
Inflammation as Therapeutic Objective in Stroke
16
Current Pharmaceutical Design, 2008, 14, 3549-3564 3549 1381-6128/08 $55.00+.00 © 2008 Bentham Science Publishers Ltd. Inflammation as Therapeutic Objective in Stroke Joaquín Jordán 1, * , Tomás Segura 2 , David Brea 3 , Maria F. Galindo 4 and José Castillo 3 1 Grupo de Neurofarmacología. Departamento de Ciencias Médicas. Facultad de Medicina. Universidad Castilla-La Mancha. Centro Regional de Investigaciones Biomédicas. Spain; 2 Servicio de Neurología. Complejo Hospitalario Uni- versitario de Albacete. Albacete. Spain; 3 Department of Neurology, Clinical Neuroscience Research Laboratory, Hospi- tal Clínico Universitario de Santiago de Compostela, University of Santiago de Compostela. Spain and 4 Unidad de Neu- ropsicofarmacología Translacional. Complejo Hospitalario Universitario de Albacete. Albacete. Spain Abstract: Ischemic stroke is the most frequent cause of persistent neurologic disability in modern Western societies. Al- beit it is still not clear whether inflammation is merely an epiphenomenon or rather has a disease-promoting function, ac- cumulating evidence implicates inflammation in many forms of acute neurodegenerative disorders including ischemia. The immune cell influx during a neuropathological event is thought to be elicited by glial cells, especially microglia. This article reviews the cellular and molecular pathways involved in stroke-induced inflammatory response in the CNS. We fo- cused on how CNS innate immune cells including microglia and macrophages play integral roles in receiving and propa- gating inflammatory signals, and how activated microglia secrete a wide range of factors. We present the relevance of the expression of adhesion molecules after ischemia including selectin, immunoglobulin superfamily, integrins, and the role of inflammatory mediators such as cytokines, chemokines and matrix metalloproteinases. Further, we explore the role of transcription factors in inflammation, and the function of immunomodulation and innate and adaptive immunity in brain ischemia, focusing on immunosupression therapies for acute stroke. Although several approaches for anti-inflammatory treatment have proven effective in animal models, clinical trials of immune system modulation therapy after stroke have not yet proved successful. There is still much to be done in order to translate interesting findings into therapies, but un- doubtedly studying the cellular and molecular pathways may not only improve our understanding of inflammatory mechanism but also serve as a basis for designing effective therapies. 1. INTRODUCTION Stroke is the second leading cause of death and the bur- den of disease in high income countries [1]. Stroke occurs due to a loss of blood supply to part of the brain, initiating the ischemic cascade. As oxygen or glucose becomes de- pleted in ischemic brain tissue, the production of high energy phosphate compounds such as adenosine triphosphate (ATP) fails leading to failure of energy dependent processes (such as ion pumping) necessary for tissue cell survival. This sets off a series of interrelated events that result in cellular injury and death by necrosis. Dead cells by necrosis produce the release of all cytoplasmatic content into the extracellular space activating the corresponding inflammatory response. The central nervous system (CNS) has for long been re- garded as an immune privileged organ, with the blood–brain barrier (BBB) tightly regulating the influx of immune cells and mediators from the vascular compartment to the brain parenchyma [2]. Inflammation is generally a beneficial re- sponse of an organism to infection but, when prolonged or inappropriate, it can be detrimental. Neuronal loss in acute (e.g. stroke and head injury) and chronic [e.g. multiple scle- rosis and Alzheimer's disease] CNS diseases has been asso- ciated with inflammatory processes systemically and in the *Address correspondence to this author at the Grupo de Neurofarmacologia, Departamento de Ciencias Médicas, Facultad de Medicina, Universidad Castilla-La Mancha. Centro Regional de Investigaciones Biomédicas, Avda Almansa, 14, 02006-Albacete, Spain; Tel: 34-967599200; Fax: 34-967- 599327; E-mail: [email protected] brain. Brain inflammation is characterized by activation of microglia and astrocytes, expression of key inflammatory mediators, but limited invasion of circulating immune cells. Inflammation induces rapid expression of key inflammatory mediators -cytokines, chemokines and prostaglandins- which in turn up-regulate adhesion molecules, increase permeabil- ity of the BBB, facilitating invasion of peripheral immune cells, induce release of potentially toxic molecules and com- promise brain cells. Because the BBB is disrupted after stroke, the immune system comes into contact with CNS antigens, in both the brain and periphery [3]. In recent years, major advances in the study of the role of the immune system and inflammation in brain ischemia have been done. There are many evidences that inflammation and immune response play an important role in the outcome of ischemic stroke patients, and they have been associated to larger brain damage. However, on the other hand, these mechanisms might be necessary for the resolution of dead cells and the initiation of repairing mechanisms. In this work we review the inflammatory response to brain ischemia, the role of innate immunity, and the potential role of endogenous anti-inflammation and immune-modulation processes. 2. CELLULAR INFLAMMATORY RESPONSE The CNS innate immune cells including microglia and macrophages play integral roles in receiving and propagating inflammatory signals. Microglia are a highly responsive population of cells with a well established role in regulating the immune surveillance of the nervous system [4,5]. Found
Transcript of Inflammation as Therapeutic Objective in Stroke

Current Pharmaceutical Design, 2008, 14, 3549-3564 3549
1381-6128/08 $55.00+.00 © 2008 Bentham Science Publishers Ltd.
Inflammation as Therapeutic Objective in Stroke
Joaquín Jordán1,*, Tomás Segura
2, David Brea
3, Maria F. Galindo
4 and José Castillo
3
1Grupo de Neurofarmacología. Departamento de Ciencias Médicas. Facultad de Medicina. Universidad Castilla-La
Mancha. Centro Regional de Investigaciones Biomédicas. Spain; 2Servicio de Neurología. Complejo Hospitalario Uni-
versitario de Albacete. Albacete. Spain; 3Department of Neurology, Clinical Neuroscience Research Laboratory, Hospi-
tal Clínico Universitario de Santiago de Compostela, University of Santiago de Compostela. Spain and 4Unidad de Neu-
ropsicofarmacología Translacional. Complejo Hospitalario Universitario de Albacete. Albacete. Spain
Abstract: Ischemic stroke is the most frequent cause of persistent neurologic disability in modern Western societies. Al-
beit it is still not clear whether inflammation is merely an epiphenomenon or rather has a disease-promoting function, ac-
cumulating evidence implicates inflammation in many forms of acute neurodegenerative disorders including ischemia.
The immune cell influx during a neuropathological event is thought to be elicited by glial cells, especially microglia. This
article reviews the cellular and molecular pathways involved in stroke-induced inflammatory response in the CNS. We fo-
cused on how CNS innate immune cells including microglia and macrophages play integral roles in receiving and propa-
gating inflammatory signals, and how activated microglia secrete a wide range of factors. We present the relevance of the
expression of adhesion molecules after ischemia including selectin, immunoglobulin superfamily, integrins, and the role
of inflammatory mediators such as cytokines, chemokines and matrix metalloproteinases. Further, we explore the role of
transcription factors in inflammation, and the function of immunomodulation and innate and adaptive immunity in brain
ischemia, focusing on immunosupression therapies for acute stroke. Although several approaches for anti-inflammatory
treatment have proven effective in animal models, clinical trials of immune system modulation therapy after stroke have
not yet proved successful. There is still much to be done in order to translate interesting findings into therapies, but un-
doubtedly studying the cellular and molecular pathways may not only improve our understanding of inflammatory
mechanism but also serve as a basis for designing effective therapies.
1. INTRODUCTION
Stroke is the second leading cause of death and the bur-den of disease in high income countries [1]. Stroke occurs due to a loss of blood supply to part of the brain, initiating the ischemic cascade. As oxygen or glucose becomes de-pleted in ischemic brain tissue, the production of high energy phosphate compounds such as adenosine triphosphate (ATP) fails leading to failure of energy dependent processes (such as ion pumping) necessary for tissue cell survival. This sets off a series of interrelated events that result in cellular injury and death by necrosis. Dead cells by necrosis produce the release of all cytoplasmatic content into the extracellular space activating the corresponding inflammatory response.
The central nervous system (CNS) has for long been re-garded as an immune privileged organ, with the blood–brain barrier (BBB) tightly regulating the influx of immune cells and mediators from the vascular compartment to the brain parenchyma [2]. Inflammation is generally a beneficial re-sponse of an organism to infection but, when prolonged or inappropriate, it can be detrimental. Neuronal loss in acute (e.g. stroke and head injury) and chronic [e.g. multiple scle-rosis and Alzheimer's disease] CNS diseases has been asso-ciated with inflammatory processes systemically and in the
*Address correspondence to this author at the Grupo de Neurofarmacologia,
Departamento de Ciencias Médicas, Facultad de Medicina, Universidad
Castilla-La Mancha. Centro Regional de Investigaciones Biomédicas, Avda
Almansa, 14, 02006-Albacete, Spain; Tel: 34-967599200; Fax: 34-967-
599327; E-mail: [email protected]
brain. Brain inflammation is characterized by activation of microglia and astrocytes, expression of key inflammatory mediators, but limited invasion of circulating immune cells. Inflammation induces rapid expression of key inflammatory mediators -cytokines, chemokines and prostaglandins- which in turn up-regulate adhesion molecules, increase permeabil-ity of the BBB, facilitating invasion of peripheral immune cells, induce release of potentially toxic molecules and com-promise brain cells. Because the BBB is disrupted after stroke, the immune system comes into contact with CNS antigens, in both the brain and periphery [3].
In recent years, major advances in the study of the role of the immune system and inflammation in brain ischemia have been done. There are many evidences that inflammation and immune response play an important role in the outcome of ischemic stroke patients, and they have been associated to larger brain damage. However, on the other hand, these mechanisms might be necessary for the resolution of dead cells and the initiation of repairing mechanisms. In this work we review the inflammatory response to brain ischemia, the role of innate immunity, and the potential role of endogenous anti-inflammation and immune-modulation processes.
2. CELLULAR INFLAMMATORY RESPONSE
The CNS innate immune cells including microglia and macrophages play integral roles in receiving and propagating inflammatory signals. Microglia are a highly responsive population of cells with a well established role in regulating the immune surveillance of the nervous system [4,5]. Found

3550 Current Pharmaceutical Design, 2008, Vol. 14, No. 33 Jordán et al.
scattered throughout the CNS, microglia constitute 5–15% of the total brain cell population and form a meshed network able to detect and react to modifications of the local envi-ronment [6]. In the mature brain, microglia typically exist in a resting state characterized by ramified morphology, and monitor the brain environment. The CNS endogenous mi-croglia share many properties with macrophages, having developed from a common precursor cell [7-9]. Early in fetal development, monocyte-like cells infiltrate the CNS and develop into parenchymal microglia. In the adult, parenchy-mal microglia in contrast to perivascular “microglia” are not frequently repopulated by fresh monocytes [10].
Microglia are primarily involved in immune surveillance [6, 11], but when activated have macrophage-like capabili-ties including phagocytosis, inflammatory cytokine produc-tion, and antigen presentation [12]. Normally these neuroin-flammatory changes are transient with microglia returning to a resting state as the immune stimulus is resolved. Aging or neurological disease, however, may provide a brain envi-ronment where microglia are more “reactive or primed” to a peripheral immune challenge [13]. In response to certain cues such as brain injury or immunological stimuli, however, microglia are readily activated. Activated microglia secrete a wide range of factors, some of which can actively trigger apoptosis in neuronal cell cultures. At the same time, micro-glia are also reported to increase neuronal survival through the release of trophic and anti-inflammatory factors.
Features common to microglia and systemic macro-phages include the expression of innate immune receptors and the ability to phagocyte pathogens, cells or cellular de-bris [7, 14]. Microglia is activated within minutes of ische-mia onset and produces a plethora of inflammatory media-tors, which exacerbate tissue damage [15]. Furthermore, mi-croglia responses are segregated not only respect to time, but some extend also spatially. After focal cortical ischemia, there is a second involvement of the ipsilateral thalamus at-tributable to retrograde degeneration of thalamo-cortical pro-jection fibres [16]. The PET tracer
11C-PK11195 binds to the
peripheral benzodiazepine receptor, which is abundant on brain-derived activated microglia, and that has been em-ployed in experimental and clinical studies addressing ischaemia-related inflammation, has shown that microglial activation becomes significant after a few days of stroke and persists for over 30 days, with a peak around 2 weeks for stroke onset [17]. Notably, the spatial distribution of
11C-
PK11195 binding evolves to include not only the core but also the rescued penumbra where inflammation may be sec-ondary to selective neuronal damage [18].
Recently it has been shown that patients with acute stroke had significantly better outcome with minocycline treatment compared with placebo [19], by involving minocycline anti-inflammatory properties, especially its ability to suppress activation of microglia, are likely to contribute to cytoprotec-tion in the CNS [20].
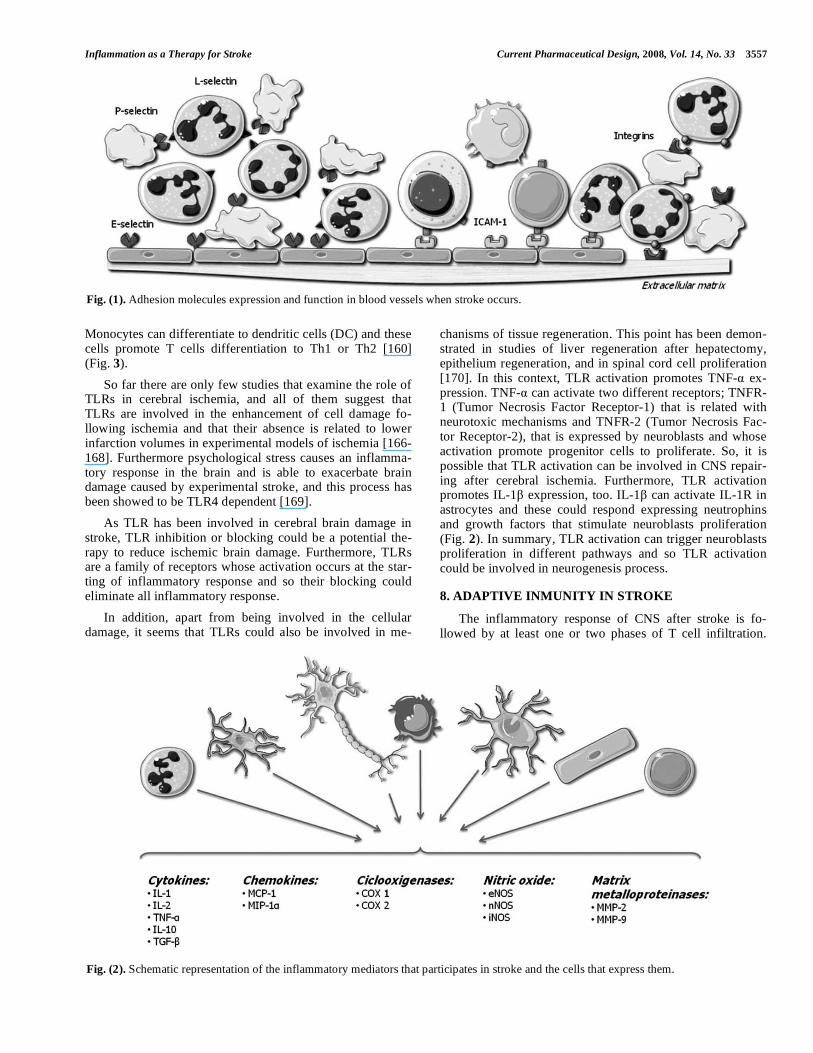
3. EXPRESSION OF ADHESION MOLECULES AF-TER ISCHEMIA
Inflammation after stroke involves leukocytes infiltration in brain parenchyma, specially neutrophils, that contribute to cerebral damage after ischemia [21] through reperfusion or
secondary injury mechanisms. Infiltration through the endo-thelium involves rolling, adhesion and transendothelial mi-gration of leukocytes. Therefore, adhesion molecules in leu-kocytes and endothelial cells are key molecules that contrib-ute to cerebral damage. Adhesion molecules that participate in this process are classified in three types; selectins, the superfamily of immunoglobulin and integrins.
3.1. Selectins
L-selectin, E-selectin and P-selectin are glycoproteins involved in the initial interaction between leucocytes and endothelial cells in the periphery of the infarct. They interact with P-selectin glycoprotein ligand (PSGL1) and other gly-cosylated ligand [22], to induce a transitory and reversible action that leads to other secondary cellular interactions me-diated by a different group of adhesins. As a consequence of the activation of adhesins, there is a recruitment of leuco-cytes, taking place their aggregation and adhesion to the vas-cular wall in a later stage. Such changes are responsible for the obstruction of the microvascularization and for the phe-nomenon of ‘no-reflux’. The conversion of the endothelium into a prothrombotic state, the production of free radicals and the rise in vascular permeability are other factors responsible for cellular damage mediated by inflammation.
At the individual level, P-selectin is found in platelets [23, 24] and endothelial cells, being its counter receptor, which contains the oligosaccharide sialyl Lewis X, present in leukocytes [25]. The existence of stores of P-selectin in the cytoplasm of endothelial cell [26] allows its mobilization to the cell surface within minutes from the activation of endo-thelial cells by thrombin, complement, and histamine [27].
Knowledge about the role of P-selectin has been pro-vided from both experimental models and clinical studies. From clinical studies it has been stated that surface expres-sion of P-selectin on platelets is related to clinical worsening after acute ischemic stroke, and a positive correlation be-tween P-selectin expression and NIHSS scores has been ob-served [28]. Studies on experimental models of cerebral ischemia have revealed that the lack of P-selectin or its blockage with monoclonal antibodies [29] or inhibitors [30, 31] was associated with improved neurological outcome and reduced cerebral infarct volumes.
The role of E-selectin and L-selectin in cerebral ischemia is not so clear. E-selectin can be found in endothelial cells and leukocytes, playing an important task in the develop-ment of inflammation in vivo [32]. E-selectin is also up-regulated in microvessels of animal models of ischemia, 2 hours after reperfusion [32,33]. L-selectin is found in endo-thelial cells and leukocytes mediating on leukocyte rolling, but its role in brain ischemia is less clear.
3.2. Anti-Selectin Treatment Possibilities
Studies in P-selectin knockout mice, and alternatively functional blockade of P-selectin with a monoclonal anti-body in wild-type mice, have shown decreased infarct vol-umes after transient permanent middle cerebral artery occlu-sion (MCAO) respect to controls. Furthermore, administra-tion of sCRsLex, an inhibitor of selectin-mediated platelet-leukocyte interactions, reduces infarct volumes in experi-mental models. However results varied from focal to global

Inflammation as a Therapy for Stroke Current Pharmaceutical Design, 2008, Vol. 14, No. 33 3551
experimental models of ischemia, since P-selectin blockade or deficiency was associated to better outcome and smaller infarcts in focal ischemia, while animals treated with anti-bodies against P-selectin showed reduced survival periods in models of gobal ischemia [34].
Knowledge about E-selectin and L-selectin is not such clear, therefore treatments against these selectins are less studied. Although it has been shown that E-selectin can in-duce immune tolerance and reduce injury upon intranasal administration to animals [35]. By the other hand, L-selectin treatments with L-selectin antibody has been shown ineffec-tiveness in animal models of stroke [36].
3.3. Immunoglobulin Superfamily
The immunoglobulin superfamily includes 5 members: intercellular adhesion molecule-1 (ICAM-1), intercellular adhesion molecule-2 (ICAM-2), vascular adhesion molecule-1 (VCAM-1), platelet–endothelial cell adhesion molecule-1 (PECAM-1), and the mucosal vascular addressing cell adhe-sion molecule 1 (MAdCAM-1). All of them are expressed on activated endothelial cells, mediating in the adhesion of leu-kocytes to endothelia, and creating stronger attachments than selectins [37].
ICAM1 is constitutively expressed, although its expres-sion in cerebral microvascular endothelial cells is increased by IL-1 , TNF- , lipopolysaccharide (LPS) [38], as ob-served in in vitro models of ischemia-like insults [39]. There are some other evidences that indicate its role in cerebral ischemia. Thus, increased expression of ICAM-1 has been found in areas of cerebral ischemia in rats submitted to MCAO [40]. Knock-out mice for ICAM-1 gene presented less cerebral damage than wild-type mice subjected to cere-bral ischemia [23]. The implication of ICAM-1 in cerebral ischemia has also been demonstrated in clinical studies, and a rise in serum concentrations of soluble adhesion molecules has been found after cerebral ischemia. Soluble ICAM-1 concentrations are elevated [41] and peak in patients within 24 hours of acute ischemic stroke, being these levels corre-lated with the infiltration of polimorphonuclear leukocytes [42]. Furthermore, ICAM-1 expression in brain microvessels is significantly increased in the cerebral infarcts of patients who die from ischemic stroke [43].
Localized ICAM-1 expression has been found in his-tological studies of normal human carotid bifurcation [44], a region of high-risk for the development of atherosclerotic plaque, while endothelial ICAM-1 expression is increased in symptomatic versus asymptomatic carotid plaque [45]. Pa-tients with carotid atherosclerosis who may be described as “prestroke stage” have raised concentrations of soluble (s)ICAM-1 [46]. This is in concordance with elevated solu-ble ICAM-1 levels that were found in patients with stroke risk factors [41].
Little is known about other intercellular adhesion mole-cules such as ICAM-2, VCAM-1, or PECAM-1. ICAM-2 may serve to stimulate the ligand binding of ICAM-1 to CD11b/CD18 [29]. Significantly, ICAM-2 is expressed not only by endothelial cells but also by resting and activated platelets [29]. In contrast to the other 2 integrin ligands, ICAM-1 and ICAM-3 (CD50), which are not found on plate-
lets, ICAM-2 may be involved in the mediation of leukocyte-platelet interactions in the microvessels during inflammation and thrombosis. The role of VCAM-1 in stroke is controver-sial. While some authors have described an increase in VCAM-1 mRNA after cerebral ischemia [29], others have failed to observe significant changes [47].
PECAM-1 is expressed at constant levels and plays addi-tional roles in attaching endothelial cells among them, and in negotiating with leukocytes [37]. These data support the role of cell adhesion molecules in the tight adhesion of leuko-cytes after cerebral ischemia, including the unique role of ICAM-2 in leukocyte-platelet interactions.
3.4. Immunoglobulin Superfamily Treatment Possibilities
A few studies have evaluated the possible treatment for ischemic stroke blocking receptors of the immunoglobulin superfamily.
Anti-ICAM1 antibody treatment has been shown to di-minish cerebral damage [32], neutrophil accumulation, apop-tosis and neurological deficits [49] in animal models of cere-bral ischemia. Furthermore, experimental studies performed on cultured endothelial cells have demonstrated an increase in the expression of ICAM-1 in cells subjected to ischemia [38].
Although ICAM-1 involvement in cerebral ischemia has been demonstrated in vivo, in vitro and in clinical studies, clinical trials with anti-ICAM-1 antibody have showed the existence of side effects and no improvement in outcome at 90 days, compared to control patients. The causes of these negative results could be that the antibody was obtained from mouse, and that included patients were not recanalized.
In a study of global cerebral ischemia in rats, leukotriene receptor antagonist improved neurological deficits and re-duced neuron death by inhibiting the ischemia-induced upregulation of VCAM-1 in the hippocampus of ischemic rats [50]. However, another study showed that treatment with VCAM-1 antibodies did not have any effect on stroke out-come suggesting that VCAM-1 may not play a significant role in ischemic brain injury [51].
3.5. Integrins
Integrins are another family of adhesion molecules that consist of heterodimeric membrane glycoproteins, with a common subunit and a variable subunit, that play a role in cell-cell and in cell-extracellular matrix interactions. There are three subfamilies of subunits, denoted 1–3. Members of the 1 subfamily bind collagen, laminin and fibronectin and are involved in the structure of the extrace- llular matrix. 2 integrins (CD18) are involved in leukocyte cell adhesion, and 3 integrins, also known as the cyto- adhesins, include the platelet glycoprotein IIb/IIIa ( IIb/ 3) and the vitronectin receptor ( v/ 3), which are factors in-volved in clot generation and stabilization. At the level of the basal lamina, integrins link endothelial cells to components of the extracellular matrix, such as laminin and collagen while in the brain, integrins join the endothelial cells, astro-cytes, and basal lamina that comprise the blood-brain barrier, being crucial for the maintenance of the integrity of the cere-bral microvasculature [52]. Leukocyte integrins are activated

3552 Current Pharmaceutical Design, 2008, Vol. 14, No. 33 Jordán et al.
by chemokines, cytokines, and other chemoattractants. In order to bind leukocytes to activated endothelium, integrins must be expressed on the cell surface, to be able to recognize endothelial cell adhesion molecules. Although leukocyte rolling is believed to be mediated primarily by P-selectin and E-selectin [27], the resulting firm cohesion to the vascular endothelium requires the expression of ICAM-1 in endothe-lial cells, and the interaction with the leukocyte integrin CD11b/CD18 [53]. In vitro studies have revealed that hy-poxia causes an increase of neutrophil CD11b expression compared to normoxia, and that this injury was protected by aprotinin because of the reduction of the upregulation of neutrophil CD11b [54].
3.6. Anti-Integrin Therapy as Treatment to Reduce
Ischemic Stroke Damage
Furthermore, in in vivo studies conducted on rats sub-jected to MCAO, administration of anti-CD11b or anti-CD18 monoclonal antibodies reduced infarct volumes and apopto-sis, and was associated to decreased accumulation of neutro-phils [55,56].
Despite the positive experimental data, when this ap-proach was performed in clinical studies, anti-integrin thera-pies with antibodies against CD11/CD18 in acute stroke pa-tients resulted negative, being these studies terminated pre-maturely due to a lack of effect on predetermined endpoints [57, 58].
4. INFLAMMATORY MEDIATORS
4.1. Cytokines
Most inflammatory reactions are mediated by cytokines which may potentiate ischemic brain injury. Cytokines are a group of small glycoproteins that play a significant role as activators of adhesion molecules. In the brain there are di- fferent cell types capable to secrete cytokines such as; mi-croglia, astrocytes, endothelial cells and neurons. In addition, it has been shown that peripherally derived cytokines are involved in brain inflammation. Thus, peripherally derived mononuclear phagocytes, T-lymphocytes, natural killer (NK) cells and PMN’s, produce and secrete cytokines and might contribute to inflammation of the CNS. Cytokines are up-regulated in the brain in response of a variety of stimulus including ischemia, being IL-1, interleukin-6 (IL-6), TNF- , interleukin-10 (IL-10) and TGF- , the most studied cytoki-nes related to inflammation in stroke.
Interleukine-1 (IL-1)
Members of the family of IL-1 family are expressed at low level or undetectable levels in healthy brain but their expression is rapidly up-regulated by ischemia. IL-1 has two isoforms, one called IL-1 and the second one named IL-1 . These two isoforms and its endogenous inhibitor, IL-1 recep-tor antagonist (IL-1ra) are the most studied ones in experi-mental stroke. IL-1 acts through two different receptor types (type I and II) [50, 60]. Type I receptor can be found on a variety of cell types and it binds to both IL-1 forms. On the contrary, type II receptor can be found on the cell surface of neutrophils, type B lymphocytes and macrophages and binds IL-1 with higher affinity [61].
Both IL-1 and IL- are synthesized as precursor proteins that lack a leader sequence. As a precursor IL-1 is fully active, whereas IL-1 is inactive and needs to be cleaved to mature IL-1 by the cysteine-aspartate protease caspase 1 [62].
IL-1 is produced in the CNS by various cell types such as; microglia, astrocytes, neurons and endothelium [63]. It has been shown that IL-1 mRNA is expressed following several brain injury types including kainite excitotoxicity [64] and LPS [65]. Furthermore, rising of mRNA within 15–30 minutes after ischemia has been demonstrated [66] which leads to an increase in protein a few hours later [67]. Twenty minutes after transient global cerebral ischemia in rats, IL-1beta mRNA and protein expression were increased not only during early reperfusion (1 h), but also at later times (6–24 h) indicating a biphasic expression [68].
There are studies that correlate an increase on the levels of IL-1 after ischemia and a worsening of the infarct sever-ity. However, evidences that IL-1 is neurotoxic are also con-siderable though controversial. IL-1 injected into a healthy brain does not cause any overt damage. Likewise, IL-1 added directly to pure neurons in culture does not cause death. Most IL-1 effects have been described in astrocytes. IL-1 promotes astrocytes proliferation and activation, which leads to astrogliosis [69].
IL-1 in the Treatment of Stroke
IL-1 is a potent pyrogen and increased body temperature exacerbates after experimental injury and worsen prognosis in acute stroke patients [70]. All studies have demonstrated an association between IL-1 upregulation and the infarct size increment, or a bad outcome, however they only show the existence of such association yet they did not comment any therapeutic possibilities using IL-1 as therapeutic tar-get. In this context other studies have introduced some con-troversy to this issue pointing to certain neuroprotective role of IL-1 [71, 72]. Thus, pretreatment of cultures of mouse primary cortical neurons with IL-1 or IL-1ß showed an at-tenuation of neurotoxicity induced by NMDA. Such neuro-protection mediated by IL-1 resulted inhibited when a neu-tralizing antibody to neuronal growth factor (NGF) was used [71]. Additionally, treatment of rat primary cortical neuron cultures with IL-1ß attenuated neuronal death induced by exposure to excytotoxic amino acids (glutamate, NMDA, AMPA, and kainate) [72]. The neuroprotective effects at-tributed to IL-1ß seem to be partially mediated by induction of NGF. In that study [72], IL-1ß was added to culture media both before (pretreatment) and after (post-treatment) expo-sure to excitatory amino acids. It was not determined whether pretreatment alone would have conferred greater neuroprotection nor if post-treatment alone would have ex-acerbated neurotoxicity. The common factor in both studies appears to be that the neuroprotection afforded by IL-1 de-pends on exposure of cultures to IL-1 prior to injury (pre-treatment). Other studies have demonstrated that intraven-tricular injection of recombinant IL-1B after MCAO in-creases the formation of brain edema, the volume of the size and the influx of neutrophils [73]. Those authors demon-strate that these phenomena happen when IL-1 is adminis-tered to rats [73], and observed that IL-1 deficient mice pre-sented smaller infarcts in comparison with wild type mice.

Inflammation as a Therapy for Stroke Current Pharmaceutical Design, 2008, Vol. 14, No. 33 3553
Furthermore, overexpression or treatment with IL-1ra re-duced the size of the infarcts and the severity of neurologic deficits [74, 75] while IL-1ra deficient mice exhibited a dra-matic increase in ischemic damage [76].
Interleukin 6 (IL-6)
IL-6 is a plethoric cytokine with several detrimental ef-fects which may contribute to early inflammatory injury in the brain. IL-6 is involved in the regulation of neuronal apoptosis [77]. IL-6 is up-regulated following cerebral ischemia [78]. Different studies suggest that IL-6 has detri-mental effects in cerebral ischemia. Thus, raised plasma con-centrations of IL-6 are a powerful predictor for early neuro-logical deterioration [79] and are associated with greater infarct volumes [80] and bad outcome [81]. Furthermore, as demonstrated by our group, the association between IL-6 and early neurological worsening, prevails without regard to the initial size, topography, or mechanism of the ischemic in-farction [81].
Tumor Necrosis Factor- (TNF- )
In the CNS, the pro-inflammatory cytokine TNF- is considered the principal mediator of neuroinflamattion that elicits a cascade of cellular events culminating in neuronal death. TNF- orchestrates a diverse array of functions in-volved in immune surveillance and defense, cellular homeo-stasis, and protection against certain neurological insults [82]. TNF- is upregulated in the brain after ischemia. Induc-tion of TNF- mRNA has been proved to happen in ischemic cortex after both permanent [83, 84] and transient MCAO [78] in rats. Barone et al., [85] have reported that following MCAO, the induction of TNF- was associated with exacer-bation of neurological deficits and infarct size. Analysis of the temporal profile of mRNA expression of cytokines in ischemic rats have revealed that the up-regulation of TNF- mRNA is proportional to IL-1 [86] and IL-6 [87] up-regula- tion. Initial increases are seen 1-3 h after ischemia onset [83], and have a two-phase pattern of expression with a second peak at 24-36 h [88, 89]. In clinical studies it has been shown that TNF- is upregulated in the brain tissue of patients with acute cerebral infarction [90], and appears sequentially in the infarction core and peri-infarct areas before it is expressed in the contralateral hemisphere and other remote brain areas [91]. Concentration of TNF- in cerebrospinal fluid (CSF) are increased in patients with acute ischemic stroke [79], including those with pronounced white matter lesions [92]. Serum concentrations of TNF- are also increased in most studies with acute ischemic stroke patients [79, 93] and raised TNF- concentrations in plasma of patients suffering from lacunar infarctions are associated with early neurologic deterioration and poor functional outcome [94].
TNF- in the Treatment of Stroke
There are a considerable number of studies about the neurotoxicity of TNF- but its role is still controversial [95]. Some of these studies support the deleterious effects of TNF-
in experimental stroke studies. Inhibition of TNF- reduces ischemic brain injury [96], while administration of recombi-nant TNF- protein after stroke onset worsens ischemic brain damage [85]. TNF- binding protein, an endogenously pro-duced inhibitor of TNF- signaling, is a soluble protein pro-duced by cleavage of the TNF- extracellular binding do-
main of its membrane-bound receptor [97]. The action of TNF- can be blocked using specific TNF- neutralizing antibody (TNF- ab) or TNF- binding protein, that binds TNF- and prevents it from interacting with its receptors. Administration of TNF- ab or TNF- binding protein [98] after cerebral ischemia has demonstrated to be beneficial [99]. TNF- deficient mice showed a clear reduction of the infarction, compared to wild-type mice, while infusion of TNF- worsen infarct volume in focal cerebral ischemia [85]. However, TNF- may also protect the brain under cer-tain circumstances. TNF- appears to be involved in the ischemic tolerance process [100], since TNF-receptor defi-cient mice present larger infarcts than wild-type ones [101].
The biological actions of TNF- are mediated through two distinct cell surface receptors, receptor 1 (TNFR1, p55) and TNF receptor 2 (TNFR2, p75), to which it exhibits fairly equal affinity. Most effects induced by TNF- are mediated by TNFR1, which contains a death domain (DD) that inter-acts directly with TNFR1 and may act as a bifurcation point for signaling related to cell death or cell survival. The differ-ential patterns of localization of TNF receptors on neuronal or glial cells, their expression profile and activational state on these cells, and the down-stream effectors that they acti-vate, are thought to play a critical role in determining if TNF- will have protective or cytotoxic role [102].
Ischemic preconditioning causes up-regulation of neu-ronal TNFR1, and intracerebral administration of TNFR1 antisense oligodeoxynucleotide, which causes a reduction in TNFR1 expression, inhibits the ischemic preconditioning-induced protective effect, suggesting that TNFR1 up-regulation is implicated in the phenomenon of ischemic tol-erance [101]. Finally preconditioning with TNF- also ap-pears to be neuroprotective in ischemic cerebral injury. In-tracisternal administration of TNF- significantly reduced infarct size and decreased microglial activation in a MCAO model of cerebral ischemia [103].
Interleukin 10 (IL-10)
Interleukin-10 is an anti-inflammatory cytokine, mainly secreted by lymphocytes and monocytes/macrophages, which acts by inhibiting IL-1 and TNF- , and by suppressing cytokine receptor expression and receptor activation as well. IL-10 is synthesized in the CNS and is up-regulated in ex-perimental stroke [104]. In acute ischemic stroke, elevated concentrations of IL-10 in CSF have also been found [105]. Clinical data have shown that subjects with reduced produc-tion levels of IL-10 have an increased risk of stroke, support-ing a protective role for this cytokine [93]. In addition, we have demonstrated that low plasma concentrations of IL-10 (<6 pg/ml) are associated with clinical worsening, independ-ently of hyperthermia, hyperglycemia or neurological condi-tion on admission [106].
IL-10 could be a Potential Anti-Inflammatory Therapy for Stroke
Since IL-10 has been shown as an anti-inflammatory cytokine, exogenous administration of this cytokine could be a possible therapeutic strategy to reduce brain damage after stroke. This strategy was showed to be a good approach in animal models. IL-10–deficient mice have an increased stroke lesion size after MCAO [107], and administration

3554 Current Pharmaceutical Design, 2008, Vol. 14, No. 33 Jordán et al.
[108] and gene transfer of IL- 10 [109] in models of cerebral ischemia seems to have beneficial effects independently of stroke subtype [110].
Transforming Growth Factor- (TGF- )
Increased expression of TGF- mRNA has been shown in ischemic tissues 1-6 hours after ischemic insult in rodent models [111], remaining elevated up to 15 days [112]. Such expression may coincide with the influx of monocytes and macrophages and with microglial proliferation in injured tissues [113]. In this context, some experimental studies have tried to elucidate the potential neuroprotective or neurotoxic role of TGF- in ischemic stroke.
TGF- could be a Neuroprotectant Drug in Stroke
Overexpression of TGF- using an adenoviral vector resulted in mouse brain protection from ischemic stroke and in a reduction of the accompanying inflammatory response [114]. Another study has shown that TGF- protect cultured neurons from ischemia-like insults [115]. In addition, TGF- has demonstrated its neuroprotective role when administered before ischemic insult [116]. Other authors have reported both beneficial and insignificant effects when TGF- is ad-ministered after an ischemic insult [117]. It has been pointed out that results could be dependent on the TGF- administra-tion site since one study showed that TGF- reduced infarct volume when administered into the penumbra area of rats, 1 hour after MCAO, while its neuroprotective effect was ab-sent when injected in the core area of the infarct [118].
It has been proposed that TGF- could be neuroprote- ctant by blocking apoptosis, or that participates in the reco- very of ischemic stroke because its effect is visible in the penumbra area and it is present in the recovery phase of some CNS diseases [119].
4.2. Chemokines
Chemokines are a class of cytokines, generally small ones, which incite neutrophils and macrophages to migrate toward the source of the chemokine. They play important roles in cellular communication and inflammatory cell re-cruitment. Expression of chemokines after cerebral ischemia is thought to be deleterious by increasing leukocyte infiltra-tion [120]. In this context, levels of a variety of chemokines such as monocyte chemoattractant protein-1 (MCP-1), IL-8 and macrophage inflammatory protein-1 (MIP-1 ), have been found to increase in animal models of ischemia, and its inhibition or deficiency has been associated with reduced injury [121]. MCP-1 is a potent chemoattractant of mono-cytes and its expression induces an increment of monocytes infiltration in cerebral parenchyma after ischemia. A signifi-cant increase of MCP-1 levels in CSF was found in patients with acute ischemic stroke [122].
In addition to their chemotactic properties, it has been found that chemokines affect the permeability of the blood brain barrier (BBB) and thus, addition of MCP-1 enhanced 17-fold the permeability of the barrier in an in vitro model, suggesting their implication in the opening of the BBB in cerebral ischemia [123]. Finally it has been proposed that chemokines could have an important role in homing stem cells to injured regions and could also be involved in marrow derived stromal cell migration into ischemic brain [124].
On the other hand, chemokines are also signaling mole-cules that down regulate microglia activity. On such mole-cule is fractalkine (CX3CL1), which is primarily expressed by neurons and which has been shown before to inhibit se-cretion of pro-inflammatory cytokines by activated microglia [125]. After transient focal cerebral ischemia, fractalkine-deficient mice had a reduction in infarction size and lower mortality rate, when compared to wild-type littermates [126]. Fractalkine acts through its G-protein-coupled receptor CX3CR1 and may participate in the activation and chemoat-traction of microglia into the infarcted tissue. Fractalkine contributes to the control of leukocyte trafficking from blood vessels into the injured area. After ischemia, fractalkine im-munoreactivity was strongly increased in morphologically intact cortical neurons of the ischemic penumbra and its syn-thesis was also induced in endothelial cells of the infarcted area after ischemia. CX3CR1 expression was detected in the activated microglial cells of the ischemic tissue after ische-mia, and became strongly up-regulated in macrophages/ phagocytic microglia inside the infarted tissue after ischemia [127]. Indeed, Lavergne et al., proposed that the extra adhe-sion of monocytes observed in individuals carrying rare al-leles of CX3CR1 may favour mechanisms leading to stroke [128].
4.3. Cyclooxygenase (COX) and their Possible role in
Stroke Therapy
There are two isoforms of COX. COX-1 is constitutively expressed in many cells types, including microglia and leu-kocytes during brain injury [129]. COX-2 is constitutively expressed in excitatory neurons, whereas in many organs its expression is regulated by a variety of stimuli, such as in-flammatory mediators or mitogens [130].
It has been shown that COX-1 deficient mice have in-creased vulnerability to focal brain ischemia [131], although COX-1 inhibition increased the number of healthy neurons in hippocampus in transient global ischemia [132]. These discrepancies could be due to differences in the focal versus the global ischemic models.
COX-2, the rate-limiting enzyme of prostanoid synthesis, is associated with the production of free radicals and toxic prostanoids and is induced during inflammation and cerebral ischemia. COX-2 is up-regulated 12-24 hours after ischemia [133] and it is expressed in neurons and vascular cells in the border of the ischemic territory [134] and in other cerebral zones including remote regions from the infarct [135]. It has been proposed that COX-2 metabolites are deleterious in cerebral ischemia. Furthermore, COX-2 inhibitors treatment has been demonstrated to improve neurological outcome after cerebral ischemia [134,136], and COX-2 deficient mice have reduced injury after NMDA exposure [131], whereas COX-2 overexpression exacerbates brain injury [137].
4.4. Nitric Oxide (NO) and Nitric Oxide Synthase (NOS)
and their Potential as Therapeutic Targets
NO is an important signaling molecule involved in physiological processes such as neuronal communication, host defense, and regulation of the vascular tone [138]. This relatively stable gas readily diffuses into cells and cell mem-branes where it reacts with molecular targets. NO is synthe-

Inflammation as a Therapy for Stroke Current Pharmaceutical Design, 2008, Vol. 14, No. 33 3555
sized by nitric oxide synthetase (NOS). There are three known isoforms of NOS; 1) neuronal NOS (nNOS, NOS I), localized in particular groups of neurons; 2) inducible NOS (iNOS, NOS II), that is induced during pathological states, associated to inflammation, and 3) endothelial NOS (eNOS, NOS III), mainly found in endothelial cells, [139]. eNOS and nNOS are constitutively expressed and are regulated by in-tracellular calcium while iNOS is inducible, and it is not regulated by intracellular calcium.
NO may cause DNA damage in cerebral ischemia trough the formation of peroxynitrite [140], but its presence at nor-mal levels is also important. The beneficial or detrimental effects of this molecule will depend on where and when is expressed [141]. After induction of ischemia, vasodilator effect of NO produced by eNOS, is beneficial because in-duces vasodilatation and limits blood flow reduction [142]. However, when ischemia has been developed, NO produced by iNOS contribute to brain injury [141].
iNOS is expressed in the postischemic brain, reaching its peak level in infiltrating cells 48 hours from the onset. It has been demonstrated that its expression is detrimental and therefore its inhibition has yield in reduced infarct volumes and reduced neurological deficits [143, 144]. Furthermore, knock-out mice for iNOS gene have smaller infarcts than wild type mice when they are submitted to MCAO [143]. All these studies demonstrate the deleterious role of iNOS in cerebral ischemia, providing evidence that iNOS could be a therapeutic target for ischemic stroke.
4.5. Matrix Metalloproteinases (MMPs)
These proteases are a family of over 20 endopeptidases and during the development they play a central role in the modulation of extracellular matrix allowing neurite out-growth and cellular migration. MMPs are responsible for remodeling the extracellular matrix and that can degrade all its constituents. MMPs are secreted as pro-enzymes that need to be activated. Tissues contain inhibitors to their ac-tion, such as 2-macroglobulin and tissue inhibitors of me- talloproteinase.
MMP-2 (gelatinase A) and MMP-9 (gelatinase B) have been implicated in cerebral ischemia [145]. Elevated MMP-9 levels were found in brain tissue and in serum from patients with acute ischemic stroke [146], and had been claimed to be responsible for the rupture of the BBB, leading to the devel-opment of vasogenic edema and facilitation of hemorrhagic transformation of infarctions [147, 148].
The relationship between inflammatory response and expression of the MMPs is being revealed nowadays. Both IL-6 and TNF- are cytokines capable of expressing MMP-9. The gene promoter region of MMP-9 contains a union place for the activated protein 1 (AP-1) and for the NF- B that respond to a large number of inflammatory stimulants. The immediate response genes (c-fos and c-jun) form the heterodimer AP-1 that activates the MMP-9 gene [149].
MMPs as Possible Therapeutic Agents or Targets
Inhibition of MMPs in experimental models of ischemia have been shown to reduce infarct size and brain edema [150]. MMP-9 deficient mice had smaller infarcts than wild-type controls subjected to ischemia [151]. Since transplanted
mice with bone marrow obtained from MMP-9 deficient mice had smaller injury than mice transplanted with cells containing MMP-9, it is considered that such deleterious effects are due to MMP-9, derived from peripheral inflam-matory cells.
However despite its deleterious effects, it is thought that MMP has potential beneficial effects in ischemic stroke, since its elevation in later phases of cerebral ischemia seems to be related to plasticity and recovery. Since, although an early increase in MMPs has been associated with the BBB breakdown and aggravation of ischemic injury, delayed ex-pression MMPs in the peri-infarct cortex has been associated with neurovascular remodeling and stroke recovery [152]. MMP-9 is associated with several growth factors such as vascular endothelial growth factor (VEGF), which is in-volved in angiogenesis. Furthermore, increased ischemic injury was observed at 14 days after MCAO, in rats treated with MMP inhibitors [152].
5. ROLE OF TRANSCRIPTION FACTORS IN IN-FLAMATION
Largely, inflammation promotes an orchestral response involving the rapid upregulation and activation of a variety of genes. Cerebral ischemia induces massive changes in gene expression. Recent studies have shown that transcription factors like P53, peroxisome proliferator-activated receptors (PPARs), interferon regulatory factor (IRF)-1, signal trans-ducer and activator of transcription (STAT)-3 and nuclear factor (NF)-kappaB promote inflammatory gene expression and thus precipitates severe neuronal damage.
The p53 tumor suppressor gene is a sequence-specific transcription factor that activates the expression of genes engaged in promoting growth arrest or cell death in response to multiple forms of cellular stress [153]. In the mature ner- vous system, numerous studies indicate that p53 plays a key role in neuronal death following certain number of insults including ischemia. PPARs are ligand-activated transcription factors of the nuclear hormone receptor superfamily. The 3 PPAR isoforms (alpha, delta/beta and gamma) are known to control many physiological functions including glucose ab-sorption, lipid balance, and cell growth and differentiation. Of interest, PPAR-gamma activation was recently shown to mitigate the inflammation associated with chronic and acute neurological insults. After focal ischemia, PPAR-gamma expression was observed to be increased in the brain, espe-cially in the peri-infarct area. Nuclear factor kappa-B (NFkB) is one of the most important transcription factors playing a pivotal role in mediating inflammatory responses to a variety of signals, including inflammatory cytokines, bacterial and viral products, oxidative stress, hypoxia-reoxygenation and irradiation.
As the transcription of inflammatory genes is the first step of any inflammatory cascade, therapies that target the pro-inflammatory transcriptional events will potentially curtail inflammation at the very beginning of the signalling process.
5.1. Transcription Factors as Neuroprotectant Targets in Stroke
Transcription factors are being studied as molecular tar-gets for therapeutic repair, since they intricately regulate a

3556 Current Pharmaceutical Design, 2008, Vol. 14, No. 33 Jordán et al.
variety of genes that modulate cellular functions. Transcrip-tional activation can be viewed as a double-edged sword since individual transcription factors can induce either neu-roprotective or neurotoxic genes. p53-deficient mice show reduced neuronal death after ischemia [154] and the an-tisense knockdown of p53 resulted in a significant increase in neuronal survival after ischemia [155]. Moreover, inhibi-tion of NF B signaling pathway has been associated with alteration in activity-dependent synaptic plasticity, suggest-ing that that the NF B signaling pathways are actively in-volved in modulation of important neuropsychological func-tions such as synaptic remodeling and plasticity [156].
PPAR-alpha and the PPAR-gamma agonists protect against stroke and that this beneficial outcome is associated with improved endothelial relaxation, reduced oxidative stress and decreased VCAM1 and ICAM1 expression. In adult rodents, pre-treatment with rosiglitazone or pioglita-zone one day prior to ischemia resulted in decreased micro-glial activation and macrophage infiltration, as well as de-creased expression of proinflammatory COX2, iNOS and IL-1 mRNA in the ischemic hemisphere [157]. Rosiglitazone pre-treatment in both rats and mice also significantly de-creased the infarct volume following focal ischemia and this effect was completely reversed when a specific PPAR-gamma antagonist, GW9662 was administered prior to thia-zolidinediones treatment [157]. Finally, mild hypothermia, a small decreases in brain temperature to 30-34 degrees, which inhibits inflammation after experimental stroke and brain inflammation, and results in reduced inflammatory cell infil-trate [158].
6. IMMUNOMODULATION IN STROKE
Since inflammation has an important role at all stages of the disease process and it is produced by the immune system, immune system could be a therapeutic target in the treatment of cerebrovascular disease. Secondary brain damage origi-nated by inflammation is influenced by innate and adaptive immune responses. Innate immunity represents a fast but relatively blunt inflammatory and toxic response to invading microorganisms but also interacts with several modified en-dogenous antigens. Adaptive immunity is much more spe-cific than innate immunity but may take several days or even weeks to be fully mobilized. It involves a stochastic rea- rrangement process in immunoblasts, leading to generation of a large number of T and B cell receptors and immuno- globulins, which can recognize foreign antigens.
After stroke organism suffers a series of changes some of them produce inflammation and other of them try to contain the inflammatory response. This response is known as im-munomodulation. Evidences that immunomodulation takes part in stroke is the high rate of infections in stroke patients. This process should be studied and its knowledge could help to establish new immunomodulatory therapies. In this part we review the role of the immune system and the immuno-modulation process in stroke.
7. INNATE INMUNITY IN STROKE
Innate immunity represents the first line of defense against pathogens and does not require prior exposure to foreign antigens to be activated. Macrophages, neutrophils,
dendritic cells, NK cells, are the cell types that constitute the innate immune system, which in the CNS includes microglia and perivascular macrophages. On the other hand, the induc-tion of adaptive immunity requires some signals provided by the innate immune system to facilitate the expansion of anti-gen specific T and B lymphocytes, which are important for the production of antibodies and for the formation of long-living memory cells. Unlike adaptive immunity, in which a huge number of potential antigens can be recognized by T and B cells, in the innate immune system, cells must recog-nize their cognate antigens by using a predetermined subset of germline encoded receptors. Because of their limited ex-pression of receptors, cells of the innate immune system may not be able to recognize every possible antigen. Thus, these cells focus on a few highly conserved structures, expressed by large groups of microorganisms. The conserved structural patterns are called pathogen-associated molecular patterns (PAMPs), calling pattern recognition receptors (PRRs) to the receptors of the innate immune system that recognize these structures.
The family of the Toll-like receptors (TLR) is the best characterized kind of PRRs in mammalian species. Although exact gene numbers may differ from one specie to another, it is likely that most mammals have 10 to 15 TLRs. TLRs de-tect multiple PAMPS [159]. TLR4 detects lipopolysaccha-ride (LPS), TLR2 detects bacterial lipoproteins and lipo-teichoic acids, TLR5 detects flagellin, TLR9 detects un-methylated CpG DNA of bacteria and viruses, TLR3 detects double stranded RNA, and TLR7 detects single-stranded viral RNA [160].
TLRs also detect endogenous ligands that might signal other dangerous conditions, such as the presence of degrada-tion products from macromolecules, products from prote-olytic cascades, intracellular components of broken cells and products from genes that are activated by inflammation, such as hyaluronan, fibrinogen, fibronectin, HSP60, HSP70, and others [159-164].
TLR signalling pathway has been studied above all in TLR2 and TLR4 and it has been demonstrated that is very similar to IL-1 receptor pathway. In Fig. (2) is showed a schematic representation of TLR pathway and the principal effects of its activation. TLR pathway is activated when en-dogenous or exogenous ligands interact with their receptor. This receptor has an adaptor protein named as MyD88. MyD88 interact with the kinase IRAK-1 (IL-1R associated kinase) and IRAK-2 that activates another adaptor protein, TRAF6 (TNF Receptor Activated Factor-6). This adaptor protein activates NF- B because it allows I B degradation. Therefore, NF- B is translocated to the nucleus and activates the transcription of several genes such as IL-1, TNF- , IL-6, iNOS, COX-2, ICAM-1, VCAM-1… This process starts inflammatory response [165].
TLRs have been detected in CNS, such as TLR2, TLR3, TLR4 and TLR9. All of them are expressed by microglia, astrocytes express TLR2, TLR3 and TLR4 and neurons ex-press TLR2 and TLR4 [166]. Furthermore other cells from immune system express TLRs in the CNS, such as mono-cytes, neutrophils, basophils, eosinophils, NK cells… Mono-cytes are important cells in the immune system that express TLRs and contribute to generate adaptive immune response.
Inflammation as a Therapy for Stroke Current Pharmaceutical Design, 2008, Vol. 14, No. 33 3557
Monocytes can differentiate to dendritic cells (DC) and these cells promote T cells differentiation to Th1 or Th2 [160] (Fig. 3).
So far there are only few studies that examine the role of TLRs in cerebral ischemia, and all of them suggest that TLRs are involved in the enhancement of cell damage fo- llowing ischemia and that their absence is related to lower infarction volumes in experimental models of ischemia [166-168]. Furthermore psychological stress causes an inflamma-tory response in the brain and is able to exacerbate brain damage caused by experimental stroke, and this process has been showed to be TLR4 dependent [169].
As TLR has been involved in cerebral brain damage in stroke, TLR inhibition or blocking could be a potential the- rapy to reduce ischemic brain damage. Furthermore, TLRs are a family of receptors whose activation occurs at the star- ting of inflammatory response and so their blocking could eliminate all inflammatory response.
In addition, apart from being involved in the cellular damage, it seems that TLRs could also be involved in me-
chanisms of tissue regeneration. This point has been demon-strated in studies of liver regeneration after hepatectomy, epithelium regeneration, and in spinal cord cell proliferation [170]. In this context, TLR activation promotes TNF- ex-pression. TNF- can activate two different receptors; TNFR-1 (Tumor Necrosis Factor Receptor-1) that is related with neurotoxic mechanisms and TNFR-2 (Tumor Necrosis Fac-tor Receptor-2), that is expressed by neuroblasts and whose activation promote progenitor cells to proliferate. So, it is possible that TLR activation can be involved in CNS repair-ing after cerebral ischemia. Furthermore, TLR activation promotes IL-1 expression, too. IL-1 can activate IL-1R in astrocytes and these could respond expressing neutrophins and growth factors that stimulate neuroblasts proliferation (Fig. 2). In summary, TLR activation can trigger neuroblasts proliferation in different pathways and so TLR activation could be involved in neurogenesis process.
8. ADAPTIVE INMUNITY IN STROKE
The inflammatory response of CNS after stroke is fo- llowed by at least one or two phases of T cell infiltration.
Fig. (1). Adhesion molecules expression and function in blood vessels when stroke occurs.
Fig. (2). Schematic representation of the inflammatory mediators that participates in stroke and the cells that express them.

3558 Current Pharmaceutical Design, 2008, Vol. 14, No. 33 Jordán et al.
Surprisingly, whether T cells play a beneficial or detrimental role in these processes is still controversial. T-cells are dif-ferentiated in two different subsets, T-helper1 (Th1) and T-helper2 (Th2). Th1 cells secrete proinflammatory cytokines, including interleukin-2 (IL-2), IL-12, interferon-gamma, and tumor necrosis factor-alpha, that may play a key role in the pathogenesis of stroke, whereas CD4
+ Th2 cells may play a
protective role through anti-inflammatory cytokines such as IL-4, IL-5, IL-10, and IL-13 (Fig. 3). Therefore, Th1 cells promote inflammation and the following secondary brain damage and Th2 cells promote anti-inflammatory responses reducing secondary brain injury. However, inflammation could be necessary to remove cell debris from necrotic tissue that dies after ischemia. Therefore, Th1-Th2 balance could be the clue of a regulated inflammatory response. It has been demonstrated that stroke induces a Th1/Th2 shift in mice. This phenomenon could be a neuroprotective response of the brain to reduce inflammatory response [171].
T-cells should be considered as therapeutic targets for ischemic stroke. However, because infection is a leading cause of mortality in the postacute phase of ischemic stroke, and considering the anti-inflammatory role of Th2 cells, treat-ment targeting T-cells should be carefully designed to reduce deleterious and enhance protective actions of T-cells [172].
9. STROKE INDUCES IMMUNODEPRESSION
As it has been already exposed, innate and adaptive im-munity play an important role in the outcome following cerebral ischemia. Regarding to this, we must consider the existence of an important balance between inflammation and immunodepression. The unbalance of this system involves an increase of secondary damage to the tissue caused by in-flammatory damage or by infection.
Eighty-five percent of all stroke patients have relevant complications, being infection the most frequent one, to such an extent that infection is the leading cause of death in pa-tients suffering from stroke. To support this, it has been shown that after cerebral ischemia, mice develop sponta- neous pneumonia and septicemia due to apoptotic loss of lymphocytes, shift of Th cells to Th2 cytokine production, and lost of monocytes [173]. There are some evidences, such as infection rate, showing that stroke induces immuno- depression, however the mechanisms and the signaling that down-regulate immune responses after ischemia remains unclear. It has been proposed that pro-inflammatory cytoki-nes produced by damaged brain tissue can directly lead to hypothalamic-pituitary axis and CNS activation, resulting in immune dysfunction [174].
Although immunodepression has been related with se- condary brain damage, it could also be a neuroprotective mechanism to reduce brain damage. Therefore, depletion of circulating T-cell populations and suppression of IFN , mechanisms of immunodepression in stroke, might counter-act the inflammatory brain after stroke. Indeed, stroke sup-press autoaggressive Th1 responses. Furthermore, brain epi-topes that usually are inaccessible to immune system are exposed after the disruption of the BBB, in stroke. That may induce to the immune system to attack the CNS. Therefore,
immunodepression could be a neuroprotective mechanism to avoid autoimmunity [174].
10. IMMUNE SYSTEM MODULATION
There are two major ways to manipulate immune system: immunosuppression with different drugs and immunization. Immunization can either be active, in which immune re-sponse is induced through exposure to an antigen, or passive, in which preformed antibodies are administered directly.
10.1 Immunosupression Therapies for Acute Stroke
Since inflammation is deleterious in acute stroke, immu-nosupression could be beneficial for acute ischemic stroke patients. The immunosuppressive drugs available today in-clude; anti-inflammatory corticosteroids, cytotoxic drugs such as azothioprine and cyclosphosphamide, fungal and bacterial derivates inhibiting T-cell activation such as cy-closporine A and rapamycin and non-conventional immuno-suppressive drugs whose immunosupressor role has been recently known. In this context statins and Granulocyte Col-ony Stimulating Factor (G-CSF) could be promising immu-nomodulators therapies.
G-CSF as Immunomodulator
There is compelling evidence that G-CSF exerts im-munoregulatory effects in adaptive immunity. G-CSF en-hances the total lymphocyte count in both bone marrow and peripheral blood and increases CD3
+, CD4
+ and CD8
+ cells
as well as CD3-CD16
+CD56
+ NK cells, while the increase in
CD4+ and CD8
+ T cells results from CD45RO
+ memory T
cells and from cells expressing the CD38 activation marker 2. G-CSF not only alters T cell numbers, but also T cell functions through the shift of T cell subsets in both bone marrow and peripheral blood. G-CSF polarizes T cell differ-entiation from Th1 to Th2 cells and induces Th2 responses with the production of IL-4 and IL-10 [175-177] accompa-nied by a decrease in production of IFN- and IL-2 [178], thereby suppressing T cell proliferative responses to alloge-neic stimulation [177]. In addition, G-CSF increased the production of TGF- [179], and decreased the production of TNF- . G-CSF treatment elevates a CD4
+CD25
+ T cell sub-
set that constitute functional regulatory T cells [180]. Since these cells act secreting anti-inflammatory cytokines they have a role as immunomodulators.
Furthermore, G-CSF treatment induces tolerogenic dendritic cells in the peripheral blood. These cells are poor stimulators of Th1 cells but they are good stimulators of Th2 cells which are the T cells that express anti-inflammatory mediators [181].
Statins could have a Role as Immunomodulators
There are some evidences indicating that statins have some beneficial effects independent of cholesterol reduction. Statins inhibit recruitment and activation of immune-competent cells, such as macrophages, and they inhibit the IFN- -induced expression of class II major histocompatibil-ity complexes (MHCII) on antigen-presenting cells. MHCII are required for antigen presentation and T-cell activation via the T-cell receptor (TCR). TCR activation may trigger both proliferation and differentiation of T cells and influences

Inflammation as a Therapy for Stroke Current Pharmaceutical Design, 2008, Vol. 14, No. 33 3559
their effector function, such as the release of cytokines. Cy-tokines released by activated T cells induce further T-cell proliferation and differentiation, APC activation and B-cell antibody production. The ability of statins to downregulate expression of MHCII may lead to decrase Th1 differentiation and activation in vivo and thus inhibit the release of pro-inflammatory cytokines. This suggest that statins may be useful to reduce inflammatory response after stroke [182].
10.2. Immunization and Induction of Tolerance
Pre-exposition of the brain to a short ischemic event can result in subsequent resistance to severe ischemic injury [183]. This phenomenon has been named as preconditioning, and it has been described to be present in several organs, especially heart and brain. Preconditioning proportions ischemic tolerance and it has been observed in humans in clinical practice. Indeed, less severe strokes have been de-scribed in patients with prior ipsilateral transient ischemic attacks (TIA) within a short period of time [184].
Although nature of induced protection and the underlying molecular mechanisms of preconditioning remain poorly understood, several mechanisms for the induction and main-tenance of tolerance in the brain have been postulated [185]. Several works have tried to solve this question, pointing some of them to different mechanisms that involve in some extent inflammatory mediators such as NF- B [186] and their activators signals like proinflammatory cytokines, neu-rotrophic factors and neurotransmitters [187].
Numerous studies have also demonstrated that precondi-tioning of the brain with a variety of potentially harmful stimuli, induces a profound tolerance to a subsequent epi-sode of ischemia. A brief ischemic insult can also induce ischemic tolerance in the spinal cord [188] and other organs, such as the heart [189], liver [190] or kidneys [191]. The exact molecular mechanisms underlying delayed ischemic tolerance are not well understood, but requirements for de novo protein synthesis [192], activation of the proinflamma-tory transcription factor NF- B [186], and induction of in-flammatory cytokines such as TNF, IL-1, IL-6 and IL-8 have been demonstrated.
Since administration of lipopolysacharide (LPS) can in-duce ischemic tolerance [193], Karikó et al. [194] have de-veloped an hypothetic model to explain this phenomenon. They hypothesized that tolerance is dependent on inhibition of TLR and cytokine signaling pathways, suppressing in this way the inflammatory response to ischemia. When an ischemic event takes place, the ischemic cascade involves TLR activation and cytokine expression, activating inflam-mation, among other mechanisms. Furthermore, TLR and cytokine signaling, trigger other pathways to increase ex-pression of signaling inhibitors, decoy receptors and anti-inflammatory cytokines. These pathways induce immune suppression and when an ischemic event occurs, inflamma-tory inhibitors are present, reducing inflammatory response and the subsequent secondary cell death.
Alternatively, brain preconditioning could be the conse-quence of immunomodulation strategies. Following stroke, novel CNS antigens are exposed to the immune system that could increase cellular damage. Therefore, strategies to re-
duce this response have been proposed. Animals can acquire tolerance to a variety of vascular antigens including myelin-related proteins and E-selectin, before MCAO surgery is performed. These animals experience smaller infarcts than animals tolerized to an irrelevant antigen [35, 195].
Ischemic tolerance is an endogenous mechanism that evolves to protect the organism from ischemia, being conse-quently a good example of immunomodulation.
11. CONCLUSIONS
Cerebral ischemia triggers a very important inflammatory response, which has been associated to an increase in brain damage and poor outcome in stroke patients. Therefore, anti-inflammatory therapies should be considered to reduce brain damage. However, inflammation is necessary to initiate the processes of neovascularization and regeneration. Therefore, “contained” inflammatory response might be necessary and beneficial after stroke. It is possible that optimal treatment of inflammation in stroke include an inhibition in the acute phase and activation in later phases to promote neovasculari-zation, plasticity and regeneration.
Furthermore, the latest advances in the immunomodula-tion field, together with the advances on the knowledge of the ischemic tolerance phenomenon and the role of innate immunity in such phenomenon could open a possibility for the application of immunomodulatory therapies prior to a stroke insult to prepare the organism to can stand better the inflammatory response when stroke occurs.
In summary, inflammation has been associated to an in-crease in brain damage in stroke patients. Furthermore, in-flammation is necessary to activate repairing mechanisms. Therefore, it is necessary a strict control of inflammatory response after stroke to reduce brain damage without inhibi-tion of the repairing mechanisms.
REFERENCES
[1] Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJ. Global
and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet 2006; 367: 1747-57.
[2] Perry VH. A revised view of the central nervous system microenvi-
ronment and major histocompatibility complex class II antigen presentation. J Neuroimmunol 1998; 90: 113-21.
[3] Kaur C, Ling EA. Blood brain barrier in hypoxic-ischemic condi-tions. Curr Neurovasc Res 2008; 5: 71-81.
[4] Aloisi F. Immune function of microglia. Glia 2001; 36: 165-79.
[5] Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci 1996; 19(8): 312-8.
[6] Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. Gan WB: ATP mediates rapid microglial response to local brain in-
jury in vivo. Nat Neurosci 2005, 8: 752-758.
[7] Ling EA, Wong WC. The origin and nature of ramified and amoe-
boid microglia: a historical review and current concepts. Glia 1993; 7: 9-18.
[8] Schmidtmayer J, Jacobsen C, Miksch G, Sievers J. Blood mono-
cytes and spleen macrophages differentiate into microglia-like cells on monolayers of astrocytes: membrane currents. Glia 1994; 12:
259-67.
[9] Streit WJ, Graeber MB. Heterogeneity of microglial and perivascu-
lar cell populations: insights gained from the facial nucleus para-digm. Glia 1993; 7: 68-74.
[10] Hickey WF, Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science
1988; 239: 290-2.

3560 Current Pharmaceutical Design, 2008, Vol. 14, No. 33 Jordán et al.
[11] Nimmerjahn A, Kirchhoff F, Helmchen F: Resting microglial cells
are highly dynamic surveillants of brain parenchyma in vivo. Sci-ence 2005, 308: 1314-8.
[12] Garden GA, Moller T: Microglia biology in health and disease. J Neuroimmune Pharmacol 2006, 1: 127-37.
[13] Perry VH, Newman TA, Cunningham C: The impact of systemic
infection on the progression of neurodegenerative disease. Nat Rev Neurosci 2003, 4: 103-12.
[14] Nakajima K, Kohsaka S. Microglia: activation and their signifi-cance in the central nervous system. J Biochem 2001; 130: 169-75.
[15] Banati RB, Gehrmann J, Kreutzberg GW. Early glial reactions in ischemic lesions. Adv Neurol 1996; 71: 329-36; discussion 336-7.
[16] Schroeter M, Zickler P, Denhardt DT, Hartung HP, Jander S. In-
creased thalamic neurodegeneration following ischaemic cortical stroke in osteopontin-deficient mice. Brain 2006; 129: 1426-37.
[17] Price CJ, Wang D, Menon DK, Guadagno JV, Cleij M, Fryer T, et al. Intrinsic activated microglia map to the peri-infarct zone in the
subacute phase of ischemic stroke. Stroke 2006; 37: 1749-53.
[18] Baron JC. How healthy is the acutely reperfused ischemic penum-
bra? Cerebrovasc Dis 2005; 20 Suppl 2: 25-31.
[19] Lampl Y, Boaz M, Gilad R, Lorberboym M, Dabby R, Rapoport A, et al. Minocycline treatment in acute stroke: an open-label, evalua-
tor-blinded study. Neurology 2007; 69(14): 1404-10.
[20] Jordan J, Fernandez-Gomez FJ, Ramos M, Ikuta I, Aguirre N,
Galindo MF. Minocycline and cytoprotection: shedding new light on a shadowy controversy. Curr Drug Deliv 2007; 4: 225-31.
[21] Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell Signal 2001; 13: 85-94.
[22] McEver RP, Cummings RD. Perspectives series: cell adhesion in
vascular biology. Role of PSGL-1 binding to selectins in leukocyte recruitment. J Clin Invest 1997 Aug 1; 100(3): 485-91.
[23] Connolly ESJ, Winfree CJ, Springer TA, Naka Y, Liao H, Yan SD, et al. Cerebral protection in homozygous null ICAM-1 mice after
middle cerebral artery occlusion. Role of neutrophil adhesion in the pathogenesis of stroke. J Clin Invest 1996; 97: 209-16.
[24] Moore KL, Eaton SF, Lyons DE, Lichenstein HS, Cummings RD, McEver RP. The P-selectin glycoprotein ligand from human neu-
trophils displays sialylated, fucosylated, O-linked poly-N-acetyllactosamine. J Biol Chem 1994; 269: 23318-27.
[25] Huang J, Kim LJ, Mealey R, Marsh Jr HC, Zhang Y, Tenner AJ, et
al. Neuronal protection in stroke by an sLe(x)-glycosylated com-plement inhibitory protein. Science 1999; 285: 595-9.
[26] Stenberg PE, Shuman MA, Levine SP, Bainton DF. Redistribution of alpha-granules and their contents in thrombin-stimulated plate-
lets. J Cell Biol 1984; 98: 748-60.
[27] Diacovo TG, Roth SJ, Buccola JM, Bainton DF, Springer TA.
Neutrophil rolling, arrest, and transmigration across activated, sur-face-adherent platelets via sequential action of P-selectin and the
2- integrin CD11b/CD18. Blood 1996; 88: 146-57.
[28] Cha JK, Jeong MH, Kim EK, Lim YJ, Ha BR, Kim SH, et al. Sur-face expression of P-selectin on platelets is related with clinical
worsening in acute ischemic stroke. J Korean med sci 2002; 17: 811-6.
[29] Mayadas TN, Johnson RC, Rayburn H, Hynes RO, Wagner DD. Leukocyte rolling and extravasation are severely compromised in
P-selectin deficient mice. Cell 1993; 74: 541-54.
[30] Huang J, Choudhri TF, Winfree CJ, McTaggart RA, Kiss S, Mocco
J, et al. Postischemic cerebrovascular E-selectin expression medi-ates tissue injury in murine stroke. Stroke 2000; 31: 3047-53
[31] Mocco J, Choudhri T, Huang J, Harfeldt E, Efros L, Klingbeil C, et
al. HuEP5C7 as a humanized monoclonal anti-E/P-selectin neurovascular protective strategy in a blinded placebo-controlled
trial of nonhuman primate stroke. Circulation Research 2002; 91: 907-14.
[32] Zhang RL, Chopp M, Zhang ZG, Phillips ML, Rosenbloom CL, Cruz R, et al. E-selectin in focal cerebral ischemia and reperfusion
in the rat. J Cereb Blood Flow Metab 1996; 16: 1126-36.
[33] Zhang R, Chopp M, Zhang Z, Jiang N, Powers C. The expression
of P- and E-selectins in three models of middle cerebral artery oc-clusion. Brain Res 1998; 785: 207-14.
[34] Lehmberg J, Beck J, Baethmann A, Uhl E. Effect of P-selectin
inhibition on leukocyte-endothelium interaction and survival after global cerebral ischemia. J Neurol 2006; 253: 357-63.
[35] Chen Y, Ruetzler C, Pandipati S, Spatz M, McCarron RM, Becker
K, et al. Mucosal tolerance to E-selectin provides cell-mediated protection against ischemic brain injury. Proc Natl Acad Sci USA
2003; 100: 15107-12.
[36] Yenari MA, Sun GH, Kunis DM, Onley D, Vexler V. L-selectin
inhibition does not reduce injury in a rabbit model of transient focal cerebral ischemia. Neurol Res 2001; 23: 72-8.
[37] Frijns CJM, Kappell LJ. Inflammatory cell adhesion molecules in
ischemic cerebrovascular disease. Stroke 2002; 33: 2115-22.
[38] Hess DC, Zhao W, Carroll J, McEachin M, Buchanan K. Increased
expression of ICAM-1 during reoxygenation in brain endothelial cells. Stroke 1994; 25: 1463-8.
[39] Stanimirovic D, Shapiro A, Wong J, Hutchison J, Durkin J. The induction of ICAM-1 in human cerebromicrovascular endothelial
cells (HCEC) by ischemia-like conditions promotes enhanced neu-trophil/HCEC adhesion. Journal of Neuroimmunology 1997; 76:
193-205
[40] Zhang RL, Chopp M, Chen H, Garcia JH. Temporal profile of ischemic tissue damage, neutrophil response, and vascular plugging
following permanent and transient (2H) middle cerebral artery oc-clusion in the rat. J Neurol Sci 1994; 125: 3-10.
[41] Shyu KG, Chang H, Lin CC. Serum levels of intercellular adhesion molecule-1 and E-selectin in patients with acute ischaemic stroke. J
Neurol 1997; 244: 90-3.
[42] Bitsch A, Klene W, Murtada L, Prange H, Rieckmann P. A longi-
tudinal prospective study of soluble adhesion molecules in acute stroke. Stroke 1998; 29: 2129-35.
[43] Lindsberg PJ, Carpe?n O, Paetau A, Karjalainen-Lindsberg ML,
Kaste M. Endothelial ICAM-1 expression associated with inflam-matory cell response in human ischemic stroke. Circulation 1996;
94: 939-45.
[44] Endres M, Laufs U, Merz H, Kaps M. Focal expression of intercel-
lular adhesion molecule-1 in the human carotid bifurcation. Stroke 1997; 28: 77-82.
[45] DeGraba TJ, Siren AL, Penix L, McCarron RM, Hargraves R, Sood S, et al. Increased endothelial expression of intercellular ad-
hesion molecule-1 in symptomatic versus asymptomatic human ca-rotid atherosclerotic plaque. Stroke 1998; 29: 1405-10.
[46] Hwang SJ, Ballantyne CM, Sharrett AR, Smith LC, Davis CE,
Gotto AM, Jr., et al. Circulating adhesion molecules VCAM-1, ICAM-1, and E-selectin in carotid atherosclerosis and incident
coronary heart disease cases: the Atherosclerosis Risk In Commu-nities (ARIC) study. Circulation 1997; 96: 4219-25.
[47] Vemuganti R, Dempsey RJ, Bowen KK. Inhibition of intercellular adhesion molecule-1 protein expression by antisense oligonucleo-
tides is neuroprotective after transient middle cerebral artery occlu-sion in rat. Stroke 2004; 35: 179-84.
[48] Zhang RL, Chopp M, Li Y, Zaloga C, Jiang N, Jones ML, et al. Anti-ICAM-1 antibody reduces ischemic cell damage after tran-
sient middle cerebral artery occlusion in the rat. Neurology 1994; 44: 1747-51.
[49] Bowes MP, Zivin JA, Rothlein R. Monoclonal antibody to the
ICAM-1 adhesion site reduces neurological damage in a rabbit cerebral embolism stroke model. Exp Neurol 1993; 119: 215-9.
[50] Zhang LH, Wei EQ. Neuroprotective effect of ONO-1078, a leu-kotriene receptor antagonist, on transient global cerebral ischemia
in rats. Acta Pharmacol Sin 2003; 24: 1241-7.
[51] Justicia C, Martin A, Rojas S, Gironella M, Cervera A, Panes J, et
al. Anti-VCAM-1 antibodies did not protect against ischemic dam-age either in rats or in mice. J Cereb Blood Flow Metab 2006; 26:
421-32.
[52] Haring HP, Akamine P, Habermann R, Koziol JA, Del Zoppo GJ. Distribution of integrin-like immunoreactivity on primate brain mi-
crovasculature. Journal of Neuropathology and Experimental Neu-rology 1996; 55: 236-45.
[53] Springer TA. Adhesion receptors of the immune system. Nature 1990; 346: 425-34.
[54] Harmon D, Lan W, Shorten G. The effect of aprotinin on hypoxia-reoxygenation-induced changes in neutrophil and endothelial func-
tion. Eur J Anaesthesiol 2004; 21: 973-9.
[55] Zhang ZG, Chopp M, Tang WX, Jiang N, Zhang RL. Postischemic treatment (2-4 h) with anti-CD11b and anti-CD18 monoclonal anti-

Inflammation as a Therapy for Stroke Current Pharmaceutical Design, 2008, Vol. 14, No. 33 3561
bodies are neuroprotective after transient (2 h) focal cerebral
ischemia in the rat. Brain Res 1995; 698: 79-85.
[56] Prestigiacomo CJ, Kim SC, Connolly ES, Jr., Liao H, Yan SF,
Pinsky DJ. CD18-mediated neutrophil recruitment contributes to the pathogenesis of reperfused but not nonreperfused stroke. Stroke
1999; 30: 1110-7.
[57] Krams M, Lees KR, Hacke W, Grieve AP, Orgogozo JM, Ford GA. Acute Stroke Therapy by Inhibition of Neutrophils (ASTIN):
an adaptive dose-response study of UK-279,276 in acute ischemic stroke. Stroke 2003; 34: 2543-8.
[58] Becker K, Kindrick D, Relton J, Harlan J, Winn R. Antibody to the alpha4 integrin decreases infarct size in transient focal cerebral
ischemia in rats. Stroke 2001; 32: 206-11.
[59] Dinarello CA. Interleukin-1 and interleukin-1 antagonism. Blood
1991; 77: 1627-52.
[60] Dripps DJ, Brandhuber BJ, Thompson RC, Eisenberg SP. Interleu-kin-1 (IL-1) receptor antagonist binds to the 80-kDa IL-1 receptor
but does not initiate IL-1 signal transduction. J Biol Chem 1991; 266: 10331-6.
[61] Koga S, Ogawa S, Kuwabara K, Brett J, Leavy JA, Ryan J, et al. Synthesis and release of interleukin 1 by reoxygenated human
mononuclear phagocytes. J Clin Invest 1992; 90: 1007-15.
[62] Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard
AD, Kostura MJ, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature
1992; 356: 768-74.
[63] Rothwell NJ. Functions and mechanisms of interleukin 1 in the brain. Trends Pharmacol Sci 1991; 12: 430-6.
[64] Minami M, Kuraishi Y, Satoh M. Effects of kainic acid on messen-ger RNA levels of IL-1 beta, IL-6, TNF alpha and LIF in the rat
brain. Biochem Biophys Res Commun 1991; 176: 593-8.
[65] Buttini M, Boddeke H. Peripheral lipopolysaccharide stimulation
induces interleukin-1 beta messenger RNA in rat brain microglial cells. Neuroscience 1995; 65: 523-30.
[66] Buttini M, Sauter A, Boddeke HW. Induction of interleukin-1 beta
mRNA after focal cerebral ischaemia in the rat. Brain Res Mol Brain Res 1994; 23: 126-34.
[67] Davies CA, Loddick SA, Toulmond S, Stroemer RP, Hunt J, Rothwell NJ. The progression and topographic distribution of inter-
leukin-1beta expression after permanent middle cerebral artery oc-clusion in the rat. J Cereb Blood Flow Metab 1999; 19: 87-98.
[68] Haqqani AS, Nesic M, Preston E, Baumann E, Kelly J, Stanimi-rovic D. Characterization of vascular protein expression patterns in
cerebral ischemia/reperfusion using laser capture microdissection and ICAT-nanoLC-MS/MS. FASEB J 2005; 19: 1809-21.
[69] John GR, Chen L, Rivieccio MA, Melendez-Vasquez CV, Hartley
A, Brosnan CF. Interleukin-1beta induces a reactive astroglial phe-notype via deactivation of the Rho GTPase-Rock axis. J Neurosci
2004; 24: 2837-45.
[70] Azzimondi G, Bassein L, Nonino F, Fiorani L, Vignatelli L, Re G,
et al. Fever in acute stroke worsens prognosis. A prospective study. Stroke 1995; 26: 2040-3.
[71] Carlson NG, Wieggel WA, Chen J, Bacchi A, Rogers SW, Gahring LC. Inflammatory cytokines IL-1 alpha, IL-1 beta, IL-6, and TNF-
alpha impart neuroprotection to an excitotoxin through distinct pathways. J Immunol 1999; 163: 3963-8.
[72] Strijbos PJ, Rothwell NJ. Interleukin-1 beta attenuates excitatory
amino acid-induced neurodegeneration in vitro: involvement of nerve growth factor. J Neurosci 1995; 15: 3468-74.
[73] Yamasaki Y, Matsuura N, Shozuhara H, Onodera H, Itoyama Y, Kogure K. Interleukin-1 as a pathogenetic mediator of ischemic
brain damage in rats. Stroke 1995; 26: 676-80; discussion 681.
[74] Yang GY, Zhao YJ, Davidson BL, Betz AL. Overexpression of
interleukin-1 receptor antagonist in the mouse brain reduces ischemic brain injury. Brain Res 1997; 751: 181-8.
[75] Relton JK, Martin D, Thompson RC, Russell DA. Peripheral ad-
ministration of interleukin-1 receptor antagonist inhibits brain damage after focal cerebral ischemia in the rat. Exp Neurol 1996;
138: 206-13.
[76] Pinteaux E, Rothwell NJ, Boutin H. Neuroprotective actions of
endogenous interleukin-1 receptor antagonist (IL-1ra) are mediated by glia. Glia 2006; 53: 551-6.
[77] Herrmann O, Tarabin V, Suzuki S, Attigah N, Coserea I, Schneider
A, et al. Regulation of body temperature and neuroprotection by endogenous interleukin-6 in cerebral ischemia. J Cereb Blood Flow
Metab 2003; 23: 406-15.
[78] Wang X, Yue TL, Barone FC, White RF, Gagnon RC, Feuerstein
GZ. Concomitant cortical expression of TNF-� and IL-1 mRNAs follows early response gene expression in transient focal ischemia.
Mol Chem Neuropathol 1994; 23: 103-14.
[79] Vila N, Castillo J, Davalos A, Chamorro A. Proinflammatory cyto-kines and early neurological worsening in ischemic stroke. Stroke
2000; 31: 2325-9.
[80] Castillo J, Rodriguez I. Biochemical changes and inflammatory
response as markers for brain ischaemia: molecular markers of di-agnostic utility and prognosis in human clinical practice. Cere-
brovasc Dis 2004; 17 Suppl 1: 7-18.
[81] Smith CJ, Emsley HC, Gavin CM, Georgiou RF, Vail A, Barberan
EM, et al. Peak plasma interleukin-6 and other peripheral markers of inflammation in the first week of ischaemic stroke correlate with
brain infarct volume, stroke severity and long-term outcome. BMC Neurol 2004; 4: 2
[82] Sriram K, O'Callaghan JP. Divergent roles for tumor necrosis fac-
tor-alpha in the brain. J Neuroimmune Pharmacol 2007; 2: 140-53.
[83] Liu T, Clark RK, McDonnell PC, Young PR, White RF, Barone
FC, et al. Tumor necrosis factor- expression in ischemic neurons. Stroke 1994; 25: 1481-8.
[84] Buttini M, Appel K, Sauter A, Gebicke-Haerter PJ, Boddeke HW. Expression of tumor necrosis factor alpha after focal cerebral
ischaemia in the rat. Neuroscience 1996; 71: 1-16.
[85] Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN, Lysko PG, Feuerstein GZ. Tumor necrosis factor-alpha. A mediator
of focal ischemic brain injury. Stroke 1997; 28: 1233-44.
[86] Liu T, Mc Donnell PC, Young PR, White RF, Siren AL, Hallen-
beck JM, et al. Interleukin-1 mRNA expression in ischemic rat cortex. Stroke 1993; 24: 1746-51.
[87] Schindler R, Mancilla J, Endres S, Ghorbani R, Clark SC, Di-narello CA. Correlations and interactions in the production of inter-
leukin-6 (IL-6), IL-1, and tumor necrosis factor (TNF) in human blood mononuclear cells: IL-6 suppresses IL-1 and TNF. Blood
1990; 75: 40-7.
[88] Murakami Y, Saito K, Hara A, Zhu Y, Sudo K, Niwa M, et al. Increases in tumor necrosis factor-alpha following transient global
cerebral ischemia do not contribute to neuron death in mouse hip-pocampus. J Neurochem 2005; 93: 1616-22.
[89] Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activa-
tion of the peripheral immune system. J Cereb Blood Flow Metab 2006; 26: 654-65.
[90] Tomimoto H, Akiguchi I, Wakita H, Kinoshita A, Ikemoto A, Nakamura S, et al. Glial expression of cytokines in the brains of
cerebrovascular disease patients. Acta Neuropathol 1996; 92: 281-7.
[91] Sairanen T, Carpen O, Karjalainen-Lindsberg ML, Paetau A, Tur-
peinen U, Kaste M, et al. Evolution of cerebral tumor necrosis fac-tor-alpha production during human ischemic stroke. Stroke 2001;
32: 1750-8.
[92] Tarkowski E, Rosengren L, Blomstrand C, Wikkelso C, Jensen C,
Ekholm S, et al. Early intrathecal production of interleukin-6 pre-dicts the size of brain lesion in stroke. Stroke 1995; 26: 1393-8.
[93] van Exel E, Gussekloo J, de Craen AJ, Bootsma-van der Wiel A, Frolich M, Westendorp RG. Inflammation and stroke: the Leiden
85-Plus Study. Stroke 2002; 33: 1135-8.
[94] Castellanos M, Castillo J, Garcia MM, Leira R, Serena J, Chamorro A, et al. Inflammation-mediated damage in progressing lacunar in-
farctions: a potential therapeutic target. Stroke 2002; 33: 982-7.
[95] Hallenbeck JM. The many faces of tumor necrosis factor in stroke.
Nat Med 2002; 8: 1363-8.
[96] Yang GY, Gong C, Qin Z, Ye W, Mao Y, Bertz AL. Inhibition of
TNFalpha attenuates infarct volume and ICAM-1 expression in ischemic mouse brain. Neuroreport 1998; 9: 2131-4.
[97] Dawson DA, Martin D, Hallenbeck JM. Inhibition of tumor necro-
sis factor-alpha reduces focal cerebral ischemic injury in the spon-taneously hypertensive rat. Neurosci Lett 1996; 218: 41-4.

3562 Current Pharmaceutical Design, 2008, Vol. 14, No. 33 Jordán et al.
[98] Lavine SD, Hofman FM, Zlokovic BV. Circulating antibody
against tumor necrosis factor-alpha protects rat brain from reperfu-sion injury. J Cereb Blood Flow Metab 1998; 18: 52-8.
[99] Wang CX, Shuaib A. Involvement of inflammatory cytokines in central nervous system injury. Prog Neurobiol 2002; 67: 161-72.
[100] Ginis I, Jaiswal R, Klimanis D, Liu J, Greenspon J, Hallenbeck JM.
TNF-alpha-induced tolerance to ischemic injury involves differen-tial control of NF-kappaB transactivation: the role of NF-kappaB
association with p300 adaptor. J Cereb Blood Flow Metab 2002; 22: 142-52.
[101] Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, et al. Altered neuronal and microglial responses to excitotoxic
and ischemic brain injury in mice lacking TNF receptors. Nat Med 1996; 2: 788-94.
[102] Dopp JM, Mackenzie-Graham A, Otero GC, Merrill JE. Differen-tial expression, cytokine modulation, and specific functions of
type-1 and type-2 tumor necrosis factor receptors in rat glia. J Neu-roimmunol 1997; 75: 104-12.
[103] Nawashiro H, Tasaki K, Ruetzler CA, Hallenbeck JM. TNF-alpha
pretreatment induces protective effects against focal cerebral ischemia in mice. J Cereb Blood Flow Metab 1997; 17: 483-90.
[104] Strle K, Zhou JH, Shen WH, Broussard SR, Johnson RW, Freund GG, et al. Interleukin-10 in the brain. Crit Rev Immunol 2001; 21:
427-49.
[105] Tarkowski E, Rosengren L, Blomstrand C, Wikkelso C, Jensen C,
Ekholm S, et al. Intrathecal release of pro- and anti-inflammatory cytokines during stroke. Clin Exp Immunol 1997; 110: 492-9.
[106] Vila N, Castillo J, Davalos A, Esteve A, Planas AM, Chamorro A.
Levels of anti-inflammatory cytokines and neurological worsening in acute ischemic stroke. Stroke 2003; 34: 671-5.
[107] Grilli M, Barbieri I, Basudev H, Brusa R, Casati C, Lozza G, Ongini E. Interleukin-10 modulates neuronal threshold of vulner-
ability to ischaemic damage. Eur J Neurosci 2000; 12: 2265-72.
[108] Spera PA, Ellison JA, Feuerstein GZ, Barone FC. IL-10 reduces rat
brain injury following focal stroke. Neurosci Lett 1998; 251: 189-92.
[109] Ooboshi H, Ibayashi S, Shichita T, Kumai Y, Takada J, Ago T,
et al. Postischemic gene transfer of interleukin-10 protects against both focal and global brain ischemia. Circulation 2005; 111: 913-9.
[110] Kim JS, Yoon SS, Kim YH, Ryu JS. Serial measurement of inter-leukin-6, transforming growth factor-beta, and S-100 protein in pa-
tients with acute stroke. Stroke 1996; 27: 1553-7.
[111] Klempt ND, Sirimanne E, Gunn AJ, Klempt M, Singh K, Williams
C, et al. Hypoxia-ischemia induces transforming growth factor beta 1 mRNA in the infant rat brain. Mol Brain Res 1992; 13: 93-101.
[112] Wienner C, Gehrmann J, Lindholm D, Topper R, Kreutzberg GW,
Hossmann KA. Expression of transforming growth factor-beta1 and interleukin-1 beta mRNA in rat brain following transient fore-
brain ischemia. Acta Neuropathol 1993; 86: 439-46.
[113] Tilg H, Trehu E, Atkins MB, Dinarello CA, Mier JW. Interleukin-6
(IL-6) as an anti-inflammatory cytokine: Induction of circulating IL-1 receptor antagonist and soluble tumor necrosis factor receptor
p55. Blood 1994; 83: 113-8.
[114] Pang L, Ye W, Che XM, Roessler BJ, Betz AL, Yang GY. Reduc-
tion of inflammatory response in the mouse brain with adenoviral-mediated transforming growth factor-ss1 expression. Stroke 2001;
32: 544-52.
[115] Lu YZ, Lin CH, Cheng FC, Hsueh CM. Molecular mechanisms responsible for microglia-derived protection of Sprague-Dawley rat
brain cells during in vitro ischemia. Neurosci Lett 2005; 373: 159-64.
[116] Mori E, Del Zoppo GJ, Chambers JD, Copeland BR, Arfors KE. Inhibition of polymorphonuclear leukocyte adherence suppresses
no-reflow after focal cerebral ischemia in baboons. Stroke 1992; 23: 712-8.
[117] Gross CE, Bednar MM, Howard DB, Sporn MB, MacMillan V. Transforming growth factor-beta1 reduces infarct size after ex-
perimental cerebral ischemia in a rabbit model. Stroke 1993; 24: 558-62.
[118] Johnston RE, Dillon-Carter O, Freed WJ, Borlongan CV. Trophic
factor secreting kidney cell lines: In vitro characterization and functional effects following transplantation in ischemic rats. Brain
Res 2001; 900: 268-76.
[119] Benveniste EN. Cytokine actions in the central nervous system.
Cytokine Growth Factor Rev 1998; 9: 259-75.
[120] Emsley HC, Tyrrell PJ. Inflammation and infection in clinical
stroke. J Cereb Blood Flow Metab 2002; 22: 1399-419.
[121] Garau A, Bertini R, Colotta F, Casilli F, Bigini P, Cagnotto A, et al. Neuroprotection with the CXCL8 inhibitor repertaxin in tran-
sient brain ischemia. Cytokine 2005; 30: 125-31.
[122] Losy J, Zaremba J. Monocyte chemoattractant protein-1 is in-
creased in the cerebrospinal fluid of patients with ischemic stroke. Stroke 2001; 32: 2695-6.
[123] Stamatovic SM, Shakui P, Keep RF, Moore BB, Kunkel SL, Van Rooijen N, et al. Monocyte chemoattractant protein-1 regulation of
blood-brain barrier permeability. J Cereb Blood Flow Metab 2005; 25: 593-606.
[124] Wang L, Li Y, Chen J, Gautam SC, Zhang Z, Lu M, et al. Ischemic
cerebral tissue and MCP-1 enhance rat bone marrow stromal cell migration in interface culture. Exp Hematol 2002; 30: 831-6.
[125] Ré DB, Przedborski S. Fractalkine: moving from chemotaxis to neuroprotection. Nat Neurosci 2006; 9: 859-61.
[126] Soriano SG, Amaravadi LS, Wang YF, Zhou H, Yu GX, Tonra JR, Fairchild-Huntress V, Fang Q, Dunmore JH, Huszar D, Pan Y.
Mice deficient in fractalkine are less susceptible to cerebral ische-mia-reperfusion injury. J Neuroimmunol 2002; 125: 59-65.
[127] Tarozzo G, Campanella M, Ghiani M, Bulfone A, Beltramo M.
Expression of fractalkine and its receptor, CX3CR1, in response to ischaemia-reperfusion brain injury in the rat. Eur J Neurosci 2002;
15: 1663-8.
[128] Lavergne E, Labreuche J, Daoudi M, Debré P, Cambien F, Deterre
P, Amarenco P, Combadière C; GENIC Investigators. Adverse as-sociations between CX3CR1 polymorphisms and risk of cardiovas-
cular or cerebrovascular disease. Arterioscler Thromb Vasc Biol 2005; 25: 847-53.
[129] Schwab JM, Beschorner R, Meyermann R, Gozalan F, Schluesener HJ. Persistent accumulation of cyclooxygenase-1-expressing mi-
croglial cells and macrophages and transient upregulation by endo-thelium in human brain injury. J Neurosurg 2002; 96: 892-9.
[130] Smith WL, Marnett LJ. Prostaglandin endoperoxide synthase:
structure and catalysis. Biochim Biophys Acta 1991; 1083: 1-17.
[131] Iadecola C, Sugimoto K, Niwa K, Kazama K, Ross ME. Increased
susceptibility to ischemic brain injury in cyclooxygenase-1-deficient mice. J Cereb Blood Flow Metab 2001; 21: 1436-41.
[132] Candelario-Jalil E, Gonzalez-Falcon A, Garcia-Cabrera M, Alvarez D, Al-Dalain S, Martinez G, et al. Assessment of the relative con-
tribution of COX-1 and COX-2 isoforms to ischemia-induced oxi-dative damage and neurodegeneration following transient global
cerebral ischemia. J Neurochem 2003; 86: 545-55.
[133] Planas AM, Soriano MA, Rodriguez-Farre E, Ferrer I. Induction of cyclooxygenase-2 mRNA and protein following transient focal
ischemia in the rat brain. Neurosci Lett 1995; 200: 187-90.
[134] Nogawa S, Zhang F, Ross ME, Iadecola C. Cyclo-oxygenase-2
gene expression in neurons contributes to ischemic brain damage. J Neurosci 1997; 17: 2746-55.
[135] Sairanen T, Ristimaki A, Karjalainen-Lindsberg ML, Paetau A, Kaste M, Lindsberg PJ. Cyclooxygenase-2 is induced globally in
infarcted human brain. Ann Neurol 1998; 43: 738-47.
[136] Sugimoto K, Iadecola C. Delayed effect of administration of COX-2 inhibitor in mice with acute cerebral ischemia. Brain Res 2003;
960: 273-6.
[137] Dore S, Otsuka T, Mito T, Sugo N, Hand T, Wu L, et al. Neuronal
overexpression of cyclooxygenase-2 increases cerebral infarction. Ann Neurol 2003; 54: 155-62.
[138] Llorens S, Jordán J, Nava E. The nitric oxide pathway in the car-diovascular system. J Physiol Biochem 2002; 58: 179-88.
[139] Nathan C. Inducible nitric oxide synthase: what difference does it
make? J Clin Invest 1997; 100: 2417-23.
[140] Cui J, Holmes EH, Greene TG, Liu PK. Oxidative DNA damage
precedes DNA fragmentation after experimental stroke in rat brain. Faseb J 2000; 14: 955-67.
[141] Iadecola C. Bright and dark sides of nitric oxide in ischemic brain injury. Trends Neurosci 1997; 20: 132-9.
[142] Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, et al.
Enlarged infarcts in endothelial nitric oxide synthase knockout

Inflammation as a Therapy for Stroke Current Pharmaceutical Design, 2008, Vol. 14, No. 33 3563
mice are attenuated by nitro-L-arginine. J Cereb Blood Flow Metab
1996; 16: 981-7.
[143] Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME. Delayed
reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J Neurosci 1997;
17: 9157-64.
[144] Parmentier S, Bohme GA, Lerouet D, Damour D, Stutzmann JM, Margaill I, et al. Selective inhibition of inducible nitric oxide syn-
thase prevents ischaemic brain injury. Br J Pharmacol 1999; 127: 546-52.
[145] Rosenberg GA, Navratil M, Barone F, Feuerstein G. Proteolytic cascade enzymes increase in focal cerebral ischemia in rat. J Cereb
Blood Flow Metab 1996; 16: 360-6.
[146] Clark AW, Krekoski CA, Bou SS, Chapman KR, Edwards DR.
Increased gelatinase A (MMP-2) and gelatinase B (MMP-9) activi-ties in human brain after focal ischemia. Neurosci Lett 1997; 238:
53-6.
[147] Castellanos M, Leira R, Serena J, Pumar JM, Lizasoain I, Castillo J, et al. Plasma metalloproteinase-9 concentration predicts hemor-
rhagic transformation in acute ischemic stroke. Stroke 2003; 34: 40-6.
[148] Montaner J, Alvarez-Sabin J, Molina C, Angles A, Abilleira S, Arenillas J, et al. Matrix metalloproteinase expression after human
cardioembolic stroke: temporal profile and relation to neurological impairment. Stroke 2001; 32: 1759-66.
[149] Kusano K, Miyaura C, Inada M, Tamura T, Ito A, Nagase H, et al. Regulation of matrix metalloproteinases (MMP-2, -3, -9, and -13)
by interleukin-1 and interleukin-6 in mouse calvaria: association of MMP induction with bone resorption. Endocrinology 1998; 139:
1338-45.
[150] Pfefferkorn T, Rosenberg GA. Closure of the blood-brain barrier by matrix metalloproteinase inhibition reduces rtPA-mediated mor-
tality in cerebral ischemia with delayed reperfusion. Stroke 2003; 34: 2025-30.
[151] Asahi M, Sumii T, Fini ME, Itohara S, Lo EH. Matrix metallopro-teinase 2 gene knockout has no effect on acute brain injury after fo-
cal ischemia. Neuroreport 2001; 12: 3003-7.
[152] Zhao BQ, Wang S, Kim HY, Storrie H, Rosen BR, Mooney DJ,
Wang X, Lo EH. Role of matrix metalloproteinases in delayed cor-tical responses after stroke. Nat Med 2006; 12: 441-5.
[153] Gomez-Lazaro M, Fernandez-Gomez FJ, Jordán J. p53: twenty five
years understanding the mechanism of genome protection. J Physiol Biochem 2004; 60: 287-307.
[154] Crumrine RC, Thomas AL, Morgan PF. Attenuation of p53 expres-sion protects against focal ischemic damage in transgenic mice. J
Cereb Blood Flow Metab 1994; 14: 887-91.
[155] Jordán J, Galindo MF, González-García C, Ceña V. Role and regu-
lation of p53 in depolarization-induced neuronal death. Neurosci-ence 2003; 122(3): 707-15.
[156] O'Mahony A, Raber J, Montano M, Foehr E, Han V, Lu SM, Kwon
H, LeFevour A, Chakraborty-Sett S, Greene WC. NF-kappaB/Rel regulates inhibitory and excitatory neuronal function and synaptic
plasticity. Mol Cell Biol 2006; 26: 7283-98.
[157] Tureyen K, Kapadia R, Bowen KK, Satriotomo I, Liang J, Fein-
stein DL, Vemuganti R. Peroxisome proliferator-activated receptor-gamma agonists induce neuroprotection following transient focal
ischemia in normotensive, normoglycemic as well as hypertensive and type-2 diabetic rodents. J Neurochem 2007; 101: 41-56.
[158] Yenari MA, Han HS. Influence of hypothermia on post-ischemic inflammation: role of nuclear factor kappa B (NFkappaB). Neuro-
chem Int 2006; 49: 164-9.
[159] Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol 2005; 17: 1-14.
[160] Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol 2004; 5: 987-95.
[161] Akira S, Sato S. Toll-like receptors and their signalling mecha-nisms. Scand J Infect Dis 2003; 35: 555-62.
[162] Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins
linking innate and acquired immunity. Nat Immunol 2001; 2: 675-80.
[163] Medzhitov R. Toll-like receptors and innate immunity. Nat. Rev. Immunol 2001; 1: 135-45.
[164] Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Im-
munol 2003; 21: 335-76.
[165] Nguyen KB. Critical role for STAT4 activation by type 1 interfer-
ons in the interferon-[gamma] response to viral infection. Science 2002; 297: 2063-6.
[166] Ziegler G, Harhausen D, Schepers C, Hoffmann O, Rohr C, Prinz
V, et al. TLR2 has a detrimental role in mouse transient focal cere-bral ischemia. Biochem Biophys Res Commun 2007; 359: 574-9.
[167] Cao CX, Yang QW, Lv FL, Cui J, Fu HB, Wang JZ. Reduced cerebral ischemia-reperfusion injury in Toll-like receptor 4 defi-
cient mice. Biochem Biophys Res Commun 2007; 353: 509-14.
[168] Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain
I. Toll-like receptor 4 is involved in brain damage and inflamma-tion after experimental stroke. Circulation 2007; 115: 1599-608.
[169] Caso JR, Pradillo JM, Hurtado O, Leza JC, Moro MA, Lizasoain I.
Toll-Like Receptor 4 Is Involved in Subacute Stress-Induced Neu-roinflammation and in the Worsening of Experimental Stroke.
Stroke 2008.
[170] Zhang Z, Schluesener HJ. Mammalian toll-like receptors: from
endogenous ligands to tissue regeneration. Cellular and Molecular Life Sciences (CMLS) 2006; V63: 2901-7.
[171] Prass K, Meisel C, Hoflich C, Braun J, Halle E, Wolf T, et al. Stroke-induced immunodeficiency promotes spontaneous bacterial
infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J Exp Med
2003; 198: 725-36.
[172] Hendrix S, Nitsch R. The role of T helper cells in neuroprotection and regeneration. J Neuroimmunol 2007; 184: 100-12.
[173] Dirnagl U, Klehmet J, Braun JS, Harms H, Meisel C, Ziemssen T, et al. Stroke-induced immunodepression: experimental evidence
and clinical relevance. Stroke 2007; 38: 770-3.
[174] Chamorro A, Urra X, Planas AM. Infection after acute ischemic
stroke: a manifestation of brain-induced immunodepression. Stroke 2007; 38: 1097-103.
[175] Klangsinsirikul P, Russell NH. Peripheral blood stem cell harvests
from G-CSF-stimulated donors contain a skewed Th2 CD4 pheno-type and a predominance of type 2 dendritic cells. Exp Hematol
2002; 30: 495-501.
[176] Agnello D, Mascagni P, Bertini R, Villa P, Senaldi G, Ghezzi P.
Granulocyte colony-stimulating factor decreases tumor necrosis factor production in whole blood: role of interleukin-10 and prosta-
glandin E(2). Eur Cytokine Netw 2004; 15: 323-6.
[177] Rutella S, Pierelli L, Bonanno G, Sica S, Ameglio F, Capoluongo
E, et al. Role for granulocyte colony-stimulating factor in the gen-eration of human T regulatory type 1 cells. Blood 2002; 100: 2562-
71.
[178] Pan L, Delmonte J, Jr., Jalonen CK, Ferrara JL. Pretreatment of donor mice with granulocyte colony-stimulating factor polarizes
donor T lymphocytes toward type-2 cytokine production and re-duces severity of experimental graft-versus-host disease. Blood
1995; 86: 4422-9.
[179] Hirayama Y, Sakamaki S, Matsunaga T, Kuroda H, Kusakabe T,
Akiyama T, et al. Granulocyte-colony stimulating factor enhances the expression of transforming growth factor-beta mRNA in CD4-
positive peripheral blood lymphocytes in the donors for allogeneic peripheral blood stem cell transplantation. Am J Hematol 2002; 69:
138-40.
[180] Zou L, Barnett B, Safah H, Larussa VF, Evdemon-Hogan M, Mot-
tram P, et al. Bone marrow is a reservoir for CD4+CD25+ regula-tory T cells that traffic through CXCL12/CXCR4 signals. Cancer
Res 2004; 64: 8451-5.
[181] Xiao BG, Lu CZ, Link H. Cell biology and clinical promise of G-CSF: immunomodulation and neuroprotection. J Cell Mol Med
2007; 11: 1272-90.
[182] Palinski W. Immunomodulation: a new role for statins? Nat Med
2000; 6: 1311-2.
[183] Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe
M, et al. 'Ischemic tolerance' phenomenon found in the brain. Brain Res 1990; 528: 21-4.
[184] Weih M, Kallenberg K, Bergk A, Dirnagl U, Harms L, Wernecke
KD, et al. Attenuated stroke severity after prodromal TIA: a role for ischemic tolerance in the brain? Stroke 1999; 30: 1851-4.

3564 Current Pharmaceutical Design, 2008, Vol. 14, No. 33 Jordán et al.
[185] Dirnagl U, Simon RP, Hallenbeck JM. Ischemic tolerance and
endogenous neuroprotection. Trends Neurosci 2003; 26: 248-54.
[186] Blondeau N, Widmann C, Lazdunski M, Heurteaux C. Activation
of the nuclear factor-kappaB is a key event in brain tolerance. J Neurosci 2001; 21: 4668-77.
[187] Mattson MP, Culmsee C, Yu Z, Camandola S. Roles of nuclear
factor kappaB in neuronal survival and plasticity. J Neurochem 2000; 74: 443-56.
[188] Zvara DA, Colonna DM, Deal DD, Vernon JC, Gowda M, Lundell JC. Ischemic preconditioning reduces neurologic injury in a rat
model of spinal cord ischemia. Ann Thorac Surg 1999; 68: 874-80.
[189] Murry CE, Jennings RB, Reimer KA. Preconditioning with ische-
mia: a delay of lethal cell injury in ischemic myocardium. Circula-tion 1986; 74: 1124-36.
[190] Tsuyama H, Shimizu K, Yoshimoto K, Nezuka H, Ito H, Yama-
moto S, et al. Protective effect of ischemic preconditioning on he-patic ischemia-reperfusion injury in mice. Transplant Proc 2000;
32: 2310-3.
[191] Toosy N, McMorris EL, Grace PA, Mathie RT. Ischaemic precon-
ditioning protects the rat kidney from reperfusion injury. BJU Int 1999; 84: 489-94.
[192] Currie RW, Ellison JA, White RF, Feuerstein GZ, Wang X, Barone FC. Benign focal ischemic preconditioning induces neuronal Hsp70
and prolonged astrogliosis with expression of Hsp27. Brain Res 2000; 863: 169-81.
[193] Tasaki K, Ruetzler CA, Ohtsuki T, Martin D, Nawashiro H, Hal-
lenbeck JM. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontane-
ously hypertensive rats. Brain Res 1997; 748: 267-70.
[194] Kariko K, Weissman D, Welsh FA. Inhibition of toll-like receptor
and cytokine signaling--a unifying theme in ischemic tolerance. J Cereb Blood Flow Metab 2004; 24: 1288-304.
[195] Becker KJ, McCarron RM, Ruetzler C, Laban O, Sternberg E, Flanders KC, et al. Immunologic tolerance to myelin basic protein
decreases stroke size after transient focal cerebral ischemia. Proc Natl Acad Sci USA 1997; 94: 10873-8.




















