
Activation of the ciliary neurotrophic factor (CNTF) signalling pathway in cortical neurons of...
11
Activation of the ciliary neurotrophic factor (CNTF) signalling pathway in cortical neurons of multiple sclerosis patients Ranjan Dutta, 1 Jennifer McDonough, 1, Ansi Chang, 1 Lakshman Swamy, 1 Alan Siu, 1 Grahame J. Kidd, 1 Richard Rudick, 2 Karoly Mirnics 3 and Bruce D. Trapp 1 Department of Neurosciences, Lerner Research Institute, Cleveland, 2 Mellen Center for Multiple Sclerosis Treatment and Research, Cleveland Clinic Foundation, OH, USA and 3 Department of Psychiatry, Kennedy Center for Human Development, Vanderbilt University, Nashville, TN, USA Present address: Oak Clinic for MS Research at Kent State University, Kent, OH, USA Correspondence to: Bruce D. Trapp, PhD, Department of Neurosciences/NC30, Lerner Research Institute, Cleveland Clinic, 9500 Euclid Ave, Cleveland, OH 44195, USA E-mail: [email protected] Neuronal and axonal degeneration results in irreversible neurological disability in multiple sclerosis (MS) patients. A number of adaptive or neuroprotective mechanisms are thought to repress neurodegeneration and neurological disability in MS patients. To investigate possible neuroprotective pathways in the cerebral cortex of MS patients, we compared gene transcripts in cortices of six control and six MS patients. Out of 67 transcripts increased in MS cortex nine were related to the signalling mediated by the neurotrophin ciliary neurotrophic factor (CNTF). Therefore, we quantified and localized transcriptional (RT-PCR, in situ hybridiza- tion) and translational (western, immunohistochemistry) products of CNTF-related genes. CNTF-receptor complex members, CNTFRa, LIFRb and GP130, were increased in MS cortical neurons. CNTF was increased and also expressed by neurons. Phosphorylated STAT3 and the anti-apoptotic molecule, Bcl2, known down stream products of CNTF signalling were also increased in MS cortical neurons. We hypothesize that in response to the chronic insults or stress of the pathogenesis of multiple sclerosis, cortical neurons up regulate a CNTF-mediated neuroprotective signalling pathway. Induction of CNTF signalling and the anti-apoptotic molecule, Bcl2, thus represents a compensatory response to disease pathogenesis and a potential therapeutic target in MS patients. Keywords: multiple sclerosis; gene expression; CNTF; neuroprotection Abbreviations: MS = Multiple sclerosis; CNTF = ciliary neurotrophic factor; LIF = leukaemia inhibitory factor; FDR = false discovery rate Received March 16, 2007 . Revised July 20, 2007 . Accepted August 6, 2007 Introduction Multiple sclerosis (MS) affects more than 1.5 million people worldwide and is the major cause of non-traumatic neurological disability in young adults in North America and Europe (Weinshenker, 1996; Noseworthy et al., 2000). Approximately 85% of MS patients initially have a relapsing-remitting disease course (RR-MS) characterized by alternating periods of neurological disability and recovery (Weinshenker, 1996; Noseworthy et al., 2000). Within 25 years a large majority of RR-MS patients exhibit a secondary-progressive disease course (SP-MS) character- ized by steadily increasing and irreversible neurological disability (Weinshenker et al., 1989; Noseworthy et al., 2000). Relapses are caused by inflammatory demyelination of white matter, while permanent disability results from neurodegeneration. Axons are transected during inflamma- tory demyelination (Ferguson et al., 1997; Trapp et al., 1998), but this axonal loss often remains clinically silent due to the ability of the brain to compensate for neuronal loss. Continuous neurological decline during secondary progressive MS (SP-MS) is thought to occur when the brain can no longer compensate for additional axonal or neuronal loss (Trapp et al., 1998). Activation of new cortical areas (Reddy et al., 2000; Rocca et al., 2003) doi:10.1093/brain/awm206 Brain (2007), 130, 2566^2576 ß The Author (2007). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For Permissions, please email: [email protected] by guest on June 18, 2014 http://brain.oxfordjournals.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Activation of the ciliary neurotrophic factor (CNTF) signalling pathway in cortical neurons of...

Activation of the ciliary neurotrophic factor (CNTF)signalling pathway in cortical neurons of multiplesclerosis patientsRanjan Dutta,1 Jennifer McDonough,1,� Ansi Chang,1 Lakshman Swamy,1 Alan Siu,1Grahame J. Kidd,1
Richard Rudick,2 Karoly Mirnics3 and Bruce D.Trapp
1Department of Neurosciences, Lerner Research Institute, Cleveland, 2Mellen Center for Multiple SclerosisTreatment andResearch, Cleveland Clinic Foundation,OH,USA and 3Department of Psychiatry, Kennedy Center for Human Development,Vanderbilt University, Nashville, TN, USA�Present address: Oak Clinic for MS Research at Kent State University, Kent, OH, USA
Correspondence to: Bruce D.Trapp, PhD, Department of Neurosciences/NC30, Lerner Research Institute,Cleveland Clinic, 9500 Euclid Ave, Cleveland, OH 44195, USAE-mail: [email protected]
Neuronal and axonal degeneration results in irreversible neurological disability in multiple sclerosis (MS)patients. A number of adaptive or neuroprotective mechanisms are thought to repress neurodegenerationand neurological disability in MS patients. To investigate possible neuroprotective pathways in the cerebralcortex of MS patients, we compared gene transcripts in cortices of six control and six MS patients. Out of67 transcripts increased in MS cortex nine were related to the signalling mediated by the neurotrophin ciliaryneurotrophic factor (CNTF).Therefore, we quantified and localized transcriptional (RT-PCR, in situ hybridiza-tion) and translational (western, immunohistochemistry) products of CNTF-related genes. CNTF-receptorcomplex members, CNTFRa, LIFRb and GP130, were increased in MS cortical neurons. CNTF was increasedand also expressed by neurons. Phosphorylated STAT3 and the anti-apoptotic molecule, Bcl2, known downstream products of CNTF signalling were also increased in MS cortical neurons. We hypothesize that inresponse to the chronic insults or stress of the pathogenesis of multiple sclerosis, cortical neurons up regulatea CNTF-mediated neuroprotective signalling pathway. Induction of CNTF signalling and the anti-apoptoticmolecule, Bcl2, thus represents a compensatory response to disease pathogenesis and a potential therapeutictarget in MS patients.
Keywords: multiple sclerosis; gene expression; CNTF; neuroprotection
Abbreviations: MS=Multiple sclerosis; CNTF=ciliary neurotrophic factor; LIF= leukaemia inhibitory factor; FDR= falsediscovery rate
Received March 16, 2007. Revised July 20, 2007. Accepted August 6, 2007
IntroductionMultiple sclerosis (MS) affects more than 1.5 million peopleworldwide and is the major cause of non-traumaticneurological disability in young adults in North Americaand Europe (Weinshenker, 1996; Noseworthy et al., 2000).Approximately 85% of MS patients initially have arelapsing-remitting disease course (RR-MS) characterizedby alternating periods of neurological disability andrecovery (Weinshenker, 1996; Noseworthy et al., 2000).Within 25 years a large majority of RR-MS patients exhibita secondary-progressive disease course (SP-MS) character-ized by steadily increasing and irreversible neurological
disability (Weinshenker et al., 1989; Noseworthy et al.,2000). Relapses are caused by inflammatory demyelinationof white matter, while permanent disability results fromneurodegeneration. Axons are transected during inflamma-tory demyelination (Ferguson et al., 1997; Trapp et al.,1998), but this axonal loss often remains clinically silentdue to the ability of the brain to compensate for neuronalloss. Continuous neurological decline during secondaryprogressive MS (SP-MS) is thought to occur when the braincan no longer compensate for additional axonal orneuronal loss (Trapp et al., 1998). Activation of newcortical areas (Reddy et al., 2000; Rocca et al., 2003)
doi:10.1093/brain/awm206 Brain (2007), 130, 2566^2576
� The Author (2007). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For Permissions, please email: [email protected]
by guest on June 18, 2014http://brain.oxfordjournals.org/
Dow
nloaded from

compensate for damage caused by inflammatory demyeli-nation of white matter (Pantano et al., 2002). Othercompensatory mechanisms include remyelination(Lassmann et al., 1997), redistribution of sodium channelson demyelinated axons (Waxman, 2006), and expression ofneurotrophic factors by immune and CNS resident cells(Martino, 2004).Ciliary neurotrophic factor (CNTF) is a member of the
four alpha-helical cytokine family including interleukin 6(IL-6), leukaemia inhibitory factor (LIF), interleukin 11(IL-11), oncostatin M (OSM), cardiotrophin-1 (CT-1) andcardiotrophin-like cytokine (CLC) (Weisenhorn et al.,1999). CNTF plays an important role in brain developmentand injury (Stockli et al., 1991; Winter et al., 1995;Lee et al., 1997; Dallner et al., 2002) and enhances survivalof dorsal root ganglia, hippocampal (Ip et al., 1991), striatal(de Almeida et al., 2001) and retinal neurons (Petersonet al., 2000). CNTF null mice display progressive lossof motor neurons and reduced muscular strength (Masuet al., 1993), while CNTFRa null mice die within 24 hof birth because of motor neuron deficits (DeChiara et al.,1995).CNTF signals through the tripartite complex of CNTFRa,
LIFRb and GP130 and activates several downstreamsignalling cascades including JAK/STAT and NFkB (Tagaet al., 1989; Taga and Kishimoto, 1997; Heinrich et al.,1998; Middleton et al., 2000). Activation of STATs andNFkB, in turn, increases transcription of neuroprotectivemolecules including FGF, EGF and IGF1. Two to threepercent of the human population is CNTF deficient due toa frameshift mutation in exon 2 (Takahashi et al., 1994;Thome et al., 1997). MS patients carrying this nullmutation in the CNTF gene show earlier disease onset(Giess et al., 2002b) and faster neurological decline than MSpatients with CNTF. CNTF is also increased in CSF fromMS patients as they recover from acute exacerbations(Massaro et al., 1997; Massaro, 1998) and studies supporta role for CNTF in oligodendrocyte survival and immunesuppression in MS animal models (Linker et al., 2002;Kuhlmann et al., 2006). It is possible, therefore, that CNTFdelays neurological decline in MS patients. To investigatethe molecular mechanisms that contribute to neuronalsurvival in the cerebral cortex of MS patients, we comparedgene transcripts in motor cortex of MS and controlpatients. We recently quantified and localized genetranscripts which were reduced in MS cortex (Duttaet al., 2006). In this report, we describe significant increasesin mRNA’s encoding proteins associated with CNTFsignalling. Furthermore, these mRNAs and their transla-tional products are enriched in cortical neurons. Wepropose that a CNTF-mediated neuroprotective responseis active in MS cortex and this response is part of theendogenous defense mechanisms mounted by the brain tocombat the progressive neurological disability observed inMS patients.
Material and methodsTissue and microarray analysisSix MS and six control brains were obtained from ClevelandClinic and CNMD Brain Bank, University of Pittsburgh, and havebeen characterized previously (Mirnics et al., 2000; Dutta et al.,2006). Brains were removed according to a rapid autopsyprotocol, sliced into 1-cm thick slices, and either fixed in 4%paraformaldehyde for morphological studies or rapidly frozen forbiochemical analysis. Sections (14-mm thick) from frozen blocks ofthe motor cortex were cut and immunostained for myelinproteolipid protein (PLP) & MHC Class II as described previously(Trapp et al., 1998). Blocks without demyelination or otherpathology indicted by increased activation of MHC class IIpositive cells were sectioned as described later for preparation ofRNA and protein. Fixed blocks from the contralateral motorcortex were similarly characterized for demyelination and otherfrank pathology and used for immunocytochemical and in situhybridization studies. Demographics of the MS and controlpatients were described previously and the tissue analysed herehave been used to describe decreases in mitochondrial andGABA gene transcripts in MS cortex (Dutta et al., 2006). Detailsof RNA isolation, labelling and microarray analysis have beendescribed previously (Dutta et al., 2006). Briefly, non-lesion motorcortex samples from six MS patients and six individuals withoutneurological disease were compared on Affymetrix U133A andU133B microarrays. Among 555 significantly altered transcripts,488 were decreased and 67 were increased in the MS samples(P50.05, 1.5-fold). We used a permutation-based false discoveryrate (FDR) analysis to determine whether transcript changeswere due to inherent biological differences between controland MS tissue or by ‘chance’. The FDR was only 5.9% (Duttaet al., 2006).
Quantitative RT-PCRCortical gray matter was separated from subcortical white matterby scoring the block at the white matter/gray matter juncture witha scalpel prior to cutting multiple 60-mm thick sections in acryostat. RNA was isolated from the cortical sections as describedand used for microarray and RT PCR experiments (Dutta et al.,2006). RNA from the six MS samples and six control samples wasreverse transcribed as described previously (Dutta et al., 2006).Gene-specific PCR was performed using SYBR Green I kit and aRoche Lightcycler (Roche Diagnostics, Indianapolis, IN) andstandardized to 18S RNA (Ambion Inc., Austin, TX). Primersequences are appended (Supplemental Data I). Each sample wasrun in triplicate with melting curve analysis to detect primerdimer artefacts. Normal distribution of the data was confirmedusing a Shapiro–Wilk test (Analyse-It Software, Leeds, UK). DeltaCt values were subsequently used to determine relative expressionchanges. The data was compared by the Student’s t-test.
Western blotsFor protein analysis, 60 mm thick frozen cortical sections from fiveMS and five control brains were homogenized in proteinextraction buffer, separated on 4–12% NuPage Bis–Tris gels(Invitrogen, Carlsbad, CA) and transferred to PVDF membranes(Invitrogen Inc., Carlsbad CA) as described previously (Duttaet al., 2006). After blocking (5% non-fat milk), membraneswere placed in primary antibodies overnight, incubated in
Activation of CNTF signalling pathway in cortical neurons of MS patients Brain (2007), 130, 2566^2576 2567
by guest on June 18, 2014http://brain.oxfordjournals.org/
Dow
nloaded from

horseradish peroxidase-tagged secondary antibodies, treated withSupersignal Pico detection reagent (Pierce Inc., Rockford, IL)and then exposed to Kodak X-Omat film (Kodak Inc., Rochester,NY). Membranes were stripped and subsequently re-probedwith glyceraldehyde-3-phosphate dehydrogenase (GAPDH).The average intensity for each band was quantified with Image J(NIH, Bethesda, MD). Differences between control and MSsamples were compared by the Student’s t-test.
AntibodiesThe antibodies used in the study are commercially available,well characterized and include CNTF, GAPDH, MAP2 andPLP (Chemicon Inc., Temecula, CA), CNTFRa (R&DBiosystems, Minneapolis, MN), SOCS3, LIFRb and GP130(Santa Cruz Biotechnologies, Santa Cruz, CA), STAT3 andpSTAT3 (Cell Signaling Technology, Danvers, MA), Bcl2(AbCam Inc., Cambridge, MA), GFAP and MHC Class II(Dako, Carpentenia, CA).
ImmunocytochemistryFree-floating sections (30-mm thick) were cut from paraformalde-hyde-fixed blocks, rinsed, boiled for antigen retrieval andimmunostained for CNTF (1:100), SOCS3 (1:200), LIFR (1:250),GP130 (1:250) and STAT3 (1:100) as described previously (Trappet al., 1998) using the avidin–biotin (ABC) procedure anddiaminobenzidine (DAB). Frozen (14-mm thick) sections wereair dried for 30–60min, fixed for 10min in 95% ethanol and usedto localize CNTFRa (1:200), pSTAT3 (1:50) and Bcl2 (1:200). Fordouble-labelling, free-floating sections were pretreated as describedearlier, incubated with primary antibodies for 5 days and thenincubated with Texas Red- and FITC-conjugated secondaryantibodies for 2 h. Sections were rinsed, mounted with vectashield(Vector Labs, Burlingame, CA) and examined in a Leica confocalmicroscope (Leica Microsystems, Exton, PA).
In situ hybridizationParaffin-embedded sections from control and MS motor cortexwere hybridized with riboprobes specific to CNTFRa, Bcl2, CNTFand proteolipid protein as described previously (Dutta et al., 2006).Riboprobes were generated from PCR products using gene-specificprimers for CNTFRa, Bcl2 and a combination of primers for CNTF(all sequences appended in Supplementary Data 1). For cellularresolution of mRNA expression, sections were dipped in auto-radiographic NTB2 emulsion (Kodak, Rochester, NY), counter-stained with hematoxylin-and-eosin and examined by dark-fieldand bright-field microscopy. Bright-field images were used toquantify mRNA abundance. Grains per neuronal perikaryal areawere quantified for at least 30–35 upper motor neurons in at leastsix sections from two control and two MS motor cortices. Normaldistribution of the data was confirmed using a Shapiro–Wilk test(Analyse-It Software, Leeds, UK). Statistical significance (P50.05)was determined by the Student’s t-test.Tissue sections were processed for the combined localization of
CNTF mRNA and the astrocytic marker, glial fibrillary acidicprotein (GFAP) immunoreactivity. Paraffin-embedded sectionswere processed overnight at 4�C in rabbit anti-GFAP (1:250),rinsed in RNAse-free PBS, incubated in biotinylated goat anti-rabbit IgG, and processed for the localization of antibody bindingwith the avidin–biotin technique and diaminobenzidine (DAB) as
the chromogen. Sections were subsequently dried and processedfor CNTF cRNA hybridization as described earlier. The relativelocalizations of silver grains and immunoprecipitate of GFAP wereassessed by bright-field microscopy.
ResultsMyelinated, non-lesion motor cortex samples from six MSpatients and six individuals without neurological diseasewere obtained at autopsy and compared on AffymetrixU133A and U133B microarrays (Dutta et al., 2006).Statistical significance was determined by permutation-based FDR analysis. Among the 555 significantly alteredtranscripts, 488 were decreased and 67 were increased inthe MS samples (1.5-fold, P50.05) (Dutta et al., 2006).Nine of the 67 increased gene transcripts were related toCNTF signalling (Fig. 1A). CNTF-related genes increased inMS cortical microarrays were also increased when measuredby RT-PCR. Computer generated hierarchical clusteringof transcripts, gene accession numbers, P-values and foldchanges of microarray and RT-PCR measurements areshown in Fig. 1A. Location of these transcripts withregards to CNTF signalling pathways is shown schematicallyin Fig. 1B.
CNTF receptorsCNTF signals through the tripartite receptor complex whichincludes CNTFRa, GP130 and LIFbR. We comparedtranscriptional and translational products of these threegenes in control and MS motor cortex. There was asignificant increase in levels of CNTFRa mRNA (1.82-foldby microarray, P50.05; 1.90-fold by RT-PCR, P50.05,Fig. 2A) and CNTFRa protein (1.52-fold; P50.0008,Fig. 2B) were significantly increased in MS cortex.mRNAs encoding LIFbR (+1.6-fold P= 0.067) and GP130(+1.2-fold, P= 0.61) were also increased in microarrays butdid not reach statistical significance (Supplemental Data,Fig. A). However, when measured by RT-PCR, LIFRb andGP130 mRNAs were increased by 2.14-fold (P50.05) and2.65-fold (P50.005), respectively, in MS cortex (Fig. 2A).Translational products of these transcripts were alsosignificantly increased (LIFbR 1.31-fold, P50.005; GP1301.40-fold, P50.0001) when measured by western blots(Fig. 2B). We next determined the immunocytochemicaldistribution of CNTF-receptor subunits. CNTFRa (Fig. 2Cand D), LIFbR (Fig. 2E and F) and GP130 (Fig. 2G and H)were highly enriched in neurons in both control (Fig. 2C, Eand G) and MS cortex (Fig. 2D, F and H). Upon activation,LIFbRs are translocated from the nucleus to the cytoplasm(Gouin et al., 1999; Gardiner et al., 2002). In contrast tocontrol sections where most LIFbR immunoreactivitylocalized to neuronal nuclei (Fig. 2E inset), significantLIFbR was detected in the perinuclear cytoplasm andproximal dendrites of neurons in MS cortex (Fig. 2F inset).CNTFRa mRNA was localized and quantified by in situhybridization using radiolabelled probes. mRNA encoding
2568 Brain (2007), 130, 2566^2576 R. Dutta et al.
by guest on June 18, 2014http://brain.oxfordjournals.org/
Dow
nloaded from
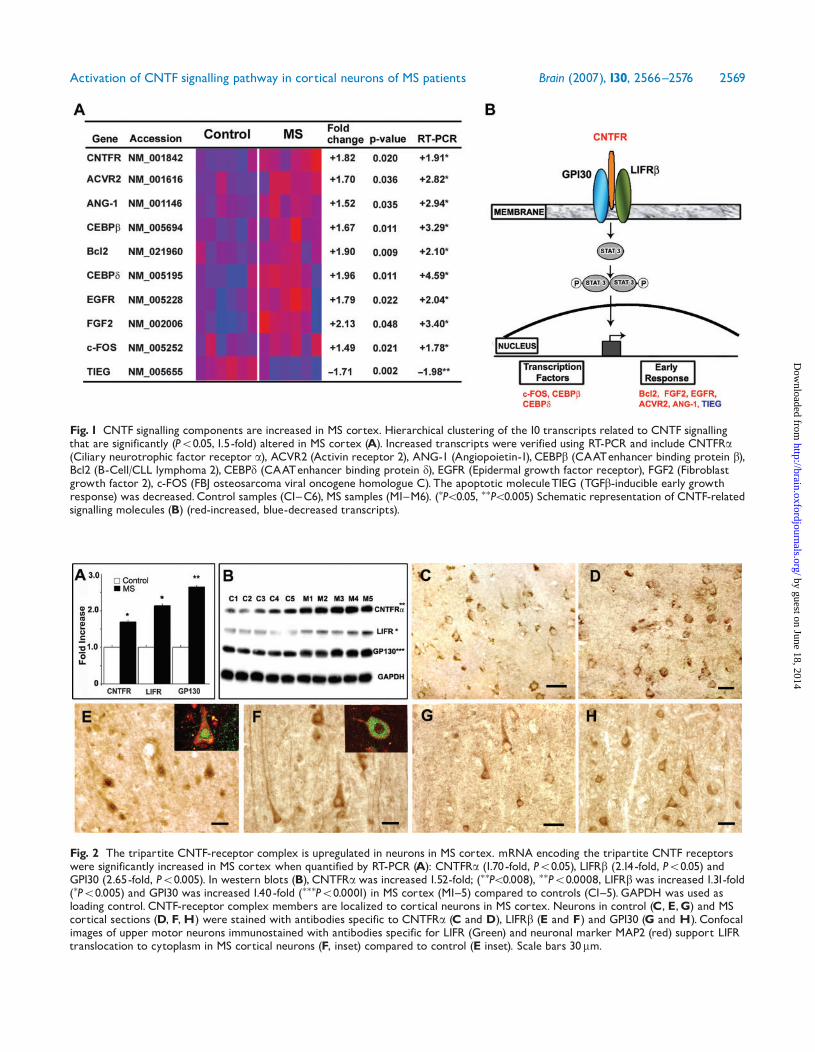
Fig. 2 The tripartite CNTF-receptor complex is upregulated in neurons in MS cortex. mRNA encoding the tripartite CNTF receptorswere significantly increased in MS cortex when quantified by RT-PCR (A): CNTFRa (1.70-fold, P50.05), LIFRb (2.14-fold, P50.05) andGP130 (2.65-fold, P50.005). In western blots (B), CNTFRawas increased 1.52-fold; (��P50.008), ��P50.0008, LIFRbwas increased 1.31-fold(�P50.005) and GP130 was increased 1.40-fold (���P50.0001) in MS cortex (M1^5) compared to controls (C1^5).GAPDH was used asloading control. CNTF-receptor complex members are localized to cortical neurons in MS cortex. Neurons in control (C, E,G) and MScortical sections (D, F,H) were stained with antibodies specific to CNTFRa (C and D), LIFRb (E and F) and GP130 (G and H). Confocalimages of upper motor neurons immunostained with antibodies specific for LIFR (Green) and neuronal marker MAP2 (red) support LIFRtranslocation to cytoplasm in MS cortical neurons (F, inset) compared to control (E inset). Scale bars 30mm.
Fig. 1 CNTF signalling components are increased in MS cortex. Hierarchical clustering of the 10 transcripts related to CNTF signallingthat are significantly (P50.05, 1.5-fold) altered in MS cortex (A). Increased transcripts were verified using RT-PCR and include CNTFRa(Ciliary neurotrophic factor receptor a), ACVR2 (Activin receptor 2), ANG-1 (Angiopoietin-1), CEBPb (CAATenhancer binding protein b),Bcl2 (B-Cell/CLL lymphoma 2), CEBPd (CAATenhancer binding protein d), EGFR (Epidermal growth factor receptor), FGF2 (Fibroblastgrowth factor 2), c-FOS (FBJ osteosarcoma viral oncogene homologue C). The apoptotic moleculeTIEG (TGFb-inducible early growthresponse) was decreased.Control samples (C1^C6), MS samples (M1^M6). (�P50.05, ��P50.005) Schematic representation of CNTF-relatedsignalling molecules (B) (red-increased, blue-decreased transcripts).
Activation of CNTF signalling pathway in cortical neurons of MS patients Brain (2007), 130, 2566^2576 2569
by guest on June 18, 2014http://brain.oxfordjournals.org/
Dow
nloaded from
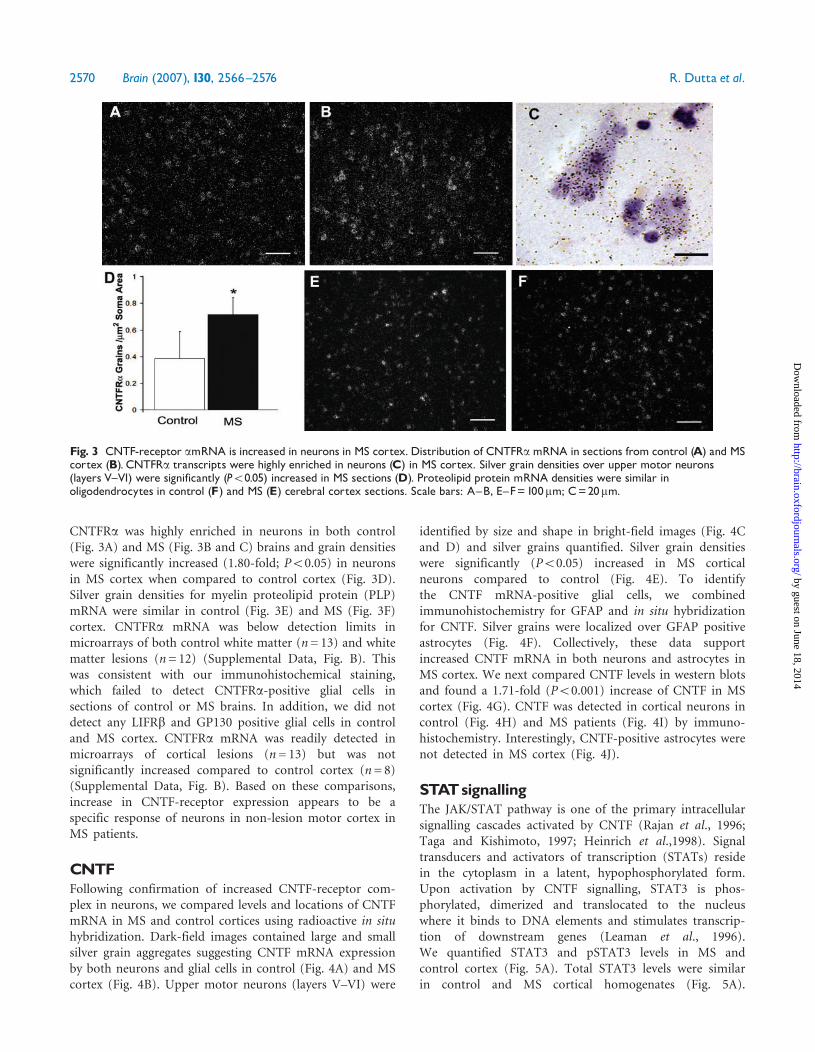
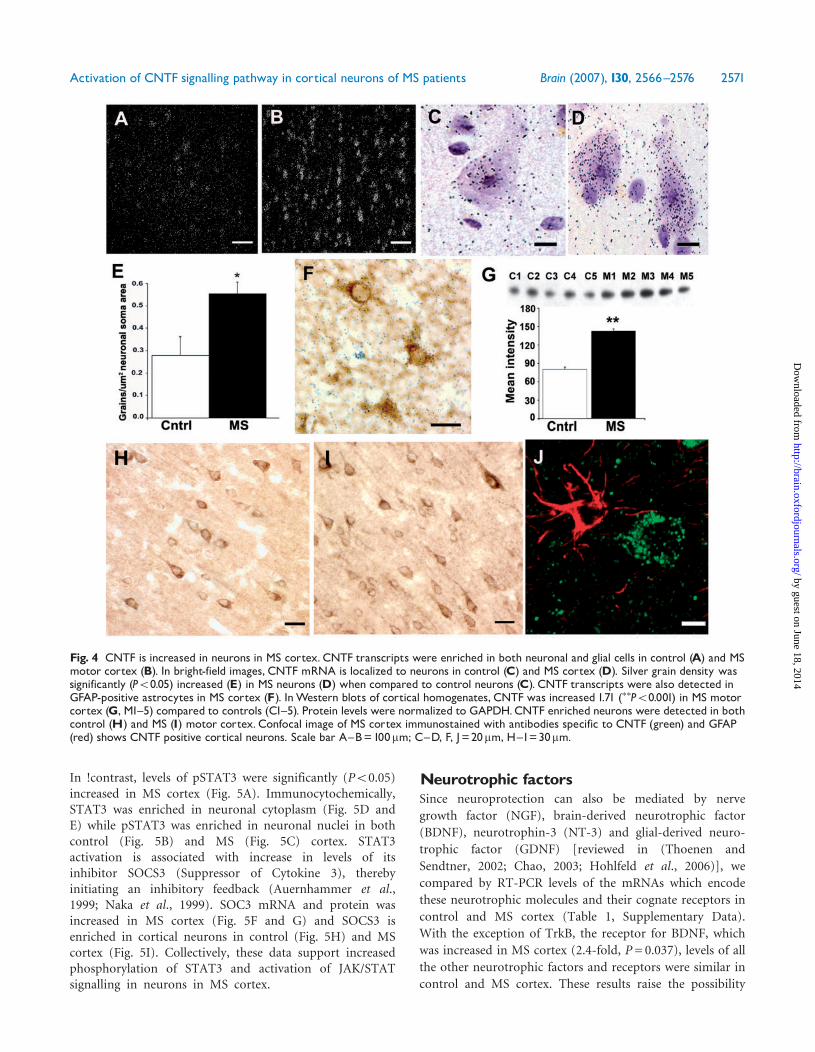
CNTFRa was highly enriched in neurons in both control(Fig. 3A) and MS (Fig. 3B and C) brains and grain densitieswere significantly increased (1.80-fold; P50.05) in neuronsin MS cortex when compared to control cortex (Fig. 3D).Silver grain densities for myelin proteolipid protein (PLP)mRNA were similar in control (Fig. 3E) and MS (Fig. 3F)cortex. CNTFRa mRNA was below detection limits inmicroarrays of both control white matter (n= 13) and whitematter lesions (n= 12) (Supplemental Data, Fig. B). Thiswas consistent with our immunohistochemical staining,which failed to detect CNTFRa-positive glial cells insections of control or MS brains. In addition, we did notdetect any LIFRb and GP130 positive glial cells in controland MS cortex. CNTFRa mRNA was readily detected inmicroarrays of cortical lesions (n= 13) but was notsignificantly increased compared to control cortex (n= 8)(Supplemental Data, Fig. B). Based on these comparisons,increase in CNTF-receptor expression appears to be aspecific response of neurons in non-lesion motor cortex inMS patients.
CNTFFollowing confirmation of increased CNTF-receptor com-plex in neurons, we compared levels and locations of CNTFmRNA in MS and control cortices using radioactive in situhybridization. Dark-field images contained large and smallsilver grain aggregates suggesting CNTF mRNA expressionby both neurons and glial cells in control (Fig. 4A) and MScortex (Fig. 4B). Upper motor neurons (layers V–VI) were
identified by size and shape in bright-field images (Fig. 4Cand D) and silver grains quantified. Silver grain densitieswere significantly (P50.05) increased in MS corticalneurons compared to control (Fig. 4E). To identifythe CNTF mRNA-positive glial cells, we combinedimmunohistochemistry for GFAP and in situ hybridizationfor CNTF. Silver grains were localized over GFAP positiveastrocytes (Fig. 4F). Collectively, these data supportincreased CNTF mRNA in both neurons and astrocytes inMS cortex. We next compared CNTF levels in western blotsand found a 1.71-fold (P50.001) increase of CNTF in MScortex (Fig. 4G). CNTF was detected in cortical neurons incontrol (Fig. 4H) and MS patients (Fig. 4I) by immuno-histochemistry. Interestingly, CNTF-positive astrocytes werenot detected in MS cortex (Fig. 4J).
STATsignallingThe JAK/STAT pathway is one of the primary intracellularsignalling cascades activated by CNTF (Rajan et al., 1996;Taga and Kishimoto, 1997; Heinrich et al.,1998). Signaltransducers and activators of transcription (STATs) residein the cytoplasm in a latent, hypophosphorylated form.Upon activation by CNTF signalling, STAT3 is phos-phorylated, dimerized and translocated to the nucleuswhere it binds to DNA elements and stimulates transcrip-tion of downstream genes (Leaman et al., 1996).We quantified STAT3 and pSTAT3 levels in MS andcontrol cortex (Fig. 5A). Total STAT3 levels were similarin control and MS cortical homogenates (Fig. 5A).
Fig. 3 CNTF-receptor amRNA is increased in neurons in MS cortex. Distribution of CNTFRamRNA in sections from control (A) and MScortex (B). CNTFRa transcripts were highly enriched in neurons (C) in MS cortex. Silver grain densities over upper motor neurons(layers V^VI) were significantly (P50.05) increased in MS sections (D). Proteolipid protein mRNA densities were similar inoligodendrocytes in control (F) and MS (E) cerebral cortex sections. Scale bars: A^B, E^F=100mm; C=20mm.
2570 Brain (2007), 130, 2566^2576 R. Dutta et al.
by guest on June 18, 2014http://brain.oxfordjournals.org/
Dow
nloaded from
In !contrast, levels of pSTAT3 were significantly (P50.05)increased in MS cortex (Fig. 5A). Immunocytochemically,STAT3 was enriched in neuronal cytoplasm (Fig. 5D andE) while pSTAT3 was enriched in neuronal nuclei in bothcontrol (Fig. 5B) and MS (Fig. 5C) cortex. STAT3activation is associated with increase in levels of itsinhibitor SOCS3 (Suppressor of Cytokine 3), therebyinitiating an inhibitory feedback (Auernhammer et al.,1999; Naka et al., 1999). SOC3 mRNA and protein wasincreased in MS cortex (Fig. 5F and G) and SOCS3 isenriched in cortical neurons in control (Fig. 5H) and MScortex (Fig. 5I). Collectively, these data support increasedphosphorylation of STAT3 and activation of JAK/STATsignalling in neurons in MS cortex.
Neurotrophic factorsSince neuroprotection can also be mediated by nerve
growth factor (NGF), brain-derived neurotrophic factor
(BDNF), neurotrophin-3 (NT-3) and glial-derived neuro-
trophic factor (GDNF) [reviewed in (Thoenen and
Sendtner, 2002; Chao, 2003; Hohlfeld et al., 2006)], we
compared by RT-PCR levels of the mRNAs which encode
these neurotrophic molecules and their cognate receptors in
control and MS cortex (Table 1, Supplementary Data).
With the exception of TrkB, the receptor for BDNF, which
was increased in MS cortex (2.4-fold, P= 0.037), levels of all
the other neurotrophic factors and receptors were similar in
control and MS cortex. These results raise the possibility
Fig. 4 CNTF is increased in neurons in MS cortex. CNTF transcripts were enriched in both neuronal and glial cells in control (A) and MSmotor cortex (B). In bright-field images, CNTF mRNA is localized to neurons in control (C) and MS cortex (D). Silver grain density wassignificantly (P50.05) increased (E) in MS neurons (D) when compared to control neurons (C). CNTF transcripts were also detected inGFAP-positive astrocytes in MS cortex (F). InWestern blots of cortical homogenates, CNTF was increased 1.71 (��P50.001) in MS motorcortex (G, M1^5) compared to controls (C1^5). Protein levels were normalized to GAPDH.CNTF enriched neurons were detected in bothcontrol (H) and MS (I) motor cortex. Confocal image of MS cortex immunostained with antibodies specific to CNTF (green) and GFAP(red) shows CNTF positive cortical neurons. Scale bar A^B=100mm; C^D, F, J=20mm, H^I=30mm.
Activation of CNTF signalling pathway in cortical neurons of MS patients Brain (2007), 130, 2566^2576 2571
by guest on June 18, 2014http://brain.oxfordjournals.org/
Dow
nloaded from

that activation of CNTF signalling is the major neuropro-
tective pathway induced in cortex. The significance of
increased TrkB remains to be determined.
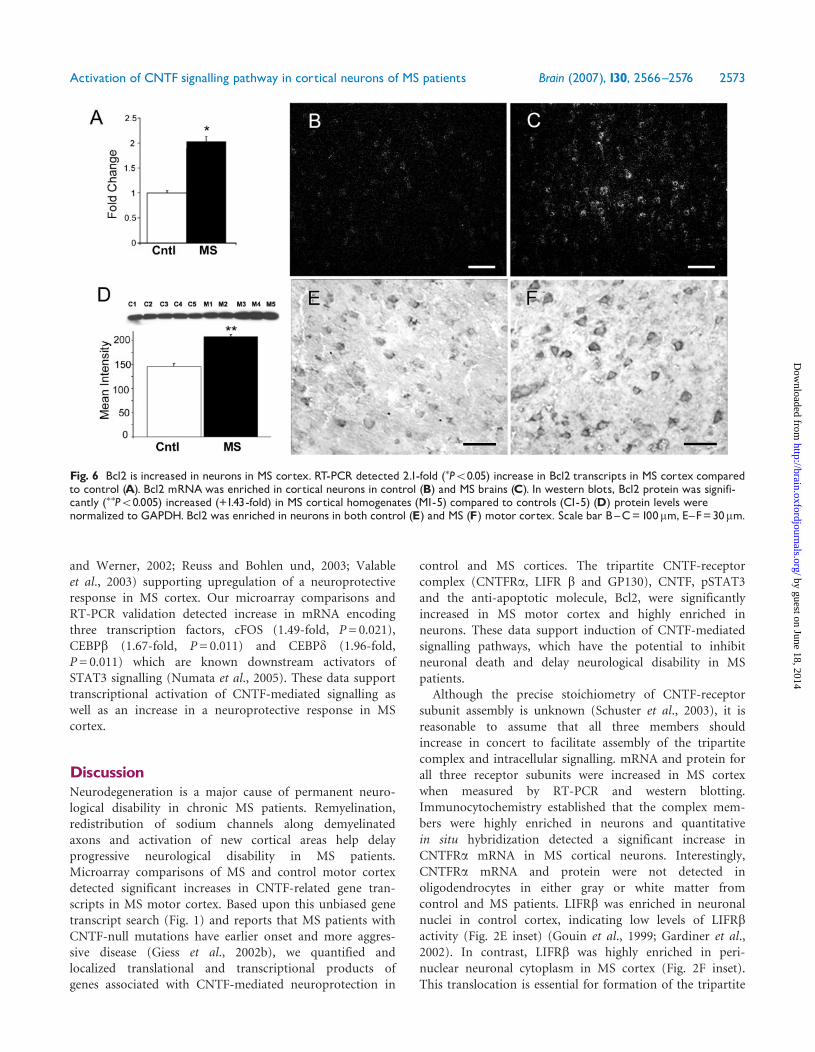
Transcription factors andneuroprotective genesIf CNTF is inducing a neuroprotective signalling cascade, itshould result in transcription of neuroprotective molecules.Our microarray comparison detected a 1.9-fold increase inmRNA encoding the anti-apoptotic molecule Bcl2 in MScortex. This increase in Bcl2 transcripts was confirmed byRT-PCR (2.1-fold; P50.05) (Fig. 6A). In situ hybridizationstudies localized most Bcl2 mRNA to neurons and supportsincreased neuronal Bcl2 transcripts in MS cortex (Fig. 6C)when compared to control cortex (Fig. 6B). Significantincreases in Bcl2 protein (1.43-fold; P50.008) were also
detected in MS cortex (Fig. 6D). Similar to Bcl2 mRNA,most Bcl2 protein was detected in neurons in both control(Fig. 6E) and MS (Fig. 6F) cortex. We detected significantdecrease by RT-PCR (1.98-fold, P= 0.0001) in expression ofTGF-b inducible early growth response transcript (TIEG)mRNA in MS cortex. This result provides further evidenceof an anti-apoptotic response in MS cortex as increasedexpression of TIEG is associated with increased apoptosis(Ribeiro et al., 1999). These data support the induction ofa Bcl2-mediated anti-apoptotic pathway in neurons in themotor cortex of MS patients. This anti-apoptotic responsewas also coupled with increased expression of severalneuroprotective molecules including FGF2 (fibroblastgrowth factor 2) (3.4-fold by RT-PCR, P= 0.050), ACVR2(Activin receptor 2) (+2.82, P= 0.045), ANG-1(Angiopoietin-1) (+2.94, P= 0.040) and epidermal growthfactor receptor (EGFR) (2.04-fold, P= 0.050) (Alzheimer
Fig. 5 STAT3-signalling pathway is activated in cortical neurons of MS patients. (A) Phosphorylated STAT3 (pSTAT3) was increased MScortical homogenates (M1^M3) when compared to controls (C1^C3).Total STAT3 was similar in MS and control samples.GAPDH was usedas protein loading control. �P50.05. Cortical neurons express pSTAT3 and STAT3 (B^E). Control (B,D) and MS (C, E) motor cortexsections were stained with pSTAT3 (B,C) and STAT3 (D, E) antibodies. pSTAT3 is enriched in neuronal nuclei (B,C) while total STAT3 isenriched in neuronal cytoplasm (D, E). SOCS3 is increased in MS cortex (F^I). RT-PCR detected a 3-fold (P50.05) increase in SOCS3transcripts in MS cortex compared to control (F). SOCS3 protein was also increased significantly in MS cortical homogenates (M1-3)compared to controls (C1-3) (G). Protein levels were normalized to GAPDH. SOCS3 is enriched in neurons in both control (H) and MS (I)motor cortex. Scale bar=30mm.
2572 Brain (2007), 130, 2566^2576 R. Dutta et al.
by guest on June 18, 2014http://brain.oxfordjournals.org/
Dow
nloaded from
and Werner, 2002; Reuss and Bohlen und, 2003; Valableet al., 2003) supporting upregulation of a neuroprotectiveresponse in MS cortex. Our microarray comparisons andRT-PCR validation detected increase in mRNA encodingthree transcription factors, cFOS (1.49-fold, P= 0.021),CEBPb (1.67-fold, P= 0.011) and CEBPd (1.96-fold,P= 0.011) which are known downstream activators ofSTAT3 signalling (Numata et al., 2005). These data supporttranscriptional activation of CNTF-mediated signalling aswell as an increase in a neuroprotective response in MScortex.
DiscussionNeurodegeneration is a major cause of permanent neuro-logical disability in chronic MS patients. Remyelination,redistribution of sodium channels along demyelinatedaxons and activation of new cortical areas help delayprogressive neurological disability in MS patients.Microarray comparisons of MS and control motor cortexdetected significant increases in CNTF-related gene tran-scripts in MS motor cortex. Based upon this unbiased genetranscript search (Fig. 1) and reports that MS patients withCNTF-null mutations have earlier onset and more aggres-sive disease (Giess et al., 2002b), we quantified andlocalized translational and transcriptional products ofgenes associated with CNTF-mediated neuroprotection in
control and MS cortices. The tripartite CNTF-receptorcomplex (CNTFRa, LIFR b and GP130), CNTF, pSTAT3and the anti-apoptotic molecule, Bcl2, were significantlyincreased in MS motor cortex and highly enriched inneurons. These data support induction of CNTF-mediatedsignalling pathways, which have the potential to inhibitneuronal death and delay neurological disability in MSpatients.
Although the precise stoichiometry of CNTF-receptorsubunit assembly is unknown (Schuster et al., 2003), it isreasonable to assume that all three members shouldincrease in concert to facilitate assembly of the tripartitecomplex and intracellular signalling. mRNA and protein forall three receptor subunits were increased in MS cortexwhen measured by RT-PCR and western blotting.Immunocytochemistry established that the complex mem-bers were highly enriched in neurons and quantitativein situ hybridization detected a significant increase inCNTFRa mRNA in MS cortical neurons. Interestingly,CNTFRa mRNA and protein were not detected inoligodendrocytes in either gray or white matter fromcontrol and MS patients. LIFRb was enriched in neuronalnuclei in control cortex, indicating low levels of LIFRbactivity (Fig. 2E inset) (Gouin et al., 1999; Gardiner et al.,2002). In contrast, LIFRb was highly enriched in peri-nuclear neuronal cytoplasm in MS cortex (Fig. 2F inset).This translocation is essential for formation of the tripartite
Fig. 6 Bcl2 is increased in neurons in MS cortex. RT-PCR detected 2.1-fold (�P50.05) increase in Bcl2 transcripts in MS cortex comparedto control (A). Bcl2 mRNAwas enriched in cortical neurons in control (B) and MS brains (C). In western blots, Bcl2 protein was signifi-cantly (��P50.005) increased (+1.43-fold) in MS cortical homogenates (M1-5) compared to controls (C1-5) (D) protein levels werenormalized to GAPDH. Bcl2 was enriched in neurons in both control (E) and MS (F) motor cortex. Scale bar B^C=100mm, E^F=30mm.
Activation of CNTF signalling pathway in cortical neurons of MS patients Brain (2007), 130, 2566^2576 2573
by guest on June 18, 2014http://brain.oxfordjournals.org/
Dow
nloaded from

complex and mandatory for CNTF signalling. LIFRbtranslocation from nucleus to cytoplasm provides anatomi-cal support for increased CNTF signalling in MS corticalneurons. Increased CNTF-receptor subunit expression andtranslocation of LIFRb subunits to the neuronal cytoplasmmay be the result of increased CNTF expression astranscriptional and translational products of CNTF weresignificantly increased in MS motor cortex and detected incortical neurons. Detection of CNTF mRNA in astrocytesand cortical neurons of MS patients raises the possibility ofboth an autocrine and a paracrine mode of CNTF action inMS cortex.Activation of the CNTF tripartite receptor complex is
known to activate several signalling pathways includingJAK/STAT, NFkB and MAP kinase [Reviewed in (Segal andGreenberg, 1996)]. In stable non-differentiating cells, CNTFpreferentially activates STAT3 compared to other membersof the STAT family (Stahl et al., 1995). We provideevidence for STAT3 activation in neurons in the motorcortex of MS patients. While STAT3 levels were similar inhomogenates from control and MS cortex, its phosphoryla-tion state was significantly increased in MS samples. Wefurther demonstrate that STAT3 is highly enriched inneurons and more importantly that pSTAT3 is enriched inneuronal nuclei in MS cortex. One of the primary targetsassociated with activation of STAT3 is its inhibitor SOCS3(Auernhammer et al., 1999; Naka et al., 1999). To providefurther evidence of STAT3 activation, we quantified levelsof SOCS3 in MS cortex. Increased levels of SOCS3 proteinwas detected in MS cortical neurons. Our data, therefore,provides evidence that known downstream components ofSTAT3 and CNTF signalling are activated in MS cortex. Itshould be stressed, however, that these data are correlativein nature and we recognize the possibility that STAT3 couldbe activated by multiple signalling pathways.We also quantified mRNAs encoding NGF, BDNF, NT-3
and GDNF. CNTF was the only neurotrophin increased inMS cortex. Using RT-PCR, mRNA encoding TrkB, thereceptor for BDNF, was increased MS cortex. TrkB waspreviously detected in neurons close to MS plaques(Kerschensteiner et al., 1999) and BDNF expression wasshown to be increased in inflammatory white matter lesionsin MS brains (Kerschensteiner et al., 1999). Since oursamples were collected from non-inflamed regions of MScortex, it was not surprising that BDNF levels were notincreased. While the relevance of increased TrkB expressionand identification of a possible source of its ligand remainsto be determined, it is possible that TrkB signalling mayalso act in parallel with CNTF signalling in MS corticalneurons.Essential to a CNTF-mediated neuroprotective response
is the expression of neuroprotective molecules. Transcriptsencoding three transcription factors, cFOS (Kuroda et al.,2001; Kelly et al., 2004) CEBPb and CEBPd (Cantwell et al.,1998;Weihua et al., 2000) that are activated during STAT3signalling (Numata et al., 2005) were increased in
microarrays of MS cortex. More importantly, transcriptsencoding three neuroprotective molecules Bcl2, FGF2 andANG-1 were also significantly increased in MS corticalmicroarrays. Because of its anti-apoptotic function, wequantified and localized Bcl2 transcriptional and transla-tional products. Bcl2 protein and mRNA were significantlyincreased in homogenates from MS cortex and was shownto be highly enriched in neurons by immunocytochemistryand in situ hybridization. Quantification of silver graindensities detected a significant increase of Bcl2 transcriptsin MS cortical neurons compared to control corticalneurons. We previously reported an increase in apoptoticneurons in lesions in MS cortex (Peterson et al., 2001).Induction of a Bcl2-mediated anti-apoptotic pathway mayinhibit neuronal apoptosis in non-lesion cortex in MSpatients.
A role for CNTF-mediated neuroprotection in MSpatients was previously proposed based upon earlierdisease onset and more severe symptoms in MS patientswith CNTF null mutations (Giess et al., 2002b). Hoffmanand colleagues (Hoffmann and Hardt, 2002), however,failed to detect differences in disease onset or progressionin MS patients with CNTF-null mutations and suggestedthat other neurotrophic factors may compensate for loss ofCNTF. Our data describing upregulation of endogenousCNTF and CNTF signalling components in MS corticalneurons provide additional support for CNTF-mediatedneuroprotection in chronic MS patients. Significantincreases in CSF levels of CNTF in MS patients recoveringfrom relapses suggest that this neuroprotective pathwayis operational during early stages of MS. mRNA encodingCNTF have been reported increased following spinalcord injury (Widenfalk et al., 2001). As in MS, ALSpatients with CNTF null mutations have earlier onset anda more aggressive disease (Giess et al., 2002a). Exogenousdelivery of CNTF has also been associated with decreaseddegeneration of striatal neurons and cortical-striatal circuitsin animal models of Parkinson’s and Huntington’sdisease (Emerich et al., 1997). CNTF, therefore, appearsto have a neuroprotective role in several neurodegenerativediseases.
While the therapeutic potential for CNTF in CNSdiseases has been clear for sometime, systemic delivery ofCNTF as a therapeutic is complicated by its short half-lifeand its inability to readily pass the blood–brain barrier.In an ALS clinical trial, systemic delivery of CNTF atrelatively high doses (>5mg/kg of body weight) caused anumber of side effects including aseptic meningitis,respiratory failure and hepatic infections (Miller et al.,1996). It was recently shown that at the concentrations usedin the ALS trial, CNTF can activate the IL-6Ra (Schusteret al., 2003), raising the possibility that these side effectswere not due to interaction of CNTF with its cognatereceptor. Development of human CNTF variants thatinteract specifically with the cognate CNTF-receptor com-plex may provide a more effective mechanism for testing
2574 Brain (2007), 130, 2566^2576 R. Dutta et al.
by guest on June 18, 2014http://brain.oxfordjournals.org/
Dow
nloaded from

the efficacy of systemic CNTF therapies. The data describedhere raises the possibility that therapeutic enhancement ofendogenous CNTF signalling pathways as an additionaltherapeutic approach for increasing neuronal survival inMS and other neurodegenerative diseases.
Supplementary materialSupplementary material is available at Brain online.
AcknowledgementsThis work was supported by the NIH (NINDS) grants RO1NS35058 and PO1 NS38667 (B.D.T). Authors would like tothank Thalia Torres for her excellent technical help with thein situ hybridization.
ReferencesAlzheimer C, Werner S. Fibroblast growth factors and neuroprotection.
Adv Exp Med Biol 2002; 513: 335–51.
Auernhammer CJ, Bousquet C, Melmed S. Autoregulation of pituitary
corticotroph SOCS-3 expression: characterization of the murine SOCS-3
promoter. Proc Natl Acad Sci USA 1999; 96: 6964–9.
Cantwell CA, Sterneck E, Johnson PF. Interleukin-6-specific activation of
the C/EBPdelta gene in hepatocytes is mediated by Stat3 and Sp1. Mol
Cell Biol 1998; 18: 2108–17.
Chao MV. Neurotrophins and their receptors: a convergence point for
many signalling pathways. Nat Rev Neurosci 2003; 4: 299–309.
Dallner C, Woods AG, Deller T, Kirsch M, Hofmann HD. CNTF and
CNTF receptor alpha are constitutively expressed by astrocytes in the
mouse brain. Glia 2002; 37: 374–8.
de Almeida LP, Zala D, Aebischer P, Deglon N. Neuroprotective effect of a
CNTF-expressing lentiviral vector in the quinolinic acid rat model of
Huntington’s disease. Neurobiol Dis 2001; 8: 433–46.
DeChiara TM, Vejsada R, Poueymirou WT, et al. Mice lacking the CNTF
receptor, unlike mice lacking CNTF, exhibit profound motor neuron
deficits at birth. Cell 1995; 83: 313–22.
Dutta R, McDonough J, Yin X, et al. Mitochondrial dysfunction as a cause
of axonal degeneration in multiple sclerosis patients. Ann Neurol 2006;
59: 478–89.
Emerich DF, Winn SR, Hantraye PM, et al. Protective effect of
encapsulated cells producing neurotrophic factor CNTF in a monkey
model of Huntington’s disease. Nature 1997; 386: 395–9.
Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute
multiple sclerosis lesions. Brain 1997; 120: 393–9.
Gardiner NJ, Cafferty WB, Slack SE, Thompson SW. Expression of gp130
and leukaemia inhibitory factor receptor subunits in adult rat sensory
neurones: regulation by nerve injury. J Neurochem 2002; 83: 100–9.
Giess R, Holtmann B, Braga M, et al. Early onset of severe familial
amyotrophic lateral sclerosis with a SOD-1 mutation: potential impact
of CNTF as a candidate modifier gene. Am J Hum Genet 2002a; 70:
1277–86.
Giess R, Maurer M, Linker R, et al. Association of a null mutation in the
CNTF gene with early onset of multiple sclerosis. Arch Neurol 2002b;
59: 407–9.
Gouin F, Couillaud S, Cottrel M, Godard A, Passuti N, Heymann D.
Presence of leukaemia inhibitory factor (LIF) and LIF-receptor chain
(gp190) in osteoclast-like cells cultured from human giant cell tumour
of bone. Ultrastructural Distribution Cytokine 1999; 11: 282–9.
Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L.
Interleukin-6-type cytokine signalling through the gp130/Jak/STAT
pathway. Biochem J 1998; 334: 297–314.
Hoffmann V, Hardt C. A null mutation in the CNTF gene is not
associated with early onset of multiple sclerosis. Arch Neurol 2002; 59:
1974–5.
Hohlfeld R, Kerschensteiner M, Stadelmann C, Lassmann H, Wekerle H.
The neuroprotective effect of inflammation: implications for the therapy
of multiple sclerosis. Neurol Sci 2006; 27 (Suppl 1): S1–7.
Ip NY, Li YP, van de Stadt I, Panayotatos N, Alderson RF, Lindsay RM.
Ciliary neurotrophic factor enhances neuronal survival in embryonic rat
hippocampal cultures. J Neurosci 1991; 11: 3124–34.
Kelly JF, Elias CF, Lee CE, et al. Ciliary neurotrophic factor and leptin
induce distinct patterns of immediate early gene expression in the brain.
Diabetes 2004; 53: 911–20.
Kerschensteiner M, Gallmeier E, Behrens L, et al. Activated human T cells,
B cells, and monocytes produce brain-derived neurotrophic factor
in vitro and in inflammatory brain lesions: a neuroprotective role of
inflammation? J Exp Med 1999; 189: 865–70.
Kuhlmann T, Remington L, Cognet I, et al. Continued administration of
ciliary neurotrophic factor protects mice from inflammatory pathology
in experimental autoimmune encephalomyelitis. Am J Pathol 2006; 169:
584–98.
Kuroda H, Sugimoto T, Horii Y, Sawada T. Signaling pathway of ciliary
neurotrophic factor in neuroblastoma cell lines. Med Pediatr Oncol
2001; 36: 118–21.
Lassmann H, Bruck W, Lucchinetti C, Rodriguez M. Remyelination in
multiple sclerosis. Mult Scler 1997; 3: 133–6.
Leaman DW, Leung S, Li X, Stark GR. Regulation of STAT-
dependent pathways by growth factors and cytokines. FASEB J 1996;
10: 1578–88.
Lee MY, Deller T, Kirsch M, Frotscher M, Hofmann HD. Differential
regulation of ciliary neurotrophic factor (CNTF) and CNTF receptor
alpha expression in astrocytes and neurons of the fascia dentata after
entorhinal cortex lesion. J Neurosci 1997; 17: 1137–46.
Linker RA, Maurer M, Gaupp S, et al. CNTF is a major protective factor in
demyelinating CNS disease: a neurotrophic cytokine as modulator in
neuroinflammation. Nat Med 2002; 8: 620–4.
Martino G. How the brain repairs itself: new therapeutic strategies in
inflammatory and degenerative CNS disorders. Lancet Neurol 2004; 3:
372–8.
Massaro AR. Are there indicators of remyelination in blood or CSF of
multiple sclerosis patients? Mult Scler 1998; 4: 228–31.
Massaro AR, Soranzo C, Carnevale A. Cerebrospinal-fluid ciliary
neurotrophic factor in neurological patients. Eur Neurol 1997; 37:
243–6.
Masu Y, Wolf E, Holtmann B, Sendtner M, Brem G, Thoenen H.
Disruption of the CNTF gene results in motor neuron degeneration.
Nature 1993; 365: 27–32.
Middleton G, Hamanoue M, Enokido Y, et al. Cytokine-induced nuclear
factor kappa B activation promotes the survival of developing neurons.
J Cell Biol 2000; 148: 325–32.
Miller RG, Bryan WW, Dietz MA, et al. Toxicity and tolerability of
recombinant human ciliary neurotrophic factor in patients with
amyotrophic lateral sclerosis. Neurology 1996; 47: 1329–31.
Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P. Molecular
characterization of schizophrenia viewed by microarray analysis of gene
expression in prefrontal cortex. Neuron 2000; 28: 53–67.
Naka T, Fujimoto M, Kishimoto T. Negative regulation of cytokine
signaling: STAT-induced STAT inhibitor. Trends Biochem Sci 1999; 24:
394–8.
Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple
sclerosis. N Engl J Med 2000; 343: 938–52.
Numata A, Shimoda K, Kamezaki K, et al. Signal transducers and
activators of transcription 3 augments the transcriptional activity of
CCAAT/enhancer-binding protein alpha in granulocyte colony-stimulat-
ing factor signaling pathway. J Biol Chem 2005; 280: 12621–9.
Pantano P, Iannetti GD, Caramia F, et al. Cortical motor reorganization
after a single clinical attack of multiple sclerosis. Brain 2002; 125:
1607–15.
Activation of CNTF signalling pathway in cortical neurons of MS patients Brain (2007), 130, 2566^2576 2575
by guest on June 18, 2014http://brain.oxfordjournals.org/
Dow
nloaded from

Peterson JW, Bo L, Mork S, Chang A, Trapp BD. Transected neurites,
apoptotic neurons and reduced inflammation in cortical MS lesions.
Ann Neurol 2001; 50: 389–400.
Peterson WM, Wang Q, Tzekova R, Wiegand SJ. Ciliary neurotrophic
factor and stress stimuli activate the Jak-STAT pathway in retinal
neurons and glia. J Neurosci 2000; 20: 4081–90.
Rajan P, Symes AJ, Fink JS. STAT proteins are activated by ciliary
neurotrophic factor in cells of central nervous system origin. J Neurosci
Res 1996; 43: 403–11.
Reddy H, Narayanan S, Arnoutelis R, et al. Evidence for adaptive
functional changes in the cerebral cortex with axonal injury from
multiple sclerosis. Brain 2000; 123: 2314–320.
Reuss B, Bohlen und HO. Fibroblast growth factors and their receptors in
the central nervous system. Cell Tissue Res 2003; 313: 139–57.
Ribeiro A, Bronk SF, Roberts PJ, Urrutia R, Gores GJ. The
transforming growth factor beta(1)-inducible transcription factor
TIEG1, mediates apoptosis through oxidative stress. Hepatology 1999;
30: 1490–7.
Rocca MA, Mezzapesa DM, Falini A, et al. Evidence for axonal pathology
and adaptive cortical reorganization in patients at presentation with
clinically isolated syndromes suggestive of multiple sclerosis.
Neuroimage 2003; 18: 847–55.
Schuster B, Kovaleva M, Sun Y, et al. Signaling of human ciliary
neurotrophic factor (CNTF) revisited. The interleukin-6 receptor can
serve as an alpha-receptor for CTNF. J.Biol Chem 2003; 278: 9528–35.
Segal RA, Greenberg ME. Intracellular signaling pathways activated by
neurotrophic factors. Annu Rev Neurosci 1996; 19: 463–89.
Stahl N, Farruggella TJ, Boulton TG, Zhong Z, Darnell JE, Jr,
Yancopoulos GD. Choice of STATs and other substrates specified by
modular tyrosine-based motifs in cytokine receptors. Science 1995; 267:
1349–53.
Stockli KA, Lillien LE, Naher-Noe M, et al. Regional distribution,
developmental changes, and cellular localization of CNTF-mRNA and
protein in the rat brain. J Cell Biol 1991; 115: 447–59.
Taga T, Hibi M, Hirata Y, et al. Interleukin-6 triggers the association
of its receptor with a possible signal transducer, gp130. Cell 1989; 58:
573–81.
Taga T, Kishimoto T. Gp130 and the interleukin-6 family of cytokines.
Annu Rev Immunol 1997; 15: 797–819.
Takahashi R, Yokoji H, Misawa H, Hayashi M, Hu J, Deguchi T. A null
mutation in the human CNTF gene is not causally related to
neurological diseases. Nat Genet 1994; 7: 79–84.
Thoenen H, Sendtner M. Neurotrophins: from enthusiastic expectations
through sobering experiences to rational therapeutic approaches. Nat
Neurosci 2002; 5 (Suppl): 1046–50.
Thome J, Nara K, Foley P, et al. Ciliary neurotrophic factor (CNTF)
genotypes: influence on choline acetyltransferase (ChAT) and acetylcho-
line esterase (AChE) activities and neurotrophin 3 (NT3) concentration
in human post mortem brain tissue. J Hirnforsch 1997; 38: 443–51.
Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal
transection in the lesions of multiple sclerosis. N Engl J Med 1998; 338:
278–85.
Valable S, Bellail A, Lesne S, et al. Angiopoietin-1-induced PI3-kinase
activation prevents neuronal apoptosis. FASEB J 2003; 17: 443–5.
Waxman SG. Ions, energy and axonal injury: towards a molecular
neurology of multiple sclerosis. Trends Mol Med 2006; 12: 192–5.
Weihua X, Hu J, Roy SK, Mannino SB, Kalvakolanu DV. Interleukin-6
modulates interferon-regulated gene expression by inducing the ISGF3
gamma gene using CCAAT/enhancer binding protein-beta(C/EBP-beta).
Biochim Biophys Acta 2000; 1492: 163–71.
Weinshenker BG. Epidemiology of multiple sclerosis. Neurol Clin 1996;
14: 291–308.
Weinshenker BG, Bass B, Rice GP, et al. The natural history of multiple
sclerosis: a geographically based study. I. Clinical course and disability.
Brain 1989; 112: 133–46.
Weisenhorn DM, Roback J, Young AN, Wainer BH. Cellular aspects of
trophic actions in the nervous system. Int Rev Cytol 1999; 189: 177–265.
Widenfalk J, Lundstromer K, Jubran M, Brene S, Olson L. Neurotrophic
factors and receptors in the immature and adult spinal cord after
mechanical injury or kainic acid. J Neurosci 2001; 21: 3457–75.
Winter CG, Saotome Y, Levison SW, Hirsh D. A role for ciliary
neurotrophic factor as an inducer of reactive gliosis, the glial response
to central nervous system injury. Proc Natl Acad Sci USA 1995; 92:
5865–9.
2576 Brain (2007), 130, 2566^2576 R. Dutta et al.
by guest on June 18, 2014http://brain.oxfordjournals.org/
Dow
nloaded from




















