
Age-related accumulation of Reelin in amyloid-like deposits
20
Neurobiology of Aging 30 (2009) 697–716 Age-related accumulation of Reelin in amyloid-like deposits Irene Knuesel a,∗ , Myriel Nyffeler b , Cecile Morm` ede c , Mary Muhia b , Urs Meyer b , Susanna Pietropaolo b , Benjamin K. Yee b , Christopher R. Pryce b,1 , Frank M. LaFerla d , Aline Marighetto c , Joram Feldon b a Institute of Pharmacology and Toxicology, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland b Laboratory of Behavioral Neurobiology, ETH Zurich, Schwerzenbach, Switzerland c Laboratory of Cognitive Neurosciences, UMR5106, University of Bordeaux 1, Talence Cedex, France d Department of Neurobiology and Behaviour, University of California, Irvine, USA Received 26 February 2007; received in revised form 23 July 2007; accepted 13 August 2007 Available online 27 September 2007 Abstract Accumulating evidence suggest that alterations in Reelin-mediated signaling may contribute to neuronal dysfunction associated with Alzheimer’s disease (AD), the most common form of senile dementia. However, limited information is available on the effect of age, the major risk factor of AD, on Reelin expression. Here, we report that normal aging in rodents and primates is accompanied by accumulation of Reelin-enriched proteinous aggregates in the hippocampal formation that are related to the loss of Reelin-expressing neurons. Both phenomena are associated with age-related memory impairments in wild-type mice. We provide evidence that normal aging involves loss of Reelin neurons, reduced production and elimination of the extracellular deposits, whereas a prenatal immune challenge or the expression of AD-causing gene products, result in earlier, higher, and more persistent levels of Reelin-positive deposits. These aggregates co-localize with non-fibrillary amyloid-plaques, potentially representing oligomeric A species. Our findings suggest that elevated Reelin plaque load creates a precursor condition for senile plaque deposition and may represent a critical risk factor for sporadic AD. © 2007 Elsevier Inc. All rights reserved. Keywords: Mus musculus; Rattus norvegicus; Callithrix jacchus; Hippocampus; Stratum lacunosum-moleculare; Entorhinal cortex; Piriform cortex; Episodic- like memory; Radial arm maze; GABAergic interneurons; PolyIC; Neuroinflammation; 3xTg-AD mice; SynGAP; GFAP; Dab1; F4/80; Immunohistochemistry; Alzheimer’s disease 1. Introduction Reelin is a large extracellular glycoprotein secreted by Cajal-Retzius cells and mediating proper positioning of neurons during development through the activation of the apolipoprotein E receptor 2 (ApoER2) and very-low- density lipoprotein receptor (VLDLR, D’Arcangelo et al., 1995, 1999; Hiesberger et al., 1999; Howell et al., 1997; Trommsdorff et al., 1999). Transduction of the signal involves the interaction of the adapter protein Dab1 with the intracel- lular NPxY motiv of these receptors, resulting in tyrosine ∗ Corresponding author. Tel.: +41 44 635 55 97; fax: +41 44 635 68 74. E-mail address: [email protected] (I. Knuesel). 1 Present address: NOVARTIS Pharma AG, Basel, Switzerland. phosphorylation of Dab1, activation of Src family of non- receptor tyrosine kinases, and triggering a downstream cytosolic kinase cascade beginning with the activation of phosphatidylinositol-3-kinase (PI3K) and ending with the inhibition of glycogen synthase kinase 3 (GSK3; for recent review, see Herz and Chen, 2006). The signal is terminated with Reelin targeted to the lysosome and Dab1 degraded by the proteasome (Arnaud et al., 2003; Bock et al., 2004; Morimura et al., 2005). In the adult brain, Reelin expression is maintained by GABAergic interneurons and glutamater- gic pyramidal neurons in layer II of the entorhinal cortex (Alcantara et al., 1998; Miettinen et al., 2005; Pesold et al., 1998; Ramos-Moreno et al., 2006) and the same signaling cascade is used in adult synapses to modulate neuronal func- tion and synaptic plasticity by regulating glutamate receptor activity through the phosphorylation of intracellular tyrosine 0197-4580/$ – see front matter © 2007 Elsevier Inc. All rights reserved. doi:10.1016/j.neurobiolaging.2007.08.011
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Age-related accumulation of Reelin in amyloid-like deposits

A
AmRarpac©
KlA
1
botd1Ttl
0d
Neurobiology of Aging 30 (2009) 697–716
Age-related accumulation of Reelin in amyloid-like deposits
Irene Knuesel a,∗, Myriel Nyffeler b, Cecile Mormede c, Mary Muhia b, Urs Meyer b,Susanna Pietropaolo b, Benjamin K. Yee b, Christopher R. Pryce b,1,
Frank M. LaFerla d, Aline Marighetto c, Joram Feldon b
a Institute of Pharmacology and Toxicology, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerlandb Laboratory of Behavioral Neurobiology, ETH Zurich, Schwerzenbach, Switzerland
c Laboratory of Cognitive Neurosciences, UMR5106, University of Bordeaux 1, Talence Cedex, Franced Department of Neurobiology and Behaviour, University of California, Irvine, USA
Received 26 February 2007; received in revised form 23 July 2007; accepted 13 August 2007Available online 27 September 2007
bstract
Accumulating evidence suggest that alterations in Reelin-mediated signaling may contribute to neuronal dysfunction associated withlzheimer’s disease (AD), the most common form of senile dementia. However, limited information is available on the effect of age, theajor risk factor of AD, on Reelin expression. Here, we report that normal aging in rodents and primates is accompanied by accumulation ofeelin-enriched proteinous aggregates in the hippocampal formation that are related to the loss of Reelin-expressing neurons. Both phenomenare associated with age-related memory impairments in wild-type mice. We provide evidence that normal aging involves loss of Reelin neurons,educed production and elimination of the extracellular deposits, whereas a prenatal immune challenge or the expression of AD-causing generoducts, result in earlier, higher, and more persistent levels of Reelin-positive deposits. These aggregates co-localize with non-fibrillarymyloid-plaques, potentially representing oligomeric A� species. Our findings suggest that elevated Reelin plaque load creates a precursor
ondition for senile plaque deposition and may represent a critical risk factor for sporadic AD.2007 Elsevier Inc. All rights reserved.
eywords: Mus musculus; Rattus norvegicus; Callithrix jacchus; Hippocampus; Stratum lacunosum-moleculare; Entorhinal cortex; Piriform cortex; Episodic-ike memory; Radial arm maze; GABAergic interneurons; PolyIC; Neuroinflammation; 3xTg-AD mice; SynGAP; GFAP; Dab1; F4/80; Immunohistochemistry;
prcpirw
lzheimer’s disease
. Introduction
Reelin is a large extracellular glycoprotein secretedy Cajal-Retzius cells and mediating proper positioningf neurons during development through the activation ofhe apolipoprotein E receptor 2 (ApoER2) and very-low-ensity lipoprotein receptor (VLDLR, D’Arcangelo et al.,
995, 1999; Hiesberger et al., 1999; Howell et al., 1997;rommsdorff et al., 1999). Transduction of the signal involveshe interaction of the adapter protein Dab1 with the intracel-ular NPxY motiv of these receptors, resulting in tyrosine
∗ Corresponding author. Tel.: +41 44 635 55 97; fax: +41 44 635 68 74.E-mail address: [email protected] (I. Knuesel).
1 Present address: NOVARTIS Pharma AG, Basel, Switzerland.
bMig(1cta
197-4580/$ – see front matter © 2007 Elsevier Inc. All rights reserved.oi:10.1016/j.neurobiolaging.2007.08.011
hosphorylation of Dab1, activation of Src family of non-eceptor tyrosine kinases, and triggering a downstreamytosolic kinase cascade beginning with the activation ofhosphatidylinositol-3-kinase (PI3K) and ending with thenhibition of glycogen synthase kinase 3� (GSK3�; for recenteview, see Herz and Chen, 2006). The signal is terminatedith Reelin targeted to the lysosome and Dab1 degradedy the proteasome (Arnaud et al., 2003; Bock et al., 2004;orimura et al., 2005). In the adult brain, Reelin expression
s maintained by GABAergic interneurons and glutamater-ic pyramidal neurons in layer II of the entorhinal cortexAlcantara et al., 1998; Miettinen et al., 2005; Pesold et al.,
998; Ramos-Moreno et al., 2006) and the same signalingascade is used in adult synapses to modulate neuronal func-ion and synaptic plasticity by regulating glutamate receptorctivity through the phosphorylation of intracellular tyrosine
6 logy of
re
ladmsiato1laseIcce�1wsasola
Flpiatdl2iArlaibafa
2
2
TA
G
1
2
3
4
5
6
R
98 I. Knuesel et al. / Neurobio
esidues (Beffert et al., 2005, 2006; Chen et al., 2005; Weebert al., 2002).
In line with its critical role in synaptic transmission andearning and memory, several recent findings suggest thatlterations in Reelin signaling may contribute to neuronalysfunction associated with Alzheimer’s disease (AD). Thisost common type of dementia is characterized by progres-
ive cognitive decline and two histopathological hallmarksncluding amyloid-beta (A�) plaques, which are caused byn imbalance in A� metabolism, and neurofibrillary tangleshat result from abnormal phosphorylation and aggregationf Tau (Glenner and Wong, 1984; Grundke-Iqbal et al.,986). Reelin binding to its receptors potently downregu-ates the activity of GSK3�, a major Tau kinase, and as
result mutant mice with defects in the Reelin signalinghow increased levels of hyperphosphorylated Tau (Beffertt al., 2004; Hiesberger et al., 1999; Ohkubo et al., 2003).n addition, the Reelin receptors are also targets of theommon ApoE isoform �4, a major genetic risk factor asso-iated with sporadic AD (Corder et al., 1993; Schmechelt al., 1993). The finding that recombinant ApoE �3 and4 inhibit Reelin binding by 50–60% (D’Arcangelo et al.,999) indicates that lipid-associated ApoE could competeith Reelin for lipoprotein receptor activation. As a con-
equence, impaired ApoE receptor signaling may result indysbalance in cholesterol homeostasis, which has been
hown to profoundly affect the production and traffickingf A� (Simons et al., 1998) and hypothesized to under-ie accelerating synaptic loss and onset of dementia (Herznd Chen, 2006; Raber et al., 1998; Weeber et al., 2002).
wCd1
able 1nimals employed
roup Species Strain Genotype Treatment Breeding
Mice C57B6 Jico Wild type None Janvier, LGenest-SFrance
C57B6J SynGAP+/+ None ETH ZurSynGAP+/−
C57B6J Wild type PrenatalPolyI:C orvehicleexposure
ETH Zur
129/C57B6J 3xTg-AD None ETH ZurNon-Tg controls
Rats Wistar WIST:Hanlbm None ETH Zur
Monkeys Callithrixjacchus
None ETH Zur
AnthropoUniversi
AM: radial arm maze task; IHC: immunohistochemistry; C: quantification of Ree
Aging 30 (2009) 697–716
urther support for a direct link between Reelin and amy-oid precursor protein (APP) processing has recently beenroved by Hoe and colleagues by demonstrating that Reelinncreases the intracellular interaction of Dab1 with ApoER2nd APP and promotes their cleavage, resulting in reducedoxic A� production (Hoe et al., 2006a). Moreover, recentata from AD patients reporting altered Reelin expressionevels in both CSF and cortical tissue (Botella-Lopez et al.,006; Saez-Valero et al., 2003) point to profound alterationsn Reelin processing and signaling potentially underlyingD-related neuronal dysfunction. Although Reelin, ApoE
eceptors, and APP functionally converge at synaptic sitesikely joining forces to control glutamate receptor activitynd regulate neurotransmission, cognition and memory, its currently unknown whether Reelin expression is affectedy age; the major risk factor of AD. We therefore set outcomprehensive investigation on Reelin expression in dif-
erent species during normal and pathological forms ofging.
. Methods and materials
.1. Animals
All procedures were approved by the local authorities and
ere performed in accordance with the European Communityouncil Directive of 24 November 1986 (86/609/EEC). Sixifferent groups of animals were employed (Table 1). Group: Adult (4–5 months), middle-aged (11–14 months), and oldColony Age Number Experiment
eaint Isle,
4–5 months 7 ♂ Behaviouraltesting (RAM)
11–14 months 10 ♂20–23 months 16 ♂ Reelin-IHC: C/P
ich 3 months 4 ♂, 3 ♀ Reelin-IHC: C/P6 months 6 ♀12 months 4 ♂, 4 ♀24 months 8 ♂ per genotype
ich 6 months 10 ♂, 10 ♀ Reelin-IHC: C/P
5 ♂, 5 ♀ich 15 months 8 ♂ Reelin-IHC: P
10 ♂ich 6 months 3 ♂ Reelin-IHC
20 months 3 ♂ich 1 years 1 ♀ Reelin-IHC
logyty Zurich
10 years 1 ♀lin-pos neurons; P: quantification of Reelin-pos plaques.

logy of
((kc1gataodh(laieETih2gfibhk2
2
a(smaclths(asttadtdp4fd
2
sa0ssg1ratspso4tstisPro(52ipfaOrdDtI(aa5C
2
stwi
I. Knuesel et al. / Neurobio
20–23 months) wild-type mice were obtained from JanvierFrance) and caged in groups of 4–5 in a climatized animal-eeping room maintained under a 12 h reversed light–darkycle and ad lib food and water (University of Bordeaux, France). For the behavioral tests, the animals were sin-le caged and introduced to a progressive food deprivationnd maintained at about 90% of their free feeding weighthroughout the experiments. Group 2: Synaptic Ras-GTPasectivating protein, SynGAP+/+ and SynGAP+/− mice werebtained from heterozygous breeding pairs, genotyped asescribed (Vazquez et al., 2004) at the age of 3 weeks andoused in same sex groups of 4–5 in a specified pathogen freeSPF) animal facility (ETH Zurich, Schwerzenbach, Switzer-and) under 12 h reversed light–dark cycle and ad lib foodnd water. Group 3: Offspring born to PolyIC- (5 mg/kg,njected at gestational days 9 or 17) or vehicle-treated moth-rs were weaned and sexed at 3 weeks and housed at theTH Zurich as described (Meyer et al., 2006). Group 4:he offspring of triple transgenic Alzheimer’s mice harbor-
ng two transgenes (encoding APPSwe, and tauP301L) on aomozygous PS1M146V knock-in background (Oddo et al.,003) and the non-transgenic control mice were housed inroups of 4–5 in the SPF facility as described above. Con-rmation of the genotypes was done with postmortem tailiopsies as described (Oddo et al., 2003). Groups 5/6: Theousing conditions for rats and common marmoset mon-eys were as described (Nyffeler et al., 2007; Pryce et al.,005).
.2. Behavioral testing
The construction and dimension of the eight-arm radialrm maze (RAM) have been described in detail elsewhereMarighetto et al., 1999). The task consisted of concurrenterial alternations assessing episodic-like memory perfor-ance. Each subject was individually assigned six adjacent
rms which were grouped into three pairs (A, B and C), eachonsisting of a baited and unbaited arm. For each pair, theocation of the food reward alternated between successiverials. A total of 14 daily sessions were performed (2 forabituation and 12 for memory tests). Each memory test ses-ion consisted of 23 trials: 3 acquisition trials for each pairwith both arms rewarded), followed by 20 memory test tri-ls (Fig. 1A). On each trial, two adjacent arms were openedimultaneously. A choice was considered to be made whenhe subject had reached the food located at the end of the arm;his also triggered the closure of the door to the alternativerm. After the subject returned to the central platform, theoor was closed and the trial ended. Subjects were confinedo the centre during a 10 s inter-trial interval. The level ofifficulty of a given trial depended on the number of inter-
osed trials, resulting in retention intervals ranging from 0 to(Fig. 1A). The percentage of correct choices was recordedor each session and averaged by blocks of three consecutiveays or by trials of the same level of retention interval.
Z(ws
Aging 30 (2009) 697–716 699
.3. Immunohistochemistry
Animals were deeply anesthetized with an overdose ofodium pentobarbital and perfused through the ascendingorta with a fixative containing 4% paraformaldehyde in.15 M phosphate buffer, pH 7.4. After postfixation in theame fixative overnight, the brains were cryoprotected in 30%ucrose, frozen, and stored at −80 ◦C (groups 1, 5, 6). Mice ofroups 2–4 were perfused with the above fixative containing5% picric acid. These brains were processed for antigen-etrieval immunohistochemistry as described (Fritschy etl., 1998). For all groups, free-floating sections (40–50 �mhick) were cut coronally on a sliding microtome. Randomampled serial sections were collected throughout the hip-ocampal formation and stored at −20 ◦C in cryoprotectantolution until further processing. Sections were incubatedvernight at room temperature (immunoperoxidase; IP) or◦C (immunofluorescence, IF) in the primary antibody solu-
ions (Table 2) diluted in PBS containing 2% normal goaterum and 0.3% Triton X-100. Sections were then washedhree times with PBS. For the IP stainings, a 1 h incubationn biotinylated secondary antibodies (Jackson ImmunoRe-earch Laboratories Inc., West Grove, PA) diluted 1:500 inBS was performed at room temperature, followed by threeinses in PBS, incubation with Vectastain Kit (Vector Lab-ratories; Burlingame, CA) and the 3,3-diaminobenzidineDAB; Sigma–Aldrich Inc., St. Louis, MO) staining for–10 min as described (Meyer et al., 2006; Nyffeler et al.,007; Pryce et al., 2005). Counterstaining of Reelin-IP label-ng was done with 0.25% Cresyl violet according to standardrotocols. For double or triple IF, sections were incubatedor 30 min at room temperature in corresponding secondaryntibodies coupled to Alexa488 (Molecular Probes, Eugene,R), Cy3 or Cy5 (Jackson ImmunoResearch Laborato-
ies), washed again, mounted on gelatinized glass slides, airried, and coverslipped with FluorSaveTM (Calbiochem, Saniego, CA) mounting medium. Control stainings included
he incubation of the sections with 1 �g/ml mouse or rabbitgG (Jackson ImmunoResearch), rabbit pre-immune serumdiluted at 1:500), or the secondary goat-anti-mouse or rabbitntibody (coupled to HRP, Alexa488, or Cy3; diluted 1:500)lone. For the nuclear labeling, sections were incubated formin in 10 nM SYTOX green nucleic acid stain (Invitrogen,arlsbad, CA) dissolved in PBS.
.4. Data analysis
All quantitative analyses were done on one randomlyelected hemisphere and performed blind to the age, geno-ype, or treatment classification of the animals. IP stainingsere analyzed with bright-field microscopy and digital
mages acquired using a color digital camera (AxioCam,
eiss, Germany) controlled by AxioVision 4.1 softwareZeiss). Exposure times were set so that pixel brightnessas never saturated, and were held constant during acqui-
ition of all images for each experiment. IF double and
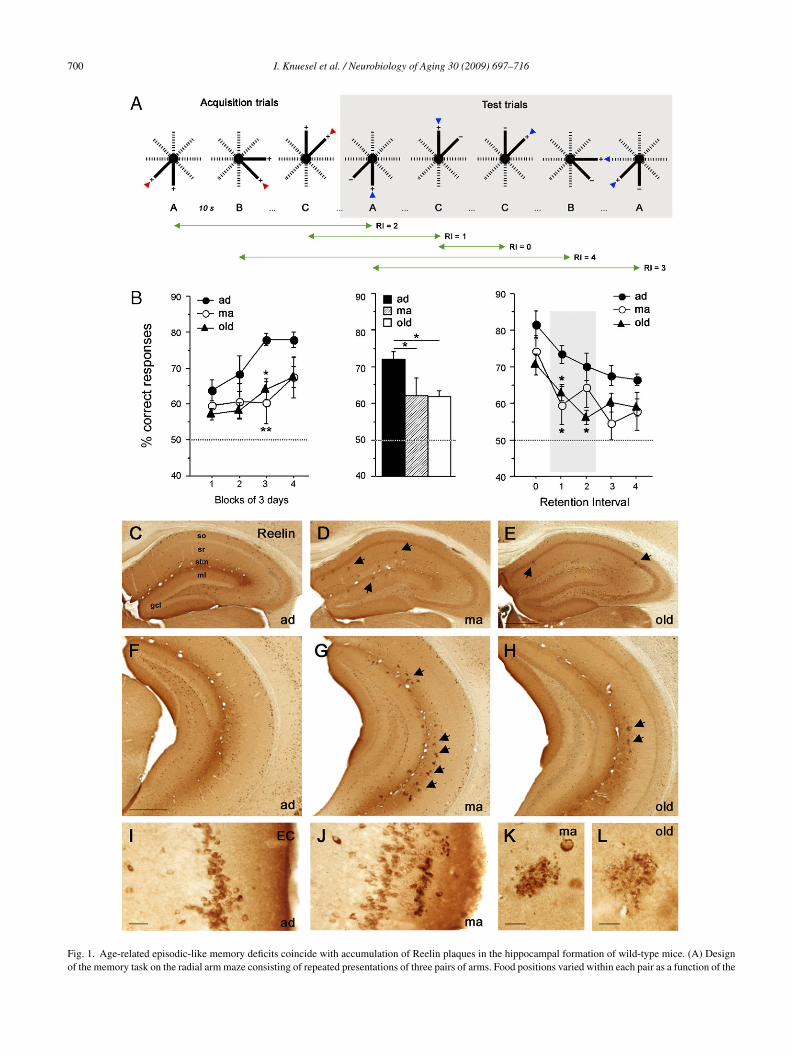
700 I. Knuesel et al. / Neurobiology of Aging 30 (2009) 697–716
Fig. 1. Age-related episodic-like memory deficits coincide with accumulation of Reelin plaques in the hippocampal formation of wild-type mice. (A) Designof the memory task on the radial arm maze consisting of repeated presentations of three pairs of arms. Food positions varied within each pair as a function of the

I. Knuesel et al. / Neurobiology of Aging 30 (2009) 697–716 701
Table 2List of antibodies
Target protein Species Dilution Protocol Source Number
Rodent Reelin (aa164–496, clone G10) Mouse 1:1000 IP/IF Chemicon International, Temecula, CA MAB5364Human ApoER2 Rabbit 1:500 IP/IF Santa Cruz Biotech. Inc., Santa Cruz, CA sc-20746Mouse Dab1 Rabbit 1:1000 IP/IF Chemicon International, Temecula, CA AB5840Mouse Fe65 Rabbit 1:1000 IP/IF Affinity BioReagents Inc., Golden, CO PA1-752Rat SynGAP Rabbit 1:500 IF Affinity BioReagents Inc., Golden, CO PA1-046Cow GFAP Rabbit 1:5000 IF DakoCytomation, Glostrup, Denmark Z0334Mouse F4/80 (clone CI:A3-1) Rat 1:100 IF Abcam plc, Cambridge, UK ab6640Oligomer species (A11) Rabbit 1:50 IP/IF BioSource Intl. Inc., Camarillo, CA AHB0052Human �-amyloid (1-40/42) Rabbit 1:500 IP/IF Chemicon International, Temecula, CA AB5076Human �-amyloid (1-17; clone 6E10) Mouse 1:500 IP Chemicon International, Temecula, CA MAB1560Rodent �-amyloid (all isoforms) Rabbit 1:300 IP/IF Chemicon International, Temecula, CA AB5571Human Tau (phospho T205) Rabbit 1:2000 IP/IF Abcam plc, Cambridge, UK ab4841M IFM IF
I
t(oce5fiossifS
2p
wbmatfo
dtigtvis∑
p
2(
tm(FtC
p(momRpcbRfhppa
ouse Laminin (all isoforms) Rabbit 1:5000ouse Fibronectin Rabbit 1:200
P: immunoperoxidase staining; IF: immunofluorescence staining.
riple labelings were visualized by confocal microscopyLSM-510 Meta, Zeiss) using a 40× (NA 1.3) or 100×bjective (NA 1.4) and sequential acquisition of separatehannels. The pinhole aperture was set to 1.0 Airy unit forach channel. Stacks of consecutive optical sections (6–12;12 × 512 pixel, spaced 1 �m in z) were acquired at a magni-cation of 0.11–0.22 �m/pixel. For visual display, Z-sectionsf both channels were summed and projected in the z dimen-ion (maximal intensity) and merged using the image analysisoftware Imaris (Bitplane, Zurich, Switzerland). Cropping ofmages, adjustments of brightness and contrast were identicalor each labeling and done using Adobe Photoshop (Adobeystems, San Jose, CA).
.4.1. Volume analysis and quantification of Reelinlaques (groups 1–4)
Volumetric analysis of the hippocampus was performedith StereoInvestigator software (version 6.50.1, Micro-rightfield, Colchester, VT). Every section per series waseasured providing an average of seven to nine sections per
nimal. The subfield areas were delineated with a 2.5× objec-ive (NA 0.075). The volumes were estimated according to theormula V =
∑Atnom × 1/ssf, where
∑A is the summed areas
f the delineated subfields (CA, DG, hilus; separately for
(itl
receding visit. Red arrowheads indicate the chosen arms during acquisition, blue aB) Statistical analysis of the mean performance on the radial arm maze (RAM) tasice across 12 days of training, yielded a significant effect of age (F2,30 = 3.3, p < 0
ver days (middle graph) was significantly different between adult (ad, 72.0 ± 2.1%)ice. Analysis of the mean performances using RI as within-subject factor yieldeI (F4,120 = 23.6, p < 0.0001). ANOVA of the RI separately revealed a significant< 0.05). All values are given as mean ± S.E.M. *p < 0.05; **p < 0.01, statistical sihance level. (C–K) Reelin-immunoperoxidase (IP) staining of the dorsal (C–E) arain sections obtained from ad (3 months; C, F and I), ma (12 months; D, G,eelin-immunoreactivity (IR) is evident in neuronal soma and the neuropil throug
ound in plaque-like accumulations (arrows), restricted to the dorsal CA stratum oriippocampus, they are strongly enriched in stratum lacunosum-moleculare (slm) anlaques in old versus ma subjects. (I and J) High magnification pictures of the laterredominantly found in layers II and I. Age-related plaques are found exclusivelynd are characterized by a granular rather than fibrillary appearance. Scale bars: (E
Chemicon International, Temecula, CA AB2034Chemicon International, Temecula, CA AB2033
orsal and ventral hippocampus), tnom is the nominal sectionhickness of 40 �m (groups 2–4) or 50 �m (group 1), and ssfs the section sampling fraction (1/4, group 1; 1/9, group 3; 1/8roups 2 and 4). Plaques were counted exhaustively withinhe delineated hippocampus using a marker tool of StereoIn-estigator. For the analysis, the total number of plaques wasncluded; estimated for the dorsal and ventral hippocampuseparately according to the formula Ptot =
∑P × 1/ssf, where
P is the sum of plaques, as well as the total number oflaques per unit volume (Ptot/voldhip or vhip (mm3)).
.4.2. Stereological estimation of Reelin neuronsgroups 1–3)
The total number of Reelin neurons was determined alonghe whole septo-temporal axis of the hippocampus on high
agnification images composed using MosaicTM softwareExplora Nova, Universal Laboratory Imaging, LaRochelle,rance). The same sections as described above (starting at
he beginning of the hippocampal formation) were included.ell counts were done in CA (including so, sr, slm) and DG
ml) using NIH ImageJ software by applying a threshold tonclude only immunolabeled cells in the analysis. A mask ofhe cells in the CA or DG, respectively, was created by out-ining the corresponding area and excluding particles smaller
rrowheads show the correct choices during test trials. RI: retention interval.k, expressed as % correct choices of the three different groups of wild-type.05) and blocks (F3,90 = 11.6, p < 0.001). The mean performance collapsedand middle-aged (ma, 62.2 ± 4.2%; p = 0.04) or old (61.7 ± 1.8%; p = 0.02)
d an age × RI interaction (F8,120 = 2.0, p < 0.05) and a significant effect ofeffect of age for the RI = 1 (F2,30 = 3.7, p < 0.05) and RI = 2 (F2,30 = 4.0,
gnificance based on Fisher’s LSD post hoc analysis. Dotted lines representnd ventral hippocampus (F–H) and entorhinal cortex (I and J) in coronal
J and K), and old (24 months; E, H and L) wild-type mice. In ad mice,hout the hippocampus and cortex. In ma and old mice, Reelin-IR is also
ens (so) and radiatum (sr) and outer DG molecular layer (ml). In the ventrald DG ml (arrows in D–H). Note the reduction in both Reelin neurons and
al entorhinal cortex (EC) of representative ad and ma mice. Reelin-IR wasin layer I. (K and L) Plaques are found in close vicinity to Reelin neuronsand F) 500 �m, (I) 50 �m, and (K and L) 20 �m.

7 logy of
tnaCt(
2
wCplv(tessmhmS
3
3ph
iesMdniateddaia2mmottbo
s0pAtpiemmFbcanotie
iamnniir2oiC(Iwngitam(
pmpvEfdod
02 I. Knuesel et al. / Neurobio
han 80 and larger than 500 pixels2. For the analysis, the totalumber of cells was included, estimated again for the dorsalnd ventral hippocampus separately according to the formulatot =
∑C × 1/ssf, where
∑C is the sum of cells, as well as
he total number of cells per unit volume (Ctot/voldhip or vhipmm3)).
.5. Statistical analysis
Data were analyzed using analysis of variance (ANOVA)ith the statistical software StatView version 5.0 (Abacusoncepts, Inc., Berkeley, CA). Fisher’s LSD post hoc com-arisons were performed whenever appropriate. Age (threeevels), genotype (controls, mutants), and treatment (PolyIC,ehicle), were the main between-subjects factors, while axisdorsal, ventral), subfields (CA, DG), blocks of behavioralraining sessions (four levels), and retention interval (five lev-ls) were included as main within-subjects factor. The factorex (groups 2 and 3) was dropped to enhance statistical powerince it did not yield a significant outcome. Pearson’s productoment correlations were performed between the immuno-
istochemical variable (ratio cells/plaque) and the behavioraleasures (mean performance in the RAM) using StatView.tatistical significance was set at p < 0.05.
. Results
.1. Age-related episodic-like memory deficits arearalleled by accumulation of Reelin plaques in theippocampal formation of wild-type mice
Epidemiological evidence suggests that prodromalmpairments in multiple cognitive domains includingxecutive functioning, episodic memory and perceptualpeed occur several years before AD diagnosis (Collie and
aruff, 2000). Qualitatively, these impairments are notifferent from the cognitive impairments observed duringormal aging, suggesting continuity rather than discontinu-ty from normal senescence to preclinical AD (Backman etl., 2004). Similarly, structural and functional alterations ofhe hippocampal formation which is critically involved in thencoding, storage and retrieval of episodic memory occururing normal human aging and are evident long before theiagnosis of AD (Small et al., 2000). Here, we used the radialrm maze to assess potential episodic-like memory deficitsn normal aged wild-type mice using a “concurrent seriallternations” task (Mingaud et al., 2005; Mormede et al.,006). Three groups of mice were employed: adults (ad, 4–5onths), middle-aged (ma, 11–14 months), and old (20–23onths, Table 1). The location of the food reward on a pair
f presented adjacent arms varied according to an alterna-
ion rule between successive trials (Fig. 1A). The mouse hado remember which of the two arms of a given pair hadeen visited in a particular trial until the next presentationf this pair in order to alternate its choices among succes-rooa
Aging 30 (2009) 697–716
ive presentations. The retention interval (RI) ranged fromto 4 (Fig. 1A), depending on the number of trials inter-
osed between two successive presentations of the same pair.nimals were tested for 12 consecutive days starting with
hree acquisition trials, followed by 20 test trials per day. Theerformance, expressed as % correct choices, significantlyncreased across training days in ad subjects and to a lesserxtent in ma and old mice (Fig. 1B, left). The overall perfor-ance was significantly better in ad (72.0 ± 2.1%) than ina (62.2 ± 4.2%; p = 0.04) and old (61.7 ± 1.8%; p = 0.02,ig. 1B, middle) mice. When the performance was averagedy trials of the same RI, the analysis revealed that the per-entage of correct responses decreased with increasing RI inll groups (Fig. 1B, right). The ma and old subjects displayedormal immediate memory (RI = 0) but very poor retentionf previous events as soon as RI was increased by one orwo interposed trials, indicating that the aged mice exhib-ted a selective and specific deficit of memory for recentpisodes.
In view of the role of Reelin and its receptor ApoER2n modulating LTP and learning (Beffert et al., 2005; Qiu etl., 2006; Weeber et al., 2002), we determined whether theemory impairments in aged wild-type mice were accompa-
ied by alterations in localization and distribution of Reelineurons in the hippocampal formation (Table 1). Reelin-mmunoreactivity (IR) was detected in both the neuropil andnterneuron somata throughout the forebrain (Fig. 1C–K), aseported in adult and aged wild-type mice (Miettinen et al.,005; Ramos-Moreno et al., 2006). Representative picturesf ad, ma and old mice show the pattern of Reelin neuronsn the hippocampal formation, predominantly localized inA stratum oriens (so), radiatum (sr), lacunosum-moleculare
slm), DG molecular layer (ml), hilus, and layers I andI of the entorhinal cortex. A prominent somatic labelingas also evident in all layers of the neocortical areas. Theeuropil staining was clearly differentiated from the back-round (i.e. corpus callosum) and was especially prominentn the ventral slm of ad subjects. Although the distribu-ion of Reelin-positive cells did not change during aging,
reduction of the number of these cells in so, slm, andl was evident, particularly in the ventral hippocampus
Fig. 1H).A major feature not seen in ad subjects were the Reelin-
ositive plaque-like aggregates in the neuropil of ma and oldice, highly restricted to distinct layers of the hippocam-
al formation including the dorsal subiculum, CA so, sr, andentral CA slm and outer ml of the DG (arrows in Fig. 1D,, G and H). In addition, layer I of the entorhinal and piri-
orm cortex contained Reelin plaques. Except for a few singleeposits in the somatosensory cortex and caudate putamen inld subjects, no other brain region examined contained theseeposits. In order to rule out that these aggregates simply
eflect non-specific antibody labeling, we performed a seriesf control stainings of the same brain sections using mouser rabbit IgG, rabbit pre-immune serum, or the secondaryntibodies alone (Supplementary Fig. 1). None of these
logy of
seansliCarSdwah
3cm
ptanSgovtapFoolfti(ttamctpsroCyrntc
ccIcira
3b
tTatecStGtsAwRcbpeptatm
aaetflwispsKpamtw
I. Knuesel et al. / Neurobio
tainings produced any immunoperoxidase reactivity on thextracellular aggregates. With immunofluorescent secondaryntibodies, only the intracellular age-pigment lipofuscin wason-selectively labeled by the secondary antibodies (data nothown). These data confirm that the Reelin-IR in extracellu-ar deposits is not a staining artifact but show that Reelins specifically localized to these aging-related aggregates.loser examinations of these accumulations revealed a tightssociation with Reelin-expressing neurons and a granularather than fibrillary appearance at 1 year of age (Fig. 1K).imilar appearances albeit often associated with a moreiffuse and irregular shape was evident in two years oldild-type mice (Fig. 1L). These results suggest that Reelin
ccumulates in amyloid-like aggregates specifically in theippocampal formation of aged wild-type mice.
.2. Neuronal loss and increase in plaque load bothontribute to the memory deficits observed in wild-typeice
We quantified the number of both Reelin neurons andlaques in the dorsal and ventral hippocampus obtained fromhe behaviorally tested animals by stereology. The statisticalnalysis confirmed the profound loss of Reelin-expressingeurons in the old compared to ma and ad subjects (p < 0.01).eparate comparisons for the subregions yielded significantroup differences for the dorsal (ad versus old, ma versusld: p < 0.05) and ventral hippocampus (ad versus old, maersus old: p < 0.01; Fig. 2A). Similarly, statistical evalua-ions of the plaques yielded significant differences betweend and ma mice for both the dorsal and ventral parts (all< 0.01) but not between ad and old subjects (all p > 0.05,ig. 2B), reflecting reduced production and eliminationf plaques in a subgroup of old subjects. However, a fewld mice were characterized by a marked increased plaqueoad (outlier in Fig. 8A), suggesting that these individualsail to remove the protein aggregates. By expressing thewo variables as a ratio, cells/plaques, we obtained signif-cant reductions in ma compared to ad mice in the dorsalp < 0.05) and between both aged groups compared to ad inhe ventral hippocampus (all p < 0.01, Fig. 2C), supportinghe view that the combination of cell loss and plaqueccumulation may account for age-related hippocampalemory impairments. This hypothesis was supported by the
orrelational analysis between the ratio of cells/plaques andhe mean performance on the RAM revealing a significantositive relationship (df = 31, r = 0.46, p = 0.006). A similarignificant relationship was evident when comparing theatio of cells/plaques and the mean performance focusingn RI 1 and 2 (Fig. 2D, df = 31, r = 0.499, p = 0.003).orrelational analysis of the three age groups separatelyielded a statistical significant relationship for ma (df = 8,
= 0.66, p = 0.03) and old (df = 14, r = 0.63, p = 0.007), butot for the ad subjects (df = 5, r = 0.09, p = 0.86). However,wo individual outliers were responsible for the significantorrelation (Fig. 2E). This is reminiscent of the variability inhtlp
Aging 30 (2009) 697–716 703
ognitive decline among elderly subjects and confirms thatognitive aging is not strictly linked to chronological age.ndeed, removing these two outliers from the analysis did nothange the overall correlation (df = 29, r = 0.444, p = 0.011),ndicating that the morphological deficit may indeed rep-esent a good predictor of the cognitive performance indulthood.
.3. Reelin plaques are extracellular deposits invadedy activated microglia and astrocytes
Degenerating tissue and deposition of insoluble pro-ein aggregates are stimulants of inflammatory responses.o determine whether Reelin plaques are associated withctivated microglia and reactive astrocytes, we performedriple-fluorescence labeling using GFAP and F4/80 as mark-rs for astrocytes and activated microglia, respectively, inombination with Reelin-antibodies and the nuclear dyeytox green on brain sections of ma behaviorally naıve wild-
ype mice (Fig. 3A–H). We found a local increase in bothFAP and F4/80-IR in layers with high plaque load, par-
icularly prominent in slm. The astrocytic processes closelyurrounded but did not co-localize with the plaques (Fig. 3D).ctivated microglia were found in high densities around andithin the plaques, as indicated by the co-localization ofeelin and F4/80 (Fig. 3H, arrowheads). Moreover, microgliao-localized with Reelin in degenerating neurons, as showny the presence of condensed and shrunken Sytox green-ositive nuclei (Fig. 3E–H, arrows). The inset in H shows annlarged view of a degenerating cell within a Reelin-positivelaque. These results confirm the extracellular localization ofhese plaques and further suggest that their presence triggerslocal neuroinflammatory response that might contribute to
he elimination of these proteinous deposits in old wild-typeice.Reelin, via its interaction with ApoER2 and subsequent
ssociation of Dab1 and Fe65, modulates APP processingnd secretion of A� peptides in neuronal cell cultures (Hoet al., 2006a,b). Here, we tested whether the expression ofhese proteins are affected by the reduction of Reelin. Double-uorescent labeling on hippocampal brain sections of maild-type mice showed that Reelin plaques (Fig. 3I) differed
n terms of the size of individual aggregates. ApoER2, whichhowed a strong clustered IR throughout the hippocam-al formation (Fig. 3J–L), was particularly enriched aroundmaller, potentially developing aggregates (Fig. 3, inset in
is enlarged in L) and formed there larger clusters com-ared to the plaque-free neuropil. The bigger Reelin-positiveggregates were not co-stained for ApoER2. ApoER2-IR wasuch weaker in neocortical and other brain areas as compared
o the hippocampus (data not shown). Both Dab1 and Fe65ere highly enriched in large Reelin plaques throughout the
ippocampal formation (Fig. 3M–P). These results indicatehat altered levels of Reelin are associated with the accumu-ation of signaling proteins involved in APP trafficking androcessing.
704 I. Knuesel et al. / Neurobiology of Aging 30 (2009) 697–716
Fig. 2. Neuronal loss and increase in plaque load contribute to the episodic-like memory deficits observed in aged wild-type mice. (A) Statistical analysis of thenumber of Reelin-expressing neurons revealed a significant axis × age interaction (F2,30 = 4.3, p < 0.05), and significant main effects of age (F2,30 = 8.5, p < 0.01)and axis (F1,30 = 145.9, p < 0.001). The interaction reflects the more pronounced neuronal loss in ventral than in dorsal subfields of old compared to ma and admice. (B) Similarly, statistical evaluations of the plaques yielded an age × axis interaction (F2,30 = 5.9, p < 0.01) and a significant effect of axis (F1,30 = 50.9,p ues in thr RI = 1s
3c
Rn
ce
< 0.001). (C) Group comparisons of the ratio of Reelin-positive cells/plaqatio cells/plaques and the mean performance in the memory task includingignificance based on Fisher’s LSD post hoc analysis.
.4. Reelin plaques are found in aged Wistar rats andommon marmoset monkeys
To determine whether the pronounced accumulation ofeelin plaques accompanying neuronal loss was a phe-omenon unique to mice or a more general morphological
asas
e dorsal and ventral hippocampus. (D and E) Correlational analyses of theand 2. Values are given as mean ± S.E.M. *p < 0.05, **p < 0.01, statistical
haracteristic of normal aging, we performed a qualitativevaluation of rat and monkey brain tissue obtained from adult
nd aged subjects processed for Reelin immunohistochemi-try (Fig. 4). A similar Reelin-IR was evident in rat tissues described in mice and was restricted to interneurons ino, sr, and slm, as well as DG gcl, ml, and hilus (Fig. 4A).
I. Knuesel et al. / Neurobiology of Aging 30 (2009) 697–716 705
Fig. 3. Reelin plaques are extracellular accumulations invaded by activated microglia and astrocytes. (A–H) Representative high-magnification images oftriple-immunofluorescence (IF) stainings of the ventral slm of 12-month-old wild-type mice using mouse anti-Reelin antibodies in combination with antibodiesraised against markers of astrocytes (GFAP) and activated microglia (F4/80). Nuclei are counterstained with SytoxTM green. Note the close association ofboth astrocytes and activated microglia with Reelin plaques (D and H). Co-localization of Reelin and F4/80 is evident within the plaque-like accumulations(arrowheads in E, F and H). Arrows and the inset in H point to condensed nuclei, invaded by activated microglia and likely representing degenerating Reelin-positive cells. (I–P) Members of the amyloid precursor protein signaling pathway are localized to Reelin-positive hippocampal plaques in wild-type mice.Representative images of double-IF stainings of the ventral hippocampus using mouse anti-Reelin and antibodies raised against the Reelin receptor ApoER2(I–L), and the intracellular adaptor proteins Dab1 (M and N) and Fe65 (O–P). (I) Reelin plaques differ in size and shape: the arrow points to large accumulations;arrowheads to smaller Reelin aggregates. (J and K) Clusters of ApoER2 are in immediate contact with the Reelin plaques (green), predominantly with thesmaller aggregates (inset in K is enlarged in L). (M–P) Immunostaining of the intracellular adaptors Dab1 and Fe65 in the hippocampus reveals extracellulara P). Sca
Wtcs
ccumulations highly co-localizing with the Reelin plaques (insets in N und
e did not detect Reelin plaques in 6-month-old rats buthe dorsal and ventral hippocampus of 2-year-old subjectsontained Reelin-positive aggregates in the neuropil of so,r, and slm (high magnification in Fig. 4D). However, at
2por
le bars: (A, E, M and O) 15 �m; (I) 10 �m; (L) 5 �m.
years of age the ventral slm did not contain numerouslaques but was characterized by strikingly low numbersf Reelin-expressing neurons, suggesting a pronounced age-elated loss of GABAergic cells (Fig. 4B, arrows). In addition,

706 I. Knuesel et al. / Neurobiology of Aging 30 (2009) 697–716
Fig. 4. Reelin plaques are found in aged Wistar rats and common marmoset monkeys. Low and high magnification images of coronal brain sections takenfrom 6- (A) and 20-month (B–D) old rats, and 1- (G–H) and 10-year-old monkeys (F) visualized with Reelin immunohistochemistry. (A) No Reelin plaquesare observed in 6-month-old rats. (B–D) Reelin plaques and Reelin expressing cells with dystrophic neurites (arrows in C) are found in the hippocampus of20-month-old rats. Note the loss of Reelin neurons in slm in the ventral hippocampus (arrows in B) of the 20-month-old subject. (E) Schematic representationof the monkey hippocampus. (F) Weak immunoreactivity of Reelin-expressing cells is evident in 10-year-old marmoset brain sections but large extracellularplaques are found in both sr and slm (arrows). (G–H) Aggregated Reelin-IR is present both intracellularly and extracellularly on hippocampal brain sectionsof the 1-year-old monkey. Scale bars: (A) 200 �m; (C and H) 20 �m; (E) 500 �m; (G) 10 �m.

logy of
m(wAmRhiobTmen
3R
siwdchw1sl(rtsvmerFamuptdaro(nR2cnln
3an
iesYceivpIotmm(ntdoaacyiTdCRhpmcogm
3c
dtIA(ci
I. Knuesel et al. / Neurobio
any Reelin neurons with dystrophic neurites were foundFig. 4C). A similar Reelin-IR pattern as described in rodentsas evident in the 1- and 10-year-old monkey hippocampus.lthough the intensity of Reelin-IR in neuronal soma wasuch weaker compared to rodent tissue (Fig. 4F and H),eelin plaques were clearly identifiable in sr and slm of theippocampus of both the 1- and 10-year-old monkey (arrowsn F). Higher magnification revealed intracellular aggregatesf Reelin throughout the hippocampal formation (Fig. 4G)ut not in cortical or non-cortical brain areas (data not shown).hese results suggest that Reelin plaques represent a generalorphological phenomenon, related to the loss of Reelin-
xpressing interneurons in the hippocampal formation duringormal aging.
.5. Heterozygous SynGAP knockout mice have lesseelin neurons and develop fewer plaques
To directly test the hypothesis of a predictive relation-hip between Reelin-expressing neurons and plaques, wenvestigated in mice with neurodevelopmental abnormalitieshether an early loss of GABAergic interneurons affects theevelopment of Reelin plaques during aging. We employed aohort of SynGAP (synaptic Ras-GTPase activating protein)eterozygous knockout (SG+/−) mice and their littermateild-type controls (SG+/+) at the age of 3 (ad), 6 and2 (ma), and 24 months (old). The complete loss of theynaptic Ras/Rap inhibitor SynGAP leads to early apoptoticoss of GABAergic neurons and death 2–3 days after birthKnuesel et al., 2005). Here, we investigated whether neu-onal loss was also evident in adult SG+/− mice and affectedhe Reelin-expressing class of GABAergic interneurons. Thetereological estimates of Reelin neurons in the dorsal andentral hippocampus of ad, ma, and old SG+/+ and SG+/−ice revealed that SG+/− mice had significantly less Reelin-
xpressing cells in the hippocampus than SG+/+ (p = 0.003),egardless of age (ad, p = 0.05; ma, p = 0.007; old, p = 0.03,ig. 5B). The effect was particularly prominent in CA sond slm (Fig. 5A and C) and affected the ventral subfieldore than the dorsal. Double-immunofluorescence staining
sing Reelin and SynGAP antibodies on old SG+/+ hippocam-al brain sections confirmed the co-localization of thesewo proteins in both healthy (Fig. 5D–F, arrows) as well asegenerating GABAergic interneurons (arrowheads). Evalu-tion of Reelin plaques in this cohort of mice (Fig. 5G–K)evealed significantly lower levels in the ventral hippocampusf 12-month-old SG+/− compared to age-matched controlsp < 0.05), whereas the difference in the dorsal subfields didot reach statistical significance (Fig. 5I). The number ofeelin plaques was not different between genotypes in either4-month-old or less than 6-month-old mice (Fig. 5L), indi-
ating that the lower plaque load in 12-month-old mice wasot due to an earlier and faster accumulation of extracellu-ar aggregates but related to the reduced number of Reelineurons in SG+/− mice.tcat
Aging 30 (2009) 697–716 707
.6. Prenatal immune challenge results in earlierccumulations of Reelin plaques and loss of Reelineurons
To determine whether a prenatal immune challenge lead-ng to cytokine-associated fetal brain inflammation (Meyert al., 2006) and changes in the expression levels ofeveral immune modulators in adulthood (Meyer, Nyffeler,ee, Feldon, Knuesel, unpublished data) exerts long-termonsequences on the development of Reelin plaques, wevaluated the plaque-load using Reelin immunohistochem-stry in adult wild-type mice prenatally exposed to eitherehicle (CON) or the synthetic viral mimic polyriboinosinic-olyribocytidilic acid (PolyIC; Meyer et al., 2005, 2006).n line with our previous observations, CON subjects hadnly few plaques at 6 months of age (Fig. 6A). In contrast,he hippocampus and entorhinal cortex of PolyIC-exposedice contained greater numbers of Reelin plaques, againost prominent in the ventral CA so, sr, slm, and DG ml
Fig. 6B and D), as well as layer I of the lateral entorhi-al cortex. Cresyl violet counterstainings confirmed thathe Reelin plaques were extracellular (data not shown) asescribed in wild-type subjects. In line with the previousbservations in mice and rats, the increase in plaques wasccompanied by a loss of Reelin neurons in CA so, slm,nd DG ml, as shown by unbiased stereological quantifi-ation. Statistical analyses of the number of Reelin neuronsielded significant differences between the CA and DG areas,rrespective of the septo-temporal axis (Fig. 6E, p < 0.05).he number of plaques was significantly increased in bothorsal and ventral hippocampus in PolyIC compared toON subjects (p < 0.05). Correspondingly, the ratio betweeneelin-positive cells and plaques combined for the wholeippocampus dropped significantly in PolyIC subjects com-ared to CON (p = 0.015), similar to the comparison betweena and old wild-type mice (Fig. 2C). These results indi-
ate that a prenatal immune challenge accelerates the lossf Reelin neurons and the accumulation of Reelin aggre-ates in the hippocampal formation of adult wild-typeice.
.7. Reelin plaques are highly increased in AD mice ando-localize with non-fibrillary species of amyloid-beta
Reelin-IR has been detected in plaques of APP/PS1ouble-transgenic AD mice (Wirths et al., 2001) and linkedo APP processing and A� production (Hoe et al., 2006a).n view of the prominent accumulations of protofibrillary� plaques in 12-month-old triple transgenic AD mice
3xTg-AD, Oddo et al., 2003), we evaluated whether Reelino-localizes with A� plaques in this animal model of AD. Wenvestigated brain tissue obtained from 15-month-old non-
ransgenic control (CON) and 3xTg-AD mice. In CON, aomparable accumulation pattern and load of plaques as inge-matched C57Bl6 wild-type mice was evident. In con-rast, 3xTg-AD mice showed a pronounced increase in Reelin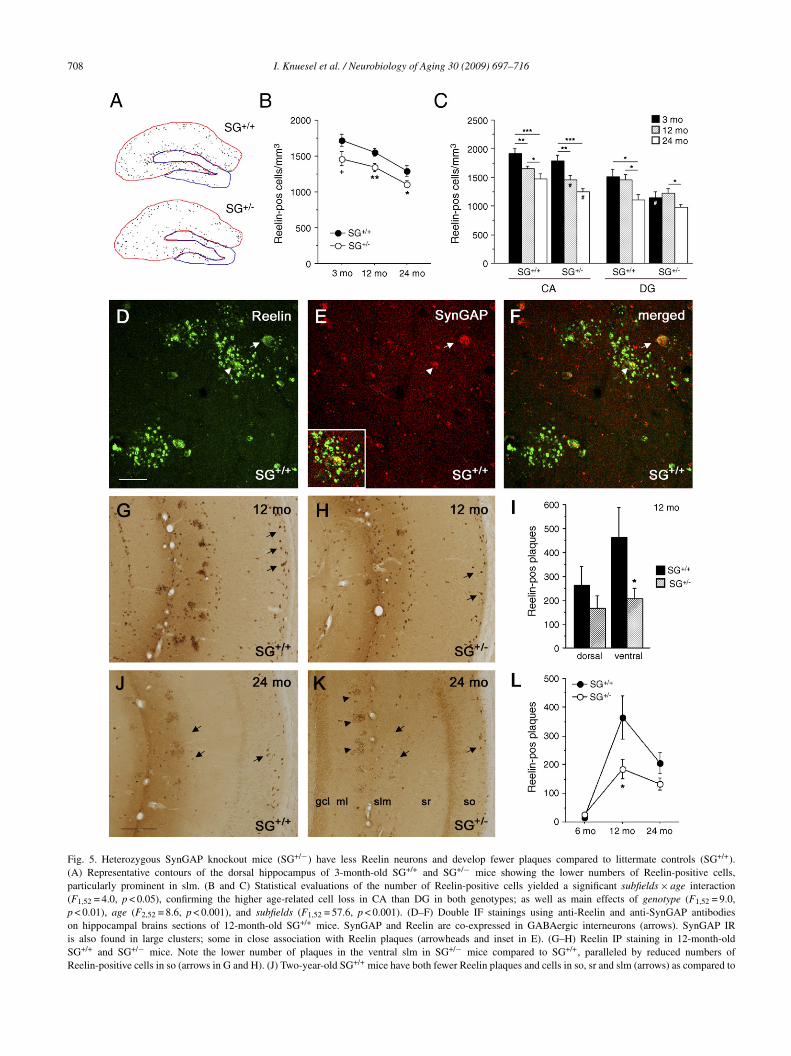
708 I. Knuesel et al. / Neurobiology of Aging 30 (2009) 697–716
Fig. 5. Heterozygous SynGAP knockout mice (SG+/−) have less Reelin neurons and develop fewer plaques compared to littermate controls (SG+/+).(A) Representative contours of the dorsal hippocampus of 3-month-old SG+/+ and SG+/− mice showing the lower numbers of Reelin-positive cells,particularly prominent in slm. (B and C) Statistical evaluations of the number of Reelin-positive cells yielded a significant subfields × age interaction(F1,52 = 4.0, p < 0.05), confirming the higher age-related cell loss in CA than DG in both genotypes; as well as main effects of genotype (F1,52 = 9.0,p < 0.01), age (F2,52 = 8.6, p < 0.001), and subfields (F1,52 = 57.6, p < 0.001). (D–F) Double IF stainings using anti-Reelin and anti-SynGAP antibodieson hippocampal brains sections of 12-month-old SG+/+ mice. SynGAP and Reelin are co-expressed in GABAergic interneurons (arrows). SynGAP IRis also found in large clusters; some in close association with Reelin plaques (arrowheads and inset in E). (G–H) Reelin IP staining in 12-month-oldSG+/+ and SG+/− mice. Note the lower number of plaques in the ventral slm in SG+/− mice compared to SG+/+, paralleled by reduced numbers ofReelin-positive cells in so (arrows in G and H). (J) Two-year-old SG+/+ mice have both fewer Reelin plaques and cells in so, sr and slm (arrows) as compared to

I. Knuesel et al. / Neurobiology of Aging 30 (2009) 697–716 709
Fig. 6. Prenatal immune challenge results in earlier accumulations of Reelin plaques and loss of Reelin neurons. Representative images of coronal brain sectionsprocessed for Reelin immunohistochemistry taken from 6-month-old wild-type mice prenatally exposed to either vehicle (CON) or the synthetic viral mimicpolyriboinosinic-polyribocytidilic acid (PolyIC), a synthetic analogue of double-stranded RNA. (A and B) The IP labeling shows the early accumulation ofplaques in the ventral slm (arrows) of PolyIC subjects while in CON only a few Reelin plaques are found at this age. (C and D) Higher magnification of the DGshows the presence of Reelin plaques in the most outer region of the molecular layer. Note the concomitant reduction of Reelin-positive cells. (E) Statisticalanalyses of the number of Reelin neurons yielded a significant subfield × treatment interaction (F1,26 = 5.6, p = 0.025), reflecting the selective neuronal loss inCA in the PolyIC as compared to CON; and significant main effects of axis and subfield (both F1,26 > 131, p < 0.0001). For the plaques, significant main effectsoPp
1o(pmp
f treatment (F1,26 = 5.03, p = 0.03) and axis (F1,26 = 12.1, p = 0.002) were found. TolyIC subjects compared to CON (p = 0.015). All values are given as mean ± S.Eost hoc analysis. Scale bars: (A) 500 �m; (C) 50 �m; (E) 20 �m.
-year-old SG+/+ mice. (K) A reduction of Reelin-positive cells is also evident in 2-f plaques and reduced Reelin-immunoreactivity in the neuropil of the slm. ReelinI) Statistical evaluation of Reelin plaques in the dorsal and ventral hippocampus in< 0.05) and a main effect of axis (F1,14 = 12.9, p < 0.01). (L) Comparison of age-reice. All values are given as mean ± S.E.M. +p = 0.05, *p < 0.05, **p < 0.01, #p < 0.
ost hoc analysis. Scale bars: (D) 30 �m; (J) 200 �m.
he ratio between Reelin-positive cells and plaques dropped significantly in.M. *p < 0.05; ***p < 0.001, statistical significance based on Fisher’s LSD
year-old SG+/− mice compared to the 1-year-old subjects. Note the absenceplaques are mostly localized in the outer layer of the DG ml (arrowheads).SG+/+ and SG+/− mice revealed a axis × genotype interaction (F1,14 = 5.7,lated plaque changes (averaged for dorsal and ventral) in SG+/+ and SG+/−05 for genotype comparisons, statistical significance based on Fisher’s LSD

710 I. Knuesel et al. / Neurobiology of Aging 30 (2009) 697–716
Fig. 7. Reelin plaques are highly increased in Alzheimer’s disease mice and co-localize with non-fibrillary species of amyloid-�. Representative contours ofthe dorsal (upper drawings) and ventral (lower drawings) hippocampus of 15-month-old non-transgenic (CON) and triple-transgenic (3xTg-AD) mice. Reddots represent the individual Reelin plaques. (C–I) Double IF staining of the dorsal CA1 sr of 3xTg-AD mice using anti-Reelin combined with anti-human A�
and anti-A� oligomer species antibodies. (C–E) A�-IR is present in both fibrillary plaques (arrow) and Reelin neurons. (F–H) A�-IR is also present in Reelinplaques, likely representing non-fibrillary forms of extracellular plaques, as supported by the co-localization of Reelin with the A11 anti-A� oligomer-specificantibody (I). (J) Rodent-specific fibrillary A� plaques (arrow) are also found in 3xTg-AD mice, preferentially localized around the Reelin plaques (enlargedview in inset). (K) A selective association of Reelin- and A�-IR is also evident in CON wild-type littermates. (L) Single-labeled A� oligomer-specific depositsare found in neocortical layer I. (M) Statistical evaluation of the Reelin plaques in the dorsal and ventral hippocampus of CON and 3xTg-AD mice revealeda main effect of genotype (F1,16 = 7.4, p = 0.015) and axis (F1,16 = 24.6, p < 0.0001). Separate comparisons for the dorsal and ventral hippocampus yieldeds 1), andV d L) 20
p(cptT�
scp
ignificant differences between CON and 3xTg-AD in the ventral (**p = 0.0alues are given as mean ± S.E.M. Scale bars: (C, F and I) 10 �m; (J, K an
laques, especially in the dorsal and ventral CA sr and slmFig. 7A and B). The statistical analysis yielded a signifi-ant genotype effect (p < 0.01) and confirmed again the more
ronounced effect in ventral (p < 0.01) as compared withhe dorsal hippocampus (p = 0.052, Fig. 7M) in 3xTg-AD.o examine whether the Reelin plaques co-localized with-amyloid plaques in 3xTg-AD, we performed double-IFHbfip
a close to significant difference in the dorsal subfields (+p = 0.05, Fig. 7L).�m.
taining using antibodies against Reelin and human A�. Weonfirmed the presence of the characteristic fibrillary A�laques as described (Oddo et al., 2003; Fig. 7D, arrow).
uman A�-IR was present in Reelin-expressing neurons,ut no co-localization of the two markers was found inbrillary plaques (Fig. 7C–E). However, some A�-IR wasresent in non-fibrillary plaques, largely co-localizing with
logy of
RRrpfiaIdfiwgtAaAcatcisptp
4
rRnaWRtirad
(rCeM
Fp7a
I. Knuesel et al. / Neurobio
eelin-IR (Fig. 7F–H). Double-IF using antibodies againsteelin and A� oligomer species (A11, Kayed et al., 2003)
evealed a highly similar IR pattern, suggesting that plaquesositive for human A� and Reelin represent soluble, non-brillary amyloid deposits (Fig. 7I). While we observed anlmost complete overlap between Reelin- and A� oligomer-R, single-labeled A�-containing oligomeric species wereetected outside the hippocampal formation (Fig. 7L), con-rming the specificity of the antibody stainings. To determinehether Reelin is also localized in endogenous A� aggre-ates, we next stained the sections with antibodies againsthe rodent �-amyloid and Reelin (Fig. 7J). Rodent-specific�-IR was localized in fibrillary amyloid deposits, closely
ssociated with the Reelin deposits (arrow in J) in 3xTg-D mice. Due to the high sequence similarity and potential
ross-reactivity between the rodent and human-specific A�ntibodies, we also stained control sections of wild-type lit-ermates. We observed a selective association and partialo-localization between Reelin and A�-positive aggregatesn the hippocampus of control mice (Fig. 6K). These data
uggest that the accumulation of Reelin in non-fibrillarylaques may promote the aggregation of A� species; poten-ially creating a nucleation site for fibrillary amyloid A�laques.RpiC
ig. 8. Comparison of the Reelin plaque load during normal aging (A), in animalsrenatal immune challenge (C), and neurodegenerative disorders (D). All values are5th (top) and 25th percentile (bottom) and the median line. The upper and lower erbove the 90th and below 10th percentile are represented as black dots.
Aging 30 (2009) 697–716 711
. Discussion
We report the novel finding that normal aging inodents and primates is accompanied by accumulation ofeelin-positive deposits and reduction of Reelin-expressingeurons in the hippocampal formation. Both phenomenare associated with episodic-like memory impairments.e provide evidence that normal aging involves loss ofeelin-positive neurons, reduced production, and elimina-
ion of the extracellular deposits, whereas prenatal brainnflammation or the expression of AD-causing gene productsesults in earlier, higher, and more persistent levels of Reelinggregates, potentially contributing to progressive cognitiveecline (Fig. 8).
The distribution of Reelin-IR described hereFigs. 1 and 4) is in accordance with previous studies,evealing its expression in GABAergic interneurons, adultajal-Retzius cells, and pyramidal cells in layer II of thentorhinal cortex in rodents and humans (Abraham andeyer, 2003; Alcantara et al., 1998; Miettinen et al., 2005;
amos-Moreno et al., 2006; Riedel et al., 2003). Therominent loss of Reelin neurons in CA so, slm and DG mls in line with reductions in the GABAergic markers GAD67,albindin, and Somatostatin in the aged rat hippocampuswith genetically induced neurodevelopmental abnormalities (B), followingdisplayed in box plot graphs. The dimensions of the box are defined by the
ror bars indicate the 90th and 10th percentile, respectively. All observations

7 logy of
(sSfeCc
∼scoBncbtssitvmha2ivaWrbia
epoIt2tOhaeac(Mtwttow
ad2sdmisaspehfFsnted
olnsbeutirpegp
riatpibtmvcdAtai
12 I. Knuesel et al. / Neurobio
Potier et al., 1994; Shetty and Turner, 1998), and Somato-tatin/NPY in APP/PS1 transgenic mice (Ramos et al., 2006).ubfield- and layer-specific reductions have been reportedor Parvalbumin, Calretinin, and Calbindin, as well as a mod-rate (Riedel et al., 2003) to severe loss of Reelin-positiveajal-Retzius cells (Baloyannis, 2005) and layer II pyramidalells in the AD entorhinal cortex (Chin et al., 2007).
Hippocampal GABAergic interneurons represent only10% of the total hippocampal population, but their exten-
ively ramified axons innervate several thousand pyramidalells, thereby playing a major role in synchronizationf neuronal activity and network oscillations (Freund anduzsaki, 1996). They are also the target of several subcorticaleurotransmitter systems, including septal GABAergic andholinergic, as well as monoaminergic projections fromrainstem areas. Although the cholinergic components con-act all neuronal types in the hippocampus, the GABAergiceptohippocampal fibers terminate on interneurons exclu-ively (Freund and Buzsaki, 1996). A loss of Reelin cellsn hippocampal subfields is expected to profoundly affecthe afferent subcortical systems and alter hippocampal acti-ity and function. A deficit in cholinergic neurotransmissionay underlie the episodic-like memory impairments reported
ere (Fig. 1), in line with the reduction of cholinergic activityccompanying normal aging and AD (Sarter and Bruno,004). Complementary glutamatergic changes are likelynvolved in age-related memory impairments, potentiallyia reduced Reelin-mediated modulation of NMDA receptorctivity and LTP (Beffert et al., 2005; Chen et al., 2005;eeber et al., 2002). It is conceivable that age-related
eductions of Reelin indirectly affect cognitive performancesy favoring the production of soluble A� species, shown tompair LTP (Rowan et al., 2005; Walsh et al., 2002; Wang etl., 2002).
To the best of our knowledge, no data are available on thexistence of Reelin-positive accumulations in the hippocam-al formation of wild-type mice, rats, non-human primatesr humans. Three studies described Reelin- and ApoER2-R, respectively, in neuritic compartments of A� plaques inransgenic AD mice (Miettinen et al., 2005; Motoi et al.,004; Wirths et al., 2001). It is unclear, however, whetherhey were associated with non-fibrillary or fibrillary plaques.n the other hand, our observations fit very well with earlyistological descriptions of age-associated morphologicalnd pathological changes in rodent brains reporting the pres-nce of granular/fibrillary deposits in the hippocampus ofged C57Bl/6, as well as in adult immunologially defi-ient athymic nude and senescence accelerated prone miceAkiyama et al., 1986; Jucker et al., 1994a; Lamar et al., 1976;
andybur et al., 1989). Several lines of evidence suggesthat these non-characterized, proteinous deposits are identicalith the Reelin aggregates described here: (1) The descrip-
ion of their morphology and layer-specific accumulation inhe hippocampal formation are in full agreement with ourbservations. (2) In line with our findings, these depositsere found in close association with astrocytes. (3) Jucker
t
pR
Aging 30 (2009) 697–716
nd colleagues report a highly comparable time course ofeposit formation with a peak at 18 months, a decline at4 months, and high variability at 30 months, likely repre-enting the few subjects with persistent high plaque load asescribed here. (4) We also share the observation that femaleice are more affected than males. The immunohistochem-
cal findings described by Jucker and colleagues includedeveral extracellular matrix proteins, including Laminin B2,
110 kDa Laminin-binding protein, as well as Heparin-ulfate-proteoglycan (Jucker et al., 1994b). Interestingly,roteolytic activity of Reelin on adhesion molecules of thextracellular matrix (including Laminin and Fibronectin)as been described (Quattrocchi et al., 2002). However, weailed to see any immunoreactivity of Laminin isoforms oribronectin within the Reelin plaques (data not shown). Sinceome of the antibodies might have labeled these structureson-specifically (M. Jucker, personal communication), fur-her proteomic characterizations are needed to identify thexact composition of these aging-related extracellular Reelineposits.
No immunohistochemical evidence for co-localizationf Reelin and A� plaques in AD has been provided,ikely because of the selective association of Reelin withon-fibrillary plaques (Fig. 7). Altered expression and glyco-ylation patterns of Reelin in CSF and cortical extracts haveeen reported in AD (Botella-Lopez et al., 2006; Saez-Valerot al., 2003). A 180-kDa Reelin fragment is preferentiallyp-regulated, while full-length and other forms remain unal-ered in control and AD subjects (Botella-Lopez et al., 2006),ndicating that the proteolytic processing alone cannot beesponsible for these alterations. The different glycosylationattern of the 180-kDa Reelin fragment in AD (Botella-Lopezt al., 2006) and our data obtained from 3xTg-AD mice sug-est that the increase in Reelin in AD may be linked to itsresence in soluble, non-fibrillary A� plaques.
The characterization of Reelin plaques in wild-type miceevealed accumulations of ApoER2 in potentially develop-ng plaques, and the intracellular adaptor molecules Dab1nd Fe65 in large extracellular aggregates (Fig. 3), indicatinghat aging-induced Reelin reduction may provoke abnormalrotein processing and degradation of downstream signal-ng proteins in wild-type mice. This hypothesis is supportedy studies in transgenic animals showing ectopic accumula-ions of Dab1 in Reelin-, ApoER2-, and VLDLR-deficient
ice (Rice and Curran, 2001; Trommsdorff et al., 1999). In-itro studies showed that internalization of Reelin-ApoER2omplexes is required for endocytosis and proteasomal degra-ation of Dab1 (Bock et al., 2004; D’Arcangelo et al., 1999).direct role of Reelin in APP processing is demonstrated by
he increased co-immunoprecipitation of Dab1 with ApoER2nd APP and decreased A� production (Hoe et al., 2006a),ndicating that reduced Reelin levels might favor the produc-
ion of neurotoxic A� species.Although we do not know yet whether the loss of Reelin-ositive neurons is a direct cause or a consequence of theeelin aggregates, several lines of evidence suggest that

logy of
tddciamwontr(ri
vpmstcmesnolitiodsIim(Tpame(otmlietR2
bm
of2omsfpaAa(pmwitobsRrwraifhbpaoatwsttip
pappsfwRtaT
I. Knuesel et al. / Neurobio
hese two phenomena are related: Staining with the nuclearye, Sytox green, and the microglial marker F4/80 revealedegenerating Reelin cells within plaques, as indicated by theondensed nuclei (Fig. 3). Plaque production decreased dur-ng aging after reduction of Reelin neurons in both micend rats (Figs. 1 and 4). SynGAP heterozygous knockoutice have fewer Reelin cells and develop fewer plaques thanild-type littermates (Fig. 5). These findings extend previ-us observations in mutant SynGAP mice showing increasedeuronal apoptosis during late embryonic and early postna-al development (Knuesel et al., 2005). These mice also haveeduced LTP and perform poorly in spatial memory tasksKim et al., 2003; Komiyama et al., 2002), indicating that neu-odevelopmental abnormalities affecting Reelin expressionmpair cognitive performance in adulthood.
The association of Reelin plaques with astroglia and acti-ated microglia (Fig. 3) indicates activation of inflammatoryrocesses. The observation of reduced plaque load in oldice and rats compared to ma wild-types (Figs. 1, 4 and 5)
uggests a beneficial immune response potentially limitinghe progression of neurodegeneration. On the other hand,hronic activation of inflammatory pathways can be detri-ental and promote degeneration, as suggested by increased
xpression of inflammatory mediators in postmortem AD tis-ue (Akiyama et al., 2000). However, it is unclear whethereuroinflammation represents a cause or consequence ofngoing disease processes. Based on genetic and epidemio-ogical studies demonstrating a link between polymorphismsn cytokines and other immune molecules with AD, as well ashe beneficial effect of anti-inflammatory drugs, respectively,nflammation has been considered a critical driving forcef AD (for review, Wyss-Coray, 2006). We have recentlyemonstrated that a prenatal immune challenge results inignificant increases in fetal brain cytokine levels, includingL1�, IL6, IL10, and TNF�, neuromorphological alterationsn juvenile and adult brains, and multiple behavioral abnor-
alities in the adult offspring, including cognitive deficitsMeyer et al., 2005; Meyer et al., 2006; Nyffeler et al., 2006).he findings presented here demonstrate that early mani-ulations of inflammatory pathways can change the coursend severity of neurodegenerative processes in wild-typeice. In particular, prenatal PolyIC exposure provokes an
arlier and much higher accumulation of Reelin plaquesFigs. 2 and 6) and suggests that the long-term consequencesf an early immune challenge favor the development of pro-einous aggregates in the hippocampal formation of wild-type
ice. We also report accelerated loss of Reelin neurons fol-owing PolyIC exposure. Considering the moderate cell lossn ma mice, these findings suggest that the prenatal PolyICxposure affects the survival of Reelin neurons in the postna-al brain, in line with our previous observation of a reducedeelin-IR in the hippocampus of juvenile mice (Meyer et al.,
006).A marked and prolonged increase in Reelin plaques num-er was found in 3xTg-AD mice (Fig. 7). These transgenicice are characterized by early intraneuronal accumulation
cfnc
Aging 30 (2009) 697–716 713
f A�, deficits in LTP and learning and memory before theormation of fibrillary plaques and tangles (Billings et al.,005; Oddo et al., 2003). Immunotherapy results in clearancef intracellular A� and improvement in cognitive perfor-ance (Billings et al., 2005; Oddo et al., 2004), implicating
oluble/intraneuronal rather than fibrillary A� as causativeactors of cognitive impairments. Although cognitive deficitsersist or worsen, intraneuronal A� is relatively transientnd declines with disease progression in humans and animalD models (Wirths et al., 2004). The reduction in Reelin
nd concomitant accumulation in non-fibrillary A� plaquesFig. 7H and I) in 3xTg-AD mice reported here provides arobable explanation for the exacerbating memory impair-ents. Furthermore, the finding that Reelin co-localizedith oligomeric A� species in the hippocampus but not
n other brain areas (Fig. 7L) confirms the specificity ofhe immunostaining and indicates that Reelin itself formsligomeric aggregates that are recognized by the A11 anti-ody. However, further biochemical evidence is needed toubstantiate these findings. Interestingly, we also observedeelin deposits that were surrounded by rodent-specific fib-
illary A� plaques in 3xTg-AD mice (Fig. 7J). Althoughe cannot rule out completely the possibility of human and
odent A�-specific antibody crosstalks, the observation ofpartial co-localization of Reelin plaques with rodent A�
n CON mice (Fig. 7K) indicates that Reelin deposits mayavor the development of fibrillary amyloid plaques. Thisypothesis is in line with the novel role of Reelin in the adultrain which point to a critical involvement of this signalingathway in the trafficking and processing of APP (Hoe etl., 2006a). On the other hand, is also conceivable that theverexpression of mutant human APP in the 3xTg-AD micend the extracellular accumulation of A� peptides induceshe aggregation of Reelin. Based on our findings in agedild-type rodents and marmoset monkeys we consider this
cenario unlikely. However, we cannot rule out that underhese non-physiological conditions in transgenic mice, pro-ein processing, degradation and clearance is likely severelympaired and may initiate a vicious cycle of extracellularrotein aggregations involving both Reelin and A� peptides.
In conclusion, our data revealed a strikingly consistentattern of age-related loss of Reelin-expressing neuronsnd concomitant accumulation in extracellular amyloid-likelaques in the hippocampal formation (Fig. 8). We pro-ose a model postulating that normal forms of aging involveuccessful elimination of plaques, whereas persons at-riskail to do so. This hypothesis is supported by the outlierild-type mice, characterized by unchanging high levels ofeelin plaques. Factors able to modulate course and magni-
ude of the plaque load include impaired immune responsesnd mutations favoring the production of toxic A� peptides.he reduced plaque number in animals with lower Reelin
ell levels suggests a link between neuronal loss and plaqueormation. Our results indicate that dysfunctional Reelin sig-aling may represent a causative factor in creating a precursorondition for senile plaque deposition in sporadic AD.
7 logy of
D
ohs
A
3taBm(Hth
A
f2
R
A
A
A
A
A
B
B
B
B
B
B
B
B
C
C
C
C
D
D
F
F
G
G
H
H
H
14 I. Knuesel et al. / Neurobio
isclosure statement
The authors declare that they have no competing financialr personal interests and that none of the author’s institutionsave contracts relating to this research through which it maytand to gain financially now or in the future.
cknowledgements
The present study was supported by SNF Grant Nr.100AO-100309 and Grants of the Swiss Federal Insti-ute of Technology Nr. TH-9/04-2 and TH-22/04-3. Were extremely grateful to Jean-Marc Fritschy and Mary. Kennedy for their support and critical reading of theanuscript, as well as providing us the SynGAP mutant mice
MBK); to Liz Weber, Corinne Sidler, Peter Schmid, andannes Sigrist for their excellent technical support; and to
he Animal Services Department Schwerzenbach for animalusbandry and care.
ppendix A. Supplementary data
Supplementary data associated with this article can beound, in the online version, at doi:10.1016/j.neurobiolaging.007.08.011.
eferences
braham, H., Meyer, G., 2003. Reelin-expressing neurons in the postnataland adult human hippocampal formation. Hippocampus 13, 715–727.
kiyama, H., Barger, S., Barnum, S., Bradt, B., Bauer, J., Cole, G.M.,Cooper, N.R., Eikelenboom, P., Emmerling, M., Fiebich, B.L., Finch,C.E., Frautschy, S., Griffin, W.S., Hampel, H., Hull, M., Landreth, G.,Lue, L., Mrak, R., Mackenzie, I.R., McGeer, P.L., O’Banion, M.K.,Pachter, J., Pasinetti, G., Plata-Salaman, C., Rogers, J., Rydel, R., Shen,Y., Streit, W., Strohmeyer, R., Tooyoma, I., Van Muiswinkel, F.L., Veer-huis, R., Walker, D., Webster, S., Wegrzyniak, B., Wenk, G., Wyss-Coray,T., 2000. Inflammation and Alzheimer’s disease. Neurobiol. Aging 21,383–421.
kiyama, H., Kameyama, M., Akiguchi, I., Sugiyama, H., Kawamata, T.,Fukuyama, H., Kimura, H., Matsushita, M., Takeda, T., 1986. Peri-odic acid-Schiff (PAS)-positive, granular structures increase in the brainof senescence accelerated mouse (SAM). Acta Neuropathol. (Berl) 72,124–129.
lcantara, S., Ruiz, M., D’Arcangelo, G., Ezan, F., de Lecea, L., Curran, T.,Sotelo, C., Soriano, E., 1998. Regional and cellular patterns of ReelinmRNA expression in the forebrain of the developing and adult mouse.J. Neurosci. 18, 7779–7799.
rnaud, L., Ballif, B.A., Cooper, J.A., 2003. Regulation of protein tyro-sine kinase signaling by substrate degradation during brain development.Mol. Cell. Biol. 23, 9293–9302.
ackman, L., Jones, S., Berger, A.K., Laukka, E.J., Small, B.J., 2004. Mul-tiple cognitive deficits during the transition to Alzheimer’s disease. J.Intern. Med. 256, 195–204.
aloyannis, S.J., 2005. Morphological and morphometric alterations ofCajal-Retzius cells in early cases of Alzheimer’s disease: a Golgi andelectron microscope study. Int. J. Neurosci. 115, 965–980.
effert, U., Durudas, A., Weeber, E.J., Stolt, P.C., Giehl, K.M., Sweatt, J.D.,Hammer, R.E., Herz, J., 2006. Functional dissection of Reelin signaling
H
Aging 30 (2009) 697–716
by site-directed disruption of Disabled-1 adaptor binding to apolipopro-tein E receptor 2: distinct roles in development and synaptic plasticity.J. Neurosci. 26, 2041–2052.
effert, U., Weeber, E.J., Durudas, A., Qiu, S., Masiulis, I., Sweatt, J.D., Li,W.P., Adelmann, G., Frotscher, M., Hammer, R.E., Herz, J., 2005. Mod-ulation of synaptic plasticity and memory by Reelin involves differentialsplicing of the lipoprotein receptor Apoer2. Neuron 47, 567–579.
effert, U., Weeber, E.J., Morfini, G., Ko, J., Brady, S.T., Tsai, L.H., Sweatt,J.D., Herz, J., 2004. Reelin and cyclin-dependent kinase 5-dependentsignals cooperate in regulating neuronal migration and synaptic trans-mission. J. Neurosci. 24, 1897–1906.
illings, L.M., Oddo, S., Green, K.N., McGaugh, J.L., LaFerla, F.M., 2005.Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 45, 675–688.
ock, H.H., Jossin, Y., May, P., Bergner, O., Herz, J., 2004. Apolipoprotein Ereceptors are required for Reelin-induced proteasomal degradation of theneuronal adaptor protein Disabled-1. J. Biol. Chem. 279, 33471–33479.
otella-Lopez, A., Burgaya, F., Gavin, R., Garcia-Ayllon, M.S., Gomez-Tortosa, E., Pena-Casanova, J., Urena, J.M., Del Rio, J.A., Blesa, R.,Soriano, E., Saez-Valero, J., 2006. Reelin expression and glycosylationpatterns are altered in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A.103, 5573–5578.
hen, Y., Beffert, U., Ertunc, M., Tang, T.S., Kavalali, E.T., Bezprozvanny,I., Herz, J., 2005. Reelin modulates NMDA receptor activity in corticalneurons. J. Neurosci. 25, 8209–8216.
hin, J., Massaro, C.M., Palop, J.J., Thwin, M.T., Yu, G.Q., Bien-Ly, N.,Bender, A., Mucke, L., 2007. Reelin depletion in the entorhinal cortexof human amyloid precursor protein transgenic mice and humans withAlzheimer’s disease. J. Neurosci. 27, 2727–2733.
ollie, A., Maruff, P., 2000. The neuropsychology of preclinical Alzheimer’sdisease and mild cognitive impairment. Neurosci. Biobehav. Rev. 24,365–374.
order, E.H., Saunders, A.M., Strittmatter, W.J., Schmechel, D.E., Gaskell,P.C., Small, G.W., Roses, A.D., Haines, J.L., Pericak-Vance, M.A., 1993.Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’sdisease in late onset families. Science 261, 921–923.
’Arcangelo, G., Homayouni, R., Keshvara, L., Rice, D.S., Sheldon, M.,Curran, T., 1999. Reelin is a ligand for lipoprotein receptors. Neuron 24,471–479.
’Arcangelo, G., Miao, G.G., Chen, S.C., Soares, H.D., Morgan, J.I., Curran,T., 1995. A protein related to extracellular matrix proteins deleted in themouse mutant reeler. Nature 374, 719–723.
reund, T.F., Buzsaki, G., 1996. Interneurons of the hippocampus. Hip-pocampus 6, 347–470.
ritschy, J.M., Weinmann, O., Wenzel, A., Benke, D., 1998. Synapse-specific localization of NMDA and GABA(A) receptor subunits revealedby antigen-retrieval immunohistochemistry. J. Comp. Neurol. 390,194–210.
lenner, G.G., Wong, C.W., 1984. Alzheimer’s disease: initial report ofthe purification and characterization of a novel cerebrovascular amyloidprotein. Biochem. Biophys. Res. Commun. 120, 885–890.
rundke-Iqbal, I., Iqbal, K., Tung, Y.C., Quinlan, M., Wisniewski, H.M.,Binder, L.I., 1986. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc.Natl. Acad. Sci. U.S.A. 83, 4913–4917.
erz, J., Chen, Y., 2006. Reelin, lipoprotein receptors and synaptic plasticity.Nat. Rev. Neurosci. 7, 850–859.
iesberger, T., Trommsdorff, M., Howell, B.W., Goffinet, A., Mumby, M.C.,Cooper, J.A., Herz, J., 1999. Direct binding of Reelin to VLDL receptorand ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 andmodulates tau phosphorylation. Neuron 24, 481–489.
oe, H.S., Magill, L.A., Guenette, S., Fu, Z., Vicini, S., Rebeck, G.W.,
2006b. FE65 interaction with the ApoE receptor ApoEr2. J. Biol. Chem.281, 24521–24530.oe, H.S., Tran, T.S., Matsuoka, Y., Howell, B.W., Rebeck, G.W., 2006a.Dab1 and Reelin effects on APP and ApoEr2 trafficking and processing.J. Biol. Chem. 281, 35176–35185.

logy of
H
J
J
K
K
K
K
L
M
M
M
M
M
M
M
M
M
N
N
O
O
O
P
P
P
Q
Q
R
R
R
R
R
R
S
S
S
I. Knuesel et al. / Neurobio
owell, B.W., Hawkes, R., Soriano, P., Cooper, J.A., 1997. Neuronal positionin the developing brain is regulated by mouse disabled-1. Nature 389,733–737.
ucker, M., Walker, L.C., Kuo, H., Tian, M., Ingram, D.K., 1994a. Age-related fibrillar deposits in brains of C57BL/6 mice. A review oflocalization, staining characteristics, and strain specificity. Mol. Neu-robiol. 9, 125–133.
ucker, M., Walker, L.C., Schwarb, P., Hengemihle, J., Kuo, H., Snow,A.D., Bamert, F., Ingram, D.K., 1994b. Age-related deposition of glia-associated fibrillar material in brains of C57BL/6 mice. Neuroscience60, 875–889.
ayed, R., Head, E., Thompson, J.L., McIntire, T.M., Milton, S.C., Cot-man, C.W., Glabe, C.G., 2003. Common structure of soluble amyloidoligomers implies common mechanism of pathogenesis. Science 300,486–489.
im, J.H., Lee, H.K., Takamiya, K., Huganir, R.L., 2003. The role of synap-tic GTPase-activating protein in neuronal development and synapticplasticity. J. Neurosci. 23, 1119–1124.
nuesel, I., Elliott, A., Chen, H.J., Mansuy, I.M., Kennedy, M.B., 2005. Arole for synGAP in regulating neuronal apoptosis. Eur. J. Neurosci. 21,611–621.
omiyama, N.H., Watabe, A.M., Carlisle, H.J., Porter, K., Charlesworth, P.,Monti, J., Strathdee, D.J., O’Carroll, C.M., Martin, S.J., Morris, R.G.,O’Dell, T.J., Grant, S.G., 2002. SynGAP regulates ERK/MAPK signal-ing, synaptic plasticity, and learning in the complex with postsynapticdensity 95 and NMDA receptor. J. Neurosci. 22, 9721–9732.
amar, C.H., Hinsman, E.J., Henrikson, C.K., 1976. Alterations in the hip-pocampus of aged mice. Acta Neuropathol. (Berl) 36, 387–391.
andybur, T.I., Ormsby, I., Zemlan, F.P., 1989. Cerebral aging: a quantitativestudy of gliosis in old nude mice. Acta Neuropathol. (Berl) 77, 507–513.
arighetto, A., Etchamendy, N., Touzani, K., Torrea, C.C., Yee, B.K., Rawl-ins, J.N., Jaffard, R., 1999. Knowing which and knowing what: a potentialmouse model for age-related human declarative memory decline. Eur. J.Neurosci. 11, 3312–3322.
eyer, U., Feldon, J., Schedlowski, M., Yee, B.K., 2005. Towards animmuno-precipitated neurodevelopmental animal model of schizophre-nia. Neurosci. Biobehav. Rev. 29, 913–947.
eyer, U., Nyffeler, M., Engler, A., Urwyler, A., Schedlowski, M., Knuesel,I., Yee, B.K., Feldon, J., 2006. The time of prenatal immune challengedetermines the specificity of inflammation-mediated brain and behav-ioral pathology. J. Neurosci. 26, 4752–4762.
iettinen, R., Riedel, A., Kalesnykas, G., Kettunen, H.P., Puolivali, J., Soini-nen, H., Arendt, T., 2005. Reelin-immunoreactivity in the hippocampalformation of 9-month-old wildtype mouse: effects of APP/PS1 genotypeand ovariectomy. J. Chem. Neuroanat. 30, 105–118.
ingaud, F., Mormede, C., Pallet, V., Jaffard, R., Higueret, P., Marighetto,A., 2005. Beneficial effects of vitamin A enriched diet on working mem-ory in aged mice. SfN abstract, No. 199/20.
orimura, T., Hattori, M., Ogawa, M., Mikoshiba, K., 2005. Disabled1 reg-ulates the intracellular trafficking of reelin receptors. J. Biol. Chem. 280,16901–16908.
ormede, C., Mons, N., Jaffard, R., Marighetto, A., 2006. Functionalalterations in prefronto-hippocampal circuits in a mouse model of aging-related decline in short-term memory. SfN abstract, No. 812/13.
otoi, Y., Itaya, M., Mori, H., Mizuno, Y., Iwasaki, T., Hattori, H., Haga,S., Ikeda, K., 2004. Apolipoprotein E receptor 2 is involved in neuriticplaque formation in APP sw mice. Neurosci. Lett. 368, 144–147.
yffeler, M., Meyer, U., Yee, B.K., Feldon, J., Knuesel, I., 2006. Mater-nal immune activation during pregnancy increases limbic GABA(A)receptor immunoreactivity in the adult offspring: implications forschizophrenia. Neuroscience 143, 51–62.
yffeler, M., Zhang, W.N., Feldon, J., Knuesel, I., 2007. Differential
expression of PSD proteins in age-related spatial learning impairments.Neurobiol. Aging 28, 143–155.ddo, S., Billings, L., Kesslak, J.P., Cribbs, D.H., LaFerla, F.M., 2004. Abetaimmunotherapy leads to clearance of early, but not late, hyperphospho-rylated tau aggregates via the proteasome. Neuron 43, 321–332.
S
Aging 30 (2009) 697–716 715
ddo, S., Caccamo, A., Shepherd, J.D., Murphy, M.P., Golde, T.E., Kayed,R., Metherate, R., Mattson, M.P., Akbari, Y., LaFerla, F.M., 2003. Triple-transgenic model of Alzheimer’s disease with plaques and tangles:intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421.
hkubo, N., Lee, Y.D., Morishima, A., Terashima, T., Kikkawa, S.,Tohyama, M., Sakanaka, M., Tanaka, J., Maeda, N., Vitek, M.P., Mitsuda,N., 2003. Apolipoprotein E and Reelin ligands modulate tau phos-phorylation through an apolipoprotein E receptor/disabled-1/glycogensynthase kinase-3beta cascade. FASEB J. 17, 295–297.
esold, C., Impagnatiello, F., Pisu, M.G., Uzunov, D.P., Costa, E., Guidotti,A., Caruncho, H.J., 1998. Reelin is preferentially expressed in neuronssynthesizing gamma-aminobutyric acid in cortex and hippocampus ofadult rats. Proc. Natl. Acad. Sci. U.S.A. 95, 3221–3226.
otier, B., Krzywkowski, P., Lamour, Y., Dutar, P., 1994. Loss of calbindin-immunoreactivity in CA1 hippocampal stratum radiatum and stratumlacunosum-moleculare interneurons in the aged rat. Brain Res. 661,181–188.
ryce, C.R., Feldon, J., Fuchs, E., Knuesel, I., Oertle, T., Sengstag, C.,Spengler, M., Weber, E., Weston, A., Jongen-Relo, A., 2005. Postnatalontogeny of hippocampal expression of the mineralocorticoid and gluco-corticoid receptors in the common marmoset monkey. Eur. J. Neurosci.21, 1521–1535.
iu, S., Korwek, K.M., Pratt-Davis, A.R., Peters, M., Bergman, M.Y., Wee-ber, E.J., 2006. Cognitive disruption and altered hippocampus synapticfunction in Reelin haploinsufficient mice. Neurobiol. Learn. Mem. 85,228–242.
uattrocchi, C.C., Wannenes, F., Persico, A.M., Ciafre, S.A., D’Arcangelo,G., Farace, M.G., Keller, F., 2002. Reelin is a serine protease of theextracellular matrix. J. Biol. Chem. 277, 303–309.
aber, J., Wong, D., Buttini, M., Orth, M., Bellosta, S., Pitas, R.E., Mahley,R.W., Mucke, L., 1998. Isoform-specific effects of human apolipopro-tein E on brain function revealed in ApoE knockout mice: increasedsusceptibility of females. Proc. Natl. Acad. Sci. U.S.A. 95, 10914–10919.
amos, B., Baglietto-Vargas, D., del Rio, J.C., Moreno-Gonzalez, I.,Santa-Maria, C., Jimenez, S., Caballero, C., Lopez-Tellez, J.F., Khan,Z.U., Ruano, D., Gutierrez, A., Vitorica, J., 2006. Early neuropathol-ogy of somatostatin/NPY GABAergic cells in the hippocampus of aPS1xAPP transgenic model of Alzheimer’s disease. Neurobiol. Aging27, 1658–1672.
amos-Moreno, T., Galazo, M.J., Porrero, C., Martinez-Cerdeno, V.,Clasca, F., 2006. Extracellular matrix molecules and synaptic plastic-ity: immunomapping of intracellular and secreted Reelin in the adult ratbrain. Eur. J. Neurosci. 23, 401–422.
ice, D.S., Curran, T., 2001. Role of the reelin signaling pathway in centralnervous system development. Annu. Rev. Neurosci. 24, 1005–1039.
iedel, A., Miettinen, R., Stieler, J., Mikkonen, M., Alafuzoff, I., Soini-nen, H., Arendt, T., 2003. Reelin-immunoreactive Cajal-Retzius cells:the entorhinal cortex in normal aging and Alzheimer’s disease. ActaNeuropathol. (Berl) 106, 291–302.
owan, M.J., Klyubin, I., Wang, Q., Anwyl, R., 2005. Synaptic plasticitydisruption by amyloid beta protein: modulation by potential Alzheimer’sdisease modifying therapies. Biochem. Soc. Trans. 33, 563–567.
aez-Valero, J., Costell, M., Sjogren, M., Andreasen, N., Blennow, K.,Luque, J.M., 2003. Altered levels of cerebrospinal fluid reelin in fron-totemporal dementia and Alzheimer’s disease. J. Neurosci. Res. 72,132–136.
arter, M., Bruno, J.P., 2004. Developmental origins of the age-relateddecline in cortical cholinergic function and associated cognitive abilities.Neurobiol. Aging 25, 1127–1139.
chmechel, D.E., Saunders, A.M., Strittmatter, W.J., Crain, B.J., Hulette,C.M., Joo, S.H., Pericak-Vance, M.A., Goldgaber, D., Roses, A.D., 1993.Increased amyloid beta-peptide deposition in cerebral cortex as a con-
sequence of apolipoprotein E genotype in late-onset Alzheimer disease.Proc. Natl. Acad. Sci. U.S.A. 90, 9649–9653.hetty, A.K., Turner, D.A., 1998. Hippocampal interneurons expressing glu-tamic acid decarboxylase and calcium-binding proteins decrease withaging in Fischer 344 rats. J. Comp. Neurol. 394, 252–269.

7 logy of
S
S
T
V
W
W
W
W
W
16 I. Knuesel et al. / Neurobio
imons, M., Keller, P., De Strooper, B., Beyreuther, K., Dotti, C.G., Simons,K., 1998. Cholesterol depletion inhibits the generation of �-amyloidin hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 95, 6460–6464.
mall, S.A., Nava, A.S., Perera, G.M., Delapaz, R., Stern, Y., 2000. Eval-uating the function of hippocampal subregions with high-resolutionMRI in Alzheimer’s disease and aging. Microsc. Res. Technol. 51,101–108.
rommsdorff, M., Gotthardt, M., Hiesberger, T., Shelton, J., Stockinger,W., Nimpf, J., Hammer, R.E., Richardson, J.A., Herz, J., 1999.Reeler/Disabled-like disruption of neuronal migration in knockout micelacking the VLDL receptor and ApoE receptor 2. Cell 97, 689–701.
azquez, L.E., Chen, H.J., Sokolova, I., Knuesel, I., Kennedy, M.B., 2004.
SynGAP regulates spine formation. J. Neurosci. 24, 8862–8872.alsh, D.M., Klyubin, I., Fadeeva, J.V., Cullen, W.K., Anwyl, R., Wolfe,M.S., Rowan, M.J., Selkoe, D.J., 2002. Naturally secreted oligomers ofamyloid beta protein potently inhibit hippocampal long-term potentia-tion in vivo. Nature 416, 535–539.
W
Aging 30 (2009) 697–716
ang, H.W., Pasternak, J.F., Kuo, H., Ristic, H., Lambert, M.P., Chromy,B., Viola, K.L., Klein, W.L., Stine, W.B., Krafft, G.A., Trommer,B.L.,2002. Soluble oligomers of beta amyloid (1–42) inhibit long-term poten-tiation but not long-term depression in rat dentate gyrus. Brain Res. 924,133–140.
eeber, E.J., Beffert, U., Jones, C., Christian, J.M., Forster, E., Sweatt,J.D., Herz, J., 2002. Reelin and ApoE receptors cooperate to enhancehippocampal synaptic plasticity and learning. J. Biol. Chem. 277,39944–39952.
irths, O., Multhaup, G., Bayer, T.A., 2004. A modified beta-amyloid hypothesis: intraneuronal accumulation of the beta-amyloidpeptide—the first step of a fatal cascade. J. Neurochem. 91, 513–520.
irths, O., Multhaup, G., Czech, C., Blanchard, V., Tremp, G., Pradier, L.,
Beyreuther, K., Bayer, T.A., 2001. Reelin in plaques of beta-amyloidprecursor protein and presenilin-1 double-transgenic mice. Neurosci.Lett. 316, 145–148.yss-Coray, T., 2006. Inflammation in Alzheimer disease: driving force,bystander or beneficial response? Nat. Med. 12, 1005–1015.




















