
Advanced Oxidation Protein Products as Novel Mediators of Inflammation and Monocyte Activation in...
10
of December 7, 2013. This information is current as 1, 2 Monocyte Activation in Chronic Renal Failure Novel Mediators of Inflammation and Advanced Oxidation Protein Products as Tilman Drüeke and Béatrice Descamps-Latscha Sandrine Canteloup, Jean-Michel Dayer, Paul Jungers, Khoa, Chantal Capeillère-Blandin, Anh Thu Nguyen, Véronique Witko-Sarsat, Miriam Friedlander, Thao Nguyen http://www.jimmunol.org/content/161/5/2524 1998; 161:2524-2532; ; J Immunol References http://www.jimmunol.org/content/161/5/2524.full#ref-list-1 , 12 of which you can access for free at: cites 38 articles This article Subscriptions http://jimmunol.org/subscriptions is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/ji/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/cgi/alerts/etoc Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved. Copyright © 1998 by The American Association of 9650 Rockville Pike, Bethesda, MD 20814-3994. The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on December 7, 2013 http://www.jimmunol.org/ Downloaded from by guest on December 7, 2013 http://www.jimmunol.org/ Downloaded from
Transcript of Advanced Oxidation Protein Products as Novel Mediators of Inflammation and Monocyte Activation in...

of December 7, 2013.This information is current as
1, 2Monocyte Activation in Chronic Renal FailureNovel Mediators of Inflammation and Advanced Oxidation Protein Products as
Tilman Drüeke and Béatrice Descamps-LatschaSandrine Canteloup, Jean-Michel Dayer, Paul Jungers,Khoa, Chantal Capeillère-Blandin, Anh Thu Nguyen, Véronique Witko-Sarsat, Miriam Friedlander, Thao Nguyen
http://www.jimmunol.org/content/161/5/25241998; 161:2524-2532; ;J Immunol
Referenceshttp://www.jimmunol.org/content/161/5/2524.full#ref-list-1
, 12 of which you can access for free at: cites 38 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 1998 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on Decem
ber 7, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on D
ecember 7, 2013
http://ww
w.jim
munol.org/
Dow
nloaded from

Advanced Oxidation Protein Products as Novel Mediators ofInflammation and Monocyte Activation in Chronic RenalFailure1, 2
Veronique Witko-Sarsat,* Miriam Friedlander, † Thao Nguyen Khoa,*Chantal Capeillere-Blandin,‡ Anh Thu Nguyen,* Sandrine Canteloup,* Jean-Michel Dayer,§
Paul Jungers,* Tilman Drueke,* and Beatrice Descamps-Latscha3*
We previously demonstrated the presence of advanced oxidation protein products (AOPP), a novel marker of oxidative stress inthe plasma of uremic patients receiving maintenance dialysis. The present study in a cohort of 162 uremic patients showed thatplasma concentrations of AOPP increased with progression of chronic renal failure and were closely related to advanced glycationend products (AGE)-pentosidine (r 5 0.52,p < 0.001), taken as a marker of AGE. In vivo, the relevance of AOPP and AGE-pentosidine in monocyte-mediated inflammatory syndrome associated with uremia was evidenced by close correlations betweenAOPP or AGE-pentosidine and monocyte activation markers, including neopterin, IL-1R antagonist, TNF-a, and TNF solublereceptors (TNF-sR55 and TNF-sR75). To determine the mechanisms by which AOPP and AGE could be directly involved inmonocyte activation, AOPP-human serum albumin (HSA) and AGE-HSA were produced in vitro by treating HSA with oxidantsor glucose, respectively. Spectroscopic analysis confirmed that AOPP-HSA contains carbonyls and dityrosine. Both AOPP-HSAand AGE-HSA, but not purified dityrosine, were capable of triggering the oxidative burst of human monocytes in cultures. TheAOPP-HSA-induced respiratory burst was dependent on the chlorinated nature of the oxidant and on the molar ratio HSA/HOCl.Collectively, these data first demonstrate that AOPP act as a mediator of oxidative stress and monocyte respiratory burst, whichpoints to monocytes as both target and actor in the immune dysregulation associated with chronic uremia.The Journal ofImmunology,1998, 161: 2524–2532.
U remic patients suffer from a dysregulation of the immunesystem, characterized by the paradoxical coexistence ofa clinically and biologically evident state of immunode-
ficiency and activation of immunocompetent cells, including T andcells, and of monocytes (reviewed in Ref. 1). Substantial evidenceaccumulated in recent years has implicated activated monocyte-derived proinflammatory cytokines in such immunologic disorders(2–4). The presence of a chronic inflammatory state has also beenwidely documented in end-stage renal disease patients receivingmaintenance hemodialysis (recently reviewed in Ref. 5). It is com-monly attributed to the constantly renewed activation of circulat-ing neutrophils and monocytes following blood passage throughdialysis circuits and subsequent generation of activated comple-ment components due to contact of plasma with bioincompatiblemembranes and/or transfer of endotoxins from the dialyzate to the
blood compartment. This conjunction leads to 1) a massive gen-eration of reactive oxygen species (ROS),4 e.g., O2
2, H2O2,1O2,
and OH2, and chlorinated oxidants such as HOCl by activatedneutrophils; and 2) an increased production of cytokines, e.g.,IL-1b and TNF-a, by activated monocytes. Recent studies suggestthat both the oxidative stress induced by ROS and the proinflam-matory effects of these cytokines are reinforced by profound de-fects in antioxidant (6, 7) and anticytokine systems (3, 4) andlargely contribute tob2m amyloid arthropathy (8, 9) and acceler-ated atherosclerosis (10, 11), which remain leading causes of mor-bidity and mortality in dialysis patients (12).
The exquisite vulnerability of proteins to ROS is now well doc-umented (13–16). Oxidation of amino acid residues such as ty-rosine, leading to the formation of dityrosine, protein aggregation,cross-linking, and fragmentation, is an example of ROS-mediatedprotein damage in vitro. In contrast, evidence for the presence ofsuch oxidatively damaged proteins in vivo and their possible clin-ical significance was still lacking until recently (15, 17). Indeed, inthe search for whether such protein oxidative damage could reflectthe dialysis-associated oxidative stress, we were able to isolate andcharacterize dityrosine-containing protein cross-linking productsin the plasma of dialysis patients, which we designated advancedoxidation protein products (AOPP) (17).
The contribution of uremia per se to the chronic inflammatorystate has been suggested, and consistent evidence has been af-forded that both monocyte activation and a defect in antioxidant
*Institut National de la Sante et de la Recherche Medicale, Unit 90 and Departmentof Nephrology, Necker Hospital, Paris, France;†University Hospitals, Cleveland, OH44106;‡Centre National de la Recherche Scientifique, Unite de Recherche Associe´e400, Paris, France; and§Division of Immunology and Allergy, University Hospital,Geneva, Switzerland
Received for publication February 12, 1998. Accepted for publication April 27, 1998.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby markedadvertisementin accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by the Extramural Grant Program, Baxter Corporation(B.D.-L. and M.F.) and in part by National Institutes of Health Grant DK-45619(M.F.) and Grant AOA94047 from the De´legation a la Recherche Clinique, AP-Hopitaux de Paris (P.J.).2 A portion of this work was presented at the 29th Annual Meeting of the AmericanSociety of Nephrology, New Orleans, LA.3 Address correspondence and reprint requests to Dr. Beatrice Descamps-Latscha,Institut National de la Sante et de la Recherche Medicale, Unit 90, Hopital Necker,161 rue de Sevres, 75015 Paris, France. E-mail address: [email protected]
4 Abbreviations used in this paper: ROS, reactive oxygen species; AOPP, advancedoxidation protein products; AGE, advanced glycation end product; HSA, human se-rum albumin; MDA, malondialdehyde; GSH-Px, glutathione peroxidase; IL-1Ra, in-terleukin-1 receptor antagonist.
Copyright © 1998 by The American Association of Immunologists 0022-1767/98/$02.00
by guest on Decem
ber 7, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
systems occur early in the course of chronic renal failure and grad-ually increase with its progression to end-stage renal disease (4, 7).Interest has focused on the role of “uremic toxins” generated dur-ing the course of chronic renal failure, some of which havingknown effects on neutrophil and monocyte functions (18, 19).Among these, growing efforts are being devoted to the potentialtoxicity of advanced glycation end products (AGE) (19–24).
The formation of AGEs has been widely documented duringdiabetes and aging and has been held to be responsible for tissuedegradation. The presence of increased plasma levels of AGEs hasbeen observed in dialysis patients, independently of diabetes, andit is likely that, like ROS, AGEs contribute tob2m deposits (21,22). Interestingly, the hypothesis that oxidative stress is implicatedin AGE formation might also be relevant to the uremic toxicitysyndrome (24, 25). More recently, we reported that the well-char-acterized AGE-pentosidine accumulates with progression ofchronic renal failure in close relationship with neopterin, a well-characterized monocyte activation marker (26).
In the search for whether AOPP could act as mediators in themonocyte-mediated inflammatory process associated with chronicrenal failure and participate in the monocyte activation state, wedetermined 1) in vivo, AOPP levels in the plasma of uremic pa-tients with varying degrees of renal failure in relationship withimmunologic markers; and 2) in vitro, the potency of AOPP toinduce the monocyte respiratory burst.
Materials and MethodsPatients
One hundred and sixty-two nondialyzed chronic renal failure patients atvarious stages of renal failure were enrolled in the study after giving in-formed consent. They included mild renal failure defined by creatinineclearance (Ccr) of 41 to 80 ml/min (n 5 73), moderate renal failure withCcr of 20 to 40 ml/min (n 5 53), and advanced renal failure with Ccr,20ml/min (n 5 36). Primary renal diseases are indicated in Table I. Patientssuffering from diabetes mellitus, systemic lupus erythematosus, malignanttumors, or acute infection or receiving immunosuppressive therapy at thetime of blood sampling were excluded from the study. Controls consistedof 31 healthy adults recruited among blood donors from our blood trans-fusion center.
Blood collection, plasma isolation, and monocyte preparation
Venous blood (5–10 ml) was collected in standard sterile polystyrene vac-uum tubes, with 5 mM EDTA. After centrifugation (6003 g for 10 min),the plasma was stored in 500-ml aliquots at270°C until use. Assays wereconducted on duplicate samples thawed once.
To investigate the effect of AOPP on monocyte respiratory burst, mono-cytes were isolated from blood (20 ml) of normal volunteers by a two-stepprocedure involving erythrocyte sedimentation on dextran followed by leu-kocyte-rich plasma layer sedimentation on Nycoprep (1.068 solution; Ny-comed Pharma, Oslo, Norway) according to the supplier’s instructions.
Determination of AOPP
AOPP were determined in the plasma using the semiautomated methodpreviously devised in our laboratory (17). Briefly, AOPP were measured by
spectrophotometry on a microplate reader (model MR 5000, Dynatech,Paris, France) and were calibrated with chloramine-T (Sigma, St. Louis,MO) solutions that in the presence of potassium iodide absorb at 340 nm(27). In test wells, 200ml of plasma diluted 1/5 in PBS was placed on a96-well microtiter plate (Becton Dickinson Labware, Lincoln Park, NJ),and 20ml of acetic acid was added. In standard wells, 10ml of 1.16 Mpotassium iodide (Sigma) was added to 200ml of chloramine-T solution(0–100mmol/liter) followed by 20ml of acetic acid. The absorbance of thereaction mixture is immediately read at 340 nm on the microplate readeragainst a blank containing 200ml of PBS, 10ml of potassium iodide, and20 ml of acetic acid. The chloramine-T absorbance at 340 nm being linearwithin the range of 0 to 100mmol/liter, AOPP concentrations were ex-pressed as micromoles per liter of chloramine-T equivalents.
Spectroscopic analysis of AOPP-HSA and of dityrosine preparedin vitro
Human serum albumin (HSA; type V; Sigma) was exposed to HOCl(Fluka, Buchs, Switzerland) as described previously (17). Briefly, HOClstock solution (100 mM) was freshly prepared in PBS, and the concentra-tion was measured by spectrophotometry using a molar extinction coeffi-cient of 350 M21 cm21 at 290 nm at pH 12. Various concentrations ofoxidants of HOCl were added to HSA at the indicated HSA/HOCl molarratio. The AOPP-HSA preparation was incubated for 30 min at room tem-perature and then dialyzed overnight against PBS and tested for AOPPcontent. AOPP-HSA were also prepared by exposing purified HSA (100mg/ml) to chloramine-T or hydrogen peroxide (H2O2) at the indicated con-centrations and was dialyzed overnight against PBS. Dityrosine synthesiswas adapted from the Anderson method (28). Briefly, 270 mg of tyrosinewas dissolved in 250 ml of 0.2 M borate buffer, pH 9.5, in the presence of6 mM H2O2. The reaction was started by the addition of 6.3 mg of horse-radish peroxidase and performed for 18 h at 37°C. Then the mixture wasconcentrated almost to dryness in a rotatory evaporator under vacuum at35°C. The brown powder was suspended in water acidified by concentratedHCl. A precipitated was removed by filtration through a glass filter funnel(G4; Millipore, Bedford, MA). Further separation from unreactedL-ty-rosine and impurities was performed by chromatography on a cation ex-changer. The brown solution was then applied to a fibrous cellulose phos-phate (Sigma; 50–100mm) column (1.5, 25 cm) equilibrated with 0.2 Nacetic acid. After washing, the elution was performed with 0.5 M NaCl in0.2 N acetic acid. Fractions of 3 ml were collected and evaluated afterdilution in 0.1 N NaOH spectrophotometrically at 280 and 315 nm. Fluo-rescence detection (lex 5 320 nm, lem 5 410 nm) was also monitored.Fractions exhibiting dityrosine fluorescence were pooled and concentratedby lyophilization. The concentrate was solubilized in distilled water, fil-trated, then loaded on a Dowex 50W-X8 (Bio-Rad) column previouslysoaked in 1 M HCl. The column was extensively washed with water toremove all NaCl and acetic acid. The dityrosine was subsequently elutedwith 2 M ammonium hydroxide. Fluorescent fractions were concentrated ina rotatory evaporator under vacuum. The final fraction was reprecipitatedseveral times in methanol-ether and stored under ether. Finally, a slightlyyellow dityrosine was obtained (yield, 12%) and characterized by UV spec-tra, fluorescence emission, and magnetic resonance spectroscopy (29, 30).
Spectroscopic analyses on HSA, AOPP-HSA, and purified dityrosinewere performed on a Kontron SF 25 spectrophotometer; UV and visiblespectra were recorded in PBS at pH 7.5 at room temperature. Absorptionand emission spectra were recorded in denaturing 6 M urea buffer at pH7.5. Protein-bound dityrosine production was assayed in plasma or HSAsamples by fluorescence measurements after dilution of the sample in 20mM phosphate buffer, pH 7.5, in the presence of 7 M urea (10). After a30-min incubation, the fluorescence emission spectra of dityrosine wasrecorded from 550 to 350 nm following excitation at 320 nm using aKontron SF 25 spectrophotometer (Kontron, Zurich, Switzerland) and wasmeasured at its maximum at 410 nm. The assay was calibrated by meansof external standardization using a calibration curve generated in the sameurea medium with authentic dityrosine. Its concentration was monitoredspectrophotometrically at 315 nm, E5 5 mM21 cm21 at pH 7.5 (29, 30).
Determination of carbonyl residues
Carbonyl residues were determined as previously described (31) using di-nitrophenylhydrazine. Briefly, samples were submitted to 10 mM dinitro-phenylhydrazine in 2.5 M HCl for 1 h, followed by deproteinization with20% TCA. The pellet was washed three times in ethanol/ethyl acetate andsolubilized in guanidine 6 M. The carbonyl concentration was measured byspectrophotometry at an OD of 370 nm withe370 5 22 mM21 cm21. Theprotein concentration was determined in parallel using OD at 280 nm inreference to BSA.
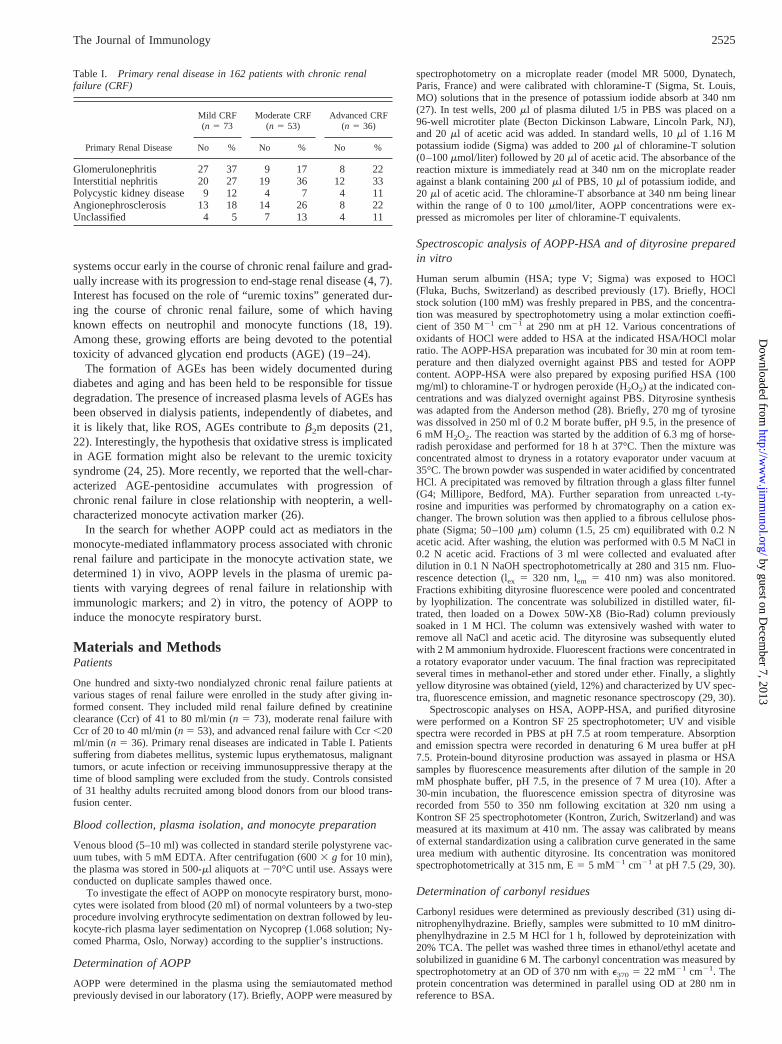
Table I. Primary renal disease in 162 patients with chronic renalfailure (CRF)
Primary Renal Disease
Mild CRF(n 5 73
Moderate CRF(n 5 53)
Advanced CRF(n 5 36)
No % No % No %
Glomerulonephritis 27 37 9 17 8 22Interstitial nephritis 20 27 19 36 12 33Polycystic kidney disease 9 12 4 7 4 11Angionephrosclerosis 13 18 14 26 8 22Unclassified 4 5 7 13 4 11
2525The Journal of Immunology
by guest on Decem
ber 7, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

AGE preparation and determination
The AGE-HSA used in vitro was prepared by incubating HSA (type V;Sigma; 50 mg/ml) with 500 mM glucose in PBS for 65 days at 37°C understerile conditions. The AGE-pentosidine, as a marker of nonenzymatic gly-cation of proteins, was measured using a modification of the method de-scribed by Odetti et al. (32). Briefly, plasma proteins or AGE-HSA wereprecipitated on TCA. The pellets were hydrolyzed in 2 ml of 6 N HCl anddissolved in 250ml 0.01 M heptafluorobutyric acid (Sigma). The equiva-lent of 4 mg of plasma protein was injected into an HPLC system (WatersDivision of Millipore, Marlborough, MA). A 25-3 0.46-cm C18 Vydactype 218TP (10mm) column was used (Separations Group, Hesperia, CA).HPLC was programmed with a linear gradient of 10 to 17% acetonitrilefrom 0 to 35 min. Pentosidine was eluted at approximately 30 min asmonitored by fluorescence excitation at 335 nm and emission at 385 nm.Pentosidine prepared according to the method of Sell and Monnier (33)was used as standard, and results were given in picomoles per milligram ofprotein.
Measurement of thiobarbituric acid-reacting substances
Thiobarbituric-reacting substances, including malondialdehyde (MDA),were determined using a commercially available kit according to the sup-plier’s instructions (Sobioda, Grenoble, France). Briefly, tetraethoxypro-pane was used as standard; each molecule of tetraethoxypropane hydro-lyzes to yield one molecule of MDA under assay conditions. One hundredmicroliters of plasma was mixed with a mixture of thiobarbituric and per-chloric acid and boiled for 1 h, then butanol (Carlo Erba, Milan, Italy) wasadded. Tubes were vortexed and centrifuged to extract MDA. Fluorometricmeasurements (excitation at 532 nm and emission at 553 nm) were per-formed on supernatants, using a Kontron SFM 25.
Determination of glutathione peroxidase (GSH-Px) activity
The level of selenium-dependent plasma GSH-Px was determined accord-ing to the method of Anderson (34) and expressed as micromoles ofNADPH oxidized per milliliter.
Measurement of cell activation markers, cytokines, and cytokineinhibitors
Commercially available kits were used for measuring plasma levels ofneopterin (monocyte activation marker; RIA, Behring Diagnostic, Rueil-Malmaison, France), IL-1R antagonist (IL-1Ra; ELISA Quantikine, R&DSystems, Minneapolis, MN), soluble CD23 (B cell activation marker;ELISA, BioSource Europe, Fleurus Belgium), and TNF-a (ELISA, Bio-Source). Plasma levels of TNF soluble receptors, TNF-sR55 and TNF-sR75, were determined as described previously (35) with specific mAbsprovided by Dr. H Gallati (F. Hoffmann-La Roche, Basel, Switzerland).The plasma level of soluble CD25 (T cell activation marker) was deter-mined with a kit (EIA, Cobas Core, sIL-2R, Hoffmann-La Roche) providedby M. E. Nobile (Roche, Neuilly, France).
Measurement of monocyte respiratory burst
The capacity of AOPP-HSA or AGE-HSA to activate the monocyte respi-ratory burst was measured by chemiluminescence, using dimethylbiacri-dinium (lucigenin) as the chemoluminigenic substrate. In this system thereductive dioxygenation of lucigenin that yields luminescence is strictlyNADPH-oxidase dependent (36). One hundred microliters of monocytesuspension (23 105/ml) was automatically injected into tubes containing100 ml of HBSS (basal activity), the tested preparations at the indicatedconcentrations (native HSA, AOPP-HSA or AGE-HSA, or human AB se-rum-opsonized zymosan (23 109 yeast cell wall units/ml), or PMA (Sig-ma). Chemiluminescence production was measured in duplicate in a lumi-
nometer (LB953 Berthold, Wildbad, Germany), and luminescenceintensity was expressed in counts per minute.
Statistical analysis
The data were analyzed using standard statistical methods (Statistica Soft-ware, Tulsa, OK). Differences between means were evaluated using Stu-dent’s paired or unpairedt test where appropriate or ANOVA for compar-ing more than two groups. Relationships between variables were testedusing simple (Pearson’s r correlation coefficient) or multiple linear regres-sion analysis as indicated. All values are reported as the mean6 SEM.Statistical significance was set atp , 0.05.
ResultsAOPP accumulate in the course of chronic renal failure
The mean plasma level of AOPP was significantly higher inchronic renal failure patients than in healthy control subjects (5262 vs 296 5 mmol/l; p , 0.001). AOPP concentrations increasedover a nearly threefold range from the incipient to the advancedstage of chronic renal failure (p , 0.001; Table II). An inverserelationship between AOPP levels and creatinine clearance wasseen (r5 20.46,p , 0.001; Fig. 1). However, no significant effectof the underlying nephropathy on circulating levels of AOPP wasidentified (Table III).
AOPP are accurate markers of oxidative stress in chronic renalfailure
The mean plasma level of MDA, the lipid peroxidation product,was significantly higher at the incipient stage of chronic uremiathan in controls, but remained stable during the progression ofrenal failure (Table II). Thus, no relationship was found betweenAOPP and MDA levels (r 5 0.16,p 5 NS). In contrast, the plasmalevel of GSH-Px, a major antioxidant enzymatic system, decreasedsignificantly with the progression of renal failure, reaching half itsinitial level at an advanced stage of chronic renal failure (Table II;p , 0.001). An inverse relationship was found between AOPP andGSH-Px levels (r5 20.34,p , 0.001). However, this correlation
FIGURE 1. Influence of chronic renal failure on the plasma level ofAOPP in 162 patients. Exponential growth correlation was used as a best-fit model. The correlation coefficient (r) between creatinine clearance val-ues and AOPP concentrations was significant at the indicatedp value.
Table II. Influence of the progression of chronic renal failure (CRF) on plasma levels of AOPP,malondialdehyde, and glutathione peroxidase in 162 patients with CRFa
Healthy Subjects(n 5 31)
Mild CRF(n 5 73)
Moderate CRF(n 5 53)
Advanced CRF(n 5 36)
AOPP (mmol/L) 296 4.9 426 2.6*** † 516 3.0*** 72 6 4.4*** §
Malondialdehyde (mmol/L) 2.56 0.17 3.06 0.08*† 3.06 0.05 3.26 0.09Glutathione peroxidase (IU/ml) 716 2.6 656 2.0 496 2.1*** ‡ 346 1.7**§
a Means6 SEM. Statistical significance at *p , 0.05, **p , 0.01; ***p , 0.001 when compared with†healthy subjects,‡mild CRF patients, and§moderate CRF patients.
2526 ADVANCED OXIDATION PROTEIN PRODUCTS IN CHRONIC UREMIA
by guest on Decem
ber 7, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
was not significant after these two parameters were adjusted forcreatinine clearance (r 5 0.16,p 5 NS).
Both AOPP and AGE-pentosidine are closely related to themonocyte activation state in chronic renal failure patients
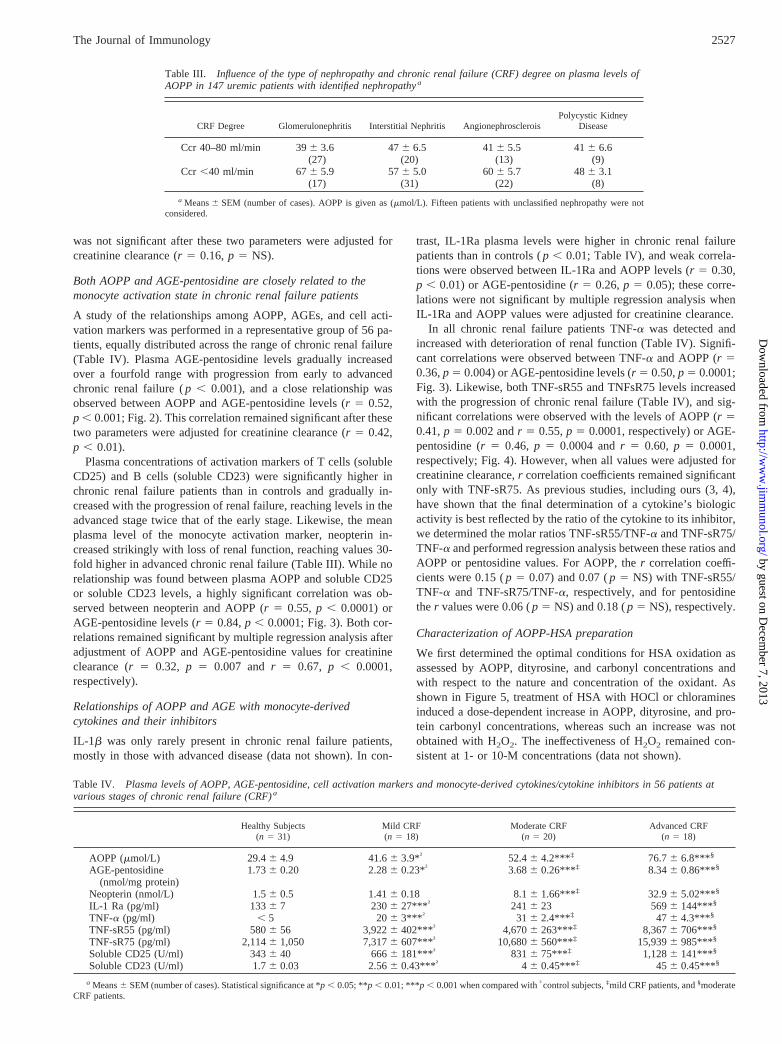
A study of the relationships among AOPP, AGEs, and cell acti-vation markers was performed in a representative group of 56 pa-tients, equally distributed across the range of chronic renal failure(Table IV). Plasma AGE-pentosidine levels gradually increasedover a fourfold range with progression from early to advancedchronic renal failure (p , 0.001), and a close relationship wasobserved between AOPP and AGE-pentosidine levels (r 5 0.52,p , 0.001; Fig. 2). This correlation remained significant after thesetwo parameters were adjusted for creatinine clearance (r 5 0.42,p , 0.01).
Plasma concentrations of activation markers of T cells (solubleCD25) and B cells (soluble CD23) were significantly higher inchronic renal failure patients than in controls and gradually in-creased with the progression of renal failure, reaching levels in theadvanced stage twice that of the early stage. Likewise, the meanplasma level of the monocyte activation marker, neopterin in-creased strikingly with loss of renal function, reaching values 30-fold higher in advanced chronic renal failure (Table III). While norelationship was found between plasma AOPP and soluble CD25or soluble CD23 levels, a highly significant correlation was ob-served between neopterin and AOPP (r 5 0.55, p , 0.0001) orAGE-pentosidine levels (r5 0.84,p , 0.0001; Fig. 3). Both cor-relations remained significant by multiple regression analysis afteradjustment of AOPP and AGE-pentosidine values for creatinineclearance (r 5 0.32, p 5 0.007 andr 5 0.67, p , 0.0001,respectively).
Relationships of AOPP and AGE with monocyte-derivedcytokines and their inhibitors
IL-1b was only rarely present in chronic renal failure patients,mostly in those with advanced disease (data not shown). In con-
trast, IL-1Ra plasma levels were higher in chronic renal failurepatients than in controls (p , 0.01; Table IV), and weak correla-tions were observed between IL-1Ra and AOPP levels (r 5 0.30,p , 0.01) or AGE-pentosidine (r 5 0.26,p 5 0.05); these corre-lations were not significant by multiple regression analysis whenIL-1Ra and AOPP values were adjusted for creatinine clearance.
In all chronic renal failure patients TNF-a was detected andincreased with deterioration of renal function (Table IV). Signifi-cant correlations were observed between TNF-a and AOPP (r 50.36,p 5 0.004) or AGE-pentosidine levels (r 5 0.50,p 5 0.0001;Fig. 3). Likewise, both TNF-sR55 and TNFsR75 levels increasedwith the progression of chronic renal failure (Table IV), and sig-nificant correlations were observed with the levels of AOPP (r 50.41,p 5 0.002 andr 5 0.55,p 5 0.0001, respectively) or AGE-pentosidine (r5 0.46, p 5 0.0004 andr 5 0.60, p 5 0.0001,respectively; Fig. 4). However, when all values were adjusted forcreatinine clearance,r correlation coefficients remained significantonly with TNF-sR75. As previous studies, including ours (3, 4),have shown that the final determination of a cytokine’s biologicactivity is best reflected by the ratio of the cytokine to its inhibitor,we determined the molar ratios TNF-sR55/TNF-a and TNF-sR75/TNF-a and performed regression analysis between these ratios andAOPP or pentosidine values. For AOPP, ther correlation coeffi-cients were 0.15 (p 5 0.07) and 0.07 (p 5 NS) with TNF-sR55/TNF-a and TNF-sR75/TNF-a, respectively, and for pentosidinether values were 0.06 (p 5 NS) and 0.18 (p 5 NS), respectively.
Characterization of AOPP-HSA preparation
We first determined the optimal conditions for HSA oxidation asassessed by AOPP, dityrosine, and carbonyl concentrations andwith respect to the nature and concentration of the oxidant. Asshown in Figure 5, treatment of HSA with HOCl or chloraminesinduced a dose-dependent increase in AOPP, dityrosine, and pro-tein carbonyl concentrations, whereas such an increase was notobtained with H2O2. The ineffectiveness of H2O2 remained con-sistent at 1- or 10-M concentrations (data not shown).
Table III. Influence of the type of nephropathy and chronic renal failure (CRF) degree on plasma levels ofAOPP in 147 uremic patients with identified nephropathya
CRF Degree Glomerulonephritis Interstitial Nephritis AngionephroscleroisPolycystic Kidney
Disease
Ccr 40–80 ml/min 396 3.6 476 6.5 416 5.5 416 6.6(27) (20) (13) (9)
Ccr ,40 ml/min 676 5.9 576 5.0 606 5.7 486 3.1(17) (31) (22) (8)
a Means6 SEM (number of cases). AOPP is given as (mmol/L). Fifteen patients with unclassified nephropathy were notconsidered.
Table IV. Plasma levels of AOPP, AGE-pentosidine, cell activation markers and monocyte-derived cytokines/cytokine inhibitors in 56 patients atvarious stages of chronic renal failure (CRF)a
Healthy Subjects(n 5 31)
Mild CRF(n 5 18)
Moderate CRF(n 5 20)
Advanced CRF(n 5 18)
AOPP (mmol/L) 29.46 4.9 41.66 3.9*† 52.46 4.2*** ‡ 76.76 6.8*** §
AGE-pentosidine(nmol/mg protein)
1.736 0.20 2.286 0.23*† 3.686 0.26*** ‡ 8.346 0.86*** §
Neopterin (nmol/L) 1.56 0.5 1.416 0.18 8.16 1.66*** ‡ 32.96 5.02*** §
IL-1 Ra (pg/ml) 1336 7 2306 27*** † 2416 23 5696 144*** §
TNF-a (pg/ml) , 5 206 3*** † 316 2.4*** ‡ 476 4.3*** §
TNF-sR55 (pg/ml) 5806 56 3,9226 402*** † 4,6706 263*** ‡ 8,3676 706*** §
TNF-sR75 (pg/ml) 2,1146 1,050 7,3176 607*** † 10,6806 560*** ‡ 15,9396 985*** §
Soluble CD25 (U/ml) 3436 40 6666 181*** † 8316 75*** ‡ 1,1286 141*** §
Soluble CD23 (U/ml) 1.76 0.03 2.566 0.43*** † 4 6 0.45*** ‡ 456 0.45*** §
a Means6 SEM (number of cases). Statistical significance at *p , 0.05; **p , 0.01; ***p , 0.001 when compared with†control subjects,‡mild CRF patients, and§moderateCRF patients.
2527The Journal of Immunology
by guest on Decem
ber 7, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
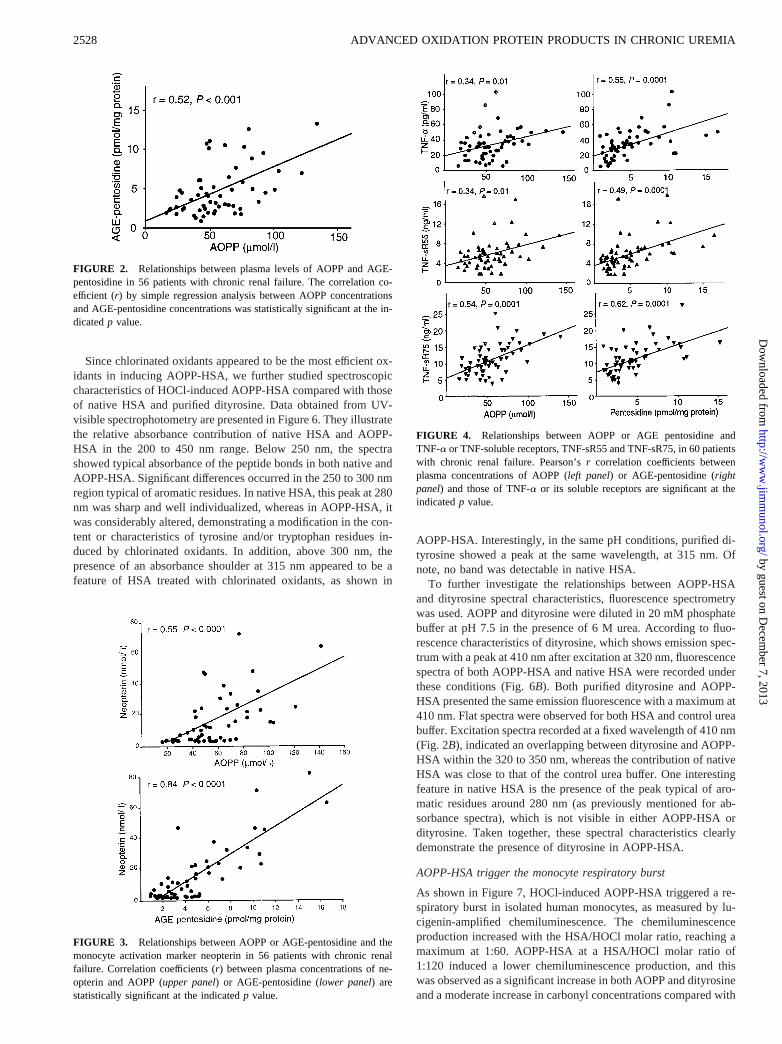
Since chlorinated oxidants appeared to be the most efficient ox-idants in inducing AOPP-HSA, we further studied spectroscopiccharacteristics of HOCl-induced AOPP-HSA compared with thoseof native HSA and purified dityrosine. Data obtained from UV-visible spectrophotometry are presented in Figure 6. They illustratethe relative absorbance contribution of native HSA and AOPP-HSA in the 200 to 450 nm range. Below 250 nm, the spectrashowed typical absorbance of the peptide bonds in both native andAOPP-HSA. Significant differences occurred in the 250 to 300 nmregion typical of aromatic residues. In native HSA, this peak at 280nm was sharp and well individualized, whereas in AOPP-HSA, itwas considerably altered, demonstrating a modification in the con-tent or characteristics of tyrosine and/or tryptophan residues in-duced by chlorinated oxidants. In addition, above 300 nm, thepresence of an absorbance shoulder at 315 nm appeared to be afeature of HSA treated with chlorinated oxidants, as shown in
AOPP-HSA. Interestingly, in the same pH conditions, purified di-tyrosine showed a peak at the same wavelength, at 315 nm. Ofnote, no band was detectable in native HSA.
To further investigate the relationships between AOPP-HSAand dityrosine spectral characteristics, fluorescence spectrometrywas used. AOPP and dityrosine were diluted in 20 mM phosphatebuffer at pH 7.5 in the presence of 6 M urea. According to fluo-rescence characteristics of dityrosine, which shows emission spec-trum with a peak at 410 nm after excitation at 320 nm, fluorescencespectra of both AOPP-HSA and native HSA were recorded underthese conditions (Fig. 6B). Both purified dityrosine and AOPP-HSA presented the same emission fluorescence with a maximum at410 nm. Flat spectra were observed for both HSA and control ureabuffer. Excitation spectra recorded at a fixed wavelength of 410 nm(Fig. 2B), indicated an overlapping between dityrosine and AOPP-HSA within the 320 to 350 nm, whereas the contribution of nativeHSA was close to that of the control urea buffer. One interestingfeature in native HSA is the presence of the peak typical of aro-matic residues around 280 nm (as previously mentioned for ab-sorbance spectra), which is not visible in either AOPP-HSA ordityrosine. Taken together, these spectral characteristics clearlydemonstrate the presence of dityrosine in AOPP-HSA.
AOPP-HSA trigger the monocyte respiratory burst
As shown in Figure 7, HOCl-induced AOPP-HSA triggered a re-spiratory burst in isolated human monocytes, as measured by lu-cigenin-amplified chemiluminescence. The chemiluminescenceproduction increased with the HSA/HOCl molar ratio, reaching amaximum at 1:60. AOPP-HSA at a HSA/HOCl molar ratio of1:120 induced a lower chemiluminescence production, and thiswas observed as a significant increase in both AOPP and dityrosineand a moderate increase in carbonyl concentrations compared with
FIGURE 2. Relationships between plasma levels of AOPP and AGE-pentosidine in 56 patients with chronic renal failure. The correlation co-efficient (r) by simple regression analysis between AOPP concentrationsand AGE-pentosidine concentrations was statistically significant at the in-dicatedp value.
FIGURE 3. Relationships between AOPP or AGE-pentosidine and themonocyte activation marker neopterin in 56 patients with chronic renalfailure. Correlation coefficients (r) between plasma concentrations of ne-opterin and AOPP (upper panel) or AGE-pentosidine (lower panel) arestatistically significant at the indicatedp value.
FIGURE 4. Relationships between AOPP or AGE pentosidine andTNF-a or TNF-soluble receptors, TNF-sR55 and TNF-sR75, in 60 patientswith chronic renal failure. Pearson’sr correlation coefficients betweenplasma concentrations of AOPP (left panel) or AGE-pentosidine (rightpanel) and those of TNF-a or its soluble receptors are significant at theindicatedp value.
2528 ADVANCED OXIDATION PROTEIN PRODUCTS IN CHRONIC UREMIA
by guest on Decem
ber 7, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
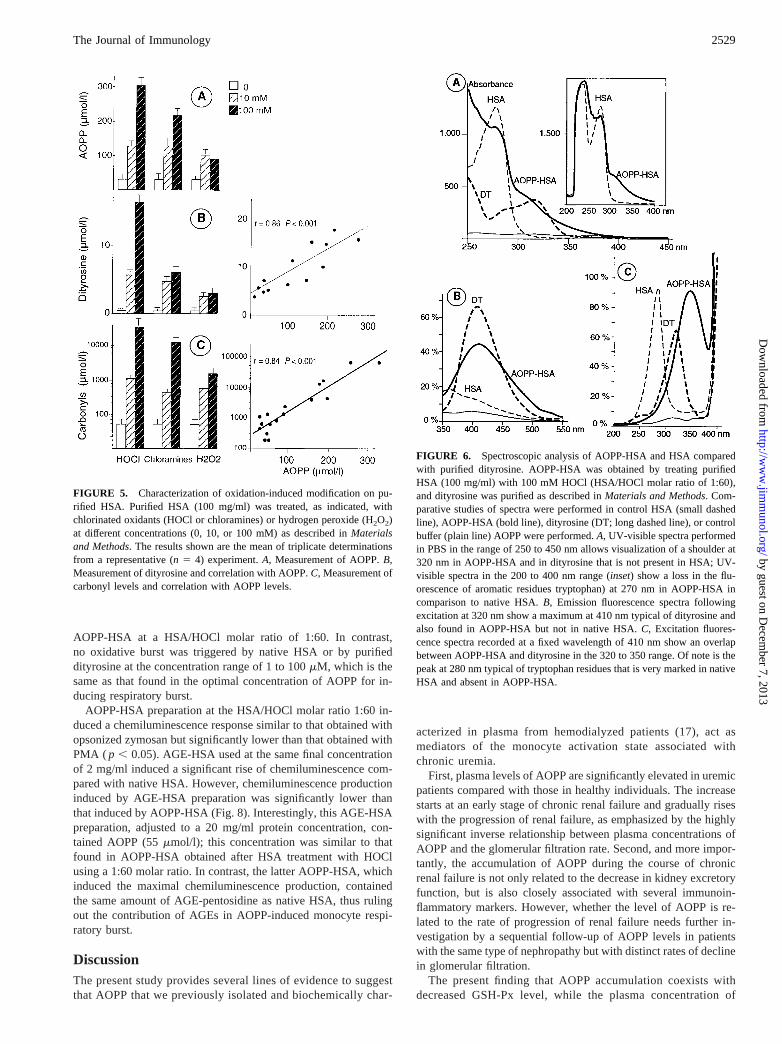
AOPP-HSA at a HSA/HOCl molar ratio of 1:60. In contrast,no oxidative burst was triggered by native HSA or by purifieddityrosine at the concentration range of 1 to 100mM, which is thesame as that found in the optimal concentration of AOPP for in-ducing respiratory burst.
AOPP-HSA preparation at the HSA/HOCl molar ratio 1:60 in-duced a chemiluminescence response similar to that obtained withopsonized zymosan but significantly lower than that obtained withPMA ( p , 0.05). AGE-HSA used at the same final concentrationof 2 mg/ml induced a significant rise of chemiluminescence com-pared with native HSA. However, chemiluminescence productioninduced by AGE-HSA preparation was significantly lower thanthat induced by AOPP-HSA (Fig. 8). Interestingly, this AGE-HSApreparation, adjusted to a 20 mg/ml protein concentration, con-tained AOPP (55mmol/l); this concentration was similar to thatfound in AOPP-HSA obtained after HSA treatment with HOClusing a 1:60 molar ratio. In contrast, the latter AOPP-HSA, whichinduced the maximal chemiluminescence production, containedthe same amount of AGE-pentosidine as native HSA, thus rulingout the contribution of AGEs in AOPP-induced monocyte respi-ratory burst.
DiscussionThe present study provides several lines of evidence to suggestthat AOPP that we previously isolated and biochemically char-
acterized in plasma from hemodialyzed patients (17), act asmediators of the monocyte activation state associated withchronic uremia.
First, plasma levels of AOPP are significantly elevated in uremicpatients compared with those in healthy individuals. The increasestarts at an early stage of chronic renal failure and gradually riseswith the progression of renal failure, as emphasized by the highlysignificant inverse relationship between plasma concentrations ofAOPP and the glomerular filtration rate. Second, and more impor-tantly, the accumulation of AOPP during the course of chronicrenal failure is not only related to the decrease in kidney excretoryfunction, but is also closely associated with several immunoin-flammatory markers. However, whether the level of AOPP is re-lated to the rate of progression of renal failure needs further in-vestigation by a sequential follow-up of AOPP levels in patientswith the same type of nephropathy but with distinct rates of declinein glomerular filtration.
The present finding that AOPP accumulation coexists withdecreased GSH-Px level, while the plasma concentration of
FIGURE 5. Characterization of oxidation-induced modification on pu-rified HSA. Purified HSA (100 mg/ml) was treated, as indicated, withchlorinated oxidants (HOCl or chloramines) or hydrogen peroxide (H2O2)at different concentrations (0, 10, or 100 mM) as described inMaterialsand Methods. The results shown are the mean of triplicate determinationsfrom a representative (n 5 4) experiment.A, Measurement of AOPP.B,Measurement of dityrosine and correlation with AOPP.C, Measurement ofcarbonyl levels and correlation with AOPP levels.
FIGURE 6. Spectroscopic analysis of AOPP-HSA and HSA comparedwith purified dityrosine. AOPP-HSA was obtained by treating purifiedHSA (100 mg/ml) with 100 mM HOCl (HSA/HOCl molar ratio of 1:60),and dityrosine was purified as described inMaterials and Methods. Com-parative studies of spectra were performed in control HSA (small dashedline), AOPP-HSA (bold line), dityrosine (DT; long dashed line), or controlbuffer (plain line) AOPP were performed.A, UV-visible spectra performedin PBS in the range of 250 to 450 nm allows visualization of a shoulder at320 nm in AOPP-HSA and in dityrosine that is not present in HSA; UV-visible spectra in the 200 to 400 nm range (inset) show a loss in the flu-orescence of aromatic residues tryptophan) at 270 nm in AOPP-HSA incomparison to native HSA.B, Emission fluorescence spectra followingexcitation at 320 nm show a maximum at 410 nm typical of dityrosine andalso found in AOPP-HSA but not in native HSA.C, Excitation fluores-cence spectra recorded at a fixed wavelength of 410 nm show an overlapbetween AOPP-HSA and dityrosine in the 320 to 350 range. Of note is thepeak at 280 nm typical of tryptophan residues that is very marked in nativeHSA and absent in AOPP-HSA.
2529The Journal of Immunology
by guest on Decem
ber 7, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
malondialdehyde remains stable, supports the contention thatAOPP are more accurate markers of oxidative stress than lipidperoxidation products (17). The occurrence of oxidative stressin chronic renal failure patients whether on dialysis or not hasbeen suggested in studies showing an imbalance between oxi-dant and antioxidant systems and their cofactors (6, 7). Recentreports have stressed the role of such an oxidative stress in theaccelerated atherosclerosis process associated with end-stagerenal disease (12).
To determine the role of AOPP in uremia-associated immunedysregulation, we analyzed their relationships with cell activationmarkers, with emphasis on monocytes as a potential source ofoxidants and proinflammatory cytokines. While no relationshipwas found between AOPP and T or B cell activation markers, aclose correlation was observed between AOPP and neopterin, themonocyte activation marker. This selective relationship betweenAOPP and monocyte activation was further established with pos-itive correlations between AOPP and TNF-a and its soluble re-ceptors, and, to a lesser degree, with IL-1Ra, although these cor-relations tended to be of only borderline significance when valueswere corrected for creatinine clearance. These latter findings sug-gested that the relationship was not related to cytokine or cytokineinhibitor biologic activities, and this was further evidenced by theabsence of correlation between AOPP and the TNF-sR55/TNF-aor TNF75/TNF-a molar ratios, which may better reflect the bio-logic activity of TNF-a (3, 4).
Of note, in another state of profound immune dysregulation, inHIV patients, we observed very high plasma levels of AOPP. Inthis clinical condition, characterized by a pronounced oxidativestress in the absence of renal failure, AOPP was an exquisitemarker of oxidative stress correlating tightly with the degree ofmonocyte activation (37).
FIGURE 7. Effect of HSA-AOPP preparations on monocyte respiratoryburst measured by lucigenin-amplified chemiluminescence (CL). HSA wasoxidized at different HSA/HOCl molar ratios to induce differential oxida-tive modification. The preparations were then adjusted to a protein con-centration of 20 mg/ml, and the level of oxidation was evaluated by mea-suring AOPP, dityrosine, and carbonyl concentrations. These AOPP-HSApreparations (25ml at 20 mg/ml) were then used to stimulate isolatedmonocytes (100ml at 2 3 105 cells/ml) in the presence of 25ml of HBSSand 100ml lucigenin. Dityrosine used at a concentration of 5mmol/l didnot trigger the respiratory burst (the same result was obtained with dity-rosine concentrations ranging from 5–25mmol/l). Chemiluminescence pro-duction (measured in counts per minute) was recorded for 60 min. Resultsfrom triplicate determinations of a representative experiment are shown(n 5 5).
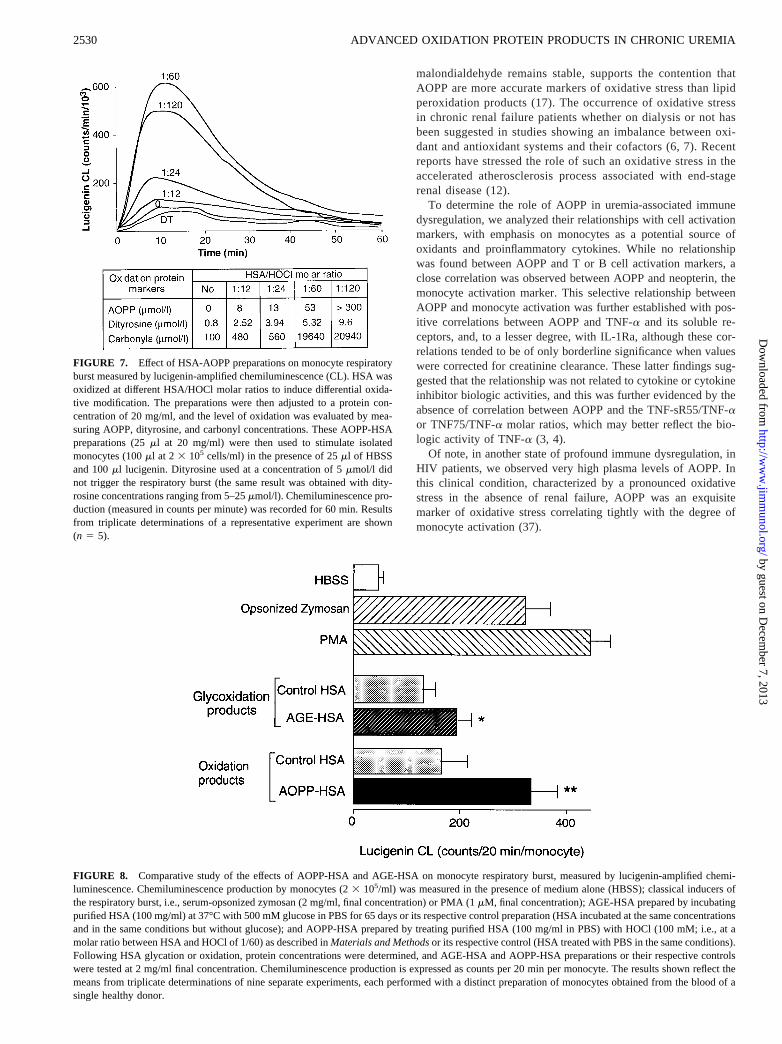
FIGURE 8. Comparative study of the effects of AOPP-HSA and AGE-HSA on monocyte respiratory burst, measured by lucigenin-amplified chemi-luminescence. Chemiluminescence production by monocytes (23 105/ml) was measured in the presence of medium alone (HBSS); classical inducers ofthe respiratory burst, i.e., serum-opsonized zymosan (2 mg/ml, final concentration) or PMA (1mM, final concentration); AGE-HSA prepared by incubatingpurified HSA (100 mg/ml) at 37°C with 500 mM glucose in PBS for 65 days or its respective control preparation (HSA incubated at the same concentrationsand in the same conditions but without glucose); and AOPP-HSA prepared by treating purified HSA (100 mg/ml in PBS) with HOCl (100 mM; i.e., at amolar ratio between HSA and HOCl of 1/60) as described inMaterials and Methodsor its respective control (HSA treated with PBS in the same conditions).Following HSA glycation or oxidation, protein concentrations were determined, and AGE-HSA and AOPP-HSA preparations or their respective controlswere tested at 2 mg/ml final concentration. Chemiluminescence production is expressed as counts per 20 min per monocyte. The results shown reflect themeans from triplicate determinations of nine separate experiments, each performed with a distinct preparation of monocytes obtained from the blood of asingle healthy donor.
2530 ADVANCED OXIDATION PROTEIN PRODUCTS IN CHRONIC UREMIA
by guest on Decem
ber 7, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

Another important aspect of the present study was to furtherinvestigate the relationship between AOPP and advanced glyco-sylation proteins as assessed by AGE-pentosidine. We and otherinvestigators have previously documented that chronic renal fail-ure is associated with increased AGE formation independent ofdiabetes or aging (24–26). In the present study, the close correla-tion observed between plasma AGE-pentosidine and AOPP dem-onstrates that this relationship already exists in uremic patients notyet undergoing dialysis and further suggests that AGE and AOPPmay share common mechanisms of formation and/or common bi-ologic activities in vivo. Moreover, several studies pointed to theinvolvement of oxidative pathways in the formation of AGE, no-tably in patients with end-stage renal failure (24, 38).
Our previous observations demonstrated that, in vivo, AOPPresult from oxidant-induced protein cross-linking and correlatewith dityrosine levels. To determine the mechanisms by whichAOPP could be involved in monocyte activation, the potentialproinflammatory effects of AOPP-HSA produced in vitro by ex-posing HSA to chlorinated oxidants were studied. As evidenced inthe case of carbonyl and dityrosine levels in oxidant-treated HSA,chlorinated oxidants appear very efficient in inducing AOPP. Spec-troscopy studies reveal that AOPP-HSA could be identified as fol-lows: in UV-visible spectra, by perturbation of the aromatic resi-due band associated with a large band at 320 nm, the latterproperty being used for routine quantification of AOPP; in fluo-rescence spectra by the loss of tryptophan and tyrosine emissionand the appearance of the dityrosine contribution clearly visualizedin the excitation spectrum. Therefore, the spectral measurement ofAOPP, which is maximum at 320 nm, is the result of an overlap-ping between chromophores, among which dityrosine is identifiedas a predominant component.
We then demonstrated that AOPP-HSA, but not purified dity-rosine, possesses the ability to activate isolated monocytes in vitro,emphasizing three remarkable features of the induced respiratoryburst: 1) its induction is dependent on the chlorinated nature of theoxidant, i.e., HOCl and chloramine, but not H2O2; 2) its intensityis directly related to the level of protein oxidation as defined by themolar ratio of protein to oxidant and ascertained by dityrosine andcarbonyl measurements; and 3) it is related to the protein cross-linking structure, since purified dityrosine alone has no effect onmonocytes.
In the past, studies on the interactions between proteins andoxidants have focused on the structural changes induced by oxi-dants generated by water pulse radiolysis, including superoxideand hydroxyl radicals (13, 14). Such studies have demonstratedthat structural modifications of proteins (selective loss of an aminoacid, fragmentation, or aggregation) were highly dependent on thenature of the oxidant. Since AOPP formation is optimal with chlo-rinated oxidants, it is interesting to speculate that its formation invivo might result from enzymatic activity of phagocyte-derivedMPO (39). Interestingly, although the best-characterized productof MPO is HOCl, MPO-derived enzymatic activity can also gen-erate dityrosine (16), a compound that correlates with AOPP lev-els, and aldhehyde compounds, which may exert a potent biologiceffect at the site of inflammation (40). MPO can also convertL-serine to glycolaldehyde, which mediates the formation of car-boxymethylysine, described as a byproduct of protein glycation(41). The pathophysiologic relevance of MPO-derived chlorinatedoxidant is also illustrated by its role in the formation of oxidativelymodified low density lipoproteins (42).
Another important aspect of the present study was to furtherinvestigate the relationship between AOPP and AGEs. BothAOPP-HSA and AGE-HSA triggered monocyte NADPH oxidaseactivation, leading to superoxide anion production. Of note, while
AOPP-HSA did not contain AGE-pentosidine, significant concen-trations of AOPP were found in AGE-HSA preparations. Takentogether, these data suggest that AGE-proteins contain some struc-tural motif that results from an oxidative process and support thehypothesis that AGE-mediated biologic activities might depend ontheir level of oxidation (43). The interaction between AGEs andmacrophages is now well established. Macrophages may internal-ize AGE-modified proteins via a specific receptor, or RAGE (21,44), which is also expressed by endothelial cells (45). Interest-ingly, the macrophage RAGE can be up-regulated by TNF-a (21,46). How macrophages process AOPP-HSA remains elusive, buttaking into account a possible structural resemblance betweenAGE and AOPP, the involvement of a similar receptor-mediatedprocess is plausible.
In conclusion, AOPP may represent a novel class of proinflam-matory mediators acting as a mediator of oxidative stress andmonocyte respiratory burst. The monocyte is thus, at the sametime, the elective cellular target of AOPP and a potential source ofoxidants inducing AOPP.
AcknowledgmentsWe are grateful to Dr. P. Soubiran (Laboratoire d’Analyses MedicalesSpecialise en Immunologie, Nice, France) for measurement of plasma lev-els of soluble CD25, and to Dr. M. Thevenin, Ph.D. (Laboratoire de Bio-chimie A, Necker Hospital, Paris, France), for determination of plasmaGSH-Px activity. We thank Francoise Tresset for skillful technical assis-tance, Mathilde Labrunie, M.S., for statistical analysis, and Dr. M. WebbM.D. (St. Thomas Hospital, London, U.K.), for critical advise on themanuscript.
References1. Descamps-Latscha, B. 1993. The immune system in end-stage renal disease.
Curr. Opin. Nephrol. Hypertens. 2:883.2. Dinarello, C. A. 1992. Cytokines: agents provocateurs in hemodialysis?Kidney
Int. 41:683.3. Pereira, B. J. G., L. Shapiro, A. J. King, M. E. Falagas, J. A. Strom, and
C. A. Dinarello. 1994. Plasma levels of IL1b, TNFa and their specific inhibitorsin undialyzed chronic renal failure, CAPD and hemodialysis patients.Kidney Int.45:890.
4. Descamps-Latscha, B., A. Herbelin, A. T. Nguyen, P. Roux-Lombard, J. Zingraff,A. Moynot, C. Verger, D. Dahmane, D. de Groote, P. Jungers, and J. M. Dayer.1995. Balance between IL-1b, TNF-a, and their specific inhibitors in chronicrenal failure and maintenance dialysis: relationships with activation markers of Tcells, B cells, and monocytes.J. Immunol. 154:882.
5. Descamps-Latscha, B., and P. Jungers. 1996. Immunological and chronic inflam-matory abnormalities in end stage renal disease. InReplacement of Renal Func-tion by Dialysis.C. Jacobs, C. M. Kjellstrand, K. M. Koch, and J. F. Winchester,eds. Kluwer, Dordrecht, Boston, p. 1083.
6. Richard, M. J., J. Arnaud, C. Jurkovitz, T. Hachache, H. Meftahi, F. Laporte,M. Foret, A. Favier, and D. Cordonnier. 1991. Trace elements and lipid peroxi-dation abnormalities in patients with chronic renal failure.Nephron 57:10.
7. Ceballos-Picot I, V. Witko-Sarsat, M. Merad-Boudia, A. T. Nguyen, M. Theve-nin, M. C. Jaudon, J. Zingraff, P. Jungers, and B. Descamps-Latscha. 1996. Glu-tathione antioxidant system as a marker of uremia progression and oxidativestress in chronic renal failure.Free Radical Biol. Med. 21:845.
8. Van Ypersele de Strihou, C., M. Jadoul, J. Malghem, B. Maldague, and J. Jamart.1991. Effect of dialysis membrane and patient’s age on signs of dialysis-relatedamyloidosis.Kidney Int. 39:1012.
9. Capeillere-Blandin, C., T. Delaveau, and B. Descamps-Latscha. 1991. Structuralmodifications of humanb2 microglobulin treated with oxygen-derived radicals.Biochem. J. 277:175.
10. Linder, A., B. Charra, D. J. Sherrard, and B. H. Scribner. 1974. Acceleratedatherosclerosis in prolonged maintenance hemodialysis.N. Engl. J. Med. 290:698.
11. Maggi, E., R. Bellazzi, F. Falaschi, A. Frattoni, G. Perani, G. Finardi, A. Gazo,M. Nai, D. Romanini, and G. Bellomo. 1994. Enhanced LDL oxidation in uremicpatients: an additional mechanism for accelerated atherosclerosis?Kidney Int.45:876.
12. Becker, B. M., J. Himmerlfarb, W. L. Henrich, and R. M. Hakim. 1997. Reas-sessing the cardiac risk profile in chronic hemodialysis patients: a hypothesis onthe role of oxidant stress and other non-traditional cardiac risk factors.J. Am. Soc.Nephrol. 8:475.
13. Davies, K. J. 1987. Protein damage and degradation by oxygen radicals.I. General aspects.J. Biol. Chem. 262:9895.
2531The Journal of Immunology
by guest on Decem
ber 7, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

14. Davies, K. J., and A. L. Goldberg. 1987. Oxygen radicals stimulate intracellularproteolysis and lipid peroxidation by independent mechanisms in erythrocytes.J. Biol. Chem. 262:8220.
15. Dean, R. T., S. Fu, R. Stocker, and M. J. Davies. 1997. Biochemistry and pa-thology of radical-mediated protein oxidation.Biochem. J. 324:1.
16. Heinecke, J. W., W. Li, H. L. Daehnke, and J. A. Goldstein. 1993. Dityrosine, aspecific marker of oxidation, is synthesized by the myeloperoxidase-hydrogenperoxide system of human neutrophils and macrophages.J. Biol. Chem. 268:4069.
17. Witko-Sarsat, V., M. Friedlander, C. Capeillere-Blandin, T. Nguyen-Khoa,A. T. Nguyen, J. Zingraff, P. Jungers, and B. Descamps-Latscha. 1996. Advancedoxidation protein products as a novel marker of oxidative stress in uremia.KidneyInt. 49:1304.
18. Vanholder, R., S. Ringoir, A. Dhondt, and R. M. Hakim. 1991. Phagocytosis inuremic and hemodialysis patients: a prospective and cross sectional study.KidneyInt. 39:320.
19. Ritz, E., R. Deppish, and P. Nawroth. 1994. Toxicity of uraemia–does it come ofAGE? Nephrol. Dial. Transplant. 9:1.
20. Makita, Z., R. Bucala, E. J. Rayfield, E. A. Friedman, A. M. Kaufman,S. M. Korbet, R. H. Barth, J. A. Winston, H. Fuh, K. R. Manogue, and A. Cerami.1994. Reactive glycosylation endproducts in diabetic uraemia and treatment ofrenal failure.Lancet 343:1519.
21. Miyata, T., R. Inagi, Y. Iida, M. Sato, N. Yamada, O. Oda, K. Maeda, and H. Seo.1994. Involvement ofb2-microglobulin modified with advanced glycation endproducts in the pathogenesis of hemodialysis-associated amyloidosis: inductionof human monocyte chemotaxis and macrophage secretion of tumor necrosisfactor-a and interleukin-1.J. Clin. Invest. 93:521.
22. Niwa, T., S. Miyazaki, T. Katsuzaki, N. Tatemichi, Y. Takei, T. Miyazaki,T. Morita, and Y. Hirasawa. 1995. Immunohistochemical detection of advancedglycation end products in dialysis-related amyloidosis.Kidney Int. 48:771.
23. Friedlander, M. A., Y. C. Wu, A. Elgawish, and V. M. Monnier. 1996. Early andadvanced glycosylation end products: kinetics of formation and clearance in peri-toneal dialysis.J. Clin. Invest. 97:728.
24. Monnier, V. M., D. R. Sell, R. H. Nagaraj, S. Miyata, S. Grandhee, P. Odetti, andS. A. Ibrahim. 1992. Maillard reaction-mediated molecular damage to extracel-lular matrix and other tissue proteins in diabetes, aging, and uremia.Diabetes41(Suppl. 2):36.
25. Miyata, T., Y. Wada, Z. Cai, Y. Iida, K. Horie, Y. Yasuda, K. Maeda,K. Kurokawa, and C. Van Ypersele de Strihou. 1997. Implication of an increasedoxidative stress in the formation of advanced glycation end products in patientswith end-stage renal failure.Kidney Int. 51:1170.
26. Friedlander, M. A., V. Witko-Sarsat, A. T. Nguyen, Y. C. Wu, M. Labrunie,C. Verger, P. Jungers, and B. Descamps-Latscha. 1996. The advanced glycationendproduct pentosidine and monocyte activation in uremia.Clin. Nephrol. 45:379.
27. Witko, V., A. T. Nguyen, and B. Descamps-Latscha. 1992. Microtiter plate assayfor phagocyte-derived taurine-chloramines.J. Clin. Lab. Anal. 6:47.
28. Anderson, S. O. 1966. Covalent cross-links in a structural protein, resilin.ActaPhysiol. Scand. 263:1.
29. Bayse, G. S., A. W. Michaels, and M. Morrisson. 1972. The peroxidase-catalyzedoxidation of tyrosine.Biochim. Biophys. Acta 284:34.
30. Amado, R., R. Aeschbach, and H. Neukom. 1984. Dityrosine: in vitro productionand characterization.Methods Enzymol. 107:377.
31. Resnik, A. Z., and L. Packer. 1994. Oxidative damage to proteins: spectropho-tometric method for carbonyl assay.Methods Enzymol. 233:357.
32. Odetti, P., J. Fogarty, D. R. Sell, and V. M. Monnier. 1992. Chromatographicquantitation of plasma and erythrocyte pentosidine in diabetic and uremic sub-jects.Diabetes 41:153.
33. Sell, D. R., and V. M. Monnier. 1989. Structure elucidation of a senescencecross-link from human extracellular matrix: implication of pentoses in the agingprocess.J. Biol. Chem. 264:21597.
34. Anderson, M. E. 1985. Determination of glutathione and glutathione disulfide inbiological samples.Methods Enzymol. 113:548.
35. Roux-Lombard, P., L. Punzi, F. Hasler, S. Bas, S. Todesco, H. Gallati,P. A. Guerne, and J. M. Dayer. 1993. Soluble tumor necrosis factor receptors inhuman inflammatory synovial fluids.Arthritis Rheum. 36:485.
36. Allen, R.C. 1986. Phagocytic leukocyte oxygenation activities and chemilumi-nescence: a kinetic approach to analysis.Methods Enzymol. 133:449.
37. Vilde J. L., V. Witko-Sarsat, D. Salmon-Ceron, J. Saba, C. Mandet, andB. Descamps-Latscha. 1995. Persistence of active oxidation of serum proteins inpatients receiving dapsone for secondary prophylaxis of pneumocystosis. InAb-stracts of the 35th Interscience Conference on Antimicrobial Agents and Che-motherapy.ICAAC, San Francisco, p. 237.
38. Kawamura, M., J. W. Heinecke, and A. Chait. 1994. Pathophysiological concen-trations of glucose promote oxidative modification of low density lipoprotein bya superoxide-dependent pathway.J. Clin. Invest. 94:771.
39. Klebanoff, S. J. 1969. Myeloperoxidase-halide-hydrogen peroxide antibacterialsystem.J. Bacteriol. 95:2131.
40. Anderson, M. M., S. L. Hazen, F. F. Hsu, and J. W. Heinecke. 1997. Humanneutrophils employ the myeloperoxidase-hydrogen peroxide-chloride system toconvert hydroxy-amino acids into glycolaldehyde, 2-hydroxypropanal, and acro-lein: a mechanism for the generation of highly reactivea-hydroxy anda, b-un-saturated aldehydes by phagocytes at sites of inflammation.J. Clin. Invest. 99:424.
41. Leeuwenburgh, C., J. E. Rasmussen, F. F. Hsu, D. M. Mueller, S. Pennathur, andJ. W. Heinecke. 1997. Mass spectrometric quantification of markers for proteinoxidation by tyrosyl radical, copper, and hydroxyl radical in low density lipopro-tein isolated from human atherosclerotic plaques.J. Biol. Chem. 272:3520.
42. Daugherty, A., J. L. Dunn, D. L. Rateri, and J. W. Heinecke. 1994. Myeloper-oxidase, a catalyst for lipoprotein oxidation, is expressed in human atheroscle-rotic lesions.J. Clin. Invest. 94:437.
43. Miyata, T., K. Maeda, K. Kurokawa, and C. van Ypersele de Strihou. 1997.Oxidation conspires with glycation to generate noxious advanced glycation endproducts in renal failure.Nephrol. Dial. Transplant. 12:255.
44. Vlassara, H., M. Brownlee, and A. Cerami. 1988. Specific macrophage receptoractivity for advanced glycosylation end products inversely correlates with insulinlevels in vivo.Diabetes 37:456.
45. Schmidt, A. M., R. Mora, R. Cao, S. D. Yan, J. Brett, R. Ramakrishnan,T. C. Tsang, M. Simionescu, and D. Stern. 1994. The endothelial cell binding sitefor advanced glycation end products consists of a complex: an integral membraneprotein and a lactoferrin-like polypeptide.J. Biol. Chem. 269:9882.
46. Vlassara, H., L. Moldawer, and B. Chan. 1989. Macrophage/monocyte receptorfor nonenzymatically glycosylated protein is upregulated by cachectin/tumor ne-crosis factor.J. Clin. Invest. 84:1813.
47. Witko-Sarsat, V., and B. Descamps-Latscha. 1997. Advanced oxidation proteinproducts: novel uraemic toxins and pro-inflammatory mediators in chronic renalfailure?Nephrol. Dial. Transplant. 12:1310.
2532 ADVANCED OXIDATION PROTEIN PRODUCTS IN CHRONIC UREMIA
by guest on Decem
ber 7, 2013http://w
ww
.jimm
unol.org/D
ownloaded from




















