
Ascorbate protects liver from metabolic disorder through ...
Upload
uni-erlangenCategory
view
0download
0
Activation-induced NKT cell hyporesponsiveness protectsfrom �-galactosylceramide hepatitis and is independent ofactive transregulatory factors
Markus Biburger*,† and Gisa Tiegs†,‡ ,1
*Laboratory for Experimental Immunology and Immunotherapy, Nikolaus-Fiebiger-Center for Molecular Medicine,Medical Department III, University Hospital Erlangen, Germany; ‡Center of Internal Medicine, Division ofExperimental Immunology and Hepatology, University Medical Centre Hamburg-Eppendorf, Hamburg, Germany;and †Institute of Experimental and Clinical Pharmacology and Toxicology, University of Erlangen-Nuremberg,Erlangen, Germany
Abstract: NK T (NKT) cells, unique lymphocytesexpressing features of NK and T lymphocytes, canspecifically be activated with the glycolipid antigen�-galactosylceramide (�-GalCer). In humans andmice, this activation provokes pronounced cyto-kine responses. In C57BL/6 mice, �-GalCer injec-tion additionally induces NKT-mediated liver in-jury, representing a model for immune-mediatedhepatitis in humans. However, a single �-GalCerpretreatment of mice prevented NKT-mediatedliver injury, cytokine responses (systemically andlocally in the liver), and up-regulation of hepato-cellular Fas upon �-GalCer rechallenge. As �-Gal-Cer is used as a NKT cell-activating agent in clini-cal trials, an investigation of tolerance inductionappears crucial. We demonstrate that �-GalCertolerance does not depend on Kupffer cells, IL-10,Caspase-3-mediated apoptosis, or CD4�CD25� Tregulatory cells (Tregs), which are crucial in othermodels of immunological tolerance. Amending rel-evant, earlier approaches of others, we coculti-vated highly purified, nontolerized and tolerizedliver NKT cells ex vivo and could convincinglyexclude the relevance of transdominant NKTTregs. These results strongly suggest �-GalCer-in-duced tolerance to be exclusively caused by NKTcell intrinsic hyporesponsiveness. Tolerized miceshowed specific diminishment of the intrahepaticCD4� NKT cell subpopulation, with the CD4– pop-ulation largely unaffected, and revealed down-modulation of �-GalCer-specific TCR and the NKTcostimulator glucocorticoid-induced TNFR-re-lated protein on liver NKT cells, whereas inhibi-tory Ly49I was increased. In conclusion, �-GalCertolerance could serve as a model for the frequentlyobserved NKT cell hyporesponsiveness in tumorpatients and might help to develop strategies fortheir reactivation. Conversely, approaches to ren-der NKT cells hyporesponsive may constitute newtherapeutic strategies for diseases, where aberrantNKT cell activation is causally involved. J. Leukoc.Biol. 84: 264–279; 2008.
Key Words: lymphocytes � natural killer T cells � tolerance � liverimmunology � anergy � animal models
INTRODUCTION
NK T (NKT) cells are a unique subpopulation of T cells, whichin their majority, express NK surface markers such as NK1.1,IL-2Rß, and to some extent, Ly49 family members and revealthe Thy1high, CD44high, CD45RBhigh phenotype of activated Tcells (defined in C57BL/6 mice; see ref. [1]). The predominantpopulation of NKT cells expresses a TCR repertoire with aninvariant V�14-J�18 TCR� chain in mice or V�24-J�18 inhumans and a restricted TCRß repertoire. This TCR mediatesrecognition of glycolipid antigens presented by the MHC classI-like molecule CD1d [1], such as isoglobotrihexosylceramide,which has for some time been considered to be the physiolog-ical antigen [2] (an assumption that is challenged by recentpublications; refs. [3, 4]), or synthetic �-galactosylceramide.This surrogate antigen had been developed by Kirin BreweryCo., Ltd. (Tokyo, Japan) for cancer treatment [5], as NKT cellshave been suggested to exert antitumor responses in severalcancer types (reviewed in refs. [6, 7]). In fact, NKT cellactivation has already been analyzed in clinical trials as atherapeutic approach for cancer treatment [8–12]. Also, NKTcells are believed to be involved in the prevention of autoim-munity (see refs. [13, 14]). However, besides such beneficialeffects, there is in fact cumulating evidence that NKT cellsplay pivotal roles in the onset of pathological processes. Theyappear to be involved in several murine disease models such asatherosclerosis [15, 16], allergen-induced airway inflammationand asthma [17–19], oxazolone-induced ulcerative colitis [20],antibody-induced joint inflammation and arthritis [21, 22], anddepending on experimental design and mouse strain, alsopristane-induced lupus [23] or experimental autoimmune en-
1 Correspondence: Center of Internal Medicine, Division of ExperimentalImmunology and Hepatology, University Medical Centre Hamburg-Eppendorf,Hamburg, Martinistr. 52, 20246 Hamburg, Germany. E-mail: [email protected]
Received June 6, 2007; revised February 29, 2008; accepted March 11,2008.
doi: 10.1189/jlb.0607352
264 Journal of Leukocyte Biology Volume 84, July 2008 0741-5400/08/0084-264 © Society for Leukocyte Biology

cephalomyelitis [24]. They are also suggested to participate inthe onset of several hepatic disorders in man such as immu-nopathogenesis of chronic hepatitis C virus-induced hepatitis[25], intrahepatic bile duct lesions in primary biliary cirrhosis[26], and cirrhosis progression in chronic viral hepatitis [27].Also, two well-established murine models of immune-mediatedhepatitis induced by injection of Con A [28] or �-galactosyl-ceramide (�-GalCer) [29, 30] are strictly NKT cell-dependent[31, 32].
Several reports describe an impairment of NKT cell cytokineresponses to �-GalCer restimulation after a preceding �-Gal-Cer injection [33–37]. Also, other processes associated withNKT cell activation, such as, e.g., TCR down-modulation, havebeen reported to fail upon �-GalCer reinjection [36]. Consid-ering the Janus-like capability of NKT cells to inhibit oraugment pathologic processes [13] and our observation that asingle �-GalCer pretreatment ameliorated liver injury upon�-GalCer rechallenge, we were interested in the mechanismsof �-GalCer-induced immunosuppression and protection fromliver injury. Because of our recent identification of T regulatorycells (Tregs), Kupffer cells (KCs), and IL-10 as importantfactors in a prima facie similar model of immunological toler-ance induction by Con A [38], we also wanted to characterizetheir potential role in �-GalCer tolerance. Here, we demon-strate that neither these factors nor caspase-3-mediated apo-ptosis were relevant for �-GalCer tolerance in terms of pre-vention of cytokine production and liver injury. In contrast, wecould prove this tolerance to be based on a passive mechanism,i.e., activation-induced hyporesponsiveness, and disclosetolerization-induced changes among the intrahepatic NKT cellpopulations as well as phenotypic changes.
MATERIALS AND METHODS
Mice
Male C57BL/6 wild-type or IL-10�/� [39] mice (8–12 weeks) were obtainedfrom Harlan-Winkelmann (Borchen, Germany), Elevage Janvier (Le Genest-Saint-Isle, France), or animal facilities of the University of Erlangen-Nurem-berg (Germany) and were maintained under controlled conditions (22°C, 55%humidity, 12 h day/night rhythm) and fed standard laboratory chow. All micereceived human care according to the guidelines of the National Institutes ofHealth (Bethesda, MD, USA) and the legal requirements in Germany.
Animal treatments
�-GalCer was kindly provided by Kirin Brewery Co., Ltd. Directly before i.v.injection, the stock solution of 200 �g/ml in vehicle (0.5% w/v polysorbate-20)was diluted in pyrogen-free saline to achieve (if not, different doses arementioned for particular experiments) a dose of 1 �g per mouse in 200 �l. Forhepatocyte-specific transcription inhibition with D-galactosamine (GalN),galactosamine-hydrochloride (Carl Roth GmbH, Karlsruhe, Germany) wasadministered i.p. in pyrogen-free saline (70 mg/ml) at 200 �l/20 g mouse 30min prior to i.v. injection of 200 ng �-GalCer. The effect of exogenous IL-10on �-GalCer hepatitis was analyzed by i.v. injection of 1 �g recombinantmurine (rm)IL-10 (Peprotech, London, UK) 30 min prior to injection of 200 ng�-GalCer. For KC depletion, 100 �l liposome-encapsulated dichloromethyl-ene-bisphosphonate (Clodronate liposomes, derived from Dr. Nico van Rooi-jen, Vrije Universiteit, Amsterdam, The Netherlands) was injected i.v. 48 hbefore �-GalCer rechallenge as described previously [40]. Dichloromethylene-bisphosphonate, for their preparation, was a gift of Roche Diagnostics (Mann-heim, Germany). Efficiency of KC depletion was verified by immunohistology(not shown).
For inhibition of caspase-3-like caspases, the irreversible inhibitors Z-Val-Ala-Asp(O-methyl)-fluoromethyl ketone [zVAD(OMe).fmk; R&D Systems,Wiesbaden, Germany] or Z-Val-Ala-fluoromethyl ketone (zVAD) fmk (Bachem,Weil am Rhein, Germany) were reconstituted in DMSO and diluted in pyrogen-free saline to the indicated doses directly before injection in a total volume of200 �l per 20 g mouse with a final concentration of �4% DMSO. Applicationregimens were 5 mg/kg z-VAD(OMe).fmk i.p. 20 min prior to treatment with 1�g �-GalCer or alternatively, 10 mg/kg z-VAD.fmk i.v. 20 min prior to�-GalCer treatment (200 ng) with an additional i.p. injection of 5 mg/kg 6 hthereafter. Activity of z-VAD.fmk had been verified before [30].
In vivo depletion of CD4�CD25� Tregs was achieved by i.v. injection of 300�g rat anti-mouse CD25 mAb (clone PC-61.5) purified from hybridoma su-pernatant using Thiophilic-Superflow resin (BD-Clontech, Heidelberg, Ger-many) 1 day prior to �-GalCer injection. Efficiency of Treg depletion wasverified by flow cytometry. Therefore, splenocytes were stained with anti-CD4and anti-CD25 mAb. CD25 detection was carried out with clone 7D4, whichrecognizes another CD25 epitope than PC-61.5 to prevent false-negativestaining of cells in PC-61.5-injected mice caused by epitope-masking.
For in vivo neutralization of TNF-� or IFN-�, mice were injected i.v. with300 �g IgG, purified from sheep anti-mouse TNF-� polyclonal antiserum [41]with 75 �l polyclonal rabbit anti-TNF-� antibody IP-400 (Genzyme, Cam-bridge, MA, USA) or with 200 �l rabbit anti-mouse IFN-� serum [42], one-halfhour prior to treatment. Corresponding amounts of normal rabbit serum (SigmaChemical Co., St. Louis, MO, USA) or purified, total IgG from normal sheepserum (Sigma Chemical Co.) were used as negative controls.
Sampling of material
Mice were lethally anesthetized (150 mg/kg i.v. methohexital�15 mg/kgheparin). Cardiac blood was withdrawn for analysis of plasma transaminasesand cytokines. Livers were excised, frozen in liquid nitrogen, and stored at–20°C for preparation of RNA and subsequent real-time RT-PCR.
Analysis of liver injury
Hepatocyte damage was assessed 16–17 h after �-GalCer treatment by mea-suring plasma enzyme activities of alanine-aminotransferase (ALT) and aspar-tate-aminotransferase (AST) using the automated COBAS Mira system (RocheDiagnostics).
Cytokine quantification
Sandwich ELISAs for murine plasma cytokines were performed using Nunc-ImmunoTM 96-well flat-bottom MaxisorbTM microtiter plates (Nunc GmbH,Wiesbaden, Germany). Antibodies were purchased from BD-PharMingen (SanDiego, CA, USA) for IL-2, IL-4, IL-6, and IL-10. Streptavidin-peroxidase waspurchased from Roche Diagnostics. IFN-�, TNF-� and TGF-ß were quantifiedusing DuoSetTM ELISA-Development systems from R&D Systems, togetherwith tetramethyl benzidine substrate-reagent set (BD-PharMingen) as peroxi-dase chromogen according to the manufacturer’s instructions. For a singleexperiment, cytokine concentrations were measured in plasma of �-GalCer-tolerized or control mice using the BD Cytometric Bead Array™ (BD Bio-sciences, San Jose, CA, USA).
RNA isolation and real-time RT-PCR forcytokine mRNAs
Isolation of RNA from liver tissue was carried out using the Total-RNAisolation kit (Macherey-Nagel, Duren, Germany). mRNA was transcribed intocDNA using SuperScriptTM II RNase H– RT, oligonucleotides, and oligo-(dT)primers from Invitrogen (Karlsruhe, Germany). Real-time RT-PCR was per-formed using a LightCycler rapid thermal cycler system (Roche Diagnostics)and LightCycler-FastStart DNA Master SYBR-Green I (Roche Diagnostics),according to the manufacturer’s instructions. Primer pairs were used as de-scribed previously [30]. In addition, for Fas quantification, we used 5�-Fas:5�-CGCTGTTTTCCCTTGCTGCA-3� and 3�-Fas: 5�-ACTGAGGTAGTTT-TCACTCCA-3�. To confirm amplification specificity, melting curves of PCRproducts were analyzed. Relative mRNA levels were calculated by means of2�CP (�CP�difference of crossing-points of test and respective control sam-ples, as extracted from amplification curves by the LightCyclerTM software)after normalization with respect to -actin mRNA levels.
Biburger and Tiegs NKT cell anergy protects from �-GalCer hepatitis 265

Isolation and flow cytometric analysis of livermononuclear cells (MNCs)
To isolate hepatic MNCs, livers were passed through 100 �m nylon meshes,and hepatocytes were removed by centrifugation (800 g, 20 min) in isotonic37% Percoll solution (Amersham-Biosciences, Freiburg, Germany) containing100 U/ml heparin. Erythrocytes were lysed in 139 mM NH4Cl, 19 mM Tris. Forflow cytometry using a standard protocol, including preblocking FcRs, typi-cally, 4 105 liver leukocytes were stained using anti-mouse-CD16/32 mAb(“Fc-block”; clone 93, eBioscience, San Diego, CA, USA), FITC- or cychrome-labeled anti-mouse-CD3ε mAb (clone 145-2C11), FITC- or PE-labeled anti-mouse-NK1.1 (clone PK136), PE-labeled anti-mouse-CD25 (clone PC-61.5)mAb, PE-labeled anti-mouse-Ly49I (all BD-PharMingen), biotinylated or PE-labeled anti-mouse glucocorticoid-induced TNFR-related protein (GITR; cloneYGITR 765, BioLegend, San Diego, CA, USA), PE- or Tricolor-labeled anti-mouse-CD4 (clone RM4-5, Caltag, Hamburg, Germany), anti-mouse-CD25-PEmAb (clone 7D4, Miltenyi Biotec, Bergisch Gladbach, Germany), and FITC-,PE-, or CyChrome-conjugated streptavidin (Jackson Immunoresearch, WestGrove, PA, USA). For preparation of �-GalCer/CD1d tetramers to enablestaining of �-GalCer-specific TCR, biotinylated rmCD1d (kindly provided byDirk Busch, Institute of Medical Microbiology, Immunology and Hygiene,Technical University of Munich, Germany) was loaded with �-GalCer for 20 hat room temperature. Subsequently, CD1d was incubated in molar excess atroom temperature for 8 h with PE-conjugated streptavidin for labeling andtetramerization with stepwise addition of the streptavidin. Data were recordedand analyzed using a three-color FACScanTM flow cytometer (BD Biosciences)with BD Biosciences CellQuestTM software.
Purification of CD3�� NK1.1� NKT cells andCD4� CD25� Tregs
For purification of CD3�NK1.1� NKT cells, liver MNCs were stained withanti-CD3ε and anti-NK1.1 mAb and sorted in the Cell Sorting Core Facility ofthe University of Erlangen-Nuremberg using a MoFlo™ cell sorter (DakoCy-tomation GmbH, Hamburg, Germany; purity, �95%). For Treg purification, acombined sorting procedure was carried out using magnetic bead separation(CD4�CD25� Treg isolation kit, mouse, Miltenyi Biotec) and subsequent FACSsorting as described previously [38].
Ex vivo assays
Cells were isolated as described above, seeded in 96-well cell-culture plates,and stimulated with 15 ng/ml �-GalCer or a corresponding volume of vehicleas negative control. iGB3 (Alexis Corp., Lausen, Switzerland) was solubilizedin chloroform:methanol (2:1), dried, resuspended in methanol, and diluted inmedium to reach a final concentration of 15 �g/ml with methanol remainingbelow 1%. In assays using liver MNCs (0.8–2105 cells/well), no additionalAPCs had to be added; in experiments with purified NKT cells (0.8105
cells/well), bone marrow-derived dendritic cells (DCs; 0.4105 cells/well;kind gift from Carsten Wiethe Department of Dermatology, University Hospitalof Erlangen-Nuremberg, Erlangen) were used as �-GalCer-presenting cells. Inall assays analyzing potential suppressive effects of Tregs or cells from �-Gal-Cer-pretreated mice, close cell–cell contact was facilitated by using round-bottom plates. Cytokine concentrations in the supernatant were determined byELISA.
Statistical analysis
All data are expressed as mean � SEM (if n�3). For calculation of statisticalsignificance, data were analyzed using Student’s t-test if two groups werecompared or the Bonferroni test if several groups were tested against oneanother. P � 0.05 was considered significant.
RESULTS
Prevention of �-GalCer-induced liver injury andcytokine production upon tolerance induction
To analyze the potential of NKT cell activation to induce a stateof immunological tolerance, C57BL/6 mice were pretreated
with 1 �g �-GalCer and challenged 3 or 8 days thereafter. The�-GalCer-induced cytokine response and/or the induction ofhepatic injury were used in this study as readout for the abilityof NKT cells to be activated efficiently in vivo. In comparisonwith control mice, mice pretreated with �-GalCer at Days –3 or–8 revealed significantly reduced liver injury upon �-GalCerchallenge (Fig. 1A). Reduced liver injury was also found in anexperiment where only 50 ng �-GalCer had been used forpretreatment (data not shown). �-GalCer injection is wellknown to induce pronounced production of a broad range ofTh0, Th1, and Th2 cytokines. To analyze the effect of �-GalCerpretreatment, we measured cytokine responses in plasma andon the mRNA level in liver tissue of pretreated and controlmice at 1.5 h or together with liver transaminases 16–17 hafter rechallenge, i.e., at time-points where peak levels ofintrahepatic mRNA and maximum plasma concentrations forseveral cytokines are detectable [30]. Early plasma levels ofIFN-�, IL-2, IL-4, and TNF were decreased significantly bymore than 80% (Fig. 1B) in �-GalCer-pretreated mice. Thiscorresponded well to significantly diminished mRNA levels inthe liver at that time-point (Fig. 1C). Also, 16–17 h after�-GalCer rechallenge diminished IFN-�, TNF-�, and IL-6responses were measured in liver and plasma (not shown). Incontrast, pretreatment induced a tendency to increased IL-10plasma levels at both time-points and significantly increasedintrahepatic IL-10 mRNA 1.5 h after rechallenge (Fig. 1, B andC). Pretreatment had no relevant influence on TGF-ß produc-tion (not shown). It is worth mentioning that a BD Biosciencesmultiplex array analysis of a single experiment also revealedsignificantly reduced plasma levels of IL-1, GM-CSF, andCXCL-1 in �-GalCer-pretreated mice (data not shown). More-over, we found significantly reduced intrahepatic Fas mRNAexpression upon �-GalCer rechallenge (measured 16 h afterrechallenge) in tolerized mice in comparison with mock-pre-treated mice (Fig. 1D).
Modulation of NKT cell numbers/frequencies
Previous reports had documented NKT cells to become de-pleted or undetectable shortly after �-GalCer injection intomice, followed by recurrence and modulation of the NKTpopulation size within the following days. As disappearance ofNKT cells would result in a tolerance-like phenotype, wecharacterized NKT cell numbers and frequencies in livers of�-GalCer- and vehicle-treated animals. After 3 days, the num-ber of CD3�NK1.1� NKT cells in the liver of �-GalCer-pretreated mice even exceeded that of vehicle-treated controlmice. As shown in Figure 1E for two representative experi-ments, 8 days after pretreatment, the intrahepatic number ofNKT cells was more than two-thirds of the NKT cells in thecontrol group.
For the 8-day time-point, the modulation of the NKT popu-lation was analyzed in more detail by characterizing NKT cellsalso with regard to �-GalCer/CD1d-tetramer staining. Wefound the frequency of NKT cells among intrahepatic lympho-cytes on average to be reduced in tolerized mice by abovetwo-fifths for CD3�NK1.1�-defined NKT cells (the averageNKT cell frequency was 7.8% in tolerized compared with14.3% in control mice in nine experiments) or above one-halffor NKT cells being defined by �-GalCer/CD1d-tetramer stain-
266 Journal of Leukocyte Biology Volume 84, July 2008 http://www.jleukbio.org

ing (5.6% compared with 10.1% in six experiments; forchanges in NKT frequency, see also Fig. 7). However, as aresult of increased total numbers of intrahepatic lymphocytesin tolerized mice, the total number of hepatic CD3�NK1.1�
NKT cells was measured to be reduced by only approximatelyone-fifth in tolerized mice (the average NKT cell number intolerized mice calculated from mean values of nine experi-ments was 4.8105 cells compared with 6.2105 cells incontrol mice) and the total number of tetramer-positive cells byapproximately one-third (3.4105 cells compared with5.1105 cells in six experiments). This demonstrates that highnumbers of NKT cells are still present in the liver of tolerizedmice, and �-GalCer pretreatment does not result in an exten-sive depletion of NKT cells.
Fas down-modulation in tolerance is coupled toreduced TNF-� responses
As we had shown before that Fas ligand (FasL) expression onNKT cells was modulated by �-GalCer-induced TNF-� [30],we were interested in whether Fas expression and correspond-ingly, its reduced expression in tolerized mice were also asso-ciated with respective TNF-� responses. Injection of �-GalCerto C57BL/6 mice induced significant up-regulation of Fasexpression in the liver within a few hours, as detected byquantification of relative mRNA levels by real-time RT-PCR(Fig. 2). Hepatocyte-specific gene expression is studied fre-quently by application of GalN, which specifically inhibitstranscription in hepatocytes by depletion of the hepatocellularpool of uracil nucleotides [43]. Pretreatment of mice with GalN
(700 mg/kg) 30 min prior to �-GalCer injection (200 ng/mouse)abolished �-GalCer-induced Fas up-regulation, suggestingthat Fas was mainly expressed by hepatocytes. Pretreatment ofmice with the TNF-�-neutralizing antibody (0.3 mg/mouse IgGpurified from sheep anti-mouse TNF-� polyclonal antiserum
Fig. 1. A single pretreatment with �-GalCerimpaired susceptibility to �-GalCer rechallengein vivo. (A) Pretreatment with 1 �g �-GalCerprotected mice from liver injury upon rechal-lenge with 1 �g �-GalCer 3 or 8 days thereafterin comparison with vehicle controls, as assessedby plasma ALT activity. Reduced susceptibilityto rechallenge was reflected in reduced plasmacytokine concentrations in plasma of �-GalCer-pretreated mice 1.5 h after rechallenge (B) andreduced relative intrahepatic cytokine mRNAexpression at the same time-point as measuredby real-time RT-PCR (C). Tolerance inductionwith respect to plasma ALT levels was detected
in each of eight independent experiments (n�3); reduced cytokine responses at 1.5 h were found in two independent experiments (n�4). (D) Tolerized micerevealed significantly reduced intrahepatic Fas mRNA expression upon �-GalCer rechallenge 8 days after pretreatment. Relative mRNA expression wasquantified by RT-PCR using liver samples that had been taken 16 h after rechallenge in parallel with cardiac blood withdrawal for ALT measurement. Thisimpaired expression was detected in each of three independent experiments. The graph depicts the summary of these experiments (ntotal�9). (E) In �-GalCer-and vehicle-treated mice, intrahepatic CD3�NK1.1� NKT cell population sizes were quantified by FACS analysis. At both time-points where tolerance inpretreated mice had been demonstrated, i.e., 3 days or 8 days after �-GalCer treatment, large NKT cell populations were detected in the livers, and their sizewas increased significantly after 3 days and moderately decreased after 8 days. The graphs represent the summary of two experiments per time-point (n�4).All results are expressed as mean � SEM; *, P � 0.05, versus vehicle control.
Fig. 2. �-GalCer-induced intrahepatic Fas expression is mainly restricted tohepatocytes and dependent on TNF-�. To characterize the effect of �-GalCertreatment on Fas expression in the liver, C57BL/6 mice were treated withvehicle as a negative control or with 200 ng �-GalCer. To test for Fasexpression by hepatocytes and its potential association with TNF-�, some micehad been pretreated with GalN or anti-TNF-� antibody 30 min prior to�-GalCer injection. After �-GalCer application (4.5 h), mice were killed, andthe liver was excised for RT-PCR analysis (n�3; mean�SEM; *, P� 0.05, vs.vehicle control; #, P�0.05, vs. only �-GalCer-treated group).
Biburger and Tiegs NKT cell anergy protects from �-GalCer hepatitis 267
[41]) instead of GalN prior to �-GalCer injection had virtuallythe same effect, clearly indicating that Fas up-regulation onhepatocytes is mediated by �-GalCer-induced TNF-�. Thus,down-modulated, intrahepatic Fas expression in tolerized miceupon rechallenge was probably not an independent effect butrather reflected diminished TNF-� responses under these con-ditions.
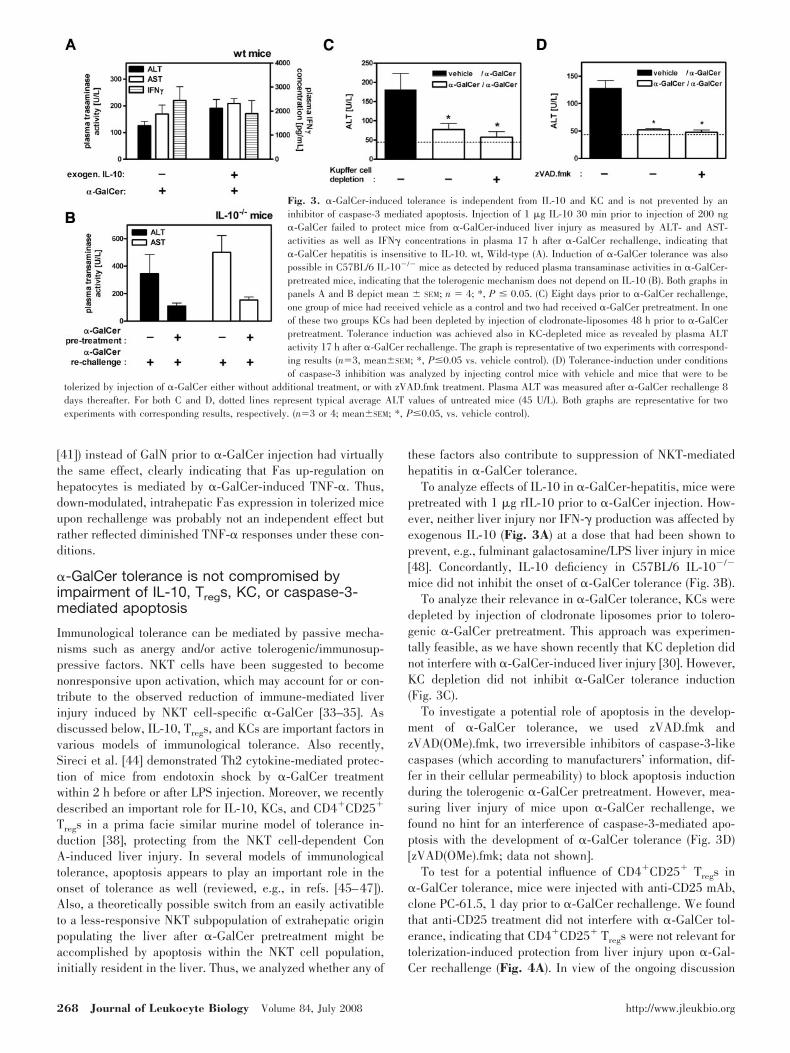
�-GalCer tolerance is not compromised byimpairment of IL-10, Tregs, KC, or caspase-3-mediated apoptosis
Immunological tolerance can be mediated by passive mecha-nisms such as anergy and/or active tolerogenic/immunosup-pressive factors. NKT cells have been suggested to becomenonresponsive upon activation, which may account for or con-tribute to the observed reduction of immune-mediated liverinjury induced by NKT cell-specific �-GalCer [33–35]. Asdiscussed below, IL-10, Tregs, and KCs are important factors invarious models of immunological tolerance. Also recently,Sireci et al. [44] demonstrated Th2 cytokine-mediated protec-tion of mice from endotoxin shock by �-GalCer treatmentwithin 2 h before or after LPS injection. Moreover, we recentlydescribed an important role for IL-10, KCs, and CD4�CD25�
Tregs in a prima facie similar murine model of tolerance in-duction [38], protecting from the NKT cell-dependent ConA-induced liver injury. In several models of immunologicaltolerance, apoptosis appears to play an important role in theonset of tolerance as well (reviewed, e.g., in refs. [45–47]).Also, a theoretically possible switch from an easily activatibleto a less-responsive NKT subpopulation of extrahepatic originpopulating the liver after �-GalCer pretreatment might beaccomplished by apoptosis within the NKT cell population,initially resident in the liver. Thus, we analyzed whether any of
these factors also contribute to suppression of NKT-mediatedhepatitis in �-GalCer tolerance.
To analyze effects of IL-10 in �-GalCer-hepatitis, mice werepretreated with 1 �g rIL-10 prior to �-GalCer injection. How-ever, neither liver injury nor IFN-� production was affected byexogenous IL-10 (Fig. 3A) at a dose that had been shown toprevent, e.g., fulminant galactosamine/LPS liver injury in mice[48]. Concordantly, IL-10 deficiency in C57BL/6 IL-10�/�
mice did not inhibit the onset of �-GalCer tolerance (Fig. 3B).To analyze their relevance in �-GalCer tolerance, KCs were
depleted by injection of clodronate liposomes prior to tolero-genic �-GalCer pretreatment. This approach was experimen-tally feasible, as we have shown recently that KC depletion didnot interfere with �-GalCer-induced liver injury [30]. However,KC depletion did not inhibit �-GalCer tolerance induction(Fig. 3C).
To investigate a potential role of apoptosis in the develop-ment of �-GalCer tolerance, we used zVAD.fmk andzVAD(OMe).fmk, two irreversible inhibitors of caspase-3-likecaspases (which according to manufacturers’ information, dif-fer in their cellular permeability) to block apoptosis inductionduring the tolerogenic �-GalCer pretreatment. However, mea-suring liver injury of mice upon �-GalCer rechallenge, wefound no hint for an interference of caspase-3-mediated apo-ptosis with the development of �-GalCer tolerance (Fig. 3D)[zVAD(OMe).fmk; data not shown].
To test for a potential influence of CD4�CD25� Tregs in�-GalCer tolerance, mice were injected with anti-CD25 mAb,clone PC-61.5, 1 day prior to �-GalCer rechallenge. We foundthat anti-CD25 treatment did not interfere with �-GalCer tol-erance, indicating that CD4�CD25� Tregs were not relevant fortolerization-induced protection from liver injury upon �-Gal-Cer rechallenge (Fig. 4A). In view of the ongoing discussion
Fig. 3. �-GalCer-induced tolerance is independent from IL-10 and KC and is not prevented by aninhibitor of caspase-3 mediated apoptosis. Injection of 1 �g IL-10 30 min prior to injection of 200 ng�-GalCer failed to protect mice from �-GalCer-induced liver injury as measured by ALT- and AST-activities as well as IFN� concentrations in plasma 17 h after �-GalCer rechallenge, indicating that�-GalCer hepatitis is insensitive to IL-10. wt, Wild-type (A). Induction of �-GalCer tolerance was alsopossible in C57BL/6 IL-10�/� mice as detected by reduced plasma transaminase activities in �-GalCer-pretreated mice, indicating that the tolerogenic mechanism does not depend on IL-10 (B). Both graphs inpanels A and B depict mean � SEM; n � 4; *, P � 0.05. (C) Eight days prior to �-GalCer rechallenge,one group of mice had received vehicle as a control and two had received �-GalCer pretreatment. In oneof these two groups KCs had been depleted by injection of clodronate-liposomes 48 h prior to �-GalCerpretreatment. Tolerance induction was achieved also in KC-depleted mice as revealed by plasma ALTactivity 17 h after �-GalCer rechallenge. The graph is representative of two experiments with correspond-ing results (n�3, mean�SEM; *, P�0.05 vs. vehicle control). (D) Tolerance-induction under conditionsof caspase-3 inhibition was analyzed by injecting control mice with vehicle and mice that were to be
tolerized by injection of �-GalCer either without additional treatment, or with zVAD.fmk treatment. Plasma ALT was measured after �-GalCer rechallenge 8days thereafter. For both C and D, dotted lines represent typical average ALT values of untreated mice (45 U/L). Both graphs are representative for twoexperiments with corresponding results, respectively. (n�3 or 4; mean�SEM; *, P�0.05, vs. vehicle control).
268 Journal of Leukocyte Biology Volume 84, July 2008 http://www.jleukbio.org

regarding a functional inactivation of CD4�CD25� Tregscaused by anti-CD25 mAb-induced CD25 down-regulation orTreg depletion (discussed below), we characterized the effectsof anti-CD25 mAb treatment on CD25� or FoxP3� T cellpopulations. Efficient disappearance of CD4/CD25 double-positive cells within 1 day after PC-61.5 injection was verifiedby FACS analysis (Fig. 4B). Also, functionality of PC-61.5treatment in our hands had been proven earlier in a model ofCon A tolerance [38]. However, staining of transcription factorFoxP3 showed that the frequency of FoxP3-positive Tregs wasvirtually unchanged, indicating that PC-61.5 treatment did notcause Treg depletion but rather down-modulation of CD25surface expression. In an additional experiment, we analyzedthe susceptibility of NKT cell activation by �-GalCer to sup-pression by CD4�CD25� Tregs. Ex vivo cocultivation of FACS-purified NKT cells together with CD4�CD25� Tregs (purifiedby magnetic bead sorting plus FACS sorting; 98% purity),even at a 1:1 ratio, revealed no inhibitory effect of Tregs on�-GalCer-induced cytokine secretion of NKT cells (Fig. 4C).This insusceptibility of NKT cells to immunosuppression byTregs further supports the notion that CD4�CD25� Tregs areprobably not involved in �-GalCer tolerance.
Analysis of �-GalCer tolerance in ex vivo assays
Immunological tolerance may be caused by a complex networkof factors in an entire organism, be locally limited mainly toone organ, or occur on a single cell basis.
To investigate whether �-GalCer tolerance might be detect-able in the isolated population of hepatic leukocytes or wouldalternatively need factors deriving from other cells, liver MNCsfrom tolerized or control mice were isolated and restimulated invitro. In fact, liver MNCs from vehicle-pretreated mice showed
pronounced cytokine production upon �-GalCer restimulationin vitro, whereas MNCs from �-GalCer-pretreated mice re-vealed largely diminished production of these cytokines(IFN-�, IL-2, IL-4, and TNF-�; data not shown). Also, in initialexperiments with restimulation by the ligand iGB3 that pref-erentially binds to the V7 chain in contrast to �-GalCer,which has higher affinity to V8.2, we found a tendency toreduce IFN-� production by MNCs from �-GalCer-tolerizedmice (258�148 pg/ml vs. 22�13 pg/ml for vehicle-pretreatedvs. �-GalCer-pretreated mice; mean�SEM; n�6; data wereobtained from two independent experiments).
To exclude the possibility that reduced cytokine productionmight simply be caused by the observed differences in NKTcell frequencies within the liver MNC populations of �-GalCer-and vehicle-pretreated mice, hepatic NKT cells from mice ofboth groups were purified by FACS sorting (purity, 95%) andstimulated with �-GalCer using mouse DCs as APCs. Also,with these highly purified NKT cells, we found pronouncedcytokine production by nontolerized NKT cells, whereas NKTcells from �-GalCer-pretreated mice revealed significantlylower cytokine responses to �-GalCer restimulation (Fig. 5A).
To investigate the possibility that NKT cells from tolerizedmice might have developed active tolerogenic features (suchas, e.g., an increased fratricidal cytotoxicity), purified NKTcells from nontolerized and tolerized mice were cocultured at a1:1 ratio. In fact, tolerized NKT cells did not suppress cytokineproduction by nontolerized NKT cells upon �-GalCer stimula-tion (Fig. 5B), thereby excluding �-GalCer-induced develop-ment of transactive, tolergenic properties among intrahepaticNKT cells as a cause of impaired responses to restimulation.
To analyze if �-GalCer tolerance might be mediated by any
Fig. 4. CD4� CD25� Tregs are not essen-tial for �-GalCer tolerance. (A) Treg de-pletion by i.p. injection of 300 �g anti-CD25 mAb 1 day prior to rechallenge didnot inhibit tolerance, as reflected in therelative suppression of plasma ALT activ-ities in tolerized mice (n�4; mean�SEM;*, P�0.05, vs. nontolerized control). (B)i.p. injection of anti-CD25 mAb PC61.5resulted in efficient down-regulation of CD25on the cell surface of CD4�forkhead box P3(FoxP3)�CD25� Tregs (upper panels) with-out depletion of these cells (as judged bytheir FoxP3 expression, lower panels)within 1 day. The effects of anti-CD25 in-jection were analyzed by triple staining ofperipheral blood leukocytes with anti-CD4,anti-FoxP3, and anti-CD25 mAb 24 h afterPC-61.5 injection. CD25 detection was car-ried out with clone 7D4, which recognizesanother CD25 epitope than PC-61.5 to pre-vent false-negative staining of cells in PC-61.5-injected mice caused by epitope mask-ing. The cells depicted in these panels hadbeen gated for being viable lymphocytes,according to light-scatter characteristics,and being CD4-positive. FSC, Forward-scatter. (C) Purified NKT cells and splenic
Tregs were incubated alone (8104 cells per well) or together at a 1:1 ratio in the presence of �-GalCer (15 ng/ml) presented by DCs (4104/well). Cytokineconcentrations were measured in duplicate after 3 days of culture.
Biburger and Tiegs NKT cell anergy protects from �-GalCer hepatitis 269

other �-GalCer-induced, transactive mechanisms (such as reg-ulatory cells, suppressive cytokines) in the liver, we testedwhether hepatic MNCs from tolerized mice were able to imposeimmune suppression on MNCs from nontolerized mice.Whereas the cytokine response of tolerized MNCs to �-GalCerrestimulation in vitro was diminished by 95% (as measuredby quantifying IFN-� production during 3 days of culture),these tolerized MNCs could not suppress MNCs from nontoler-ized mice upon coculture in a 1:1 ratio (data not shown),similar to the results with purified NKT cells. This indicatedthat the observed disability to respond efficiently to �-GalCerrestimulation is also not caused by NKT cell-independent,transactive suppression factors among liver MNCs, whichcould have evolved upon �-GalCer pretreatment but probablyby intrinsic impairment of NKT cell responses.
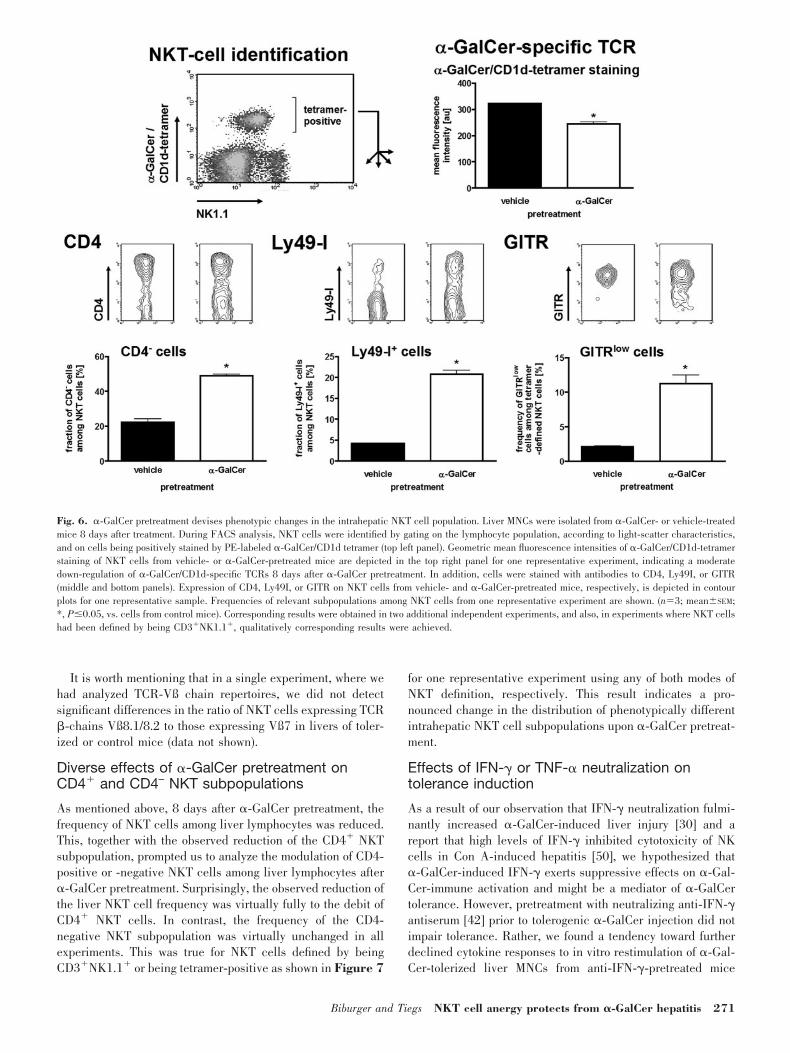
Tolerance-associated phenotypic changes in theintrahepatic NKT cell population
To identify possible causes of �-GalCer nonresponsiveness, wecharacterized changes of some surface markers of NKT cells,identified by being positive for CD3 and NK1.1 or for �-Gal-Cer/CD1d tetramers, 8 days after �-GalCer tolerization by flowcytometry. One prominent effect of �-GalCer pretreatment onthe NKT population was a down-modulation of NK1.1 with thepresence of NK1.1low NKT cells still after 8 days (data notshown). However, as this NK1.1 down-regulation has beendemonstrated in a large number of reports, we did not focusfurther on these results. NKT cells in the liver are known to bealmost exclusively CD4� or double-negative. In control mice,CD4� NKT cells represented the largely predominant popula-tion with more than two-thirds of liver NKT cells. In contrast,only about one-half of hepatic tetramer-positive NKT cells from�-GalCer-tolerized mice showed CD4 surface expression (Fig.6). Measuring the geometric mean of �-GalCer/CD1d-tetramerstaining, we found that expression of the �-GalCer-specificTCR was down-regulated significantly in the intrahepatic NKTpopulation of tolerized mice (Fig. 6). In addition, we analyzedthe expression of GITR, which had been shown to act as anefficient costimulator for NKT cells [49]. In nontolerized mice,virtually all intrahepatic NKT cells revealed strong staining forGITR (Fig. 6), whereas only a minority of less than 10% ofclassical T cells, probably CD4� CD25� Tregs, which expresshigh amounts of GITR (cited in ref. [49]), was GITR� (data notshown). In livers of tolerized mice, the fraction of NKT cellsthat lacked pronounced surface expression of this costimulatorwas increased significantly (Fig. 6). This effect was found onlyfor NKT cells but not for conventional T cell populations inlivers of tolerized mice (data not shown). In contrast to CD4,TCR, and GITR down-regulation, �-GalCer-pretreated miceshowed a significantly higher fraction of NKT cells that ex-pressed Ly49I, i.e., a representative inhibitory receptor (Fig. 6).
It is important to mention that qualitatively identical resultswere received when expression of these markers was investi-gated using the classical CD3�NK1.1� phenotype for NKTcell characterization. Quantitatively, the phenotypic changesdescribed above were typically found to be equally or evenmore pronounced in analyses based on CD3�NK1.1� (data notshown).
Fig. 5. �-GalCer nonresponsiveness of purified liver NKT cells from �-Gal-Cer-pretreated mice is nontransferable to naıve cells. NKT cells were preparedseparately from three tolerized and three nontolerized control mice 8 days afterpretreatment and cultured together with DCs for antigen presentation (4104
cells/well) in the presence or absence of 15 ng/ml �-GalCer. (A) DCs culturedalone, with or without �-GalCer, as well as NKT cells (8104 cells/well) in theabsence of �-GalCer revealed no relevant cytokine production. Upon �-GalCerstimulation for 3 days, NKT cells from nontolerized mice readily producedIFN-�, IL-2, IL-4, and TNF-�, whereas those from �-GalCer-pretreated micebarely responded (n�3; mean�SEM; *, P�0.05; one of two independentexperiments is shown). (B) NKT cells were cultured separately at a density of8 104 cells/well. In addition, they were cocultured at equal numbers (i.e.,8�8104/well) in six different combinations (with respect to the mice fromthat had been isolated; mean�SEM; *, P�0.05, vs. NKT cells from controlmice; #, values below detection limit).
270 Journal of Leukocyte Biology Volume 84, July 2008 http://www.jleukbio.org
It is worth mentioning that in a single experiment, where wehad analyzed TCR-Vß chain repertoires, we did not detectsignificant differences in the ratio of NKT cells expressing TCR-chains Vß8.1/8.2 to those expressing Vß7 in livers of toler-ized or control mice (data not shown).
Diverse effects of �-GalCer pretreatment onCD4� and CD4– NKT subpopulations
As mentioned above, 8 days after �-GalCer pretreatment, thefrequency of NKT cells among liver lymphocytes was reduced.This, together with the observed reduction of the CD4� NKTsubpopulation, prompted us to analyze the modulation of CD4-positive or -negative NKT cells among liver lymphocytes after�-GalCer pretreatment. Surprisingly, the observed reduction ofthe liver NKT cell frequency was virtually fully to the debit ofCD4� NKT cells. In contrast, the frequency of the CD4-negative NKT subpopulation was virtually unchanged in allexperiments. This was true for NKT cells defined by beingCD3�NK1.1� or being tetramer-positive as shown in Figure 7
for one representative experiment using any of both modes ofNKT definition, respectively. This result indicates a pro-nounced change in the distribution of phenotypically differentintrahepatic NKT cell subpopulations upon �-GalCer pretreat-ment.
Effects of IFN-� or TNF-� neutralization ontolerance induction
As a result of our observation that IFN-� neutralization fulmi-nantly increased �-GalCer-induced liver injury [30] and areport that high levels of IFN-� inhibited cytotoxicity of NKcells in Con A-induced hepatitis [50], we hypothesized that�-GalCer-induced IFN-� exerts suppressive effects on �-Gal-Cer-immune activation and might be a mediator of �-GalCertolerance. However, pretreatment with neutralizing anti-IFN-�antiserum [42] prior to tolerogenic �-GalCer injection did notimpair tolerance. Rather, we found a tendency toward furtherdeclined cytokine responses to in vitro restimulation of �-Gal-Cer-tolerized liver MNCs from anti-IFN-�-pretreated mice
Fig. 6. �-GalCer pretreatment devises phenotypic changes in the intrahepatic NKT cell population. Liver MNCs were isolated from �-GalCer- or vehicle-treatedmice 8 days after treatment. During FACS analysis, NKT cells were identified by gating on the lymphocyte population, according to light-scatter characteristics,and on cells being positively stained by PE-labeled �-GalCer/CD1d tetramer (top left panel). Geometric mean fluorescence intensities of �-GalCer/CD1d-tetramerstaining of NKT cells from vehicle- or �-GalCer-pretreated mice are depicted in the top right panel for one representative experiment, indicating a moderatedown-regulation of �-GalCer/CD1d-specific TCRs 8 days after �-GalCer pretreatment. In addition, cells were stained with antibodies to CD4, Ly49I, or GITR(middle and bottom panels). Expression of CD4, Ly49I, or GITR on NKT cells from vehicle- and �-GalCer-pretreated mice, respectively, is depicted in contourplots for one representative sample. Frequencies of relevant subpopulations among NKT cells from one representative experiment are shown. (n�3; mean�SEM;*, P�0.05, vs. cells from control mice). Corresponding results were obtained in two additional independent experiments, and also, in experiments where NKT cellshad been defined by being CD3�NK1.1�, qualitatively corresponding results were achieved.
Biburger and Tiegs NKT cell anergy protects from �-GalCer hepatitis 271

[0.7�0.3 ng/ml vs. 15.4�7.2 ng/ml IFN� by cells from �-Gal-Cer- vs. vehicle-pretreated, anti-IFN-�-injected mice(P�0.05) compared with 1.7�0.9 ng/ml vs. 15.0�6.3 ng/ml in�-GalCer- vs. vehicle-pretreated control serum-injected mice(P 0.05); n�3]. However, these changes were associated witha further reduction of NKT cell frequencies among liver MNCsin these mice (58.6% �-GalCer-induced reduction of NKTfrequencies among liver MNCs in anti-IFN-�-injected micecompared with 23.2% reduction in control serum-injectedmice, P�0.05), indicating that the onset of NKT cell nonre-sponsiveness, as reflected in cytokine responses to restimula-tion, was not crucially affected by IFN-� neutralization.
As we and several others have described previously theimportant role of TNF-� in NKT cell function, we wonderedwhether TNF-� might also play a role in the induction of NKTcell hyporesponsiveness. In vivo analysis of �-GalCer-inducedtolerance in anti-TNF-�-pretreated mice was not feasible, ascirculating anti-TNF-� antibodies would probably also affectthe outcome of �-GalCer rechallenge several days after pre-treatment. Thus, we analyzed the responsiveness of liver MNCsfrom mice of the different pretreatment groups in vitro. Micewere preinjected with rabbit control serum or polyclonal anti-TNF-� serum (IP-400, Genzyme) one-half hour prior to injec-tion of the vehicle or �-GalCer. Similar to the anti-IFN-� test,we found a tendency to an even more pronounced �-GalCer-induced reduction of the NKT cell frequency upon anti-TNF-�pretreatment (55.5% �-GalCer-induced reduction of NKT fre-quencies among liver MNCs in anti-TNF-�-injected mice com-pared with 47.2% reduction in control serum-injected mice,P 0.05; n�6; summary of two experiments). In contrast tomice tolerized under conditions of IFN-� neutralization, the invitro IFN-� response of MNCs from anti-TNF-�-pretreated,�-GalCer-tolerized mice to �-GalCer restimulation was in-creased in comparison with control serum-pretreated, tolerizedmice [2.6�1.2 ng/ml vs. 9.4�2.8 ng/ml (P 0.05) by cellsfrom �-GalCer- vs. vehicle-pretreated, anti-TNF-�-injectedmice compared with 1.1�1.0 ng/ml vs. 10.7�2.3 pg/ml in�-GalCer- vs. vehicle-pretreated, control serum-injected mice(P�0.05)]. However, this anti-TNF-�-induced increase in�-GalCer-induced hyporesponsiveness was not significant(P 0.05 between �-GalCer-pretreated control and �-GalCer-
pretreated anti-TNF-� mice), indicating that TNF-� might beinvolved to some degree but is probably not a major factor inthe development of �-GalCer-induced hyporesponsiveness ofNKT cells.
DISCUSSION
In this study, we demonstrated that already a single pretreat-ment with �-GalCer rendered mice tolerant to �-GalCer re-challenge with respect to cytokine production (except for IL-10) and liver injury. The finding that �-GalCer treatmentresults in a “blocked” response to restimulation has gainedparticular importance, as �-GalCer has already been used inclinical studies as an anti-tumor agent, and NKT cells appearto be involved in the onset of several diseases as mentionedabove. If homologous effects should also apply to humans, itwill be important to comprehend the mechanism of �-GalCer-induced tolerance to find strategies for retaining or re-estab-lishing the ability of NKT cells to be effectively activated toconfer antitumoral activity. Inversely, intentional suppressionof NKT cell activation per se or its physiological effects in vivomay enable treatment of NKT-mediated diseases.
Potential mechanisms of tolerance
The mechanism by which the first �-GalCer injection mightcause tolerance with respect to elementary NKT cell activationand/or NKT-mediated disease induction could be persistingdepletion of NKT cells, induction of “active” immunologictolerance conferred by regulatory cells or immunomodulatorycytokines, or induction of activation-induced nonresponsive-ness.
In addition to mechanisms that directly modulate immuneactivation, reduced cytotoxic susceptibility may contribute toprotection from liver injury.
In the context of �-GalCer tolerance, reduced expression ofintrahepatic Fas in �-GalCer-challenged, tolerized mice com-pared with �-GalCer-treated, control mice may be associatedwith reduced sensitivity of hepatocytes, consistent with studiesreporting diminished �-GalCer- or GalN/�-GalCer-induced
Fig. 7. �-GalCer pretreatment down-mod-ulates the frequency of the intrahepaticCD4� but not CD4– NKT subpopulations.Intrahepatic MNCs isolated from or vehicle-or �-GalCer-treated mice 8 days after treat-ment were incubated with anti-CD4 mAbtogether in addition to �-GalCer/CD1d tet-ramer (left panel) or CD3 and NK1.1 (rightpanel) for NKT cell identification. Thegraphs depict the frequency of the CD4�
and CD4– NKT cells among liver lympho-cytes revealed. Whereas the significantlydiminished frequency of NKT cells amongliver lymphocytes of �-GalCer-pretreatedmice was largely based on a reduction ofCD4� NKT cells, the frequency of the CD4-negative NKT subpopulation was virtuallyunchanged in all experiments. This was true for NKT cells defined by being tetramer-positive or CD3/NK1.1 double-positive. The figure shows the results of onerepresentative experiment of four independent tests for each of both NKT definitions (n�4; mean�SEM; *, P�0.05).
272 Journal of Leukocyte Biology Volume 84, July 2008 http://www.jleukbio.org

liver injury in Fas-deficient lpr�/� mice [32, 51]. As we coulddemonstrate that Fas up-regulation by hepatocytes upon�-GalCer injection is presumably mediated by �-GalCer-in-duced TNF-�, the diminished intrahepatic Fas expression intolerized mice probably reflects their diminished TNF-� re-sponse to �-GalCer rechallenge. This suggests that protectionfrom liver injury, besides the more general aspect of immunesuppression, includes an aspect of reduced susceptibility ofhepatocytes to Fas/FasL cytotoxicity. Both aspects are inter-connected by modulation of TNF-� expression in �-GalCertolerance.
Recently, also Parekh et al. [35] had investigated effects of�-GalCer injection in mice, describing induction of nonrespon-siveness as an outcome of this treatment. Whereas in theirimportant manuscript, the authors mainly characterized NKTcell activation with respect to cytokine production and prolif-eration, in this work, we extended our analysis about physio-logical consequences by characterizing effects of tolerizationon �-GalCer-induced liver injury. Moreover, whereas Parekhet al. [35] used splenic NKT cells in the majority of experi-ments, we focused on intrahepatic NKT populations. Also, inthis regard, our work accounts for completing the comprehen-sion of �-GalCer tolerance, as presumably, there are profounddifferences between splenic and hepatic NKT cells (see, e.g.,refs. [52–54]).
Intrahepatic NKT cell numbers
As NKT cells are the critical cell population that mediates�-GalCer-induced liver injury and cytokine production, theirabsence could mimic a tolerogenic phenotype. It is well knownthat mouse NKT cells become transiently undetectable in theliver rapidly after activation by Con A [31], anti-CD3 [55], or�-GalCer [29, 56–59] treatment. Originally, this phenomenonwas attributed to activation-induced death of NKT cells [55,56]. However, more recently, it has been shown that theiractivation is associated with transient down-modulation of theTCR complex and NK1.1, both of which are used for flowcytometric characterization [57–59], and that actually NKTcells are not persistently depleted. These results, indicatingthat �-GalCer injection does not result in pronounced, persis-tent depletion of the intrahepatic overall NKT cell populationcould be reproduced in our experiments. Although the numberof liver NKTs was somewhat decreased later on at Day 8 afterpretreatment, at Day 3, when pretreated mice were also toler-ized against �-GalCer-induced liver injury, the intrahepaticNKT cell number of �-GalCer-pretreated mice had even ex-ceeded that of vehicle-treated control mice. This increase inintrahepatic NKT cell numbers at Day 3 after �-GalCer injec-tion corresponds to recently described kinetics of NKT cellexpansion in spleen and/or liver, peaking �3 days after treat-ment, before returning to approximate normal levels [36, 37,57, 58]. In particular, Uldrich et al. [37] have shown that theexpansion of NKT populations in spleen and liver is dependenton costimulatory signaling by CD40 and CD28, whereas thesubsequent contraction of these populations requires the pro-apoptotic Bcl-2 family member Bim. This expansion of theintrahepatic NKT population at Day 3, i.e., at a time-pointwhere we had observed �-GalCer-hepatitis protection, ex-
cluded pronounced, �-GalCer-induced depletion of NKT cellsas the main cause for this tolerance.
Active immune modulation in�-GalCer tolerance?
Immunomodulatory leukocytes and immunosuppressive cyto-kines are important factors in a large number model of activeimmunological tolerance. Upon analysis of the cytokine re-sponses to �-GalCer in glycolipid- or vehicle-pretreated mice,we had found that in contrast to most other cytokines, there wasa tendency to increase production of IL-10 in tolerized mice,one of the most prominent inhibitory cytokines. Moreover,IL-10 was recently found to be important for NKT cell activa-tion-induced protection from experimental autoimmune en-cephalomyelitis [60]. IL-10 also plays a role in the develop-ment of cellular tolerance, as it has been demonstrated to beimportant for the differentiation of Tr1 Tregs (reviewed, e.g., inref [61]). However, the missing interference of exogenous IL-10on �-GalCer-induced liver injury as well as the possibility toinduce tolerance in IL-10�/� mice, demonstrated in this work,suggest that this cytokine is probably not an essential mediatorof �-GalCer tolerance. This was further supported by theobservation that multiple vaccination of C57BL/6 mice with�-GalCer-loaded DCs caused vast expression of IL-10 uponsubsequent �-GalCer injection but did not affect liver injury orproinflammatory cytokine production (Biburger, Tiegs, Man-fred Lutz, and C. Wiethe, unpublished data).
KCs are not only responsible for phagocytic removal of dyingcells but also for induction and maintenance of tolerance, ashas been shown, e.g., in rat models of hepatic [62] or cardiac[63, 64] allotransplantation. They have been described to in-duce apoptosis of liver-infiltrating, high-affinity CD8� T cells[65] and can produce immunoregulatory factors such as IL-10[66] as well as TNF-�, IL-6, TGF-� and -, NO, and reactiveoxygen species [67]. However, we found that KC depletion byclodronate liposomes prior to tolerogenic �-GalCer pretreat-ment did not affect �-GalCer-induced tolerance to �-GalCer-mediated hepatitis, thereby excluding an important role ofintrahepatic macrophages in the development of �-GalCertolerance.
CD4�CD25� Tregs are well known for their ability to conferimmunological tolerance in humans and several mouse models.A common method for characterization of potential involve-ment of CD4�CD25� Tregs in immunological processes in miceis the functional impairment of their regulatory function byinjecting anti-CD25 mAb such as clone PC61.5. The preven-tion of CD4�CD25� Treg function upon this anti-CD25 treat-ment is attributed in the literature to depletion (e.g., ref. [68]),down-regulation of CD25 associated with the functional inac-tivation of CD4�CD25� Tregs (e.g., refs. [69, 70]), or both [71].Identification of CD25 down-regulation as a result of anti-CD25 treatment in the recent literature has been enabled as aconsequence of the identification of FoxP3 as an additional,relevant, CD25-independent Treg marker. In agreement withseveral reports, we also detected disappearance of CD25� cellsamong CD4 lymphocytes without significant loss of CD4�FoxP3�
cells within the first day after PC61.5 treatment (without ex-tending this analysis to longer periods). This supports thecumulating assessment in the literature of impairment of the
Biburger and Tiegs NKT cell anergy protects from �-GalCer hepatitis 273

regulatory capacities of CD4�CD25� Tregs by anti-CD25 treat-ment (which we had also observed in a model of Con Atolerance [38]) to be caused by functional inactivation associ-ated with CD25 down-regulation rather than (or at least parallelto) Treg depletion.
Recently, Ly et al. [72] described that protection from type1 diabetes by �-GalCer-activated, invariant NKT cells requiresthe activity of CD4�CD25� Tregs and suggested that Tregswould regulate activation and anergy induction of NKT cells.The latter notion was based on data revealing significantlyreduced IL-2 responses of splenocytes from �-GalCer-pre-treated mice versus vehicle-pretreated mice to in vitro �-Gal-Cer restimulation. These in vitro responses were claimed to berestored when CD4�CD25� Tregs had been inactivated in the�-GalCer-pretreated mice by anti-CD25 treatment. However,we do not fully agree with this interpretation, as the respectiveexperiments reveal such an effect for IL-2 but for neitherIFN-� nor IL-4. In addition, the discussed in vitro IL-2 pro-duction of splenocytes from �-GalCer-pretreated mice doesnot, in fact, appear to be caused by specific NKT cell activationat all, as the in vitro IL-2 production in response to �-GalCerrestimulation is virtually identical to that in response to vehiclein the anti-CD25- or control IgG-treated group, respectively.Thus, it rather appears that in splenocytes from �-GalCer-pretreated mice, there is no specifically �-GalCer-inducedIL-2 production at all (the same appears to be true for IFN-�),indicating that NKT cells are fully nonresponsive with regardto IL-2 production, independent from the presence or absenceof Treg activity.
This interpretation—together with our results that pretreat-ment with PC61.5 did not interfere with development of toler-ance in vivo in our experiments, and purified Tregs did notsuppress NKT cell activation, even under close cell–cell con-tact—prompts us to suppose that Tregs are not critically in-volved in �-GalCer tolerance. Recently, in agreement with ourresults in mice, Jiang et al. [73] provided evidence that incontrast to conventional T cells, which are susceptible to CD4�
CD25� regulatory cell suppression, freshly isolated, humanV�24� NKT cells were able to produce cytokines in thepresence of CD4�CD25� Tregs [73]. It is not impossible,however, that as suggested by Ly et al. [72] (see above), Tregsmight modulate the overall outcome of NKT cell activation invivo (with regard to, e.g., cytokine production and activation ofT, B, and NK cells), presumably by indirect effects such asmodulation of third-party cells, like APCs.
It should be noted that our experiments, like those in allother reports based on techniques such as anti-CD25 treatmentor Treg purification using positive selection for CD25� cells, donot account for a potential contribution of CD25-negative Tregs.
We had performed in vitro assays with liver MNCs to testwhether any other active regulatory factors might have beenevoked in this population by the tolerizing �-GalCer pretreat-ment in vivo. However, whereas liver MNCs from tolerizedmice revealed a pronounced suppression of cytokine responsesto restimulation, cytokine production by nontolerized MNCswas not affected by coculture with respective cells from �-Gal-Cer-tolerized mice in a 1:1 ratio. This suggests that �-GalCer-induced tolerance is not based on active, transferable factorsbut rather on a passive effect such as hyporesponsiveness. In
principle, apparent overall hyporesponsiveness of the entiretyof NKT cells might be caused by development of an activelysuppressive subpopulation among activated NKT cells, by aphenotypic switch in the resident population, or repopulationwith different NKT populations that could restrain activationof, in principle activatable, NKT cells. Parekh et al. [35] hadsuggested nonresponsiveness of NKT cells as the basis ofobserved deficiencies in proliferation and cytokine productionafter �-GalCer pretreatment, which would not enclose by-stander suppression. This was assessed by coculture of spleno-cytes from �-GalCer-pretreated and naıve mice, in principle,similar to coculture of liver MNCs, as accomplished in thiswork. However, the frequency of NKT cells among liver MNCsused in our study is below 20% and is even much lower amongsplenocytes used by Parekh et al. [35] and in addition, differsbetween splenocytes from tolerized and nontolerized mice(3.4–3.9% among B220-negative splenocytes from naıve miceand 1.6–1.7% from mice pretreated 1 month earlier with�-GalCer [35]). In their physiological environment in vivo (e.g.,as a result of local accumulation at relevant venues), immuno-regulatory leukocytes may be able to exert efficient suppres-sion, even if being present at rather low frequencies in therespective total organ/compartment. In vitro, however, such lowfrequencies might possibly be insufficient to reveal significantsuppression. Thus, it cannot be excluded that a NKT cellsubpopulation, which might have developed active, tolerogenicfeatures upon �-GalCer pretreatment, would have been ignoredin these in vitro coculture assays as a result of the already lowfrequency of NKT cells and their even lower frequency in thetolerized group. This was the reason for us to cocultivate highlypurified NKT cells from tolerized mice with those from non-tolerized mice to assess a potential influence of conceivableregulatory NKT cells. These analyses represent an amendmentto the important experiments of Uldrich et al. [37] and Parekhet al. [35]: Uldrich and coworkers [37] used purified NKT cellsfrom tolerized or nontolerized mice together with DCs but didnot analyze potential transdominantly suppressive NKT cellsby cocultivating NKT cells from both groups together. In-versely, Parekh et al. [35], in addition, cocultivated NKT cellsfrom both groups together but apparently only in the form ofsplenocytes with rather low and varying NKT frequencies asdiscussed above.
The results of our coculture experiments with highly purifiedNKT cells from nontolerized and tolerized mice convincinglydemonstrated that �-GalCer pretreatment does not induce atransactive, suppressive capacity among liver NKT cells.
Activation-induced nonresponsiveness
Taken together, the in vivo and ex vivo results clearly demon-strate that in contrast to Con A tolerance [38], �-GalCer-induced protection from liver injury and cytokine boost upon�-GalCer restimulation is not mediated by active tolerogenicfactors but independent from such factors by activation-in-duced nonresponsiveness of liver NKT cells. Thus, in sum-mary, features of �-GalCer-induced, long-term protection from�-GalCer hepatitis appear to be fundamentally different fromprotection from Con A hepatitis [38] (see comparison in Table1) as well as those of Th2-dependent, �-GalCer-induced pro-tection from LPS shock [44] and experimental autoimmune
274 Journal of Leukocyte Biology Volume 84, July 2008 http://www.jleukbio.org

encephalomyelitis [60] or Treg-dependent protection from type1 diabetes by �-GalCer-activated, invariant NKT cells [72].Our results with IL-10 knockout mice and KC depletion alsoshow that these factors are not only dispensable as activefactors but are also not relevant in the process of NKT aner-gization.
Concomitants and potential elicitors ofhyporesponsiveness induction
Upon analysis of phenotypic changes of the intrahepatic NKTpopulation during tolerance, we found that 8 days after initial�-GalCer injection, the frequency of CD4� NKT among intra-hepatic lymphocytes was reduced significantly, whereas thefrequency of CD4– NKT cells remained unchanged. This sug-gested that there may be a pronounced modulation in theallocation of different NKT subpopulations in the liver. Thiscould be caused by differences in migration/retention as aresult of divergent equipment with chemokine receptors or bydistinct responses with respect to �-GalCer-induced expan-sion/contraction. The observation that in the state of �-GalCer-induced tolerance, the size of the CD4-negative populationremains largely unaffected may be of pronounced importancefor the attempts to use �-GalCer as an antitumoral agent: In as.c. sarcoma model, using sarcoma cell line MCA-1, Crowe etal. [52] demonstrated that among all NKT cell subsets tested,the CD4– fraction of liver-derived NKT cells was mostly, if notcompletely, responsible for NKT-mediated tumor rejection.Also, in a lung metastases model using B16F10 melanomacells, the authors found the hepatic CD4– NKT subpopulationto be an efficient mediator of �-GalCer-induced antitumorimmunity, whereas other NKT subsets, including hepaticCD4� NKT cells, were shown to be less potent [52].
In addition to the modulation of CD4�/� NKT cell popu-lations in tolerized mice, we detected reduced expression of�-GalCer-specific TCR on NKT cells. In a state of tolerance, 8days after �-GalCer injection, an increased proportion of NKTcells also failed to express high amounts GITR on their surface,which has been shown to be a potent costimulatory moleculefor NKT cells and to be temporarily up-regulated for 3–4 daysafter an �-GalCer encounter [49].
As an interesting side-effect, the observation that GITR issubstantially expressed by virtually all NKT cells (at least prior
to tolerization), but only a small fraction of conventional Tcells, may qualify this molecule as a novel marker for NKTcells, which in combination with additional markers, may aidand ameliorate NKT cell detection and identification. In com-parison with the GITR staining using DTA-1 mAb, as shown inthe work of Kim et al. [49], staining with the anti-GITR cloneYGITR 765 used in this work appeared to facilitate distinctdiscrimination between GITR-positive and -negative cells (asnoticed for differentiation between conventional T cells andNKT cells in liver MNC preparations; not shown).
It is conceivable that in �-GalCer-tolerized mice, GITR–/low
NKT cells and also, in particular, cells with reduced TCR mayhave an increased threshold for activation. In contrast to down-modulation of TCR and costimulatory GITR, expression ofinhibitory Ly49I was increased on liver NKT cells in tolerizedmice.
In summary, these data suggest that �-GalCer-induced non-responsiveness is accompanied and probably caused to somedegree by variations of the intrahepatic repertoire of NKTsubpopulations that may differentially respond to �-GalCerand a shift of the balance between stimulatory and inhibitoryreceptors in favor of NKT cell suppression, as depicted in themodel in Figure 8. As none of the observed phenotypicchanges appears to affect the NKT cells as a whole, it istempting to speculate that to some degree, different subpopu-lations might be modulated by different mechanisms. However,this does not exclude the possibility that persistent changes ofintracellular signaling pathways might contribute to �-GalCernonresponsiveness.
In the literature, there are some discrepancies with respectto �-GalCer-induced modulation of inhibitory receptors onNKT cells. Ota et al. [74] reported an increased expression ofinhibitory members of receptor families CD94/NKG2 (mainlyNKG2A) and Ly49 (without distinguishing among Ly49-A, -C,-I, or -G2) on hepatic NKT cells 5–7 days after �-GalCerinjection. Moreover, the authors suggested an important role forinteraction between NKG2A and its ligand Qa-1b in negativelyregulating NKT responses to rechallenge subsequent to �-Gal-Cer pretreatment [74]. In contrast, Uldrich et al. [37] foundvirtually no modulation of inhibitory Ly49 receptors (measur-ing expression of Ly49-A, -C/I, or -G2) on splenic NKT cells 1or 2 weeks after �-GalCer injection and even detected apersistent down-modulation of an inhibitory CD94/NKG2A (asmeasured by using antibodies against CD94, NKG2A, orNKG2A/C/E) receptor that persisted for at least 2 weeks onthese cells. In the present work, we detected a significantincrease in the frequency of Ly49-I-expressing liver NKT cells8 days after �-GalCer injection and could thus substantiate theresults of Ota et al. [74] on Ly49-A/C/I/G2. However, whereasOta et al. [74] reported an IFN-�-mediated up-regulation ofNKG2A ligand Qa-1b as a relevant factor for down-modulationof NKT cell responsiveness, in our hands, IFN-� neutralizationduring �-GalCer pretreatment did not interfere with its tolero-genic effect. Yet, these results are not contradictory, as in ourexperiments, IFN-� activity was neutralized only at the time oftolerance induction, and tolerized cells were restimulated invitro, whereas Ota et al. [74] mainly characterized effects ofpersistent IFN-� neutralization, e.g., by using IFN-��/� miceor in vitro experiments in the presence of neutralizing antibod-
TABLE 1. Comparison of Relevant Factors in �-GalCer and ConA Hepatitis and Tolerance Models
Liver injury/tolerancemodel:
Con A �-GalCer
Effect of IFN-� neutralization on liverinjury protection aggravation
Suppressive/protective effect of IL-10 Yes NoImportance of KC in tolerance Yes NoImportance of CD4�CD25� Tregs in
tolerance Yes NoDevelopment of nonresponsiveness upon
tolerance induction Noa Yes
aWith the exception of IL-2, which is poorly expressed in Con A-tolerizedmice upon restimulation, independent from Tregs, KC, or IL-10 [38].
Biburger and Tiegs NKT cell anergy protects from �-GalCer hepatitis 275

ies. Thus, �-GalCer-induced IFN-� may actually transientlymodulate the balance between stimulatory and inhibitory NKsignals [50, 75] and thereby, contribute to down-modulation ofNKT cell responsiveness (e.g., by interaction of IFN-�-inducedQa-1b with �-GalCer-up-regulated NKG2A [74]) but is prob-ably not an important factor for the initiation of NKT cellanergy.
Fujii et al. [76] had described that in contrast to injection offree �-GalCer, an injection of �-GalCer-loaded DCs does notinduce hyporesponsiveness but rather, supports NKT cell ac-tivation. This had led to the conclusion that the outcome of aninitial activation of NKT cells—anergy or pronounced capa-bility of restimulation—may depend on the type of �-GalCer-presenting cells; e.g., in the liver, the organ with the largestNKT population, the vast majority of cells that present previ-ously injected �-GalCer to liver-resident or circulating NKTcells may probably be hepatocytes. They are known to expressCD1d [77] but are clearly no highly specialized APCs. Incomparison with DCs, hepatocytes have been shown to evokeonly a limited repertoire of NKT cell responses to �-GalCer[78]. Thus, upon �-GalCer injection, a large number of NKTcells might actually be stimulated under suboptimal conditionssuch as inappropriate costimulation. Activation by �-GalCerwithout appropriate costimulation, which has been shown tomodulate NKT cell responses [37], might render NKT cellsnonresponsive, analogous to the well-known anergy of conven-tional T cells.
As TNF-� was pivotal in �-GalCer-induced, NKT-mediatedliver injury and appeared to play a contrary role to IFN-� [30],we wondered whether TNF-� neutralization might affect notonly hepatocyte damage [30] but also the onset of hyporespon-siveness. In addition, we had found that �-GalCer-inducedFasL up-regulation on NKT cells was reduced under conditionsof TNF-� neutralization [30], indicating a feedback signalingof �-GalCer-induced TNF-� on NKT cells. TNF-� neutraliza-tion during �-GalCer tolerization, to some extent but not sig-
nificantly, abrogated hyporesponsiveness induction, as re-vealed by increased cytokine responses of respective liverMNCs upon �-GalCer rechallenge. This indicates that TNF-�might act auxiliary to a limited degree but is also not anessential factor for the development of NKT cell hyporespon-siveness upon �-GalCer injection.
Implications for NKT cell investigation and theirtherapeutic use
There is compelling evidence in the literature that NKT cellscan inhibit or augment pathologic processes by activation orsuppression of immunologic processes in disease- and stage-specific manners (reviewed in ref. [13]). Our results about�-GalCer-induced liver injury [30] and �-GalCer-induced pro-tection from hepatitis induction (this work) now round off thesefindings by demonstrating that both outcomes are not excludingone another but can appear within the same disease model. Up-or down-modulation of NKT-based immune responses withtheir different effects on pathology can be two faces/phases ofthe same process, and primary activation is followed by a stateof hyporesponsiveness.
The phenomenon of �-GalCer-induced nonresponsivenessraises the necessity to thoroughly investigate NKT cell activa-tion states in all experimental approaches comprising repeatedcycles of NKT cell stimulation. It is necessary to show whetherNKT cells are predominantly activated or rather renderedhyporesponsive by these procedures, as this will be crucial tocorrectly judge whether NKT cells act as promoters or sup-pressors in the respective model and at the relevant time-points.
As mentioned above, �-GalCer has been used in clinicaltrials for cancer treatment [8–12]. If �-GalCer-induced non-responsiveness to subsequent �-GalCer applications shouldalso occur in humans, this would bear important implicationsfor the use of �-GalCer as a therapeutic agent for tumortreatment. In addition, �-GalCer tolerance might serve as a
Fig. 8. Schematic model of tolerance induction by �-GalCer pretreatment. An initial injection of �-GalCer induces pronounced cytokine production by NKT cellsincluding IFN-�, IL-4, and TNF-�, and the latter is an important mediator of liver injury [30]. This initial activation leads to phenotypical changes includingdown-regulation of activating receptors and up-regulation of inhibitory receptors on single cells or within the entire population with respect to changes infrequencies of the respective positive or negative NKT cell subsets. This, possibly together with additional modifications of signaling pathways, may render NKTcells nonresponsive to restimulation, resulting in reduced cytokine production and liver injury. NKT cells are represented as segments of cells to refer to the factthat variations in surface expression may take place on individual cells [such as for: TCR (glycolipid-specific TCR with invariant v� chain) expression] and/or bychanges in the ratios of positive/negative cells (such as for CD4), e.g., by alterations in their individual homing and/or proliferation.
276 Journal of Leukocyte Biology Volume 84, July 2008 http://www.jleukbio.org

model for the frequently found nonresponsiveness of NKT cellsin tumor patients (reviewed in ref. [79]), which might be causedby repeated contact with NKT cell-activating tumor antigens.Thus, improved knowledge of underlying mechanisms andidentification of methods that overcome �-GalCer tolerancemight help to develop strategies for reactivation of hyporespon-sive NKT cells against tumor entities.
If our observation that �-GalCer treatment provokes differ-ential modulation of total numbers of NKT subsets in mice alsoholds true for humans, this may also be highly relevant forNKT-based tumor therapy, as diverse NKT subpopulationshave also been described repeatedly in humans to exert anti-thetic effects such as anti-tumor immunosurveillance or im-mune suppression.
NKT cells do not appear to have only beneficial effects suchas suppression of autoimmunity or tumor repression. In fact,there is cumulating evidence that NKT cells may participate inthe onset of a remarkable number of experimental pathologicalprocesses such as atherosclerosis, airway inflammation,asthma, ulcerative colitis, arthritis, lupus, experimental auto-immune encephalomyelitis, and the onset of several hepaticdisorders as described above [15–30]. Injection of free �-Gal-Cer as a strategy for NKT cell decommissioning bears the riskof disadvantageous side-effects by NKT cell activation. How-ever, as anergy induction is mainly unaffected by neutraliza-tion of the key cytokine TNF-� (or IFN-� as well), �-GalCertreatment with simultaneous, anticytokine treatment might an-ergize NKT cells without respective cytokine-mediated side-effects.
Hence, we suggest the induction of persistent nonrespon-siveness of NKT cells as a potential therapeutic approach fordiseases or disease models, where inadequate NKT cell acti-vation is involved in pathogenesis.
ACKNOWLEDGMENTS
This work was supported by the Deutsche Forschungsgemein-schaft (DFG) grants TI 169/6-3 and -4. The perfect technicalassistance of Sonja Heinlein and Andrea Agli is gratefullyacknowledged. We thank Kirin Brewery Co., Ltd. (Japan), forproviding �-GalCer and Prof. Dirk Busch (Institute of MedicalMicrobiology, Immunology and Hygiene, Technical Universityof Munich, Munich, Germany) for �-GalCer/CD1d tetramers.
REFERENCES
1. Bendelac, A., Rivera, M. N., Park, S. H., Roark, J. H. (1997) MouseCD1-specific NK1 T cells: development, specificity, and function. Annu.Rev. Immunol. 15, 535–562.
2. Zhou, D., Mattner, J., Cantu III, C., Schrantz, N., Yin, N., Gao, Y., Sagiv,Y., Hudspeth, K., Wu, Y. P., Yamashita, T., Teneberg, S., Wang, D.,Proia, R. L., Levery, S. B., Savage, P. B., Teyton, L., Bendelac, A. (2004)Lysosomal glycosphingolipid recognition by NKT cells. Science 306,1786–1789.
3. Speak, A. O., Salio, M., Neville, D. C., Fontaine, J., Priestman, D. A.,Platt, N., Heare, T., Butters, T. D., Dwek, R. A., Trottein, F., Exley, M. A.,Cerundolo, V., Platt, F. M. (2007) Implications for invariant natural killerT cell ligands due to the restricted presence of isoglobotrihexosylceramidein mammals. Proc. Natl. Acad. Sci. USA 104, 5971–5976.
4. Porubsky, S., Speak, A. O., Luckow, B., Cerundolo, V., Platt, F. M., Grone,H. J. (2007) Normal development and function of invariant natural killerT cells in mice with isoglobotrihexosylceramide (iGb3) deficiency. Proc.Natl. Acad. Sci. USA 104, 5977–5982.
5. Kobayashi, E., Motoki, K., Uchida, T., Fukushima, H., Koezuka, Y. (1995)KRN7000, a novel immunomodulator, and its antitumor activities. Oncol.Res. 7, 529–534.
6. Brutkiewicz, R. R., Sriram, V. (2002) Natural killer T (NKT) cells andtheir role in antitumor immunity. Crit. Rev. Oncol. Hematol. 41, 287–298.
7. Swann, J., Crowe, N. Y., Hayakawa, Y., Godfrey, D. I., Smyth, M. J. (2004)Regulation of antitumor immunity by CD1d-restricted NKT cells. Immu-nol. Cell Biol. 82, 323–331.
8. Okai, M., Nieda, M., Tazbirkova, A., Horley, D., Kikuchi, A., Durrant, S.,Takahashi, T., Boyd, A., Abraham, R., Yagita, H., Juji, T., Nicol, A.(2002) Human peripheral blood V�24� V11� NKT cells expand fol-lowing administration of �-galactosylceramide-pulsed dendritic cells. VoxSang. 83, 250–253.
9. Giaccone, G., Punt, C. J., Ando, Y., Ruijter, R., Nishi, N., Peters, M., vonBlomberg, B. M., Scheper, R. J., van der Vliet, H. J., van den Eertwegh,A. J., Roelvink, M., Beijnen, J., Zwierzina, H., Pinedo, H. M. (2002) Aphase I study of the natural killer T-cell ligand �-galactosylceramide(KRN7000) in patients with solid tumors. Clin. Cancer Res. 8, 3702–3709.
10. Nieda, M., Okai, M., Tazbirkova, A., Lin, H., Yamaura, A., Ide, K.,Abraham, R., Juji, T., Macfarlane, D. J., Nicol, A. J. (2004) Therapeuticactivation of V�24�V11� NKT cells in human subjects results in highlycoordinated secondary activation of acquired and innate immunity. Blood103, 383–389.
11. Ishikawa, A., Motohashi, S., Ishikawa, E., Fuchida, H., Higashino, K.,Otsuji, M., Iizasa, T., Nakayama, T., Taniguchi, M., Fujisawa, T. (2005) Aphase I study of �-galactosylceramide (KRN7000)-pulsed dendritic cellsin patients with advanced and recurrent non-small cell lung cancer. Clin.Cancer Res. 11, 1910–1917.
12. Chang, D. H., Osman, K., Connolly, J., Kukreja, A., Krasovsky, J., Pack,M., Hutchinson, A., Geller, M., Liu, N., Annable, R., Shay, J., Kirchhoff,K., Nishi, N., Ando, Y., Hayashi, K., Hassoun, H., Steinman, R. M.,Dhodapkar, M. V. (2005) Sustained expansion of NKT cells and antigen-specific T cells after injection of �-galactosyl-ceramide loaded maturedendritic cells in cancer patients. J. Exp. Med. 201, 1503–1517.
13. Wilson, S. B., Delovitch, T. L. (2003) Janus-like role of regulatory iNKTcells in autoimmune disease and tumor immunity. Nat. Rev. Immunol. 3,211–222.
14. Hammond, K. J., Kronenberg, M. (2003) Natural killer T cells: natural orunnatural regulators of autoimmunity? Curr. Opin. Immunol. 15, 683–689.
15. Tupin, E., Nicoletti, A., Elhage, R., Rudling, M., Ljunggren, H. G.,Hansson, G. K., Berne, G. P. (2004) CD1d-dependent activation of NKTcells aggravates atherosclerosis. J. Exp. Med. 199, 417–422.
16. Nakai, Y., Iwabuchi, K., Fujii, S., Ishimori, N., Dashtsoodol, N., Watano,K., Mishima, T., Iwabuchi, C., Tanaka, S., Nakayama, T., Taniguchi, M.,Miyake, S., Yamamura, T., Kitabatake, A., Joyce, S., Van Kaer, L., Onoe,K. (2004) Natural killer T cells accelerate atherogenesis in mice. Blood104, 2051–2059.
17. Lisbonne, M., Diem, S., de Castro Keller, A., Lefort, J., Araujo, L. M.,Hachem, P., Fourneau, J. M., Sidobre, S., Kronenberg, M., Taniguchi, M.,Van Endert, P., Dy, M., Askenase, P., Russo, M., Vargaftig, B. B.,Herbelin, A., Leite-de-Moraes, M. C. (2003) Cutting edge: invariant V�14NKT cells are required for allergen-induced airway inflammation andhyperreactivity in an experimental asthma model. J. Immunol. 171,1637–1641.
18. Akbari, O., Stock, P., Meyer, E., Kronenberg, M., Sidobre, S., Nakayama,T., Taniguchi, M., Grusby, M. J., DeKruyff, R. H., Umetsu, D. T. (2003)Essential role of NKT cells producing IL-4 and IL-13 in the developmentof allergen-induced airway hyperreactivity. Nat. Med. 9, 582–588.
19. Araujo, L. M., Lefort, J., Nahori, M. A., Diem, S., Zhu, R., Dy, M.,Leite-de-Moraes, M. C., Bach, J. F., Vargaftig, B. B., Herbelin, A. (2004)Exacerbated Th2-mediated airway inflammation and hyperresponsivenessin autoimmune diabetes-prone NOD mice: a critical role for CD1d-dependent NKT cells. Eur. J. Immunol. 34, 327–335.
20. Heller, F., Fuss, I. J., Nieuwenhuis, E. E., Blumberg, R. S., Strober, W.(2002) Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis,is mediated by IL-13-producing NK-T cells. Immunity 17, 629–638.
21. Kim, H. Y., Kim, H. J., Min, H. S., Kim, S., Park, W. S., Park, S. H.,Chung, D. H. (2005) NKT cells promote antibody-induced joint inflam-mation by suppressing transforming growth factor 1 production. J. Exp.Med. 201, 41–47.
Biburger and Tiegs NKT cell anergy protects from �-GalCer hepatitis 277

22. Chiba, A., Kaieda, S., Oki, S., Yamamura, T., Miyake, S. (2005) Theinvolvement of V�14 natural killer T cells in the pathogenesis of arthritisin murine models. Arthritis Rheum. 52, 1941–1948.
23. Singh, A. K., Yang, J. Q., Parekh, V. V., Wei, J., Wang, C. R., Joyce, S.,Singh, R. R., Van Kaer, L. (2005) The natural killer T cell ligand�-galactosylceramide prevents or promotes pristane-induced lupus inmice. Eur. J. Immunol. 35, 1143–1154.
24. Jahng, A. W., Maricic, I., Pedersen, B., Burdin, N., Naidenko, O., Kro-nenberg, M., Koezuka, Y., Kumar, V. (2001) Activation of natural killer Tcells potentiates or prevents experimental autoimmune encephalomyelitis.J. Exp. Med. 194, 1789–1799.
25. Durante-Mangoni, E., Wang, R., Shaulov, A., He, Q., Nasser, I., Afdhal,N., Koziel, M. J., Exley, M. A. (2004) Hepatic CD1d expression inhepatitis C virus infection and recognition by resident proinflammatoryCD1d-reactive T cells. J. Immunol. 173, 2159–2166.
26. Harada, K., Isse, K., Tsuneyama, K., Ohta, H., Nakanuma, Y. (2003)Accumulating CD57� CD3� natural killer T cells are related to intrahe-patic bile duct lesions in primary biliary cirrhosis. Liver Int. 23, 94–100.
27. De Lalla, C., Galli, G., Aldrighetti, L., Romeo, R., Mariani, M., Monno, A.,Nuti, S., Colombo, M., Callea, F., Porcelli, S. A., Panina-Bordignon, P.,Abriganani, S., Casorati, G., Dellabona, P. (2004) Production of profibroticcytokines by invariant NKT cells characterizes cirrhosis progression inchronic viral hepatitis. J. Immunol. 173, 1417–1425.
28. Tiegs, G., Hentschel, J., Wendel, A. (1992) A T cell-dependent experi-mental liver injury in mice inducible by concanavalin A. J. Clin. Invest.90, 196–203.
29. Osman, Y., Kawamura, T., Naito, T., Takeda, K., Van Kaer, L., Okumura,K., Abo, T. (2000) Activation of hepatic NKT cells and subsequent liverinjury following administration of �-galactosylceramide. Eur. J. Immunol.30, 1919–1928.
30. Biburger, M., Tiegs, G. (2005) �-Galactosylceramide-induced liver injuryin mice is mediated by TNF-� but independent of Kupffer cells. J. Im-munol. 175, 1540–1550.
31. Takeda, K., Hayakawa, Y., Van Kaer, L., Matsuda, H., Yagita, H.,Okumura, K. (2000) Critical contribution of liver natural killer T cells toa murine model of hepatitis. Proc. Natl. Acad. Sci. USA 97, 5498–5503.
32. Nakagawa, R., Nagafune, I., Tazunoki, Y., Ehara, H., Tomura, H., Iijima,R., Motoki, R., Kamishohara, M., Seki, S. (2001) Mechanisms of theantimetastatic effect in the liver and of the hepatocyte injury induced by�-galactosylceramide in mice. J. Immunol. 166, 6578–6584.
33. Singh, N., Hong, S., Scherer, D. C., Serizawa, I., Burdin, N., Kronenberg,M., Koezuka, Y., Van Kaer, L. (1999) Cutting edge: activation of NK Tcells by CD1d and �-galactosylceramide directs conventional T cells tothe acquisition of a Th2 phenotype. J. Immunol. 163, 2373–2377.
34. Burdin, N., Brossay, L., Kronenberg, M. (1999) Immunization with�-galactosylceramide polarizes CD1-reactive NK T cells towards Th2cytokine synthesis. Eur. J. Immunol. 29, 2014–2025.
35. Parekh, V. V., Wilson, M. T., Olivares-Villagomez, D., Singh, A. K., Wu,L., Wang, C. R., Joyce, S., Van Kaer, L. (2005) Glycolipid antigen induceslong-term natural killer T cell anergy in mice. J. Clin. Invest. 115,2572–2583.
36. Ikarashi, Y., Iizuka, A., Koshidaka, Y., Heike, Y., Takaue, Y., Yoshida,M., Kronenberg, M., Wakasugi, H. (2005) Phenotypical and functionalalterations during the expansion phase of invariant Va14 natural killer T(Va14i NKT) cells in mice primed with �-galactosylceramide. Immunol-ogy 116, 30–37.
37. Uldrich, A. P., Crowe, N. Y., Kyparissoudis, K., Pellicci, D. G., Zhan, Y.,Lew, A. M., Bouillet, P., Strasser, A., Smyth, M. J., Godfrey, D. I. (2005)NKT cell stimulation with glycolipid antigen in vivo: costimulation-de-pendent expansion, Bim-dependent contraction, and hyporesponsivenessto further antigenic challenge. J. Immunol. 175, 3092–3101.
38. Erhardt, A., Biburger, M., Papadopoulos, T., Tiegs, G. (2007) IL-10,regulatory T cells, and Kupffer cells mediate tolerance in concanavalinA-induced liver injury in mice. Hepatology 45, 475–485.
39. Kuhn, R., Lohler, J., Rennick, D., Rajewsky, K., Muller, W. (1993)Interleukin-10-deficient mice develop chronic enterocholitis. Cell 75,263–274.
40. Schumann, J., Wolf, D., Pahl, A., Brune, K., Papadopoulos, T., vanRooijen, N., Tiegs, G. (2000) Importance of Kupffer cells for T-cell-dependent liver injury in mice. Am. J. Pathol. 157, 1671–1683.
41. Gantner, F., Leist, M., Lohse, A. W., Germann, P. G., Tiegs, G. (1995)Concanavalin A-induced T-cell-mediated hepatic injury in mice: the roleof tumor necrosis factor. Hepatology 21, 190–198.
42. Kusters, S., Gantner, F., Kunstle, G., Tiegs, G. (1996) Interferon � playsa critical role in T cell-dependent liver injury in mice initiated byconcanavalin A. Gastroenterology 111, 462–471.
43. Keppler, D. O., Pausch, J., Decker, K. (1974) Selective uridine triphos-phate deficiency induced by D-galactosamine in liver and reversed by
pyrimidine nucleotide precursors. Effect on ribonucleic acid synthesis. J.Biol. Chem. 249, 211–216.
44. Sireci, G., La Manna, M. P., Di Sano, C., Di Liberto, D., Porcelli, S. A.,Kronenberg, M., Dieli, F., Salerno, A. (2007) Pivotal advance: �-galacto-sylceramide induces protection against lipopolysaccharide-induced shock.J. Leukoc. Biol. 81, 607–622.
45. Pinkoski, M. J., Green, D. (2002) Dying for acceptance: apoptosis intolerance. Curr. Opin. Organ Transplant. 7, 2–6.
46. Ferguson, T. A., Stuart, P. M., Herdon, J. M., Griffith, T. S. (2003)Apoptosis, tolerance and regulatory T cells—old wine, new wineskins.Immunol. Rev. 193, 111–123.
47. Chiffoleau, E., Walsh, P. T., Turka, L. (2003) Apoptosis and transplanta-tion tolerance. Immunol. Rev. 193, 124–125.
48. Louis, H., Le Moine, O., Peny, M. O., Gulbis, B., Nisol, F., Goldman, M.,Deviere, J. (1997) Hepatoprotective role of interleukin 10 in galac-tosamine/lipopolysaccharide mouse liver injury. Gastroenterology 112,935–942.
49. Kim, H. J., Kim, H. Y., Kim, B. K., Kim, S., Chung, D. H. (2006)Engagement of glucocorticoid-induced TNF receptor costimulates NKTcell activation in vitro and in vivo. J. Immunol. 176, 3507–3515.
50. Dong, Z., Zhang, C., Wei, H., Sun, R., Tian, Z. (2005) Impaired NK cellcytotoxicity by high level of interferon-� in concanavalin A-inducedhepatitis. Can. J. Physiol. Pharmacol. 83, 1045–1053.
51. Fujii, H., Seki, S., Kobayashi, S., Kitada, T., Kawakita, N., Adachi, K.,Tsutsui, H., Nakanishi, K., Fujiwara, H., Ikarashi, Y., Taniguchi, M.,Kronenberg, M., Ikemoto, M., Nakajima, Y., Arakawa, T., Kaneda, K.(2005) A murine model of NKT cell-mediated liver injury induced by�-galactosylceramide/d-galactosamine. Virchows Arch. 446, 663–673.
52. Crowe, N. Y., Coquet, J. M., Berzins, S. P., Kyparissoudis, K., Keating, R.,Pellicci, D. G., Hayakawa, Y., Godfrey, D. I., Smyth, M. J. (2005)Differential antitumor immunity mediated by NKT cell subsets in vivo. J.Exp. Med. 202, 1279–1288.
53. Johnston, B., Kim, C. H., Soler, D., Emoto, M., Butcher, E. C. (2003)Differential chemokine responses and homing patterns of murine TCR � NKT cell subsets. J. Immunol. 171, 2960–2969.
54. Eberl, G., Lees, R., Smiley, S. T., Taniguchi, M., Grusby, M. J., Mac-Donald, H. R. (1999) Tissue-specific segregation of CD1d-dependent andCD1d-independent NK T cells. J. Immunol. 162, 6410–6419.
55. Eberl, G., MacDonald, H. R. (1998) Rapid death and regeneration of NKTcells in anti-CD3ε- or IL-12-treated mice: a major role for bone marrow inNKT cell homeostasis. Immunity 9, 345–353.
56. Leite-de-Moraes, M. C., Herbelin, A., Gouarin, C., Koezuka, Y., Schnei-der, E., Dy, M. (2000) Fas/Fas ligand interactions promote activation-induced cell death of NK T lymphocytes. J. Immunol. 165, 4367–4371.
57. Wilson, M. T., Johansson, C., Olivares-Villagomez, D., Singh, A. K.,Stanic, A. K., Wang, C. R., Joyce, S., Wick, M. J., Van Kaer, L. (2003) Theresponse of natural killer T cells to glycolipid antigens is characterized bysurface receptor down-modulation and expansion. Proc. Natl. Acad. Sci.USA 100, 10913–10918.
58. Crowe, N. Y., Uldrich, A. P., Kyparissoudis, K., Hammond, K. J., Hay-akawa, Y., Sidobre, S., Keating, R., Kronenberg, M., Smyth, M. J., God-frey, D. I. (2003) Glycolipid antigen drives rapid expansion and sustainedcytokine production by NKT cells. J. Immunol. 171, 4020–4027.
59. Harada, M., Seino, K., Wakao, H., Sakata, S., Ishizuka, Y., Ito, T., Kojo,S., Nakayama, T., Taniguchi, M. (2004) Down-regulation of the invariantV�14 antigen receptor in NKT cells upon activation. Int. Immunol. 16,241–247.
60. Singh, A. K., Wilson, M. T., Hong, S., Olivares-Villagomez, D., Du, C.,Stanic, A. K., Joyce, S., Sriram, S., Koezuka, Y., Van Kaer, L. (2001)Natural killer T cell activation protects mice against experimental auto-immune encephalomyelitis. J. Exp. Med. 194, 1801–1811.
61. Roncarolo, M. G., Gregori, S., Battaglia, M., Bacchetta, R., Fleischhauer,K., Levings, M. K. (2006) Interleukin-10-secreting type 1 regulatory Tcells in rodents and humans. Immunol. Rev. 212, 28–50.
62. Sun, Z., Wada, T., Maemura, K., Uchikura, K., Hoshino, S., Diehl, A. M.,Klein, A. S. (2003) Hepatic allograft-derived Kupffer cells regulate T cellresponse in rats. Liver Transpl. 9, 489–497.
63. Kamei, T., Callery, M. P., Flye, M. W. (1990) Kupffer cell blockadeprevents induction of portal venous tolerance in rat cardiac allografttransplantation. J. Surg. Res. 48, 393–396.
64. Squiers, E. C., Salomon, D. R., Pickard, L. L., Howard, R. R., Pfaff, W. W.(1990) Abrogation of the induction of portal venous tolerance in a cardiactransplant model resulting from Kupffer cell inhibition by gadolinium.Transplantation 50, 171–173.
65. Kuniyasu, Y., Marfani, S. M., Inayat, I. B., Sheikh, S. Z., Mehal, W. Z.(2004) Kupffer cells are required for high affinity peptide-induced dele-tion, not retention, of activated CD8� T cells by mouse liver. Hepatology39, 1017–1027.
278 Journal of Leukocyte Biology Volume 84, July 2008 http://www.jleukbio.org

66. Knolle, P., Schlaak, J., Uhrig, A., Kempf, P., Meyer zum Buschenfelde,K. H., Gerken, G. (1995) Human Kupffer cells secrete IL-10 in responseto lipopolysaccharide (LPS) challenge. J. Hepatol. 22, 226–229.
67. Knolle, P. A., Gerken, G. (2000) Local control of the immune-response inthe liver. Immunol. Rev. 174, 21–34.
68. Stephens, L. A., Gray, D., Anderton, S. M. (2005) CD4�CD25� regulatoryT cells limit the risk of autoimmune disease arising from T cell receptorcrossreactivity. Proc. Natl. Acad. Sci. USA 102, 17418–17423.
69. Kohm, A. P., McMahon, J. S., Podojil, J. R., Begolka, W. S., DeGutes, M.,Kasprowicz, D. J., Ziegler, S. F., Miller, S. D. (2006) Cutting edge:anti-CD25 monoclonal antibody injection results in the functional inacti-vation, not depletion, of CD4�CD25� T regulatory cells. J. Immunol.176, 3301–3305.
70. Fecci, P. E., Sweeney, A. E., Grossi, P. M., Nair, S. K., Learn, C. A.,Mitchell, D. A., Cui, X., Cummings, T. J., Bigner, D. D., Gilboa, E.,Sampson, J. H. (2006) Systemic anti-CD25 monoclonal antibody admin-istration safely enhances immunity in murine glioma without eliminatingregulatory T cells. Clin. Cancer Res. 12, 4294–4305.
71. McNeill, A., Spittle, E., Backstrom, B. T. (2007) Partial depletion ofCD69low-expressing natural regulatory T cells with the anti-CD25 mono-clonal antibody PC61. Scand. J. Immunol. 65, 63–69.
72. Ly, D., Mi, Q. S., Hussain, S., Delovitch, T. L. (2006) Protection from type1 diabetes by invariant NK T cells requires the activity of CD4�CD25�
regulatory T cells. J. Immunol. 177, 3695–3704.
73. Jiang, S., Game, D. S., Davies, D., Lombardi, G., Lechler, R. I. (2005)Activated CD1d-restricted natural killer T cells secrete IL-2: innate helpfor CD4� CD25� regulatory T cells? Eur. J. Immunol. 35, 1193–1200.
74. Ota, T., Takeda, K., Akiba, H., Hayakawa, Y., Ogasawara, K., Ikarashi,Y., Miyake, S., Wakasugi, H., Yamamura, T., Kronenberg, M., Raulet, D.H., Kinoshita, K., Yagita, H., Smyth, M. J., Okumura, K. (2005) IFN-�-mediated negative feedback regulation of NKT-cell function by CD94/NKG2. Blood 106, 184–192.
75. Zhang, C., Zhang, J., Sun, R., Feng, J., Wei, H., Tian, Z. (2005) Opposingeffect of IFN� and IFN� on expression of NKG2 receptors: negativeregulation of IFN� on NK cells. Int. Immunopharmacol. 5, 1057–1067.
76. Fujii, S., Shimizu, K., Kronenberg, M., Steinman, R. M. (2002) ProlongedIFN�-producing NKT responses induced with �-galactosylceramide-loaded DCs. Nat. Immunol. 3, 867–874.
77. Mandal, M., Chen, X. R., Alegre, M. L., Chiu, N. M., Chen, Y. H.,Castano, A. R., Wang, C. R. (1998) Tissue distribution, regulation andintracellular localization of murine CD1 molecules. Mol. Immunol. 35,525–536.
78. Trobonjaca, Z., Leithauser, F., Moller, P., Schirmbeck, R., Reimann, J.(2001) Activating immunity in the liver. I. Liver dendritic cells (but nothepatocytes) are potent activators of IFN-� release by liver NKT cells.J. Immunol. 167, 1413–1422.
79. Seino, K., Motohashi, S., Fujisawa, T., Nakayama, T., Taniguchi, M.(2006) Natural killer T cell-mediated antitumor immune responses andtheir clinical applications. Cancer Sci. 97, 807–812.
Biburger and Tiegs NKT cell anergy protects from �-GalCer hepatitis 279
Copyright © 2022 FDOKUMEN




















