
Adenovirus‐mediated inhibition of SPARC attenuates liver fibrosis in rats
12
THE JOURNAL OF GENE MEDICINE RESEARCH ARTICLE J Gene Med 2008; 10: 993–1004. Published online 9 July 2008 in Wiley InterScience (www.interscience.wiley.com) DOI: 10.1002/jgm.1228 Adenovirus-mediated inhibition of SPARC attenuates liver fibrosis in rats Alejandra M. Camino 1† Catalina Atorrasagasti 1† Daniela Maccio 1 Federico Prada 2 Edgardo Salvatierra 2 Miguel Rizzo 1 Laura Alaniz 1,3 Jorge B. Aquino 1 Osvaldo. L. Podhajcer 2,3 Marcelo Silva 1 Guillermo Mazzolini 1,3 * 1 Gene Therapy Laboratory, Liver Unit, School of Medicine, Austral University, Buenos Aires, Argentina 2 Gene Therapy Laboratory, Fundaci´ on Instituto Leloir, Buenos Aires. Argentina 3 CONICET (Consejo Nacional de Investigaciones Cient´ ıficas y T´ ecnicas) *Correspondence to: Guillermo Mazzolini, Liver Unit, School of Medicine. Austral University, Avenida Presidente Per´ on 1500 (B1629ODT) Derqui-Pilar, Buenos Aires, Argentina. E-mail: [email protected] † Both investigators shared first authorship. Received: 12 December 2007 Revised: 13 May 2008 Accepted: 15 May 2008 Abstract Background The interaction between fibrogenic cells and extracellular matrix plays a role in liver fibrosis, yet the mechanisms are largely unknown. Secreted protein, acidic and rich in cysteine (SPARC) is a matricellular glycoprotein that is expressed by hepatic stellate cells and is overexpressed in fibrotic livers. We investigated the in vivo role of SPARC in experimentally induced liver fibrosis in rats. Methods A recombinant adenovirus carrying antisense SPARC was constructed (AdasSPARC). Advanced liver fibrosis was induced in Sprague- Dawley rats by prolonged intraperitoneal administration of thioacetamide. Animals received injections of AdasSPARC or Adβ gal (control adenovirus) via the tail vein and directly into the liver 1 week after the first dose. The pathological changes in liver tissues and indices of fibrosis were assessed at eight weeks. Expression of SPARC, transforming growth factor (TGF)-β and α-smooth muscle actin were evaluated by quantitative real-time polymerase chain reaction, western blotting, enzyme-linked immunosorbent assay and immunohistochemistry. Results Hepatic SPARC expression significantly increased during the development of liver fibrosis. AdasSPARC markedly attenuated the development of hepatic fibrosis in rats treated with thiocetamide, as assessed by decreased collagen deposition, lower hepatic content of hydroxyproline and less advanced morphometric stage of fibrosis. AdasSPARC treatment reduced inflammatory activity (Knodell score) and suppressed transdifferentiation of hepatic stellate cell to the myofibroblasts like phenotype in vivo. Furthermore, in vitro inhibition of SPARC on hepatic stellate cells decreases the production of TGF-β . Conclusions This is the first study to demonstrate that knockdown of hepatic SPARC expression ameliorates thioacetamide-induced liver fibrosis in rats with chronic liver injury. SPARC is a potential target for gene therapy in liver fibrosis. Copyright 2008 John Wiley & Sons, Ltd. Keywords Secreted protein, acidic and rich in cysteine; gene therapy; antisense; liver fibrosis; extracellular matrix; hepatic stellate cells. Introduction Liver cirrhosis is characterized by an excessive and altered accumulation of hepatic extracellular matrix (ECM), which finally leads to disorganization of parenchymal architecture and liver failure [1,2]. Following sustained liver injury of any etiology such as viral, toxic, autoimmune, metabolic or genetic Copyright 2008 John Wiley & Sons, Ltd.
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Adenovirus‐mediated inhibition of SPARC attenuates liver fibrosis in rats

THE JOURNAL OF GENE MEDICINE R E S E A R C H A R T I C L EJ Gene Med 2008; 10: 993–1004.Published online 9 July 2008 in Wiley InterScience (www.interscience.wiley.com) DOI: 10.1002/jgm.1228
Adenovirus-mediated inhibition of SPARCattenuates liver fibrosis in rats
Alejandra M. Camino1†
Catalina Atorrasagasti1†
Daniela Maccio1
Federico Prada2
Edgardo Salvatierra2
Miguel Rizzo1
Laura Alaniz1,3
Jorge B. Aquino1
Osvaldo. L. Podhajcer2,3
Marcelo Silva1
Guillermo Mazzolini1,3*
1Gene Therapy Laboratory, LiverUnit, School of Medicine, AustralUniversity, Buenos Aires, Argentina2Gene Therapy Laboratory,Fundacion Instituto Leloir, BuenosAires. Argentina3CONICET (Consejo Nacional deInvestigaciones Cientıficas y Tecnicas)
*Correspondence to:Guillermo Mazzolini, Liver Unit,School of Medicine. AustralUniversity, Avenida PresidentePeron 1500 (B1629ODT)Derqui-Pilar, Buenos Aires,Argentina.E-mail: [email protected]
†Both investigators shared firstauthorship.
Received: 12 December 2007Revised: 13 May 2008Accepted: 15 May 2008
Abstract
Background The interaction between fibrogenic cells and extracellularmatrix plays a role in liver fibrosis, yet the mechanisms are largely unknown.Secreted protein, acidic and rich in cysteine (SPARC) is a matricellularglycoprotein that is expressed by hepatic stellate cells and is overexpressedin fibrotic livers. We investigated the in vivo role of SPARC in experimentallyinduced liver fibrosis in rats.
Methods A recombinant adenovirus carrying antisense SPARC wasconstructed (AdasSPARC). Advanced liver fibrosis was induced in Sprague-Dawley rats by prolonged intraperitoneal administration of thioacetamide.Animals received injections of AdasSPARC or Adβgal (control adenovirus)via the tail vein and directly into the liver 1 week after the first dose. Thepathological changes in liver tissues and indices of fibrosis were assessed ateight weeks. Expression of SPARC, transforming growth factor (TGF)-β andα-smooth muscle actin were evaluated by quantitative real-time polymerasechain reaction, western blotting, enzyme-linked immunosorbent assay andimmunohistochemistry.
Results Hepatic SPARC expression significantly increased during thedevelopment of liver fibrosis. AdasSPARC markedly attenuated thedevelopment of hepatic fibrosis in rats treated with thiocetamide, asassessed by decreased collagen deposition, lower hepatic content ofhydroxyproline and less advanced morphometric stage of fibrosis. AdasSPARCtreatment reduced inflammatory activity (Knodell score) and suppressedtransdifferentiation of hepatic stellate cell to the myofibroblasts likephenotype in vivo. Furthermore, in vitro inhibition of SPARC on hepaticstellate cells decreases the production of TGF-β.
Conclusions This is the first study to demonstrate that knockdown ofhepatic SPARC expression ameliorates thioacetamide-induced liver fibrosis inrats with chronic liver injury. SPARC is a potential target for gene therapy inliver fibrosis. Copyright 2008 John Wiley & Sons, Ltd.
Keywords Secreted protein, acidic and rich in cysteine; gene therapy; antisense;liver fibrosis; extracellular matrix; hepatic stellate cells.
Introduction
Liver cirrhosis is characterized by an excessive and altered accumulation ofhepatic extracellular matrix (ECM), which finally leads to disorganization ofparenchymal architecture and liver failure [1,2]. Following sustained liverinjury of any etiology such as viral, toxic, autoimmune, metabolic or genetic
Copyright 2008 John Wiley & Sons, Ltd.

994 A. M. Camino et al.
abnormalities, hepatic stellate cells (HSC) become acti-vated and transdifferentiate into myofibroblasts [3].These myofibroblasts are characterized by excessive colla-gen synthesis, decreased release of collagen degradingmatrix metalloproteinases (MMPs) and an enhancedexpression of inhibitors of the MMPs [1,4]. Therefore,HSC play an essential role in the pathogenesis of liverfibrosis and seem to be the primary target cells for inflam-matory stimuli [1,5,6]. Consequently, activated HSC arethe prominent goal for antifibrotic therapies.
Secreted protein, acidic and rich in cysteine (SPARC),also termed osteonectin or BM-40, is a secretedmultifunctional matrix-associated glycoprotein involvedin a wide number of biological processes [7,8]. SPARChas the ability to interact with several extracellular matrixcomponents, bind and modulate the activity of specificgrowth factors, and regulate matrix metalloproteinaseexpression and activity [8–11]. Moreover, the productionof SPARC is increased at sites of wound healing [12,13]and tissue remodelling [14,15]. High levels of SPARCare also found in several malignant tumors of epithelialand non-epithelial origin [16–19] and suppression ofSPARC expression in human melanoma cells abrogatedtheir tumorigenicity [18].
Recent evidence indicates that SPARC, transientlyexpressed by interstitial fibroblasts at sites of tubulo-interstitial injury and fibrosis, is also involved in thepathogenesis of renal fibrosis [20,21]. Furthermore, over-expression of SPARC has been observed in the settingof pulmonary injury and fibrosis [22,23]. Bleomycin-induced lung fibrosis was shown to be significantlyreduced in SPARC-null mice in comparison with similarlytreated wild-type mice [23]. It was recently reportedthat SPARC is highly expressed in fibroblasts frompatients with scleroderma [24,25]. Specific inhibition ofSPARC by small interfering RNA decreased expressionof collagen and attenuated the profibrotic stimulusof exogenous transforming growth factor (TGF)-β1 innormal human fibroblasts cultured in vitro [24]. In fibroticlivers, there is an increased in TGF-β1 expression levelsspecifically in HSC [26], and an important role of thiscytokine in activation of HSC and in the genesis ofhepatic fibrosis was demonstrated [26,27]. In addition,many different strategies of molecular therapy havefocused on the inhibition of TGF-β effects, includinggene transfer of truncated TGF-β receptor type II oradministration of a soluble TGF-β type II receptor[28,29].
SPARC expression has been reported in severalfibrotic liver diseases, such as cirrhosis [30,31], biliaryatresia [32] and hepatocellular carcinoma [19,33].Under nonpathological conditions, SPARC is expressedconstitutively by hepatocytes [30,31,33]. In addition,both human and rat activated HSC over-express SPARC[30,31,33]. Noteworthy, a recently reported analysis ofrat HSC proteins revealed that SPARC is highly expressedin activated HSC [34].
In the present study, we assessed the role of SPARC onthe process of liver fibrogenesis in rat liver fibrosis induced
by thioacetamide (TAA). The results demonstrated thatSPARC inhibition mediated by an adenovirus encodinga SPARC antisense mRNA ameliorated hepatic fibrosisin vivo. Our results suggest SPARC as a potential targetfor antifibrotic treatments.
Materials and methods
Generation of recombinant adenoviralvectors
AdasSPARC, a first-generation replication-defective ade-novirus was constructed and produced as previouslydescribed [35]. Briefly, a 1.7-kb Sa/I fragment containingthe coding sequence of human SPARC and a 527-bp SalIfragment containing the bacterial β-galactosidase genewere cloned in pADPSY-LTRSVpolyA vector to generateadenoviral vectors carrying SPARC cDNA in antisenseorientation (AdasSPARC) or Adβgal, respectively. AdasS-PARC and Adβgal were expanded in 293 cells, purifiedby cesium chloride density gradient, desalted using aPD-10 Sephadex G-25 column (Amersham Biosciences,Piscataway, NJ, USA), and stored at −80 ◦C. The con-centration of recombinant vector was expressed as 50%tissue culture infectious doses (TCID50) per milliliter [36].The total viral particle : infective viral particle ratio was10. Schematic representation of the recombinant aden-ovirus encoding for a SPARC antisense mRNA is shown inFigure 1A.
Figure 1. Construction of the recombinant adenovirus AdasS-PARC and experimental protocol. (A) Schematic representationof the recombinant adenovirus AdasSPARC. This vector carriesthe coding sequence of human SPARC cDNA in antisense underthe control of RSV promoter. (B) Time schedule of TAA adminis-tration, gene transfer, and sample analysis. Adenoviruses wereadministered simultaneously with the initiation of TAA treat-ment and one week later. Samples were harvested at day 10 andat week 7
Copyright 2008 John Wiley & Sons, Ltd. J Gene Med 2008; 10: 993–1004.DOI: 10.1002/jgm

SPARC and liver fibrosis 995
Animal model and gene deliveryprotocols
For the present study, male Sprague-Dawley ratsweighting 250–300 g (purchased from Biofucal, BuenosAires, Argentina) were used. Animals received foodad libitum and were maintained at Austral UniversityLaboratory Animal Unit. Cirrhosis was induced byintraperitoneal (i.p.) administration of TAA (Sigma, StLouis, MO, USA), diluted in saline (200 mg/kg bodyweight), three times a week for 7 weeks, as describedpreviously [37,38]. Rats were injected with 500 µl(5 × 109 TCID50) AdasSPARC, Adβgal or saline via thetail vein at day 0, simultaneously with the first dose ofTAA, and by laparotomy directly into liver parenchymaat day 7 (Figure 1B). Six animals were allocated to eachtreatment group. Liver tissue was obtained for analysis atweeks 2 and 7. All procedures were performed accordingto the Guide for the Care and Use of Laboratory Animalof the US National Research Council and were approvedby the School of Biomedical Sciences, Austral University.
Efficiency of adenoviral-mediated geneexpression
Immunohistochemistry for β-galactosidase was per-formed 48 h after injection of 5 × 109 DICT50 of Adβgalvia the tail vein and at day 10, after a second similardose of Adβgal was injected directly into the liver. Frozenliver sections were treated with 0.03% hydrogen peroxidein alcohol for 20 min to block endogenous peroxidaseand incubated with a primary rabbit anti-rat antibodyagainst β-galactosidase (kindly provided by Dr A. Suburo,Austral University) for 24 h at 4 ◦C. Antibody bindingwas detected by biotinylated goat anti-rabbit serum andavidin-biotin-peroxidase complex (Vectastain ABC Elite;Vector Labs., Burlingame, CA, USA). Peroxidase activ-ity was detected using 3,3-diaminobenzidine (DAB) withnickel enhancement as described previously [39].
Cell culture and treatments
The experiments were performed using the HSC lineCFSC-2G (kindly provided by M. Rojkind, Albert EinsteinCollege of Medicine, New York, NY, USA) [40]. Cellswere cultured in minimum essential medium (MEM)supplemented with 10% bovine fetal serum and non-essential amino acids for 24 h, after which the mediumwas replaced for a serum-free MEM. Treatments werecarried out 12 h later. HSC were transduced with eitherAdβgal or AdasSPARC using different multiplicities ofinfection for 5 days. Supernatants were collected atdifferent time points to measure TGF-β production.
Quantitative real-time polymerasechain reaction (PCR) for assessment ofhepatic SPARC mRNA expression
Liver tissue from rats injected with saline or with5 × 109 TCID50 of AdasSPARC or Adβgal for 2 and7 days was homogenized with Polytron (Janke & KunkelIKA-WERK, Staufen im Breisgau, Germany) and totalRNA was extracted by using Tri Reagent (Sigma-AldrichCo., St Louis, MO, USA). Five micrograms of RNAwere reverse transcribed with 200 U of SuperScriptII Reverse Transcriptase (Invitrogen, Carlsbad, CA,USA) using 500 ng of Oligo (dT) primers. cDNAswere subjected to semiquantitative PCR. Each 25-µlreaction volume contained 1 unit Taq DNA polymerase(Invitrogen), 1 × PCR reaction buffer (20 mM Tris-HCl,pH 8.4, and 50 mM KCl), 1.5 mM Mg2Cl, 0.4 µM ofeach specific primer for SPARC, SPARC sense (5′-CCACTCGCTTCTTTGAGACC-3′) and SPARC antisense(5′-TAGTGGAAGTGGGTGGGGAC-3′), 200 µM of dNTPs.PCR conditions were: 90 s at 94 ◦C and then 40 cycles of30 s at 94 ◦C, 30 s at 60 ◦C and 30 s at 72 ◦C. The averageof triplicate data of each sample was used to calculate therelative change in gene expression after normalizationtarget concentration to cDNA. cDNA was quantifiedusing the OligoGreen Single Stranded Quantification kit(Invitrogen) according to the manufacturer’s instructions.
ELISA assay of TGF-β
Passaged CSFC-2 cells were serum-starved for 24 h priorto the assay. Conditioned media were collected andcentrifuged at 5000 g for 10 min at 4 ◦C. The supernatantswere analysed for the active form of TGF-β1 by ELISA(R&D Systems Minneapolis, USA.) following the protocolprovided by the manufacturer. This immunoassay systemwas chosen for its sensitive and specific detection ofbiologically active TGF-β1. To determine the amount oftotal TGF-β1 in the conditioned media, samples werepretreated with 1M HCl for 15 min at room temperatureprior to neutralization with 1M NaOH, as indicated by themanufacturer. This procedure converted all latent TGF-β1 to the active form. Finally, the reaction was stoppedand the optical density of each well was determined at540 nm or 570 nm within 30 min.
Immunofluorescence assays
Dual immunofluorescence for identification of SPARCand α-smooth muscle actin (SMA)-positive cells wasperformed in liver samples at week 7. For immunoflu-orescence microscopy, 5-µm sections were deparaffinizedand rehydrated. Sections were pre-heated with buffercitrate in a microwave protocol. Slides were blockedwith 10% of goat serum in phosphate-buffered saline(PBS)–0.2% Tween for 60 min at room temperature.Then, samples were incubated overnight at 4 ◦C in a
Copyright 2008 John Wiley & Sons, Ltd. J Gene Med 2008; 10: 993–1004.DOI: 10.1002/jgm

996 A. M. Camino et al.
humid chamber with a polyclonal rabbit anti-SPARC anti-body (32 µg/ml; kindly provided by Dr Helen Sage,Washington University) and a monoclonal mouse anti-α-SMA antibody (1 : 1000; Dako, Denmark). After threewashes with PBS, bound antibodies were detected withRodamine-conjugated goat anti-mouse immunoglobu-lin (Ig)G (Jackson ImmunoResearch, West Grove, PA,USA) and flourescein isothiocyanate-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch). Both secondaryantibodies were diluted 1 : 40 in PBS–0.2% Tween andincubated for 2 h at 37 ◦C. Images were captured from aNikon E800 microscope (Nikon, Tokyo, Japan) coupledto a CCD camera. Control experiments without primaryantibody showed only a faint background staining (datanot shown).
Immunohistochemistry formyofibroblast like cells
After fixation in 10% phosphate-buffered formalin,liver samples were washed twice with water, storedin 70% ethanol (4 ◦C) and embedded in paraffin.For immunohistochemical analysis, 5-µm sections weredeparaffinized and rehydrated. Endogenous peroxidasewas blocked with peroxidase blocking agent complex(Vectastain ABC Elite; Vector Laboratories, Burlingame,CA, USA). For detection of activated HSC, tissue sectionswere incubated with mouse anti-α-SMA monoclonalantibody (Dako, Copenhagen, Denmark; diluted 1 : 50)overnight at 4 ◦C in PBS containing 1% Tween-20 and1% bovine serum albumin. Slides were incubated withperoxidase-linked biotinylated goat anti-mouse secondaryantibodies for 60 min, washed and further incubatedwith labelled polymer for 10 min at room temperature.Sections were washed twice with PBS, incubated withDAB substrate chromogen for 8 min, washed with water,incubated with DAB enhancer (Innovex Biosciences;Richmond, VA, USA) for 5 min, and washed with waterprior to counterstaining with hematoxylin. A control forimmunostaining specificity in which the primary antibodywas substituted with non-immune mouse serum, wasnegative. Quantitative analysis of immunohistochemicalstaining for anti-α-SMA was performed by computerizedmorphometric analysis (CMA). Approximately 200–300light microscope fields (×400) per specimen werecaptured and analysed using a colour threshold detectionsystem developed in Matlab 6.0 (MathWorks, Inc., Natick,MA, USA). The results obtained were expressed as apercentage of positive area.
Western blot analysis
Liver tissue from rats injected with saline or with5 × 109 TCID50 of AdasSPARC or Adβgal for 10 days washomogenized with Polytron (Janke & Kunkel IKA-WERK,Staufen im Breisgau, Germany) and centrifugated twiceat 4 ◦C for 20 min at 10 000 g in lysis buffer with protease
inhibitors (50 mM Tris(hydroxymethyl)aminomethane-HCI buffer, pH 7.4, containing 0.1% Tween-20, 150 mM
NaCl, 10 µg/ml aprotinin, 5 µg/ml pepstatin, 5 µg/mlleupeptin, 1 mM phenyl-methylsulfonyl fluoride and25 µg/ml E64). After centrifugation, cleared tissuelysates were stored at −80 ◦C until analysis. Forimmunoblotting, 20 µg of total protein was loadedand separated on 10% reduced SDS-PAGE gels andtransferred to polyvinylidene difluoride membranes asdescribed previously [41]. Rat SPARC was detected byusing rabbit polyclonal anti-SPARC antibody (diluted1 : 500). Blots were then developed with horseradishperoxidase-conjugated goat anti-rabbit IgG (JacksonImmunoResearch) diluted 1 : 3000 in blocking buffer[41]. Protein loading and transfer was monitored bystaining the membranes with Ponceau Red beforestaining with antibodies. Measurement of total proteinconcentration was performed using Bradford assay [42].Bands intensities were measured by densitometer analysisusing the ImageJ, version 1.33 software (NationalInstitutes of Health, Bethesda, MD, USA).
Assessment of liver inflammatoryactivity and fibrosis
The Knodell score was used to grade the severityof the necroinflammatory process and fibrosis [43].Additionally, liver fibrosis was also semiquantitativelyassessed by trichromic staining in 5-µm liver sectionsusing the METAVIR score [44]. Microphotographs frompicrosirius red staining were taken under Nomarski light(differential interference contrast). To assess collagenfiber maturation and the degree of packaging, liversections were examined with polarized light microscopyusing an Olympus BX60 microscope (Olympus, Tokyo,Japan) configured for observation in reflected lightsimple polarized light mode. Quantitative analysis wasperformed by CMA of samples stained with Sirius red.For CMA, whole liver samples were analysed with theexception of large centrilobular veins and large portaltracts (≥150 µm). Two hundred light microscope images(×200) per specimen were captured and analysed using acolour threshold detection system developed in Matlab 6.0(MathWorks, Inc.). The results obtained were expressedas a percentage of positive area. Fibrosis assessment wasperformed blind by an experienced pathologist.
Additionally, collagen deposition was measured byhydroxyproline assay as detailed previously [45]. Hydrox-yproline content was quantified colorimetrically in dupli-cate from 0.2-g liver samples at 557 nm from a standardcurve with the amino acid alone and against a blankreagent. The results obtained were expressed as mg/g ofliver tissue.
Statistical analysis
Data are expressed as mean ± SD or mean ± SEM whenappropriate. Statistical analysis was performed using
Copyright 2008 John Wiley & Sons, Ltd. J Gene Med 2008; 10: 993–1004.DOI: 10.1002/jgm
SPARC and liver fibrosis 997
Student’s t-test. p < 0.05 was considered statisticallysignificant.
Results
In vivo gene transfer of liver tissue byadenovirus
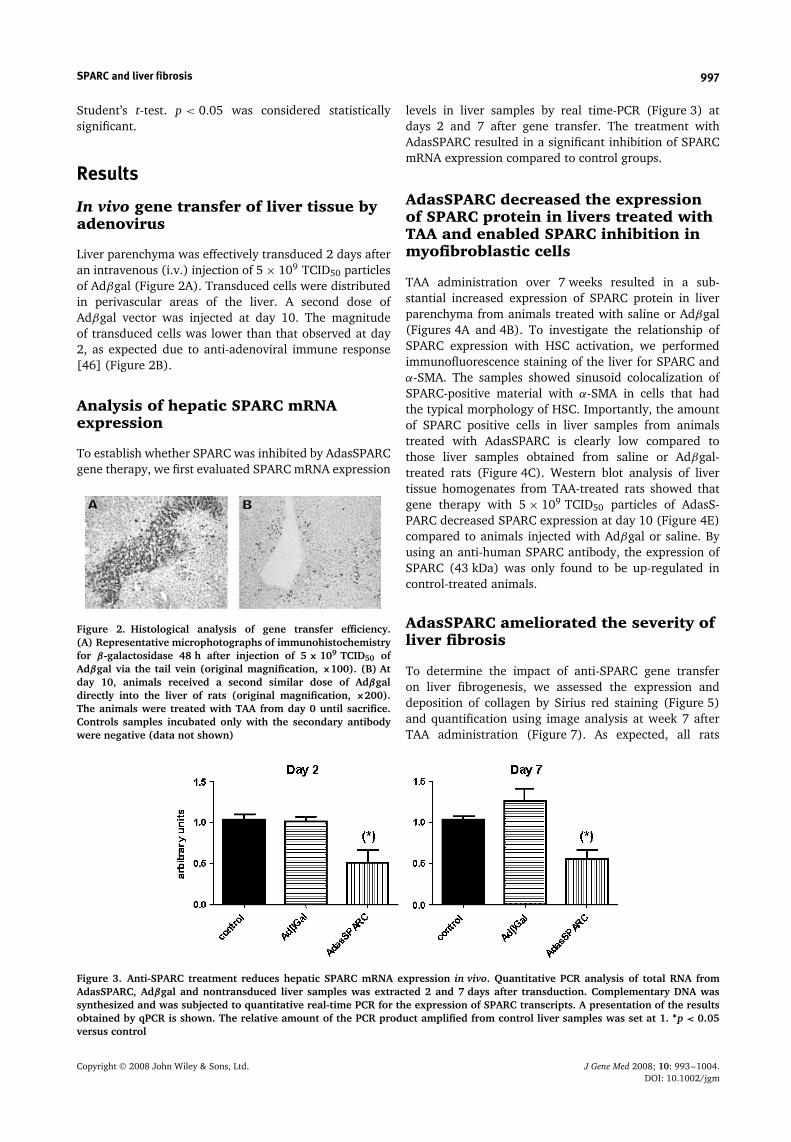
Liver parenchyma was effectively transduced 2 days afteran intravenous (i.v.) injection of 5 × 109 TCID50 particlesof Adβgal (Figure 2A). Transduced cells were distributedin perivascular areas of the liver. A second dose ofAdβgal vector was injected at day 10. The magnitudeof transduced cells was lower than that observed at day2, as expected due to anti-adenoviral immune response[46] (Figure 2B).
Analysis of hepatic SPARC mRNAexpression
To establish whether SPARC was inhibited by AdasSPARCgene therapy, we first evaluated SPARC mRNA expression
Figure 2. Histological analysis of gene transfer efficiency.(A) Representative microphotographs of immunohistochemistryfor β-galactosidase 48 h after injection of 5 × 109 TCID50 ofAdβgal via the tail vein (original magnification, ×100). (B) Atday 10, animals received a second similar dose of Adβgaldirectly into the liver of rats (original magnification, ×200).The animals were treated with TAA from day 0 until sacrifice.Controls samples incubated only with the secondary antibodywere negative (data not shown)
levels in liver samples by real time-PCR (Figure 3) atdays 2 and 7 after gene transfer. The treatment withAdasSPARC resulted in a significant inhibition of SPARCmRNA expression compared to control groups.
AdasSPARC decreased the expressionof SPARC protein in livers treated withTAA and enabled SPARC inhibition inmyofibroblastic cells
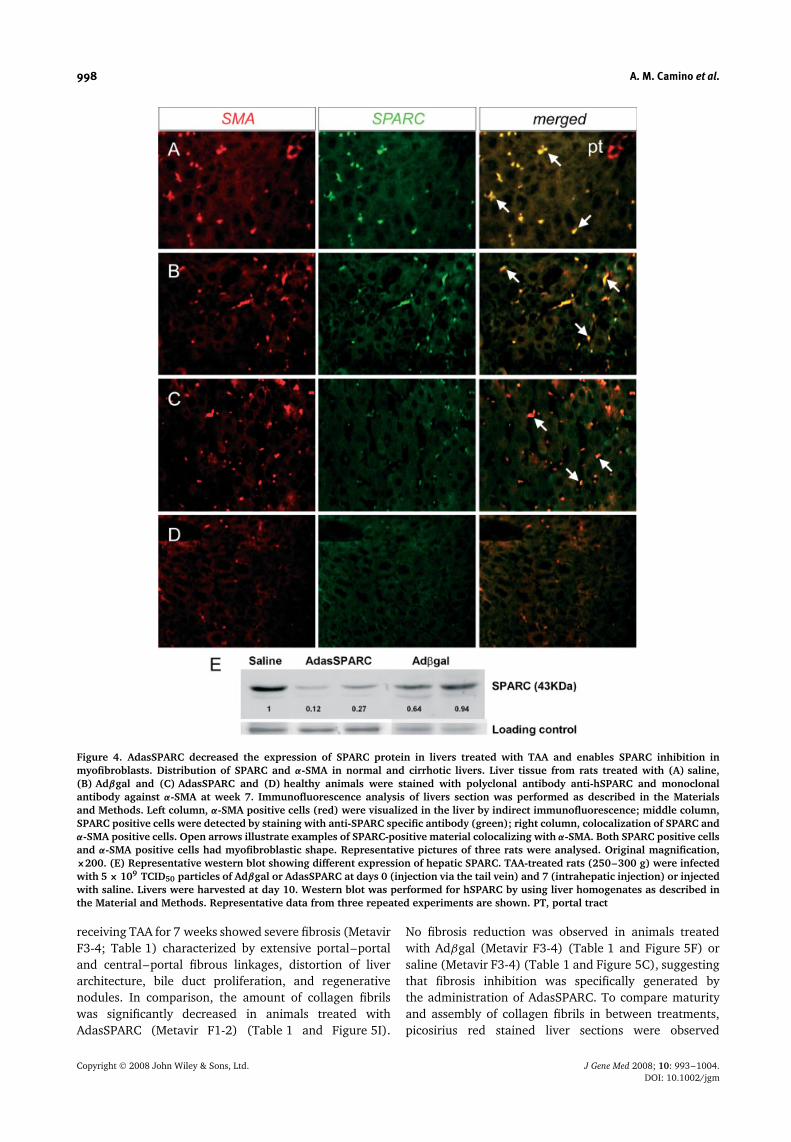
TAA administration over 7 weeks resulted in a sub-stantial increased expression of SPARC protein in liverparenchyma from animals treated with saline or Adβgal(Figures 4A and 4B). To investigate the relationship ofSPARC expression with HSC activation, we performedimmunofluorescence staining of the liver for SPARC andα-SMA. The samples showed sinusoid colocalization ofSPARC-positive material with α-SMA in cells that hadthe typical morphology of HSC. Importantly, the amountof SPARC positive cells in liver samples from animalstreated with AdasSPARC is clearly low compared tothose liver samples obtained from saline or Adβgal-treated rats (Figure 4C). Western blot analysis of livertissue homogenates from TAA-treated rats showed thatgene therapy with 5 × 109 TCID50 particles of AdasS-PARC decreased SPARC expression at day 10 (Figure 4E)compared to animals injected with Adβgal or saline. Byusing an anti-human SPARC antibody, the expression ofSPARC (43 kDa) was only found to be up-regulated incontrol-treated animals.
AdasSPARC ameliorated the severity ofliver fibrosis
To determine the impact of anti-SPARC gene transferon liver fibrogenesis, we assessed the expression anddeposition of collagen by Sirius red staining (Figure 5)and quantification using image analysis at week 7 afterTAA administration (Figure 7). As expected, all rats
Figure 3. Anti-SPARC treatment reduces hepatic SPARC mRNA expression in vivo. Quantitative PCR analysis of total RNA fromAdasSPARC, Adβgal and nontransduced liver samples was extracted 2 and 7 days after transduction. Complementary DNA wassynthesized and was subjected to quantitative real-time PCR for the expression of SPARC transcripts. A presentation of the resultsobtained by qPCR is shown. The relative amount of the PCR product amplified from control liver samples was set at 1. ∗p < 0.05versus control
Copyright 2008 John Wiley & Sons, Ltd. J Gene Med 2008; 10: 993–1004.DOI: 10.1002/jgm
998 A. M. Camino et al.
Figure 4. AdasSPARC decreased the expression of SPARC protein in livers treated with TAA and enables SPARC inhibition inmyofibroblasts. Distribution of SPARC and α-SMA in normal and cirrhotic livers. Liver tissue from rats treated with (A) saline,(B) Adβgal and (C) AdasSPARC and (D) healthy animals were stained with polyclonal antibody anti-hSPARC and monoclonalantibody against α-SMA at week 7. Immunofluorescence analysis of livers section was performed as described in the Materialsand Methods. Left column, α-SMA positive cells (red) were visualized in the liver by indirect immunofluorescence; middle column,SPARC positive cells were detected by staining with anti-SPARC specific antibody (green); right column, colocalization of SPARC andα-SMA positive cells. Open arrows illustrate examples of SPARC-positive material colocalizing with α-SMA. Both SPARC positive cellsand α-SMA positive cells had myofibroblastic shape. Representative pictures of three rats were analysed. Original magnification,×200. (E) Representative western blot showing different expression of hepatic SPARC. TAA-treated rats (250–300 g) were infectedwith 5 × 109 TCID50 particles of Adβgal or AdasSPARC at days 0 (injection via the tail vein) and 7 (intrahepatic injection) or injectedwith saline. Livers were harvested at day 10. Western blot was performed for hSPARC by using liver homogenates as described inthe Material and Methods. Representative data from three repeated experiments are shown. PT, portal tract
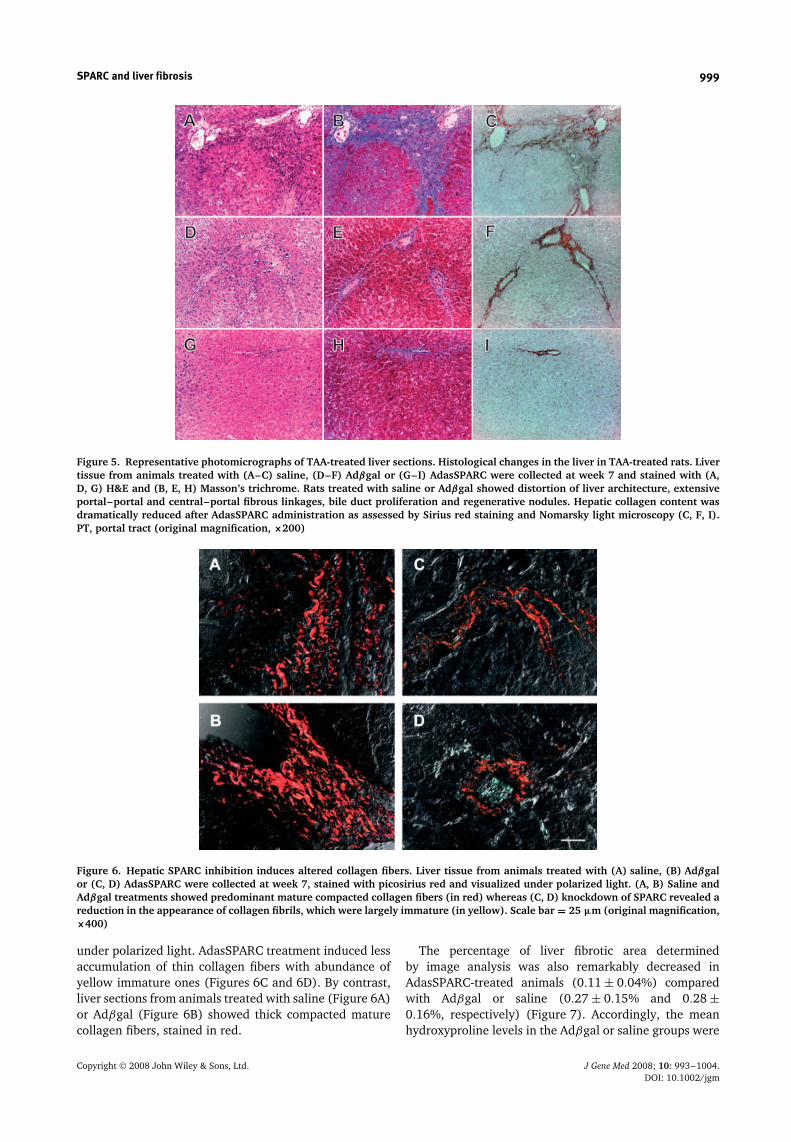
receiving TAA for 7 weeks showed severe fibrosis (MetavirF3-4; Table 1) characterized by extensive portal–portaland central–portal fibrous linkages, distortion of liverarchitecture, bile duct proliferation, and regenerativenodules. In comparison, the amount of collagen fibrilswas significantly decreased in animals treated withAdasSPARC (Metavir F1-2) (Table 1 and Figure 5I).
No fibrosis reduction was observed in animals treatedwith Adβgal (Metavir F3-4) (Table 1 and Figure 5F) orsaline (Metavir F3-4) (Table 1 and Figure 5C), suggestingthat fibrosis inhibition was specifically generated bythe administration of AdasSPARC. To compare maturityand assembly of collagen fibrils in between treatments,picosirius red stained liver sections were observed
Copyright 2008 John Wiley & Sons, Ltd. J Gene Med 2008; 10: 993–1004.DOI: 10.1002/jgm
SPARC and liver fibrosis 999
Figure 5. Representative photomicrographs of TAA-treated liver sections. Histological changes in the liver in TAA-treated rats. Livertissue from animals treated with (A–C) saline, (D–F) Adβgal or (G–I) AdasSPARC were collected at week 7 and stained with (A,D, G) H&E and (B, E, H) Masson’s trichrome. Rats treated with saline or Adβgal showed distortion of liver architecture, extensiveportal–portal and central–portal fibrous linkages, bile duct proliferation and regenerative nodules. Hepatic collagen content wasdramatically reduced after AdasSPARC administration as assessed by Sirius red staining and Nomarsky light microscopy (C, F, I).PT, portal tract (original magnification, ×200)
Figure 6. Hepatic SPARC inhibition induces altered collagen fibers. Liver tissue from animals treated with (A) saline, (B) Adβgalor (C, D) AdasSPARC were collected at week 7, stained with picosirius red and visualized under polarized light. (A, B) Saline andAdβgal treatments showed predominant mature compacted collagen fibers (in red) whereas (C, D) knockdown of SPARC revealed areduction in the appearance of collagen fibrils, which were largely immature (in yellow). Scale bar = 25 µm (original magnification,×400)
under polarized light. AdasSPARC treatment induced lessaccumulation of thin collagen fibers with abundance ofyellow immature ones (Figures 6C and 6D). By contrast,liver sections from animals treated with saline (Figure 6A)or Adβgal (Figure 6B) showed thick compacted maturecollagen fibers, stained in red.
The percentage of liver fibrotic area determinedby image analysis was also remarkably decreased inAdasSPARC-treated animals (0.11 ± 0.04%) comparedwith Adβgal or saline (0.27 ± 0.15% and 0.28 ±0.16%, respectively) (Figure 7). Accordingly, the meanhydroxyproline levels in the Adβgal or saline groups were
Copyright 2008 John Wiley & Sons, Ltd. J Gene Med 2008; 10: 993–1004.DOI: 10.1002/jgm

1000 A. M. Camino et al.
Table 1. Effect of gene therapy with AdasSPARC on the severity of necroinflammatory activity and fibrosis
Saline (n = 6) Adβgal (n = 6) AdasSPARC (n = 6)
Periportal ± bridging necrosis 2.67 ± 1.37 3.80 ± 0.45 1.75 ± 1.39a
Intralobular degeneration and focal necrosis 3.17 ± 1.17 3.40 ± 1.34 2.50 ± 0.93Portal inflammation 3.67 ± 0.52 3.60 ± 0.55 1.00 ± 0.00b
Fibrosis 3.33 ± 0.52 3.00 ± 0.00 0.88 ± 0.35b
Knodell score 12.83 ± 1.94 13.80 ± 1.64 6.13 ± 1.81b
Metavir score F3 (n = 4), F4 (n = 2) F3 (n = 4), F4 (n = 2) F1 (n = 5), F2 (n = 1)
ap < 0.005 respect to Adβgal. bp < 0.0001 respect to saline and Adβgal.
higher than in the AdasSPARC-treated animals (76 ±11 mg/g, 76 ± 26 mg/g and 52 ± 22 mg/g, respectively)(Figure 7). The data suggest that the administration ofAdasSPARC significantly attenuated the severity of liverfibrosis.
AdasSPARC decreases necroin-flammatory activity induced by TAA
Hematoxiline-eosin-stained samples of AdasSPARC-treated animals (Figure 8A) showed a lower necroin-flammatory activity index (Knodell score) (6.13 ± 1.81,p < 0.0001) than livers from Adβgal (13.80 ± 1.64)(Figure 8B) and saline-treated animals (12.83 ± 1.94).AdasSPARC-treated rats showed lower amounts of peri-portal, bridging and focal necrosis, although this didnot reach significant differences (Table 1). On the otherhand, a significant reduction of periportal inflammationand fibrosis was found after anti-SPARC gene therapycompared to saline and Adβgal groups.
Inhibition of SPARC expression in livertissue reduced the amount ofmyofibroblastic cells
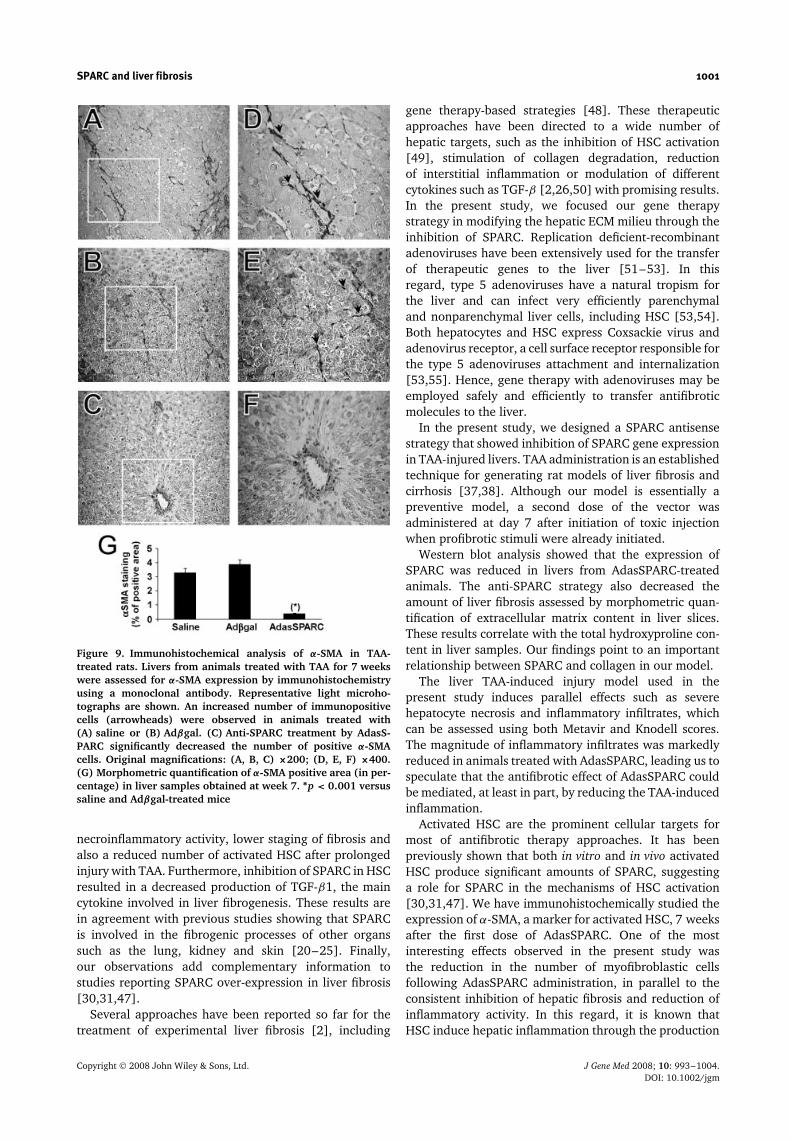
At week 7, animals treated with AdasSPARC (Figure 9C)displayed a significant reduction in α-SMA staining com-pared to those treated with saline (Figure 9A) or Adβgal(Figure 9B). Quantitative analysis of myofibroblastic cellsshowed that the percentage of positive area in theAdasSPARC group was significantly lower (p < 0.001)than saline and Adβgal-treated rats (0.37 ± 0.05% versus3.2 ± 0.3% and 3.85 ± 0.31%, respectively) (Figure 9G).These results suggest that inhibition of SPARC expressionby AdasSPARC may be involved in the reduction of thenumber of myofibroblastic cells, resulting in an attenua-tion of the hepatic fibrogenic process induced by TAA.
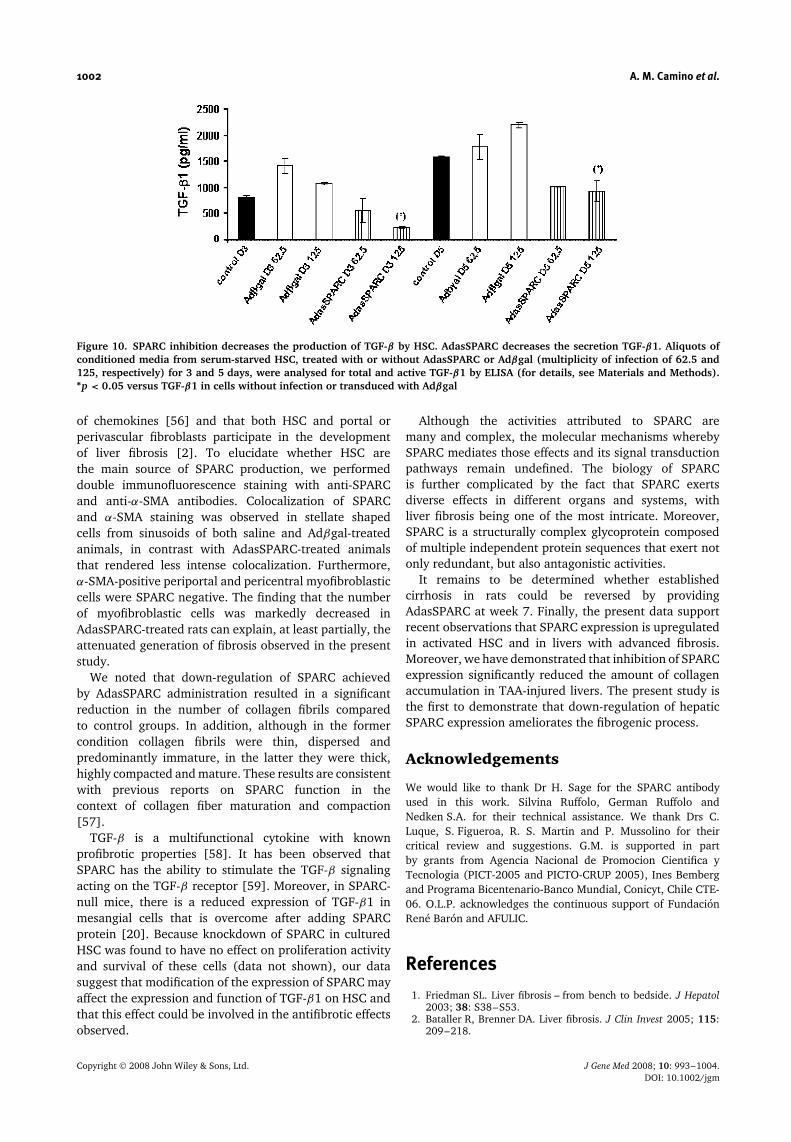
Effect of SPARC knockdown on TGF-β1production by cultured HSC
We next examined TGF-β1 protein concentrations in HSCculture by ELISA. The levels of TGF-β1 protein wereobserved to increase at baseline and with culture timeon plastic dishes in nontransfected HSC. Infection withAdasSPARC in cultured HSC led to a significant decrease
Figure 7. Histomorphometric analysis of liver fibrosis andhydroxyproline quantification. Preventive gene therapy ofTAA-treated rats with AdasSPARC significantly reduces theamount of collagen in the liver as determined by the contentof hydroxyproline (white bars). Ratio of hydroxyproline content(mg) and hepatic weight (g). The percentage of fibrosis areain the liver by morphometric computarized analysis of Siriusred staining was remarkably decreased in AdasSPARC-treatedanimals (black bars). The values are mean ± SD obtainedfrom six rats in each group. MCA, morphometric computerizedanalysis; HYP, hydroxyprolin. ∗p < 0.05 versus Adβgal andsaline
Figure 8. Detection of inflammatory infiltrating cells in liverparenchyma of TAA-treated rats. Seven weeks after TAA treat-ment, livers from animals treated with saline, (A) AdasSPARCor (B) Adβgal were stained with H&E. Hepatocyte necrosis andintense inflammatory infiltrate (arrows) was observed predom-inantly around the portal space in control animals. Infiltrationby inflammatory cells was markedly attenuated in AdasS-PARC-treated rats. PT, portal tract. Original magnification, ×200
in secreted TGF-β1 levels compared to those infected withAdβgal, or uninfected with adenovirus (Figure 10).
Discussion
In the present study, the results obtained suggest thatSPARC plays a central role in liver fibrogenesis because theadministration of AdasSPARC vector efficiently preventedfibrogenesis in an experimental model of liver fibrosis.Thus, animals treated with AdasSPARC showed decreased
Copyright 2008 John Wiley & Sons, Ltd. J Gene Med 2008; 10: 993–1004.DOI: 10.1002/jgm
SPARC and liver fibrosis 1001
Figure 9. Immunohistochemical analysis of α-SMA in TAA-treated rats. Livers from animals treated with TAA for 7 weekswere assessed for α-SMA expression by immunohistochemistryusing a monoclonal antibody. Representative light microho-tographs are shown. An increased number of immunopositivecells (arrowheads) were observed in animals treated with(A) saline or (B) Adβgal. (C) Anti-SPARC treatment by AdasS-PARC significantly decreased the number of positive α-SMAcells. Original magnifications: (A, B, C) ×200; (D, E, F) ×400.(G) Morphometric quantification of α-SMA positive area (in per-centage) in liver samples obtained at week 7. ∗p < 0.001 versussaline and Adβgal-treated mice
necroinflammatory activity, lower staging of fibrosis andalso a reduced number of activated HSC after prolongedinjury with TAA. Furthermore, inhibition of SPARC in HSCresulted in a decreased production of TGF-β1, the maincytokine involved in liver fibrogenesis. These results arein agreement with previous studies showing that SPARCis involved in the fibrogenic processes of other organssuch as the lung, kidney and skin [20–25]. Finally,our observations add complementary information tostudies reporting SPARC over-expression in liver fibrosis[30,31,47].
Several approaches have been reported so far for thetreatment of experimental liver fibrosis [2], including
gene therapy-based strategies [48]. These therapeuticapproaches have been directed to a wide number ofhepatic targets, such as the inhibition of HSC activation[49], stimulation of collagen degradation, reductionof interstitial inflammation or modulation of differentcytokines such as TGF-β [2,26,50] with promising results.In the present study, we focused our gene therapystrategy in modifying the hepatic ECM milieu through theinhibition of SPARC. Replication deficient-recombinantadenoviruses have been extensively used for the transferof therapeutic genes to the liver [51–53]. In thisregard, type 5 adenoviruses have a natural tropism forthe liver and can infect very efficiently parenchymaland nonparenchymal liver cells, including HSC [53,54].Both hepatocytes and HSC express Coxsackie virus andadenovirus receptor, a cell surface receptor responsible forthe type 5 adenoviruses attachment and internalization[53,55]. Hence, gene therapy with adenoviruses may beemployed safely and efficiently to transfer antifibroticmolecules to the liver.
In the present study, we designed a SPARC antisensestrategy that showed inhibition of SPARC gene expressionin TAA-injured livers. TAA administration is an establishedtechnique for generating rat models of liver fibrosis andcirrhosis [37,38]. Although our model is essentially apreventive model, a second dose of the vector wasadministered at day 7 after initiation of toxic injectionwhen profibrotic stimuli were already initiated.
Western blot analysis showed that the expression ofSPARC was reduced in livers from AdasSPARC-treatedanimals. The anti-SPARC strategy also decreased theamount of liver fibrosis assessed by morphometric quan-tification of extracellular matrix content in liver slices.These results correlate with the total hydroxyproline con-tent in liver samples. Our findings point to an importantrelationship between SPARC and collagen in our model.
The liver TAA-induced injury model used in thepresent study induces parallel effects such as severehepatocyte necrosis and inflammatory infiltrates, whichcan be assessed using both Metavir and Knodell scores.The magnitude of inflammatory infiltrates was markedlyreduced in animals treated with AdasSPARC, leading us tospeculate that the antifibrotic effect of AdasSPARC couldbe mediated, at least in part, by reducing the TAA-inducedinflammation.
Activated HSC are the prominent cellular targets formost of antifibrotic therapy approaches. It has beenpreviously shown that both in vitro and in vivo activatedHSC produce significant amounts of SPARC, suggestinga role for SPARC in the mechanisms of HSC activation[30,31,47]. We have immunohistochemically studied theexpression of α-SMA, a marker for activated HSC, 7 weeksafter the first dose of AdasSPARC. One of the mostinteresting effects observed in the present study wasthe reduction in the number of myofibroblastic cellsfollowing AdasSPARC administration, in parallel to theconsistent inhibition of hepatic fibrosis and reduction ofinflammatory activity. In this regard, it is known thatHSC induce hepatic inflammation through the production
Copyright 2008 John Wiley & Sons, Ltd. J Gene Med 2008; 10: 993–1004.DOI: 10.1002/jgm
1002 A. M. Camino et al.
Figure 10. SPARC inhibition decreases the production of TGF-β by HSC. AdasSPARC decreases the secretion TGF-β1. Aliquots ofconditioned media from serum-starved HSC, treated with or without AdasSPARC or Adβgal (multiplicity of infection of 62.5 and125, respectively) for 3 and 5 days, were analysed for total and active TGF-β1 by ELISA (for details, see Materials and Methods).∗p < 0.05 versus TGF-β1 in cells without infection or transduced with Adβgal
of chemokines [56] and that both HSC and portal orperivascular fibroblasts participate in the developmentof liver fibrosis [2]. To elucidate whether HSC arethe main source of SPARC production, we performeddouble immunofluorescence staining with anti-SPARCand anti-α-SMA antibodies. Colocalization of SPARCand α-SMA staining was observed in stellate shapedcells from sinusoids of both saline and Adβgal-treatedanimals, in contrast with AdasSPARC-treated animalsthat rendered less intense colocalization. Furthermore,α-SMA-positive periportal and pericentral myofibroblasticcells were SPARC negative. The finding that the numberof myofibroblastic cells was markedly decreased inAdasSPARC-treated rats can explain, at least partially, theattenuated generation of fibrosis observed in the presentstudy.
We noted that down-regulation of SPARC achievedby AdasSPARC administration resulted in a significantreduction in the number of collagen fibrils comparedto control groups. In addition, although in the formercondition collagen fibrils were thin, dispersed andpredominantly immature, in the latter they were thick,highly compacted and mature. These results are consistentwith previous reports on SPARC function in thecontext of collagen fiber maturation and compaction[57].
TGF-β is a multifunctional cytokine with knownprofibrotic properties [58]. It has been observed thatSPARC has the ability to stimulate the TGF-β signalingacting on the TGF-β receptor [59]. Moreover, in SPARC-null mice, there is a reduced expression of TGF-β1 inmesangial cells that is overcome after adding SPARCprotein [20]. Because knockdown of SPARC in culturedHSC was found to have no effect on proliferation activityand survival of these cells (data not shown), our datasuggest that modification of the expression of SPARC mayaffect the expression and function of TGF-β1 on HSC andthat this effect could be involved in the antifibrotic effectsobserved.
Although the activities attributed to SPARC aremany and complex, the molecular mechanisms wherebySPARC mediates those effects and its signal transductionpathways remain undefined. The biology of SPARCis further complicated by the fact that SPARC exertsdiverse effects in different organs and systems, withliver fibrosis being one of the most intricate. Moreover,SPARC is a structurally complex glycoprotein composedof multiple independent protein sequences that exert notonly redundant, but also antagonistic activities.
It remains to be determined whether establishedcirrhosis in rats could be reversed by providingAdasSPARC at week 7. Finally, the present data supportrecent observations that SPARC expression is upregulatedin activated HSC and in livers with advanced fibrosis.Moreover, we have demonstrated that inhibition of SPARCexpression significantly reduced the amount of collagenaccumulation in TAA-injured livers. The present study isthe first to demonstrate that down-regulation of hepaticSPARC expression ameliorates the fibrogenic process.
Acknowledgements
We would like to thank Dr H. Sage for the SPARC antibodyused in this work. Silvina Ruffolo, German Ruffolo andNedken S.A. for their technical assistance. We thank Drs C.Luque, S. Figueroa, R. S. Martin and P. Mussolino for theircritical review and suggestions. G.M. is supported in partby grants from Agencia Nacional de Promocion Cientifica yTecnologia (PICT-2005 and PICTO-CRUP 2005), Ines Bembergand Programa Bicentenario-Banco Mundial, Conicyt, Chile CTE-06. O.L.P. acknowledges the continuous support of FundacionRene Baron and AFULIC.
References
1. Friedman SL. Liver fibrosis – from bench to bedside. J Hepatol2003; 38: S38–S53.
2. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 2005; 115:209–218.
Copyright 2008 John Wiley & Sons, Ltd. J Gene Med 2008; 10: 993–1004.DOI: 10.1002/jgm

SPARC and liver fibrosis 1003
3. Reeves HL, Friedman SL. Activation of hepatic stellate cells – akey issue in liver fibrosis. Front Biosci 2002; 7: D808–D826.
4. Schuppan D, Ruehl M, Somasundaram R, Hahn EG. Matrix asa modulator of hepatic fibrogenesis. Semin Liver Dis 2001; 21:351–372.
5. de Leeuw AM, McCarthy SP, Geerts A, Knook DL. Purified ratliver fat-storing cells in culture divide and contain collagen.Hepatology 1984; 4: 392–403.
6. Pinzani M. Liver fibrosis. Springer Semin Immunopathol 1999;21: 475–490.
7. Termine JD, Kleinman HK, Whitson SW, Conn KM, McGar-vey ML, Martin GR. Osteonectin, a bone-specific protein linkingmineral to collagen. Cell 1981; 26: 99–105.
8. Mann K, Deutzmann R, Paulsson M, Timpl R. Solubilization ofprotein BM-40 from a basement membrane tumor with chelatingagents and evidence for its identity with osteonectin and SPARC.FEBS Lett 1987; 218: 167–172.
9. Barker TH, Baneyx G, Cardo-Vila M, et al. SPARC regulatesextracellular matrix organization through its modulation ofintegrin-linked kinase activity. J Biol Chem 2005; 280:36483–36493.
10. Brekken RA, Sage EH. SPARC, a matricellular protein: at thecrossroads of cell-matrix communication. Matrix Biol 2001; 19:816–827.
11. Sage EH, Bornstein P. Extracellular proteins that modulatecell–matrix interactions. SPARC, tenascin, and thrombospondin.J Biol Chem 1991; 266: 14831–14834.
12. Reed MJ, Puolakkainen P, Lane TF, Dickerson D, Bornstein P,Sage EH. Differential expression of SPARC and thrombospondin1 in wound repair: immunolocalization and in situ hybridization.J Histochem Cytochem 1993; 41: 1467–1477.
13. Bradshaw AD, Reed MJ, Sage EH. SPARC-null mice exhibitaccelerated cutaneous wound closure. J Histochem Cytochem2002; 50: 1–10.
14. Bradshaw AD, Sage EH. SPARC, a matricellular protein thatfunctions in cellular differentiation and tissue response to injury.J Clin Invest 2001; 107: 1049–1054.
15. Lane TF, Sage EH. The biology of SPARC, a protein thatmodulates cell–matrix interactions. FASEB J 1994; 8: 163–173.
16. Porte H, Chastre E, Prevot S, et al. Neoplastic progression ofhuman colorectal cancer is associated with overexpression ofthe stromelysin-3 and BM-40/SPARC genes. Int J Cancer 1995;64: 70–75.
17. Porte H, Triboulet JP, Kotelevets L, et al. Overexpression ofstromelysin-3, BM-40/SPARC, and MET genes in humanesophageal carcinoma: implications for prognosis. Clin CancerRes 1998; 4: 1375–1382.
18. Ledda MF, Adris S, Bravo AI, et al. Suppression of SPARCexpression by antisense RNA abrogates the tumorigenicity ofhuman melanoma cells. Nat Med 1997; 3: 171–176.
19. Lau CP, Poon RT, Cheung ST, Yu WC, Fan ST. SPARC and Hevinexpression correlate with tumour angiogenesis in hepatocellularcarcinoma. J Pathol 2006; 210: 459–468.
20. Francki A, Bradshaw AD, Bassuk JA, Howe CC, Couser WG,Sage EH. SPARC regulates the expression of collagen type Iand transforming growth factor-beta1 in mesangial cells. J BiolChem 1999; 274: 32145–32152.
21. Pichler RH, Hugo C, Shankland SJ, et al. SPARC is expressed inrenal interstitial fibrosis and in renal vascular injury. Kidney Int1996; 50: 1978–89.
22. Kuhn C, Mason RJ. Immunolocalization of SPARC, tenascin, andthrombospondin in pulmonary fibrosis. Am J Pathol 1995; 147:1759–1769.
23. Strandjord TP, Madtes DK, Weiss DJ, Sage EH. Collagenaccumulation is decreased in SPARC-null mice with bleomycin-induced pulmonary fibrosis. Am J Physiol 1999; 277:L628–L635.
24. Zhou X, Tan FK, Guo X, et al. Small interfering RNA inhibition ofSPARC attenuates the profibrotic effect of transforming growthfactor beta1 in cultured normal human fibroblasts. ArthritisRheum 2005; 52: 257–261.
25. Zhou X, Tan FK, Reveille JD, et al. Association of novelpolymorphisms with the expression of SPARC in normalfibroblasts and with susceptibility to scleroderma. ArthritisRheum 2002; 46: 2990–2999.
26. Gressner AM, Weiskirchen R, Breitkopf K, Dooley S. Roles ofTGF-beta in hepatic fibrosis. Front Biosci 2002; 7: d793–807.
27. Parsons C, MT, RA R. Molecular mechanisms of hepaticfibrogenesis. J Gastroenterol Hepatol 2007; 22(Suppl 1):S79–S84.
28. Qi Z, Atsuchi N, Ooshima A, Takeshita A, Ueno H. Blockade oftype beta transforming growth factor signaling prevents liverfibrosis and dysfunction in the rat. Proc Natl Acad Sci USA 1999;96: 2345–2349.
29. Yata Y, Gotwals P, Koteliansky V, Rockey D. Dose-dependentinhibition of hepatic fibrosis in mice by a TGF-beta solublereceptor: implications for antifibrotic therapy. Hepatology 2002;35: 1022–1030.
30. Blazejewski S, Le Bail B, Boussarie L, et al. Osteonectin (SPARC)expression in human liver and in cultured human livermyofibroblasts. Am J Pathol 1997; 151: 651–657.
31. Frizell E, Liu SL, Abraham A, et al. Expression of SPARC innormal and fibrotic livers. Hepatology 1995; 21: 847–854.
32. Lamireau T, Le Bail B, Boussarie L, et al. Expression of collagenstype I and IV, osteonectin and transforming growth factor beta-1 (TGFbeta1) in biliary atresia and paucity of intrahepatic bileducts during infancy. J Hepatol 1999; 31: 248–255.
33. Le Bail B, Faouzi S, Boussarie L, et al. Osteonectin/SPARC isoverexpressed in human hepatocellular carcinoma. J Pathol1999; 189: 46–52.
34. Kristensen DB, Kawada N, Imamura K, et al. Proteome analysisof rat hepatic stellate cells. Hepatology 2000; 32: 268–277.
35. Alvarez MJ, Prada F, Salvatierra E, et al. Secreted proteinacidic and rich in cysteine produced by human melanomacells modulates polymorphonuclear leukocyte recruitment andantitumor cytotoxic capacity. Cancer Res 2005; 65: 5123–5132.
36. Reed LJ, Muench H. A simple methods of estimating 50percentage points. Am J Hyg 1938; 27: 493–497.
37. Muller A, Machnik F, Zimmermann T, SchubertH. Thioacetamide-induced cirrhosis-like liver lesions inrats – usefulness and reliability of this animal model. Exp Pathol1988; 34: 229–236.
38. Oren R, Dotan I, Papa M, et al. Inhibition of experimentallyinduced cirrhosis in rats by hypothyroidism. Hepatology 1996;24: 419–423.
39. Shu SY, Ju G, Fan LZ. The glucose oxidase-DAB-nickel methodin peroxidase histochemistry of the nervous system. NeurosciLett 1988; 85: 169–171.
40. Greenwel P, Rubin J, Schwartz M, Hertzberg E, Rojkind M. Liverfat-storing cell clones obtained from a CCl4-cirrhotic rat areheterogeneous with regard to proliferation, expression ofextracellular matrix components, interleukin-6, and connexin43. Lab Invest 1993; 69: 210–216.
41. Ledda F, Bravo AI, Adris S, Bover L, Mordoh J, Podhajcer OL.The expression of the secreted protein acidic and rich in cysteine(SPARC) is associated with the neoplastic progression of humanmelanoma. J Invest Dermatol 1997; 108: 210–214.
42. Bradford MM. A rapid and sensitive method for the quantitationof microgram quantities of protein utilizing the principle ofprotein-dye binding. Anal Biochem 1976; 72: 248–254.
43. Knodell RG, Ishak KG, Black WC, et al. Formulation andapplication of a numerical scoring system for assessinghistological activity in asymptomatic chronic active hepatitis.Hepatology 1981; 1: 431–435.
44. Beaussier M, Wendum D, Fouassier L, et al. Adaptative bile ductproliferative response in experimental bile duct ischemia. JHepatol 2005; 42: 257–265.
45. Woessner JF Jr. The determination of hydroxyproline in tissueand protein samples containing small proportions of this iminoacid. Arch Biochem Biophys 1961; 93: 440–447.
46. McConnell MJ, Imperiale MJ. Biology of adenovirus and itsuse as a vector for gene therapy. Hum Gene Ther 2004; 15:1022–1033.
47. Nakatani K, Seki S, Kawada N, et al. Expression of SPARC byactivated hepatic stellate cells and its correlation with the stagesof fibrogenesis in human chronic hepatitis. Virchows Arch 2002;441: 466–474.
48. Prieto J, Qian C, Hernandez-Alcoceba R, et al. Gene therapy ofliver diseases. Expert Opin Biol Ther 2004; 4: 1073–1091.
49. Olaso E, Ikeda K, Eng FJ, et al. DDR2 receptor promotes MMP-2-mediated proliferation and invasion by hepatic stellate cells.J Clin Invest 2001; 108: 1369–1378.
50. Dai WJ, Jiang HC. Advances in gene therapy of liver cirrhosis: areview. World J Gastroenterol 2001; 7: 1–8.
51. Wilson JM. Adenovirus-mediated gene transfer to liver. Adv DrugDeliv Rev 2001; 46: 205–209.
Copyright 2008 John Wiley & Sons, Ltd. J Gene Med 2008; 10: 993–1004.DOI: 10.1002/jgm

1004 A. M. Camino et al.
52. Ilan Y, Saito H, Thummala NR, Chowdhury NR. Adenovirus-mediated gene therapy of liver diseases. Semin Liver Dis 1999;19: 49–59.
53. Yu Q, Que LG, Rockey DC. Adenovirus-mediated gene transferto nonparenchymal cells in normal and injured liver. Am JPhysiol Gastrointest Liver Physiol 2002; 282: G565–G572.
54. Garcia-Banuelos J, Siller-Lopez F, Miranda A, Aguilar LK,Aguilar-Cordova E, Armendariz-Borunda J. Cirrhotic rat liverswith extensive fibrosis can be safely transduced with clinical-grade adenoviral vectors. Evidence of cirrhosis reversion. GeneTher 2002; 9: 127–134.
55. Inagaki Y, Kushida M, Higashi K, et al. Cell type-specificintervention of transforming growth factor beta/Smad signalingsuppresses collagen gene expression and hepatic fibrosis in mice.Gastroenterology 2005; 129: 259–268.
56. Maher JJ, Lozier JS, Scott MK. Rat hepatic stellate cells producecytokine-induced neutrophil chemoattractant in culture andin vivo. Am J Physiol 1998; 275: 847–853.
57. Bradshaw AD, Puolakkainen P, Dasgupta J, Davidson JM,Wight TN, Sage EH. SPARC-null mice display abnormalities inthe dermis characterized by decreased collagen fibril diameterand reduced tensile strength. J Invest Dermatol 2003; 20:949–55.
58. Framson PE, Sage EH. SPARC and tumor growth: where theseed meets the soil? J Cell Biochem 2004; 92: 679–690.
59. Schiemann B, Neil J, Schiemann W. SPARC inhibits epithelialcell proliferation in part through stimulation of the transforminggrowth factor-beta signaling system. Mol Biol Cell 2003; 14:3977–3988.
Copyright 2008 John Wiley & Sons, Ltd. J Gene Med 2008; 10: 993–1004.DOI: 10.1002/jgm




















