
A Subchronic Exposure to Trichloroethylene Causes Lipid Peroxidation and Hepatocellular...
10
TOHCOLOGKAL SCIENCES 43, 145-154 (1998) ARTICLE NO. T X 9 8 2 4 5 6 A Subchronic Exposure to Trichloroethylene Causes Lipid Peroxidation and Hepatocellular Proliferation in Male B6C3F1 Mouse Liver Stephen R. Channel,* John R. Latendresse,f Jay K. Kidney,* John H. Grabau.t John W. Lane,t Linda Steel-Goodwin,* and Matthew C. Gothaus§ *Toxicology Division, Armstrong Laboratory, •fMantech Environmental Technology, Inc., and fGEO-CENTERS, INC., Wright-Patterson Air Force Base, Ohio 45433; and §77ie Medical College of Ohio, Toledo, Ohio 43614 Received October 29, 1997; accepted March 19, 1998 A Subchronic Exposure to Trichloroethylene Causes Lipid Per- oxidation and Hepatocellular Proliferation in Male B6C3F1 Mouse LiveT. Channel, S. R., Latendresse, J. R., Kidney, J. K., Grabau, J. H., Lane, J. W., Steel-Goodwin, L., and Gothaus, M. (1998). Toxicol. Sri. 43, 145-154. The common groundwater contaminant trichloroethylene (TCE), when given by oral gavage, can produce free radical spe- cies during metabolism. Furthermore, TCE end-stage metabolites, trichloroacetic acid and dichloroacetic acid, cause lipid peroxida- tion in mouse liver. The time courses of lipid peroxidation, free radical generation, and 8-hydroxydeoxyguanosine (8OHdG) for- mation were used to assess the level of oxidative stress in the liver of B6C3F1 mice dosed orally once daily, 5 days a week for 8 weeks at 0, 400, 800, and 1200 mg/kg TCE in corn oil. Peroxisomal proliferation, cell proliferation, and apoptosis were evaluated at selected times during the study. Lipid peroxidation, as measured by thiobarbituric acid-reactive substances (TBARS), was signifi- cantly elevated at the two highest dose levels of TCE on days 6 through 14 of the study. 8OHdG levels were statistically significant in the 1200 mg/kg/day group on days 2, 3,10, 28, 49, and 56 only. The highest measured free radical load, 307% of oil control, occurred at day 6. A significant increase in cell and peroxisomal proliferation was observed during the same time period in the 1200 mg/kg/day group. Necrosis or an increase in apoptosis was not observed at any dose. The temporal relationship between oxidative stress and cellular response of proliferation, both of which occur and resolve within the same relative time period, suggests that TCE-induced mitogenesis may result from alteration in the liver microenvironment which offers a selective advantage for certain hepatocyte subpopulations. Trichloroethylene (TCE) is a widely used industrial solvent and degreasing agent. Because it is commonly detected as a groundwater contaminant, its potential to adversely affect hu- man health has been studied and debated for many years. The primary concern about TCE environmental exposures arises from the National Toxicology Program (NTP) studies that have shown TCE to be a rodent hepatocarcinogen when given in high doses by corn oil gavage (NCI, 1976; NTP, 1990). Con- sistent species differences have been reported. Mice, especially the B6C3F1 strain, develop hepatocellular neoplasms while rats do not (NTP, 1988). Two major TCE metabolites, chloral hydrate (CH) and trichloroacetic acid (TCA), as well as a reported minor metabolite dichloroacetic acid (DCA), also produce liver tumors in mice (Herren-Freund et al, 1987; Daniel et al, 1992; DeAngelo et al, 1991). CH has been found to be genotoxic in a variety of in vitro systems, whereas TCA, DCA, and the parent compound, TCE, are recognized as non- genotoxic carcinogens in the mouse (reviewed in ECETOC, 1994). Tumor dose-response characteristics and species-spe- cific metabolic differences in terms of TCA and/or DCA pro- duction may account for TCE's tumorigenicity (Larson and Bull, 1992a; Templin et al, 1993; reviewed in Goeptar et al, 1995). Significant histopathologic differences have been ob- served between TCA- and DCA-induced liver lesions follow- ing chronic exposure (Bull et al., 1990; Pereira, 1996), leading to the suggestion that each may produce liver tumors by distinct mechanisms. Bull et al. (1990) reported that TCA, unlike DCA, caused large dose-related hepatocellular accumu- lations of lipofuscin, a lipid membrane peroxidation by-prod- uct. Furthermore, TCA chronic treatment resulted in sustained peroxisomal proliferation, whereas this effect was transitory for DCA (Bull et al, 1993). TCA and DCA have been shown to cause oxidative stress in rodents. Both increase the formation of thiobarbituric acid- reactive substances (TBARS) in a dose response manner in mouse liver following a single oral dose, suggesting that each was capable of yielding a radical species which could initiate lipid peroxidation (Larson and Bull, 1992b). Recently, CH and TCA were shown to generate free radicals and to induce lipid peroxidation in male B6C3F1 mouse liver microsomes (Ni et al, 1996). This laboratory has shown that carbon-centered free radicals are produced in B6C3F1 liver slices exposed to the parent compound TCE (Steel-Goodwin et al, 1996). The purpose of this study was to evaluate dose-response and temporal characteristics of selected biological endpoints in the 145 1096-6080/98 by guest on July 24, 2016 http://toxsci.oxfordjournals.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of A Subchronic Exposure to Trichloroethylene Causes Lipid Peroxidation and Hepatocellular...

TOHCOLOGKAL SCIENCES 43 , 145-154 (1998)
ARTICLE NO. TX982456
A Subchronic Exposure to Trichloroethylene Causes Lipid Peroxidationand Hepatocellular Proliferation in Male B6C3F1 Mouse Liver
Stephen R. Channel,* John R. Latendresse,f Jay K. Kidney,* John H. Grabau.t John W. Lane,tLinda Steel-Goodwin,* and Matthew C. Gothaus§
*Toxicology Division, Armstrong Laboratory, •fMantech Environmental Technology, Inc., and fGEO-CENTERS, INC.,Wright-Patterson Air Force Base, Ohio 45433; and §77ie Medical College of Ohio, Toledo, Ohio 43614
Received October 29, 1997; accepted March 19, 1998
A Subchronic Exposure to Trichloroethylene Causes Lipid Per-oxidation and Hepatocellular Proliferation in Male B6C3F1Mouse LiveT. Channel, S. R., Latendresse, J. R., Kidney, J. K.,Grabau, J. H., Lane, J. W., Steel-Goodwin, L., and Gothaus, M.(1998). Toxicol. Sri. 43, 145-154.
The common groundwater contaminant trichloroethylene(TCE), when given by oral gavage, can produce free radical spe-cies during metabolism. Furthermore, TCE end-stage metabolites,trichloroacetic acid and dichloroacetic acid, cause lipid peroxida-tion in mouse liver. The time courses of lipid peroxidation, freeradical generation, and 8-hydroxydeoxyguanosine (8OHdG) for-mation were used to assess the level of oxidative stress in the liverof B6C3F1 mice dosed orally once daily, 5 days a week for 8 weeksat 0, 400, 800, and 1200 mg/kg TCE in corn oil. Peroxisomalproliferation, cell proliferation, and apoptosis were evaluated atselected times during the study. Lipid peroxidation, as measuredby thiobarbituric acid-reactive substances (TBARS), was signifi-cantly elevated at the two highest dose levels of TCE on days 6through 14 of the study. 8OHdG levels were statistically significantin the 1200 mg/kg/day group on days 2, 3,10, 28, 49, and 56 only.The highest measured free radical load, 307% of oil control,occurred at day 6. A significant increase in cell and peroxisomalproliferation was observed during the same time period in the 1200mg/kg/day group. Necrosis or an increase in apoptosis was notobserved at any dose. The temporal relationship between oxidativestress and cellular response of proliferation, both of which occurand resolve within the same relative time period, suggests thatTCE-induced mitogenesis may result from alteration in the livermicroenvironment which offers a selective advantage for certainhepatocyte subpopulations.
Trichloroethylene (TCE) is a widely used industrial solventand degreasing agent. Because it is commonly detected as agroundwater contaminant, its potential to adversely affect hu-man health has been studied and debated for many years. Theprimary concern about TCE environmental exposures arisesfrom the National Toxicology Program (NTP) studies that haveshown TCE to be a rodent hepatocarcinogen when given in
high doses by corn oil gavage (NCI, 1976; NTP, 1990). Con-sistent species differences have been reported. Mice, especiallythe B6C3F1 strain, develop hepatocellular neoplasms whilerats do not (NTP, 1988). Two major TCE metabolites, chloralhydrate (CH) and trichloroacetic acid (TCA), as well as areported minor metabolite dichloroacetic acid (DCA), alsoproduce liver tumors in mice (Herren-Freund et al, 1987;Daniel et al, 1992; DeAngelo et al, 1991). CH has been foundto be genotoxic in a variety of in vitro systems, whereas TCA,DCA, and the parent compound, TCE, are recognized as non-genotoxic carcinogens in the mouse (reviewed in ECETOC,1994). Tumor dose-response characteristics and species-spe-cific metabolic differences in terms of TCA and/or DCA pro-duction may account for TCE's tumorigenicity (Larson andBull, 1992a; Templin et al, 1993; reviewed in Goeptar et al,1995). Significant histopathologic differences have been ob-served between TCA- and DCA-induced liver lesions follow-ing chronic exposure (Bull et al., 1990; Pereira, 1996), leadingto the suggestion that each may produce liver tumors bydistinct mechanisms. Bull et al. (1990) reported that TCA,unlike DCA, caused large dose-related hepatocellular accumu-lations of lipofuscin, a lipid membrane peroxidation by-prod-uct. Furthermore, TCA chronic treatment resulted in sustainedperoxisomal proliferation, whereas this effect was transitoryfor DCA (Bull et al, 1993).
TCA and DCA have been shown to cause oxidative stress inrodents. Both increase the formation of thiobarbituric acid-reactive substances (TBARS) in a dose response manner inmouse liver following a single oral dose, suggesting that eachwas capable of yielding a radical species which could initiatelipid peroxidation (Larson and Bull, 1992b). Recently, CH andTCA were shown to generate free radicals and to induce lipidperoxidation in male B6C3F1 mouse liver microsomes (Ni etal, 1996). This laboratory has shown that carbon-centered freeradicals are produced in B6C3F1 liver slices exposed to theparent compound TCE (Steel-Goodwin et al, 1996).
The purpose of this study was to evaluate dose-response andtemporal characteristics of selected biological endpoints in the
145 1096-6080/98
by guest on July 24, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from
146 CHANNEL ET AL.
TABLE 1Study Design
Treatment
Water controlCorn oil controlTCE 400 mg/kg/dayTCE 800 mg/kg/dayTCE 1200 mg/kg/day
No. of mice
77777
Time points
1111111111
Total no. of mice
7777!n11ii
mouse liver for mice dosed 56 days with TCE given by corn oilgavage. Elevations in endpoints including peroxisome prolif-eration and lipid peroxidation have been demonstrated else-where using in vitro or following acute in vivo TCE, or itschloroacetic acid metabolite, exposures (Bull et at, 1990,1993; Elcombe et ai, 1984). Cell proliferation for subacuteTCE gavage exposure has not been adequately characterized.We report the only instance to date where these putativemechanisms in the pathway toward TCE-induced hepatocarci-nogenesis are examined in the whole animal given the parentcompound under a dosing regimen known to produce tumors ina chronic bioassay. Furthermore, we characterize the extentand time course of hepatocellular proliferation, apoptosis, and
indicators of oxidative stress, as well as other relevant patho-logic observations.
MATERIALS AND METHODS
Chemical and dosing solutions. Trichloroethylene (99.5+% without an-tioxidant additives) was obtained from Aldrich Chemical Company (Milwau-kee, WI, Lot No. MF 01428EF). Gavage solutions were prepared in corn oil(Mazola, Best Foods, Somerset, NJ). Fresh dosing solutions were preparedeach week, analyzed by gas chromatography immediately following prepara-tion, and again at week's end to ensure potency. All gavage solutions werestored at 4°C.
Animals, housing, and husbandry. Male hybrid mice (B6C3Fl/CrlBR)were maintained under a program of care accredited by the American Asso-ciation for the Accreditation of Laboratory Animal care. Twelve-week-old, 25-to 30-g mice were obtained from the Portage, Michigan, production facility ofCharles River Laboratories (Wilmington, MA). Upon receipt, the mice werehoused five mice per polycarbonate cage on hardwood chip bedding and heldin isolation from other rodents for 7 days. The mice were housed in sanitizablerooms providing 10-15 complete fresh air changes per hour while maintainingan air temperature of 20 ± 2°C and 45 ± 5% relative humidity. Electronicallycontrolled full spectrum fluorescent light was provided on a 12:12-h light:darkcycle. Fresh conditioned (reverse osmosis) water and certified rodent chow(No. 5002, Purina Mills, Inc., St. Louis, MO) were available ad libitum.
Study Design. Table 1 summarizes the study design and number of ani-mals per dose group. One week prior to the study, mice were implanted withsubcutaneous magnetic identification micro-transponders (BMDS, Biomedic
0 5 10 15 20 25 30 35 40
0.2
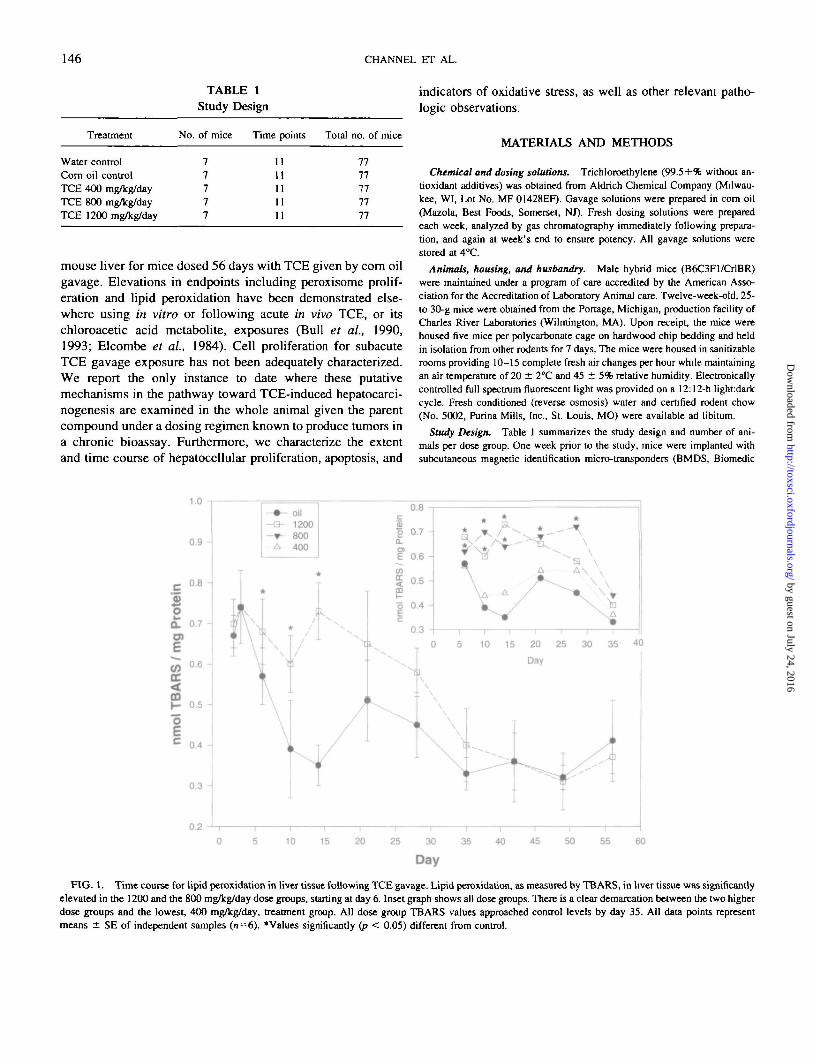
FIG. 1. Time course for lipid peroxidation in liver tissue following TCE gavage. Lipid peroxidation, as measured by TBARS, in liver tissue was significantlyelevated in the 1200 and the 800 mg/kg/day dose groups, starting at day 6. Inset graph shows all dose groups. There is a clear demarcation between the two higherdose groups and the lowest, 400 mg/kg/day, treatment group. All dose group TBARS values approached control levels by day 35. All data points representmeans ± SE of independent samples (n=6). *Values significantly (/? < 0.05) different from control.
by guest on July 24, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from
TCE INDUCES OXIDATTVE STRESS AND CELL PROLIFERATION IN VIVO 147
Data Systems, Maywood, NJ) as per the manufacturer's instructions. Micewere then randomly assigned to treatment groups and were housed in the sameroom under the conditions stated above. To conform to the weekly dosingregiment used in the previous studies conducted under the direction of the NTP(NCI, 1976; NTP, 1990), gavage was performed 5 days a week (but for only8 weeks in our study), using 20-gauge feeding needles fitted to glass syringes.Concentrations of TCE in corn oil, formulated to deliver 0, 400, 800, and 1200mg/kg/day in a total volume of 0.5 ml, were administered by gavage beginningat 0800 h each dosing day. Animal weights were recorded at the study start andweekly thereafter. The concentrations were adjusted weekly based on the meanbody weight of each treatment group.
Liver harvest and sample preparation. Liver samples were obtained onstudy days 2, 3, 6, 10, 14, 21, 28, 35, 42, 49, and 56, starting at 1300 h eachday. The harvest procedure was tightly controlled to standardize the timebetween last dosing and tissue collection. With the exception of the day 6 timepoint, all samples were scheduled to avoid sampling on a Monday following 2days of off-dose time in order to minimize that source of variability. Mice fromeach treatment group were randomly selected and weighed. Thirty minutesprior to sacrifice, an intraperitoneal (ip) injection of Af-f-butyl-a-phenylnitrone(PBN), 250 mg/kg, was administered to trap radical species for electronparamagnetic resonance spectroscopy. Preliminary work in this laboratory haddetermined that this dose of PBN is nontoxic and does not interfere withsubsequent analyses. This was confirmed by including in the analysis threemice from a control group (treated with water) that were not injected withPBN. Euthanasia was accomplished by CO2 asphyxiation. The liver wasquickly removed and washed in ice cold Dulbecco's phosphate-buffered saline(DPBS, Gibco BRL, Grand Island, NY) (pH 7.4), and a 2-mm-thick cross-section of the entire median lobe was placed in 10% neutral buffered formalin.The remaining liver from each animal was immediately snap-frozen in liquidnitrogen and apportioned for subsequent analyses. Liver tissue was not"pooled" since our process ensured that each animal contributed sufficientliver for all biochemical and histopathological assays. All reagents used insubsequent analyses were obtained from Sigma Chemical Co. (St. Louis, MO)
280 -
260 -
240
220 -
200 -
_ 180
2 160
O 140 H
S? 120 -
100
80 -
60 -
40 -
20 -
0
7I 1 f
10 20 30
Days40 50 60
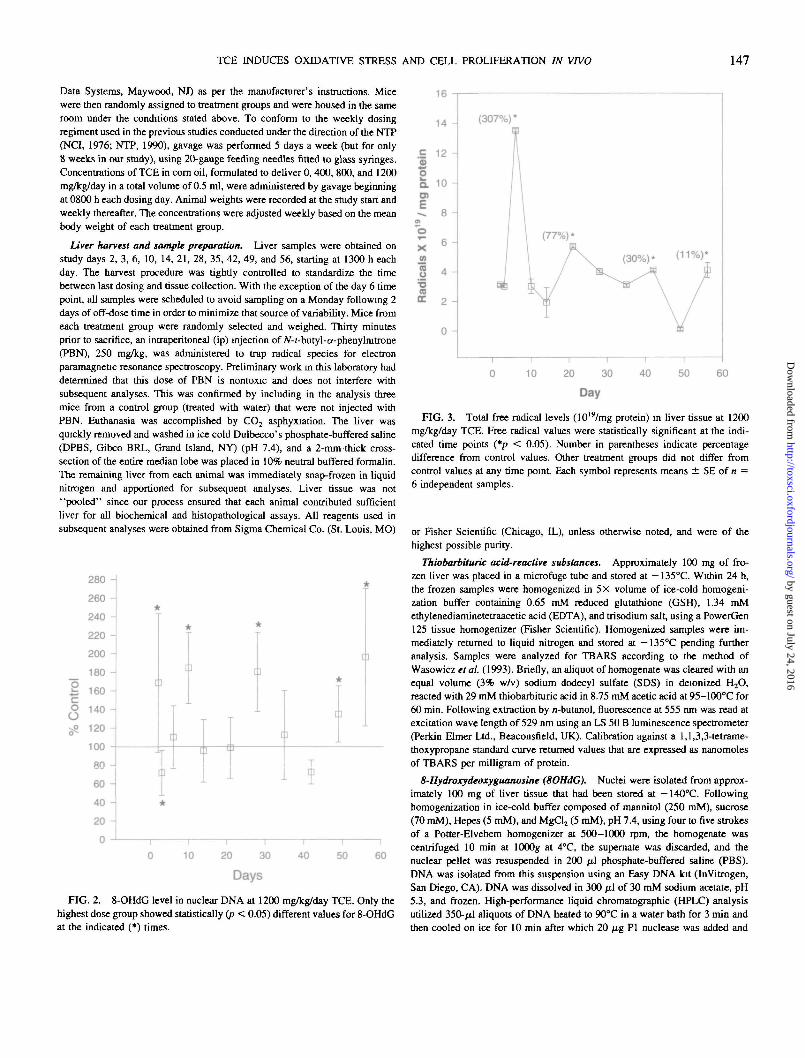
FIG. 2. 8-OHdG level in nuclear DNA at 1200 mg/kg/day TCE. Only thehighest dose group showed statistically (p < 0.05) different values for 8-OHdGat the indicated (*) times.
0 -
FIG. 3. Total free radical levels (1019/mg protein) in liver tissue at 1200mg/kg/day TCE. Free radical values were statistically significant at the indi-cated time points (*p < 0.05). Number in parentheses indicate percentagedifference from control values. Other treatment groups did not differ fromcontrol values at any time point Each symbol represents means ± SE of n =6 independent samples.
or Fisher Scientific (Chicago, IL), unless otherwise noted, and were of thehighest possible purity.
Thiobarbituric acid-reactive substances. Approximately 100 mg of fro-zen liver was placed in a microfuge tube and stored at -135°C. Within 24 h,the frozen samples were homogenized in 5X volume of ice-cold homogeni-zation buffer containing 0.65 mM reduced glutathione (GSH), 1.34 mMethylenediaminetetraacetic acid (EDTA), and trisodium salt, using a PowerGen125 tissue homogenizer (Fisher Scientific). Homogenized samples were im-mediately returned to liquid nitrogen and stored at -135°C pending furtheranalysis. Samples were analyzed for TBARS according to the method ofWasowicz et al. (1993). Briefly, an aliquot of homogenate was cleared with anequal volume (3% w/v) sodium dodecyl sulfate (SDS) in deionized H2O,reacted with 29 mM thiobarbituric acid in 8.75 mM acetic acid at 95-100°C for60 min. Following extraction by n-butanol, fluorescence at 555 nm was read atexcitation wave length of 529 nm using an LS 50 B luminescence spectrometer(Perkin Elmer Ltd., Beaconsfield, UK). Calibration against a 1,1,3,3-tetrame-thoxypropane standard curve returned values that are expressed as nanomolesof TBARS per milligram of protein.
8-Hydroxydeoxyguanosine (80HdG). Nuclei were isolated from approx-imately 100 mg of liver tissue that had been stored at -140°C. Followinghomogenization in ice-cold buffer composed of mannitol (250 mM), sucrose(70 mM), Hepes (5 mM), and MgCl2 (5 mM), pH 7.4, using four to five strokesof a Potter-Elvehem homogenizer at 500-1000 rpm, the homogenate wascentrifuged 10 min at lOOOg at 4°C, the supernate was discarded, and thenuclear pellet was resuspended in 200 /J.1 phosphate-buffered saline (PBS).DNA was isolated from this suspension using an Easy DNA kit (InVitrogen,San Diego, CA). DNA was dissolved in 300 /il of 30 mM sodium acetate, pH5.3, and frozen. High-performance liquid chromatographic (HPLC) analysisutilized 350-^1 aliquots of DNA heated to 90°C in a water bath for 3 min andthen cooled on ice for 10 min after which 20 jig PI nuclease was added and
by guest on July 24, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

148 CHANNEL ET AL.
BC
M
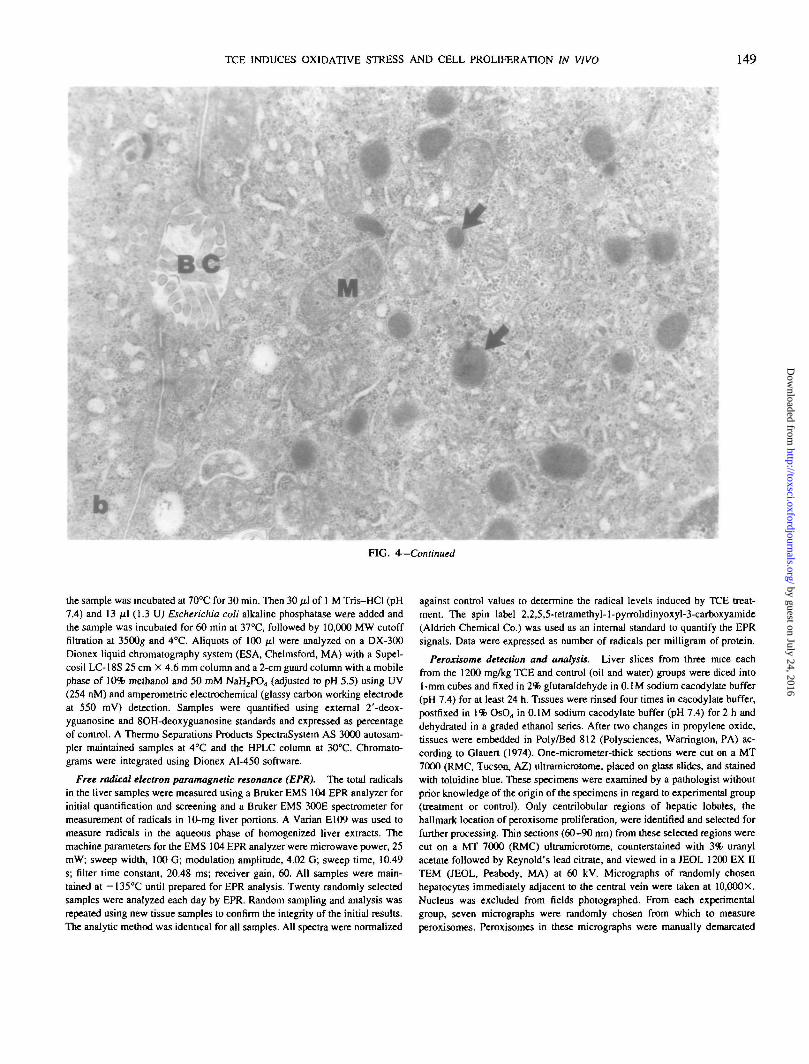
FIG. 4. Day 10. Electron photomicrographs of peroxisomes in hepatocytes from a corn oil control mouse (a) and a 1200 mg/kg TCE-exposed mouse (b).Note the numerous membrane-delimited electron dense peroxisomes (arrows) in the treatment specimen (b) compared to the control (a). BC, bile canaliculus;M, mitochondria. X 22,500.
TABLE 2Perojdsomes"
Exposure day Group Percent peroxisomal area6 Number of peroxisomesc Peroxisomal sizerf
14
1200 mg/kg TCECorn oilWater
1200 mg/kg TCECom oilWater
1200 mg/kg TCECom oilWater
2.87 ± 0.301.16 + 0.15"1.41 ±0.22
6.39 ± 0 . « /1.38 ± 0.21'1.16 ±0.39*
4.69 ± 0.22**0.77 ± 0.08'0.88 ± 0.03'
48.3 ± 6.918.7 ±2.0*17.1 ± 1.8'
64.2 ± 5.220.6 ± 1.9"14.6 ±4.1*
48.2 ± 3.409.5 ± 0.9*10.2 ± 1.9*
6.05 ± 0.436.19 ±0.208.69 ± 2.30
10.04 ± 1.1406.65 ± 0.7307.76 ± 1.90
9.84 ± 0.938.10 ± 0.689.50 ± 2.04
" Data are given as means ± SE; n = 3 per group.6 Calculated using 100 X peroxisomal area/total cytoplasmic area examined.c Number of peroxisomes per 10 mm2 of cytoplasm.d Calculated using total peroxisomal area/total number of peroxisomes; units are 104 nom2.' Significantly (p < 0.05) different from 1200 mg/kg TCE.•^Significantly (p < 0.05) different from day 6.* Significantly (p < 0.05) different from day 10.
by guest on July 24, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from
TCE INDUCES OXIDATIVE STRESS AND CELL PROLIFERATION IN VIVO 149
BCM
4
FIG. 4—Continued
the sample was incubated at 70°C for 30 min. Then 30 /xl of 1 M Tris-HCl (pH7.4) and 13 /il (1.3 UJ Eschcrichia coli alkaline phosphatase were added andthe sample was incubated for 60 min at 37°C, followed by 10,000 MW cutofffiltration at 350Og and 4°C. Aliquots of 100 ^1 were analyzed on a DX-300Dionex liquid chromatography system (ESA, Chelmsford, MA) with a Supel-cosil LC-18S 25 cm X 4.6 mm column and a 2-cm guard column with a mobilephase of 10% meihanol and 50 mM NaH2PO4 (adjusted to pH 5.5) using UV(254 nM) and amperometric electrochemical (glassy carbon working electrodeat 550 raV) detection. Samples were quantified using external 2'-deox-yguanosine and 8OH-deoxyguanosine standards and expressed as percentageof control. A Thermo Separations Products SpectraSystem AS 3000 autosam-pler maintained samples at 4°C and the HPLC column at 30°C. Chromato-grams were integrated using Dionex AI-450 software.
Free radical electron paramagnetic resonance (EPR). The total radicalsin the liver samples were measured using a Bruker EMS 104 EPR analyzer forinitial quantification and screening and a Bruker EMS 300E spectrometer formeasurement of radicals in 10-mg liver portions. A Varian E109 was used tomeasure radicals in the aqueous phase of homogenized liver extracts. Themachine parameters for the EMS 104 EPR analyzer were microwave power, 25mW; sweep width, 100 G; modulation amplitude, 4.02 G; sweep time, 10.49s; filter time constant, 20.48 ms; receiver gain, 60. All samples were main-tained at — 135°C until prepared for EPR analysis. Twenty randomly selectedsamples were analyzed each day by EPR. Random sampling and analysis wasrepeated using new tissue samples to confirm the integrity of the initial results.The analytic method was identical for all samples. All spectra were normalized
against control values to determine the radical levels induced by TCE treat-ment The spin label 2,2,5^-tetramethyi-l-pyrrolidinyoxyl-3-carboxyamide(Aldrich Chemical Co.) was used as an internal standard to quantify the EPRsignals. Data were expressed as number of radicals per milligram of protein.
Peroxisome detection and analysis. Liver slices from three mice eachfrom the 1200 mg/kg TCE and control (oil and water) groups were diced into1-mm cubes and fixed in 2% glutaraldehyde in 0.1 M sodium cacodylate buffer(pH 7.4) for at least 24 h. Tissues were rinsed four times in cacodylate buffer,postfixed in 1% OsO4 in 0.1 M sodium cacodylate buffer (pH 7.4) for 2 h anddehydrated in a graded ethanol series. After two changes in propylene oxide,tissues were embedded in Poly/Bed 812 (Polysciences, Warrington, PA) ac-cording to Glauert (1974). One-micrometer-thick sections were cut on a MT7000 (RMC, Tucson, AZ) ultramicrotome, placed on glass slides, and stainedwith toluidine blue. These specimens were examined by a pathologist withoutprior knowledge of the origin of the specimens in regard to experimental group(treatment or control). Only centrilobular regions of hepatic lobules, thehallmark location of peroxisome proliferation, were identified and selected forfurther processing. Thin sections (60-90 run) from these selected regions werecut on a MT 7000 (RMC) ultramicrotome, counterstained with 3% uranylacetate followed by Reynold's lead citrate, and viewed in a JEOL 1200 EX DTEM (JEOL, Peabody, MA) at 60 kV. MicrogTaphs of randomly chosenhepatocytes immediately adjacent to the central vein were taken at 10.000X.Nucleus was excluded from fields photographed. From each experimentalgroup, seven micrographs were randomly chosen from which to measureperoxisomes. Peroxisomes in these micrographs were manually demarcated
by guest on July 24, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

150 CHANNEL ET AL.
and then quantified using computer-based morphometric analysis (Quantimet570c, Leica, Inc., Deerfield, IL). Peroxisomal proliferation end points quanti-fied were peroxisomal area to cytoplasmic area, the number of peroxisomes perunit area of cytoplasm, and the average peroxisomal size.
Detection of proliferating cell nuclear antigen (PCNA). PCNA is presentin all stages of the cell cycle except GO and is a well-established marker of cellproliferation used extensively to demonstrate cell growth (Kurki el al., 1986;Bravo and MacDonald-Bravo, 1987; Garcia et al., 1989; Hall et al., 1990;Bolton et al., 1992; Sarli et al., 1995). The liver specimens were fixed in 10%neutral buffered formalin for 15 h, dehydrated in ethanol and Hemo-De(Fisherbrand, Fisher Scientific, Pittsburgh, PA), and paraffin embedded at lessthan 60"C using a Histomatic MVP (Fisher Scientific). Four-micrometer-thicksections were cut, mounted on ChemMate Plus slides (BioTek Solutions, SantaBarbara, CA), and air dried. Sections were deparaffinized in xylene and thenrehydrated through a graded ethanol series to PBS buffer (pH 7.4).
Immunohistochemical processing of the sections was performed on a Tech-Mate 1000 (BioTek Solutions, Santa Barbara, CA) automated immunostainingsystem using a modification of the method of Foley et al. (1991). Thisautomated procedure allowed all the specimens to be processed and stained atone time using batch prepared reagents which minimized the inherent proce-dural variability of manual immunostaining processes. For antigen retrieval,sections were microwaved in citrate buffer (BioTek Solutions) and thenreacted with hydrogen peroxide (3% for 20 min) and blocked with normalhorse serum (BioTek Solutions). Slides were then incubated overnight at roomtemperature with mouse monoclonal antibody (PCNA-Ab-1, Oncogene Sci-ence, Cambridge, MA). Detection of PCNA-positive nuclei was performedusing biotinylated horse anti-mouse (BioTek Solutions) secondary antibodyand avidin-biotin complex (ABC kit, BioTek Solutions) (20 min incubationfor each, at room temperature), followed by incubation in hydrogen peroxide/diaminobenzidine (BioTek Solutions) for 21 min. Tissues were counterstainedwith hematoxylin. Positive and negative tissue controls (neonatal and adultmouse liver, respectively) as well as antibody binding controls were includedin each procedure.
Quantitative image analysis was performed using a Quantimet 570c ImageAnalysis System (Leica, Inc., Deerfield, IL). Digital color images of liversections immunohistochemically labeled for PCNA were analyzed to detectpositive and negative hepatocytc nuclei. Detection criteria were feature coloridentification between brown (positive) and blue (negative) nuclei as well asnuclear morphological characteristics (area, fullness, aspect ratio, and round-ness). Serial images were analyzed from each liver section by digitizingadjacent microscopic fields along a linear path that transversed the sectionalong its greatest width. Data was collected in this manner for a minimum of18 fields per liver section.
In situ detection of apoptosis. Apoptosis was detected using the Apoptagin situ apoptosis kit (Oncor, Gaithersburg, MD) which extends and labelsfragmented DNA by incorporating deoxyribonucleotide triphosphate. Follow-ing deparaffinization in xylene and graded ethanol, slides were rinsed indistilled/deionized water and processed according to the kit manufacturer'sprotocol. This procedure was automated using a BioTek Tech Mate 1000 tissueprocessor equipped with an in situ oven (BioTech Solutions). Followingapoptotic tagging, the specimens were counterstained with hematoxylin for 1min, dehydrated in graded ethanol to xylene, and coverslipped using PermounLEstrous-phase rat uterus with significant endometrial gland involution wasused as a positive control. Liver and uterus incubated without terminal deoxy-nucleotidyl transferase served as negative controls. Apoptosis was quantifiedusing a single liver section from the median lobe, based on the number ofpositively labeled cells per 10 mm2 of tissue in combination with the morpho-logical criteria for apoptosis (Columbano et al., 1985). A single apoptotic bodywas considered to be one cell; two or more apoptotic bodies clustered closelytogether also were considered as derived from one cell. All positive stainingcells meeting the morphological criteria were counted manually. Liver areawas determined using a Quantimet 570c image analysis system (Leica, Inc.,Deerfield, IL).
Protein. Protein from all samples was determined using the BCA proteinassay (Pierce, Rockford, IL) with bovine serum albumin as a standard.
Statistics. Unless otherwise stated, data were plotted and treatment differ-ences determined by one-way analysis of variance (ANOVA) using SigmaStatand SigmaPlot software (Jandel Scientific, San Rafael, CA). Body weightswere analyzed using a repeated measures (time) one factorial (treatment)ANOVA with missing values. If found to be significant, appropriate post hoccomparisons such as Dunnett's method or Student Neuman-Keuls were ap-plied. For analysis of peroxisomal area to total cytoplasmic area and numberof peroxisomes per unit area of cytoplasm, a two factorial analysis of variance(treatment and day) was used, applying Levene's criteria to test the assumptionof equality of variances.
RESULTS
Body weight and grosspathology. Weight gain was notadversely affected by TCE dosing, at any concentration, overthe time course of this study. Moreover, at necropsy, grossobservation did not reveal any relative differences in body fatamong treatment animals compared to water controls. No grosslesions were observed in any group.
Lipid peroxidation. Lipid peroxidation, as measured byTBARS, was significantly elevated above oil controls in the1200 mg/kg dose group from day 6 to day 14 and from day 6to day 28 in the 800 mg/kg group (Fig. 1). A dose-responsetrend was evident over the same period (Fig. 1, inset). Theincrease for only the two highest dose groups suggests that forthis endpoint 400 mg/kg/day was a no-effect level. By day 35all treatment groups had TBARS values not statistically dif-ferent from the oil control mice. In addition, TBARS values inthe control animals decreased by half rapidly from a peak atday 6 to reach and maintain relatively steady levels from day10 to study end.
8OHdG. Elevations in 8OH-2'-deoxyguanosine levelswere modest. The greatest increase was noted in the1200 mg/kg/day treatment group, being 196% of oil con-trols on day 56 (Fig. 2). Levels fluctuated throughout thestudy, averaging 129% of control for the 1200 mg/kg/daygroup, with statistically significant elevation (p < 0.05)noted on days 2, 10, 28, 49, and 56 and depression on day3. No statistically significant effects were observed at lowerdoses.
Free radical EPR. The TCE-induced radicals, measuredby subtraction of the EPR signal of lyophilized liver of thecontrols from the TCE group, were significantly elevated onlyin the 1200 mg/kg dose group. The greatest elevation centeredon day 6 (307% of controls) with modest increases seen ondays 21, 42, and 56 (77, 30, and 11% of controls, respectively)(Fig. 3). The results of the initial assessment were confirmed byrepeating the analysis on random samples of frozen liver fromthe same animals (data not shown).
Peroxisomal proliferation. Only the 1200 mg/kg/daydose group was evaluated for peroxisome proliferation. Assummarized in Table 2, the ultrastructural data shows a
by guest on July 24, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

TCE INDUCES OXTOATTVE STRESS AND CELL PROLIFERATION IN VIVO 151
£ 80 H
go<D 40 ->
'55o
0 -
Day
FIG. 5. Cell proliferation time course. TCE gavage caused a significant(p < 0.05) burst of cell proliferation centered at day 10 of treatment in the 1200mg/kg/day group. Each symbol in large graph represents mean of a minimumof 18 microscopic fields examined by image analysis as described underMaterials and Methods. Inset graph shows mean ± SE for n = 6 independentsamples. A dose-response trend is evident at day 10.
treatment and time effect for percent peroxisomal area, a"treatment only" effect for number of peroxisomes and noeffect for peroxisomal size. Hepatocytes examined fromcorn oil control rats were no different from those from watercontrol rats for all the peroxisomal parameters, thus dis-counting a vehicle effect. At the highest dose level in thisstudy, TCE gavage significantly increased the percent per-oxisomal area of total cytoplasmic area compared to vehicle(corn oil) controls at days 6, 10 (Fig. 4), and 14. Moreover,total peroxisomal area per unit area of cytoplasm (percent-age peroxisomal area) peaked at exposure day 10. For alldays (6, 10, and 14), TCE treatment resulted in a signifi-cantly higher number of peroxisomes per unit area of cyto-plasm than either corn oil or water treatment alone.
Cell proliferation and apoptosis. Oral gavage of TCE incorn oil caused a significant burst of cell proliferation in theliver centered around day 10 on dose (Fig. 5). A dose-responsetrend is evident (Fig. 5, inset); however, only the 1200 mg/kgdose group showed a significant increase in cell proliferation(p < 0.05 ). PCNA-positive cells, as well as mitotic figures,were present in centrilobular, midzonal, and periportal regionswith no apparent predilection for a particular lobular distribu-tion (Fig. 6). Cytotoxicity manifested as hepatocellular necro-sis was not observed in any dose group. There were no signif-icant differences in apoptosis between treatment and controlgroups (data not shown).
DISCUSSION
Repeated oral gavage with an appropriate dose of TCEclearly precipitates several events that are temporally related.The parameters suggesting a state of oxidative stress—lipidperoxidation (Fig. 1), excess free radical production (Fig. 3),and peroxisomal proliferation (Table 2)—all center aroundtreatment days 6-14. Interestingly, cell proliferation was ele-vated in this same period (Fig. 5); however, apoptosis or cellnecrosis was absent. Cell proliferation resulting from chemicalexposures in the liver is often classified as either cytotoxic ormitogenic (Goldsworthy et al., 1991; Butterworth ef al, 1992).Cytotoxicants can produce necrosis and regenerative growth.Sustained cell proliferation resulting from regenerative growthmay increase the frequency of spontaneous mutations or fixmutations prior to DNA repair (Goldsworthy et al, 1993). Incontrast, hepatic mitogens produce a transient increase in he-patocyte proliferation in the absence of hepatocellular cytole-thality (Butterworth et al., 1992). Because microscopic exam-ination revealed no hepatic necrosis in this study and theproliferative response was transient (Fig. 5), the data supportthe conclusion that TCE in this study acted as a hepatocellularmitogen. Mitogenic agents may provide a selective growthadvantage to certain cell populations by direct growth stimu-lation and/or by suppression of "normal" hepatocyte prolifer-ation (Columbano et al., 1991; Goldsworthy et ai, 1993). Theabsence of increased apoptosis over background, itself negli-gible, with any treatment level in this study suggests that TCEexposure provides some transitory growth advantage for cer-tain cell populations within the liver.
The role of free radicals and oxidative stress in tumorpromotion has been well reviewed (Kozumbo et al., 1985;Thrush and Kensler, 1991; Janssen et al., 1993). Studies havedemonstrated that reactive oxygen species may alter cellulargrowth regulation by disrupting signal transduction moleculesand receptors without directly attacking nuclear DNA, thusacting as "epigenetic" carcinogens when tumors result (re-viewed in Van Der Vliet and Bast, 1992; Burdon, 1995; Byc-zkowski and Channel, 1996; Corcoran et al, 1994). With thesole exception of CH, TCE and its other principal metabolites,TCA and DCA, are repeatedly nonmutagenic and thus areclassed as epigenetic carcinogens (ECETOC, 1994). Commonto this epigenetic class is a group of "cancer-promoter" com-pounds that induce hepatocellular peroxisomal proliferation(Ashby et al., 1994). Our findings confirm other reports ofTCE-induced peroxisomal proliferation (Elcombe et al, 1985).
Agreeing with its epigenetic classification, the modest, andinconsistent, elevations in 8-OHdG we report suggest thatTCE-induced oxidative stress does not target nuclear materialdirectly. This is entirely concordant with TCE's reported lackof genotoxicity (ECETOC, 1994). We observed no time-coursepattern in this endpoint as we did with TBARS, peroxisomeand cell proliferation, and free radical load. In fact, high data
by guest on July 24, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

152 CHANNEL ET AL.
FIG. 6. Day 10. Photomicrograph of immunolabeled proliferating cell nuclear antigen [PCNA-positive hepatocellular nuclei (solid arrows)] from a 1200mg/kg TCE-exposed mouse. Immunolabeled nuclei are present in periportal [near bile duct (BD)], midzonal [between BD and central vein (CV)], andcentrilobular (near CV) regions of the hepatic lobule. Note the mitotic figures (open arrows). Diaminobenzidine-hematoxylin stained. X333.
variability and minimal differences from control values suggestthat nuclear DNA oxidative stress resulting from a clearlytumorigenic dose of TCE may be of questionable biologicalimportance. This is consistent with reports of prolonged (71-day) drinking water exposures to the two metabolites TCA andDCA, which produced no significant elevation of 8-OHdGlevels (Parrish et al., 1996). Interestingly, the same studyreported significant increase in 8-OHdG levels in controlB6C3F1 mice as a function of age. Additionally, while an acutesingle dose of TCA or DCA can elevate B6C3F1 mouse liverTBARS (Austin et al., 1996), a 14 day course 1 g/liter TCA orDCA. in drinking water for 14 days does not (Austin et al.,1995). This is consistent with our findings of an "accommo-dation" to the effects of TCE dosing as measured by TBARSsince clearly TBARS returned to control levels by the end ofour study. The rapid decrease in control animal TBARS fromday 6 to day 10 is not mirrored in the dosed animals. This isconsistent with the suggestion that TCE generates an oxidativeburden, albeit temporary, that may overwhelm cellular com-pensatory mechanisms.
Taken together, the time-course data suggest that the oxida-tive stress produced by TCE in the liver may be associated withevents that could result in an initial ' 'burst'' of cell prolifera-tion. After a 2- to 3-week period an accommodation processoccurs, likely the result of alterations in enzyme levels -adequately demonstrated for TCA and DCA exposures withinthis time frame (Austin et al., 1995). For longer exposuretimes, as in our study, a "steady state" apparently developswherein homeostatic mechanisms are able to compensate forthe continued oxidative challenge. It is reasonable to speculatethat under these conditions a selection process is occurring,wherein certain hepatocyte subpopulations eventually respondto the alterations in regulatory signal transduction pathwaysinduced by the chronically altered tissue microenvironment.Given the general understanding of the role of radical speciesin tumor promotion (Kozumbo et al, 1985; Thrush and Ken-sler, 1991), this would be similar to the pleiotropic effects fromcarbon tetrachloride which produces lipid peroxidation (Slater,1984), activates phospholipase A2 (Glende and Pushpendran,1986), and induces the transcription factor AP-1 (Kaplan and
by guest on July 24, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

TCE INDUCES OXIDATTVE STRESS AND CELL PROLIFERATION IN VIVO 153
Novak, 1994), depending on dose. With no compensatoryincrease in apoptosis (as we report here for TCE) the net effectwould be tissue growth and a higher probability that subse-quent chemical challenge would select for growth advantagefrom the phenotypically diverse expanded cell population.
The initial cellular response may be a characteristic of theB6C3F1 mouse liver that helps explain the marked species andstrain susceptibility to TCE-induced hepatic tumorigenesis.There is mounting evidence that the unique genetic makeup ofthe B6C3F1 strain may predispose it to a high spontaneous rateof liver tumor formation (Drinkwater and Ginsler, 1989;Drinkwater et al, 1989; Pugh and Goldfarb, 1992) and thatepigenetic chemicals may produce tumors by selection of in-trinsically unique cellular phenotypes (Pitot et al., 1991). In thepresent study, we cannot determine whether the hepatocytesthat result from the initial period of TCE-induced cell prolif-eration are the subpopulation that subsequently progress toneoplasia. What is certain is that such an early expansion of thetotal exposed cell population can only increase the probabilityof selection for "abnormal" phenotype. If indeed this is thecase, our study suggests that the state of continued oxidativestress caused by TCE oral gavage may provide the hepaticmicroenvironment necessary for that selection to occur.
ACKNOWLEDGMENTS
This research was conducted under both the Strategic Environmental Re-search and Development Program (CU-115) and the Air Force Office ofScientific Research (Environmental Initiative). The animals used in this studywere handled in accordance with the principles stated in the Guide for the Careand Use of Laboratory Animals, prepared by the Committee on Care and Useof Laboratory Animals of the Institute of Laboratory Animals Resources,National Research Council, Department of Health and Human Services, Na-tional Institute of Health Publication No. 86-23, 1985, and the Animal WelfareAct of 1966, as amended. The authors would like to thank all the personnelfrom the testing and pathology branches for their technical assistance, and Ms.Susan Godfrey and Drs. Darrol Dodd, Jeffery English, and Janucz Byczkowskifor their editorial and scientific reviews.
REFERENCES
Ashby J., Brady, A.., Elcombe, C. R., Elliot, B. M., Ishmael, J., Odum, J.,Tugwood, J. D., Kettle, S., and Purchase, I. F. H. (1994). Mechanisticallybased human hazard assessment of peroxisome proliferator-induced hepa-tocarcinogenesis. Hum. Exp. Toxicol. 13(Suppl 2, si—si 17.
Austin, E., Parrish, J. M., Kinder, D. H., and Bull, R. J. (1996). Lipidperoxidation and formation of 8-hydroxydeoxyguanosme from acute dosesof halogenated acetic acids. Fund. Appl. Toxicol.-31, 77-82.
Austin, E. W., Okita, J. R., Okita, R. T., Larsen, J. L., and Bull, R. J. (1995).Modification of lipoperoxidative effects of dichloroacetate and trichloroac-etale is associated with peroxisome proliferation. Toxicology 97, 59-69.
Bravo, R., and Macdonald-Bravo, H. (1987). Existence of two populations ofcyclin/proliferating cell nuclear antigen during the cell cycle: Associationwith DNA replication sites. J. Cell Biol. 105, 1549-1554.
Bolton, W. E., Mikulka, W. R., Healy, C. G., Schmittling, R. J., and Kenyon,N. S. (1992). Expression of proliferation associated antigens in the cell cycleof synchronized mammalian cells. Cytometry 13, 117-126.
Burdon, R. (1995). Superoxide and hydrogen peroxide in relation to mamma-lian cell proliferation. Free Rod. Biol. Med. 18, 775-794.
Bull, R. J., Sanchez, I. M., Nelson., M. A., Larson, J. L., and Lansing, A. J.(1990). Liver tumor induction in B6C3F1 mice by dichloroacetate andtrichloroacetate. Toxicology 63, 341-359.
Bull, R. J., Templin M., Larson, J. L., and Stevens, D. K. (1993). The role ofdichloroacetate in the hepatocarcinogenicity of trichloroethylene. Toxicol.Lett. 68,203-211.
Butterworth, B. E., Popp, J. A., Conolly, R. B., and Goldsworthy, T. L (1992).Chemically induced cell proliferation in carcinogenesis. IARC Sci. PubL116, 279-305.
Byczkowski, J. Z., and Channel, S. R. (1996). Chemically induced oxidativestress and tumorigenesis: Effects on signal transduction and cell prolifera-tion. Toxic Subst. Mechan. IS, 101-128.
Columbano, A., Ledda-Columbano, G. M.., Coni, P., Faa, G., Liguori, C ,Santacruz, G., and Pani, P. (1985). Occurrence of cell death (apoptosis)during involution of liver hyperplasia. Lab. Invest. 51, 670-675.
Corcoran, G. B., Fix, L., Jones, D. P., Moslen, M. T., Nicotera, P., Pberhaim-mer, F. Z., and Buttyan, R. (1994). Apoptosis: molecular control point intoxicity. Toxicol. Appl. Pharmacol. 128, 169-181.
Columbano A., Ledda-Columbano, G. M., Pichiri-Coni, G., Curte M., andPani, P. (1991). Differential effect of compensatory cell proliferation andmitogen-induced direct hyperplasia on hepatocarcinogenesis in the rat. InChemically Induced Cell Proliferation: Implications for Risk Assessment(B. E. Butterworth and T. J. Slaga, Eds.), pp. 217-225. Wiley-Uss, NewYork.
Daniel, F. B., DeAngelo, A. B., Stober, J. A., Olson, G. R., and Page, N. P.(1992). Hepatocarcinogenicity of CH, 2-chloroacetaldehyde, and dichloro-acetic acid in the male B6C3F1 mouse. Fundam. Appl. Toxicol 19, 159-168.
Drinkwater, N. R., and Ginsler, J. J. (1989). Genetic control of hepatocarci-nogenesis in C57BL/6J and C3H/HeJ inbred mice. Carcinogenesis 7, 1701—1707.
Drinkwater, N. R., Hanigan, M. H., and Kemp, C. J. (1989). Genetic deter-minants of hepatocarcinogenesis in the B6C3F1 mouse. Toxicol. Lett. 49,255-265.
DeAngelo, A. B., Daniel, F. B., Stober, J. A., and Olson, G. R. (1991). Thecarcinogenicity of dichloroacenc acid in the male B6C3F1 mouse. Fundam.Appl. Toxicol. 16, 337-347.
European Centre for Ecotoxicology and Toxicology of Chemicals (ECETOC)(1994). Trichloroethylene: Assessment of Human Carcinogenic Hazard.Technical Report No. 60, ISSN-O773-8O72-6O. Brussels, Belgium.
Elcombe, C. R., Rose, M. S., and Pratt, I. S. (1984). Biochemical, histological,and ultrastructural changes in rat and mouse liver following the administra-tion of trichloroethylene: possible relevance to species differences in hepa-tocarcinogenicity. Toxicol. Appl Pharmacol. 79, 365-376.
Foley, J. F., Dietrich, D. R,, Swenberg, J. A., and Maronpot, R. R.. (1991).Detection and evaluation of proliferating cell nuclear antigen (PCNA in rattissue by an approved immunohistochemical procedure. J. Histotech. 14,237-241.
Garcia, R. L., Coltrera, M. D., and Gown, A. M. (1989). Analysis of prolif-erative grade using anti-pcna/cyclin monoclonal antibodies in fixed, embed-ded tissues. Am. J. Pathol. 134, 733-739
Glauert, A. M. (1974). Embedding. In Practical Methods in Electron Micros-copy (A. M. Glauert, Ed.), Vol. 3, pp. 123-176. North Holland, Amsterdam.
Glende, E. A., Jr., and Pushpendran, K. (1986). Activation of phospholipaseA2 by carbon tetrachloride in isolated rat hepatocytes. Biochem. Pharmacol35, 3301-3307.
Goeptar, A. R., Commandeur, J. N. M., van Ommen, B., van Bladeren, P. J.,
by guest on July 24, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from

154 CHANNEL ET AL.
and Vermeulen, N. P. E. (1995). Metabolism and kinetics of trichloroeth-ylene in relation to toxicity and carcinogenicity. Relevance of the mercap-turic acid pathway. Chem. Res. Toxicol. 8, 3—21.
Goldsworthy, T. L., Morgan, K. T., Popp, J. A., and Butterworth, B. E. (1991).Guidelines for measuring chemically-induced cell proliferation in specificrodent target organs. In Chemically Induced Cell Proliferation: Implicationsfor Risk Assessment (B. E. Butterworth and T. J. Slaga, Eds.), pp. 253-284.Wiley-Liss, New York.
Goldsworthy, T. L., Butterworth, B. E., and Maronpot, R. R. (1993). Concepts,labeling procedures and design of cell proliferation studies relating tocarcinogenesis. Environ. Health Perspect. 101, 59-66
Hall, P. A., Levison, D. A., Woods, A. L., Yu, C.-C. W., Kellock, D. B.,Watkins, J. A., Barnes, D. M., Gillett, C. E., Camplejohn, R., Dover, R.,Waseem, N. H., and Lane, D. P. (1990). Proliferating cell nuclear antigen(pcna) immunolocalization in paraffin sections: An index of cell prolifera-tion with evidence of deregulated expression in some neoplasms. /. Pathol.162, 285-294.
Herren-Freund, S. L., Pereira, M. A., Khoury, M. D., and Olson, G. (1987).The carcinogenicity of trichloroethylene and its metabolites trichloroaceticacid and dichloroacetic acid in mouse liver. Toxicol. AppL Pharmacol. 90,
183-189.
Janssen, Y. M. W., Van Houten, B., Porm, P. J. A., and Mossman, B. T.(1993). Biology of disease: Cell and tissue responses to oxidative damage.
Lab. Invest. 69, 261-274.
Kaplan, D. J., and Novak, R. F. (1994). Induction of the transcription factorAP-1 by CC14 in rat hepatic nuclear extracts as assessed by gel shift assays:Evidence for a possible AP-1 binding site in human calpain. Toxicologist 14,
442.
Kozumbo, W. J., Thrush, M. A., and Kensler, T. W. (1985). Are free radicals
involved in tumor promotion? Chent-Biol. Interact. 54, 199-207.
Kurki, P., Vanderlaan, M., Dolbeare, F., Gray, J., and Tan, E. M. (1986).Expression of the proliferating cell nuclear antigen (PCNA)/cyclin during
the cell cycle. Exp. Cell Res. 166, 209-219.
Larson, J. L., and Bull, R. J. (1992a). Species differences in the metabolism oftrichloroethylene to the carcinogenic metabolites trichloroacetate and di-
chloroacetate. Toxicol. Appl. Pharmacol. 115, 278-285.
Larson, J. L., and Bull, R. J. (1992b). Metabolism and lipoperoxidation activityof trichloroacetate and dichloroacteate in rats and mice. Toxicol. Appl.Pharmacol. 115, 268-277.
Ni, Y-C, Wong, T-Y., Loyd, R. V., Heinze, T. M., Shelton, S., Casciano, D.,Kadlubar, F. F., and Fu, P. P. (1996). Mouse liver microsomal metabolismof chloral hydrate, trichloroacetic acid, and trichloroethynol leading toinduction of lipid peroxidation via a free radical mechanism. Drug Metab.Dispos. 24, 1-10.
National Cancer Institute (NCI) (1976). Carcinogenesis Bioassay of Trichlo-
roethylene, National Cancer Institute, CAS No. NCI-CG-TR-2, Washington,DC.
National Toxicology Program (NIP) (1988). Toxicology and CarcinogenesisStudies of Trichloroethylene in Four Strains of Rats. NTP Technical ReportNo. 273. Research Triangle Park, NC.
National Toxicology Program (NTP) (1990). Carcinogenesis Studies of Tri-chloroethylene (without Epichlorohydrin) in F344/N rats and B6C3F1Mice, NTP Technical Report No. 243. U.S. Department of Health andHuman Services, Bethesda, MD.
Parrish, J. M., Austin, E. W., Stevens, D. K., Kinder, D. H., and Bull, R. J.(1996). Haloacetate-induced oxidative damage to DNA in the liver of maleB6C3F1 mice. Toxicology 110, 103-111.
Pereira, M. A. (1996). Carcinogenic activity of dichloroacetic acid and trichlo-roacetic acid in the liver of female B6C3F1 mice. Fundam. Appl. Toxicol.31, 192-199.
Pitot, H. C , Dragan, Y. P., Rizui, T. A., Hally, J. R., and Campbell, H. A.(1991). Chemicals, cell proliferation, risk estimation, and multistage carci-nogenesis. In Chemically Induced Cell Proliferation: Implications for RiskAssessment (B. E. Butterworth and T. J. Slaga, Eds.), pp. 517-532. Wiley-Liss, New York.
Pugh, T. D., and Goldfarb, S. (1992).Growth kinetics of microscopic hepato-cellular neoplasms in carcinogen-resistant and carcinogen-responsive strainsof mice. Cancer Res. 52, 280-284.
Sarh, G., Benazzi, C , Preziosi, R., and Marcato, P. S. (1995). Assessment ofproliferative activity by anti-pcna monoclonal antibodies in formalin-fixedparaffin-embedded samples and correlations with mitotic index. Vet. Pathol.32, 93-96.
Slater, T. (1984). Free-radical mechanisms in tissue injury. Biochem. J. 222,1-15.
Steel-Goodwin, L., Pravecek, T. L., and Carmicheal, A. J. (1996). Tricholoro-ethylene metabolism in vitro: An epr/spin trapping study. Human Exp.Toxicol, 15(11), 878-884.
Templin, M. V., Parker, J. C, and Bull, R. J. (1993). Relative formation ofdichloroacetate and trichloroacetate from trichloroethylene in male B6C3F1
mice. Toxicol Appl. Pharmacol. 123, 1-8.
Thrush, M. A., and Kensler, T. W. (1991). An overview of the relationshipbetween oxidative stress and chemical carcinogenesis. Free Rad. Biol. Med.
10, 201-209.
Van Der Vliet, A., and Bast, A. (1992). Effect of oxidative stress on receptorsand signal transmission. Chem.-Biol. Interact. 85, 95—116.
Wasowicz, W., Neve, J., and Peretz, A. (1993). Optimized steps in fluoromet-ric determination of thiobarbituric acid-reactive substances: Importance ofextraction pH and influence of sample preservation and storage. Clin. Chem.39, 2522-2526.
by guest on July 24, 2016http://toxsci.oxfordjournals.org/
Dow
nloaded from




















