
AD-752582 REPORT ON ELECTRONIC AND IONIC ... - DTIC
118
BC ,-- — •• » »-.TTS^; AD-752582 REPORT ON ELECTRONIC AND IONIC REACTIONS IN ATMOSPHERIC GASES R. H. Neynaber, et al Gulf Radiation Technology Prepared for: Defense Nuclear Agency July 1972 DISTRIBUTED BY: Knn National Technical Information Service U. S. DEPARTMENT OF COMMERCE 5285 Port Royal Road, Springfield Va. 22151
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of AD-752582 REPORT ON ELECTRONIC AND IONIC ... - DTIC

BC ,-- — ■••■» ■»-.TTS^;
AD-752582
REPORT ON ELECTRONIC AND IONIC REACTIONS IN ATMOSPHERIC GASES
R. H. Neynaber, et al
Gulf Radiation Technology
Prepared for:
Defense Nuclear Agency
July 1972
DISTRIBUTED BY:
Knn National Technical Information Service U. S. DEPARTMENT OF COMMERCE 5285 Port Royal Road, Springfield Va. 22151

k'u^SPIipjf^WiHWMIRhNiiMiiiMRl^Ji.^
w 00
w 0$ *ß ib* Q
'mm' *s <
1 1 f »
OAF RAZXATIOM
SNA 29^F
Gulf-«T-A12209
REPORT ON
ELECTRONIC AND IONIC REACTIONS IN ATMOSPHERIC GASES
R. H. Neynaber, J. A. Rutherford, and D. Ä. room
^
Prepared under
Contract DNA00W2-C-G25*
for the
Defense Nuclear Agency
'..-* - '-- LJ (wrt^feu
Approved for public release; distribution unlimited,
NATIO.NAi Tfc( HN'CAI INFORMATION SERVICE
July 1972
m
GULF RADIATION TECHNOLOGY A DIVISION OF GULF ENERGY & ENVIRONMENTAL SYSTtMS COMPANY
P.O. 30X 608. SAN O'EGO. CALIFORNIA 92 112
f/«'\ m masramt**

n i n^PHqW^QVin BSffi! r^TT^wpppuppftyi ;.r-^WPP^M W" •i"W!P,.«!|i."i',,M?w™
UNCLASSIFIED Security CI«»»tfic»lion
DOCUMENT CONTROL DATA R&D (Sacutlly rlaatllleallan ol ml», body at ama tract and Inditing annotation »mi bo tntarad trhan Ihm onrill f»yorf I» clammtllad)
i omeiNA TINO »c Ti vi TV < Corpora lo auoSot)
Gulf Energy & Environmental Systems Company P. 0. Box 81608 San Diego, California 92138
la. RtlPORT ncuKiiy CHHIHC»TION
Unclassified 26. 9KOUP
1 «tPOUT TITLE
REPORT ON ELECTRONIC AND IONIC REACTIONS IN ATMOSPHERIC GASES
4 DCICitiPTivE KOTII (Typo ol rope*! and Inclualra data»)
Final Report 5 »u THomii (Html naato. rnlddla Initial, laal namta)
R. H. Neynaber, J. A. Rutherford, and D. A. Vroom
• REPORT OA TE
July 1972
7«. TOT»». NO. OF PACE*
150
76. NO. OF KtFI
19 •*. CON TRACT OR ORANT NO
DNA001-72-C-025H 6. PROJEC T NO
NWED: XA.01
«• Task & Sub task: D010
* Work Unit: 34
M. l"-HIH»TO«'l REPORT NUMBERItl
Gulf-RT-A12209
>6. OTHER REPORT NOI1I (Any othor numbara that may 6« amtlanod thla ramoti)
DNA 2944F
10 OlfTRieuTION STATEMENT
Approved for public release; distribution unlimited.
II SuPPLfMCNTARV NOVE* 11 IPONSORINO MILITARY ACTIVITY
Director Defense Nuclear Agency Washington, D. C 20305
13 ABtTRAC T
The effect of addition of internal energy to the ion or neutral reactant in an ion-neutral process has been investigated. Changes in the cros3 section for certain processes are reported and a change in the state of excitation of the final product can in some cases be postulated. The following reactions have been considered:
N+ + 02 •* N + 02+
N^ + 02 - N2 + 02+
N2+ + 0 ■* N0+ + N
0 + N -+ NO + N
N+ + N. •* N + N„+ 2 i
N+ + 0,, ■*■ 0 + N + 0+
N+ + 0 + N0+ + 0
Where the cross section was found to increase with addition of internal energy to el eher the ion or the neutral, an approach to near resonance was, in some cases, found to have occurred. In such cases one or the other of the product particles may be pro- duced in an excited state, when the internal energy change is small compared to the ene*-g; defect f-,r the reaction, little or no change in tne cross section is observed.
s -\s
DD.ISM 473 UNCLASSIFIED
5*run^> Cl*tttfic«t.on

rT^FF*W*"p^;=«™*'' ^'.'»»j'."«..».^""«.!..^iwwpj"-~—f*T—»■ ipqppwwajgHgsgw^gyj^pwwgffwipgragw^"^
•■ifWBlWBftH fl^Pff^*^
UNCLASSIFIED Security CUssliication
KEY wonot
Ion-Neutral Reactions
Charge Exchange
Positive Ions
Metallic Ions
Negative Ions
Excited Stites in Ions
Excited Stp.tes in Neutrals
Cross Sections
Atmospheric Ion Chemistry
Aeronomy
Ion Molecule Reactions
Crossed Beams
U>
• OLI
UNCLASSIFIED Security Cltttifiration

rrw-**r~xr™?***<x!i
DNA 29MF
GULF RADIATION TECHNOLOGY
Gu1f-RT-A12209
REPORT ON
ELECTRONIC AND IONIC REACTIONS IN ATMOSPHERIC GASES
This work was supported by the Defense Nuclear Agency under NWED; Subtask HD010-31»
Work done by:
A. E. Kristensen J. K. Layton R. H. Neynaber J. A. Rutherford D. A. Vroom
Report written ry:
R. H. Neynaber J. A. Rutherford D. A. Vroom
Prepared under
Contract DNA001-72-C-0251»
for the
Defense Nuclear Agency
1 n
Approved for public release; distribution unlimited.
Gulf Radiation Technology Project O'lS1» July 1972
GULF RADIATION TECHNOLOGY A DIVISION OF GULF ENERGY & ENVIRONMENTAL SYSTEMS COMPANY
P 0 BOX 608. SAN DIEGO. CALIFORNIA 92112

!.^P.".'-r>ire^»»7*WP!p,> ■U.J.J'JP 1,11.1** "T i «.Mi» CTiw ■i'"*'-'*,gF*3g|5w*'*Ty^*s<'^'i-*""^-,'<'-t"-^-'»^.-i'j.-?! "■■-"■^yw^a.^py^nm«!
ABSTRACT
The effect of addition of internal energy to the ion or neutral reac-
tant in an ion-neutral process has been Investigated. Changes in the cross
section for certain processes are reported and a change in the state of ex-
citation of the final product can in some cases be postulated. The follow-
ing reactions have been considered:
0+ + N2 ■+ N0+ + N
N+ + N2 -* N + N2+
N+ + 0- -»■ 0 + N + 0+
N+ + 02 -»■ N0+ + 0
N+ + 02 ■> N + 02+
N2+ + 02 - N2 + 02
+
N2+ + 0 ■+ N0+ + N
Where the cross section was found to increase with addition of internal
energy to either the ion or the neutral, an approach to near resonance was,
in some cases, found to have occurred. In such cases one or the other of
the product particles may be produced in an excited state. When the inter-
nal energy change is small compared to the energy defect for the reaction,
little or no change in the cross section is observed.
iii

CONTENTS
1. IN1RODUCTION 1
2. EXPERIMENTAL METHODS 5
2.1 Primary and Secondary Ion Systems 5
2.2 Neutral Beam Formation and Measurement 8
2.3 Extrapolation Procedure 11
3. RESULTS AND DISCUSSION 15
3.1 Reaction of 0 with N_ to Form N0+ .... 15 +
3.2 Charge-Transfer Reaction of N with N- 16
3.3 Reactions of N with 0- 22
3.4 Charge Transfer from N* to 0. 29
3.5 Charge Transfer Between H and 0 36
3.6 Reaction of N with 0 to give NO 37
REFERENCES A3
APPENDIX A - FORMATION OF MAGNESIUM IONS BY CHARGE TRANSFER 45
APPENDIX B - EFFECT OF METASTABLE 0+(9D) ON REACTIONS OF 0+
WITH NITROGEN MOLECULES 57
APPENDIX C - FORMATION OF SODIUM IONS BY CHARGE TRANSFER 63
APPENDIX D - FORMATION OF CALCIUM IONS BY CHARGE TRANSFER 71
APPENDIX E - FORMATION OF IRON IONS BY CHARGE TRANSFER 89
APPENDIX F - NEGATIVE ION REACTIONS WITH NEUTRAL OZONE 103
Preceding page blank

ILLUSTRATIONS
Figure
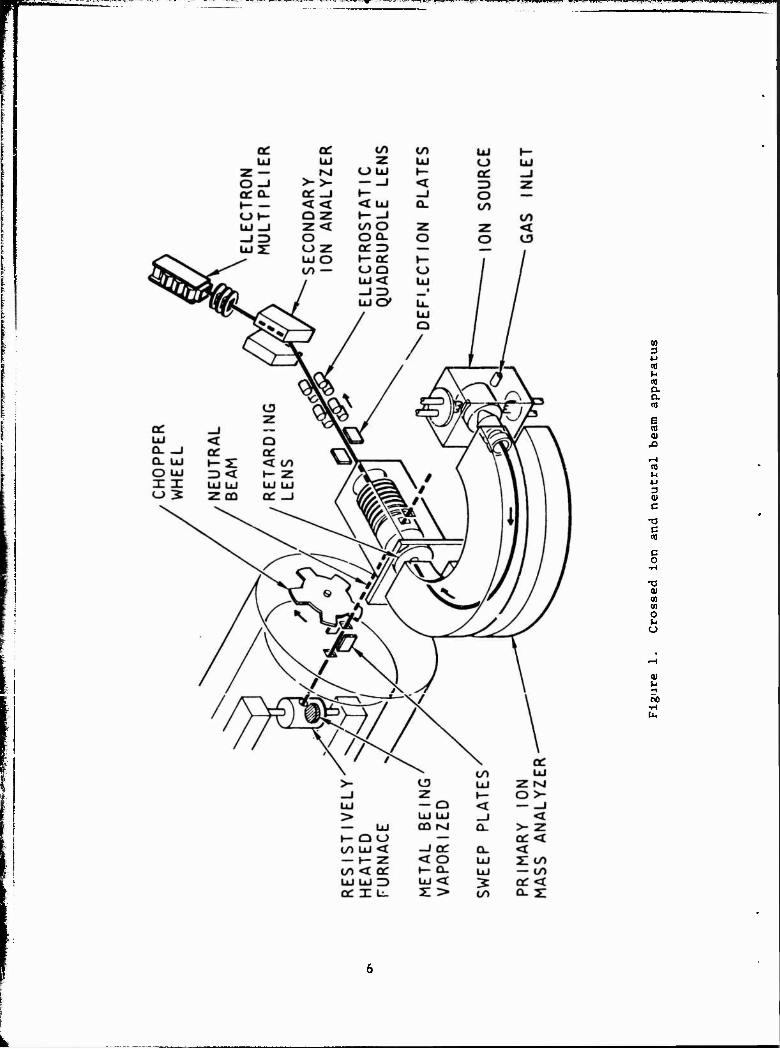
1 Crossed ion and neutral beam apparatus 6 + +
2 Reaction cross section for 0 + N -*■ NO + N as a function
of the energy in the center of mass system 17
3 Charge-transfer cross sections for N ions incident on
neutral nitrogen molecules as a function of the energy of
the incident ion 20
4 Charge-transfer cross sections for N ions incident on
neutral nitrogen molecules without vibrational excitation
and on neutral argon atoms as a function of the energy in
the center of mass of each system 21
5 Reaction cross section for N ions incide..L on 0_ to form
0 as a function of the energy in the ceiter cf mass and of
the incoming ion energy 24
6 Reaction cross sections for N ions incident on 0_ to form + +
0 and NO as a function of the energy in the center of
mass and of the incoming ion energy 26
7 Charge-transfer cross sections for N? ions incident on
neutral oxygen molecules as a function of the energy of the
incident ion 10
8 Dependence of the charge-transfer cross section for
N_ + 0„ ■*■ N_ + 0. upon the Ion-source electron energy . . 32
9 Charge-transfer crosp sections for N_ ions incident on
neutral oxygen molecules as a function of the energy of the
incident ion 33
10 Square root of the cross section for the reactions
H+ + 0 ♦ K + 0+ and H + 0 -* H + 0 + 0+ 38 +
11 Reaction cross section for N_ ions incident on neutral 0
to form NO as a function of the energy of the lncide.it
Ion 41
vi

naww^wos^sr mi.n ■i.ijwfPMtfi»!.—i- m'J4,j.,uu ...i.njmi....i. .-.LJ^-J.U, umwwmmammmm
Table
LIST OF TABLES
1 Calculation of the Population of the Vlbratlonal Levels of
Nitrogen as a Function of Temperature 9
2 Calculation of the Population of the Vibrational Levels of
Oxygen as a Function of Temperature 10
3 Measured Cross Sections for the Reaction N + N -*■ N + N . 18
4 Measured Cross Sections for the Reaction N + 0. -*■ 0 +
Products 25
+ + 5 Measured Cross Sections for the Reaction N + 0 -*■ NO +0 . 27
6 Measured Cross Sections for the Reaction N + 0- ■* N + 0 . 28
7 Measured Cross Sections for the Reaction N„ + 0_ -*■ N? + 0
when the 0. is not Vibrationally Excited 34
8 Measured Cross Sections for the Reaction N~ + 0_ ■+■ N + 0„
when the 0. is Vibrationally Excited 35
9 Measured Cross Sections for the Reaction H + 0 -► H + 0 .. 39
vii

^^^^-^ F-WWWW SSMSMM
INTRODUCTION
The transmission of radio and radar signals through the atmosphere de-
pends, in large measure, on the free electron density in the ionosphere.
The density of these free electrons is a function of both the amount of
ionization that is occurring and the magnitude of the rate coefficients for
the various mechanisms available for electron loss. In the normal atmos-
phere, ionization is produced primarily by photoionization processes initi-
ated by solar radiation. During periods of increased solar activity, the
amount of ionization and, hence, the free electron density are increased,
resulting in degradation of radio and radar equipment performance.
Artificial disturbances of the ionosphere, such as those resulting
from nuclear weapons detonation, greatly increase the free electron density.
The resultant degradation in radio and radar signals can seriously affect
the operation of military communications systems. Since the Nuclear Test
Ban Treaty nas eliminated atmospheric testing of nuclear weapons, the de-
gree to which artificial disturbances of this nature will influence elec-
tromagnetic transmission cannot be ascertained. Therefore, considerable
effort has been expended on laboratory and theoretical studies of atmos-
pheric ionization that would be expected from detonating weapons and on the
deionization processes that return the atmosphere to normal.
Part of the overall interest in the upper atmospheric process is re-
flected in the Defense Nuclear Agency (DNA) Reaction Rate Program. This
program, which supports research in the study of atmospheric deionization,
involves i_n situ measurements of natural disturbances in the atmosphere
(e.g., polar cap absorption events), theoretical studies and laboratory
measurements of pertinent reaction rates, cross sections, and other fea-
tures of the interactions that can occur among electrons, ions, and neutral
species in the upper atmosphere. Current progress in the field is reported
in the DNA Reaction Rate Handbook.

r~ - ^y- g.^«^^.^^^.«.^.'-^^..?^-.^. m*m- 'L-ii. >i nwq . ■■»■«i, -■■ »P u"w '^u; ..>■ ' ^"«—^ ' '.w.1-'. w^"-■■* ' °a....jnmiwy^y
The program under way in the Gulf Radiation Technology (Rad Tech)
laboratory (under Contract DNA001-72-C-0254) examines one phase of the
deionization problem: reactions of atmospheric ions with neutral species.
The primary objective of this phase of the ion-neutral reaction program is
the study of charge-transfer and ion-molecule processes. Since the most
probable cause of the loss of electrons in the upper atmosphere is dissoci-
ative recombination, it is important to know (1) the molecular ions that
are present, (2) their cioss sections for formation, (3) the effects of in-
ternal energy of the reactants on their rate of formation, and (4) the fate
of any excess energy that results from their formation. The present program
places special emphasis on studies of problems posed by items 3 and 4 above.
In the past, these aspects of atmospheric deionization have received very
little attention in the energy regime above thermal.
The major goal of the current program was to devise a means for intro-
ducing internal energy into the neutral and ionic primary particles and to
investigate the effect of this internal energy on the overall reaction
probability for formation of certain ionic species and the state of excita-
tion of the final products. Processes that were studied in this manner
include:
0+ + N_ ■*■ N0+ + N
N+ + N -► N + N*
N+ + 0„ -*■ 0 + N + 0+
N+ + 0- ■+ N0+ + 0
N+ + 0- ■*■ N + 0*
N2 + °2 " N2 + °^
A further program conducted during the past year was the development
of a source of oxygen atoms for use in our crossed ion-modulated beam

.■ v. ji.o^v*u.U.."raprr7 n IH"|!IWJUU»MI|W*!*SW
apparatus. In this study, we used both rf excitation of oxygen containing
molecules and thermal dissociation of molecular oxygen to form the 0 atoms.
The degree of dissociation attained in these beams was determined by meas-
uring signals Ironi the following reaction, for which the cross section is
known:
H+ + 0 ■* H + 0+
Using the 0 atom beam, the reaction
N* + 0 -+ N0+ + N
was studied.
The final task completed during the past year was the co-authorship of
a revision of a chapter in the DNA Reaction Rate Handbook entitled "Labora-
tory Methods of Obtaining Reaction Rate Data."
The following sections of this report describe the apparatus used foi
the experimental measurements, the results of the measurements, and a dis-
cussion of the results. Reprints of papers published during the past year
and preprints of papers written for future publication are included as
appendices.

iFU ■-ji.-iiuji.'A.." 'l^M'M'^,l^'^«'^4W'»i>T«l»wy<^M^^ i,u „mmnmiiinj
EXPERIMENTAL METHODS
The cross-ion-modulated neutral beam apparatus has been described in (2-4)
the literature. Thus, only an abbreviated description is given herein.
Also included is a brief description of the extrapolation procedure, de-
veloped in our laboratory, for extending the measured cross-section curves
to thermal energies.
2.1 PRIMARY AND SECONDARY ION SYSTEMS
Figure 1 shows a schematic of the experimental apparatus. The primary
ions are extracted from an electron-bombardment source and mass-analyzed at
an energy of 75 eV in a 180 magnetic-mass spectrometer. After analysis,
ehe ions pass through an aperture in an iron plate that shields the magnetic
field of the mass analyzer from the succeeding regions of the apparatus.
The ions are then retarded or accelerated to the desired collision energy,
and pass through a field-free region before intersecting the neutral beam.
Collimating apertures ensure that, from purely geometrical considerations,
all primary ions pass through the modulated neutral beam. The beam is modu-
lated at 100 Hz by mechanical chopping.
Secondary ions resulting from collisions between the primary ions and
npufral snoripe arp pvfrart*»H along rhp direction of rhp primary ion hpam
by an electric field cf approximately 2 V/cm. The ions then ente^ an elec-
tric field in which thexr energy is increased to 1650 eV. Penetration of
this accelerating field into the interaction region is reduced by the use of
a double-grid structure.
After acceleration, the ions pass through an electrostatic quadrupole
lens that forms the entrance slit for a 60 -sector magnetic-mass spec-
trometer. The mass-selected ions impinge on the first dynode of a 14-stage
CuBe electron multiplier. For all ions formed by charge transfer cr ion-
molecule reatiors, the most abundant isotope is used when making measurecents.
Preceding page blank
WigWffUI .PJMII.LHW**
«I 3 4-1
CO U CO a a.
0>
CO u i-1 3 <D C
•v c
c o
T3
<n tn o
u

,^r*\fu*w** '■-■Lvwi*^1 ■>. gw^-™-*™r™:.... j«j«^i,_i.wii>Mw^u*jffqwET«r»w*«J'fc.iSff ■%*' " ■" ■ ■-■ ■.'■.^PJ'.1 »I W*f ■ ■-'■'■'■'■».y i»'W»*N^ ■,-.'E*»^***-'-^WW*
The cross sections are corrected for the isotope effect created by collect-
ing only those having this mass. The output from the multiplier passes suc-
cessively through a preamplifier, a 100-Hz narrow-band amplifier, and a
phase-sensitive detector, and is then integrated. The output is presented
on a chart recorder.
The primary ion beam intensity is measured at the interaction region
with a Faraday cup, which can be moved into the collision region when de-
sired. The primary ion energy is determined from retarding potential meas-
urements. All surfaces at the interation region and the Faraday cup are
coated with alkadag, and the interaction region is normally maintained at
120 C to minimize suface-charging.
Because, in the present work, interest extended to small-collision
energies, only weak extraction fields at the collision region were used.
As a result, the secondary ions were not collected with 100% efficiency.
Obtaining absolute cross sections for production of various secondary ions
required, therefore, determination of their overall detection efficiency.
This latter consideration was governed by a number of factors, including
the multiplier gain and the efficiency of transmission of the secondary ions
from the interaction region to the multiplier.
The multiplier-amplifier-recorder gain is measured by modulating the
primary ions prior to their entry into the collision region. The ion cur-
rent signal is first measured with the movable Faraday cup and, after tra-
versing the secondary mass spectrometer multiplier-amplifier system, by ehe
remrd.r. Primary ±nn transmission through the second isass spectrcmeLCL i=>
92%. Typical gains for the entire system are of the order of 10 output
volts on the recorder per ampere of incoming current. In practice, gains
are measured for each product ion.
The major experimental uncertainty is associated with the collection
efficiency for the secondary ions. Collection fields large enough to ensure
total collection of the secondary ions cannot be used because of the influ-
ence these fields exert on the motion of low-energy primary ions. While
measurements of the variation of collection efficiency with the strength of
the extraction field can readily be made at high primary ion energies, the

resu?..» are not necessarily relevant to the low-energy regime, where the
dynamics of the ion-neutral process may be different. Interpretations of
the present data obtained using weak collection fields is based on the as-
sumption that, for primary ion energies above a few eV, the secondary ions
are collected with nearly equal efficiency, and that, as a result, the col-
lection efficiency is independent of both the nature and the energy of the
primary ions. This assumption implies that the energy defect in the reac-
tion is not large, since energy not expended in excitation of the products
must appear as kinetic energy and, therefore, would influence the collection
efficiency.
2.2 NEUTRAL BEAM FORMATION AND MEASUREMENT
">.. 2.1 Beams of Stable Gases
Beams of ground state N» and 0. are formed by effusion from a small
hole in a room-temperature tungsten or iridium tube cortaining the gas
(Figure 1). Beam density is determined using the cosine law of molecular
effusion where the pressure in the tungsten source, the area of the aper-
ture in the tube, and distance from the aperture to the interaction region
are known factors.
Trie vibrational energy in the neutral beam can be changed by resistive
heating of the tungsten source. Temperatures as high as 2800 K have been
used for this vibrational excitation, with the temperature being monitored
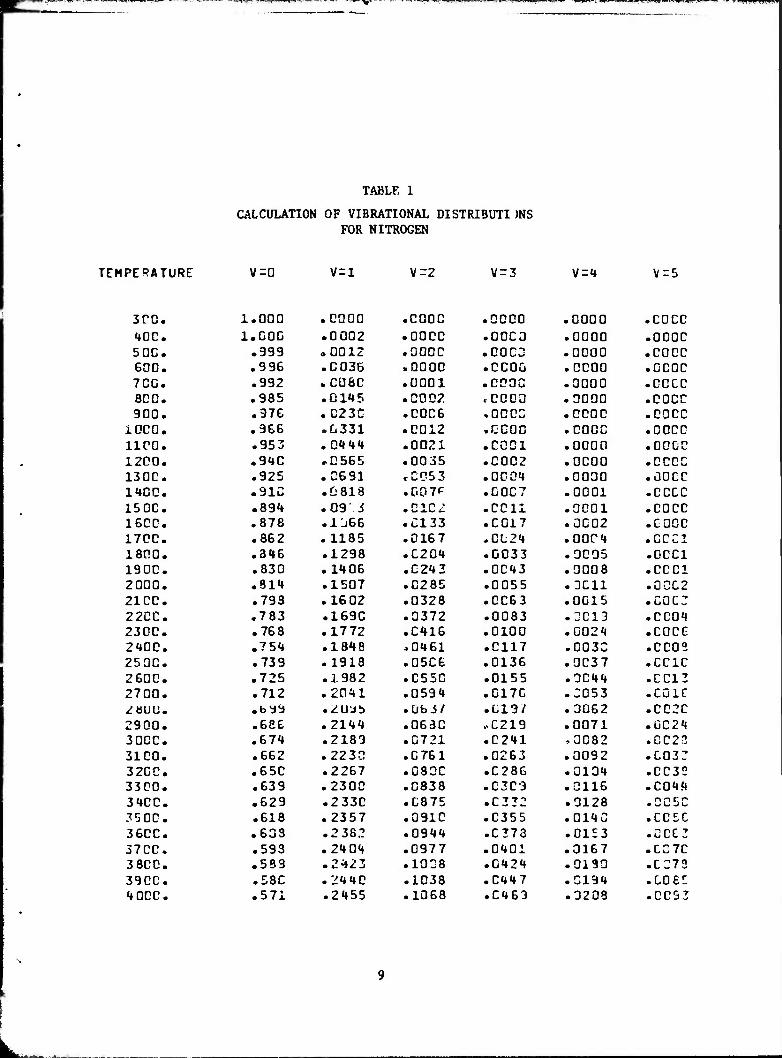
by an optical pyrometer. Tables 1 and 2 summarize the results of calcula-
tions of the vibraticr.al population cf the ground electronic state? of N
and 0_, respectively, as a function of the temperature of the gas. The beam
density of the neutral flux due to temperature changes are corrected when
effects of vibrational excitation of the neutral species are being studied.
More extensive excitation of N and 0- molecules used rf techniques.
In these studies, the furnace source was replaced by a glass U-shaped dis-
charge tube with a small hole in the bottom of the U-bend. Electrodes
attached to the outside of each leg of the U served to introduce the rf
power into the gas.
»; wm i PMBV1
TEMPERATURE
3TC. toe. 5 0C. 60C. 7CC. 8C0. 900.
iOCO. 11C0. 1200. 130C. 1400. 150C. 1600. 1700. 1800. 1900. 2000. 21CC. 2 2CC. 2300. 2100. 2500. 2600. 2700. ^»UU.
2900. 30CC. 3100. 3200. 3300. 31CC. 35 00. 36CC. 37CC. 3800. 3900. 10CC.
TABLE 1
CALCULATION OF VIBRATIONAL DISTRI3UTI )NS FOR NITROGEN
V-0 V=l \-2 V-3 V=1 \j-5
1.000 .0000 .0000 .0000 .0000 .COCO 1.C0C .0002 .0000 .0000 .0000 .0000 .999 .0012 .0000 .0003 .0000 .0000 .996 .0035 »0000 .CCOQ .0000 .OCOC .992 .0080 .0001 .0000 .3000 .GCCC .985 .0115 .0002 rCOOO .0000 .COCO .976 .C23G .0006 .0003 .0000 .coco .966 .0331 .0012 .0000 .0000 .coco .953 .01*»«» .0021 .0001 .0000 .0000 .910 .0565 .0035 .0002 .0000 .0000 .925 .0631 rC053 .0001 .0030 .ÜOCC .913 .0818 .007P .0007 .0001 • coco .891 .09 ■ S .0102 .0011 .0001 .coco .878 .1J66 .0133 .0017 .0002 .0000 .862 .1185 .0167 ,CL21 .0001 .GCC1 .316 .1298 .0201 .0033 .3005 .0001 .830 .1106 .0212 .0013 .0008 .CCC1 .814 .1507 .0285 .0055 .3011 .0002 .793 .1602 .0328 .0063 .0015 .C0C3 .783 .1690 .0372 .0083 .3013 .0001 .768 .1772 .0116 .0100 .0021 .coco .75«» .1818 -0161 .0117 .0033 .coos .739 .1913 .0506 .0136 .0037 .CC1C .725 .3.982 .0550 .0155 .3011 .0013 .712 .2011 .059«» .0170 .3053 .cuie • by9 • üUSb .übJ/ .019/ .3062 .0020 .686 .2111 .0630 »0219 .0071 .0021 .674 .2183 .0721 .0211 ,3082 .0022 .662 .2230 .0761 .0263 .0092 .0032 .650 .2267 .0800 .0286 .0131 .0032 .639 .2300 .0838 .0303 .0116 .001.1 .629 .2330 .0875 .0232 .9128 .0050 .618 .2357 .0910 .0355 .0113 • L L -j u
.633 .2 382 .0911 .0273 .0153 .GCC3
.593 .2101 .0977 .0101 .3167 .0370
.539 .2123 .1008 .012«» .3190 .0273
.58C .2110 .1038 .0117 .3131 .008!!
.571 .2155 .1068 .0163 .3203 .0093

-i«B^K .._■**■ J "JIM»*.».1* "
TABLE 2
CALCULATION OF VIBRATIONAL DISTRIBUTIONS FOR OXYGEN
TEMPERATURE V=0 V=l V-2 V = 3 V=4 VrS
300. .999 .0005 .COCO .DC CO .0000 .cooo too. .996 .0035 .0000 .0000 .0000 .cooo 500- .989 .0109 .0001 .0000 .0000 .cocc 600. .977 .0227 .0006 .COCO .0000 .0000 700. .960 .0383 .0016 .0001 .0000 .cocc 800. .9*0 .0560 .00 35 .0002 .0000 • cccc 90C. .913 .0749 .0063 .0006 .0001 .cooc 1000. .895 .0938 .0102 .0011 .0001 .0000 HOC. .871 .1120 .0149 .0020 .0003 .ooco 1200. .817 .1292 .0203 .0033 .0005 .0001 1300. .823 .1451 .0263 .GC49 .0009 .C0C2 1400. .799 .1595 .0326 .0063 .0015 .0003 15 00. .776 .1726 .0392 .0091 .0022 .C0C5 1600. .754 .1842 .0459 .C117 .0030 .0008 1700. .733 .1945 .0526 .0145 .0041 .L012 1800. .713 .2035 .0592 .0176 .0053 .0016 1900. .693 .2115 .0657 .C203 .3C67 .C022 2000. .675 .2184 .0719 .0241 .0082 .C028 2100. .657 .2243 .0779 .D275 .0098 • CG36 220G. .640 .2295 .0836 .0309 .0116 .0014. 2300. .624 .2339 .0890 .C344 .0135 .C0E1 2100. .608 .2377 .0942 .0379 .0154 .0064 2500. .591 .2409 .0990 .0413 .0174 .0075 2600. .580 .2436 .1036 .0447 .0195 .0086 2700. .567 .2458 .1079 .0480 .0216 .C098 zauu. .554 .2476 .1120 .C513 .0237 .CHI 2900. .542 .2491 .1158 .0545 .0259 .C12F 3000. .531 .2503 .1133 .0576 .0281 .0138 3100. .52C .2512 .1227 .0606 .0302 .C152 3200. .51C .2519 -1258 .G635 .0321 .0167 3300. .500 .2524 .1287 .0663 .0345 .0182 3400. .491 .2 527 .1314 .0691 .0366 .0196 3500. .482 .252!' .1340 .0717 .0387 .C211 3600. .473 .2528 .1364 .0743 .0103 .C226 3700 . .465 .2527 .1336 ,0767 .0429 .C212 3800. .457 .2 525 .14:7 .0791 .0119 .C257 39CC. .450 .2522 .1427 .081«* .0169 .C272 4000. .443 .2513 .1445 .0836 .0133 .C287
10

affigagBBBC
For both the thermal and rf excitation experiments, care was taken to
eliminate any charged particles that might be emitted by the source. E^c-
trostatic sweep plates were used for this purpose.
2.2.2 0 Atom Beam Sources
Two methods were used to form the 0 atom beam. The first of these was
dissociation of molecules containing oxygen atoms in an rf discharge, both
0 and C0„ were used; both cases resulted in the production of 0 atoms.
The major problem associated with this type of source is the lack of
knowledge of other species that may be present in the neutral beam. The rf
excitation is not specific and can produce both atoms and molecules in
metastable states with unknown concentration.
Thermal dissociation of oxygen in an iridium tube furnace was the sec-
ond technique used to produce 0 atoms. Thermal methods of dissociation
eliminate the possibility of the presence of excited 0 ana 0 atoms in the 9 3
beam, but result in low beam densities (^10 atoms/cm ) because of the low
pressures required in the source for sufficient dissociation.
An ion-probing technique was used to obtain the degree of dissociation
present in thermally produced beams. This technique uses the near-resonant
asymmetric charge-transfer reaction
H + 0 ■*■ H + 0+
The results for this reaction, together with the degree of dissociation in
the o atom beam determined using this reaction, are discussed in Section 3.5.
2.3 EXTRAPOLATION PROCEDURE
The lowest ion energies that can be obtained in our apparatus are 1 eV
or greater. The charge-exchange processes in the energy range trom thermal
to several eV is of considerable interest. An extrapolation procedure pre-
viously developed in our laboratory was used to obtain cross sections and
rate coefficients in this region. This procedure combines the energy de-
pendence of the Rapp and Francis resonant charge-transfer theory with (9)
the Gioumousis and Stevenson complex formation model.
11

mwjuiMWHASM'1 ■wp'V"- ^ mmppwpBipoa—w —-"uiu»y--"»■ ^*m**m ««■mpmR jiiw.il.imji-Uli» i "»"I"
The general formula developed for the total charge-exchange cross sec-
tion takes into account the probability that charge exchange occurs both
when a complex is formed and when i. is not. The formula also corrects for
nonrectilinear orbit when complex formation does not occur.
The total charge-exchange cross section, o, is given by
a = fa. for 2a7 <_ o_ (1)
or by
•H)° 2 + al for 2a1 > o. (2)
where f is the fraction of collisions resulting in complex formation that
decays into the change-exchange channel, and a is the Gioumousis and (9) Stevenson complex formation model cross section, represented by
■*m in (3)
where e is the electronic charge, a is the polarizability of the neutral,
and E is the barycentric interaction energy. Also, a , which is given by
1 o 1 + fey (4)
represents the Rapp ariu ricuicia resonant charge-exchange formula modified to
take into account the curved orbits of the reactants. The Rapp and Francis
formula (8)
has the form:
„1/2 - A a = A o
B logE (5)
where A and B are constants.
The extrapolation is carried ouf by fitting the modified Rapp and
Francis formula (Equation 4) to the high-energy portion of the measured
data, which is in the region in which complex formation should be negligible.
12

w\ji,r i»|w.» KF-i"'.»:iHL,WM y TKnTBTJi-fiw»>,mvg«i¥iupmiM i.n.*"*jf»«,vi ■^■flffWpgWWI^WW ■'■>■ ■'■■.■^■■f.im» *-L1-" W-'"> T!ww»r*'*_«*«■m&^Jfm^mjQ
The calculated curve is then extended to lower energies using the test
given by Equations 1 and 2 to evaluate where the measured data deviate
from the form of Equation 4. In Lhis manner, the contribution of complex
formation is determined, and the calculated curve can be extended to ener-
gies below those measured. A computer program has been developed tc per-
form the calculations.
The extrapolation technique was developed by assuming that the rela-
tive abundance of the various products emerging from the capture-formed
complex is independent of the relative knietic energy of the reactants;
that is, f remains constant. If this condition is not met, the technique
does not yield valid rate coefficients at near-thermal energies. In general,
the extrapolation technique is valid if the calculated cross-section curve
fits the experimental data to the lowest energy of measurement. This com-
parison is made for all data given in this report.
13

qSMI.MV, J. ■..*' J'ffll*«
3. RESULTS AND DISCUSSION
3.1 REACTION OF 0+ WITH N TO FORM NO+
The reaction of 0 with N2 to form NO as a function of the excitation +
in the 0 primary ion was studied in a previous phase of the program. The
results of this study were published recently, and a reprint is included in
this report as Appendix B. Briefly, the results of this study showed that + 4 +
for ground-state atomic oxygen ions (0 S), the reaction to form NO pro-
ceeds with a large cross section over the energy range from 1 to 15 eV. + 2
Excited ions (0 D), on the other hand, appear to have a very low proba-
bility of forming N0+.
Study of the reaction of 0 with N? to form NO was extended during
the last contract period. The reaction probability was studied as a func-
tion of the vibrational energy of the N_. Both the thermal and rf dis-
charge methods were employed to excite the N_. With the thermal method, it
was possible to populate the v = 1 and v = 2 levels of the N_ to a signifi-
cant extent. Table 1 gives the calculated N. vibrational distribution for
2700°K as being approximately 71% in v = 0, 20% in v = 1, and 6% in v = 2.
The vibrational distribution in the N„ molecules emerging from the rf (1 0^
source is not easily determined. Methods discussed by Schmeltekopf et al.,
coupled with our ion-probing techniques, can be used to obtain this infor-
mation. In the present work, however, obtaining this vibrational distribu-
tion was not attempted. The rf discharge may also produce N_ molecules i-.
the metastable (A £ ) state. We used NO (ionization potential, 9.27 eV) u
to prooe the beam for the presence of this species. Since the N metastable 3 +
(A I ) state has an ionization potential of 9.41 eV, charge exchange with u +
the NO would be expected. No indication of metastable N_ could be found.
The experimental results for the reaction
Preceding page blank
0+ + N2 -* N0+ + N (6)
15

HJHi. nrJtl^-^gi.«»Li.i,j.. l^l-l.-iiinj HM|Jii» ■■ .-■- u».««^WfH;'."..wy|;fl.l.Uimi|;kV, .JMJWll. |ll^ilJ..!lJ,^pjl.»IJWAJtJHlll,,IWIIP!.i..WWH""R'V*-**"llll. "^g
show an increase in the cross section at low interaction energies (below + 4
5 eV) for both the rf and thermally heated N„ beam when only 0 ( S) parti-
cles were present in the ion beam. This increase in cross section with
increasing vib rational temperature has also been observed at low-impact
energies by Neynaber and at thermal energies by Schmeltekopf et al.
One possible explanation for the increase is that the additional energy
contained in the neutral reactant assists in the formation of the activated
complex necessary for the reaction to proceed.
The experimental results for the reaction of Equation 6 (which is exo-
thermic by 1.1 eV for ground-state particles) are given in Figure 2 for the
rf excitation of the N_. Both rf-on and rf-off cases are illustrated. The
vibrational excition of the N. increases the cross section below 5 eV in
the center of mass and decreases it above this energy. As mentioned above,
this increase in cross section at low energies is consistent with the ex-
perimental results of Neynaber and those of Schmeltekopf and with the theo- (12)
retical calculations of O'Malley.
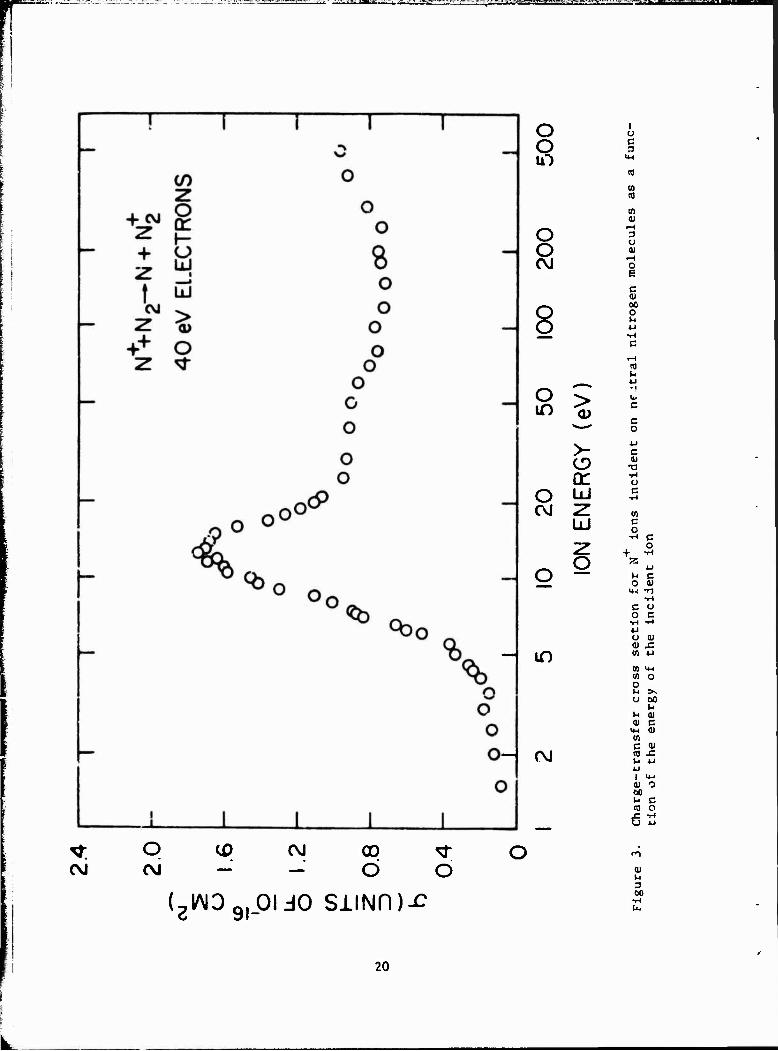
3.2 CHARGE-TRANSFER REACTION OF N+ WITH N0 4.
Preliminary results of a study of the charge-transfer reaction
N+ + K2 - N + N* (7)
(13) were discussed in a prior report. The charge-tiansfer cross-section
data for ground-state N and N0 in the v = 0 level are given in Table 3.
For convenience, these data are displayed graphically in Figure 3. From
the diagram, it can be seen that the cross section is small and decreases
with decreasing energy in the region between 4 and 1.5 eV. This behavior
is expected since the process is endothermic by approximately 1 eV. The
rise in the cross section at about 4 eV probably results from the onset of
a second channel for reaction.
The present data are in agreement with the recent studies of Maier and (14) Murad, although the cross section discussed herein tends to a somewhat
higher absolute value. The peak value (at 8.5 eV in the center of mass)
16

3V»wswn ^E-^MWW.IWWJRWfl.W.m (
(E"J39|0I P siiun )£>
01
0 u
0» C •u CD C 01 . CJ ^-N
U-l ID M-i
J= O JJ
<H C I-"
>> CM ooz u 01 cu C -u ^ 0) CO c
*J o 01 CO X. I <*-■
c *~- O O CM
K Z 00
■U h 0)
•u o *-» CJ U-l -H C CJ 3 C X
m o u
c o
CO
05 cd
Z
+
O i-( 0) .-I CO CD
c CO o CO TH O *J M CJ
CO U
0) -H
t " CM z + T*
>-l
CO o CU u-i >
01 CO CD
01 CJ c -H 01 i-H .c
C -H CO O i—1 i-l
Tl 9 5 0 "' CO 01 01 01 CO J= >-■
H D. CO o> CO u o • U B CJ cu
01 c
C CO —I o >.
■H CC C u CD CJ CO Ji <D cn O <U CD U
CC B X>
01 u 3 00
17
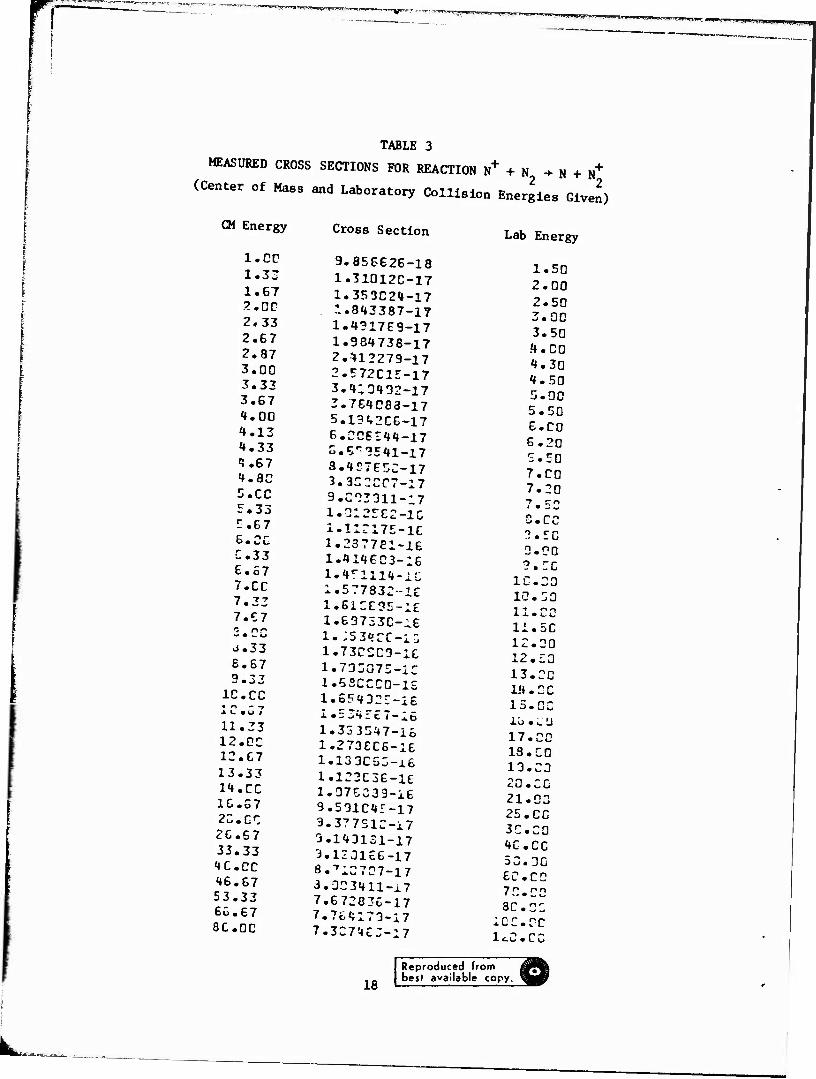
TABLE 3
MEASURED CROSS SECTIONS FOR REACTION N+ + N N + jfc
(Center of Mass and Laboratory Collision Energies Give!)
CM Energy
l.CC 1.33 1.67 2.CC 2,33 2.67 2.87 3.00 3.33 3.G7 «».00 4.13 4.33 «5.67 4.8C 5.CC 5.33 C.67 6.CC C33 E.S7 7.CC 7.33 7.€7 >- • «. <j
^,33 e.67 9.33
ic.cc
11 12 12 13 m 16. 2C. 26. 33. 46. 46. 53. 66 . 8C.
.33
.OC
.67 »33 .CC .67 .cc .57 .33 CC 67
67 OC
Cross Section
9.856626-18 1.31012C-17 1.353C24-17 1.843387-17 1.491769-17 1.984738-17 2.412279-17 2.S72C1E-17 3.413402-17 3.764C83-17 5.13t,2C6-17 6.CC6C44-17 C.6^3541-17 3.42765C-17 3.3C2CC7-17 3.C03311-17 l.oiccec-ic 1.112175-16 1.237751-16 1.414603-16 1.4T1H4-1C 1.577832-16 1.61C69S-1E 1.63733C-16 l.:S34CC-I3 1.73CCC3-16 1.703075-iC 1.5SCCC0-16 1.6?432E:-i6 1«534 3£7-16 1.333547-16 1.2736C6-16 1.133C&3-16 1.123636-16 1.376039-16 9.501C45-17 3.377S1C-17 3.143131-17 3.1Z31E6-17 8.T12727-17 3.323411-x7 7.672836-17 7.764173-17 7.3C7463-17
Lab Energy
1.50 2.GO 2.50 3.QC 3.50 4. CO 4.30 4.50 5.00 5.50 6.CO 6.20 3.50 7.CO 7.20 7.53 O p n U • L O
3.50 3.00 3.36
1C.20 10 . 5 0 11.CC 11.5C 12.00 12.30 13.CC 14. CC 15.OC
17.CO 13. CO 13. C3 20.CO 21.03 25.CC 3C.C0 4C.CC 5C.30 6C.CC 7C.CC 8C.CC
1CC.CC 1 <-C . CC
18
Reproduced from best available copy. 9

wwpppwj i .iiui.i in W>WP« HVWW J>JML- $mm mmn u*i#w*M» wwnu^f^i
TABLE 3 (Continued)
CM Energy Cross Section Lab Energy
ICO.CO 7.142880-17 150.00 12C.CC 7.485693-17 180.CG 133.33 7.637313-17 2CG.0C 166.57 7.113368-17 250.00 200.CC 8.2«»5853-17 300.CO 266.67 3.311400-17 IOC.CO 333.33 9.750685-17 500.00
—16 2 derived from our experiments s 1.7 x 10 cm , while the value at the
same interaction energy in Reference 14 is 1.0 x 10 cm .
We attempted to introduce the metastable N ( D) into the primary ion
beam to observe the effect of this species on the cross section. Although
a number of different gases containing N atoms were used to produce the N
beam, no evidence of the metastable could be found. Since several tech-
niques were unsuccessful in producing this species in the laboratory, we
conclude th^t it is not easily formed. As a consequence, it is doubtful if
N ( D) is a very common species in the upper atmosphere.
Vibrational excitation of the neutral N reactant by thermal means
appeared to have no effect on the reaction cro3s section. This behavior
is expected since no levels are close enough to cause resonance by vibra-
tional excitation to onlv the first or second vibrational levels.
Excitation of the N„ by rf did, however, produce vibrational levels
that were high enough to enhance the reaction cross section at low ener-
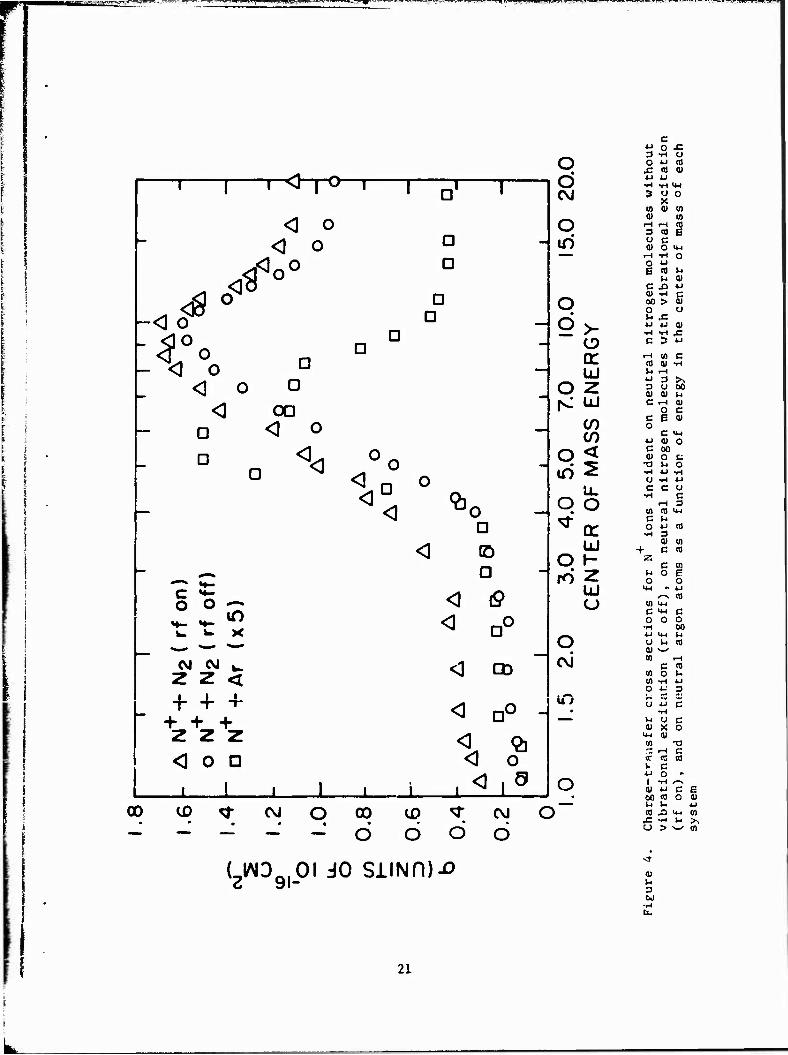
gies. Figure 4 gives the cross section data for reaction 7 over the
energy range from 1 to 20 eV in the center of mass. Both the rf-on and
rf-off cases are illustrated. The rf-on data show a larger cross section
over the total energy range with the increase being the greatest for low
interaction energies. This implies that the vibrationally excited N? reacts
more readily in tne charge-exchange process. This is probably due to the
19
__E23SES^S: .^Wt"-*''»'A*iW ^'TriTTraTTg»
o o u")
O O
o >
> o (T
o UJ OJ Z
- If)
- CO
o cvi
<£ CVJ 00 Ö Ö
LÜ
O o -
u c 3
w to
3 a a)
■H O e c 0) 00 o k<
V4
a. C
c o
c « T3
U c
to c o -H e
o f •*
4-1 ^ c o ai
-H C u o c
y (U <u je tn 4->
oo u ttl c
(2iAio9loido siiisim-c
<« 0) (I) C 0) <o .e IJ 4-1 u
I <*- <D o OO U C to O
ki 3 00
20
r t
1 \ .
!..!»FJWJMi.j4.M*.i, ■»,!,! s.-Ws .--.'«»!*«*»* Jt.],*PJ.l-L!,J*fBWW"5ff"
!
—j 1 rO-p0" 1 1 o' '
<3 o < o a
A <<f°° G
-<o<*° G
a
:^°°o G
a —
<3 o a _
< en G <3 o —
- D n *« °o G ^
< n o < * 4u <3
uo - G
< © — ^^ a
*"■* H- C H- o o *r < Q - H- « < n° k. w X ^ G
_ •»—*—'*- CVJ CVJ w
Z Z < <] OD
+ + + " + + + < a° -
z z z < <a < 0 D < o
1 I 1 1 i i .« I9
00 CD Cü O 00
- o ID d d
CJ
d (2WD9|0I dO SlINfD-O
c *J O J= 3 i-i CJ o 0 4J CD X cd CD
Ö 4J U iH -H y-t
CJ 3 o o a « to
O 01 CO iH rH Cd 3 cd e
m CJ C a» o i-i
en mol
ibrati
nter o
O oo > (u o o
o >- U U 0J •H -H j=
e> C 3 u
or •H 10 C Cd 41 -H
UJ U i-l *J 3 >> ü z 3 CJ 00 01 01 h
N LÜ C r-J 01 o c
CO BE« o
CO 4J a) O
o < e oo eu o u
lO 5 •o n o •H *J -H CJ -H 4-1
u. C C cj
o o -( 3 to cd <»-i
<fr QC C V4 out •H 3
UJ 0) CO + c cd
O H C CO
ro z ^ o e o o
LÜ /N cd
u
ions
off
gon
O CJ U CO (1) N-S
OJ OSS s
f.io
n utral
m u -u c * •H 14 cj c <u x o
u-i 01 CO T3 a i-t c <c * cd >- c 4J O -
o | 1-t ^—V
0) *-> C E « 00 co O <U
pure 4. Char
vibr
(rf
syst
21

■ ■ ™ PKnpvmnmaKCTi ■. w—->—■-«j- jf■ jij. i -rent.■.mi u n i-u^i.t LH, . ^^iiBt»i'WT»W!w^lwywT^wp^»tyym»^^
high vibrational excitation in the N», which brings the reaction closer to
resonance. The rf-on curve suggests that, for highly vibrationally excited
N«, a finite cross section for the reaction may exist at thermal energies.
(14) In their study, Maier and Murad suggest that the second channel
for reaction, which appears at about 3.5 eV in the center of mass (Figure 4),
may be due to formation of the N» product in the A or B excited states. To
test this supposition, the reaction
N+ + Ar -*■ N + ArT (8)
was studied. In this case, Ar replaces the N as the neutral reactant.
Since the iohization potential of argon (15.76 eV) is similar to that for
N9 (15.58 eV), a similar reaction threshold can be expected, although the
absolute magnitude of the cross section with argon may be smaller since the
reaction is about 0.2 eV more endothermic. The cross section for reac-
tion 8 is given in Figure 4 (note the absolute values have been multiplied
by a factor of 5 to make the scales comparable). The interesting result
of this study is that the onset of the peak for reaction 7 is at about
the same energy as that for reaction 8. If a similar mechanism is acting
in both cases, it is doubtful if the peak in reaction 7 results fron.
formation of the A or B state of N„, since similar states do not exist for +
Ar . Consequently, an alternative explanation for the peak is that the 2
peak may result from formation of the neutral N product in the excited D
state. For reaction 7, this process would be expected to have a threshold
«.»■ AAI.» 1 /. r\\1 ..U<«>. 4- ,.U~i- ,._ „U-~—..~-J TU» _-__ 1 J_rc - Ul *lU~ %*%. _/ • 4 CV | w»»Av„.« J.O «.liui. M«_ l/l'OW*. V Cfcl • 1IIC IUU1C |/LUI1UU11LCU UitiCLCULt:
between the Ar and N_ data at higher interaction energies (above 10 eV in
Figure 1) may be due to the effects of the ion-molecule reaction reported (14)
for reaction 7 by Maier and Kurad.
3.3 REACTIONS OF N+ WITH 0
The present program included studies of the effects of internal energy
in the reactants on the following processes involving N and 0 :
22

"^ ,^ . ii.-.^m^j^. vm, m
N+ + 0„ ■♦ 0 + N + 0+ (9)
N+ + 02 -» N0+ + 0 (10)
N+ + 02 ■+ N + 0* (11)
The state of excitation of the reactant 0. molecule was altered by passing the
gas through an rf discharge. The results with this discharge method were
compared with those for no discharge. For all three of the above reactions,
no effect due to discharge-induced excitation in the 0_ could be seen on
the reaction cross sections. As discussed above, we attempted to produce a
beam of N with excited states present. Thus far, we have been unable to
detect the presence of any excited N species.
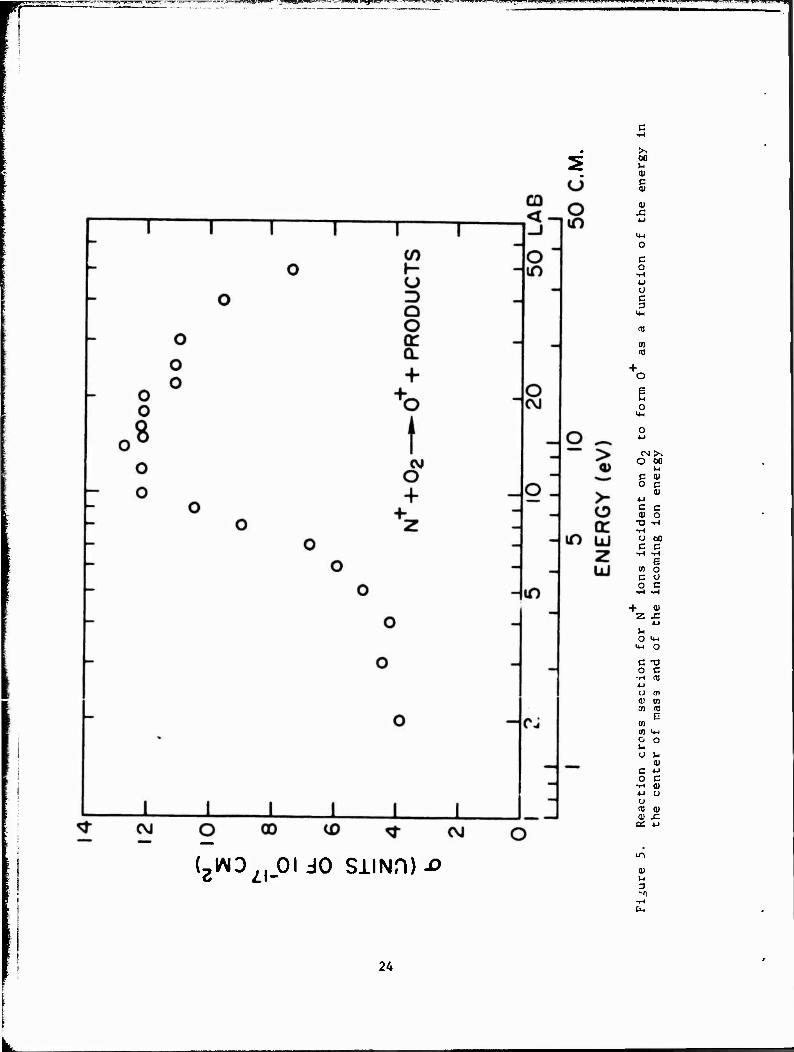
The results for the production of 0 are illustrated in Figure 5.
Tabular data are given in Table 4. Note that, in the center-of-mass system,
the cross section for the production of 0 is very small below 1.5 eV,
rises slowly to about 4 eV, and then begins to rise rapidly to a peak at
about 10 eV. If the production of 0 resulted from an ion-molecule reac-
tion in which NO was the other product, the overall process would be exo-
thermic by approximately 2.3 eV for all products in the ground state. Since
the cross section appears to be very small at low energies, this does not
seem a likely channel for the reaction. Reaction 9, a dissociative charge-
transfer process, is endothermic by 4.2 eV. Since our cross-section curve
increases sharply above this energy, we postulate that, above 4.2 eV, reac-
«-4».* n 4« .-U« «.«.•!.. «-I 1 —.- 1 JJ •-- *.!_ - J..-m.J _C /\ biuu j xo tue pnuLipai piüccoo icauxilg cu cue |ilUUULHVtl Ul \J •
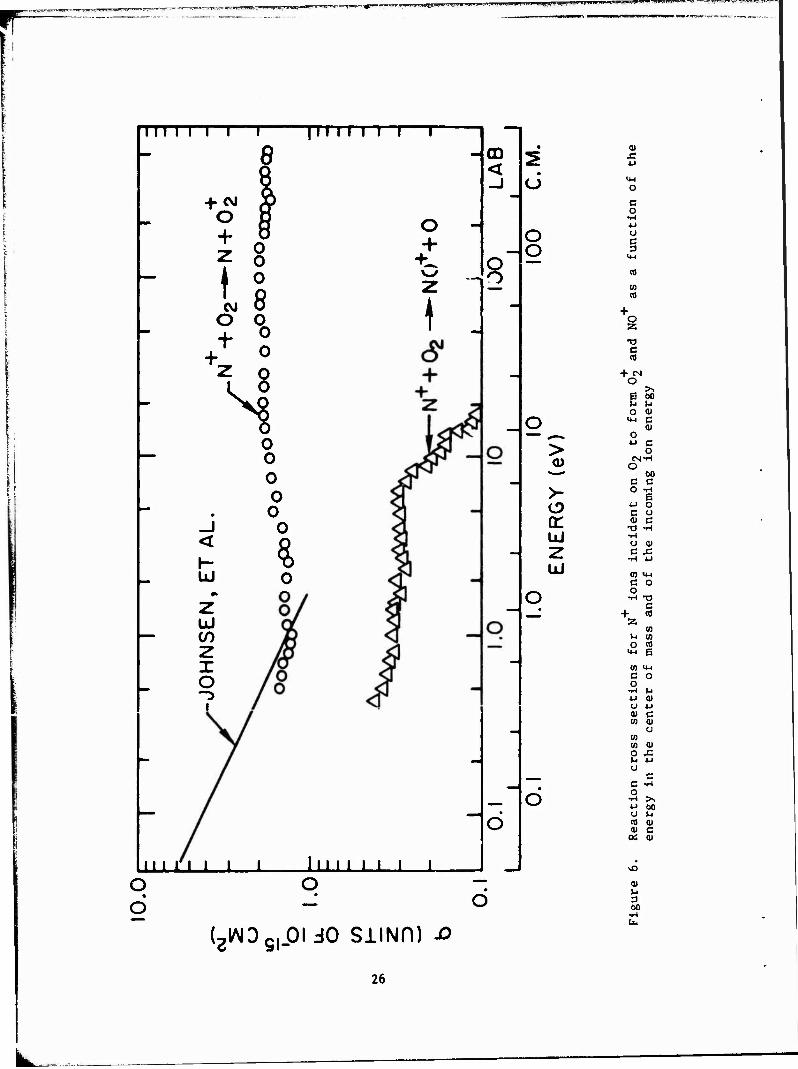
Cross-section curves for reactions 10 and 11 are given in Figure 6;
tabular data for these reactions are given in Tables 5 and 6, respectively.
For both these reactions, measured values were obtained at lower interac- (3)
tion energies than previously reported. As with reaction 9, these
cross sections are not affected by the introduction of excited states in
the 0„ beams. The deposition of the excess energy in these reactions is also +
of interest. For reaction 10, It Is energetically possible to form NO in an
excited state:
23
j^,*.'iwais.>t-^-ji,Mi'*-vitK|
u V c <u
<u
o c o
u c 3
en id
B u o
O 00 u
c <U o c
01
O 00 c c •H -H
E (fl O c o o c
+ <D Z £
4-1 M 0 <*-!
IM O
C TJ o c 4-1 CJ w ai (n a nj
G ui 01 U-l O O t-> u u
4>
O C
*J u u m ai
OS 4J
(2W0z|_0l dO SllNfl)-0 a» u 3
24

''^füMM'W* IW»Ä W+ "^^^si-jiuiipmunmiii ■.inL»i«i.m»,te^^-r-»«j i ii.iui.1 >«ui*rf'3T"*3mn!«wn*v*>w'9*9«!nHK^*«n*F
TABLE 4
MEASURED CROSS SECTIONS FOR THE REACTION N+ + 0_ + 0+ + PRODUCTS
(Center of Mass and Laboratory Er.ergies are Given)
CM ENERGY CROSS SECTION LAB ENERGY
1.39 3.3P247C-17 2.CO
2.09 4.432452-17 3.00 2.78 5.173351-17 4.00
3.48 5.041545-17 5.30 4.17 5.352657-17 6.GO 4.37 6.757513-17 7.CO 5.57 3.055573-17 3.GO 6.26 1.C51696-16 3.00 6.96 1.216523-16 1C.00 3.35 1.137623-16 12.CO 9.74 1.239593-16 14.00
ID.43 1.222CCG-1E 15.00 11.13 1.22C414-16 16.00 12.52 1.243658-16 18.30
13.91 1.228434-16 20.00 15.30 1.114733-16 22.00 17.39 1.116015-16 25.00 20.87 1.1C3333-16 30.00 27.S3 9.582357-17 46.CO 34.78 7.447742-17 50.CO
1 Repro^ii | best ava
roJ I Zm>K --*- 'min /äpn^ Nable copy. fj^jP
25
viim.w^J
in i i i i—r |ii 11 i i i—r as <
o +
-.8
u
O O
I
>
>- o UJ z UJ
O
111111 I I III I I II I I O o
(2WDg|0ld0 S1INPI) -O
26
C o 1-1 ■u u §
o z
§
8 60
o (1) c (1) o
•u ß o
CN-H o c c O -H
B •u o C U <U c
T3 i-l
U 01 n 4=
CO «4-1 C O o
c
10 CO ct) E
C0 MH c o o
u u 01 c (0 01
u CO CO 0) o x:
C -H o u oo U )-i R) 01 2 c as 01
01 u 3 00
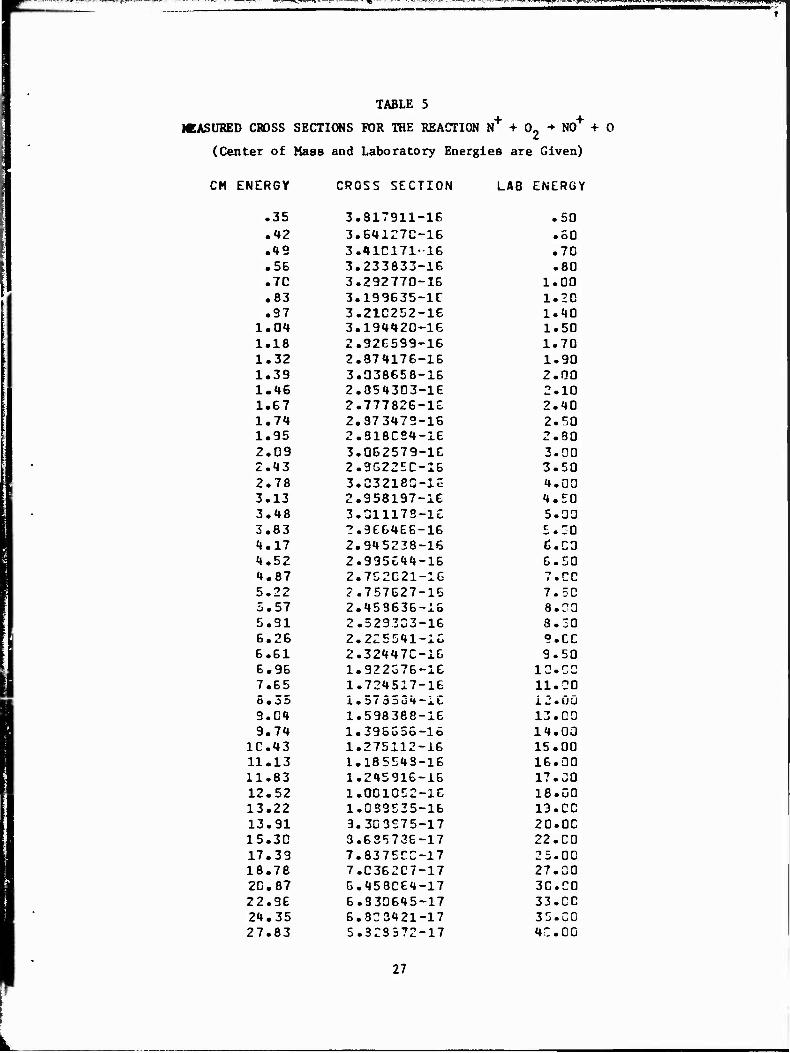
TABLE 5
MEASURED CROSS SECTIONS FOR THE REACTION N+ + 0, N0+ + 0
(Center of Mass and Laboratory Energies are Given)
CM ENERGY CROSS SECTION LAB ENERGY
.35 3.817911-16 .50
.42 3.641Z7C-16 .60
.«»9 3.410171-16 .70
.56 3.233833-16 .80
.7C 3.292770-16 1.00
.83 3.199635-1C 1.20
.97 3.21C252-16 1.40 1.04 3.194420-16 1.50 1.18 2.926599-16 1.70 1.32 2.874176-16 1.90 1.39 3.038658-16 2.00 1.46 2.854303-1E 2.10 1.67 2.777826-16 2.40 1.7«» 2.373472-16 2.50 1.95 2.318C24-16 2.30 2.09 3.062579-16 3.00 2.43 2.9622EC-16 3.50 2.78 3.032180-16 4.00 3.13 2.958197-16 4.50 3.48 3.311173-16 5.00 3.83 2.966466-16 5.30 4.17 2.945238-16 C.C3 4.52 2.995644-16 6.50 4.87 2.7C2C21-16 7.CO 5.22 2.757627-16 7.50 5.57 2.453636-15 8.00 5.91 2.529333-16 8.50 6.26 2.225541-1C 9.CO 6.61 2.32447C-16 9.50 6.96 1.922376-16 10.00 7.65 1.724517-16 11.00 5.35 I. 573504-iC 12.00 9.C4 1.598388-16 13.00 9.74 1.395656-16 14.00
1C.43 1.275112-16 15.00 11.13 1.185543-16 16.00 11.83 1.245916-16 17.00 12.52 1.001052-16 18.00 13.22 1. 059535-16 13. CO 13.91 3.303S75-17 20.00 15.30 3.635736-17 22.00 17.39 7.8375C0-17 25.00 18.78 7.C362C7-17 27.00 2D. 87 6.458C64-17 30.00 22.96 6.330645-17 33.00 24.35 6.323421-17 35.00 27.83 5.323572-17 4C.00
27
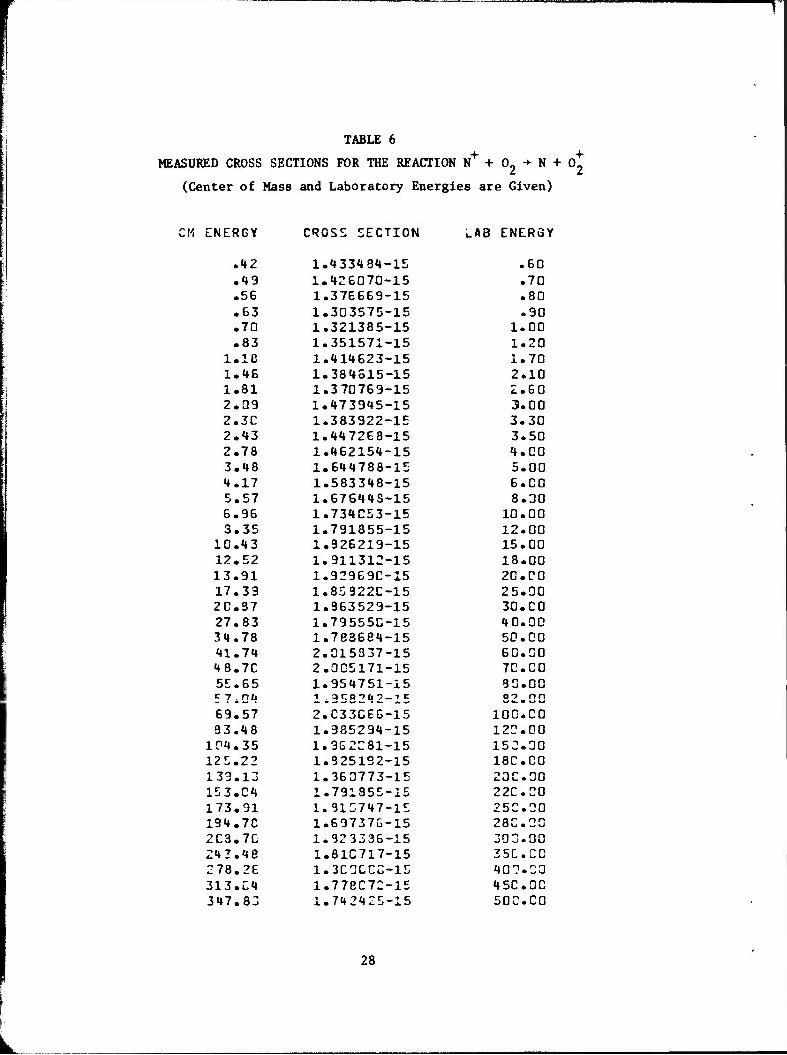
TABLE 6
MEASURED CROSS SECTIONS FOR THE REACTION N+ + 02 ■* N + 0*
(Center of Mass and Laboratory Energies are Given)
CM ENERGY CROSS SECTION i_AB ENERGY
.42 1.433484-15 .60
.49 1.426070-15 .70
.56 1.376669-15 .80
.63 1.303575-15 .90
.70 1.321385-15 1.00
.83 1.351571-15 1.20 l.ie 1.414623-15 1.70 1.46 1.384615-15 2.10 1.81 1.370769-15 2.60 2.09 1.473945-15 3.00 2.30 1.383922-15 3.30 2.43 1.447268-15 3.50 2.78 1.462154-15 4.CO 3.48 1.644788-15 5.00 4.17 1.583348-15 6.CO 5.57 1.67644S-15 8.00 6.96 1.734053-15 10.00 3.35 1.791855-15 12.00
10.43 1.826219-15 15.00 12.52 1.911312-15 18.00 13.91 1.92969C-15 20.CO 17.33 1.85322C-15 25.00 20.37 1.363529-15 30.CO 27.83 1.795550-15 40.00 34.78 1.783684-15 50.00 41.74 2.015337-15 60.00 48. 7C 2.005171-15 7C.C0 55.65 1.954751-15 90.00 5 7i04 1 * 9E82IJ 2-1S 32.00 69.57 2.C33C66-15 100.CO 93.48 1.985294-15 122.00
104.35 1.3G2C81-15 152.30 125.22 1.925192-15 18C.C0 133.13 1.360773-15 20C.00 153.C4 1.791855-15 22C.0O 173.91 1.315747-15 250.00 194.7C 1.697376-15 23C.CC 2C3.7C 1.323336-15 303.00 241. 4e 1.81C717-15 35C.CC 278.26 1.3C0CCC-15 40?.CO 313.C4 1.778C72-1E 45C.0C 347.83 1.742425-15 50C.C0
28

I ..HflMV^luai.JII
N+(3P ) + O.(xV) -v NO+(a3E+) , + 0(3P ) O / g V"l o
This process is nearly energy-resonant and may well be the favored path
since it does not require the 6-eV exothermicity of the reaction path lead-
ing to ground-state products to be transformed into kinetic energy.
The charge-transfer reaction 11, exhibits a large cross section
over the entire energy range studied. Previous publications ' show
that such behavior in charge-transfer processes is indicative of a near-
resonant reaction path. For reaction 11, near-resonance can be obtained
by a mechanism such as
N+(3P ) + O.(xV) ■* N(2D) + ot(X2n ) + 0.09 eV ° 2 g 2 g
Figure 6 also includes the recent data of Johnson, Brown, and (16)
Biondi, who have studied reactions 10 and 11 using drift-tube mass
spectrometer techniques. They obtained a total rate constant for both re- -10 3 actions of 5 x 10 cm /sec over the entire range from thermal to 1 eV.
Comparison of the sum of our two reaction cross sections with the cross sec-
tion obtained from their rate coefficients in the range of overlap shows
good agreement.
3.A CHARGE TRANSFER F10M N* TO 0.
In earlier studies of the charge-tränster reaction
N2 + °2 "" N2 + °2 (12)
in our laboratory,v the effects of vibrational excitation of the 0 on
the reaction cross section was not examined.
(13) In a previous report, we described a reinvestigation of this reac-
tion in which special attention was given to the change in cross section as
a function of the state of vibrational excitation of the N_ primary ion.
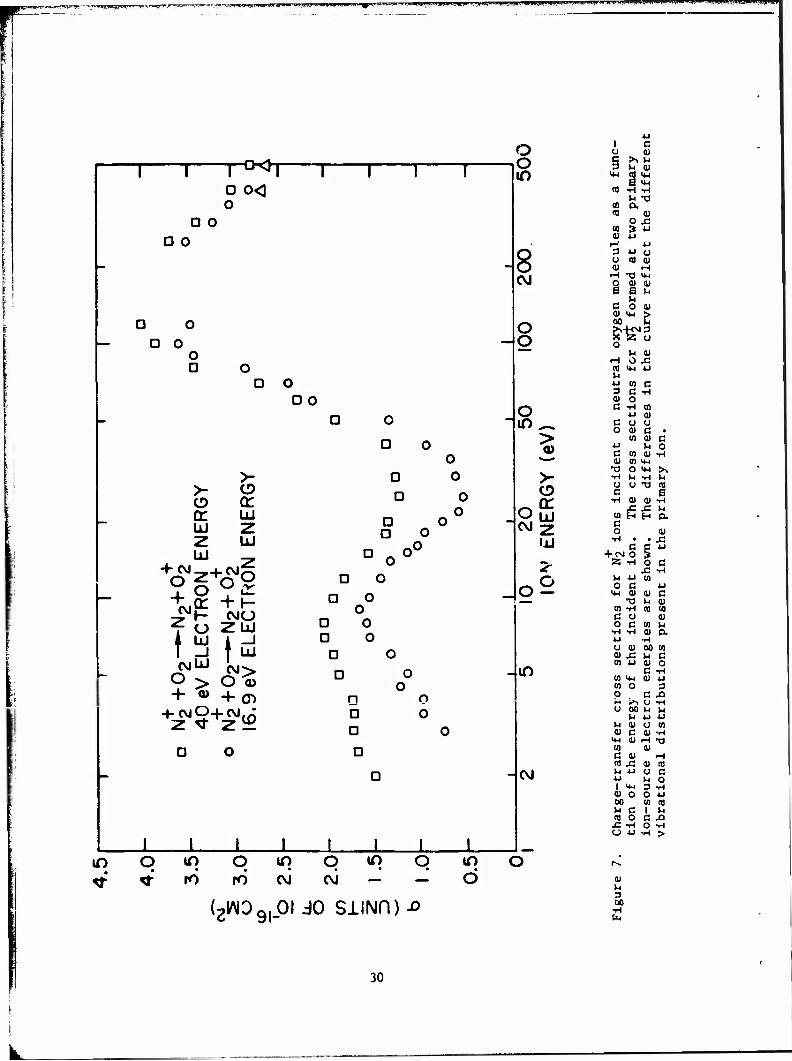
The results of this study are given in Figure 7. Two cases are represented:
29
WMUJ ajl Wm "J^ffTWU V^J^'iPUH'iyT1 ■WHMffgfBWBywrywgW WgpgBPff M p—^Bg^w HiWpWHywyfTBgPWffWIWWWPiepi'WPff'BF
D 0< O
D O D O
D O D O
O D
D O
>- a >- o a o oc «r Lü a
a o LÜ Z Z LÜ ~o LÜ -^
O Z O °
-f Ü ZLÜ
D
o D 0
o D O
0
A LÜ i -1 T -1 t LÜ WLÜ \
o > o «
□ Ü
a o D 0
o + • + <x> n n
"z" §+z^ D a
0
o o o
_ o
O O
CJ
o o
o >
>-
o£ cvJz
o2
m
C\J
in O in O if) O in O if) • <r ro ro C\J CJ — — o
(2IAIO 9|0I 30 SlINfD-O
1 C a CU
§ fr M <D
Z S 4-1 <4-l
cd -H •H M -o
a) a cd a)
o J= CO $ 4J (U 4J
i-f 4J 3 4J CJ cj a) ai <u rH
rH T3 <4-l
o <u cu 0 6 n
M e o V at 4-i 00 & 5rt? 3
o M OJ
rH O £ cd IM 4J M 4-1 CO c n c •H a) o ß -H CO
4J CJ c o CJ O CD c •
CO cu c u u o C CO CU iH a) co >M
•a o «4-1 >. •H u ■H h CJ u T3 « C 6
•H CU (U -H Ä £ U
CO H H a C o CU
1-1 . • sz a C 4J + CN O Z -H o c
x: -H M 4J CO o c 4J
<u CD CU c T3 u cu
CO -H CO CO C O HI o q CO u
•H iH 0) o. 4J ■H CJ <u OO CO CU ,C H c CO 4-1 cu o
C -H CO M-l CU 4-1 CO o 3 o c x> >-> ^ U -H CJ oo M l-l
M 4J 4J M CU CJ CO <u c CU -H
4H CD rH XI CO CU C 01 ■H CO JZ cu ro U 4-> CJ C u u o
1 >*-( 3 -H cu o O 4-1 oo co ca u c 1 u ctt O c x>
JZ -H O -rl U 4J •H >
r-
0) IJ
3 00
30

i'!*'Aiww*!>H!J*'^wi«!yüyu~- -"^ ■
(1) very low electron energies (16.9 eV) in the primary ion source, and
(2) higher electron energies (40 eV). A brief discussion of these results
is given to assist in interpreting the newer result in which the 0„ is +
vibrationally excited. Comparison of the two N_ curves in Figure 7 reveals
that the results obtained for high ion energies (above 50 eV) are similar.
Below 50 eV, however, the cross section for N, ions with very little vibra-
tional excitation (those ions forced by 16.9-eV electrons) is less than
that for ions with appreciable vibrational energy. Franck-Condon calcula- + 2
tions indicated that, below the onset of the L(A H ) state (M.6.9 eV>, all 2 u
direct ionization of N. produces ions in the zeroth and first vibrational
levels of the ionic ground state.
The probable reason for the difference in the two curves shown in
Figure 7 is that, if sufficient energy is available internally in the N„ + 4
ion, formation of the 0„(a n ) state is possible. This process, which is
nearly resonant, can be represented as
N2(X2C=3,4> + °2(X\v=0> - V^g.v^ + °2(<\> (13)
The importance of the vibrational excitation of the N_ in the charge
transfer is further demonstrated by examining the primary ion-source elec-
tron-energy dependence of Lhe cross section for 10-eV N~ ions on 0~. This
dependence (Figure 8) indicates that the cross section for the process
changes rapidly for electron energies between 16 and 19 eV. At these en-
ergies, higher vibrational levels of the ground ionic state are probably
being filled by cascade from the L(A H ) state.
The results of the experiments in which vibrational energy was added
to the neutral 0„ also showed an increase in cross section. Figure 9 is a
plot of the data for cold and hot 0?. Tabular data are given in Tables 7
and 8, respectively. Note that, for both reactions, the vibrational exci-
tation of the N„ was kept low (v = 0,1) by the use of 16.9-eV electrons to
form the ion. The effect in the present case, therefore, is that due to
vibrationally excited 0. alone. For these experiment, the 0_ gas was heated
31

. tngwMwnjs "W iwkjwm w) - a 1X41J w^wpppf^^ta
1 1 l l 1
—
+ CVI 0 >
0
0
—
+ CVI
g 0 z >
t ° I Q: CVI UJ
0 z + u
0
0 + CV1 z
z 2 0
0 0 0 0 0 0 0 0
— 0
—
— 8o 0 —
1 1 1 1 1 o 10
cvj
CVJ
O ro
00 cvj
CO cvj ^
CVI ££ UJ
CVJ CVJ
o CVJ
CD
Ld
O
\- o UJ _J UJ
- CD
o cvi
in 10 Ö
o> u w 3 O to 1 c o
■H
01
e o a 3
+CM O
CM z
CM o
+ CM> z 01
u o o .-1 u-t
c o o •H >, U 50 U M a» v at 00 a> o M o
(2W39|_0ld0 SlINfD-O
0> Jd «H 00 CD C CO -O U CO *J J= I OI+CM ooz M
5.8 <J H
x: • « >.
00
0 0) G
01 01 o c c 01 o
TJ U C *J a> a a. a* at iH a 01
01 M 3 00
32

i. ■ ■ m. p ii" * •«.» >j*, i a-mtar&rr-w P.i'pyuiJMM^W'P WI«WL".?J(
8-
^-^ 6 C\J ^ O
CD b 'o
O 4 (J) h- Z 3 3
b 2
0
N2 +02^N2 + 02
a 30C°K 02
o I600°K 02
.00
cfn
O _Oo O D
o° o oo ° o° ° O o.° .
ICRD. 'a
ÜD^ ^ D
J L
%n°D
o o
a
2 5 10 20 50 100 200 500 ION ENERGY (eV)
Figure 9. Charge-transfer cross sections for N2 ions incident on neutral oxygen molecules as a function of the energy of the incident ion. The lower set of data represents the cross section for neutral O2 with no vibrational excitation, while the upper curve gives the cross section when vibrational excitation is present in the 0_.
33

B i ■!!■ ..«HIT'I ■tf"1-"*?*"^-*-
TABLE 7
HEAS-4ED CROSS SECTIONS FOR THE REACTION»J + Oj * »2 + 0* WHEN THE 02 IS NOT VIBRATIONALLY EXCITED
(Center of of Mass and Laboratory Energies are Given)
CM ENERGY
1.6C 1.87 2.13 2.1C 2.57 3.20 3.73 f.27 1.3C 5.33 6.1C 7.«»7 8.DC 9.53 9.SC
ir .£7 11.73 13.33 1E.CC 13.67 21.33 26.67
37.33 12. £7 13.02 53.33 S1.CC
133.33 16f.CC 136.b7 213.33 2e£.£7
CROSS SECTION
7.331371-17 9.338161-17 3.S19CC1-17 1.216237-16 1.143737-16 1.313755-16 1.531533-16 1.573371-16 1.612693-16 1.533311-i6 1.115511-16 1.313301-16 1.126787-16 1.035351-16 3.1371EC-17 7.113133-17 5.91:7362-17 5.521333-17 6.0C3633-17 6.22353C-17 7.327166-17 1.323355-16 2.135121-16 2.153139-16 2.375133-16 3.125131-16 3.6C1567-1P 3.153C55-16 3.523S72-io 3.2CG+C3-1& j.053121-16 2.3C2CCG-16 2.569C73-16
LAB ENERGY
3.00 3.50 1.00 1.50 5.CO 6.0C 7.GO 3.00 9.00 10.00 12.00 l«f.0Q 15.00 16.00 18.00 20.00 22.CO 25.00 30.CU 35.CO 10.00 50.00 6C.00 70.00 30.00 90.00 100.OC 120.00 250.00 300.30 350.00 ion.00 50C.CL
Reproduced from best available copy.
34

TABLE 8
MEASURED CROSS SECTIONS FOR THE REACTION N2 + 02 WHEN THE 02 IS VIBPATIONALLY EXCITED
(Center of Mass and Laboratory Energies are Given)
N2 + ol
ENERGY CROSS SECTION LAB ENERGY
1.C7 2.761325-16 2.0C 1.33 2.175822-16 2.50 1.6D 2.6G1526-16 3.00 2.13 2.754557-16 4.00 2.67 2.9G7248-16 5.CO 3.2C 2.710317-16 6.00 3.73 2.919115-16 7.00 «♦.27 2.383629-16 8.00 4.8C 2.569162-16 9.CO 5.33 2.270422-16 10.00 6.40 2.497465-16 12.00 3.CC 2.341373-16 15.00 9.6C 2.66C651-16 IS.CO 10.67 1.962456-16 20.CO 13.33 2.322023-16 25.00 1G.0C 2.373624-16 30.00 21.33 2.919115-16 4C.C0 2C.67 4.032987-16 5C.00 32.CC 4.257C42-16 60.00 37.33 4.651597-16 73.00 12.67 6.019260-16 8C .00 48. CC 7.189671-16 ?U.CC J J • w *J 6.203187-16 103.00 64 • uC 1•U3-o/j-lb J. Z L. • 0 Li 8C.CC 3.032155-16 150.00 133.33 6.634351-16 250.30 119.33 6.930063-16 28G.00 lCutuC 5.321303-16 300.00 186.67 5.757113-16 350.00 213.33 1.653CrC-i6 400.00 266.67 '>.697426-16 500.00
Reproduced Iron best available copy. 9
35

. ...■...,» IIU«W?H»W' ijirmiHwmmi.tijw.MVM'Wlf' '•■'■ WHJH.IJW-I-^JIIH»',1
to approximately 1600 K in an Iridium furnace (no dissociation of the 0„
occurs at this temperature).
Examination of Figure 9 shows that there is an increase in the charge-
transfer cross section for vibrationally excited 0_ over the total energy
range studied. The reason for the change is probably identical to that for
adding vibraticnal energy to the N_ ions. The heated 0„ gas has some parti-
cles in high enough vibrational levels that the reaction becomes near- + 4
resonant for formation of the 0- in the excited (a n ) state. This reaction
can be written as
<(x2zg,v=0,l> + °2(x3zLv=0,l,2> * N2(XX> + °2<A.> <14>
Vibrational excitation of the 0„ may be more effective than similar excita-
tion in N- (compare Figures 7 and 9) because the 0. excitation not only de-
creases the energy defect of the reaction, but also improves the Franck-
Condon overlap between the ground-neutral and excited-ion states.
3.5 CHARGE TRANSFER BETWEEN H+ AND 0
The charge-transfer reaction
H+ + 0 ■*• H + 0+ (15)
was studied to evaluate the operation of our 0 atom sources and for use in
determining the degree of dissociation present in the 0 uc<ui arising from
either rf or thermal dissociation. Reaction 15 is particularly suited
for these tasks because it is accidentally resonant, making it easy to
study. Furthermore, cross sections for this reaction at both thermal (18)
and high energies have been derived from earlier studies. These
earlier measurements allow comparison of our results with others to check
our degree of dissociation.
Care m>ist be taken when using reaction 15 for proving the 0 atom beam
since the competing endothermic reactions
H+ + 0 -+ H + 0 + 0+ (16)
36

r»wn,n;mi!"« naanm* «".«P. i!|PPWP
and
H+ + 02 -► OH + 0+ (17)
can appreciably affect the measured cross-section curve. The first of these
competing reactions can only occur with the absorption of 5.1 eV of kinetic
energy from the system, while the second requires only 0.7 eV.
Our investigations using the 0 atom beam produced by thermal dissocia-
tion of 0- show that the cross section for reaction 15 is larger than
that of reaction 16, and that any effects from this reaction can easily
be subtracted from the overall curve measured for production of 0 .
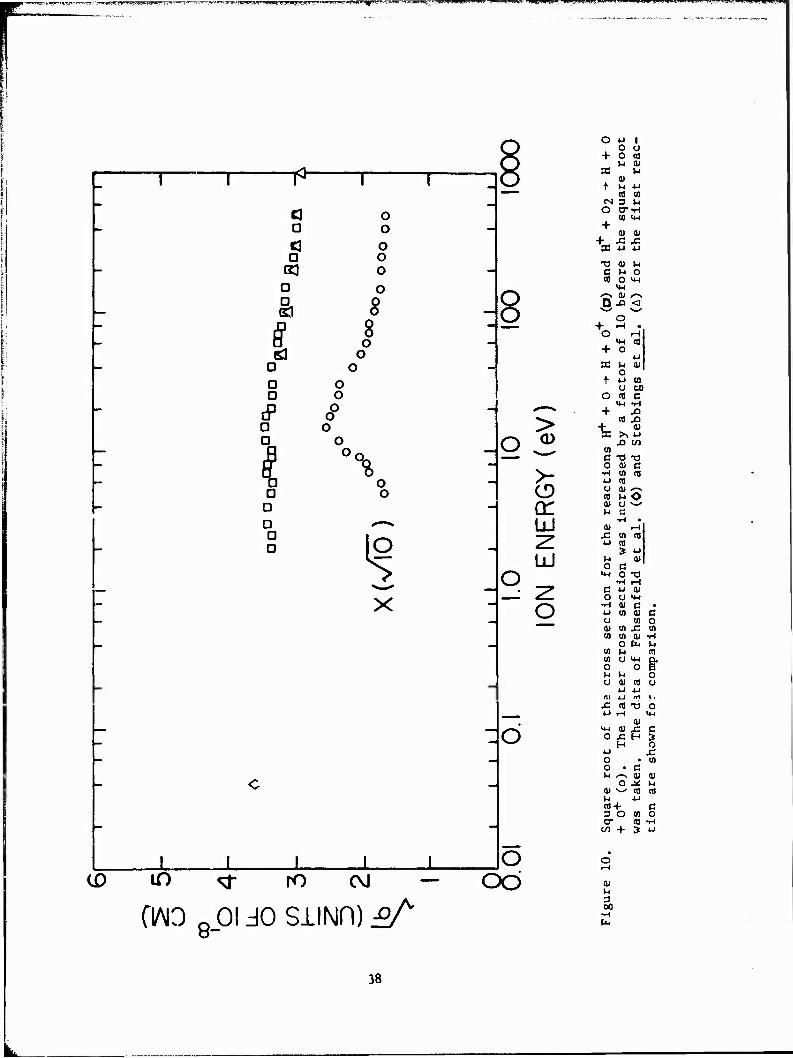
Figure 10 is a plot of the cross sections for reactions 15 and 16.
Reaction 17 was found to be insignificant. All the data shown in
Figure 10 were obtained using the thermal dissociation method. The
degree of dissociation was approximately 20%. Tabular data for reaction
15 are given in Table 9.
Using the rf discharge technique met with less success. Both 0„ and
C0_ were used in attempts to produce 0 atoms. Although higher beam densi-
ties were obtained using this technique, interpretation of the data proved
difficult perhaps because of the presence of metastable species in the beam.
Our success in measuring cross sections for the reaction of Equation 15
using 0 atoms produced by thermal dissociation has allowed us to attempt
measurements of other reactions involving 0 atoms. The following subsection
gives a report on one of these processes.
J.6 REACTION OF N* WITH 0 TO GIVE N0+
The ion-molecule reaction
N* + 0 -> N0+ + N (18)
has been studied in the energy range from 1 to 4 eV in the laboratory system.
Two difficulties were encountered during the measurements. The first was the
low density of 0 atoms in the beam coming from the Iridium furr^e. The
37
F tPTUB «?»v * fim ^oqn '■'^>»*?«ww"^^F,-
: 1 1 .*- I i
0 o a o Ö 0 D o
B3 o a 0
g 8 : ff
8 o
ö o D o D o a o
tf o° a o a o
- 1 °% - a o a o o -
1 U a a 1 ;
—
X
c
—
I I 1 1 1 CD lO ro CO
> o *>
>
cr LÜ
o ÜJ
o
:o
o oo
0AI3 8 01 dO SlINfl) £A
O «J o + o u
01 t U 4-1
to to CM 3 u O tr-H
(0 H-l + V Q)
+ A JZ 33 u 4J
•a a) n C M O a) o <4-i
IM
D X> < o
+ -i • o <M
+ O EC
+ to
c + XI
(0 XI
X> C/> CO C T3 T3 O <D C
•H CO (0 ■u cfl
4) /-N M O o *^ a
o 01 u ai x: CO CO 4-i to
3 4-1 u <u O ß
>4-i o *a
4J CD U «4-1
TJ ai c 4J co cu u co tu to jz co co at
O En
U <4-l o u
cu CO
c o
to co o >-l CJ
(II 4-1 ffl I,
X (0 T) o 4-1 t-H U-l
'S US g H O
4J X O »CO o • c hA|| tl
O -* H 0) w «J <0 »J 4-1 t0+ c 3 O CO O cr to -H w + J u
cu u p 00
38
■fr°7w> W'W-1 mm <,M*i?j*-iy"*,*n
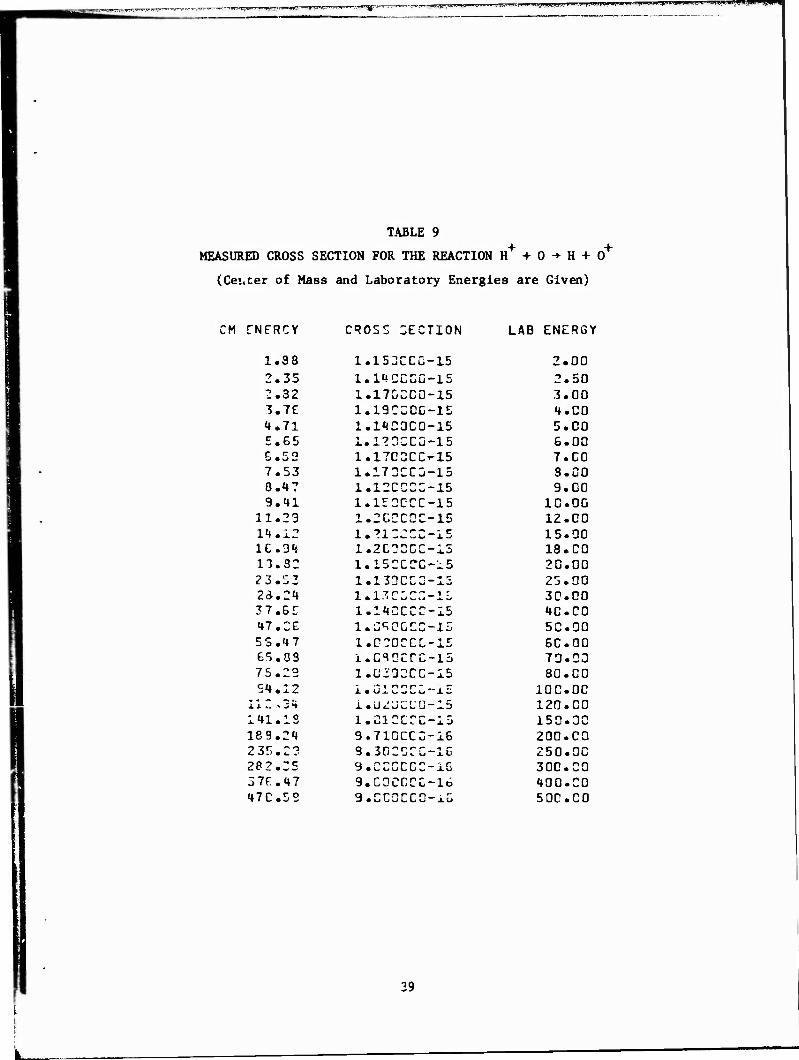
TABLE 9
MEASURED CROSS SECTION FOR THE REACTION H+ + 0 -»■ H + 0+
(Cei.cer of Mass and Laboratory Energies are Given)
CM 5NFRCY CROSS SECTION LAB ENERGY
1.38 1.153CCG-15 2.00 2.35 l.HCCCG-15 2.50 2.32 1.17CCC0-15 3.00 3.7E 1.19CC0G-15 1.C0 it.71 1.11C0C0-15 5.00 5.65 1.120CCO-15 6.00 G.53 1.17C3CC'-15 7.CO 7.53 1.173CC3-15 8.CO 8.1? 1.12CCC2-15 9.00 9.11 1.152CCC-15 10.00
11.23 1.2CCCCC-15 12.00 11.12 l.?1225C-15 15.00 ie.3t 1.2C0CCO15 18. CO 13.32 1.152CCC-15 20.00 23.ZZ X • 1JJC^W"XJ 25.00 23.21 1.13CLC2.-1L 30.00 37. 65 1.11CCCC-15 10.CO 17.26 l.S^OCCC-io 5C.00 55.17 1.C2Q2CC-15 6C.00 E5.0S i.c^ccrc-15 70.03 75.23 1.C33CCC-I5 80.00 SI.12 i.Ü12SCC-x5 IOC.00
11Z > 21 1.U^JL;L'C-15 120.00 111.13 1.C1CCCC-15 150.30 189.21 9.71GCC2-16 200.CO 235.2? 9.302CTC-16 250.OC 282.35 3 .Cui_iCCC~lG 300.CO 57F.17 9.CGCCCC-16 100.00 17C.52 9.CC3CCC-i6 50C.C0
39

second difficulty was noise associated with having the mass of the primary
ion beam (28 amu) close to that of secondary ions (30 amu). The resolution
of the secondary mass spectrometer employed for the study was sufficient to
completely separate the mass 28 peak from the mass 30 peak; nevertheless, a
certain amount of dc noise resulting from scattering of the primary mass 28
peak appeared at mass 30. (Note that the magnitude of the product 30 peak
is several orders of magnitude less than that of the primary beam at mass 28.)
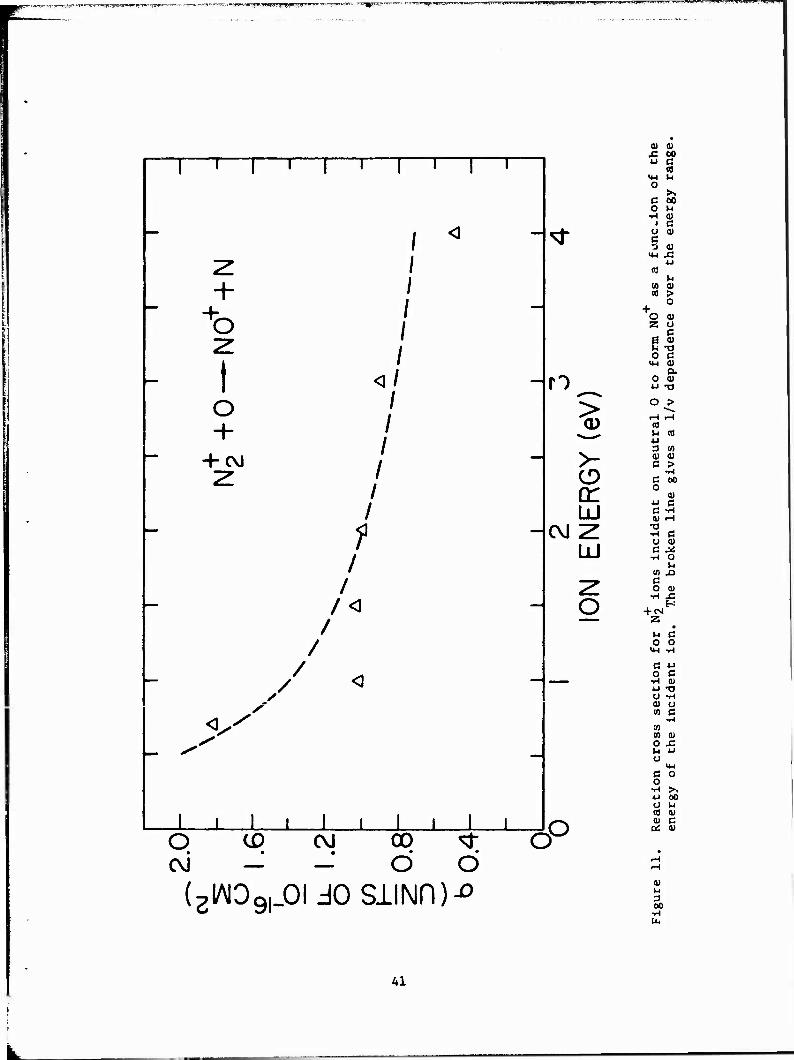
Figure 11 gives the results obtained for the reaction of Equation 18.
The results are compared to a 1/v dependence, where v is the velocity of the
center of mass. The fit to a 1/v dependence is fair, indicating that the (9)
reaction may proceed by an ion-dipole mechanism.
The cross section for the reaction of Equation 18 is smaller than might
be expected for an exothermic ion-molecule reaction. This small cross sec-
tion may indicate that the products of the reaction cannot easily absorb the
excess energy in the reaction (^3.0 eV).
Previous measurements of a thermal energy coefficient for the reaction
of Equation 18 were performed. Ferguson et al. obtained k = 2.5 x 10
cm /sec at 300 K. Extension of our measured data using the 1/v fit shown in
Figure 11 results in a rate coefficient of about 1/6 of that reported by
Ferguson et al. at the same energy.
Work on reactions involving 0 atoms is continuing.
40
1 I 1 i 1 i 1 ' 1 1
— /
< —
2 /
+ / ■
-+>v / "■
o / < 2
- t /
o 1 1
1 1
1 1
+ + 00 —
z:
f 1
1
/
/<3 /
/
• <
y
</ ^
_ ^ ■■■
1 l 1 I 1 1 1 1 1 1
-<t
-n
-C\J
>-
Q: UJ
UJ
- o
O CD 0J CO Sf • • • » «
CM — — O O
(2IAI39h0ld0SllNn)-°
o Q
V <u X 00
cd
o
c oo o u •H V
' c O CU
§ <u w
« u
« > *■ ° O tt)
53 O „ c a <u o c
p. o <u *J -a o >
(0 M A 4J 3 CO <u <u c >
•H c oo o
u c C -H (U iH
•a •H C o a) c .* •H O
CO JD a o cu •H .C
z • o o
C 4J o c
•H <J> ■M T3 a -H a) o ca e
•H (0 to cu o £.
u IM
n o o •rl >, i-i 00 O M to cu
OS
u 3 00
41

_ - _ ^ -:■ ■;^..I^-,.BH,<-»«, mar*m*mm*mmimmn!l^^^^^ n.jimmm.^.m.'J'J-."- «W».HJ1.»>-UPIIJWW«
REFERENCES
1. M. H. Bortner, Coordinator, DNA Reaction Rate Handbook, DASA-1948, General Electric Company Missile and Space Division, Philadelphia, Pennsylvania, 1967.
2. J. A. Rutherford, R. F. Mathis, B. F. Turner, and P. A. Vroom, "For- mation of Magnesium Ions by Charge Transfer," J. Chem. Phys. 55, 3785 (1971).
3. R. F. Stebbings, B. R. Turner, and J. A. Rutherford, "Low-Energy Collisions Between Some Atmospheric Ions and Neutral Particles," J. Geophys. Res. 71, 771 (1966).
4. B. R. Turner, J. A. Rutherford, and R. F. Stebbings, "Charge Transfer Reactions of Nitric Oxide and Atomic and Molecular Ions of Oxygen and Nitrogen," J. Geophys. Res. 71, 4521 (1966).
5. F. A. Wolf and B. R. Turner, "Energy Dependence of the Charge-Trans- fer Reaction in Thermal and Low-Electron-Volt Region," J. Chem. Phys. 48, 4266 (1968).
6. C. F. Giese, "Strong Focusing Ion Source for Mass Spectrometers," Rev. Sei. Instr. 30, 260 (1959).
7. M. Knudsen, "Kinetic Theory of Gases," Methuen's Monographis on Physi- cal Subjects, 3rd ed., Methuen '• Co. Ltd., London, 1950.
8. D. Rapp and W. E. Francis, "Charge Exchange Between Gaseous Ions and Atoms," J. Chem. Phys. 37, 2631 (1962).
9. G. Gioumousis and D. P. Stevenson, "Reactions of Gaseous Molecule Ions with Gaseous Molecules; V. Theory," J. Chem, Phys. 29, 294 (1958).
10. A. L. Schmeltekopf, E. E. Ferguson, and F. C. Fehsenfeld, "Afterglow Studies of the Reactions He+, He(2^S), and 0"1" with Vibrationally Ex- cited N2," J. Chem. Phys. 48, 2966 (1968).
11. R. H. Neynaber, private communication of results obtained using merg- ing beams technique under an ARPA/ONR contract.
12. T. F. O'Malley, "A Simple Model for the High Energy Reaction of 0+
Ions with N2," J. Chem. Phys. 52, 3269 (1970).
Preceding page blank
43

^^yl^^^W>^><^?TF^>•*^;,T**J^■'*^'V^'^'-^J■■^.'^■'!''* -H«""1»"--'." ' - >*•"■ '» ^'»■■■^"»■"■' "Tm.'iyfflS^iy'*
13. R. H. Neynaber, J. A. Rutherford, and D. A. Vroom, "Electronic and Ionic Reactions in Atmospheric Gases," Yearly Technical Summary Re- port, September 1970 through June 1971, Gulf Radiation Technology Report GULF-RT-A10767, July 1971.
14. W. B. Maier II and E. Murad, "Study of Collisions Between Low Energy N"1" and N£: Reaction Cross Sections, Isotopic Compositions, and Kinetic Energies of the Products," J. Chem. Phys. 55, 2308 (1971).
15. J. A. Rutherford, R. F. Mathis, B. R. Turner, and J). A. Vrooii, "For- mation of Sodium Ions by Charge Transfer," J. Chem. Phys. 56, 4654 (1972).
16. K. Johnson, H. L. Brown, and M. A. Biondi, "Ion-Molecule Reactions Involving N£, IT1", 0$, and 0+ Ions from 300°K to -vL eV," J. Chem. Phys. 52, 5080 (1970).
17. F. C. Fehsenfeld and E. E. Ferguson, "Thermal Energy Reaction Rate Constants for H+ and C0+ with 0 and NO," J. Chem. Phys. 56, 3066 (1972).
18. R. F. Stebbings, A. C. H. Smith, and H. Ehrhardt, "Charge Transfer Between Oxygen Atoms and 0+ and H+ Ions," J. Geophys. Res. 49, 2349 (1964).
19. E. E. Ferguson, F. C. Fehsenfeld, P. D. Goldan, A. L. Schmeltekopf, and H. I. Schiff, "Laboratory Measurement of the Rate of the Reaction N$ + 0 -*■ N0+ + N at Thermal Energy," Planet. Space Sei. 13, 823 (1965).
44

T^'^^'Tr7rw^?t W.W^.UM»-W^i,tjuijpiirr..X.*WJ»w^^v:»'S^"-M'..-?J *«■»;.' vfijji!!ii..i.^■ywj-».mJ"%'»y." wywpPCTWPy^w^aw^WM*»11 ■tT-^V^JWBt-IWi-.mm mm».m-Wm-M>-i* ^■\A'PJf*flP*J»?W^iii,.iJJip.J^«iMjPP!i*.IMP
APPENDIX A
FORMATION OF MAGNESIUM IONS BY CHARGE TRANSFER
45

i.i H*QNg!9*<gV^BDi >.Ti~\*- "'* ■I'-Ai ■', Ji. I ■ '«^"M ■»•■>ja'"ii«.J*il.Lilil^MM
Reprinted from:
THE J II l! R \ A I OK (II KM I (A I. PHYSICS VOLUME Si, NUMHKK S li OCTOBER I 0 ; !
Formation of Magnesium Ions by Charge Transfer*
J. A. R(ITIIKRFORI), R. I-. MATHIS, B. R- TURNKR, ANI> D. A. VFOOM
Gulf Radiation Technology, A Division of Gulf Energy and Environmental Systems, Incorporated, San Diego, California 92112
(Received 15 January 1971)
The charge transfer cross sections for several ions in collision with atoms of magnesium to form MR* have been mcasure.1 in the cn.rgy range 1-500 cV. The ions 0», Nf, N,\ NO*, (V, H«04, HJO\ N,OV, and MR* have l>ecn used. The results obtained are analyzed by considering the possibility of resonant or near resonant processes l>cing res|>onsiblc for the large cross sections obtained for most processes. When the possibility of resonance type mechanisms docs not exist, the cross sections arc found to lie con sidcrablv smaller. The effect of mctaslable stales present in the primary ion licam on the charge transfer cross section is etamined. extrapolation of the measured cross sections to thermal energies has also been performed.
I. INTRODUCTION may have a magnitude and energy dependence similar lo those found for the symmetric ease.4 In such cases,
Cnnsidcrahle effort has been devoted to the study of it appears that while other reaction channels are oflen symmetric resonant charge transfer processes in recent possible, the greatest contribution to the total cross years. These studies were, for a large part, prompted section comes from trie channel which results in by the several theories for resonant charge transfer closest energy resonance between the rcactant and such as those of Hates,' of Rapp and Francis,' and of product states. Firsov.1 More recently, it has been shown that the The reactions studied here represent a further test cross section for asymmetric rcsona.nt charge transfer of the above hypothesis since several different rcac-
Preceding page blank 47

!W»pillWl!W.>'J>"WAi^»,lJlJIIWSJl»ld«Ill!llill^ll«IIJI.II)l,l!!|IIJl'\
3786 RUTHERFORD, MATHIS, TURNER, AND VROO.I
RESISTIVELY HEATED FURNACE
METAL BEING VAPORIZED
SWEEP PLATES
PRIMARY ION MASS ANALYZER
FiO. 1. Crossed im and neutral beam apparatus.
ION SOURCE
GAS INLET
lions involving the same neutral species are considered. Furthermore, cases where no resonance and near reso- nance exist are present.
The cross sections obtained also have a practical ap- plication with regard to understanding the ionosphere since magnesium is known from rocket studies*'* to be among the more prevalent metallic ions in the altitude region ranging from 83 to 112 km. Two properties of these ions result in an atmospheric behavior different from that of the more common Oj+ and NO*. Firstly, they are atomic and hence are not removed by dis- sociative recombination; secondly, they have ionization potentials below those of all the common atmospheric constituents, and there are, therefore, no neutral species with a significant number density with which these ions may undergo chagre transfer. The metallic ions formed in the ionosphere are therefore expected to be long lived.
II. EXPERIMENTAL
Although the apparatus has been described pre- viously/-' it was felt that the experiment reported here differs sufficiently from past work to warrant a complete description of the machine and a discussion of the experimental procedure.
A. Primary and Secondary Ion Systems
A schematic of the apparatus is shown in Fig. 1. The primary ions arc extracted from an electron bombardment source and mass analyzed at an energy of 75 cV in a 180° magnetic mass spectrometer. After mass analysis, the ions pass through an aperture in an iron plate that shields the magnetic field of the mass analyzer from the succeeding regions of the apparatus. They are then retarded or accelerated to the desired collision energy. The ions next pass through a field- free region before intersecting with the neutral beam. Collimating apcratures ensure that, from purely geometrical considerations, al! primary ions pass
through the modulated neutral beam (modulated at 100 Hz by mechanical chopping). Secondary ions re- sulting from collisions between the primary ions and neutrals are extracted along the direction of the primary ion beam by an electric field of approxi- mately 2 V/cm. The ions then enter an electric field where their energy is increased to 1650 eV. Penetration of this accelerating field into the interaction region is reduced by use of a double grid structure. After ac- celeration, the ions pass through an electrostatic quadrupole lens1* which forms the entrance slit for the 60° sector magnetic mass spectrometer. The selected ions impinge on the first dynode of a 14-stage CuBc electron multiplier. For magnesium, the most abundant isotope at mass 24 was used when making measure- ments. The cross sections have been corrected for the isotope effect created by collecting only the 78.6% of the magnesium beam which has mass of 24 amu. The output from the multiplier passes successively through a preamplifier, a 100-Hz narrow-band ampli- fier, phase-sensitive detector and is then integrated. The output ib presented on a chart recorder.
The primary ion beam intensity is measured at the interaction region with a F-r^Uay cup vhich can be moved into the collision region when drs/cd. The pri- mary ion energy is determined from retarding po- tential measurements. All surfaces at the interaction region and the Faraday cup arc coated with a collidal graphite in alcohol, and the interaction region is nor- mally maintained at a temperature of 120oC to mini- mize surface charging.
Because interest in the present work extends down to small collision energies, it was necessary to use only weak extraction fields at th'* collision region. As a result, the secondary ions were not collected with 100% efficiency. To obtain absolute cross sections for production of various secondary ions, it was necessary, therefore, to determine their over-all detection effi- ciency. This latter consideration is governed by a
48

MAGNESIUM IONS BY CHARGE TRANSFER J787
number of factors including the multiplier gain n.nd the cfliciency of transmission of the secondary ions from the interaction region l.o the multiplier.
The gain of the multiplicr-amplificr-rccordcr system is measured hy modulating the primary ions prior to their entering the collision region. The ion current signal is fl!·st measured with the movable Faraday cup and then, after traversing the secondary mass spectrometer multiplier amplifier system, by the recorder. Transmission of the primary ion through the second mass spectrometer was observed to be 92%. In Table r the systt~m gain for several species of ion impinging upon the multiplier is shown.
The major experimental uncertainty is associated witl1 the collection efficiency for the secondary ions.
TAnLF. I. System gain for several incident ions.•
I on mass Ion species System gain -- ------ - ---------------------------
I n+ l.OXIO+u 2 H2+ 1.4X w+u 4 He+ 1.4Xt0m
12 c+ I.SXJO+n I •\ N+ 1.8XJ0+U \6 o+ 1.6XJO+n 18 H2o+ 1. 7X JQ+II 23 ~a+ 7. 7X IQ+II 24 .Mg+ 8. 7X 10+12
28 N2+ 1. OX fO+II 28 co+ l.IXIO+u 30 ~o+ !.OX to+u 32 (h+ !.OX!O+!: 39 K+ 5. 7X 10+12
-to Ar+ 6.0XIo+11
40 ca+ 4. 7XIO+It 44 C(h+ r,.7Xt0+lt
• For an ion ~n~r~:Y of 2.16~ V impinging on the multlpli~r.
The uncertainty arises because collection fields ilufficiently large to ensure total co~lcction of the secondary ions cannot be cmployC<"1 due to the influence these fields woulrl. exert on the motion of low-energy primary ions. While mc;•:.urements of the variation of collcdion efiicicncy with the strength of t.hc extraction field may readily be made at high primary ion energies, these results arc not necessarily relevant to the low-energy regime, where the dynamics of the Mg+ production may be different. Interpretation of the present data obt;1ined using weak collection fields is therefore based on the H.so.umption that at energies above a few electron vnlt~, the Mg-l" ions arc produced by simple electron transfer in collisions involving littlf' momentum tmnsfer and that as a result, the collection efficiency it. independent of both the nature and energy of the primary ions. It is implicit in this assumption that the ('nerv,y defect in the reaction is small, since energy not expended in excitation of the products mw;t appear as
TABU! rr. Collection em.dency or ICC:Ondary lonl produud by charge exchange at Interaction region.
Reactants
Ns+:N2 Ot+:Ot N+:Ot
Crot~aectlon• (to-" cm1)
29.0 15.5 16.5
: ::::-== ...... ,, ' Secondary ton
collection eiTICiendea" l%)
80 68 74
II
• The primary Ions were Jlroductd by bombardment with 40-tV rl«tron~<. The lon eneray uoed for thtKe mtn•urement• waa 400 eV.
b Awaumln1 eros~ 14l'Ctlon• ll~ttd (taken •• m01t rtJire!K'IItatlve of tho.e rtported In the literature).
kinetic energy and therefore would influence the collection efficiency.
A number of charge transfer reactions for which absolute cross sections had been previously determined were tabulated. Using the same experimental conditions and the detection efficiencies deduced from measurements of the primary beam, as well as the same signal strengths and detector sensitivity, the collection efficiency of our instrument is obtained by comparing our measured cross sections with the best tabulated values. The efficiencies so obtained, and illustrated in Table II, varied by 20%; this may represent a real variation in the collection eOi....ciency or simply reflect the discrepancies in the published cross-section values. In the present work, a collection efficiency of 75% is used in the evaluation of the cross sections from measurerr.!'n~ of the neutral beam density, the primary ion beam intensity, the dimensions of the neutral beam at the interaction zone, the system gain, and the recorded signal.
An over-all analysis of all possible systematic errors has been performed. From this study, a possible error of ±30% is placed on the absolute charge transfer cross sections for the higher reaction energies. This error may increase to as much as a factor 2 at the lowest impact energies.
EXIT HOLE
ALUMINA RODS
~·ro. 2. Neutral btam Knud.en celllul'l\&ce.
49
I
I I 1 i I
I

■WS?PPTf¥i3$1^*?^
3788 RUTHERFORD, MATH IS, TURNER, AND VROOM
300
250
150
o - 100
g 200 [ o H*+m—***H* a-
50
0
o o o o o •
O O D O
0 o o o o o
J_ J_
Kic. 3. Chaise transfer cross sections lor Mg*. N.\ and N* ions impinging on neutral Mg as a (unction of the energy of the incident ion.
12 5 10 20 50 100 200 500 TOO
ION ENERGY (eV)
B. Neutral Beam Formation and Measurement
The neutral atom beams originate in a heated mo- lecular effusion source (a Knudsen cell). A sketch of this cyündrically shaped furnace is shown in Fig. 2. Tantalum was used for the construction of the Knudsen cell. The cavity is surrounded by i\-in. walls and is heated with a tantalum wire filament which passes through five evenly spaced holes. The heater wires are insulated by hollow alumina tubing. A lid of tantalum is held tightly in place by screws. The tem- perature is measured with a chromel alumel thermo- couple mounted inside the furnace wall. The chromel alumel thermocouple was shown through repeated tests to be reproducible even after having been re- moved and replaced.
The cosine law of molecular eff'ision" was applied to determine the beam density knowing the equilibrium vapor pressure of the magnesium in the Knudsen cell at a particular temperature. The number density, n, in the beam at the interaction region is then computed under effusive How condition to be
n^Naa/4***, (1)
where A'0 is the number density .a the furnace, a is the an a of the aperture in the furnace, and f is the distance fron the aperaturc to the interaction region.
The vapor pressure of magnesium is greater than 1 toiT before its melting point is reached. As a conse- quence, sufficient pressure ccild be obtained in the Knudsen cell without melting the solid. Magnesium reacts slowly with air, -»Mowing the problem of oxida- tion to be kept to a minimum.
Calculation of the beam density with Eq. (1) has been used for the preliminary analysis of the data. One difficulty associated with this method is the' the temperature must be quite accurately known as the pressure is ? sensitive function of the temperature. A small temperature gradient between the location of the
thermocouple a: d the inside of the chamber could result in a large error in the calculated beam density and thus the cross section. In addition to this, any temperature gradient in the volume of the furnace containing the metal vapor would result in a corre- sponding uncertainty in the furnace pressure and thus in the neutral beam density.
To ensure that our temperature measurements were accurate and that our oven behaved correctly, a complimentary technique was employed. This method utilized neutron activation analysis to determine t he- number of atoms deposited into the collector by the neutral beam in a known time period. In the initial attempts to collect the atom beam, a small poly- ethylene cylinder, was used. It was found that the magnesium atoms did not stick in this collector but scattered away. To overcome this problem, a collector consisting of a polyethylene bag attached to the end of the polyethylene cylinder was used. This assembly was located so that all the beam passing through the collision region entered the collector, ("omrricraaily available "Baggies" were chosen as the type of |"<oly- ethylene bags since they were found to be both free of contamination and thin walled. After the deposition of approximately 100 jig of magnesium, the bag was re- moved and sent to Gulf Radiation Technology Neutron Activation Analysis Facility for measurement. This method gave results in close agreement with the density calculated from the molecular effusion considerations. It is interesting that even though magnesium is a condcnsiblc beam, several reflections were required before total sticking to a room-temperature surface could be achieved.
m. RESULTS
The cross sections for charge transfer between neutral magnesium and the nine inns investigated were determined over the energy range 1 500 cV. These cross sections arc presented here in graphical form
50

i'JWW^fr^B^-'^T^TP.u^.-»^ BjflWWIP^IWP^WW^WP^iWIPfBWiPBggy" i.iiim.i.ii.nnijyj^wBB^tm.v'^' pg^gppgiwjw^^g^'iigwii
MAGNESIUM IONS BY CHARGE TRANSFER
350
3789
FIG. 4. Charge transfer era» sections (or NTiO+, HiO*, and NO* ions impinging on neutral Mg as a function of the energy of the incident ion.
300
250
§ «J 200
To '50
100
50
0 I
D »*•••«-«o ■ n,*
*»* o
o o o a*.
a „ o o o 0°Oo, " a - _ o o o
a ua o OD o ,aac,S 2° D0
5 10 20 50 100 200 500 1000
I OH ENERGY (eV)
TABLE HI. Recombination energies and relevant energies of reactanls and products cited in the text.*
Species and ground state
Mg(3j», 'S.)
N,(X%+),..
X<2>», <S„>)
Excited neutral states and energy above ground state
(eV)
(fl'»2.-)_,8.164 (fl»n,)_, 7.353 <Ä*n,)_, 7.987
ap\'DM*),7 382
Ground ionic state and ionization potential
(eV)
(it.'Sm). 7 644
(**V)_, 15.580
(2*»,»/»,). 14.352
(Ar*n)_i, 12.053
Ion state and energy above ionic ground state
(eV)
(3p.*P>n), 4.419
(Jf«r,+)^, 0.270
(a «n.)^, 4.018
• Atomic kvtls obtain«! (torn C. Mom. Natl. Bur. Std (I. S.)( irr 467, The Rand Cnnnration. Kr<nrrh Memorandum. K.M.-S20I-ARPA. March Vol. I (1949). Mokrutar Irvrl« obtained Irom F. H- Gilmorr. "Bask Kncrxy 1967. l-rvrl ami Equilibrium I>ata for AtmosHirrir Atoms and Mnlrrulrv"
Kic. 5. Charge transfer cross sections for HrO* ions impingin» on neutral Mg as a function of the energy of the incident ion.
E u or
1«#0
120
100
80
2 60
b *»o
20
0
O HjO** at« — H,0 ♦ H ♦ atf*
O oo
10 20 50 100 200
ION ENERGY (eV)
500 1000
51

iWt'-'*^lHJJ|PX''J|U^WW:ll!4BW>*WIJW*?W'^1' VSB&: ■gwwBBWWgWpppwiWpiP^gp'gWPBBgp 11 (.1L kJJIUPI. IWWfJ^PP»
3790 RUTHERFORD, MATHIS, TURNER, AND VROOÄ*
1 £U
too
1 I i- ■ —r ■ l i t "T —i r
? 80 o ° o 0
o o 0 ou o
M
,o 60 i o
b
: o°°° 0
oo
02+ + Mg — 02 + Mg+
°0
-
20
0
10 eV Ions
X2ng a^u i i i~ 1 J— ' i ' ■ ■
12 16 20 2k 28
ELECTRON ENERGY (eV)
32 36 1*0
Fie. 6. Dependence of the charge transfer cross section upon the source electron energy for the reaction Oi++ Mg-*0,+Mg+-
since this type of representation aids the discussion of the data. In general, the reactions have been grouped such that processes which exhibit similar structure and which have comparable cross sections appear in the same figure.
Figure 3 gives the cross section for char?' transfer for N»+ and N+ ions incident on neutre^ magnesium. The symmetric resonant reaction Mg+-Mg is also shown for comparison.
The cross sections for three other ions (NiO*, HiO4", and NO*) incident on Mg are shown in Fig. 4. Like the previous three reactions, these processes have a large cross section. The smallest cross section observed was that.for (J* on magnesium. The results for HjO+ are given in Fig. 5.
Figure 6 illustrates the variation in cross section for charge transfer between <V and magnesium as the number of excited states present in the primary ion
beam is varied. Data of this type are obtained by chang- ing the energy of the electrons in the primary ion source and measuring the charge transfer cross section at a fixed ion-impact energy (in this case 10-eV ions). When the electron energy is such that no electronically excited ion states can be formed in the Oj+ beam (less than 16 eV), the cross section for charge transfer is seen to be considerably lower than when excited states are present (electron energies greater than 16 eV). Similar studies involving both 0+ and NO*, which have metastable ionic states, showed little dependence of the cross section on primary ion source electron energy. From this we can conclude that the cross sec- tions for charge exchange must be similar for both the ground and metastable ionic states.
The cross sections for charge exchange between the ground state of 0»+ and the metastable (V state with Mg are shown in Fig. 7. The curve for the ground
er
175
150
125
100
75
"b 50
25
* A
o OO.
a o °°o
o o 0 0 0
O o
A 0|* *» Mt-0,» Ma* <(«•«•«> ° O C
O 0»* ♦ 1 -0, ♦ Ha» UllIlM«)
a »t* » W - Ot» Mt* Hmil mm*
a o
12 5 10 20 50 100 200 500 1000
ION ENERGY (eV)
Fie. 7. Charge-transfer cross sections involving excitation for Oi* ions with Mg as a function of the incident ion energy. The cross section is shown for ground-state (V ions, for (V ions in the mixture of states (composite) which results when 40-eV electrons impinged on O» to form the 0>+ .and for the excited states in the composite alone, i.e., after the contribution of the ground-state ions in the composite has been removed.
52

^PBrcgpmg^3n~>f.PUji(J0'W^9£l -IIJI» j i>ui iwi-i'11-!"« «wwwmipwi
MAGNESIUM IONS BY CHARGE TRANSFER 3791
state is obtained using ions formed with 16-eV electrons. The curve for the composite beam of ground and metastable ions is obtained using 40-eV electrons in the ion source. In order to determine the cross section for the excited state, the ratio of the ground to meta- stable state concentration in the beam must be known. This ratio is determined in a separate experiment using a technique previously developed in this laboratory," which showed that for 40-eV electrons impacting on Oj, 32% of the ions formed are in metastable states.
IV. DISCUSSIOH
In attempting to interpret the results obtained for the charge transfer reactions, the major difficulty arises in accounting for the excess energy released. In the discussion of the results given below an attempt has been made to correlate the magnitude of the charge transfer cross section with the formation of excited states of the products of the collision. The resultant processes should be considered as only possible mecha- nisms. Recombination energies and energy levels quoted in the discussion below are given in Table III.
In the symmetric charge transfer process between Mg and Mg*, exact resonance must exist and as a consequence a large cross section would be expected. In Fig. 3, the results for this process show that the cross section is indeed large. The slope of the curve is similar to that predised by Rapp and Francis1 but the magnitude of the cross section is twice the predicted results. Previous resonant charge transfer measure- nun ts for Li*+Li" and Cs*+Cs " have also been found to give cross sections larger than predicted by the theory.
The magnitudes of the cross sections for the reaction of NV" and N* on Mg arc both similar to that for the symmetric case. Such a result is indicative of a near resonance mechanism for the reaction which would probably leave the Nj or N in an excited state. Two po5iible nearly resonant routes for electron capture to form excited Ni states are
Kt+iX JZ.-),-, + Mg(3i*. '5.)— N,(«' «2.)_
+ Mg+(35,,5,/,) + AE(0.042eV) and
N,*(X *V),x.+Mg(3**, •£)—Ni(Ä »n,)_
+ Mg+(3j, *Sui) - A£(0.051 eV).
The possible mechanisms available for the reaction of N* with Mg are less complex since fewer possible states of the product N atom exist in the energy regime of interest. One possible mechanism which is nearly resonant is
*♦(»/>,)+Mg(M >5.)-»N(«ZV)
+ Mg* (ip, »/>,„•) + A£(0.087 eV).
This reaction results in both products being left in
excited states. It is interesting to note that if the N neutral formed is in the excited (lO§/i°) state the energy released in the neutralization process is 12.15 eV. This energy is very similar to the ionization potential Oj and the reaction of Oj* on Mg might therefore be expected to be similar to that given above. This reac- tion will be discussed below.
The cross sections for charge exchange of NtO*, HtO*, and NO* in collisions with magnesium can be seen in Fig. 4 to be smoothly increasing with decreasing ion c.iergy. Comparison of these results with those for Nj* and N* in Fig. 3 shows that the cross sections are of the same order of magnitude, indicating that pro- cesses having a small energy defect may again pre dominate. For the cases of NjO* and HjO*. possible reactions are difficult to determine because of the lack of spectroscopic information available on the location of the excited states of these species.
The reaction of NO* with magnesium ,nay be of importance in the upper atmosphere because of the large concentration of NO4". As for the reactions discussed above, charge exchange between Mg and NO+ is exo- thermic, in this case by 1.61 eV. Electronic levels that will allow near resonance do not exist in cither the product ion or neutral, and vibrational excitation of the neutral NO ground state may therefore be expected. Since the internuclear distance in NO+ is less than in NO, the Franck-Condon transition zone for processes leading to production of NO from NO* overlaps many vibrational levels of the neutral product. Such a con- figuration favors the formation of products with con- siderable vibration excitation.
The recombination energy for HjO* is about 7 eV* and as a consequence this process would \r endo- thermic for ground-state ions. Figure 5 shows that the measured cross section is both appreciable and exhibits the shape expected for a near resonant process. This observation can be justified by either the presence of excited ions in the primary beam or a slightly higher recombination energy for the H|0*.
The case of O* on Me differs from that fnr the other atomic ion, N+, in that no energy level combination in the secondary neutral or ion will allow near resonance. This fact probably accounts for the low measured cross section. (Data with poor signal-to-noise ratio suggest a CTOss-section value of '.ess than 20X 1ft-1* cm'.)
The results for Oi+ on Mg, seen in Fig. 7, again indicate that near resonant processes may be important. The cross sections for both the ground and excited states show a rapid increase with decreasing ion energy. If the O»* ground-state species «combines to give Oj in the ground state, the recombination energy is nearly equal to that for recombination of N* ground state to form N(*ZW). This suggests that the mecha- nisms for the Oj* ground state recombination could be
0,*(X »II,) + Mg(3j», »S„)--0, (X %-)
+ Mg*(3>, */»,*•) + A£(0.0 eV).
53

^..Mm-i.,min.m"jitim^^i'-w^i.'.'!iwm^mmW!Wffmi
3792 RUTHERFORD, MATHIS, TURNER, AND VROOM
TAELE TV. Calculated anas sections and rate ayHkienU.
300*K 600*K 120011
Croat Rate Croat Rate Cross Rate section coefficient section oocffiocot section coefficient XIO» xio» xio» xio» xto« X10»
Reaction cm> cnrVsec tor cm*/aec cm« cnrysec
Mg++Mg 63.2 SI 0 560 63.9 50.2 81 0 N*+Mg 14.3 12.2 14.4 17.3 14.1 24.0 fV(composite)+Mg 27.4 20.1 21.5 22.4 17.7 26.0 o,+(jf>r,-)+Mg 16.8 12.3 13.2 13.8 11.1 16.3 N,++Mg« 10.7 7.24 11.3 10.8 12.1 16.3 NO*+Mg 10.8 8.11 12.7 13.4 13.6 20.4 IW+Mg 13.4 21.7 31.8 31.1 31.3 43.2
1 The reliability of the N't' ♦ Ms rviraisolation is not as high as for the other reaction* shown.
For the excited oxygen molecular ion (oTJ.), sufficient excess energy exists to cause dissociation of the resultant oxygen molecule. Since many dissociative curves exist through which such a process might pro- ceed, this may be a probable mechanism, and could help to explain why the excited state has a much larger cross section for charge transfer than the ground state.
The charpr transfer processes described above may be broken into two groups, those involving molecular iens and those involving atomic ions. Consideration of the results shows that all the molecular species give cross sections for reaction whose energy dependence exhibits the behavior that might be expected for resonant or near resonant processes. Processes with a small energy defect can easily be realized with molecular species because of the multitude of vibration rotation levels which lie above every electronic state. From this point of view, molecules can be considered as possessing a near continuum of states available for reaction.
For the two atomic species studied, one case of near resonant behavior and one case of nonrcsonant behavior was observed. X* charge exchange with Mg has a large cross section and the mechanism proposed tor this reaction shows that near resonance can be obtained by assuming both the N and Mg* arc formed in excited states. For atomic oxygen no such combina- tion of states exists and the cross section for the process is small.
It is interesting to note that low excited ionic states exist for magnesium. The presence of such a low-lying state allows the X* charge exchange to occur. Work similar to that dcscril>cd for magnesium has been done in our laboratory ust.ig sodium as the neutral target. In this case, the sodium ground-state ion, since it is devoid of loosely bound electrons, has no low-lying levels. As a consequence, near resonance cannot be obtained for cither N* or O*. Our experiments have shown that the cross sections for N* and O* on Na
are small. These results will be the subject of a future publication.
A. Extrapolation
Using our experimental technique, interaction ener- gies of less than I eV are difficult to obtain. Neverthe- less, considerable interest in reactions of this nature lies in the energy range from thermal to 1 or 2 eV. In order to obtain cross-section values in this region, an extrapolation method devcioped in this laboratory has been used.1* This extrapolation method combines the energy dependence of the Rapp Francis1 resonant charge transfer theory with the energy dependence of the Gioumousis-Stevenson'* complex formation model.
The general formula devcioped for the total charge exchange cross section takes into account the proba- bilities for charge exchange to occur both when a complex is formed and when it is not, and also corrects for nonrectilincar orbit when complex formation docs not occur. The method can be formulated as follows. The total charge exchange cross section a will be given
o-=/»i when 2o-|<<rj (2) or by
*= (/— iVi+o-i when 2a,>at. (3)
Here / is the fraction of collisions resulting in complex formation which decays into the charge exchange channel. at is the Gioumousis Stevenson complex formation model cross section and is represented by
a,-a-(2A»//-)'", -4)
in which e is the electronic charge, « is the polariza- bility of the neutral, and F. is the baryecntric inter- action energy. Also, o-|, given by
»i = »a[. + (V4o-.)"], (5)
54

v**mv*^K ( ^aig^iai^jijfiPWJBUt.J»-'PL''t-»waallt
lJ^*-^'' -IJ'J1^. w^^K^mtl^^^
MAGNESIUM IONS BY CHARGE TRANSFER 3793
represents the Rapp-Francis resonant charge exchange formula modified to take account of the curved orbits of the reactants. The form of the Rapp-Francis formula is
otm=A~B\agE, (6)
where A and B are constants. The extrapolation is carried out by fitting the
modified Rapp-Francis formula [Eq. (5)] to the high-energy portion of the measured data, that is, in the region where complex formation should be negligible. The calculated curve is then extended to lower energies using the test given with Fqs. (2) and (3) to evaluate when the measured data deviate from the form of Eq. (5). In this manner the con- tribution of complex formation is determined and the calculated curve can be extended to energies below those measured. A computer program has been de- veloped to perform the calculations.
The extrapolation technique was developed by assuming that the relative abundance of the various products emerging from the capture-formed complex is independent of the relative kinetic energy of the reactants; that is,/ remains constant. If this condition is not met, the technique will not give valid rate coefficients at near thermal energies. In general, whether or not the extrapolation technique is valid can be teen by observing if the calculated cross-section curve fits the experimental data to the lowest energy of measurement. In the present studies, the extrap- olation is found to be valid for all systems measured except HjOf and HiO+ on magnesium.
Table IV gives the results obtained for the ex- trapolation of the N*, N,+, NO+, and NjO* to thermal
energies. Cross sections and rate constants are given for three temperatures.
ACKNOWLEDGMENTS
The authors wish to acknowledge the benefit of discussions with our colleagues, particularly James K Layton, Dr. R. F. Stebbings, and Dr. J. W. McGowan. Special recognition is due Dr. D. M. J. Compton for his suggestion that activation analysis be used to determine beam densities.
•This work was supported by the Defense Atomic Support Agency under Contract DASA01-69-C-0044.
11). R. Bates, in Atomic and Mokcular Processes, edited bv D. R. Bates (Academic, New York, 1962).
» I). Rapp and W. E. Francis, J. Chem. Phvs. 37, 2631 (1962t '0. B. Firsov, Zh. Eksp. Tcor. Fit 21, 100 (1951). ' K. F. Stebbings and J. A. Rutherford, J. Gcophvs. Res. 73,
1035(1968). 4 R. S. Narcisi and A. B. Bailev, J. Geophvs. Res. 70, 3687
(1965). • R. A. Goldberg and L. J. Blumle, J. Geophvs. Res. 75, 133
(1970). 1 R. F. Stebbings, B. R. Turner, and J. A. Rutherford, J.
Geophys. Res. 71, 771 (1966). »B. R. Turner, J. A. Rutherford, and R. F. Stebbings, 1.
Geophys. Res. 71, 4521 (1966). • B. R. Turner and J. A. Rutherford, J. (Jeophys. Res. 73, 6751
(1968). ■ C. F. Geise, Rev. Sei. Instr. 30, 260 (1959). " M. Knudsen, Ann. Physik 28, 75 (1909); 28, 999 (1909). » B. R. Turner, J. A. Rutherford, and I). M. J. Compton,
J. Chem. Phys. 48, 1602 (19685; R. F. Mathis, B. R. Turner, and J. A. Rutherford, ibid. 49, 2051 (1968).
u D. C. Lorents, G. Black, and O Heinz, Phys. Rev. 137, 1049 (1965).
"L. I- Marino, A. C. H. Smith, and E. Caplingcr, Phvs. Rev. 128, 2243 (1962).
»F. A. Wolf and B. R. Turner, J. Chem. Phvs 48, 4226 (1968). ™G. Gioumousis and I). P. Stevenson, J. ("hem. Phvs. 29,
294(1958).
55

wlfr^ljUWi^H-J^j^Jj^'l'tfj;'■uumu^i i,•»»^yT'.T'^-r MJ.^wuy.'i, IIJ.IJUHüMJISPIJP'^!
APPENDIX B
EFFECT OF METASTABLE 0+(2D) ON REACTIONS OF 0+ WITH NITROGEN MOLECULES
Preceding page blank
57

BR5SISPW»«®
Reprinted from:
THE JOURNAL OF CHEMICAL PHYSICS VOLUME 55. NUMBER 12 IS DECEMBER 1971
Effect of Metastable O+(*£>) on Reactions of 0+ with Nitrogen Molecules*
J. A. RUTHERFORD AND D. A. VBOOH
Gulf Radiation Technology, A Dfaision of Gulf Energy and Environmental Systems Company, San Diego, California 92112 (Received 26 July 1971)
Laboratory measurements indicate that the metastable ions of O* in reaction with N» in u.e low-energy range (14 eV) react to form principally N«+, while the ion-molecule reaction to form NO* has a very small probability. The ground-state O* ion reacts mainly to form NO4". The abundance of metastable O* *D ions was determined using the observation that 0*(*ö) +N» has a small cross section for forming N0+. The ion energy dependence for both reactions has been measured within the energy range 1.0-500 eV.
The ion-molecule reaction most widely dealt with by aeronomists has been that involving O4" in colli- sion with Ni to form NO1". The effect of excited states of O4" on the rate of this reaction has been open to question. Dalgarno1 has pointed out that in the F region O+CD) ions are lost principally by col- lisions with neutral O» and Nj. An indication that the excited states may not be of importance in the formation of NO*" was reported by Stebbings el a/.* who noted there was little change in the cross sec- tion for Cr*"-r-Nx—»NO++N when excited states of O4" ions were introduced into the beam. The results re- ported here will show that the cross section for the reaction involving the excited atomic ion FJO*"(*Z)) + Nj—►NO++N3 is in fact very small. Furthermore, we will indicate that the principal channel for these reactants results in production of Ni+ which is prob- ably formed in the (4 *n«, t= 1) state.
The apparatus employed for the cross-section mea- surements has been described previously.*-* In this instrument, the primary ions are formed in an elec- tron bombardment ion source, the electron energy of which can be carefully controlled. These ions are ex- tracted from the source, mass analyzed, and acceler- ated or retarded to the desired energy. This ion beam then crosses a modulated neutral beam (modulated at 100 Hz), in this case N». The products of collisions between the ion and neutral beams are extracted along the direction of the primary ion beam, accelerated, focussed. and mass analyzed in a second mass spec- trometer. The selected ions are then detected using an electron multiplier coupled with lock-in amplifier techniques.
The neutral beam is formed by effusion from a room-tcmperalurc orifice and modulated by mechan- ical chopping. The neutral beam density is determined by measuring the pressure in the neutral source with a differential pressure manometer and calculating the effusion from this source under known geometrical conditions.
The ratio of the metastable electronic states to the ground slate can be varied by careful control of the energy of the ionizing electrons in the primary ion source. When the electron energy L below the thresh- old for excited-state formation, no excited states can be formed and the resultant beam will be composed
entirely of ground-state ions. As the ionizing electron energy is increased metastable ions will appear in the beam.
Two reactions have been studied in detail here. These are
0++N»->NO++N (1) and
0,"-r-Nr-K)T-N,+. (2)
The effects of metastable O4" ions in the primary ion beam can be seen for Reaction (1) from Fig. 1. Here the relative probability for forming NO4" in collisions
14 «V IONS 0**>*2~N0*i-N
26 28 30 32 3« 36 SOURCE ELECTRON ENERGY (tV>
40
FIG. 1. Graphic 1 representation of the decrease in the relative cross section for formation of NO* ions from ()*" impinging on Ni as a function of the ion source electron energy. The ion- neutral interaction energy is 14 eV. The open circles represent the case for pure O, in the source while the open triangles rep- resent the case ior an Oi/He mixture.
between 04" and Nj is plotted as a function of the electron energy used to form the O* ion. Two cases are shown: one where pure oxygen is used in the ion source and the other where the source contained 1% (h in pure helium. The latter case has been included since it gives an enhancement in the number of meta- stable 0+ ions in the beam and therefore better illus- trates our conclusions. This enhancement arises because helium ions formed in the source will undergo a dis- sociative charge-transfer process with ()j to yield the metastable 0*(*D). Note that the NO* data in Fig. 1 have been normalized to a value of 10 at 21 eV. This is below the threshold of 22 eV required for produc- tion of 0+(JO) from molecular oxygen if the forma
Preceding page blank 5622
59

REACTIONS OF 0+ WITH NITROGEN MOLECULES 5623
tion of 0+(*D) from pair production is neglected. This latter process is known to have a small cross section. Examination of the curves shows that for the formation of N0+, the relative cross section normalized on the total 0+ current starts to decrease
TABLE I. Cross section for 0++Nr-»NO++N.
Laboratory energy (eV)
1.2 1.4 1.5 1.7 2 0 2.2 2.5 2.7 3.0 3.2 3.5 4.0 4.5 5.0 6.0 7.0 8.0 0.0
10.0 11.0 12.0 13.0 14.0 15 n 16.0 17 0 18.0 19.0 20.0 21 0 22.0 ZVO 24 0 25 0 26 0 27 0 28 0 30 0 32 0 34 0 35.0
Center-of-mase Cross section kinetic energy (units of
(eV) 10-» cm*)
0.76 0.89 0.95 1.08 1.27 1.40 1.59 1.72 1.91 2.04 2.23 2.55 2.86 3.18 3.82 4.45 5.09 5.73 6.36 7.00 7.64 8.27 8.91 9.55
10.18 10.82 11.45 12.09 12.73 13.36 14.00 14.64 15.27 15.91 16.55 17.18 17.82 19.09 20 36 21 64 22 27
1.3 1.2 1.3 1.6 1.5 1.7 1.8 2.0 2.1 2.2 2.6 2.8 2 9 3.0 3.4 3 6 4.0 3.8 4.3 4.6 4.2 4.4 4.3 4.0 3.8 3.5 3.2 2.7 2.2 1.8 1.5 1.3 1.0 0.80 0.65 0.53 0.44 0.34 0.22 0.15 0.15
in the cast- of pure oxygen above the threshold for 0+('D) indicating thai the presence of this excited state depletes ihe number of ground state particles available for reaction. This same result is seen for the helium ovygen mixture evrept »hat the effect is nol Mt-n unlil slightly higher in energy due to the
-i 1 r-
14 tv IONS
0*+N,- 0*Nj
o
b
HELIUM OXYGEN MIXTURE
k A 4 * °
PURE OXYGEN
a&A
22 26 28 30 32 34 36 SOURCE aECTRON ENERGY (eV)
38 40
FIG. 2. Observed cross section for the charge-transfer reaction 0++Ni~0+Ni+ as a function for the ion source electron energy. The ion-neutral interaction energy is 14 eV. The open triangles represent the case for pure Oj in the source while the open circles represent the case for an Oj/He mixture.
excited state not being formed in significant concen- tration until above the ionization threshold for helium.
The electron energy dependence for the reaction O^+Ni—»O+Nj* is given in Fig. 2. The cross sec- tion shown in this figure (the observed cross section) is obtained using the total 0+ current. An effect op- posite to that seen in Fig. 1 is observed here. In the curve for pure Oi it is noted that the observed cross section for forming Nj^ is very small for electron energies below the 0*(SD) threshold and then in- creases as the number of excited states is increased. The effect is the same for the oxygen-helium mix- tures below the threshold for ionization of helium but is more pronounced above this point as more excited states are then present. It must be noted that all the data shown in the figures are for a fixed ion-neutral interaction energy of 14 eV and illustrate the change which this cross section undergoes as the number of excited states in the beam is varied. An
40r
30-
20
10
T r
V 1 1 1 1 1 r
,o o 'o oi
>o0#P' .o«*>
0* ta+Nj— O+Nj
o
b
JL _L_ 05 5 10 20 W
ION ENERCV itv) 100 200 500
Fir. .1 Charge transfer crow «return f<vr 0'"/)H\\ .»!• V as a function of ihr ion energy in I he lalmrali ry . *\< n,
60

5624 J. A. RUTHERFORD AND D. A. VROOM
interaction energy of 14 eV was chosen because the ion-molecule cross-section curve peaks at this value.
Examination of the figures allows the following con- clusions to be drawn:
0+(45)+Nr-»NO+-r-N,
0+(«S)+N,*-»0+N1+,
0+(*ö)-fN1-O+N,+.
(3)
(4)
(5)
That is, Fig. 1 shows that NO*" is formed below the threshold for excited O4* ions [[Reaction (3)]. Simi- larly, Fig. 2 shows that below the O+CD) threshold very little Nj+ is formed [Reaction {4)3, but above the onset of the excited state, Ni+ is readily formed [Reaction (5)]. Using the above information coupled with previously obtained results, we will show that
O+COJ+N^NOH-N. (6)
Assume that the cross section for this process is small compared to that for Reaction (3). Now since the observed cross section a for any process can be writ- ' n as
»-2>./.. (7)
where an is the cross section for reactant ions in state n present in fractional abundance /„ we can write our total cross section for formation of NO"1' as
a = <T('.<;, /i'.<5,+a,*D; fi'n-- (8)
Now since we have assumed that O-(,B)«<T(*S), the
presence of this species in the total 0+ beam can be shown to lower the observed cross section by a per- centage equal to its fractional abundance. Examina- tion of Fig. 1 therefore indicates for electrons of 40 eV, that the excited-state concentration is 20% of the total beam for pure ()- and 37.5% for the oxygen/ helium mixture. As discussed above. Fig. 2 shows that the charge transfer process between O*" ions and N}
goes primarily with the excited state [Reactions (4) and (5)]. I'sinu the fractional abundances determined above, it is therefore possible to correct the observed cross sections for Reactions (5) obtained using the total O* beam intensity. For Reaction (5) [()^(JZ)) + N, «O+N..-] we ge'. for 14 eV ions colliding with the neutral partii'e. a cross section of 29.5X10"" cm1
for the pure O» case and a cross section of 30.6X 10~w cm* for the Oj/helium mixture.
Using a totally different technique developed pre- viously in this laboratory,4 we have determined the fractional abundance of 0+('Z)) from the attenuation of an O1" beam in another gas to be 24%. This value was obtained using 40-eV electron energy in pure Oj. Using this value for the fractional abundance, we have obtained a cross section for Reaction (5) at 14 eV of 26.8X10-'* cm1. The difference between this value and those determined above is within experi- mental error and therefore indicates that our assump- tion of a small cross section for Reaction (6) is valid.
Using the above-determined fractional abundances, it is possible to obtain cross-section curves for the separate ion states for each reaction that is found to proceed with large probability. The charge ex- change cross section leading to production of Nj+ is given in Fig. 3. The values given by this figure are slightly iower at low energies than those reported previously.1 Cross sections for the ion-molecule re- action producing NO+ are given in Table I.
Examination of Fig. 3 shows that the cross section for the reaction of O+CD) with Nj remains large down to the lowest interaction energy studied. The energy a lüabie in charge transfer with 0+(JD) is 16.1 eV. 'inis energy is almost exactly that required for formation of the N5+ in the (A 'II«, v=l) state. This reaction is therefore a source of excited Ni+ ions in the F region where the 0+ concentration is high. Decay of the Ni+(/f HI.) state produces Mcincl ra- diation. A discussion of how this reaction may lead to an enhancement of the Meinel radiation observed from airglow has been given by Wallace and Braid- foot.*
* Work supported by the Defense Nuclear Accncv under Contract DASA01 69 C 0044.
1 A. Dalgamo and M. B. McFJroy, Planetary Space Sei. 11, 727 (1963).
1 R. F. Stebbinus, B. R. Turner, and J. A. Rutherford. J. Geophys. Res. 71, 771 (1966).
:j. A. Kuthcnorci, R. F. Mat his, H. K. turner, and l>. A. Vroom, J. Chem. Phys. 55, 3785 (1971).
• B. R. Turner, J. A. Rutherford, and J). M. J. Compton. J. Chem. Phys. 48, 1602 (1968).
' I.. W&llace and A. L. Broadfoot. Manctary Space Sei. 17, 975 (1969).
61

^^^m^TV^-m^mv
APPENDIX C
FORMATION OF SODIUM IONS BY CHARGE TRANSFER
Preceding page blank
63

t.wawt*W-TfflCT^g,,'g^^,,^wl!W!"^.JW^"ll'>4P,«''l-«»g^fWW
Ref>rinted from:
THE JOURNAL OF CHEMICAL PHYSICS VOLUME 56, NUMBER 4 I MAY 1972
Formation of Sodium Ions by Charge Transfer*
J. A. RuTHESrOKD, R. F. MATHlS.f B. k. TURNU, AND D. A. VKOOM
Gulf Radiation Technology, A Division of Gulf Energy and Environmental Systems, San Vitgo, California 9Z11Z
(Received 21 January 1971)
Cross sections have been measured in the energy range 1-500 eV for the charge transfer of several ions with neutral sodium. The ions studied are 0+ N+, Nt+, NO*, 0,+, H,0+, H|0\ N,0*, and Na+. The mag- nitude of these cross sections can be related to the availability of reaction paths which have a small internal energ> defect. The smaller the energy defect the larger the cross section. Where possible the measured cros.- sections have been extrapolated to thermal energy. Comparison is made with other experiments.
I. INTRODUCTION
Sodium ioi- have been shown in rocket studies to be one of the more prevalent metallic species in the upper atmosphere.1 Since this ion is monatomic, recombination is slow. In addition its ionization potential (5.138 eV) is significantly below that of the more common at- mospheric species, and it cannot therefore be readily destroyed by charge transfer. As a consequence, metallic ions of this type may be expected to be long lived in the upper atmosphere. Information on charge transfer reactions leading to formation of sodium ions from the neutral species may therefore be important to an understanding of atmospheric deionization.
The results presented in this paper represent the second part of an experimental program in which asymmetric charge transfer processes leading to produc- tion of metallic ions are being studied. In cases where the ionization potential of the metal is lower than or equal to that required to form the primary ion, charge transfer to the ground state will be either exothermic or resonant In the first publication in this series* it was shown that even though several exothermic channels for reaction may be open at all interaction energies, the greatest contribution to the total cross section comes from the channel which results in closest energy resonance between reactant and product states. Similarly, if no channel allows near-energy resonance, then the charge transfer cross section is found to be small. The results to be presented here lead to the same rftnrhKtnnc
II. EXPERIMENTAL
A detailed description of the crossed beam apparatus employed for the charge transfer measurements between ions and metal vapors has been given previously.'
The neutral beam is generated in a resistively heated Knudsen cell. The major difficulty encountered in these experiments is the determination of the neutr.il beam density. Two methods were employed. First, the density was calculated from the system geometry, the size of the port of egress in the Knudsen cell, the temperature of the cell, and the vapor pressure of sodium.' This technique has been employed in our previous experi- ment and demands accurate values in the vapor pressures as a function of temperature. The second
Preceding page blank
method involved running the neutral beam for a known time and collecting all the sodium which passes through the collision region in a liquid nitrogen cooled vessel (liquid nitrogen coo'ing was required to ensure com- plete collection). The amount deposited was then determined using activation analysis.1 For sodium the beam densi- ies calculated from the two methods did not agree, the neutron activation analysis gave the higher density. This difference i a factor of 2) was attributed to inaccuracy in determination of the sodium vapor pressure due to oxide layer contamination in either our cell or that used to determine the published vapor pressure. The beam densities used here are therefore those determined by activation analysis.
The possible error which may exist in our absolute cross sections is greater than in the previously reported Mg results and arises due to uncertainty in the neutral beam density. For these experiments we estimate that our values may be in error by as much as 40% at high energy, increasing to 60% at the lowest energies. It should be noted that this is an error in the absolute magnitude. The reproducibility in the measured energy dependence was better than 15%.
III. RESULTS
Charge transfer cross sections for reactions between nine ions and neutral sodium have been measured in the energy range 1-500 eV. The cross sections are presented in graphical form since this representation aids the interpretation and discussion of the data. Processes which exhibit similar structure and which have similar cross sections have been grouped, where possible, in the same figure.
The resonant charge transfer cross section for sodium ions on sodium is given in Fig. I together with the data for H,0, H,0\ and NV)*- Ail of the processes have cross sections, which are similar in magnitude and which show an increase in cross section with decreasing energy down to the lowest measured energies. The results for the symmetric resonant process, Na*"+N*a, could not be determined below about 30 eV primary ion energy as separation of the fast primary from the s'ow secondary ions could not be achieved at lower energies.
Figure 2 gives the charge transfer cross sections for NV and NO ions impinging on neutral sodium. Here
654
65

"IJWUWM^JM-. i^*"» npEEQ^pRB^pannpR* ""?"»5"? iq^^mpnijJWM-.'^vjiupp .aii.uijiJiflp
FORMATION OF SODIUM IONS BY CHARGE TRANSFER 4655
300
2SD •
100 ■
ISO
»•- »So
O HJONHO— MjütHo'
© «"oo, «*til
20 SO 100 ION ENERGY Itvl
FIG. 1. Charge transfer cross sections for Na+, H»0+, HW3+, and NtO+ ions impinging on neutral Na as a function of the energy of the incident ion.
again the cross sections increase with decreasing ion energy. The magnitude of the cross sections for these two ions on neutral sodium are, however, considerably smaller than those for the three polyatomic species shown in Fig. 1. Possible reasons for this difference will be given in the following section.
As in the case of the magnesium results reported earlier,' the possible effects of metastable species in the primary ion beam were investigated. Figure 3 shows the variation in the charge transfer cross section for 50 eV Oj+ impinging on Na as the electron energy in the ion source was varied. For electron energies of less than 16 eV, no excited electronic states can exist in the beam and the cross sections measured are for the 0|+(X 71,) ground state. Increasing the energy above 16 eV introduces some excited states and the cross section for Na+ production can be seen to increase. Above about 30 eV the ratio of ground to metastable ion states remains nearly constant, and the cross section therefore becomes a constant with respect to the electron energy.
a>
O NO*»NO'»I.J»Ne<
*A„
A A 1« 4
O 0 o
iO 20 SO 100 ZOO SCO COO ION ENERGY UV>
Kic. 2. Charge transfer cross sections for SV and NO* ions impinging on neutral Niui function of the energy of the incident ion.
w 1 1 I 1 1 1 l— T T ■ i i i r ■ i i — r r I 1
«V • Na — 0,-N«"
40 ION ENEMY 40«V Oooooo o o o O 1
JO I * O
o
-
20 o o
o o
•
10
0 ' i i i i 1 1 1 1 1 1 1 1 1 1 ■1 1 ..!.. 0 2 4 « S 10 12 M N) W2O22 24 2S2SSO32MMM40
ELECTRON ENERGY I.V)
FIG. 3. Dependence of the charge transfer cross section upon the sou--* electron energy for the reaction 0,++Na—»Oi+Na*. The Oi+ had a kinetic energy of 100 eV.
By keeping the electron energy below 16 eV, the cross section for the ground state Oj+ alone can be determined. Using the data obtained from variation of the primary ion source energy it is possible to obtain cross sections for charge transfer from the metastable states provided the ratio of ground to metastable states is known. This information is obtained in a supple- mentary experiment. The technique employed here was developed previously in this laboratory.* The per- centage of metastable states in a beam of Oj+ formed with 40 eV electrons (the energy used to determine the composite curve given in Fig. 4) was found to be 32%. Using this percentage, the composite curve, and the information given in Fig. 3, the metastable state charge transfer cross sections could be obtained. The results are given together with the ground state curve in Fig. 4.
The NO4 is also known to have a metastable state. In these experiments, NjO was used to produce the NO*
«40
-r ....._,
i20
too A A
eo
60
o
b o o
°00°o
40 o oc °
20
ft . I 1 1
a 0,' (EXCITED) • No — Oj . NO-
o 0,* (COMPOSITE). Na — O, • Na*
□ 0,* (GROUND STATE). No-Oj . Na*
°o ,
20 SO
ION ENERGY !,vl
J i '00 200
o o
O O
FIG. 4. Charge transfer cross sections for different states of excitation of the 0%* ions with Na as a function of the incident ion energy. The cross section is shown for ground slate fV ions, for Oi* ions in the mixture of slates (composite) which result«, when 40 eV electrons impinged on Oi to form the fV, and for the excited states alone.
66

-- miwti'Ff"»1**! mi HI p i ipji i ij 11 nil n:^m-**xmm<ti^'m"---***^!*'*mww!ßi
4656 RUTHERFORD, MATHIS, TURNER, AND VROOM
TABLE I. Recombination energies and relevant energies of reactants and products cited in the text.*
Excited neutral states Ground ionic state Ion state and energy Species and and energy above and ionization above ionic ground
ground state ground state (eV) potential (eV) state (eV)
NaOt.'S,«) WS), 5.13« N, (Jf'V) (»'A-h 8-89 (X'V), 15.58 NO(jrm) (o«n), ~o (x>2;+), 9.27 o,(jr«2,-) (Bi.-), 6.12 am,), 12.06 (a*u.), 4.04
■Atomic energy levels obtained from C. E. Moore. Natl. Bur. Std. Atom» and Molecules." The Rand Corp.. Research Memorandum (U.S.) Cirr. No. 4*7 (1949). Vo). I. Molecular Ieveb obtained from F. RM 5201-ARPA. March 1967. R. Gilmore. "Basic Energy Level and Equilibrium Data for Atmospheric
primary beam. Previous experiments in this laboratory have shown that NO4- ions produced from NjO contain a very low percentage of rnetastable ions.
Attempts were also made to measure the charge transfer between the atomic ions 0+ and N+ and atomic sodium. The cross sections were found in both cases to be very small. A possible explanation for the absence of reaction will be given in the following section.
IV. DISCUSSION
All the charge transfer processes reported here are exothermic or energy resonant for transfer of an electron from the ground state of neutral sodium into the lowest available state of the neutralized ion. In interpreting the charge transfer results obtained, an attempt has been made to account for the excess energy available. In this regard a correlation between the size of the cross section, its energy dependence, and the final states of the products, has been made. Recombination energies and atomic and molecular energy levels used in the discussion are given in Table I.
The four largest cross sections measured for charge e. hange with neutral sodium are given in Fig. 1. Of these the largest process, over the energy range considered, is the resonant charge transfer process Na and Na+. The energy dependence of the other three cross sections is essentially that which would be pre- dicted for mechanisms with small energy defects.1
With species such as HjO+ HjO+, and N,^, it is not possible to say in what final slates the polyatomic species will be produced due to a lack of information on these states. The possibility of the excess energy residing as internal energy in the sodium ion produced can be discounted as the lowest excited stak of Na+
lies approximately 23 eV above the ground state. The charge transfer cross section for Mi+ on sodium,
shown in Fig. 2, is much smaller than that for Na with the polyatomic ions. The excess energy available in the reaction, 10.44 eV, is sufficient to dissociate the Ni into two ground state nitrogen atoms. Another possible mechanism in%*o!ves formation oi the N'j molecule in a highly excited str'e such as the w'A..
Either reaction path leads to a near-resonant mecha- nism. Since the dissociation energy of N», 9.76 eV, is very nearly equal to that available from the reaction, 10.44 eV, dissociation of the molecule would not be expected to be probable without a large shift in the bond length of the neutral prior to forming the dis- sociative state, i.e., the repulsive curves leading to the dissociation limit at 9.76 eV will pass through the Franck-Condon region at an energy greater than 10.44 eV. As a consequence, dissociation can only occur through violation of the Fr .nek-Condon prin- ciple. This violation of the Franck-Condon principle tends to favor the second mechanism outlined above for the Ni+4-Na reaction. This process can be repre- sented as
N,+(Jf'2,+)^+Na(3,;*5,/,)-eN,(w'A.)fc.1o
+Na+(2/>«, •50)+A£(0.02 eV).
The charge transfer cross section between NO* and Na is also small, see Fig. 2. The excess energy, in this case, 4.13 eV, is not sufficient to dissociate the molecule and in fact lies about 0.6 eV, below the first molecular excited state. The only possible reaction mechanisms which lead to near resonance here arc ormation of the ground mo' cular state in a high
vibrational level or formation of the a'll excited state. The latter process is endothermi by approximately 0.6 eV and also involves a large shift in internuclear distance between the ionic "täte and ihe final state. The formation of the highly excited vibrational ground state does not invoive as large a change in internuclear distance and is therefore the more probable reaction mechanism.
In Fig. 4, the charge transfer cross sections for the ground and excited oxygci ions ire shown. Again the shape of both curves is consistent with a mechanism having a small energy defect. The excess energy available from reaction of sodium with both the ground Oj+ state, 6.92 eV, and the excited state 10.96 eV, is sufficient to dissociate the resultant oxygen molcrults (A>=5.12 eV). For the ground state Oj+ ion, the
67
r»g!^»sg3P»I7-g»»ji»ag>^^ JijMjtumm""•," ' ' _'" ' " -., . ~. "gg^
FORMATION OF SODIUM IONS BY CHARGE TRANSFER
TABLE II. Calculated crou sections «ml rate coefficient«.
4657
300°K 600°K 1200,K
Cross section Rate coefficient Cross section Rate coefficient Cress section Rate coefficient Reaction X10»cm» XlCcm'/sec XlOUcm" X10"cm«/sec X10»cm» X10»cmV«ec
Nt++Na 24.5 18.9 13.9 15.2 8.33 12.8
0,+(X«n,)+Na 18.5 13.8 13.1 13.9 9.78 14.7
Oi+(a«n.)+Na 26.2 19.6 23.6 24.9 21.2 31.8
H#++Na 31.9 27.4 32.1 39.1 31.4 54.1
N,0++Na 28.7 20.2 27.1 26.9 25.2 35.6
dissociation products resulting from charge transfer must both be in the ground state. This process can be represented as
0,+(X*n,)+Na(3*, *$,,,)- *20(»/>)+Na(2fVS.)
+1.80 eV.
The two oxygen atoms would be expected to carry away equal amounts of the excess energy.
For the metastable 0»+ more energy is available and as a consequence the products of a dissociative process may be in excited states. Oxygen atoms in the excited lD and '5 states as well as the ground *P state are possible products. This density of reaction channels is greater than for the ground state process and may explain the greater cross section for this species.
The charge exchange cross sections for 0++Na and N++Na art as stated above, very small, and no data were obtained for these processes. This result may be attributed to the lack of energy levels available in either product to make near resonance possible. A similar result was found previously for the reaction of 0+ with magnesium.' For N+ on magnesium, energy levels allowing a near-resonance mechanism exist, and the cross section was found to be significantly larger.
In general, when molecular specie *r» being con- sidered, the possibility of near resonance will generally exist for an exothermic prxess. The multitude of bound electronic states nth their associated vibra- tional and rotational levels coupled with dissociation continuum makes near resonance possible for all exothermic processes. This general conclusion is well demonstrated here and in previously reported charge transfer processes.1 s
Other measurements of charge transfer cross sections uetween atmospheric ions and sodium have been re- ported. Henderson et al* have obtained results for Ni+ and 0»+ on sodium in the energy range 25-200 eV. The energy dependence of their cross sections is similar to ours, but the absolute values tend to be about an order of magnitude lower. Peterson7 has also measured Nt+-lS'a charge exchange. Once again, the energy dependence is similar in the two experiments but his absolute values are about a factor of 2 larger
than ours. It should be noted that both Henderson el al. and Peterson used hot wire detectors to obtain their beam densities.
The results reported here have been obtained for primary ion energies ranging from about 1 to 500 eV energy. Much of the interest in charge transfer cross sections between atmospheric ions and metallic species is in the energy range from thermal to 1 or 2 eV. In order to extend our measured charge transfer cross sections to lower energies, an extrapolation technique has been developed and has been described fu'ly in a previous publication.*
One assumption inherent in the extrapolation pro- cedure is that the relative abundances of the various products emerging from the capture-formed complex are independent of the relative kinetic energy of the reactants. Failure of this criterion invalidates the derived rate coefficients. In general, whether or not the extrapolation technique is valid can be seen by ob- serving if th" calculated cross section fits the experi- mental data to the lowest energies considered. A com- puter program has been developed to perform the calculations. Results for the five ion species which were found to extrapolate are given in Table II. Both cross sections and rate coefficients are presented.
Thermal energy rate coefficients have been obtained for charge transfer from Nj+, 0»+, and NO! to Na bv Farragher et al* using a flowing afterglow technique. For N,+ and Oi+ they obtained values of 5.8X10-'° cmVsec anc" 6.7XIO"10 em'/sec, respectively, while for NO1", values of about an order of magnitude less were obtained. The value for Nj+ and Oi+ are between a factor of 2 and 3 lower than those given in Table II.
ACKNOWLEDGMENTS
The authors would like to thank both Dr. R. H. Neynaber and Professor R. F. Stebbings for their interest in the experiments and their helpful comments on the manuscript.
•This work was supported by the Defense Nuclear Agency under Contract No. DASA01-69-C-0044.
t Present address: Dept. of Physics, University of Missouri at Roll». Roll«, Mo.
68

Huf* Jt-PYPf WM1* äW«SW; HM.I«# iwiwpp^iinpn jMfjnS pBu^wwpp^i *^JI •^^^^^^W^WBR^PWiippiiWI
4658 RUTHERFORD, MATHIS, TURNER, AND VROOM
1R. S. Nardii and A. B. Bailev, J. Geophys Res. 70, 3687 (1965).
'J. A. Rutherford, R. F. Mathis, B. R. Turner, «nd D. A. Vroom, J. Chem. Phys. SS, 3785 (1971).
»R. E. Honig, UCA Rev. 23, 567 (1962) * B. R. Turner, J. A. Rutherford, and D. M. J. Compton, J.
Chem. Phys. 48, 1602 (1968); R. F. Mathis, B. R. Turner, and J. A. Rutherford, ibid. 49, 2051 (1968).
• See, for example, Ref. 2, R. F. Stebbings, B. R. Turner, and
J. A. Rutherford, I Geophys. Res. 71, 771 (1966) and B R. Turner, J. A. Rutherford, and R. F. Stebbings, ibid 71, 4521 (1966).
•W R. Henderson, J. E. Mentall, and W. L. Fite, J. Chem. Phys. 46, 3447 (1967).
»I. R. Peterson (prjvau ommunication). Wolf and B. R. Tuni-r, J. Chem. Phys. 48,4226 (1968).
•A. L. Farragher, J. A. Peden, and W. L. Fite, J. Chem. Phvs. 50,287(1969).
69

T-Fri" im'.,*«;'i»-A^^jf,jjui^K»|p|pp||
APPENDIX D
FORMATION OF CALCIUM IONS BY CHARGE TRANSFER
Preceding page blank
71

w^uim w,,vmmjumdw*imJLIJPIIvmim-***^^».*? .^ ^j ^^^^lll*^w*l|uwj»fit|P||Jt]H|lWWJ'J'111 mi'-" lewwt.11«!^IIPW-J1 ''*-''."JPJi'^^^ftJlW.p^'i*1 »iwp*w^Jwgw»gWffBIP^pHgBy,*|iJ'!i..IWJWiPWPSjtgli«fW31gi|Mlj»EHWP
FORMATION OF CALCIUM IONS BY CHARGE TRANSFER*
J. A. Rutherford, R. F. Mathis, B. R. Turner, and D. A. Vroom
Gulf Radiation Technology A Division of Gulf Energy & Environmental Systems
San Diego, California 92112
ABSTRACT
Charge transfer cross sections have been measured
in the energy range from 1 to 500 eV for nine common atmos-
pheric ions in collision with neutral calcium atoms. The
ions studied are O , N , N NO , O HO , HO , + + * I & 5
NO , and Ca . In all cases the cross sections are found
to be large indicating near resonant processes. Mechanisms
consistent with this observation are given. Extrapolation
of the measured cross sections to thermal energies has
also been carried out.
This worn was supported by the Defense Nuclear Agency under Contract No. DASA01-69-C-0044.
Preceding page blank
73

^^^^^^.^^^^^^WBW^^^y^r^^
I. INTRODUCTION
The experiments discussed represent the third in a series of studies
undertaken to investigate the importance of charge transfer as a method
of producing metallic ions in processes related to aeronomy and the low
energy aspects of asymmetric charge transfer. Studies involving magnesium 2
and sodium have been reported previously.
Calcium is one of the more dominant metallic species present in the
upper atmosphere. Its presence has been observed in rocket experiments 4 5
and in studies of optical emissions from the atmosphere. ' The low
ionization potential of this species (6. 113 eV) coupled with the fact that it
is atomic, make the normal processes for deionization in the atmosphere
(dissociative recombination and charge transfer) ineffective. As a con-
sequence, calcium ions vill be long-lived and therefore may have an
influence on the chemistry of the upper atmosphere greater than the con-
centration of this species would suggest.
II. EXPERIMENTAL
The experiments were performed using the tandem mass spectrometer
apparatus described in detail previously. In this instrument, a modulated
neutral beam of the metallic species is interposed between two mass spectro-
meters. The first of these spectrometers produces the primary ion beam
while the second is used to analyze the products of reaction. The primary
ion source is constructed in such a manner that the energy of the electron
beam can be regulated with enough precision to allow control of the state
74
^mjimttuH

fsifpnRiMpMiviHPiniiipq
of excitation of the ions in the beam.
The neutral beam was formed in a heated cell as described in
reference 1. The vapor pressure in the cell was generally of the order
50 microns. At the pressures used in the metal vapor cell, dimerization
of the metal atoms is not expected to be a problem. In spite of this,
during the course of the experimental investigations attempts were made
to see charge-transfer to dimers in the beam. No evidence of such
species was found. As in the previous experiments, both activation
analysis and molecular effusion techniques were used to determine the
neutral beam density. For calcium, these two methods agreed to within
1%.
Other possible experimental errors have been discussed in reference 1.
The total error is expected to be similar here, that is ± 30% at high impact
energies rising to a factor of ± 2 at the lowest energies.
HI. RESULTS
Charge transfer cross sections have been measured for collisions
between nine ions and neutral calcium in the energy range from 1 to 500 eV.
These cross sections are presented here in graphical form. Unless
otherwise noted, the primary ions were formed using 4Ü eV electrons.
In Fig. 1 are shown the charge transi'er cross sections for Ca , HO , + + 2
NO and N incident on Ca atoms. The results for the symmetric Ca on
Ca are shown only for interaction energies above 30 eV since the fast
primary and slow secondary calcium ions cannot be separated below this
energy.
Cross sections for HO and NO on Ca are given in Fig. 2 while those
for N and O are given in Fig. 3. It should be noted that the O cross
section is for the ground state of the primary ion only. A beam of pure
75

r-«j«M.m^g«q^g^pjwsTqBPpngyp-yyüwigipnn»j '■»■■'T'i'!" -'!"l<mg»^*'-T -u .miW'JLWP^WW^Wi^pg^aig^WWjy
ground state O is obtained by keeping the electron energy in the primary
ion source below the threshold energy for formation of metastable states.
When the electron energy was raised above the threshold for metastable
state production, only a small charge in the charge transfer cross sections
was observed. The NO ion is also kr own to possess metastable states.
In these experiments the concentration of metastables was kept low. As
a consequence, the cross section curve for NO on Ca in Fig. 2 may be
taken to be that for the ground state of the ion.
For O a large effect due to metastable states was observed. In
Fig. 4 the results obtained for the charge transfer cross section at one
ion energy (100 eV) are given as a function of the number of metastable
ions in the beam. The number of metastables is varied by changing the
primary ion source electron energy. Below 16 eV, no metastable O
ions may be formed and Fig. 4 shows a cross section of about 55 XlO"*" cm
As the electron energy is increased the cross section is seen to rise reach-
ing a limiting value above 30 eV. This rise is due to the metastables in
the beam. In Fig. 5 the charge transfer cross section for ground state O + 2
on Ca is shown. This curve is obtained using O ions formed by electrons
with energies less than 16 eV. The composite curve, also shown in Fig. 5
is obtained using O ions formed with 40 eV electrons. In order to obtain
the curve for the excited state alone it is necessary to have the ground state
cross section, the composite curve and the percentage of metastable ions
in the beam. This latter value is determined in a subsidiary experiment.
The cross section for the excited state alone is also shown in Fig. 5.
IV. DISCUSSION
Mechanisms postulated to explain the experimental results obtained
must in some manner account for all the energy released in the reaction. 1 2 Previous experiments in this series ' have shown that where excited
76

»rTTi.*". '-'.'-"■■■ ■-"MJ...,^ „„^^yg.^^y^^^jjr,! i um i^,!^, ^ ..I ,j vmijffngii^uJimv .lum, pBmpp)jni|| HPUIHP.JIWIM-1I ■»I'-.-^IH, I
states of the products allow near resonant reactions, the cross section
for the process is largest at low energies and decreases as the energy
of the ions increases. When no excited states can give a resonant
condition, the cross section was found to be small.
For calcium, all the cross sections were found to be large and product
states giving resonant channels are therefore postulated to be the main
reaction paths. Table 1 contains the states of the reactant and products
used in the discussion together with their energies. The reaction paths
suggested below are to be considered only as possible explanations for
the results.
Figure 1 gives the results obtained for the symmetric charge transfer
process of Ca + Ca. This reaction must have exact energy resonant for
ground state Ca and as a consequence a large cross section could be
expected. The charge transfer cross sections for HO , NO (Fig. 1) + 5 1
and HO (Fig. 2) on Ca are of comparable size to the symmetric case.
The energy dependence and magnitude of these cross sections is that
which would be predicted for reaction channels having small energy defect.
For these polyatomic species it is difficult to ascertain what the states of
the products might be. For HO two possible mechanisms are suggested, 2 +
however. One of these involves production of the Ca in an excited state
and the other leads to dissociation of the polyatomic molecule. These
reactions can be written as
H00+ + Ca(1S )^H00 + Ca+(5s,2S ) + 0.039 eV ,
and
H00+ + Ca^S )^H + OH + Ca+(4s,2SJ + 1. 39 eV .
2 O £
The excess energy in the last reaction could go into kinetic energy of the
neutral produc*s.
77
tmmmmmmm

■g
C nJ +J y to <u
o CO « 'a * Li *
g x K di
a >
r-i tu u
ß rt 01 &) •rt 00 «H
CD ß
.2 »
3 2 o u
«
W CQ <:
CO a o
3 cr <u v
o 3 O a
"S «}
4) -t-j «I CO
<u
U X 0)
c o
a O
•r«
4) > O
d
00
ß 0)
2 1 ß 3 O h Ü
>
nt
ß 0)
•4->
O a. ß o ni N
ß O
> 4>
00 .2 Ki a)
ß m
•o S ü ß 2 *» ™ u U « M y **
r i rt *> W *» > • o
JO nl
•Ü v fi ♦* «a j2 CO °>
.2 -o 3 5 J 00
on Is-
CM
2 "■ QO CO O
O CO "^°» o **■ cP co A b,
04 O»
CO in
(X vT in ^<
GO
^
CO CM
CO
CO • m co CO r~ sO ^H
■^ N£) o
■-I CM a * * C4 o~* ■» O l—l
CO 5T co
% 1
co a, •—I CM <M <M X X
CM*
,00
NO
I
< 3 W
CO
CQ
CO
CM co
CO --. .—,
a
U
a CM
co «X
CM
CM c I 00 W J rO x X o CM 2 O x c o
<M
CQ
M "" 2 ^
ß c • •< o o
vO
>« o
t>
78

njji 14 WA iveCPnPVWPmsv
The reaction of Ca with N o is probably dissociative in nature since
the exothermicity of the reaction (6. 78 eV) is much greater than the
dissociation energy (1. 68 eV). The H.
since this species exist only as an ion.
dissociation energy (1. 68 eV). The HO reaction will also be dissociative
The charge transfer cross section for N on calcium, shown in Fig. 1,
is somewhat smaller than that for the polyatomic species but is still large.
The excess energy available in this reaction (9.47 eV) can go to producing 7
N in an excited state. Several N states lie in the correct energy region. 1 1_- 3 - 1 Among these are the a H,a 1) ,B'S and the W A states. The 0 g u u u
several reaction paths available may account for the large charge transfer
cross section for N on calcium. to
The charge transfer cross rection for NO on calcium is smaller than
that for N on calcium. Consideration of the energies of the possible m
states available in the products shows that a near resonant channel exists
if the Ca is formed in the (4p, P,#r>) state. This reaction may be written
as
NO+(X12+) + Ca(1SQ)—NO(X2ri) + Ca+(4p, 2P°/2) + 0.003 eV .
The energies of the states involved are given in Table I. The fact that
there is only one possible near resonant channel may account for this cross
section being smaller than that for the N case. It is of interest to note that
for this reaction, where only one reaction path may exist, the experimental
scatter in the data is less than for the case of N where many paths may
exist. Although no concrete evidence of how the presence of these different
paths leads to increased scatter in the results exists, one can postulate
that the different possible channels have different reaction probabilities
as a function of interaction energy thereby leading to structure in the curve.
This structure appears in our data as scatter.
For O in the ground state charge exchanging with calcium, the cross
section values are similar, at low energies, to those for the NO reaction.
79

laS^B^BBi^HHH
Here again a single reaction path, leading to very close energy resonance,
exists. The reaction is represented as
0+(4S°) + Ca^S )-~0{3P) + Ca+(5p,2P.) + 0.001 eV . o t + 12
In previous studies involving reactions of O with Mg and Na the
charge transfer cross sections were small. In these forme r cases no
set of product states allowed a near resonant mechanism.
Near resonance also exists for the reaction of N with calcium. In
this case the energy defect is larger than for the O reaction and this
may be one of the factors which causes the N cross section to be smaller
than that for O at low energies. The reaction may be written as
N+(3P) + Ca^S )—N(4S ) + Ca+(4f,2F°) - 0.016 eV . o o
The results for O on Ca, seen in Fig. 5 again indicate that near
resonant processes may be important. For the ground state two near
resonant processes are given
and
C>2(X2ng) + Ca(1So)-02(B32J + Ca+(V) - 0. 1 eV ,
c£(X2n ) + Ca^S )—•20(3P) + CaVs,) + 0.835 eV . 2 g o t
Of these two the second is the more probable since the excess energy can
easily be absorbed by the two ground state O atoms. For the excited
state of O only a dissociative process is suggested. If both the O atoms
are produced in the ground state the reaction may b° written as
CV (a4II ) + CafS )^20(3P) + Ca+(2S J + 4. 873 eV . 2 u o £
The excess energy here is great enough that one of the O atoms may be
produced in an excited state such as D or possibly even S. It should
be noted that if dissociation does occur in these reactions, oxygen atoms
with kinetic energies well above thermal are produced.
Using our experimental technique, interaction energies of less than
1 eV are difficult to obtain. In many applications the interest in charge
transfer reactions lies in the energy range from thermal to 1 or 2 eV.
In order to obtain cross section values in this region, an extrapolation
80
. . , .^Jto-HJM.-,

i.i i i iii — i mi mump »in " i'.m;'imBiiii»i."."'ij-»ni imwm.in, WJI 1 tmmmmm RKKJUl!l'.WiiMAU.MUU#.|W>!
g method developed in this laboratory has been used. This technique
assumes that the relative abundance of the various products emerging
from the interaction complex remains fixed. If this condition is not met,
the technique will not give valid rate coefficients near thermal energies.
In general, whether or not the extrapolation procedure is valid can be
seen by observing if the calculated cross section fits the experimental
data to the lowest energy of measurement. In this regard, we have
arbitrarily set an average limit on this deviation of ± 10% for the fit of
the experimental and calculated cross sections at the lowest measured
energies. We also require that no deviations larger than this occur over
the intermediate energy range between the lowest energy points and the
high energy region where the calculation is normalized to the measured
cross sections. In the present studies, the extrapolation procedure is?
found to be valid for all systems. The cross sections and rate coefficients
obtained for three different temperatures are given in Table II.
ACKNOWLEDGEMENT
The authors would like to thank both Dr. R. H. Neynaber and Professor
R. F. Stebbingt for their interest in the experiments and their helpful
comments on the manuscript.
81

gpmmBm-' W*®*?Gtmmß. i^RJlPff^HnlW*
C
4) o u
Li
X) a d m C O
u
en en O u o
TJ 4>
3 y
O
<
o o o
c u 0) 4)
•r-< ai U "^.
,3 CO
41 £ 0 ü
O o —4
4> o •*-» 1—1
id X
o o o
u 41
0) to 0 u u
o <J U o
c o
•rj (M U C 4>
to <» H B) O O
u *
q 4) H o -
4) o U o
•—I i> (
+-* »■ m «
c
C £ 41 «ir M 2 o ~" b u
4!
.•"I
<T-
00 in
ro
a* o
00
rO
r3 u
IN
CO
a*
U
0
o CO
00
<N
R.L Q.I
CO
in
OJ
in rO
<N
CO
<N
6
CO
<N
00
r-
r- <N
U
M
X
- 2-L.0.1
in no
n
U
o
■* •*
CO 00
o
vO
CO
rO
O
m
U
O Z
t~ r- el • in 00
! *6
e<j in ■*
in m
m NO r-
in in <£ ■*
<N o- O » • •
o
U
oo
00 in
it U
I..S !M
00 in
PO
-O
U
82

REFERENCES
Present address: Dept. of Physics, University of Missouri at Rolla,
Rolla, Missouri.
1. J. A. Rutherford, R. F. Mathis, B. R. Turner, and D. A. Vioom,
J. Chem. Phys. 55, 3785 (1971).
2. J. A. Rutherford, R. F. Mathis, B. R. Turner, and D. A. Vroom,
J. Chem. Phys. 56, 4654 (1972).
3. R. S. Narcisi and A. B. Bailey, J. Geophys. Res. 70, 3687 (1965).
4. A. L. Broadfoot, Planet. Space Sei. 15, 503 (1967).
5. H. S. Hoffman and M. S. Longmire, Nature 218, 858 (1968).
6. B. R. Turner, J. A. Rutherford and D. M. J. Compton, J. Chem.
Phys. 48, 1602 (1968), and R. F. Mathis, B. R, Turner and
J. A. Rutherford, J. Chem. Phys. 49, 2051 (1968).
7. F. R. Gilmore, J. Quant. Spectry. Radiative Transfer £, 369 (1965).
8. F. A. Wolf and B. R. Turner, J. Chem. Phys. 48, 4226 (1968).
83
PW «mmm tpM HWj HP j IIU ' ?'W i ■ IB^WWF wygwyjpiy^B p»
1 1 1 1 1
- <p a < o o a
<! o 0 a o
- o<3 0 a
O O a o< 0
— cx3 o
o a
ö3 o a o
- <P o D -
0 o a <
«o O a 0 O
o 0 o O
a
a + 4- o o o o
- 0 ,~
o o a O + + To - a ^oo<;
o O O O
a a
+ CM CM + O I Z CM
Y ♦ I T " o o ? - o o D
o o □ V + + 3
o 0 a + o o + Ta CM CM +CM u x z z o a < o O a
1 1 1 » 1
o 8
O
-8
-8
8
_ o
- m
> o cr UJ z UJ
Z o
- CVJ
8 in
8 vr
8 8 8
u
c c o 00 C
-i-i
% ■a E I-
CO
C O
+ «M z TO
+ * o
M #
Z CO C
+ ".2
T3 + y tt c u H
tu £ -c O *J "•<« 0) O
" 00 ^-» U O 4, <u c CO m
(0 j)
O «->
w o ^1
CO
C nl fi
(uiobs9Hoi)«o
M to
u o
u 3 00
84

üiBijngwpwsipifppwBf^wP - ^WHOTBHj» PSVpm V» I WWWW'SWflWS»
! I r " 1 1 1
-
□ a a
o o
o o
-
a
a o
o o
a
a o
o
D
D o o
o
- a
□ o
o
-
a o + a
D
a
o o
o o
x + + o o +
?! O T o o
++ + a
a D
o o
o o I z
a o
1 1 1 1 1 1 o m to
8 ro
O «n CM
8 CM
o «n 8 O
m
8
8 CM
-8
o $ > o cr UJ z
O UJ CM
z o
- m
(0
U -«
u *-> 3 0) C C o 00 c
•f-l
00 c a, £
c o
o
C m
+ m
°«§
°J •H
CO u C C O •■*
en m ro O u u
(tuobs on-ö
o >> 00 u a c
1 c 4» o 00 -H
«d c i =•
3 00
91-
85
k-~-J" "—*■"**• gjJäatj^ÜilfclfciBi;

I^pws^^? pilTOWiy^ij»im»Jii.w|i IIIH.MHU _ii|jBiju.imiiwii|.jmw^«^ ujuui immMKn STSJ II ^-.MJUqiM
1
««
<3 <3
3 <3
<
< o < o
< o < o
<3 o
! o o o o o
o LÜ
o g o o z o o
o QC o
o ■K, + o O o o o + + 0 o z o » t 0 o O
V o o + + + + D o z
<3 o
m
8 CM
8
«n %
z UJ
X
CM
J I-
o CM
Z o
m
O CM
O CO
o 10
o o CM
(wo bs oi) -° 91-
m d n) Ü ~* n) h
3 4> ß
C o
c ■r"l 00 c
T3 C «J
c o
2 S rt$ O
•»-4
■B ? O —■
<« U m C 0) -,-t ß .1
00 an
O «, ** c « V ** 4)
». . *C 0)
1 .2 00 U V. C rt 3
J3 ^ U rt
4J
3 00
||AHU|

<i^L«*4.^«'w*-wwj|iJ*p!i|M w> JH, ^ $gz&®l®w*m™i*0mmmG>w W? *u w jw J'«
i 1
r- 1 ' 1 r 1 -1— 1 1 1 —r—
- o —
— —
—
o
o
o
—
— o —
—
0 o
o o
+ o
<-> +
CM o » o u
z o > 0)
—
+ + CM o 8
—
0
-
i 1 1 1 1 1 1 1 1 1 i
o
If) to
-8_
if)
> 4>
_ u' rr CVJ UJ
z UJ
o o oj a:
h- o UJ
m UJ
_ o
m
O O O
o CD
O § o c\J
■:
(UJO bs 01)-° * 91- '
c o *H •
■4-* > U 01 oi
^-4 o 01 o a; o VM
u o 3 >s O on U) 14
C o
01
•r-l 01
>- u u -#-• id 01
b C
O, rt 01 TJ x; rt
x: 0+ M a. U a
01 c x: o H *-> U <U+ tn rt tn U rn O + h (M u o o>
VI t 01 rt c () <a u + *-> i + r\J
0) o bfl >H c R) o x: •»-• u o <l> nJ ,r; 41 <■' l-l
Vl 01 0 X 4) o u t. o i> »w
T3 >» »-« Ml (Li t> u< 01 4» r Q 41
TP
01 I. 3 00
87
lfatifräW**7iHi.1*. «fc«^^..
«EBW«W?Hfl«(Bp^^PHJ«qpwa»5W"!<!W*
I
1 1 + 3
1 1 1 1
o *o ♦ 0 — ü OJ
u ♦ O 0
i *M
CM O 1 a o <
o - »
* o o
o ♦ a O <J — Ü *"»
o o ♦ LÜ a o < ♦ UJ H
— —. h- c/> D o < — o V) Q LU o
£L z
o D o <
O 2E _ X O o: C i o o _
LU u o *•"* *** + W + M ♦w o O O a o <
D o <
D
o
o
o <
<
— D
D
o
o o <
< ■—
o
o o
o
— o —
1 1 1 1 1 1 o o o o o m o m IO CM CM i^m
o o
o
I § (iuobs9l„oi)-°
o + <M™ ö M 0 O o u <u M
4-* JZ 4J u s «J cu 1 4->
CO 0> o o co
C
1 "0 C >
m o 3 •r-l o o
+ <M*« *r O 00
u c 0)
CD 0 X X «44 •* O V4 c
o o * CO 4->
CM c 0 t—4
3 o CO CD
•f-i 4-1 4> «J CO »4
-4-» -r4
O •r4
u c o . o c
•r4 4->
•£ CO •r 4)
«4-1
0 *3 -■* 4_1
> co CO V CO
4-*
o m
a> o 4->
ca *->
CO CO o u s ** til -H
>- e> a:
CO
4-»
C
u
■C
c u
O 0) ° »,
LU u H ^, 4) J5
z 0) »44
CO U d)
o LU «4-1 •r4
T3 >• 00
£ *4 rt o 4J SH
CM Z h W ^4
o O <*4 8 *" c
CO C O o
0J
2 o o -r4 4->
•I-*
«» u 4-» X „ 1) co 0)
■a CO CO Z E o C 4-> t,
m •r-*
c.2 -r4
.. C a> 4* ti i; DO c nJ
0 O «M - 0
u X a>
O <-\ •" ■H U o 4->
U U -D CM i d O ft)
DO u
3 ■« 00
m <fl " a U
CO «J
c E
in
i)
DO
88
-^gj--j'- --

(PHBWBWPSW St. J K ■,'.. IAU. w*M wwujHBgi
APPENDIX E
FORMATION OF IRON IONS BY CHARGE TRANSFER
89
i TlDMIJMiiihTiTrtt

FORMATION OF IRON ION5 BY CHARGE TRANSFER
J. A. Rutherford and D. A. Vroom
Gulf Radiation Technology A Division of Gulf Energy & Em -onmental Systems
San Diego, California 92112
ABSTRACT
Reactions of iron atoms with common atmospheric ions have been
studied in the energy range from 2 to 500 eV. The -ons considered are
H , O , N , N , NO , O , H20 and HO . With the exception of one
proton transfer reaction, all of the processes studied involved charge
transfer. All but one of the charge transfer reactions were found to
have large cross sections over the total energy range and probably
proceed by near resonant energy paths. Where possible the measured
cross sections have been extrapolated to thermal energies.
INTRODUCTION
Iron is known to be an important minor constituent of the upper 1 2
atmosphere; ' its presence being accounted for by the ablation of
meteors. Even though this species is in low concentration, it can have
an important effect on the chemistry of the upper atmosphere. Its low 3
lonization potential (7. 870 eV) allows it to undergo charge exchange
with all common atmospheric ions. Iron ions, once formed in the
This work was supported by the Defense Nuclear Agency under Contract No. DASA01-69-C-0044.
Preceding page blank w
j^^.:..^.^^^^ | -nfflgEUffi--| n minrriliM

j.iiiiajHiu ^twwm5ijjiuL'iiii.ipi
upper atmosphere, should have a long lifetime since radiative recombi-
nation (a process known to have small cross sections) is probably the
only significant loss mechanism.
The results reported here represent the fourth part in a series
of experiments to look at asymmetric charge transfer processes involving 4
metal atoms. Previous sections have dealt with reactions of magnesium, 5 6 sodium and calcium interacting with common atmospheric ions. These
previous studies have shown the importance of near resonant reaction
paths in low energy charge transfer. The results in this present publi-
cation support this observation.
EXPERIMENTAL
The apparatus and experimental techniques employed for measure-
ment of charge transfer processes between ions and metal vapor have 4
been described previously. Briefly the apparatus consists of two mass
spectrometers operating in tandem. The modulated beam of metal vapor
is interposed between the two spectrometers. The first mass spectrometer
is used to analvze the primary ion beam while the second analyzes the
modulated beam of secondary ions produced by collisions between the
primary ion beam and the metal vapor.
The low vapor pressure of iron compared with the previously
studied metal vapors required a molecular effusion source capable of
withstanding high temperatures. This cell was constructed from a high
purity alumina crucible surrounded by tungsten foil. Passage of current
through the foil served to heat the crucible. The temperature was monitored
using an optical pyrometer. Pure iron wire placed in the crucible was
used as the source cf iron vapor. Pressures of the order of 50 microns
were generally used in the heated crucible. The neutral beam was formed
by effusion of the iron vapor from a small hole in the side of the crucible.
92
jLgtumäg^MUtaimäVitiM^^

At ins pressures used in the metal vapor cell, dimerization of the metal
atoms is not expected to be a problem. In spite of this, during the course
of the experimental investigations, attempts were made to see charge -
transfer to dimers in the beam. No evidence of such species was found.
4-6 As in the previous metal atom studies, two methods were used
to obtain the neutral beam density; molecular effusion calculations and
activation analysis. The errors in both methods are larger here than
in the previous studies. The molecular effusion method suffers from
errors in temperature measurement due to use ->f the optical pyrometer
while the neutron activation technique is troubled by trace quantities of
manganese. Errovs of up to ± 30% can exist for both methods. The two
methods gave, however, iron beam densities that agreed to within 1. 5%.
The possibility of a larger systematic error in the absolute cross sections 4-6
than that reported for the other metal atoms data exists here because
of the increased uncertainty in the neutral beam density. The maximum
possible error here is ±50% at high impact energies increasing to as
much as a factor of 2. 2 at the lowest energies.
RESULTS AND DISCUSSION
The cross sections for charge transfer, and in one case, proton
transfer, between neutral iron atoms and several ions were determined
over the energy range from 2 to 500 eV. An electron energy of 40 eV
was used to form the primary ions. These cross sections are presented
here in graphical form since this type of presentation aids in the
discussion of the data.
The cross sections for the charge transfer of neutral iron with the + + + +
ions HO , O , O and H are shown in Fig. 1, while those for charge 2 2
transfer with N«, NO and N are shown in Fig. 2. The charge transfer
93
w Jt'fr-nft --4- i^-'-T1—Tg"4ir-- -~-;ir. if~, ■

gjWgjBTiiWüfUlMiPl BH^.".». i mmw. | ll ^||HIJg|H.WWtJ..fiJPL.UI. JM^g,-! 3BT*
and proton transfer reactions involving HO are shown in Fig. 3.
The cross section curves shown in Figs. 1 and 2 are all very
similar in that they show an increase in magnitude with decreasing ion
energy. This increase is similar for all seven species shown. In
addition to this, the absolute magnitudes of the cross section curves are
all quite Jarge and do not show a large variation from species to species.
.-16 For example, at 10 eV all the cross sections lie between 10 and 35 xlO cm'
while at 500 eV the total range of cross sections is between 5 and 30 X 10 2 + + cm . It is interesting to note in this regard, that H and O (see Fig. 1)
which have similar ionization potentials, have cross section curves that
are essentially the same.
The explanation for the similarity in the magnitude of the cross
sections for all species in Fig. 1 and 2 probably apices due to the multi-
plicity of states in which the iron ion may be formed by the charge trans-
fer process. Previous studies of charge transfer with metal atoms in 4-6
this laboratory have shown that only for cases of very near resonance
are the cross sections large at low energies. That is, in cases where
formation of the product particles in specific states can result in a small
energy defect, the cross section is large. Iron, with its multitude of
excited ion states allows a near resonance situation to exist for all the
reactions shown in Figs. 1 and 2. Near resonance can, in some cases,
also arise when the resultant neutral particle is left in an excited state.
In our experiment, the state of excitation of the products could not be
determined.
The cross section curves for the reaction of iron with H O (see
Fig. 3) differ from those discussed above. The charge transfer process
leading to production of the neutral product HO and H can be seen from
the figure to reach a maximum at about 7 eV ion energy and then decrease
to lower energies. Such behavior is indicative of a non-resonant process.
94
MMMlrtllitittlMiiriMilMitfitMiliiiMiifcriii n nmm mit m—iwiiw mirninr ■ iflinwtfiWWrft r

|jl||piPH^p;^jaWPBBWW^?^g'^WBP!Sg!W>WPjWW>gp^?f>MPi4J' Wjiypimmau^j». . »i; ' ji'«jpiw»iv,.j|ixi«i^ji,iy^w»^it^i>i^;Tii'™»i4ji*it.-. . i.i v-'>^-»*^rwww,"">»wwigi"»f>»<T«iB'
+ 7 The recombination energy of HO is thought to be about 7 eV and the
it
reaction is therefore endothermic by about 0.87 eV. The cross section
curve is therefore what would be expected in this case.
The proton-transfer cross section shown in Fig. 3 decreases
rapidly with increasing ion energy over the whole range considered. This
behavior is what would be expected for an exothermic ion-molecule
reaction. The excess energy for this reaction can come from the FeH
bond formed. As a consequence, when one considers both the charge
transfer and proton transfer processes, it can be seen that a large cross
section for destruction of HO exist over the entire energy range studies.
The effects of long-lived excited states in the primary ion beam were
investigated for cases where these species are known to exist (O , O + 2
and NO ). In all cases very little change was observed in the cross section
curve when excited states were introduced into the primary ion beam. The
relative independence of the cross section on the state of excitation of
the incident ion is not surprising since the multitude of states available
in the iron ion makes near resonance possible for these species also.
Considerable interest in charge transfer cross sections for the energy
range from thermal to 1 or 2 eV exists. Our measurements cover only the
top end of the range. In order to obtain values for lower interaction energies
an extrapolation procedure has been developed. This procedure, which is Q
described iuiiy in another publication has been used to extend the iron
charge transfer cross sections to lower energies.
The extrapolation procedure operates on the assumption that the
relative abundance of the various products emerging from the reaction
~i:ays constant over the total energy range from thermal to 500 eV. Failure
of this condition invalidates the rate coefficients obtained at low energies
from the calculation. In general, whether or not the extrapolation procedure
is valid can be seen by observing if the calculated cross section fits the
95
^^tÜMtttltlttämtlSmatttttmam^mmmmmmtmmmi<i« 11 ilraHiMin i i wmtätttUKm— nr

..iMJWMIiliU'jmW'W'J!1- PI'W'ilip.'I'MJJ'li' 11 . . I I Ü I I TIT*"W! 1 WWffi.1 JJSHiJWK?*
experimental data to the lowest energy of measurement. In this regard,
we arbitrarily have set an average limit on this deviation of ±10% for the
fit of the experimental and calculated cross sections over the lowest
measured energy points. We also require that no deviations larger
than this occur over the intermediate energy range between the lowest
energy points and the high energy region where the calculation is normalized
to the measured cross sections. Results for the charge transfer reactions
between iron and the ions studied for which the extrapolation is valid
are given in Table I. Both cross sections and rate coefficients are
given.
ACKNOWLEDGEMENT
The authors would like to thank Dr. R. H. Neynaber for his
continuing contributions to the research and for his comments on the
manuscript.
96
tfjjjjjjjlijfllffljjjffi^ nrnauiWHr i • --umaf15*'--' *4

"SWPSPPWW tüiiHM* '«wBüpjt^B.miiiian.iflHw .Mpa*W.P^U(iJV^;W N" ll!»|I'Pl^WB?l«P»k^*J»» - -Ji.mfÄPW1w*P1H?!fIä4Ä|J'*aP!Wt!»'
C 01
tu o U B «
S
(0 C o
•»-4 +-> y u
w tn ui O
ü XI 0)
•—< 3
u
w
*J U s «> v es
• r*
sO co —i in ■«J*
a E t>- •* P4 oo •
o o •
ro
8 u
^2 <£ •
t^ oo vO CT» mi CO **4
» 2 w «3 x
o Ä ! o o OJ ß ■—< O
•43 N
2 S lg ü o*
t^ o O in ro o
«3 l~ in ^H CO in r» <r (0 o -t O -" u X ü *
-M U C 4) v to
•f* ^^ co 04 i^
" «*'- ■* ■* (M ^ •* 00 00 *H P 1) O 0
• PO in f—i
• <M
ro t^ •
CO
u2 o
o> «-* *>
0
a x «
o o \0
0
* p y c
in
(M
0^ O
in o ■0«
00
od
in 04 •
H to —• r~\ ca M O 0 "■ h X ü
•y o 5 4) « IQ
•i-<
y ^T~~ o^ N£)
.n °° ? S S ü
in
•* 1—*
C4 •
^4 • in
ü o i—I
« 2
X «5 x
v-/ o o ro c
o
y £ oo u 0 O o oo r- oo 00 •*
CO a « • • • • « ,„ «n vO 00 r- vO -* oo o (A .-H M ~H ro •—* »-4 01 0) o o -1
,*? x u
V C ti Ui 0 0) 4) 4> 4> h V
f* (K tu Ui ■l- h y + flj + + + + + + 0 1) + + + + 0* 0 + <M Ol *
Ä 2 0 2 2 0 X
97
irrtüffJ-IWlr i« fWtii a if

Mi-Mfflpwi tmwgB**&m&sz*mm
REFERENCES
1. R. S. Narcisi and A. D. Bailey, J. Geophys. Res. 70, 367o (1965).
2. W. Swider, Jr., Planet. Space Sei. 17, 1233(1969).
3. C. E. Moore, Nat. Std. Ref. Data Ser., Nat. Bur. Stds.
(US), 34, 1970.
4. J. A. Rutherford, R. F. Mathis, B. R. Turner and D. A. Vroom,
J. Chem. Phys. 55, 378J (1971).
5. J. A. Rutherford, R. F. Mathis, B. R. Turner and D. A. Vroom,
J. Chem. Phys. 56, 4654 (1972).
6. J. A. Rutherford, R. F. Mathis, B. R. Turner, and D. A. Vroom,
submitted for publication in J. Chem. Phys.
7. B. R. Turner and J. A. Rutherford, J. Geophys. Res. 73, 6751 (1968).
8. F. A. Wolf and B. R. Turner, J. Chem. Phys. 48, 4226 (1968).
98
BMHaBMMliiiüimrUMlMiiinmiiri n iiinmmiiDiin

lÄJ^-lWt4^W«,U,l%iLJ^Jpi^yi!*y(L ^WEMB ^B ijSMBMWW-^ juqjjiff." "- 'f "-".'JJ-J^i^ f^WV«!li WiilULUMWWWmiüJ' fP5(BWW*»?p"BBS?
+ O
. CM I
£
CM O
rO T
CM + <M I O <3 O
<1
<
-<
O OD > OD
+
O
I + +
ff + x t
O D
X o
<J
<
<
<
o° o o o
o
3 o OD o OD
o ff
o O o OD
i o o
o 0
SD OD OD OD
O OD o O
o <D o D>
>° ft , D> D>
DO DO
D D
D
o o
1 1 1 ro O
OJ O in lO
(uuo bs 01)-° v 91-
O O o
o o
c o u
(6 u *-> 3 V c c o 00 ß
ÖD ß
0,
00 fi O
*0 e n)
>
to +0
>- + *• A e? o fc et: ■»<
Lü + 'S o g ac -H •*• u ss fc ß o ■«* z: tt» ~ ° 0) X ß ** o - 0 vi
••* O
ü >> 2 Ö0 W n 09 ß
\n o »
« X H ** 0) t«
vi O
ß s 2 -2 M «
(\J 00 vi *« _ «1 *
•C 09 U os
5 "H
J) h 3 M
99

->,%WM.JUIHHII ;iis»p«»i«'giBT-"ww»»»aij»yw»j^^ ' - ; ■.'»■» *"-'»g»-M.i"' y^m^'w,^
1 1 1 1 1 1
<J o D „_
<J o D
<3 o D < o D
<3 o B —
<1 o G <1 o D < 8 D —
< o D < o D < o D
- < o D —
< o D
< o D < o D
- <« °0 8 —
<3 o a <3 o D + \>
' > 8
o r-i &>
D + + O
f
+£ ^™
< <3
o o
a CM a z +
0 o f n t LD <D t
< o £ <
< <]
o 0
Li_ +
+ fvi
+ £ ++
Z z 2 - <J 0
< o D
1 1 1 1 1 1 m ro
O ro CO
O CVJ
io m
O O g o o m
o o CO
O a> m ^
>- o a: LÜ
CM m
- o
m
CJ
O
(O
C o u
<6 u 3 ß c O
ß
(30 c
c o
c
O
ß
z * M a,
^* -r-t
c ß
o ,H
53 « u -ß
01 o u
oo v C v
(wo bs oi)-°
0 to 43 ß **
U O
» ß V o 00 Tj
2 ß
li 3 00
100
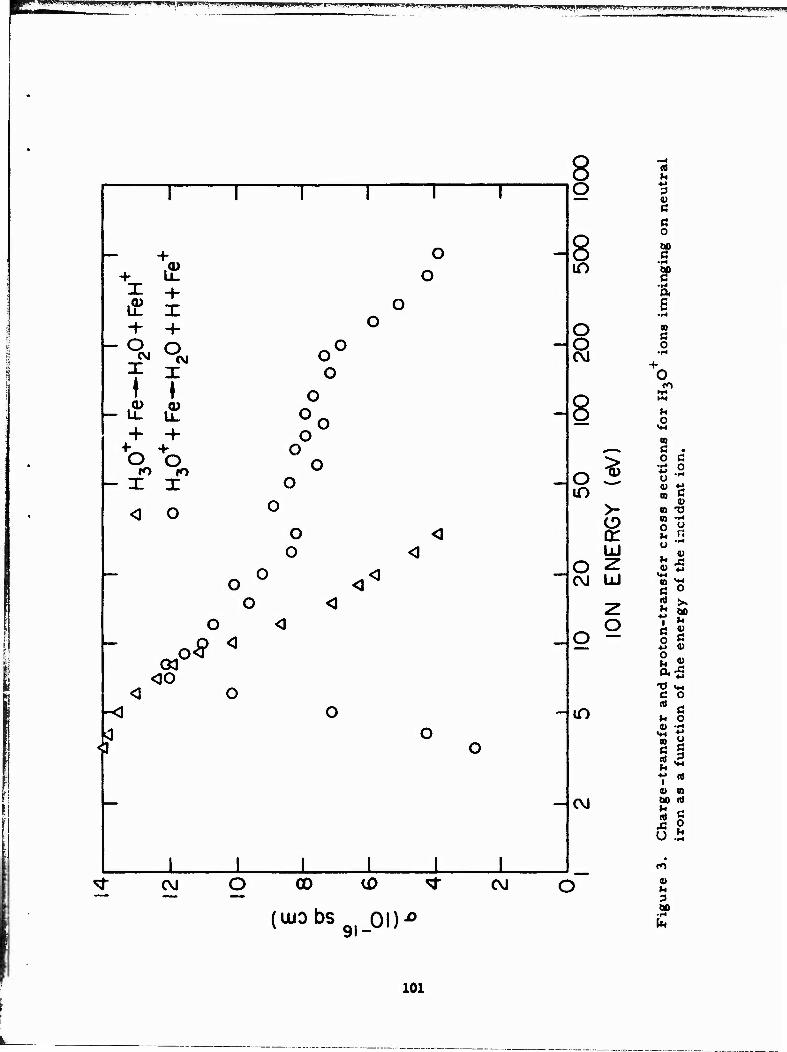
* iy»^.~KIULlll,^-j]|^JpiL^OJBIJ4pi|l'. ljS.I.Jj^jai!J|.^UJ,J.!!J,JiWtf*W|JLW ■p iwjwgy PWEJH P ■■»*'. ?' g;"' i wpp ■ qgw ■IHüR»^^ ptua jüjgH^ygr
.0)
iff X + + o
CNJ o CNJ
X X ! ♦ .* 0) u_ u_ + +
4- + Ü O
K) IO X X
o o
o o,
o
<J o
CM <io
O o
O <3 O <
O <J
0$ <
-<
8 >4
O 3 —i «
C C o
8 £ m
* ft 6
o (0 c o o
CM + O
ro
8 o ^^~ <n
OB
sect
ioi
snt
ion
.
o cr LU
o 2 <\J LU
o2
- m
- CM
CM CO CO CM
(Luobs9i_oi)-°
a> m *a a> ■•■■ 0 u * .2
**< . S o (4 M
I ß o
u c 4)
2 s a, JC
c o
H O fl> -M
c s
v * V « 00 rt
nt C
U .tJ
v
DO
U.
101

MGGE&fUltiwmwW.!n!a 'M\\W*.':1 n.wi|..^,(,wujJW.WJ«! jiij^iujPHMi. ■■AJjPJJ>_I^JaüMtfWg'JB4W-..i1'-'- -' 'l'Tg?irr™l<^.'WP-^ry^:?-7y^?»««q^
fa2s^.
APPENDIX F
NEGATIVE ION REACTIONS WITH NEUTRAL OZONE
Preceding page blank 103

TanfJP^'W.!! [ill ,t IJ-BljiyflJglWUU^^^Ji U I.«
NEGATIVE ION REACTIONS WITH NEUTRAL OZONE*
J. A. Rutherford, B. R. Turner, and D. A. Vroom
Gulf Radiation Technology A Division of Gulf Energy 8c Environmental Systems
San Diego, California 92112
ABSTRACT
Charge transfer reactions between O , O , OH and NO and
O have been studied to obtain cross sections for formation of O .
The energy range of the ions extended from 1 eV - 500 eV. The
accepted value for the electron affinity of O has been checked by
looking at the above charge transfer process as well as F , Cl ,
and CO with O . The results show that the electron affinity of O
must be close to accepted value of approximately 2 eV. A further
result of this study is that the electron affinity of N00 must be similar
to that of O .
Preceding page Wank
*This work was supported by the Defense Nuclear Agency under Contract No. DASA01-69-C-0044.
105
feäMäBf-^- v --- .-Tfgji -

3HW». *.'.'■*■, \ <■ -.. u-> -: ., jMipjuuuikjf JWU'. A, L.uii^i.ij^puij,ty gwmBfqppyafg^gapgg.«-,JJ'JHA-U'-T. pnrag wgg
INTRODUCTION
Ozone is known to be an important constituent of the lower atmosphere
and, as a consequence, its chemistry is of interest. One aspect of the
problem is the conjecture that the negative ion of this species is an
important intermediate in the formation of long-lived negative ions in the
D region. Previous investigations of reactions of ions with this species
have been restricted to thermal energy studies with O and O using flowing
2 afterglow techniques. The study of negative ion reactions is always hampered
by the lack of knowledge of the electron affinity of many of the species. One
aspect of the present study has been to try and establish with more reliability
these important constants.
The present investigation has also pointed out the importance of near
resonance in low energy negative ion charge transfer processes. As in
3 4 previous studies ' using positive ions the results obtained indicate that
unless a reaction channel which allows all the excess energy in the reaction
to be absorbed by the products the charge transfer cross section will be
small at iow energies.
APPARATUS
A detailed description of the apparatus in the configuration employed
for positive ion-neutral metal vapor charge transfer reactions has been
given in a previous publication. The reactions investigated here involve
106
I fetat^s^

the formation of negative ions. In general the only apparatus changes
required for such studies are a reversal of the magnetic and electric fields
employed. The major experimental modification here was the construction
of an ozone generator capable of producing pure ozone and the installation
of a sample handling system which did not lead to decomposition of this
unstable species. The generation and handling system will be described
in detail after a brief outline of the apparatus.
The instrument consists of tandem mass spectrometers with the
neutral beam interposed at right angles. Primary ions are federated by
electron impact in the ion source of the first mass spectrometer, a 180-deg
magnetic sector- After mass analysis, the primary ions are accelerated
or retarded to the desired interaction energy and allowed to interact with
the neutral beam. Product ions formed due to reaction between the primary
ions and the neutral beam are extracted from the interaction region along
the direction o.c the primary beam by a weak electric field. Collection
efficiencies of about 70% are usually obtained. These ions are then
accelerated, focused and mass analyzed in a 60-deg magnetic sector, and
finally detected with a Be-Cu electron multiplier. The neutral beam effuses
from a small hole in a room-temperature iridium tube located in a separate-
ly pumped chamber of the apparatus. The beam passes from this source
chamber, through an intermediate chamber which serves to collimate the
beam and into the experimental region. There it is mechanically chopped
and allowed to interact with the primary ion beam. The modulation of the
107

neutral beam allows selective amplification and phase-sensitive detection.
The neutral beam density is determined from the pressure of ozone
in the tube, and the geometry of the apparatus. The ozone pressure was
determined using a differential pressure measurement. This method
which assures that the pressure measured is that at the point of effusion
from the tube, requires that the gas be pure. As is shown below, our beam
was composed of pure O .
oxygen flow were then terminated and, after evacuation of system, the U-tube
*
Ozone was produced using an electric discharge in a low-pressure flow I
of pure oxygen. A schematic representation of the generation system which
5 _ 1 is similar to that employed by Griggs and Kaye is given in Fig. 1. Pure
oxygen (Lände ultrahigh purity dry) at a pressure of about 0. 1 Torr flowed >
from a leak valve through a stainless steel tube and irito a liquid-nitrogen- i -. a
cooled glass U-tube. This U-tube had a collector protruding from the bottom j
i to accumulate the liquid ozone. The U-tube was connected to a silica-gel
i filled ozone storage container, also constructed of glass. The connection
between the U-tube and the storage container is made using stainless steel
i fittings. The ac potential required to produce the discharge in the U-tube f
was applied using a neon sign transformer between this fitting and ground |
potential (stainless steel inlet tube and fitting used for cr»«necting the U-tube
and inlet).
Ozone trapped at the bottom of the U-tube was allowed to collect until |
a sufficient supply for a day's experiments was obtained. The discharge and J 1
108
i
i

m^^^^mmm^^'™&"^mmmmmmmfl''mmmM*m*m*!memmim mmmmT^mmmmwv
warmed to room temperature. During the warming period, the o-zoue
vaporized and transferred to the dry-ice-cooled silica gel in the storage
contained. The ozone was stored until needed in the cooled silica gel.
To obtain ozone at a pressure sufficient to produce an adequate beam in
the crossed beam apparatus, the silica gel was warmed slightly by re-
placing the dry ice with an alcohol slush bath. Ozone from the storage
container was allowed into the neutral beam source through a stainless
steel leak valve and stainless steel tube.
Care was taken to keer impurities in the ozone beam to a minimum.
Apart from the use of pure 0_ gas for formation of ozone, the generation
system including the silica gel storage vessel was pumped and outgassed
before the oxygen discharge was initiated. After sufficient ozone was
formed, the generator was again pumped to remove any excess O . This
last operation was performed prior to transfer of the ozone to the silica gel.
To test the purity of the ozone prepared in this manner, the resultant
beam was probed using O ions. The cross section for the reaction
0~ + 0_—-O" + O + 0„ was found to be small while the corresponding Ct 3 u &
fast slow slow fast
process 0„ + O^'O + O is know to be large. Fast O ions crossing fast slow fast slow
our ozone beam did not produce an appreciable quantity of slow O ions.
Since 0„ is the most probable impurity in our ozone beam, the dearth of
O" ions produced in the above reaction indicates an absence of this species.
109

|jjgFiM*gBpw,J.H,» H;I.I wij .A ji 7M!<-Vl^MI*^:JWI*PW!r^^jW'y"t';.' i' ■!! .L'-V ■■■' w;.-W yjpc !'■■■• «jM^^yg^^"-^»»!!^!*wff^^^^ji^fijw^ i ?J*v?*v fMyJPJHW, ^ipqjj^^-yM^^yywBIL»^. ypqtfflffgqBgj«
RESULTS
Reactions of ozone with O", O" OH~, NO" F~, Cl", O" and CO" & 2 3 3
have been investigated. Those processes which were found to have readily-
measurable cross sections have been studied in detail while for those for
which the reaction probability is small, only upper limits on the cross
sections are given.
The ions for which reactions with ozone have been studied in detail
are O , O , OH and NO . In all cases the major reaction channel is
non-dissociative charge transfer to form O . The cross section data
obtained for the first three of these reactions are given in Fig. 2 while
those for NO on ozone are presented in F. g. 3.
The symmetric charge transfer reaction O on O has only been
studied at high interaction energies. Experimental Mmitations, arising
from the inability of our secondary mass spectrometer to distinguish
between fast primary and slow secondary O , prevented low-energy
values being obtained. The resonant charge transfer cross section was
-16 2 found to vary between 6 and 4x 10 cm in the range between 3 50
and 400 eV.
The upper limits for charge transfer reacti-ws of the ions F , Cl
and CO? with ozone are given in Table I.
The charge exchange cross section for the process O + NO -~0 + NO 3*32
was remeasured here. The results which agree with our previous measure-
ments are given in Fig. 3.
110

y^jwl'^»^^i.T«^i^Mj|)^^''^4W^^£'yy^^
The nature of the errors present in our experiment have been dis-
3 cussed previously. The magnitude of the error in absolute cross sections
given here depend upon the species being studied. For O , O , OH and
O charge exchange with ozone, the possible error on the values reported
are ±25% at impact energies above 30 eV and may increase to as much as
±40% at the lowest ion energies. For F , Cl and CO charge exchange
with ozone, only upper limits on the cross section are given. For these
measurements the primary beams of F , Cl and CO used were small,
and no detectable signal could be found at only impact energy. The actual
cross sections for these charge exchange processes may be much lower
than the upper limits given.
For NO charge transfer with neutral ozone and the reverse reaction
of O on NO , experimental difficulties were encountered due to the
similarity in mass of the primary and secondary ions (NO is 46 amu and
O 48 amu). In our apparatus, the secondary ions are extracted in the
same direction as the primary ions and both beams therefore enter the
secondary mass .spectrometer. These two beams will have, in general,
different masses and different energies (i.e., the primary ions will have
the energy with which they passed through the collision region plus the
energy due to the accelerating lenses after the collision region, while the
secondary ions have only the energy due to the accelerating region). Under
these conditions if the primary ion has a mass similar to but slightly less
than that of the secondary ion, there will be a range of primary ion energies
111

Vtyte&llWr j*g's^rw?y'w«wiy«.wj.a»f*'*^'V'».. " ^■fft^j^^w/iwiy^wkP!ww^^^j^^^i'w?)<BPg^w^^ yw^swpjjp^™
when both the primary ion and the secondary ions are in focus in the
secondary mass spectrometer. Under these conditions the DC signal due
to the primary ion beam overloads the amplifier system to such an extent
that the secondary ion signal cannot be measured. This situation occurs
for the reaction of NO on O in the primary ion energy range from 10 to A 3
100 eV. For O on NO , a related but less severe problem occurs in
the ion energy range from 12 to 30 eV. Here, the resolution of the primary
mass spectrometer is such that above 12 eV a small amount of NO is
present in the O, beam making measurement impossible up to an energy
of 30 eV where the secondary mass spectrometer is able to separate the
fast from the slow NO particles. The amount of NO present in the
primary ion beam above 12 eV is large compared to the secondary ion signal
but is very small compared to the O and as a consequence does not interfere
with measurement of the O charge transfer with NO_. 3 *
DISCUSSION
Table II lists the electron affinities used to calculate the energetics
of the reactions studied. The electron affinity of O is listed at 1. 9 eV.
o This value .vhich was first obtained by Wood and D'Orazio is in good
agreement with the recent photodetachment value of 2. 09 eV given by
9 Sinnott and Beaty but is lower than earlier accepted values which placed
this constant at about 3 eV. The choice of approximately 2 for the
electron affinity of O can be justified by considering the magnitude of
112

m^gv^n^w^^l'^^<!fW^^^'i!^^^f^^n'r^'^^K'i?
the charge transfer cross sections measured here for reactions where the
other component has a known electron affinity.
A possible reason for the uncertainty which has existed and still in
some instances does exist concerning the value of the adiabatic electron
affinity of species such as O and NO may be that tri „omic moleciil »s
can undergo changes in geometry between the negative ion and neutral
ground state. A significant change in geometry can lead to a vertical
electron affinity which is appreciably higher than the adiabatic value since
the product will be formed in a high vibratiLonal level. In many cases,
earlier experimental va. ues reported for electron affinities have been high
and have tended to become lower as measurements have improved. The
values for the molecular species listed in Table II are the currently accepted
values and, in general, tend to be lower than those reported in earlier
publications.
If changes in geometry take place when going from the neutral to
negative ion of such a molecule as O , they would tend to lower the
symmetric charge transfer cross section. The ctiss section values measured
for O on O here are lower than one would normally expect for symmetric
charge transfer between two atomic species where geometry is not a
-15 2 14 consideration. H + H has a cross section of approximately 4x 10 cm
at 200 eV as opposed to approximately 4x10 cm for O on O . The
structure of O ir. the gas phase is known with considerable accuracy the
o 15 0-0-0 bond angle being 116 47' dfc 2'. For the negative ion, the bond angle
113

is less well known, the value being predicted to be 110 ±5 by Smith.
This prediction has been supported by the solid phase ESR results of
17 Tagaya and Takeshi. The low value of the O on O cross section obtained
here may indicate the difference in angle may be greater than 6 but may
also be the result of the fact that the O primary ion is formed in high
vibrational levels which would tend to make this cross section smaller than
one would expect for a resonant process.
In Fig. 2, the data for the three species in Table II which have electron
affinities lower than that of O , namely, O , O and OH , are presented. 3 2
All three cross sections are large, the one for OH on O being the largest.
This latter case has the closest energy balance (see Table II). All three
reactions are exothermic and it is probable that the excess energy in the
reactions may be absorbed into vibrational energy of the products. For O
on ozone the excess energy is approximately 1. 5 eV which is equal to the
O bond energy (D (O -O ); 1.5 eV). Here the energy may be taken up
by vibrationally excitation of both the O and O products or partially by
formation of the On in the excited (a A ). This latter state lies 0.98 eV 2 g
above the neutral ground state.
For the reactions O + O and O + O , thermal energy values of the 3 m >
2 rate coefficients have been measured previously. The measured points are
included in our figure and a smooth interpolation of our data to these results
is shown. For OH + O,, no thermal energy values has been determined.
The extension of our data was made using the extrapolation procedure
114

«nil»,™, nojiiuf ••m ^a^mmfrr *i^Bij$inw*-W**MW-*^™'-'-t#^m?^JL'&l9äK'J'" »■'■miJWJiWI£l
———— -*— 1-—'
12 - previously employed. It is of interest to note that for O + 0 and
O + O the extrapolation procedure gave thermal energy rate constants
that were higher by approximately a factor of 2 compared to the measured
values shown in the figure.
The electron affinities of F ! and Cl are greater than that for O
and as a consequence the processes are endothermic. The cross sections
for charge exchange with these species is uierefore small as indicated
in Table I.
The reaction of NO with O is of interest since the electron affinity « 3
of N00 is in doubt. Fehsenfeid et al. have established that the NO electron
affinity should be greater than 1. 8 eV since the reaction
OH + NO.—NO + OH
13 is known to proceed at thermal energies with a large rate coefficient.
Our results here indicate that the electron affinity of this species must
be close to that of ozone since both the forward and reverse negative
reactions with these two species are found to proceed (see Fig. 3) with
nearly equal cross section at the lowest energies measured.
In the above reactions, the cross sections are seen to be appreciable
when the reaction is exothermic. The largest cross section at low energies
is for the OH on O which has the smallest energy detect. This result is
consistent with previous positive ion charge transfer " reactions which
show that if near energy resonance exists, the process proceeds with a
high probability.
115

!j,HHWpi..),j,»»VI«-, .■»>■■'*■ ^ViT.wlfMWjj^.jii^,^^ ■,:, „n, im'lHuw
REFERENCES
1. E. E. Ferguson, Rev. of Geophys. 5, 305 (1967).
2. F. C. Fehsenfeld, A. L. Schmeltekopf, H. I. Schiff, and E. E. Ferguson, Planet. Space Sei. 15_, 373 (1967).
3. J. A. Rutherford, R. F. Mathis, B. R. Turner and D. A. Vroom, J. Chem. Phys. 55, 3785 (1971).
4. J. A. Rutherford, R. F. Mathis, B. R. Turner, and D. A. Vroom, J. Chem. Phys., May 1, 1972.
5. M. Griggs and S. Kaye, Rev. Sei. Instr. 39, 1658 (1968).
6. H. I. Schiff, private communication.
7. J. A. Rutherford and B. R. Turner, J. Geophys. Res. 72, 3795 (1967).
8. R. H. Wood and L. A. D'Orazio, J. Phys. Chem. 69, 2562 (1965).
9. G. Sinnott and E. C. Beaty, in the "Abstracts of Papers of the Vllth International Conference on the Physics of Electronic and Atomic Collisions," Amsterdam, July, 1971, North Holland Publishing Co., p. 176.
10. See, for example, R. C. Curran, J. Chem. Phys. 35, 1849 (1961), H. O. Pritchard, Chem. Rev. 52, 529 (1953), and N. S. Buchel'nikova Usp. fiz. nauk. 65, 531 (1958).
11. F. R. Gilmore, "Basic Energy-Level and Equilibrium Data for Atmos- pheric Atoms and Molecules, " Rand Corp. Memorandum RM-5201-ARPA, March 1967.
12. F. A. Wolf, and B. R. Turner, J. Chem. Phys. 48, 4226 (1968).
13. F. C. Fehsenfeld, E. E. Ferguson and D. K. Böhme, Planet. Space Sei. 17_, 1759 (1969).
14. D. G. Hummer, R. F. Stebbings and W. L. Fite, Phys. Rev. 119, 668 (1960).
116

^gggsraa'g''^^ m>m}wjmjyem>mMiJ'' ■.-' ■ ^ i iwpwwii, |i. ^r>"
REFERENCES (contd)
15. T. Tanakaand Y. Morino, J. Mol. Spectrosc. 33, 538 (1970).
16. P. Smith, J. Phys. Chem. 60, 1471 (1956).
17. K. Tagaya and M. Takeshi, J. Phys. Soc. Japan 23, 70(1967).
117

JijpW'WaJJWMiP-^^^ JWMi.-^i^i^.ijiL i«^^M^jp_i^jj^^iiigj,.j^
TABLE I
Cross section upper limits for some negative ion
charge transfer reactions with ozone in the
impact energy range from 1 to 10 eV.
Reaction
F + O •—F + O"
Cl" + Cv—a + O" 3 3
CO" + O_—CO„ + O + O" 3 3 2 3
Cross section upper limit
(cm )
2 x 10 -17
2 X 10 ■17
2 X 10 -16
118

'™M,PW
1 *
f
1
t • V - i
TABLE n
Electron affinities of the atoms and molecules studied.
* Electron Affinity
I Species (eV)
O 1.478*
F 3.5b
Cl 3.7b
i ' |. OH 1.83c
02 0.43d
N02 -2.0e
03 1.9f
I | R. S. Berry, J. E. Mackie, R. L. Taylor, and R. Lyncl:. 11 J. Chem. Phys. 43, 3067 (1965).
"Bond Energies, Ionisation Potentials and Electron Affinities, " V. I. Vedeneyev, L. V. Gurvich, V. N. Kondrat'yev, V. A. Medvedev and Ye. L. Frankevich, eds., St. Marten's Press, New York (1966).
| CL. M. Branscomb, Phys. Rev. 148, 11 (1966).
I R. Celotta, R. Bennett, J. Hall, J. Levine, and M. W. Siegel, Bull. Am. Phys. Soc. 16_, 212 (1971).
This work.
fRef. 8.
• f 119
*
||^j^gümWPpit?BffglBPWHWB^Wg* mm* immmmmw ■pppupjpji ■BIP» ipM!iJpq|^p^^*qiipppp9AP^P9H|pBPJP|0PH
O
<d h
< C
—■ 00
— ÜJ c ÜJ V)U o CJ — N < Z UJ Ü oc 0>-oc o C£Z> • H- QQI- ~* </> ÜJ < 4)
COH- OC h Ixi OC< UJ 3
00 -Hl Z O QL
o w-iZ In tN( Q UJ UJ o <Ul-
120

^ip^af^^mmw^m^rsvfiw- w*jimy <H !?n^PQIRP «PW - mnpinQR?; ■ II-JW . I «q^pK^P i ji.j««.«--«wij!i.ijipnmp
r~i—!—rn~n—m—r~i—i—i—rq
CM o
O
o o o
o o
I
O •
O -
+ o t
o + "I
>
>-
a:
UJ
CO co <
O Lu • O
UJ
yj o
CM I o o
D
i i i I I I I I I I I—L ^N"On00N\DlA-J^
o o o
e oo v C v oo"^ c v
p x 3C oo
S o
o ^
o 3 u rt O «
« S CD c « O A Ü H y «» . 8 >* M CO
<o *•*
4-> C a] v £ » *0 a» ß nS
•" E « •C *2
i u O «a O
Co
C i ö » fl) -W »3 •■-<
C fl) I4 .r«
Q> +j . 1 CO
-0 <• v «D
«J, * E rj
«1 m r
(WD g.OI dOSUNfUf/V
o v
o ° -Hi *n »I 4) ~
IB K
2 °
.c d 9
o «
2 -
H 41 flJ C 3 ** Of C to o
M
V U 3 00
«I PJ-4
C° * « O n">-2 •5 o j, rt rt h h .>
S, • at O* rt-"« « «
X « 1» £ • • > CcO V 2 « £ S - c*ü£ « ■ • ^
.S -• ><~ »-r ° M c c S .2 ? o C *" o •»*
£. " £ 1« M rt *>
** »< * 2 o o u 2
a» C *• •O «I i < S u * * -^ h
0) 4> O O
u<
121
jags BBgMB ugww.1 .* i' y ly.L"1... i^^^^ff^yuBa?; WKKWWWPW^SBWIBB» PMninR H^^HPW
id
a o •H
> •w*
id
4) c
■«->
•o c id
en O •o ß id
61
2 ß v «
XI ■ ß O
u id V u •o V «J id o
T3 ß
A u *» v » ß
■0 ß * o -g
** o « ß m ■■-■
ü ^
00
ß V
(0 in <r to cvj
(2W09hOIJO SlINO)^ In 3 00
122
*




















