genuine samples ranging from
0°
1 06 samples examined by O liveri3 ranged from 0° to
S oc. Chem . Ind 1 89 7 , 1 6 , 3 09 .
2 S oc. Chem . I nd . , 1 886 , 5 , 3 03 .
S oc. Chem. Ind . , 1 89 4 , I 3 , 4 5 .

OLIVE OIL .
All fixed Oils likely to be added as adulterants would increase the réfraction
,except neat ’s-foot
,arachis , lard , and tallow oils .
The unsapo nifiable matter of genuine Olive Oil contains phytosterol ,but no cholesterol . I t does not exceed 1 . 5 in normal olive oils ofthe first and second pressing. In Ol ive residuum Oils (les hu i les degrignon d
’
olives), however , and also in certain Olive oils of the thirdpressing
,larger quantities , even as much as of unsaponifiable
matter have been found by the writer , derived , according to M illiau ,
from the shell of the olive nut . Any excess would probably includehydrocarbons from mineral or rosin Oil
,or wax alcohols from sperm
or some other marine animal Oil .Co t tonseed Oi l , unless i t has been rendered insensitive by heating ,would be detected by Halphen
’s colour test (page 1 3 the result of
which must,however
,be interpreted in conjunction with the quantita
tive values,especially the sp . gr .
,refractive power
,iodine value ,
thermal tests,and the titer test of the mixed fatty acids , all of which
would be raised by the addition of cottonseed oil . Some of thesemight be adjusted by the addition of a third oil , but the fraud would bedetected by an abnormali ty s omewhere if a complete examinationwere made . The addition of mineral Oil or sperm oil , for instance ,would increase the percentage of unsaponifiable matter . O thercolour tests which may be applied are those with Silver ni trate (Bechi ,Mi lli au) and nitric acid (see under Cottonseed As somegenuine olive oils have been found to give a brown colour in Bechi ’stest and a yellowish-brown colour with nitric acid
,the results of these
colour tests must be used with caution . A mixture of cottonseed andlard oils could be made having the normal sp . gr. and iodine value ofOl ive Oil
,but the oleo—refractometer
,the ti ter test of the mixed fatty
acids,and the iodine value of the liquid fatty acids
,would detect
adulteration with such a m ixture .Sesame o i l would be detected by the very delicate furfuraldehydetest (p . Some genuine olive oils have been found to give apale violet or rose-red colouration in thi s test
,but i t i s unlikely for
error to occur if proper consideration be given to the quanti tativevalues . Sesame oil
,if present
,would affect the quantitative values
very much in the same way as cottonseed oil .Arach is o i l can be detected in olive oil by Bellier ’s test (p . 99)and estimated by Renard ’s process (p . The ordinary quantitative values of this oil are little different from those of olive Oil . Some

1 1 8 MODE S OF EXAMINING FATS , OILS , AND WAXE S .
indications of i ts presence might be Obtained by an abnormally highiodine value
,but this would be very uncertain (recorded values for
arachis oil,
to 1 05 ; for olive oil , to I t may be notedthat sesame oil i s frequently mixed with arachis oil
,and the two may
,
therefore,occur together . Arachidic acid occurs in rape and mustard
oils,but these would betray their presence by lowering the saponifica
tion value and raising the iodine value and thermal values of olive oil .Poppy o i l or ma ize o i l added to olive oil would affect the sp . gr .
,
iodine values,and thermal values much in the same way as cottonseed
oil,but they would not alter the titer test of the mixed fatty acids
,nor
increase the percentage of solid fatty acids as cottonseed oil would do .
Poppy oil has a high iodine value and dries strongly . Maize oil i ssaid to give a pecul i ar red co lour when shaken with nitric acidwhich i s qui te different from the colour obtained with cottonseed oil .Lard o i l in most of its quantitative values closely resembles olive oil
,
and would not be detected by the ordinary tests . The odour of th esample when warmed might reveal its presence
,but the detection of
cholesterol in the unsaponifiable matter by Bomer’s test (see under
Cholesterol ”) would be the best evidence of the presence of lard oil .I t might also be worth while to look for stearic acid in the mixed fattyacids by Hehner and M itchell ’s process , since olive oil contains nostearin . Lard oil would increase the percentage of solid fatty acids .Fish and o th er marine an imal O i ls would probably show themselves by the taste
,the smell on warming the sample
,and the red
colour produced on heating the oil with caustic soda solution . Mostfish oils would raise the sp . gr . and iodine value , but they would beespecially identified by the insoluble bromoglyceride formed by addingbromine to the solution of the oil in ether and acetic acid in Hehner andM itchell ’s process (p . L inseed o i l would
,of course
,give ' a
similar precipitate .The characters of commercial olive Oil must depend to some extentupon whether
,in the process of expression or extraction , the Ol ive
kernels have been crushed and the olive-kernel oi l included .
OLIVE-KERNEL OIL .
This oil was formerly believed to be quite different in propertiesfrom ordinary olive oil (the oil of the mesocarp), having a sharpandbitter taste
,a dark green or brown colour , and being readily soluble in

1 20 MODE S OF EXAMINING FATS , OILS,AND WAXE S .
amined by I tallie,was a pale yellow thin oil
,having an acrid taste and
composed of palmi tin,Olein
,and linolin
,with traces of volatile acids
,
leci thin and phytosterol . A plant, Camellia oleifera , closely allied to
the tea plant,i s largely cultivated in China for the sake of the pale
bland oil obtained from its seeds,which i s said to be a very good lub
ricant for delicate machinery . I t i s dangerous for use as a food,unless
refined , owing to the presence of saponin . The seeds of the JapaneseCamelli a japoni ca also yield an oil which is said to excel as a lubricant .
I I . RAPE OIL GROUP .
B lack Mustard Seed Oi l . Radish Seed Oi l .Eruca Sativa S eed Oi l . Rape Oi l (Colza Oi l).Indian Mustard S eed Oi l . Ravi son Oi l .Jamba Oi l . Wh i te Mustard Seed Oi l .
BLACK MUSTARD OIL .
O
WHITE MUSTARD OIL .
(See also p . These oils,Obtained
,respectively
,from the seeds
of B rassi ca (S inapi s) nigra and Brassi ca (S inapi s) alba,resemble
rape oil in composition and general characters . The resemblance isclosest in the case of white mustard oil
,that of black mustard being
higher than rape oil in sp . gr . and iodine value . A sample of crudemustard-husk Oil (by-product from the manufacture of mustardfrom black and white seed mixed) examined by Archbutt gave thefollowing results :
S p . gr. at
Viscosity at I 5 . 5°
Saponification valueIodine value (Wijs) .
Unsaponifiable matter,
Free (ole ic) acid ,Maumené thermal test (50 grm . O i l 1 0 c .c . 97%
A sample of mustard-husk oil examined by Hehner and Mi tchellgave of brominated glyceride insoluble in etherMustard oil i s used for soap-making , burn ing , and lubricating .
In drying properties i t resembles rape.
oi l , and contains arachidicacid .
I (See p .
1 Archbut t . J . S oc. Chem. Ind . , 1 89 8 , 1 7 , 1 009 .
Practically the same as
that of rape o i l.
1 73 1
3 8
7s . o°
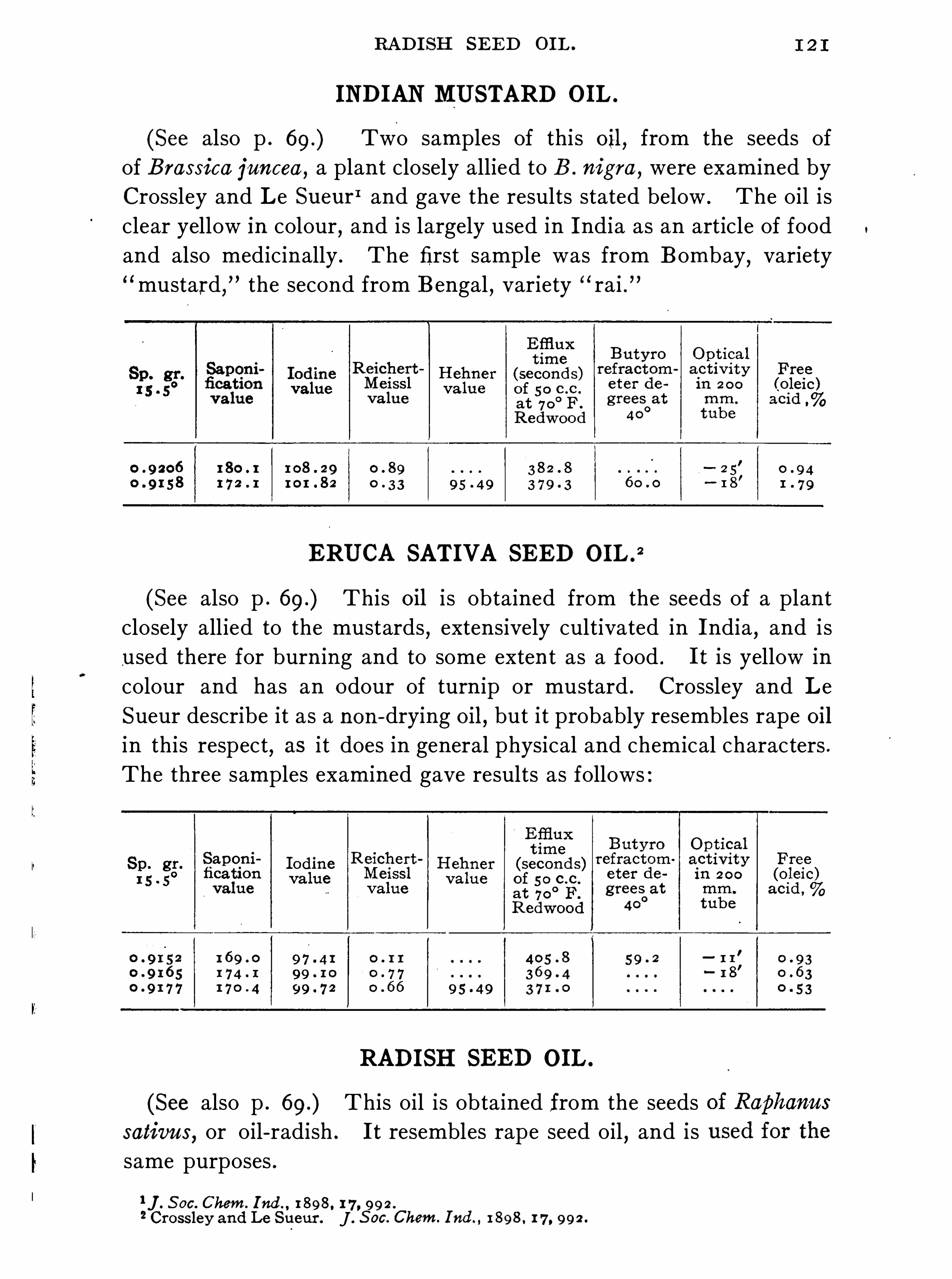
RADI SH SEED OIL .
INDIAN MUSTARD OIL .
(See also p . Two samples of this Oil, from the seeds ofof B rassi ca juncea ,
a plant closely allied to B . nigra , were examined byCrossley and Le Sueur I and gave the results stated below . The oil i sclear yellow in colour
,and is largely used in India as an article of food
and also medicinally . The first sample was from B ombay,variety
“mustard ,” the second from B engal , variety “ rai .”
ERUCA SATIVA SEED OIL?
(See also p . This Oil i s obtained from the seeds of a plantclosely allied to the mustards
,extensively cultivated in India
,and i s
used there for burning and to some extent as a food . I t i s yellow incolour and has an Odour of turnip or mustard . Crossley and LeSueur describe it as a non-drying Oil , but i t probably resembles rape Oilin this respect
, as i t does in general physical and chemical characters .The three samples examined gave results as follows :
RADISH SEED OIL .
(See also p . This oil i s obtained from the seedssativus
,or oil-radish . I t resembles rape seed oil
,and is
same purposes .S oc. Chem. Ind 1 89 8 , 1 7 , 9 9 2 .
2Crossley and Le S ueur . J. S oc. Chem. Ind . , 1 89 8 , 1 7 , 9 9 2 .

1 2 2 MODE S OF EXAMINING FATS,OILS , AND WAXES .
RAPE OIL . COLZA OIL .
(See also p . This oil i s obtained from the seeds of severalvarieties of Brassi ca campestri s
,of the order Cruciferae , cultivated ex
tensively in France , Germany , Austri a-Hungary , Roumania , S . Russia,
India,China
,and Japan . The oils yielded by the diff erent varieties
of seed,though botanically quite distinct
,are similar in their chief
physical and chemical characters,and are not distinguished com
mercially , being all sold as rape or colza Oil . IRape oil i s Obtained from the crushed seed by expression or by extraction with solvents . The crude Oil i s yellowish-brown or brownishgreen in colour
,has a peculiar odour and somewhat pungent taste
,
and contains foreign matters which separate to some extent by keepingthe Oil
,but cannot be wholly removed by passive treatment . These
lessen the combustibili ty,cause much smoke during the burning
,and
also tend to promote decomposition of the oil,with liberation of free
acid . To remove them,the crude or “ brown rape oil” is usually re
fined by agitating i t,while warm
,with from 0 .5 to -Of strong
sulphuric acid ; and after the foreign matters and suspended acid havesubsided
,the oil i s washed by agitation with steam and hot water .
Thi s process i s simple and rapid , but i t has the disadvantage thatsome hydrolysi s of the esters takes place
,increasing the amount
of free fatty acid in the Oil,which is detrimental to i t as a lubricant ;
the refined Oil i s also liable to retain traces of free sulphuric acid . RapeOil intended for use as a lubricant is
,therefore
,preferably refined with
fuller ’s earth . Refined rape oil i s pale yellow in colour and has acharacteristic taste and smell .The following results were Obtained with a consignment of Chineserape seed oil
,before and after refining on a large scale .
S p . gr. at
S aponificat ion valueIodine valueMaumené thermal valueUnsaponifiable matter,Free (Ole ic) ac id ,
1 For furt her part iculars see Archbut t and Deeley . Lubricat ion and Lubricant s , 1 0 7 .

1 24 MODE S OF EXAMINING FATS , OILS , AND WAXE S .
stirring,and then for 3 hours longer, stirring at intervals . I t re
mained clear and fluid. Some of the same oil,previously frozen
,
having been added,the Oil was kept in ice for 3 hours longer , with
occasional stirring,but the frozen oil slowly melted . The temperature
was then gradually reduced to - 1 0 to and the Oil became veryturbid
,but after remaining for 2 hours at thi s temperature
,with
stirring,i t did not lose its fluidity . After still further reducing the
temperature to and stirring,the Oil solidified in about half an
hour .The chief physical and chemical constants of rape oil are given onp . 69 , and the oleo-refractometer value on p . 44 . Some constants ofa number Of Indian crude rape oils expressed from different varietiesof pure seeds have been determined by Crossley and Le Sueur I andare given in the table on p . 1 26 . The following results by
'
Archbutt
were Obtained with rape oil extracted from the seed by ether in thelaboratory
Thefollowing are some observed data from the mixed fatty acidsof rape oil
Sp . gr. at
Sp . gr. at
S olid ifying-po int colza o i l
(titer test) rape Oil
Refractive index at 6Iod ine value of mixed fatty acidsIodine value of liqu id fatty ac ids
S oc. Chem. Ind . , 1 89 8 , 1 7 , 9 89 .

RAPE OIL . COLZA OIL . 1 25
Assay of Commerc ia l Rape Oi l .—Owing to the enormous extentto which rape oil i s used for lubricating and burning
,the estimation
of free acid i s of great importance (see under “Olive O il ,” p .
The method is described on p . 9 .
According to Archbutt and D eeley,
1 commercial refined rape Oil
contains on an average about of free acid calculated as oleic ac id ,ranging from 1 % to about but seldom exceeding 3 78
samples,all representing large contracts
,gave the following results :
Number Of sample s Free (o le ic) ac id,
Average,
I t i s evident from these figures that carefully refined rape oil shouldnot contain more than 3 of total acidity . The traces of free sulphuricacid in 3 samples of rape oil refined with this acid were determined andfound to range from to from which it i s concludedthat the percentage of free mineral acid in refined rape Oil Should notexceed of H, SO 4 ,
which is equivalent to of Oleic acid .
Rape oil i s subjec t to numerous adulterations,the more importan t
of which can be detected with tolerable certainty .
1 Lubricat ion and Lubricants , p . 2 1 1 .
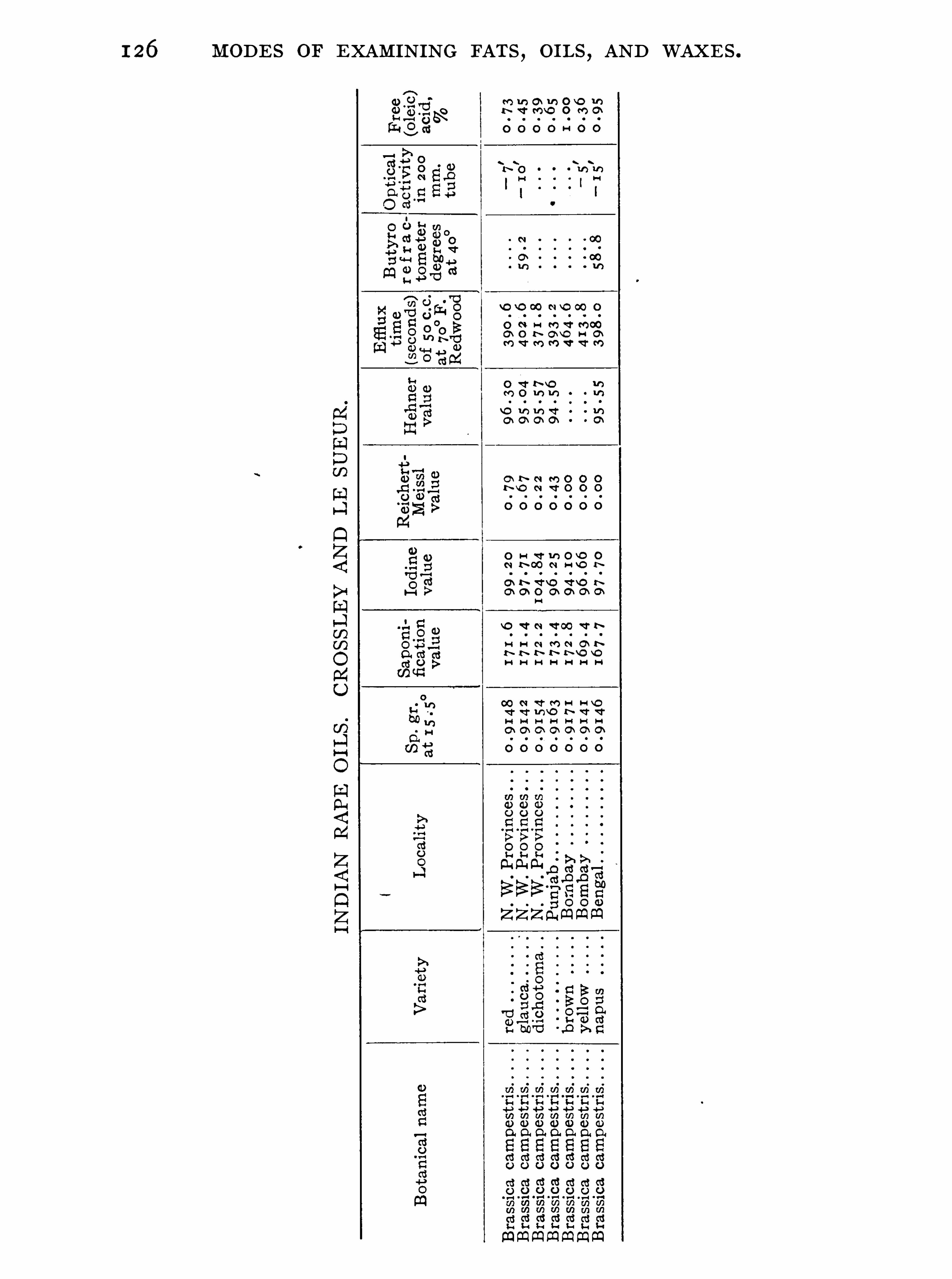
1 26 MODE S OF EXAMINING FATS, OILS , AND WAXE S .

1 28 MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
of i t may possibly contain even 50% of these oils , while the smelland colour will be little affected . S eed and nut oils deteriorate rape oilby increasing its gumming properties
,with the exception of arachis oil
and cocoanut olein,and the addition of either of these i s improbable .
Arachis oil could be detected as in olive oil (page due allowancebeing made for the arachidic acid naturally present in rape Oil i tself(see p . and cocoanut "olein would be indicated by the raisedsaponification value and reduced iod ine value of the sample.
The vi scosi ty of rape Oi l i s a valuable indication of its puri ty , as i ti s moderately constant and exceeds that of any oil likely to be used asan adulterant . The sample should always be compared with a Specimen Oi rape oil known to be genuine
,or with pure glycerol diluted to
sp . gr . which at I 5 .5° has the same viscosity as average rape Oil .
The time of efllux of 50 c .c . from Redwood ’s viscometer at 70° F .
should not be less than 3 70 seconds , and ranges from this up to about4 1 5 seconds ? The number recorded by Crossley and Le Sueurfor B ombay rape oil i s exceptionally high . The lowered viscosity ofan adulterated oil could be corrected bythe addition of castor or blownOil
,but
.
these would raise the sp . gr . and acetyl value . Heavy mineraloil would be found in the unsaponifiable matter .The saponifi cation value of genuine rape Oil ranges from to
A value in excess of would indicate the presence of ravision or other more strongly drying Oil . A lower value thanwould indicate the presence of an unsaponifiable oil or sperm oil , orboth . Refined rape oil has been frequently adulterated with a speciallypurified m ineral oil . This addition interferes with the burningqualities of the oil
,causing it to smoke and form much deposi t on the
wick .
The i odine value of rape oil ranges from 9 7 to 1 05 being Slightlyless than that of cotton or sesame Oil
,and considerably below that of
the more strongly drying oils . This test i s useful for the detection ofravison Oil
,which has a higher iodine value than rape oil ?
The Maumene’
thermal value , or rise of temperature on mixinggenuine rape oil (50 grm .) with sulphuric acid ( 1 0 c .c .) containing9 7% of HZSO ranges from about 58° to An abnormally high
1 Thie se numbers refer to an inst rument wh ich de livers 5 0 of wa t er a t 7 0
°F . in
secon s .
2Mi lrath (Zei tsch . afientl . Chem 1 9 0 7 , 1 9 , 3 7 1 ) obta ined th e fo llowing re sul t s W i th 3sample s of Au st rian rape o i l : S p . g r . , t o refract i on a t t o 6 . 9 ;
a t to ac id va lu e , t o sa p on ifi ca t ion valu e ,t o i od i ne va ue ,
t o Th e se are except ionally high iod ine va lu es .

RAPE OI L . COLZA OIL .
I
result indicates ravison or other more strongly drying oil,and a low
figure indicates mineral or Sperm oil . Hebner and M i tchell ’s brominethermal test (p . 60) may be used for the same purpose .The melting and solidifying-points of the mixed fatty acids
of rape oil are raised by cottonseed and lowered by many other oils,
such as ravison,linseed
,or fish Oils .
The unsaponifiable matter should not exceed In the expressedoil i t i s usually near I % ,
but in rape Oil extracted from the seed bypetroleum spirit some allowance must be made for residual hydrocarbons . If more than 2% of unsaponifiable matter be found , i t shouldbe purified by resaponification ; and if still in excess of the purifiedproduct should be further examined to ascertain whether mineral orrosin Oil
,cholesterol from animal oils
,or\wax alcohols from sperm oi l
are present . In genu ine rape oil,the unsaponifiable matter consists
mainly of phytosterol .A simple and useful ox idati on test may be made by exposing 1 grm .
of the sample on a watch-glass to the air in a water-oven at 1 00° forabout 1 6 hours , side by side with a sample of known purity ; bothsamples being contained in watch-glasses of the same curvature . On
examining the condition of the oils when cold,genuine rape oil of the
best quality for lubricating will be found to be still quite fluid whencaused to flow by inclining the glass
,and will not have dr i ed ; inferior
samples will have dried at the edges or have crept up and formed dryspots on the sides of the glass
, and most rape oils will have thickenedmore or less considerably . Livache’s test may also be used .
An abnormally low sp . gr . and viscosity of extracted rape oil i ssometimes due to incomplete expulsion of the petroleum spiri t usedin the extraction process . Such oil will have an abnormally lowflashing
-point. When tested in the Pensky—Martens or Gray closed- testapparatus
,normal rape Oil usually flashes at 4 1 0° to 450° F . ( 2 1 0
° toThe writer has occasionally met with samples Of extracted oi l
flashing at 1 80° F. and losing about 1 in weight in 1 hour when 1 grm .
of the oil was heated in a platinum dish in the water-oven .
Va lenta’s ace t i c ac id tes t (p . 62) gives very characteristic indications in the case Of rape oil and may be found useful in certaincases .Halph en
’s co lour test for cottonseed Oil and the furfural test
for sesame oil should not be omitted . They may be relied upon togi ve negative indications wi th genuine rape oil . Both tests are very
Vol. I I .
—9

1 30 MODE S OF EXAMINING FATS,OI LS
,AND WAXE S .
delicate and must only be used as confirmatory evidence . Theamount of foreign oil present must be calculated from the quantitativevalues. Press-bags which have been used for cottonseed and afterward for rape seed may be the cause of traces of colour in Halphen
’s
test .Ravison and cottonseed O i ls are two of the commonest adulterantsof rape oil . B oth raise the sp . gr .
,saponification value and Maumené
thermal value , and lower the viscosity . Ravison oil raises the iodinevalue , and lowers the m . p . of the fatty acids . Cottonseed oil does notappreciably affect the iodine value of the oil
,but i t raises the iodine
value Of the liquid fatty acids and rai ses also the m . p . of the oil and ofi ts mixed fatty acids . Cottonseed oil can only be added to refined rapeoil ; if added to the crude oil , i t causes i t to become red when refinedwith sulphuric acid . B oth ravison and cottonseed Oils ' are mores trongly drying oils than rape .L inseed o i l i s a very Objectionable adulterant of rape oil . I t maybe added before refining or by crushing the seeds together . It causessuch a marked effect in‘
raising the sp . gr .,iodine value
,thermal values
with sulphuric acid and bromine,in lowering the vi scosity of the oil
and the.
m . p . of the mixed fatty acids,and In Increasmg the tendency
of the oil to oxidize,that even a small admixture cannot fail to be de
tected. Linseed oil and fish oils are especially identified by means ofB ehner and M i tchell ’s bromo-glyceride test .Fi sh o i ls are recogni sable by their peculiar taste and odour onwarming
,also by the colourations developed with caustic soda and sul
phuric acid . They lower the viscosi ty in a marked degree , and affectthe quantitative values much in the same way as linseed oil . Train oi l
i s said to be best detected by agitating 1 00 drops of the oil with 1 ofsulphuric acid
,when the depth of the red colouration will follow the
proportion of the adulterant present .Hedge-mustard o i l may be used for adulterating rape oil , which i tclosely resembles . The most characteristic test i s said to be theproduction of a green colour when the oil i s treated with a quanti ty ofalcoholic potash insufficient for complete saponification , and thefiltered liquor strongly acidified with hydrochloric acid .
JAMBA OIL .
The oil described under this name in the table on p . 69 i s a kind ofrape oil which is occasionally exported from Kurrachi . I t closely

MODE S OF EXAMINING FATS, OILS , AND WAXE S .
BRAZIL-NUT OIL.
(See also p . This Oil i s Obtained from the B razil nuts ofcommerce
,the produce of Bertholleti a excelsa , a tree which flourishes
In northern Brazil and Venezuela . I t i s a pale yellow oil,Odourless
and of pleasant taste,but easily turning rancid . I t i s used for culinary
purposes when fresh,also for burning and soap-making .
CAMELINE OIL . GERMAN SESAME OIL.
(See also p . This oil i s obtained from the seeds of Camelinasativa (Myagrum sativum),
“ Gold of P leasure,a plant of the order
Crucif erce . According to Schaedler , i t has a golden yellow colour , aSharp
,peculiar taste and smell
,and dries slowly . I t i s used for burning
and for making soft soap . The cold-pressed oil i s sometimes alsoused as an edible oil . Cameline oil i s said to be used as an adulterantof rape oil
,but would be readily detected by its higher sp . gr .
,iodine
value and Maumené value . I t is li able to be contained in linseed oilfrom East Indian seed
,and may account for the low iodine value and
inferior drying properties of some samples of that oil . I
COTTONSEED OIL .
(See page Cottonseed oil is now expressed 111 enormousquantities in the United S tates
,on the continent of Europe
,and in
Great Britain,from the seeds of the diff erent varieties of the cotton
plant ?Crude co t tonseed o i l has a sp . gr . ranging from
I t contains in solution,often to the extent of I 070, a characteristic colour
ing matter,which gives i t a ruby-red colour
,sometimes so intense as to
appear nearly black . The crude oil gives a bright red colourationwith strong sulphuric acid (page The soap from crude cottonseedoil rapidly oxidises on exposure to air with production of a fine purpleor violet-blue colouration .
3 This test is characteristic . The col1 Lewk ow i t sch , Techno lo g o f Oils , I I , 43 .
2 S ee Lewk owi t sch . O ils ,Fa t s and Waxe s , I I , 1 43 .
3 Cot t onseed blue i s stat ed by Kuh lmann t o h ave th e compos it ion Of 017 H24 04. I t is
amorph ou s ; readi ly de st roy ed by oxid ising agen t s ; inso luble in water , d ilu t ed acids , and
alka lies ; sparing l soluble in carbon d isu lph ide and ch loroform , bu t more read ily in a lcoh o l
and e th er ; and disso lve s W i th pu rple colour in s trong sulphuric acid . Th e unoxid isedco lour ingmat t er of co t ton seed o i l has been examined by J. Longmore , wh o , in a commun i ca
t ion t o A llen , sta t ed tha t i t i s a pungent go lden y e llow product , in soluble in wa t er ,
bu t so luble in a lcoh o l and alka line so lu t ions , and precipitat ed from ‘
the la t ter on add it ionof acids . I t dy e s well and perfect ly fast on both wool and silk.

COTTONSEED OIL .o 1 33
ouring matter causes the oil to produce stains , and i t is removed byagitating the crude oil at the ordinary temperature with 1 0 to 1 5% ofsolution of sodium hydroxide of sp . gr . when the alkali combineswith the colouring matter and the free fatty acids of the oil . Themixture becomes filled with black flocks which deposit on standing ,
I
and leave the oil but slightly coloured . The loss from refining is usuallyfrom 4 to 7 . 5 but occasionally amounts to 1 2 or 1 5 . Hence it i sdesirable , before purchasing crude cottonseed oil for refining , to asoertain by a laboratory experiment what the percentage of loss i s likely tobe . Frequently the treatment with alkali is only carried far enough toremove the greater part of the colouring matter
,the oil being
,thenboiled with a solution of bleaching powder and subsequently treatedwith dilute sulphuric acid . This method of treatment is economical
,
but the oil acquires an unpleasant taste and smell which cannot be removed . Hence chemical bleaching Cannot be used for the oil which isrequired for edible purposes .Refined co t tonseed o i l i s of a straw or golden-yellow colour
,or
occasionally nearly colourless . The sp . gr. usually ranges fromto and the solidifying point from 1
° to By subjectionto cold and pressure a certain proportion of “ stearine ” i s separated
,
the m . p . of the residual oil being correspondingly lowered . Thisrefined oil i s usually almost free from acid
,and
,when properly pre
pared , i s of pleasant taste . I t i s extensively employed for edibleand culinary purposes . I t i s now substi tuted for olive oil in someof the liniments of the Uni ted S tates Pharmacopoei a
,but its principal
applications are in soap-making and the manufacture of facti ti ousbutter.
The solid esters of cottonseed oil consist mainly of palmi tin,with a
li ttle stearin , the liquid contains olein and linolin .
Cottonseed oil i s characterised by the high m . p . of its mixed fattyacids and by the colour tests described below . In the elaidintest i t gives an orange-coloured semi-fluid mass . I t i s not i tself veryliable to sophistication
,owing to its cheapness
,but it i s frequently em
ployed to adulterate other oils . Most oils l ikely to be added to cotton
1 Th e deposit thu s formed , cons ist ing of colourin and albuminou s mat t ers , alka li , andpart ially sap on ifi ed o i l , i s t echn ically called“muc iIag e . It i s decomposed wi th a s li gh t
excess o f acid , and th e result in dark-co lou red gr ease i s h eat ed t o a t emperature of 1 2 0°
( 2 5 0° F .) with concent rat ed su phuric acid , wh ich renders in soluble th e co louri ng mat ters ,
e tc . , wh ile th e impure fat ty acids rise t o th e surface . On d ist illing th ese with su perh eat eds team , a m ixture of fat ty acids i s obta ined ,
which i s separat ed into s t eari c and o le i c ac i dsby pre ssure . Th e
“co t tonseed st earin " thu s obta ined i s employ ed for maki ng soap and
composite candles and for variou s adul terat ions .

1 34 MODE S OFIEXAMINING FATS , OILS , AND WAXE S .
seed oil would lower the m . p . of the fatty ac ids,linseed Oil and whale
oil would be found by Hehner and M itchell ’s bromo-glyceride test.For the detection of maize oil see under “Maize Oil.”For the detection of cottonseed oil in other Oils
,Halphen
’s colour
test generally sufli ces , and a determination of the sp . gr.,iodine value
,
Maumené thermal test,and melting or solidifying-point of the mixed
fatty acids will generally enable the proportion present in a mixture tobe determined.The unsaponifiable matter i s usually near to I % and containsphytosterol . The rise of temperature of 50 grm . of the oil with 1 0 c .c .of sulphuric acid (97% H, SO4) i s about 75° to The viscosity at1 5 .5
° is about 3 4 that of refined rape oil at the same temperature .The following are some observed analytical data from the mixedfatty acids of cottonseed oil
Sp . gr. at
Sp . gr. at
S o lid ify ing-po int (titer test)Lewkow itsch .
Refractive index at 600Iod ine value of m ixed fatty ac idsIodine value of liqu id fatty acids
Co ttonseed s tearin (cotton oi l stearin) (see p . 1 33) i s , properlyspeaking
,the solid fat separated from cottonseed Oil by cooling and
pressing . It is a pale yellow fat,of butter-like consistency , and
largely employed for the manufacture of butter substitutes . Thearticle known in commerce as “ cottonseed stearin ” i s usually impurestearic acid from cottonseed oil
,obtained by the method given in the
foot-note on page 1 33 . The crude oil expressed from decorticatedcottonseed i s sometimes very rancid and semi-solid at the ordinarytemperature from the separation of sol id fatty acids in the free state .By pressure i t would yield a product similar to that obtained bydistillation .
1 Natural refined o i l4 1 Part ly
“demargari nated
1 Winter o il.

1 36 MODE S OF EXAMINING FATS,OI LS
,AND WAXE S .
on cottonseed cake may give the colour indications of cottonseed Oil .Thus lard from the fat of pigs
,and butter from the milk of cows fed
on cottonseed cake may give the test and yet be quite free fromcottonseed oil .S i l verNi trate Test—This test
,originated by B echi
,
I depends uponthe presence in cottonseed oil of a substance which gives a brownprecipitate with silver nitrate . I t may be applied to the oil or to themixed fatty acids therefrom . Several modifications are in use . Themethod recommended by an I talian Government Commission in
which is substantially that of B echi,requires the two following
reagentsA . S ilver nitrate
,1 grm . ; alcohol (98% by volume) 200 c .c . ; ether,
40 c .c . ; nitric acid , grm .
B . Amyl alcohol,1 00 c .c . ; rape Oil, 1 5 c .c .
1 0 c .c . of the Oil to be examined are m ixed in a test- tube with 1 c .c .of reagent A ,
and then shaken with 1 0 c .c . of reagent B . The mixturei s next divided into two equal portions
,one of which is immersed in
boiling water for 1 5 minTITes . The heated sample is then removed fromthe water-bath , and i ts colour compared with the unheated half .Presence of cottonseed oil i s indicated by the reddish-brown colourationof the heated portion . Onl y the purest alcohol should be used
,and
the rape oil used should be cold drawn,and only slightly coloured ;
i t should be filtered in a hot-water oven before preparing the reagent .To guard against errors from impurity of the materials
,a blank test
Should be instituted Side by side with the actual test .The part played by the rape oil in
O
this test i s explained,according
to B echi,by the fact that whereas fresh cottonseed oils give the silver
nitrate indication without rape oil,old and rancid samples or their
mixed fatty acids do not interact unl ess thi s oil be added . Manychemists consider the addition of rape oil unnecessary . Thus , in theofficial method of the Swiss Society of Analysts3 a single reagent i s used ,which is prepared by dissolving 1 grm . of silver nitrate in 5 c .c . ofwater , adding 200 c .c . of alcohol
, 40 c .c . of ether and c .c . of nitricacid 1 0 c .c . of the oil are heated for 1 5 minutes in boiling waterwith 3 c .c . of this reagent , and it is said that 1 % of cottonseed oil , i fpresent , will be detected . Petkow
,4 who recommends this method ,
1 Chem . Zei t. , I I , 1 3 2 8 .
2 S e e A na ly s t , 1 7 0 .
S u i sse de Chim . et ISharm . , 3 5 , 448 .
4 Zei t. ofientl . Chem. , 1 9 0 7 , 1 3 , 2 1 .

COTTONSEED OIL .
states that the sensitiveness of B echi ’s test depends upon the relativeamount of silver nitrate used .
M illiau I prefers to operate upon the mixed fatty acids , but in preparing these regard must be had to the fact that prolonged heating ofthe acids at 1 00
° or washing them by boiling with water must beavoided
,as both cause a loss of the reacting body ?
This is probably the reason why some chemists have concludedthat Milliau ’
s test i s less delicate than B echi ’s . The following is themethod of procedure recommended by Archbutt : Approximately 5grm . of the oil are saponified , and the fatty acids are liberated from thesoap solution by dilute sulphuric acid in a separating funnel and dissolved by shaking with about 70 c .c . Of ether . The ethereal solution ,
after drawing Off the aqueous liquid,i s well washed with small quanti
ties of cold water and poured through a dry filter into a ‘dry flask .
The ether is distilled Off,and the fatty acids are heated on the steam
bath for about 5 to 1 0 minutes to drive off the remaining traces ofether and water
,and at once dissolved by pouring 20 c .c . of absolute
alcohol into the flask . The solution is transferred to a 1 -in . diametertest-tube and raised to boiling
,then 2 c .c . of a '
30% aqueous solution ofsilver nitrate are added and the test-tube is shaken and held
l
over awhite tile . In the presence of 5% of cottonseed oil a characteristicbrown turbidity i s produced almost immediately . If there be no immediate colouration
,the solution i s kept under observation for a
minute or two at boiling heat by moving the tube to and fro from thetile to the flame
,and if only 2% of cottonseed oil be present a distinct
brown colouration will be Obtained,though more . slowly developed .
This test has been examined by a large number of chemi sts and isknown to be given . only by cottonseed , kapok , and baobab oils . Todistinguish the two latter from cottonseed oil
,see under “Halphen
’s
Colour Test .” Some genuine rape oils appear to reduce the Silvernitrate very slightly
,but the reaction takes place slowly and the colour
produced is blackish,while with cottonseed oil i t i s brown . I t has
also been observed that some olive oils give a brown colour in B echi ’stest . Fats which have been exposed to the air or have become rancidmay reduce the silver solution owing to the presence of aldehydiccompounds . Thus B evan3 found that lard which had been exposedto the air for some days gave Bechi ’s test
,while some taken from the
1 J . Amer . Chem . S oc. , 1 893,
1 5 , 1 5 3 .
2 Archbu t t and Deeley . ubrica t ion and Lubricant s , p . 2 9 0.
3 Ana lys t , 1 89 4 , 1 9 , 88 .

1 38 MODE S OF EXAMINING FATS, OILS , AND WAXE S .
interior of the mass had no reducing property . I t has also been Shownthat genuine butter and lard from animals fed on cottonseed Oil maygive this reaction . On the other hand
,B echi
’
s test , like Halphen’s ,
i s not given by cottonseed Oil which has been heated to and allcottonseed oils do not respond to the test to the same extent . Therefore
,thi s test i s no more certain than Halphen
’s and can only be used
as an auxiliary to the quanti tative reactions .I t should be noted that Tortelli and Ruggeri I state that by applyingthis test to the fatty acids from the lead soaps soluble in ether
,cotton
seed oil which has been heated to250° long enough not to respond to
the ordinary testmay still be detected . 5 grm . of the oil are saponifiedwith 30 c .c . of alcoholic potassium hydroxide (60 grm . of the hydroxidein 1 000 c .c . of 90% alcohol), the solution exactly neutralised with 1 0%acetic acid and poured in a thin stream into a hot solution about 300c .c . in volume containing 5 grm . of lead acetate . The washed anddried lead soaps are warmed for about 20 minutes with anhydr ousether under a reflux condenser
,and the ethereal solution
,when cold
,is
filtered into a separating funnel and decomposed by Shaking withdilute hydrochloric acid . After separating the aqueous liquid andwell washing the ethereal solution with cold water
,the ether i s dis
tilled off and the residual fatty acids are dissolved in 1 0 c .c . of 90%alcohol and I c .c . of a 5% aqueous solution of silver nitrate . Theliquid i s transferred to a test-tube and placed in water at 70° toI t i s stated that 1 % of cottonseed oil can be detected in olive oil byheating for 2 minutes
,and by heating for several hours 1 0% of oil
which had been heated to 250° for 20 minutes could still be
detected .
N i tri c Acid Test—This test is given in the form recommendedby Lewkowi tsch ? A few c .c . of the oil are vigorously shaken inthe cold with an equal volume of nitric acid of sp . gr . and thenallowed to stand . Cottonseed oil gives an immediate coffee-browncolouration
,which becomes very intense on standing
,and mixtures of
other oils with cottonseed give a similar brown colour . S tronger acidgives less definite results . The only advantage this test has overHalphen
’s test i s that the brown colour i s still given by cottonseed oil
which has been heated so as to no longer respond to the latter test ; onthe other hand
,Lewkowi tsch states that he has met with many Ameri
1 Anna li . di l. Laboratoria . Chim. de Gabelle.. 1 900 , 4 , 249 .
2 Oi ls , Fat s and Waxes , I I , 1 6 3 .

1 40 MODE S OF EXAMINING FATS,OILS , AND WAXE S .
pale yellow or golden-yellow colour and an odour of maize meal ormalt . I t i s a semi-drying oil
,rather more strongly drying than cotton
seed o il,
I but much less so than linseed oil . I t i s,therefore
,unsuitable
either for lubricating or for mixing with paint . It i s used to some extent as an edible oil and for burning
,but its proper use i s for soap
making . I t makes an excellent soft soap,pale
,and as free as can be
from obj ectionable odour .The Sp . gr . of maize oil ranges from to at I t
solidifies at 1 0°
(Schaedler) below 20°
36°
(Hopkins3 ) ;but deposits solid fat on standi ng , even at the ordinary temperature(Lewkowitsch4) . I t dissolves in 50 volumes of absolute alcohol at
The absolute viscosi ty at 1 5 6° i s (viscosi ty of cottonseedoil
,to The oil does not form a solid elaidin . In
Maumené’s test
,the ri se of temperature ranges from about
‘
8 1 toMaize oil i s chiefly composed of Olein and linolin
,with a small pro
portion of saturated esters .6 The high Reichert value,
found byVulte and G ibson7 proves the presence of volatile acids
,and may help
in the detection of maize oil in mixtures . The unsaponifiable matterranges from about to and contains
,according to G ill and
Tufts ,8 sitosterol , the acetate of which melts at As the phytosterol acetate prepared from cottonseed Oil phytosterol was found tomelt at 1 20° to G ill and Tufts have proposed to make use of thisdifference for the detection of maize oil in cottonseed oil . Fromto of lecithin has been found in maize oil .Maize oil i s more likely to be used as an adulterant of other oils thanto be itself adulterated . I t gives no colouration with Halphen
’s
reagent or with furfural ; the presence of cottonseed oil or sesame oilcould
,therefore
,readily be detected
,unless the cottonseed oil had been
heat'ed . In the latter case , the raised m . p . and solidifying-point( titer test) of the mixed fatty acids would indicate cottonseed oil .Fish oils would be indicated by the bromoglyceride test and theodour on warming the sample .The following are some data obtained by examination of the mixedfatty acids from maize oil
1 Archbu t t , J. S ocuChem. I nd . , 1 89 9 , 1 8 , 3 46 .
2 J . S oc. Chem . I nd . , 1 89 2 , 1 1 , 5 04 .
Amer . Chem. S oc. , 1 8 8 , 20, 9 48 .
4 O ils , Fa t s and Waxe s , I , 1 3 1 .
5 Archbu t t , S oc. Chem . I nd . , 1 89 9 , 1 8, 3 46 .
6J . Amer . C m . S oc. , 1 89 8 , 20, 9 48 .
Amer . Chem . S oc. , 1 9 00 , 2 2 , 4 5 3Amer. Chem . S oc. , 1 9 03 , 2 5 , 2 5 1 .

SE SAME OIL .
W infield .
Lewkow i tsch .
Iod ine value of the liqu id fatty ac ids
PUMPKIN-SEED OIL .
(See p . This Oil i s largely used for culinary purposes inAustri a and Hungary , and ranks there next to olive oil in price . I t i sobtained from the seeds of the common gourd or pumpkin , Cucurbi tapepo. The cold-expressed oil prepared by Poda I was greenish , withfaint red fluorescence ; that prepared by roasting and subsequent hotexpression
,as on a commercial scale
,was brownish-green with deep
red fluorescence . I t easily becomes rancid , and has considerabledrying properties . As a result of the examination of several commercial samples
,as well as of genuine samples expressed by himself ,
Poda gives the following limits for the genuine oil :
Saponification value —1 90 . 2
Bute -refractometer,25
° —7 2 . 5
M . p . of fatty ac ids,commenced .
M . p . of fatty ac ids, ended
- 29 .8
Linseed,sesame
,cottonseed
,and rape oils are said to be used
adulterants .
SESAME OIL . TEEL OIL . GINGILI OIL .
(See p . S esame oil,sometimes called benne oil
,but distinct
from the oil of ben or behen,i s pale yellow
,usually of a deeper hue
than almond oil,nearly Odourless
,and has a bland and agreeable taste .
That expressed from the seeds congeals at about but that extracted by solvents at about I t i s used as an edible oil , in cookery ,and in the manufacture of margarine
,i t being compulsory in Germany
and Austria to add 1 0% of i t to butter substi tutes to facilitate theirdetection when used to adulterate butter . In B elgium , 5% must beadded (Lewkowi tsch). Sesame oil i s used in pharmacy and perfumery
,for soap-making
,and for adulterating almond and Olive oils . I t
1 Zei tsch , Nahr. Genussm. , 1 898 , 6 2 5 .

1 42 MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
is also commonly mixed with arachis Oil. I t dries more strongly thanrape oil
,but much less than cottonseed oil
,and does not readily turn
rancid .
“ German sesame oil ” is a name sometimes given to cameline oil .Sesame oil contains olein , linolin , palmitin , and stearin , but i ts composition i s not fully known . The unsaponifiable consti tuents , amounting to about to (Lewkowi tsch), include phytosterol , to
of a strongly dextrorotatory substance,
“ sesamin,
” and a phenolic body
,
“ sesamol,
” which gives a brilliant red colouration withfurfural and hydrochloric acid
,by means of which sesame oil can be
identified . Sesamol exists in the oil as a complex compound,from
which i t i s liberated by an acid ?
Sesame oil i s dextrorotatory , and in the absence of castor , croton ,and rosin Oi ls
,thi s property may assi st in i ts detection . T he following
observations have been published :
B ishOp ,2 6 samples
Rakusin,3 3 samples .
Utz,4 3 samples
Sprinkmeyer and Wagner,5 3 samples
The chief physical and chemical constants of thi s oil are given onpage 70 and the oleo-refractometer value on page 45 .
The mixed fatty acids have given the following figures
Solidify ing-po int (titer test) .
Refractive index at 60°Iod ine value of m ixed fatty ac idsIod ine value of liqu id fatty ac ids
1 Compare Villavecchia and Fabris , Zei t. ungem. Chem 5 0 ; ,Bomer Zei t. Nahr .
Genns sm . 1 89 9 , 2 , 7 05 ; Kre is , Chem. Zei t. 1 9 03 . 2 7 , 1 03 0 an 5 6° Canzoneri and
Perc iabo sco , Gazzetta , 2 5 3 ; and Malagn in i and Armanni , Chem. Zei t. , 884 .
2.J P harm. Chim . , 1 88 7 . 3 00 .
3 Chem. Zei t. , 1 9 06 , 3 0, 1 43 .
‘ P harm. Zei t 4 5 , 49 0 .
l5 Zei t. Nahr. Genussm. , 1 9 05 , 1 0, 3 4 7 .

MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
emulsion will become pink while being shaken . Villavecchia andFabris found this colouration to be caused by furfural
,produced by
the action of the acid on the sugar,and they have modified the test
by using a solution of furfural instead of sugar . As furfural i tselfgives a violet colouration with hydrochlori c -aci d, a very small quantityonly must be used .
A 2% solution of furfural in alcohol is prepared . c .c . of thissolution i s placed in a test- tube
,1 0 c .c . of hydrochloric acid and
1 0 c .c . of the oil are added,the tube is then corked
,Shaken for half a
minute and allowed to stand . If only 1 of sesame oil be present,the
acid which separates has a pink colouration ; with a strong rosered colour i s Obtained . This test i s recommended , as i t i s simplerthan that with sugar
,and half a minute ’s shaking is quite sufli cient .
Wauters suggests pouring the oil on the reagent,and says that less than
1 can be detected by a crimson colour at the point of contact .Lehnk ering ,
I in exam in ing a series of pure sesame oils,found some
which,while of normal iodine value and refractive index
,gave only
feeble colours in the furfural test,not more than as much colour
as was given by the oils which reacted most strongly . O ils extractedfrom the seeds with ether gave colours ranging in intensity from 5 to 8on the same scale .Rancid Oils may give a brown i sh tint in the Baudouin test , which willmask the reaction when only small amounts of sesame oil are present .Sprinkmeyer
2 found that rancid cottonseed oil containing sesame oilgave no red colour unless at least 1 7% of sesame oil were present .This shows the importance o f using fresh cottonseed oil in testingmargarine
,which
,according to German law
,must contain sufli cient
sesame oil to give a distinct red colouration when 0 .5 c .c . of the clearmelted fat i s mixed with 9 .5 c .c . of cottonseed oil and shaken withhydrochloric acid and furfural as directed above . Kreis3 states thatrancid sesame oil gives a less intense colouration than fresh oil . Am
buhl obtained an indigo-blue colour in applying the B audouin testto some old rancid sesame oil . Kreis4 thinks this must have been amixture of the red colour due to furfural with the green colour obtainedin B i shop ’s test . B ishops found that fresh sesame oil gave nocolour when shaken with times i ts volume of hydrochloric acid1 Zei t. o
'
fientl . Chem 1 9 03 , 9 , 4 3 6 .
2-Zei t. Nahr . Genussm. , 1 9 08 , 1 5 , 2 0.
3 Chem . Ze i t . , 1 9 08 , 23 , 8 7 .
4 Chem . Zei t. . 1 89 9 , 23 , 80 2 .
P harm . Chim . , 1 889 , 20, 2 44 .

SE SAME OIL . 1 45
but if exposed to air and light for a few days i t coloured the acidgreen . O il which had been exposed for years coloured the acid almostblue
,and a blue colouring matter separated on standing
,the acid be
coming green again . This reaction may , therefore , modify the colourobtained in the B audouin test with old sesame oils , but i t i s stated thatthe oil which separates from the green or blue-coloured hydrochloricacid will give the red colour on being shaken with hydrochloricacid and furfural .Several observers have found that in applying the furfural test tocertain olive oils of undoubted puri ty the acid liquid assumes a violetcolouration after a Short time ; da S ilva I found Douro olive oil gave thiscolour . It has also been observed that some genuine I talian , Tun i sianand Algerian olive oils give a rose colouration
,Similar to that produced
by about 5% of sesame oil ?'
According to M illiau ,this i s caused by a
colouring matter derived from the aqueous part of the pulp of the fruit,
and if the test -be applied to the mixed fatty acids instead of to theoriginal oil any possibili ty of error i s obviated . Therefore
,Milliau ’
s
modification shoul d be adopted in cases where doubt exists as to thecause of the colouration .
So ltse in’s Test . —The Oil i s mixed with an equal volume of stan
nous chloride solution,German Pharmacopoeia strength , shaken
vigorously (once only), and placed in boiling water . A red coloura
tion is produced in the presence of sesame oil . This reaction is saidto be more delicate than B audouin ’s
, and to be specially applicable tobutters and margarines artifically coloured with coal-tar dyes, whichare reduced and rendered colourless . As the delicacy of the reactioni s impaired if the liquids remain too long in contact without separating ,Soltse in recommends diluting the oil with twice its volume of petroleumspirit , adding half the volume of stannous chloride , Shaking well , andstanding the tube in water at aboutToch er’s Tes t . —I 5 c .c . of the oil are Shaken for about 30 secondswith a freshly made
,practically colourless solution of 1 grm . of pyro
gallol in 1 5 c .c . of concentrated hydrochloric acid Theaqueous liquid is drawn off through a wet filter-paper and heatedfor 1 5 minutes on a water-bath . In the presence of sesame oil i t becomes coloured reddish-purple
,appearing red by transmi tted
,and blue
by reflected light . The test is very delicate,and will readily detect 2%
1 Bull . S oc. Chim . , 1 89 8 , 1 88 .
2 V11lavecch ia and Fabris , gei t. angew. Chem. , 1 89 2 , 5 09 .
Vo l. I I .
—1 0

1 46 MODE S OF EXAMINING FATS , OILS,AND WAXE S .
of sesame oil in rape or olive oil . B ellier says this reaction is not givenby certain genuine olive oils which give a red colour in the furfuraltest ?Adulteration with rape oi l would lower the sp . gr . and saponificationvalue of sesame oil and the melting and solidifying-points of the fattyacids .P oppyseed oi l woul d raise the iodine value and thermal tests , andalso the refractometer numbers . It would lower the melting andsolidifying-points of the mixed fatty acids .Cottonseed oi l
,unless i t has been altered by heating
,would be in
dicated by Halphen’s test . I t woul d raise the melting and solidifying
points of the mixed fatty acids and would tend to raise the iodinevalue of the liquid fatty acids
,sesame oil acids absorbing from
to and cottonseed oil acids from to of iodine .Cottonseed oil would increase the ri se of temperature in the thermaltests . I t would not materially alter the other values . The mixedfatty acids of cottonseed oil were found by Farnsteiner2 to containrather more linolic acid than those of sesame Oil
,he having obtained
tetrabromides corresponding with of linolic acid from theformer and to from the latter . “
But a larger number ofsamples need investigating .
Arachi s oi l would be detected and determined by isolating i tsarachidic acid .
SOJA-BEAN OIL .
This oil i s obtained from soja or soy beans , the seeds of S oja j aponi ca(S oja hi spida), a plant native to China , Manchur ia , Korea , and Japan ,
but also grown elsewhere . I t has marked drying properties and , according to D e Negri and Fabris
,readily solidifies . The values on p .
70 are b ased upon the resul ts of Morawski and S tingl , De Negri andFabris
,and Shukoff . Four commercial samples of Chinese bean oil
examined by Korentschewsk i and Zimmermann,3 one obtained direc t
from the factory in Kharbin,gave the following results
,some of which
are quite different from those previously recorded by the other observers . The oil is described as dark brown
,having a faint odour
suggesting tung oil,and a bland taste .
1 Ann . Chem. A na l . , 1 89 9 . 4 , 2 1 7 .
2 Zei t. Nahr. C enu ssm 1 89 9 . 2 . 1 .
3 Chem. Zei t 1 9 0 5 , 29 , 7 7 7 .

1 48 MODE S OF EX AMINING FATS , OILS,AND WAXES .
of unsaponifiable matter which , as in the case of maize oil , containssi tosterol . I About 2% of lecithin was also found . The vi scosityof the oil at 20° was times that of rape oil at the same temperature .The refractive index at 20
° was D e Negri2 obtained thefollowing results from the mixed fatty acids :
So lid ify ing-po intIod ine value
IV.
’ LINSEED OIL GROUP .
P ine Nut Oi lPo
gy seed 011.
Sa ower Oi l.Sunfl ower Oi l .Tung Oi l .Walnut Oi l . Nut Oi l .
CANDLE NUT OIL .
(See also p . This oil i s obtained from the seed-kernels ofAleuri tes moluccana (A . tri loba), a tree which flourishes over the wholeof the South Sea I slands . The cold-pressed oil i s almost colourless ,or slightly yellow
,of an agreeable flavour and smell
,and i s used as an
edible oil . The hot-pressed ,
Oil has a brownish-yellow colour andunpleasant flavour
,and is used for technical purposes?» I t dr ies less
rapidly than linseed oil and is used for mixing paints and making oil ~varnishes .4 I t i s Obtainable in enormous quanti ties , and may beemployed as an adulterant ~
of linseed oil (Lewkowi tschS). Thepublished iodine and saponification values of this oil vary considerably,as IS shown in the following table
1 Burian , M ona tsh . , 1 89 7 , 1 8 , 5 5 1 .
2Chem. Zei t. , 1 89 8 , 22 , 9 7 6 .
3 S cha edler , Techno logi e der Fet t e . p . 6 63 .
°1 S p on’
s Ency cloped ia . IV , 1 3 9 3 .
5 Technology of O ils , I I , 6 9 .

CEDAR NUT OI L .
0 . 9 20a , 0 . 9 2 6b
1 84a , 1 8 7 .4b1 3 6 . 3 a , 1 3 9 . 3 bRe ich ert -Me issl valu e
Ace t y l va lu eUnsapon ifiable mat t er. 0 5 3
M i xed F a tty A cids .
So lid ifyi ng-po intS o lid ify ing-po int (t iter t e st ) .Iod in e val t 4 2 . 7a , 1 44 . 1 bIod ine va lu e o f liqui d fat t y
acids .
Walker and Warburton6 obtained from 7 . 28 to of brominatedglycerides by Hebner and M i tchell ’s process from the sample examinedby Lewkowi tsch .
A sample of the commercial oil,described as Lumbang oil
,recently
exam ined by the revi ser,was light brown in colour
,had an unpleasant
and somewhat pungent odour,and gave the following results :
Sp . gr . at
Saponification valueIod ine value
The oil dried nearly as rapidly as linseed Oil,and had about half
the viscosi ty of rape oil at 1 5 . I t contained 2 of free (oleic)acid . The saponifiéation and iodine values agree with those observedby Lewkowitsch and Kassler .
CEDAR NUT OIL .
(See p . The commercial oil i s expressed from the seeds ofthe S iberian cedar , Pinus cembra; i t i s golden-yellow ,
and of agreeable ,though somewhat rancid taste . I t contains the glycerides of linolic
1 0es terr . Chem . Zei t 1 89 8 , 1 , 2 0 2 .
2Techno logy of 0118 , I I , 6 7 .
3 5 ei fenseider Ze i t . ; F arben-Ze i t. , 1 9 03 . 8 , 3 5 9 .
4Zei t. Nahr . Genussm . , 1 9 03 , 1 0 2 5 .
1
5 B u ll . Imp . I ns t , 1 9 0 7 , 5 , 1 3 5 .
“Ana ly s t, 1 9 0 2 , 2 7 , 2 3 7 .
Ext ract ed 1 1 8 5 7Extract edfrom th e seeds from kerne ls
by (a) pe t roleum s pirit , (b)
e th er .
o f nu t s o f A .
m o luccana
( S ou th S eaIsland s) .
Oi l from Ext ract Ext ractF ij i . ed b y ed from
e th er th e k erfrom n e ls o fseeds of Ch ineseA . mo luc A .
cana triloba .from th e
Came
ro on s .

1 50 MODES OF EXAMINING FATS , OI LS , AND WAXE S .
and oleic acids,the former predominating
,a very li ttle linolenic acid
i s also present ; among the solid fatty acids palmitic acid has beenidentified
,and there i s also present a considerable proportion of
volatile fatty acids .A specimen of this oil examined by von SchmoellingI gave thefollowing results :
Sp . gr . at 1 5°
So lid ify ing-po intSaponificat ion value .
Iodine value (Waller) .
Hehner valueVo latile fatty ac idsFree fatty ac idsNeutralization valueUnsaponifiable matter
,
Maumené test (Archbutt’
s method)L iqu id fatty ac ids, (Muter’s method)Iod ine value of liqu id fatty ac ids .
Cedar nut oil is used in S iberia as an edible oil , and is said to betechnically of value as a fairly rapid drying oil of pale colour . VonSchmoelling states , however, that the “ varnish” produced by heatingthe oil with 5% of manganese borate for 4 hours to 1 40° or 1 50° tooktwice as long to dry on glass as l inseed oil varnish similarly prepared
,
and the product was very viscid and resembled a blown oil . Thelast-mentioned charact eristic , together with the high price of the oilwould
,he thinks
,prevent i t from being used in the manufacture of
varnish .
HEMPSEED OIL.
(See also p . This oil i s obtained from the seeds of the hempplant
,Cannabi s sativa . The expressed oil i s at first greenish or
brownish-yellow,deepening in colour on exposure to the air . I t
consists of the esters of liquid and solid fatty acids,the former composed
approximately,according to Hazura and Grii ssner, of 70% of linolic ,
1 5% of linolenic and isolinolenic and 1 5% of oleic acids ; the solidfatty acids are said to be palmitic and oleic .Hempseed oil has a high iodine value and is a strongly drying oil
,
though i t dries less rapidly than linseed oil . I t i s used for making1 Chem . Zei t. . 1 9 00 , 24 , 8 1 5 .

1 52 MODE S OF EXAMINING FATS,OILS
,AND WAX E S .
POPPYSEED OIL . POPPY OIL.
(For constants see p . This oil 15 expressed from the seeds ofthe opium poppy
,P apaver somniferum
,the yield ranging from about 4 1
to about according to the variety . The seed is black,brown
,
yellow,or white , the latter being considered the richest . The plant
i s extensively cultivated in Egypt,Asia Minor
,Persia
,India
,and
China ; i t is also grown in France and Germany .
The cold—drawn oil extracted by the first pressing (hui le blanche)i s straw yellow in colour
,limpid
,and almost odourless ; i t has a pleasant
almond-like flavour,and being Slow to become rancid i s largely used
on the continent of Europe as a salad oil and also as an adulterant ofol ive oil . Owing to its drying properties and pale colour
,which i t
retains,i t is in demand for the manufacture of artists ’ pigments
,the
sun-bleached oil being used for white pigments and the unbleachedbut pale coloured oil for coloured pigments ? The inferior varietiesof poppy Oil
,obtained by a second pressing (hui le rouge) and from in
ferior seed,are used for soap-making and burning .
In a sample of oil from genuine poppy-seed,Tolman and Munson
found of solid fatty acids . The li quid fatty acids were foundby Hazura and Grii ssner2 to consist , approximately , of oleic acid 30l inolic acid 65 l inolen ic and i solinolenic acid The calculatediodine value of such a mixture i s 1 58 , which approximates to the actualiodine value of the liquid fatty acids
,found by Tortell i and Ruggeri
to be and by Tolman and Munson,
Hebner and
M i tchell obtained no insoluble brominated glyceride from 4 samples ofpoppyseed oil .Utz3 has stated that practically all the commercial poppy oilsexamined by him contained more or less (up to of sesame oil ,not added as an adulterant
,sesame seed and oil being dearer than those
of the poppy,but due to careless methods of manufacture , the two
kinds of oil being expressed in the same works . The sesame oil i sdetected by its lower iodine value , and by the colour tests of Soltseinand B audouin . Owing to this admixture , Utz believes the iodinevalue of genuine poppyseed oil
,usually stated as 1 30 to 1 4 1 , has been
generally understated . The oil which he extracted by petroleumether from three varieties of seed gave the followmg results :1 Lo t t er , Chem . Zei t '
1 89 4 , 1 8 , 1 69 6 .
2M ona tsh . Chem . , 1 888 , 9 , 1 80 .
3 Chem . Zei t. , 1 9 03 , 2 7 , 1 1 7 6 .

SAFFLOWER OI L .
O i l from
Ind ian poppyseed
A specimen of commercial poppy oil which probably contained lessthan 5% of sesame Oil had an iodine value of and another
,
which gave only faint indications of sesame oil by the colour tests ;absorbed of iodine . I t may be mentioned
,however
,that
Tolman and Munson obtained iodine values of and
from cold-drawn poppy oil expressed from seed which they state wasidentified as that of P . somnifera .
Utz confirms B ishop ’s statement that pure poppy oil i s inactive,
and suggests that the sample examined by Crossley and Le Sueurwhich had a rotation of +4’ may have contained sesame oil .The viscosity of poppyseed Oil at 70° F. is about two- thirds that ofrefined rape oil at the same temperature . 4 samples tested by Crossleyand Le Sueur required from'
254 to 259 seconds for the outflow of50 c .c . from Redwood ’s viscometer
,water at 70° F. requ i ring sec
onds from the same instrument .O ther results for poppy oil are
SAFFLOWER OIL .
(For constants see p . Safllower or saffron oil i s the productof the seeds of the safflower (saffron) plant , Carthamus tinctorius , whichis being increasingly cultivated in the Caucasus and in Turk estan ;Iit was at one time extensively cultivated all over India
,and is
.
stillgrown to some extent in that country . The oil is obtained by expression or extraction
,the yield being about I t has a pleasant
taste,especially when Obtained from the husked seeds , when it is of
1 Ty la ik ow . J. S oc . Chem . Ind 1 9 02 , 2 1 , 864 .

1 54 MODE S OF EXAMINING FATS,OILS , AND WAXE S .
better quality than that obtained from the unhusked . seeds . I t has abright yellow colour, and a taste very similar to that of sunflower Oil .It is a good drying oil . The viscosity of the samp l es examined byCrossley and Le SueurI was about two-th irds that of refined rape oil ,50 c .c . requiring from 243 to 294 seconds to flow from Redwood’s viscometer at 70° F . The same observers Obtained butyro-refractometerreadings at 40° from toAccording to Le Sueur
,
2 the insoluble fatty acids of safllower oilconsist of about 1 0% of solid acids (palmitic and stearic) and 90% ofli quid fatty acids (Oleic and linolic). No linolenic acid is present .
SUNFLOWER OIL .
(For constants see p . This oil i s expressed from the seeds ofthe sunflower
,Hcli anthus annuus
,which is widely grown in southern
Russia . The cold-pressed oil i s pale yellow and of pleasant,mild
taste and Odour . I t i s used for culinary purposes,also as the vegetable
Oil inmargarine,and has been detected as an adulterant of olive oil .
Oil of the second pressing , which is more coloured , i s used as a lampoil
,and in the manufacture of varnishes as a substi tute for linseed oil ,
but i t dries much more slowly than the latter . I t i s also used for soapmaking .
Sunflower oil contains the glycerides of palmitic , oleic , and linolicacids
,and probably also of linolenic and isolinolenic acids in small
proportion . Tolman and Munson Obtained andrespectively
,of solid fatty acids from 2 samples
,which also gave the
following results : iodine values , and 1 04 .1 ; iodine values of theliquid fatty acids
,and Tortelli and Ruggeri found the
iodine value of the liquid fatty acids from a sample of oil whichabsorbed 1 3 7% of iodine . A sample examined by Jean3 contained0 . 7 2% of unsaponifiable matter .
TUNG OIL. CHINESE WOOD OIL . JAPANESE WOOD OIL.
WOOD OIL .4
(For constants see p . Tung oil i s derived from the seeds ofAleuri tes cordata (Elaeococca vernici a) a plant which is grown exten
S oc. Chem. Ind . , 1 89 8 , 1 7 , 9 3 2 .
S oc. Chem. I nd . , 1 9 00 , 1 9 , 1 04 .
11 A nn . Chim. Anal . , 1 9 0 1 , 6 , 1 66 .
4 Th e t erm wood o i l”i s also appli ed to Gurj un balsam . an o leoresm obta i ned from th e
East Ind ies .

1 56 MODE S OF EXAMINING FATS,OI LS
,AND WAXE S .
and the molecular weight in benzene solution ascertained by thefreezing-point method showed that
‘
while the fatty acids of the originaloil have a molecular weight of to those of the polymeri sedoil gave values ranging from to In a further experiment ,in which the oil was heated for several hours in a sealed tube at 300 to
the iodine value of the fatty acids was reduced to 68 . 5 and"themolecular weight increased to showing that in this case polymerisation had proceeded still further . Polymerised tung oil i sinsoluble in the usual solvents
,and hence the molecular weight of the
polymerized oil itself could not be determined .
I odine also has a remarkable action on this oil . Jenkins foundthat if a saturated solution of iodine in chloroform or carbon disulphidebe dropped upon the oil i t immediately solidifies i t . Bromine hasno such action . If 1 grm . of tung oil be dissolved in
‘
5 c .c . ofchloroform
,m ixed with 5 c .c . of a saturated solution of iodine in chloro
form and stirred,the whole may become suddenly converted into a
j elly in a few seconds . Some samples take much longer,and may need
gentle warming ; some refuse to gelatinise unless a larger quanti ty(2 to 4 grm .) of the oil be taken .
Tung oil dri es very rapi dly . 0 . 5 grm . was exposed to the air in awater-oven at about 1 00° on a watch glass . In about 3 m inutes adry ring formed round the spot of oil
,and in 3 hours the oil was dry
throughout and had gained 1 . 56% in weight . Raw linseed oilsimilarly treated had just begun to dry at the edges in 3 hours , and hadgained 0 92% in weight (Archbutt) .
5 grm . of tung Oil stirred in the cold with 2 c .c . of carbon disulphideand 2 c .c . of sulphur chloride for 90 seconds suddenly stiffened to athick and sticky jelly
,not nearly so hard as the product ob tained in a
similar manner from either castor or linseed oil ?In the elaidin test
,Jenkins obtained a brownish-red product ,
consisting of an oily layer resting on a nearly solid portion . Whenstirred up
,the whole would scarcely flow .
In the Valenta test, using glacial acetic acid , Jenkins found theturbidity temperatures of two samples to be 44° and respectively .
S trong sulphuri c aci d immediately solidifies tung oil , forming ablack clot
,but by mi xing 1 0 grm . of the oil with 40 grm . of olive oil ,
Jenkins found i t possible to measure the rise of temperature , and aftermaking a correction for the heat developed by the olive oil obtainedi 1 Jenkins . J. S oc. Chem. Ind . , 1 89 7 , 1 6, 1 9 5 .

WALNUT OIL . 1 57
specific temperature reacti ons of 298 and 330 with two diff erentsamples .N i tric aci d rapidly converts tung oil into a tough mass
,which
darkens and becomes more brittle : on standing .
The vi scosi ty seems to be very variable , possibly due to a partialpolymerisation of some samples . Two samples tested by Jenkinsrequired
,respectively
,1 433 and 858 seconds for the outflow of 50 c .c .
from Redwood’s viscometer at 60° F .
Halphen ’s reacti on for cottonseed oil gives no colouration with gen
uine samples of tung oil .Commercial samples of tung oil have been found to contain fromto of unsaponifiable matter and from to of
free (olei c) aci d.
The mixedfatty aci ds have been found to solidi fy at toand to have an iodine value of 1 50 to 1 59 .
Tung oil is said to cause severe ulcers when brought into contactwith the sk in
,
I and its use in cosmetic and similar preparations (forwhich patents have been taken out) should be prohibited .
B lakeman2 has patented as a drying oil a mixture of 85 parts of anon-drying oil such as cottonseed oil ” and 1 5 parts of tung oil mi xedwith a drier .
WALNUT OIL . NUT OIL .
(For constants see p . This oil i s obtained by expression fromthe kernels of the walnut
,Juglans regi a, which , if the nuts have been
kept for 2 to 3 months after they have been gathered , yield from 63 toif kept too long
,the oil expressed from the kernels may be rancid .
The fir st-expressed,
“ virgin,
” Oil is almost colourless,with faint
odour and not unpleasant flavour ; i t i s largely used in some parts ofEurope as a substitute for olive Oil for edible purposes . The secondpressed or “fire-drawn oil is greenish-coloured
,with an acrid flavour .
B eing an excellent drying oil,forming a varnish which is pale in colour
and less liable to crack than linseed oil varn ish,walnut oil is much used
for preparing artists ’ colours . I t is also a good lamp oil , and can beused. for making soft soap .
Walnut oil contains the esters of myristic,lauric
,oleic , linolic ,
linolenic and isolinolenic acids . According to Hazura and Grii ssner ,
1 Hertkorn . Chem. Zei t. , 1 9 03 , 27 , 63 5 .
2 U. S . Pa tent , 7 6 7 68 2 , 1 9 04 .
Q

MODES OF EXAMINING FATS , OILS , AND WAXE S .
80% of the liquid fatty acids consist of linolic acid , 1 3% of linoleni cand isolinolenic acids
,and 7% of Oleic acid . The calculated iodine
value of this mixture of liquid acids is 1 87 (Hebner and Mi tchell) ;t he iodine value of the liquid fatty '
acids determined by Tortelli andRuggeri was found to be the oil from which they were obtainedhavmg an Iodine value of5 samples of cold-pressed B ulgarian walnut oil examined by Petkow’
gave results as follows :Sp . gr. at 1 5
°
Refractometer reading, 40
° 67-68
Iodine valueA sample from the Punj ab examined by Crossley and Le Sueur gavea butyro-refractometer reading of at and 50 c .c . . required23 2 seconds to flow from Redwood ’s viscometer at 70° F.
A sample of black-walnut oil from J. nigra examined by Kebler2was a straw yellow coloured oil
,having an agreeable walnut-like
odour and taste,and gave the following results :
Sp . gr. at 1 5°
The o il became turbid at
Ac idSaponification valueIodine value . .
Hebner valueRe ichert-Me issl valueM . p . of fatty acids
Confirmation of the high Reichert—Meissl value of this sample is required
,seeing that ordinary walnut oil has no Reichert—Meissl value .
walnut oil i s l iable to adulteration with other oils , most of whichwould lower the iodi ne value . Poppy oil would not appreciably alterthe ordinary constants
,and B ellier3 has proposed a test for this oil
based upon the difference in solubili ty of the solid fatty acids in 70%alcohol at 1 7° to 1 9
° in presence of a definite amount of potassiumacetate . 1 c .c . of the oil i s warmed in a test- tube with 5 c .c . of a solution of 1 6 grm . potassium hydroxide in 1 00 c .c . of 9 1 to 93% alcohol ,until a clear solution is obtained
,a similar test being simultaneously
made with a walnut oil of known puri ty . The tubes are then closedand kept at about 70° for 30 minutes . A quanti ty of dilute acetic acid( 1 volume of glacial acetic acid and 3 volumes of water) exactly1 Zei t. Nahr. Genussm. , 1 9 01 . 4 , 8 2 8.
F rank,Ins t. , 1 9 0 1 , 1 5 1 , 3 9 4 .
3 A nn . Ch i m . A na l . , 1 9 05 , 1 0, 5 2 .

1 60 MODE S OF EXAMINING FATS , OILS , AND WAXE S .
mm . tube,caused a rotation ranging from 7 6
° to 9 . Dowzard1
gives + 8° to Rakusin2+8° to +8 Lythgoe
3 found the optical rotation of 44 samples of genuine castor oil to range from 23 4
° to(Ventzke , 200 mm .
,these numbers fall within the limits
given by Deering and Redwood,whose values correspond with
2 1 . 9° to 28° Ventzke .
Castor oil consists mainly of triricinolein,the glyceride of two iso
meric hydroxylated acids,ric inoleic and isoricinole ic . Small quantities
of tristearin and of the glyceride of dihydroxystearic acid are also present . Palmi tin and olein are absent . Lewkowitsch4 points out thatless saturated fatty acids than ricinoleic must also be present
,otherwise
the iodine value of castor oil would not exceed about 6 7 .
Crude R icino le ic Acid , HC , 8H3 3O3 ,may be prepared from castor
Oil by the method employed for the preparation of oleic acid from oils ;or castor oil may be saponified, and the soap fractionally precipitatedwith calcium chloride . The first third should be rejected . The laterfractions are purified by crystallisation from alcohol
,and decomposed
by dilute hydrochloric acid .
Ricinoleic acid is a thick oily liquid,which solidifies below It i s
insoluble in water,but i s miscible in all proportions with alcohol and
ether . The alcoholic solution has an acid reaction,an unpleasant ,
persistent,acrid taste
,and does not oxidise in the air . Like oleic
acid,i t combines with the atoms of bromine
,and by treatment wi th
nitrous acid i s gradually converted into a stereo—i somer,ricinela’idic
acid,a body crystalli sing from alcohol in white needles
,melting at
and forming an additive compound with Br , . When heated withphosphorus
,iodine
,and water
,ricinoleic acid yields an iodo- acid ,
which by the action of nascent hydrogen (hydrochloric acid and zinc)i s converted into stearic acid .
When distilled in a partial vacuum,ricinoleic acid yields enanthal
or normal heptoic aldehyde, C6H, 3
.CHO ,and an acid of the acrylic
series . This behaviour may be used for the detection of castor oil .For thi s purpose the sample should be saponified , and the fatty acidsliberated and rapidly distilled in a small retort . The distillate i sShaken with a saturated solution of acid sodium sulphite
,the resultant
crystals pressed,dissolved in a solution of sodium carbonate , and the
1 Chemi st and Druggi s t, 1 9 0 1 , 5 8, 3 2 5 .
2 Chem . Zei t. , 1 9 06 , 3 0, 1 4 3 .
8J . Amer . Chem. S oc. , 1 9 0 5 , 2 7 , 888 .
4 O ils , Fat s and Waxe s , I I , 3 2 6 .

CAS TOR OI L .
liquid distilled in a current of steam . The enanthal will collect onthe surface of the distillate as a highly refractive liqu id , of peculiararomatic odour
,boiling at Enanthal is also produced by sub
jecting the alkali-metal salts of ricinoleic acid to dry distillation ,but
if sodium hydroxide be present in addition ,sodium sebacate is formed
,
and methyl-hexyl carbinol and methyl-hexyl ketone are found in thedistillate .Ricinoleic acid forms a series of salts
,many of which are soluble in
,
and may be crystallised from ,alcohol or ether .
The oleo-refractometer value of castor oil i s given in the table onp . 44 . The bute -refractometer readings of the 44 samples examinedby Lythgoe averaged at and at 2 the variations fromthese numbers being slight and not exceeding :t o . 5 . Lythgoe givesthe values for each o . 5
° from 1 5° to 3 5°
The following are some results of examination of the mixed fattyacids of castor oil (see also p . 7 1 )
Sp . gr. at
Sp . gr. at 980—99
0
So lid ifying-po intRefractive index atI odine value of m ixed fatty acidsIodine value of liqu id fatty ac ids
COMMERCIAL CASTOR OIL .
The peculiar physical characters of pure castor oil distinguish i tsharply from most other oils
,but i t is liable to adulteration with oils ,
such as cottonseed or rape oils,blown oils
,rosin oil
,etc .
,which may
be lower in price,for the detection of which the following tests may be
used .
The sp. gr . of genuine castor oil i s exceptionally high,and should
lie between and at Adulteration with any othernatural fixed oil or mixture of oils would lower the sp . gr .
,and although
this might be adjusted by the addition of rosin o il,which often has asp . gr . as high as the presence of the latter would be easilydetected by an estimation of the unsaponifiable matter, which in
V0]. I I .
—1 1

1 62 MODE S OF EXAMINING FATS , OI LS,AND WAXES .
genuine castor oil i s usually less than 0 . B lown rape or cottonseed oil might be added wi thout altering the sp . gr.
,and without
causing any large increase of the unsaponifiable matter, but these oilswould be detected by some of the following tests .The vi scosi ty of castor oil at the ordinary temperature exceeds that ofall other natural fixed oils
,but is approached by that of rosin oil and
may be exceeded by that of blown oil . Twenty-three samples ofIndian castor Oil tested by Deering and Redwood in the Redwoodviscometer required from 1 1 60 to 1 1 90 seconds for the outflow of50 c .c . at 1 00° F . The absolute viscosity
,determined by Archbutt
and Deeley,was found to be at 1 00° F .
,at 1 50° F .
,and
at 2 1 2° F ?
The solubi li ty in alcohol of castor oil i s greater than that of anyOil likely to be used as an adulterant . Absolute alcohol dissolvescastor oil in every proportion ; 90% alcohol (sp . gr . dissolvesless , 1 volume of castor oil requiring from to volumes of90% alcohol at 20° accordi ng to experiments by I tallie , 2 or from 3 to ‘
4
volumes according to Dowzard .3 A genuine sample tested by Arch
butt dissolved perfectly in 2 volumes of 90% alcohol at 23 samplesof Indian castor O ils examined by Deering and Redwood were completely soluble in 3 volumes of alcohol of sp . gr . 30 at 1 5 Fromthese results i t appears that castor oil
,if genuine
,should dissolve
completely in 3 volumes of 90% alcohol at I t i s usual,however ,
to employ 5 volumes of alcohol , as recommended in the Bri ti sh Pharmacopoeia . Archbutt and Deeley4 found that when onl y 5% ofei ther rape
,blown rape
,cotton seed
,poppy
,maize
,or curcas oils
were mixed with castor oil, 5 volumes of 90% alcohol gave a strongly
turbid mixture at which deposited a small quantity of oil onstanding . The following test
,originally recommended by FinkenerS
(who used a Slightly stronger alcohol), may therefore be employed withconfidence as a rapid method of assay : Measure exactly 1 0 c .c . ofcastor oil in a graduated
,stoppered test cylinder
,add 50 c .c . of alcohol
(sp . gr . and well mix . If genuine,a clear and bright solutio n
will be obtained at If as little as 5% of foreign oil be present, th eliquid wi ll remain strongly turbid
,even on warming to Oleic
aci d would not be detected by the alcohol test , but i t can be estimated1 Lubricat ion and Lubricant s , p . 1 6 7 .
2 Lew k ow i t sch , Oils , Fat s and Waxes , I I , 3 3 0.
3 S oc. Chem. I nd . , 1 9 0 1 , 20, 3 7 0 .
4 ubrica t ion and Lubricant s , p . 3 1 9 .
S oc. Chem. Ind. , 1 88 7 . 6 , 1 48 .

1 64 MODE S OF EXAMINING FATS,OILS , AND WAXE S .
tallow,or lard oil
,is present
,a clear mixture of the three being readily
obtained . Pure castor oil mixes with refined rosin oil in all proportions .The acetyl value (see p . 3 2) of castor oil to exceedsthat of any other known oil , and i s one of themost valuable indicationsof i ts purity . Al though blown oils also have high acetyl values (about45 to 65) they do not nearly approach castor oil in this respect , andthe detection of 1 0% of blown oil in castor oil is possible . Grapeseed oil
,if added to castor oil
,would lower the sp
'
. gr .The Hehner value (95 . 3 to 95 . and the i odine value would belowered by adulteration with blown oil ; the Rei chert-Mei ssl value
,
Maumene’
thermal value,and saponificati on value would
,on the other
hand,be raised . Refined rosin oi l
,which has been extensively
employed for the adulteration of castor oil , neutralises no alkali , oronly a trifling quanti ty
,and would
,therefore
,lower the sapOnification
value .
The high opti cal rotati on caused by castor oil has already beenreferred to and would be lowered by most other natural fixed oils
,
except croton oil and laurel oil (Rakusin). I t would not be availablein the presence of rosin oi l
,which i s strongly dextrorotatory . Some
samples of sesame Oil have a marked rotatory power (see p .
The refractometer would also be useful . Lythgoe mentions an instancein which 50% of cottonseed oi l was found in a sample of “ castor oil ”from Massachusetts . This was detected at first by the low opticalactivi ty of the sample
,and was confirmed by the low refractive power
and sp . gr . and the high iodine value,also by Halphen
’
s colourreaction .
Among other possible adulterants,poppyseed oi l would lower
the sp . gr .,acetyl value
,and viscosi ty
,and would raise the iodine value ;
lard oi l would lower the Oleo-refractometer value,sp . gr .
,viscosi ty ,
acetyl,and iodine values ; coconut oi l would raise the saponification
value and would lower the sp . gr .,Hehner value and iodine value ;
and seal oi l would lower the sp . gr .,viscosity and acetyl value , and
would raise the iodine value . S esame oi l would be detected by thecharacteri stic colour tests for this oil .The formation of sebacic acid
,when the sample is distilled alone or
with a quantity of alkali insuffi cient for its complete saponific ation ,
may 'be employed as a test for foreign fixed oils in castor oil .When castor oil i s distilled at 300° until i t has lost from 5 toof i ts weight
,a yellowish-brown Oil with green fluorescence remains ,

CAS TOR OIL .
which ismiscible in all proportions with mineral oils,ceresin or vaseline
,
and is not soluble in alcohol or acetic acid . The name “floricin
” hasbeen given to this product . According to Fendler
,
I i t contains theester of undecylenic acid . A similar product is obtained by heatingthe oil in an autoclave to 260
°—300
° under a pressure of 4 to 6 atmospheres for about 1 0 hours ?Es t ima t ion of Cas tor Oi l . —Lane ,3 having found that leadricinoleate is insoluble in petroleum ether , has devi sed the followingmethod Of estimating castor oil in mixtures
,soaps
,and alizarin oils .
The liquid acids in these cannot be estimated by the lead- soap and
ethylic ether method,because lead stearate and palmi tate are soluble
in a solution of lead rincinoleate in ethylic ether .From 3 to grm . of the oil or fatty acids are taken . If thesample be sulphonated (Turkey-red oil), a quantity suffi cient to yieldthis amount of fatty acid must be first boiled for about 2 hours withdilute hydrochloric acid ( 1 : with frequent shaking
,until desulphona
tion is complete,the acid liquid being then poured into a separating
funnel,shaken with ether
,and the ethereal solution washed with water
until free from acid . The ether having been distilled off,the fatty
matter is weighed and saponified with alcoholic potass ium hydroxide ,a drop of phenolphthalei n added
,the solution rendered slightly acid
with acetic acid and then very faintly pink with N 1 0 sodium hydroxide .
The liquid is then slowly poured into a boiling m ixture of 200 c .c .of water and 30 c .c . of a 1 0% solutionOf lead acetate contained in a500 c .c . Erlenmeyer flask
,the boiling is kept up ' for 5 or 6 minutes ,
and the liquid is then cooled by rotating the fl ask under a stream ofrunning water
,the movement being continued until the lead soaps
adhere to the sides of the flask and the aqueous solution is clear . If i tremain milky
,desulphonation was not complete , and the test must
be repeated . The aqueous solution is poured off,or filtered if necessary ,
the flask is then heated on the steam-bath until the contents are melted ,again cooled
,and any remaining water shaken out .
The flask containing the dry lead salts i s heated,and about 1 0
c .c . of petroleum spirit are added,which usually mixes with the
soaps and renders them more fluid .
°
75 to 80 c .c . more petroleumspirit are added
,and the solution is boiled under a reflux condenser .
I t i s then poured into a stoppered,graduated
, 500 c .c . cylinder of thin1 Deu tsch . pharm . Ges . B er. . 1 9 04 , 1 4 , 1 3 5 .
2 Eng . P a t . 2 4 9 3 5 , 1 9 0 5 .
S oc. Chem. Ind 1 90 7 , 2 6 , 5 9 7 .

1 66 MODE S OF EXAMINING FATS,OILS , AND WAXE S .
glass,having a stop-cock just below the 250 c .c . mark , or into a 500
c .c . graduated flask . The flask containing the salts i s repeatedlyrinsed with petroleum ether
,boiling each time
,and transferring as
much as possible of the lead soaps to the cylinder or flask,using about
200 to 2 25 c .c . of petroleum spiri t altogether . The ethereal solutionis then diluted nearly to the 500 c .c . mark wi th petroleum spiri t boilingat 28° to the whole is boiled for 1 minute (neglect of this detailwill cause an inaccurate result) and allowed to stand in a cool place forat least 3 hours , and preferably all night
,in order that the lead ricin
oleate , etc . , may completely separate .If the sample contains 80% or more of castor oil , petroleum spiri tof 38° to 40° b . p . i s used for rinsing and washing the flask ; if under
that of 28° to 30° b . p . i s used,the percentage of castor oil
present being judged,approximately
,by the fact that when under
80% the soaps will di ssolve in the hot spiri t of 30° b . p .,while if over
80% a semi-fluid mass remains which is more perfectly extractedby the solvent of higher b . p . I t i s essential , however , that the greaterpart of the spiri t used for the dilution Should be of the lower b . p . Thelead ricinoleate is precipi tated from this
,on cooling
,as a characteristic ,
flocculent mass resembling aluminum hydroxide,lead stearate and
palmitate forming white,Slowly subsiding powders .
Af ter the complete separation of the insoluble lead salts , the liquidi s dili ited to exactly 500 c .c . , shaken and allowed to settle . 250 c .c .are then drawn off
,fil tered if necessary
,distilled down to 75 or 80 c .c . ,
Shaken in a separating funnel wi th 1 0 c .c . of 1 0% acetic acid , and washedwith water until the washings are perfectly neutral to phenolphthalei nand made alkaline by one drop of N 1 0 sodium hydroxide . The solution i s then distilled from a 200 c .c . Erlenmeyer flask until most of thepetroleum spiri t has been expelled
,mixed with 50 c .c . of neutral
alcohol,and ti trated with N/ I o sodium hydroxide . The volume of
decinormal alkali used X 0 . 0282,gives the equivalent weight of oleic
acid,and if we assume that the other oils in the mixture are vegetable
oils,such as olive
,maize
,etc .
,which contain approximately 80% of
liquid acids,the result X—‘QOBQ represents oil other than castor oil . If the
admixed oil can be identified,however
,i ts percentage of liquid acids
should,of course
,be substi tuted for 80 . The weight thus Obtained ,
multiplied by 2,subtracted from the weight of oil or mixed fatty acids
taken,multiplied by 1 00 and d ivided by the weight taken , gives the
perbentage of castor oil in the sample . If the mixed fatty acids are

1 68 MODE S OF EXAMINING FATS,OILS , AND WAXE S .
may be distingu i shed as olive Turkey-red oil and cottonseed Turkeyred oil
,respectively .
When castor Turkey-red Oi l i s shaken with water and ether,
the sulphonated fatty acids are dissolved by the water and the nonsulphonated acids and unaltered glycerides by the ether ; the formermay be caused to separate from the aqueous portion by saturating itwith common salt or sodium sulphate
,and the latter can be recovered
by evaporating off the ether . The solubility of Turkey -red oil inwater is due to the presence of the sulphonated acid .
The proportion of fatty matter present in alizarin oil varies considerably . I t may be as low as 40 , and occasionally reaches 65the usual proportion being about 50 The amount required shouldbe specified by purchasers and controlled by analysis .Turkey—red oil
,if properly prepared from pure castor Oil
,when
largely diluted , even with hard water, will bear the addition of ammonium hydroxide to alkaline reaction without showing any turbidi tyon standing for several hours . A turbidity or precipitate denotesthe presence of solid fats
,and indicates the employment of either
impure castor oil e .g .
,castor oil foots) or of rape , cottonseed , olive ,
or other oil containing stearin or palmitin .
The sp . gr . of Turkey-red Oil i s very variable . According to Wilson,
1
the sp . gr . of a 45% oil ranges from toThe analysis of Turkey-red Oil may have as its object the estimation of ( 1 ) the amount and nature of the fatty matter
,alkali
,etc .
,
contained in the sample,and ( 2) the source of the fatty matter and the
presence or absence of adulterants .In the estimation of the total fatty matter i t i s customary todecompose the sulphonated oils by boiling with dilute hydrochloricacid . Lewkowitsch2 recommends B enedi kt
’
s method . About 4
grm . of the sample are accurately weighed into a porcelain basin ,and 20 c .c . of hot water are added gradually . Should the liquid beturbid
,ammonia is run in until i t i s faintly alkaline to phenolphthalei n .
A clear solution will thus be obtained . 1 5 c .c . of dilute sulphuricacid (equal volumes of strong acid and water) are next added and anaccurately weighed quanti ty
,about 1 0 grm .
,of paraffin wax . The
mixture is heated until a clear oily layer floats upon the surface . I t
i s then made quite cold,the solidified cake i s removed
,carefully dried ,
S oc. Chem . I nd 1 89 1 , 1 0, 2 6 .
2 O ils . Fa t s and Waxe s , I I I , 1 5 8 .

ALIZARIN OIL . TURKEY-RED OIL .
and weighed . From the weight,that of the added paraffin wax is
deducted,and the remainder represents the total fatty matter in the
quantity taken .
According to Herbig ,I the simplest method is that of F inster-B reinl .
30 grm . of the Oil are weighed into a flask of 2 1 0 c .c . to 250 c .c . capacity,
having a long narrow neck graduated in tenths of a c .c . 1 00 c .c . ofwater and 2 5 c .c . of sulphuric acid (62° B = 1 L753 sp . gr .) are addedand the mixture is heated until the oily layer which forms i s perfectlytransparent . A hot solution of sodium chloride or sulphate is thenpoured into the flask to bring the O ily layer into the neck
,and after
standing half an hour the volume i s read off. Each 1 c .c .=3% of
fatty matter,but as the average sp . gr . of the oil i s the resul t
should be multiplied by this factor .In a later paper2 Herbig recommends the following method . 1 0
grm . of the oil are heated in a flask with 50 c .c . of water,until dissolved
,
and the solution is then mixed with 25 c .c . of dilute hydrochloric acid and boiled for 3 to 5 minutes . When cold
,the contents of
the flask are transferred to a separating funnel with water and wellshaken with about 200 c .c . of ether . The aqueous layer is drawn off,and the ethereal solution washed with three successive quantities ofcold water . The greater part of the ether is distilled off
,the residue
transferred to a beaker and the rest of the ether allowed to evaporatespontaneously . The residual oil is heated for 1 or 2 minutes over afree flame until bubbles cease to appear
,then dried for 30 m inutes at
1 05°and weighed . The aqueous extract and washings
,if mixed and
heated to expel the dissolved ether,may be used for estimation of the
fatty sulphuric acid and glycerol .To estimate the total f ree aci d
,Wilson dissolves 5 to 7 grm . of the
oil in alcohol or alcohol-ether and titrates with N 2 alcoholic potassiumhydroxide
,keeping the temperature low and stirring rapidly so as to
avoid local excess of alkali,owing to the great tendency of the esters
in all Turkey-red oils to undergo hydrolysis . The amount of freeacid , calculated as ricinoleosulphuric acid , in a 45 oil may range from2 2 to 30 070. This method is intended for Turkey-red oils preparedwith sodium hydroxide or potassium hydroxide
,but good results may
be obtained even in the case of ammonia Turkey-red oils . Obviously ,the figure obtained gives no idea as to the percentage of total fattyJ . S oc. Chem . Ind . , 1 9 02 , from Chem . Rev. F ett Hare
2 Chem . Rev. F ett-Harz 1 3 , 1 8 7 , 2 1 1 and 2 4 1 ; ab s . in ]. S oc. Chem . I nd . , 1 9 06 .25 , 1 009 .

1 70 MODES OF EXAMINING FATS,OILS , AND WAXE S .
acids actually present,owing to the great diff erence in the molecular
equivalents of the diff erent acids . If this information be desired, thepercentage of neutral oil should be estimated and the fatty acidsgot by difference .For the estimation of the neutral oi l
,Lewkowi tschI dissolves
30 grm . of the sample in 50 c .c . of water, adds 20 c .c . of ammonia and30 c .c . of glycerin , and shakes twice with ether, using 1 00 c .c . eachtime . The ethereal solution i s washed with water to remove tracesof soap
,run through a dry filter into a tared flask
,the ether distilled
off,and the residual oil dr ied at 1 00° and weighed .
Scheurer-Kestner,
2 after pointing out the different shades producedin dyeing and printing by the sulphonated and non-sulphonated fattyacids
,respectively
,proposed a method of roughly estimating these
volumetrically by ti tration with standard ammonia solution,dependent
Upon the fact that litmus becomes blue when the sulphonated acids
are neutralised,while phenolphthalei n does not become reddened
until the neutralisation of the whole of the fatty acids present i s completed . In a particular sample he found the molecular weight of themixed non—sulphonated acids 47 2 and that of the sulphonated acids402 . The following are the molecular weights of some of the pureacids which may be present
Ricino le ic 298
D i-ricinole ic 5 78
Ricino leosulphuric . 3 78
Di-ricinoleosulphuric 658
Jouillard3 says thi s method gives inaccurate results , especiallyas diricinoleosulphuric acid is almost invariably present . He advi sesan estimation of the molecular weight of the fatty acids soluble andinsoluble in water
,by Raoult’s method
,taking care
,in separating these
by shaking with water and ether as already described,that the whole
of the water-soluble acids are extracted . Jouillard also recommendsan estimation of the glycerol and sulphuric acid in the sulphonated oil .For the estimation of fatty-sulphuric aci d, Herbig (loc. ci t.)boils 4 grm . of the oil with 30 c .c . of dilute hydrochloric acidfor 40 minutes , removes the oil by shaking with ether , and estimatesthe SO 3
in the aqueous liquid by precipitation with barium chloride .From the weight Obtained should be deducted the amount of SO 3
1 Oils , Fa t s and Waxes , I I I . 1 5 9 .
2 Compt. rend . , 1 1 2 , 1 5 8 and 3 9 5 .
3 B ull. S oc. Chim . , 1 89 1 , 6 , 6 3 8 .

1 72 MODE S OF EXAMINING FATS,OILS , AND WAXE S .
CROTON OIL .
(See also p . Croton oil i s obtained by expression or extractionwith alcohol from the seeds of Croton Tiglium,
the yield being about53 to I t i s a brownish-yellow to dark reddish-brown
,viscid
Oil, with disagreeable Odour and acrid burn ing taste (B riti sh Pharma
capa i a), intensely purgative and vesicatory .
The lighter varieties darken very much with age . Croton oildiffers from castor Oil in being soluble in petroleum spiri t . I t hasslight drying power and forms no elaidin with n itrous acid . I t i scomposed of the glycerides of tiglic acid and other higher homologuesof oleic acid
,and of stearic
,palmitic
,myristic
,l auric
,caproic
,butyric
,
and acetic acids . Oleic acid itself has not been identified in the O il .Dunstan and B oole I have shown that the vesicating consti tuent i s aneutral , resinous substance of empirical formula C I 3
H, gO4 ,which
forms but a small proportion of the so—called “ croton-oleic acid ” fromwhich it i s obtained .
Croton oil i s more strongly dextrorotatory than castor Oil . Rakusin?
using a Soleil-Ventzke instrument with 200 mm . tube , obtainedthe following values :
Croton oil,
toCastor oil
,8 0
° toThe discrepancies in the analytical data for croton oil as obtainedby different Observers are probably largely dependent upon the methodby which the oil was obtained . Thus
, Javillier3 prepared three samples : .
the first by simple expression,the second by extraction with ether
,and
the third by digestion at 75° with 95% alcohol ; the first two methodsbeing those described by the French Codex of 1 884 . The yield andcharacter of the products are shown in the following table :
Yie ld 3 8% 1 2%Co lou r Pa le . Ligh t brown . Very dark brown .
S o lubili ty ( 1 vo l. o f 011 + 2 vo ls . S oluble a t 7 5°S o luble at 7 5
° S oluble in th e co ld .
abso lu t e alcoh o l) .
S o li d ifica t ion t emperatu re 8°
Iod ine numberS ap on ifi ca t ion valu eAc id valu e
1 P roc. Roy . S oc. , 1 89 5 , 5 8 , 2 3 8 .
2 Chem . Zei t. , 1 9 06 , 3 0, 1 4 3 .
3 J . P harm . Chi m. , 1 89 8 , 7 , 5 24 .

CURCAS OI L . 1 73
The acid value was estimated by dissolving the oil in ether andtitrat ing directly with N/ 1 0 alcoholic potassium hydroxide .
Duliere I states that croton o il Obtained by extraction wi th petroleumSpiri t or by cold expression has the same characteristics as the offi cialoil
,but that Oils prepared by hot expressi on or by extraction of the
non-decorticated seeds with ether,difl er from i t in colour
,acidity
,
and degree of solubili ty in absolute alcohol,although the chief chemical
constants are ‘
practically identical . He gives the following table ofconstants for this oil :
Sp . gr. a t
Sp . gr. at 1 00°
So lubility in 9 2% alcoho l 1 in 63Critical temperature (Grismer)C leo -refractometer degrees , 2 2° + 3 5Butyro
-refractometer degrees,27
°
Acid value (Burstyn)Saponification valueRe ichert M e issl valueAcetyl value (Lewkow i tsch) 38 . 64S olidifying-po int of m ixed fatty ac ids to 1 6 . 7
°
Two samples of croton Oil examined by Lewkowitsch2 gave thefollowing results
S aponification value .
Acety l valueTh e same observer3 found 0 5 5 of unsaponifiable matter in severalsamples of croton oil and to as the ti ter test of t he mixedfatty acids .Adulteration of croton o il with castor oil would be detected by theincreased acetyl value . Hydrocarbons
,which are said to be frequently
added (D uliere), would lower the saponification value and increase th epercentage Of unsaponifiable matter .
CURCAS OIL .
(See p . This is from the seeds of the “ physic nut or purgingnut ” tree
,J atropha curcas
,a plant chiefly cultivated in the Portuguese
colonies,and especially in the Cape Verde I slands . I t i s yellowish
brown in a crude state,pale yellow when refined
,has a faint unpleasant
odour,and powerful purgative properti es
,1 0 drops having a greater
purgative effect than a tablesp‘oonful of castor oil (Klein) . I t i s onlyslightly soluble in alcohol , 1 00 volumes of absolute alcohol at 1 5 . 5
°
dissolving about volumes (Archbutt4), and freely soluble inA nn . de P harm . de Louva in, 1 89 9 , 2 2 9 and 2 7 8 .
2 A naly s t , 1 89 9 , 24 , 3 1 9 .
3 Oils , Fa t s and “faxe s ,I I , 1 84 .
S oc. Chem . I nd . , 1 009 .
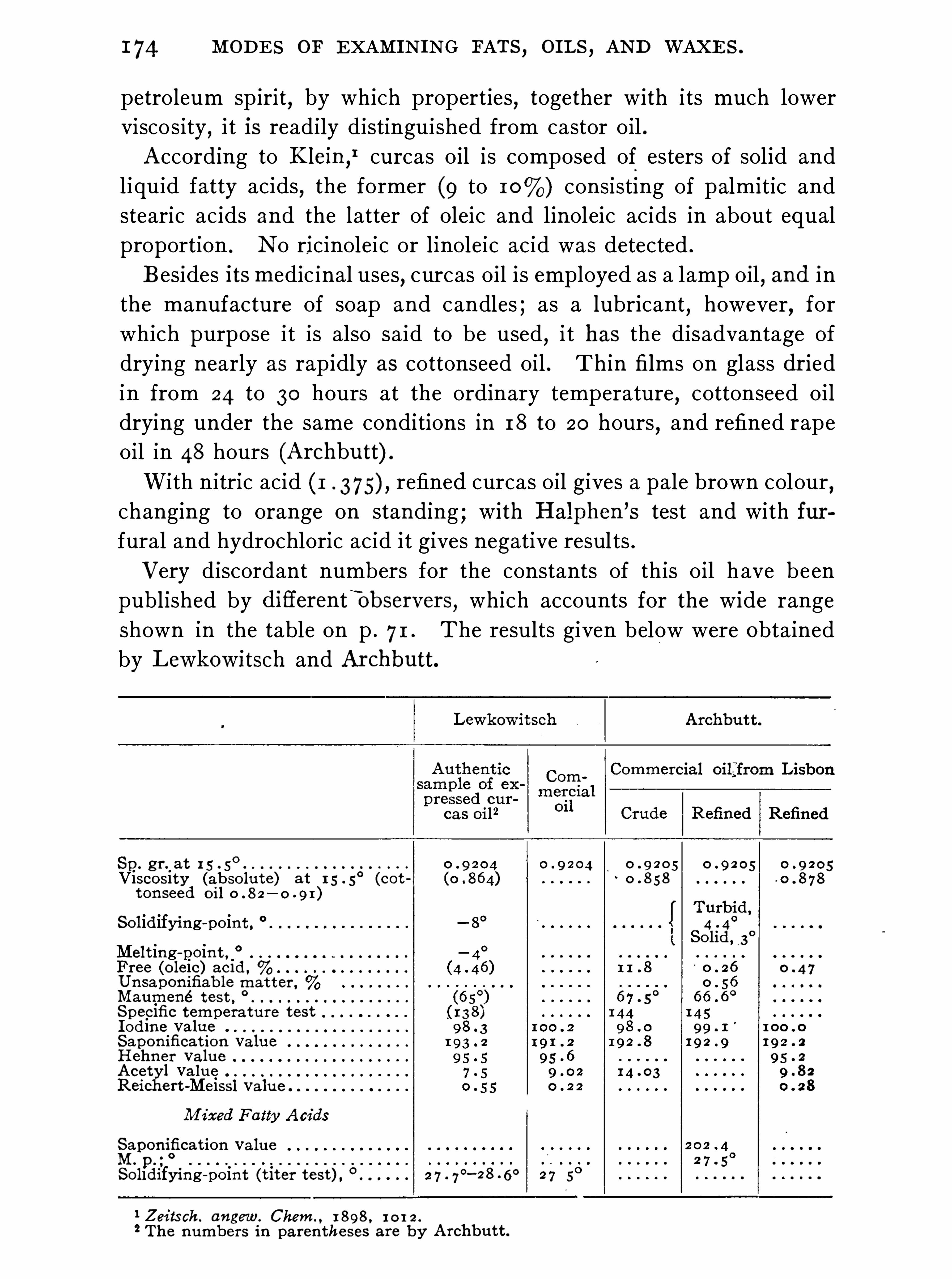
1 74 MODE S OF EXAMINING FATS , OILS , AND WAXE S .
petroleum spirit,by which properties
,together with i ts much lower
visco sity,i t is readily distinguished from castor oil .
According to Kle in,
I curcas oil i s composed of esters of solid andliquid fatty acids
,the former (9 to consisting of palmitic and
stearic acids and the latter of oleic and linoleic acids in about equalproportion . No ricinoleic or linoleic acid was detected .
B esides i ts medic inal uses,curcas oil i s employed as a lamp oil
,and in
the manufacture of soap and candles ; as a lubricant , however, forwhich purpose i t is also said to be used
,i t has the disadvantage of
drying nearly as rapidly as cottonseed oil . Thin films on glass dr iedin from 24 to 30 hours at the ordinary temperature , cottonseed oildrying under the same conditions in 1 8 to 20 hours
,and refined rape
oil in 48 hours (Archbutt).Wi th nitric acid ( 1 refined curcas oil gives a pale brown colour
,
changing to orange on standing ; with Halphen’s test and with fur
fural and hydrochloric acid i t gives negative resul ts .Very discordant numbers for the constants of this oil h ave beenpublished by diflerent '
I
Observers,which accounts for the wide range
shown in the table on p . 7 1 . The results given below were Obtainedby Lewkowi tsch and Archbutt.
S p . gr . at 1 5 . 5°
5Viscos ity (abso lut e) a t 1 5 . 5° (co t
t onseed o i l 0 . 8 2—0 . 9 1 )
So lid ifyi ng—po in t .Melt ing-p0in t ,Free (o le ic) ac id ,
Un sapon ifiab le mat t er ,Maumené t e st ,S pec ifi c t emperature t e stIod ine valu eS ap on ificat ion valu eHehner valueAce t 1 va lue 1 4 -03Re ic ert -Me issl value
S aponificat ion valueM. pS olidify i ng-po in t ( t it er t est) . 2 7 7
°- 28 . 6°
2 7 5°
1 Zei ts ch . angew. Chem . , 1 89 8 , 1 0 1 2 .
2 Th e numbers in parenthe ses are by Archbut t .

1 76 MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
VI . CACAO BUTTER GROUP .
Bassia Ta llow . Laure l Oi l .B orneo Tallow (Tangk awang Fat). Mafura Ta llow .
Cacao Bu tter. Nu tmeg Bu tter .Ch inese Vege table Ta llow (S t illing ia Tal Palm Oi l .
low). Phu lwara Butter.Co tton Oi l “S tearine . P iney Tallow (Ma labarGoa Bu tter (Kokum Butter). Ta llow).
Sh ea Bu tter (Galam Bu tter).
BASSIA TALLOW .
(For constants see p . This fat,as met with in commerce , i s
a m ixture of the fats obtained from the seeds of Bassi a longifol‘iaand of B . latifoli a
,the former being termed Mohwah butter or Mowrah
seed oi l, and the latter Mahua butter or I llipe’ bu tter . I t i s used in the
manufacture of soap and candles .
BORNEO TALLOW . (TANGKAWANG FAT.)
(For constants see p . This fat is expressed from the seedsof several different kinds of plants
,belonging to the order D iptero
carpacece , notably S horea stenoptera,a native of Northwest Borneo .
I t i s a greenish-yellow fat,gradually changing to white
,resembling
cacao butter in consistency and taste . I t has a crystalline structure ,and is covered with fine needles of stearic acid . I t melts at 35° toAccording to Geitel‘, I i t consists chiefly of the esters of stearic and olei cacids
,but Klimont2 has i solated from it oleodistearin and oleodi
palmitin . The commercial fat has been found to contain from 8 to1 0% of free (stearic) acid . I t i s used in Europe in the manufactureof soap and candles .
CACAO BUTTER . OIL OF THEOBROMA .
(See also page This-oil IS expressed from the beans or seedsof the cacao tree
,Theobroma cacao
,from which ordinary cocoa is
obtained,and must not be confused with cocoanut oil from Cocos
nucifera . I t i s Obtained in large quantities as a b y-product in themanufacture of chocolate .
prak t. Chem . , 3 6 , 5 1 5 .
2M ona tsh . Chem . , 1 9 04 , 2 5 , 9 2 9 .

CACAO BUTTER .
The percentage of cacao butter in the roasted nibs or beans has beencarefully estimated by Davies and McLellan1 by extraction of thepowdered nibs with petroleum spirit . B eans from Ecuador
,
Venezuela , Dutch Guiana , Brazil , the West Indies , Africa , andCeylon were examined . The average percentages of fat in thediff erent sorts ranged from to the average of the wholebeingCacao butter i s a yellowish solid
,gradually turning white on keeping .
At the ordinary temperature it may be broken into fragments,but
softens in the hand and melts in the mouth . I t fuses between 30 and34
°
(rarely at to a transparent yellowish liquid , which congealsagains
.
at 20 . the temperature rising to about I t has an agre eable odour
,tastes like chocolate
,and does no t readily become rancid .
Lewkowitsch,
2 however,has shown that cacao butter
,if exposed to the
combined action of sunshine,air
,and moisture for a few days
,becomes
bleached and rancid like any other fat . It dissolves in 20 parts of hotalcohol
,separating almost completely on cooling
,and is also dissolved
by ether and ethyl acetate . I t i s largely used as the fat in the manufacture of chocolate creams
,and in pharmaceutical preparations and
cosmetics .Cacao butter chiefly consists of the propenyl esters of stearic , palmitic ,oleic
,and linolic acids . C . Kingzett3 Obtained from cacao butter an
acid ' of the formula C64H1 2 802 ,which he named theobromic acid ,
but neither Traub4 nor GrafS were able to find any fatty acids of highermolecular weight than arachidic . Hehner and M itchell6 found 40%of stearic acid ; Farnste iner7 obtained 59 . 7% of saturated acids , 3 1 2%
of Oleic acid,and 6 . 3% of other acids . Klimont8 believes these acids
to exist mainly in the' form of mixed esters . Matthes and Rohdich9have found in the unsaponifiable matter two phytosterols , identical ,respectively
,with the sitosterol and stigmasterol isolated by Windhaus
from calabar bean fat .The following results have been obtained by examination of the
mixed fatty acids of cacao butter (see also p .
S oc. Chem . I nd 1 9 04 , 2 48 1 .
S oc . Chem . I nd . , 1 89 9 , 1 5 5 6 .
Chem . S oc. , Tran s 1 8 7 8 , 3 3 , 3 8 .
4 A rch . P harm 2 1 , 1 9 .
5 I bi d . , 26 , 8 3 0 .
6 A na ly s t , 1 89 6 , 1 , 3 2 1 .
7 Zei ts ch . Nahr . enu ssm . , 1 89 9 , 2 , 1 .
8 M ona tsh . , 1 9 0 2 , 23 , 5 1 ; 1 9 0 5 . 26 , 5 6 3 .
9 B er . , 1 9 08 , 4 1 , 1 9 .
Vo l. 11 ,
—1 2

1 78 MODE S OF EXAMINING FATS , OILS , AND WAXES .
Solidifying-po int (titer test) 48 3—49 2 Lewkowitsch .
Refractive index at 60° 1 .422 Thoerner.
Neutralisation value 1 90 Thoerner.
3 2 .6 De Negri andFabris
, Thoerner.
Cacao Bu tter Adu lterants .—The adulterants which should be
looked for in cacao butter ‘are numerous,and include cocoanut and
palm-nut oils,and the “ stearines ” prepared from them (a mixture of
2/3 palm-nut “ stearine ” and coconut stearine ” i s said to be afavourite substitute), tallow and lard , stearic acid , sesame and othervegetable oils
,beeswax and parafli n wax . For the detection of these
adulterants the most important estimations are the i odine value ,
saponificati on value,Rei chert-Mei ssl value
,aci d value
,and sp. gr.
,to
gether with the m. p . of the fat and i ts mixedfatty acids , or the ti tertest of the latter .Coconut and palm-nut oi ls and “ stearines would lower theiodine value and sp . gr .
,raising at the same time the saponification
value and Reichert-Meissl value . They would also lower the ti tertest of the fatty acids .Tallow ,
beyond somewhat lowering the Sp . gr .,would cause scarcely
any change in the other values . I t may be detected by BjOrklund ’s
test (see p . 1 79) and the phytosterol acetate test (p . the latterof which would also have to be applied to for the detection of lard ,although the presence of lard would have some tendency to raise theiodine value .S teari c acid would be detected by an estimation of the acid valuewhich
,in genuine cacao butter
,does not usually exceed about
Most vege table o i ls would lower the sp . gr . of the fat and the titertest of its mixed fatty acids
,and woul d raise the iodine value ; cotton
seed and sesame oils would be detected by their characteristic colourindications
,arachis oil by i ts arachidic acid
,etc .
Beeswax and parafi'
in wax would be easily detected ; the formerwould raise the acid value and m . p . of the fat
,lowering at the same
time the iodine and saponification values , and both beeswax andparaffi n wax would increase the amount of unsaponifiable matter ,which in genuine cacao butter i s quite small .

MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
Pure cacao butter 1 0—1 5Cacao butter+ 5% beef tallow
Lewkowi tsch I found that cacao butter containing as much as 1 0%of tallow will dissolve in 2 parts of ether at although requiring ali ttle longer than the genuine butter does
,and that the chief indication
to be relied upon is not so much the time required for crystalli sationto commence
,as this varies with diff erent samples of cacao butter ,
but the characteristic way in which genuine cacao butter crystalli sesas compared with adulterated samples . Wi th genuine samples ,distinct tufts of crystals appear at the bottom and sides of the tube ,whereas 5% and more of tallow are recognised by flocks separatingfrom the chilled solution .
A method for the detection of coconut oil in butter, a pplicable
also to lard and cacao butter,has been described by L . Robin ,
2 basedupon the almost complete solubili ty of coconut oil fatty acids in alcoholof 56 5% strength and the very slight solubility of the fatty acids frombutter and cacao butter . 5 grm . of the fat are saponified by boiling forabout 5 minutes with 25 c .c . of alcoholic potassium hydroxide , the operation being conducted in a flask graduated at 1 50 c .c . attached to areflux condenser . After cooling
,suffi cient water is added to reduce
the alcoholic strength to 56 . and a volume of N 2 hydrochloricacid (prepared with alcohol) suffi cient to exactly neutrali sethe alkali and liberate the fatty acids from the soap . The volumeof acid required is ascertained by a previous titration . The contentsof the flask are then made up to 1 50 c .c . at 1 5° with alcohol ,well mixed
,allowed to stand for at least half an hour , and filtered .
50 c .c . of the filtrate are titrated with N/ 1 0 alkali , using phenolphthalein as indicator , and the result calculated to c .c . per 1 grm . of fatrepresents fatty aci ds soluble in 56 . 5% alcohol . The author of theprocess states that if even 5% of coconut oil i s present in cacao butter ,the “ alcohol-soluble ” number will be not less than 3 , while the ratio ofthe alcohol—soluble number to the saponification value of the fat willbe less than 60 . A ratio of 45 to 60 corresponds with the presence of
S oc. Chem . I nd . , 1 89 0 . 1 8, 5 5 7 .
2 A nn . de Chim . A na l . , 1 9 06 , 1 1 , 4 5 4 , and 1 2 , 1 8 1 .

CHINE SE TALLOW .
5 to 1 0% of coconut oil , a ratio of 3 5 to 45 with 1 0 to and a ratio
Robin ’s analytical results are summarised in the following table
For the detection of coconut oil by means of the “ ethyl ester value(Hanus and Stekl ’s method) see under “ Coconut O il , ” p . 1 87 .
CHINESE VEGETABLE TALLOW (STILLINGIA TALLOW).
(For constants see p . This fat is obtained from the fruits of avariety of plants
,the most important of which is the Chinese tallow
tree,S ti llingia sebifera (Croton sebiferum I t is largely employed
in the manufacture of soap and candles .The fat is found as a coating about 0 5 mm . thick on the seeds , fromwhich it is melted by steam heat . The seeds themselves contain astrongly drying oil (stillingia Oil) of quite a different character fromthe fat which coats them
,and some of this i s liable to be contained in
the commercial tallow . Its presence would be Shown by its h igh iodinevalue and rotatory power on polari sed light .Commercial Chinese vegetable tallow is a white or greenish fat
,
without taste or smell , the analytical values of which vary considerablyowing to its being Obtained from different plants and prepared indifferent ways . I t is believed to consist of esters of palmitin and olein ,
and these exist,according to Klimont
,
1 partly as the mixed esteroleodipalmi tin . A sample tested by B ehner and M i tchell containedno stearin . Valenta
,by the lead-salt-ether method
,obtained 3 5 56 070
of oleic acid and of palmitic acid .
COTTON OIL “STEARINE .
This is the name given to the soft solid fat that separates when cottonseed oil i s chilled . I t is utili sed in the manufacture of margarine and
1 Monatsh . Chem. , 1 903 , 24 , 408 .

MODE S OF EXAMINING FATS,OI LS
,AND WAXE S .
of soap . I t consists chiefly of palmitin and a little stearin,with Olein
and linolin (see “Cottonseed
GOA BUTTER (KOKUM BUTTER , MANGOSTEEN OIL).
(For constants see p . This fat is expressed from the seeds ofthe East Indian plant
, Garcinia indi ca . I t i s used locally as a food ,while the commercial product i s manufactured into soap . It consistschiefly of the mixed glyceride
,oleo-distearin (Heise).
LAUREL OIL.
(For constants see p . This i s a butter-like fat of greeni shyellow colour
,slightly bitter taste
,and peculiar aromatic odour ,
obtained from the berries of the laurel tree,Laurus nobi li s , which yield
about I t consists largely of trilaurin,with probably a small
amount of olein and linolin . I t i s employed in the preparation ofveterinary medicines .
MAFURA TALLOW .
(For constants see p . This is a light yellow fat obtained fromthe seeds of Mafurei ra oleifera . I t consists of glycerides of solid fattyacids (7 1 and Of liquid fatty acids (23 (De Negri and Fabris).It i s used in the manufacture of soap and candles .
N.UTMEG BUTTER .
(For constants see p . This is a yellow tallow-like fat obtainedfrom the seeds of the Myri sti ca oflicinali s , the yield being about 20 to25 AS expressed
,i t consists of a fixed oil with 8 to 1 0% of an es
sential oil (nutmeg oil). The glyceridic portion is composed chieflyof myristin and olein
,with a small proportion of butyrin (Jean).
The fat is used in the manufacture of perfumes and for medicinalpreparations .
PALM OIL.
(See also page Palm oil is the product of several species ofpalm
,but particularly of Elai s guineeni s and E . melanocca. The

1 84 MODE S OF EXAMINING FATS,OILS , AND WAXE S .
The following results have been obtained by examination of themixed fatty acids Of palm Oil :
Sp . gr. at 98° Allen .
S p . gr. at Archbutt .
So lidifying—po int (titer test) 3 5 . 8°—47 Various .
usually 44°—45
°
Iodine value 53 -3—58 -9
Iodine value of liquid fatty acids 95—99
Commercial Palm Oi l .
Palm oil as met with in commerce varies greatly in quality . I t
almost always contains more or less water and solid impurities . Someof the irregular oils Odcasionally contain 25 or but the usualrange is from 2 to while most of the regular oil does not containmore than 5 or
3
I t i s usual to sell palm oil on the assumption that2% of such foreign matters are present ; any excess over this i s allowedfor .Water i s best estimated by exposing 1 0 grm . of the sample to atemperature of 1 1 0° for an hour or two
,and noting the loss of weight
(see If the residual Oil be then dissolved in warm petroleumSpiri t
,the solid impuri ti es will settle to the bottom ,
and can be filteredoff
,washed with a little ether
,dried
,removed from the filter
,and
weighed . After weighing, the residue may be ignited , when the ashwill indicate with sufficient accuracy the proportion of sand and
mineral matters,and loss of weight will give that of the organi c matter .
In many cases the water can be estimated with suffi cient accuracyby noting the measure of the aqueous layer which separates whenthe undried sample is dissolved in petroleum spirit
,or simply kept
melted in a graduated tube immersed in hot water .Palm oil is not
,as a rule
,adulterated with other fats
,but it frequently
contains a large proportion offreefatty acids . The free acid raises thesolidifying-point of the oil
,and causes i t to have a corrosive action
11 4 sample s , see p . 1 86 , omi t ing two very ac id and ranc id o ils which absorbed on lyand re spect ive ly .
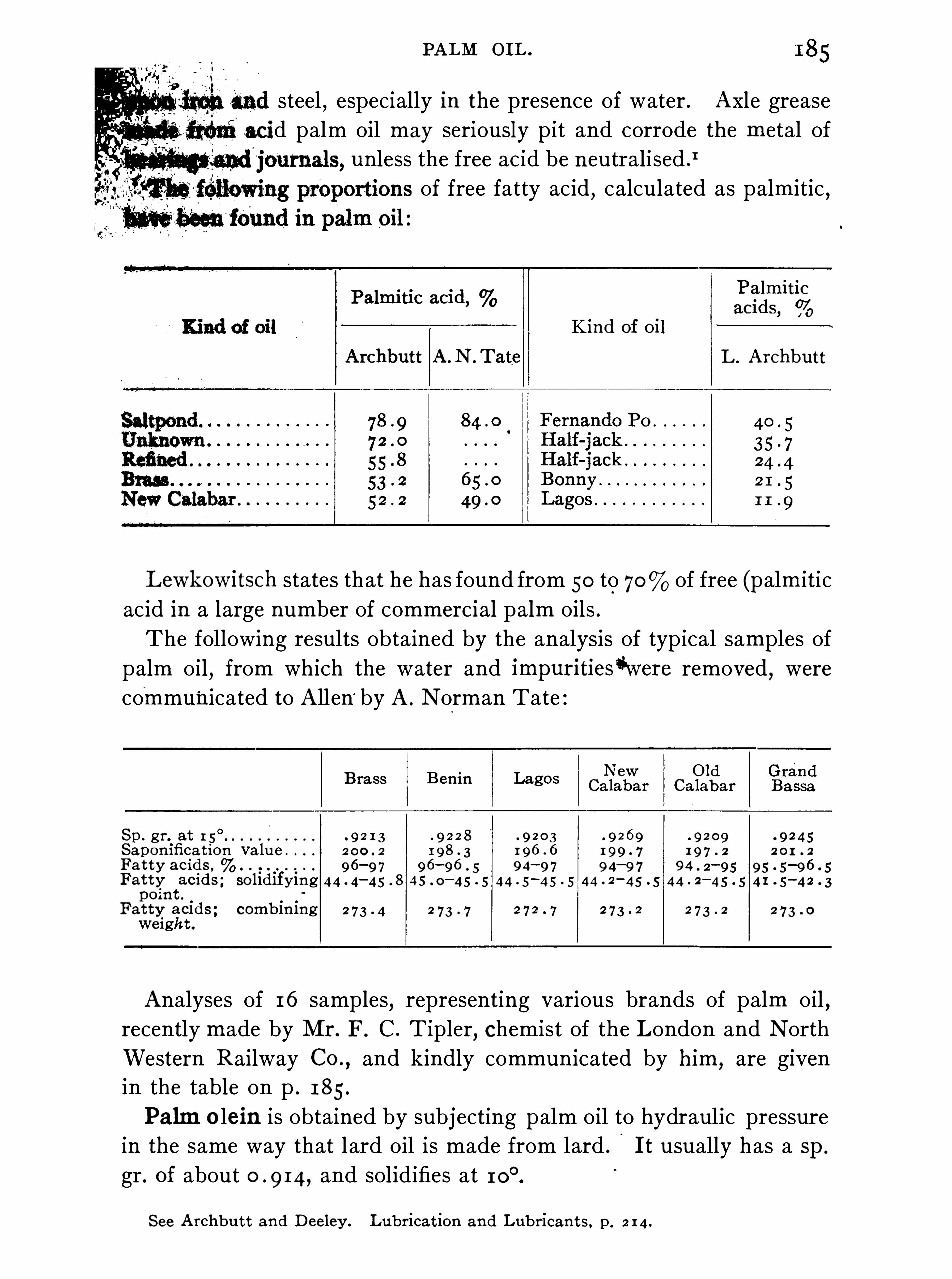
PALM OI L .
steel,especially in the presence of water . Axle grease
d palm oil may seriously pit and corrode the metal ofunless the free acid be neutrali sed ?
of free fatty acid,calculated as palmitic
,
Kind of o i l
L . Archbutt
Lewkowi tsch states that he has found from 50 to 70% of free (palmiticacid in a large number of commercial palm oils .The following results obtained by the analysis of typical samples ofpalm oil
,from which the water and impuri ties 'hvere removed , were
communicated to Allen by A . Norman Tate :
S p . g r at 1 5°
. 9 2 1 3 . 9 2 2 8 . 9 203 . 9 2 6 9 . 9 2 09 . 9 24 5S ap on lfica t ion va lu e . 1 9 7 . 2 2 0 1 . 2
Fa t t y amd s . 9 6—9 7 9 6
-9 6 -5 9 4
—9 7 9 4
—9 7 9 4 -2
—9 5 9 5 -s
-9 6 -s
Fat tyt
ac id s ; so lidify ing —45 . 8 4 5 . 0
—4 5 . 5
—45 . 5 4 1 . 5
p omFat ty acids combin ing 2 7 3 . 7we i ght .
Analyses of 1 6 samples,representing various brands of palm oil ,
recently made by Mr . F . C . Tipler, chemist of the London and North
Western Railway Co .,and kindly communicated by him
,are given
in the table on p . 1 85 .
Palm o le in is obtained by subjecting palm oil to hydraulic pressurein the same way that lard oil i s made from lard . I t usually has a sp .
gr. Of about and solidifies atS ee Archbu t t and Deeley . Lubricat ion and Lubricant s , p . 2 1 4 .

MODE S OF EXAMINING FATS,OILS , AND WAXE S .

MODE S OF EXAMINING FATS , OILS , AND WAXE S .
higher than that of themajority of vegetable fats . Allen observed arange of from to at M ; Crossley and Le Sueur Obtaine d values from to at 1 00
°
Coconut oil has a peculiar and highly complex chemical composition . I t i s chiefly composed of laurin and myristin
,but contains also
Six other glycerides,including caproin
,caprylin
,caprin
,palmitin
,
stearin and olein ? Very little stearin i s present ; Lewkowi tsch foundonly of stearic acid? Hehner andM itchell none . The volatileacids are chiefly capric and caprylic .Coconut oil i s used for candle and soap-making . I t i s an excellentilluminant
,emitting no smoke
,and is largely used for making night
lights . I t forms a hard and white soap,the aqueous solution of which
is not readily precipitated by common salt ; hence this soap is availablefor use with sea-water (mari ne soap).Coconut oil and the “ stearine ” made from it are also used as sub
stitutes for and adulterants of butter,lard
,and cacao butter . By
treatment with alcohol and animal charcoal a neutral coconut oili s produced
,which is sold under such names as “ vegetable butter
,
”
“vege taline , lactine ,
” “nucoline
,
” “ laureol,etc . When well
prepared , these products are white , of about the consistence of butter ,Of agreeable
,sweet flavour
,and
,according to Jean
,3 free from ten
dency to become rancid . Coconut oil i s frequently used in the preparation of margarine .
Coconut “ Ole ine i s used as a lubricant , usually as an ingredient ofblended oils .The data given below and on p . 7 2 have been obtained fromthe mixed fatty acids of coconut oil
Sp . gr. at 98° Allen.
S o lidifying-po int (titer test) 2 1 . 2°—25 . 2
° Lewkow itsch .
Refractive index at Thoerner.
Saponification value 2 58 Thoerner
Mean combining we ight 1 96—204 Alder a bt .
Iodine value 8 . 4—9 3 Variou s .
1 S ee Ulzer , Chem. Rev. F ett Harz I nd . , 6 , 2 03 ; B lumenjeld and S e idel , J . S oc. Chem. Ind
9 1 4 ; Jensen , Zei tsch . Narh. Genussm . , 1 9 05 1 0, 26 5 ; Haller and You ssoufianC0
2
m t. rend 803 ; Pau lm ey er . J . S oc. Chem . Ind . , 1 9 0 7 ,
i ls , Fa t s and axe s I I . 5 1 8 .
3 Jou r. S oc . Chem . I nd . ,

COCONUT OIL . 1 89
Coconut Oil i s alleged to be liable to adulteration with suet,beef
marrow,and other animal greases
,as also with almond oil and wax .
These would be detected by the reduced sp . gr . at the temperatureof boiling water and the reduced saponification and Reichert-Meisslvalues . Indeed
,there is no addition likely to be made in practice ,
excepting that of palm-nut oil,which
,if in notable proportion
,would
not be detected by these tests . The same methods,if used with
discretion,will equally serve to estimate the approximate proportion
of the adulterant . P alm-nut oi l cannot be detected by the aboveor any other satisfactory method , but as it i s employed for the samepurposes as coconut oil , the substitution has little practical importance .
Hanus and Stek lI have proposed a new constant for the detectionof coconut Oil in other oils and fats
,which they name the “ ethyl
ester value .” 5 grm . of the melted and filtered fat are weighed into anErlenmeyer flask of about 200 c .c . capacity ( 1 4 cm . high and 7 cm . wide)heated for 1 5 m inutes in a thermostat at then rapidly mixed with30 c .c . of N 1 0 alcoholic potassium hydroxide
,vigorously shaken
until quite clear,and again heated in the thermostat until the lapse
of 1 0 minutes from the time of adding the alkali . To the liquidis next added 2 c .c . of dilute sulphuric acid of such strength as to exactlyneutralise the 30 c .c . of alkali , sufficient water to make the volumeup to 1 40 c .c . , and a few fragments of pumice stone . The flask is fittedwith a cork and bulb tube
,connected to an inclined condenser 70 cm .
long,and the liquid rapidly distilled . The first 30 c .c . of alcoholic
distillate are collected in a graduated cylinder,and the next 1 00 c .c .
of aqueous distillate in a 1 00 c .c . flask . The distillation should befinished within 25 minutes . The latter (aqueous) fraction i s rinsedinto an Erlenmeyer flask
,suffi cient alcohol being used to bring the
esters into solution,the free fatty acids are neutralised
,and the esters
are saponified by heating for about 45 minutes under a reflux condenser with 40 c .c . of N 2 alcoholic potassium hydroxide . Whencold
,the excess of alkali i s titrated with N 1 0 hydrochloric acid , the
result giving the number of c .c . of N/ 1 0 alkali required to saponifythe respective esters from 5 grm . of fat . The “ ethyl-ester values ”of the following fats were determ ined :
1 Zei tsch . Nahr. Genussm . , 1 9 08 . 1 5 , 5 7 7 .

1 90 MODE S OF EXAMINING FATS , OILS , AND WAXES .
Coconut o il, 5 samplesPalm-nut o i l
,1 sample
Cows’ butter,1 5 samples
Margarine, 5 samples
Lard, 4 samples
Cacao butter, 3 samples
I t appears from these results that the method is capable of detectinga small percentage of coconut oil in margarine
,lard
,or cacao butter
,
but not less than 1 5% could be detected in cows ’ butter . If coconutoil alone be present
,the numbers afford a means of approximately
estimating the proportion,but the presence of palm-nut oil would
upset the calculation .
Coconu t“o le ine ” and coconut
“stearine are products oh
fained by' submitting coconut Oil to hydraulic pressure . The
following figures,obtained in Allen ’s laboratory from samples supplied
to him by Price ’s Patent Candl e Company , show the relative physicaland chemical characters of the two products
Sp . gr. at
O O O O O O O O O O O O O O O O O O O O O O O O O O O O O
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
SachsI obtained the following values from a sample of commercialhard coconut “ stearine
,
” said to be' a favourite substitute for cacaobutter .
Sp . gr. at 1 00°
M . pS olid ifying-poiS aponification valueIodine valueRe ichert-M e issl value
1 Chem. Rev. F ett-Harz-Ind . , 1 9 08 , 1 5 , 9 , 3 0 .
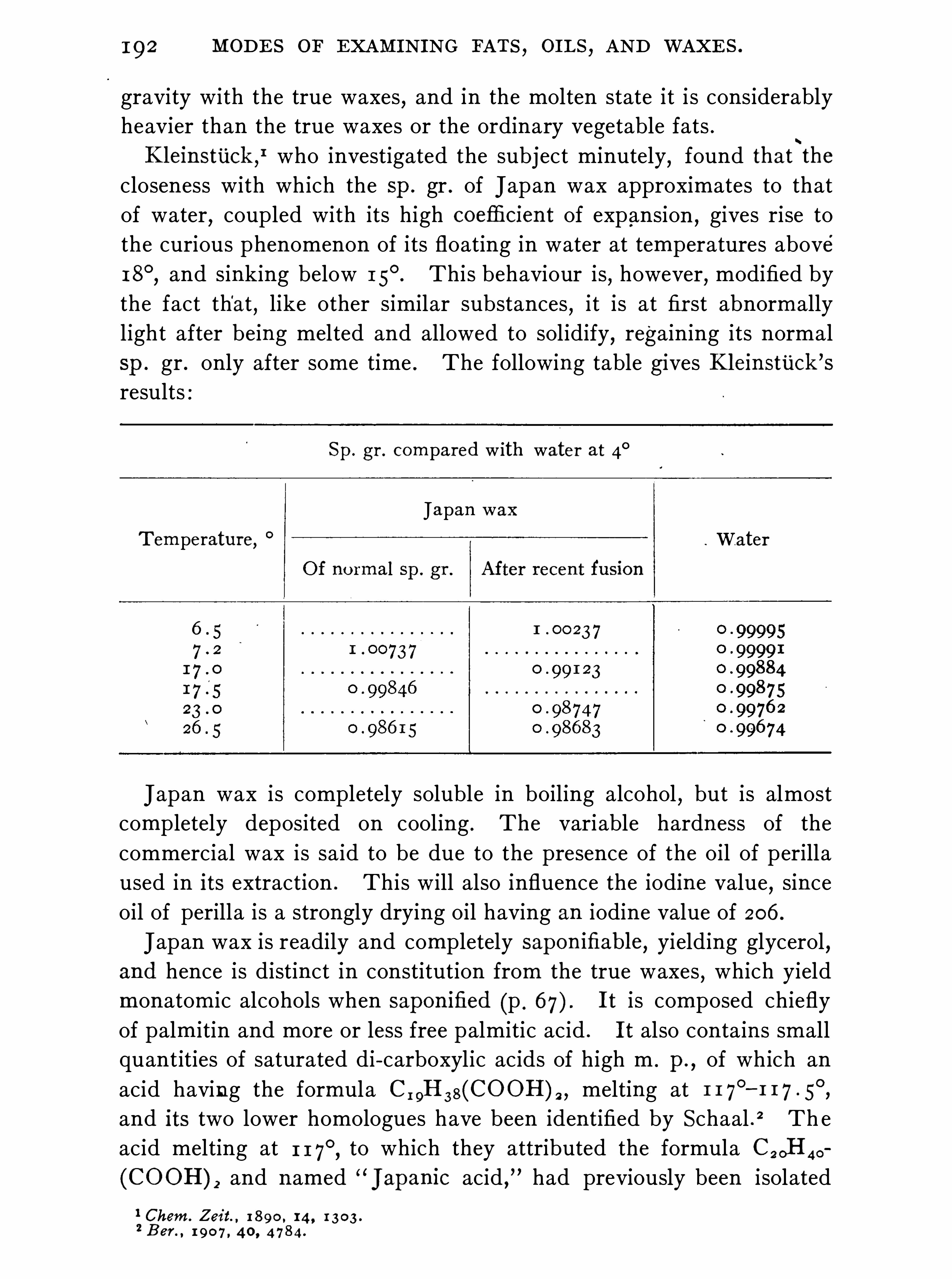
1 92 MODE S OF EXAMINING FATS , OILS,AND WAXE S .
gravity with the true waxes , and in the molten state i t i s considerablyheavier than the true waxes or the ordinary vegetable fats .Kleinstii ck ,
I who investigated the subject minutely,found that
l
the
closeness with which the sp . gr . of Japan wax approximates to thatof water
,coupled with its high coefl
‘i cient of expansion
,gives rise to
the curious phenomenon of i ts floating in water at temperatures aboveand sinking below This behaviour is
,however
,modified by
the fact that,like other similar substances
,i t i s at fir st abnormally
light after being melted and allowed to solidify,regaining its normal
sp . gr . only after some time . The following table gives Kleinst ii ck ’s
results :
Sp . gr . compared with water at 4°
Japan wax
T emperature,
Water
Japan wax is completely soluble in boiling alcohol,but is almost
completely deposited on cooling . The variable hardness of thecommercial wax is said to be due to the presence of the oil of perillaused in i ts extraction . This will also influence the iodine value , Sinceoil of perilla is a strongly drying oil having an iodine value of 206 .
Japan wax is readily and completely saponifiable , yielding glycerol ,and hence is distinct in constitution from the true waxes , which yieldmonatomic alcohols when saponified (p . I t i s composed chieflyof palmitin and more or less free palmitic acid . I t also contains smallquantities of saturated di-carboxylic acids of high m . p .
,of which an
acid having the formula C 1 9H
3 8(COOH) melting at 1 1 7°
and i ts two lower homologues have been identified by Schaal ? Th eacid melting at to which they attributed the formula C , oH4o
(COOH), and named “
Japanic acid ,” had previously been isolated1 Chem. Zei t 1 89 0 , 1 4 , 1 3 03 .
2 B er. . 1 9 0 7 . 40. 4 7 84 .

JAPAN WAX . 1 93
by Gei tel and Van der Want I and found to exist in combination withpalmitic acid as a mixed glyceride . The latter observers also founda small quantity of oleic acid
,and about 5 to 6% of soluble fatty acids
which,in their opinion
,had been produced by the ac tion of the oxidis
ing agents used to bleach the wax .From 5 . 4 to of free (palmitic) acid have been found in com
mercial Japan wax . Ahrens and Hett found from to inwaxwhich they extracted in the laboratory .
The following results have been obtained by the examination ofJapan wax and its fatty acids
,in addition to those given in the table on
P 7 2
Unsaponifiable matter, 7 to Ge i te l and Van
der Want .G lycero l, Allen .
Glycero l, M itchell.Fatty acids
,inso luble
, Geitel and Van
Fatty acids , so luble , der Want.Fatty ac ids , so luble , Allen .
P roperti es of Insoluble Fatty Acids .
S p . gr. at 98 to Allen .
M . p 56° to 5 7
° Allen .
S olidifying-po int 53° to 56 . 5
° Allen .
Combining we ight to Allen .
That the consti tution of Japan wax is peculiar i s evident from thestudy of the products Of i ts saponification ,
and i s shown also by itshigh sp . gr . both in the solid and liquid state
,in which characters i t
diff ers widely from themajori ty of solid fats . The sp . gr . of the insolubleacids
,considered in conjunction with their mean combining weight
,
renders i t doubtful whether ' these fatty acids really consist of palmiticacid
,and it may be noted that Hehner and M itchell in working out
their process for the estimation of stearic acid in fats found that thefatty acids prepared from Japan wax
,whil e possessing apparently
the properties of palmitic acid,prevented the crystalli sation of stearic
acid in an anomalous manner . The percentage of glycerol producedby saponification of the wax , as estimated by the permanganateprocess in two of the samples examined by Allen
,i s notably in excess
of that yielded by tripalmitin,especially in the sample which gave
prakt. Chem. , 1 9 00 , 6 1 , 1 5 1 .
Vo l . II .
—1 3

1 94 MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
the highest result,the glycerol from which sample was estimated
several times with great care . Whether this high proportion was realor due to some unusual consti tuent which rendered the estimationby permanganate inaccurate was not ascertained . Any considerableproportion of a diglyceride containing palmitin and a dibasic acidwould raise the proportion of glycerol and would also ‘
explain therelatively low combining weight of the insoluble fatty acids .Japan wax is stated to be frequently adulterated with water; withwhich i t is capable of forming a sort of emulsion when the two areagitated together a little above the m . p . of the wax .
La Wall 1 found a number of samples adulterated with 20'to 25%
of starchy matter . The sp . gr . of the adul terated wax was only Slightlyhigher than that of the genuine article . Such adulteration wouldbe readily detected by means of ether in which the wax would dissolveleaving the starch .
The addition of tallow would be detected by the lowered m . p . andincreased iodine value ; in fact , the characteristic properties of Japanwax would render the detection of adulteration easy .
MACASSAR OIL .
(For constants see page Macassar oil is a soft fat , forming60 to 70% of the seed-kernels of S chlei chera trijuga , a tree growing inIndia and the East Indies . I t i s used locally for cooking
,illum inating ,
and medicinal purposes,and has long been esteemed in Europe as a
valuable hair restorer . I t consists of esters of lauric,palmitic. arachidic
and oleic acids,with small quantities of acetic and butyric acids . I ts
odour is largely due to the presence of a small amount toof hydrocyanic acid .
Wijs found of unsaponifiable matter in this fat . Of thenon-volatile fatty acids , 45% were saturated and 55% unsaturated(liquid) acids ; the latter had an iodine value of v . I tallie foundthe sp . gr . of the mixed insoluble fatty acids at
MYRTLE WAX.
(For constants see page This i s a greenish-white fat , ofwax-like appearance
,separated from the berries of different kinds of
Myrica (M . cerifera ,M . caroli ensi s , I t contains myristin , palmi
1 Amer.J. P harm . , 69 , 1 8 .
2 A na ly s t, 1 89 6 , 2 1 , 3 28 .

1 96 MODE S OF EXAMINING FATS,OI LS
,AND WAXE S .
I t i s worthy of notice that all the fatty acids of which the esters aresaid to be present contain an even number of carbon atoms . Thesame remark applies to coconut oil
,which has a very similar composi
tion (see page but usually contains a larger proportion of lowerfatty acids . Thus
,the saponification value of palm-nut oil i s usually
about 247 , but differs somewhat with the mode of preparation . If
i t be extracted from the palm-kernels by a sol-vent instead of by pressure
,the proportion Of higher fatty acids i s increased
,and the m . p .
and saponification value of the product are respectively raised andlowered in proportion . Palm-nut oil i s stated to be sometimes adulterated with
,or substituted by
,lard or tallow
,coloured with turmeric
and scented with orri s root . Wi th modified figures for the saponification value and distillate-acidi ty
,the method of examining coconut oil
for such adulterants fully applies to palm-nut oil .Palm-nut oil i s largely used. for soap-making
,mixed with other fats .
The commercial oil contains free fatty acids,sometimes in very large
proportion . Valenta found from 7 to 58% in different samples .The mixed fatty acids of palm-nut oil have given the followingresults (see also page 7 2)
Solidifying-po int (titer test)Refractive index at 60°Iod ine value
Sachs 1 obtained the following results by,examination of palm-nut
stearine,
” which i s used,in admixture with other vegetable fats
,
as a substitute for cacao butter °
S p . gr. at 1 00°
So lidifying-po intM . p .
S aponification ValueIod ine valueRe ichert-Me issl value
M ixed Fatty Acids .
M . pS o lidifying-po intMean mo lecu lar we ight
1 Chem. Rev. F ett-Harz-Ind . , 1 9 08 , 1 5 , 9 , 3 0.

LARD OIL .
VIII . LARD OIL GROUP .
Lard Oi l . Tallow Oi l.
Neatsfoots Oi l. Egg Oi l .
LARD OIL.
(See p . Lard,especially the softer kind
,subjected to hydraulic
pressure yields a considerable quantity of fluid called “ lard oil ,”or “ lard olein , ” while the solid portion constitutes “pressed lard ,” or“ lard stearin .
”Consequently , the m . p . and other characters of
lard oil depend much on the temperature at which the pressing i sconducted
,winter—pressed lard oil naturally containing less of the
solid constituents of lard than that expressed at a higher temperature .Prime lard Oil is a nearly colourless
,pale yellow or greenish coloured
oil,having but little odour
,and composed of the esters of chiefly Ole ic ,
stearic,and palmitic acids
,with some linolic and perhaps linolenic
acids . It usually thickens at about and becomes solid ta butsome samples exhibit wide departures from these limits . A specimenof pure winter—pressed Oil examined by Henry began to deposi t flakes at
was thick at and solid at 1 I t did not remelt completelyuntil the temperature reached On the other hand , a sampletested by Duyk
I began to crystalli se at Commercial lard oilvaries in character from the nearly neutral sweet oil above describedto the acid
,rancid
,and off ensive-smelling lard oils of deep brown
colour called “Extra NO .
“Extra No . and “
Extra No . 3”
(Schweitzer and Lungwitz) .
In many of its indications lard oil closely resembles olive oil,which i t
simulates in its behaviour with nitric acid,the elaidin-test
,and the
temperature produced by strong sulphuric acid .
Lard oil i s extensively employed as a lubricant . The chief adulterants affect its viscosity and non-drying characters
,and therefore its
value for lubricating . Lard oil is often employed in lighthouse andsignal lamps
,and a small percentage of free acid or of cottonseed oil
affects,injuriously
,i ts quality for these purposes .
The acidi ty of lard oil , calculated as oleic acid , should not exceedOf 47 samples tested by Jenkins? 40 satisfied this condition ,
and the average acidity of the whole was1 B u ll. de l
’
Assoc. B eige , 1 9 0 1 . 1 5 , 1 8 .
2 Pri vate commun icat ion .

MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
1 0 samples contained between 0 and I % .
30 samples contained between 1 and5 samples contained between 2 and2 samples contained between 4 and
Of 1 4 samples examined by Archbu tt,1 sample contained1 1 samples contained from to2 samples contained
4 samples examined by Tolman and Munson contained from toand 4 by Sherman and Snell from to of free oleic acid .
The sp. gr . of genuine American lard oil at ranges fromto according to Schweitzer and Lungwitz , I and the results
published by other chemists fall within these limits . Of 47 samples ofcommercial lard Oil examined by Jenkins and believed to be genuine
,
only one sample had a higher sp . gr . the remainder rangedfrom to the average being Adulterants
,such as
cottonseed oil,maize oil
,and fish oils
,would raise the sp . gr .
The oleo-refractometer is a valuable instrument for examining lardoil
,the recorded devi ation caused by which ranges from 1
° to 5 .
All fixed oils likely to be added as adulterants,except arachis
,neatsfoot ,
and tallow oils,would increase the refraction (see page
The average vi scosi ty of commercial lard oil i s about the sameas that of olive oil
,but i t varies between rather wide limits . The
efli ux time of 45 samples examined by Jenkins ranged from 356 to534 seconds for 50 c .c . at 1 5 .5
° from Redwood ’s viscometer,the
average being 43 7 seconds . ‘
Olive oil from the same viscometer at1 5 .5
° required 426 seconds . The majority of the samples fell with ina narrower range
,as is shown below .
6 requ ired from 3 56—3 99 seconds .
9 requ ired from 400—42 2 5 seconds .
1 7 requ ired from 427—449 seconds .
7 requ ired from 45 1—466 seconds .
3 requ ired from 47 7—495 seconds .
3 required from 508—534 seconds.
The Maumené thermal value (50 c .c . of oil and 1 0 c .c . of 9 7%sulphuric acid) ranges from about 40° to practically the sameas in the case of olive oil . This is , therefore , a valuable test , since mostyils likely to be added as adulterants would increase the temperatureindication .
S oc. Chem. I nd 1 89 5 , 1 4 , 1 2 9 .

MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
Sp . gr. at
Butyro-refractometer r e a d i n g at
1 5 5°
Saponification valueIod ine valueIodi ne value of J estimated .
L iqu id fatty acids 1 calculated .
S o lid fattyM . p . of m ixed fattyFree (ole ic) acid
,
Cottonseed oil,unless i t has been heated
,would be detected by
Halphen’s colour test ; sesame oi l by the furfural test . Vegetable oils
as a class would be detected by the phytosterol acetate test . Somevegetable oils would be indicated by the appearance of a well-definedband in the absorption spectrum
,near the line B . Genuine lard oil
gives no absorption bands .For the detection of arachi s oi l
,Renard ’s process must be used
(see under Arachis Hehner and M i tchell ’s bromoglyceride testwould prove the presence offi sh oi ls or linseed oi l .
The oxidation test described under “O live Oil i s usefully ap
plied to lard Oil intended for lubricating .
NEATSFOOT OIL .
(See also p . Neatsfoot oil i s obtained by boiling the feet ofoxen in water until all the Oil has risen to the surface . I t i s usually thecustom in rendering establishments to use the whole leg below the knee ,and no doubt the majori ty of the neatsfoot oil of (American) commercei s made in this manner (Lythgoe). The commercial Oil also often includes that from the feet of sheep and horses . 1 0 neatsfoot yieldfrom 2 to pints of oil .Pure neatsfoot oil has a pale golden-yellow colour, a no t unpleasantodour of beef fat
,and slowly deposi ts “ stearine ” on standing . The
portion which remains fluid at a low temperature is used as a lubricantfor clock s . The commercial oil i s largely used for leather dressing andto some extent as a lubricant
,chiefly in admixture with mineral oils .
Neatsfoot oil i s composed chiefly of olein ,with some palmitin and
stearin . No ester of a fatty acid less saturated than oleic i s present in

NEATS FOOT OIL .
Shelbourn). The unsaponifiable matterconsists chiefly of cholesterol and a pig
with bone oil,fi‘
sh,seed
,and
e ign matters soon afterand if preserved underoil occurs . Excessivedue either to adultera
en made by Costewho prepared a number of samples in the laboratorydifferent breeds of oxen and from a calf ’s feet . A sum
mary o f their results is given in the following table , together with someresults by other authori ties with commercial oils believed to be genuine :
—1 2
amd u sed )
M ixed F a tty A cids
The oleo-refractometer should be of great‘value in examining samples
of neatsfoot oil . The presence of seed oils and fish oils would bereadily detected by its means . Sheep ’s-foot oi l i s the standard oilused
“
in this instrument .Observation of sp . gr . and iodine value
,together with the Maumené
test , would serve to detect many of the most objectionable adulterants .Rape oil would reduce the saponification value . Fish oils and linseedoil would be shown by the bromo-glyceride test . M ineral and rosinoil would be found in the unsaponifiable matter . B one oil would be
S oc. Chem. Ind . , 1 9 03 , 22 , 7 7 5 .

MODE S OF EXAMINING FATS,OILS , AND WAXE S .
detected,most likely
,by the ash
,arid probably would increase the
amount Of free fatty acid . Vegetable Oils,as a class
,would be found
by the phytosterol acetate test .
TALLOW OIL .
(See p . Tallow oil,or tallow “ olein
,i s obtained by submit
ting tallow to hydraulic pressure,and its characters difler , as in the
case of lard Oil , according to the temperature at which it has beenexpressed . I t i s largely used as a lubricating oil , especially in admixturewith mineral lubricating oils . “
Ox” oil Should be tallow oil expressed
from beef tallow .
“Animal Oil might contain the fat of other animals .The name “ tallow oil ” i s sometimes incorrectly applied to crude oleicacid
,and care has to be taken that such oil i s not inadvertently pur
chased for l ubricating purposes .G ill and Rowe l give analytical constants of 3 samples of tallow oil ,
as follows :Sp . gr. at 1 00°
T iter test 3 5° to 3 7 . 5
°
Maumené test ( 1 00% HzSO4 used) 3 5
°
Iodine value to 56 . 7Iodine value of m ixed fatty acids to
Two samples Of refined animal oil examined by Archbutt gavethe following numbers :
S p . gr. at
Relat ive effi ux t ime (seconds) at
(Refined rape o il,600—630 seconds)
Free (o le ic) acidMaumené test (97% H2$O 4 used)S aponification valueIodine value
On cooling to 50° F no crystals formed in 3 hours , but on loweringthe temperature to 46° F . crystalli sation commenced
,and S lowly
continued until the oil ceased to flow .
8 samples of animal oil,believed to be genuine
,examined by Dunlop
,
2
had iodine values ranging from to and sp . grs . ( 1 5 . from'
Amer . Chem . S oc. , 1 9 02 , 24 , 466 .
2 A na ly s t. 1 9 0 7 . 3 2. 3 1 9 .

MODE S OF EXAMINING FATS,OI LS
,AND WAXE S .
temperatures,gives the Hager-Salkowski indication for cholesterol ,
and yields a solid elaidin . According to Kitt , the oil i s composedchiefly of the ester of oleic acid wi th palmitic and stearic acids .It contains cholesterol and lecithin . The results obtained by the abovenamed chemists are given in the following table :
Sp . gr. at 1 5°
Solid ifying-po int .M . pSaponification valueIodine valueHebner valueRe ichert-Me issl valueFree (ole ic) acidCholestero l,
Mixed F atty Acids .
Saponification valueIodine value
IX . TALLOW AND BUTTER GROUP .
Beef Fat . Horse Fat .Bu tte r Fat. Lard .
Bone Fat . Mutton Fat .Tallow .
BEEF FAT.
(See also p . B eef fat i s more solid than lard , though itdiff ers In consistency as well as in
‘
chemical composition with the partof the animal from which it is obtained . It i s largely used in the preparation of oleomargarine for the margarine industry
,and also as an
adulterant of lard (see under “Tallow
BUTTER FAT.
(S ee special article .)
BONE FAT. BONE GREASE . BONE TALLOW .
(See p . Bone fat i s obtained by boiling bones with water andskimming the oil ; by steam ing bones in close digesters ; or by ex

BONE FAT. B ONE GREASE . B ONE TALLOW .
solvents . I t is chiefly used for soap and candle
ranges in colour from drab to deep brown,has a character
frequently contains a large proportion of free fatty acids,
contains lime soaps in solution,besides more 01° less calcium
sand,dirt
,and water . I ts fatty acids usually solidify at
about the same temperature as those of lard,though the best samples
approach ordinary tallow in this respect . These variations in qualityl’argely depend upon the kind of bones the fat is obtained from
,the
length of time they have been kept before being treated for the extraction of the fat
,the process of extraction employed
,etc . Bullocks ’
hollow shank bones yield the best fat (Carpenter).In 1 0 samples of commercial bone fat analysed by ValentaI the ashranged from to water from to except in one veryimpure
,nearly black sample
,which contained 6 . the total fatty
ac ids ranged from to the free fatty acids from tothe iodine value from 48 to the saponification value from
200 to 207 ; and the m . p . of the fatty acids from toAccording to Shukoff
,
2 the following varieties are recogni sed inRussian commerce : B enzine bonefat (S t. Petersburg), a very pure fat ,containing from to of water ; benzine bonefatfromS . Russi a
,
usually very impure,containing water and impuri ti es 3 to free acids
30 to titer test 40 to benzinehorse-bonefat, containing 3 to 4% ofwater and impurities
,titer test whi te natural bone fat (S t . Peters
burg) from the gelatine factories , containing 0 .3 to of water andimpurities
,20 to 30% of free fatty acids , ti ter test 40° to 4
In 3 79 samples of bone fat examined by Schestakoff3 the free fattyacids ranged from toMarrow fat from ox bones
,prepared by Dunlop4 was light yellow
,
resembled hard lard in consistence,and had the following character
istics : iodine value,
butyro-refractometer reading at 2
saponification value , free fatty acid,
For the valuation of bone fat, Shukoff and Schestakoff recommend
the following procedure :5Water.
-Dry 5 grm . at 1 00° to 1 1 0° until constant in weight ; owing
to . the tenacity with which the lime soaps retain water,over 24 hours ’
1 Zei ts . Chem. Ind 1 88 26 5 .
2 Chem. Rev. F ett-Harz nd . , 1 9 0 1 . 8 , 2 2 9 .
3 I bid . , 1 9 02 . 9 , 1 80 .
4 A na ly s t , 1 90 7 , 3 2 , 3 1 8.
5 Chem. Rev. F ett-Harz-Ind . . 5 , 5—8 and 2 1
—23 .

MODE S OF EXAMINING FATS , OI Ls , AND WAXE S .
drying may be required . The water can also be estimated bydifference .Fat , and Non
-fat ty Impur i ti es .—
1 0 grm . are gently melted on thewater-bath
,and heated for about an hour with 3 to 5 drops of hydro
chloric acid,with frequent stirring
,to decompose the lime soaps . The
fatty matter i s then dissolved out with 40 c .c . of petroleum spirit ,which is poured through a tared filter-paper into a weighed flask . Theinsoluble matter i s rinsed on to the fil ter
,well washed with petroleum
spiri t,dried
,and weighed . The fatty matter is estimated by dis
tilling off the solvent and drying at 1 00 to 1 1 0° until constant .
Ash .—This is estimated by careful combustion of a weighed
quanti ty of the fat . The calcium in the ash,existing chiefly as car
bonate and Oxide,i s estimated by titration
,and the corresponding
amount of lime soaps calculated from the result , 260 being taken as theaverage molecular weight of the fatty acids . When sand
,calcium
phosphate,etc .
,are present
,a quantitative analysi s of the ash may be
necessary,but this is seldom required .
Unsapon ifiable Ma t ter.—When the bone fat i s intended for soap
making,the unsaponifiable matter should be estimated , as any
amount in excess of that natural to the fat,say must be regarded
as an impurity .
Ti ter Test .—The titer test of the mixed fatty acids is estimated by
Dalican’s process
,as in the case of tallow and other fats .
HORSE FAT.
(See also p . The fat of the horse is light or dark yellow incolour
,and varies in consistency according to the part of ~ the animal
i t has been obtained from . I t consists of the esters of oleic and linoleicacids ( the latter consti tuting about 1 0% of the total fatty acids), and ofsaturated fatty acids of which palmitic acid is probably the chief constituent . Horse fat i s sometimes used as an adulterant of lard andtallow .
The following results of examination of horse fat and oil have beenpublished by Dunlop 1
1 Ana ly st, 1 9 0 7 , 3 2 , 3 1 8 .

208 MODES OF EXAMINING FATS,OILS
,AND WAXES .
TALLOW .
(See also page Tallow is the fat of certain ruminant animals,
separated from the enveloping membrane of the tissue by the processof melting out or “ rendering .
” Tallow is classed commercially as
“ beef and “mutton ” tallow,but each of these may comprise the fat
of other animals besides the ox and sheep .
Pure tallow is white and almost tasteless,but much of that in com
merce has a yellow colour and a disagreeable rancid flavour .In chemical composition
,tallow is composed essentially of the gly
cerides of palmi tic,stearic
,and oleic acids
,but these do not wholly
exist as simple esters,as was formerly believed . Hansen
I has i solatedfrom beef and mutton tallow palmito-distearin
,stearo-dipalmitin ,
oleo-dipalmitin and Oleo-palmito-stearin . On the other hand,
BOmer2 has found about 1 1 /2% of tristearin in beef tallow , 4 to 5%
in pressed beef tallow,and 3% in mutton tallow . According to
Farnsteiner3 the unsaturated acids include a small amount of linolenicacid . Hehner and Mi tchell
,4 in a sample of beef “ stearine ” of
iodine value found of stearic acid . In several samplesof beef tallow
,LewkowitschS found from 2 1 to 2 2% of stearic acid .
The following table of results by Hehner and M i tchell shows the percentage of stearic acid
,etc .
,found in the fat from diff erent parts of a
Scotch sheep 1 8 months old
0 0 0 0 0 0 0
O O O O O O
The ham fat was fluid,and that from the breast was almost fluid ,
at the ordinary temperature .
1 A rch. Hy g . , 1 9 02 , 42 , 1 .
2 Zei tsch . Nahr. Genussm . , 1 90 7 , 14 , 9 0 .
3 Ibi d . , 1 89 9 , 2 , 1 .
4 A na ly s t , 1 89 6 , 2 1 3 2 8 .
5 O ils . Fat s and axes , I I , 6 3 9 .
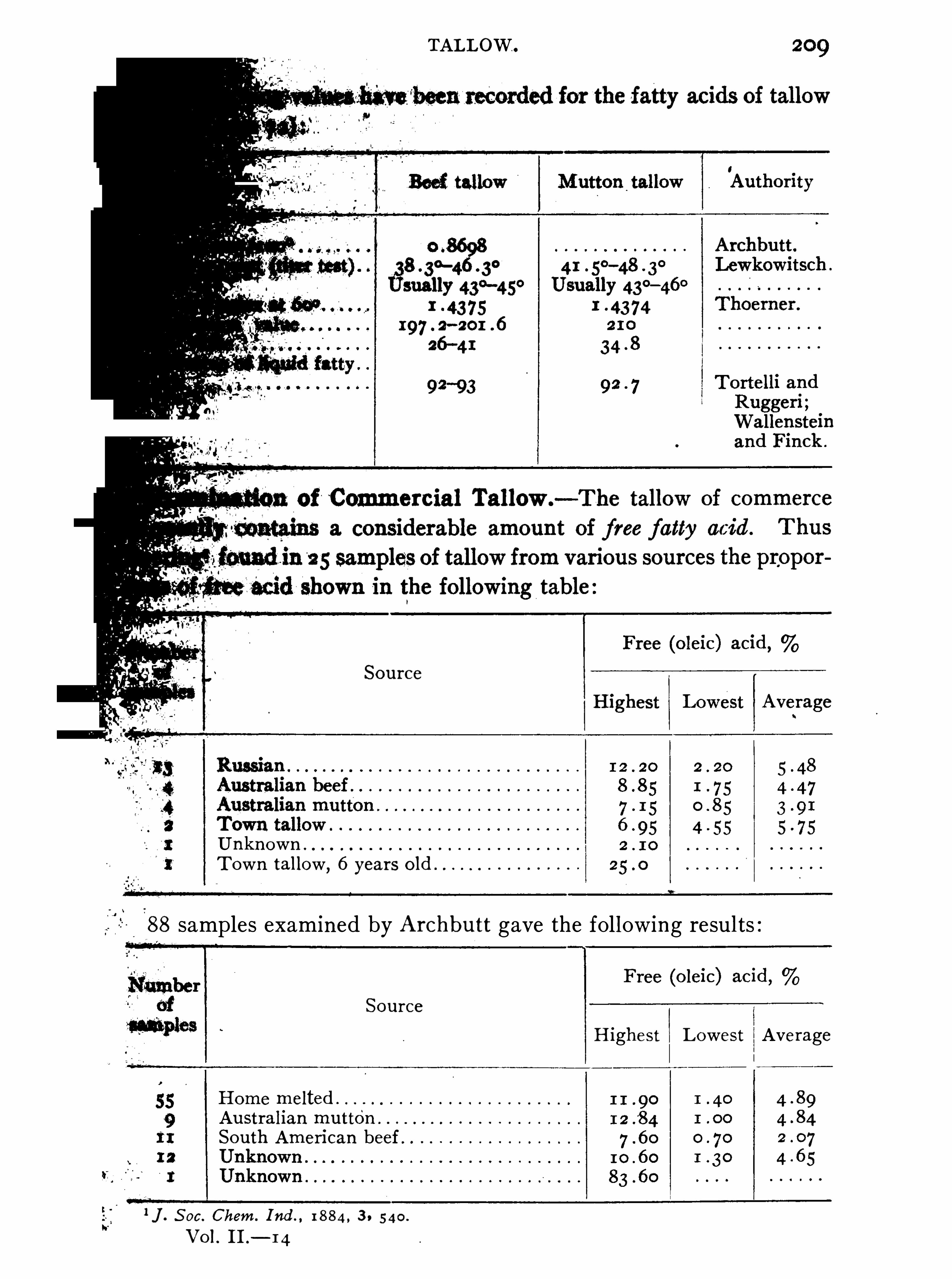
TALLOW.
0 0 0 0 0
Source
Unknown .
Town tallow,6 years Old
88 samples examined by Archbutt gave the following results
Source
Highe st Lowest Average
Home me lted .
AustralianSouth Ameri can beef
S oc. Chem . Ind . , 1 884 , 3 , 5 40 .
Vol. I I .
—1 4

2 1 0 MODE S OF EXAMINING FATS,OILS , AND WAXE S .
2 2 7 samples of tallow , supplied to a specification limiting the free(oleic) acid to contained a m inimum of a maximum of
and anj
average of of free (oleic) acid ?
The free acid in 36 samples of Australian tallow examined byNorman Tate ranged from to and in 2 7 7 samples reportedby Schestakoff2 the free acid found was as follows :
Large proportions of free acid may be due to adulteration of thetallow with wool-grease acids or stearic acid from cottonseed oil ,but they are usually due to hydrolysis of the tallow itself having occurredprevious to the rendering of the fat . From whatever cause produced ,free acid is objectionable and depreciates the value of the tallowto the candle and soap-maker
,besides unfitting i t for use as a lubricant .
Tallow frequently contains more or less water,infusible matters ,
and mineral impurities,and has been occasionally purposely adul
terated with starch,china clay
,whiting
,barium sulphate , etc . Fats
of greater fusibility,especially bone fat
,may be present, and wool
grease acids and cottonseed stearine ” have been extensively used .
Cakes of tallow are said to have been met with the interior of whichconsisted of inferior fats .The presence of water
,starch
,and insoluble substances generally
can be detected and their proportion roughly ascertained by meltinga fair sample of the tallow in a graduated cylinder heated in a waterbath , and reading off the volume of impurities which settle out . Theinsoluble matter in samples of tallow representing large lots is usuallyunder and the water rarely exceeds to 1 .5 Water can beaccurately estimated as described under “ Lard .
” Insoluble impuri ti es
can be estimated by dissolving 1 0 to 20 grm . of the tallow in etheror petroleum spiri t
,filtering through a tared filter-paper , well washing
the paper and contents with the solvent to remove fat,drying at
and weighing . The residue on the filter may be examined under themicroscope
,when starch
, gelatinous matter, or fragments of ti ssue will1 Archbu t t and Dee ley . Lubrica t ion and Lubricant s ,
p . 2 1 2 .
2 Chem . Rev. F ett—Ha rz-Ind . , 1 9 02 , 9 , 1 80 .

2 1 2 MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
has been observed that tallow which has not been washed and pur ifiedand which , therefore , contains particles of blood , etc . , acquires alight brown colour when agi tated in a melted state with 1 /5 of i ts volumeof nitric acid (sp . gr . L . Mayer
I recommends an examinationof the “ oleine ” obtained by allowing the melted tallow to crystalli sefor 1 8 hours at 35° and then squeezing the liquid portion throughfilter cloth . The iodine value of this should not exceed 55 if the tallowbe genuine
,but in presence of cottonseed oil or stearine a much higher
value will be obtained . A more scientific test would be the estimation of the i odine value of the liquidfatty acids , those of tallow absorbing92 to 93% of iodine , while the liquid acids from cottonseed oil absorbnearly Vegetable Oils and fats generally would be detectedby the phytosterol acetate test (pageTallow has been occasionally met with which has been largelyadulterated with the distilledfatty aci dsfromwool grease . Mayer2 hasdescribed a sample which consisted almost exclusively of such fattyacids . It had a very high acid value
,smelt strongly of wool grease ,
yielded only of glycerol on saponification ,and when the aqueous
solution of the soap was shaken with ether and the ethereal solutionseparated and evaporated
,a considerable amount of unsaponifiable
matter containing cholesterol was obtained which gave a violetcolouration
,changing to blue
,when evaporated with concentrated
hydrochloric acid and ferric chloride . Mayer states that 5% of woolgrease can be detected in tallow by this method . The fatty acidsseparated from the soap formed in the above process turned yellow ina few days
,and after several months had acquired a deep orange
yellow tint .The presence of bonefat would be indicated by an excessive amountof ash , containing calcium phosphate . In 5 samples of bone fatexamined by Valenta the amount of ash ranged from toaveraging Genuine tallow leaves a mere trace of ash . Tallowcontain ing bone fat would most probably be very acid .
Horse fat would tend to make tallow yellow in colour and soft .I t would raise the iodine value and would also lower the titer test ofthe mixed fatty acids . Horse fat contains no stearin . I t has markeddrying characters
,and the intense yellow colour of ethereal solutions of
the unsaponifiable matter appear to be characteristic of thi s fat .31 Di ngln poly t. J . , 1 883 , 24 7 , 3 0 5 .
2 Loc. cat .3 Dunlo p . A na ly s t , 1 9 0 7 , 3 2 , 3 1 7 .

CODLIVER OIL . 2 1 3
mes added to soft tallow and usuallythe saponification value , can be es
unsaponifiable matter , which , in genuineThe same process would indicate
of rosin oi l and also wool fat.and coconut Oi ls , besides raising the s
'
aponification
increase the Reichert-Mei ssl value and reduce the Hehner
and frequent adulteration of tallow some yearscandle manufacturers to adopt a process of
assaying samples for the relative proportions of Oleic and solid fattyaci ds . This they effect by Dalican
’
s method,which consists in esti
mating the solidifying point of the mixed fatty acids by the “ titer test ”(see p . 5 The lowest permissible solidifying point of the acids i soften fixed at corresponding to a mixture of oleic and solid fattyacids in equal proportions . The following table by F . Dalican showsthe approximate yield of solid fatty acids (
“ stearic acid ”) from 1 00
parts of tallow . The corresponding oleic acid may be found by subtracting the percentage of solid acids from
The titer test of tallow used for making railway wagon axle greaseshould not fall below 4 1 °
X. WHALE OIL GROUP .
Codliver and All ied O i ls . Herring Oi l.Shark-liver Oi l . S eal Oi l .
Menh aden Oi l . Whale Oi l .Sardine Oi l . Japan Fi sh Oi l . Porpo i se Oi l .
CODLIVER OIL .
(For constants see S trictly speaking, codliver Oil i s the oilobtained from the liver of the cod
,Gadus morrhua, but the closely
analogous oils obtained from the livers of other Species of Gadus and of

MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
the Gadidae family,such as the ling
,coalfish
,hake
,haddock
,and
whiting,are frequently mixed with codliver oil and cannot in the
present state of our knowledge be distinguished from it .The best Norwegian codliver OilI i s extracted from the fat livers ofthe cod caught in the early part of the winter fisheries in the LofodenI slands . At this season of the year cod is about the only fish caughtin that locality
,and there is little opportunity of mixing other fish oils
with i t . The Finmark oil i s more liable to be mixed,as the cod caught
there are accompanied by large numbers of haddock,ling
,and other
fish ? Newfoundland codliver oil i s considered inferior to Norwegianfor medicinal purposes
,and is sometimes largely adulterated with
menhaden and seal oil .3 Lewkowi tsch states that commercial Coastcod oil ” is a liver Oil which may have been obtained from any fishwhich the trawlers ’ nets bring up from the open sea ; i t may , therefore ,contain oil from the shark
,dogfish , etc . , in addition to those mentioned
in the first paragraph .
Several qualities of codliver oil are recognised in commerce : pale ,used only in medicine ; light brown , an after-yield , of inferior quality ,but still largely used in medicine ; and dark brown , or tanners ’ oil ,obtained by roughly boiling down the livers remaining from the foregoing processes .The purest codliver oil has a pale yellow colour , and i s never quitecolourless unless artificially bleached . I t i s limpid
,has a slight
odour and taste,and a faint acid reaction . If prepared at a high
temperature,or if the livers be allowed to partially putrefy
,the acid
reaction is more decided and the colour pale or dark brown , thedarkest varieties being transparent only in thin layers
,and having a
repulsive,fishy odour
,and bitterish
,acid taste .
The composition of codliver oil is very complex and not yet fullyknown . By fractionating the methyl esters ,
of the fatty acids invacuo
,Bull4 found that about 80% distilled over below and from
these he obtained the saturated acids,myristic and palmitic , with a
small quantity of stearic acid,the unsaturated acids
,oleic and erucic ,
and two new acids,to one of which he gave the formula C I 6H3 0
O 2 , andto the other C 20H3 30 2 . The latter he named gadolini c acid; i t is said ~
to occur in large quanti ty . By the sodium- salt-ether method Bull has
1 U . S . Consu lar Report No . 1 843 , Jan . 6 . 1 9 04 .
2Mann . P ha rm . Jou r . , 1 9 03 . 7 1 , 840.
3 S age . Chemi s t and Drugg i s t , 1 9 03 , 62 , 5 7 1 .
‘1 Chem . Zei t. , 1 89 9 , 23 , 9 9 6 and 1 04 3 . B eri chte , 1 906 , 3 9 , 3 5 7 0.

2 1 6 MODE S OF EXAMINING FATS,OI LS
,AND WAXES .
but if an iodide has been added i t will be found in the incombustibleresidue . The usual proportion of iodine in iodi sed codl iver oil i saboutA ferrated codliver oil i s also employed
,containing about 1 % of
ferrous oleate .Good medi cinal codliver oi l should deposit no solid fat at 0° (Pharm.
Germ) , but a granul ar crystalline deposit i s Often produced on coolingoils of the lower qualities .The Bri ti sh Pharmacopoeia describes codliver oil as pale yellow
,
with a slight fishy,but not rancid odour . I t states that it i s the oil
extracted from the fresh liver of the cod , Gadus morrhua, by the applica ~tion of a temperature not exceeding 1 80° F . and from whichsolid fat has been separated by filtration at about 23° F I t
states that no solid fat should separate on exposure of the oil for 2hours to a temperature of 3 2° F . but no test is given by which theoil from Gadus morrhua can be distinguished from allied Oils ?As previ ously stated
,the “
codliver oil ” of commerce is in practiceobtained from several members of the Gadidae
,or cod family ; and , as
long as i t i s produced from these fish solely,li ttle exception can be
taken . The livers of various other fish are,however
,apt to be em
ployed,and the detection of the substi tution is very diffi cult . Adul
teration with fish oils , such as menhaden oil , with blubber oils , such asporpoise
,seal
,and whale oils
,and with mineral and rosin oils i s also
practised .
The constants of a number of fish-liver oils other than codliver oil ,many of which were prepared from the fresh livers by Thomson andDunlop and Were , therefore , undoubtedly genuine , are given in thetable on page 2 2 1 . Thomson and Dunlop
,
2 who have studied thequestion carefully
,are of opinion that
,so far as present knowledge goes ,
taking into consideration the wide limits '
of variation of cod oils and thesmall amount of inf ormation we possess of the others
,coalfish , haddock ,
hake,ling
,whiting
,and Skate liver oils are practically undistinguishable
from each other and from codliver oil by chemical or physical tests .They also believe the detection of seal oil to be almost impossible bychemical methods
,and whale oil difli cult . The smaller yield of bromi
nafed glycerides , however , enables seal and whale Oils to be detected
1 The Un it ed S tat e s Pharmaco os ia.
de scri bes codliver p i l as a fi xed oi l , obta ined fromth e fre sh liver of Gadus morrhua mm , and of
.
o th er spe01e s o f Gadus .
"
2 Papers read before th e Assoc iat ion of Publi c Analy st s of S cot land .June . 1 9 05 . and Jan . ,
1 906 .

CODLIVER OIL . 2 1 7
ally be found useful in detecting some of the others,such
The liver oils,such as shark and dogfish , which
S of unsaponifiable matter may be detected bymeans of this characteristic
,if present in sufficient proportion , and
il can be easily detected , even in small proportion , by meanssaponification and Reichert values .
The sp. gr . of codliver oil ranges from about to atthe darker varieties being generally the heavier . The Uni ted S tatesPharmacopoeia demands a sp . gr . of to at whichcorresponds with abou t to at The Oil from fish
allied to the cod is sometimes of a slightly higher sp . gr . Thus , thatprepared in Grimsby from a mixture of the livers of cod
,haddock ,
ling,and whiting has a sp . gr. of while the product Obtained
in Aberdeen from haddock livers has a sp . gr . of i s somewhatless Vi scous
,and develops more heat with sulphuric acid than the
other varieties of codliver oil . A sample of very much decomposed(44% free acid) brown codliver Oil examined by Bull had a sp . gr .of I t i s evident that the Sp . gr . affords no reliable indicat ionof the presence of other fish oils in codliver oil .The i odine value (Wijs) usually ranges from about 1 54 to 1 70 ,
but a value as high as 1 8 1 has been recorded (Wijs). Wi th referenceto some of the very low numbers , below 1 54, which have been published ,i t may be noted that values estimated by the Hii bl method are from6 to 1 0% lower than those by the Wijs method for this oil , also that theoil from decomposed livers is lower in iodine value than that from freshlivers .The saponificati on value ranges from about 1 79 to 1 90 . A lowervalue might be dueto shark liver or dogfish l iver Oil, if accompanied byan excessive amount of the kind of unsaponifiable matter characteristicof those oils
,or it might indicate the presence or rosin or mineral oil ,
which would be found wholly in the unsaponifiable matter . An
abnormally high value might be due to porpoise oil .The refractometer is of limited use , except as a rapid sorting test ,
and in this respect i t is inferior to the iodine value . DowzardI foundthe refraction of I 3 samples of Newfoundland and Norwegian and 1
sample ofEnglish codliver oil to range from +43 .5 to +4 when testedin Amagat and Jean ’s oleo-refractometer at while 3 samples of paleseal oil ranged from to and he gave a table showing that1 Pharm. J . , 1 89 8 , 5 3 2 .
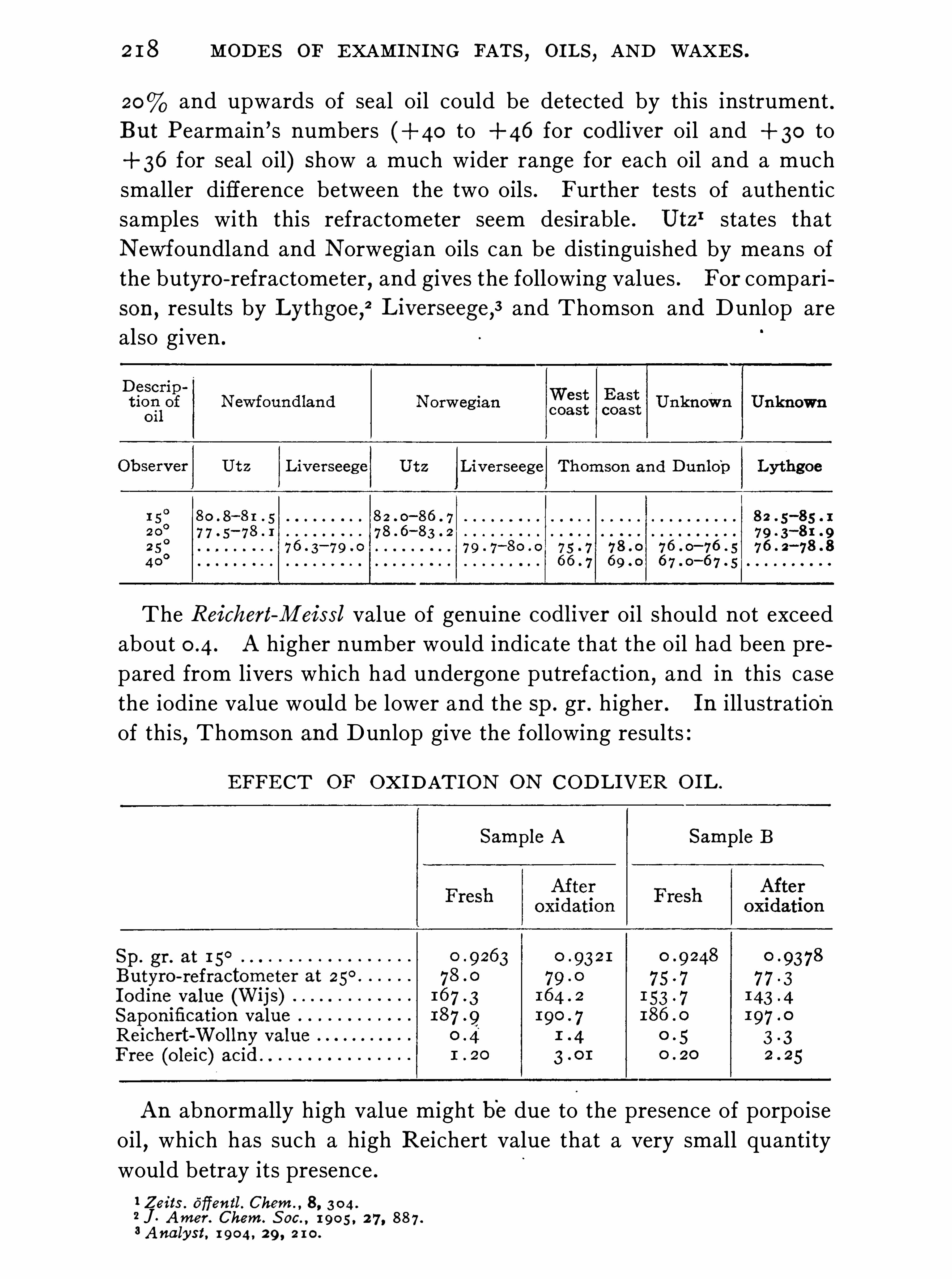
2 1 8 MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
20% and upwards of seal oil could be detected by this instrument .But Pearmain ’s numbers to +46 for codliver oil and + 30 to+ 36 for seal oil) Show a much wider range for each oil and a muchsmaller difference between the two oils . Further tests of authenticsamples with this refractometer seem desirable . UtzI states thatNewfoundland and Norwegian oils can be distinguished by means ofthe bute -refractometer
,and gives the following values . For compari
son, results by Lythgoe? L iverseege ,3 and Thomson and Dunlop are
also given .
The Rei chert-Mei ssl value of genuine codliver oil Should not exceedabout A higher number would indicate that the oil had been prepared from livers which had undergone putrefaction
,and in this case
the iodine value woul d be lower and the sp . gr . higher . In i llustrationof this
,Thomson and Dunlop give the following results
EFFECT OF OXIDAT ION ON CODLIVER O IL .
Sample A Sample B
Sp . gr. at 1 5°
Butyro-refractometer at 25
°
Iodine value (Wijs)S aponification valueRe ichert-Wollny valueFree (o le ic) acid
An abnormally high value might be due to the presence of porpoiseoil
,which has such a high Reichert value that a very small quantity
would betray its presence .1 Zei ts . afien tl . Chem . , 8 , 3 04 .
2 J. Amer. Chem. S oc. , 1 90 5 , 2 7 , 88 7 .
3 A naly s t , 1 9 04 , 29 , 2 1 0.

MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
1 .
“ If 1 drop of the oil be dissolved in 20 drops of chloroform andthe solution shaken with 1 drop of sulphuric acid
,the solIItion will ac
quire a violet-red tint,rapidly Changing to rose-red and
,finally
,
brownish-yellow .
”
(In the German Pharmacopoeia the oil i s directedto be dissolved in carbon disulphide instead of chloroform .)
2 .
“If a glass rod moistened with sulphuric acid be drawn through
a few drops of the Oil on a porcelain plate,a violet colour will be
produced .
”
3 .
“If 2 or 3 drops of fuming nitric acid be allowed to flow along
side of 1 0 or 1 5 drops of the oil , contained in a watch-glass , a red colourwill be produced at the point of contact . On stirring the mixture witha glass rod
,the colour becomes bright rose-red
,soon changing to lemon
yellow (distinction from seal oi l,which Shows at first no change of
colour,and from otherfi sh oi ls
,which become at first blue and after
wards brown and yellow).In regard to the above tests
,i t may be remarked that the violet
colouration produced by sulphuric acid is characteristic , not of codliver oil alone
,but Of liver oils generally . Rancid oils do not give the
Violet colour,but only the red . Thomson and Dunlop have obtained
the violet colour with porpoise and seal O ils which they extracted themselves from the blubbers and
,therefore
,the test is not exclusively
Characteristic even of liver 0115 . I t i s,in fact
,really of very little use
,
and,as Lewkowitsch has pointed out
,i t depends entirely upon the
presence in the oil of impurities which the modern processes of extraction and refining tend to eliminate
,SO that the purer the Oil the
less marked the indication becomes . As regards the’ test with fumingnitric acid
,Tolman has shown that it i s liable to give misleading results
with perfectly genuIne codliver oils of American origin . A variety ofother colour tests have been proposed
,but as they are more likely to
mislead than to afford reliable information respecting the genuinenessof codliver oil i t i s not thought worth while to describe them .
Sk a te-l iver o i l , obtained from Raia bati s, has been proposed as asubstitute for codliver oil . I t i s bright or golden yellow in colour ,i s neutral in indication
,and has a slightly fishy Odour and taste . I t
darkens but little under the influence of chlorine,and is said to give
an odour of valeric acid when heated with a solution of alkali . A
sample exam ined by Thomson and Dunlop gave the results Shown inthe table on p . 22 1 .


2 22 MODE S OF EXAMINING FATS,OI LS
,AND WAXES .
SHARK-LIVER OIL. SHARK OIL .
(See also pages 73 and The shark oil known in commerceis chiefly obtained from the liver of the basking shark (Cetorhinus(S elache) max imus), chiefly caught off the coast of Norway , but thedogfish and several allied fish also contribute to it .Shark oil has been largely employed in tanneries and as a substi tutefor codliver oil
,but in England i t is now almost disused .
Owing to frequent adulteration , the physical and chemical characters of Shark oil have been mi sstated by many authorities . Thus it hasbeen alleged to be of very low sp . gr . ,
a character in all probabilityreally due to the presence of a large proportion of mineral oil or similaradulterant . Whether or not these oils of low sp . gr . were uniformlyadulterated is no longer of practical interest
,as oil of such character
i s not now met with . The “ Shark oil ” usually indicated 40° to 42° onCasartelli
’
s oleometer,and the analogous “
African fish oil ” 48° to
50°
(see foot-note , pageShark oil i s peculiar in yielding a very notable proportion of unsaponi
fiable matter , consisting in great part of cholesterol . If the sample besaponified in the usual way, and the aqueous solution of the soapagitated with ether
,the separated ethereal layer leaves on evaporation
a nearly colourless crystalline mass , which , if dissolved in boiling alcohol ,deposits abundant plates of cholesterol
,which yield the characteristic
colour-reactions .Analyses of a number of shark-liver and some other oils are givenin the following table . I t will be noticed that the percentage ofunsaponifiable matter i s irregular .Shark oil has been found to yield from 20 to 2 2% of an insolublebrominated ester by Hehner and M itchell ’s process
,and the “ bromide
value ” of a specimen examined by Procter and B ennett was found tobe 1 6 .5
—much lower than that of codliver oil .In regard to the three last-named oils in the following table , the dog
fish ” i s probably Acanthias vulgari s , the piked dogfish , a small sharkfrequently seen in shoal water on B ritish coasts . The “
crampfish
may be one of the electric rays , Torpedo . The “ sunfish ” may bethe basking shark
,which floats with its dorsal fin above the water
like the true sunfish,Orthagori scus mola; the latter is not a shark .

MODE S OF EXAMINING FATS , OILS,AND WAXES .
MENHADEN OIL .
(See p . This oil i s obtained from Alosa menhaden,a North
American fish allied to the herring . It i s derived from the wholebody of the fish
,by boiling with water and pressing . I t resembles
codliver oil in many respects,and is used to adulterate Newfound
land codliver oil (Sage). It i s chiefly employed for degras and asa currying oil . Sometimes i t i s added as an adulterant to linseed oil .Some constants and variables of menhaden oil are given in the followingtable :
Authority
Description
Sp . gr. at
S p . gr. at
Butyro-refractometer, 25°
Butyro—refractometer
, 40°
IValenta test, 0
Free (ole ic) acid ,Unsaponifiable matter
,
Iodine valueSaponification value
Adulteration with mineral or rosin oil would be detected by estimating the unsaponifiable matter , which in g enuine menhaden oildoes not exceed about
SARDINE OIL . JAPAN FISH OIL.
(See p . 7 S ardine oil i s obtained from species of sardines belonging to the family Clupeidce . According to a recent paper by T sujimoto
,
2
Japanese sardine oil i s obtained from Clupanodon melanosti cta T . andS .
,and does not possess the low iodine value commonly attributed to it .
Three authentic samples examined by him had (Wijs) iodine values ofand respectively (see table below) . These samples
were greenish-brown to reddish-brown in colour,and all deposited
1 Th e acid u sed gave 6 5 ° with bu t t er fat , and 9 4° t o 9 6° with codliver o il.2 J . College of E ngi neeri ng . Toky o Imp . Un iversit y , 1 906 , 4 , 1 .
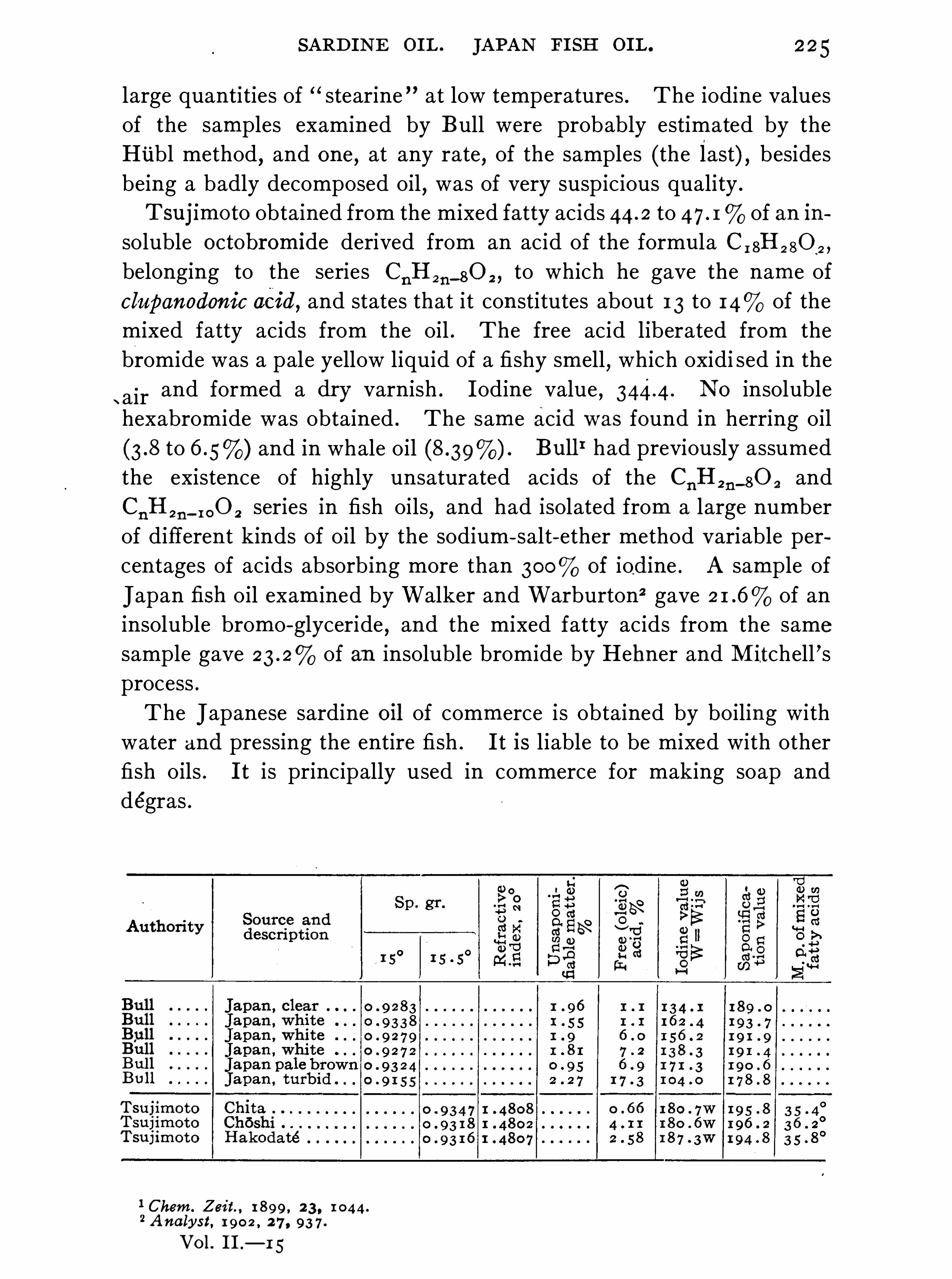
SARDINE OI L . JAPAN FI SH OI L.
large quantities of “ stearine at low temperatures . The iodine valuesof the samples examined by B ull were probably estimated by theHubl method
,and one , at any rate , of the samples (the last), besides
being a badly decomposed oil,was of very suspicious quality .
T sujimoto obtained from the mixed fatty acids to of an insoluble octobromide derived from an acid of the formulabelonging to the series to which he gave the name ofclupanodonic acid
,and states that i t constitutes about 1 3 to 1 4% of the
mixed fatty acids from the oil . The free acid liberated from thebromide was a pale yellow liquid of a fishy smell
,which oxidi sed in the
\air and formed a dry varnish . Iodine value,
No insolublehexabromide was obtained . The same acid was found in herring oilto 6 .5 and in whale oil (8 .39 BullI had previously assumed
the existence of highly unsaturated acids of the CnH, n_ 8O , and
series in fish oils,and had isolated from a large number
of diff erent kinds of oil by the sodium-salt—ether method variable percentages of acids absorbing more than 300% Of iod ine . A sample ofJapan fish oil examined by Walker and Warburton2 gave of aninsoluble bromo—glyceride
,and the mixed fatty acids from the same
sample gave 2 of an insoluble bromide by Hebner and M itchell ’sprocess .The Japanese sardine Oil of commerce i s obtained by boiling withwater and pressing the entire fish . I t is liable to be mixed with otherfish oils . I t i s principally used in commerce for making soap anddegras .
Tsuj imo to Ch itaTsu j imoto
TSUJlmOtO Hakodate
1 Chem. Zei t. , 1 89 9 , 23 , 1 044 .
2 A naly st , 1 9 02 . 2 7 , 9 3 7 .
Vo l. I I .
—1 5
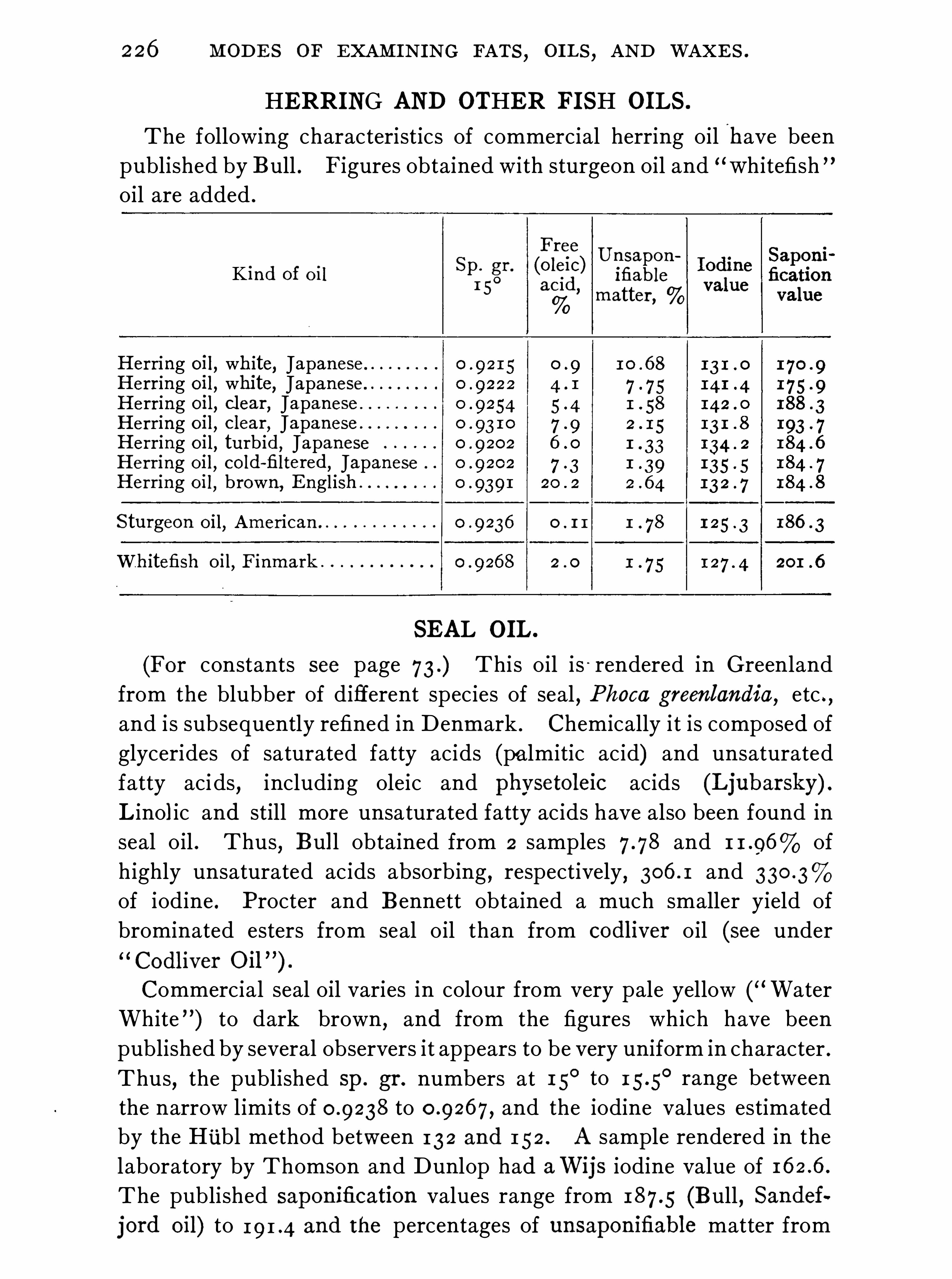
226 MODE S OF EXAMINING FATS,OI LS , AND WAXE S .
HERRING AND OTHER FISH OILS .
The following characteristics of commercial herring oil h ave beenpublished by Bull . Figures obtained with sturgeon oil and Whitefishoil are added .
FreeUnsaponSp . gr (o le ic)K ind of 011
1 5°
ac id Ifiable
matter,
Herring oi l, wh ite , JapaneseHerring o i l, whi te , JapaneseHerring Oil
,clear, Japanese .
Herring o i l,clear
,Japanese
Herring o i l, turbid , JapaneseHerring o i l
,cold—fi ltered , Japanese
Herring o i l,brown, English
S turgeon o il,American
Wh itefish o il,Finmark 2 . 0
SEAL OIL.
(For constants see page This oil i s - rendered in Greenlandfrom the blubber of different species of seal , Phoca greenlandia, etcand i s subsequently refined in Denmark . Chemically i t i s composed ofglycerides of saturated fatty acids (pa lmitic acid) and unsaturatedfatty aci ds
,including oleic and phy setoleic acids (Ljubarsky).
Linolic and still more unsaturated fatty acids have also been found inseal oil . Thus , Bull obtained from 2 samples and ofhighly unsaturated acids absorbing , respectively , and 330 .3%of iodine . Procter and B ennett obtained a much smaller yield ofbrominated esters from seal oil than from codliver oil (see underCodliver
Commercial seal oil varies in colour from very pale yellow (“WaterWhite ”) to dark brown , and from the figures which have beenpublished by several observers i t appears to be very uniform in character .Thus
,the published sp . gr. numbers at 1 5° to range between
the narrow limits of to and the iodine values estimatedby the Hubl method between 1 3 2 and 1 5 2 . A sample rendered in thelaboratory by Thomson and Dunlop had aWijs iodine value ofThe published saponification values range from (Bull , Sandefjord Oil) to and the percentages of unsaponifiable matter from
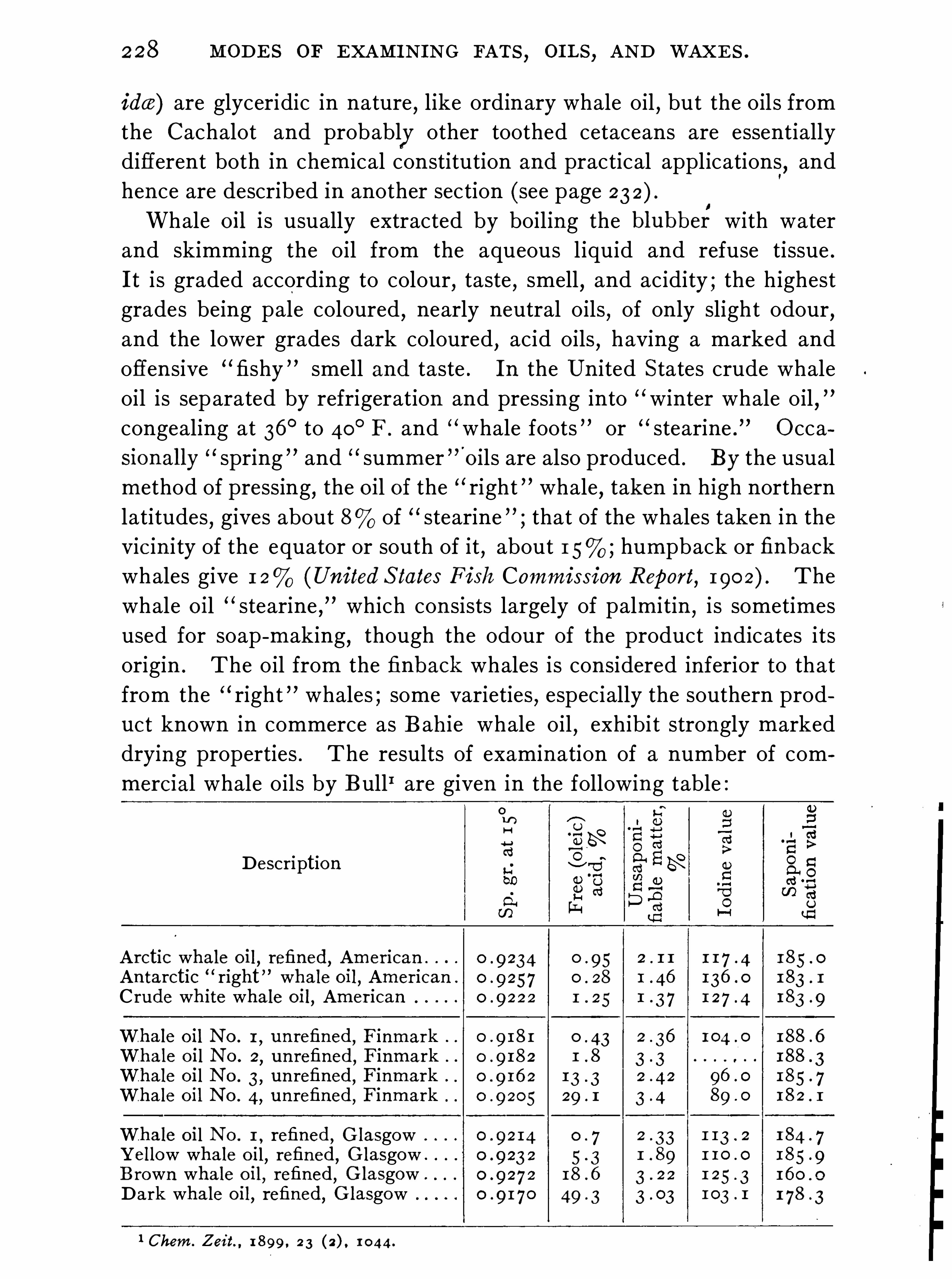
2 28 MODE S OF EXAMINING FATS , OI LS,AND WAXE S .
idce) are glyceridic in nature , like ordinary whale oil , but the oils fromthe Cachalot and probably other toothed cetaceans are essentiallydifferent both in chemical constitution and practical applications
,and
hence are described in another section (see page 23Whale Oil i s usually extracted by boiling the blubberf with waterand skimming the oil from the aqueous liquid and refuse tissue .It i s graded according to colour
,taste
,smell
,and acidity ; the highest
grades being pale coloured,nearly neutral oils
,of only slight odour
,
and the lower grades dark coloured,acid oils
,having a marked and
offensive “ fishy ” smell and taste . In the Un i ted S tates crude whaleoil i s separated by refrigeration and pressing into “winter whale oil
,
”
congealing at 36° to 40° F . and “whale foots ” or stearine . Occasionally
“ spring and “ summer Oils are also produced . By the usualmethod of pressing
,the Oil Of the “ right ” whale
,taken in high northern
latitudes,gives about 8% of stearine ” ; that of the whales taken in the
vic ini ty of the equator or south of it,about humpback or finback
whales give 1 2% (Uni ted S tates Fi sh Commi ssi on Report, Thewhale Oil “ stearine
,
” which consists largely of palmi tin,i s sometimes
used for soap-making,though the odour of the product indicates i ts
origin . The oil from the finback whales i s considered inferior to thatfrom the “ right whales ; some varieties , especially the southern produot known in commerce as B abie whale oil
,exhibi t strongly marked
drying properties . The results of examination of a number of commercial whale oils by B ull 1 are given in the following table :
0InH
"J
Description
Arctic whale o i l, refined, American .
Antarctic “right ” whale o i l, American . 0 . 28
Crude wh ite whale o i l,American 1 . 25
Whale o il No . I , unrefined ,F inmark
Whale o i l NO . 2 , unrefined , F inmark o 9 1 8 2 3 . 3Whale O i l No . 3 , unrefined , Finmark 0 9 1 62Whale o i l NO . 4 , unrefined ,Finmark 0 9 205
Whale o i l NO . 1, refined , G lasgow 0 7Ye llow whale o i l,refined , Glasgow .
B rown whale o i l,refined , Glasgow . 1 60 0
0 9 3Dark whale Oi l, refined, G lasgow 1 78 3. 9 1 70 4
1 Chem. Zei t. . 1 89 9 , 2 3 1 044 .

WHALE OI L . 2 29
oil is variable . I t i s composedd and unsaturated acids
,some of the latter being
Hebner ' and M i tchell Obtained 25 and Walkermean of of brominated ester from 2
the sodium-salt-ether method,found a smaller
unsaturated acids in whale oil than in most otherimoto found of an acid of the C
nH
, 4_ SO ,
The a mount of unsaponifiable matter i s variable , but does notexceed This and the much higher sp . gr . readily distingu ishordinary whale oil from the sperm oils . Adulteration with mineral orrosin oil would , of course , increase the percentage of unsaponifiablematter . Rosin oil i s the most likely adulterant . A sample of “ whaleoil ” supplied for oil-tempering steel
,examined by the reviser
,had a
sp . gr . of at requIred only of potash for saponification
,and contained of unsaponifiable matter of sp . gr .
easily soluble in acetone .The recorded sp . grs . of genuine whale oil range from (Liverseege) to (Bull) ; the saponification values from 1 88 to 1 94
(Schweitzer and Lungwitz), the low value of 1 60 Obtained by Bull being exceptional . The range of iodine values is wide , 89 to I 36 in thesamples examined by Bull
,and there was also a fairly wide range in
the percentages of highly unsaturated acids which he found by thesodium-salt-ether method .
8 samples of whale oil examined by Milrath 1 in the butyro—refractometer gave readings of to at 2 5° and to atThe mixed fatty acids of whale oil have been found to possess thefollowing characteristics
Authority
1 00°
Sp . Gr . at1 00
0 Archbutt .
So lidifying-po int (titer test) 2 2 . 9°—23 . 9
° Lewkow itsch .
Butyro-refractomete r reading, 40° 43 3 L iverseege .
Iod ine value S chwe itze r and
Lungwitz .
Whale oil i s liable to adulteration with seal oil,which so nearly re
sembles whale oil that it cannot be detected by chemical means , except1 Zei tsch . afientl. Chem. , 1 90 7 . 1 9 , 3 7 1 .

230 MODE S OF EXAMINING FATS,OILS
,AND WAXES .
perhaps by a lowering of the “ brom ide value (see under “ Codliver
Whale Oil i s used as an illuminant and to some extent as a lubricant .The lower grades are used in leather manufacture and for temperingsteel .
PORPOISE OIL .
(See also p . 7 Commercial porpoise oil i s derived not only fromthe black porpoise
,Phocxna communi s
,usually caught off the coast of
Denmark and in the Mediterranean and B lack Sea near Trebizond ,but also largely from the beluga or white whale
,Delphinapterus leucas ,
caught in the White Sea, the S t . Lawrence,and on various parts of the
Canadian coasts . The oils from the grampus or killer whale ,Orca gladiator, and the various species known as blackfish , especiallyGlobicephalus melas , also rank as “ porpoise oil .”Porpoise oil is prepared in much the same manner as whale oil . In
some instances,Oil of a superior quali ty drains from the blubber at the
ordinary temperature,but the greater part i s obtained by boiling the
tissue with water. The oil is pale yellow to brown in colour and , according to Schaedler
,i s composed of the glycerides of valeric , palmitic ,
stearic, physetoleic , and oleic acids .
The liquid “ oleine ” obtained from the soft fat of the head and jawby exposing the fat to a low temperature and straining off the oil whichremains fluid
,contains a much larger percentage of valerin and of un
saponifiable matter than the body oil . In the case of G. melas , the massof fat taken from the head has the shape of a half watermelon , and theliquid oil obtained from it i s known as “melon oil .” These j aw oilsare specially prepared in America for lubricating watches and otherdelicate mechanisms
,and command a high price . The body oils are
also used for lubricating .
Porpoise oil i s remarkable for the large proportion. of valerin whichi t contains . A sample examined by Allen yielded of volatilefatty acids
,having a mean combining weight of (C SHmO 2
Chevreul,the original discoverer of valeric acid which be
i solated from porpoise oil and called “phocenic acid , preparedbarium salts of volatile fatty acids equivalent to 3% of valericacid
,so that the composition of the Oil i s evidently very variable .
From a sample of oil from Globi cephalus melas Chevreul preparedbarium salts corresponding to of valeric acid
,besides a con

232 MODE S OF EXAMINING FATS , OILS , AND WAXES .
siderable proportion of spermaceti . Hence the oils from the Delphinidae appear to form an intermediate group between those of thesperm whales and the whalebone whales .The different percentages of volatile acid found is no doubt due tothe diff erence in composition between the body oils and the j aw oils .Steenbuch found valeric acid to consti tute 1 0% of the soluble acids ofthe body oil and 26% of the acids from the j aw oil . Moore obtainedvolatile acids equivalent to and 24 .30% of valeric acid from twosamples of j aw oil
,but only from a sample of body oil .
Owing to its peculiari ty of composition , porpoise oil has a highsaponification value and Reichert-Meissl value . I t i s saponified withgreat facili ty by aqueous potash
,the product being coloured reddish
brown . Wi th the elaidin test,porpoise oil gives but little solid elaidin .
The results of examination of a number of samples Of these oil s aregiven in the table on p . 23 1 .
XI . SPERM OIL GROUP .
Sperm Oi l . Arct ic Sperm Oi l . Bo ttlenose Oi l .Dolph in Oi l .
SPERM OIL .
(See pp . 73 and Sperm oil proper is obtained from thehead-cavities and blubber of the cachelot or sperm whale (Physetermacrocephalus) . Several other of the toothed whales (Odontoceti )yield allied products
,and the Oil from one of these
,namely
,the doegling ,
or bottlenose whale (Hyperoodon rostratum) i s known under the nameof Arctic sperm oil .Sperm oil on cooling readily deposi ts crystalline scales of spermaceti .This i s removed by filtration
,but unl ess the operation be conducted
at a very low temperature a portion of the wax is liable to remaIn In
solution .
In the oil refineries at San Francisco the crude sperm oil is separatedby refrigeration and pressmg into :
‘
1 .
“winter sperm oil,congealing
below 38° F the yield being about 2 .
“ spring sperm oil ,congealing at 50° to 60° F.
, 3 .
“ taut-pressed oil,
” melting at 90°to 95° F . , and 4 .
“ crude spermaceti,
” melting at 1 1 0° to 1 1 5° F .
1 1 % (United S tates Fi sh Commission Report ,

SPERM OIL . 233
Spei rn oil i s a thin yellow liquid , and when of good quality i s nearlyfree from odour . Inferior specimens have an unpleasant fishy smelland taste . I ts sp . gr . is very low , ranging between 5 and
Sperm oil i s one of the most valuable oils in commerce . I t hasbeen found preferable to any other fixed oil for lubricating the spindlesof cotton and woo llen mills and for light machinery generally
,owing to
its l impidity and freedom from tendency to “ gum .
”
Some indication of the peculiar composition of sperm oil was givenin 1 823 by Chevreul . Chevreul
’s observations seem to have been
wholly forgotten until Allen some years since called attention to theunique constitution of sperm oil .Sperm oil gives on saponification products very different from thoseyielded by ordinary oils . When saponified with potassium hydroxideit forms potassium oleate and monohydric alcohols , the nature ofwhich is at present unknown . By agitating the aqueous solution ofthe resultant soap with ether
,the higher alcohols are dissolved
,and
may be recovered by evaporating the solvent . The fatty acids maybe isolated by acidifying the soap solution and again shaking withether . From the residual liquid glycerol can be obtained
,though in
much smaller proportion than from most other O ils and fats . Theexistence of glycerol has been proved by Fendler2 and Dunlop
,3
whose results are given in the following table :
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0
The higher alcohols,i solated in the manner described above
,form a
pale yellow,solid
,semi-crystalline substance
,the m . p . ofwhich depends
on the completeness with which the oil had been previously purified1 Dealers in sperm and similar o ils commonly u se a special hy drome t er , devised by
Casart e lli , on th e sca le of which wat er i s 0°and rape o i l S perm 011 s tand s at 44
°
to 46°and south ern wha le o i l at abou t 24
°on th e same sca le .
2 Chem. Zei t. , 1 9 05 , 29 , 5 5 5 .
a J. S oc. Chem . I nd . , 1 9 08 . 2 7 , 64 .

234 MODES OF EXAMINING FATS,OILS
,AND WAXES .
from spermaceti . They are insoluble in water,but readily soluble in
alcohol and ether,and are volatile apparently without change in a
vacuum , condensing as a perfectly colourless liquid , of sp . gr . atwhich solidifies on cooling to a crystalline mass . The ether
residue from sperm oil i s apparently a mixture of homologous alcoholswhich
,according to Lewkowi tsch I belong for the most part , if not
wholly , to the ethylene series . Dunlop has published the followingresults of examination of the unsaponifiable matter (wax alcohols , etc .)from six authentic samples Of sperm oil from which the spermaceti hadbeen removed :
Description M . p
1 a . Cachalot o i l from head-matter1 b . Cachalot Oil from body—matter2a . Cachalot o i l from head-matter2b . Cachalot o il from body-matter3 . Arctic sperm o i l
4 . Arctic sperm o i l . .
5 . Southern sperm o il
6 . Southern sperm o i l
The following table contains some further results given by the mixedalcohols from Southern sperm (S), Arctic sperm (A), and genuinecommercial oil (C) the origin of which was unknown
Sp . gr. at
Sp . gr. at O . 827 IC
So lidifying-po int ,M . p .
,0
Iodine value 65 -3°
Acetyl saponification value
1 J. S oc. Chem . Ind . , 1 89 2 ,1 1 , 1 3 4 .
-5
23—24—2423
47 -0
47 -0
45 -7
39
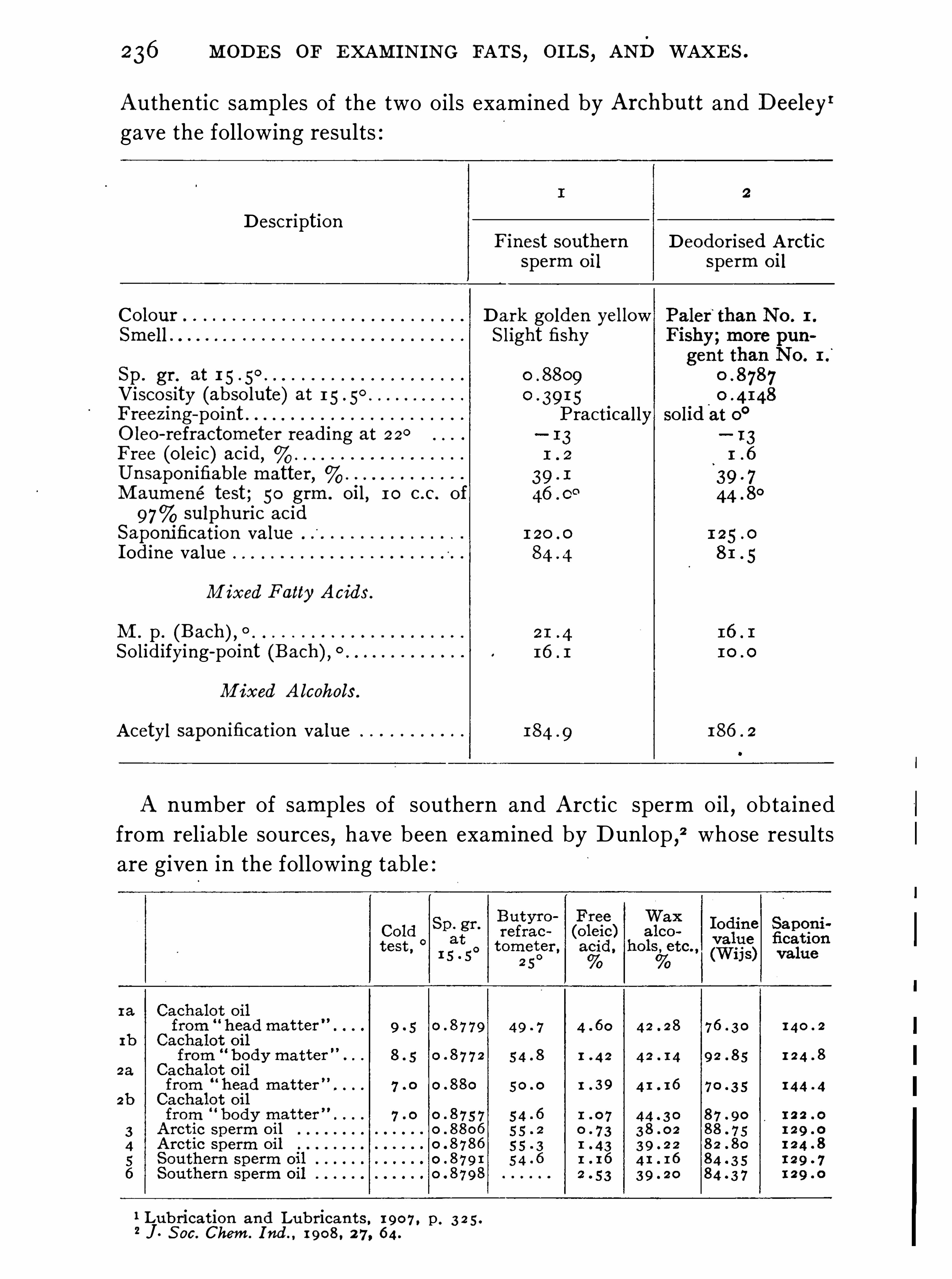
236 MODE S OF EXAMINING FATS , OI LS , AND WAXES .
Authentic samples of the two oils examined by Archbutt and DeeleyI
gave the following results
DescriptionFinest southern Deodorised Arctic
sperm o i l sperm o i l
Co lour Dark go lden yellowSmell . S light fishySp . gr. at
Viscosity (abso lute) at 1 5 5°
Freezing-po int .
O leo-refractometer reading at 2 2°Free (Ole ic) acid ,Unsaponifiable matter
,
Maumené te st ; 50 grm . Oi l,1 0 c .c . o f
97% sulphuric ac idSaponification valueIodine value
M ixed F atty Acids .
M . p . (Bach) ,So lid ifying-po int (Bach), 0
M ixed A lcohols .
Acetyl saponification value
A number of samples of southern and Arctic sperm oil,obtained
from reliable sources,have been examined by Dunlop? whose results
are given in the following table :
1 4 2
5 0 0 I 3 9 I 44 -4
1 Lubricat ion and Lubricant s , 1 90 7 . p . 3 2 5 .
2 J. S oc. Chem. Ind . , 1 9 08 , 2 7 , 64 .

SPERM OIL .
The oils numbered 1 and 2,from 2 animals
,represent the Oil after
removal of the spermaceti from the “ head ” and “ body ” “matter
,
respectively ; these Oils are not usually kept separate in practice , butare mixed together , and hence the differences in the iodine and saponiflcation values
,though noteworthy
,are not of practical importance .
The sp. gr . of genuine commercial sperm Oil ranges from toat In the absence of mineral oil
,Dunlop considers that a
figure within 5 and will generally indicate a pure oil . Adulteration with any other fixed oil would raise the gravity, but this couldbe corrected by the addition of light mineral oil .The saponificati on value of genuine sperm oil appears to range fromabout 1 20 to 1 3 7 . I t would be raised by the addition of a fixed oiland lowered by that of mineral oil
,but a mixture of the two might
be added having the same saponification value as sperm oil .The nature and estimation of the saponification
-products affordthe most satisfactory means of detecting adulterations of Sperm oil ,which
,when genuine
,yields from 60 to 63% of insoluble fatty acids ,
and 3 7 to 42% of ether-residue consisting of higher alcohols . No
other animal or vegetable Oil,except shark-liver oil and Oils from allied
Cetacea (e . g .
,bottlenose oil), i s known to yield more than 2% to ether ,
and,with few exceptions (e . g .
,porpoise oil and some varieties of
whale oil), all other fixed oils yield fully 95% of insoluble fatty acids ,and from 1 0 to 1 2% of glycerol . Hence , in a case of adulteration ofsperm oil with any other fatty oil
,estimation of the ether-residue will
detect the admixture and approximately estimate the proportion .
Some specimens of shark-liver oil yield a considerable proportion ofether-residue
,and hence if shark oil be present the ether process will be
rendered inaccurate . Genuine shark—liver oil has a comparativelyhigh sp . gr .
,to and has a very high halogen-a bsorption ,
besides giving a well-marked Violet colouration and great increase oftemperature with strong sulphuric acid .
The foregoing process,if used without discretion
,would fail in the
case of a mixture of mineral oil and a fatty oil in certain proportions ,but a careful consideration of the results and further examination ofthe products will allow of such amixture being readily distinguishedfrom sperm o il . Thus from an inspection of the figures in the following table i t appears that while the saponification-products yielded bysperm oil wou ld be approximately Simulated by those given by a judicious mixture of mineral oil with rape oil
,in the latter case the sum of

238 MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
the fatty acids and ether-residue would be several units less than1 00 ,and there would be a larger proportion of glycerol produced .
B esides . the ether-residue would probably be insoluble in cold rectifiedSpiri t (see below), and to Obtain a mixture of the same sp . gr . as spermoil
,so very light a mineral oil would require to be used that i t would
necessarily be liquid,even at and would have so low a flashing-point
that i t could without diffi culty be detected in,and even distilled out
of,th e original oil or the ether—residue . Sperm oil does not flash
below 400° F .
Product s o f th e sap on ificat ion of 1 00 part s o f o i l
E ther-residue
Although mineral Oils are practically insoluble in rectified alcohol,
Nash1 has shown that a solution of sperm-oil alcohols in absolutealcohol
,and even in alcohol of sp . gr .
,unless much diluted
,
dissolves mineral oil freely . Absolute alcohol must,therefore
,not be
used in testing the unsaponifiable matter for mineral oil ; but if theunsaponifiable matter from 5 grm . of a sperm-oil sample be normal inamount and completely soluble in .50 c .c . (not less) of cold alcohol of
sp . gr .,the sample i s most probably genuine . Dunlop states
,
however,that even under these conditions a considerable amount of
mineral oil may be dissolved and lost sight of . The following tests formineral oil are
,however
,available :
Ho lde’s Test—6 to 8 drops of the oil are boiled in a test-tube for2 minutes with 5 c .c . of N 2 alcoholic potas sium hydroxide , and to thesoap solution thus prepared distilled water i s added very gradually ,well mixing after each addition
,until from 0 .5 to 1 5 c .c . have been
added altogether . If mineral oil be absent,the solution remains clear ,
even when mixed with the maximum quantity of water, but the presenceof even 1 % of mineral oil i s said to cause the formation of a turbidity .
1 Analyst, 1 904 , 29 , 3 .

240 MODE S OF EXAMINING FATS , OILS,AND WAXE S .
As whale oils were found by Procter and B ennett to yield from 2 7 to3 7% of brominated esters , a greater difference between the yield fromoil No . 2 and mixture No . 5 might have been expected . But
further experiments are needed . Procter and B ennett themselvesobtained of brominated esters from the sperm oil which theytested .
In examining sperm oil the i odine value i s not so useful or so neces
sary a test as in the case of other oils . The recorded values have afairly wide range
,Dunlop ’s figures ranging from to (Wijs),
but as these extreme values were obtained from head-matter Oil andbody-matter oil
,respectively , and not from the mixed Oil, they must be
regarded as abnormal . A range from 80 to 90 would include mostgenuine commercial sperm oils
,though Bull records a value as low as
(probably obtained by the Hiibl method) for an Arctic spermoil containing of unsaponifiable matter and having a saponification value of Dunlop has pointed out that there is a relationship between the iodine value of the wax alcohols and that of the oil .This will be seen by comparing the” iodine numbers given in the tableon p . 234 with those in the table on p . 236 from the same oils . Thediff erence in the amount of iodine absorbed by diff erent oils Would ,therefore
,appear to be due partly to the variable composition of the
alcohols .Sperm Oil has a much lower Viscosity than most fixed oils
,and this
physical property in absolute measure is given on p . 236 . Theefli ux time of 50 c .c . of sperm oil from Redwood ’s viscometer has beenstated as follows : at 60° F .
,1 7 7 to 20 1 seconds ; at 70° F .
,1 3 7 to 1 64
seconds . Southern sperm oil seems to be rather lower in viscosi ty thanArctic sperm
,but more samples need testing .
The colour indication with sulphuric acid (page 4 1 ) is often a usefultest for the purity of sperm oil . The genuine oil gives a brown colouration ,
becoming somewhat darker with a tinge of violet on stirring .
Shark-liver oil gives a well-marked Violet colour when tested in thesame manner
,the tint changing to red or reddish-brown on stirring .
ARCTIC SPERM OIL. DOEGLING OI L . BOTTLENOSE OIL .
(See also page 7 S everal species of toothed cetaceans yield anoil analogous to that obtained from the cachelot or sperm whale . Thechief of these in economic importance is the product from the doegling

ARCTIC SPERM OI L . 24 1
or bottlenose w hale (Hyperoodon rostratum),I which is known in com
merce as “Arctic sperm oil .”Bottlenose oil deposits more or less spermaceti when cooled
,but the
yield is not nearly so large as that obtained from the head matter andoil of the sperm whale
,though of good quality and high m . p . B ottle
nose oil often has a more or less unpleasant odour,but this pecul iarity
,
together with the small proportion of free acid present in the crude oil,
can be removed to a great extent by agitation with a solution of sodiumcarbonate or by analogous treatment . The refined Oil i s straw-yellow .
The chemical constitution of doegling oil was first pointed out byScharling (Jour. P rakt. Chem.
,who found it to consist essentially
of the ester Of a higher monatomic alcohol,dodecyl doeglate , C H2 5
C , 9H
3 9O 2 and hence to yield on saponification dodecyl alcohol and
doeglic acid . Further investigation on this point is desirable . Bullthinks that Scharling ’
s doeglic acid (C 1 9H
3 6O 2) must have been amixture of gadolinic acid (C 20H3 80 2) (see
“under Cod-liver Oil ”)and oleic acidThe wax alcohols
,etc .
,obtained from bottlenose Oil by agitating
the aqueous solution of the saponified oil with ether and separatingand evaporating the ethereal solut ion ,
have s imi lar characters to theproduct obtained in a s imilar manner from sperm oil (see underSpermThe mixed fatty acids prepared by Allen from several specimens of bottlenose oil were found to have a sp . gr . of theircombining weights ranging from 2 75 to 294 .
Bottleno se oil presents the closest resemblance to sperm oil . In i tsSp . gr . Vi scosity
,solubili ty in acetic acid
,saponification
equivalent , and behaviour with strong sulphuric acid and the elai dintest , i t presents no tangible difference from Sperm oil . On saponifica
tion it yields from 6 1 to 65 of fatty acids,and from 3 7 to 4 1 of ether
residue , in this respect simulating true sperm oil in the closest manner .The only differences observed by Allen in the course of a series of verycareful comparative examinations of sperm and doegling oils have beenthe slight tendency of the latter to gum or thicken on exposure , and thesomewhat higher m . p . of the fatty acids from sperm oil . I t has ,therefore
,been convenient to treat them together in the preceding
pages under the “Examination of Commercial Sperm O il .”
1 Th ere h as been much confusion re spect ing th e bo t t leno se . at least e ight d ifferent wh a lesand dolph i ns havi ng been designat ed by that name .
Vo l. I I .
—1 6

242 MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
DOLPHIN OIL .
The oil from the blubber of the blackfish,Globi cephalus melas , i s
Ci tron-yellow in colour,deposits spermaceti when cooled
,and contains
a large proportion of the glyceride of valeric acid . I t presents manypoints of resemblance with porpo1se oil (see under “Porpoise
X II . BEESWAX GROUP . SOLID WAXES .
Beeswax . Ch ine se Insect Wax .
Carnauba Wax . Spermacet i .
W oo l Fat . Woo l Wax .
BEESWAX?(See also table on page 7 B eeswax is the material Of which thehoneycomb of bees i s composed . To obtain the wax the honey isdrained off
,the comb expressed
,melted in water
,the impurities
allowed to subside,and the wax allowed to cool or run into suitable
moulds . About 1 pound of wax i s obtained from 20 pounds of honey .
In the process,as described by Hirschel? the bulk of the wax is first
separated by treatment with hot water and straining from dead bees ,etc .
,and the residue pressed in layers with straw in a filter-press . The
pressed residue is again boiled with water and pressed,after which there
still remains 1 0 to 1 5% of wax in the press-cake . This is extractedwith petroleum spirit and known as “ ex tracti on wax
,
” the former being“pressed wax .
”
3 samples of genuine “ extraction wax ” examinedby Hirschel
,previous to bleaching and refining
,were dark brown
,soft
,
greasy substances,of unpleasant odour . They had higher acid
values than normal beeswax to and lower “ ratio numbersto also much higher iodine values to the
m . p . to and sp . gr . at 1 5° to though lowerthan
,did not differ greatly from those of ordinary beeswax . All three
samples gave faint indications for rosin,and in Weinwurm
’s test (page
257) behaved as if containing about 5% of parafli n wax .
Ye l lowWax .—Normal beeswax is a tough
,compact
,solid substance ,
K
of a yellowish or brownish colour,with a slight lustre and a finely
granular fracture . I ts taste is faint and slightly balsamic , and th eodour is honey-like and characteristic . I t does not feel greasy tothe touch .
S
1 B ibliographie s re la t ing t o bee swax and t o waxe s used for adu lterat ing i t are given inJ. S oc . 7 5 6 , 7 5 7 .
2 Chem . Zei t. , 1 9 04 , 28 , 2 1 2 .

MODE S OF EXAMINING FATS , OI LS,AND WAXE S .
soluble in warm methylic alcohol and ether . Meli ssi c aci d, C
resembles cerotic acid in appearance,but crystalli ses more readily
and melts at I t i s readily soluble in hot ethyl alcohol,chloro
form,petroleum spiri t
,and carbon disulphide
,but almost insoluble in
warm methyl alcohol and ether . For the properties of these acidsand their derivatives and the method of separating them in a purestate from beeswax
,the papers by Marie should be consulted . The
proportion of crude cerotic acid existing in beeswax in the free stateusually ranges from 1 2 toMyri c in i s the chief constituent of beeswax insoluble in alcohol .
I t i s a solid,wax- like body
,melting at On saponification ,
i tyields a palmitate
,myricyl alcohol
,and a small quanti ty of soap from
an acid of the oleic series . Hence myricin has essentially the con.
stitution of myricyl palmitate .Myricy l a lcoho l , C 3 °H6 I . OH
,may be prepared by heating myricin
or beeswax itself in a closed.
vessel for an hour or 2 with excess ofalcoholic potassium hydroxide
,nearly neutralising the excess with
acetic acid (using phenolphthalei n as an indicator), and precipitatingthe turbid liquid with excess of lead acetate . The precipitate , consisting of a mixture of lead soaps and myricyl alcohol
,i s washed
,
dried,and exhausted with hot ether or petroleum Spirt in a Szombathy
tube . On evaporating the solvent,the wax-alcohol is Obtained in
white glittering crystals,which may be purified by washing with cold
alcohol and recrystallisation from ether . I t may also be prepared ina similar manner from carnaiiba wax .
Myricyl alcohol is a crystalline silky substance,melts at 85° to 86°
to a colourless liquid,and solidifies to a fibrous °mass at about 1
°
lower . It i s insoluble in water , scarcely soluble in cold alcohol , ether ,or benzene
,and but little in cold chloroform ; I t dissolves readily in
boiling alcohol,ether
,chloroform
,benzene
,and petroleum spiri t .
When fused with potassium hydroxide,or heated to 2 20° with potash
lime as long as hydrogen is evolved,i t i s converted into potassium
melissate , KC 3 OH5 902 ,which
,on solution in water and treatment
with an acid,gives meli ssic acid .
Wh i te or B leach ed Wax.—By exposure to moisture , air , and
l ight,beeswax becomes decolouris ed . I t i s usually exposed to sunlight
in thin cakes,but the bleaching is a slow process . In order to expose
as large a surface as possible , Ramboe I has proposed to break the wax1 Chem. Zei t. , 1 89 6 . 20, 1 004 .

obules by emulsifying it with hot water and pouringcold water to which a little oil Of turpentine has been
added . Wax thus subdivided,if previously mixed with bleached wax
,
can be bleached by sunlight in 3 or 4 days . The addition“of bleached
wax to the yellow wax is found to greatly reduce the time required
B eeswaxmay also be bleached by cautious treatment with chromic ornitric acid ; but chlorine cannot be advantageously employed owing tothe formation of chlorinated substitution-products which give rise tohydrochloric acid when the wax is burnt . I t may also be bleached byboiling it with a dilute solution of potassium dichromate and sulphuricacid . The wax thus treated has a greenish colour from the presenceof chromium compounds
,which it holds very persistently
,but which
may be removed by boiling the product one or more times with asolution of oxalic acid . Permanganate bleaching is also employed .
It is not every kind of wax which can be efl’ectually bleached . Thepresence of a small proportion of fatty matter appears to facilitate the
The eff ect of bleaching on the constants of beeswax has been studiedby Buisine
,
I B erg? and others . The aci d value is raised,
' least bynatural
,most by chromic acid bleaching
,to a suffi cient extent to lower
the ratio number and suggest adulteration with stearic acid . Thus ,Buchner has recorded the following numbers for chemically bleachedwax : acid value
,to saponification value , to
ratio number,
to The saponificati on value of bleached wax isalways a few units higher than that of the yellow wax . Berg found thei odine value lowered by the natural and permanganate methods ofbleaching
,but
,curiously
,raised by chromic acid bleaching
,except
in the case of I talian waxes,which had their iodine values lowered in
all cases . D ieterich found the iodine value of bleached wax toBuckner
’s number is slightly lowered by natural and permanganate
bleaching ; chromic acid sometimes raises it considerably, sometimes does not alter it
,or even lowers i t in the case of I tal ian waxes .
Natural and permanganate bleaching either do not affect or slightlyraise the refractive power of the wax ; treatment with chromic acid orany method of bleaching I talian wax lowers the refractive power .Chromic acid frequently raises the m . p . ; other processes lower it a1 B u ll . S oc. Chim 1 890 , 4 , 46 5 , in which de ta iled resu lt s are given of y ellow waxe s b efore and aft er bleach ing by several me thod s ; a lso Campt. rend . , 1 89 1 , 1 1 2 , 7 3 8 .
2Chem. Zei t. , 1 9 02 . 26 , 605 .

246 MODE S OF EXAMINING FATS , OILS , AND WAXE S .
trifle . A Morocco wax bleached by chromic acid had i ts m . p . raisedfrom 64 5
° to Medicus and Wellenste inl found the m . p .
of a wax raised from to by chromic acid bleaching,that of
the mixed free fatty acids being raised from 68° to and their acidvalue from to An ultimate analysis gave C ,.8HSGO 2
or C 2 9Hn 2 for the yellow wax and C 2 4
H4gO , or C 2 5
HSOO 2 for the
bleached wax . They attribute the increased acidity to the splittingup of the acids with formation of acids of lower molecular weight .White wax is not completely soluble in chloroform (D ieterich) .
Ana lys is of Genu ine Beeswax .—The proportion of crude ceroti c
acid in beeswax can be ascertained by titration with standard acid andphenolphthalei n in the usual way
,but
,owing to the very high combin
ing weight of the acid,the operation must be conducted with extreme
care . Hehner2 recommends that alcoholic potassium hydroxideshould be used
,and that i t should be prepared from pure material
and from spiri t which has been redistilled from potassium hydroxide .I t should be about N/3 - that is
,1 c .c . should correspond to to
c .c . of N/ 1 acid . The alkali Should be standardised several timeswith the acid
,and the results should not differ by more than c .c .
of standard alkali for each 1 0 c .c . of acid used . 5 grm . of the waxshould be heated in a flask wi th 50 c .c . of me thylated spiri t which hasbeen redistilled from sodium hydroxide . When the wax is perfectlymelted
,an alcoholic solution of phenolphthalei n is added in nb t too
small an amount . The indicator must not be acid , as is frequentlythe case
,but must preiriously have been rendered pink by addition of
alkali in faint excess . The standard solution of alcoholic alkalii s then added drop by drop
,the liquid being kept well agitated until
the pink colour becomes permanent,when the volume employed
is observed . The combining weight of cerotic acid being 4 1 0 (basedon Brod i e ’s formula , C 2 7H5 4O , . As a matter of fact
,the mean mo
lecular weight of the free acid in beeswax has been found by Hebnerto be a volume of standard alkali corresponding to 1 c .c . ofnormal acid represents grm . of cerotic acid . The percentageof cerotic acid may be found by multiplying the percentage ofpotassium hydroxide required for neutralisation by As thevolume of standard alkali required by 5 grm . of wax amounts to only afew cubic centimetres
,a very finely graduated burette should be
employed .
1 Zei t. Nahr . Genussm. , 1 902 , 5 , 1 09 2 .
2Ana ly s t, 1 88 3 , 8 , 1 6 .

248 MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
responds to grm . of myricin . Hubl found 20 samples of yellowwax to require from to of potassium hydroxide for the saponification of the myricin
,which figures correspond to proportions of that
substance varying from 88 to These results ful ly confirm thoseof Hehner
, who found in sixteen samples of yellow English wax proportions of saponifiable substance expressed as myricin ranging from
to the average being and in 24 samples of commercial bleached wax proportions ranging from to the averagebeingWhen estimated volumetrically by the above method
,the free acid
expressed as cerotic acid and the esters expressed as myricin togetherusually amount to somewhat more than the average being
,ac
cording to Hehner, It is evident
,therefore
,that wax requires
more alkali for saponification than would be required for a mixture ofpure cerotic acid and myricin .
The results above recorded prove that genuine beeswax i s of approximately constant composition . Hebner’s experiments Show thatthe proportion the cerotic acid bears to the myricin in English beeswax (unbleached) averages 1 : while Hii bl finds ‘
ratios varyingfrom 1 : to 1 in English bleached wax Hehner found anaverage ratio of 1 :Adu ltera t ions of Beeswax.
—Commercial beeswax is liable to contain a number of adulterants
,among which the following are recorded
Water ; mineral matters, as kaolin , gypsum . barium sulphate , and yellow ochre ; sulphur ; starch and flour ; resinous substances , as colophony ,galipot
,and burgundy pitch ; fatty substances , as stearic acid , stearin ,
Japan wax,and tallow ; paraffin and ozokerite, ; and vegetable waxes , as
carnaiiba wax . Spermaceti i s also said to have been used.
Water has been met with in beeswax to the extent of being purposely introduced . I t may be detected and estimated as describedunder “Lard .
”
Mineral matters may be detected and estimated by igniting thewax . They will also remain insoluble on dissolving the sample inturpentine
,chloroform
,or benzene . As much as 1 7% of yellow
ochre has been found in unbleached beeswax .
S tarch and flo ur will be left undissolved on treating the wax withwarm turpentine . The liquid may be fil tered , the residue washed witha li ttle ether
,and examined under the microscope with solution of
i odine . 60% of starch has been met with . Small quantities of starch

BEE SWAX .
nine wax that has been rolled or pressed,the
being dusted over wi th flour to prevent the wax from
Sulphur has been found as an adulterant of unbleached wax . I t
d by boiling the sample with a weak solution of soda,
acetate to the cooled liqu id,when a black or brown
if sulphur be present .of the above-named impuri ties or ad
mixtures shoul d be melted over water , and the molten wax fil teredthrough paper and dried in the water-oven before proceeding with the
As a useful prelimi nary test,the sample may be dissolved in chloro
form,in which genuine yellow wax is completely soluble . Incomplete
solubili ty would indicate the presence of parafli n,ceresin
,carnai iba wax
or wool wax (D ieterich) . White wax,however
,even when genuine
,i s
not completely soluble in chloroform .
J . Werder (Chem. Zei t .
,1 898, 2 2
, 38 , 59) finds that the Zeissbutyro
-refractometer may advantageously be employed in the examination of di fferent kinds of wax
,especially when the amount of material
at disposal is very limited,and that the indications obtained with it are
quite as valuable as in the case of oils and fats . Owing to the h igh m . p .
of the wax,i t i s necessary to work at a higher temperature than
usual,preferably 66° to and then to reduce the results to the
normal temperature,
AS shown in the annexed table,the figures
given by genuine beeswax vary from 426° to 4 the great majori ty
of specimens falling between 44° and 4 and i t seems to make little orno difference to the refractive power whether they are tested before orafter bleaching . samples 1 9 to 24 had previously been examinedchemically
,and had been rejected on the ground of their abnormal
acid and ester values,which were as follows
Number of sample Acid Value

250 MODES OF EXAMINING FATS,OILS
,AND WAXE S .
No . 24 is a product called Glanzwachs , obtained by adding someof the mixture of stearic and palmitic acids as used in the manufactureof stearin candles (No . 28) to a genuine wax , thi s being a form ofadulteration commonly employed in Switzerland .
Refract ive Power of Different Kinds of Wax.
Sample
1 . B leached , from2 . B leached ,
from T urkey3 . Bleached , from Mo ldavia4 . Yellow, from Egypt5 . Yellow,
from Monte Christo . f
6 . Ye llow,from France .
7 . Ye llow,from Savoy
8 . Ye llow ,from California .
9 . Ye llow,from North Africa .
1 0. Yellow,from M assowah . .
1 1 . Ye llow ,from I taly
1 2 . Ye llow ,from I taly rent samples
1 3 . Yellow,from I taly
1 4 . Ye llow,from M exico
1 5 . Ye llow,from M exico
1 6 . Ye llow ,from Syria
1 7 . Ye llow,from Casablanca .
1 8 . Ye llow ,from Smyrna
1 9 . B leached , in ch ips (professedly20 . Wh ite church candles (pro fessedly genu ine)2 1 . Wh ite church candles (profe ssedly genu ine)2 2 . Wh ite church candles (professedly genu ine)23 . Ye llow wax, source unknown24 . Wax adu lterated with No . 28
25 . Paraffi n
26 . Ceresin .
27 . Tallow28 . S tearin candle material .
29 . Carnauba wax . .
30. Japan wax
d ifferent samples .
The sp . gr . of beeswax is a useful indication of the presence offoreign admixtures . Great discrepancies occur in the recorded sp . gr .of possible adulterants of beeswax
,as determined by various Observers ,
the differences being probably due to the faulty methods of observation .
The subject has been investigated by W . Chattaway in Allen ’slaboratory
,with the following results :
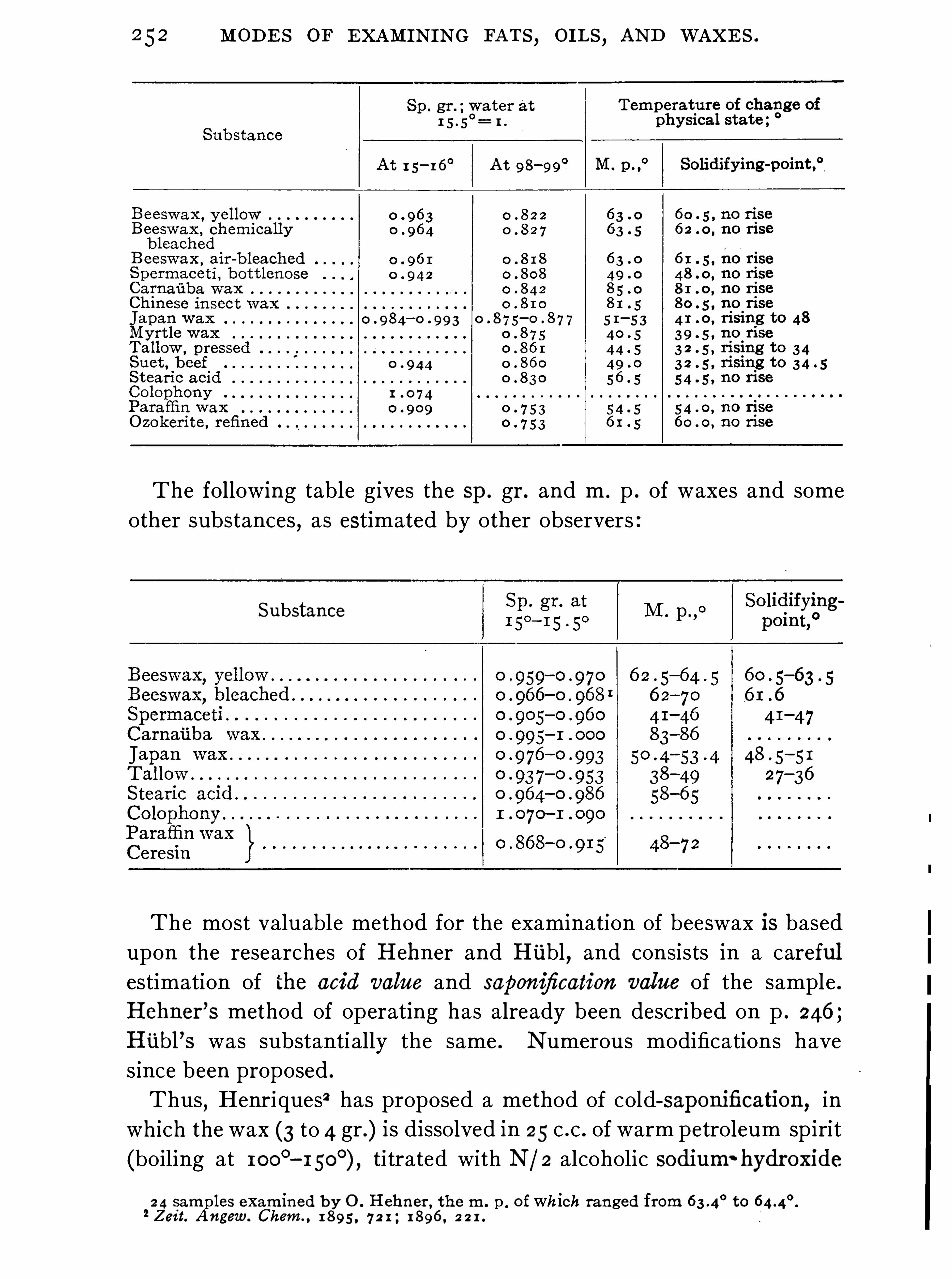
MODE S OF EXAMINING FATS , OILS , AND WAXE S .
0 0 0 0 0 0 0 0 0 0 0 0
0 . 9 84—0 . 9 9 3 0
0 0 0 0 0 0 0 0 0
The following table gives the sp . gr . and m . p . of waxes and someother substances
,as estimated by other observers
Substance
O O O O O O O O O O O O O O O O O O O O
0 0 0 0 0 0 0 0 0 0 0 0
O O O O O O O O O O O O O O O O O O O O O
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0
O O O O O O O O O O O O O O O O O O O O O O
The most valuable method for the examination of beeswax is basedupon the researches of Hebner and Hubl
,and consists in a careful
estimation of the acid value and saponificati on value of the sample .Hehner’s method of operating has already been described on p . 246 ;
Hi'
Ibl’s was substanti ally the same . Numerous modifications have
since been proposed .
Thus,Henriques2 has proposed a method of cold-saponification, in
which the wax (3 to 4 gr .) i s dissolved in 25 c .c . of warm petroleum spiri t(boiling at 1 00° ti trated with N/ 2 alcoholic sodium~ hydroxide
24 samples exam ined by O . Hehner , th e m . p . o f which ranged from to2Zei t. A ngew. Chem. , 1 89 5 , 7 2 1 ; 1 89 6 , 2 2 1 .

BEE SWAX . 2 53
made with 96% alcohol for estimating the acid value , thenmixedwith N 1 alcoholic sodium hydroxide and allowed to stand for 24 hoursin the cold , the saponification value being estimated by titrating theexcess of alkali . There appears to be no advantage in this process ,and it i s slower than the ordinary method . The acid values estimated by it are lower than those estimated by Hehner ’s method(D ieteri ch , Buchner) . Cohn 1 finds 3 hours ’ boiling with alcoholicpotassium hydroxide under a reflux condenser necessary to ensurecomplete saponification ,
but others have shown 1 hour to be suffi cientif strong enough alcohol (96 to 98 be used . Buchner? has proposed touse a Szombathy extractor as reflux condenser , the concentration ofthe alkali resulting from its use helping the saponification . E ichorn3has suggested using amyl alcohol instead of ordinary alcohol
,in order
to carry out the neutralisation and saponification at a higher temperature . B erg4 states that different waxes require diff erent times for saponification ; German seldom less than 2 hours , usually 3 to 3 1 2 hours ;Morocco
,about 5 hours ; East Af rican , up to 6 hours ; Chinese and
Tonking,up to 8 hours . He thinks every wax contains lactonic
anhydrides which are difli cult to saponify , differing from Buchner , whobelieves these substances are of only rare occurrences . In the revi ser ’sexperience , the following method of operating usually gives goodresults :8 grm . of the wax are warmed with 70 c .c . of neutralised rectifiedalcohol until melted
,then mixed with neutralised phenolphthalei n
solution and carefully titrated with N 2 alcoholic potassium hy
droxide . This gives the acid value . For ascertaining the saponifi
cation value, 5 grm . of wax are boiled under a reflux condenser
for at least 1 hours with 25 c .c . of N 2 alcoholic potassiumhydroxide , made with alcohol of 95 to 96% strength , and thentitrated with N/ 2 hydrochloric acid and phenolphthalei n in theusual way .
The difference between the acid and saponification values wastermed by Hiibl the “ ester value,” and he found the rati o, 2
5
0
2
1332532,genuine beeswax ranged from to Adulteration with unsaponifiable matters (parafli n ,
ceresin) lowers the saponification value without disturbing this ratio ; acid substances (stearic acid , resin) , raise the
i
1 Zei t. afientl . Chem. , 1 904 , 1 0, 404 ,
2 Chem. Zei t. . 1 9 0 5 , 29 , 3 2 .
8 Zei t. ana l . Chem. , 1 9 00. 3 9 , 640.
‘Chem. Zei t. , 1 9 07 , 3 1 , 5 3 7 .
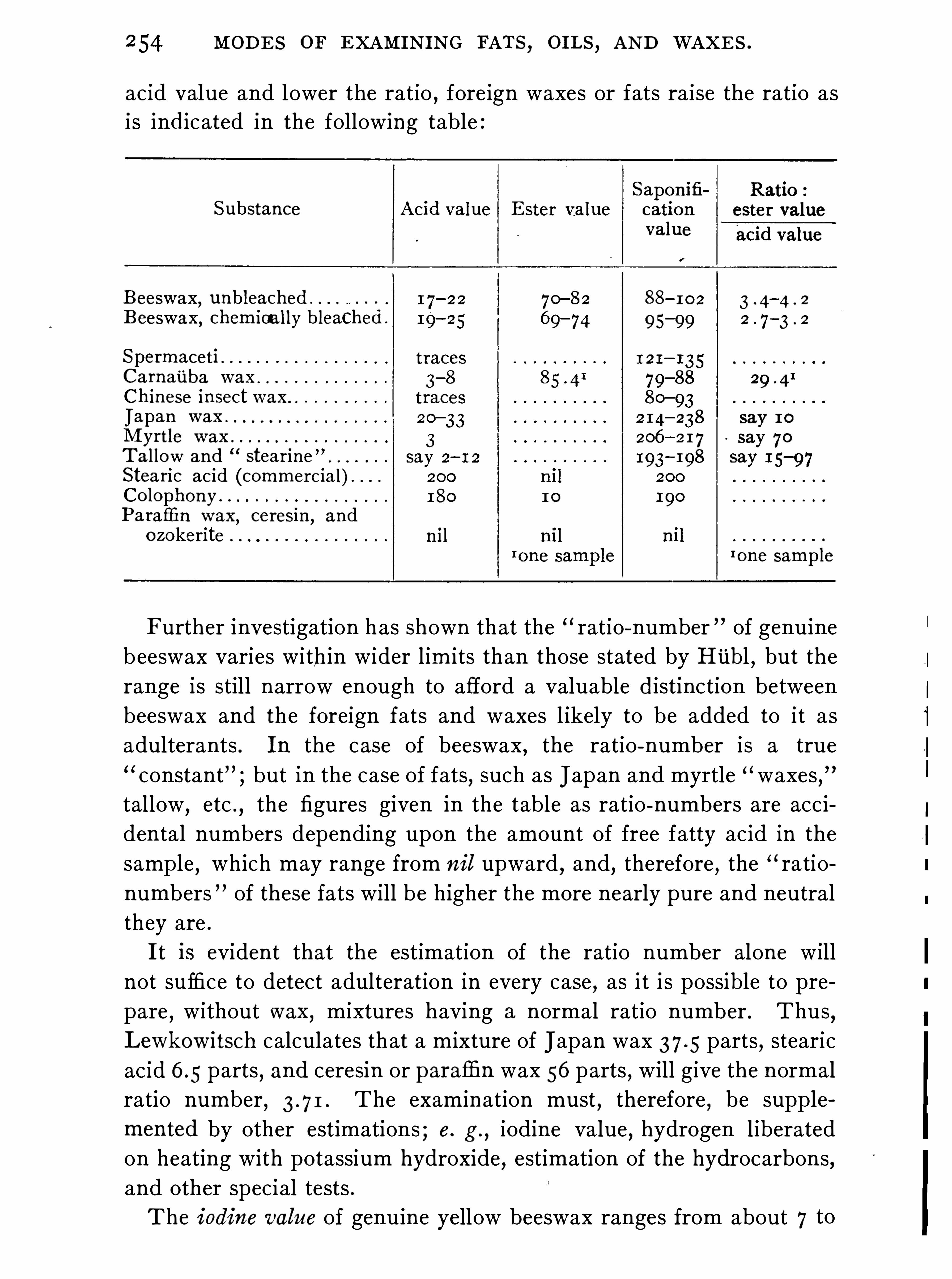
MODE S OF EXAMINING FATS,OILS , AND WAXE S .
acid value and lower the ratio , foreign waxes or fats raise the ratio asi s indicated in the following table :
S ubstance
B eeswax,unbleached ” 88—1 02 3 . 4
—4 2
B eeswax , chem ica lly bleached. 95—99 2 7
—3 2
Carnai‘
Iba wax
Ch inese insect waxJapan wax . .
Myrt le wax
T allow and stearineS tearic ac id (commercial) .
Co lophony .
Parafli n wax,ceresin
,and
ozokeriteI one sample
Further investigation has shown that the “ ratio-number of genuinebeeswax varies within wider lim i ts than those stated by Hubl , but therange is still narrow enough to afford a valuable distinction betweenbeeswax and the foreign fats and waxes likely to be added to it asadulterants . In the case of beeswax
,the ratio-number is a true
“ constant” ; but in the case of fats, such as Japan and myrtle “waxes ,”tallow
,etc .
,the figures given in the table as ratio-numbers are acci
dental numbers depending upon the amount of free fatty acid in thesample
,which may range from ni l upward
,and
,therefore
,the “ ratio
numbers ” of these fats will be higher the more nearly pure and neutralthey are .I t is evident that the estimation of the ratio number alone willnot suffice to detect adulteration in every case
,as i t i s possible to pre
pare,without wax
,mixtures having a normal ratio number . Thus ,
Lewkowitsch calculates that a m ixture of Japan wax parts , stearicacid 6 .5 parts , and ceresin or parafli n wax 56 parts , will give the normalratio number
,The examination must
,therefore , be supple
mented by other estimations ; e . g .
,iodine value
,hydrogen liberated
on heating with potassium hydroxide,estimation of the hydrocarbons ,
and other special tests .The i odine value of genuine yellow beeswax ranges from about 7 to

256 MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
Buchner regards a wax as genu i ne In this respect when i t shows nodeposit of stearic acid after I to 2 hours .A quantitative modification of the test (making i t applicable torosin as well as fatty acid) has since been proposed by Buchner :15 grm . of the sample are treated with 1 00 c .c . of neutrali sed 80% alcohol
,and the flask and contents are weighed . The alcohol i s gently
boiled for 5 minutes , with frequent agitation . The flask and contentsare then cooled
,and the solution made up to its original weight by the
addition of 80% alcohol , well mixed , corked , and allowed to stand for1 2 hours (Berg). The solution is then filtered through a ribbed fil ter ,and 50 c .c . of the filtrate are ti trated with N/ 1 0 potassium hydroxidewith phenolphthalei n . The figures thus obtained are known asBuchner numbers . Some resul ts with waxes
,fats
,and mixtures are
given in the following table :
Substance
B eeswax, yellow
Beeswax,wh ite
Palm wax
Carnai iba wax
Japan wax
S tearin (from tallow)Co lophony .
S tearic acidM ixtures giving no rmal rat io numbers
1 . S tearic acid,stearin and ceresin
2 . S tearic acid,Japan wax and ceres in
3 . Ros in, stearin and ceresinGenuine beeswax+ 25% mixture NO . 1 .
Genu ine beeswax+ 50% m ixture No . 1 .
According to Bohrisch and Richter? th e Buchner’s number ofgenuIne beeswax may range from 2 to 6 . These authors , who fullyexamined 73 samples of beeswax from different parts of Germany ,found 38 samples adulterated ; of these , 34 contained paraffi n andceresin and 4 contained stearic acid , tallow or carnatiba wax .
Colophony or rosin,like stearic acid
,if added to beeswax increases
the acid value ; and although the acid value of rosin i s somewhat lowerthan that of stearic acid
,i ts greater solubili ty in alcohol causes i t to
increase the Buchner number in greater proportion . Colophony is1 Chem. Zei t. , 1 89 5 , 1 9 , 1 4 2 2 .
2 P harm. Centralh . . 1 9 06 .4 7 , 2 0 1 . etc .

BEE SWAX .
easily detected in beeswax by means of the Li ebermann-S torch reacti on ;
i t may be estimated by applying Twichell ’s process to the mixture ofrosin and fatty acids Obtained by extracting the adulterated samplewith boiling 80% alcohol and fil tering when cold .
According to WeinwurmI2 or 3% of ceresin or paraffi n , or 5% of
rosin,may be detected as follows : 5 grm . of filtered wax are saponified
in 25 c .c . of N/ 2 alkali and the alcohol removed . 20 c .c . of glycerolare run in
,the whole warmed in the water-bath till solution is eff ected
,
and 1 00 c .c . of boiling water added . Pure wax gives a clear,trans
parent,or translucent solution
,through which ordinary printed matter
may be read with ease . 5% of ceresin or rosin yields a cloudy liquid ,and the print i s no longer legible ; 8% of ceresin causes a decided precipitate . If the solution be clear
,or
,if i t be opaque
,2% of
ceresin is added to another sample of the wax,and the saponification
repeated,when from the appearance of the soap solution the presence
or absence of either impurity may be deduced .
R . Henriques2 reports favorably on the above process , but simplifiesi t by applying the Leffmann-B eam alkali-glycerol method for saponifyIng . A piece of wax about the size of a pea is boiled in a test-tube for3 or 4 minutes with 5 c .c . of alkali glycerol (see p . The solution
,
which i s at first quite clear,becomes gradually Cloudy . After boiling
for about the time mentioned,the oil collects in a layer and the under
lying fluid becomes clear . The bubbles of the boil ing mass also nowbecome smaller and the glycerol commences to distil . As soon as thispoint is reached the heating is discontinued . The fluid is now pouredinto another test-tube
,in order to separate i t from the unsaponified
portion ; an equal weight of hot water is added , and the liquid boiledand allowed to cool . In the case of pure wax the solution will be eitherquite clear and transparent
,or at any rate suffi ciently translucent to
allow of large printed matter being read through it,as described by
Weinwurm . Should,however
,on the contrary
,as much as 5% of
foreign hydrocarbons be present,the fluid will be quite opaque . Wi th
an admixture of only 3% of ceresin or paraffi n ,the indication is un
certain , and the further treatment recommended by Weinwurm tomeet such cases should be followed .
Lewkowitsch3 states that he can recommend Wéinwurm ’s test for
pure beeswax , but that turbidity of the solution does not necessarily1 Analyst. 1 89 7 , 22 , 242 .
2 I bid , 1 89 7 , 2 2 , 2 9 2 .
2 Oils ; Fa t s and Waxes , I I , 7 7 2 .
Vol . I I .
- I 7

2 58 MODES OF EXAMINING FATS , OILS,AND WAXE S .
prove the presence of ceresin or parafi n wax , since i t is also producedby carnai iba wax and insect wax .Parafiin, ceresin,
and ozokeri te are the only adulterants of beeswaxwhich tend to reduce in a notable degree the saponification value .They also reduce the sp . gr . in a marked manner
,bu t thi s indication
has li ttle more than a quali tative value . In a sample consisting solelyof beeswax and hydrocarbon wax the proportion of the former may bededuced with considerable accuracy from the results of the saponification
,each of potassium hydroxide required representing 3%
of beeswax in the sample .Estimati on of Fatty A lcohols and of Parafiin and Ceresin.
—Anadulteration of beeswax with less than 6% of ceresin or parafi n cannotbe detected with certainty by any of the ordinary methods , because therelations between the free fatty acid and saponifiable and unsaponi
fiable matters in genuine beeswax vary within somewhat widel imits .Werder I has proposed to saponi fy 2 grm of the sample of waxwith alcoholi c potass ium hydroxide
,dry on sand
,extract the dry
mixture of soap,sand
,etc .
,with pure dry ether in a Soxhlet extractor and
Weigh the mixture of alcohols and hydrocarbons . B ut as the residueof unsaponifiable matter obtained from 20 samples of genuine beeswax in th is manner ranged from to i t i s evident thatat least 4 .5% of paraffin wax or wax alcohol s could be added to somesamples without detection .
A preferable method is a direct estimation of the hydrocarbonspresent
,and a method for doing this has been worked out by A . and P .
Buisine? based upon an Observation of Dumas and S tas . The wax issaponified with potassium hydroxide and heated with potash lime , bywhich treatment the higher alcohols are converted into their corresponding fatty acids with evolution of hydrogen , the volume of which servesas a measure of their amount , while the hydrocarbons present are leftunattacked and can be extracted from the residue with solvents . Thefollowing modification of the process has b een described by Mangold :32 to 1 0 grm . of the wax are melted in a porcelain basin and intimatelymixed
,by stirring
,with an equal weight of finely powdered caustic
potash . The saponified mass , when cold , i s powdered in a mortar ,and intimately mixed with 3 grm . of a mixture ( 1 part potassium1 Chem . Zei t. , 24 , 9 6 7 .
2 B ull . S oc. Chim . , 1 89 0 , 3 , 5 6 7 .
3 Chem. Zei t. . 1 89 1 , 1 5 , 7 9 9 .

260 MODE S OF EXAMINING FATS,OI LS , AND WAXE S .
If a measurement of the hydrogen is not required,the powdered
mixture of saponified wax and alkali lime may be transferred to asimple boiling tube , which is closed by a rubber stopper and a shortpiece of bent glass tube
,supported vertically in a bath of oil or melted
carnai iba wax , and heated to 2 50° until no more gas is evolved ; the
temperature i s then raised for a short time to Evolution of gasi s ascertained by attaching a short length of rubber tubing to the glasstube and immersing the free end in water
,but the rubber tube must not
be left permanently attached,otherwise water may be sucked back and
cause an explosion . When gas-evolution has ceased,the tube and
contents are allowed to cool somewhat and the contents,while still
hot , are removed as far as possible to a basin by means of a pointedglass rod . If allowed to become quite cold
,the mixture sets hard and
cannot be removed . The bottom of the boiling tube is finally brokenout and what adheres to the Sides scraped off as far as possible . Themixture is then placed in a paper extraction thimble with alternatelayers of powdered pumice
,the tube and basin are rinsed out with dry
ether , which is poured through the thimble in the Soxhlet ’s tube , and thecontents are then thoroughly extracted with dry ether for 4 or 5 hours .The (turbid) ethereal extract will contain a li ttle soap . I t i s firstshaken in a separating funnel wi th hydrochloric acid to decomposecalcium soaps
,and after drawing this off and washing with water i t
i s Shaken with dilute p otassium hydroxide solution containing alittle alcohol in order to remove fatty acids . After again washing withwater , the ether is distilled Off and the hydrocarbons weighed .
The proportion of hydrocarbons which has been found in genuinebeeswax by Buisine
,Mangold
,Kebler, and Ahrens and HettI
has ranged from to 1 7 .5 1 5 samples of apparently genuinecommercial yellow beeswax examined by Archbutt were found tocontain from 1 2 to of hydrocarbons ; 6 obviousl y adul teratedsamples contained from to 5 The process i s quite easy towork with a little practice
,but is evidently not capable of detecting
with certainty less than about 6% of hydrocarbons , owing to the comparative wide variation in the amount obtained from genuine samples .S permaceti i s not usually an adulterant of beeswax , but occasionallyits substi tution will be profitable and may be practi sed . I t i s the only °
adulterant which would cause the sample to Show less free acid , andyet require an increased proportion of alkali for its saponification ,
1 Zei t. éfientl. Chem 5 , 9 1 .

BEE SWAX .
at the same time yielding glycerol and reducing the sp . gr . and m . p .
In the absence of carnauba wax , a direct indication of the presence andproportion of spermaceti may be obtained from an estimation of them . p . of the higher alcohols of the sample .From an inspection of the table on page 2 59 , i t appears that carnauba
wax requires for complete saponification a proportion of alkali not verydifferent from that required by beeswax
,but i s distinguished from the
latter by the smaller (but very variable) proportion of alkali requiredby the free acid . An admixture of carnaiiba wax wlll be furtherindicated by the increased sp . gr . and higher m . p . of the sample .Another proof of the presence of carnai iba wax is obtainable byremoving free acid by alcohol and alcoholic potassium hydroxide
,
saponifying the separated neutral wax,precipitating the solution with
lead acetate,and exhausting the precipitate with petroleum spiri t
,
and decomposing the lead soap with Hot hydrochloric acid . B eeswax,when thus treated
,yields a product which is chiefly palmitic acid (m . p .
while the product similarly Obtained from carnaiiba wax islargely cerotic acid (m . p . ,
Among the hydrocarbons isolated from the wax may be foundunchanged cholesterol
,if the sample had been adulterated with wool
wax , since Lewkowi tsch I has shown that cholesterol i s practicallyunchanged by heating for 2 hours with soda-lime at 2 and that ofthe total alcohols of wool wax 80% were recovered unchanged . In
presence of wool wax,cholesterol may also be looked for in the un
saponifiable matter . Some results of examination of the unsaponifiablematter from waxes are given in the following table (Archbutt andDeeley)
Sp . gr. at
Iodine valueSaponification value of mixed acetates
1 J . S oc. Chem. Ind . , 1 89 6. 1 5 , 1 4 .

MODE S OF EXAMINING FATS , OI LS , AND WAXE S .
For the detection of artificial colouring matters, Lemaire I recommendsthe following tests :
‘
A small fragment of the wax is dissolved in chloroform , and 2 or3 drops of hydrochloric acid are added to the solution . Theproduction of a rose-red colour indicates artificial colouring matter .Another portion i s saponified by boiling with caustic soda solution ,then treated hot with excess of hydrochloric acid . If a fugitive rosered colour be obtained
,which turns green on adding excess of ammoni a ,
the wax is artificially coloured . Another piece of the wax i s melted ina capsule with saturated boric acid solution ; on evaporating to drynessthe residue acquires a reddish colour with wax containing added colouring matter .D i eterich2 has shown that the analytical values of beeswax from
combsfive years old diff ered very little from those of new beeswax ; thechief differences were in the colOur
,and the sp . gr . and m . p . of the
waxes . Thus,the fresh wax was nearly colourless
,had the highest
sp . gr . and the highest m . p . (65° to The Old wax was dark
brown,had the lowest sp . gr . and lowest m . p . (63
° to 63 .
Hehner”sMethodfor the Analysi s of Complex Candle-mixtures .
3—Theestimation of the percentage of beeswax in so-called wax candl es hasassumed importance since
,in 1 904, the College of R ites of the-Roman
Catholic Church prescrib ed the use of candl es containing definitepercentages of beeswax for certain ritual and altar purposes . Suchcandl es may contain
,b esides b eeswax
, parafli n (ceresine , ozok erite),stearine (commercial stearic acid), and spermaceti.Hebner’ s method for the analysis of such mixtures depends primarilyupon the approximate constancy of composition of normal beeswax.Thus
,24 samples of good commercial b leached wax were found by
Hehner to contain free acid,calculated as cerotic acid4 ranging from
1 5 5 to 1 7 6% and averaging 1 6 6% or almost exactly one-sixth of thewax
,and saponifiable esters , calculated as myricyl palmitate , ranging
from 85 . 2 to and averaging or very nearly timesthe acidity expressed as above . The free acid extracted from a smallnumber of samples was - found to have a molecular weight of 407(C 2 7H5 40 2
Upon the basi s of these figures,the composition of a mixture con
S oc. Chem. Ind . , 1 904 , 2 3 , 840.
2Chem. Zei t. , 1 9 0 7 , 3 1 , 9 8 7 .
2From a paper by O t to Hebner read before th e S oc ie ty of Pubic Analy st s .
4 Brod ie ’
s formu la , Cz7H54Oz.

264 MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
funnel is placed in a large vessel of water at about 35 ° and left for anhour or two for the ether to rise . The aqueous portion is then drawnoff into another funnel and shak en twice more with ether
,after which
it should b e clear or nearly so. Alcohol must not b e added at thisstage to promote separation of the ether
,as partial saponification of the
esters would occur and lead to inaccurate results . The etherealsolution is rej ected as it i s separated
,and not washed with water.
To the soap solution is now added hydrochloric acid in excess,by
which the soap is decomposed and a layer of ether containing thel ib erated fatty acid rises . This is allowed to separate thoroughly
,
and the lower aqueous layer is drawn Off and rej ected. The fattyether
,having b een washed carefully with hot water several times
,i s
evaporated,and the residual fatty acids dr ied and accurately weighed ;
they are then very carefully titrated with alcoholic potassium hydroxidesolution
,which must not b e stronger than N/3 , owing to the high
molecular weight of the cerotic acid. Care must also b e tak en thatthe alcohol in which the acid is dissolved b efore titration is exactlyneutral ised and that the alk aline solution is k ept free from carbonateand accurately standardised.
The only acids which can compose the material titrated are the freeacids of the wax
,with a mean molecular weight of about 407 , and the
“ stearine,
” with a molecular weight of 27 2 j : I . The molecular weightgives
,therefore
,the proportion of crude cerotic acid in the mixture.
In a separate portion of the original wax mixture the total acidityis now estimated as usual
,and also the ester value . The molecular
weight of the free acids b eing known,the actual percentage of free
acids in the mixture can b e calculated,and as the percentage of real
wax acid of molecular weight 407 in these free acids is also k nown ,the percentage of free wax acid in the mixture can also b e calculated.
Multiplying this by 6,the amount of b eeswax in the mixture is arrived
at ; and sub tracting it from the total acid, the percentage of “ stearineis ob tained. Multiplying the wax acid by 5 3 , the ester portion of theb eeswax
,expressed in terms of myricine, results ; this sub tracted from
the total saponifiable matter gives the measure of any spermaceti thatmay have been present. A calculation reducing this remainder fromterms of myricine , with an equivalent of 676 , into terms of spermaceti,with an equivalent of about 480, results in a close approximation ofthe actual percentage of spermaceti. This remainder, if any , i spai
‘afli n. The presence of paraffi n
,if more than 4 or is always

BEE SWAX . 265
seen during the saponification for the estimation of the ester value .The paraffi n is the only material estimated by diff erence.Of other acid substances which would interfere with the processthere might be present traces of mineral or oxalic acid from ' thebleaching processes. These can easily be tested for and
,if necessary
,
removed by melting some of the sub stance with boiling water andtesting the aqueous solution . Resin acids do not mix to any practicable amount with candle materials. The bee stops all crevices of thehive with various resins collected from trees and buds
,called propoli s .
All honeycomb is more or less contaminated with propolis,but the
latter separates from the molten wax and is not an ingredient ofcommercial wax . Carnauba wax has an exceedingly small acidvalue
,which need not be tak en into consideration .
The following results of analyses of known mixtures Show the degreeof accuracy ob tainable :
0 0 0 0 0
When the candle mixture is free from spermaceti the total saponifiable matter
,calcul ated as myricine , should be 5 3 times the amount
of wax acid found. How nearly this is the case is shown by thefollowing analyses of candle material acknowledged to be free fromspermaceti :
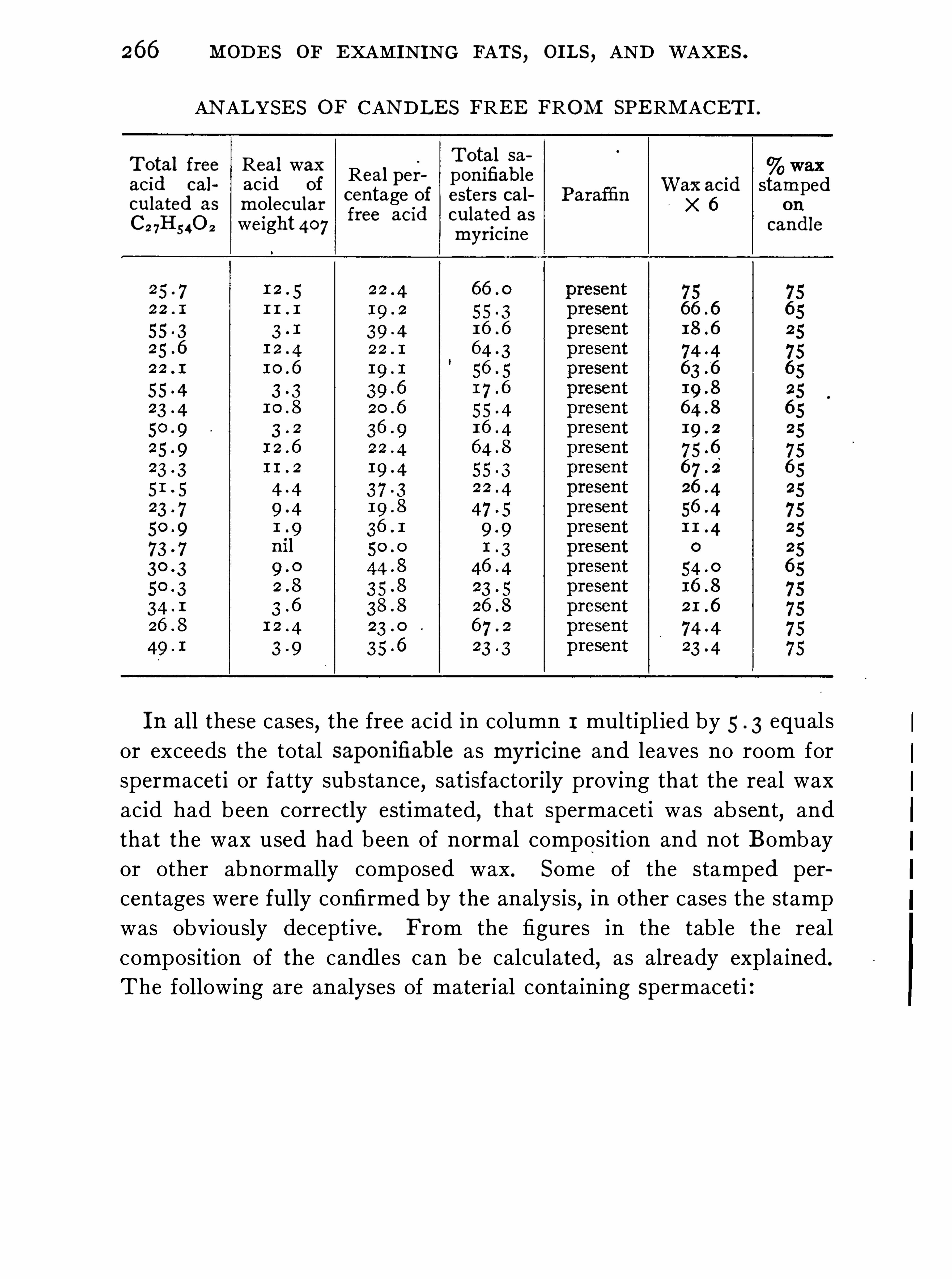
266 MODE S OF EXAMINING FATS , OILS , AND WAXES .
ANALYSES OF CANDLES FREE FROM SPERMACETI .
T otal saReal per ponifiable
centage of esters cal Paraffi nfree acid culated as
myricine
Total free Real waxacid cal acid of
culated as molecular
C 2 7H5402 we ight 407Wax acid stamped
candle
In all these cases,the free acid in column I multipl ied by 5 . 3 equals
or exceeds the total saponifiable as myricine and leaves no room forspermaceti or fatty sub stance
,satisfactorily proving that the real wax
acid had been correctly estimated,that spermaceti was ab sen t
,and
that the wax used had been of normal composition and not Bombayor other abnormally composed wax . Some of the stamped percentages were fully confirmed by the analysis
,in other cases the stamp
was obviously deceptive. From the figures in the table the realcomposition of the candles can be calculated
,as already explained.
The following are analyses of material containing Spermaceti :

268 MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
material from the alcohol is again effected. Once more oxidationwith chromic acid in glacial acetic acid i s carried out
,and practical ly
the whole of the wax alcohol will now have been converted into itscorresponding fatty acid . By treatment with alcoholic potassiumhydroxide , dilution of the soap solution and shak ing out with ether, aseparation of the paraffi n is effected, while the fatty acid is separated fromthe soap solution and its molecular weight ascertained. An insightinto both the acid and the wax-alcoholic portions of the sub stance isthus ob tained
,and any parafli n present is separated and estimated.
The same process is applicable to more complex mixtures ; but thereare many practical difli culties caused by the sparing solub il i ty ofmyricyl alcohol in ether and the fact that the soaps of the higheracids are so l i ttle soluble in water and form such very viscid solutions .Further investigation of this process is proceeding in Hebner’s laboratory.
Ind ian Beeswax (Ghedda Wax).
Ch inese Beeswax.
This wax,though of good quali ty and colour
,yields analytical
values which diff er very materi ally from those obtained with Europeanwaxes . I t i s softer and more plastic than normal beeswax . The acidvalues are very low
,and the ester values high
,with the resul t that
the ratio-numbers range from to the mean being about 1 2 .
According to Hooper ,I Ghedda wax i s derived from three species of
bees , Apis dorsata , A . indi ca,and A . florea , but chiefly from A .
dorsata . The analytical characters of the waxes are Shown in thefollowing table
A. florea
( 5 samples) AV .
1 I nd i an Agri c. Ledger , 1 9 04. 7 3—1 00 ; S oc. Chem. Ind . , 1 9 04 . 2 3 , 8 2 8 .

CHINESE BEESWAX .
Buchner I i s of opinion that Ghedda wax is a true beeswax , differingfrom the European kind quanti tatively but not qualitatively . Thus
,
he obtained by analysis of a sample,cerotic acid
,palmitic acid
,
melissyl alcohol,calculated from the hydrogen evolved in
Buisine’s process
,hydrocarbons
,Hooper says Indian
wax i s rarely adulterated,and as there is a large quantity of i t pro
duced,analysts must be on their guard against mistaking specimens of
this wax for adulterated beeswax .
Buchner2 obtained the following results by examination of severalsamples of Indian and Chinese beeswax .
0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0
Hooper (loc. ci t. above) also describes the wax produced by theDammar or Kota bees
,Melipona (Tri gona) Species . These very small
stingless insects produce a sticky,dark coloured wax
,having a m . p . of
70 . acid value,
saponification value , ester value,
ratio number, 4 .3 ; and iodine value , The product more nearly
resembles the propolis of honey bees than true wax,from which i t
differs largely in chemical and physical characters .Very similar to Indian beeswax from A . dorsata i s Annamese bees
wax , which has been examined by B ellier .3 The commercial wax i s
grayish-yellow,not homogeneous
,and appears to have been kneaded
by hand into prismatic cakes which were found to contain water ,insoluble in benzene
,and of ash . After melting and
straining,i t resembles European beeswax in general appearance , but its
chemical and physical characters are similar to those of the Indian wax ,as shown below .
1 Chem. Zei t. , 1 0 5 , 29 , 3 2 . and 1 906 , 30, 5 28 .
2Zei t. 6 entl. hem. , 3 , 5 7 0 .
3 A nn. h im . anal . appl . , 1 9 06 . 1 1 , 3 66 .

2 70 MODE S OF EX AMINING FATS,OILS
,AND WAXE S .
M .pAcid value'
.
Saponification valueEster value .
Rat io number .
Iodine value .
Hydrogen liberated at 250° by potassa
lime , per grm . of wax c .c. at 0° and 760 mm .
Hydrocarbons 1 0 5
CARNAUBA WAX . CARNAHUBA WAX .
(See pp . 73 , 26 1 and This is a very hard,sulphur-yellow
or yellowish-green substance,which coats the leaves of a palm
,
Coperni cia cerifera , the carnatiba tree of B razil . The leaves aredetached
,beaten
, and the dust,amounting to about 50 gr . per
leaf,collected and melted into a mass . The brittle
,lustrous wax thus
obtained has a sp . gr . of melts at 84° to and dissolves inalcohol and boiling ether . On ignition
,i t leaves a small quanti ty of
ash,which often contains iron oxide .
Carnatiba wax has a very complex composition . I t has beeninvestigated by Berard
,S tory-Maskelyne
,Piverling , and very thor
oughly by S tii rck e , I who found it to consist mainl y of myricyl cerotate .In the alcoholic extract Of t he wax , Berard found free cerotic acid ,since confirmed by Hehner and Hubl ; S tory-Maskelyne and S tii rck e ,in the same solution
,found free myricyl alcohol . S tiircke Obtained
from carnaiiba wax the following substances : a crystalline parafiinoidhydrocarbon melting at about ceryl alcohol, C 2 7H 5 5OH ,
a crystalline substance melting at these two fractions did not exceed 1 1 / 2
to 2 Myri cyl alcohol was found to the extent of about45 a dihydri c alcohol
,melting at and converted on
heating wi th soda-lime into an acid melting at and having thecomposition , an aci d of the formula C 2 3
H4 7 . COOH,
melting at 7 2 . i someric with lignoceric acid ; ceroti c acid, the chiefacid of carnauba wax
,melting at or an acid i someric therewith ; ‘a
hydroxyacid of the formula yielding onheating with soda lime the acid C , 9
H3 8 melting at
Allen and Thomson2 obtained and Archbutt ofunsaponifiable matter (alcohols , etc .) fromcarnati ba wax . Lewko
1 Annalen , 223 , 2 8 3 .
2Chem. News . 1 88 1 . 43 , 2 6 7 .

2 72 MODE S OF EXAMINING FATS , OILS,AND WAXES .
E . Valenta has found carnauba wax in a number of commercialceresins and parafii ns which were characterised by their high m . p .
and great hardness . I t i s employed to impart these properties and togive a peculiar lustre to the wax . Valenta gives the following -figuresshowing the influence of carnauba wax
,melting at 8 on the m . p . of
mixtures containing i t .
M . of substance or m ixture
Thes e results show a very marked increase in the m . p . of the substances by the addition even of 5% of carnauba wax . Further additions increase the m . p . in a diminished ratio .
The proportion of carnauba wax existing in admixture wi th theforegoing substances
,or with Japan wax
,can be ascertained by esti
mating the percentage of potash required for the neutralisation of thefree acid an d for the saponification of the esters of the sample , and bythe estimation of the unsaponifiable matter .Carnauba wax is bleached for candle-making‘ by fil tration throughanimal charcoal
,or by hydrogen peroxide or potassium bichromate .
Candl es are seldom made of carnauba wax alone , but of a mixturecontaining 20 to 30% of stearine and ozokerite . “
Brilli ant paraffin ”i s a mixture of paraffi n wax
, 7 carnauba wax,25
“B rilli ant
gelatin,
” used for finishing leather,i s prepared by adding a liquid
containing water,potassium carbonate
,and carnauba wax to a solution
of gelatin . Carnauba wax is also used in making special varni shes ,and in the manufacture of phonograph cylinders .
CHINESE INSECT WAX .
(See p . In Western China,
2 not far from the Thibetanfrontier
,an evergreen tree
,Ligustmm lucidum,
grows . Early‘ 1 J . S oc. Chem. Ind . , 1 89 4 , 1 3 ,
S oc. Chem . I nd . , 1 89 2 , 1 1 , 282 .

SPERMACETI .
in the spring , numerous brown pea-shaped scales containing the larvaof the wax insect
, Coccus pela, appear on its boughs and twigs .These scales are gathered
,wrapped in packages
,conveyed about 200
miles to Chia—ting, the centre of the industry , made up into smallpackets with leaves
,and suspended under the branches of a species of
ash . The insects on emerging from the packets creep up to the leavesof the ash tree s , and afterward descend to the twigs and brancheson which the wax is deposited by the males. After 1 00 days
,the
depos i t i s complete,and the branches are then cut down
,the wax
scraped off,and what remains on the twigs i s separated by boiling
with water,which destroys the insects and necessitates a fresh supply
of larva in the next year from outside districts . A pound of larvascales will produce 4 or 5 pounds of wax .
The product i s a clear white,highly crystall ine
,brittle wax
,called
from its appearance “ vegetable spermaceti .” It consists,principally
,
of ceryl cerotate . It i s chiefly used in China for coating the exteriorsof candles made of animal and vegetable tallow
,also as a sizing for
paper and cotton goods,for imparting a gloss to silk
,and as a furniture
polish .
I
SPERMACETI
(See also table on p . 7 Spermaceti exists in solution in the oilfrom the sperm whale
,bottlenose whale
,dolphin
,and allied cetaceans
,
but not in the oil from the whalebone whales . It i s present mostabundantly in the oil from the head cavities
,and is commonly stated
to be a special product thereof . This ' i s an error,the oil from the
blubber also depositing spermaceti on cooling,and in practice the head
and blubber oils are treated together .Crude spermaceti forms crystalline scales of a yellowish or browni shcolour . I t i s purified by fusion
,pressure
,and boiling with a solution
of potash,to remove adhering oil and neutralise traces of acid . In
practice , the complete removal of the oil i s not aimed at , as a small proportion is found to conf er desirable properties on the product . I t is
then remelted and cast into cakes .As thus obtained
,spermaceti i s a snow-white or ‘
transparent substance of marked crystalline structure . It fuses at 43° to The
Chem. Ind . , 1 89 7 , 1 6 , 685 .
2 e fi gure commonly sta t ed as th e m . p . of sp ermace t i .rea lly refers to th e soli d i fy i ngo int as de t erm ined by th e t it er t e st . Th e spermacet i from bo t t leno se o i l me lt s a t a sensibly1gh er t empera ture than that from t ru e sperm o i l.
Vol . I I .
—1 8

2 74 MODE S OF EXAMINING FATS,OILS , AND WAXES .
sp . gr . at the ordinary temperature i s commonly between andbut diff ering statements are made
,probably owing to difficul ty
attending the estimation , in consequence of the crystalline structureof the substance .
‘
Much more trustworthy estimations can be madeof the sp . gr . in the molten condition
,which ranges between and
at a temperature of 98° to 99° (water at 1 5 .5°
Spermaceti i s insoluble in water,but dissolves in boiling alcohol
,
ether , chloroform ,carbon disulphide
,and fixed and volatile oils .
Cold alcohol dissolves the adhering oil only . From its solution in hotalcohol or ether it separates in crystalline form
,and
,after repeated
purification in this manner,the m . p . reaches to and the
crystals consi st of pure cetin .
Cetin or Cety l Pa lm i tate , i s the chief constituent of spermaceti
,which
,in addition
,contains certain homologous
ethers . Thus,on saponification i t yields
Acids Alcoho ls
Q Q Q Q Q Q Q Q Q Q Q
0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0
Cety l A lcoho l , C 1 6H 3 3.OH ,
may be obtained in a state of approximate purity by saponifying spermaceti previously crystalli sed fromhot alcohol . On evaporation of its ethereal solution , cetyl alcohol remains as a white or yellowish-white , tasteless, 1nodorous , crystallinemass
,melting at 49 . When carefully heated i t distils without de
composition at about and is volatile with the vapour of water.
I t i s quite insoluble in water , but readily soluble in alcohol , ether, andpetroleum spirit .When heated with potash-lime to a temperature of 250
°
cetyl alcohol is converted into potassium palmitate,with evolution of
hydrogen .
Cetyl alcohol heated with glacial acetic acid forms cetyl acetate ,C I 6H3 3
.C 2H302 ,a crystalline substance melting at 2 2
° to andboiling at 200° under a pressure of 1 5 mm .
The proportion of potassium hydroxide required for the saponification of spermaceti i s about corresponding to a saponification

2 76 MODE S or EXAMINING FATS , OILS,AND WAXES .
sample , the exceptionally high iodine value is accounted for by thepresence of more sperm oil than usual
,owing to the low temperature
and presSure employed in its preparation .
The behaviour on saponification , low acid and iodine values,to
gether with its physical characters,amply suffi ce to identify Spermaceti
and to detect any admixture . The most likely adulterants arestearic and palmitic acids
,stearin
,tallow
,and paraffin wax .
Palmi ti c and steari c aci d will be detected and determined by estimating the free acid of the sample by titration with standard alkali andphenolphthalein
,any proportion of acid less than 1 % being neglected .
An admixture of beeswax would somewhat increase the acidity of thesample . Added fatty acids may also be detected by melting thesample in a test-tube immersed in boiling water
,agitating with 2
volumes of ammonia of Sp . gr.
,and
'
allowing the whole to cool .If the Spermaceti be pure
,i t will rise to the surface and leave the am
mon ia nearly or entirely clear ; but if adulterated with stearic acid , athick white emulsion will be formed
,which retains the spermaceti if the
proportion of the adulterant be large,but allows it to rise and form a
separate layer if the steari c acid is present only in moderate amount .1 % of the adulterant is said to be recognisable by this test . Dunl opfound i t rel iable
,down to about but with smaller quantities the
test appeared somewhat uncertain .
Tallow and stearin are recognisable in Spermaceti by the iodinevalue being in excess of the numbers given above ; by the change in thefracture
,feel
,and appearance of the sample ; and by the tall owy smell
produced on heating. They will also be indi cated by the results of thesaponification of the sample . In presence of ei ther adulterant the percentage of alkali required for saponification will be increased , thesaponification
-equivalent correspondingly lowered,while the ether
extract will be diminishedand the percentage of fatty acids increasedalmost in direct proportion to the extent of the adulteration . Thesaponification
-equivalent of Spermaceti averaging about 438 and thatof tallow about 288
,each unit per cent . of the adulterant will reduce
the saponification-equivalent by 1 .5 . Thus,if a sample be found to
require of‘
potassium hydroxide for saponification ,correspond
ing to an equivalent of 380, the proportion of tallow may be assumed tobe
(438—38°)X 2

SPERMACETI .
If free fatty acids are present , together with neutral fats , the samemethod of calculation will show approximately the sum of the twoadulterants and
,the fatty acids having been previously estimated
,
the proportion of fats can be ascertained ; or, preferably, the fatty acidsmay be previously estimated in the same portion of the sample
,
and only the additional quantity of alkali required for the saponification of the neutral fat taken into account in the calculation . Theether-residue from genuine
' spermaceti being at least and fromfatty acids and neutral fats practically ni l, . the percentage of suchadulterants can be ascertained with accuracy . Each uni t of etherresidue obtained
,represents approximately
,2% of real spermaceti in
the sampleParafiin dimin ishes notably the sp . gr . of the sample , yields 1 00%of ether-residue
,neutralises no alkali
,and cannot , by admixture with
any proportion of fatty acid or fat , be made to give results on saponification similar to those yielded by genuine spermaceti . Thus , amixture of equal parts of paraffin and tallow will yield 50% of etherresidue
,but the saponification-equivalent will be about 5 76 . Paraffin
can be detected by Holde ’s test,as in the case of sperm oil
,as little
as 3 .5% being capable of detection according to Dunlop . Smallerquantities may
,however
,be detected by boiling the unsaponifiable
matter with acetic anhydride and observing the behaviour of the solution . If the spermaceti is genuine
,the solution rema ins clear on
cooling , but if paraffin wax is present , i t becomes turbid , owing toseparation of the latter . As l ittle as 1 of paraffi n wax in spermacetican be detected in this manner (Dunlop).


280 MODE S OF EXAMINING FATS , OILS , AND WAXE S .
Browne (J . Amer . Chem. S oc.,1 899 , 2 1
,807) gives the following
composition :
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
O O O O O O O O O O O O O O O O
0 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
O O O O O O O O O O O O O O O O O O O O
O O O O O O O O O O O O O O O O O O O O O O O
O O O O O O O O O O O O O O O O O O O O O O O O O O
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
According to Duclaux (Compt. rend,1 886
,1 02 , butter fat
contains from 2 to of caproic and from to ofbutyric acid .
Lewkowitsch (O ils , Fats and Waxes , 4th Ed .,668) only finds
of stearic acid in the insoluble fatty acids of a butter fat giving aReichert-Meissl value of Hehner and Mi tchell also found verysmall proportions of stearic acid
,and in some cases none . Some ob~
servers have found a 'mounts over 6% but in view of the above , theseresults must be accepted with reserve . James Bell (The Chemi stry ofFoods
,2, 44) designated a mixed ester found by him as glyceryl
palmito-butyrate ; A . W . B lyth and Robertson (Proc. Chem. S oc.,
1 886,
designated an ester examined by them as glyceryl stearopalmito-butyrate .Exam inat ion of Butter Fat—The examination of butter fat isundertaken for the purpose of detecting adulteration with foreign fats .Many substances have been described as being used for suchpurpose
,but the fats used are practically confined to lard
,oleo products
,
and coconut oil . Cottonseed o il,stiffened .with beef stearin
,and
cottonseed stearin,and other vegetable oils are also used
,but these
latter are very easy of detection . The adulteration may consist of theentire substitution of margarine (oleomargarine) for the butter, inwhich case no diffi culty is experienced in detection , or one or more ofthe above-mentioned foreign fats may be used, when small SOphistications (under are often difli cult to certify , especially if a judiciousmixture has been used . Coconut Oil probably seldom finds its way

BUTTER FAT.
into butter directly,but as a constituent of substitutes . I t i s therefore
absolutely necessary not to rely on any one test,the use Of two O’r three
reliable methods,however
,being usual ly quite sufli cient to detect the
most skilful adulteration .
During the last few years,many methods have arisen for the purpose
of detecting one or other form of adulteration in butter . As, however,
many of these either fail to effect the required result , or are no advanceon existing methods
,only such are here given as are easy of application
,
of real utility,and which
,with one exception
,are used fairly generally .
In all forms of butter fat analy sis,reference to limits for true butter fat
is an absolute necessity ; a selection of the results obtained by variousobservers with each method is therefore givenThe methods here described are those which directly or indirectly
Obtain values for : ( 1 ) The refractive index of the fat ; (2) the content Of volable fatty acids soluble and insoluble in water ; (3) thesp . gr. Of t he fat ; (4) the mean molecular weight Of the total fattyacids ; (5) the mean molecular weight of the soluble and insolublefatty acids .
1 . Th e Re fract ive Index of th e Fat .—This test is of considerablevalue for quickly detecting very flagrant adulteration ; i t breaks downin however the case of the more skilful forms of scientifically mademixtures . This will be understood when one considers that coconut andpalm-kernel Oils give a lower figure than butter fat
,while beef fat
,lard ,
and other adulterants give higher figures . It is obvious,therefore
,that
mixtures can be made to give the same reading as genuine butter fat .Notwithstanding th is
,the test is so qul ckly and easily performed , and
so Often affords immediate indication as to the direction of the adulteration
,that it should not be neglected .
S everal different forms of instrument are made for measuring therefractive index of fats .The Zeiss Butyro-refractometer is by far the most convenient , re
quiring only about 5 drops of the fat ; and reading with extreme delicacy . The scale is an arbitrary one
,and may be converted to re
refractive index (ND) if required . I t shoii ld be noted that free fattyacids tend to reduce the reading
,but the amount usually contained in
butter fat is too small to produce any effect . Much confusion hasarisen owing to there not being any fixed temperature at which tomake the Observation
,and various workers have unfortunately chosen
different temperatures . The most used temperature is and it

282 MODE S or EXAMINING FATS , OILS , AND WAXE S .
i s hoped that this will become universal . Readings taken at othertemperatures may be converted fairly well by subtracting or addingof a scale division for every degree rise or fall in temperature
the refraction being reduced as the temperature rises . In order thatthe reading may be taken at any temperature and quickly convertedto the equivalent at the standard temperature
,Leach and Lythgoe
have devised a special Sl ide rule to perform the calculation,and also to
convert the Zeiss scale to ND or vi ce versa if required . They take
account of the fact that the correction is not the same for all fats .R ichmond (Analyst, 1 907 , 3 2 , 44) criticises this rule adversely and pointsout sources of error . To avoid chance Of error i t i s therefore better totake the reading at the temperature required
,in which case the
Zeiss water-heating apparatus should be used , by the use of which theprisms of the instrument may be brought to the required temperaturein a few minutes , and maintained within a few tenths Of a degree for aconsiderable time . Richmond (i bi d .) gives the following method forthe preparation Of a correction chart
“In the centre Of a Sheet of squared paper
,at least 20 units by 1 2 ,
lay out vertically the 3 5° l ine , dividing it into 1 00 parts . At the 1 09line draw a line perpendicular to this on both sides and lay out temperatures 1
° unit ; at the 24 line draw a similar line , laying outtemperatures 5 unit . Join the corresponding temperatures extending them to zero . These will be the temperature lines . On the1 09 line find a point 8 .5 units to the right ; j oin this and 1 00 on the3 5
° l ine,extending i t across the sheet ; draw through each 5 on the 3 5
line lines of refraction parallel to this . TO correct readings,find cor
responding temperature and refraction lines . The correction is thenumber of units between lines corresponding to the temperature readand the temperature to which it is to be corrected
,measured hori
zontally.
The following figures have been recordedNorwegian (Rifle), (December) to (June) at meanto 4 1 95Russian (Lewin), to 42 .0 atBritish (Thorpe) 3 7 1 samples from various farms and colleges .to at 45°
Dutch (Bemelmanns), stall-fed cows (November) to(September) , and for cows kept in the open field toDutch (Fritzsche), to at
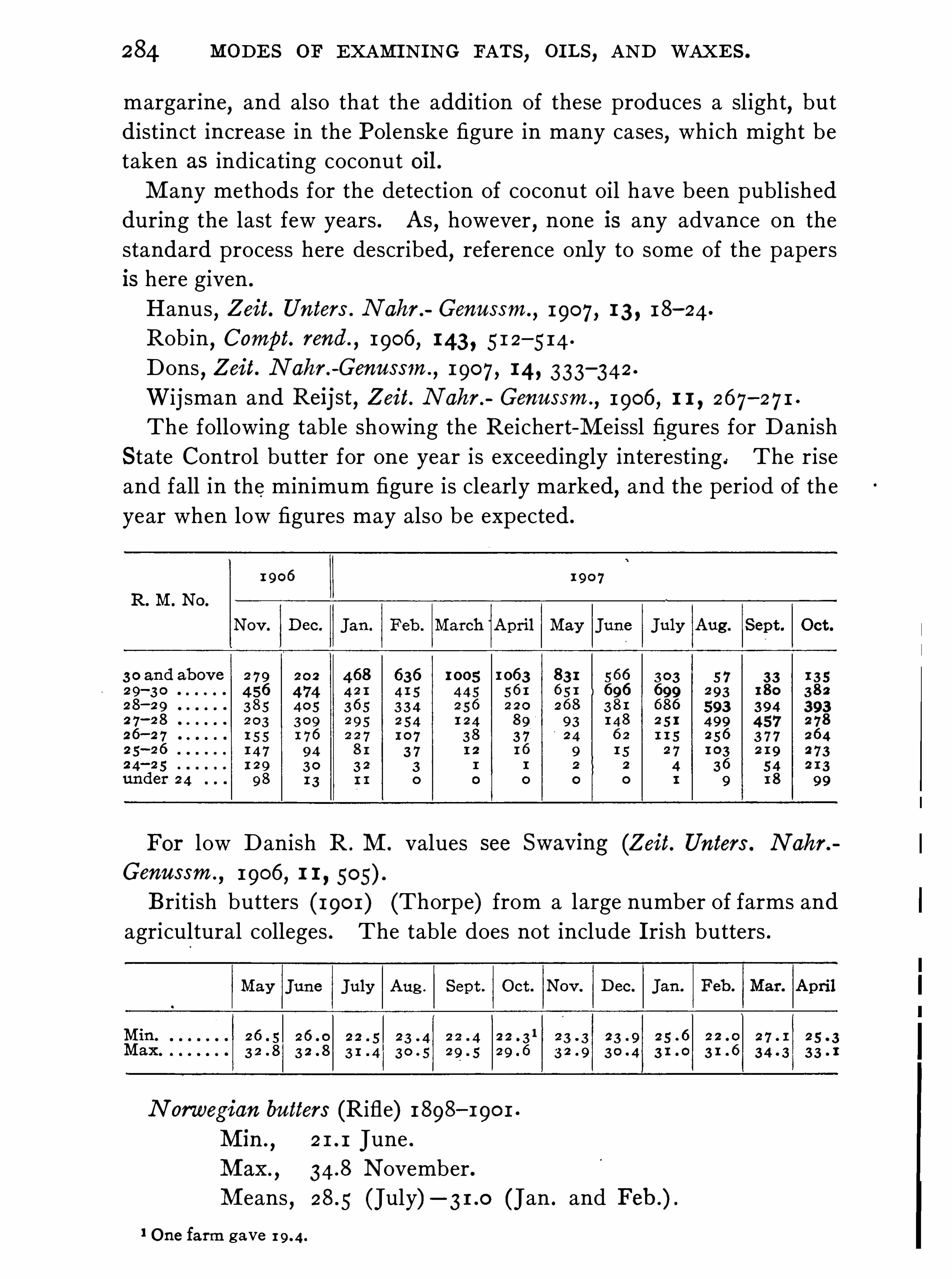
284 MODE S OF EXAMINING FATS,OILS , AND WAXE S .
margarine,and also that the addition of these produces a slight
,but
distinct increase in the Polenske figure in many cases,which might be
taken as indicating coconut Oil .Many methods for the detection of coconut oil have been published
during the last few years . As,however
,none is any advance on the
standard process here described,reference only to some of the papers
is here given .
Hanus,Zei t. Unters . N ahr . Genussm. ,
1 907 , 1 3 , 1 8—24.
Robin,Compt. rend
,1 906, 1 43 , 5 1 2
—5 1 4 .
Dons,Zei t. N ahr .
—Genussm.
, 1 907 , I 4 , 333—342 .
Wij sman and Reijst, Zei t. N ahr . Genussm.,1 906 , 1 1 , 267
—2 7 1 .
The following table showing the Reichert-Meissl figures for DanishState Control butter for one year is exceedingly interesting. The riseand fall in the minimum figure is clearly marked
,and the period of th e
year when low figures may also be expected .
1 90 7
0 0 0 0 0 0
0 0 0 0 0 0
0 0 0 0 0 0
0 0 0 0 0 0
0 0 0 0 0 0
For low Danish R . M . values see Swaving (Zei t. Unters . Nahr.
Genussm.,1 906, I I ,
British butters ( 1 901 ) (Thorpe) from a large number of farms andagricultural colleges . The table does not include Irish butters .
N orwegian butters (Rifle) 1 898—1 90 1 .
Min .,
June .Max
, November.Means
, (July) (Jan . and1 One farm gave

BUTTER FAT.
German butters (Ludwig), for 1 1 0 samples , 1 906 .
Dutch butters (Bemelmanns), 1 905 .
S tall -fed, (Sept ) (March) .
In open fields, (Oct .) (March) .
Note the low figure for the exposed cattle .Australian andN ew Zealand (Theodor), 1 903—04.
26 .5 These butters are usually distinguished by highvalues .
Canadian (Theodor),The Reichert-Meissl figure is undoubtedly depressed in butter de
rived from cows that have been poorly fed ' and badly housed ,especially in low temperatures . I t i s undoubtedly these causes whichled to the low figures so common in S iberian butter, and which broughtthem under grave suspicion . The figure al so Shows a regular fallduring the lactation period in individual cows
,so that, when calving
takes place almost entirely at one period, as for instance in Ireland ,periods of low Reichert-Meissl values will be found . To obviate th is ,calving should be spread over the whole year . This is well seen in thefollowing table (Handby Bal l , Analyst, 1 907 , 3 2 ,
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
3 . Spec ific Gravi ty of Butter Fat—This was suggested byJames Bell
,who showed that melted butter fat is of sensibly h igher
sp . gr. than lard or margarine . Bell took the sp . gr . of the fat at1 00
° F. (3 7 . by means of a Sp . gr . bottle furnished with a thermom
eter, and his figures express the sp . gr. at 1 00° F as compared with thatof water at the same temperature . Some chemists take the sp . gr .at 1 00° and thereby greatly diminish the sensitiveness of the test , inthat the difference between butter fat and most fats likely to be usedas adulterants is not so great at the high temperature , owing to thegreater coeffi cient of expansion of butter fat .Skalweit (J . S oc. Chem. Ind.
,1 894, 1 3 , 54) finds that the differences
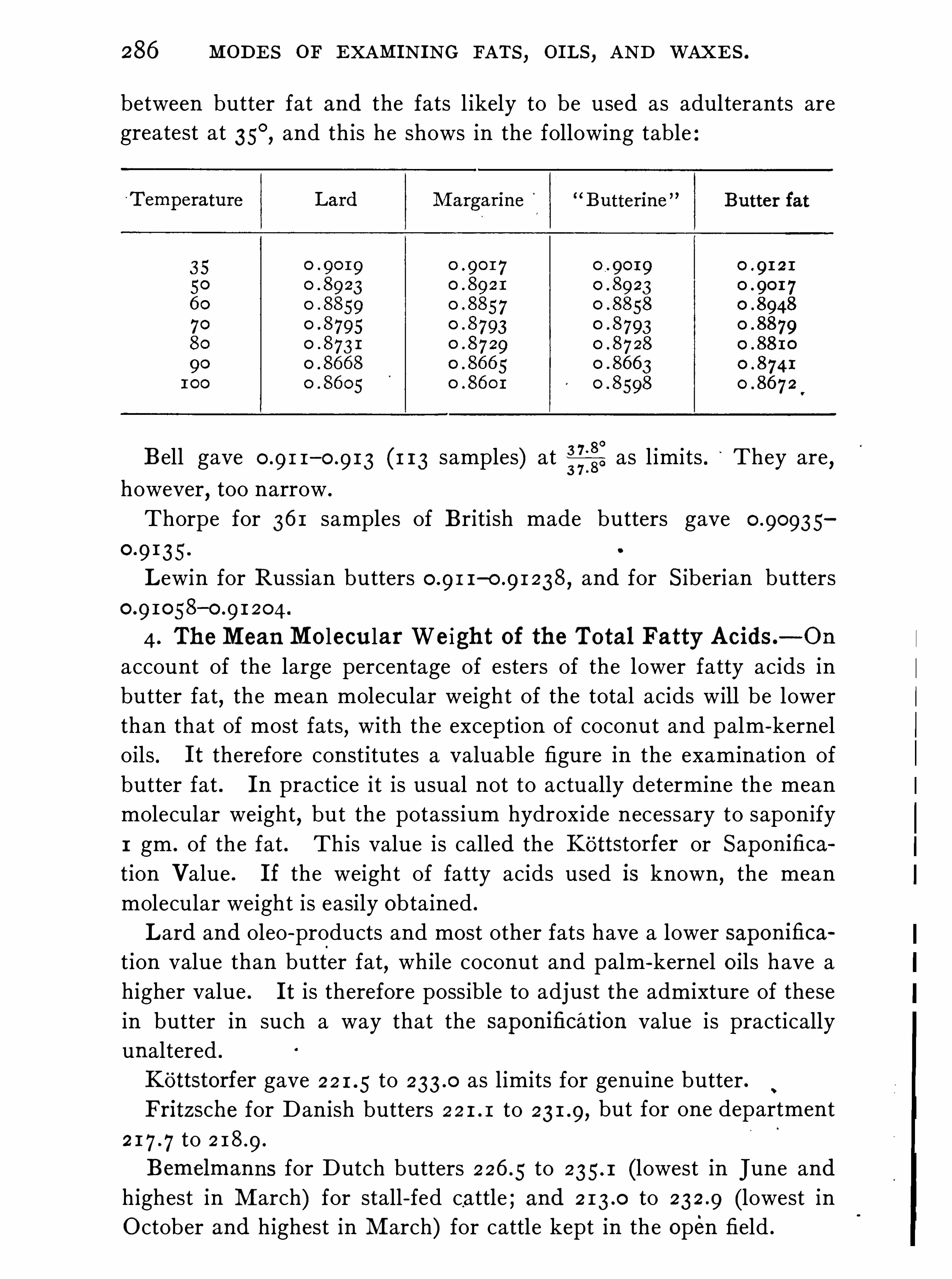
286 MODE S OF EXAMINING FATS , OILS , AND WAXE S .
between butter fat and the fats l ikely to be used as adulterants aregreatest at 3 and this he shows in the following table
Bell gave ( 1 1 3 samples) at as limits . They are,
however,too narrow .
Thorpe for 36 1 samples of British made butters gave 5
Lewin for Russian butters and for S iberian butters
4 . Th e Mean Mo lecu lar W e igh t of th e Total Fatty Aci ds .—Ou
account of the large percentage of esters of the lower fatty acids inbutter fat
,the mean molecular weight of the total acids will be lower
than that of most fats,with the exception of coconut and palm-kernel
oils . I t therefore constitutes a valuable figure in the examination ofbutter fat . In practice i t is usual not to actually determine the meanmolecular weight
,but the potass ium hydroxide necessary to saponify
1 gm . of the fat . This value is called the KOttstorfer or Saponification Value . If the weight of fatty acids used is known , the meanmolecular weight is easily Obtained .
Lard and oleo-products and most other fats have a lower saponification value than butter fat
,while coconut and palm-kernel Oils have a
higher value . It is therefore possible to adjust the admixture of thesein butter in such a way that the saponification value is practicallyunaltered .
KOttstorfer gave to 233 .0 as limits for genuine butter .Fri tzsche for Danish butters to 23 1 9 , but for one departmentto
B emelmanns for Dutch butters to (lowest in June andhighest in March) for stall-fed cattle ; and to (lowest inOctober and highest in March) for cattle kept in the Open field .
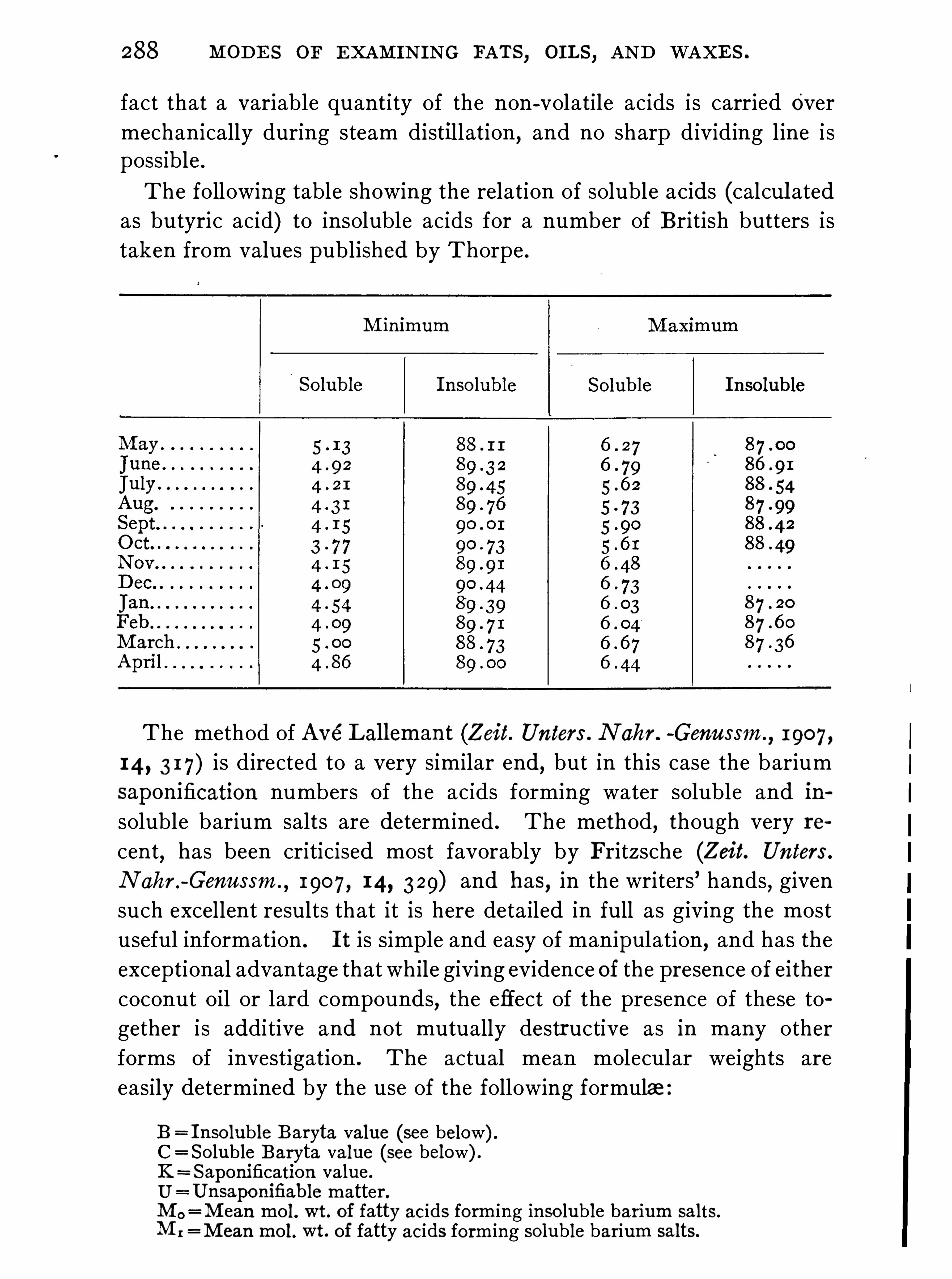
288 MODE S OF EXAMINING FATS,OILS
,AND WAXES .
fact that a variable quantity of the non-volatile acids i s carried Overmechanically during steam distillation
,and no sharp dividing line i s
possible .The following table showing the relation of soluble acids (calcul atedas butyric acid) to insoluble acids for a number of British butters istaken from values published by Thorpe .
M inimum
March
The method of Avé Lallemant (Zei t. Unters . N ahr.-Gennssm.
,1 907 ,
1 4 , 3 1 7) i s directed to a very similar end, but in this case the bariumsaponification numbers of the acids forming water soluble and insoluble barium salts are determined . The method , though very recent
,has been criticised most favorably by Fritzsche (Zei t. Unters .
N ahr .-Gennssm.
, 1 907 , I 4, 3 29) and has , in the writers ’ hands , givensuch excellent results that it is here detailed in full as giving the mostuseful information . I t is simple and easy of manipulation , and has theexceptional advantage that while giving evidence Of the presence of ei thercoconut oil or lard compounds
,the effect of the presence of these to
gether i s additive and not mutually destructive as in many otherforms of investigation . The actual mean molecular weights areeasily determined by the use of the following formula :
B = Inso luble Baryta value (see below).C = So luble Baryta value (see below).K=Saponification value .
U Unsaponifiable matter.
Mo=Mean mo l. wt . of fatty acids forming inso luble barium salts .
M I=Mean mol. wt . of fatty acids forming soluble barium salts .

BUTTER FAT.
S total fatty acids .
So we ight of acids forming inso luble barium salts .
S ; J acids forming soluble barium salts.
MOX ES o 1 000.
76-7
S = I
S x= S—So .
S o i s directly estimated by drying and weighing insolublebarium soaps and igniting .
As values for M I Avé Lallemant givesFor butter fat ,For lard
,28 1 I
For coconut o il,
1 45 . 8
The following figures are given by him
ButterButter+ 1 0% lard .
ButterButter+ 1 0% coconut o i lButter+ 1 0% lard .
He states that butter has always a negative value for bwhile for a number of other fats i t i s always positive and not less than
The writers are of the opinion that this formula must not be adhered to too strictly
,as in certain cases with mixtures of butter fat
and coconut oil negative values are obtained for b +c), but insuch mixtures they have always found the value for b to exceed
while in cases Of butter fat which give only a very small negative value for b +c) the value for c is always well belowI t must be borne carefully in mind that the values obtained in themethods given above have a distinct connection one with
,
another .This is perhaps best illustrated by a study of the following table dueto Thorpe .
1 Th e fi gures for a , b and 0 do not correspond , as they do not belong t o th e same sample .
Vo l. I I .
- 1 9
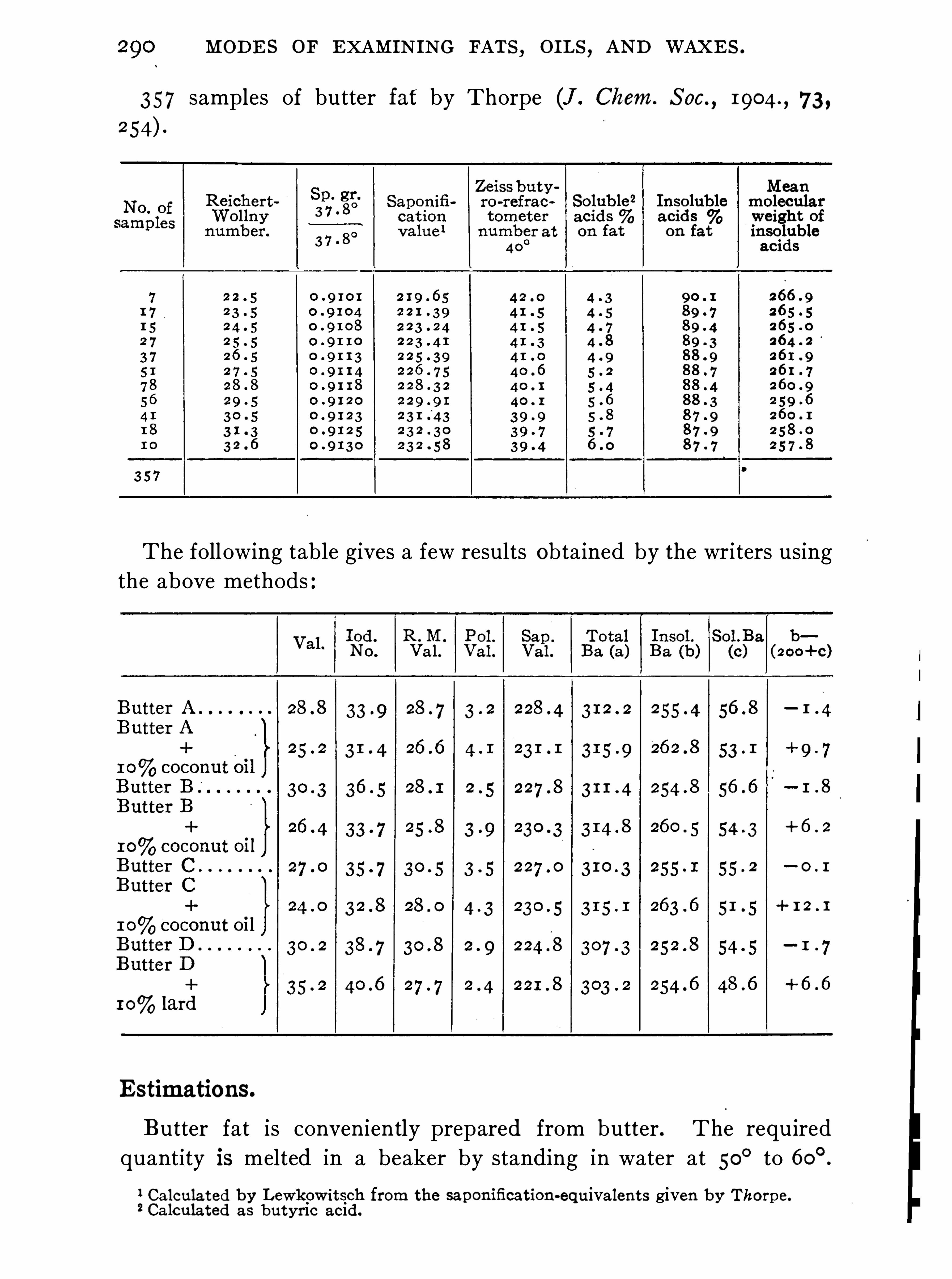
MODE S OF EXAMINING FATS , OILS , AND WAXES .
3 5 7 samples of butter fat by Thorpe (J. Chem. S oc. , 7 3 ,
254)
The following table gives a few results Obtained by the wri ters usingthe above methods
3 1 1 4
_ O . I
+ 1 2 . I
1 0% lard
Estimations.
Butter fat is conveniently prepared from butter. The requiredquantity is melted in a beaker by standing in water at 50° to
1 Calcula t ed by Lewk owi t sch from the saponificat ion-eq uivalent s given by Thorpe .
2 Ca lculated as but y ric acid .

292 MODE S OF EXAMINING FATS , OILS,AND WAXE S .
apparatus i s withdrawn from its case by tak ing hold of the base plateor the telescope carrier—never the telescope i tself—and placed in a
convenient position for looking into the telescope . The illuminationmay be supplied either by the daylight coming in through a window or
by the light of a lamp .
The refractometer can be used In conjunction with any k ind of
FIG . 9 .
heating appliance which affords a current Of water Of constant temperature and uniform speed . Connection with the refractometershould always be established so that the water enters at D and flowsOff at E .
Appli cati on of the S ample of Butter to theP ri sms .
—The prism casing isopened out by revolving the screw-head
,F,clockwise , giving it a half
turn until checked,when one half (B) of the prismcasing can simply be

BUTTER FAT. 293
hinged down . The post H keeps B in the position shown in thefigure . The surfaces of the prisms and of the metal parts must now becleaned with scrupulous care
,soft clean linen and a little alcohol or
ether being best for the purpose .A small quantity of the sample o f butter to be tested is then melted
down in a little spoon and poured upon a small filter of blotting-paper,
held in one hand . The first two or three drops of clear butter fat percolating the filter are applied to the surface of the folding prism ,
indoing which it i s expedient to tilt up the apparatus with the left hand
,
so as to bring the surface in question to an approximately horizontalposition . The filtering of the sample of butter is not absolutely mecessary. The quantity required for the test may be taken up by means Ofa glass rod
,but the precaution of carefully rounding the ends of the rod
and of avoiding impurities floating on the surface of the melted buttershould not be disregarded .
The observer now presses the component B against A and turns thescrew-head
,F,in the reverse direction to contact with another stop
,
whereby B is secured against dropping back and close adhesion of thetwo prism surfaces is effected as well . The apparatus is at the sametime replaced on its base plate .The mirror
, J , should be arranged in such a position that the borderline appears distinctly
,which may necessitate slight shifting or turning
of the entire apparatus . The draw at the ocular Should also be adjusted so that the scale can be seen distinctly .
The first thing necessary is to ascertain that the entire space betweenthe prism surfaces is uniformly packed with butter fat . For that purpose the small image of the prism surface
,situated about I cm . in
front of the ocular,is scanned with a magnifier (or with the naked eye),
held at the requisite distance from the ocular . In this way minute airbubbles with in ‘
the stratum o f fat,which would prejudice the sharpness
of the border-line,will be readily detected .
If a current of water of constant temperature has previously cirenlated for a time through the prism body
,the border-line will quickly ,
generally in about a minute,assume a fixed position and attain the
maximum of sharpness . Both being obtained,the appearance of the
border-line (whether colourless or , if coloured , the tint) also the positionof the border-l ine relatively to the scale are noted
, and at the same timethe temperature registered by the thermometer read off .
Integral divisions of the scale are read off immediately in the field

294 MODE S OF EXAMINING FATS , OI LS , AND WAXES .
of View , tenths of a division being determined by the aid of the microm’
eter screw (G in Fig . 9) in the following manner . The border-line i sadjusted by means of the screw upon a division o f the scale
,when the
micrometer drum will indicate the number of tenths to be added to thenumber of integral parts of the scale appertaining to the particulardivision . If a number of tests are being made in immediate succession , a little practice , with the help of an assistant to attend to themelting and hand the small samples of butter
,will make it easy to
carry out the refractometrical test Of from 25 to 30 samples of butterin the space of 1 hour.Wollny has devised a special thermometer which indicates thehighest admissible values for genuine butters at temperatures varyingfrom 30° to A standard fluid is provided for the purpose of testingthe adjustment of the ocular scale and directions are given for resettingthe Scale when necessary .
The use of sodium-light illumination greatly sharpens theborder-line .
2 . The Rei chert-Mei ssl-P olenske Method—In this process the sa
ponification is conducted according to themethod devised by Leffmann
and B eam . 5 grm . of the fat and 20 grm . of glycerol are weighedinto a 300 c .c . flask and 2 c .c . Of 50% sodium hydroxide solutionadded (made by dissolving good sodium hydroxide in an equalweight of water and allowing to stand till clear). The flask is heatedover a flame with constant Shaking , till i t clears suddenly . Coolthe soap and add 1 00 c .c . of recently well-boiled distilled water , tillsolution of the soap is effected . grm . of powdered pumice siftedthrough muslin (the grade and quantity are important) is added andthen 40 c .c . of sulphuric acid solution . (20 c .c . of sulphuric acid diluted to c .c . , and the solution adjusted so that 3 5 c .c . neutralise2 c .c . of the sodium hydroxide solution .) The flask is at once connected to the condenser , and heated with a small flame till the insolubleacids are completely melted ; the flame i s then increased and 1 1 0 c .c .distilled in 1 9 to 2 1 minutes . Condenser water should be from 1 8° toand dimensions of apparatus exactly as shown in Fig . 1 0 . When
1 1 0 c .c . have distilled , the flame is removed and a 2 5 c .c . cylinderplaced under the condenser to catch any drops . After mixing the contents Of the 1 1 0 c .c . flask
,they are filtered and 1 00 c .c . titrated with
N 1 0 alkali,using 0 . 5 c .c . of a 1 alcoholic solution of phenolphthalei n
as indicator . This number of c .c . increased by after subtrac

296 MODES OF EXAMINING FATS , OILs,AND WAXE S .
Rather varying results have been Obtained by various Observers , butthe following table will be found a very fair guide
Re ichert-Me issl New Buttervalues values3 2 3 5
3 1 3 2
3° 3 .0
2928
27 2 . 426
25 1 . 8
24 1 723 1 .6
A “New Butter Value exceeding by 0 .5 c .c . the figure corresponding with the Reichert-Meissl value found
,indicates the presence of
coconut or palm-kernel Oil or coconut stearine .”If an approximate idea of the amount of coconut oil present i s
desired , Polenske ’s original table (Arbei l aus dem Kai serl . Gesund. ,
1 904 , 20, 543) should be consulted (reproduced in Lewkowitsch : O ils ,Fats and Waxes
,Vol . i i
,p . 702 , 4th
It has been stated that the presence of oily drops in the first distillate(after cooling at is distinctive Of coconut oil
,but this is undoubtedl y
fallacious . Many chemists also employ only 90 c .c . of water for solution of the soaps and 50 c .c . Of sulphuric acid solution (25 c .c . sulphuricacid to c .c . with water).For the Reichert-Wollny method see Margarine .3 . S pecific Gravi ty of the Fat.—This is best determined in a 25
grm . bottle . The dry melted fat is run into the bottle a few degreesbelow the temperature at which the gravity is to be Observed
,great care
being taken that no air bubbles are introduced at the same time . Thestopper is screwed gently home and the bottle immediately immersedin water of the desired temperature for 45 minutes . The test is thenfinished in the usual manner . For conven ience
,especially if many
determinations are made,a bath regulated to the temperature by means
of a thermostat may be used .
4 . S aponificati on Value (Kottstorfer Value) .
—The saponification
value is defined as “ the number of mg . of potassium hydroxide , whichis necessary to completely saponify 1 grm . of fat .”(Saponification equivalent is the number o f grm . of butter fat saponi
fied by (i . e .,one equivalent) of potassium hydroxide . It i s obvious
that this is merely another mode of expressing the same resul t , and

BUTTER FAT. 297
the one figure can easily be calculated from the other . Kottstorfer’s
original mode of expression is now more generally used .)I
Solutions required : 1 . N/ 2 hydrochloric acid accurately prepared .
2 . Alcoholic potassium hydroxide approximately N/2 strength . 3 .
1 alcoholic solution Of phenolphthalein .
The potassium hydroxide solution is prepared by dissolving 1 7 to 20grm . of stick potassium hydroxide (purified by alcohol) in the smallestpossible quantity of water
,and then making up to 500 c .c . with alcohol
of not less than 94% (by weight). The solution is allowed to standover-night
, and the clear liquid syphoned off for use . If the alcohol ispure
,the solution will be colourless
,or nearly so (i t i s advisable
to test the alcohol before making up the solution and rejecting any
which gives more than a very pale colour when boiled with a strongsolution of sodium hydroxide).The test i s carried out as followsAbout 2 grm . of the clear melted and filtered fat are weighed into a
200 c .c . Jena flask,and an accurately measured quantity of the alco
holic potassium hydroxide solution run in from a 25 c .c . pipette . A
like quantity of the same solution is run from the same pipette inexactly the same way into a clean 200 c .c . flask . The flasks areconnected to reflux condensers and heated in a water-bath so that thealcohol gently boils ; this is continued for 30 minutes . The flask containing the butter fat should be shaken occasionally , particularly atthe commencement . The flasks are removed from the bath
,20 drops
of phenolphthalei n solution added to each,and the contents titrated
while hot with the N/ 2 acid .
If F = grm. of fat taken.
X = c .c . of acid requ ired in the contro l experiment.Y= e .c . Of ac id requ ired to neutralise the excess of alkali in the test.
and saponification equ ivalent
5 . The Barium Method of Avé-Lallemant. —I .9 to grm . of thefiltered butter fat are saponified with 2 5 c .c . of approx . N/2 alcoholicsodium hydroxide (carefully standardised), boiling for 30 minutes ;while still warm , ti trate with N/ 2 hydrochloric acid tophenolphthalei n .
The alcohol is removed as completely as possible by boiling and blowing air into the flask . The soap is dissolved in 1 50 to 1 80 c .c . of hotrecently boiled distilled water
,in a 250 c .c . flask . S tand on the water

298 MODE S OF EXAMINING FATS , OILS,AND
,WAXES .
bath 5 minutes , and add 50 c .c . of approximately N 5 barium chloridesolution (25 grm . crystalli sed barium chloride in Allowto remain I 5 minutes on the water-bath to cause the insoluble bariumsalts to coalesce . Cool , fill to the mark with water and filter off 200
c .c . into a beaker,beat this to nearly boiling on a sand-bath
,add 1 c .c .
hydrochloric acid and 1 0 c .c . of approx . N/ 1 sulphuric acid . Fil terthe barium sulphate on a gooch crucible , wash till free from chlorides ,and finally with two quantities of 1 0 c .c . Of warm alcohol . Dry toconstant weight . Increase the weight of barium sulphate found by2 and calculate to BaO (BaSO4X 0.65 7 I = barium oxide). Sub
tract th is last from the barium oxide value of the barium chloridesolution (which must be standardised in an exactly similar way) .This gives the barium oxide value Of the acids forming insoluble bariumsalts . Calculate th is to 1 grm . Of fat= insol . barium oxide value (b).The saponification value is also calculated as barium oxide (KHO X
=barium oxide) for I grm . of fat = total barium oxide value (a),then a—b = sol . barium oxide value (C), and so find b (200+C).
After much experience with this method,the writers advise the use of
5 grm . of fat for th e test . After saponification and removal Of thealcohol
,the soaps are made up to 250 c .c . with water at and 1 00
c .c . pipetted Off at that temperature for the estimation Of the bariumoxide
,the test being finished as above . By this means a more ac
curate figure is Obtained for a .
”
Qual i tative Tes ts .
The above quanti tative examinations may be supplemented whendesirable by one or more qualitative tests . These are useful moreespecially as confirmation of the presence or nature of some adulterantindicated by the quantitative figures . Some of them are sometimesused as sorting ” tests .
I . Mi croscopi cal Appearance .
—When genuine butter i s examined ina thin film
,under a low power ( X 25 to 50 diam .) between crossed
nicols,i t appears as a homogeneous mass
,but if i t has been melted ,
bright specks or patches,or sometimes even crystals , giving a play of
colours with a selenite plate , may be noticed . Margarine (oleo lmargarine) has a similar appearance to butter which has been meltedand reset .This test may be used for rough sorting out purposes, setting asidefor further examination all samples showing crystalline structure . I t

300 MODES OF EXAMINING FATS , OILS , AND WAXE S .
and palm-kernel Oils,will lower the temperature . The test
,therefore
,
if carefully performed , is of real value , especially as confirmatory ord iagnostic of adulteration by lard products .Many methods of carrying out the test have been proposed
,but the
following is simple and easy and quite suffi cient . A long thin testtube , preferably of Jena glass , about 0 .5 in . in diameter , and suffi
ciently long to take in the scale of a thermometer up to is markedaccurately at 3 and 6 c .c . with lines all round the tube . A thermometerwith very small bulb is fixed in the tube by means of a cork
,so that the
bulb is Opposite the 3 c .c . l ine . The carefully dried butter fat (filterpaper pellets should be shaken up with it beforehand) i s measured inat 2 7 to 29
° till the bottom of the meniscus coincides with the 3 c .c .line . Absolute acetic acid (Kahlbaum ’
s i s to be preferred) i s thenrun in until the 6 c .c . line is reached (the acid should be measured at adefinite temperature , say. 1 5 to The thermometer is insertedand the fat dissolved by shaking in water at about The tube isthen withdrawn
,and the contents allowed to cool in the air
,shaking
gently,and holding the tube in a good light . Immediately the faintest
turbidity is noticed the temperature is read . The tube is then slightlywarmed and a fresh reading obtained . The end point is quite sharp ,and consecutive readings should scarcely differ.I t must be carefully borne in mind that every Operator should obtain
figures for himself for pure butters,as the least change
,especially in
the acid,produces a change in the results . Operating in the above
manner,the writers have found butter to vary between 2 5° and
mostly between 3 7° and The greatest variations were foundin Danish S tate Control butters . 1 0% Of lard produces a rise ofabout 7°4 . Halphen’
s test for cottonseed oi l and cottonseed stearine may becarried out as follows : 2 to 3 c .c . of the melted fat are di ssolved in anequal volume of amyl alcohol in a test-tube
,2 to 3 c .c . of a 1 % solu
tion of sulphur in carbon disulphide added,and the tube is placed
in a boiling water-bath for 20 minutes . In the presence of cottonseedoil
,or cottonseed “ stearine ” a characteristic crimson colour is pro
duced . This test is capable of detecting less than I t is possibleto treat cottonseed oil so as to evade th is test , but this is not usuallydone . The test is applicable to ' the acids from cottonseed oil .The test will detect 1% of cotton seed Oil if the heating be done inclosed test-tubes .

BUTTER FAT. 30 1
5 . Baudou in’s Test f or S esame Oi l.—Take 1 0 c .c . of the melted
fat in a test-tube,add 2 drops of a 2% alcoholic solution of furfural ,
and 1 0 c .c . of concentrated hydrochloric acid . Shake for a minute .In the presence of sesame oil the aqueous layer will be a crimson colour .The test is sensitive to but certain azo dy es interfere with thedelicacy .
Sprinkmeyer (Zei t. Unters . N ahr. u . Genussm. , 1 908 , 1 5 , 20—2 1 )states that rancid cottonseed Oil prevents the red colouration unless1 7% of sesame Oil is present .6 . Phytosterol and Phytosteryl Acetate Test f or Vegetable F ats and
Oi ls , Including Coconut Oi l.—Boil 50 grm . Of the clear fat with
7 5 c .c . of 95% alcohol . Cool and pour Off the alcohol and repeat theextraction with a further 75 c .c . The combined extracts , which willcontain the bulk of the cholesterol and phytosterol
,and some fat are
transferred to a porcelain basin , and an excess of solid sodium hydroxidehaving been added
,evaporated , stirring occasionally . After the
bulk of the alcohol has gone,add more than suflficient sodium
hydrogen carbonate to convert the sodium hydroxide to carbonate ,then sand and carry to dryness . Grind up the dry residue in the dishand extract with light petroleum . The residue from the ether istreated with 5 c .c . of approx . N/ 2 alcoholic sodium hydroxide andagain evaporated to dryness with sand . Re-extract with petroleumether
,evaporate
,and take up with the smallest possible quantity of
absolute alcohol . (If much coloured , boil first with a small quantityof 95% alcohol and a little finely divided animal charcoal , fil ter, andevaporate to dryness). Allow to crystallise and examine microscopically . The residue is converted into acetate and recrystallised in theusual manner .Juck enack and Pasternack (Zei t. Unters . N ahr . u . Genussm.
,1 904 ,
7 , 1 93—2 1 4) give from I I 3 . 2
° to (corr .) as the m . p . of thecholesteryl acetate of pure butter (after 5 crystalli sations), and from
to (corr .) for that obtained from butters adulterated withvarying quantities of coconut oil .I t has been proved that animals fed on Oil cakes sometimes producea butter fat giving indications of the oil present in the cakes , notablyby Halphen
’s test
,but phytosterol has not been found in butter fat
from animals so fed .
7 . H inks ’ Test (Analyst, 1 907 , 3 2 ,—This is a valuable qualita
tive test for coconut and palm-kernel oils . It i s based on the fact that

302 MODE S OF EXAMINING FATS , OILS, AND WAXES .
the latter contain a fat which crystalli ses from alcohol in a characteristic form . To one who has once become familiar with the shape ofthe crystals
,the test is valuable and reliable
,but without this experi
ence , and if not carried out exactly as recommended it may prove misleading . The writers have been able to detect 2 .5% of coconut Oil inthis way .
The test is carried out as follows5 c .c . of the clear fat are dissolved in twice their volume Of ether , ina wide test-tube
,and packed in ice . After 30 minutes (much solid fat
will have separated) the whole mass is thrown on a plaited filter . Thefiltrate i s evaporated in a basin and heated on a boiling water-bath .The residual fat is poured into a test-tube
,and 3 to 4 times its volume
Of alcohol (96 to 9 7% by volume) added . Boil,when solution will be
effected . The tube is then kept in water at 5° for 1 5 minutes , and thealcoholic layer is then rapidly filtered into another tube
,which is then
kept at 0° for 2 or 3 hours .After this time a portion is withdrawn by a glass tube
,dropped on a
slide,covered without pressure
,and immediately examined at a magni
fication of 200 to 300 diameters . (The examination must be quicklycarried out
, as the crystals are Soon redi ssolved as the liquid warms .In hot weather a cooled stage i s necessary
,conveniently made by plac
ing a flat piece Of ice contained in a petri dish under the slip .)Butter crystallises in round granular masses
,but if coconut or palm
kernel oil i s present,numerous fine feathery crystals will be seen as
well . Lard,however
,produces crystals which are not unlike those
from coconut and palm-kernel oils .
Butter.
The examination of butter itself,apart from the special examination
Of the butter fat,consists usually Of the estimation of water, fat ,
curd,and salt . By
“ curd ” is usually meant the solids-not-fat , withoutthe mineral constituents
,which are usually included under the general
term “ salt .” For special‘
purposes, the actual percentage of proteins i sestimated and also the actual percentage of salt as sodium chloride .B esides these
,an investigation into the nature Of the colouring matter
and preservative ( if any) present , is Often necessary .
Such an examination is of value for the purpose of ascertainingwhether a butter i s properly made
,whether i t has been properly worked

MODE S OF EXAMINING FATS,OI LS , AND WAXE S .
Refraction (Zeiss) not to be above 48 at 31 00
°
1 5°
NO preservatives , except common salt and boron mixtures , whichshall not be present in greater quantity than reckoned as boricacid .
Uni ted S tates of Ameri ca—M inimum limit for fat and inrenovated and process butter
,not more than 1 6% of water .
Reichert-Meissl value not less than 24 .
Sp . gr . at not less than but there are differentregulations in different S tates .England
—Maximum limi t for water 1 6% and in milk-blendedbutters 24
1 . Water.—Apart from the examination of the fat
,thi s estimation
is the most important . The pe rcentage of water in butter variesnaturally for many reasons . The method of churning
,and especially
the temperature of churning,i s a most important factor in determining
the quanti ty of water left in the worked butter . Taking 60° F. as
roughly the correct churning temperature,then temperatures decidedly
above,or decidedly below this
,will result in the inclusion of too much
water . At elevated temperatures a very large quanti ty of water can beworked into and retained by the butter
,as is found in the case of Irish
“ pickled ” butters,and butters so made are often quite firm
,and do not
even appear moist . The addition Of salt tends to produce a drierbutter
,though the appearance of a salt butter would lead to an Op
posite conclusion , seeing that moisture exudes from salt butters ” insmall drops when cut . There is no diffi culty
,in properly managed
churning operations,in keeping to a fairly constant water content
,and
for t his reason , in most countries a maximum percentage for water i sei ther legally or tacitly enforced .
I t seems to be generally agreed that butters containing 1 3 to 1 4% ofwater have the best flavour.Canadi an Butters .
—T he D ept . Of Agriculture gives as theaverage of a large number . Theodor
,in 1 903
—04, gives toN ew Zealand.
— ( I 905) Average Theodor gives to( 1 903Australian to (Theodor).Dani sh—From 1 89 7 to 1 900 , of butter contained betweenand Of water .
Sp . gr . not to be below 5 at

BUTTER FAT . 305
From 1 897 to 1 904, in butters supplied to Canada by Denmark , asteady increase from to was found by the Dept . of Agricul
I ri sh Fi rkin Butter .
—Twooney (Report of Dept . Committee , 1 906 ,
England) gives the following percentages of butters containing morethan 1 6% of water .
Season 1 902—3 samples)Season 1 903
—4 samples)
Season 1 904—5 samples) 7 .
Season 1 905—6 samples)
2 . Curd .—Under this term is usually included the total solids-not
fat,less the ash . I t i s rather a variable figure
,and depends very much
on the method of and care In making . The curd is likely to be muchhigher in the case Of butters made from whole milk than from cream
,
but as the former is scarcely made to-day,certainly not for the market
,
i t does not much concern the analyst . In properly made butters thecurd varies from very small amounts to 2 .5 The higher limit israre and probably to is most usual . As thi s estimation ischiefly Of value for the detection Of the addition of condensed milk ,casein , etc . , i t i s far preferable to actually determine the protein . Whenthis estimation gives more than Of casein
,i t may be taken as
almost certain that addition of m ilk products has been made . In suchcases there i s usually an excess of water
,and if whole or dried milk
has been worked in,an estimable amount of lactose will probably be
found .
3 . Ash .—This term usually includes salt and preservative
,as well as
mineral constituents of the original cream . I t is sometimes necessaryto actually estimate the sodium chloride present . In fresh buttersonly a trace of chlorides will be found
,while in salt butters even 1 2
may occur,the quantity Of salt present being almost entirely con
trolled by the taste Of the consumer .The careful estimation of the various constituents of curd and ashis only necessary when forms of adulteration are suspected whichare not easily apparent . The ques tion of preservatives and colouringmatters is now practically determined by the law of the countryconcerned .
The following table gives a number of values for water , curd , salt ,etc . , for various butters
Vo l. I I .
—20
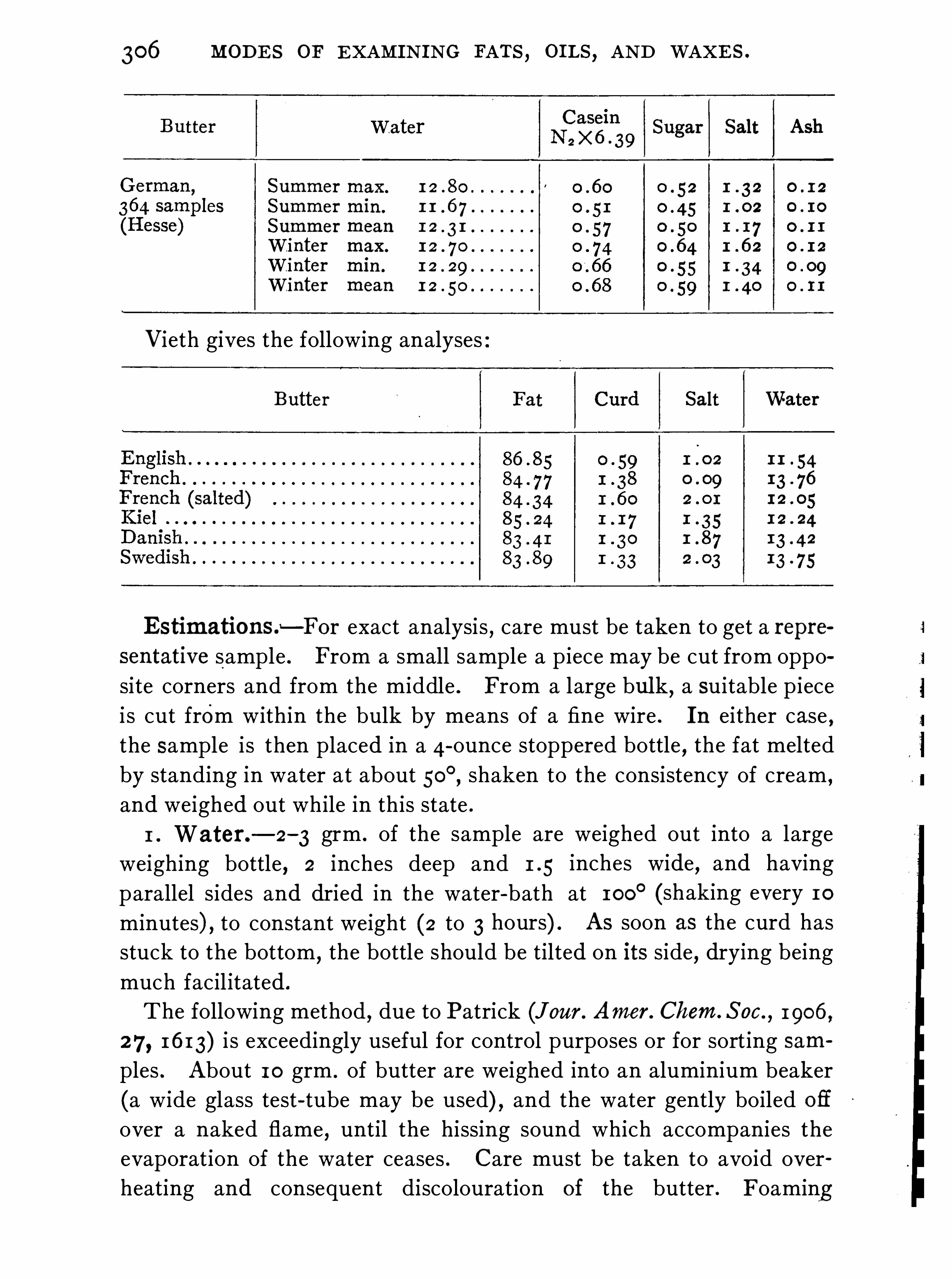
306 MODE S OF EXAMINING FATS , OILS , AND WAXE S .
Water
Summer max.
Summer min. 1 1 6 7Summer mean
Winter max. 1 2 70Winter min. 1 2 29Winter mean 1 2 50
Vieth gives the following analyses
Butter
Estimations .- For exact analysi s
,care must be taken to get a repre
sentative sample . From a small sample a piece may be cut from opposite corners and from the middle . From a large bulk
,a suitable piece
i s cut from within the bulk by means of a fine wire . In either case ,the sample i s then placed in a 4-ounce stoppered bottle , the fat meltedby standing in water at about shaken to the consistency of cream ,
and weighed out while in this state .I . Water.
—2- 3 grm . of the sample are weighed out into a largeweighing bottle , 2 inches deep and 1 .5 inches wide , and havingparallel sides and dried in the water-bath at 1 00
°
(shaking every 1 0minutes), to constant weight (2 to 3 hours). As soon as the curd hasstuck to the bottom
,the bottle should be tilted on its side , drying being
much facil itated .
The following method,due to Patrick (Jour . Amer. Chem. S oc. ,
1 906 ,
2 7 , 1 6 1 3) i s exceedingly useful for control purposes or for sorting samples . About 1 0 grm . of butter are weighed into an aluminium beaker(a wide glass test-tube may be used), and the water gently boiled Oflover a naked flame
,until the hissing sound which accompanies the
evaporation of the water ceases . Care must be taken to avoid overheating and consequent discolouration Of the butter . Foaming

308 MoDE s -
OF EXAMINING FATS , OILS,AND WAXE S .
proceeded with . If a filter has been used,this i s dropped in before
digestion . If the fat i s not thus removed,serious charring will take
place .4 . Lactose .
—This is never estimated directly,but always by
difference , unless adulteration with sugar is suspected , when the waterextract of the curd may be examined polarimetrically or with Fehling ’ssolution to confirm the result by difference .5 . To tal Ash .
—Gently ignite the solid-not-fat in a crucible at aslow a temperature as possible or chlorides will be appreciably volatilised .
6 . Sa lt—This i s best estimated in another sample of butter . 1 0
grm . of the butter are weighed into a cylinder, 5 c .c . Of chloroform
added and suffi cient water to make with the water in the,
butter 50 c .c .M ixed well
,without vigorous shaking
,and allowed to settle
,or
,better ,
separated by rotation . 1 0 c .c . of the aqueous layer 2 grm . of butter)are placed in a white porcelain dish
,20 c .c . of water added , the whole
roughly neutralised to neutral litmus-paper,and titrated with N/ 1 0
silver nitrate,using a potassium chromate indicator . The exact
strength of the silver solution should be ascertained against a knownweight of sodium chloride .If the solution i s carefully neutralised before titration
,the following
preservatives,up to the strengths given
,do not interfere
B oric acid,
sodium fluoride,
salicylic acid,and
,B
naphthol,sufficient to saturate the aqueous layer .
Co lou r ing Ma tters .—For the S imple distinction between annatto
and coal-tar colours,Doolittle ’s method is recommended (U. S . Dept.
Agri c. Bur . of Chem. Bul . 65 , About 2 c .c . of the melted andfiltered fat are dissolved in a li ttle ether
,in each of 2 test-tubes . Into
one is poured an equal volume Of dilute ( 1 3) hydrochloric acid , andinto the other an equal volume of dilute ( 1 1 0)potassium hydroxide .The mixtures are shaken well and allowed to stand .
A yellow aqueous layer in alkali tube indicates annatto .
A reddish aqueous layer in acid tube indicates azo-dyes .For the systematic examination for colouring matters , the followingmethod of Leeds (Analyst, 1 887 , 2 2 , 1 50) should be used
1 00 grm . of butter are dissolved in 300 c .c . petroleum ethersp . gr .) in a separating funnel , the curd and water drawn off , and theether washed several times wi th water . The ethereal solution is keptat 0° over-night ; i t i s then poured off from separated glycerides andshaken wi th 50 c .c . N 1 0 alkali . The aqueous layer is separated and

w.BUTTER FAT .
blue to S ame
Same with N02 N0 change .f u m e s a n dodour o f burntsugar
B lue , changing Green to y e llowat once to d i rt y i sh -green .
Part ly d e c o l Deco lourised No change .
ouri sed
S ame
P ink and dered , y e llow co lou risededges
AS slight differences of opinion as to the colours may occur , it i sadvisable where possible to check them with the actual dye .A m ixture of two dyes i s not diff erentiated
,the predominating dye
rule .5 method (A . O .A . C .) for azo colours : Spread a few dropsfied fat upon a porcelain surface and add a pinch of fuller ’sthe presence of various azo-dyes
,a pink to red colouration
309
The precipitateion of the colourTo identify
of alcohol and testtaining the concen

MODE S OF EXAMINING FATS , OI LS, AND WAXES .
will be produced in a few minutes . Some samples of fuller ’s earthact more readily than others .Special Oil-soluble
,azo-colours (Sudans) are now much used . See
A . O .A . C . methods,1 908 , p . 1 95 .
Palm Oi l . -Recent legislation,especially in the United States
,
against the sale of butter substitutes containing special colouringmatter
,has led to the use of highly coloured Oils , such as palm Oil and
fixed oil of mustard . The detection of palm oil has been speciallystudied by Crampton and S imons (J. Amer. Chem. S oc.
,1 905 , 2 7 ,
who have found that tests used for detection of rosin Oil are applicable . The following descriptions are from Leffmann and B eam ’
s
S elect Methods in F ood Analysi s , 2d Ed .,238 .
Success in the application of the tests depends On several pointsThe samples must be kept in a cool dark place until used
,fil tered at
a temperature not above the heating must be as brief as possible,
th e testing made promptly thereafter,and the reagents must be pur e
and colourless .Halphen ’
s Method—1 00 c .c . of the filtered fat are dissolved in300 c .c . of petroleum spirit and shaken out with 50 c .c. of potassiumhydroxide solution The water is drawn Off
,made distinctly
acid with hydrochloric acid,and shaken out with 1 0 c .c . carbon
tetrachloride . The latter is drawn off and part of it tested by adding2 c .c . of a mixture of 1 part Of crystallised phenol in 2 parts of carbontetrachloride with 5 drops of hydrobromic acid (sp . gr .
,The
test is best performed in a porcelain basin and the contents mixed bygentle agitation . Palm Oil gives almost immediately a bluish-green .
Li ebermann-S torch Method—1 0 c .c . of th e filtered fat are shakenwith an equal volume of acetic anhydride
,1 drop of sulphuric acid
(sp . gr .,
is added,and the mixtur e Shaken for a few seconds . If
palm Oil is present the heavier liquid that separates on standing willbe blue with a tint of green (see p .
Preservatives .—Butter i s examined systematically for preservatives
in the following way :About 50 grm . are placed in a long tube , 25 c .c . of chloroform addedand mixed with the butter . 1 00 c .c . of sodium hydrogencarbonate are added and the whole gently mixed . Vigorous shakingmust be avoided . The tube i s stood upright until the aqueous layerhas separated
,or it may be rotated . The aqueous layer i s used as
follows :

3 1 2 MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
benzoic acid . can be detected in this manner,but owing to the
solubili ty of benzoic acid in fat,the following alternative method i s
recommended for detecting smaller quantities :About 1 0 grm . of the butter are boiled for 30 minutes with 1 0 c .c . ofalcohol
,and 1 to 2 drops of di lute sulphuric acid
,the mixture well
cooled , and after pouring Off the alcohol into a separator, diluted withwater and a few drops of dilute sulphuric acid added
,extracted with
ether,and finished as above .
Fluorides .
—I o c .c . of the aqueous solution are evaporated to dryness
in a platinum crucible and igni ted gently . The ash i s moistenedwith a few drops of sulphuric acid
,and the crucible closed with a
watch-glass,coated with parafli n wax , having some scratches made
through the wax . Cold water or ice i s placed in the watch-glass andthe crucible stood on a hot plate for 2 hours . In the presence Of
fluorides the scratches will be found to be etched on the glass .If a systematic investigation is not needed
,i t may be noted that
B ori c aci d may be detected as described above,using the water
which separates from the butter on melting .
S ali cyli c acid,if present to the extent of may be detected by
Shaking the melted butter with ferric chloride solution .
fl-naphthol, if present to the extent of may be detected bymixing the butter
,rendered alkaline by a few drops of sodium car
bonate solution,with the diazo-emulsion .
B oric acid may be estimated in the following way : (Richmond andHarrison
,Analyst, 1 902 , 2 7 , 1 79 , and Richmond and Mi ller, Analyst,
25 grm . of butter and 1 0 to 1 5 c .c . Of chloroform are placed in astoppered cylinder and sufficient water added to make the total quantity of water present 25 c .c . (the butter as an average may be taken tocontain 3 .5 c .c . Of water). The substances are gently mixed andallowed to stand until separation occurs
,or centrifuged . 20
x
c .c . of theaqueous layer (containi ng the boric acid of 20 grm . of butter) are pipettedinto a 300 c .c . flask
,1 0 c .c . of a solution of phenolphthalei n in
50% alcohol) added . The mixture i s boiled,and titrated while boiling
with N/ 1 0 sulphuric acid until colourless , and then with N 1 0 sodiumhydroxide until faintly pink . 25 c .c . of glycerol or 2 grm . of mannitolare added and the liquid again titrated until pink . Then if x c .c . ofalkali used for the final titration and y c .c . required by the glycerolor mannitol used :

OLEOMARGARIN MARGARINE .
(ao—y) X 0 .0062 X 5 boric acid in The factor i s givenhere
,but it is advisable to ascertain this against the alkali used.
For the estimation of salicylic acid , see Revis and Payne (Analyst,1 907 , 3 2 ,
The question Of rancidity of butter as a whole i s the one whichchiefly concerns the analyst . The causes of this change cannot in thepresent state of our knowledge be set forth in any definite pronouncement . I t has been attributed largely to the growth of micro-organismsand moulds
,but light and oxygen play a considerable part in the chain
of factors tending to this end .
In the case Of butter fat,Laxa (Arch . f. Hyg .
,1 902 , 4 1 , 1 1 9) has
certainly shown that species of Oi di um,P eni ci llium,
and Mucor,also
Baci llus fluorescens li quefaci ens , eflect hydrolysis of the fat , thusforming fatty acids . The volatile fatty acids are in their turn furtherattacked . It seems that the method of attack is enzymic in i ts nature .While In certain forms of rancidi ty free fatty acids make their appearance and can be estimated in the usual way (page i t must not besupposed that rancidity 1s correctly measured 1n all cases by the freefatty acidity :Solste in (Chem. Rev. Fett. u . Harz. Ind .
,1 905 , 1 2 , 1 7 7) has shown
that the products causing the characteristic eff ect of rancidity can bedistilled in a current of steam
,and finds that the products from rancid
lard give strong aldehydic indications,but aldehydes hardly occur in
the case of rancid butter . For the detection of rancidity he recommends the application of Weiman ’s phosphoric reagent to the distillate .In conclusion
,i t may be said that taste and smell are , so far , the best
indicators of rancidity .
Oleomargar in . Margar ine .
Substitutes for butter are largely_ sold under the above names .
The better varieties contain as their base “ oleo-Oil,
” a productof beef fat . The best portions of the fat are taken from thenewly-killed animal
,chilled quickly
,and rendered at a low tempera
ture . The product,which is called “Premier Jus
,
” i s allowed to setslowly to a granular condition
,and then
,after placing in bags , sub
mi tted to hydraulic pressure . The soft portion of the oil i s expressed ,producing “ oleo-Oil.” The m . p . Of the “ oleo-oil ” can be adjusted tothe time of year
,by regulating the pressure employed .
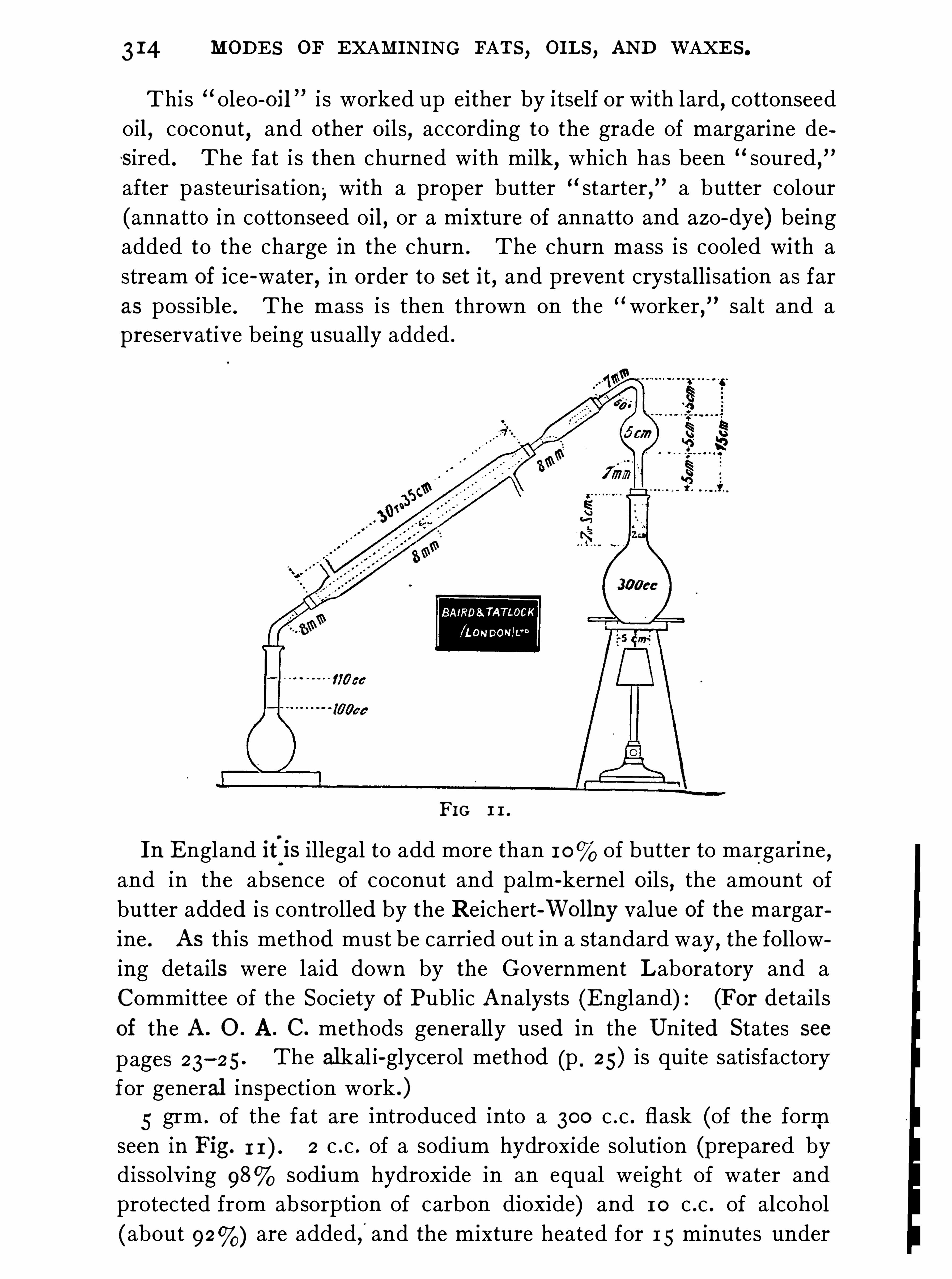
3 14 MODES OF EXAMINING FATS,OILS , AND WAXE S .
This “ oleo-Oil i s worked up either by itself or with lard,cottonseed
oil , coconut , and other oils, according to the grade of margarine des ired . The fat i s then churned with milk
,which has been “ soured
,
”
after pasteurisation, with a proper butter “ starter,
” a butter colour(annatto in cottonseed oil , or a mixture of annatto and azo-dye) beingadded to the charge in the churn . The churn mass i s cooled with astream Of ice-water , in order to set i t , and prevent crystalli sation as faras possible . The mass i s then thrown on the “worker
,
” salt and apreservative being usually added .
FIG 1 1 .
In England itiis illegal to add more than 1 0% of butter to margarine ,and in the absence of coconut and palm-kernel Oils , the amount ofbutter added i s controlled by the Reichert-Wollny value Of the margarine . As this method must be carried out in a standard way , the following details were laid down by the Government Laboratory and aCommittee of the Society Of Public Analysts (England) : (For detailsOf the A. O . A . C. methods generally used in the United S tates seepages 23—2 5 . The alkali-glycerol method (p . 25) i s quite satisfactoryfor general inspection work .)
5 grm . of the fat are introduced into a 300 c .c . flask (of the formseen in Fig. 2 c .c . of a sodium hydroxide solution (prepared bydissolving 98% sodi um hydroxide in an equal weight of water andprotected from absorption of carbon dioxide) and 1 0 c .c . of alcohol(about 92 are added, and the mixture heated for 1 5 minutes under

3 1 6 MODE S OF EXAMINING FATS , OILS , AND WAXE S .
Austro—Hungary .
—M ilk and cream may be churned with margarine,
but not more than 1 00 pints by weight of milk (or a correspondi ngamount of cream) to every 1 00 pints by weight of fats not butter fat .In Austria
,sesame oil to 1 0% by weight must be added . In Hun
gary grm . of dimethylamino-azo-benzene must be added to every1 00 kilos of margarine .B elgium
—Margarine may contain 1 0% of butter and at least 50parts of sesame oil to be added during churning and 2 parts Of drystarch to parts of fat .Sweden—Margarine must contain 1 0% sesame oil .Denmark .
—B utter fat in margarine not to exceed Sodiumchloride to be the only preservative and 1 0% sesame oil to be present .France .
—Not more than 1 0% of butter fat to be present .Germany
—M ilk and cream may be used as in Austro-Hungary .
German law insists that margarine shall contain so much sesame oil thata distinct red colour is produced when 0 .5 c .c . of the clear meltedfat i s mixed with 9 .5 c .c . of cottonseed Oil
,and the mixture shaken
with an equal volume of hydrochloric acid and a few drops Of 2%alcoholic furfural solution .
I taly .
—Margarine is not to be coloured like butter .England
—Margarine is not to contain more than I 6% of water,or more than 1 0% of butter fat .
Addendum to page 3 1 0.
Messrs . S . P . and S . S . Sadtler in a private communication to theAmerican editor report the following results of the application Of theLiebermann-Storch test to samples of oils that" may b epresent in commercial butter substi tutes or in similar substances :
Colour obtai ned
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
O O O O O O O O O O O O O O O O O O O O O O O O O O O O
Mustard Oil was easily detected by the test noted by Tolman andMunson ; namely, in saponifying with alcoholic solution of alkali , apiece of bright silver is put into the liquid . The metal will be tarnished if mustard oil is present . Mustard oil may be an adul terant ofrape Oil and thus be found in oils adul terated with the latter .

LARD.
BY C . AINSWORTH MITCHELL , B . A . (OXON), F . I . C .
(See p . Lard is the fat of the pig,melted and strained to separate
ti ssue and impuri ties . That known as “ bladder-lard ” or leaf lard isusually prepared solely from the omentum or fat surrounding the kidneys . Keg-lard is made from the fat of the entire animal
,and usually
melts between 28 and and solidifies between 24 and hence itmelts at a lower temperature than that from the omentum
,which has a
m . p . of 42 to 4 and alone has the right to be called lard . The mixedfat from the entire animal would be more appropriately termed “ hogdripping . In the American trade lard is classified into the followinggrades (Wiley ; Bull . 1 3 , U. S . Dept. Agriculture) :
1 . Neutra l lard , rendered from the perfectly fresh leaf Of the pig at atemperature between 40° and I t contains about of freefatty acids . The best quality i s used solely for making oleomar
garine , while a second quality, rendered from the back fat , i s bought byconf ectioners .
2 . Leaf lard Obtained by rendering the residue left from 1 atsteam-heat under pressure .3 . Cho ice Ke ttle-rendered Lard—Choi ce lard—This consists offat rendered in steam-j acketed open kettles from portions of the leafand back fat not used in I .
4 . Prime steam lard , rendered by direct steam heat , mainly fromthe fat of the head
,heart
,and small intestines
,though it may also con
sist of the fat from any part Of the animal .5 . Guts , a low quality, which may be derived from any part of theanimal except the heart and lungs .Compos i t ion of Lard .
—Lard contains the glycerides of stearic ,palmitic
,myristic
,lauric
,Oleic
,and linoleic acids , while Farnsteiner
(Zei t. Untersuch N ahr . Genussm.,1 899 , 2 , 1 ) has also detected traces
of linolenic acid .

MODE S OF EXAMINING FATS,OILS , AND WAXES .
The proportion of stearin di ffers with the origin of the lard . ThusHehner and Mi tchell (Analyst, 1 896 , 2 1 , 3 26) found the amounts Ofstearic acid yielded by fat from different parts of the same pig to rangefrom in the flare lard fatty acids to 9% in head fat fatty acids .These diff erences appeared to be chiefly due to variations in theamounts of the liquid fatty acids . The characteristic diff erences in theform of the crystals obtained from beef fat and lard in the Belfield test(see below) were attributed by Hehner and M itchell to a larger proportion of stearic acid in the former ; but Kreis and Hafner (Zei t. Untersuch N ahr. Genussm.
,1 904, 7 , 64 1 ) have found that the difference is
due to the lard crystals consisting of a mixed ester,heptadecyl-di
s tearin (m . p . 50 .5° and whereas beef and mutton fat crystals
both consist of palmito-distearin (m . p . 50.5° and
Exam ination of Commercia l Lard .—The Analyti cal values or
dinarily obtained in the examination of pure lard are shown in thetable on page 7 2 . There are , however , cases of frequent occurrencein which it i s difli cult to decide whether a sample IS genui ne though abnormal in its values
,or whether i t has been skilfully adulterated .
Taking into consideration the natural variations in fat from differentpigs and from different parts of the same pig
,a sample giving abnormal
values can only be regarded as suspicious,unless the presence of an
adulterating substance be detected by special tests .I odine Value .
—Lards of American origin are , as a rule , characterisedby a considerably higher iodine value than lards of European origin .
Thus of 1 00 samples of American lard examined by Voigtlander(Zei t. angew . Chem.
,1 898 , 85 7) no fewer than had an iodine
value of 6 1 to 66 , while in 4 1 cases the value exceeded 64 . On theother hand
,D ieterich who examined 11 2 samples of German lard ,
obtained iodine values of 48 to 53 with while only in 2 cases wasthe value “as high as 64 .
An estimation of the iodine value and of the stearic acid by Hebnerand M i tchell ’s method (p . 393) in the mixed fatty acids of a lard may ,when considered together
,sometimes give useful indications of adultera
tion with beef fat and a vegetable oil . Samples of lard , believed to begenuine
,examined by Hehner and M i tchell gave the followmg results :
Iodine values ,
S tearic acid , 6—7A high iodine value (e . g .
,6 in conjunction with a high proportion

MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
a trace to A rapid method Of ascertaining the amount of waterwas based by Polenske (Arb. a . d . Kai serl . Gesundhei tsamte, 1 907 , 2 5 ,
505) on the temperature at which the melted fat becomes turbid .
This has recently been confirmed by Fi scher and Schellens (Zei t.Untersuch . N ahr . Genussm.
,1 908, 1 6
,who obtained the fol
lowing results in substantial agreement with the figures of PolenskeWater
, %T urbidityOf the samples of German lard examined by Fi scher and Schellens ,none contained more than Of moisture
,and
,in their opinion
,
therefore , lard should not show a turbidity temperature exceeding 7The method i s not applicable to the determination of moisture intallow or beef fat .In the case of lard adulterated with water
,the latter may be esti
mated by heating 1 0 grm . of the sample at 1 1 0° until no more globulesof water are seen
,and ascertaining the loss In weight . This form of
adulteration i s no longer of frequent occurrence .De tec t ion of Vege table Oi ls .
-When a consideration of theanalytical values (notably the iodine value) of a sample of lard indicatesthe probable presence of a vegetable oil
,further evidence may be
Obtained from the iodine value of the liquid fatty acids,as mentioned
above .The phytosteryl acetate test (see page 30 1 ) may then be used forfurther proof
,and special tests may be applied for the detection of the
vegetable Oils most likely to be present . These are cottonseed oi l
and cottonseed “stearin ,
”sesame oi l
,maize oi l
,arachi s oi l and coconut
Cottonseed O i l and stearine may.
be detected by the silver nitratetest , Halphen
’s test and the nitric acid test (seeCottonseed Oi l, page
S esame Oi l may be detected by the furfural test,Soltsien
’s test and
Tocher ’s test (see S esame Oi l , pageMa ize Oi l . —NO distinctive colour test for this oil has been discovered .
Its presence in lard will be indicated by the phytosteryl acetate test ,the high iodine value of the liquid fatty acids
,a high yield of linolic
tetrabromide on brominating the liquid fatty acids,and the negative
results of characteristic tests for the other Oils . The Reichert-Meisslvalue may also afford confirmatory evidence (see Maize Oi l, pageArachi s Oi l i s best detected by a determination of the arachidic acidby Renard ’s method (see Arachi s Oi l, page

LARD . 3 2 1
Coconu t o i l will be indicated by the increased sapon1fication valueand the Reichert-Meissl value (see also Coconut Oi l , page 1 87 , andButter
,page
In drawing conclusions as to the adulteration Of a lard with cottonseed or sesame oil
,i t s hould be remembered that indications given by
the special colour tests may possibly be due to the pigs having been fedupon cottonseed or sesame oil-cake . Thus
, Soltsien (Chem. Zentralbl . ,
1 90 1 , I , 539) found that manyAmerican lards gave a faint colouration inHalphen
’s test for cottonseed oil similar to that which would have been
produced by an addition of about 1 % Of that oil ; while Dunlop(J . S oc. Chem. Ind. , 1 906 , 2 5 , 459) found that fat from the back andShoulder of a pig fed upon cottonseed cake gave indications in thetest corresponding to no less than 1 095. The iodine value of the fat ,however
,was quite normal . (See p .
Beef Fat and Other An ima l Fa ts—Some indications of thepresence of beef or mutton stearin in lard may be afforded by a determination of the m . p . of the fatty acids and of the proportion of stearicacid which they contain
,especially when considered in conjunction
with the iodine value and the results of special tests for vegetable Oils .Crystalli sati on of the Fat. —A qualitative test for the presence of beeffat was described by B elfield (Analyst, 1 888
,I 3 , When the fat
i s dissolved in ether,and the solution allowed to evaporate spontane
ously in a test-tube closed with a little cotton-wool , crystals are Obtainedwhich , examined under the microscope , appear broad with chiselshaped ends in the case of lard
,but needle-shaped and grouped
in fan-like bunches when derived from beef or mutton fat . Kreisand Hafner (supra) have shown that the difference is due to the crystalsconsisting of different mixed glycerides . In the case of the flare lardrecrystallisation as advocated by S tock (Analyst, 1 894 , 1 9 , 2) tends togive crystals approximating in general form to those obtained frombeef fat (Hehner and M itchell
,supra) .
When a mixture of lard and beef fat i s crystallised in this way , theform of the crystals is intermediate between those from the respectiveingredients
,though approximating more toward the form of the lard
crystals .Again
,Dunlop has shown (loc. ci t.) that when the “ plumose
crystals from beef or mutton fat are recrystallised , they finally Shownumerous individual
,flat
,chisel-ended crystals closely resembling
those Obtained in a first crystallisation from lard . Moreover, sinceVo l. I I .
-2 1

3 2 2 MODE S OF EXAMINING FATS, OILS
,AND WAXE S .
the first crystals from beef fat are more soluble than the fir st crystalsfrom lard
,recrystallisation of a mixture of the two from ether (as in
S tock ’s method) will not effect a concentration of the beef stearin .
S tock’
s P rocess (Analyst, 1 894 , 1 9 , 2) i s a quantitative applicationof the B elfield test . The deposits obtained on crystallising the fatunder fixed conditions are washed with definite quantities Of ether at adefinite temperature
,and weighed . The results are then compared
with those Obtained under the same definite conditions from standardmixtures Of lards Of different m . p. with diff erent proportions of beefstearin .
In the ligh t of the experiments of B ehner and Mi tchell (Analyst,1 896 , 2 1 , 3 28) and of Dunlop (loc. further investigation of thenature and behaviour of the deposits from mixed fat appears necessarybefore trust can be placed in the results of this test .With regard to the original qualitative test
,Dunlop has shown that
i t is necessary to examine the crystals under a magnification Of 300 to400 diameters to see the form of individual crystals , and not to relysolely on the fan-like grouping of the bunches of crystals
,as seen under
a magnification of 1 00 diameters as a proof of the presence of beefstearin .
At i ts best,the test can , as yet , only be regarded as affording con
firmatory evidence of adulteration ; and , as was pointed out by Hebner(Analyst, 1 902 , 2 7 , the occurrence of the “ beef-form ” of crystalsshould only be regarded as a proof Of the presence of beef or muttonfat when the presence of a vegetable Oil has been detected and the lardhas a high iodine value .

3 24 MODE S OF EXAMINING FATS , OILS,AND WAXE S .
from mixed and unmixed seed from various sources . I t has beenshown that linseed from different localities carefully freed from foreignseed yields an oil quite equal to that from B altic seed . The superiorityof B altic oil i s entirely due to the more careful harvesting . The fore ign seeds usually aff ect the drying qualities of the Oil . Linseed variesin size and colour . The usual colours are a purplish-brown , and areddish-brown
,but a white variety i s known . This variety i s grown
in some parts of the North-West Provinces of India,but no care i s
taken to keep the strain pure . According to Church , I the seed contains 45% of oil , the product Obtained from a picked sample beingnearly colourless .Leather (loc. ci t.) describes—experiments carried out in India withregard to the efl
’ect of transference of rich seed to localities which
usually produce a seed Of low Oil content ; the results show a reductionin the percentage yield under these conditions .Prepara t ion of Oil from Seed .
—The percentage of oil obtainedfrom a given variety of seed cannot be said to be constant , as the yieldi s influenced by conditions of soil
,climate, ripeness , seasons , etc .
Schindler and Waschata2 found in 43 samples Of linseed oil fromto of oil
,an average of The following table ,
showing the Oil contents of seed from different sources,i s due to them .
O O O O O O O O O O O O O O O O O O O O O O O O O
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
O O O O O O O O O O O O O O O O O O O O O O O O O O O
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
Extraction with ethyl ether yields larger quantities of crude oil thanis Obtained by using petroleum ether .Leather3 has examined seeds of Indian origin , and by a processof double extraction using ethyl ether has found the oil contents of different seeds vary from 3 to The double extraction con
1 Ch em is try of Paint s and Pa int ing , 1 9 0 1 .
2 B ened ikt and Ulze t . Analy se der F ette , etc. , 1 908 .
S oc. Chem. Ind . , (Abst ract ) 1 9 0 7 , 26 , 6 2 2 .

LINSEED OIL . 3 25
S isted in first extracting the bulk of the O il,then drying and
,crushing
the residue before a second extraction . This method gave values 3%higher than by single extraction .
According to Audes,
I linseed contains 8% Of water, 33% Oi l
,25%
of albuminoid matter,traces of tannin
, 4 to 5% of ash . When linseedis cultivated for Oil only , a larger yield is Obtained than when fibrehas been desired—in the latter case the seed is not ripened sufficiently
,
and therefore does not possess the full oil content .The seed is kept from 2 to 6 months before pressing
,as new seed
yields a turbid and more viscous product containing much mucilage .The oil expressed from American and Russian seed usually containsa larger proportion of moisture than Calcutta and East Indian seedwhich is invariably full-grown and matured
,and consequently gives a
greater yield Of oil .The extraction of Oil from seed is carried out by one Of the followingmethods :
1 . Co ld pressure, y1e ld 20%
2 . Ho t pressure , yie ld3 . Extraction with various so lvents , yield 3 2% -
33
1 . Cold pressure gives a light coloured golden-yellow product , whichis not unpleasant to the taste . In some parts Of Russia
,India
,and
Germany i t i s considered edible ; further i t contains less solid glyceridesand consequently is a better drying oil .
2 . Hot pressure gives a darker coloured product , having an acridflavour
,which renders it only suitable for technical purposes .
3 . Extracti on ,which is not extensively practised in England .
M itarewski2 states that oil extracted by the usual solvents , benzene ,carbon disulphide
,carbon tetrachloride and naphtha
,lacks the
characteristic smell of l inseed or pressed Oil. In the case of carbondisulphide extraction
,the oil has a strong smell of garlic , and a dark
reddish colour,while Oil extracted by other solvents i s of a yellowish
green colour . Solvents do not cause any determinable alteration inthe Oil or cake
,except carbon disulphide
,which increases the quantity
of volatile acids—hence high saponification values result . Toch3 statesthat the solvent is liable to extract proteids from the seed , thusdecreasing its value as a food
,and he advises examination of oil for
1 Vege table Fat s and Oils , 1 9 02 .
2 J . S oc. Chem . Ind 1 9 06 , 2 5 , 8 1 8 .
3 Technology of Mixed Pa int s , 1 9 0 7 .

3 26 MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
nitrogen in order to ascertain whether it i s extracted or,as he terms
i t,
“ new process ” oil .According to Petit
,
I extraction yields a product containing all thefats present in the seed
,some of which do not dry .
Fassbender and Kern2 have shown that i t i s possible to obtain a pureoil from an impure seed , as the cake retains a large proportion Of theforeign oils .Linseed Cak e .
—T he press cakes Obtained by the pressing of linseedfurn ish a very valuable cattle food which retains about 1 0% of theOil . The cake obtained by the extraction process is of littl e or novalue as food .
According to the researches of Dunston,3 Henry
,
3 ' 4 and Auld , 3 ' 4
l inseed cake contains a cyanogenetic glucoside “l inamarin” togetherwith an enzyme which occurs naturally in the flax plant and in linseed .
In the ordinary process of hot pressing the temperature is \high enoughto destroy the enzyme
, ,but with cold pressure the enzyme is active , and
in the presence of water acts on the glucoside with the production ofhydrocyanic acid . I t is suggested that cases Of cattle poisoning previously considered to be due to the presence of foreign seeds mightbe explained by this enzyme action .
Refining—The varieties of linseed oil usually recognised in com
merce are Raw,
” “Refined
,
” and “B oiled .
” The latteri s dealt with in a subsequent chapter .Raw linseed Oil varies in colour from amber-yellow to yellowishbrown and has frequently a greenish fluoresence , and a somewhatsweet
,sickly odour . In the refining process these two character
istics are removed . According to Toch (loc. the colouring ofl inseed Oil is usually due to chlorophyll
,or
,as occasionally happens
when a reddish cast is Observed,to erythrophyll ; and bleaching de
pends on the conversion of these colouring matters into yellowxanthophyll .For ordinary refining
,the process generally adopted i s that of
agitating the raw Oil after a preliminary settling and heating , whichcauses the coagulation of impur ities
,with 1 %—2% of sulphuric acid
(sp . gr . in lead-lined tanks . The charred mass formed bythe acid carries down the bulk Of the impurities
,and after again being
1 Whit e Lead and Zinc Paint s , 1 9 0 7 .
2 Zei t. angew . Chem . , 1 89 7 , 3 3 1 .
3 P roc. Roy . S oc. , 1 9 06 ,B
, 7 8 , 1 4 5 .
4 J. S oc. Chem . I nd . , 1 9 08 , 2 7 , 4 2 8 .

MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
stands its colour much better on heating,hence the preference for
B altic Oil in varnish-making ; also it is “ harder ” and does not tend todeposi t mucilage or break on standing . For varnish -making it isessential that the oil should be free from mucilage
,which
,as previ
ously stated , i s deposited on standing . Metallic lead appears to hastenthe deposition
,hence the practice of storing oil in lead-lined vessels .
Thompson 1 has investigated this question,and has found that the
formation of mucilage or coagulation of oil i s due to the presence ofphosphates and sulphates . Thompson has examined the “mucilage
,
”
spawn,
” or break,
” and after removal of Oil by petroleum-etherfound i t to contain of ash
,which
,on analys i s
,showed
CaO ,MgO ,
P 205 , and traces of sulphate . It will beseen that storage effects the removal of those substances which renderoil unsuitable for varnish-making . (See paragraph on Ash .)Niegmann
2 states that linseed oil to be used for varnish,boiled oil
,
l inoleum,etc .
,should not form flocks when quickly heated to boiling
in a test-tube . Also that only Oils which do not give any deposit onstanding for a long time
,which assume a clear greenish colour on
boiling,and do not Show turbidity on standing when exposed to air
for a month should be used . (See Lippert , Zei tsch . angew. Chem.,
Raw oil , intended for making pale boiled oil or varnish , should nothave a sp . gr . below otherwise i t will probably contain a proportion of other seed oils which will impair its drying qualities ; 3%of such admixture i s the maximum allowable in linseed to be used forproducing oil for varnish work .
In practice , the best Oil i s that which dries most perfectly , but therapidity of drying and the condition of the ultimate product are important factors to be considered in judging i ts quality . Thus the driedOil may be tough , very elastic , hard and brittle , or rotten . An oil giving a hard product is to be preferred
,as elastici ty can be readily
imparted in the after-treatment if required .
Extraction from Paint —Linseed oil (both raw and boiled) i sthe most generally employed vehicle for paint . For high-class work ,as in artists ’ colours
,poppy seed and walnut seed oil are frequently
used . It can be readily extracted therefrom by the use of any ordinary ‘
solvent . In some cases extraction may not be complete owing to
1 J. Amer . Chem . S oc. , 1 9 03 , 2 5 , 7 1 6 .
2 Chem . Ze i t. , 1 9 05 , 29 , 46 5 .

LINSEED OIL .
di fi cult to free the solution in ether fromthe finely divided pigment being held inils
, a centrifuge must be used , and this is
Of linseed oil is b est carried out by shaking the paintthen allowing the pigment to settle
,and evaporating
clear solution in a tared vessel . I t is to be notedes of oil required by diff erent pigments to produce a
rs considerably .
substitutes for linseed oil in generalfor painting work have been
patented,but these have not so far met with approval .
In America,according to Holley and Ladd , I cottonseed oil , although
seldom found in house paints , i s often used in barn paints .Compos i t ion .
—The elementary composition of linseed oil i s givenby different authorities as below .
Compo sitionSource Observer
Co ld drawn 78 1 1 1 0 96 1 0 93 }S chadler7 5 2 7 1 0 98 1 3 85
7 7 5 7 1 1 3 1 1 1 0 C loez .
7 5 2 1 1 0 7 1 1 4 08 Williams,
2
Mean of 2 es
timations .
Extracted from Balt ic seed by pe tro 76 24 1 0 63 1 3 I 3 B earn.
leum ether.
1 Holley and Ladd . Mixed Pa in t s , e t c . , 1 9 08 .
2 A na ly s t. 1 89 8 , 23 , 2 5 3 .
3 29
ace between the Oil and the pigment , cf .
Chem. Ind .,1 907 , 26
,who found a
fatty matter in white lead,probablywhite lead .
be distilledoil extracted . I t must be pointed outto obtain values from an extracted Oiloriginal Oil used
,cf . Boettinger , Chem.
c. Chem. Ind.,1 895 , 1 4, 1 69 . Also see

330 MODE S OF EXAMINING FATS , OILS,AND WAXE S .
Church (loc. ci t.) states that oil extracted by carbon disulphide contains more oxygen and less carbon than oil obtained by pressure .The chemical composition of linseed oil is not completely understood . The following figures are quoted
,but the reader i s referred to
Lewkowitsch (loc. ci t.) for a résumé of present views together with thereferences to original papers .Linseed Oil contains :
1 0%—1 5% glycerides of solid fatty acids ;i . e .
,stearic
,palmitic
,and myristic .
85%—90% l iquid glycerides .B azura and Grussner state that the fatty acids from the liquidglycerides consist Of
O le ic acidL inole ic acidL ino lenic acidI solino len ic ac id
Fahrion gives the followingUnsaponifiable matterCombined fatty acids—palmitic and myristic acidsO le ic acidL ino le ic acidL ino lenic ac1d
SeeFokin . Chem. Centr .
,abstr .
,1 902 , 2 , 8 , 60 1 .
Tolman and Munson . J . Amer. Chem. S oc. , 1 903 , 25 , 960.
Fahrion . Zei ts . angew . Chem.,1 903 , 1 1 93 .
Fahrion . I bid. ,1 904 , 1 484 .
Hallei' . Compt. rend ,1 906 , 1 46 , 259 .
Fokin . J. S oc . Chem. Ind. 1 906 , 25 , 93 5 .
B edford . Ueber die unges '
attigten Satiren des L einols ,” InauguralDi ssertati on ,
Halle a/s.,1 906 .
Erdmann and B edford . B er .,1 909 , 42 , 1 3 24 .
B edford and R ispe . Mid , 1 334 .
Specific Gravi ty .—The sp . gr . i s usually at bu t may
range between and depending on source , age , exposure ,and method of refining .
See table of constants,and general chapter on sp . gr . B allantyne1
has investigated the influence of exposure to light and air on linseed oil .1 J. S oc. Chem . Ind . , 1 89 1 . 1 0, 3 0.
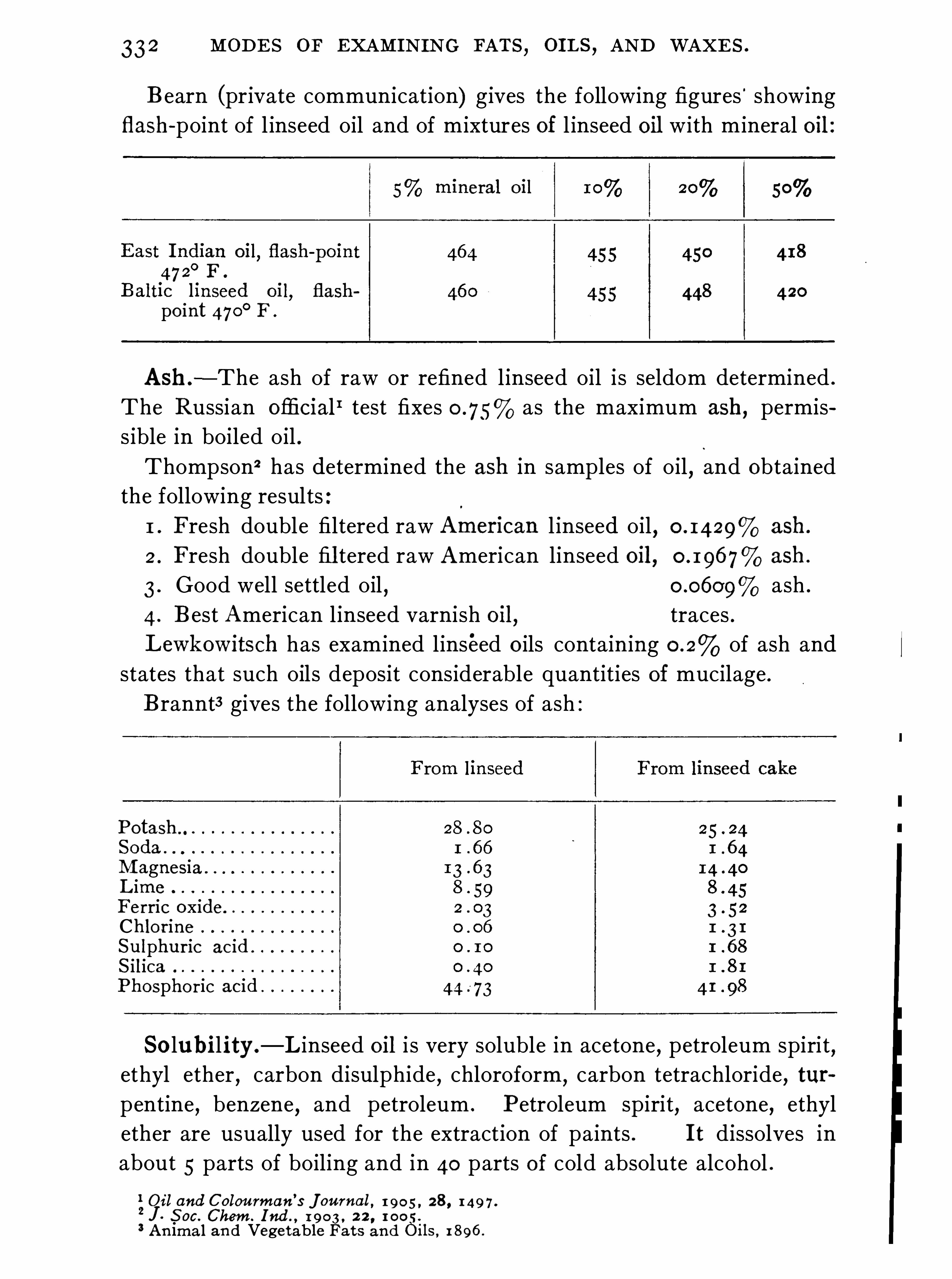
33 2 MODE S or EXAMINING FATS,OILS , AND WAXE S .
B earn (private communication) gives the following figures ‘ showingflash-point of l inseed oil and of mixtures Of l inseed Oil with mineral Oil :
47 2° F .
po int 470° F .
Ash .—The ash of raw or refined linseed oil i s seldom determined .
The Russian ofli cial I test fixes as the maximum ash , permissible in boiled oil .
5
Thompson2 has determined the ash in samples of oil,and Obtained
the following results1 . Fresh double filtered raw Amer1can l inseed oil , ash .
2 . Fresh double fil tered raw American linseed Oil,
ash .
3 . Good well settled oil,
ash .
4 . B est American linseed varnish Oil,
traces .L ewkowitsch has examined lins 'eed Oils containing of ash andstates that such oils deposit considerable quantities Of mucilage .B rannt3 gives the following analyses of ash :
PotashS oda
MagnesiaL ime
Ferric oxide .
Ch lorineS u lphuric ac idS ilicaPhospho ric acid
So lubi l i ty .—Linseed Oil i s very soluble in acetone
,petroleum spiri t ,
ethyl ether,carbon disulphide
,chloroform
,carbon tetrachloride , tur
pentine,benzene
,and petroleum . Petroleum spirit
,acetone , ethyl
ether are usually used for the extraction of paints . I t dissolves inabout 5 parts of boiling and in 40 parts Of cold absolute alcohol .
1 Oi l and Colou rman’
s J ournal , 1 9 0 5 , 28 , 1 49 7 .
2 J . S oc. Chem . Ind . , 1 00 53 An imal and Vegetable Fat s and Oi ls , 1 89 6 .

LINSEED OIL . 333
Free Fatty Ac i ds .—The amount of free fatty acids present in alinseed
Oil is low ,usually ranging from 0 .5 to
NordlingerI and Thomson
,and B allantyne2 have , however , ex
amined samples containing free fatty acids ranging from toAccording to Lippert
,3 “ livering of paints is due to the
presence of free fatty acids . The proportion of free acids increasewith age . A sample Of raw B altic linseed oil (33 years Old) examinedby Bearn ,
contained 5% free fatty acids . The sample had a veryrancid taste . (See note on linseed soap stock .)Unsapon ifiable Mat te r .—Thomson and B allantyne4 found oilfrom various sources to contain to WilliamsS foundto in raw Oil
,and 1 .3 to 2 .3% in an Oil boiled at a high temperature .
He is of the opinion that any Oil containing more than 2 .5% mustbe considered as adulterated . He also states that low temperatureboiling does not affect the amount of unsaponifiable matter .Fendler6 states that the normal amount of unsaponifiable matterdoes not exceed and that blowing or oxidation does not increasethe amount . Linseed Oil obtained by pressure does not containmore unsaponifiable matter than that obtained by extraction . Herecommends the determination of the mineral matter by the iodineabsorption of the unsaponifiable matter , and states that the unsaponifiable matter of linseed Oil i s usually solid . According to Niegeman
,
the iodine value of the unsaponifiable matter does not vary greatly ;further
,the value is not decreased by exposure of the unsaponifiable
matter to air in the dark,though light is fatal . When dried to a skin ,
no iodine value is Obtained .
Niegeman ’ found in 1 8 samples a maximum value of and aminimum of —an average of 1 .3 5 the average being exceededin 7 samples . In view of these figures
,he Suggests that i t is unfair
to condemn an oil solely because its unsaponifiable matter exceeds
Thoms and Fendler8 fix as limit,and state that exposure to
light and air,or even breaking the oil
,does not increase this value .
The value is only increased when the linseed Oil has been dried to a
1 J . S oc. Chem. Ind 1 889 , 8 , 806 .
2 I bi d . , 1 8 1 , 1 0, 2 3 6 .
3 Zei tsch . angew. Chem. , 1 89 7 , 7 7 9 .
4 J . S oc. Chem . I nd . , 1 89 1 , 1 0, 3 3 6 .
5 I bid . , 1 89 8 , 1 7 , 3 05 .
6 B er . , I 04 , 3 7 . 2 9 4 .
7 Chem. a t , 1 9 04 , 28 , 9 7 .
8 End , 1 9 06 , 30, 83 2 .

334 MODE S OF EXAMINING FATS, OILS , AND WAXE S .
varnish , and there is a reduced iodine value . The proportion Of
unsaponifiable matter is not less in extracted oil than in oil Obtained bypressure (confirming Fendler) thus in pressed Oil
,and in
Oil extracted by ether from the same seed .
Thomson and DunlopI found
,in 5 samples , unsaponifiable matter
ranging from to averageAs the result of I 50 estimations B earn found an average of
unsaponifiable matter ; only in rare cases did the figure exceedSapon ifi cat ion Va lue .
—Wright and M itchell2 quote values rangingfrom 1 83 to 2 2 1 , while Lewkowitsch3 gives 1 90 toIodine Value .
-Many Of the earliest recorded iodine values are fartoo low
,viz . , about 1 50. This is due to the fact that the proper condi
tions for the estimation had not been ascertained . By the adoption ofproper methods perfectly satisfactory results are obtained
,and this
value is one of . the most characteri stic tests for identification purposes , though discretion must be used in interpreting low figures .Linseed oil (raw and refined) has been found by various workersto have an iodine value varying from 1 65 to 200
,the average values
being 1 75 to 1 90 . These values are higher th an those of any fatty Oilother than perilla oil .The highest iodine value on record appears to be that Obtained byThomson and Dunlop
,4 who found to be the value of a sample
of Oil expressed from R iga seed . The following figures due to Thomson and Dunlop Show that an apparent relationship exists betweenthe iodine value and the refractive index .
Sourceat 25
°
L inseed o i l (Riga)L inseed o i l (S t .L inseed Oi l (North American)S kate liver Oi lLinseed o i l (Calcutta)Haddock liver O i lLinseed o il, river plateWh iting liver Oi l
The above figures Show a close resemblance between liver Oilsand linseed Oil . Thomson and Dunlop consider that Wijs ’ method1 Ana ly st, 1 9 06 , 3 1 , 28 1 .
1 An imal and Vegetable F ixed Oils and Fats , e tc . , 1 903 .
3 Ch emical Technology and Ana ly s is of Oils and Fat s , 1 904 .
A naly st , 1 906 , 3 1 , 28 1 .

336 MODE S OF EXAMINING FATS,OI LS , AND WAXE S .
The eff ect of boiling on the iodine value is shown in the section onboiled oil .3 . Linseed Oil which has been extracted from a paint i s frequentlyfound to have low iodine values . B oettinger
1 found that linseed oil,
having an original iodine value of altered when ground withwhite lead—after 1 7 days—to and to after 2 months .Whiting and ochre showed the same changes .The iodine value of a sample i s a fairly good indication as to itsdrying properties . Fox has shown that the oxygen absorption Of an oilbears a relationship to the iodine value . This relationship i s clearlyshown in the ozone figures quoted on page 339 .
See paragraph on sp . gr. for relationship between sp . gr . and iodinevalue . Also see Williams (J. S oc. Chem. Ind.
,1 900, 1 9 ,
For influence Of atmospheric oxidation on constants see Shermanand Falk (J. Amer. Chem. S oc.
,1 901 , 23 , 1 56 , 1 905 , 2 7 ,
Brom ine Values .—McIlhiney
2 gives the following as being ordinaryaverage figures for linseed oil :
B rom ine substitutionvalue
less than 7
McIlhiney states that a low addition figure may be caused by rosin ,rosin Oil
,benzene
,or mineral oils
,which usually have figures below I 5 ,
or by the presence of some other seed Oil,e . g .
,corn or cottonseed oil
,
which have figures Of 73 and 63 , respectively , or by boiled oil (Oldfashioned boiling).Turpentine i s the only adulterant causing an addition figure higherthan 1 1 0 .
B romine Substi tuti on—A higher figure than 7 indicates turpentine ,rosin
,rosin oil
,benzene
,or heavy petroleum . M ineral acid would
also raise the figure,but this could be detected by high free acid
value .Inso luble Bromides .
—B ehner andM i tchell,Analyst, 1 898 , 23 , 3 1 0 ,
have made a number of experiments confirming those of B azura andothers as to the oxidation and bromination products of linseed andother drying Oils . For details of these see p . 3 55 .
1 Chem . Zei t 1 89 8 , 22 , 1 02 . Al so Chem. Zei t. , 1 89 8 , 22 , 5 5 8 .
2 Parker Mc Ilh iney ,Report on L inseed Oi l t o Comm issioner of Agricu lture , New York
S tat e , 1 9 0 1 , or see J. Amer. Chem . S oc. , 1 89 9 , 2 1 , 1 084 . E t i bid . , 1 9 02 , 24 , 1 1 09 .

LINSEED OIL . 33 7
1 90 1 , 1 4 , 3 59) has modified,Hehner
applied same to the detection Of drying
this test for the detection of raw linseeds . Lewkowi tsch2 prefers to bromi
prepared with due precautions against
des see Proctor and B ennett,J. S oc.
importance in the examination ofon Adulteration .
page 3 50 for values .m . p . of the linseed Oil fatty acids
1 7 to whilst the solidifying-point7-5
°
to to as the titer test of
at there are 98% fatty acids present in the fat ;refractive index at 60°
A . Pegami4 have determined the molecular equ iva
fatty acids in linseed o il,and give the following
L inseed Oi l,fresh
co ld-drawn
acid va lue (3 )saponification valuecalculated from acidvalue ( 5)
calcu lated from saponification value
difference between the recorded and calculated molecular weightsbed by the authors to lactones . In order to avoid this error ,
2 , 9 , 1 8 2 , 2 04 , 205 .

338 MODE S OF EXAMINING FATS , OILS , AND WAXE S .
they hydrate the lactones by boiling the fatty acids with an excess ofalkali , and titrate the excess .Refrac t ive Index .
—Owing to the fact that the refractive indexOf l inse ed Oil does not differ greatly from that of other O ils
,i ts value
as a means of detection of adulterants is somewhat limited . Rosin,
rosin Oil,and mineral oil raise the refractive index
,but i t must be
borne in m ind that oxidation has the same effect . WegerI found
(using a Zeiss refractometer) that a sample of raw linseed oil gave areading of while
,after treatment with 5% of manganese-lead
resinate,this was raised to 858 ° and
,after heating for 1 hour,
toSjollema
2 prefers the refractive index to the iodine value as ameans Of detection of adulteration
,and points out that the reading
is lowered by the presence Of fatty acids . Sjollema suggests a correetion of scale division for each degree centigrade when the temperature is not and confirms the fact that the refractive index isincreased by oxidation .
Harvey,3 as a result of the examination (using an Abbé refractom
eter) of 2 7 samples of linseed oil , obtained at 20° values varying fromto
Proctor and Holmes,4 using a Zeiss refractometer
,found linseed oil
with an iodine value of 1 74 had refractive index of atalso linseed oil having iodine value 1 64 had refractive index(see “
B lownSee also Thomson and Dunlop5 whose values are quoted in paragraphon Iodine Values .Po lar ime tr ic Ro ta t ion .
—Linseed oil i s (at any rate for allpractical purposes) Optically inactive
,and therefore adulteration by
rosin oil can be detected by its rotatory power .B ishop
,
6 using a Laurent saccharimeter—20 cm . tube—found—0 .3
° rotation by a sample of linseed oil .Thoerner7 found no rotation at 50Filsinger8 advises the use of a filtered solution of the Oil in chloroform or alcohol
,and gives the following figures as the result of the
1 Zei tsch . angew . Chem 1 89 9 , 2 9 7 .
2 J . S oc. Chem . I nd . , 1 9 03 , 2 2 , 9 6 7 . (Abst ract .)3 S oc. Chem . I nd . , 1 9 05 , 24 , 7 1 8 .
4 bi d . , 1 9 0 5 , 24 , 1 2 89 .
6 A na ly s t , 1 9 06 , 3 1 , 28 3 .
S oc. Chem. I nd 9 9 0.
7 Chem . Zei t. , 1 89 4 , 1 1 5 4 .
8 Chem . Ze i t. , 1 89 4 , 1 00 5 .

MODE S OF EXAMINING FATS,OI LS
,AND WAXE S .
During the year 1 909 the imports of Soya bean to England becameconsiderable in quantity
,over tons being received . This has
been crushed in quanti ty for the first time in England, and in view of
the prevailing difference in the prices of Soya bean Oil and linseed Oilthe adulteration of the latter by the former is not improbable and suchadulteration must be sought for . The drying qualiti es Of l inseed oil
would of course be impaired by such adul teration .
The following qualitative tests may be applied for the indicationof adulterants
,though their absolute reliablity cannot be vouched
for . A quantitative exam ination i s always advised .
Fi sh oils may Often be detected by Odour. The delicacy of this test i sincreased by rubbing the warm oil on the hands
,and
,according to
Brannt,
of fish oil can be detected by this means . Fi sh oiladulteration can also be recognised by a peculiar scum which risesto the surface when such oils are boiled . B earn blows steam throughthe warm oil and states that 2% of fish Oil can be recognised by thefishy smell of the issuing steam .
The sulphuric acid colour test is a useful indication of the purityof linseed oil . With a genuine sample
,a dark brown clot i s formed ;
if rosin Oil or fish Oil be present,a reddish-brown spot quickly forms
,
which,in the former case
,retains its red tint for a long time
,while a
peculiar scum forms over it . This test i s also applicable to the detection of rosin oil in boiled linseed Oil .Fish Oi ls may also be detected by the darkening produced by passinga rapid stream of chlorine through the oil or by the reddish colourproduced by boiling the Oil with alcoholic Sodium hydroxide . As atest for cod oil
,which i s not unfrequently used in‘ the case of linseed
Oil intended for the preparation of printing ink,A . Morell recommends
the following test : 1 0 grm .
"of the oil are well agitated with 3 ‘grm .
of nitric acid,and the whole left to stand . Wi th pure linseed
oil the colour changes during the stirring to sea-green,afterward
becoming dirty greenish-yellow,while the acid assumes a light yellow
colour. In presence Of 5% Of cod oil,after standing some time ,
the oil i s said to acquire a dark brown colour,and the acid is tinged
orange or dark yellow,according to the proportion of the adulterant
present . A similar test has been described by Conrath for thedetection Of rosin Oil .
Japan wood oil is distinguished by the very hard black clot i t giveswith sulphuric acid
,and by yielding a highly coloured semi-solid

LINSEED OI L .
product with the Elai din test . If heated for a short time to aboutthe O il becomes a transparent jelly
,the change occurring either at once
or on cooling .
Cottonseed can be detected by the Halphen colour reaction (see pageRosin (colophony) can be detected by the L iebermann-S torch
reaction . If the sample i s very dark,Lewkowitsch recommends the
extraction of the colophony with alcohol and the testing of this extract .L ippert
I i s of opinion that the Liebermann-S torch reaction is notconclusive enough
,but certainly ought not to be omitted in a qualita
tive examination .
The following statement shows how the diff erent constants areaffected by various adulterants
,and will be of value in the examination
of a suspected Oil .
Effect of Adu l terat ion on Characteris t ics of Linseed Oi l .
Sp . Gr .—~ M ineral and foreign seed oils are lighter
,while rosin and
rosin oils are heavier ; thus , by a jud1c1ous m i xture of each class ofadulterant
,extensive adulteration can be effected without alteration of
the sp . gr . A mixture of mineral and rosin oil may be used ,rosin itself being sometimes also added . The mineral Oil i s usually oflow sp . gr . 5 as the heavier oils are too greasy . The-rosinoil employed for adulterating linseed oil is free from smell even whenheated
,but has a peculiar taste which is not masked by the linseed Oil .
Tung oil may be added as its sp . gr. i s higher than that of linseed oil ;it would
,however
,be detected by the bromide test .
Fi lm Tes t .—By means of this test non-drying oils can be detectedif present in suffi cient quantity
,the extent of drying varying with the
extent of adulteration . Rosin oil causes linseed Oil to remain “ tacky ”and prevents its ever becoming hard .
If the hydrocarbons are volatile,they may be removed by distillation
in steam .
Solidificat ion-po int .—The solidifying pOint of pure linseed oil i sgiven on ' page 70 ,
and samples containing other seed O ils solidify at ahigher temperature . The same remark applies to the relative fusibility of the fatty acids , those prepared from cottonseed Oil having anexceptionally high m . p .
1 Chem. Rev. F ett-Harz-Ind . , 1 9 0 5 , 1 2 , 4 .

342 MODE S OF EXAMINING FATS, OILS , AND WAXES .
Maumené and Brom ine Thermal Values—The values obtainedfor linseed oil are not characteri stic enough to be relied on as definiteindications of adulteration
,because some fish O ils have both high iodine
and thermal values .Refractometer Reading—By thi s means rosin and rosin oil andmineral oils may be detected .
Saponification Value .—Low saponification values may be due to
adulteration by mineral oils,rosin oil
,or turpentine . Rape oil i s in
dicated by a low value . The small proportion usually present (bycontamination in seed) i s not detected .
Unsapon ifiable Matter.—Adulteration by mineral Oil and rosin
oil will be detected by this estimation . Volatile hydrocarbons,such as
benzene,turpentine
,though unsaponifiable , will not be
'
determined
as such .
Iodine Value .—Lewkowitsch considers that if the iodine value of a
linseed Oil i s below 1 70 i t can be justly presumed that adulteration hastaken place
,ei ther in the seed itself before the oil was expressed or in
the Oil , but insists on the recognition of the fact that a low iodine valuei s (under certain circumstances) quite consistent with puri ty . See
paragraph on Iodine Value . A high iodine value is not in itself a proofof the puri ty
,since fish oils
,rosin oils
,and even drying Oils may be pres
ent in considerable quantities,and yet give figures quite within the
range of ordinary practice . The tendency of foreign seed Oils i s toreduce the iodine value
,while fish Oils may scarcely have any effect
at all . (See Thomson and Dunlop ’s figures in section on IodineValues .) The phytosteryl acetate test is reliable for the detection offish (liver) or blubber Oils .Brom ine Values .
—Turpentine will cause a sample of linseed Oil togive high figures
,while rosin and rosin Oi l will be indicated by a low
bromine absorption and addition value , and a high bromine substitution value .Inso luble Brom ide Test—This test i s of great value in the de
tection of adulteration , as will be appreciated by an examination of thetable given on page 3 55 , where i t i s evident that many of the adulterantsyield no insoluble bromides
,or in any case much small er quantities
than pure linseed Oil . The behaviour of insoluble bromides on heating i s of importance . Fi sh Oils which give high yields of insolublebromides can be distinguished from linseed oil by their diff erent behaviour on heating . The insoluble linolenic hexabromides from

MODE S OF EXAMINING FATS , OILS, AND WAXE S .
ing converted into a tough or hard varni sh is shared by all fatty Oils ofvegetable or animal origin . The transformation may be very slow
,
but i t ultimately takes place . This tendency is much enhanced byheating the oil while passing a current of air through or over i t (see“B oiled By continued boiling the Oil becomes very thick , andmay be drawn out into elastic threads
,wh ich are very sticky but do
not produce a greasy stain on paper . This product i s used in themanufacture of printing ink . The drying of linseed oil i s facilitatedby the use Of siccatives .When spread in a film
,linseed oil dries to a substance known as
linoxyn . Linoxyn is a neutral substance insoluble in ether or ordinarysolvents . The constitution of linoxyn has not yet been ascertained
,
and i t was formerly considered to be the final oxidation productof linseed Oil . Reid
,
I however,has Shown that on long exposure
,from
2 to 5 years , depending on various conditions, i t i s transformed , firstinto a semi-fluid
,and finally into a vi scous fluid
,which he terms “ su
peroxidised linseed Oil .” The product is much darker in colour thanordinary linseed Oil , i s heavier than water , almost completely solublein alcohol
,soluble to a considerable extent in water
,and strongly
acid,forming solid compounds with most basic pigments . All -drying
oils yield linoxyn,the amount varying with the amount of linolenic or
linoleic acid present . L ewkowitsch has proved that if linseed oil iskept protected from light
,moisture
,and air , i t keeps indefinitely .
The chemical changes which occur . in the boiling and drying oflinseed oil are very imperfectly understood . According to Mulder
,
part of the linolin is decomposed during the boiling , with formation oflinoleic anhydride
,or a more highly oxidised body
,such as hydroxy
linoleic acid . According to Fox,the oxidation products are formed
from the acids and the glycerol is decomposed into acids of the acrylicseries
,forming the irritating vapours which always accompany oil-boil
ing . Acetic and formic acids are prom inent constituents of thesevapours
,and carbon dioxide and water are also present . The state
ments of Mulder and Fox are probably too sweeping . Allen isolatedof nearly pure glycerol from the products of the saponification
of linseed oil which had been boiled by the steam process . B auerand Hazura consider Mulder ’s ‘explanation Of the drying of linseedOil to be only partially correct . They investigated the subject andarrived at the following conclusions :1 J. S oc . Chem . Ind . , 1 89 4 . I 3 , 1 0 20 .

LINS EED OI L .
linolenic acid an oil contains , the more rapidly it
ly that the linolenic acids are the firstthat linoleic acid plays a subordinate part
,
cottonseed Oils would have better drying
oxidation are not merely additive compounds,
heir oxygen as hydroxyl groups . This is inbehaviour of unsaturated compounds in general .salts is similar to that of the acids themselves .exposure to air at ordinary temperatures
,or by
the fatty acids are oxidised withsolid
,insoluble in ether
,but reconverted
heating with alkali .4 . The drying properties Of Oils depend upon the presence Of linoleic
,
linolenic,and i solinolenic acids , as Oleic acid forms no solid oxidation
products . During the drying Of linseed oil,only the glyceryl of the
non-“drying esters is oxidised,as i s shown by the very small quantitiesof carbonic
,formic
,and acetic acids formed by passing pure air through
pumice soaked in linseed oil . The samples of linseed Oil which werestill in the first stage of oxidation
,as shown by their being still soluble
in ether,contained and of free acid . The substance insol
uble in ether , called by Mulder linoxyn , produced by the oxidation oflinseed Oil
,i s an ester tegmed hydroxylinolin . The drying properties
of an oil appear to be in direct ratio to the proportion of the glyceridesof linolenic and linoleic acids present .The change of composition undergone by 1 00 grm . of linseed and
poppy oils by exposure to air during 1 8 months was found by ClOez tobe as follows :
L inseed O i l POppy Oi l
Compo s ition of original o i l. .
Compo sition after 1 8 month s .
Difference .
" 0 76 + 1 3 98

346 MODE S OF EXAMINING FATS , OILS , AND WAXE S .
The quantity of oxygen absorbed was greater than that given Off
in the form of carbon dioxide and water,and the oil finally showed a
considerable increase in weight .Wi lliams
I gives the following table showing the change takingplace in the elementary composition of linseed Oil on boiling .
9 I4 l s l é l 7 l s
NOS . 1 and 2,raw linseed oil ; NO . 3 , moderately stout boiled
oil ; Nos . 4 to 1 0,solid oil , as used in manufacture of linoleum ; No . 4 ,
NO . 5 , Nos . 6 to 9 , and No . 1 0,made by different processes .
According to Reid,
“ an increase in weight of 1 0% i s Observed inlinoleum manufacturef This does not represent the actual gain inweight
,as there is loss of volatile acids
,carbon dioxide
,etc . Toch3
states that he Obtained 1 9% oxygen absorption and that the quantityof carbon dioxide obtained never exceededSabin4 records results of experiments on the oxidation of linseed oil
,
in which it was found that air drawn through a series of flasks wettedwith linseed Oil dried the
r
oil as follows : after a day or two the oilin the first flask began to bleach
,and in due course dried
, as was shownby the formation of a film of linoxyn
,and then dried still further as
indicated by the shrivelling and wrinkling of the film . During thewhole of this time (about 1 0 days) the oil in the other flasks was notacted on . Then the Oil in the second flask bleached and dried
,then
the third and so on . In 2 months,the whole series was dry . The
removal Of ozone from the air i s suggested as an explanation of theseresults .Shearman and Falk5 have investigated the effect Of atmospheric
oxidation on the constants of linseed Oil .
1 Ana ly st, 1 89 8 , 23 , 2 5 3 .
2 S oc. Chem. I nd . , 1 89 8 , I 7 , 7 5 .
0
3 och . Ch em . and Techno logy of Mixed Pa i nt s and P i gment s , 1 90 7 .
4 J. S oc. Chem . I nd . , 1 9 06 , 25 , 5 7 8 .
5 J. Amer. Chem . S oc. , 1 903 , 25 , 7 1 1 .

348 MODE S or EXAMINING FATS,OI LS
,AND WAXE S .
the oxidation of vegetable drying O ils and has shown that the veloci ty Ofoxidation can be expressed mathematically .
I t i s of interest to note that the effi ciency of a drier frequently decreases after a certain quantity has been added .
Influence o f Ligh t on Dry ing .—According to Cloez
,the maximum
amount of oxidation takes place with colourless glass,but with blue
,
red,green
,or yellow
,a longer time is required the nearer the colour
approaches the yellow .
A . Genthe I has studied the efl ect Of light on the process of drying ,and has shown that :
1 . Little or no oxidation takes place when flasks of brown glass areused and that the presence or absence Of light greatly influences therate of absorption of oxygen .
2 . The full rate of absorption does not develop immediately . Thereis an induction period of about 2 hours , after which' the absorption rateincreases .3 . Using a Uviol lamp and vessels of “Uviol glass , the same degree of oxidation was Obtained in 1 day as required in parallel ex‘
periments 8 to 1 0 days in daylight and 50 days in dark .
4 . The addition of siccatives caused no acceleration in the “Uviolexperiments .5 . Using resinates
,oleates
,and linoleates as siccatives the accelera
tion was 1 0 to 1 5 better in the case of the linoleates than the other two .
6 . Violet rays apparently have the best drying effect .7 . That linseed oil first forms a primary catalyser and then acts asan accepter
,and siccatives are to be regarded as pseudocatalysers ,
which have only a stimulative eff ect upon the formation of the primaryauto-catalyser . This latter has not been isolated .
8 . The results can be expressed by O stwald ’s equation for autocatalysis . The amounts of oxygen absorbed by linseed oil in dryingin the dark average 23% at ordinary temperatures , and at9 While the amounts absorbed on exposure to Uviol light were 2and respectively . The volatile products formed in the dryingprocess are equal to 1 5% of the weight of Oil .The effect of light on the rate of drying of linseed oil i s of technicalimportance
,as also is the effect of different coloured rays on the colour
of the dried film ; especially is this the case when the oil i s mixed withwhite lead . The writer i s at present investigating the latter point .1 Zei tsch . angew. Chem. , 1 9 , 208 7 .

LINSEED OIL . 349
This question was brought to his notice by the yellowing of white-leadpainting which had dried in a room lighted only from a green-glassdome . The paint became yellowish after drying . Trial showed thatthe white colour Of the paint was restored by exposure to sunlight .Peti t states that white lead and linseed oil dried in the dark assumes ayellowish tint
,while white lead and poppy Oil do not behave in thi s
manner . The writer has confirmed Petit ’s Observation on linseed oil,
but has invariably found that the yellow colour i s removed byexposure to light ; in fact , the change is reversible and appears to goon indefinitely .
Influence of Tempera ture .—Chevreu l has shown that linseed oil
dries more rapidly at 25°—28° than at 1 5°—1 8°M itarewsk i I states that when Oil is stored the temperature influencesthe degree of oxidation
,and this i s accelerated by storage at
Exposure to light for 26 days had no eff ect .Influence of Storage .
—Fahrion considers that in oil wh ich i s keptthe unsaturated acids polymerise
,and that these complex bodies ab
sorb oxygen rapidly . Chevreul has , however , found that short boilingyields a better drying oil than one which has been boiled for a longerperiod . I t has been shown that polymerisation does not occur as suggested by Fahrion .
Re lat ionsh ip between Drying Properties and Iod ine Va lues .
The iodine absorption of an Oil stands in close relationship to theoxygen absorption
,and consequently is indicative of the drying power .
This relationship is clearly Shown in paragraph on Ozone Absorption ,better agreement being Obtained than with oxygen values .Fish and liver Oils although having h igh iodine values do not drylike linseed oil and therefore the iodine value cannot be accepted asan absolute indication Of drying power .
Characterist ics of Linseed Oi l.
Sp . gr
So lidification-po intM . p .
Flash-po intAsh
Free fatty acidsUnsaponifiable matterSaponification value
1 J . S oc. Chem. Ind . , (abstra ct ) , 1 9 06 , 25 , 8 1 8 .
at 1 5°
stearine depo sits at —25°
1 6 to 20°
450~ -5OO
O F . (close), 258° open
from traces to ( limit)-5% -
4%up to average1 83—22 1 mgrm . KOH,
average1 9 2

3 50 MODE S OF EXAMINING FATS,OILS
,AND WAXE S .
Re ichert-M e issl valueIodine valueBromine values
O O O O O O O O O O O O O O
Inso luble brom idesMaumené test. .
B romine thermal valueRefractive indexOptical rotationO leo refractometerButyromet er .
Viscosity
For characteristic constants of various varieties of seed,see works
Of Lewkowitsch,Wright and M i tchell
,B enedikt and Ulzer
,and other
standard works ; also Wijs , loc. G ill and Lamb,Analyst, 1 900, 24,
97 ; Thomson and Dunlop , Analyst, 1 906 , 3 1 , 28 1 ; P . McIlhiney,
loc. ci t.
Sp . gr
Solid ifying-po intT iter T est
1 7°
Under 1 3°
Inso luble fatty acids unsaponifiable matter 95Neutralisation value , mg . KOH 1 96
—20 1Mean mo lecular we ight 283Iodine value . 1 70
—200Acetyl value . 8 . 5
Refracti ve index 1 . 4546 at 60°
Lino leic Acid .
Linoleic acid was isolated by Sch iller in the following mannerLinseed oil was saponified with
‘
solution of sodium hydroxide , and thesoap purified by repeatedly salting out . The aqueous solution of thesoap was then precipitated by calcium chloride . From the well-washedprecipitate the calcium linoleate was dissolved out by ether. The
1 7 5—1 90, sometimes h igher
1 05—1 1 5
1 00—1 1 0less than 723 average 25
90 1 4529-8—33
at 1 5°
Practically ni l.
+ 48 to + 54 degrees84—90
'at 20
°
second at 70° F .
Linseed Oi l Fatty Acids .
. at
at
at 99°
water atat 1 00°
water at 1

3 52 MODE S OF EXAMINING FATS,OILS
,AND wAX E s .
O
ist which diff er in yielding a solid and a liquid bromo-derivative . Thesolid tetrabromide is soluble in ether
,alcohol
,benzene
,chloroform
,
and glacial acetic acid,but only sparingly soluble in petroleum
.
ether .On reduction by zinc and alcoholic
‘
hydrochloric acid it yields linoleicacid .
Lino leates .-The salts of linoleic acid are diffi cult to obtain pure .
They are white,amorphous (except in the case of the zinc salt), bodies
which become coloured on exposure to air,and are soluble in alcohol
and ether . Potassium and sodium linolates containing an excess ofalkali absorb oxygen rapidly and become yellow and dry when exposedin a finely divided state to the air
,dissolve in water with dark
brownish-red colour,and give
,on addition of hydrochloric acid
,a
brown greasy resin . The ethereal solution of lead linoleate , whenevaporated on a glass plate
,leaves a white amorphous residue of lead
hydroxylinoleate . The acid separated from this salt by hydrogen sulphide and dissolved in alcohol remains on evaporation as a nearlycolourless viscid mass
,which becomes blood-red without change of
composition when heated to 1 00° or treated with acids or alkalies .
The colourless alcoholic solution of hydroxylinoleic acid is notaltered by alkali carbonates at the boiling heat
,but caustic alkalies
turn it red even at ordinary temperatures .
Lino len ic Ac id.
Linolenic acid has been prepared by reduction of its hexabromideobtained from linseed Oil .The acid is a nearly colourless Oil having a sp . gr . at of
O .92 28 .
I Linolenic acid rapidly absorbs oxygen on exposure to theair
,becom ing brown in colour . The hexabromide melts at
1 79°
B edford (loc . ci t .) is of the Opinion that two isomerides Of linolenicacid exist
,one which yields a solid hexabromide , melting at 1 79
°
I 80°,the other a liquid tetrabromide . The existence Of isolinolenic
acid is debatable,its existence as a glyceride has been suggested by
Hazura,but B edford considers that the liquid tetrabromide of lino
lenio acid has been mistaken by Hazura for the soluble isol inolenichexabromide .
1 Hehner and Mitch e ll , A na ly s t . , 1 89 8 , 23 , 3 1 3 .

LINSEED OIL .
Misce l laneous Notes .
Linseed Oil has the property Of dissolving sulphur,and at 7 7° F .
dissolves at 3 20° F . On cooling the solutions, sul
phur is deposited slowly .
The Offi cial Balsamum sulphuri s is prepared by boiling linseed Oil
with of sulphur . A dark vi scous mass i s obtained .
The absorption of phosphorus by linseed Oil has been investigatedby Katz .Ni trous acid does not give a solid elai din with linseed oil .Ni tric acid interacts with linseed Oil—the rate depending on thestrength Of acid employed . A moderately strong acid converts linseedOil into a vi scid mass insoluble in petroleum ether
,while fuming nitric
acid inflames i t . For method of detection Of linseed Oil : in walnutOil
,see Halphen
,Bull . S oc. Chem. , 1 905 , 3 3 , 5 7 1 .
BOILED LINSEED OIL .
When linseed Oil i s heated,i ts drying properties are considerably
increased if,during the heating
,certain substances known as “ driers ”
are present .The earli est process consisted in heating the Oil by fire in
‘
a boilerto a temperature of 2 1 0 to 260° in the presence of driers . This processhas been almost entirely superseded by newer processes
,which have
the meri t of yielding products paler in colour and requiring a lowertemperature for their application . Among these processes may bementioned that of heating the Oil to about in a steam-jacketed
,
agitated vessel,whilst in some cases air i s blown through . In the
Hartley-B lenk insop process a still lower temperature is employedby using a soluble drier (manganese linoleate).
According to various writers,i t i s a somewhat common practice to
add soluble driers to unboiled linseed Oil,and to sell the mixture as
“ boiled oil,
” which is known in the trade as bung oil .” Such Oil
has exactly the same constants as unboiled Oil, but does not dry so wellas oil which has been boiled . Among painters and others usingboiled oil there is considerable doubt frequently expressed as towhether Oils made by the newer processes are as good as those produced when only lead driers were used in the boiling . Boiling increasesthe sp . gr . and viscosi ty of linseed oil , the extent of each increase beingdependent on the extent of boiling and method used . Fire boiling issaid to produce the most viscid product .
Vol. I I .
—23

3 54 MODE S OF EXAMINING FATS , OILS,AND
'
WAX E S .
The colour of boiled Oil varies according to the method and timeof manufacture
,and also the original colour of the Oil used . I t i s a
common practice among Oil refiners to use their dark oil for boiling purposes . Boiled Oil i s of a darker colour than raw Oil
,and i s of a red
dish-brown Shade . Lead driers are said to produce a dark coloured Oil.Boiled Oil can be distinguished from unboiled by the presence Ofa metallic residue (due to driers) when ignited , and also by followingcharacteristics .Sp . Gr.
—o .945 ; according to McIlhiney, the sp . gr . may beas high as but i s seldom overIodine Value .
-Willi ams gives the following table showing thedecrease in the iodine value which takes place on boiling .
Thin Thin S tout Very stoutIodine value
,
The above figures are probably too high,as the Hubl solution was
allowed to act for 1 8 to 20 hours . See section on Drying of Linseed forchanges in ul timate composition which take place on boiling .
Lewkowitsch has shown that i t i s necessary to remove the metalfrom a boiled Oil by treatment with mineral acids before determiningthe iodine value
,otherwise the value Obtained will be too high .
McIlhineyI has modified his process for the estimation of bromine
absorption in the case Of boiled Oil and linseed oil in presence of driers .(See original paper .)The nature of the driers added to linseed oil can be generallyinferred from an examination of the ash left on burning a quantity ofthe sample
,a little at a time
,in a porcelain dish . The residue should
be specially tested for lead,copper
,zinc
,iron
,manganese
,and borates .
Sulphates , acetates , borates and other salts may be detected byagitat ing the original oil with a solution Of sodium carbonate , separating the aqueous portion
,and examining the solution in the usual way ,
or by boil ing 25 grm . of the sample with dilute hydrochloric acid forhalf an hour wi th constant stirring, allowing to separate into 2 layers ,and syphoning off the acid layer , and testing by ordinary methods formetals (lead, manganese) and acids (boric , oxalic ,M . Kitt (Chem. Rev. Fett-Harz-Ind.
,1 901 , 8, (3) 40) gives the
following table,showing the alterations taking place on boiling
linseed Oil. The numbers (from o to 6) represent the different stagesfrom thin Oil to the consistency of india rubber .
Amer. Chem. S oc. , 1 9 02 , 1 1 09 .

3 56 MODE S or EXAMINING FATS,OILS , AND WAXES .
be again pointed out that in order to obtain figures of any value,great
care must be taken to Obtain exactly similar conditions during comparative tri als . See section on Drying of Linseed O il .The United S tates Navy Department Specification
,1 905 , demands
that boiled linseed Oil must be pure kettle-boiled oil,free from rosin .
A film left after flowing the oil over glass and allowing to drain in avertical position for 1 2 hours
,must be dry , free from tackiness in 1 2
hours at a temperature of 70° F .
For the rules of testing boiled l inseed Oil i ssued by the RussianM inister Of Marine , see J . S oc. Chem. Ind. (abstract), 1 905 , 24, 1 55 .
Uses .—Boiled Oil on drying gives a somewhat hard film
,which i s
liable to crack on exposure,and i t i s therefore the custom
,in painting
,
to add a little raw oil in order to give a more elastic and durable coat .The detection of small quantities of raw linseed oil in boiled Oil i s therefore of li ttle or no importance .Adu l tera t ion .
—Boiled oil i s adul terated in the same manner as
linseed Oil (see paragraph on Adulteration), and adulteration is morefrequent in the boiled variety . The usual adulterants are rosin
,mineral
and rosin oils,tung and fish or blubber Oils .
A l arge number of substitutes are marketed and they are usuallycompounded from the above .I t must be pointed out that boiled oil containing liquid driers maylegitimately contain a small proportion of turpentine .The following test i s frequently recommended as a rapid method ofdetection of adulteration . Add water after saponification with alcoholic potassium hydroxide : if a strong turbidi ty is produced it indicates the presence of adulterants . This test has
,however
,been
investigated by L ippertI who has proved it to be unreliable .
Lippert2 states that linseed oils boiled with lead or manganese oxidesgive only brown colourations with S torch-Morawski
’s test , while
red or blue colourations are obtained when rosinates,rosin
,or fish
Oils are present . Maize Oil gives similar reactions .Li thogi
'
aph ic Varn i shes .—If the temperature of heating of linseed
oil be raised to 2 50 to the so-called lithographic varnish is obtained .
The time of boiling determines the viscosity of the product . ‘ Driersare not used in the preparation of li thographic varni shes . Lewko
wi tsch has shown that the process taking place is one Of polymerisation ,1 Zei tsch . angew. Chem . , 1 89 7 , 6 5 5 .
1 Chem. Rev. F ett-Harz-Ind . , 1 9 0 5 , 1 2, 4 .
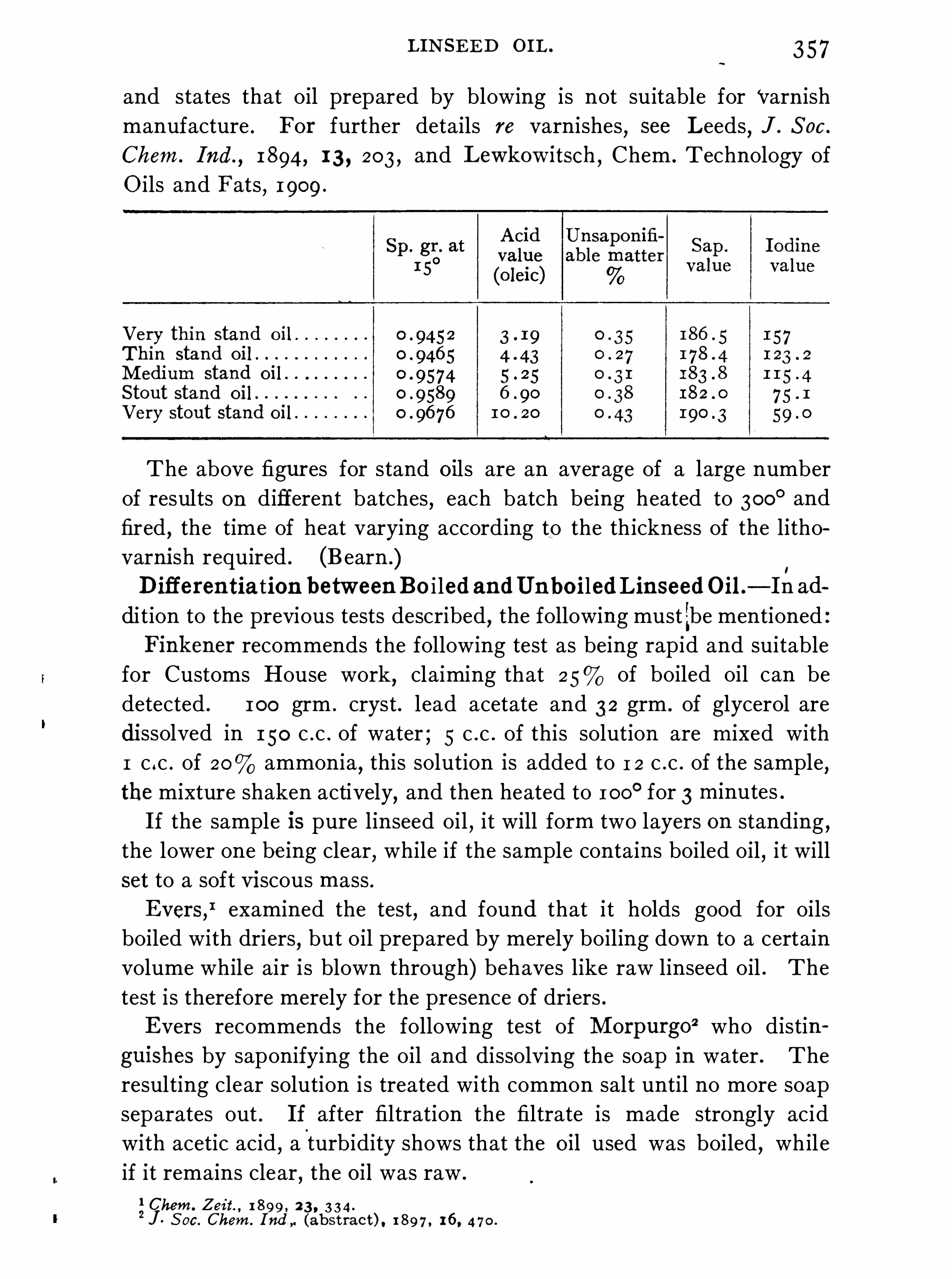
LINSEED OIL . 3 57
and states that oil prepared by blowing is not suitable for ‘varnishmanufacture . For further details re varnishes
, see Leeds,J . S oc .
Chem. Ind.,1 894 , 1 3 , 203 , and Lewkowitsch , Chem . Technology of
O ils and Fats, 1 909 .
Very th in stand o i l
Th in stand o i l
Medi um stand o i l
S tout stand o i l
Very stout stand o i l
The above figures for stand Oils are an average of a large numberof results on different batches
,each batch being heated to 300° and
fired,the time of heat varying according to the th ickness of the litho
varni sh required . (B earn .
Diff erentia tion betweenBo i led andUnbo i ledLinseedOil .—In addi tion to the previous tests described
,the following must"be mentioned
Fink ener recommends the following test as being rapid and suitablefor Customs House work
,claiming that 2 5% of boiled oil can be
detected . 1 00 grm . cryst . lead acetate and 3 2 grm . of glycerol aredi ssolved in 1 50 c .c . of water ; 5 c .c . of this solution are mixed with1 c .c . of 20% ammonia , this solution is added to 1 2 c .c . of the sample ,the mixture shaken actively, and then heated to 1 00° for 3 minutes .If the sample is pure linseed oil
,i t will form two layers on standing ,
the lower one be ing clear,while if the sample contains boiled oil , i t will
set to a soft vi scous mass .Evers ,
I examined the test,and found that i t holds good for oils
boiled with driers,but oil prepared by merely boiling down to a certain
volume while air i s blown through) behaves like raw linseed oil . Thetest i s therefore merely for the presence Of driers .Evers recommends the following test of Morpurgo
2 who distinguishes by saponifying the oil and dissolving the soap in water . Theresulting clear solution is treated with common salt until no more soapseparates out . If after filtration the filtrate is made strongly acidwith acetic acid
,a turbidity shows that the Oil used was boiled , while
if it remains clear,the Oil was raw .
1 Chem. Zei t. , 1 89 9 , 23 3 3 4 .
2 J . S oc. Chem . Ind (Abst rac t) , 1 89 7 , 1 6 , 4 7 0 .

3 58 MODE S or EXAMINING FATS , OILS , AND WAXE S .
A very useful test to distinguish between raw and pale boiled Oil i sto shake the sample with ammonium sulphide when a dark colouration is Obtained (B earn).The analysi s of a sample of boiled l inseed Oil
,which
,in addit ion to
containing various mineral additions and free fatty acids,i s also
adulterated with rosin,rosin oil
,and mineral Oil
,i s a complex problem .
The following plan is recommended : The substantial accuracy Of
the results has been established by Allen : 2 5 grm . of the sampleshould be shaken in a separator several times with dilute hydrochloricacid . The aqueous liquid
,which may contain lead
,zinc
,manganese
,
borates,and other mineral additions
,is separated from the oily layer,
and the latter i s washed by agitation with water till the washings nolonger redden li tmus . The Oil is then treated with rectified spirit , andthe free fatty and resin acids titrated with standard alkali and phenolphthalei n . The neutral point having been reached
,the alcoholic
layer i s separated from the residual Oil,which consists of neutral fatty
oil and hydrocarbon oils of the original sample . These may beseparated in the usual manner . The alcoholic solution is then concentrated , water added , and any globules of oil dissolved by agitatingwith petroleum spiri t . After separation from the aqueous liquid
,and
evaporation Of the solvent,the small residue of neutral oils may
'
beweighed
,and the amount found added to the main portion . The
aqueous solution i s then acidified with dilute hydrochloric or sulphuric acid , when an oily layer i s obtained , consisting of the free fattyand rosin acids of the original sample
,together with such additional
amount as may have been formed by the decomposition of metallicsoaps in the first stage of the process . This i s separated from theaqueous liquid , washed with a li ttle water , and filtered through wetpaper . On subsequently drying the fil ter in the water-oven , the fattyacids pass through
,and can be collected in a small tared beaker , the
portion remaining on the filter being dissolved in ether . After weighing the fatty acids in the beaker
,1 grm . i s treated by Twi tchell’s proc
ess for the separation of fatty and rosin acids . The amount of rosinthus found
,subtracted from the mixed fatty and resin acids , gives that
Of the fatty acids alone . By agitating the original sample with alcohol ,separating the solution from the undissolved Oil
,and titrating the
former with standard alkali,the sum of the fatty and rosin acids
originally existing in the oil can be ascertained .
Driers .—Among the siccatives formerly in use were l itharge ,

3 60 MODE S OF EXAMINING FATS , OILS , AND WAXE S .
remaining except at a red heat,when most of the lead volatilises with
it as lead chloride .The analytical examination of a drier is in i tself Of li ttle value as anindication Of i ts properties .Patent driers under fancy names are frequently met with . The number Of such materials is legion
,and many are worthless products con
taining large quantities of inert substances which have no siccativeproperties and merely act as diluents .
References to Li terature Relating toThorpe . J . S oc . Chem . Ind .
,1 890, 9 , 628 .
Hartley . J . Chem. S oc . , 1 893 , 63 , 1 29 .
Weger . Zei ts . ang . Chem .,1 896 , 53 1 .
Weger . I bid .,1 897 , 40 1 , 542 , 560 .
Lippert . Zei tsch . angew . Chem.,1 900 ,
1 33 .
Lippert . Chem . Zei t ., 1 903 , 1 6 , 365 .
Fawsitt . J . S oc . Chem. Ind.,1 903 , 2 2 , 538 .
Endemann and Paisley . Amer . Chem. Jour . , 1 903 , 29 , 68 .
Lippert . Zei t . f . angew . Chem.,1 905 , 94 .
Tauber . Chem. Zei t.
,1 906 , 1 25 2 .
Meister . Fa'
rber-Zei t.,1 908 , 1 53 .
Lewk owitsch . Loc . ci t .
, 3 , 1-06 .
Fokin . J . S oc . Chem. Ind .
,1 907 , 26 , 1 1 49 .
When a drying Oil containing manganese oxide in solution is dissolvedin an equal volume Of benzene and agitated with air in a closed vessel ,rapid absorption of oxygen takes place
,especially at a temperature of
40 to If the supply of air is repeatedly renewed , the liquid becomes thick
,and on distilling Off the solvent a residue is obtained
which solidifies on cooling to a dry and perfectly elastic solid . By
limiting the oxidation,various intermediate products are Obtainable .
The last product i s characterised by its elasticity and i ts insolubili tyin water , alcohol , and ether . I t i s almost instantly saponified bysodium hydroxide in the cold ; and on subsequent separation of thefatty acids
,i t i s found that the solid acids have undergone no alter
ation,while the liquid fatty acid has been converted into viscous
products,characteri sed by their solubili ty in water and by the salts
which they form .
For later investigation of above,see Dunlap and Shenk , J . Amer.
Chem. S oc ., 1 903 , 826 .

BLOWN OILS .
BLOWN OILS , OXIDISED OILS , BASE OILS, OR
“SOLUBLECASTOR OILS .
”
Various products known by these or similar names are manufactured by blowing a stream of air through fatty oils . The Oils whichlend themselves most readily to the treatment are cotton , rape , and linseed oils
,but the process is also carried out with Olive
,lard
,ground-nut
,
and some fish oils,the latter being almost entirely used in the leather
industries .Cottonseed
,rape
,and linseed oils are the most usual oils used .
B lown cottonseed oil and rape Oil are respectively known as lardine andrapine . The oil is usually heated by a steam coil at the commencement of the blowing to a temperature of though this i s not strictlynecessary
,at least with certain oils : care must be exercised to avoid
too high a temperature (above except in certain cases in whicha temperature of 1 1 0 to 1 1 5
° i s employed . The process usually lastsfrom 1 2 to 48 hours , according to the nature of the Oil under treatment ,the character of the product desired
,and the size and power of the
apparatus . A considerable rise in temperature takes place , and provision has to be made to prevent this becoming too great . Thequality of the product can be varied by arresting the process at anyparticular stage .The blown oil is usually of a clear yellow colour
,with a disagreeable
smell and taste suggesting its origin .
The temperature of working has some influence on the rate ofoxidation
,the action being hastened by high temperatures ; the
products obtained from high temperature oxidation are usually darkerin colour and have a more pronounced odour that those prepared atlower temperature .The perfect miscibility Of such Oils with heavy mineral Oils givesthem an advantage over castor oil in the manufacture of lubricatingmixtures for heavy machinery
,hence the term “ soluble castor oil . ”
Opinions are divided as to the suitability of blown oils as lubricants .They are stated to be useful on account of their high sp . gr . and
viscosity . Some authorities,however
,look on them with Suspicion , as
they object to the low flash-point , and the liabili ty of such oils to “ gum .
”
Mineral and castor oils are mutually Soluble only to a very limitedextent , but by addition Of some other oil , such as tallow oil , perfectunion can be obtained . When the oxidation of cottonseed oil i s

362 MODE S OF EXAMINING FATS,OILS , AND WAXE S .
pushed to an extreme,the product has a sp . gr . of and is not
readily miscible with heavy mineral Oils . B lown Oils yield sebacicacid on dry distillation
,and contain but an insignificant proportion
of unsaponifiable matter . The Odour, taste , and colour-reaction ofthe oil with sulphuric acid will afford an indication of i ts origin
,and
more definite information can be obtained by an examination of thephysical and chemical characters of the fatty acids produced by itssaponification .
O ils,on being blown
,increase in viscosity
,and in the case of
linseed oil the oxidation is continued until the oil has become solid forlinoleum-making . The solidified oil obtained is heavier than water .References for linoleum : P inette
, Chem. Zei t.,
1 896 , I 6 , 28 1 ;
Reid,J . S oc . Chem. I nd .
,1 896 , 1 5 , 75 ; Lewkowi tsch , Analyst, 1 902 ,
2 7 , 1 40 ; Ingle , J . S oc. Chem. I nd .
, 1 904 , 23 , 1 1 97 .
Leeds I gives the results of the examination of two oxidised linseedoils prepared by blowing linseed Oil with oxygen in a jacketed pan .
The oxidised oils are more readily soluble in alcohol than the originalOils . B enedikt and Ulzer2 give the following table as showing thesolubili ty of 1 part of blown oil in alcohol .
Cottonseed o i l d isso lved inB lown cottonseed o i l ( lab
oratory sample), parts abso lute alcohol atB lown cottonseed o i l (commercial sample),
Fox and B aynes,3 Thomson and B allantyne
,4 Chapman and Rolfe ,s
have examined a number of blown oils .Fox (Oi l and Col . Journ .
,1 887 , 8 , 549) has publi shed the following
figures,showing the changes produced in Oilsby blowing with air :
l B efore Aft er B efo re After I B efore Aft er
At a more recent date,Lewkowi tsch has made a thorough investiga
tion into the blowing of Oils,and tables A
,_
B,C,and D are extracted
from his paper in the Analyst, 1 902 , 2 7 , 683 .
1 J . S oc. Chem . I nd 1 89 0 , 9 , 84 7 .
2 Ze i tsch . angew . Chem 1 88 7 , 2 4 5 .
3 A na ly s t , 1 88 7 , 1 2 , 3 3 .
4 J . S oc. Chem . I nd . , 1 89 2 , 1 1 , 5 06 .
5 Chemi cal News , 1 89 4 , 2 .
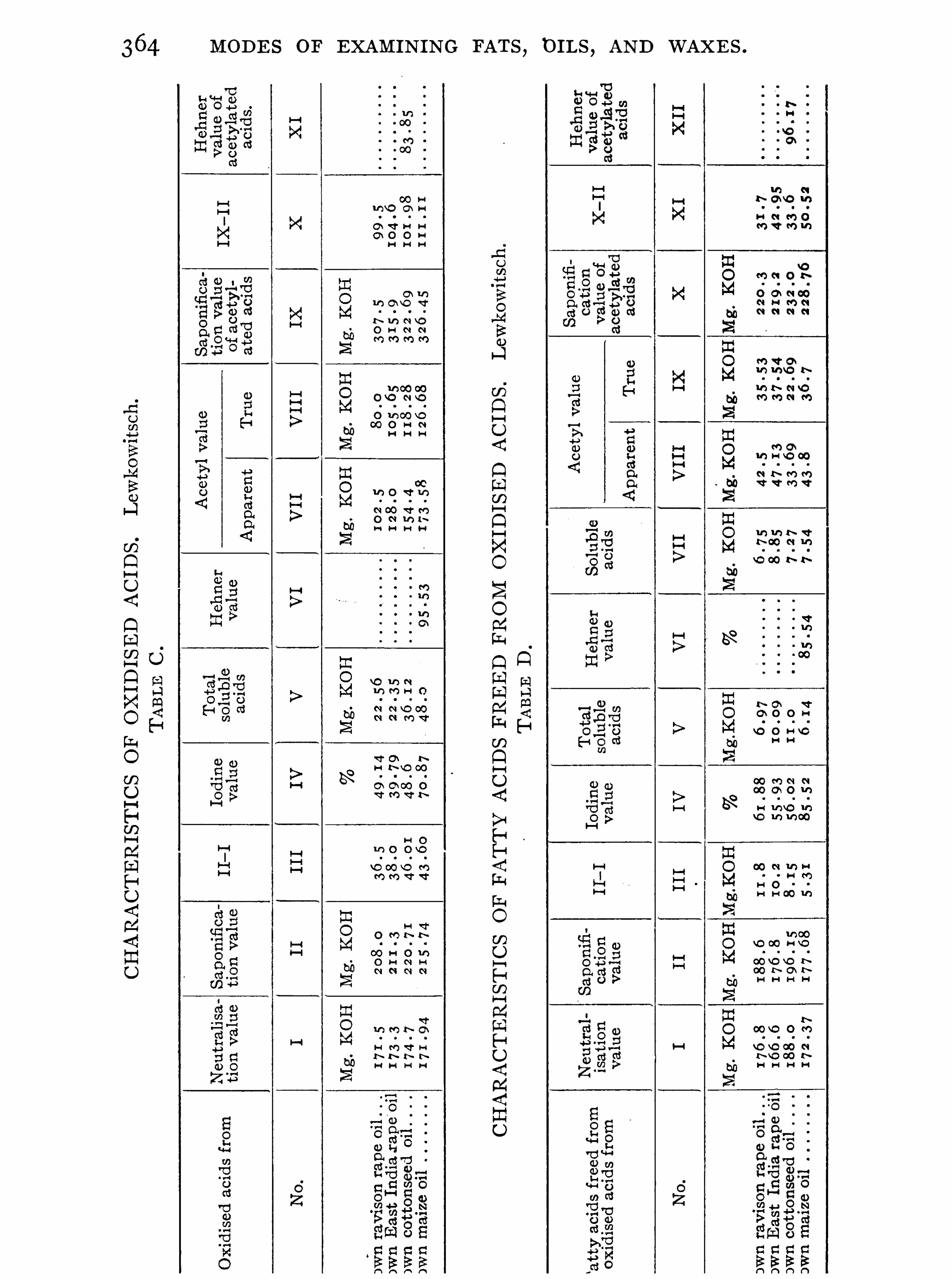
MODES OF EXAMINING FATS , OILS , AND WAXE S .

BLOWN OILS . 365
The investigations show that blowing results in the following changes :The sp . gr . is increased and the free acid and saponification valuesare also increased . The saponification values of the oxidised acidsare higher than the neutrali sation value
,thus pointing to the presence
of lactones . Saponification of the blown Oil i s more difficult than thatof the original Oil . Soluble acids are formed , hence the Hehner valueis low . The iodine values decrease , and the oils acquire a considerable acetyl value . The oxidised acids are insoluble in petroleumether
,but separation by this solvent is not complete . There does not
appear to be any numerical connection between the acetyl value andthe oxidised acids .Lewkowitsch also gives the following table as the result of heatinglinseed and cottonseed oil to while air was blown through .
TABLE SHOWING CHANGES IN BLOWING OF LINSEED AND
COTTONSEED O ILS .
IxflvkoudnuflL
Oi l blown at
-9 2 9
1 9 4 .
0 1
1 7
200 2 1 2 .
1 2 5 2 2 5
1 1 7
Proctor and Holmes have investigated the process of the oxidation ofOils by blowing . The method adopted was that of blowing air through1 00 c .c . of Oil contained in a test-tube , which was heated to 1 00
° bymeans of a water-bath . At intervals samples were drawn ,
and the

366 MODE S OF EXAMINING FATS, OILS , AND WAXE S .
refractive index and iodine value determined . The following are themost important results and conclusions :Wi thout exception
,on blowing any oil
,the sp . gr. and refractive
index increase as oxygen is absorbed,and at the same time the iodine
value,which is a measure of unsaturated linkages
,diminishes as these
become saturated with oxygen . In no case,however
, was i t foundthat in 24 hours ’ blowing the Oil at all approached complete saturation ,and even in commercial blown and oxidised Oils the iodine value showsthat saturation is far from complete .The process of oxidation is a somewhat complicated one . An in
teresting peculiari ty which is striking in several Oils (especially Moellerscodliver and pale seal) i s that the iodine value remains practicallyunaffected during the first three or four hours of blowing
,though the
rise of sp . gr . and refractive index shows that oxygen is being absorbed .
This,of course
,indicates that the saturated linkages which are measured
by the iodine absorption are still unopened,and that oxygen
._
is temporarily retained merely in solution or in some other way than directlinking on the carbon chain . Usually
,when a drop in the iodine
value does set in,i t proceeds rap idly for some time , as if the oxygen
previously absorbed now took i ts place in the broken linkage . In
other cases,as in castor Oil
,littleor no change takes place in any Of the
constants during the first three hours of blowing , while in the secondthree it i s relatively rapid .
Polymerisation does . unquestionably take place , aff ecting the refraction and sp . gr. ,
but probably not affecting the iodine value or perhapsthe refractive constant
,in which the natural relation between refrac
tive power and density is taken into account .The following table gives the figures before and after the experiments . For intermediate values
,see original paper . (J. S oc. Chem.
Ind .
,1 905 , 24 ,
The only oil in which the dispersion is probably high is whale oil,
and constants are given m a table,Of which the original di spersion was
ri s ing on blowing to the other constants being quite normal .The refractive indices and dispersions of three other whale oils givenin Table XX of paper are very constant
,beginning at to and
ending in all cases at so that i t i s evident that high dispersion i snot a usual characteristic .N ote.
-All O ils were blown at and measurements taken at Iod inevalues by Hanus method . Refractive index measured by a Ze iss refractometer.

368 MODE S OF EXAMINING FATS , OI LS , AND WAXE S .
hydroxy-fatty acids at the line of junction . As the non-volatile acidsare all contained in the petroleum layer
,i t i s unnecessary to again
shake out the aqueous layer with petroleum spirit . After running Offthe lower liquid the petroleum layer i s withdrawn from above
,leaving
the hydroxy-fatty acids in the funnel . If the quanti ty is considerable,
i t may enclose unoxidised fatty acids ; and it is therefore advisableto dissolve the mass in a dilute solution of soda or ammonia
,and re
peat the treatment with petroleum spiri t after acidifying with hydrochloric acid .
The united petroleum spiri t extracts are evaporated,and the residue
,
consisting of the unoxidised fatty acids and unsaponifiable matter , driedto constant weight ( I ). I t i s then di ssolved in 25 c .c . of 90% alcoholand titrated with seminormal alkali
,the mg . of KOH being cal
culated on the original Oil . The number thus obtained,which the
author terms the “ inner saponification value ,” furnished a measureof the non-volatile and unoxidised fatty acids .The neutral alcoholic solution is extracted with petroleum spirit
,the
extracts washed with alcohol,the petroleum spiri t evaporated
,and the
residue of unsaponifiable matter dried and weighedThe diff erence between ( 1 ) and (2) gives the quantity of nonvolatile fatty acids
,the molecular weight oi which can be calculated
from the inner saponification value .The hydroxy-fatty acids left in the separating funnel are dissolvedin hot alcohol
,the solvent evaporated
,the residue dried to constant
weight,ignited
,the ash deducted
,and the difference taken as the
hydroxy-fatty acidsThe sum of 1 + 3 gives the Hehner value .The following results were thus obtained with cottonseed Oil andthree oxidation products
,which were prepared by exposing the oil on
wash-leather for 8 and 1 2 days,respectively . The leather was cut into
fragments and extracted with cold petroleum spiri t , furnishing products A and B . The second leather still contained a considerableamount of product insoluble in petroleum spiri t , which was subse
quently extracted from it with cold ether I t was a thick yellowOil , soluble in alcohol .

BLOWN OILS .
on process i scation values .in B i s only
are both fractions of the same oxidationand the greater proportion Of unsapon ifiable matter wasby ‘the preliminary treatment with petroleum spirit which
n arrived at on this point is that during thethe unsaponifiable matter remains intact , and
oxy-fatty acids of liver Oils (Zei ts . angew . Chem.,1 89 1 ,
those of cottonseed Oil are completely soluble in ether .T he foregoing method of analysis affords a means of examining thecourse of oxidation during the drying Of linseed oil
,and is also appli
cable to the examination of unoxidised fats and O ils,as is seen in the
Saponification valueInner saponification valueHehner valueUnsaponifiable matter .
Oxy-fatty acids .
Non-vo latile fatty ac ids .
Mo lecular we ight of fatty ac ids .
From these results it i s Obvious that when ,as in the case of tallow
and Olive oil,the total saponification and inner saponification values
are nearly identical,the amount of volatile or Of oxy-fatty ac ids must
Vo l. I I .
—24

3 70 MODES OF EXAMINING FATS,OILS
,AND WAXE S .
be insignificant . Butter fat,on the other hand , by reason of i ts volatile
acids,shows a considerable difference between the two values
,
and the Reichert-Meissl value for 5 grm .) can be calculated fromthis difference . This calculated value is higher than the normal
,
owing to the fact that the Reichert value only represents a portion ofthe total volatile acids .In addition to the usual methods for determining the presence ofblown oils in lubricants
,Marcusson
I proposes to distinguish betweenrape or cottonseed oils
,using a method based on the diff erent behaviour
of the lead salts of the fatty acids from the oxidised Oils toward petroleum ether . Also by the behaviour toward petroleum ether of thefatty acids separated from the lead salts insoluble in ether . He givesthe following table :
Fatty ac ids Obtained from lead salts in ether
Coml. blown rape o i l
Coml. b lown rape o il
B lown rape o il prepared by authorComl. blown cottonseed Oi l
B lown cottonseed o i l prepared byauthor
The percentages refer to the weight of oil used for the preparationof the lead salts of the fatty acids . The fatty acids from blown cottonseed oil melted at 54 to while those from blown rape oi l were ohand semifluid .
L ewk owitsch (lot. ci t.) is of the Opinion that differentation by meansof th e m . p . of the fatty acids would yield more information than thecomparison of the solubility Of the lead salts in petroleum ether .The writer wishes to thank Mr . J . Gould B earn ,
M . Sc ., of Hull , for
k indly reading proofs and supplying certain figures which are includedin this section .
1 Chem . Rev. F ett-Harz-I nd . , 1 9 05 , 1 2 , 2 9 0 .
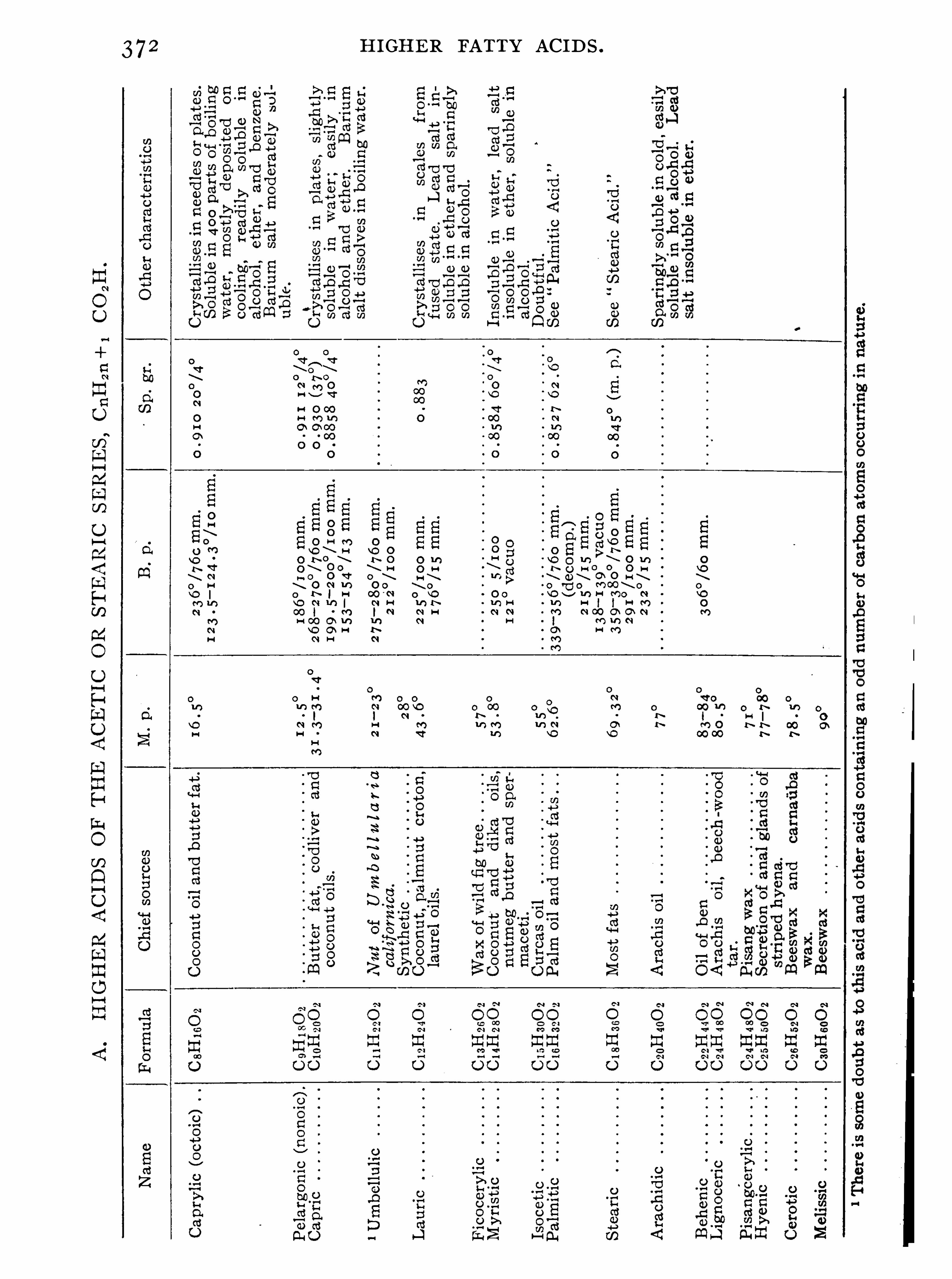
HIGHER FATTY ACIDS .


3 74 HIGHER FATTY ACID S .

3 76 HIGHER FATTY ACIDS .
ether ; lead elaidate i s , however , only very slightly soluble , like leadstearate , and this property , when utilised in separating lead salts ofthe unsaturated from those of the saturated series
,requires to be used
wi th caution .
C . The acids of the linolei c seri es form additive compounds with 4atoms of bromine and interact with a larger proportion of B ilbl’sreagent than do acids of the oleic series . They absorb oxygen fromthe air
,and oxidation of linoleic acid in the cold with dilute potassium
permanganate leads to the formation of sativic or te trahydroxystearicacid . They are not affected by nitrous acid . The lead salts aresoluble in ether .D . Linoleni c acid combines with 6 atoms of bromine or iodineand readily absorbs oxygen . Ni trous acid does not produce solidisomerides . Lead linolenate is easily soluble in ether .E . Ri cinolei c acid combines with 2 atoms of bromine
,does not
absorb oxygen on exposure to the air,i s gradually converted by ni
trous acid into a solid‘
stereo isomeride . I ts lead salt i s soluble inether . Oxidation with potassium permanganate gives two trihydroxystearic acids .Recogn i t ion and Es t ima t ion of Fa t ty Ac ids —The me thodsavailable for the detection and to some extent for the estimation Of thehigher fatty acids are based on the characters just described . In
many cases i t i s unnecessary to effect actual separation of the fattyacids in a mixture
,i t being sufficient to ascertain the joint amount
,
or to ascertain indirectly and approximately the proportion of theacids Of different origin known to be presentMe thods no t Invo l v ing S epara t ion .
—a . Free fatty acids can beaccurately estimat ed by titration in alcoholic solution with standardalkali
,using phenolphthalei n to indicate the point of neutrality .
The mode of operating is fully described on page 9 . A mixture of1 part of alcohol to 2 of amyl alcohol as solvent is recommended bySwoboda (Chem. Zei t. , 1 900 ,
24 , 285) as i t avbids the formation Of twolayers . Neutral substa nces—e . g .
,fats and hydrocarbons—do not
interfere . M ineral acids and acid salts must first be removed by agitation with water , or estimated by titration in alcoholic solution withmethyl-orange as indicator
,and resin acids must be separated or duly
allowed for . In the case of a m ixture of several fatty acids the result i sbest expressed in terms Of the principal or most characteristic acid

GENERAL PROPERTIE S .
present,and in most cases such a mode of statement gives a close ap
proximation to the total of the free fatty acids present .Conversely
,when the substance under examination consists wholly
of a mixture of fatty acids,titration with standard alkali suffices to
ascertain the mean combining weight of the mixed acids . This isfound by dividing the number Of mg . of fatty acids employed for thetitration by the number of c .c . of normal alkali required for neutralisation .
In cases of a mixture of two homologous acids,the nature Of which is
known or can be ascertained by other means,the result of the titration
gives the means of ascertaining the proportions in which the two constituent acids exist in the mixture . An example of the application ofthe method to this purpose is given on page 383 .
b. KOttstorfer’
s method (page 1 5) may be regarded as a processof approximately ascertaining the mean combining weight of the fattyacids of an Oil
,fat or wax without actually isolating them . 3 c .c . Of a
2% alcoholic solution of alkali blue 6B Meister , Lucius and Brii ning) can be substituted with advantage for phenolphthalein , whenthe oils give dark coloured solutions . This solution i s red withalkali
,blue with acid . The saponification values Obtained are fairly
constant , and are important in determining the nature Of an oil or fat ;but
,as a means of ascertaining the mean combining weight of the acids
,
the method is only applicable to O ils which yield 95% of fatty acids onhydrolysis .Tortelli and Pergami (Chem. Rev. ,
Fett. Harzind.,1 902 , 1 82) have
stated that the acids from oils and fats contain quantities of aubydrides or lactones which are not attacked by dilute alkali in the cold
,
and therefore too high a molecular weight is found . The truemolecular weight is found by heating with excess of N/ 2 alkali , andtitrating back with hydrochloric acid . The further statement ismade that in the case Of fresh samples the number Obtained is only afew units greater than the neutralisation value
,but that in old samples
the anhydride is quite important . A few of their results are tabulated .
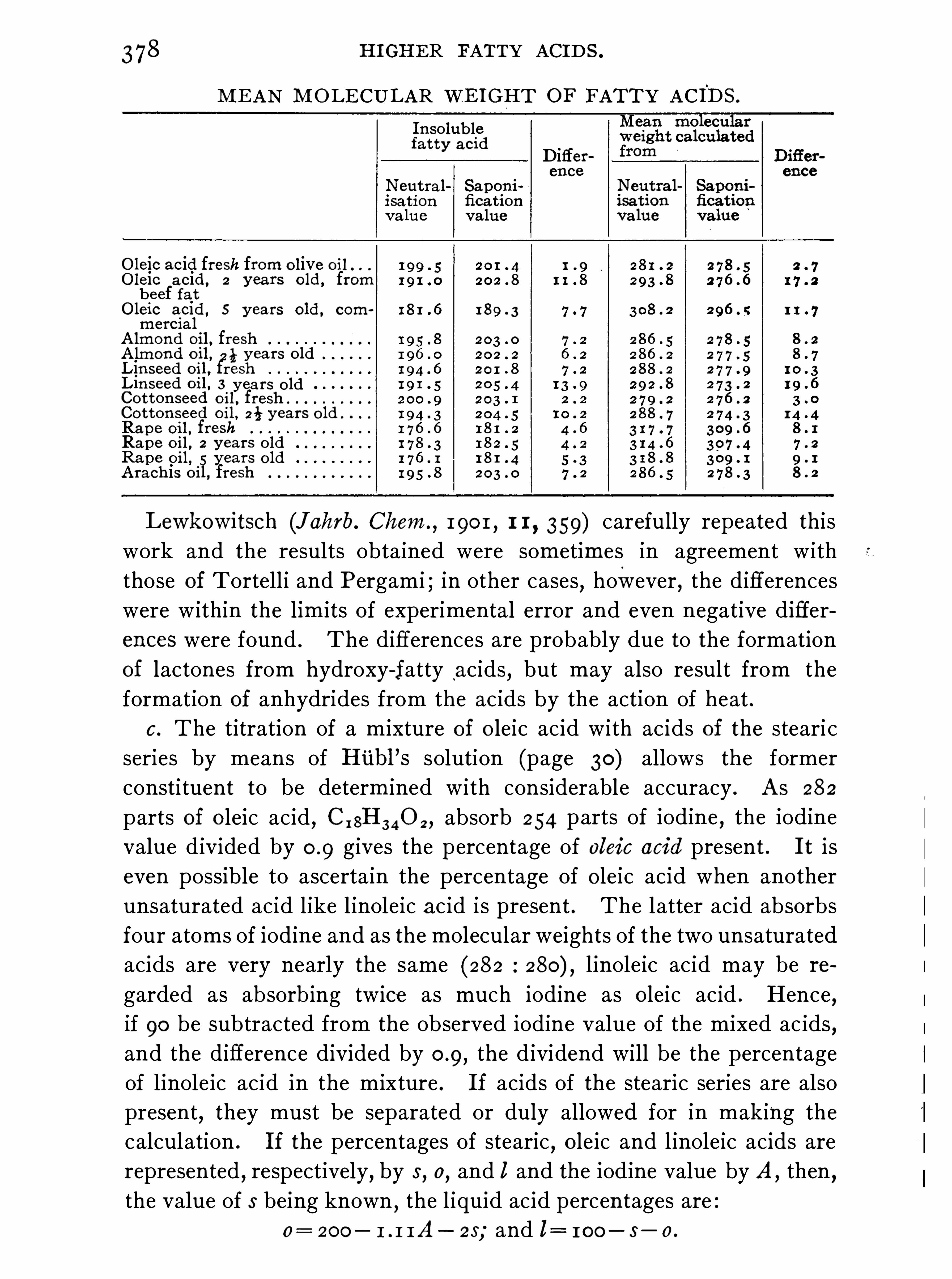
3 78 HIGHER FATTY ACIDS .
MEAN MOLECULAR WEIGHT OF FATTY ACIDS .
fInsolub l
eiat t aciy Differ
Ole ic ac id fresh from o live o i lOle ic acid , 2 y ears Old , frombeef fatOle i c acid , 5 y ears Old , commercia l
Almond oi l , fre shA lmond o i l , 2 1} y ears o ldL inseed Oi l , fre shL in seed o i l , 3 y ea rs o ldCot t onseed o i l , freshCot ton seed O i l , 25> y ears o ld .Rape o i l , fre shRape o i l , 2 y ears o ldRape o i l , y ears o ld
Arach is o i fre sh
Lewkowitsch (J ahrb. Chem.,1 90 1 , 1 1 , 3 59) carefully repeated this
work and the results obtained were sometimes in agreement withthose Of Tortelli and Pergami ; in other cases , however , the differenceswere within the limi ts of experimental error and even negative differences were found . The diff erences are probably due to the formationOf lactones from hydroxy-fatty acids , but may also result from theformation Of anhydrides from the acids by the action of heat .
c . The titration of a mixture Of Oleic acid with acids Of the steari cseries by means of Hubl ’s solution (page 30) allows the formerconsti tuent to be determ ined with considerable accuracy . As 282
parts Of Oleic acid, C 1 8H3 4
O , ,absorb 2 54 parts of iodine , the iodine
value divided by gives the percentage of olei c acid present . I t i seven possible to ascertain the percentage of Oleic acid when anotherunsaturated acid like linoleic acid i s present . The latter acid absorbsfour atoms of iodine and as the molecular weights of the two unsaturatedacids are very nearly the same (282 linoleic acid may be regarded as absorbing twice as much iodine as Oleic acid . Hence ,if 90 be subtracted from the observed iodine value of the mixed acids ,and the difference divided by the dividend will be the percentageof linoleic acid in the mixture . If acids of the stearic series are alsopresent
,they must be separated or duly allowed for in making the
calculation . If the percentages of stearic,Oleic and linoleic acids are
represented,respectively
,by s
,o,and l and the iodine value by A , then ,
the value of s being known,the liquid acid percentages are :
o= 200 1 . 1 1A 2s; and l= 1 00—s o .
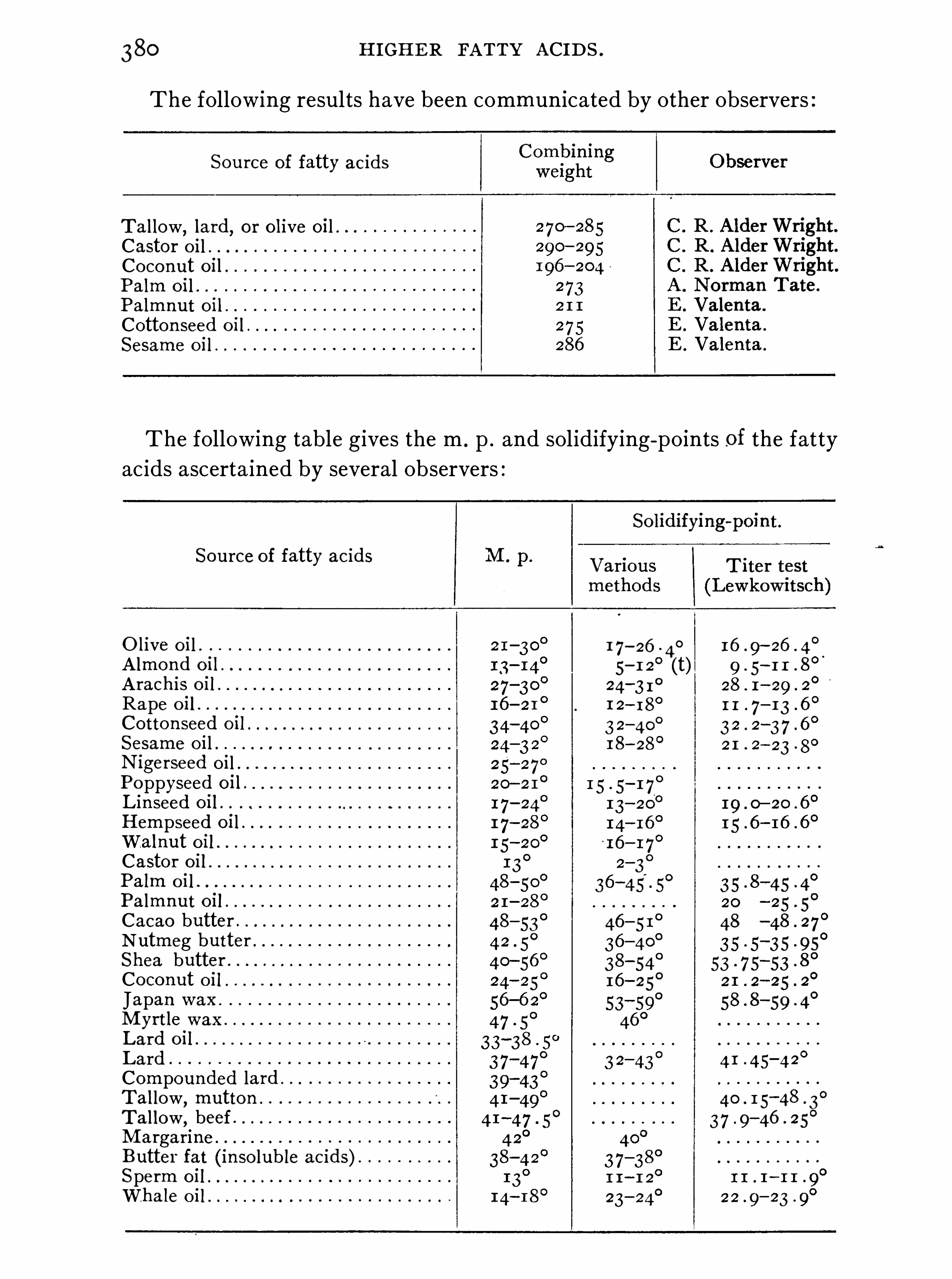
380 HIGHER FATTY ACIDS .
The following results have been communicated by other observers
Source of fatty ac ids
T allow,lard
,or Olive Oil
Castor o ilCoconut OilPalm o i l
Palmnut O i l
Cottonseed O i l
Sesame Oi l .
The following table gives the m . p . and solidifying-points of the fattyacids ascertained by several observers
Source of fatty ac ids
O live O i l . 1 7—26 . 4
°1 6 . 9Almond o i l . 5
—1 2° (t)Arach is o i l . 24—3 1
°28 1—29 2
Rape o i l . 1 2—1 8° 1 1 . 7
3 2—40
°
S esame O i l . 1 8—28° 2 1 . 2—23 . 8°
Nigerseed 011
Poppyseed O i l .
L inseed o il .
Hempseed o i l
Walnut o i lCastor o i l .
Palm o i l
Palmnut o il
Cacao butter .
Nutmeg butterS hea butter .
Coconut o i l .
Japan waxMyrt le waxLard Oil
Lard 4 1 . 45—42
°
Compounded lardTallow
,mutton 40 1 5
—48 3
°
T allow, beef . 3 7 . 9MargarineButter fat ( inso luble acids)S perm o i l .
Whale O i l .

SEPARATION .
The differences in the m . p . and solidifying-points are in greatmeasuredue to different methods of observation .
The figures have,in some instances
,considerable practical value .
Thus,the high m . p . of the fatty acids obtained on saponification
distinguishes cottonseed oil from nearly all other liquid fixed O ils Ofvegetable origin
,and enables its presence to be inferred in admixture
with other oils ; the m . p . of the acids from cacao butter i s remarkably constant
,and i s sometimes useful as a test of the purity Of the fat ;
while the solidifying-point of the acids from palm oil aff ords a practicalindication of the value of the sample to the candle manufacturer .The same remark applies to the fatty acids of tallow
,and a table has
been constructed by Dalican (page 2 1 2) by which the proportion ofOleic and solid fatty acids which a sample of tallow will yield can bededucted from the solidifying-point of the mixed acids .Separa t ion of Mixed Fatty Ac ids .
—The actual separati on ofmixed fatty acids is often a problem of extreme diffi culty
,and indeed
cannot in all cases be satisfactorily solved in the present condition Of
chemi stry . Methods for eff ecting the recognition and separation ofthe lower members of the stearic series will be found in Volume 1 .
The principles which have been applied to the fatty acids enumeratedin the tables on page 3 7 2 et seq . include the following
1 . The mixed fatty acids are well washed by agitation with hotwater , when those containing 1 0 atoms or fewer of carbon are dissolved .
This process is applied to the analysis of the fatty acids from butterfat .
2 .. The mixed fatty acids Obtained by treating the soaps with a
moderate excess of dilu te sulphuric acid are distilled with water,either
with or without the aid Of a current of steam (page Thismethod allows a more or less complete separation of the homologuesup to lauric acid from the higher members of the stearic series . Thesoluble acids Obtained in 1 are not necessarily the same as in 2 .
3 . The acids are converted into barium salts,and the precipitate
treated with water or alcohol . The barium salts of lower members upto capric acid can be dissolved out by boiling water (page4 . The alcoholic solution Of the acids i s precipitated by magnesiumacetate . By operating fractionally some useful separations may beeffected (see below).5 . The acids are converted into lead salts
,which are then treated
with ether or alcohol . An application of this principle enables oleic

HIGHER FATTY ACIDS .
acid and its homologues to be separated from the higher acids of thestearic series .6 . Fractional distillation , fractional fusion and pressure , and frac
tional solution in or crystallisation from alcohol or other solvents areother processes employed for the separation of the fatty acids .No precise method of separating oleic acid and its homologues fromlinoleic acid has hitherto been devised . Possibly one might be basedon the conversion of the acids of the Oleic series into isomers Of higherm . p . and modified properties by means of nitrous acid . Methods1,2,and 3 have already been sufl‘i ciently described , and those under 6
do not require further notice . Methods 4 and 5 , however, are described in detail below .
S eparati on of theHigher Fatty Acids of the S teari cS eri esf—T he higherhomologues of the stearic series can be separated from the lower members by treatment with hot water or distillation in a current ofsteam , and from the insoluble and non-volatile acids Of other series bytreatment of the lead soaps with ether . By proper application of thesemethods there may be Obtained a mixture of solid
,non-volatile homo
logues of stearic acid , which , according to its origin , may contain moreor less lauric , myristic , palmitic , stearic , arachidic , and other less frequently occurring acids of the series . The separat ion of these homologues i s extremely diffi cult , and a quanti tat ive est imation of severalimmediate homologues occurring in a mixture i s especially so . Ad
vantage may be taken of the limited solubili ty of arachidi c acid inalcohol to effect its separation
,as is done in Renard ’s process for the
detection of earthnut Oil (page and indeed the solubili ty of thehomologues in alcohol rapidly increases with a diminution of the number of carbon atoms in the acid . For the actual separati on Of thehigher homologues of the stearic series from each other
,however
,the
most satisfactory method is that Of Heintz (J . pr . Chem.,1 855 , 66 ,
based on fractional precipitation of the alcoholic solution of the acidswith magnesium acetate . This salt precipitates acids of the stearicseries more easily than it does oleic acid and its homologues , and ,of the different homologues of the stearic series
,those of the highest
molecular weights are thrown down first . In practice , 40 grm .
of the mixed fatty acids should be dissolved in such a proportion ofhot alcohol that nothing will separate on cooling
,even at and the
hot liquid treated with a boiling alcoholic solution Of grm . Of
magnesium acetate . The liquid is well agitated and allowed to become

384 HIGHER FATTY ACIDS .
will contain only two homologues,in which case the result Of the titra
tion not only indicates the nature of the homologues present,but in
many cases allows of their relative proportion being calculated . Thus,
if,in the course Of a systematic fractional precipitation as magnesium
salts,a fraction Of fatty acids is obtained having a mean combining
weight Of 264 .5 , i t will almost certainly consist essentially of a m ixture ofstearic and palmi tic acids
,the former of wh ich has the molecular weight
284 and the latter 2 56 , the difference being 28 . Hence every 1 %
of stearic acid in the mixture will raise the combining weight orfor every unit above 256 found for the combining weight Of the fraction3 . 5 7 of stearic acid should be calculated . As with all indirect methodsof the kind
,the results Obtained are fairly satisfactory when both
consti tuents are present in considerable proportions, but are ofli ttle value for mixtures in which one constituent very largely predominates .The titration having been completed
,the alcohol may be boiled
Off and the fatty acids again liberated and subjected to renewedfractional precipitation or crystallisation from alcohol . The productsso Obtained can be again titrated
,and thus th e progress of the
i solation and purification of the fatty acids checked in a simple andsatisfactory manner .Valuable information respecting the composit i on Of the variousfractions obtained by the precipitation as magnesium salts i s Obtainableby determining the 111 . ps . of the fatty acids . For this purpose theyshould be purified by a single crystalli sation from hot alcohol anddried by pressure between blotting-paper . Unfortunately , the m . p .
of a mixture of two or more homologous fatty acids i s not the meanof the m . ps . Of the constituent acids . The m . ps . of various mixturesof solid fatty acids have been very carefully determined by Heintz ,who has also noticed that the mixtures
,on solidifying
,crystallise in
more or less characteri stic forms or remain amorphous , accordingto the proportions in which the constituents are present . The following are some of the more important of the results of Heintz :
Vo l. 11 ,
—25
Crystalline scales.
S lender needles .
Very indistinct needles .
Amorphou s,wavy, dull.
Large crystalline lam ina .
Large crystalline lam ina .
Amorphous,lumpy .
S lender needles .
S lender needles .
Crystalline scales .
Crystalline scales .

386 HIGHER FATTY ACIDS .
Heintz also noticed that the addition of a third acid,even Of higher
m . p ., to a mixture of two homologous acids causes a lowering of the
m . p . This is shown in the following table :a:‘
30g .2
d H j:m (I)
.8 i é15
edce
Manner of 3 E Manner Of
so lidificatIOn 8 is? 1 so lidification"6
'0H790
U) pgHT } 0
53 as «3
The importance of ascertaining the solidi fying points of mixturesof fatty acids has been latterly emphasised as the data Obtained aremore reliable than those derived from m . p . observati ons . Heintz Oh ~
tained his values by the capillary method , but L . E . O . de Vi sser (Rec.
Trav. Chim.,1 898, 1 7 , 1 82
,by using larger quanti ties and only
allowing the temperature to fall slowly,Obtained the following values
for the solidifying points of mixtures of stearic and palmiti c acids :M IXTURE OF STEARIC AND PALM IT IC ACIDS .
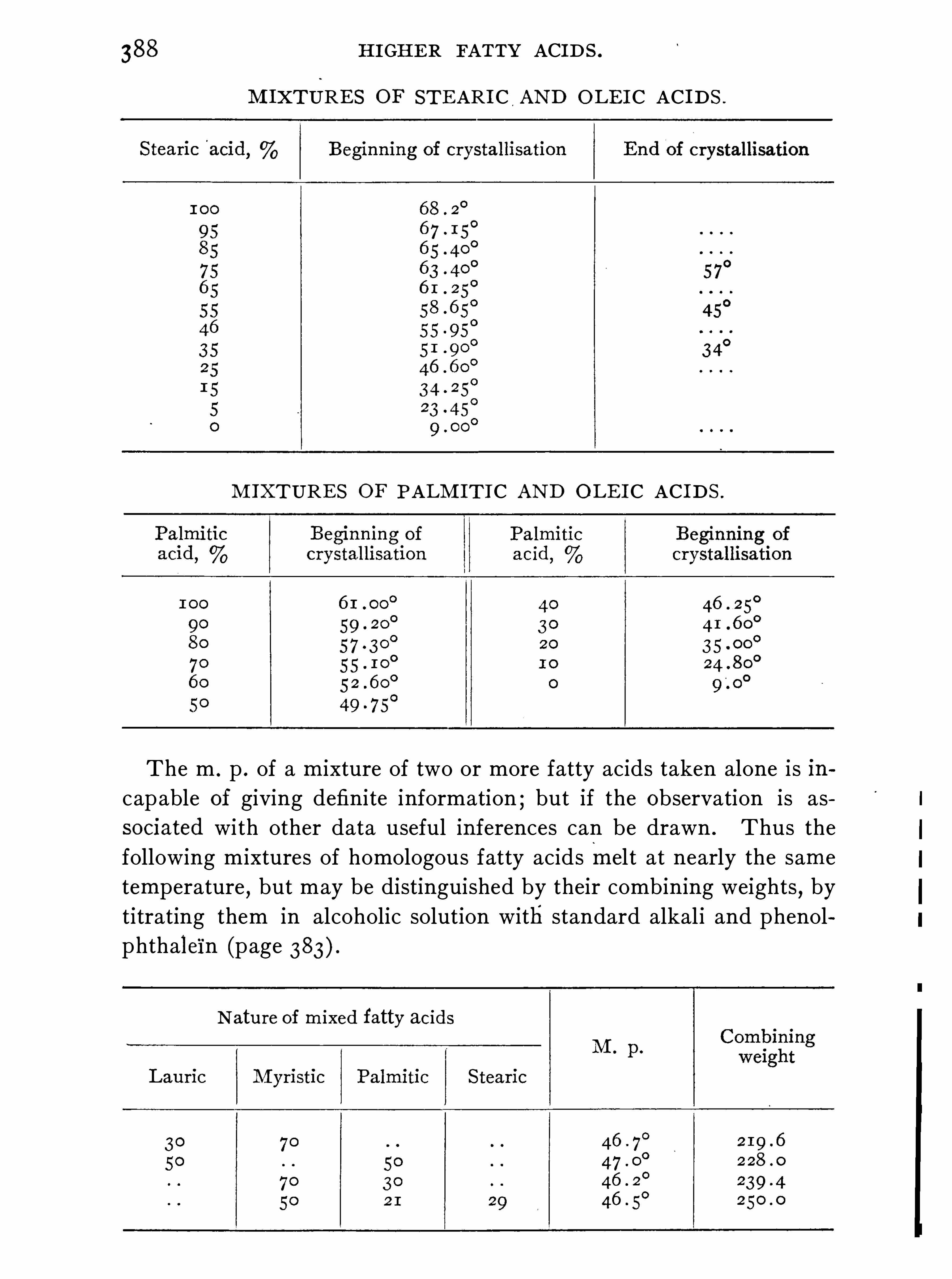
388 HIGHER FATTY ACIDS .
M IXTURES OF STEARIC AND OLEIC ACIDS .
M IXTURES OF PALM IT IC AND OLEIC ACIDS .
The m . p . of a m ixture of two or more fatty acids taken alone is incapable of giving definite information ; but if the Observation is associated with other data useful inferences can be drawn . Thus thefollowing mixtures of homologous fatty acidsmelt at nearly the sametemperature
,but may be distinguished by their combining weights
,by
titrating them in alcoholic solution with standard alkali and phenolphthalein (page
Nature of mixed fatty acids

S EPARATION OF STEARIC SERIE S .
This method Of separation must , however, be used with care , especially if more than 2 acids are present in the mixture . Kreis andHafner (B er .
,1 903 , 3 6 , 2 7 70) i solated in this way from lard an acid ,
C I 7H
3 402 ,
m . p . 5 5 but Holde,Ubbelohde and Marcusson
(B er .
, 1 905 , 3 8 , 1 250) have shown that not only this , but all the socalled daturic acids are mixtures of acids containing an even numberof carbon atoms . Erroneous conclusions from the mixed m . p . determination may arise from ,
the presence of .an acid , a ,of a higher m . p .
than acids b and c,and fractional precipitation of such a mixture may
give rise to several fractions Of approximately the same 111 . p . andcombining weight . These
,however
,may be resolved by repeated
fractionation by means of magnesium acetate or distillation in vacuo .
The method of examining fatty acids,proposed by B enedikt and
Ulzer, consisted in preparing the acetyl derivatives and then ascertain
ing the amount of alkali required for saponification .
I t was at first assumed that only hydroxylated acids (e . g .,ricinoleic
acid) form acetyl derivatives when treated in this way , but Lewkowi tsch(J . S oc . Chem. Ind.
,660) showed that saturated acids , like
capric , palmitic , stearic , and Oleic acids , give considerable acetyl values .This is due to the formation of anhydrides of these acids
,thus ,the an
hydride so formed,not being hydrolysed
,by hot water
,and even only
partially hydrolysed when titrated in cold alcohol with alkali . Lewkowi tsch recommended (J . S oc. Chem. Ind. ,
1 890 , 9 , 846) that theacetyl actually combined with the hydroxylated fatty acid be estimatedby hydrolysing with alcoholi c potassium hydroxide
,boiling off the
alcohol and after liberation of the acid with sulphuric acid , distillingOff the acetic acid and estimating its amount in the distillate . Thevalue so obtained he called the “ true acetyl value . The method devised by L ewkowitsch in 1 897 (J . S oc. Chem. Ind.
,1 897 , 1 6 ,
is now in general use ; i t is described on p . 33 .
S epara t ion of Ac ids Of the S tear ic S er ies from Fa t ty Ac ids of
Other S e ries .—The higher homologues of the stearic series of fatty
acids being solid at ordinary temperatures,while the fatty acids
'
of
other series (e . g .,Oleic
,linoleic
,ricinoleic) are liquid , a more or less
complete separation can be effected by subjecting the mixture tofiltration or pressure . The latter plan is employed with considerablesuccess on a large scale . Crystallisation from hot alcohol also servesto free the solid fatty acids from those fluid at ordinary temperatures ,

3 90 HIGHER FATTY ACIDS .
but neither plan allows of the latter being Obtained even moderatelyfree from admixed solid acids
,and such methods are quite useless for
quantitative work .
A general method by which stearic acid and its homologues may beseparated from Oleic and other liquid fatty acids
,i s based on the fact
that the lead salts of the acids of the stearic series are almost insolublein ether
,while the corresponding compounds of the other fatty acids
are soluble . S ince the lead salts Of the solid acids are not wholly insoluble in ether
,and those Of the drying fatty acids are not completely
dissolved , the results are not strictly accurate . The best method ofoperating is probably that of Muter and De Koningh . 3 grm . Of thefat should be treated
,in a flask furnished with a long tube
,with 50 c .c .
Of alcohol and a fragment of potassium hydroxide . The contents ofthe flask are boiled till hydrolysis i s complete
,when a drop of
phenolphthalein solution i s added and acetic acid until the solutioni s slightly acid . An alcoholic solution of potassium hydroxide is thenadded drop by drop until a faint permanent pink tint i s obtained ,when the liquid is slowly poured
,with constant stirring
,into a beaker
containing a boiling solution of 3 grm . of neutral lead acetate in 200 c .c .of water . The solution i s rapidly cooled and stirred at the same time ,to induce agglomeration of the precipitate
,and the clear liquid is
poured off . The precipitate is well washed,by decantation , with
boiling water and transferred to a
‘
stoppered bottle , in which it i streated with 1 20 c .c . of ether and allowed to remain 1 2 hours . (Wal ~lenstein and Finck use a Drechsel gas-washing flask having the tubeshortened about two—thirds to contain the ethereal solution
,and pass a
current of hydrogen through it for about a minute . In the case ofcolourless fats the liquid is said to remain practically colourless at theend of 1 2 hours
,but if free access of air i s permitted
,a dark yellow
solution is produced by oxidation .) Lead oleate , hypogeate , l inoleateor ricinoleate will be dissolved by the ether
,leaving lead laurate ,
myri state,palmi tate
,stearate
,and arachidate undissolved . Lead
erucate i s sparingly soluble in cold ether,but readily in hot . The con
terits of the bottle are filtered through a covered fil ter into a Muters eparating-tube (Fig . 40 c .c . of dilute hydrochloric acidadded and the tube shaken till the clearing of the ethereal solution Showsthat the decomposition of the lead soaps is complete . The aqueousliquid
,containing lead chloride and excess of hydrochloric acid , i s run
Off through the bottom tap,water added
,and agitated with the ether ,

392 HIGHER FATTY ACIDS .
Lard , Am erican (high est )Lard , AmericanLard , B erlinLard
, German
Lard , HungarianLard , Rouman ianLardLardB eef ta llowB eef t a llow , Au st ra lianB eef t allow , B erlinB eef ta llow, HungarianCo t t on seed o i lCot t on seed Oi lCot t on se ed O i l , American , wh it eCo tt onse ed o i l , American , y e llowCot ton seed o i l , Egy pt ianCo t t onse ed o i l , PeruvianMa ize o i lNig erseed o i lArach is Oi lRape o i lCoconu t o i l
Lewkowitsch,however
,recommends that the Muter tube be not
used , the ethereal solution of the fatty acids being merely filteredthrough a folded filter into a flask . The ether is evaporated and thelast traces of water are removed in a current of carbon dioxide by immersing the flask in boiling water .The method is only appro ximately accurate and possesses the disadvantage
,that the fil tration of the ethereal solution i s frequently
slow and consequently oxidation can hardly be avoided . O thersolvents have been proposed
,notably petroleum ether b . p . below
in which lead palmitate and stearate are much less soluble than inether (Twitchell , J . S oc. Chem. Ind .
,1 895 , I 4 , 5 1 6) and benzene
(Fahrnsteiner, Zei t. Unters . N ahr . Genussm,1 898 , the lead
salts of the fatty acids being insoluble in this medium at 8 to Thismethod is
,however
,not so exact as the lead salt-ether method
,and
preference should be given to the latter .Separation of the saturated acids from the unsaturated has also beenproposed by means Of sulphuric acid (Twitchell , J . S oc. Chem. Ind.
1 89 7 , 1 6, 1 002) and by means of the greater solubility of the lithiumsalts Of the unsaturated acids in alcohol (Partheil and Ferie ; Arch .
Pharm., 1 903 , 24 1 , but these methods have been adversely
criticised and appear to Offer no advantages over the older method .
v . Raumer.Wallen st e in and F inck.Wallen st e in and Finck.v . Raumer .Wa llenst e in and Finck.Wa llenst e in and Finck.Wa llen s te in and F inck.Mu t er and DeKoni ngh .v . A sbo th .
Mut er and”
DeKon ingh .Wa llenst e in and Finck.Wa llen ste in and Finck .Wallen st e in and F inck .
Mu t er and DeKon ingh .v . A sbo th .Wa llenst ein and F inck .Wallenst e in and Finck .Wa llenst e in and Finck.Wa llenst e in and F inck.Wallenst e in and F inck.Wa llenst e in and F inck.Wa llen st e in and F inck.Wallenst e in and Finck.Wa llenst e in and F inck.

E STIMATION OF STEARIC ACID . 393
Hebner and M i tchell have devised the following method Of theestimation of steari c aci d: Prepare a supply of alcohol saturatedat 0° with pure stearic acid or with stearic acid which only containstraces of palmi tic acid . D issolve from 0 .5 to 1 grm . of the mixtureof fatty acids to be examined if these are solid , or about 5 grm . i ffluid
,in about 1 00 c .c . (exact measurement i s not necessary) Of the
stearic alcohol solution . Leave this liquid in an ice-bath overnight,
agitate the mixture next morning and allow to stand in ice for a shorttime ; filter
.
off while the mixture remains in ice,wash with stearic
alcohol solution at dry and weigh . Ascertain the m . p . of theproduct which should not be much less than S ince the sidesof the interior of the flask
,as well as the residue of crystallised stearic
acid retain a small amount of the alcoholic solution,a correction
experimentally found to be grm . has to be applied,this amount
being deducted from the total weight found . In their experimentsthe authors used methylated alcohol of sp . gr . but obviouslythe exact strength is a matter of no consequence .For maintaining a constant temperature
,Hebner and Mitchell used
an ice-chest consisting of a metal box with sockets soldered to its sidesto receive clamps for holding flasks
,submerged to the neck in ice
water,in which the analyses were carried out . The metal box was
fitted in a wooden box,and the space between the metal and wood
was packed with wool and sawdust,while a cushion of wool and flan
nel was placed between the lids of the metal and wooden boxes .For the preparation of the stearic solution about 3 grm . of purestearic acid were dissolved in about a litre Of warm alcohol of sp .
gr. and the stoppered bottle containing the solution placedovernight in the ice-water (which contained lumps of ice) in the chest ,so that the bottle was submerged up to the neck . After 1 2 hoursa considerable portion of the stearic acid had crystallised out . Thesaturated mother—liquor was syphoned off wi thout removing thebottle from the ice-water . The filtering syphon consisted of a smallthistle funnel twice bent at right angles
,fitting with its straight limb
into a flask in connection with a suction pump . The bulb Of thefunnel
,which was submerged in the ice-cold solution, was covered
over with a piece of fine calico . On applying suction , a perfectly clearstearic solution was Obtained
,saturated at or rather at o . 2
°
,
which was the temperature almost constantly shown by a standardthermometer .

394 HIGHER FATTY ACIDS .
A precisely similar mode of filtration was also adopted in the quantitative experiments on mixed fatty acids , the thistle funnel used beinga miniature one
,with a bulb not larger than about 5 of an inch in
diameter .EST IMATION OF STEARIC ACID IN M ISCELLANEOUS FAT S .
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
Horse -kidne y fa tCo t t on o i l st earin e
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
o o o o o o o o o o o o o o o o o o
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
Numerous estimations of the stearic acid in butter were made .In many cases none
,or a minute quanti ty only
,was found . In some
cases phenomena of supersaturation apparently occurred . On the
first examination in the morning the solution was perfectly clear , butafter shaking the contents and allowing to s tand some time longer inthe ice
,a small but increasing quantity of crystals formed .
The method appears ~to be inapplicable to the fatty acids from Japanwax . From mixtures of these with pure stearic acid
,the latter could
only be recovered partially,and in some cases not at all .
Kreis and Hafner (Zei t. N ahr . Genussm., 1 903 , 6 , 2 2) state that thi s
method gives trustworthy results provided that not less than 0 .5 grm .
of the mixed fatty acids is taken,as stearic acid easily forms super
saturated solutions . This i s borne out by Emerson (J . Am. Chem.
S oc .
, 1 907 , 2 9 , 1 7 50) and i s said to explain the differences in thesolubili ty Obtained . Appended are resultsB ehner andM i tchell found solubility at 0° in alcohol grm .
Kreis and Hafner found solubili ty at 0° in 95% ethyl alcohol
grm .
Emerson found solubility at 0° in 95% ethyl alcohol grm .
The alcohol used by Hebner and Mi tchell , as already pointed out ,was rectified methylated spiri t .
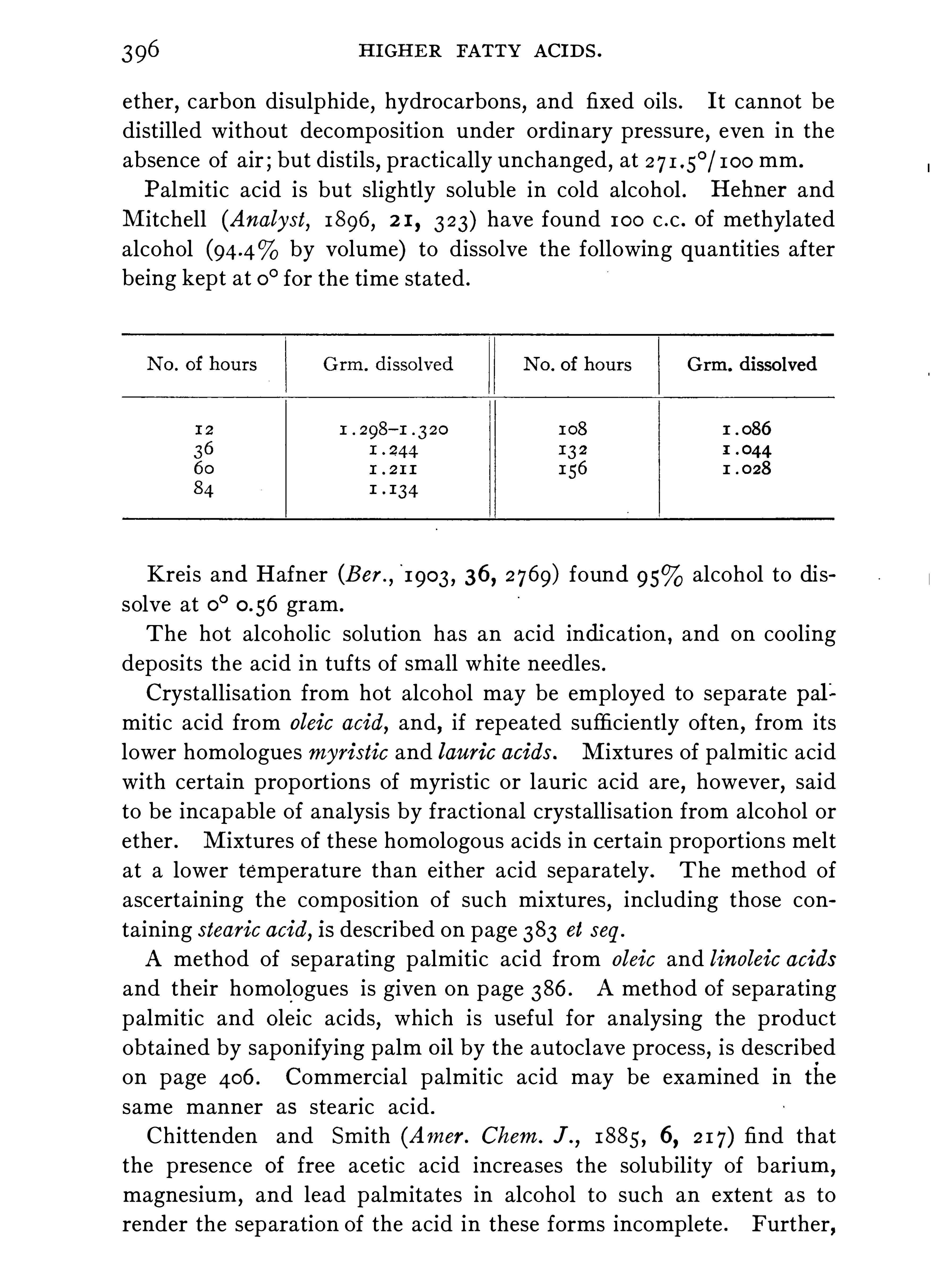
3 96 HIGHER FATTY ACIDS .
ether , carbon disulphide , hydrocarbons , and fixed oils . I t cannot bedistilled without decomposition under ordinary pressure
,even in the
absence of air ; but distils , practically unchanged , at mm .
Palmitic acid is but slightly soluble in cold alcohol . Hehner andM itchell (Analyst, 1 896 , 2 1
, 3 23) have found 1 00 c .c . Of methylatedalcohol by volume) to dissolve the following quantities afterbeing kept at 0° for the time stated .
Kreis and Hafner 3 6 , 2 769) found 95% alcohol to di ssolve at 0° gram .
The hot alcoholic solution has an acid indi cation , and on coolingdeposits the acid in tufts of small white needles .Crystallisation from hot alcohol may be employed to separate palmiti c acid from olei c acid
,and
,if repeated sufficiently often , from its
lower homologues myri sti c and lauri c aci ds . M ixtures of palmitic acidwith certain proportions of myristic or lauric acid are
,however, said
to be incapable of analysis by fractional crystalli sation from alcohol orether . M ixtures of these homologous acids in certain proportions meltat a lower temperature than either acid separately . The method ofascertaining the composition of such mixtures
,including those con
taining steari c acid,is described on page 383 et seq .
A method of separating palmitic acid from olei c and linolei c aci dsand their homologues is given on page 386 . A method Of separatingpalmitic and Oleic acids
,which is useful for analysing the product
obtained by saponifying palm Oil by the autoclave process , i s describedon page 406 . Commercial palmitic acid may be examined in the
same manner as stearic acid .
Chittenden and Smith (Amer . Chem. J .,1 885 , 6 , 2 1 7) find that
the presence Of free acetic acid increases the solubili ty of barium ,
magnesium,and lead palmitates in alcohol to such an extent as to
render the separation of the acid in these forms incomplete . Further ,

PALMI TIC ACID . 397
the precipitates undergo partial decomposition when washed,either
with water or with alcohol containing acetic acid .
Me tal l ic Palm i tates .—These present the closest resemblance to
the corresponding stearates (page 399 et and require but littleseparate description . B arium
,magnesium , and lead palmitates are
more readily soluble in alcohol,especially in presence of acetic acid
than are the corresponding stearates .Adipocere , a wax-like substance found in large quantity in corpsesburied under certain conditions
,i s said to consist largely of palmitic
acid mixed with potassium and calcium palmi tates .Aluminium palmi tate may be prepared in a manner similar to thecorresponding oleate . I t i s an elastic amorphous mass
,insoluble in
water,but dissolving in petroleum spirit and oil of turpentine to form
very viscid solutions which have found applications as varnishes . Thefilm of aluminium soap left on evaporation retains its elasticity
,and
is Odourless and impervious to water (see J. S oc.Chem. Ind., 1 882 1
,2
Aluminium palmitate has some practical interest as an ingredient of“Oil pulp ” or “ thickener .”Palmi tic Esters .
—These present a close analogy to the corresponding stearates .
‘
Glyceryl Palmi tates or Palmi tins are Obtainable synthetically bymeans similar to those employed for the preparation of the stearins .Chittenden and Smith (Amer . Chem. J 1 885 , 6, 2 1 7) have given thefollowing data
1 00 parts of,
absolut e alcoho l , at 4 1 3 5 0 2 1 0 0 00 520—2 d 1sso lve ,
Appearance of fa t deposit ed Long curved Groups of irregu larfrom a lcoh ohc so lu t i on ,n eedle s . cry stals .
Ap p eaance of fat deposit ed Warty masse s .from e th erea l so lu t ion ,
s 7 . e° ' —
4 7°
1 Krafft (B er. 1 9 03 , 3 6 , 43 4 3 ) gi ves m . p .
2 Probabl a monog l cerid e ; Grun (B er . , 1 9 0 5 , 3 8 , 2 2 84) m . p .
3 80h e ij ( ec. Trav. him . , 1 809 . 1 8 , 1 6 9 ) gi ve s m . p . gr . 0 . Gu th (Ze i t.B i ol , 1 9 02 , 44 , m . p .

398 HIGHER FATTY ACIDS .
An isomeric modification Of dipalmitin ,the B, has been Obtained ,
m . p . There was also obtained a very stable mixture of 1 partof palmitin with 3 of dipalmitin . This product crystalli sed fromalcohol in bunches of needles
,which melted at 68° to 69° and solidified
between 64° and 67°
STEARIC ACID.
C I SH3 60 2=
The glyceryl ester of thi s acid occurs extensively in nature,es
pecially in the harder fats of the animal kingdom ,such as mutton and
beef suet .Pure stearic acid may be prepared by hydrolysing tallow with potassium hydroxide
,decomposing the solution of the resultant soap with a
dilute acid,and purifying the liberated fatty acids from Oleic acid by
crystallisation from hot alcohol . The pressed crystals consist essentially of a mixture Of stearic and palmitic acids . It should bepurified by recrystalli sation , and 4 parts dissolved in such a proportionof hot alcohol that nothing will separate out on cooling to A solution of 1 part magnesium acetate in boiling alcohol is added and theliquid allowed to cool
,when magnesium stearate will separate (page
The precipitate i s filtered Off , washed with cold alcohol , boiledwith water and hydrochloric acid
,and the purity of the resultant
stearic acid proved by a careful determination of the m . p . whichshould be 2
°
(de Vi sser).The commercial product commonly termed “ stearine really consist s of a mixture of free stearic and palmitic acids
,and may be
conveniently employed for the preparation of pure stearic acid,in
stead of tallow or other fat . The “ stearine ” may be at once dissolvedin hot alcohol and the solution precipitated with magnesium acetate asabove described . Commercial stearine often contains a considerableadmixture of parafl‘in wax or other hydrocarbons , the absence of whichshould be proved before employing the substance for the preparationOf steari c acid .
Shea-butter,when obtainable
,may be conveniently employed as a
source of stearic acid , as the fatty acids produced by its hydrolysi sconsists solely of stearic and oleic acids
,which can be separated per
fectly by repeated crystallisation from hot alcohol .

400 HIGHER FATTY ACIDS .
Palmi tates and stearates may also be distinguished by the m . p .
and combining weights of the liberated fatty acids .P otassium stearate may be prepared by saturating a hot alcoholicsolution of stearic acid with alcoholic potassium hydroxide
,using
phenolphthalei n as indicator . On concentrating the solution andallowing i t to cool
,the potassium stearate crystallises in shining
needles or lamina . I t also separates on cooling a solution Of 1 part ofstearic acid and I of potassium hydroxide in 1 0 parts of water . Theopaque granules formed may be purified by crystallisation from alcohol . Or a boiling alcoholic solution of stearic acid may be mixed withan excess of a boiling aqueous solution of potassium carbonate
,the
liquid evaporated to dryness,the residue extracted with boiling alco
hol,and the filtered solution allowed to cool
,when crystals of potas
siumstearate will be deposited .
Potassium stearate dissolves in about 1 0 times its weight of waterat the ordinary temperature
,forming a mucilaginous mass . On heat
ing the solution it becomes clear,and if diluted with a large proportion
of cold water hydrogen stearate of the compositionseparates in delicate
,white
,pearly lamina
,while a basic stearate re
mains in solution . An analogous decomposition by excess of wateri s suff ered by other alkali-metal salts of the higher fatty acids
,and i s a
leading cause of their application as soaps .Ammoni um stearate i s Obtained as a crystalline mass by incor
porating strong ammonia with melted stearic acid , and keeping theproduct over sulphuric acid till the excess of ammonia has evaporated .
On further keeping in this manner,i t gradually loses ammonia .
(Wright and Thompson .)S odi um stearate resembles the potassium salt
,but i s harder . I t
IS decomposed in a similar manner,but with greater facili ty
,by excess
Of water,and is less soluble in alcohol than potassium stearate . So
d ium stearate may be separated from sodium palmitate by fractionalcrystalli sation from hot alcohol .Barium and calci um stearates are crystalline precipitates insoluble inwater . The magnesium salt is similar, but soluble in boiling alcohol .Lead stearate, as prepared by double decomposition , forms awhite amorphous powder
,melting at 1 25° to a colourless l iquid , which
solidi fies on cooling to an opaque amorphous mass . I t i s insoluble inwater
,alcohol
,ether
,or petroleum spirit . In th ese characters i t i s
Simulated by lead palmitate,myristate
,arachidate
,etc .
,but the lead

STEARIC ACID . 401
salts of Oleic acid and its homologues , as also of linoleic and ricinoleicacids
,are soluble in ether and petroleum spiri t .
Steari c Esters—Ethyl stearate i s prepared by passing hydrogenchloride into a solution of stearic acid
,in absolute alcohol . I t i s also
formed by boiling tristearin with sodium oxide,or with a quantity of
alcoholic potassium hydroxide insuffi cient for its complete saponification . Ethyl stearate is a crystalline
,easily fusible
,wax—like solid
,
m . p . 3 readily soluble in alcohol and ether , and b . p . 2 24° with
partial decomposition,b . p . in a vacuum 1 39
° or 1 54°Glyceryl stearates are Obtainable synthetically by heating together
,
under pressure , suitable proportions of stearic acid and glycerol . Products contain ing either 1
,2 , or 3 molecules of the stearic radicle are
thus Obtainable . Another method of obtaining stearin consists intesting a
,By
-tribromOpropane and sodi um stearate at 1 70—1 80° for
ten hours .Monostearin and distearin do not appear to occur naturally , but
glyceryl stearate is identical with the stearin which,in admixture
with palmitin,constitutes the less fusible portion of solid fats . For
brevi ty,this stearate i s frequently called “ stearin .
”I t i s not identi
cal with commercial “ stearine,
” which is a mixture of free stearicand palmitic acids obtained by the saponification of the neutral fats .S tearin forms white
,Shining nodules
,fine needles
,or pearly lamina
resembling spermaceti . I t i s tasteless,neutral
,and volatile almost
without decomposition in a vacuum . Heated to a high temperature ,i t decomposes and gives Off acrolein . It appears to exist in twoisomeric modifications . As crystallised from ether i t has a m . p .
of sp . gr. (Scheij , loc . If the crystals so Ob
fained be heated 4° or more above the m . p .,they are converted into
a modification which solidifies to a waxy mass at and melts at 5If the latter be reheated a few degrees above the m . p .
,the original
substance,melting at is obtained (Guth , loc.
S tearin is insoluble in water and nearly insoluble in rectified spirit .In boiling absolute alcohol i t dissolves freely
,and is deposited in flocks
on cooling . S tearin also dissolves readily in boiling ether, but theliquid retains less than on cooling . I t is readily soluble in fixedand volatile oils
,and in carbon disulphide . When heated in a vacuum ,
i t distils almost unchanged,but under the ordinary pressure i t i s decom
posed with formation of carbon dioxide,acetic acid
,water
,free carbon ,
and olefines of b . p . ranging from 1 90 toVo l . I I .
—26

402 HIGHER FATTY ACIDS .
Pure stearin does not change on exposure to air at the ordinarytemperature . When impure , i t i s liable to become rancid , apparentlyOwing to the presence of olein . S tearin readily undergoes saponification when heated with alkalis or other strong bases
,with formation
of a metall ic stearate and glycerol .
OLEIC ACID.
C 1 8H3 4O , .
Oleic acid is one of the most widely distributed fatty acids , occurring as an ester in most non-drying fixed Oils
,especially almond and
Olive Oils , and in smaller proportion also in solid fats , such as lard ,palm Oil
,butter
,and goose fat .
For the preparation of pure Oleic acid an Oil rich in Olein,as almond
or Olive Oil,i s saponified by alkali , the soap dissolved in water and
decomposed by excess of dilute hydrochloric or sulphuric acid . WhiteCastile soap may be employed as the starting-point , thus savingthe trouble of saponifying . The use Of commercial Oleic acid i s notto be recommended
,ow ing to the frequent presence of hydrocarbons .
The liberated fatty acids are separated from the aqueous liquid,and
heated for some time on the water-bath with about 1 part of finelyground lead monoxide for every 20 parts of oil taken for the operation . Excess of lead oxide should be avoided
,as it occasions the
formation of a basic oleate,which is subsequently treated with di ffi
culty . The proportion of lead oxide prescribed i s insufli cient to combine with all the fatty acid
,but the result i s merely that a portion of
the Oleic acid remains in the free state,while the more powerful
palmitic and stearic acids form lead salts .The product is next treated with about twice its volume of ether,which dissolves the lead oleate and free oleic acid
,and leaves the lead
palmitate and stearate unchanged . The solution is separated from theinsoluble salts
,and hydrochloric acid added
‘
until the aqueous liquidhas a strongly acid indication even after shaking . The lower layernow contains lead chloride
,while the ether retains the oleic acid . I t i s
separated from the acid liquid,washed by agitation with water , and
the ethereal layer removed and the ether evaporated Off as rapidly andat as low a temperature as possible .According to E . C . Saunders
,rectified spiri t (sp . gr . may be

404 HIGHER FATTY ACIDS.
rapi dly heated with excess of alkali . I ts formation is a. characteristictest for Oleic acid and its immediate homologues . To detect i t thealkaline residue should be ‘
treated with boiling water, and the liquidacidified with acetic acid
,again boiled
,and filtered hot . The fil tered
liquid will,on cooling
,deposit brilli ant needles of sebacic acid
,m . p .
and soluble in 1 000 parts of cold or 50 of boiling water .When more strongly heated with potassium hydroxide
,oleic acid yields
potass ium palmitate,oxalate and acetate
,and free hydrogen
,secondary
products being also formed . The temperature necessary for thischange is 300
° to The process i s commercially employ edfor the production Of palmitic acid . The following formula expressesthe main change which occurs :
Small quanti ties Of sebacic acid,caproic acid
,caprylic alcohol and
other compounds are also produced . The details of this process ofmanufacturing palmitic acid
,for which nearly all fatty substances ,
except mare ’s grease and suint fat , are available , have been describedby W . Lant Carpenter (J . S oc. Chem. Ind. , 1 883 , 2 , 98 . Comparealso Lewkowitsch
,J . S oc. Chem. Ind.
,1 879 , I 6,
Oleic acid combines with a molecule of bromine to form dibromostearic acid
, C 1 8H3 ,O , ,Br2 as a yellow viscous Oil having a frui t-like
odour . Oleic acid also combines in a perfectly definite manner withHii bl’s reagent , and may be estimated by that means .Ole ic acid is dissolved by concentrated sulphuric acid , a conjugateacid being formed which has been used in Turkey-red dyeing andcalico-printing .
S trong nitric acid oxidises oleic acid , acids of the acetic and oxalicseries ( including succinic acid) being formed .
By oxidation with potassium permanganate in presence of an excessof potassium hydroxide
,Oleic acid yields dihydroxystearic acid , a
crystalline compound,m . p . to and solidifying at 1 1 9
° toFor details of preparation consul t Le Sueur (Trans . Chem.
S oc. ,1 90 1 , 7 9 ,
When Oleic acid is heated to 200° or 2 1 0
° in a sealed tube withamorphous phosphorus and fuming hydriodic acid , i t assimilateshydrogen
,and i s converted into stearic acid .
When the red fumes generated by acting on nitric acid by starch orarsen ious oxide
,or by a mixture Of sulphuric acid and sodium nitri te ,
are passed for a short time into Oleic acid carefully -kept cold , the

OLE IC ACID .
l iquid gradually thickens,and in the course of an hour or so solidifies
to a crystalline mass of an isomer of oleic acid called elai dic acid . It
may be purified by agitation with boiling water , followed bv crystalli
sat ion from alcohol .E laidi c acid, C I 3H3 4O , ,
forms large pearly plates,resembling benzoic
acid,m . p . 5 1 and distilling almost unchanged . B . p .
mm ., 50 mm .
,mm . In the solid condition it is un
changed in the air, but in the fused state it readily absorbs oxygen ,becoming yellow and pasty
,and acquiring an Odour like that of poppy
,
Oi l. Wi th bromine,fused potassium hydroxide
,and phosphorus and
hydriodi c acid,elai dic acid behaves like oleic acid . Elai dic acid has a
strong acid reaction , and forms a series of well-defined salts , all ofwhich
,if neutral
,are said to be insoluble in water . S odium elaidate
crystallises from alcohol in silvery lamina,and the potassium salt in
glistening needl es . The barium and lead salts are white precipitates .The property of forming an isomer of higher m . p . under the in
fluence of nitrous acid is not peculiar to oleic acid . It is exhibitedalso by its olein , by its homologues hypogeic , deglic , and erucic acids ,by ricinoleic acid
,but not by the fatty acids characteristic Of the drying
oils .Es timation of Ole i c Acid.
—When occurring in the free state andunmixed with other acids , Oleic acid may be conveniently and accurately estimated by titration with standard alkali (page In
presence of acids of the stearic series i t may be titrated with Hubl’ssolution
,each c .c . of N/ 1 0,
iodine absorbed corresponding togrm . of Oleic acid . The estimation Of Oleic in presence of linoleicacid is described on page 3 78 .
O leic acid may be estimated gravimetrically when in admixturewith acids of the stearic series by utilising the solubility of its leadsalt in alcohol , ether , or petroleum spiri t , in the manner described forits preparation (page The best method of applying the principlefor analytical purposes i s described on page 390 .
According to F . Sear, palmitic and oleic acids can be separated byheating the mixture with excess of zinc oxide and digesting the productin the cold with carbon disulphideDavid ’s method for estimating oleic acid in the presence Of stearicacid
,described in the third edition of th is work
,is inaccurate .
A method for the approximate estimation Of oleic and solid fat tyacids in tallow is described onpage 2 1 3

406 HIGHER FATTY ACIDS .
Commerc ia l Ole ic Ac id .—Commercial Oleic '
acid i s obtained bysubjecting to hydraulic pressure the mixture Of fatty acids producedby the hydrolysi s of tallow
,palm oil
,and similar fats . The ex
pressed liquid,technically known as “ red oil
,
” contains a considerablequantity of palmi tic and stearic acids
,which separate out on keeping
the red Oil for some time at a low temperature .When fats are hydrolysed by the autoclave process
,the products
Often contain a considerable proportion of unchanged fats . In consequence of the comparative facility with which palmitin and stearinare hydrolysed
,the unaltered fat consists chiefly or wholly ”of Olein
,
which,owing to its low m . p .
,becomes concentrated in the oleic acid
expressed from the crude product . Hydrolysis under high pressurealways tends to cause more or less decomposition of the higher fattyacids
,and
,when actual distillation has been resorted to
,notable
quantities of acetic,suberic
,and sebacic acids are formed
,and the
two latter will remain with the Oleic acid,together with certain hydro
carbons,apparently belonging to the paraffi n series
,which are always
simultaneously produced .
Commercial Oleic acid,which is frequently
,but improperly
,called
oleine,
” varies considerably in properties and composition . I t i ssometimes a clear liquid
,ranging in colour from dark brown to pale
sherry,while other specimens are quite pasty from separated solid
fatty acids . By distillation in a current of steam ,oleic acid may be
obtained wholly free from colour,but possessing an acrid odour from
the presence of decomposition-products . Undistilled Oleic acidusually retains an Odour suggestive of i ts ori gin . The sp . gr . i salso variable
,ranging from about to or even more
,accord
ing to the proportions Of hydrocarbons,neutral oils
,and solid fatty
acids which happen to be present .Mineral aci ds are sometimes present in sensible quantity in commerciai Oleic acid . They rarely interfere wi th its applications ; but , ifnecessary
,may be detected and estimated as on page 7 5 , or by titrating
the alcoholic solution with alkali and methyl—orange .T he presence of an abnormal proportion of oxidation and secondaryproducts of an acid character i s indicated by agitating 50 c .c . of theoleic acid with 1 c .c . of a 1 0% solution of ammonia and 50 c .c . of water .B oth the Oleic acid and the aqueous liquid should by this means bedeprived Of any acid reaction of litmus .The presence of palmi ti c or steari c aci d in commercial Ole ic acid

408 HIGHER FATTY ACIDS .
According to All en—the hydrocarbons normally present in di stilledoleic acid range from 3 to and therefore any proportion notablyin excess of the latter figure may be attributed to an intentional sophistication of the product with mineral or shale Oil . The additionof these adulterants to Oleic acid i s extensively practised , althoughtheir presence greatly reduces the suitability of the oleic acid for oneof i ts most important applications
,which is that Of greasing wool
during the process Of sp inning . Any admixture of hydrocarbonsreduces the property of ready saponifiability for which oleic acid ischiefly valued .
The foregoing statement respecting the proport ion of unsaponifiablematter present in distilled oleic acid applies to a product obtained bysaponifying pure substances . Wool grease and the grease Obtained bytreating with acid the soapy liquors in which wool has been washedare much more impure articles . B esides the hydrocarbons formed ondistilling such g reases , the distilled product is liable to contain actualpetroleum or shale products used in the wool-spinning
,either inten
tionally or as adulterants of other oils , petroleum employed for antiseptic purposes ou the living sheep
,and cholesterol and other unsaponi
fiable matters contained in the “ suint ” or wool fat . Hence , an estimation of the “
unsaponifiable matter in such low-class oleic acidscannot be regarded as a reliable indication of the extent to which theyhave been adulterated by an actual addition of hydrocarbon Oil .
Some indication Of the origin Of the unsaponifiable matter may beobtained by treating the ether-residue with thrice its volume of rectifiedspiri t
,when the volume left iIndissolved may be regarded as ia
dicating roughly the hydrocarbons present , while the cholesterol andsolid alcohols from sperm or bottlenose oil pass into solution . (See“WoolThe following table are results Obtained by Allen from an ex
amination of specimens Of commercial Oleic acid of very differentqualities . The “ free fatty acids ” were estimated by ti tration wi thstandard alkali
,and calculated to their equivalent of Oleic acid ; but
in the case Of the semi-solid samples containing much palmitic acidthe result thus Obtained is necessarily in excess Of the truth . Thepercentage of ether-residue shows the “ hydrocarbons
,etc .
,in the
samples,while the esters were in some cases determined indirectly,
in other cases calculated from the result of KOttstorfer’s saponificationprocess , and in others deduced from the diff erence between the free
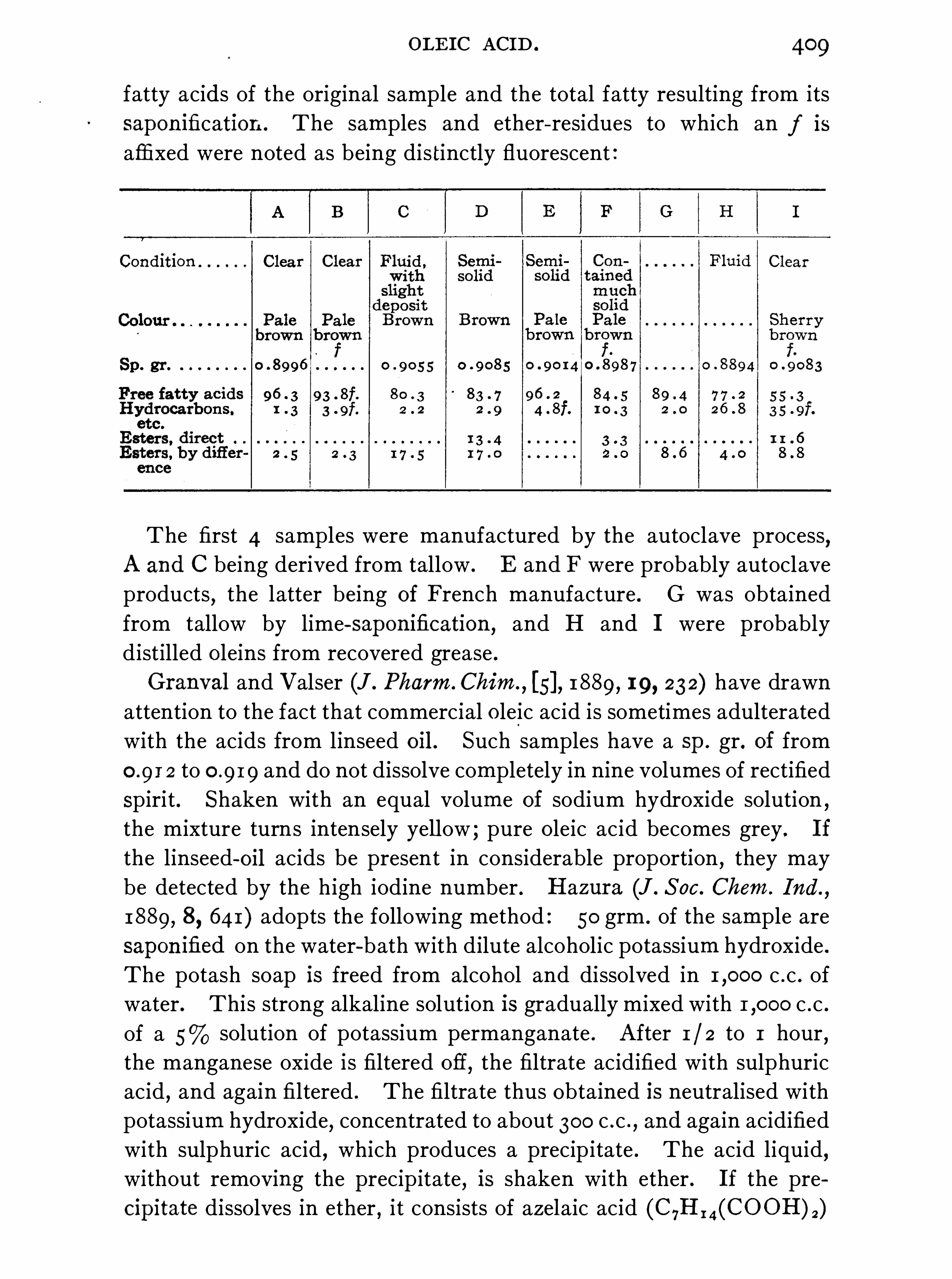
OLEIC ACID . 409
fatty acids of the original sample and the total fatty resulting from i tssaponification . The samples and ether-residues to which an f isaffi xed were noted as being dis tinctly fluorescent
Condit ion Clear
0 0 0 0 0 0 0 0
The first 4 samples were manufactured by the autoclave process,A and C being derived from tallow . E and F were probably autoclaveproducts
,the latter being Of French manufacture . G was obtained
from tallow by lime-saponification, and H and I were probablydistilled Oleins from recovered grease .Granval and Valser (J. Pharm. Chim.
,1 889 , 1 9 , 23 2) have drawn
attention to the fact that commercial Oleic acid is sometimes adulteratedwith the acids from linseed Oil. Such samples have a sp . gr. Of from
2 to and do not dissolve completely in nine volumes of rectifiedSpiri t . Shaken with an equal volume Of sodium hydroxide solution ,the mixture turns intensely yellow ; pure Oleic acid becomes grey. If
the linseed-Oil acids be present in considerable proportion , they maybe detected by the high iodine number . Hazura (J. S oc. Chem. Ind.
,
1 889 , 8 , 64 1 ) adopts the following method : 50 grm . of the sample aresaponified on the water-bath with dilute alcoholic potassium hydroxide .The potash soap is freed from alcohol and dissolved in c .c . ofwater . This strong alkaline solution is gradually mixed with c .c .of a 5% solution of potassium permanganate . After 1 2 to 1 hour ,the manganese oxide is filtered Off
,the filtrate acidified with sulphuric
acid,and again filtered . The filtrate thus Obtained is neutralised with
potassium hydroxide,concentrated to about 300 c .c . , and again acidified
with sulphuric acid,which produces a precipitate . The acid liquid ,
without removing the precipitate,is shaken with ether . If the pre
cipitate dissolves in ether , i t consists of azelaic acid

HIGHER FATTY ‘ACIDS .
and the original oleic acid is free from linseed-oil acids . If i t does notdissolve
,i t i s filtered Off
,recrystallised
' several times from water oralcohol
,with the addition of animal charcoal
,and
,after air-drying
,
its m . p . determ ined . If thi s be above linseed-oil acids areundoubtedly present .Su lpho le ic Ac id .
—When a non-drying fixed oil i s cautiously treatedwith strong sulphuric acid
,complex changes occur
,the precise nature
of which depends on the conditions of the experiment . P . Jui llard
(J . S oc. Chem. Ind .
,1 894 , 1 3 , 820) states that olein treated in the cold
with sulphuric acid yields two acids—one monobasic,the other di
basic—which appear to be addition products of sulphuric acid andolein . They are soluble in water . Oleic acid treated with sulphuricacid produces at first hydroxystearo-sulphuric acid
,C I 7H3 4 (OSO ,H)
CO ZH,from which is formed hydroxystearic acid , C I 7
H3
Me ta l l ic Olea tes .—These form a well-defined series of salts ,
many of which have received practical applications . They may beobtained by dissolving the metallic oxide of which the oleate i s requiredin warm oleic acid ; but such a method does not give compoundsof very definite composition . A preferable plan i s to precipitate anaqueous solution of sodium oleate with a neutral solution of the saltof the metal of which the oleate i s required . Zinc
,aluminium , iron ,
lead,copper
,bismuth
,and other oleates are readily Obtained in thi s
way .
These oleates are readily analysed by agitating them with ether anda dilute mineral acid
,which should be sulphuric
,hydrochloric , or
nitric,according to the metal present . The .metals pass into the
dilute acid li quid,and may be est imated by the ordinary methods of
mineral analysis . The oleic acid formed from the oleate is dissolvedby the ether
,and may be weighed after evaporating off the solvent .
Any free Oleic acid , neutral fat , or hydrocarbon (e . g .,vaseline) which
may have been present in the original substance will also be foundin the ether-residue
,and may be ascertained by the methods indicated
on page 79 et seq .
Wi th the exception of the salts of the alkali-metals,all the metallic
oleates are insoluble in water,though they dissolve in many instances
in alcohol,ether
,carbon d i sulphide , and petroleum spirit . The
calcium,magnesium
,and iron oleates also dissolve in glycerol .
P otassi um oleate i s the principal constituent of soft soap . I t i s awhite
,friable
,deliquescent substance , which with a small quantity

4 1 2 HIGHER FATTY ACIDS .
oil , and the use of a luminium soap for the purpose can onl y be regarded as an adulteration .
F erri c oleate i s dark red,but otherwise resembles the aluminium
soap .
Cupri c oleate i s a dark green , wax-l ike substance , readily Obtainedby double decomposition . It becomes quite fluid at and dissolves with green colour in all proportions of alcohol
,ether
,and fixed
oils .Lead oleate, i s the principal constituent of thelead plaster ” of pharmacy . As obtained by double decompositioni t i s a light white powder
,m . p . 80° to a yellow Oil
,and solidifying on
cooling to a brittle translucent mass . Lead oleate is quite insolublein water
,but soluble in alcohol and in ether
,especially when hot . It
is also dissolved by Oil Of turpentine and by petroleum spirit,the hot
saturated solution in the last solvent solidifying to a gelatinous masson cooling . The solubili ty of lead oleate in ether i s utili sed in analysi sfor the separation of Oleic from palmitic and stearic acids .By boiling oleic acid with water and excess of lead oxide or basiclead acetate
,a basic oleate is Obtained which is nearly insoluble in
ether .Zinc oleate i s a white unctuous powder
,soluble in carbon disulphide
and petroleum spiri t .Many of the so-called commercial “ oleates are prepared by the
use Of Castile soap instead of pure sodium oleate . They are betterdescribed at “ oleo-palmitates
,and for pharmaceutical purposes are
probably equally suitable .Ole ic Esters .
Ethyl oleate i s prepared by passing dry hydrogen chloride into asolution of Oleic acid in three times its volume of absolute alcohol .Esterification takes place very rapidly
,and the ester separates from
the liquid as an Oi ly layer . I t has a sp . gr . of at is solublein alcohol
,and i s decomposed by distillation . Nitrous acid and its
equivalents slowly convert it into the isomeric ethyl elai date .Dodecatyl oleate and its homologues are said to consti tute thegreater part of sperm and bottlenose oils .Glyceryl oleates are obtainable synthetically by heating oleic acidand glycerol together in sealed tubes at 200
° for 24 hOIIrs .
With excess of glycerol,the monolein is produced . Wi th excess of
Oleic acid,olein i s formed , and under special conditions the di oleate

OLE INS .
id to be Obtainable . Monolein and diole in are not known tonaturally
,but Olein occurs in many fixed oils , and may be
ned approximately pure by agitating Olive or almond oil with aconcentrated aqueous solution of sodium hydroxide
,which
,i t i s
saponifies the palmitin and leaves the olein mostly unchanged .
24 hours , water i s added and the soap solution separated fromily layer
,which should be washed with dilute alcohol and filtered
Igh animal charcoal . As thus prepared , Olein is a colourless,
less Oil,readily soluble in ether or in absolute alcohol
,sp . gr .
to Pure Olein is obtained when aBy-tribromopropane i sd wi th sodium oleate , it has m . p .
-5° to b . p . 23 5
° to’1 8 mm. (Guth , loc. By treatment with nitrous acid it i s
erted into solid elai din . It solidifies below can be distilled in aum
,and on exposure to air oxidises and becomes acid .

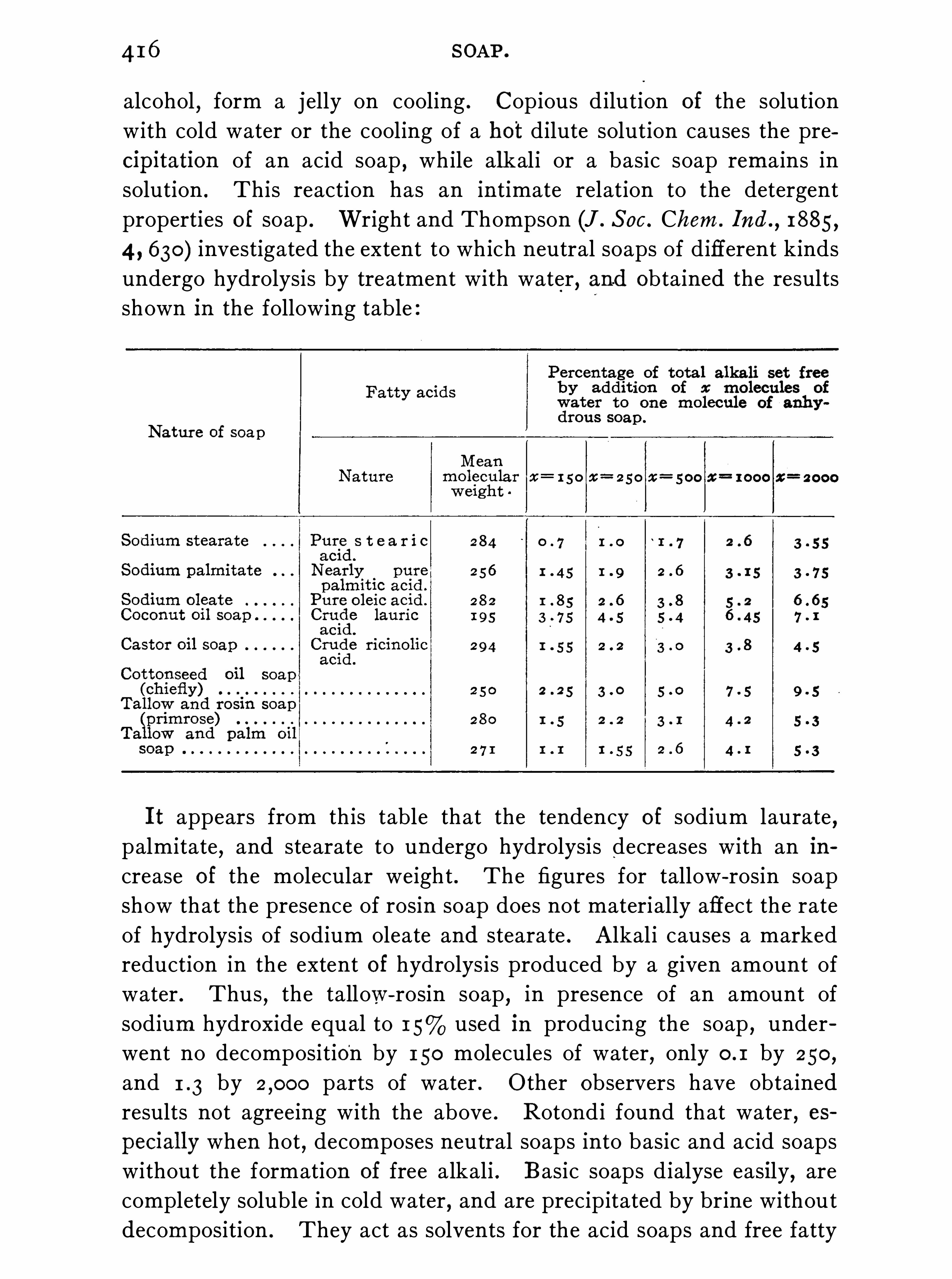
4 1 6 SOAP .
alcohol,form a jelly on cooling . Copious dilution Of the solution
with cold water or the cooling of a hot dilute solution causes the precipitation of an acid soap , while alkali or a basic soap remains insolution . This reaction has an intimate relation to the detergentproperties Of soap . Wright and Thompson (J . S oc. Chem. Ind .
,1 885 ,
4 , 630) investigated the extent to which neutral soaps of diff erent kindsundergo hydrolysis by treatment with water, and Obtained the resultsshown in the following table :
Nature of soap
2 2 5 3 0
0 0 0 0 0 0 0 0 0 0 0
I t appears from this table that the tendency Of sodium laurate ,palmitate
,and stearate to undergo hydrolysis decreases with an in
crease o i the molecular weigh t . The figures for tallow-rosin soapShow that the presence of rosin soap does not materially affect the rateof hydrolysis of sodium oleate and stearate . Alkal i causes a markedreduction in the extent Of hydrolysis produced by a given amount ofwater . Thus , the tallow-rosin soap
,in presence of an amount of
sodium hydroxide equal to 1 5% used in producing the soap , underwent no decomposition by 1 50 molecules of water , only by 250 ,
and by parts of water . O ther Observers have obtainedresults not agreeing with the above . Rotondi found that water , especially when hot , decomposes neutral soaps into basic and acid soapswithout the formation Of free alkali . Basic soaps dialyse easily , arecompletely soluble in cold water
,and are precipitated by brine without
decomposition . They act as solvents for the acid soaps and free fatty

COMMERCIAL S OAP . 4 1 7
acids,and emulsify fats without saponifying them . Carbonic acid
renders basic soaps insoluble without the formation of free alkali ;on warming the liquid re—solution takes place . Acid soaps diffusewith difficulty
,are insoluble in cold water
,and but little soluble in
water,but are soluble in warm solutions of basic soaps . Acid soaps
do not dissolve or emulsify either fatty acids or fats .The experiments of Krapps and S tern (B er.
,1 894 , 1 7 , 1 747) seem to
prove that hydrolysis increases with the molecular weight Of the fattyacids
,but th is conclusion is directly Opposed to that of Wright and
Thompson .
Many resins , especially common rosin (colophony), form soapswith alkalies . These products are no t usually commercial articles bythemselves
,but are found in large amount in cheap soaps .
The soaps of commerce may be divided broadly into 2 classeshard and soft. Hard soaps are made with solid an imal fats
,vegetable
fat Oils,or free Olei c acid and sodium hydroxide ; for soft soaps , fish oils
or vegetable drying oils are used,saponification being effected with
potassium hydroxide . Hard soaps may be thus Obtained if a solid fatis employed
,but a potassium soap is always softer than a sodium soap
produced from the same fat . The hard soaps o f commerce usuallyconsist essenti ally of sodium salts
,the excess of alkali and glycerol
having been separated,bu t with soft soaps no such separation i s at
tempted,the whole being boiled down together . Hence soft soaps are
more caustic than hard soaps and contain impurities . The solidwhite granulations
,termed “
figging , seen in soft soap consist of po tassium stearate
,and to produce them a small quanti ty of tallow is used
in the manufacture . As the figging is commonly but erroneously regarded as a proof of good quality
,i t i s sometimes imitated by an ad
mixture o i starch .
Soaps have been classified by'
W . Lant Carpenter according to theirmethod of production :
1 . Soaps produced by the direct action of fatty acids and alkali orby the decomposition of carbonates by fatty acids .
2 . Soaps produced by acting on a neutral fat by the precise quanti tyof alkali necessary for saponification ,
without the separation of anywaste liquid
,the glycerol produced by the reaction being retained “by
the soap . This Class includes (a) soaps made by the cold process , and(b) soaps made under pressure .3 . Soaps produced by the ordinary method of boiling in Open ves
VOI. I I .
—27

SOAP .
sels,working with indefinite quanti ti es of alkaline lye
,the processes
being controlled by the experience of the Operator . The soaps of thisclass may be subdivided into (a) soft soaps , in wh ich the glycerol isretained , potassium hydroxide being used ; (b) the so-called “ hydratedsoaps
,with sod ium hydroxide in which the glycerol is retained
,and of
which “marine soap ” may be taken as the type ; and (c) hard soaps ,with sodium hydroxide as a base
,in which the glycerol is eliminated
by addition of excess of brine or lye,comprising three kinds—curd
,
mottled,and yellow soaps .
The so-called “ cold process of soap-making cons ists in mixing thefat
,previously melted at as low a temperature as possible
,with just
sufficient sodium hydroxide solution (at about the same temperature)to effect complete saponification . The process has the advantage ofbeing simple
,and is Often employed for the manufacture of the cheaper
kinds of toilet soap , since the low temperature employed preventsdissipation Of the perfumes added ; but the saponification is apt to beincomplete
,the product Often containing both alkali and unsaponified
oil,besides which only the purest materials are available
,as the whole
of the glycerol and extraneous matters are retained in the final product .Transparent toilet soaps made by the cold process are liable to containa considerable proportion of alkali and sugar.
“Marine soap,
”so call ed from its property of forming a lather
with sea-water,i s made by boil ing palmnut or coconut Oil with sodium
hydroxide solution of Sp . gr . The alkali i s added graduallyuntil the presence of a faint excess is ind icated by the taste . I t isoften diffi cult to start saponification ,
but once begun i t proceedswith rapidi ty
,the mixture swell ing up almost instantaneously to
many times its volume . Additions of salt or brine,of sodium
silicate,and of sugar are often made to th is class
.
Of soap,samples
of which may contain 70% Of water .Sodium stearate suff ers no marked change in contact with 1 0 partsof water
,while potassium stearate is converted into a th ick paste or
viscid solution . Sodium and potassium palmitates closely resemblethe corresponding stearates . Sodium oleate is soluble in 1 0 parts ofwater and potassium oleate in 4 parts , forming a j elly with half thisproportion . The consistency or hardness of soap is not dependentsolely on the metal present
,but is greater in proportion to the stearin and
palmitin preexistent in the oil , and less in proportion to the olein in i t .Sodium soaps are soluble in water , but insoluble in brine and other

420 SOAP .
chloride , in quantity suffi cient to react with the free alkali which is soobjectionable an ingredient of toilet soaps . The latter may existeither as alkali or carbonate
,in addition to which there may be sul
phates , chlorides , silicates , traces of calcium ,magnesium
,aluminium
,
and iron compounds existing as impurities in the alkali used,common
salt as a result of the precipitation of the soap with brine,and
,in
transparent toilet soaps,alcohol . The use of alcohol for purifying
toilet soaps has the advantage of separating carbonates and neutralsalts
,but alkali dissolves with the soap . On subsequently evaporating
the alcohol,the soap remains as a more or less translucent mass
,the
transparency of which can be further increased by an addition ofglycerol or cane-sugar
,the latter substance sometimes being present
in large proportion in so-called “ glycerin soaps,
” from most of whichglycerol is absent .B esides the foregoing accidental impurities
,legitimate addi tions are
frequently made'
to soap . Thus , potassium and sodium carbonatesare added to cold—water soap to communicate the power of latheringr'eadily with hard water and to increase the detergent propertiesgenerally ; sodium silicate is often added to soap intended for manufacturing uses and , though obj ectionable in some cases , may be legitimate in others . Sodium aluminate i s sometimes employed ; and borax ,which possesses some detergen t properties is used . Pe troleum naphthato the extent of 1 0% i s sometimes incorporated with soap . It i s saidto increase the detergent action . A soap of th is kind
,now largely
sold,i s prepared by mixing the petroleum product with a rosin soap
mass and adding this to a common soap .
Small proportions of various substances are also added to soap ascolouring and perfuming agents . Mottling is produced by iron salts
,
ochre,ultramarine
,or even more obj ectionable matters
,such as
vermilion and copper arsenite . Such additions remain as a residue ondissolving the soap in water or spirit
,and should never exceed 1%
even in mottled soap,and should be less in other varieties . The
perfuming agents are mostly used in very small quanti ties and areineffective
,and in some of the medicated soaps the substances to which
therapeutic properties of the soap are attributed are presen t in suchsmall proportion that the same remark is applicable .Many forms of medicated soaps are now sold . Among the substancesadded are carbolic and cresylic acids
,thymol
,naphthalene and creosote
oils,petroleum
,vaselin
,camphor
,and gelatin .

AS SAY OF S OAP .
Insoluble and inert organ i c and inorganic substances are added tosoap , either with the alleged object of imparting special characters ,or manifestly to act the part of “ filling ” or adulterants . ’
Amongthese are oatmeal
,bran
,sawdust
,barium sulphate , steatite , china
clay,pipe-clay
,fuller ’s earth
,sand
,pumice-stone
,kieselguhr
,chalk
,
and whiting . Lefimann found 33% of mineral matter in a sample ofred Castile soap . The so-called “ sand soaps ” now largely used forscouring purposes are usually mixtures of common soap
,contain ing
much rosin and some free alkali , with finely pulverised quartz . Theproportion of quartz is often over D iatomaceous earth is alsoused . In a sample of a much advertised soap
,said to contain milk and
sulphur,neither of these bodies was found
,but there was much china
clay and a notable amount of free alkali .Assay and Ana lys is of Soaps .
—In analysing soaps care must betaken to obtain a fairly representative sample . In the case of hardsoaps this is best effected by cutting a transverse slice from the middleof the bar or cake . A cylinder withdrawn from a cake by means of acork-borer or cheese-sampler also affords a fairly good sample . Theouter portion o f a cake of soap may be dried out so as to be of markedlydifferent composition from the bulk of the cake and should be rejected .
In many cases it is necessary to reduce the soap to thin shavings orslices
,which should be thoroughly mixed by shaking
,and preserved in a
well-closed bottle .A comparative assay of different soaps can be effected in a use
ful and simple manner by ascertain ing what measure of a standardsolution of the sample must be added to a 50 c .c . of a very dilutesolution of calcium chloride or sulphate solution in order to obtain apersistent lather on shaking . The soap solution is made by dissolvingI O grm . of the sample in alcohol (sp . gr . filtering
,and diluting
the filtrate with the same solvent to c .c . The test is made exactly as in estimating the hardness of waters , the soap solution beingadded to the standard hard water in small quantities at a time until alather is obtained on shaking
,which remains for at least 5 minutes
when the bottle used for the operation is placed on its side . Thestandard hard water may conveniently be prepared by exactly neutralising 40 c .c . o f N/ I O sulphuric or hydrochloric acid by cautiousaddition of lime-water
,and diluting the solution to c .c . when it
will have a hardness of 1 4 degrees of Clark ’s scale .
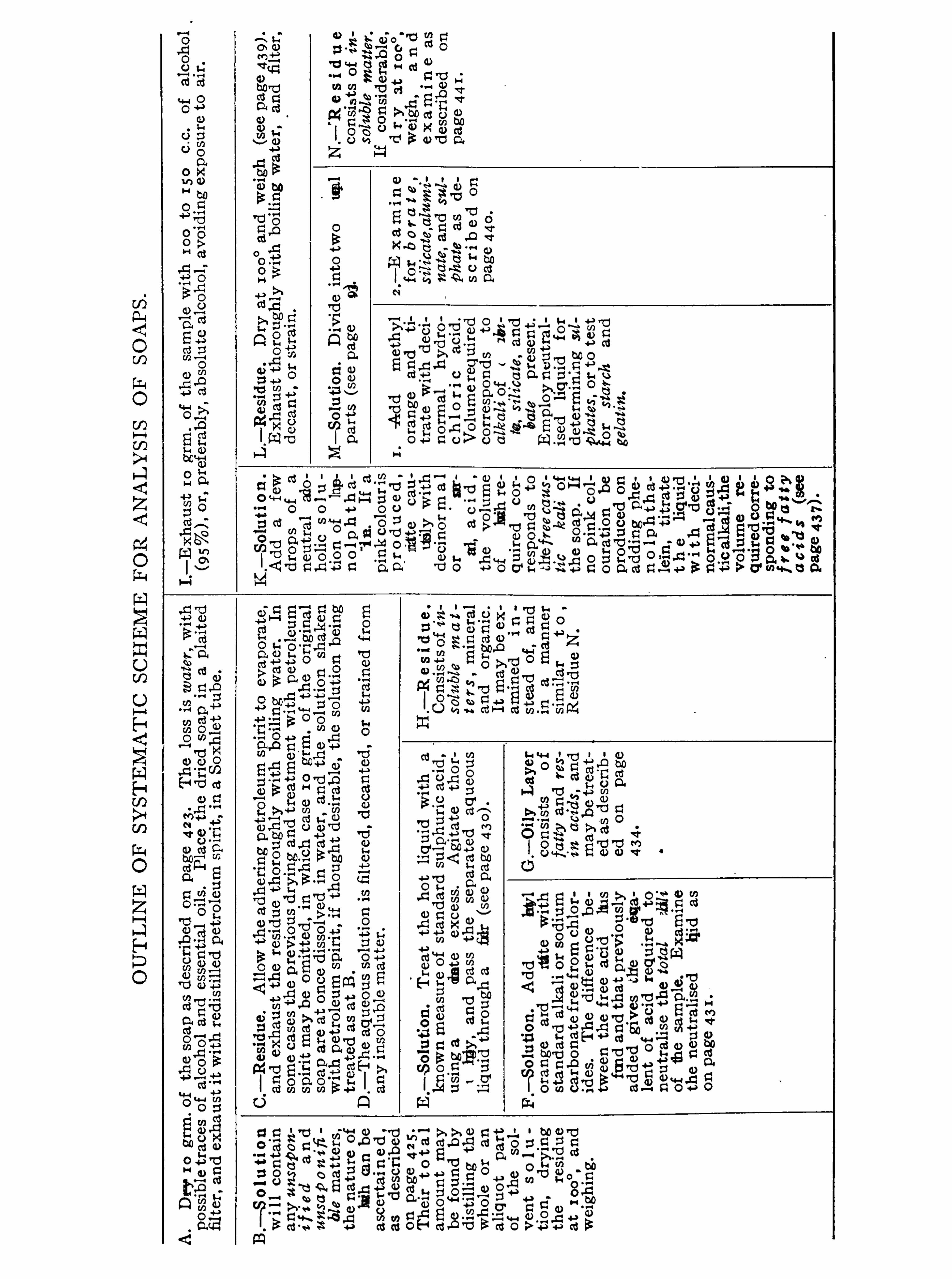
on
comp
an
zo
Aoo
Mam
a
o
n
fl
A
3893
we
.
33
p:
ou
t0
5.
8.
mpcoawou
Loo
vohm
s
d
-ou
£0
239
0
o
EEo
>
0
5
vm
o
a
.
38
3
8588
.
8
:w
E.
SEoo
p
5m
?
325
3
58
3959.
.
vo
o
su
Q
mm
u
so
fi
oo
mam
a
a
m.
52
éaa
a
Q
0
c
.
33
mo
some
s_
ow
o
zo
a
60
?
a
mo
.
wc
fimmo
k
.
o
oon
a
s
o
a
mam
mu
v
da
m“
S
0
w
a
s
?»
4om
08
mo
flag
“oncam
am.
8
o
ne
mam—Emma
hfl
950m
0
9
has“
p
czo
Em
mp
oa
ho
g
.
m«w
omwa
co
po
ntowo
v
mm
vo
cmmfioomm
o
n
58
£0
53
mo
o.
5
ao
ne
.
muo
fid
a
30
0
Q.
e
c
Q
e
a
a
es
k
-xo
cfis
ham
Emp
aco
2
C»
ao
35
0
ml.
w
as
wad
dop
a
combo
fi
nmmwoa
.
Euw
oa
bal
d
ma
250:
vowzmb
fio
a
o
ne
c
EESRm
6
38mm
23
N
o
2
820
N
580
3omma
bzo
a
0p
be
have."
E
om
mo
32
$23
30
8.
33mco
mpo
3
3mm
fiwzo
fiou
a
«map
pad
95
3
cm
25»
Son
ou
t
23
cook
s
amon
-oo.
oo
c
eo
wm
v
2a.
.
mo
wm
$3 .
wau
cm
fiuoao
p
LO
E0
Bo
b
yew
gov:
vowm
”
555m..
82
82
m
Emvcwaw
A
fip
svc
mo
EEm
5m
?
389
3
9
5owc
fio
.
a
aomou
a
3
33
1
538
v
3»
do
wn—o
wl
wad
.
Sou
fimm.
30
$80
26
mo
3
mpcoawo
too
.
A
omv
omma
cowy
“
35a
gan
ese
End
poem
zvou
o
EEOKw
msoo
svm
vop
wg
maom
may
wmwa
paw
$535
.
E
o
m
om
u
ofi
so
Lo
fi
Show
?“
.
mmoo
xo
3880
5
wmcmwn
-evhn
Gage
s
.
E
om
Emvcmpm
mo
a
Bo
cM
Aco
p
fit?
3
m
m
fix?
E
nd:
a
o
a
93
«mo
d.
.
aowaomfl.
-3
van
Ra
te
841.
.
33.
owaa
momv
$
a
13
50
95350
33mm
.
ao
wgo
wlz

S OAP .
method is to dissolve about 2 grm . of the soap in the minimum quantityof hot strong alcohol
,and to pour the liquid on a known weigh t of
clean dry sand , which is then exposed with frequent stirring to atemperature of The traces of alcohol present in transparenttoilet soaps which have been purified by solution in Spirit
,are volatilised
with the water,and if 50 or 1 00 grm . of the sample be mixed with
sand or powdered pum ice,and gradually heated in a retort to
the alcohol may be deduced from the Sp . gr . of the distillate . Thewater in soap may also be estimated rapidly
,and with ample accuracy
for most purposes,in a manner recommended by Watson Sm ith .
From 5 to I O grm . of the finely divided sample should be placed in alarge porcelain crucible
,set in a sand-bath which is heated by a small
Bunsen flame . The soap is continually stirred with a glass rod(weighed with the crucible) having a roughed and j agged end , apeculiarity which greatly facil itates the stirring and breaking up of thelumps of soap formed toward the end of the Operation . The operationis usually complete in 20 to 30 minutes , and is known to be at an endwhen a piece of plate-glass placed over the crucible (the flame beingremoved) no longer collects moisture . Care is required to preventburning of the soap
,but the odour thus developed is so characteristic
that the manipulation is easily controlled . Smith finds the resultstrustworthy to o .2s% .
The proportion of water in soap differs greatly . In the so—calleddry soaps
,
”and in some of the best kinds of curd soap
,i t does not
exceed 1 6 to 2o% ,while in inferior soaps made from coconut oil i t
sometimes reaches 70 to 8o% .
Petro leum Sp i r it So lut ion .—Under ordinary Ci rcumstances , the
material dissolved from dry soap on treatment with petroleum Spiritconsists merely of unsapom
'
fi edfats or offreefatty aci ds . Insignificantproport ions of unsaponifiable matter natural to fixed oils may also bepresent
,and nitrobenzene and essential oils used for scenting the soap
will also be dissolved . If Yorkshire grease has been used in manufacturing the soap , the residue may contain cholesterol . Cetyl alcohol
from Spermaceti and myri cyl alcohol from beeswax and .carnaiiba
wax will also be present if these waxes have been employed . If
added to the made soap,of course the unsaponified waxes will be
dissolved out,instead of simply the solid alcohols resulting from their
saponification . If the presence of waxes is suspected beforehand ,or from the amount or appearance of the residue obtained on evapo

AS SAY or SOAP . 425
rating a portion of the solution,the residual soap Should be further
exhausted with boiling toluene,which dissolves the wax—alcohols
better than petroleum Spirit .The residue from medicated soaps may also contain metallic oleatesand free carboli c and cresyli c acids , thymol, and hydrocarbons , such asvasel in and other neutral petroleum and tar products .When the nature or amount of the residue Obtained on evaporatinga small aliquot part of the petroleum spirit solution indicates thedesirability of further examining it
,the unevaporated portion should
be treated in the manner directed in the following table
SYSTEMAT IC SEPARAT ION OF UNSAPONIFIED MATTERSFROM SOAP .
Agitat e th e solut ion in pe t ro leum spirit with d ilu t e h y drochloric ac id , and separate .
b. Pe t ro leum S o lut ion . Wash fl ee from m inera l ac id by repeat edlyagita t ing with sma ll quan t it ies of wa t er . Add some a lcoh o l and
0 . Ac id S olu t ion . Exam ine for heavymeta ls Pb ,
Hg ,
Cu . Zn) and alumi num , wh ich , iffound , mu s t haveexist ed in th e soapa s olea tes . Po tassium and so diumo lea t e s may a lsohave been d issolvedif th e soap con
ta inedmuch h y dro
carbon . If m e ta lsare found at th isstage , th e amount
o f fa t t y acid s di sso lved by pe t roleum spiri t must beco rrect ed t o asoe r
ta in th e fat ty ac i d s
exist ing in th e soapin a free stat e .
Pe tro leum S olu tion . Evp a
t it rate 11 u id with s tandard a lka li and pheno lph tha le in fo r est ima t ion 0 fa tty acids (page S epara te and agita t e pe tro leumspirit severa l t ime s W i th sma ll quant it ies of sod ium h y droxideso lu t ion , separa t ing as be fore .
d . Alka line S olu t ion . Evapora t e to
sma ll bu lk , d ilu t e with th ree mea sorat e at a 1OW t emperu re s Of st rong br i ne , and fi lt er .a t ure and Observe Odou r ,
e specia lly toward th e end .We igh resi due and th en
es t ima t e unsaponified fa tb y KOtt storfer
’
s proce ss(page In absence o fwaxes , th e po tas sium h ydrox ide requ ired d ivi de db give s th e we igh totyt rue fa t s , wh ich de
ducted from wh o le res idu e ,gives that o f th e hydrocarbons , wax~ alc0hols . Ifde sired , t h e s e m a y b eiso la t ed as on page 7 8 ,
and furth er exam ined .
f. S olu t ion . Ac idu la t e with d ilu t esul ph uri c a c i d ,
a n d s e p a r a t e
lay er Of phenols .
or t it rat e port iono f d ilut ed S o
lu t ion with b rom ine , e t c . (S eep a g e 4 2 6 a n dCreo so t e Oils ,
Vo l . I I I .)
e . P r e c i p i t a t ec O n S i 5 t s O fso d ium sa lts o f
fa tty acids ex
i st ing i n t h e
soap e ith er in
th e free sta te ora s a lum in iumo r o th er m e
tallic oleat es .
Hydrocarbons , such as petroleum,vaselin
,and coal-tar Oils , are
sometimes , to a considerable extent , introduced into soap . Thoughincapable of saponification ,
they may exist in notable proportion withoutbeing suspected ; for if not used in excessive amount , and especiallyif carnauba wax be also added
,they remain in apparent solution when
the soap is dissolved in water or alcohol,and
,on decomposing the
solution with an acid , they pass wholly into the 0i layer of fatty andresin acids .Hydrocarbons may sometimes be detected by the fluorescence ex

426 S OAP .
hibited by the ethereal solution of the fatty acids . If in considerablequantity , they may be partially separated by subjecting the dry soapto a gradually increasing heat
,when the hydrocarbons will distil
,to
gether with any other volatile matter which may be present .The most satisfactory means of detecting and estimating hydrocarbons in soap is to extract them by agitating the aqueous solution ofthe sample with ether and alkali as described below . Any unsaponi
fled fat will , however , be simultaneously dissolved by the ether,and must be separated by saponifying the ether-residue with alcoholicpotash
,and again agitating the solution of the resultant soap with
ether, or the original soap may be evaporated with alcoholic potash ,and the residue d issolved in water and treated with ether .The directions given in the foregoing table do not require furthercomment , except in the case of the method indicated for the determination of phenols . Phenol and cresylic acid , and some other substances
,are dissolved on treating the soap with petroleum spiri t ,
and can be separated from the admixed fatty acids by precipitatingthe alkal ine solution with brine
,but the method is faulty for the
following reason : soaps , and especially common household and softsoaps
,are liable to contain free alkali which will react with the coal-tar
acids added to form bod ies not dissolved by petroleum spiri t , andhence the phenols obtained are only that portion not taken up by thealkal i present in the soap .
The assay of soap for the percentage of phenols and other coal-tarproducts is most conveniently and accurately effected by the followingprocess
,which was extensively used by Allen : 5 grm . weight of the
sample is dissolved in warm water with addition of from 20 to 30 c .c .of a 1 0% solution of sod ium hydroxide , according to the proportion ofphenols believed to be present . The cooled solution is then agitatedwith ether
,and the ethereal layer separated and evaporated at a low
temperature and weighed . The odour towardthe end of the evaporationand that observed on heating the residue will give considerable information as to th e nature of the admixture . Odours suggestive of gas-tarand burning gutta-percha are ' very common . The alkaline liquidseparated from the ether is then treated in a capacious separator withexcess of strong brine , which completely removes the fatty acids assodium salts
,while the phenols remain in solution . The liquid is well
agitated to cause the soap to fil ter and is then passed through a fil ter.If the soap does not coagulate , an addition of a small quantity of tallow

428 S OAP .
solution used i s that required by grm . of phenol of approximatelythe same quality as that contained in the soap .
The remaining portion of the liquid filtered from the precip i tate ofsoap may be evaporated to a small bulk
,acidified with dilute sulphuric
acid , and the separated phenols measured , but the quantity is notsufli cient to make the method satisfactory . I t i s generally betterto employ the solution for the isolation of the bromo—derivatives . For
this purpose i t is acidified with dilute sulphuric acid (without previous concentration) , and bromine-water added in slight excess . From5 to 1 0 c .c . of carbon disulphide are then added , the liquid well agitated ,and the carbon disulphide tapped off into a small beaker . Theaqueous liquid i s agitated with fresh quantities of carbon disulphide(5 of 5 c .c . each) till i t no longer acquires a red or yellow colour . Thecarbon disulphide is then allowed to evaporate spontaneously
,when a
residue is obtained consisting of the brominated derivatives of thephenols present in the soap . If crystalli sed phenol of fairly goodquality had been introduced into the soap
,the bromo-derivative is
obtained in fine long needles having very little colour,and
,if all heating
was avoided during the evaporation of the carbon disulphide,the
weight of the residue multiplied by gives a fair approximationto the amount of phenol ; but if a crude liquid article has been employed ,consisting mainly of cresyli c aci d the bromo-derivative will be deepyellow
,orange
,or red , with little or no tendency to crystallise , and the
weight will not aff ord even a rough indication of the amoun t of coaltar product present .Lewk owitsch considers the following rapid process sufli ciently ac
curate for practical purposes : A somewhat large amount of thesample
,say 1 00 grm .
,i s weighed Ofl
,dissolved in hot water
,the solution
rendered strongly alkaline with sodium hydroxide,the soap precipi
tated with sodium chloride , the curd separated and washed withstrong sodium chloride solution
,the solution boiled down of the phen
Olate to a small bulk,transferred to a stoppered measuring cylinder
of 50 or 1 00 c .c . capacity , sufficient salt added so that some remainsundissolved
,and the liquid acidified with sulphuric acid . The volume
of the separated phenols is then read Off,and the number of cubic centi
meters taken as so many grm .
The following table shows some of the results obtained in Allen ’slaboratory by the assay of representative samples of commercial carbolie soap . The descriptions given by the manufacturers a‘re strictly
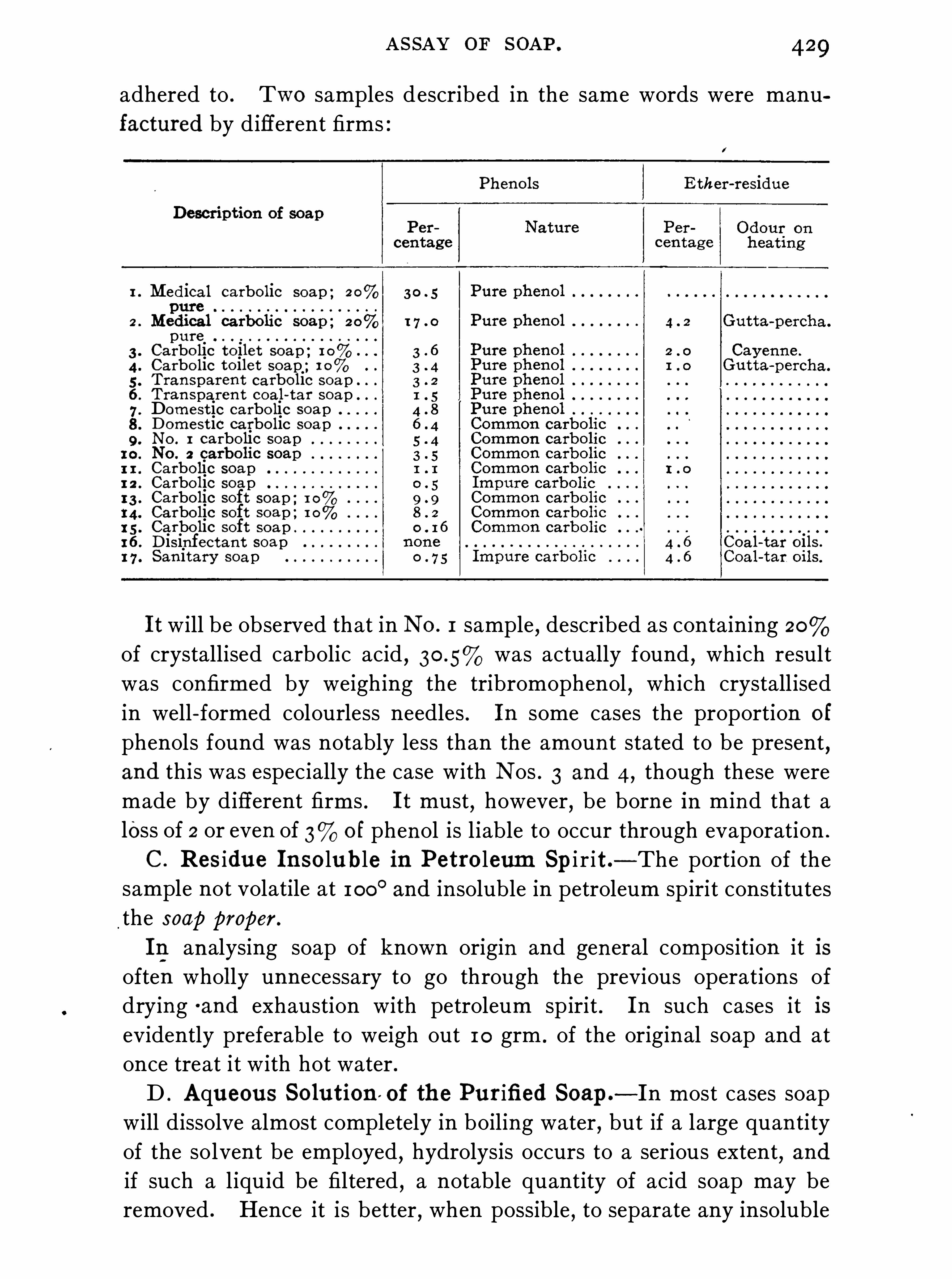
AS SAY OF SOAP .
adhered to . Two samples described in the same words were manufactured by diff erent firms
Ph eno ls E ther-residue
1 . Med ica l carbo lic soap ; 20%
2 . pu reCarbo lic t o ile t so ap ; 1 0%Carbo lic to ile t soap ; 1 0%Transparent carbo lic soa pTran sparent coal-tar soapDome st ic carbo lic soapDomest ic carbo lic soapNo . 1 carbo lic soap
0 0 0 0 0 0 0 0
Carbo lic soapCarbo lic soapCarbo lic soft soap ; 1 0%Carbo lic soft soa p ; 1 0%Carbo lic soft soapDisinfectant soap Coa l-tar o ils .
San itary soap
It will be observed that in No . 1 sample , described as containing 20%of crystallised carbolic acid
,was actually found
,which result
was confirmed by weighing the tribromophenol,which crystallised
in well-formed colourless needles . In some cases the proportion of
phenols found was notably less than the amount stated to be present,
and this was especially the case with Nos . 3 and 4 , though these weremade by diff erent firms . I t must
,however
,be borne in mind that a
loss of 2 or even of 3% o f phenol is l iable to occur through evaporation .
C . Res idue Inso luble in Petro leum Sp i r i t .—The portion of thesample not volatile at 1 00° and insoluble in petroleum spiri t constitutesthe soap proper .
In analysing soap of known origin and general composition it isoften wholly unnecessary to go through the previous operations ofdrying -and exhaustion with petroleum spirit . In such cases it isevidently preferable to weigh out 1 0 grm . of the original soap and atonce treat i t with hot water .D . Aqueous Solu t ion of th e Purifi ed Soap .
-In most cases soapwill dissolve almost completely in boiling water
,but if a large quanti ty
of the solvent be employed,hydrolysis occurs to a serious extent
,and
if such a liquid be filtered,a notable quantity of acid soap may be
removed . Hence it is better,when possible
,to separate any insoluble

430 S OAP .
matter by decantation . When the proportion of insoluble matter isinconsiderable
,there is no occasion to separate i t
,as with proper
management i t will not interfere with the subsequent Operations . An
exception occurs in the case of calcium carbonate,which
,if not
removed will neutralise acid and render the figure for the total alkal itoo high .
In many cases the aqueous solution of the soap may be advantageously agitated with ether at th is stage . Such treatment obviatesthe necessity of previously extracting the dried soap with petroleumspirit
,while i t removes hydrocarbons , unsaponified oi l
,and free fatty
aci ds in a very satisfactory manner . The ethereal layer having beenseparated (see page the aqueous liquid is again shaken with ether
,
which is separated as before . The ethereal solution may then betreated in exactly the same manner as is d irected for the petroleumspirit solution on page 425 , while the aqueous l iquid can be at oncetitrated with standard acid
,though for convenience of subsequent
manipulation of the fatty acids i t is desirable first to remove the dissolved ether by boiling the solution in a capacious flask .
E . Separat ion of Fatty Acids .—For decomposing the aqueous
solution of the soap, N 1 sulphuric acid possesses some advantages ,
and should be used in moderation,an excess of 5 c .c . beyond that
necessary to combine with alkali present being sufli cient . Wrightand Thompson prefer to substitute standard nitric acid , as it enablesthe sulphates to be estimated by barium chloride in one portion ofthe filtrate
,and the chlorides by Silver nitrate in another .
The method of manipulation for the separation of the oily layer offatty acids from the aqueous l iquid depends on circumstances .When the soap is chiefly a stearate or palmitate
,as that made from
tallow or palm oil,the liberated fatty acids are solid when cold , and
in such cases there is no better plan than to effect their precipitationin a beaker or vessel of such shape that the cake can be directly removed
,wiped with blotting-paper
,and weighed . Precipitation in a
conical flask,i s advantageous in some cases .
If the fatty acids are liquid at the ordinary temperature or form acake deficient in consistence
,a known weight of dry
,bleached bees
wax or stearic acid may be added to the hot l iquid . The fatty acidsbecome amalgamated with the melted wax
,and
,on cooling, a firm
coherent cake is formed,which may be at once wiped and weighed .
The weight of wax added (which should be about the same as that of

432
chloride, soluble fatty aci ds, glycerol, sugar, dextrin, starch , gelatin.
For the detection and estimation of these i t is necessary to operate onseparate aliquot portions of the solution .
If nitric acid has been used instead of sulphuric acid at the previousstage of the process
,the sulphates may be estimated by precipitating
an aliquot part of the solution with barium chloride .a . S odium chloride may be estimated by titration with decinormalsilver nitrate or deduced from the weight of the silver chlorideprecipitate .b. S oluble fatty aci ds rarely require estimation in soap . If theprecautions on page 430 are adopted in separating the fatty acids fromcoconut and palm nut oil soaps
,only insign ificant quantities of soluble
fatty acids will remain in the aqueous liquid . If desired,these may
be determined by distill ing the acidified solution,as described on
page 1 9 , but their amount may also be ascertained in the followingsimple manner : Titrate a certain volume of the solution with standard alkali
,using pheno lphthalei n as an indicator . Titrate another
portion of equal measure with the same alkali,using methyl-orange
to indicate the point of neutrality . The alkali consumed in the secondcase corresponds to the free mineral acid onl y
,while the difference
between this and the first estimation gives the volume of alkali required to neutral ise the soluble acids present . I c .c . of N/ 1 alkalicorresponds to grm . of capryli c acid.
All en suggested the following as a method for estimatingthe total fatty acids in coconut and palm nut oil soaps as follows :Separate the fatty acids in the ordinary manner , but in as concentrateda solution as possible . Agitate the aqueous liquid with a little ether
,
separate,and extract any d issolved fatty acids from the ether by agi
tating with dilute sodium solution . Employ the alkaline solution ohtained to neutralise the main quantity of fatty acids , and add a fewdrops of phenolphthalei n
,and then more alkali
,drop by dr op
,until
the pink colour just remains permanent . Then precipitate the hotliquid with a slight excess of magnesium sulphate
,filter
,wash with hot
water,dry the precipitate at 1 00° and weigh . Ignite the precipi tate
and weigh the residual oxide . The difference is the weight of fattyanhydrides form ing insoluble salts with magnesium . Evaporatethe filtrate
,dry the residue at and weigh . Ignite and weigh again .
The difference is the weight of fatty anhydrides forming soluble sal tswith magnesium .

AS SAY or SOAP . 433
J . A . Wilson employs the following process in the presence ofsoluble fatty acids :
I . The alkali in all forms is estimated by ti tration with standardacid in the usual manner .
2 . Another weighed quantity of the soap is decomposed in anErlenmeyer flask with a slight excess of dilute sulphuric acid
,and the
flask kept on the water-bath until the fatty acids separate quite clear .The flask is placed in ice-water to cool and then fi ltered . The fattyacids are washed 3 times successively with 250 c .c . of boi l ing water,allowed to cool each time
,and filtered . The united filtrates are
diluted to c .c .,and 500 c .c . placed in a beaker and tinted with
methyl-orange ; N/ I O alkali is then run in until the liquid acquiresthe usual colour
,after which a little phenolphthalei n is added and the
addition of standard alkali continued until a permanent pink is established . The amount used in the latter ti tration is due to soluble acidsand is calculated to caprylic acid . The fatty acids in the flask and thaton the filter are dried and weighed
,and then dissolved in alcohol and
ti trated with N 2 alkali . T he amount so used,together with that re
quired for neutralisation o f the soluble acids,deducted from the total
alkali,gives the alkali existing in forms other than as soap .
If desired , the soap may be decomposed with standard sulphuricacid , methyl-orange added , and the alkali required for neutralisationnoted ; this , deducted from the total acid used, would give the acidequivalent to the alkali existing in all forms . In th is manner areascertained
Total alkali .Combined alkaliInso luble fatty acidsSo luble fatty acid
c. Glycerol may exist in soap . In the absence of sugar , it may beestimated with considerable accuracy by the permanganate process .When glycerol is present in considerable amount in soap
,Lewkowitsch
makes the estimation by dissolving it in water,separating the fatty
matter with acid, and filtering off. The filtrate is then neutralised
with barium carbonate and boiled down to the consistency of syrup .
The residue is then extracted with a m ixture of 3 parts of 95%alcohol and I part ether , the alcoholic solution filtered and evapo
rated on the water-bath to small bulk,and finally dried under a desic
cator . The glycerol in the residue may be estimated by the acetinVo l. I I .
—28

434 S OAP .
method . A more convenient method is that of Hebner with potassiumdichromate (see under The presence of sugar rendersthe above methods wholly useless
,and one of the plans described
below must be adopted .
d . S ugar i s rarely present except in transparent toile t soaps , but inthese i t sometimes exists to the extent of 20 to 30% of the entireweight
,or in a proportion approach ing that of the anhydrous soap
present . Such soap is sometimes sold as“ glycerin soap
,
” thoughwholly destitute of glycerol .According to Donath and Mayrhofer (Zei t. anal. Chem.
,1 88 1 ,
the estimation o f sugar and glycerol may be made by addingto the solution slaked lime suffi cient to combine with the sugar and anequal quantity of washed and ignited sand
,boiling down to the con
sistency of syrup,pulverising the cooled residue and exhausting it in a
closed vessel with 80 to 1 00 c . c . of a mixture of equal parts of ether andalcohol . The glycerol will pass into solution
,and
,after cautious
evaporation of the solvent,may be estimated by methods given under
Glycerol .”Sugar may be estimated by Fehling ’s solution , after inversion,without previously separating the glycerol
,but the solution should be
dilute and the boiling very limited in duration,or the glycerol may
cause some reduction .
In an aqueous liquid containing no other bodies than sugar andglycerol
,the amount of glycerol may be deduced from the sp . gr .
,
ofthe liquid . The sugar having been previously estimated by Fehling ’ssolution or other means
,its efl ect on the sp . gr . can be readily cal
culated ; and th is being deducted from the observed sp . gr .,gives
that due to the glycerol present in the liquid . See section on Glycerol .”Organi c matters , such as starch , dextrin , gelatin , may be de
tected by special tests ; but their recogn ition i s more easy and certainin residue L
,left on treating the purified soap with alcohol .
G . Exam inat ion of th e Oi ly Layer of Fat ty Ac ids .-The
separation of the liberated fatty acids from the acidified aqueoussolution has already been described . If wax or stearic acid has beenemployed for the purpose o f obtaining a solid cake
,the further treat
ment of the fatty acids is practically lim ited to drying them anddetermining their weight . In many cases
,however
,i t is of interes t
or importance to make a further examination of the oily layer , whichin that case should be treated as described on page 2 2 .

436 S OAP .
same limitations as are stated above to apply to the saponification
equivalents .In cases in which the acids are practically insoluble in water
,a
titration in alcoholic solution with standard alkali and phenolphthalei naffords a simple and accurate means of ascertaining the proportion ofalkali exi sting in combinati onwi th thefatty and resin acids , as i t is evidentthat the amount of alkali required for neutralisation of the separatedacids must be the same as that with which they had been previously incombination .
The fact that the soaps produced by the saponification of coconutand palm nut oi ls are not readily precipitated by solution of commonsalt , may , according to W . Lant Carpenter
,be employed for detecting
the presence of these oils in soap . A sufli cient quanti ty of the soapshould be dissolved in hot water
,and the fatty acids l iberated by
acidifying the solution,and separated without Special washing or use
of ether . 1 0 grm . O f the fatty acids are treated with 39 to 40 c .c . ofN/ r sodium hydroxide or a volume just sufli cient to dissolve themcompletely . The whole is then boiled and the weight of the liquidbrought to 50 grm . by evaporation or cautious addition Of water . A
saturated solution of common salt (previously boiled with a few dropsof sodium carbonate and fil tered from any precipitate) is then run
in gradually from a burette,the liquid being constantly stirred and
kept gently boil ing . The addition is continued until the soap sud
denly precipitates , a point which is usually sharply marked . Thesoap from ordinary oils is precipitated when from 8 to 1 0 c .c . of thesalt solution has been added
,but that from coconut oil requires an
addition of more than 50 c .c . M ixtures of the fatty acids from coconut or palm nut Oil with those from other oils will of course require avolume of brine intermediate between these two limits .I . Exhaust ion of th e Soap W ith A lcoh o l . -If the original soap istolerably dry
,ordinary rectified Spiri t is usually sufli ciently strong for
the treatment at this stage ; but if the sample contain much water,absolute or nearly absolute alcohol should be used
,or the solution will
have an Objectionable tendency to gelatin ise during filtration and otherinconveniences will arise . I t i s recommended by both Leeds andWright that the portion of the soap to be treated with alcohol should bea part of that previously exhausted with petroleum spirit , but , aspointed out by C . Hope , i t i s not possible to dry soap effectually without a notable conversion of the alkali into carbonate . The treat

AS SAY OF SOAP . 43 7
ment with alcohol can be eff ected either in the Szombathy- tube , or byboiling the soap with the solvent , and filtering and washing in theusual way .
K . Exam ination of th e A lcoh o l ic So lution .—a . The estimation
of the free alkali existing in soap can be eff ected very Simply andaccurately by the method of C . Hope
,described in the table
,the error
rarely exceedi ng of the total free alkali present . The test may
be applied qualitatively by dropping an alcoholic solution of phenolphthalein onto a freshly cut surface of the soap , when a red colouration will be produced
,the intensity of which increases with the pro
portion of the alkali present . Caustic or carbonated alkali will alsobe indicated by the black or grey colouration produced by droppingmercurous nitrate on the freshly—cut surface . Each 1 c .c . of N 1
acid neutralised represents grm . of potassium oxide,
ofpotassium hydroxide
,of sodium oxide
,or o f sodium hy
droxide . Should it be desired to ascertain which is present,the method
described on page 439 must be employed .
I t is possible to have a negative alkalin ity shown at this stage . Thisresult is due to the presence of fatty acid or a diacid salt
,but acidity of
the alcohol may produce the same effect . The volume of standardalkali required to be added before a pink colour appears Should becalculated to its equivalent of olei c acid , which is stated in the analysisas existing in the free state . Any diff erence between this amount andthat found in the petroleum Spiri t solution is due to a part ial neutralisation of the free acid coexisting in the imperfectly mixed soap . Thefollowing method of treating the alcoholi c solution of a soap in such amanner as to allow of the estimation of the leading constituents ina very rapid manner has been commun icated to the author by C . Hope :2 grm . Of the soap are dissolved in hot absolute alcohol , a drop ofphenolphthalei n solution added
,and carbon dioxide passed till any
pink colouration is destroyed . The liquid is then filtered , the residue ,consisting Of total impuri ti es , washed with hot alcohol , weighed , andthen titrated with N/ 1 0 acid and methyl—orange to find the alkalinot exi sting as soap . The alcoholic soluti on is evaporated to dryness at and the residue of dry soap Weighed when constant .I t i s then ignited gently , treated with water , and the solution ti tratedwith decinormal acid and methyl-orange to find the alkali exi sting as
soap . The difference between this and the total residue before ignitiongives the fatty anhydrides , which , multiplied by gives the fatty

S OAP .
acids . The water is found with sufli cient accuracy by subtracting thesum of the weights Of the impurities and dry soap fromI t i s necessary to avoid confusion between the real alkali
,i . e .
,exist
ing in a soap in the form of potassium or sodium hydroxide,the appar
ent alkali,which corresponds to the soap
,and that corresponding to
carbonate,Sil icate
,or borate . If the estimation is made in the alco
holic solution,as recommended
,the actual hydroxide will alone be
present,the other compounds capable of neutralising acid being in
soluble in spiri t . On the other hand , the stand ard acid required toneutralise the aqueous solution of the soap (page 43 1 ) includes thatcorresponding to any soluble carbonate
,si li cate
,and borate or aluminate
in the sample .The alcoholic solution of the soap rendered neutral to phenolphthalein may be conven iently employed to estimate the alkali exi sting in combinati on wi th the fatty and resin acids of the sample . Toeffect th is
,i t i s merely necessary to add a °few drops of methyl-orange
solution to the neutralised liquid,and then at once titrate with standard
sulphuric or hydrochloric acid . The point of neutrality is sharplymarked by the production of a pink colour
,and the accuracy of the
results are all that could be desired .
In order to prevent m isunderstanding,the volumetric method Of
ascertaining the proportions of alkali existing in soap in various conditions may be recapitulated as follows :In alcoholi c soluti on of soap
— 1 . Acid required to establish neutrali ty to phenolphthalei n corresponds to free alkali , and is calculatedto oxide or hydroxide
,according to circumstances . 2 . Acid subse
quently required by same solution to produce neutrality to methylorange represents the alkali converted into soaps of fatty and resin acids .In residue insoluble in alcohol . —3 . Acid required to produce neu
trality to methyl-orange corresponds to alkali corresponding to carbonate ,si li cate
,and borate .
In aqueous soluti on of soap .
—4 . Acid required to produce neutrali ty
to methyl-orange corresponds to total alkali,whether existing as such or
converted into true soap,resin soap
,carbonate
,silicate
,borate
,alum
inate,and soluble l ime . This estimation should therefore agree
with the sum of 1 , 2,and 3 , or if any 2 of these have been deter
mined the third will be the diff erence between their sum and the totalalkaliThe volumetric estimation of the alkali in soap gives no inf orma

440 S OAP .
ment with acid,so as to Obtain a means of calculating the amount
of soluble carbonate present . This is necessary when the soap containsborate or S ilicate in addition
,but otherwise the carbonate can be de
duced with accuracy from the titration of the solution with standardacid . To ascertain the carbonate directly
,the concentrated solution
should be treated with a moderate excess of standard acid in a carbondioxide apparatus
,and the evolved carbon dioxide ascertained by the
loss of weight,precipitation as barium carbonate
,or measurement
in a nitrometer . 44 parts of carbon dioxide correspond toof potassium carbonate or 1 06 o f sodium carbonate .
1 . After expelling the last of the carbon dioxide by warming theacidified liquid
,the solution Should be divided into 2 or more
equal parts,in 1 of which the excess of acid is estimated by titrating
back with standard sodium carbonate and methyl-orange,and hence
the sum of the alkali existing in the 4 forms of carbonate, si li cate,
borate , and aluminate ascertained , while the other portion 18 examinedfor borate
,s ilicate
,and aluminate as in 2 .
The solution which has been employed for the estimation of thetotal alkali of the res idue may then be divided into 2 or moreequal parts
,which may be employed for estimating sulphates by
precipitation with barium chloride,starch by the methods described
in Volume I , and to test for gelatin by means of tannin . I f gelatinbe found
,i t is best estimated by treating another quantity of the soap
with strong alcohol and applying the Kjeldahl method to the residue .Gelatin contains about nitrogen .
2 . The other half of the aqueous solution of the residue insoluble inalcohol Should be rendered distinctly acid with hydrochloric acid , andevaporated at 1 000 in porcelain . A Slip of turmeric paper Should beimmersed in the liquid toward the end of the operation
,and allowed
to remain until the evaporation is complete . If a borate be present ,the paper will become brownish-red in colour
,and will be changed to
green,blue
,violet
,or black on addition of sodium hydroxide solution .
The residue is treated with hydrochloric acid,water added , and the
solution filtered . The residue of si li ca i s washed , dried , ignited , andweighed . As the sodium si li cate present in soap is not of constantcomposition
,though usually approximately corresponding to the
formula NazS i QO S ,i t i s not possible to deduce the amount of alkali
existing as sil icate from the weight of the Silica found ; but, in the ahsence of borates
,i t may be ascertained by estimating the carbon

AS SAY OF S OAP . 44 1
dioxide evolved on treating the aqueous solution of the residue insolublein alcohol with dilute acid . This estimation will give the means ofcalculating the alkali existing as carbonate, and the remainder of thealkali o f the residue must exist as si li cate (or aluminate).The filtrate from the Silica may be conveniently employed for es
timating sulphates by precipitation with barium chloride , or ofaluminium by precipitation with ammon ium hydroxide and of calciumin the filtrate by precipitation with ammonium oxalate . C . Hopestates that free lime is not unfrequently present in soap
,and may be
detected and estimated at this stage . I ts presence would tend toincrease the “ al kal i ” of the residue insoluble in alcohol .N.Res idue Inso luble inPetro leum Sp ir it, A lcoh o l , andWater.
After drying the residue at 1 000 and noting its weight,i t i s desirable
to exam ine i t under a low micrOSCOpic power, with a view of recognising characteristic organic structures
,which can be seen much more
distinctly after the removal of the soluble matters .Whether any further exam ination of the residue is requisite neces
sarily depends on its amount and nature and the obj ect o f the analysis .Among the various constituents of such a residue the following listcomprises those most likely to be present :
1 . Insoluble Organi c Matters,such as sawdust
,bran
,woody fibre
from oatmeal .2 . M ineral Pigments and Colouring Matters
,as red ochre , burnt
umber , various other ferruginous materials , red lead , vermilion ,Scheele ’s green , chrome green ,
ultramarine .3 . Mineral Matters used as S courers
,such as sand
,powdered quartz
,
pumice,and infusorial earth .
4 . Mineral Matters used as Adulterants or Fi llings , such as chinaclay , steatite , barium sulphate , chalk , and whiting .
The systematic recognition and estimation of these and otherpossible additions belong to inorgan ic analysis . I t is sufli cient here toindicate the following Simple method of classification with a view tofacil i tate further examination .
Organi c matters may be approximately estimated by igniting anal iquot portion o f the residue . The loss will include the volatile constituents of china clay, whiting, red ochre , etc . , as well as any vermil ion which may be present .By treatment with dilute hydrochloric acid , the original or ignitedresidue may be divided into soluble and insoluble consti tuents . The

442 S OAP .
former include whiting,chalk
,ultramarine
, Scheele ’s green , oxide ofiron
,and the greater part of the ferruginous pigments ; while barium
sulphate , steatite , sand , quartz , pumice , kieselguhr , china clay, chromegreen and vermilion are but little acted on .
Interpretat ion of th e Resu lts of Ana lys is of Soaps .- Cal
culating from the equation Of the reaction between sodium stearate ,and any strong acid , it i s found sodium stearate yields ofstearic acid . S imilarly
,the alkali used in forming the soap would b e
SO that the analysis would beS tearic ac id .
Sodium hydroxide
This statement Shows an excess of nearly owing to the hydrolysi swhich takes place . I t is evident that if the basic constituent o f a soapbe stated as anhydrous alkali
,a correction must be made in the actual
weight of fatty acid found to bring it to the corresponding quantityof anhydride . 568 parts of stearic acid correspond to 5 50 of stearicanhydride
, and the proportions of the respective anhydrides corresponding to palm itic and Ole ic acids are not very diff erent from theabove . Hence in soaps made from palm Oil
,olive Oil
,and tallow t he
necessary correction of the observed weight of fatty aci ds to thecorresponding quantity of fatty anhydrides may be made by multiplying by the factor 1 00 parts of stearic acid representing ap
proximately 97 of stearic anhydride . In the case of coconut andcastor-Oil soaps
,and many others made with mixed oils , this factor
is far from accurate,and hence it is in all cases decidedly preferable to
determine the meancombin ing weight of the isolated fatty and resinacids
,as described on page 3 7 7 , and calculate the corresponding
weight of fatty anhydride therefrom . The mean combin ing weigh tof the anhydride is always 9 less than that of the corresponding acid .
The usual figures for the fatty acids isolated from various fatty oi lsare given on page 3 78 .
Gassler Ind .,1 882
,gives the following analyses
of German resin soaps in comparison with S inclair ’s “cold-wat’er soap”
Description Of soap
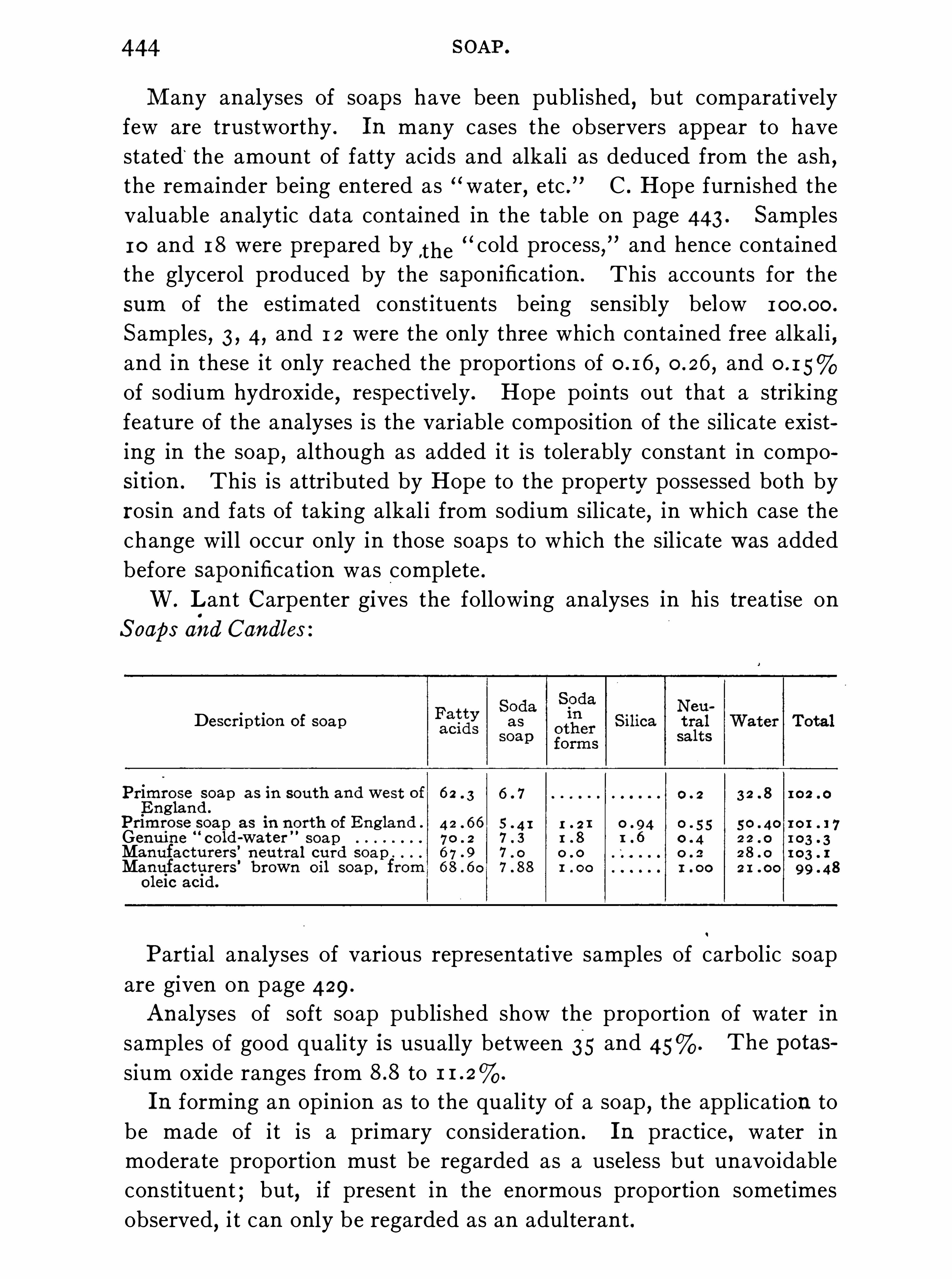
444 SOAP .
Many analyses of soaps have been published,but comparatively
few are trustworthy . In many cases the observers appear to havestated‘ the amount of fatty acids and alkali as deduced from the ash
,
the remainder being entered as “water,etc .” C . Hope furnished the
valuable analytic data contained in the table on page 443 . Samples1 0 and 1 8 were prepared by
,the “ cold process,
” and hence containedthe glycerol produced by the saponification . This accounts for thesum of the estimated const ituents being sensibly belowSamples
, 3 , 4 , and 1 2 were the only three which contained free alkali,
and in these it only reached the proportions of andof sodium hydroxide
,respectively . Hope points out that a striking
feature of the analyses i s the variable composition of the silicate existing in the soap
,although as added i t is tolerably constant in compo
si t ion . This is attributed by Hope to the property possessed both byrosin and fats of taking alkal i from sodium Silicate
,in which case the
change will occur only in those soaps to which the sil icate was addedbefore saponification was complete .W . Lant Carpenter gi ves the following analyses in his treatise on
S oaps and Candles :
Descript ion of soap
Primrose soap as in sou th and we st o f 6 2 3 6 7 0 2
England .
Primro se soap as in north of England . 4 2 6 6 5 4 1 r 2Genui ne “co ld-wat er ”
soap 7 0 2 7 3 1 8Manufacturers ' neu tra l cu rd soap . 6 7 9 7 0 0 0Manufactu rers ' brown Oi l soap , from 6 8 60 7 88 1 0
o leic acid .
Partial analyses of various representative samples of carbolic soapare given on page 429 .
Analyses of soft soap published Show the proportion of water insamples of good quali ty is usually between 35 and 45 The potassium oxide ranges from toIn forming an opinion as to the quali ty of a soap , the application tobe made of i t i s a primary consideration . In practice , water inmoderate proportion must be regarded as a useless but unavoidableconstituent ; but , if present in the enormous proportion sometimesobserved
,i t can only be regarded as an adulterant .

ANALYTIC DATA . 445
In some of the best brands of opaque toilet soap made by specialmethods
,the proporti on of water does not exceed 1 0 or but the
majori ty of the best qualities of soap,known as Marseilles
,curd
,brown
Windsor,honey
,and primrose
,contain from 1 7 to 24% of water . In
some of the transparent toilet soaps,made by solution in alcohol
,the
proportion of water i s very small (9 to but this advantage ismore than counterbalanced by the presence of 20 to 30% of sugar .Transparent soaps made in other ways
,as by the “ cold process
,
”
rarely contain half their weight of actual soap,the remainder con
sisting of water and sugar .Practically
,the proportion of alkali in a soap is the best single test
of its quality, but here again a distinction must be drawn betweenalkali exist ing in combination with fatty and resin acids
,or
,in other
words,as true soap
, and that existing in other conditions , particularlythe caustic state . Wright arranges toilet soaps in three classes
,accord
ing to the proportion the “ free ” or inorganic alkali bears to the alkaliexi sting as soap . Thus
,soaps containing less than 2 .5 parts of free
alkali for 1 00 of alkali as soap are arranged in the first class ; thosecontaining between 2 .5 and in the second
,and those containing
more than in the third class . In judging of the quality of atoilet soap , Wright also takes into account the freedom of the soapfrom adulterants
,
“filling,water
,and “ closing up ” agents
,and
from poisonous colouring matters ; as also the nature and quality of thefatty matters used as basis and their freedom from rancidity .
Although the absence of a notable proportion of “ free ” alkali i simportant in the case of toilet soaps
,owing to its powerful action on
the skin , it does not follow that a similar absence is advantageousunder other conditions . On the contrary
,for scouring and hou sehold
purposes , a limited proportion of alkali i s advantageous , and in thecase of some soaps used by manufacturers the presence of considerableproportion of alkali i s essential to success
,a solution of alkali with
sufl‘i cient soap in it to cause lathering being preferred . A neutral
soap , however pure , will for such uses be regarded as deficient in“ strength , ” and will often cause trouble through the precipitation Of
free fatty acid or acid soap in the fabric with which the soap is used .
The nature and origin of the acids are sometimes of interest injudging of the suitability of a soap for certain purposes . The presenceof rosin acids and of the acids from coconut or palm nut oil can beascertained as noted under G (p . and it is rarely of interest to in

446 SOAP .
quire further,except in the case of soap containing coal—tar bodies ,
which can be examined as described on page 426 .
The permissibility of additions to soap must be judged on themerits of each case
,but
,as a general rule
,the less extraneous matters
present the better . I t i s said that,for some purposes
,as in the treat
ment of wool and silk,a small proportion of starch is an advantage .
In contracting to supply manufacturers of textile fabrics,the soap
maker is frequently obliged to settle definitely the proportions offatty acids
,resin
,alkali
,and potato-starch which shall be present in
the soap . A soap suitable for fulling cloth and for other purposesShould not contain less than 40% of fatty acids nor more than 5%of rosin and 6 of potato-starch .
Dextrin,sugar
,starch
,Irish moss
,and gelatin are in most cases
purely adulterants,as also are kaolin
,barytes
,and other insoluble
earthy matters ; but soluble carbonates , silicates , and borates havemarked detergent properties .In a complete analysis of a soda soap
,the const i tuent s may be
s tated in the following manner :IFatty anhydrides2Soda existing as soapS ilica
2Soda existing as silicate2Sodium carbonate .
2Sod ium hydroxideSodium sulphateSod ium ch lorideCalc ium oxideFe rric oxideWater .
1 Fatty acids2 =Total detergent alkali , as sodium oxide
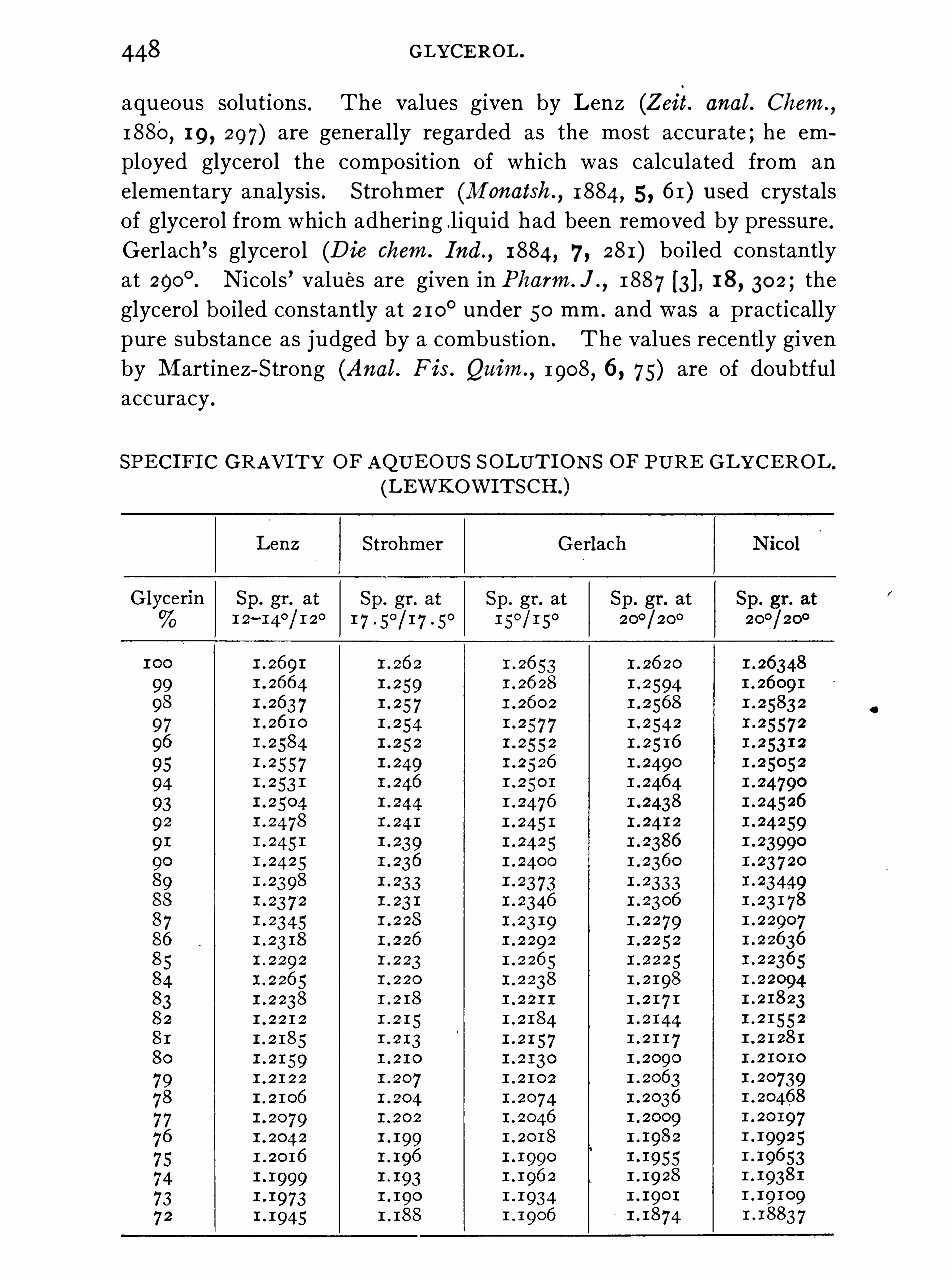
448 GLYCEROL .
aqueous solutions . The values given by Lenz (Zeit. anal. Chem.
,
1 880,1 9 , 29 7) are generally regarded as the most accurate ; he em
ployed glycerol the composition of which was calculated from anelementary analysis . S trohmer (Monatsh .
,1 884 , 5 , 6 1 ) used crystals
of glycerol from which adhering .liquid had been removed by pressure .Gerlach ’s glycerol (D ie chem. Ind.
,1 884 , 7 , 28 1 ) boiled constantly
at Nicols ’ values are given in 1 887 1 8, 302 ; the
glycerol boiled constantly at 2 1 00 under 50 mm . and was a practicallypure substance as judged by a combustion . The values recently givenby Martinez-S trong (Anal. F i s . Quim.
,1 908, 6 , 75) are of doubtful
accuracy .
SPECIFIC GRAVITY OF AQUEOUS S OLUT IONS OF PURE GLYCEROL .
(LEWKOWITSCH.)

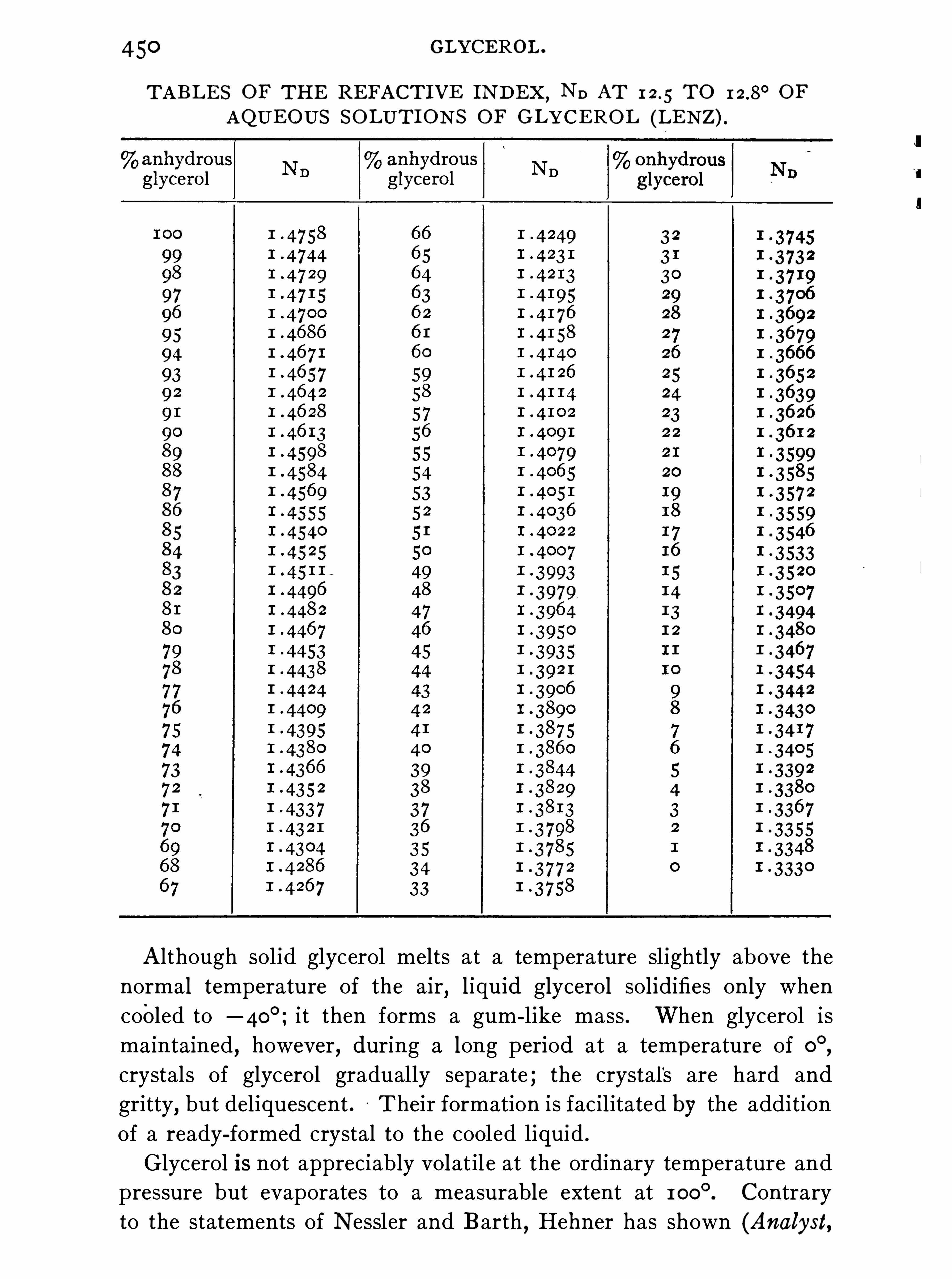
450 GLYCEROL.
TABLES OF THE REFACT IVE INDEX,ND AT TO OF
AQUEOUS SOLUT IONS OF GLYCEROL (LENZ).
Although solid glycerol melts at a temperature slightly above thenormal temperature of the air
,liquid glycerol solidifies only when
cooled to it then forms a gum-like mass . When glycerol i smaintained
,however
,during a long period at a temp erature of
crystals of glycerol gradually separate ; the crystals are hard andgritty
,but deliquescent . Their formation is facilitated by the addition
of a ready-formed crystal to the cooled liquid .
Glycerol is not appreciably volat il e at the ordinary temperature andpressure but evaporates to a measurable extent at Contraryto the statements of Nessler and Barth , Hebner has shown (Analyst,

452 GLYCEROL .
organic acids occur . The substance glycerose , obtained by theregulated oxidation of glycerol by bromine and alkali , i s exclusivelysym.
-dihydroxyacetone, (Wohl and Neuberg ,
B er .,1 900, 3 3 , 3098 and By treatment in dilute aqueous
solution with potassium permanganate , in presence of excess of alkalihydroxide , glycerol i s oxidised in a very defini te manner with formationof oxalic and carbonic acids . By treatment with potassium dichromateand sulphuric acid
,i t i s completely oxidised to carbon dioxide and
water . These changes are utili sed in the estimation of glycerol (seePage 458)When a mixture of glycerol with an aqueous solution of pure mercuric
chloride . (free from reducing substances) i s exposed to direct sunlight ,calomel i s precipitated after an interval of about 2 hours ; the liquidshows an acid indication and gives the test for an aldehyde . I t is
thought that the following action occurs
Ferric chloride behaves similarly . After the action,the transformed
glycerol has the property of dissolving the ferric hydroxide formed onadding an excess of potassium hydroxide : a carbohydrate thus appearsto be formed (Archetti , Chem. Zei t. , 1 902 , 26 ,
Glycero l Esters .—By treatment with a cold mixture of fuming ni tric
and concentrated sulphuric acid,glycerol i s converted into gly ceryl
ni trate or “nitroglycerin,
”
On mixing glycerol with strong sulphuric acid,a compound of the
formula is produced,which has
acid properties and forms soluble but unstable barium,calcium , and
lead salts .The so-called “
glycerylphosphoric acid (glycerophosphoric acid),Obtained by heating glycerol with phosphoric acid (compare Powerand Tutin
,Tmm . ,
1 905 , 87 , appears to be a mixture of a—glycerylphosphoric acid , and fi
glycerylpho sphoric acid , (Tutin and Hann ,
Tmm ., 1 906 , 89 , A somewhat differently constituted mixture
of the same acids i s obtained by the hydrolysi s of lecithin , a complexcompound of these acids with choline and the fatty acids , stearic acidand palmitic acid
,occurring in the y olk of egg and in brain ti ssue .
For the estimation of glycerophosphates , see A . Astruc , J . Pharm.,
1 898 , 7 , 5 ; A . Trillat , i bi d.,1 63 ; Imbert and Pages , i bid.
,
Glycerol dissolves large quantities of arsenious oxide to form a

DETECTI ON OF GLYCEROL .
compound of the formula C 3HSAsO
3, glyceryl arsenite , which has beenemployed by calico-printers for fixing aniline colours . It is an amberyellow
,fatty substance
,melting at 50° to a thick liquid which is
soluble in glycerol and in water,but is decomposed by excess of the
latter liquid .
When 3 parts of glycerol are heated to about 1 60° with 2 of boricacid
,glyceryl borate
, C3H5BO 3 ,
is formed,which\has been patented
as a preservative agent under the name of “ boroglyceride .”By heating glycerol with organic acids , esters are formed , having acomposition dependent on the conditions of their formation . Theseesters are generally called glycerides and are specifically designated bynames ending in in
,the mono di and tri-acetates being called
,re
spectively , monacetin , diacetin , and triacetin . S imilarly , stearic acidgives ri se to stearins
,oleic acid to Oleins
,butyric acid to butyrins
,and
so forth . The stearins,palmitins
,and Oleins have already been
described .
De tect ion of Glycero l .—When in a state of reasonable purity andconcentration
,glycerol may be recognised by its physical properties ,
no other substance likely to be met with exhibiting the combinedcharacters of a dense vi scous liquid of sweet taste and neutral reaction ;miscible with water and alcohol in all proportions ; volatile at a hightemperature ; burning with a blue flame when kindled , and leaving nocarbonaceous residue .The most characteristic property of glycerol is its behaviour whenheated in a concentrated state with potassium hydrogen sulphate ,whereby it i s converted into acrolein
, C3H4O ,with elimination of the
elements of water . The acrolei n is recognisable by its extremelypenetrating Odour , resembling that of burning fat , and its property ofcausing a flow of tears . If the vapours be passed into water
,the warm
solution wi ll be found to have the properties of an aldehyde , e . g .
, ofreducing ammoniacal s ilver nitrate
,with formation of a mirror of
metallic silver .This test is recommended by Grunhut (Zei t. anal . Che m.
,1 899 ,
38 , 3 7) as the best qualitative test for glycerol . The substancesupposed to contain glycerol is mixed with twice its weight of potassiumhydrogen sulphate and strongly heated until i t foams ; the vapours areled into a test-tube cooled with a freezing mixture . The distillatesmells distinctly ‘
of acrole‘in if glycerol is present . To confirm , add afew drops of a mixture of solutions of 3 grm . of silver nitrate in 30

454 GLYCEROL .
grm . of ammonia of 3 sp . gr . and 3 grm . of sodium hydroxide in30 grm . of water . The silver mirror Should form in the cold .
The following tests are less characteristicIf 2 drops of concentrated glycerol are treated in a dry test-tubewith 2 drops of fused phenol and the same quantity Of strong sulphuric acid and the mixture is heated very cautiously over a flameto about a brownish-yellow mass will be produced
,which
,
after cooling,dissolves in water
,to which a few drops of am
monium hydroxide have been added,with a splendid carmine-red
colouration .
According to Reichl,minute quantities of glycerol can also be
detected by boiling the solution to be examined with ' a minute quantityof pyrogallol and a few drops of sulphuric acid diluted with an equalvolume of water
,when a red colour will be produced
,changing to
violet-red on adding stannic chloride . Carbohydrates and variousalcohols give similar results .In common with other polyhydr ic alcohols
,glycerol acts on borax
to form a compound having an acid indication to litmus , whereasthe ori ginal aqueous solution of borax is alkaline . In the case ofglycerol
,glyceryl borate
, C3H5BO
3 ,i s formed
,together with sodium
metaborate NaBO , . The test may be made both in the wet and thedry way . Senier and Lowe (Trans .
,1 878 , 3 3 , 438) recommend
that the solution to be examined should be made faintly alkalineto li tmus with a dilute solution of soda
,and a bead of borax (made
by fusing the salt on a loop of platinum wire) dipped into i t . Thebead i s allowed to rest for a few minutes
,so as to allow solution to
take place on its surface,and is then held in the flame of a Bunsen
burner . A more delicate plan is to place some powdered borax in awatch-glass
,pour on it some of the faintly alkaline liquid to be tested ,
and,by means of a looped platinum wire
,introduce some of the
mixture into the flame . In either case a deep-green flame will beproduced if a moderate quanti ty of glycerol be present
,but the test
becomes indistinct if the liquid contains less than For detectingglycerol in beer
,wine
,milk
,etc .
, 50 or 1 00 c .c . of the liquid should beevaporated to dryness on the water-bath
,the residue extracted with
absolute alcohol,the solution so obtained again evaporated , and the
resultant residue moistened with a few drops of water and tested withborax as above described . Ammonium salts
,glycol , and erythri tol
give a similar indication to glycerol . Ammon ium salts may be

456 GLYCEROL .
ing glycerol by adding a solution of basic lead acetate,and subse
quently removing the excess of l ead from the filtered solution by meansof hydrogen sulphide . This method may be employed for the analysisof pharmaceutical preparations
,such as “ glycerol of tannic acid ” and
“ glycerol of gallic acid,
” and is useful as one stage of the treatment ofsoap lyes for the estimation of glycerol .Proteins and some other organic substances can often be removedcompletely by precipitating the slightly alkaline solution with zincchoride . The precipitate i s filtered Off and the filtrate renderedfaintly acid , when a further precipitation will often occur . The lasttraces of zinc may be removed from the solution by potassium ferrocyanide , which is also a very perfect precipitant of albumin .
D ilute glycerol may be further purified by evaporating off the waterat as low a temperature as possible
,and treating the residue with ab
solute alcohol, a mixture of alcohol and ether or a mixture of alcohol
and chloroform,according to circumstances . Absolute alcohol
readily dissolves glycerol,while many classes of salts (e. g .
,metallic
sulphates,phosphates
,tartrates
,etc .) are insoluble . The alkali
metal chlorides are not completely separated by alcohol alone,but a
mixture of equal volumes of absolute alcohol and dry ether leaves themundissolved . The same solvent serves to separate glycerol from sugar ,but the use of a mixture of two volumes of absolute alcohol with oneof chloroform is preferable . If the filtered solution be treated withabout twice its volume of water
,chloroform separates from the diluted
alcohol,and often carries troublesome colouring matters with it .
Any process of estimating glycerol which involves the evaporationof an aqueous or alcoholic solution and isolation of the glycerol in substance is deficient in quantitative accuracy
,as evaporation of glycerol
i n the latter end of the concentration is unavoidable,and the loss from
this cause i s Often very considerable . Even absolute glycerol i ssensibly volatile at the loss of weight varying with the mode ofheating
,the shape and material of the containing vessels
,and the sur
face exposed .
The following figures,due to Nessler and B arth (Zei t. anal . Chem. ,
1 884 , 23 , 3 2 show the rate of evaporation of glycerol under differentconditions . The experiments were made with glycerol which had beenheated for 6 hours over a water-bath at and then for 6 hourslonger in an air-bath heated at In one series of experiments theglycerol was exposed in a water-oven at 1 000 in a platinum dish 20 mm .

E STIMATION OF GLYCEROL . 45 7
high and 80 mm . diameter at the top , and 60 at the bottom ; in theother
,i t was heated in a beaker of thin glass 40 mm . high and 48 mm.
in diameterP latinum d ish G lass beaker
1 . 0 grm . lost, in first 2 hours . 46 mg . 36 mg .
grm . lost,in second 2 hours 29 mg . 1 4 mg .
grm . lo st, in th ird 3 hours . 2 1 mg . 5 mg .
Average for last 3 hours . 7 mg . 1 . 7 mg .
0 . 5 grm . lost in first 2 hours 36 mg . 45 mg .
0 . 5 grm . lost in second 2 hours 28 mg . 1 1 mg .
grm . lost in th ird 3 hours . . 23 mg . 6 mg .
Average for last 3 hours mg . 2 mg .
The following figures Show the loss of weight when heated on anopen water-bath kept briskly boiling
P latinum dish G lass beakergrm . lost, in 1 hour
,. 3 7
—39—29—30 mg . 30
—1 8 mg .
grm . lost, in 1 hour 34—29—24—30 mg . 1 1 2 mg .
O ther experiments conducted in platinum and glass vessels of various diameters showed that the loss increased with the diameter of thevessel (i . e . ,
with the surface of glycerol exposed) , and that the rate ofevaporation was less in a vessel composed of a material of low conducting power .The volatilisation of glycerol during the evaporation of an aqueousliquid may be prevented by adding an excess of lime
,which forms a
compound with it,but Clausnizer has shown (Zei t. anal . Chem. , 1 88 1 ,
20, 58) that from the product the glycerol cannot be dissolved by
absolute alcohol ; and if hydrated alcohol is employed , alkalies resultingfrom the action of the lime on phosphates may pass into the alcoholicliquid
,and carry with them substances not otherwise soluble . Even if
excess of lime be avoided,the glycerol cannot be extracted completely
from the residue by cold alcohol or ether-alcohol .
General Methods for the Estimation of Glycero l .It will be convenient first to consider the general methods used inestimating glycerol
,and then to deal later with the application of these
methods to Special cases ; as , for example , to soap lyes or commercialforms of glycerin .
Chemi cal Methods . A . Volumetric.
1 . P ermanganate Oxidati on P rocess .
—This is best carried out byBenedikt and Zsigmondy ’s modification (Chem. Zei t.
,1 885 , 9 , 975)

458 GLYCEROL .
of Wank lyn and Fox ’s method . to 0 .3 grm . of the concentratedglycerin (or a quantity of dilute glycerin corresponding to this amountand calculated approximately from the sp . gr . of the sample) i s mixedwith 250 c .c . of water in a large flask
,1 0 grm . of solid potassium
hydroxide added,and a 5% solution of potassium permanganate run in
at the ordinary temperature until the liquid ceases to be green and becomes blue or black in colour . Finely powdered potassium permanganate can be used in place of its solution . The mixture is thenboiled
,when hydrated manganese dioxide is precipitated and the
solution becomes red . A solution of sulphurous acid or of sodiumsulphite is then cautiously added
,drop by drop , until the l iqui d just be
comes colourless,and the solution then fi ltered through a fil ter suffi
ciently large to take at least half the liquid at one time . The precip itate i s thoroughly washed with hot water . The last washings sometimes become turbid owing to the formation of manganese hydroxide
,
but the turbidity disappears on adding acetic acid so as to render thesolution acid before pI ecip itating with calcium chloride . The precipitation i s effected by adding 1 0 c .c . of a 1 0% solution of calcium chlorideto the boiling liquid . The calcium oxalate i s left for some time inorder to complete the precipitation
,collected on a filter
,and
,after
washing thoroughly with hot water,i s transferred to a flask and titrated
with N 1 0 permanganate in the usual way .
I c .c . N/ 1 0 permanganate (corresponding with 5 grm .
H2C 204)= 0 .0046 grm . glycerol .In the permanganate method
,excess of sulphurous acid must be
carefully avoided,as in presence of hydrated manganese dioxide i t
destroys oxalic acid . Allen suggested the use of sodium sulphite instead oi sulphurous acid , but on adding acetic ac id before precipitation ,sulphurous acid is l iberated , which in presence of the small quantity ofmanganese dioxide which has passed through the fil ter causes the loss ofoxalic acid . There is , moreover , the danger of calcium sulphite beingprecipitated with the calcium oxalate .Herbig has therefore suggested the use of hydrogen peroxide in placeof sulphite
,and employs a smaller quanti ty of potassium permanganate .
Mangold (J . S oc . Chem. Ind .,1 89 1 , 1 0, 803) reports favourably on the
method, and recommends the following procedure : To grm . of
glycerol,dissolved in 300 c .c . of water containing 1 0 grm . potassium
hydroxide,as much of a solution containing 5% potassium perman
ganate i s added as will correspond with 1 5 times the theoretical quantity

460 GLYCEROL .
measurements must be made with the greatest care,attention being
paid to the temperature . The results upon repetiti on agree well .The method is easy and rapid . It is open to the objection that byprecipitation by lead acetate the impurities may not be perfectly removed , anything left being oxidised
,and counted as glycerol . How
ever , all higher fatty acids and all resin acids , as well as albuminoids ,sulphides , thiocyanates , and aldehydes , are completely removed , andthe lower fatty acids, such as acetic and butyric are not attacked bychromic acid . Hehner allows for variation of temperature by assuming an expansion of the dichromate solution of per Thisvalue he found for a dichromate solution
,prepared as above . Lew
kowitsch avoids a temperature correction by maintaining the solutionsat the normal temperature during titration by surrounding them witha large water-j acket .Several alterations in the procedure have been suggested byRichardsonand Jaffé (J . S oc. Chem. Ind.
,1 898 , I 7 , a stronger solution of
dichromate being used and the time of boiling much reduced .
3 . Acetin Method—The acetin method of B enedikt and Cantor(J . S oc. Chem. Ind.
,1 888
, 7 , 696) depends upon the formation oftriacetin (glyceryl triacetate) when glycerol i s heated with acetic anhydride . The triacetin i s then saponified with sodium hydroxide solution , and the amount of the latter used gives a measure of the glycerol .Lewkowitsch has shown that the method gives closely concordantresults in the case of moderately pure “ crude glycerins
,and recom
mends its adoption in all cases in which the glycerol is first i solated ina fairly pure state as in its estimation in fats and oils infra)(Lewkowitsch , Chem. Zei t.
,1 889 , I 3 , 93 , 1 9 1 , 659 ; Hehner, J . S oc.
Chem. Ind .,1 889 , 8,
S oluti ons requi red:
1 . N 2 or N/ 1 hydrochloric acid (accurately standardised) .
2 . Sodium hydroxide solution,20 grm . sodium hydroxide per
c .c . I ts strength need not be accurately known .
3 . A 1 0% sodium hydroxide solution ,Solutions 2 and 3 must be kept free from the access of carbon dioxide .
P rocess .
—1 to 1 .5 grm . of the crude glycerol, 7 or 8 grm . of acet ic
anhydride,and about 3 grm . of anhydrous sodium acetate (previously
dried in an oven) are heated from 1 to 1 .5 hours in a reflux apparatus .The mixture is allowed to cool
, 50 c .c . of water are added , and theheating is continued (still with the condenser , as triacetin is volatile in

E STIMATION OF GLYCEROL .
a current of steam) until i t begins to boil . When the O ily deposit atthe bottom of the flask is dissolved , the liquid is filtered from a whiteflocculent precipitate
,which contains most of the impurities of the
crude glycerol,allowed to cool , phenolphthalein added, and dilute
sodium hydroxide (NO . 2 solution) run in until neutral i ty is obtained .
Care must be taken not to pass that point,as triacetin i s easily
saponified .
During the operation the solution must be agitated continually,so
that the acid may not be in excess locally any longer than is unavoidable . The point Of neutrality is reached when the solution becomesreddish-yellow . I t must not be allowed to become pink . The estimation is inaccurate if the solution is more than neutralised even forthe shortest time . 25 c .c . of the strong sodium hydroxide are nowadded from a pipette . The mixture is then heated for 1 5 minutes andthe excess of alkali titrated back with normal or half-normal hydrochloric . acid . The strength of the alkali used is ascertained at thesame time by titrating another 25 c .c . measured with the same pipette .The diff erence between the titrations gives the amount of alkaliconsumed in saponifying the acetin
,and from this the quantity of
glycerol is calculated .
B . Gravimetri c Methods .
—I . Zei sel and Fanto’s method (Zei t.
landw . Versuchswesen Oest. , 1 902 , 5 , This method is based onthe fact that when glycerol is boiled with an excess of hydriodic acid(sp . gr . b . p . i t is converted quanti tatively into i sopropyliodide , which can be estimated by passing i t into a solution of silvernitrate in absolute alcohol
,and weighing the silver iodide formed .
C3H5 (OH)3+ 5HI C
3H7I 3HzO 21 2
C3H7I AgNO
3AgI C 3H7N0 3
.
Materials Requi red—1 . I t is advisable to keep a stock of hydriodic
acid Of sp . gr . containing 68% by weight of HI ; this acid canthen be suitably diluted with the aqueous glycerin or with water (3vols . hydriodic acid to 1 vol . water), so that the acid acti ng on theglycerin is of sp . gr . I t must be free from sulphur and , in ablank experiment carried out as described below
,give no precipitate
of silver iodide in the alcoholic silver nitrate solution .
2 . 40 grm . of pure silver nitrate is dissolved in 1 00 c .c . of water andmade up to 1 li tre with commercial absolute alcohol ; after 24 hoursthe solution i s filtered . The solution must be kept in the dark .

462 GLYCEROL .
3 . Red phosphorus . This must be washed with carbond isulphide,
ether, alcohol , and water , and dried in the air .The apparatus used (see Fig . 1 3) i s a modification of the well-known
Zeisel apparatus for the estimation of methoxyl . The flask,a,ca
pacity 40 c .c . , has a Side tube attached as shown , which serves to passcarbon dioxide through the apparatus . Through the condenser
, b,
circulates water maintained at 60° :L 1 0° by means of an Ehmann’
s
heating arrangement, g; the tube of the condenser is ground into theneck of flask , a
, and the joint heldin position by means of the smallSprings shown . The bulb
, c, im
mersed in water at 60 servesto count the bubbles of gas and i sfi lled to about a third with a thinmixture of red phosphorus and water(a solution of potassium arsenite canbe used in place of phosphorus
,
but the latter is preferable). TheErlenmeyer flasks
, e and f, the largerwith a mark showing 45 c .c . , thesmaller with a mark at 5 c .c . ,contain the clear alcoholic silvernitrate up to the marks aforesaid .
The glycerin is weighed into a,the
quantity taken being such as to givenot more than
.0 . 4 grm . of silver
iodide . A fragment of pumice is introduced into a,1 5 c .c . of the
hydriodic acid (sp . gr. added,and the flask immediately con
nected with the condenser and with a carbon dioxide apparatussupplying carbon dioxide
,which has been washed by passing through
dilute sodium carbonate solution . The carbon dioxide is passed atthe rate of about 3 bubbles per second . The boiling flask is immersed in a glycerin bath so that the levels inside and outside of theflask are the same ; the bath is heated by a smal l flame so as to keepthe hydriodic acid boil ing gently during the whole operation . Whenthe li quid above the precipitated silver iodide becomes clear and theoperation is complete the contents of the Erlenmeyer flasks are transferred to a large beaker ; water is added so as to make a volume of450 c .c . , and then 1 0 to 1 5 drops of dilute nitrlc acid ; the liquid is then
FIG. 1 3 .

464 GLYCEROL .
The sample i s warmed in a closed bottle by immersing in warmwater until all air-bubbles have collected at the top . The glycerol isthen allowed to cool in the cooled bottle
,preferably to the normal
temperature , and then carefully filled into the pyknometer providedwith a perforated stopper . If thi s has been pushed home
,after the
last filling , the very small drop of glycerol squeezed out is wiped off
with a linen cloth and the bottle taken out of the water-bath . The determination may be made exact to the fourth decimal if the weights arereduced to vacuum . Complicated calculation is avoided by ascertaining once for all the necessary corrections for the pyknometer whenfilled with water . Suppose the weight p has been found in air
,then the
corrected weight P will beP = p+pR.
For brass weights,the correction R for the sp . gr . likely to occur is
found from the following table
If the temperature is not one of those corresponding with the tableson page 448 , a correction of may be made for each 1 ° differenceof temperature . Lenz ’s values at 1 2 to 1 4
° have been calculated to1 5 .5
° by Richmond using this factor :' The following table shows theresult .

E STIMATION OF GLYCEROL .
’
465
Refractive Index .
-This is ascertained by one of the many forms ofrefractometer . In order to avoid the necessity of maintaining a knownconstant temperature and of accurately ascertaining the zero error ofthe instrument
,Lenz recommends that the refractive index of the
glycerol solution and of pure water he observed successively . Thefollowing table gives the differences between the refractive index ofwater and of aqueous solution of glycerol of different concentrations .
TABLE OF DIFFERENCES BETWEEN REFRACT IVE INDICES OFAQUEOUS SOLUT IONS OF GLYCEROL AND OF PURE WATER .
(ND SOLUT ION—ND WATER) (LENZ).
Degree of Accuracy of Difierent Methods for Estimating Glycerol .
F. Schulze (Chem. Zei t.,1 905 , 29 , 976) has recently made a systematic
comparison of the methods in use for estimating glycerol ; he givestables showing the results obtained with fats
,soaps
,and various com
mercial glycerins . The following are his principal conclusions :1 . The permanganate method is considered unreliable in all cases ,whether carried out by the method of B enedikt and Zsigmondy , or byHerbig or Mangold’s modifications .
2 . The acetin method failed to give concordant results . If thismethod is still to be employed
,i t i s essential that the mean of several
estimations be taken .
3 . The dichromate method gives high results as a rule . Approxi
mate values may be obtained by lowering the figures obtained byThe method is valid only -in absence of phosphoric acid .
Vo l . I I .
—30

466 GLYCEROL .
4 . Zeisel and Fanto ’s method is regarded as the most accurate for
scientific and general purposes,but i t i s too expensive for ordinary
factory working . In such cases i t should be used as a check on thedichromate method .
Lewkowi tsch (Chemi calTechnology of Fats and Oi ls) considers thatfor ascertaining the proportion of glycerol in its pure dilute aqueoussolution , oxidation methods are best , and that ei ther the permanganate ordichromate method gives good results in such cases . On the otherhand
,there is no doubt that such methods give high results with im
pure glycerin . In such cases the acetin method is preferred . TheZeisel-Panto method is said by Lewkowitsch not to give good results .Physical methods can
,of course
,be expected to give accur ate re
sults only with aqueous solutions of pure glycerol ; in presence of aknown proportion of known salts
,the influence of the latter on the
physical properties can,however
,be calculated . Several methods have
been proposed for estimating the approximate proportion of glycerolin crude glycerin by‘
making an allowance of this kind (compare forexample
,Richardson and Jaffé, loc. ci t. ; S tiefel , S eif ensiederzei tung ,
1 905 , 3 1 , but the results obtained can only be regarded asapproximations .
COMMERCIAL GLYCERIN.
Three kinds of “ glycerin ” call for consideration : 1 . Crude glycerin ;2 . di stilled or dynamite glycerin ; 3 . chemically pure glycerin.
1 CRUDE GLYCERIN.
Three kinds of crude glycerin may be distinguished : Saponification
glycerin,distillation glycerin
,soap-lye glycerin or soap glycerin .
Sapon ifi cation Glycerin .—This glycerin i s obtained by the auto
clave process Of hydrolysing fats by heating with water under highpressure
,either alone or in presence of a small proportion of lime or
magnesia . It is evaporated to a sp . gr . and is then knownas “ 28° “
raw glycerin,
” “saponifi cation glycerin or “candle glycerin .
”
I t has a sweet taste,and varies in colour from bright yellow to dark
brown . It gives but a slight precipitate with basic lead acetate ,and with hydrochloric acid should give no turbidity . The valuationof such glycerin includes the estimation of glycerol
,ash (which shoul d
not exceed 0 .3 to 0 .5 and organic impurities .

468 GLYCEROL .
when the copper, i ron , zinc, magnesium,and more or less calcium will
be dissolved as -sulphates,and can be detected in the solution by the
usual methods . The residue will contain lead sulphate,together
,
possibly , with calcium sulphate . On treating it with a hot solution ofammonium acetate
,the lead sulphate will be dissolved
,and the resul
tant solution will give a yellow precipitate with potassium chromateand a black precipitate with hydrogen sulphide .Ca lcium i s a frequent impurity occurring most commonly as
calcium oleate . I t i s most readily estimated by precipitating thediluted sample with ammonium oxalate . Precipitation in an alcoholicsolution with sulphuric acid has been recommended by Cap , but presents no advantages over the oxalate method .
A lk al in i ty in commercial glycerol i s due almost entirely to sodiumcarbonate
,and is readily estimated by titrating the diluted sample
wi th standard acid . Sulman and B erry recommend the use of litmusas an indicator
,neither phenolphthalei n nor methyl-orange giving
sharp end-points . Glycerin from soap-lyes i s purposely alkaline,
owing to the risk of concentrating it in presence of acid . Thealkalinity usually varies from 0 .5 to depending to some extenton the manner in which the lyes have been treated . In a case citedby Fleming
,in which the glycerin had been separated from the lye
by alkali instead of salt , the resulting glycerin contained 3 1 % ofsodium carbonate .Ch lor ides cannot be estimated by direct titration or precipitation with silver
,owing to the solubil i ty of silver chloride in glycerol
and the reduction of the nitrate by various impurities . The estimation is best made by allowing a weighed portion to burn away asalready described
,exhausting the carbonaceous residue with water ,
and titrating the filtered solution with N/ 1 0 silver nitrate , usingneutral potassium chromate as an indicator . Crude soap-lye glycerin susually contain from 5 to 1 0% of salt .Su lpha tes may be estimated by precipitating the diluted samplewith barium chloride . They are usually present in the product fromsoap-lyes
,and sometimes in very large amount . Glycerins Obtained
by saponifying fat with sulphuric acid are always charged with sulphates and often contain sulphi tes , thi osulphates and sulphides , the lastthree being objectionable . The milky precipitate produced on acidifying the raw product from soap-lyes sometimes contains a considerableproportion of free sulphur, the proportion amounting in some cases to

GLYCERIN . 469
40 or even 60% of the whole precipitate . Such samples will yieldobjectionable volatile sulphur compounds on distillation .
C . Ferrier (Chem. Zei t. , 1 893 , 1 6 , 1 840) proposes the followingmethodfor detecting sulphur compounds : The sample is diluted with 1 0 timesits volume of water and neutralised with hydrochloric acid . This mixture is treated at from 60° to 70° with about 3% of the carbon residuefrom the manufacture of potassium ferrocyanide (which has beenpreviously washed with dilute nitric acid and water and heated to redness in a closed crucible). One drop of the solution after treatmentwith the purified carbon residue is placed on a strip of paper saturatedwith lead nitrate . If no yellow stain appears
,the sample contains
less than part of sulphides . To detect a still smallerquantity , the sample is heated in a small flask with a few drops ofhydrochloric acid and a little sodium carbonate held over the mouthof the flask .
To detect thi osulphates and sulphi tes a few c .c . of barium chloridesolution are added to the solution of the sample and the liquid filtered .
B arium sulphite is precipitated and the thiosulphates may be found inthe filtrate
,which
,on addi tion of potassium permanganate to the
acidified solution , will become cloudy , even in the presence of onlypart of thiosulphate .
The presence of sulphite in the precipitate is proved by washing itrepeatedly with boiling water
,then adding to the remaining precipi
tate a few drops of starch and iodine solution ; in presence of sulphite‘sthe blue will gradually disappear . See also Richardson and Aykroyd(J . S oc. Chem. Ind.
,1 896 , 1 5 , 1 7 1 ) for the quantitative estimation
of these compounds .Organi c impuri ti es are estimated in soap-lye glycerin just as in
saponification glycerin .
P rotein matter, derived from the envelopes of the fat globules ,
i s nearly always present to a greater or less extent,the product from
soap-lyes containing the largest proportion,owing to the solvent action
of the alkali on the proteid matter of the fats saponified. They areobjectionable on account of the mechanical difli culties they occasionduring
'
the subsequent distillation,and the contamination of the dis
tillate with empyreumatic and coloured products . An approximateestimation of the protein matters may be made by precipitating withbasic lead acetate , and applying the Kj eldahl method . The nitrogen
,
multiplied by gives the amount of protein in the precipitate .

470 GLYCEROL .
Ros in i s a very frequent and obj ectionable impurity in the glycerinfrom soap-lyes
,but is absent from that from candle-works . A portion
of the rosin is precipitated on acidifying,but the use of basic lead
acetate is better . When rosin is present , the distillate Often has astrongly-marked fluorescence from the presence of rosin oil . Thisimpurity may be further detected and removed by agitating the samplewith ether or petroleum Spiri t
,which
,after separation and evaporation
,
leaves the rosin oil in a form recognisable by its physical characters,
taste,and odour on heating .
Higher fa tty ac ids , chiefly olei c acid, are not unfrequently presentin glycerin from soap-lyes
,even after distillation
,and are very objec
tionable in a product intended for making nitroglycerin (v. infra) .I f the amount of fatty acids be considerable
,mere dilution with water
causes their precipitation,but smaller quantities may be detected by
diluting the glycerol and passing ni trogen dioxide (NO 2) through thesample
,when a flocculent precipitate of elai dic acid (less soluble than
the original oleic acid) will be produced . Ni trogen dioxide is bestObtained by heating dry lead nitrate in a tube or small retort .Fatty acids may be detected by diluting a portion of the sample withseveral times i ts bulk of water and acidifying with hydrochloric acid .
In the presence of fatty acids the liquid becomes turbid .
By agitating glycerin with chloroform ,fatty acids
,rosin Oil
,and
some other impurities are dissolved,while certain others form a turbid
layer between the chloroform and the supernatant liquid . On separating the chloro form and evaporating it to dryness
,a residue is
obtained which may be further examined .
Lower fat ty ac ids , especially butyric and formic acids , may be notunfrequently present . The presence of free oxalic , form ic , or butyricacid in disti lled glycerol will be indicated by the acid indicat ion of thesample
,and an estimate of the amount present can be obtained by
titrating the diluted sample with standard alkali and litmus or phenolphthalei n . Butyri c acid i s sometimes present to the extent of ofthe fats saponified . Samples containing it develop an odour o f sweatwhen mixed with a few drops of dilute sulphuric acid and rubbedbetween the hands . F ormi c acid
,traces of which are often present
even in distilled glycerol,i s best detected by adding ammoniacal silver
nitrate to the diluted sample . On leaving the mixture at the ordinarytemperature for half an hour
,a black precipitate will be produced if
formic acid be present . After a longer interval,all samples of com

472 GLYCEROL .
(d) Arseni c.
-Only traces are permissible . The Gutzeit testbeing too delicate , the following should be used . The glycerin ismade very faintly alkaline by the addition of the least possible quantityof ammonia ; on adding silver nitrate no milkiness should be vis ible .An excess of ammonia must be avoided as silver arsenite is soluble inammonia .
(e) Organi c Impuri ti es .
-1 c .c . of the sample is diluted with 2 c .c .of water and mixed with a 1 0% solution of silver nitrate . The liquidshould not become black or brownish within 1 0 minutes .(f) Total non-volati le residue—ascertained as on page 467 . It
should not exceed(g) Free Acid—The glycerin must not redden blue litmus-paper .
Volatile fatty acids are detected by the production of a fruity odour(ester formation) when the sample is heated with alcohol and coneentrated sulphuric acid . 1 c .c . diluted with 2 c .c . of water should giveno precipitate on adding strong hydrochloric acid .
I t may often happen that a sample which will answer the aboverequirements is yet unsuitable for the manufacture of dynamite ; i tmust , therefore , be nitrated in the following way , which im itates thecondi tions obtaining on the large scale . A mixture of 1 part byweight of fuming nitric acid (sp . gr . 1 .5) and 2 parts of pure sulphuric acid i s prepared
,and allowed to cool in a stoppered
vessel . 3 75 grm . of the mixed acid are put into a th in-wall ed beakerof about 500 c .c . capacity, and stood in a large vessel through whicha constant current of cold water passes . .Great care must be takenthat the water does not Splash into the beaker
, to which end the leadingtube should be firmly fixed both to the tap and the basin . 50 grm .
of the glycerin are weighed out,and
,when the acids are not hotter
than from 1 2° to added drop by drop , using a thermometer as
a stirrer . The stirring must be very thorough to avoid local heating,
and the temperature must not be allowed to rise to 25° being a
safer limit . I t the temperature indicates danger,the bottom of the
beaker should be instantly perforated with the thermometer . Thesmall beaker may be weighed again to give the exact amount of thesample added
,and when the temperature of the other has fallen to
the l iquid is run out into a perfectly dry separating funnel,which may
advisedly receive a preliminary rinse with strong sulphuric acid . Thequicker the separation of the l iquids
,and the sharper the line of
demarcation between the n itroglycerin and the acids,the better is the

PURE GLYCERIN .
glycerin . The n itroglycerin is always slightly turbid,but i f it contains
flocks,or the separation is not complete in 5 or 1 0 m inutes , or if there
is a cloudy middle layer of l iquid , the glycerin must be rej ected .
With very bad samples,no separation at al l may be obtained on
standing several hours .If i t is-desired to make the test quantitative , the operation may becontinued . The acids are run off ; the nitroglycerin carefully swunground in the separator to detach drops of acid from the walls (withoutshaking it
,however), and after these drops are removed , washed with
warm (35° to water,once or twice with 20% sodium hydroxide
solution,and again with water . I t is then run into a 1 00 c .c . burette
,
or graduated tube,and when the excess of water has risen to the top
,
the volume read off . This , multiplied by gives its weight , andthe yield should be at least from 207 to 2 1 0% —the h igher the better(theory requires If preferred
,it may be weighed directly after
filtration over salt,and its sp . gr . taken . The loss in the wash
waters is insignificant .TO destroy the nitroglycerin
,i t i s best absorbed in a th in layer o f
sawdust spread in an open yard removed from any buildings,and then
set on fire with a match . I t will burn away quietly .
Owing to the danger attending th is test,many chemists d iminish the
quantity Of sample used to 1 5 grm . This is the smallest quantity thatshould be taken
, as with less than th is amount the test is quite useless .
3 . PURE GLYCERIN.
Ch emica lly pure glycerin comes on the market in three grades :Sp . gr . sp . gr . 5 , sp . gr . respectively . The “ glycerinum ”
of the Brit ish Pharmacopoeia has a sp . gr . that of the UnitedS tates Pharmacopoeia ofThe percentage of glycerol in pure glycerin can be ascertained bytaking the sp . gr. on observing the refractive index in the manner already described (page Wi th dilute solutions the percentage ofglycerol is best ascertained by means of the dichromate method ;Lewkowitsch also recommends the permanganate method for th is purpose (compare above .)Ash plus polyglycerins should not exceed ash alone shouldnot exceedAcrole in (and other reducing substances) are best detected by

474 GLYCEROL.
adding a few drops of S ilver nitrate solution to the diluted glycerin ;after stand ing 24 hours there should be no visible blackening. Thetest i s made more sensitive by us ing ammoniacal silver nitrate .Vo lati le fatty ac ids are detected as on page 470 .
Arsen ic must not exceed 1 part in The best test for arsenic is the Gutzeit test
,which is carried out as follows :
P lace in a tall test- tube about a grm . of pure zinc , 5 c .c . of dilutedsulphuric acid and 2 c .c . o f the sample . The mouth of the testtube is then covered with a tigh tly-fitting cap , made o f 3 thicknesses offilter-paper . A drop o f a 50% solution of silver nitrate i s placed on theinner surface layer and the tube allowed to stand for 1 0 minutes in thedark . If arsen ic is present
,a bright yellow stain will appear on the
filter-paper,which
,on the addition of water
,becomes black or brown .
A blank test should always be made to establish the absence of arsenicin the reagents . Sulphides (which may be detected by substitutinglead acetate for the silver nitrate in the above test) must be oxidised tosulphates be fore applying the test .The test is extremely sensitive . A less rigorous test may be made bysubstituting a drop of a saturated solution of mercuric chloride forthe silver nitrate . If no yellow colouration appears a fter 1 0 minutes
,
the sample may be considered free from arsenic .Pure glycerol does not acquire a yellow or brown colour when verygradually mixed with an equal volume of cold concentrated sulphuricacid . Sugar and certain other impurities cause a marked darkening ,or even charring
,and in presence Of any considerable quantity of
formic or oxalic acid the mixture effervesces when warmed . Oxalic
acid may be recognised more certainly by the formation of a whiteturbidity on adding calcium acetate to the diluted sample . I t is notunfrequently present in raw
,but never in distilled samples .
Pure dilute glycerol does not sensibly reduce Fehling ’s solution whenheated with it to 1 00° for a few minutes , but prolonged boiling causesprecipitation o f cuprous oxide . Dextrose and arsen i ous acid willreduce the solution even before the b . p . i s reached . Arseni coccurs in glycerin recovered from soap-lyes which have been neutralisedby crude hydrochloric acid
,owing to the fact that i t volatil ises (probably
as a compound of glycerol) when the glycerol is distilled . Cane-sugar
can be recognised by the same test , i f the sample is previously heatedto 70° or 80° for 1 0 minutes in 5 times its volume Of water and half itsvolume of strong hydrochloric acid , and the inverted solution be neu

476 GLYCEROL .
impurities which may be present, thus leaving the glycerol in acomparatively pure form .
Dis t inct ion Be tween Raw and Dis t i l led or Pure Glycerin .—The
values found for ash,organic impurities
,and the difference in behaviour
with basic lead acetate give a means of dist inguish ing between distilledand undistilled glycerin .
All so-called crude glycerin imported into the United S tates isexamined by the government chemists to ascertain whether it isreally crude or has been partially or wholly refined
,as
'
in_
the latterCase a higher rate of duty is charged . Glycerin that has been freedfrom impurities by allowing them to subside and
.
then straining andfiltering
,i s still classed as crude
,
” but if proved to have been sub
jected to further purification it is classed as “ refined .
”For practical
purposes of classification distillation is regarded as the dividing linebetween crude and refined glycerin . For th is purpose J . H . Wainwright (J. S oc. Chem. Ind. ,
1 889 , 1 1 , 1 25) attaches great importance tothe following tests :The Carbonaceo us Residue is obtained by heating 1 0 grm . of thesample in a platinum crucible till i t ignites
,when the source of heat
is removed and the sample is allowed to burn away spontaneously .
In distilled glycerin this will not be over Crude glycerin mayyield as much as The percentage of ash appears to be a lessrel iable criterion
,but should not be over
S i lver N i trate Test—5 c . c . of the sample are d iluted with 20 c .c .of distilled water
,mixed with 5 c .c . of a 2% solution of silver nitrate ,
and allowed to stand for one hour . Only a slight precipitate will beformed with distilled glycerin at the end of th is time ; whereas withcrude glycerin the precipitate is large , usually comes down at once ,and is almost alwaysflocculent.Lead Test.—The solution is prepared by boil ing 16 grm . of leadacetate and 8 grm . of lead oxide with 500 c .c . of water, and filtering .
2 volumes of th is solution are mixed with 1 volume of glycerinand 1 o f dist illed water, and allowed to stand for 1 hour. Refinedglycerin may produce a sl ight precipitate , but this is neverflocculent.Crude samples produce a more or less abundant flocculent precipitate .Wainright does not consider it safe to rely upon either of the twolast-mentioned tests alone , but if a sample
‘
will not stand both of them ,
i t is thought perfectly safe to call i t crude .Mr. Chas . C . Roberts , of the United S tates Customs Laboratorv

ESTIMATION OF GLYCEROL .
at Philadelph ia , furnishes the information that these tests are still inuse
,and in addition refined glycerin should give only inappreciable
precipitates with ammonium oxalate and barium chloride,the sample
being diluted as noted above . The reaction should be neutral orslightly acid
,and the sp . gr . at The sp . gr . i s not
,how
ever,regarded by the appraisers as an absolute requirement . Samples
not according with these requirements for “ refined glycerin ” aredutiable as “ crude .Estimation of Glycerol in Spec ia l Cases .
1 . In beer, Spiri ts and wine (see the sections on these subjects inVol .
2 . In Oi ls and fats : The estimation of glycerol in the glyceridespresent in fats and oils i s generally carried out by the acetin method .
20 grm. of the fat or oil are saponified with alcoholic potassiumhydroxide as in determin ing the Reichert value (page The soapis dissolved in a considerable volume of water
,and decomposed with
dilute sulphuric acid ; the precipitated fatty acids are filtered Off,
an excess of barium carbonate is added to the filtrate wh ich is thenevaporated on the water-bath until most of the water has been drivenoff . The residue is then extracted with a mixture of ether and alcohol
the bulk of the ether-alcohol volatilised carefully on the waterbath and the residue dried in a desiccator (preferably in a vacuum) andweighed . I t is not necessary to dry the glycerin to a constant weight ,as the amount of glycerol in i t i s determined by means o f the acetinmethod described on page 460, taking 1 to 1 .5 grm . of the crude glycerinfor the experiment .Fanto
’s Method (Zei t. angow . Chem.
,1 904 , I 7 ,
—1 0 grm . ofthe sample are saponified with 1 00 c .c . of N 2 alcoholic potassiumhydroxide
,the alcohol is partly evaporated
,1 00 c .c . of water is added ,
and the fatty acids liberated by adding acetic acid . The mixture i sthen cooled
,and the solid fatty acids which separate are collected
on a filter and thoroughly washed about 5 times with 1 5 to 20 c .c . ofboiling water ; i f the fatty acids are liquid it is advisable to add a littleparafli n wax to accelerate solidification . The aqueous filtrate and
washings are boiled down to about 60 c .c .,and when cold made up to
1 00 c .c . in a measuring flask . 5 c .c . is then taken for the estimation ofglycerol by Ze isel and Panto ’s method (see pageLewkowi tsch (Analyst, 1 903 , 28 , 1 04) found that Ze isel and Fanto ’smethod gave incorrect results when the fat was treated directly with

478 GLYCEROL .
hydriodic acid ; the modification described above of the original methodis stated by Fanto to be quite accurate . Schulze
,as already stated
,
prefers Panto ’s method to all others .3 . Glycerol in Soap .
—See under Soap .
4 . Glycerol in Soap-1yes .—A known quantity of soap-lyes is
concentrated so as to give a crude glycerin and the percentage ofglycerol in th is estimated by the acetin method (Lewkowitsch).Fanto recommends (Zei t. angow . Chem.
,1 903 , 1 6, 4 1 3) the following
modifications of Ze isel and Panto ’s method described above .20 c .c . of the lye are diluted with 2 to 3 volumes of water and anamount of s ilver sulphate added equivalent to the chlorides present.After warming on the water-bath during several minutes
,shaking well
at intervals,a hot solution of barium chloride is added so as to precipi
tate the whole of the sulphuric acid . The liquid i s fi ltered , the precipitate thoroughly washed with hot water
,and the filtrate and washings
evaporated to 80 c .c . After cooling the liquid i s made up to 1 00 c .c . ,and the glycerol in 5 c .c . estimated by the hydriodic acid method

480 CHOLE STEROL AND PHY’
I‘
OSTEROL .
Cholesterol is not acted on by dilute acids,or by concentrated
alkaline solutions even on boiling . Lewkowitsch has noted that whencholesterol is heated with soda-l ime no
,or at most very small quantities
of fatty acids are formed—an important difference from aliphaticalcohols . Chemi cally , cholesterol behaves as an unsaturated se condary alcohol .Ch o lesterol d ibrom ide , C 2 7
H46Br2 O ,
i s best prepared by dissolving 50 grm . of cholesterol in 500 c .c . of ether and
'
adding a solutionof 25 grm . of bromine in 250 c .c . of glacial acetic acid . After a shorttime the mixture sets to a mass of crystall ine needles of cholesteroldibromide . These are filtered on the pump , washed with acetic acidand then with water . The dibromide thus prepared is pure
,and if
the small quantity that remains in solution is precipitated by theaddition of water the yield is quantitative .The dibromides of the esters of cholesterol may be prepared in asimilar manner . Cholesterol dibromide prepared as above melts at
I t is readily reduced to cholesterol by the action of zinc dustand glacial acetic acid or of sodium amalgam in presence of ether .The calculated iodine-absorption of cholesterol is Lewkowitsch
obtained figures closely approximating to th is .Ch o lestery l Esters .
—Cholesteryl acetate , C 2 7H
4 5 C 2 H3O 15 best
prepared by boiling cholesterol for 20 to 30 minutes with an excessof acetic anhydride . I t crystallises from benzene in needles . I t
i s fairly soluble in ether and Slightly so in cold alcohol,but more
readily in hot . I t melts at and has a specific rotatory power :(a )DCholesteryl propi onate , C 2 7
H4 5C3HS O 18 prepared by heating
anhydrous cholesterol with half its weight of propionic anhydride onthe water-bath for half an hour . On cooling
,the propionate separates
as a white mass,and may be purified by repeatedly dissolving in
ether and reprecipating with alcohol . I t crystallises in rhombicplates something like cholesterol
,and melts at On cooling from
the melted state a play of colours i s seen about the point of solidification—violet
,blue
,green , orange , copper-red by reflected light and
the complementary colours by transmitted light . This i s verycharacteristic .Cholesteryl benzoate , C 2 7
H4 5
.C 7 H5 O 18 formed by heatingcholesterol with benzoyl chloride or benzoic anhydride , but the bestway to prepare it is to dissolve cholesterol in dry pyridine and add a

CHOLE STERYL CHLORIDE .
moderate excess of benzoyl chloride . The mixture is allowed to standovernight
,and then poured into water . The precipitated cholesteryl
benzoate is washed with a little alcohol and recrystall ised from ethylacetate or from boiling alcohol . I t is only S lightly soluble in ab solutealcohol . If crystallized from alcohol , the mother l iquors retain at 20°
grm . per 1 00 c .c . The crystals are , however , more difli cultlysoluble . The writer found that on a llowing crystals to stand inalcohol at 20° for several hours with occasional shaking onlygrm . dissolved in 1 00 c .c . Cholesteryl benzoate melts at 1 45 to 1 46
°
to a turbid liquid,which becomes clear at On cooling from
an azure—blue colour appears , and this changes to a deep bluecolour at about the point of solidification ,
which persists for a fewmoments . This behaviour is exceedingly characteristic .Cho lestery l ch lor ide , C 2 7
H4 5Cl
,was first Obtained by Berthelot by
heating cholesterol at 1 00° with strong hydrochloric acid for 1 0 hours .I t may also be prepared by the action of phosphorus pentachloride oncholesterol
,but
,according to D iels and Abderhalden
,it is most readily
obtained by the action of thionyl chloride . Cholesterol dissolves inthis substance with foam ing and the solution eventually sets to a stiffmass of cholesteryl chloride
,which may be purified by crystallisation
from ether . I t melts at and has a specific rotatory power [a]D=Many other esters of cholesterol have been prepared
,but the
only ones that need be mentioned here are the oleate and palmi tate,
as they have been found in blood serum and in some pathologicalfluids .Cholesteryl palmi tate melts at 7 7° to Cholesteryl oleate iseasily soluble in ether
,chloroform
,benzene
,and hot acetone , but in
alcohol is more difli cultly soluble than cholesterol . I t crystallises inneedles , melts at and has a sp . gr . rotatory power [a]D=I t i s insoluble in water
,but is said to possess the peculiar property
of taking up considerable quantities of water,forming a perfectly
homogeneous salve-l ike,somewhat foamy mass
,not unlike lanoline .
I t gives the cholesteryl colour indications (see later) in a modifiedmanner .The cholesteryl esters can be easily saponified by boiling with analcoholic solution of potassium hydroxide
,or
,in the cold
,by the action
of sodium ethoxide—on the ethereal solutions .D ig i tonin-c ho les teride
,C 2 7H
460 .C 5 5H 940 2 8, is prepared, accord
ing to W indaus (B er .,1 909 , 42 , by m ixing a hot solution of
'
1
Vo l . I I .
—3 1

482 CHOLE STEROL AND PHYTOSTEROL .
gm . digitonin in 1 00 c .c . 90% alcohol with a solution of gm . cholesterol in 60 c .c . of 95% alcohol . The substance is precipitated inthe crystalline form and
,after standing for 1 hour
,is filtered
,washed
with,alcohol and dried at It is easily soluble in pyridine
,but
insoluble in cold water , acetone , ether, ethyl acetate and benzene .1 00 c .c . methyl alcohol dissolves at 1 8° about gm . ; 1 00 c .c . 95%ethyl alcohol at 1 8° only gm .
,and at 78° about gm . ; 50%
boiling alcohol , gm . The dry substance is very hygroscopic andgives in a typical manner the Burchardt-Liebermann test for cholesferol . I t has no defini te m . p .
,but decomposes gradually at
Phytosterol,Coprosterol
,stigmasterol and many alcohols of other
series form similar compounds,but the esters of cholesterol do not inter
act wi th digi tonin .
Oxida t ion .—The carbon skeleton of cholesterol is extremely stable
,
and though the substance is readily attacked by oxidis ing agents,the
products are usually neutral or acid substances of the same carbon content . Oxidis ing agents usually attack the double linkage
,or both the
double link “
and the carbinol group ; but D iels and Abderhalden(B er .
,1 904 , 3 7 , 3092) found that when cholesterol is heated with
powdered copper oxide to 280 to 300° the C group is oxidisedto CO
,the ketone cholestenone be ing formed .
Ch o les tenone,C 2 7H
44O
,is,however
,more readily prepared by
a method devised by Windaus (B er .,1 906 , 3 9 , Cholesterol
d ibromide i s oxidised either by chromic acid in glacial acetic acidsolution at 70° or by means ‘ of an acid solution of potassium permanganate in the cold . The cholestenone d ibromide thus formed isreduced by means Of zinc dust and acetic acid
,and the cholestenone .
obtained in a yield of Cholestenone melts at 8 1 to andforms a hydrazone
,which crystallises in needles
,m . p . a semi
carbazone,m . p . and an
,oxime
,m . p .
Detect ion of Ch o lestero l .—When moderately pure , cholesterol iseasily recognised by its characteristic crystalline form . The sub
stance to be tested should be boiled with 90% alcohol , the solutionfiltered hot and allowed to cool slowly . Ei ther immediately on cooling,or after previous concentration
,the cholesterol will be deposited in
crystals,which
,viewed under a moderate power appear as th in , very
transparent,rhombic plates
,
-the angles of which are well defined andconstantly measure 79° and 1 00
°
The most valuable tests are,however
,the formation of the acetate ,

484 CHOLE STEROL AND PHYTOSTEROL .
colour indications are given by many derivatives and isomers of cholesterol
,and by other allied substances .
Isoch o les tero l .—This body is isomeric with ordinary cholesteroland occurs with it In wool-fat . I t has not
,however
,so far as the
writer is aware , been found in any of the organs or tissues of theanimal body .
To separate cholesterol and isocholesterol,the mixture should be
heated for 30 hours in a sealed tube to with 4 times its weightof benzoic acid or benzoic anhydride . The product is then repeatedlyboiled with rectified spirit
,when excess of benzoic acid dissolves and
the cholesteryl and incholesteryl benzoates remain . By crystallisingfrom ether , the former is obtained in shining rectangular plates andthe latter as a light crystalline powder which may be separated bydecantation and elutriation .
I socholesterol benzoate after recrystallisation from ether is obtainedin the form of minute needles melting at 1 90 toI socholesterol is Obtained by saponifying the benzoate with alcoholicsolution of potassium hydroxide . I t separates from absolute alcoholin flocks when the solution is dilute
,but a concentrated solution
solidifies as a transparent jelly . From ether i t i s deposited in needles .I t melts at 1 3 7
° to and has a specific rotatory power [ct]Dwhich is independent of the concentration of the solution .
I socholesterol gives the Sch iff’s test (page but Shows no colourchanges with sulphuric acid and chloroform or with ferric chlorideand a mineral acid . W ith the Burchardt-Liebermann test (page 483)a yellow and afterward a yellowish-red colouration appears
,with
,at
the same time,a green fluorescence . Isocholesteryl acetate is obtained
by digesting the alcohol with acetyl chloride until the evolution ofhydrogen chloride ceases
,and then heating to 1 00
° in a sealed tube .On removing the excess of acetyl chloride by evaporation
,isocholesteryl
acetate is obtained as an amorphous substance . I t i s readily solublein alcohol
,and melts below
Vegetable Ch o les tero ls .—Cholesterol is represented in the
vegetable kingdom by the isomeric substance,phytosterol , which
appears to be equally widely distributed . After the discovery of thissubstance by Hesse in calabar beans and peas in 1 878 (Ann .
,1 878 , 1 9 2 ,
many different plants were examined by various observers ; and alarge number of substances
,all very similar in properties and melting
between 1 30° and were described . A l ist is given in table I .
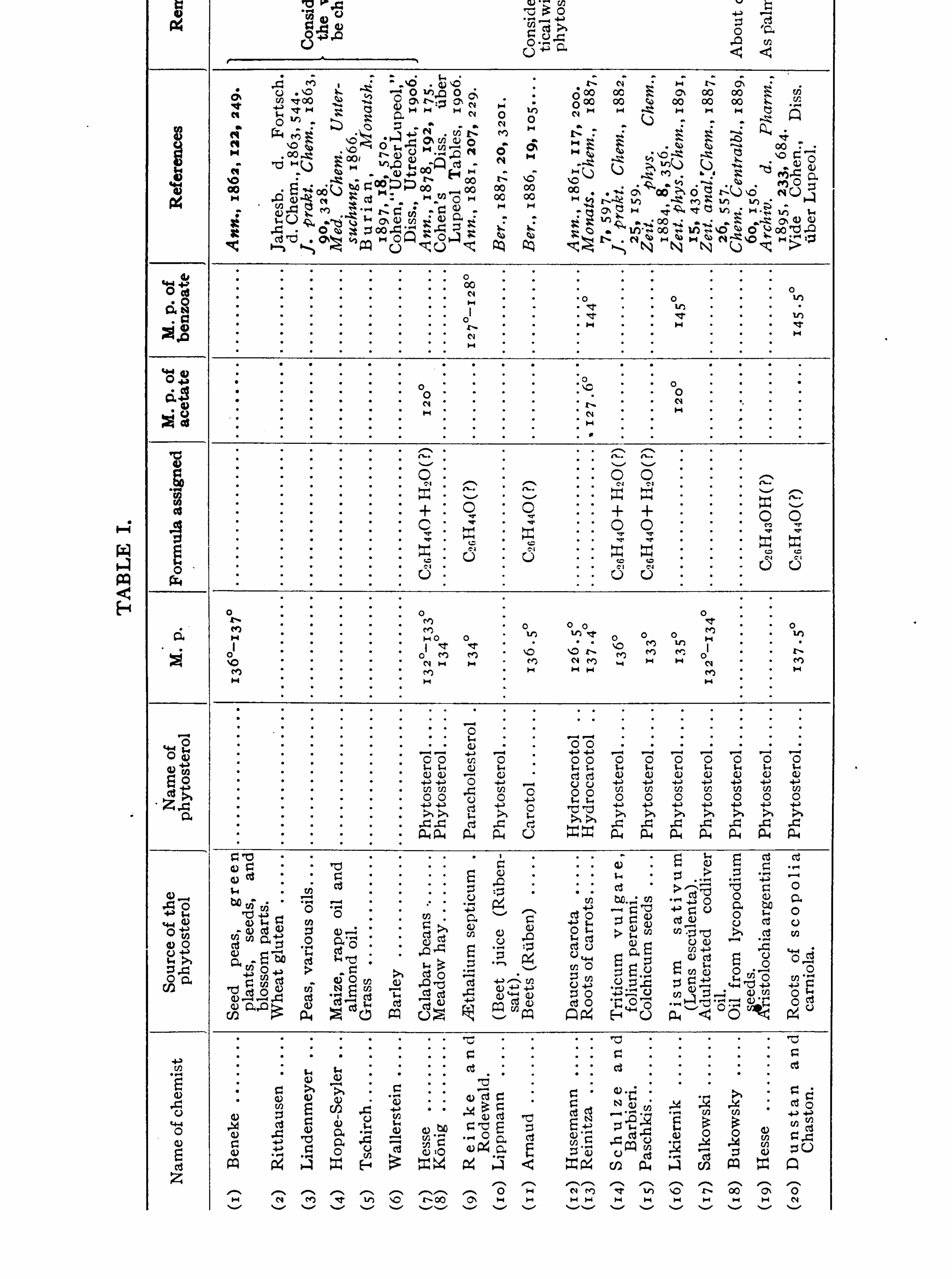
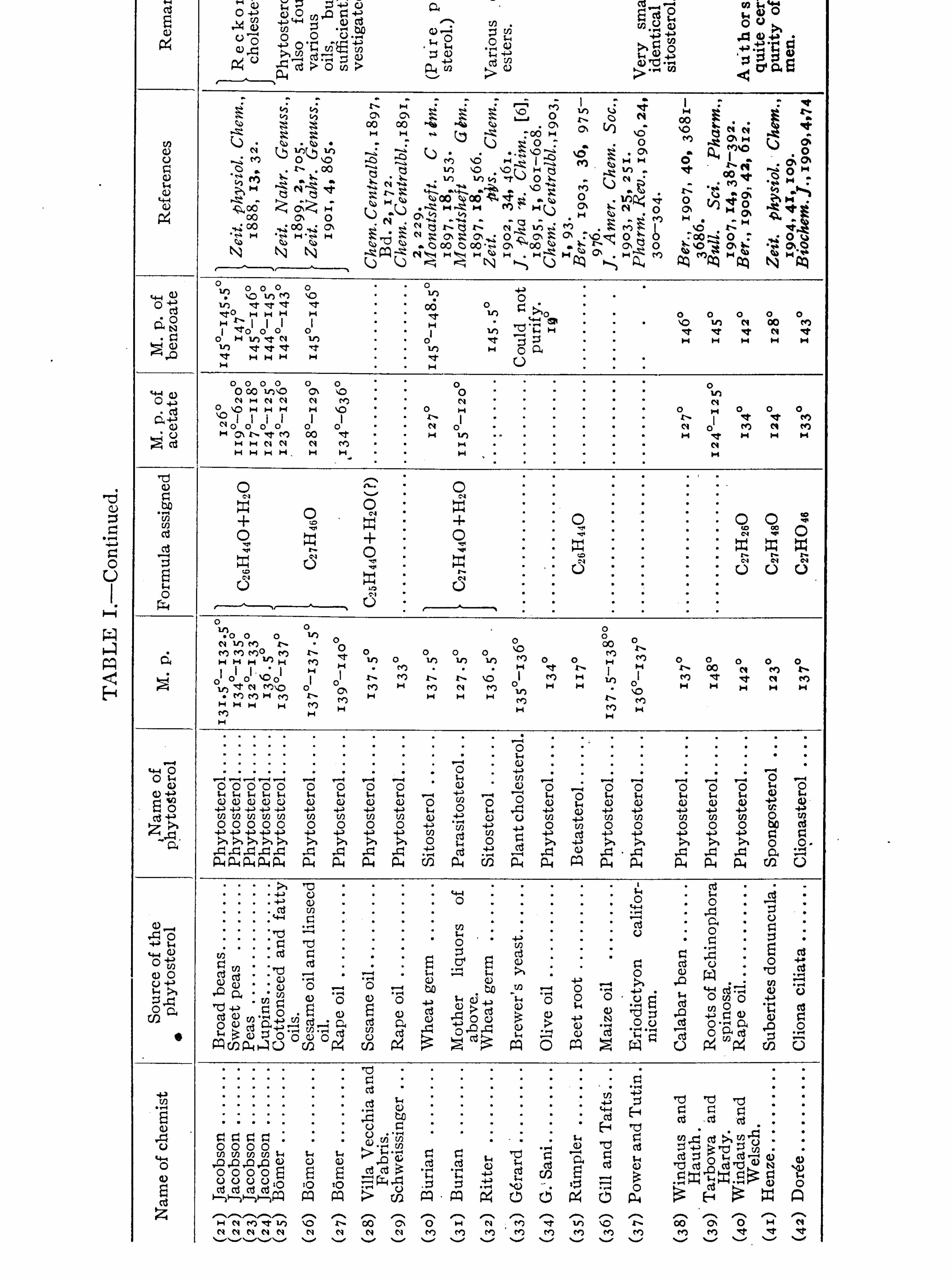
.
vomloom
n
VN-
OOOH
8M
o
§k
d§
g
.ooo-ooo.
.
.
H
mu
.
mn
.
moom
M‘
.
N
sNN
bo
kmfi
g
wo
NJ
ooo
6
3
1W5
0
0
0fl
.
0
s
.
mo
.
H
.
mOmfi
éN
o
hxs
b.
Ss
x
b
0
0
3
.
woo
la
oo
.
H
.
mowu
Su
fism”
0
$6
£695
3
Q
0
c
30
Z
.
w%..
3Mu
cou
s
a
E
0
533
«
SN
.
m
f
.
o
om.
wn
.
u
owa
$
33
mfie
SS
E
.
mmm.
mm
Ka
ma
5
3“
b
.
mfiosowé
du
n
.
N
SN
O
SRS
Q.
Es
x
b
.
NN
H
q
No
mu
m
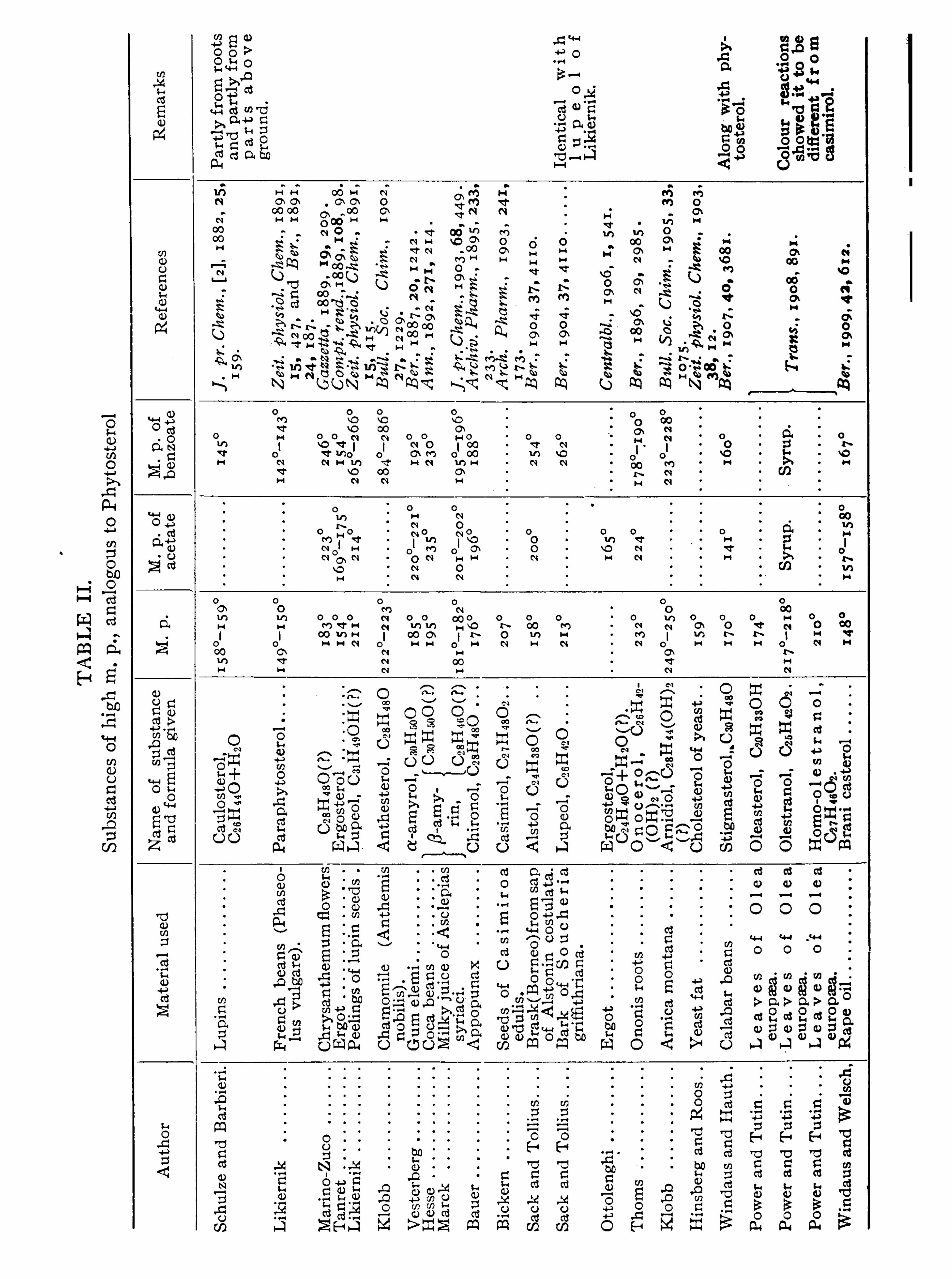

EXAMINATION OF ETHER RE S IDUE S . 489
of benzoyl chloride on a pyridine solution of the alcohol . I t crystallizesin oblong rectangular leaves and melts to a clear liquid at Whenthe melted substance cools , a play o f colours at that point of solidification is observed—yellowish -green , blue , and faint red . An account ofthe esters formed by phytosterol with the higher fatty acids will befound in a paper by R itter (Zei t. physi ol. Chem.
,1902 , 34 ,
G ill and Tufts (J . Amer. Chem. S oc.,1 903 , 25 , 25 1 , and also
Schulze andWinterstein (Zei t. physi ol . Chem.,1 905 , 43 , 3 1 6) found that
phytosterol undergoes a change on standing in the air , which causesa lowering of m . p . According to Polenske and also C . Virchow(Chem. C entr .
,1 897 , I I
,if an imals are fed with phytosterol
or foods containing phytosterol,no phytosterol is found in their fat
,nor
in the writer ’s experience,in other tissues or organs .
Isolat ion and Est imat i on of Ch o les tero l and Phytosterol .
Exam ination of Eth er Res idues .—For the separation of cholesterol
,
phytosterol,and similar subst ance from animal or vegetable matter
,
the latter must be reduced to a dry friable substance,easily capable of
extraction by ether or other solvents . Some substances may besimply dried in the oven and coarsely powdered ; others , such as organsand flesh of animals
,must be shredded and minced in a machine and
then thoroughly ground in a mortar with sand before drying . Liquids,
such as blood,containing much coagulable proteid are conveniently
mixed with sand and plaster of Paris in such quantity that the masssets solid . The solid is then powdered . This method is also veryconvenient in the case of brain
,eggs
,etc . The dried substance should
be exhausted with ether,as described on page 490 .
The ether extracts consist of fats,phosphorised fats
,such as the
leci thins , alcohols , such as cholesterol , phytosterol , etc . , and indeterminate unsaponifiable matter . I t has been stated by Dormeyer (Pfli iger
’s
Archiv , 1 906 , 6 I , 34 1—343) that the fat cannot be quantitatively
extracted from animal organs in a Soxhlet ’s apparatus with ether ,and he recommends that the tissue be subjected to artificial gastricdigest ion before extraction . In the wri ter ’s experience , however ,if the material is properly prepared for extraction by the abovemethods and the extraction be sufficiently prolonged , this diffi cultycan be overcome .The ether extract
,after distilling off the ether , is saponified by
alcoholic solution o f potassium hydroxide , the alcohol evaporated ,and the unsaponifiable matter extracted from the aqueous solution of

49o CHOLE STEROL AND PHYTOSTEROL .
the resultant soaps by agitation with ether in the manner describedon page 490 . When oils or fatty matters are to be examined
,they
may be at once saponified by alcoholic potassium hydroxide . Thech ief practical difficulty met with in th is procedure is the formationof emulsions of soap solution and ether which sometimes persist forlong periods . Further , large quantities of ether are required for complete separation of cholesterol from the soap solution . Many methodshave been proposed by various writers to overcome these and otherdifficulties and to ob tain the cholesterols in a state sufli ciently purefor weighing . A detailed critical examination of these methods hasbeen made by Ri tter (Zei t. physi ol . Chem.
,1 902 , 34, for an ac
count of which the original memoir must be consulted .
R itter recommends the following method :so grm . of fat are heated on the water-bath in a large porcelain basinwith 1 00 c . c . of alcohol and a solution of 8 grm . of sodium in I 50 c .c . of99% alcohol . When the alcohol has volatilized
, 75 grm . of sodiumchloride are added and then so much water that the greater part ofthe mass dissolves . This liquid is evaporated to dryness , first over thenaked flame
,then on the water-bath
,and finally in the drying oven
at The residue is finely powdered , placed in a paper cartridge ,and extracted with ether in a Soxhlet extractor during 9 hours . Toremove traces of soap and glycerol the ether is distilled off
,the residue
d i ssolved in as l i ttle alcohol as possible,and reprecipitated by water .
The “ cholesterol ” i s collected on a filter -and dried at The bulkof i t i s transferred to a weighed flask
,and the last adhering particles
are rinsed off with ether . The ether is evaporated and the residuedried at 1 00 to
'
The method gives satisfactory results,provided
no other unsaponifiable matter besides cholesterol or allied substancesi s present .The writer , however , prefers to use the method of saponificationby means of sodium ethoxide proposed by A . Kossel and K . Obermi iller
(Zei t. physi ol . Chem.,1 890, 1 4 , To the unevaporated ether
extract,or the dilute solution of the fat in ether
,an excess of sodium
ethoxide in concentrated alcoholic solution is added , when the fat issaponified in the cold and the soap is precipitated—often in an easilyfilterable form . After standing several hours the soap is filtered ofi
C
and thoroughly washed with a large excess of ether . The filtrate is thenrepeatedly washed in a separating funnel , first with water and then witha dilute solution of potassium or sodium hydroxide to get rid of alcohol ,

492 CHOLESTEROL AND PHYTOSTEROL .
at both acetates form isomorphous mixtures and through theaddition of phytosteryl acetate to cholesteryl acetate the m . p . o f thelatter is raised .
A good method of separating cholesterol and phytosterol has beengiven byWindaus (Chem. Zei t 1 906 , 30, 1 0 1 1 ) depending on the different solubilities of the dibromides in a mixture of ether and glacialacetic acid . The best way -o f treating any given mixture will begathered from the following description taken from his paper
I . A mixture of 4 grm . of cholesterol and 4 grm . o f phytosterol wasdissolved in 80 c .c . ether
,and 80 c .c . o f a solution of 5 grm . of bromine
in 1 00 c .c . glacial acetic acid were added,and allowed to stand at 0°
for I hour . The crystall ine precipitate (A) which formed was filteredand washed with 4 c .c . glacial acetic acid and then with 4 c .c . of 50%acetic acid . The washings were added to the main filtrate whenanother precipitate (B) was obtained . A and B
,after washing with
water and drying,weighed
,respectively
,and grm . These pre
cipitates were mixed and heated under a reflux condenser with 1 00
c .c . glacial acetic acid and 5 grm . of zinc dust for 2 hours ; the excessof zinc was filtered off and the solution treated with a large quanti tyof water . The precipitate was boiled for two hours with 1 00 c .c . of1 0% alcoholic potassium hydroxide , and the cholesterol thrown out ofsolution by the cautious addition of water . After recrystall izationfrom alcohol
,grm . of cholesterol
,melting at were recovered .
The filtrate from B,which contained the phytosterol d ibromide was
also heated for 2 hours with zinc dust,and the product treated in the
same way as cholesterol . The yield of phytosterol was grm . It
melted at 1 34 to 1 36° and its acetate at 1 26 to2 . In this experiment a mixture of 8 grm . of cholesterol withgrm . of phytosterol was‘ taken . The precipitate (A) weighed grm .
,
(B) weigh ing grm . The solution which contained the phytosterol-dibromide along with a little cholesterol-dibromide was treatedas follows : zinc dust was added , the ether distilled off , and the remainingsolution was boiled for 2 hours . The organic matter was thrown outof solution by the addition of water and taken up in ether . Theethereal solution was freed from acid by shaking with potassium hydroxide
,evaporated
,the residue acetylated by boil ing with acetic
anhydride and the acetate twice recrystallized from alcohol . I t
melted at 1 25 to 1 2 7° and weighed grm .
3 . In th is experiment a mixture of 4 grm . phytosterol and

SEPARATION OF PHYTOSTEROL FROMCHOLE STEROL . 493
grm . cholesterol was taken and treated as before . On the additionof the solution of bromine in acetic acid no precipitate was formed
,but
a precipitate (A) settled out on the further addition of 1 1 c .c . of 50%acetic acid . This weighed grm . and consisted of pure cholesteroldibromide . I t was washed with c .c . of glacial acetic and 1 1 c .c .of 50% acetic acid . On the addition of the wash liquor to the mainsolution a precipitate (B) was thrown down weighing grm .
This was not quite pure . From the filtrate about 3 grm . of phytosterylacetate was prepared .
The methods of separation of phytosterol from allied substancesof higher m . p . and from the aliphatic alcohols have not as yetbeen thoroughly worked out . Windaus and Hauth have
,however
,
recently made an important advance in thi s direction by their separation of the phytosterol obtained by Hesse from calabar beans into i tsconstituents . (Hauth , Inaugural D issertation
,Freiburg
,
Hesse ’s phytosterol melted at 1 3 2 to 1 33° and under the microscope
appeared to be perfectly homogeneous . This substance was convertedinto the acetate . 20 grm . of th is acetate was dissolved in 300 c .c .of ether and 250 c .c . of a 5% solution of brom ine in glacial acetic acidadded and the whole allowed to stand . A copious deposit of smallhard crystals separated which was washed successively with glacialacetic
,dilute acetic acid
,and water . After recrystallization from
alcohol the material melted at 2 1 1 to and had the compositionC3 0H5 0O 2 Br4 . On reduction with zinc dust and glacial acetic acid
an acetate was obtained which,after recrystallization from alcohol
,
melted at This acetate on saponification with alcoholic potassium hydroxide gave an alcohol of the formula C
3 0H
48 O ,which was
named stigmasterol, melted at and was sparingly soluble in mostsolvents
,with the exception of ether and chloroform . In ether solu
tion it had a Specific rotatory power (a )D The crystalswere very similar to those of phytosterol and showed the typicalcolour reactions of the cholesterol group .
The propi onate melted at and the benzoate atThe filtrate from the tetrabromide contained phytosterol acetatedibromide . This after reduction and saponification yielded purephytosterol . The percentage of stigmasterol in the original materialwasThe two alcohols
,stigmasterol and phytosterol , are isomorphous
and scarcely differ crystallographically .

494 CHOLE STEROL AND PHYTOSTEROL .
From this work it would seem probable that the so-called i somers oftrue phytosterol which diff er from it sligh tly in m . p . consist ofphytosterol mixed with stigmasterol or similar Substances . In the oneother case examined—the phytosterol of rape oil—th is inferenceproved correct (W indaus and Welsch , B er .
,1 909 , 42 , When
the distribution of stigmasterol and like substances in the vegetableworld has been more carefully studied
,their presence or absence may
form a useful test for the adulteration of one vegetable oil with another .For the approximate separation of the constituents of a complexether residue
,such as that yielded by “ recovered grease ” or the crude
oleic acid obtained by the distillation of such products,Schulze (J . prakt.
Chem. , 1 873 , N . F . , 7 , 1 63) has given the following method : The etherresidue is boiled for an hour or two with an equal weight of aceticanhydride . The hydrocarbons , such as petroleum ,
vaseline , cerasin ,and paraffi n
,are not dissolved , but form an oily layer on the surface of
the acetic anhydride,and may be separated while the liquid is still hot .
The acetic anhydride solution is boiled several times with water todecompose the excess of anhydride . The residue consists of acetatesof the solid alcohols
,and if boiled with suffi cient alcohol will dissolve
entirely,but on cooling the solution the cholesteryl acetate will crystal
l ize out almost completely . The acetates of the alcohol radicals formsperm oil and the waxes remain in solution
,and are precipi tated as an
oily layer by pouring the liquid into hot water . For the identificationof wax alcohols
,see article on Waxes .

496 WOOL-FAT . WOOL-GREASE . SUINT . DEGRAs (U . s .)
tion of lower saturated alcohols i s present . No glyceryl esters havebeen found in wool-fat .Darmst ’adter and L ifschutz (B er.
,1 89 7 , 29 , have reported the
isolation of the following bodies : Lah oceric aci d, C3 OH6004 ,insoluble
in water , but easily soluble in hot alcohol , . from which it crystalli ses , oncooling
,in plates of m . p . 1 03 to lanopalmi tic acid
,melting at
87 to 88° and solidifying at 83 to 85° to a lustrous crystalline mass ,and having the property of readily forming an emulsion with water ;also carnaii bi c and myri stic acids , an oily acid apparently olei c, and avolatile acid
,possibly caproi c. Among the alcohols
,separated by
absolute alcohol into several fractions,ceryl
, carnatibyl alcohol(saturated), and cholesterol were identified . The investigations ofG . de Sanctis (Chem. Zei t. , 1 895 , 1 9 , 65 1 ) point to the presence also ofpalmi ti c and ceroti c acids .The results of Lewkowitsch ’
s inquiries into the nature of wool-fat(J . S oc . Chem. Ind.
,1 892 , I I , 1 35 ; 1 896 , 1 5 , 1 4) have led him to con
clude that i t i s a true wax in the strict sense of this generic term .
Natural wool—fat resembles beeswax,i ts closest relative
,in that i t con
tains a considerable proportion of free acid and a small amount of freealcohols
,besides true waxes
,and the term wool-wax shoul d therefore
be substi tuted for wool-fat ; but considering the fact that the commercial wool-fat i s , as a rule , contaminated with fatty acids derivedfrom the soap used in scouring the wool
,i t i s more convenient to retain
the term wool—fat for the commercial product . He proposes , therefore , that the name wool-wax be given to the neutral portion of thewool-fat . This consists of a mixture of true wax and alcohols, theformer predominating considerably . The name wool-wax appears allthe more desirable
,as this neutral portion is now obtained in large
quantities,both in the anhydrous and hydrated state
,and confusion
with the crude wool-fat i s thereby avoided .
The following are the results of examinations made by Lewkowitsch ,as well as some estimations made in Allen ’s laboratory by W.
Chattaway :

WOOL-WAX (ESTERS AND FREE ALCOHOLS).
30°—30 2° 1
hydroxide for1 O . 24
I
Saponification-equivalent
Fatty acids,Alcohols
NEUTRAL ESTERS .
. 901 72
When extracted by means of solvents,wool-fat contains simply the
constituents (fatty acids , neutral esters , alcohols , and potassium saltsof lower fatty acids) natural to the wool . The following table represents the results of examination of wool-fat extracted by ether (Herbig
,J . S oc . Chem. Ind . , 1 894 , I 3 , 1 069)
Free Pe rcentage o f po ta ssiumac id h y droxide for sap on ifi
p otas cat ion on heat ing forS ource sium one h ou r
hy droxide requ ired Open flask Closed flask
1 From raw woo l-fat .
2 Prepared from lano lin .
Vo l. I I .
—3 2
Unsaponifi
able mat t er
(alcoh o ls)

498 WOOL-FAT . WOOL-GREASE . SUINT . DEGRAS (U . s .)
Wool—fat prepared by acidifying the suds obtained in the woolscouring process is of irregular composition . Potassium salts of thelower fatty acids are present in but small quantity
,since these are
removed in the first stage of the process,which consists insteeping the
wool in luke-warm water . In addition to the compounds mentionedabove as naturally present in the wool
,i t may contain unsaponified
fat and mineral oil which had been added to lubricate the wool andfatty acids derived from the soap used in scouring . The product ohtained in this way is called recovered grease , wool
-
grease, brown grease ,
and Yorkshi re grease . In the United S tates i t i s incorrectly called“ degras .” (For a description of true “ degras ” see under thathead .)The analysis of wool-fat requires a departure from the usual methods .The potassium and other mineral constituents can be estimated inthe ash obtained on ignition . On saponifying the fat and extractingthe ~
soap in the manner described below , the alcohols , includingcholesterol
,are dissolved
,recovered by evaporation of the solvent , and
examined as described under cholesterol .” By treating thesoap with acid
,the higher fatty acids will be obtained
,while the
lower fatty acids can be estimated by distillation in the usual way .
Foreign saponifiable fats will be indicated by the presence ofglycerol in the aqueous liquid separated from the fatty acids , andtheir amount will be roughly indicated by multiplying the glycerolfound by 1 0 .
Free Fa t ty Ac ids .—These are measured by treating a weighed por
tion of the fat with alcohol,and titrating with standard potassium
hydroxide in the usual manner ; the amount may be calculated fromthe mean molecular weight . Lewkowitsch (J . S oc . Chem . Ind. ,
1 892 ,
I I, 1 36) separates the free fatty acids for the estimation of the
molecular weight as follows : The amount of alkali required forneutralisation is first ascertained by titrating a small weighed quantityof the fat . A larger weighed quantity is then dissolved in alcoholand nearly neutralised with the greater part of the alkali required ,and the remainder is added cautiously until the solution becomes pinkto phenolphthalein . The mixture of neutral fat and unsaponifiable
matter,which rises to the surface
,is dissolved in ether and separated
from the soap solution,which is then repeatedly shaken out with ether .
The ethereal extracts are united and washed repeatedly with waterto remove all traces of soap . This stage of the process is very tedious

500 WOOL-FAT . WOOL-GREASE . SUINT . DEGRAS (U . s .)
hydroxide , and that , working under definite conditions , constantnumbers for these are obtained . Heating over the naked flame wasfound to effect the result much more rapidly than by means of thewater-bath
,and the action is complete at the end of one hour’s heating
in a flask provided with a vertical condenser . By reason of its convenience , this method is often employed in the commercial valuationof wool-fats . The table on page 497 shows some results obtained inthis way compared with those obtained by saponification under pressure . In the latter estimation
,2N alkali was used and the ma
terials maintained at a temperature of 1 05 toEstimati on of Unsaponifiable Matter.
-The separation of theethereal layer from the aqueous solution of saponified wool—fat andrecovered grease i s troublesome
,an intermediate stratum of a very per
sistent nature being formed . C . Rawson has suggested the followingplan :The sample is saponified in the usual way , and the resultant solutionis evaporated in a porCelain basin placed over a small flame . Towardthe end of the operation some powdered sodium hydrogen carbonatei s stirred in to neutralise the excess of alkali
,and some sand also
added . The residue is then dried at 1 00° and exhausted wi th ether ina Soxhlet tube . The ethereal solution i s then evaporated to dryness ,the residue boiled with water
,and the solution agitated with ether ; or
the ethereal solution is at once agitated with water containing a littlesodium hydroxide to dissolve any soap it may contain , and then evaporated to dryness and the residue weighed .
A more satisfactory method is that of Herbig . From 1 to 2 .5 grm .
of the fat are boiled with N 2 potassium hydroxide for an hour , theexcess of alkali neutralised wi th standard acid
,and the whole
washed into a beaker with boiling alcohol . The alcohol i s.
evaporated ,the solution heated to 70 to 7 and the fatty acids precipitated withcalcium chloride
,the amount of which has been calculated from the
saponification-equivalent . The precipitate i s filtered ofl
,well washed
with dilute alcohol ( I to and dried on the filter in vacuo . Whendry , i t i s extracted in a Szombathy extractor with freshly distilled acetone for 6 hours
,after which the acetone is evaporated
,the extract
washed with ether into a platinum basin,the ether evaporated
,and the
residue,which consists of the unsaponifiable matter and of the esters
which cannot be saponified by the ordinary process of boiling withalcoholic potash
,dried at 1 05° and weighed .

FREE FATTY ACIDS . 50 1
The chief points to be observed are the purity of the acetone— thefraction boiling between 5 and 56 .5
° being used—and thet
tem
perature at which the calc ium salts are precipitated . If too hot theyfuse
,and if too cold they become slimy
,subsequent filtration being
almost impossible in either case .It is advisable to extract the cork of the extraction apparatus withether
,alcohol
,and acetone .
For the estimation of the alcohols , free and formed by the saponification ,
it i s necessary to saponify under pressure,precipitate with
calcium chloride,and extract with acetone as described .
F . Ulzer and H . S eidel propose to ascertain , instead of the saponification-equivalent
,the total acidity number , as was recommended by
B enedikt and Mangold in the case of wax . This number is the amountof potassium hydroxide (expressed as mg . per grm .) required to neutralise the mixture of fatty acids and fatty alcohols obtained by saponifica
tion and decomposition of the soap with acid . 20 grm . of potassiumhydroxide are dissolved in 20 c .c . of water in a porcelain basin holdingfrom 3 50 to 500 c .c . , and the solution heated to boiling for about aminute
,the heating continue d on a water-bath until a thick . uniform
soap is obtained,and the basin finally placed for 2 hours in the water
oven to complete the saponification . The soap is dissolved in about250 c .c . of boiling water and decomposed with 40 c .c . of hydrochloricacid previously diluted with water . The clear fatty layer is repeatedlywashed with boiling water until the washings are free from acid , andthen dried in the water-oven . From 5 to 6 grm . of the dry mixture offatty acids and alcohols are weighed accurately and titrated with alcoholi cpotassium hydroxide with the precautions noted above in the determination of the molecular weight . The authors conclude that for thetechnical examination of a wool-fat suffi cient data are furnished by theestimation of the acid value (i . e .
,the mg . of potassium hydroxide
required to neutralise the free fatty acids of one gram), the totalacidity number
,the iodine number
,and the Reichert-Meissl number ,
together with a gravimetric determination of the unsaponifiable
matter .Lewkowitsch (J . S oc. Chem. Ind.
,1 892 , I I , 1 41 , and Chem. Anal .
of Oi ls , Fats , and Waxes) gives the following data from the analysis ofa wool-fat : The volatile acids were estimated by the Reichert processand their mean molecular weight assumed to be 1 04 (C SHM O z) .
The total free and combined fatty ac ids were well washed to free

502 WOOL-FAT . WOOL—GREASE . SUINT . DEGRAS (U . S .)
them from soluble fatty acids,and their molecular weight found
be 33 2 .
Vo latile acids from 1 grm . required,
c .c . no rmal KOH.
Free inso l . acids from 1 grm . requ ired c .c . normal KOH.
Total inso l. acids from 1 grm . required c .c . normal KOH.
Combined inso l. acids (by d ifference) . c .c . no rmal KOH.
Unsapon ifiable matter .
And therefore,
Vo latile fatty ac ids 2 I . 26%Inso luble free fatty 2 1 9 . 45%Combined insol . fatty ac ids (hydrated) 1 2
To tal unsaponifiable matter 36 . 47%
The excess over 1 00% i s of course due , in part at least , to hydrationincident to the saponification .
Lano l in .—On account of i ts property of forming readily with water
,
an emulsion,wool-fat
,purified by various patented processes
,has come
into extensive use as a basis for ointments and salves . Two preparations are recognized by the B riti sh and United S tates PharmacopoeiasAdeps Lance and Adeps Lanaz Hydrosus . I t i s commonly known aslanolin
,
”and consists of about 75 to 8b% of wool-wax with 20 to
2 5 of water . It i s usually white or slightly yellow,and of Salve
like consistence . I t does no t turn rancid . According to L iebrich ,i t should be free from all traces of chlorine
,metals
,glycerol or i ts
esters,soaps
,saline matters
,and mechanically intermixed impurities
or colouring matters,and i t Should not have any disagreeable odour .
On rubbing on blue litmus-paper no reddening should occur .D is t i l led woo l-grease i s a product ‘obtained by distilling woolfat with the aid of steam . The lighter portions
,
“ olein,
” separatedby cooling
,are used for lubricating wool
,and the more solid fractions
stearine,
” in the manufacture of soap and candles . I t has also beenused to adulterate tallow . According to Lewkowitsch (J . S oc. Chem.
Ind .,1 892 , I I
,but a small proportion of the esters originally
present in the wool- fat are found in the distilled product,the greater
portion being decomposed into fatty acids and hydrocarbons .C , 5H
3 ICOOC 1 6H3 3
= C 1 5H
3 ICOOH+ C 1 6H3m
Cetyl palmitate . Palm itic acid . Cetene .Smi th (Ann . Ch im. Phys .
,1 842 , 6 , The fatty acids
,es
pecially the higher members of the series , are further dissociated intohydrocarbons and acids of lower molecular weight . Hydrocarbons are

504 WOOL-FAT . WOOL-GREASE . SUINT . DEGRAS (U . S .)
oil constantly stirred to prevent bumping . A thermometer serves wellas a stirring rod .
The unsaponifiable oil is freed from cholesterol and other higheralcohols by boiling for an hour with 1 00 c .c . of acetic anhydride in aflask provided with a return flow condenser
,and heated over a sand
bath . Water i s added,and the solution transferred to a separatory
funnel where it i s washed with water and alcohol until the upper layeris clear and no odor of acetic acid is perceptible . The cholesterol andhigher alcohols are dissolved by the acetic anhydride
,leaving the
hydrocarbons .After submi tting the oils to this process
,an estimation of their
saponification number is made , and if more than c .c . of alcoholicpotash is used up
,the treatment with alcoholic potash and acetic
anhydride repeated .
The examination of distilled wool-grease IS conducted upon the samegeneral lines indicated in the case of wool-fat . Lewkowitsch obtainedthe following results from a sample obtained by the distillation of recovered grease , the analysis of which is stated on page 502 .
Combined fatty acids 7 . 02%Unsaponifiable 38 80%
For other analyses of distilled wool-grease see page 409 .
Alcoholic potassium hydroxide should be used in the determinationof the molecular weight .The fatty acids may also be determ ined with suffi cient accuracy bythe usual gravimetric method .
SOD OIL. DEGRAS . FRENCH DEGRAS (U . S .)
Degras is the waste fat obtained in the chamoising process andlargely used in dressing leather . The chamoising process consist s essentially in oiling the suitably prepared skins with whale or cod oil(i . e .
,the lower grades of codliver oil), stamping them in the stocks ,
and placing them in heaps , so that a fermentative change attendedwith development of heat is brought about . The process is completewhen the skins have acquired the usual yellow colour of Chamois leather .Under these conditions , oxidation of the oil takes place , and a portion ofi t combines with the skin
,from which it cannot be removed by the usual
solvents . About an equal quantity of uncombined oil i s also mechani

S OD OIL . DEGRAS . FRENCH DEGRAS (U . s .) 505
cally enclosed in the skin . After being well scraped with a bluntknife
,by which much of the excess of oil i s removed
,the Skins are
washed with lye and the emulsion treated with acid ; the fatty ’
matter
which rises to the surface is added to the oil already obtained by scraping . The product so obtained constitutes the so—called “
sod oil .”This is the method largely used in Germany and England . The following process employed in France is also used in England to a considerable extent : The treatment by oiling , stocking , and fermentingis carried out for a shorter period , SO that a larger proportion of uncombined oil remains in the skins . This i s removed by wringing orhydraulic pressing , and constitutes the “moellon ” or “ degras ” ofcommerce . The remaining uncombined oil i s removed by washingwith lye and treatment with acid
,and is usually added to the product .
The moellon of commerce is said to be invariably mixed with untreatedoils . Moellon contains less fibre
,mineral matter
,and water than
sod oil .‘Jean found that degras (moellon) contains from 1 0 to 20% of water,
and that the property of forming an emulsion with water dependsupon the presence of an oxidation product of the oil formed duringthe chamoising process . He describes it as a “ resinous substance
,
insoluble in petroleum Spirit,but soluble in alcohol and ether . It i s
saponifiable , but , unlike ordinary fat , the soap formed is not precipitated from alkaline solution by the addition of salt . The m .
p . was stated to be 65 to S imand has given it the name de’ grasformer . According to him i t i s insoluble in petroleum spirit
,benzene
,
and almost insoluble in ether . It is soluble in alkaline solutions , fromwhich it is precipitated by the addition of acid . It was also found inall animal and marine oils . Fahrion regards it as a mixture of hydroxyfatty acids and anhydrides . It i s an oxidation product , and experi ence has shown that those marine animal oils which absorb oxygenreadily are the most suitable for the preparation of degras . Fahrion
found an iodine-absorption of in degras-former . Ruhsamfound in sample No . 1 on page 5 1 0 . According to Fahrion ,
degras-former contains no nitrogen,that found by Eitner being due to
impurities .Degras-former is said not to exist in the free state in degras , butforms a part of the saponifiable matter which is readily soluble inpetroleum spirit
,in which the degras-former itself is insoluble .
B aron (Rev. Chim. Ind. ,1 89 7 , 8 , 2 25) prepares an artificial degras as

506 WOOL-FAT . WOOL-GREASE . SUINT . DEGRAS (U . s .)
follows : 1 000 kilos of neutral wool-fat (extracted by petroleum spiri t)are placed in a tinned steel vessel with 5000 kilos of cod or whale oil .The liquid is heated by a steam coil
,agitated for 3 hours , then allowed
to rest and cool for the same period,and the water withdrawn . The
water is again heated to I 50 kilos of hydrogen peroxide and 450kilos of water added
,and the whole agitated for 5 hours at a pressure
of 2 atmospheres . The resulting product is said to form an excellentmoellon , having a yellow colour and being easily emulsified and ab
sorbed by. the skins . It i s important that the wool-grease be free fromsulphuric acid
,lest thi s should di ssolve traces of iron
,and so cause
darkening of the leather .Exam ina t ion Of Degras .
—Water i s ascertained , according toS imand , by weighing 2 5 grm . of the sample in a tared porcelain basinprovided with a short thermometer as stirrer
,adding 50 to 1 00 grm . of
blubber or other oil previously dried by heating to heating themixture to the same temperature , and determining the loss in weight .Ruhsam makes the estimation by heating 2 to 3 grm . of the samplein a weighed platinum crucible until an empyreumatic odor indicatesthe complete dehydration of the fat .French degras usually contains from 1 0 to 20% of water ; sod oil
may contain as much asFree Ac id .
—Mineral acids may be detected as described on page9 . The amount i s estimated by boiling a weighed quanti ty of thesample with water and separating the watery solution
,which will con
tain the mineral acids as well as any soluble fatty acids ; the determination of the former is made by adding methyl-orange and titratingwith standard alkali until the point of neutrality i s reached . Thesoluble fatty acids are then determined by adding phenolphthalei n andtitrating a second time .Free fa t ty ac ids may be estimated in the residue insoluble in
water by di ssolving in alcohol and titrating as usual . They areusually calculated to oleic acid .
Ash .—This is estimated in the usual manner . I t should be tested
for iron . According to S imand , even as low a proportion asof ferric oxide has a distinctly injurious effect .The ash of mo ‘
ellon i s usually less than that of sod oil mayamount to severalFragmen ts of h ide may be estimated in the residue left from thesolution in petroleum spirit , which is dried , weighed , and incinerated .

508 WOOL-FAT. WOOL-GREASE . SUINT . DEGRAS (U . s .)
stance separates in flocks,which on boiling unite and adhere to the
side of the flask . The liquid is cooled,shaken out with ether
,the
ethereal solution evaporated,and the residue dried and weighed .
Jean considers that a Sp . gr . of the oil extracted from degras of lessthan indicates the presence of foreign fats
,e . g .
,wool-fat
,oleic
acid,and tallow . The sp . gr . of the oil from degras made from fish
and whale Oil i s given as to The presence of tallow isalso indicated by the higher m . p . of the fatty acids . In the examination of artificial de’ gras
,S imand takes into consideration
,in addition
to the ash and water,the following points :
1 . The degras-former,which may be derived from a small quantity
of admixed true degras or from the '
o ils .
2 . The wool-fat .3
3 . Hydrocarbons (vaseline) .
4 . Colophony .
To determine the degras-former, S imand proceeds as with genuine
degras,but substitutesEther for the petroleum spiri t
,since the wool-fat
acids are di ssolved by the former in the cold .
The estimation of the amount of wool-fat i s as yet an unsolvedproblem . The detection of cholesterol would not
,in i tself
,suffi ce
,as
i t i s a natural constituent of the fish oils used in the manufacture ofdegras . Lewkowitsch points out that by the ordinary methods ofsaponification, a portion of the wool-fat would probably be found inthe unsaponifiable portion , and that by again saponifying under pressure a definite saponification value would point to the presence ofwool-wax .
B enedikt (Anal . d . F ette n . Wachsarten) states that by es timatingthe amount of cholesteryl acetate (see page 49 1 ) a very rough approximation of the amount of wool-fat may be obtained . Wool—fat furnishes percentages of cholesteryl acetate varying from toResin may b e estimated as on page 79 , and hydrocarbons as onpage 76 .
Jean gives the following example of examinations of degras
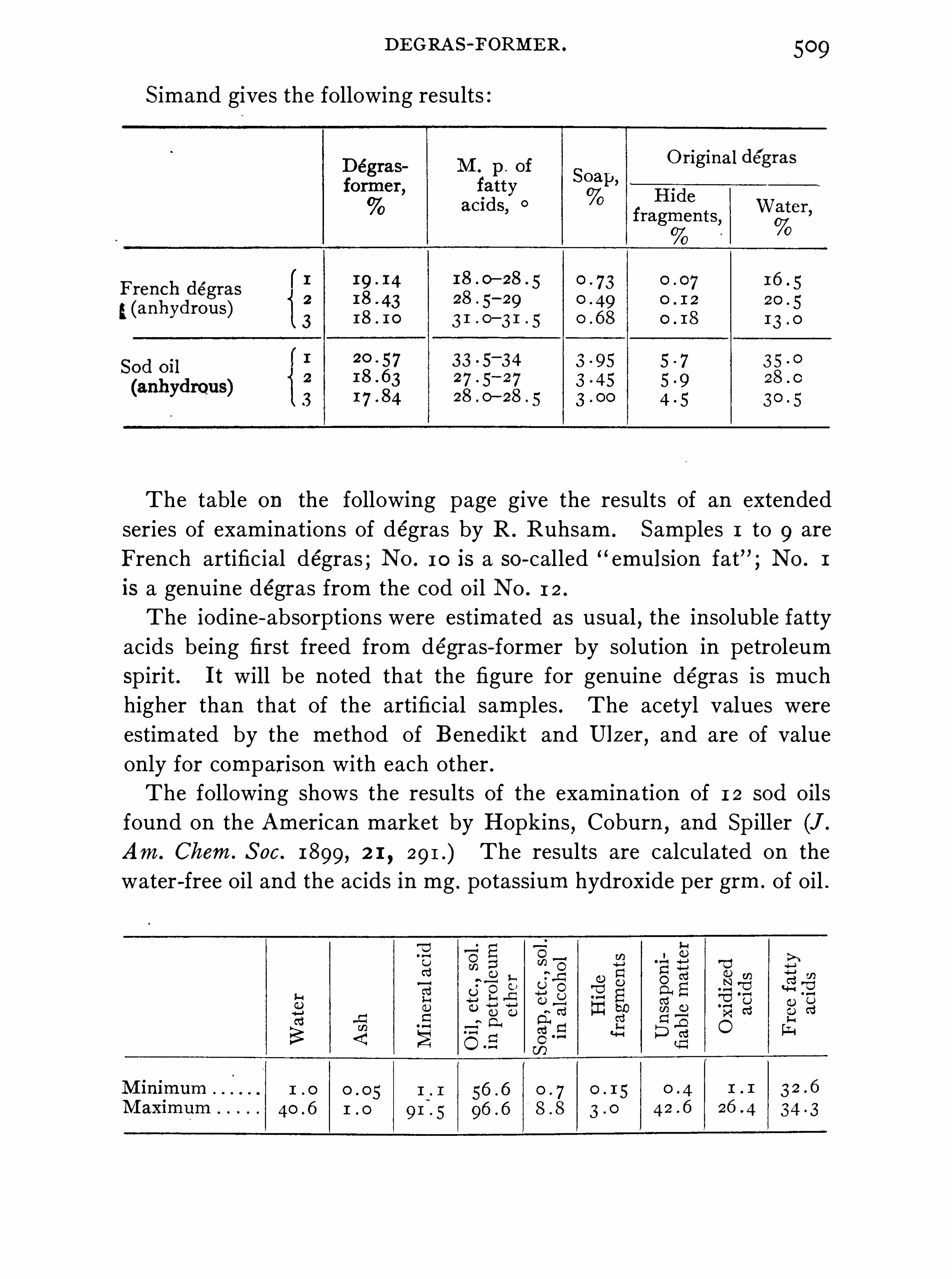
DEGRAS -FORMER .
S imand gives the following results
O riginal dégras
French degrasg(anhydrous)
Sod o i l
The table on the following page give the results of an extendedseries of examinations of degras by R . Ruhsam . Samples 1 to 9 areFrench artificial degras ; No . 1 0 is a so-called “ emulsion fat” ; No . 1
is a genuine degras from the cod oil No . 1 2 .
The iodine-absorptions were estimated as usual,the insoluble fatty
acids being first freed from degras-former by solution in petroleumSpirit . I t will be noted that the figure for genuine degras is muchhigher than that of the artificial samples . The acetyl values wereestimated by the method of B enedikt and Ulzer
,and are of value
only for comparison with each other .The following Shows the results of the examination of 1 2 sod oilsfound on the American market by Hopkins
,Coburn
,and Spiller (J .
Am. Chem. S oc. 1 899 , 2 I , The results are calculated on thewater-free oil and the acids in mg . potassium hydroxide per grm . of oil .
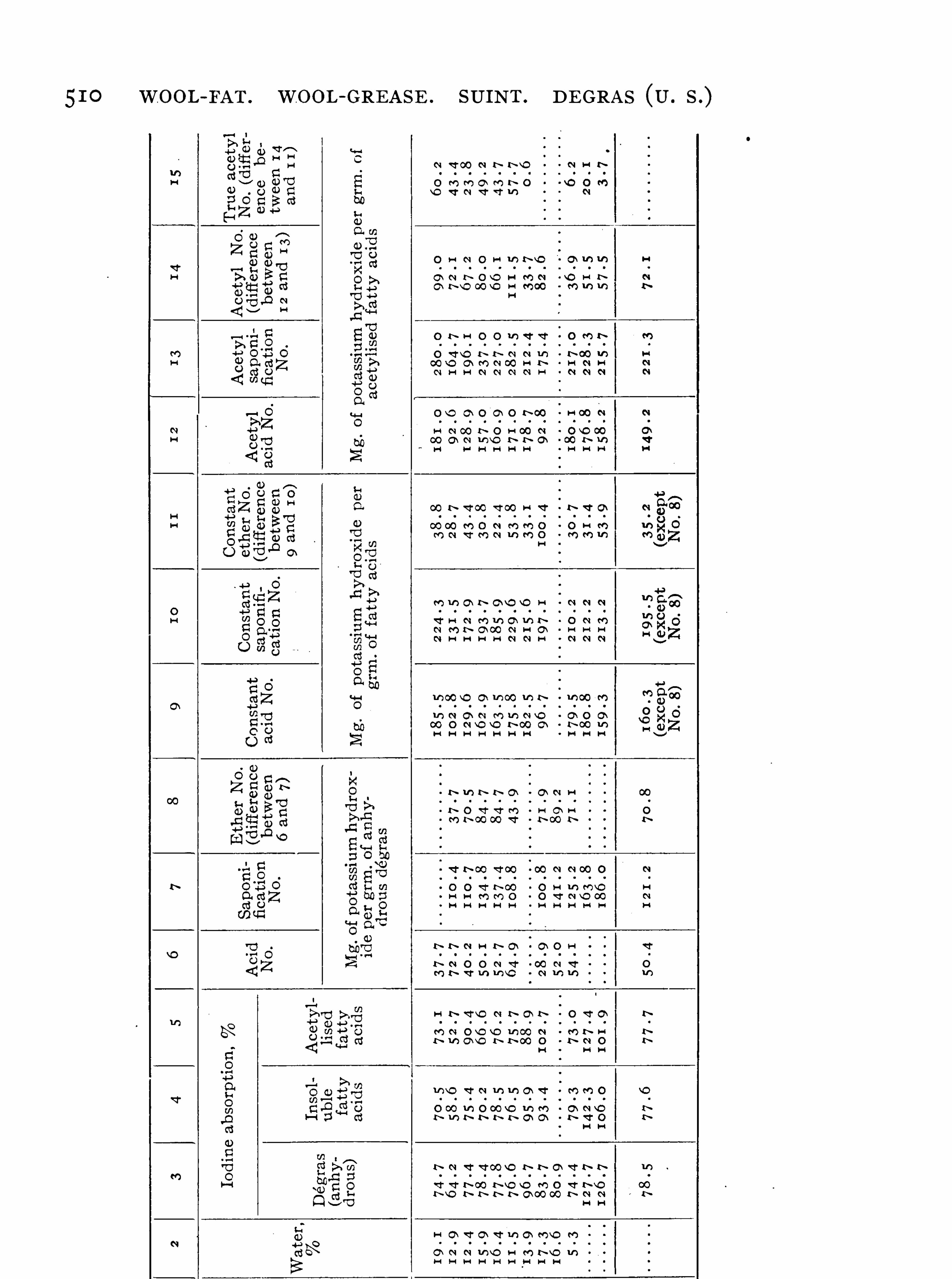
WOOL-FAT . WOOL-GREASE . SUINT . DEGRAS (U . s .)
N V OO N N N O
0 90 00 0 00 5 0O V N V ‘T V )

WOOL-FAT . WOOL-GREASE . SUINT. DEGRAS (U . s .)
are employed . In Great Britain the rating is based upon the natureof the oil , the proportion of unsaponifiable matter , and the flash test .The lowest rate is charged when an olive oil
,lard oil
,or
"‘oleine
i s used containing not more than 1 0% of unsaponifiable matter , or afish or manufactured oil containing not more than 30% of unsaponifiable matter and having a flash-point not under The highest
rate is charged in the presence of drying oils orof more than 50% of unsaponifiable matter .The “ flash-point ” of an oil intended forthis purpose may be ascertianed easily byplacing 50 c .c . in a porcelain dish or crucible ,in a sand-bath
,heating with constant churning
,
and noting the temperature at which a flashacross the surface i s produced when a smallflame is brought near .A satisfactory test of the liabili ty of an oilto inflame spontaneously may be made byMackey ’s Cloth—oil Tester ” (J . S oc . Chem.
Ind ., 1 896 , also 26 , 1 8
This consists of a water-j acketed metal oveno f the following dimensions : Outside
,8 in .
high and 6 in . diam . ; inside , 7 in . high and 4 in .
diam . The tubesAandB are 1 / 2 in . internaldiam . and 6 in . long measured from the lid .
The depth inside with the lid on is 6 in .
The lid is packed with asbestos wool,and the
tubes serve to maintain a current of air . Care should be taken that thesteam from the water jacket is neither drawn down B nor warms A .
C is a cylinder of wire gauze (24 meshes to the in .) 5 by 6 in . forminga roll 6 in . long and 1 in . diameter . In i t is placed 7 grm . ofordinary bleached cotton-wadding
,previously impregnated with 1 4 grm .
of the sample occupying the upper 4 1 / 2 in . of the cylinder .The water being brought to the b . p .
,a thermometer is inserted
in the oiled cotton contained in the gauze cylinder,which is then
placed in the bath,the thermometer being allowed to protrude through
a cork in the opening shown in the l id . The water is kept boilingand the temperature read at the end of an hour . An oil attaining atemperature of 1 00
° or over at the end of this time is to be re
garded as dangerous . The following are the results of experiments
FIG . 1 4.

CLOTH OILS .
Oi l used ximuni
y acid
Chemical examination of cloth oils is by application of principlesand methods already given . An estimation of unsaponifiable matteris important
,and if hydrocarbons are present the flash-point should be
ascertained . The iodine number will aid in the detection of drying oi ls .
The examination of commercial oleic acid is given in detail on page406 ; i t i s to be especially tested for unsaponifiable matter and for linseed-oil acids . Resin Should be looked for in the fatty acids separatedfrom the saponifiable portion as described on page 7 7 . See also under“Wool-fat ” and “Distilled Wool-grease .” Free mineral acid , whichis especially apt to be present in commercial oleic acid , is obj ectionableon account of its corrosive action on card-teeth .
In the case of “ emulsion oils ” the fatty matter may be separatedby treatment with acid and examined as above . Gelatin or gummymatters used in preparing the emulsion may be separated by additionof alcohol .
V0]. 11 .
—33


5 1 6
Codliver oi l, 73 , 2 1 3Cohesion figures, 4 1Coldwater soap, 420Co lour tests of o i ls
, 40Co lza oi l,69 , 1 2 2Co rn o i l, 70 , 1 39Cotton o i l stearin , 1 34
seed,stearin
,1 34
o i l, 70 ,
1 3 2
Crampfish o i l,223Cress-seed oi l, 70, 1 39Criti cal temperature , 63Croton oi l , 7 1 , 1 7 2Curcas oi l
, 7 1 , 1 73
DEGRAS-FORMER, 505
French, 502true
, 504“ T ' U 8 0 495Demargarinated o i ls , 1 07Digitonin—cholesteride , 48 rDoeglic acid
, 3 73Doeglin oi l, 73 , 240
Dogfish o i l,223Do lph in oi l
,242
Drying Oi ls : 3 5 , 65 : 7O ~ 3 43
EARTHNUT oi l, 69, 9 1Egg oi l
,202
Elaidic aci d, 405Ela idin reaction
, 39Elaine
, 5 1 1
E lectric conduct of o ils, 45
Eleomargaric acid, 3 74Emulsion o ils, 5 1 1Ergostero l, 488Eruca sativa seed oil
,69 , 1 2 r
Erucic acid, 3 73
Expansion coeffi c ients , 49
FAT , beef, 7 2butter, 7 2 , 279horse , 7 2, 206
mutton, 7 2 ,
207Tangkawang, 1 76woo l
, 495Fats , animal, 66, 7 2
classification of,64
composition of, 7
estimation of, 4
extraction of, 3identification of
,84purification of
, 3so lubilities of
,6 1
vegetable,66
, 7 2
Ficocerylic acid, 3 7 2
Fi rseed o i l, 70, 1 5 1
Fish o i ls,2 26
liver o i ls , 2 2 1
INDEX .
GALAM butter , 7 1 , 1 87German sesame o i l
, 70, 1 3 2
Ghedda wax,268
S ingili o i l, 70, 1 41
G lycerides , 8m ixed
,8
G lycerin , 447candle , 466
crude, 466
di sti llation , 467d istilled
, 47 1
dynamite , 47 1pure. 467
raw, 466
soap -lye , 467Glycero l, 447
detection of, 453
esters , 45 2estimation of
, 455G lycerOphosphori c acid , 45 2Goa butter
, 7 1 , 1 82
Grapeseed oi l, 7 1 , 1 75Grease
,brown
, 498
recovered , 498woo l, 498disti lled , 502Yorksh ire
, 498
Groundnut o i l, 69. 9 1
HADDOCK oi l,22 1
Hake Oi l,22 1
Halphen’s test . 1 3 5
Hazelnut Oi l, 69 , 1 05Heat of combustion, 45Hehner value , 20Hempseed oi l
, 70, 1 50
Herring o i l, 2 26
Ho i o i l,22 1
Homo-elestranol, 488
Horse fat , 7 2 , 206
Hubl ’s method , 30
Hydrocarbons in o ils, 79
Hydrocaratol, 485Hydrolysis , 1 0Hyenic acid
, 3 7 2
Hypoge ic acid, 3 73
I lli pe butter, 1 76Indian beeswax , 268
mustard oi l,69 , 1 2 1
Inso luble bromides , 28Iodine absorpti on,
26
value,29
I socetic acid, 3 7 2
I solino leni c acid, 3 74
Isoole ic acid, 3 73
I soricino leic acid,1 60
, 3 74
JAMBA o i l,69 , 1 30
Japanese wood o i l, 70, 1 54

Japan fish oi l, 73 , 2 24wax
, 7 2 , 1 9 1
Jeco leic acid, 3 73
KATCHUNG oi l, 9 1
Koettstorfer method , 1 4Kokum butter, 7 1 , 1 8 2LACTINE
,1 88
La llemanti a oi l, 70, 1 5 1
Lano lin , 502
Lanoceric acid , 496Lanopalmi ti c acid
, 496
Lard, 3 1 7ofl. 7 2 , 1 97group
,66
. 7 2
Laure l oi l, 7 1 , 1 82
Lauric acid , 3 7 2Laureo l
,1 88
Leflmann-Beam method,
L ignoceric ac id, 93 , 3 7 2
Linseed cake, 3 26
o i l. 70. 3 23bo i led, 3 53
L ino le ic acid , 3 50 , 3 74Lino len ic acid , 3 5 2 , 3 74Lithographi c varnishes
, 3 56
Livache’s method
, 3 6
Lupeol, 488MACASSAR o il
, 7 2, 1 94Mad ia o il
, 70, 1 39Mafura tallow
, 7 1 , 1 8 2
Mahua butter, 1 76Ma ize Oil
, 70, 1 39Malabar tallow
, 7 1 , 1 87Mangosteen oi l
, 7 1 , 1 82Margarine
, 3 1 3Marine an imal o i l
,6 7 , 73
soap, 4 1 8
Maumené’s test
, 58Me liss ic aci d
, 3 7 2Melon o i l
,230
Melting po ints, 5 1
Menhaden o il, 73 , 224
Moéllen, 505
Mohwah butter, 1 76Mowrah seed o il
,1 76
Mustard o i l,black
,69 , 1 20
detection of, 3 1 6
Whi te,69 , 1 20
Mutton fat, 7 2 , 207
Myric in ,244
Myricyl alcohol, 244Myristic ac id
, 3 7 2
Myrtle wax, 7 2, 1 94
N IGER o il, 70, 1 5 1seed oi l
, 70, 1 5 1
INDEX .
Nitroglycerin , 45 2uco line
,1 88
utmeg butter, 7 1 , 1 8 2Nut o i l
, 70, 1 57
OCTOIC acid, 3 5
O il, Alizarin, 1 67 lapricot kernel
,69 , 1 05
a lmond,69 , 1 02
arach is,69 , 9 1bagasses,1 1 9base
, 38, 36 1beech, 70, 1 3 1
nut, 70, 1 3 1benne
,1 41blown
, 36 1bottlenose , 73 , 240B razil nut , 70, 1 3 2brusmer
,22 1
came line, 70 , 1 3 2
candlenut, 70, 1 48
castor, 7 1 , 1 59
soluble, 36 1
cedar nut , 70, 1 48
cloth , 5 1 1
coa lfish,2 2 1
coconut, 7 2 , 1 87
codliver, 73 , 2 1 3
co lza,69 , 1 22
corn, 70, 1 39
cottonseed, 70, 1 3 2
crampfish , 2 23cress seed
, 70, 1 39curcas
, 7 1 , 1 73doegling
, 73 , 240
dogfish ,223
do lph in,242
earthnut, 69 , 9 1eruca sativa , 69 , 1 2 1gingili
, 70, 1 4 1grapeseed, 7 1 , 1 7 5groundnut, 69 , 9 1
haddock , 22 1hake
,22 1
haze lnut, 69 , 1 05he ll
,1 06
hempseed, 70, 1 50herring, 226ho i
,22 1
Ind ian mustard , 69 , 1 2 1Jamba , 69 , 1 30Japan fi sh , 73 , 224lallemant ia , 70, 1 5 1
lard , 7 2 , 1 9 7laure l 7 1 , 1 8 2
ling, 2 2 1linse ed, 70, 3 23macassar, 7 2, 1 94

5 1 8
O i l,madia
, 70, 1 39ma ize
, 70, 1 39mangosteen
, 7 1 , 1 8 2
melon,230
menhaden, 73 , 2 24
mowrah seed,1 76
mustard seed,black
,69 , 1 20
wh ite,69 , 1 20
neatsfoot, 7 2 , 200
n iger, 70, 1 5 1
seed, 70, 1 5 1
nut, 70, 1 5 7
olive,69 , 1 06kerne l, 1 1 8
oxid ised, 38palm
, 7 1 , 1 8 2
kerne l, 7 2 , 1 95
“ fi nut, 7 2 ; I QSpeach kernel
,69 , 1 05peanut
,69 , 9 1
pinenut , 70, 1 5 1plum-kerne l,69 , 1 05
poppy, 7o , 1 5 2
seed, 70, 1 5 2porpo ise , 73 , 230pumpkin—seed, 70 ,
1 4 1
radish seed,69 , 1 2 1
rape,69 , 1 2 2
rav ison ,1 3 1
red, 5 1 1
safflower, 70, 1 5 7sardine
, 73 , 2 24seal
, 73 , 226
sesame, 70, 1 4 1
German, 70, 13 2
shark-liver, 73 , 22 2skate-liver, 220, 2 2 1sod
, 504soja bean , 70, 1 46
sperm , 73 , 23°
sturgeon, 2 26sunfi sh , 223sunflower, 70, 1 54tallow , 7 2 , 202
tea seed , 69 , 1 1 9teel, 70, 1 4 1theobroma
, 7 1 , 1 76tra in, 73 , 227tung, 70, 1 54
Turkey-red , 1 6 7walnut, 70, I 5 7whale
, 73 , 2 27wheat, 70 ,
1 47Wh itefish , 2 26wh iting ,
22 1
wood,Ch inese
, 70, 1 54Japanese , 70, 1 54
O i ls,absorption spectra of
, 42base , 36 1
INDEX .
O i ls , blown , 36 1
cloth , 5 11cohes ion-figures of, 4 1co lour tests of 40compos ition
_
oi, 7
demargarinated, 1 07drymg , 3 5 , 65 , 343electric conduct of
, 45emulsion
, 5 1 1
estimation of, 4
expansion of, 49
extraction of, 3
oxid ised , 36 1purification of,6
heat of combusti on of, 45identification of
,84
marine an imal,67 , 73
me lting po int of, 5 1non—drying, 85optical activity of
, 45oxidation of
, 3 5refractive power of
, 42 , 44
sem i—dy ing, 85spec ific gravity of
, 46 , 86
so lid ifying po int o f, 5 1 , 5 5so lubilities of, 6 1temperature tests for, 58viscos ity of
, 46
O leasterol, 488
O leates , 41 0O le ic ac id , 3 73 , 402
esters , 4 1 2O le in , 502tallow,
202
O le ine , 5 1 1coconut, 1 88 , 1 90
O le ins , an imal,66
, 7 2vegetable , 64, 69O leomargarine , 3 1 3Oleorefractometer, 43O lestranol, 488
O live o i l,69 , 1 06kerne l o il, 1 1 8
o il group , 64Onocerol, 488
O xidation of o ils , 3 5Oxidi zed oi l
, 38
PALMITATES, 39 7Pa lm itic ac id
, 3 7 2, 395esters , 397
Palm kernel O i l, 7 2 , 1 95nut o i l, 7 2 , 1 95o i l, 7 1 , 1 8 2
detection of, 3 1 0
Paraphytostero l, 488
Peach kernel o i l, 69 , 1 05P eanut o i l, 69 , 9 1Phulwara butter, 7 1 , 1 87

520
Wax,Ghedda, 268
Indian,268
Japan, 7 2 , 1 9 1
myrtle , 7 2 , 1 94woo l, 496Waxes
,compos ition of
, 7liqu id
,67. 73 , 23 2
so lid,67 , 73 , 23 2Whale foots,228
o i l, 73 , 2 27




















