
CKIP-1 regulates macrophage proliferation by inhibiting TRAF6-mediated Akt activation
Upload
independentCategory
view
2download
0
The Prostate 68:965 ^ 974 (2008)
AktDown-Modulation InducesApoptosis ofHumanProstateCancerCells and SynergizesWith EGFR
TyrosineKinase Inhibitors
Claudio Festuccia,1* Giovanni Luca Gravina,2 Paola Muzi,1 DaniloMillimaggi,1
Vincenza Dolo,1 Carlo Vicentini,2 and Mauro Bologna3
1Departmentsof ExperimentalMedicine,Universityof L’Aquila, L’Aquila, Italy2Departmentsof Surgery,Universityof L’Aquila, L’Aquila, Italy
3Departmentsof Basicand Applied Biology,Universityof L’Aquila, L’Aquila, Italy
BACKGROUND. PTEN is a well-characterized tumor suppressor that negatively regulatescell growth and survival through the modulation of PI3K/Akt pathway.METHODS. In this paper, we investigated the effects of an PI3K/Akt inhibitor, perifosine, inhuman prostate cancer (PCa) cells analyzing cell proliferation, apoptosis, and the synergy withEGFR inhibitors.RESULTS. Clinically achievable concentrations of perifosine, as well as Akt gene knockdown,induced a G0/G1 arrest and apoptosis in PTEN defective PCa cells. Although PTENintroduction was able to restore the control of Akt activity and to reduce cell proliferation,the manipulation of PTEN gene was not able alone to influence apoptosis. Perifosine inducedapoptotic program also in PTEN positive cells when Akt activity was augmented by EGFsuggesting thepossibility that this drug couldbeused in combinationwithEGFR inhibitors. Thecombination treatment between erlotinib and pharmacological or molecular Akt knockdown,indeed, showed synergistic effects. This is the first demonstration that a pharmacologicalcompound against Akt activity can restore the efficacy against EGFR inhibitors in PCa and hasimportant therapeutic fallout since EGFR inhibitors have demonstrated very low effectivenessin PCa patients.CONCLUSIONS. Taken together our data have an important clinical relevance in thetreatment of advanced prostate tumors. However, further studies in the setting of combinationtherapies in advanced PCas are necessary. Prostate 68: 965–974, 2008. # 2008 Wiley-Liss, Inc.
KEY WORDS: prostate cancer; drug resistance; perifosine; PTEN; PI3K; Akt
INTRODUCTION
The majority of prostate cancer (PCa) are androgendependent and respond to androgen deprivationtherapies. Unfortunately,median time to the formationof recurrent tumors is only 24–36 months with relapseoccurring in a great majority of treated patients (forreview see Miyamoto et al. [1]). Cells of the recurrenttumors proliferate in the absence of androgen ormay be hypersensitive to low concentration of andro-gen. However, few treatments exist for this stageof disease. It is therefore important to develop alter-native therapeutic agents with improved efficacy andtolerability.
An important mechanism for increased cell survivalin advanced PCa is mediated by the phosphatidylino-sitol-3-kinase (PI3-K)/Akt (protein kinase B) signalingpathway. Phosphatase and tensin homolog deleted onchromosome 10 gene (PTEN) is a lipid phosphatase
*Correspondence to: Dr. Claudio Festuccia, Dipartimento di Medic-ina Sperimentale, Cattedra di Patologia Generale, Universitadell’Aquila, Via Vetoio, Coppito 2, 67100 L’Aquila, Italy.E-mail: [email protected] 5 December 2007; Accepted 5 February 2008DOI 10.1002/pros.20757Published online 24 March 2008 in Wiley InterScience(www.interscience.wiley.com).
� 2008 Wiley-Liss, Inc.

that negatively regulates PI3K pathway. This tumorsuppressor gene is inactivated in about 30% of primaryPCas [2,3] and in about 60% of metastatic prostatetumors [2]. We previously demonstrated that thevivo androgen ablation therapy can reinforce thePI3K/Akt pathway through several mechanismsincluding the increased PTEN loss and EGFR/Her2increased expression/activity [4]. Activated Akt, inturn, mediates anti-apoptotic signaling through theinactivation of a multitude of downstream targetsinvolved in apoptosis regulation. Blockade of thePI3K/Akt pathway leads to apoptosis of PTEN-deficient cancer cells [5]. Consequently, PI3K/Aktpathway may be a major therapeutic target [6,7] fortreatment of cancer and several novel therapies are inlate-stage phase II and phase III clinical trials [8–12].However, the manner through which PI3K pathwayblockade mediates apoptosis is currently scarcelyknown. In mammals, two major pathways can initiateprogrammed cell death: the extrinsic and intrinsicdeath pathways. The extrinsic pathway is triggered byextracellular ligands that induce oligomerization ofdeath receptors, such as Fas or other members of thetumornecrosis factor receptor superfamily, resulting inactivation of a caspase cascade leading to apoptosis.The intrinsic pathway, on the other hand, is triggered inresponse to a variety of apoptotic stimuli that inducedamagewith loss ofmitochondrialmembrane integrityand release of proapoptotic molecules.
Nevertheless, the results of a clinical trial performedon advanced prostate tumors are in disagreement withpre-clinical data indicating that this drug possessedvery little effectiveness as single therapeutic agent [11].Its clinical efficacy seems to be complicated by fatigueand gastrointestinal toxicity. However, Akt inhibitioncan represent a good therapeutical target also incombination with chemotherapic agents or anti-targetagents.
In this study, we examined the effects of a PI3K/Aktsignal pathways inhibitor, perifosine, on PCa cellshaving different Akt activity levels due in part to thePTENgene silencing/deletion. The effects of perifosinehave been previously analyzed in the PC3 PCa cellline [13,14]. In these reports, perifosine showed anti-proliferative effects triggering caspase 9 and caspase3 activation. In this paper we demonstrate thatperifosine synergizes with the EGFR tyrosine kinaseinhibitor, erlotinib inducing apoptosis also in PTENpositive PCa cells. Therefore, although Akt is knownto be a survival factor in PTEN-deficient cells andperifosine has been widely used to inhibit Akt, ourwork can be of interest for itsmechanicistic informationas well as for its therapeutic potential since advancedPCas are commonly PTEN negative and irresponsiveagainst EGFR inhibition in human patients [15].
MATERIALSANDMETHODS
Reagents
Perifosinewas supplied fromÆternaZentarisGmbH(Frankfurt am Main, Germany). All the materials fortissue culture were purchased from Euroclone (Milan,Italy). Plasticware was obtained from Nunc (Roskilde,Denmark). EGF was purchased from ImmunoToolsGmbH (Friesoythe, Germany). Erlotinib was kindlyprovided by Dr. Ken Iwata from the Osi Pharma-ceuticals (Farmingdal, NY). Antibodies were purchasedfrom Santa Cruz (Santa Cruz, CA) unless otherwiseindicated. Antibodies against phosphorylated formsof EGFR, HER2, and ERK1/2 were obtained fromBiosource International (Biosource International, CA).Anantibody recognizingboth the 46kDa (pro-form)and35 kDa (cleaved from) of caspase 9 was purchasedfrom Epitomics (Burlingame, CA). Caspase 8 inhibitor:Z-IETD-FMK was purchased from Imgenex (SorrentoValley, CA). Fas antagonist, Kp6-7 was purchased fromMerck Chemicals, Ltd (Nottingham, UK). Nok-1 anti-FasL antibodywaspurchased fromAbcam (Cambridge,UK). ELISA kit for CD178/FasL was purchased fromDiaclone (Besancon Cedex, France). Akt/protein kinaseB (PKB) kinase activity was performed using a non-radioactive assay kit (StressXpress AKT/PKB Elisa kit)which was purchased from Stressgene Bioreagents(Victoria, BC, Canada). SiRNA against caspase 8 werepurchased from Santa Cruz. SiRNAs against PTEN,Akt1, Akt2, Akt3, and Bax were purchased from NewEnglandBiolabsGmbH (Frankfurt amMain, Germany).
Cell Lines
We used two androgen-sensitive PCa cell lines(LnCaP and 22rv1) and two androgen-insensitive PCacell lines (PC3 and DU145) that were obtained fromAmerican Tissue Culture Collection (ATCC, Rockville,MD) or the German Resource Centre for BiologicalMaterial (DSMZ, Braunschweig, Germany). PC3 cellstransfected with PTEN [16] were kindly provided byDr. LeRoith (National Institute of Health, Bethesda,MD). For transfection experiments cells from themonolayers were seeded 3� 105 cells per well in six-well plates, were washedwith serum free medium andtransfected, 5 hr, with siRNAs (100 nM) incorporatedinto Lipofectamine 2000 (Invitrogen-Life Technologies,Inc.) according to themanufacturer’s instructions. Cellsmaintained in 10% FCS complete medium wereretransfected after 48 hr with half the dose of siRNA.
GrowthAssays
Cellswere seededat adensity of 2� 104 cells perdishon 50 mm Petri dishes. Cells were left to attach andgrow in 5% FCS DMEM for 24 hr. After this time, cells
The Prostate
966 Festuccia et al.

were maintained in culture medium containing thedifferent drugs used at the recommended doses. Inorder to calculate the inhibitory concentrations at 50%(IC50) of drugs, 2,500 cells were cultured in 96 wellsplates for 24–96 hr. After adhesion (16 hr), cells weregrown in the different culture conditions (see above).After 48–96 hr the cellswere exposed for 4 hr to thyazolblue (MTS; Promega,Madison,WI) and the absorbanceof the converted dye was measured at the wavelengthof 490 nm using a BioRad multiscan plate reader(BioRad, Richmond, CA). Inhibition curves weredrawn bymeans of values obtained byODpercentagesversus control for each concentration. IC50 values werecalculated by the GraFit method (Erithacus SoftwareLimited, Staines, UK) considering the slopes of inhibi-tion curves obtained for each group of tests.
The results from the combination assays wereanalyzed using the isobologram combination indexmethod of Chou andTalalay [17]. All experimentsweredone in triplicate.
In order to evaluated the effective cell proliferationwe evaluated the uptake of 3H-tymidine as follow.Cell proliferation studies by measuring the uptakeof [3H]thymidine. Briefly, cells (1–2� 104/well) weregrown overnight in 24-well plates and exposed todifferent doses of drugs or DMEM (control). Aftertreatment (24–48 h), cells were pulsed with [3H]thymi-dine (1 mCi/well) for 4–6 h, fixed (5% trichloroaceticacid), and solubilized (0.5 MNaOH) before scintillationcounting. Experiments were performed in triplicates.
Preparation of Cell Lysates andWestern BlotAnalysis
Following treatments, cells were washed with coldPBS and immediately lysed with 1 ml lysis buffer(50mMHEPES, pH7.5, 150mMNaCl, 10%glycerol, 1%Triton X-100, 1 mM EDTA, 1 mM EGTA, 50 mM NaF,1 mM sodium orthovanadate, 30 mM p-nitrophenylphosphate, 10 mM sodium pyrophosphate, 1 mMphenylmethylsulfonyl fluoride, 10 mg/ml aprotininand10mg/ml leupeptin). Lysateswere electrophoresedin SDS-PAGE, and separated proteins transferred tonitrocellulose and probed with the appropriate anti-bodies using the conditions recommended by thesuppliers.
Cell Cycle andApoptosis Analysis
Cells (1� 106) were washed in PBS and fixedfor 30 min by the addition of 1 ml of 70% ethanol.After 30 min, the cells were pelleted by centrifugation(720 g; 5 min), and resuspended in 1 ml of DNAstaining solution (PBS containing 200mg/ml RNase A,20 mg/ml propidium iodide plus 0.1% Triton X-100)and stained by incubation at room temperature for
60 min. All cells were then measured on a FACScanflow cytometer (Becton Dickinson, UK) with an argonlaser at 488 nm for excitation and analyzed usingCell Quest software (Becton Dickinson). All theflow cytometric measurements were done using thesame instrument settings, and at least 10,000 cellswere measured in each sample. Apoptotic cells weredetected by a quantifiable peak in sub-G1 phasecorresponding to the red fluorescence light emittedby subdiploid nuclei of cells, and the results wereexpressed as the percentage of death by apoptosisinduced by a particular treatment. Apoptosis was alsoevaluated by DNA fragmentation using a Titer TACScolorimetric assay kit purchased by Trevigen, Inc.(Gaithersburg, MD) and expressed as percent of cellsthat undergo apoptotic death.
Statistics
Data are expressed as the mean� SEM of at leastthree independent experiments. Statistical analysiswas performed using an unpaired Student’s t-test.A P value <0.05 was considered as significant.
RESULTS
AKTInhibition InducesAnti-Proliferativeand Pro-Apoptotic Effects in PCaCell Lines
In examining Akt phosphorylation, we observedthat PC3 and LnCaP cells have basally elevated levelsof p-Akt, DU145 cells have detectable levels of p-Aktwhereas 22rv1 cells were null. To verified if Aktinhibition was able to reduce cell growth and to induceapoptosis we examined the anti-proliferative effects ofperifosine. We observed that PTEN defective PC3and LnCaP cells resulted highly sensitive to perifosine,with an IC50 value of�5 mM.Differently PTENpositiveDU145 and 22rv1 cell lines were partially resistantto perifosine. DU145 cells presented an IC50 valueof 15 mM whereas 20 mM perifosine decreased cellsurvival by <20% in 22rv1 (Table I). In Figure 1, weshow that 30 min of treatment with perifosine caused adose-dependent loss of Akt activity (Fig. 1A) in PC3cells. Further we observed marked reduction of3H-thymidine of PC3 cells uptake (Fig. 1B) measuredafter 72 hr of perifosine treatment. These effects weredose-dependent and were associated to the reducedcell viability and an increased apoptosis (Fig. 1C).Therefore,we evaluated if anAkt gene knockdownwasable to reduce Akt expression and activation as well asthe cell proliferation and apoptosis. For this experi-ments we used a mixture of three different siRNAsversus Akt1, Akt2, and Akt3. We observed that Aktknockdown determined a reduction of Akt production(Fig. 1A) associatedwith reduced cell proliferation and
The Prostate
Perifosine Effects in Prostate Cancer Cells 967
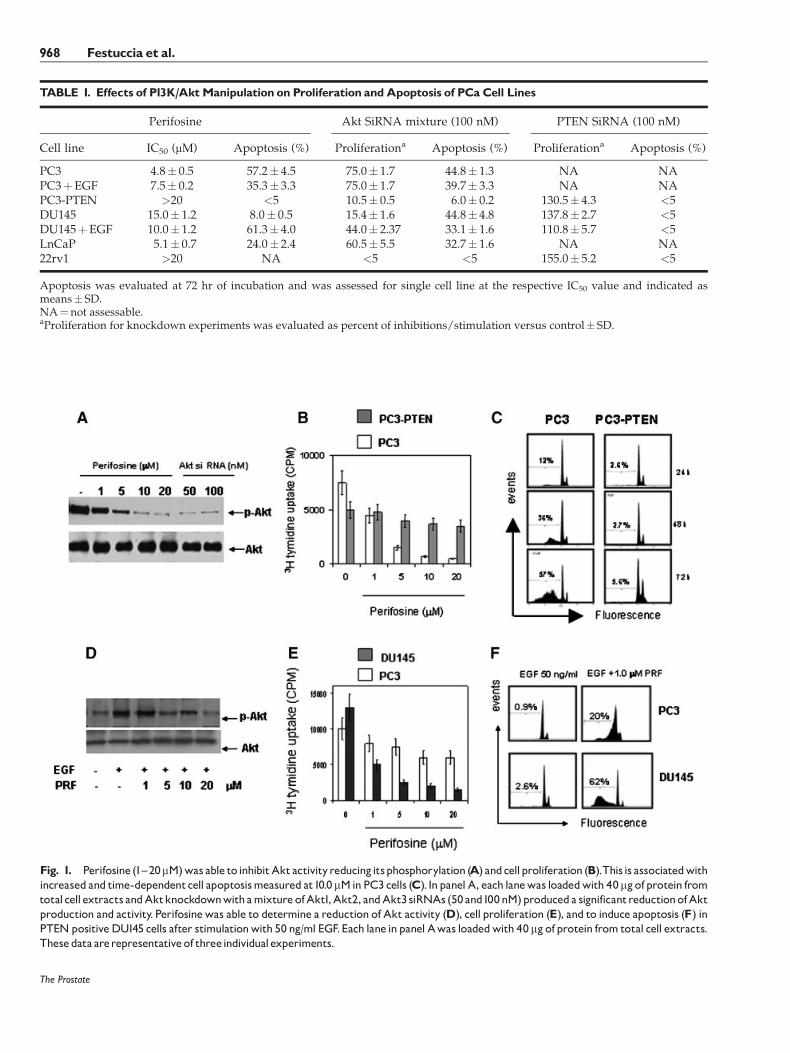
The Prostate
TABLE I. Effects of P13K/AktManipulation on Proliferation andApoptosis of PCaCell Lines
Perifosine Akt SiRNA mixture (100 nM) PTEN SiRNA (100 nM)
Cell line IC50 (mM) Apoptosis (%) Proliferationa Apoptosis (%) Proliferationa Apoptosis (%)
PC3 4.8� 0.5 57.2� 4.5 75.0� 1.7 44.8� 1.3 NA NAPC3þEGF 7.5� 0.2 35.3� 3.3 75.0� 1.7 39.7� 3.3 NA NAPC3-PTEN >20 <5 10.5� 0.5 6.0� 0.2 130.5� 4.3 <5DU145 15.0� 1.2 8.0� 0.5 15.4� 1.6 44.8� 4.8 137.8� 2.7 <5DU145þEGF 10.0� 1.2 61.3� 4.0 44.0� 2.37 33.1� 1.6 110.8� 5.7 <5LnCaP 5.1� 0.7 24.0� 2.4 60.5� 5.5 32.7� 1.6 NA NA22rv1 >20 NA <5 <5 155.0� 5.2 <5
Apoptosis was evaluated at 72 hr of incubation and was assessed for single cell line at the respective IC50 value and indicated asmeans� SD.NA¼not assessable.aProliferation for knockdown experiments was evaluated as percent of inhibitions/stimulation versus control� SD.
Fig. 1. Perifosine (1^20mM)was able to inhibitAktactivityreducingitsphosphorylation (A) andcellproliferation (B).This is associatedwithincreased and time-dependentcell apoptosismeasured at10.0mMinPC3 cells (C). InpanelA, each lanewas loadedwith 40mgofprotein fromtotalcell extracts andAktknockdownwithamixture ofAkt1,Akt2, andAkt3 siRNAs (50and100nM)produceda significantreductionofAktproduction and activity. Perifosinewas able to determine a reduction of Akt activity (D), cell proliferation (E), and to induce apoptosis (F) inPTENpositiveDU145 cells after stimulationwith 50 ng/ml EGF.Each lane inpanel Awas loadedwith 40mgofprotein from total cell extracts.Thesedata arerepresentativeof threeindividualexperiments.
968 Festuccia et al.

increased apoptosis in PTEN negative when comparedto PTENpositive cells (Table I). Conversely PTEN geneknockdown, performed in DU145 and 22rv1 cells, aswell as PTEN reintroduction in PC3 cells, was able tomodulate cell proliferation without interference withapoptosis (Table I). These data suggest that the PTENgenemanipulation is not able alone to trigger apoptoticevents whereas Akt inhibition is necessary to deter-mine both proliferation and apoptosis. A physiologicalmanner to trigger PI3/Akt pathway is the activationof EGFR by EGF. Thus, a treatment for 15 min with10 ng/ml EGF was able to induce a significantAkt activity in PTEN positive DU145 cells and thiswas blocked by perifosine pre-treatment in a dose-dependent manner (Fig. 1D). These effects wereassociated to reduced cell proliferation and viability(Fig. 1E) and to increased apoptosis (Fig. 1F). We notedthat the apoptosis induced by 1 mM perifosine wasthreefold higher in DU145 treated with 10 ng/mlEGF when compared to EGFR treated PC3 cells. Wecompared the effects of perifosine with another PI3Ksignaling pathways inhibitor, LY294002, and usingboth Akt (mixture of three Akt siRNAs) or PTEN geneknockdown (Fig. 2)with orwithout EGFaugmentation.
We observed that: (i) perifosine effects were similar toLY294002 and Akt knockdown both in term of cellgrowth inhibition and apoptosis; (ii) PTENknockdownin DU145 increased weakly cell proliferation ofDU145 cells without interference with apoptosis; and(iii) PTEN knockdown reduced the anti-proliferativeeffects of perifosine. In addition, we observed that EGFdecreased the effects of perifosine in PC3 cells suggest-ing that a further increment inAkt activitymediated byEGF in these cells seemed to be protective to perifosinetreatment.We further examined cell cycle alterations incells after exposure to perifosine. A G0/G1 cell arrest,associated to a time-dependent reduction of G2/Mphase, was observed after treatment with Perifosine inPC3 and LnCaP cells. In Figure 3A, we show the effectsof perifosine on PC3 cell cycle. The molecular analysisshows the involvement of cyclins, such as Cyclins B, D1and D2 and Cdks, such as Cdk4, 2 and 6, cyclin,regulating G1 phase. In addition Kip1/p27 and Cip1/p21WAF1 expression was also increased. Of interest,differently to LnCaP cells, increased expression ofp21WAF1 in PC3 cells occurred despite a lack offunctional p53 and this elevation (along with minimalincrease in p27KIP1) could lead to the loss of Cdkactivity/cell cycle arrest induced by this agent. Inthe following studies, we focused on revealing themechanisms underlying perifosine-induced apoptosis.
Effects of Perifosine onthe Expression of KeyMolecules Involved in the Regulation of Apoptosis
To further explore how perifosine inducesapoptosis, we next examined the effects of perifosineon the expression of several key genes involved ineither the extrinsic apoptotic pathway or the intrinsicapoptotic pathway. The proteins of the Bcl-2 familyplay critical roles in the regulation of apoptosis byfunctioning as promoters (e.g., Bax) or inhibitors (Bcl-2or Bcl-xL) of cell death process. In Figure 3B, we showthe molecular arrangements induced by perifosine inPC3 cells. Treatment of LNCaP and PC3 cells withperifosine resulted in a decreased expression of anti-apoptoticmolecules such as bcl-2, Bcl-Xl, and phospho-Bad with increased expression of pro-apoptoticproteins, such as Bax, Bak, and p14 (ARF), and releaseof mitochondrial proteins (cytochrome C, Smac/DIABLO). Control experiments were performed inPC3 cells treated with a mixture of Akt1, Akt2, andAkt3 siRNAs and were in agreement with perifosineresults (data not shown). Basally, DU145 cells were notinduced to modulate the expression of the abovementioned molecules whereas when treated with10 ng/ml EGF, DU145 cells responded to perifosineupmodulating the expression of pro-apoptotic mole-cules, except for Bax, being null for this element, and
The Prostate
Fig. 2. Analysis of apoptosis (in PC3 andDU145 cell lines treatedwith10mMofbothLY294002 andperifosine andwith100nMsiRNAofbothAktmixtureandPTEN).Theseevaluationswereperformedin thepresenceor absenceof50ng/mlEGF.
Perifosine Effects in Prostate Cancer Cells 969
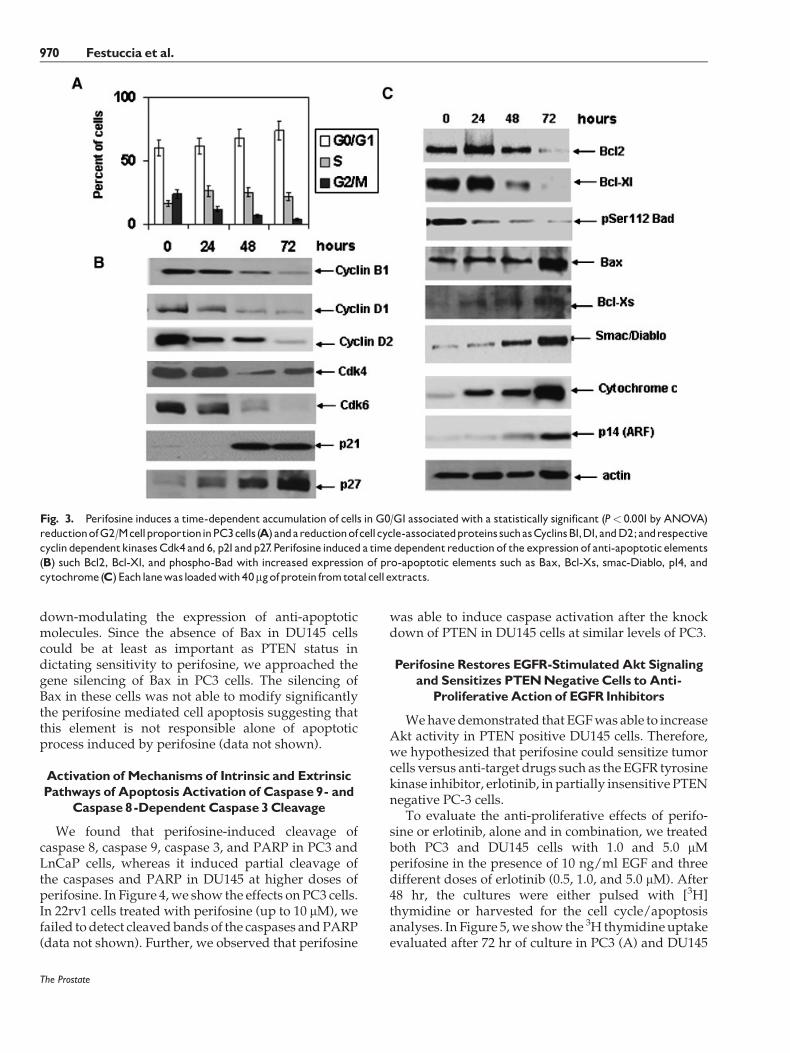
down-modulating the expression of anti-apoptoticmolecules. Since the absence of Bax in DU145 cellscould be at least as important as PTEN status indictating sensitivity to perifosine, we approached thegene silencing of Bax in PC3 cells. The silencing ofBax in these cells was not able to modify significantlythe perifosine mediated cell apoptosis suggesting thatthis element is not responsible alone of apoptoticprocess induced by perifosine (data not shown).
Activation ofMechanismsof Intrinsic and ExtrinsicPathways of Apoptosis Activation of Caspase 9- and
Caspase 8 -Dependent Caspase 3 Cleavage
We found that perifosine-induced cleavage ofcaspase 8, caspase 9, caspase 3, and PARP in PC3 andLnCaP cells, whereas it induced partial cleavage ofthe caspases and PARP in DU145 at higher doses ofperifosine. In Figure 4,we show the effects on PC3 cells.In 22rv1 cells treated with perifosine (up to 10 mM), wefailed to detect cleaved bands of the caspases andPARP(data not shown). Further, we observed that perifosine
was able to induce caspase activation after the knockdown of PTEN in DU145 cells at similar levels of PC3.
Perifosine Restores EGFR-StimulatedAkt Signalingand Sensitizes PTENNegativeCells toAnti-
ProliferativeAction of EGFRInhibitors
Wehavedemonstrated that EGFwas able to increaseAkt activity in PTEN positive DU145 cells. Therefore,we hypothesized that perifosine could sensitize tumorcells versus anti-target drugs such as the EGFR tyrosinekinase inhibitor, erlotinib, in partially insensitive PTENnegative PC-3 cells.
To evaluate the anti-proliferative effects of perifo-sine or erlotinib, alone and in combination, we treatedboth PC3 and DU145 cells with 1.0 and 5.0 mMperifosine in the presence of 10 ng/ml EGF and threedifferent doses of erlotinib (0.5, 1.0, and 5.0 mM). After48 hr, the cultures were either pulsed with [3H]thymidine or harvested for the cell cycle/apoptosisanalyses. In Figure 5,we show the 3H thymidineuptakeevaluated after 72 hr of culture in PC3 (A) and DU145
The Prostate
Fig. 3. Perifosine induces a time-dependent accumulation of cells in G0/G1associatedwith a statistically significant (P< 0.001by ANOVA)reductionofG2/McellproportioninPC3cells(A)andareductionofcellcycle-associatedproteinssuchasCyclinsB1,D1,andD2;andrespectivecyclindependentkinasesCdk4 and 6, p21andp27.Perifosine induceda time dependentreductionof the expressionof anti-apoptotic elements(B) such Bcl2, Bcl-Xl, and phospho-Bad with increased expression of pro-apoptotic elements such as Bax, Bcl-Xs, smac-Diablo, p14, andcytochrome(C)Eachlanewasloadedwith40mgofprotein fromtotalcellextracts.
970 Festuccia et al.

(B) cells as well as the apoptotic profiles by FACSanalyses (C) in PC3 cells after treatment with 1.0 mMperifosine and different doses of erlotinib. The IC50
values for erlotinibwere very different between controlcells. In facts, treatment with erlotinib alone resulted ina dose-dependent inhibition of growth with an IC50
value of 0.8 mMforDU145 cellswhereas PTENnegativePC3 cells possessed a IC50 value >10 mM, whereas theIC50 values were very similar in cultures pre-treatedwith perifosine resulting of 0.4 and 0.6 mM, respec-tively, in PC3 and DU145 cells, suggesting that dif-ferences observed in drug sensitivity were due notto a different EGFR/Her2 expression but to a dif-ferent PI3K activity. Exposure to perifosine resulted inclear synergywith a CI ranged between 0.44 and 0.60 inPC3 and 0.43 and 0.77 in DU145 cells (Table II). Wenote also that the better combination for PC3 cellswas erlotinib 1.0 mM and perifosine 5.0 mM havinga increment in the growth inhibition of 2.25 (CI¼ 0.44)whereas 0.5 mM erlotinib plus 5 mM perifosinewas the better combination for DU145 cells, having aincrement in the growth inhibition of 2.33 (CI¼ 0.43)
due to the higher effectiveness against erlotinib ofthese cells.
DISCUSSIONANDCONCLUSIONS
The behavior of many neoplasms appears to becorrelated with the expression of positive (worseprognosis) or negative (better prognosis) regulators ofthe PI3K/Akt signaling pathway. PTEN has beenfound deleted in a substantial fraction of cancers.Amplification and overexpression of Akt is frequentlyobserved in solid cancer [18,19]. Moreover, recentreports suggest that the dosage of PTEN in prostaticlesions is directly correlated to PCa progression,incidence, and overall biology [3]. Strengthening thisargument are other reports stating that prolongedandrogen-ablation therapy can lead to heightenedlevels of PI3K-signaling activity in PCa cells [4,20].Consequently, efforts to regulate signals transducedthrough the PI3K pathway may provide therapeuticinsight regarding progression and control of thedisease. Phospholipid analogues (ALK) are struc-turally novel candidate drugs with evidence of anti-neoplastic activity in both in vitro and in vivomodel systems [21]. This family includes ET-18-OCH3
(edelfosine), and the structurally simpler hexadecy-lphosphocholine (miltefosine [22]) andoctadecylpipiri-dine (perifosine [8]). Perifosine has recently enteredphase II clinical trials [8,23]. However, a phase IIstudy of perifosine in androgen independent PCa [11]showed no significant clinical activity against PCa.In this study, we first investigated the activity ofperifosine and PTEN and Akt gene manipulation in abattery of PCa cells having different PTEN expressionand Akt activity analyzing the apoptosis inductionand the synergy with EGFR inhibitors. We show thatperifosine exerts its growth inhibitory effects primarilythrough the induction of cell cycle arrest and apoptosisin PTEN defective cells and this in agreement withtwo previous reports [13,14]. Time-dependent studiesshowed that 4 hr of exposure to perifosine wassufficient to promote the arrest of cells with G1 andG2/M DNA content and relative loss in S-phasepopulation. The cell cycle analyses was in agreementwith molecular analyses of cyclins and Cdk expressionwith reduced expression of elements which determinea G0/G1 arrest including p21WAF1 and p27KIP1 havinga clear role in the G2/M progression. Importantly,perifosine inhibits the growth of PTEN defective PCacellswith an IC50 ranging 5 and 15 mMwhich arewithinthe clinically achieved and safe peak plasma concen-tration range [8,9]. These data are in agreement withaprevious paper of Patel et al. [24] performed insquamous carcinoma cells in which was demonstratedalso that the p21 induction was independent to p53
The Prostate
Fig. 4. Effects of perifosine in PC3 cells on caspase activation:perifosine induces dose-dependently the activation of caspase8 (antibody from Santa Cruz against epitope corresponding toamino acids 217^350 mapping within the caspase 8 p20 subunit ofhumanorigin) caspase9 (antibody fromEpitomics againstbothpro-and activated forms), caspase 3 (antibody from Santa Cruz againstpro and activated forms) and PARP (poly ADP-ribosepolymerase).Each lane was loaded with 40 mg of proteins and normalizedversus actin.
Perifosine Effects in ProstateCancer Cells 971

expressionleading to loss in cycline-dependent kinaseactivity and cell arrest.However,wefirst demonstratedthis in advanced PCa cells.
It is commonly accepted that the apoptotic responseof PI3K/Akt inhibition is mediated by loss of mito-chondrial membrane integrity, release of cytochromeC, and apoptosome-mediated activation of caspase 3.Whereas the precise mechanism initiating this eventhas not been directly addressed, these observations
have implied that PI3K inhibition-induced apoptosisoccurs via activation of the intrinsic mitochondrialdeath pathway.
In addition, perifosine we demonstrated thatshowed synergistic effects with erlotinib, an EGFRtyrosine kinase inhibitor. EGFR inhibition ismodulatedby Akt activity and possesses low activity in PTENdeficient carcinoma cells and this is dependent to Aktactivity [25]. Resistance to anti-EGFR therapies is an
The Prostate
TABLE II. Pharmacological Parameters of CombinationTreatmentWith Perifosine (PRF) andErlotinib (OSI-774) in PC3 andDU145Cells
Treatment A (OSI-774) Treatment B (PRF) Combination treatment
Cells Dose (mM) GI Dose (mM) GI Expected GIObserved
GI Increment (CI)
PC-3 0.5 0.97 1 0.72 0.69 0.34 2.03 (0.49)5 0.53 0.50 0.25 2.00 (0.50)
1.0 0.92 1 0.72 0.64 0.31 2.06 (0.48)5 0.53 0.45 0.20 2.25 (0.44)
5.0 0.80 1 0.72 0.52 0.25 2.08 (0.48)5 0.53 0.33 0.20 1.65 (0.60)
DU145 0.5 0.87 1 0.97 0.84 0.54 1.56 (0.64)5 0.83 0.70 0.30 2.33 (0.43)
1.0 0.52 1 0.97 0.49 0.25 1.96 (0.51)5 0.83 0.35 0.20 1.75 (0.54)
5.0 0.30 1 0.97 0.27 0.20 1.35 (0.74)5 0.83 0.13 0.10 1.30 (0.77)
Fig. 5. Dose-dependent anti-proliferative effects of Erlotinib with (B) or without (A) 1.0 mM perifosine in PC3 (A) and DU145 (B) cells.ApoptoticcelldeathevaluatedbyFACSat48hrof treatment(C)with0.5,1.0and5.0mMErlotinibinpresenceof1.0mMperifosineandcomparisonwith themaximalconcentrationofErlotinib (5.0mM)andperifosinealoneinPC3cells.
972 Festuccia et al.

emerging clinical problem and in hormone refractoryPCas EGFR tyrosine kinase inhibitors, erlotinib orgefitinib, have shown to be ineffective in clinicaltrials alone [26] or in combination therapies withchemotherapeutics [27,28]. Anti-EGFR therapies canbe influenced by the presence of PTEN mutationsas well as by the activation of alternative signalingpathways. Our findings suggest that combinationtreatment between perifosine and the EGFR tyrosinekinase inhibitor, erlotinib, may be a potential thera-peutic tool in the treatment of advanced PTENnegative, EGFR positive PCas. In our experiments,perifosine is able to restore the erlotinib effectiveness ofPTEN defective PC3 cells and synergize with erlotinibin both PTENpositive and PTENdefective cells. This isin agreement with Li et al. [29] which had previouslyshown that perfiosine was able to synergize with theanti-EGFR humanize antibody, cetuximab, in PTENnegative cells include PC3 cell line. The effects ofthis combination therapy are sustained both to theperifosine-mediated reduction of Akt activity and togefitinib-mediated reduction of ERK activity whichin turn is able to maintain the activity of Fas:FasLsystem.
In conclusion, our paper offers two major novelelements: (i) the triggering of apoptotic machinerydownstream to Fas:FasL system and (ii) the synergisticeffects of perifosine with EGFR tyrosine kinase in-hibitors such as erlotinib. These important aspects ofperifosine effectiveness are of interest both for theirmechanistic information and for their therapeuticpotential. However, further studies in the setting ofcombination therapies in advancedPCas are necessary.
REFERENCES
1. Miyamoto H, Messing EM, Chang C. Androgen deprivationtherapy for prostate cancer: Current status and future prospects.Prostate 2004;61:332–353 (Review).
2. Shukla S, Maclennan GT, Marengo SR, Resnick MI, Gupta S.Constitutive activation of P I3 K-Akt and NF-kappaB duringprostate cancer progression in autochthonous transgenic mousemodel. Prostate 2005;64:224–239.
3. Lu S, Ren C, Liu Y, Epner DE. PI3K-Akt signaling is involved inthe regulation of p21(WAF/CIP) expression and androgen-independent growth in prostate cancer cells. Int J Oncol 2006;28:245–251.
4. FestucciaC,GravinaGL,MuziP, PomanteR,VenturaL,VessellaRL, Vicentini C, BolognaM. Bicalutamide increases phopho-Aktlevels through Her2 in patients with prostate cancer. EndocrRelat Cancer 2007;14:601–611.
5. Chinni SR, Sarkar FH.Akt inactivation is a key event in indole-3-carbinol-induced apoptosis in PC-3 cells. Clin Cancer Res 2002;8:1228–1236.
6. Yang L, Dan HC, Sun M, Liu Q, Sun XM, Feldman RI. Akt/protein kinase B signaling inhibitor-2, a selective small moleculeinhibitor of Akt signaling with antitumor activity in cancer cellsoverexpressing Akt. Cancer Res 2004;64:4394–4399.
7. LuoY, ShoemakerAR, LiuX,WoodsKW,Thomas SA, de JongR.Potent and selective inhibitors of Akt kinases slow the progressof tumors in vivo. Mol Cancer Ther 2005;4:977–986.
8. Crul M, Rosing H, de Klerk GJ, Dubbelman R, Traiser M,Reichert S, Knebel NG, Schellens JH, Beijnen JH, tenBokkel Huinink WW. Phase I and pharmacological studyof daily oral administration of perifosine (D-21266) in patientswith advanced solid tumours. Eur J Cancer 2002;38:1615–1621.
9. Van Ummersen L, Binger K, Volkman J, Marnocha R, Tutsch K,Kolesar J,ArzoomanianR,AlbertiD,WildingGA.phase I trial ofperifosine (NSC 639966) on a loading dose/maintenance doseschedule inpatientswith advancedcancer.ClinCancerRes 2004;10:7450–7456.
10. Ernst DS, Eisenhauer E, Wainman N, Davis M, Lohmann R,Baetz T, Belanger K, SmylieM. 2005; Phase II study of perifosinein previously untreated patients with metastatic melanoma.Invest New Drugs 2005;23:569–575.
11. Posadas EM, Gulley J, Arlen PM, Trout A, Parnes HL, Wright J,Lee MJ, Chung EJ, Trepel JB, Sparreboom A, Chen C, Jones E,Steinberg SM, Daniels A, FiggWD, Dahut WL. A phase II studyof perifosine in androgen independent prostate cancer. CancerBiol Ther 4:1133–1137.
12. KnowlingM, BlacksteinM, Tozer R, Bramwell V, Dancey J, DoreN, Matthews S, Eisenhauer E. A phase II study of perifosine(D-21226) in patients with previously untreated metastatic orlocally advanced soft tissue sarcoma: A National CancerInstitute of Canada Clinical Trials Group trial. Invest NewDrugs 2006;24:435–439.
13. Kondapaka SB, Singh SS, Dasmahapatra GP, Sausville EA,Roy KK. Perifosine, a novel alkylphospholipid, inhibitsprotein kinase B activation. Mol Cancer Ther 2003;2:1093–1103.
14. DasmahapatraGP,Didolkar P,AlleyMC,GhoshS, Sausville EA,Roy KK. In vitro combination treatment with perifosine andUCN-01 demonstrates synergism against prostate (PC-3) andlung (A549) epithelial adenocarcinoma cell lines. Clin CancerRes 2004;10:5242–5252.
15. Canil CM, Moore MJ, Winquist E, Baetz T, Pollak M, Chi KN,Berry S, Ernst DS, Douglas L, Brundage M, et al. Randomizedphase II study of two doses of gefitinib in hormone-refractoryprostate cancer:A trial of theNationalCancer Instituteof Canada-Clinical Trials Group. J Clin Oncol 2005;23:455–460.
16. Zhao H, Dupont J, Yakar S, Karas M, LeRoith D. PTEN inhibitscell proliferation and induces apoptosis by downregulating cellsurface IGF-IR expression in prostate cancer cells. Oncogene2004;23:786–794.
17. Chou TC, Talalay P. Analysis of combined drug effects: A newlook at a very old problem. Trends Pharmacol Sci 1983;4:450–454.
18. Page C, Lin HJ, Jin Y, Castle VP, Nunez G, Huang M, Lin J.Overexpression of Akt/AKT can modulate chemotherapy-induced apoptosis. Anticancer Res 2000;20:407–416.
19. SunM,WangG, Paciga JE, FeldmanRI, YuanZQ,MaXL, ShelleySA, Jove R, Tsichlis PN,Nicosia SV, Cheng JQ. AKT1/PKBalphakinase is frequently elevated in human cancers and itsconstitutive activation is required for oncogenic transformationin NIH3T3 cells. Am J Pathol 2001;159:431–437.
20. Murillo H, Huang H, Schmidt LJ, Smith DI, Tindall DJ. Role ofPI3K signaling in survival and progression of LNCaP prostatecancer cells to the androgen refractory state. Endocrinology2001;142:4795–4805.
The Prostate
Perifosine Effects in Prostate Cancer Cells 973

21. Hilgard P, Klenner T, Stekar J, Nossner G, Kutscher B, Engel J.D-21266, a new heterocyclic alkylphospholipidwith antitumouractivity. Eur J Cancer 1997;33:442–446.
22. Sundar S, Jha TK, Thakur CP, Engel J, Sindermann H, FischerC, Junge K, Bryceson A, Berman J. Oral miltefosine forIndian visceral leishmaniasis. NEngl JMed 2002;347:1739–1746.
23. Argiris A, Cohen E, Karrison T, Esparaz B, Mauer A, Ansari R,Wong S, Lu Y, Pins M, Dancey J, Vokes E. A phase II trialof perifosine, an oral alkylphospholipid, in recurrent ormetastatichead and neck cancer. Cancer Biol Ther 2006;5:766–770.
24. Patel V, Lahusen T, Sy T, Sausville EA, Gutkind JS, SenderowiczAM. Perifosine, a novel alkylphospholipid, induces p21(WAF1)expression in squamous carcinoma cells through a p53-inde-pendent pathway, leading to loss in cyclin-dependent kinaseactivity and cell cycle arrest. Cancer Res 2002;62:1401–1409.
25. FestucciaC,MuziP,MillimaggiD,BiordiL,GravinaGL, SpecaS,Angelucci A, Dolo V, Vicentini C, BolognaM.Molecular aspectsof gefitinib antiproliferative and pro-apoptotic effects in PTEN-positive and PTEN-negative prostate cancer cell lines. EndocrRelat Cancer 2005;12:983–998.
26. Canil CM, Moore MJ, Winquist E, Baetz T, Pollak M, Chi KN,Berry S, ErnstDS,Douglas L, BrundageM, Fisher B,McKennaA,Seymour L. Randomized phase II study of two doses of gefitinibin hormone-refractory prostate cancer: A trial of the NationalCancer Institute of Canada-Clinical Trials Group. J Clin Oncol2005;23:455–460.
27. GrossM, Higano C, PantuckA, Castellanos O, Green E, NguyenK,AgusDB.Aphase II trial of docetaxel and erlotinib as first-linetherapy for elderly patients with androgen-independent pros-tate cancer. BMC Cancer 2007;7:142.
28. Salzberg M, Rochlitz C, Morant R, Thalmann G, Pedrazzini A,RoggeroE, SchonenbergerA,KnuthA,BornerM.Anopen-label,noncomparative phase II trial to evaluate the efficacy and safetyof docetaxel in combination with gefitinib in patients withhormone-refractory metastatic prostate cancer. Onkologie 2007;30:355–360.
29. Li X, Luwor R, Lu Y, Liang K, Fan Z. Enhancement of anti-tumor activity of the anti-EGF receptor monoclonal antibodycetuximab/C225 by perifosine in PTEN-deficient cancer cells.Oncogene 2006;25:525–535.
The Prostate
974 Festuccia et al.
Copyright © 2022 FDOKUMEN




















