
Chemical oxidation of trichloroethylene with potassium permanganate in a porous medium
13
Ž . Advances in Environmental Research 7 2002 217229 Chemical oxidation of trichloroethylene with potassium permanganate in a porous medium Kun-Chang Huang a, , George E. Hoag a , Pradeep Chheda a , Bernard A. Woody b , Gregory M. Dobbs b a En ironmental Research Institute, Uni ersity of Connecticut, 270 Middle Turnpike, U-210 Storrs, CT 06269, USA b United Technologies Research Center, 411 Sil er Lane, East Hartford, CT 06108, USA Accepted 30 August 2001 Abstract Ž . The extent of oxidation of dissolved-phase and pure-phase trichloroethylene TCE by potassium permanganate Ž . Ž . KMnO in a sandy aquifer matrix and the impact of reaction products H and MnO on pH, metal ion leaching 4 x and permeability of the aquifer medium near TCE source zones during KMnO flushing were investigated using 4 laboratory-scale column experiments. The results of three column experiments indicated that KMnO completely 4 dechlorinated TCE, evidenced by 100% chloride recovery, when TCE was present in dissolved phase or pure phase in the aquifer matrix. Two other column experiments were conducted to investigate the impact of H and MnO on the aquifer medium near TCE source zones. KMnO flushing of the aquifer medium with residual pure x 4 Ž . TCE showed significant decreases in the pH levels e.g. from 6.7 to 2.0 of the column effluents, and large quantities of MnO precipitates were retained in the columns. The decrease in the pH levels in the columns led to an x increase in the iron content of the column effluents. Two bromide tracer tests indicated that MnO precipitates x reduced approximately 20% of the pore space in the columns. In addition, characterization of MnO by X-ray x Ž . Ž . diffraction XRD and infrared IR spectroscopy demonstrated that the TCE KMnO reaction yielded birnessite- 4 type manganese oxide. 2002 Elsevier Science Ltd. All rights reserved. Keywords: Potassium permanganate; KMnO ; TCE; Trichloroethylene; Oxidation; Remediation; Manganese oxide 4 1. Introduction A promising alternative for the remediation of sites Ž . contaminated by trichloroethylene TCE and per- Ž . chloroethylene PCE is the injection of a perman- Corresponding author. Tel.: 1-860-486-5893; fax: 1- 860-486-5488. Ž . E-mail address: [email protected] K. Huang . ganate solution into the subsurface to chemically trans- form TCE and PCE into harmless compounds. This treatment concept has been intensely evaluated in many studies over the past decade. Aqueous batch studies have demonstrated that KMnO can completely miner- 4 alize TCE and PCE, based on the 100% recovery of Ž chlorine atoms liberated from TCE and PCE Vella . and Veronda, 1992; Truax, 1993; Huang et al., 1999 . The degradation rate of TCE and PCE by perman- ganate is influenced mainly by reactant concentration 1093-019102$ - see front matter 2002 Elsevier Science Ltd. All rights reserved. Ž . PII: S 1 0 9 3 - 0 1 9 1 01 00122-8
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Chemical oxidation of trichloroethylene with potassium permanganate in a porous medium

Ž .Advances in Environmental Research 7 2002 217�229
Chemical oxidation of trichloroethylene with potassiumpermanganate in a porous medium
Kun-Chang Huanga,�, George E. Hoaga, Pradeep Chhedaa,Bernard A. Woodyb, Gregory M. Dobbsb
aEn�ironmental Research Institute, Uni�ersity of Connecticut, 270 Middle Turnpike, U-210 Storrs, CT 06269, USAbUnited Technologies Research Center, 411 Sil�er Lane, East Hartford, CT 06108, USA
Accepted 30 August 2001
Abstract
Ž .The extent of oxidation of dissolved-phase and pure-phase trichloroethylene TCE by potassium permanganateŽ . Ž � .KMnO in a sandy aquifer matrix and the impact of reaction products H and MnO on pH, metal ion leaching4 xand permeability of the aquifer medium near TCE source zones during KMnO flushing were investigated using4laboratory-scale column experiments. The results of three column experiments indicated that KMnO completely4dechlorinated TCE, evidenced by �100% chloride recovery, when TCE was present in dissolved phase or purephase in the aquifer matrix. Two other column experiments were conducted to investigate the impact of H� andMnO on the aquifer medium near TCE source zones. KMnO flushing of the aquifer medium with residual purex 4
Ž .TCE showed significant decreases in the pH levels e.g. from 6.7 to �2.0 of the column effluents, and largequantities of MnO precipitates were retained in the columns. The decrease in the pH levels in the columns led to anxincrease in the iron content of the column effluents. Two bromide tracer tests indicated that MnO precipitatesxreduced approximately 20% of the pore space in the columns. In addition, characterization of MnO by X-rayx
Ž . Ž .diffraction XRD and infrared IR spectroscopy demonstrated that the TCE�KMnO reaction yielded birnessite-4type manganese oxide. � 2002 Elsevier Science Ltd. All rights reserved.
Keywords: Potassium permanganate; KMnO ; TCE; Trichloroethylene; Oxidation; Remediation; Manganese oxide4
1. Introduction
A promising alternative for the remediation of sitesŽ .contaminated by trichloroethylene TCE and per-
Ž .chloroethylene PCE is the injection of a perman-
� Corresponding author. Tel.: �1-860-486-5893; fax: �1-860-486-5488.
Ž .E-mail address: [email protected] K. Huang .
ganate solution into the subsurface to chemically trans-form TCE and PCE into harmless compounds. Thistreatment concept has been intensely evaluated in manystudies over the past decade. Aqueous batch studieshave demonstrated that KMnO can completely miner-4alize TCE and PCE, based on the �100% recovery of
Žchlorine atoms liberated from TCE and PCE Vella.and Veronda, 1992; Truax, 1993; Huang et al., 1999 .
The degradation rate of TCE and PCE by perman-ganate is influenced mainly by reactant concentration
1093-0191�02�$ - see front matter � 2002 Elsevier Science Ltd. All rights reserved.Ž .PII: S 1 0 9 3 - 0 1 9 1 0 1 0 0 1 2 2 - 8

( )K. Huang et al. � Ad�ances in En�ironmental Research 7 2002 217�229218
and temperature, and is independent of both ionicŽstrength and pH Yan and Schwartz, 1998; Huang and
.Hoag, 2000 . The pH does not affect the degradation ofeither TCE or PCE by permanganate oxidation. How-ever, the distribution of carboxylic acids, includingformic acid, glycolic acid, glyoxylic acid and oxalic acid,formed as the reaction intermediates is essentially de-termined by both pH and the oxidant concentration inthe reactions. The overall oxidation reaction of TCE by
Ž .KMnO is commonly written as Eq. 1 , although the4reaction involves many steps, with the initial step beingthe formation of a cyclic hypomanganate ester interme-diate that rapidly undergoes subsequent reactions viavarious pathways to yield Cl�, H�, MnO and car-2
Ž .boxylic acids Yan and Schwartz, 2000 . The carboxylicacid intermediates can be further oxidized by perman-ganate to yield CO and MnO .2 2
C HCl �2KMnO �2MnO �3Cl��H�2 3 4 2Žs .
� Ž .�2CO �2K 12Žg .
Other studies of the oxidation of TCE and PCE bypermanganate in soil matrixes have shown that bothTCE and PCE can be degraded rapidly by perman-
Žganate in various soils Gates et al., 1995; Cline et al.,1997; West et al., 1997; Schnarr et al., 1998; Tratnyek,
.et al., 1998; Siegrist et al., 1999; Huang et al., 2000 .Ž .For example Gates et al. 1995 reported more than
99% destruction of dissolved-phase TCE by KMnO in4Ž .sand and clay slurries, and Siegrist et al. 1999 re-
ported similar results in silty clay soil. Additionally, oneof the two pilot experiments conducted by Schnarr et
Ž .al. 1998 indicates �100% removal of residual pureTCE by KMnO flushing. However, a pilot scale test4
Ž .conducted by Huang et al. 2000 indicates that while
dissolved-phase TCE can be oxidized rapidly duringpermanganate flushing, the destruction of source pureTCE is a slow process, controlled by the rates of masstransfer of both aqueous permanganate and pure TCEtowards their interface. During the pilot test, it wasobserved that the MnO layers that formed above thexTCE source zones increased as the test proceeded.
ŽSimilar observations are reported in other studies Mc-Kay et al., 2000; Reitsma and Marshall, 2000; Urynowicz
.and Siegrist, 2000 . The MnO layers may act as axbarrier to the destruction of source TCE by perman-ganate flushing, and the impact of MnO precipitatesxon the effectiveness of permanganate flushing is cer-tainly an essential subject to be studied.
Permanganate reacts with many soil constituentssuch as natural organic matter, reduced metal oxidesŽ .e.g. iron, manganese, arsenic and chromium and metal
Ž 2� 2�. Žions e.g. Fe and Mn Matsubara and Nakayama,.1992; Ma et al., 1994 . These non-target species not
only consume the oxidant, but also may cause muchmore complex reaction schemes. For example while theoxidation of TCE with permanganate proceeds primar-
Žily through the oxygen donation pathway Wiberg and.Saegebarth, 1957; Yan and Schwartz, 2000 , the reac-
tions between permanganate and other organic com-pounds may involve different reaction pathways such ashydrogen atom abstraction and hydride ion abstractionŽ .Stewart, 1964; Gardner and Mayer, 1995 . Because ofthese complex and competitive reaction schemes in soilmatrixes, incomplete dechlorination of TCE and theformation of undesirable products may occur. Thus,laboratory-scale column experiments were conducted
Ž . Žto investigate: 1 the extent of oxidation of TCE in.both dissolved phase and pure phase in a sandy aquifer
Ž .matrix by KMnO ; and 2 the impact of reaction4
Table 1A partial characterization of the sandy aquifer medium used in this study
aParameters Concentration References
Ž .pH 6.5 US EPA 1980SW-846 Method 9045C
Porosity 0.30�0.38 Bowles, 1984Total Organic Carbon 1600 Perkin-Elmer, 1988
Hedges and Stern, 1984Total Inorganic Carbon 700 Same as above
Ž .Iron Fe 14 000 3050B for digestion�SW-8466010A for ICP-AES analysis
Ž .Calcium Ca 910 Same as aboveŽ .Manganese Mn 340 Same as aboveŽ .Magnesium Mg 720 Same as above
Ž .Copper Cu 19 Same as aboveŽ .Aluminum Al 13 100 Same as above
Ž .Arsenic As 2 Same as aboveŽ .Chromium Cr 30 Same as above
aAll units are in mg�kg soil, except as otherwise specified.

( )K. Huang et al. � Ad�ances in En�ironmental Research 7 2002 217�229 219
Ž � .products H and MnO on pH, metal ion leachingxand permeability of the aquifer matrix near TCE sourcezones during permanganate flushing. Additionally, themanganese oxide produced in TCE�KMnO reactions4was characterized.
2. Experimental protocol
2.1. Materials
Stainless steel-casing soil cores, taken from 15 to 28feet below ground surface from uncontaminated areasin a sandy aquifer using a Geoprobe 5400 hydraulicallypowered percussion�probing machine, were used to setup five columns for this study. The aquifer mediumconsisted primarily of medium to fine sand with occa-sional pebbles. A grain size analysis by ASTM StandardMethod D-422-63 showed that the aquifer medium
Ž .consisted of 52% medium sand 2.0�0.43 mm , 23%Ž . Žfine sand 0.43�0.08 mm , 16% coarse sand 4.8�2.0
. Ž .mm , 5% gravel 19�4.8 mm and 4% silt and clayŽ .�0.08 mm . A partial characterization of the aquifermedium for selected constituents is listed in Table 1.
ŽChemicals used in this study included KMnO Re-4.agent grade, Fisher Scientific, Pittsburgh, PA , TCE
Ž .99�% purity, Fisher Scientific , hydrazine sulfateŽN H �H SO , 99�% purity, Aldrich Chemicals, Mil-2 4 2 4
. Žwaukee, WI, USA and potassium bromide KBr, 99�.% purity, Aldrich Chemicals . KMnO solutions were4
prepared by dissolving the required quantities ofŽ .KMnO in deionized DI water. TCE solutions were4
made in Tedlar bags by mixing the required volumes ofŽ .TCE stock solutions �1060 mg�l with DI water.
TCE stock solutions were prepared in 40-ml volatileŽ .organic analysis VOA vials or 1-l volumetric flasks by
equilibrating excess pure TCE with DI water and mix-ing them overnight on a magnetic stirrer. A solution of1.9 M hydrazine sulfate was used to terminate theTCE�KMnO reaction after samples were collected. In4acidic conditions, hydrazine sulfate rapidly depleted theoxidizing strength of permanganate by reducing per-
2� � Ž .�manganate to Mn Eq. 2 . Since no MnO re-xmained in the samples after an appropriate amount ofhydrazine sulfate was added, the adsorption�bonding
Ž .of metal ions e.g. iron and manganese by MnOxprecipitates was minimized. However, since the N gas2
� Ž .�generated in the quenching reaction Eq. 2 couldpotentially purge the dissolved TCE out of the samples,sodium thiosulfate is recommended for quenching per-manganate in samples to be analyzed for VOCs.
4MnO��5N H �H SO �2H��4 2 4 2 4
4Mn2��5SO2��5N �16H O4 2Žg . 2
Ž .2
2.2. Analysis
Ž .A gas chromatograph HP GC-5890 Series II ,Žequipped with a capillary column 0.53 mm�30 m,
.J&W DB-624 , a flame ionization detector, a purgeŽ .and trap Tekmar ALS 2016 and a VOC concentrator
Ž .Tekmar LSC 2000 , was used to analyze VOCs, includ-ing TCE, in samples. Identification of VOC by-prod-ucts was performed using another gas chromatographŽ .HP GC-5890 Series II equipped with a capillary
Ž .column 0.32 mm�60 m, J&W DB-624 and a massŽ .selective detector SW-846 Method 8260B . Chloride
and bromide were analyzed using an ion chromato-Ž .graph Dionex DX 500 equipped with an ion exchangeŽ . Žcolumn Dionex Ionpac 4 mm AS9H SW-846 Method
. Ž9056 . An emission spectrometer Perkin-Elmer Plasma.40 was used to analyze metal ions including Fe andŽ .Mn SW-846 Method 6010A . KMnO concentrations4
Žwere determined using a spectrophotometer Milton.Roy Spectronic 601 at a wavelength of 526 nm. A
Ž .CHN elemental analyzer Perkin-Elmer 2400 de-termined weight percentages of organic and inorganic
Žcarbons in the aquifer medium Perkin-Elmer, 1988;.Hedges and Stern, 1984 . MnO was characterized byx
Ž .X-ray diffraction, Fourier transformed infrared FT-IRspectroscopy and iodometric titration for average oxi-
Ž .dation state Glover et al., 1989 . The XRD analysisŽwas performed with an X-ray diffractometer Scintag
.200 PDS using CuK� radiation. The beam voltage andbeam current settings were 45 KV and 40 mA, respec-tively. For FT-IR analysis, powder samples were dis-persed in KBr pellets to approximately 1�5% by weightand studied in the range of 4000�400 cm�1. The IRspectra was taken on a Nicolet SX-60 FT-IR instru-ment spectrometer using standard procedures.
2.3. Methodology
(2.3.1. Oxidation of TCE in dissol�ed phase and)pure phase
The extent of oxidation of dissolved-phase TCE andpure-phase TCE in a sandy aquifer matrix by KMnO4
Žflushing was investigated in columns 1 and 2 dissolved. Ž .phase and column 3 pure phase , at room tempera-
ture. The column system consisted primarily of a soil� Ž . Ž .�core 4.1 cm D �14.6 or 7.2 cm L , end plates,
Žinjection�sampling ports and a peristaltic pump Fig..1 . Since most KMnO might be consumed by soil4
substances before coming into contact with TCE,columns 1 and 2 were pre-flushed with a KMnO4
Ž . Žsolution �2000 mg�l for 25 h at 0.3 ml�min �1.m�day of pore velocity . This application was per-

( )K. Huang et al. � Ad�ances in En�ironmental Research 7 2002 217�229220
formed to reduce the reaction complexity in the aquifermedium so that the degree of TCE oxidation could beexamined carefully. During the pre-flushing, theKMnO concentration in the effluents of columns 14and 2 reached 1995 and 1910 mg�l, respectively, andthe pH in both columns decreased from 7.9 to 7.5.
ŽColumn 1 was then pulse-injected with 5 ml i.e. 5 ml in. Ž .20 s of TCE solution 1060 mg�l through the columnŽ .inlet Fig. 1 . In contrast, column 2 was continuously
Žand simultaneously injected with a TCE solution 250. Žmg�l at 0.15 ml�min and a KMnO solution 41804.mg�l at 0.15 ml�min at the column 2 inlet.
In column 3, the extent of oxidation of pure TCE inthe aquifer matrix by KMnO flushing was evaluated.4
� Ž .�A small hole 1.5 mm D was first made in the middleŽ . �of the column Fig. 1 . A stainless steel needle 1 mm
Ž . Ž .�D �50 mm L with three outlets, each of 1 mmŽ .D , was then inserted into the column through anadapter to facilitate the injection of pure TCE into thecolumn. A quantity of 0.32 g of pure TCE was injectedthrough the needle using a 1-ml gas-tight glass syringe,and the mass of injected TCE was determined on thebasis of the difference in mass of the syringe beforeand after the injection. This injection was followed byflushing with 1 ml of DI water to drive the TCEremaining in the needle into the column. The columnwas left to stand for 12 h and then flushed with a
Ž .KMnO solution 4080 mg�l at a relatively low flowrate4of �0.1 ml�min to avoid flushing pure TCE out of thecolumn.
During the experiments of columns 1�3, sampleswere collected periodically from the column effluentand analyzed for Cl�, pH, KMnO and VOCs including4TCE. Samples for the analysis of VOCs and Cl� were
quenched with hydrazine sulfate immediately after theywere collected. The extent of oxidation of TCE byKMnO in columns 1�3 was evaluated on the basis of4
Ž .the mass balance of chloride, determined using Eq. 3Ž . Ž . Ž .for columns 1 and 3 and Eq. 4 for column 2 .
MCl,ex Ž .P%� �100 3Ž .0.81 M �MTCE,in TCE,ex
CCl,SS Ž .P%� �100 40.81CTCE,in
where P%�percentage of TCE dechlorination; MCl,ex�mass of chloride recovered from the column efflu-ent; M �mass of TCE injected into the column;TCE,inM �mass of TCE recovered from the columnTCE,exeffluent; the factor of 0.81� the molecular weight ratioof three chlorides to TCE; C �chloride concentra-Cl,SStion in the column effluent at the steady state; andC � influent TCE concentration of column 2.TCE,in
VOC analysis was conducted immediately after hy-drazine sulfate was added to the samples to minimizethe effect of potential reactions of residual hydrazinesulfate with TCE. The quenching of VOC samples wasperformed in a 5-ml gas-tight syringe that was used tocapture the gaseous TCE that resulted from the
� Ž .�quenching reaction Eq. 2 . KMnO analysis was con-4ducted immediately after the samples were collected.Before KMnO measurement, the samples were fil-4
Žtered with syringe filters 0.45 �m, 13-mm PVDF mem-.brane, Analytical Sales and Service to remove parti-
cles. The flow rate, maintained by a peristaltic pump,was checked routinely and was readjusted whenever it
Fig. 1. Experimental apparatus of the column system.
( )K. Huang et al. � Ad�ances in En�ironmental Research 7 2002 217�229 221
Table 2Ž .Experimental conditions of columns 1�3 for investigating the oxidation of TCE in both dissolved phase and pure phase by KMnO4
in a sandy aquifer matrix
bColumn Soil bed Pore Retention Influent TCE source Flowexp. dimension volume time KMnO rate4
a Ž . Ž . Ž . Ž .I.D. �L ml min mg�l ml�minŽ .mm
1 41�146 67 223 2050 Pulse input of 0.35-ml TCEŽ .1060 mg�l
2 41�146 67 223 4080 Continuous 0.3input of TCEŽ250 mg�l�
.0.15 ml�min
3 41�72 34 262 4080 Injection of 0.13pure TCEof 0.32 g
a I.D.� inside diameter.bRetention time was estimated by dividing pore volume by flow rate.
deviated by more than 5% from its set point. Theexperimental conditions of columns 1�3 are summa-rized in Table 2.
2.3.2. Impact of H � and MnO on the source zonexsoil
2.3.2.1. Pure TCE distribution. The impact of reactionŽ � .products H and MnO on the aquifer medium nearx
TCE source zones during KMnO flushing was investi-4Ž .gated in columns 4 and 5 Table 3 . For these two
Ž .columns, two equally spaced holes ports 1 and 2 wereŽ .drilled on the sidewall of the columns Fig. 1 . Two
Ž .needles the same as the device used in column 3equipped with control valves were individually attachedto the holes for sample extraction. After ports 1 and 2and their sampling devices were made, a vacuum pumpwas used to create a 400-mmHg vacuum in the columnfor 15 min. Pure TCE was then aspirated upward intothe column until pure TCE was observed at the columnoutlet, at which time the vacuum pump was shut down.The column was maintained in a ‘closed system’ condi-tion and left to stand for 1 h. Pure TCE in the columnswas then gravity-drained for 12 h, and the mass ofresidual TCE was gravimetrically determined. Approxi-mately 7.6 and 16.3 g of pure TCE was retained incolumns 4 and 5, respectively.
2.3.2.2. Bromide tracer tests for e�aluating MnO im-xpact. After the mass of pure TCE retained in columns 4and 5 was determined, columns 4 and 5 were flushedupwards with a KMnO solution of 4 and 8 g�l, respec-4tively, at a flow rate of 0.3 ml�min. Immediately afterthe effluent was observed at the column outlet, 5 ml of
Ž .KBr solution 10 g�l was pulse-injected into thecolumn through the column inlet to determine theretention time before significant amounts of MnOxaccumulated in the column. The column was continu-ously flushed with a KMnO solution until the TCE4concentration at the outlet decreased to below 5 �g�l.Then, uncontaminated site groundwater was used toreplace the KMnO solution and to remove the KMnO4 4remaining in the columns. Another Br� tracer test wasconducted when the KMnO concentration at the out-4let decreased to below 100 mg�l. The impact of MnOxprecipitates on the permeability of the aquifer mediumwas evaluated based on the change in retention time ofthe column determined by the two Br� tracer testsŽone in the beginning and the other before termination
.of the column run . The column retention time wasŽ . Ž . Žcalculated using Eqs. 5 and 6 Levenspiel, 1972;
.Fogler, 1986 .
�Ž .tE t d tH
0 Ž .t � 5m �Ž .E t d tH
0
Ž .C tŽ . Ž .E t � 6�
Ž .C t d tH0
Ž .where t �mean column retention time min ; t� timemŽ . Ž .min ; E t � retention time distribution functionŽ �1. Ž . �min ; and C t �Br concentration in the effluentŽ .mg�l .
( )K. Huang et al. � Ad�ances in En�ironmental Research 7 2002 217�229222
Table 3�Ž .Experimental conditions of columns 4 and 5 for investigating the impact of reaction products H and MnO on the aquifer matrixx
bColumn Soil bed Pore Retention Influent Residual Flow-experiment dimension volume time KMnO pure TCE rate4
a Ž . Ž . Ž . Ž .I.D. �L ml min mg�l in column ml�minŽ . Ž .mm g
4 41�65 31 103 4000 7.6 0.35 41�65 29 97 8000 16.3 0.3
a I.D.� inside diameter.bRetention time was estimated by dividing pore volume by flow rate.
2.3.2.3. Determination of MnO accumulated inxcolumns. Flows in columns 4 and 5 were determinedwhen the KMnO concentration in the effluent de-4creased to �1 mg�l. The aquifer medium in thecolumns was then pushed out with a plunger andsub-sampled along the columns. The sub-samples wereprepared by the SW-846 3050b method for extracting
Ž . Ž .Mn II from MnO and analyzed for Mn II by thexŽ .SW-846 6010A method. The observed Mn II was used
Ž .to determine the MnO content as MnO and itsx 2Ž .distribution in the columns, assuming that all Mn II
originated from MnO .22.3.2.4. Monitored parameters and metal-leaching ex-
amination. In columns 4 and 5, parameters includingpH, Cl�, TCE and KMnO at the outlet, and KMnO4 4at ports 1 and 2 were monitored periodically. In addi-tion, before the KMnO breakthrough at the outlet of4columns 4 and 5, several samples were collected to
Ž . Žanalyze iron labeled as Fe and manganese labeledT.as Mn content in the samples after they were filteredT
with 0.45-�m syringe filters. The impact of the pro-duced H� on metal leaching from the aquifer mediumwas evaluated by correlating the variation of Fe withTthe pH value of the column effluent, since iron was
Ž .relatively abundant in the aquifer medium Table 3 .
3. Results and discussion
3.1. Oxidation of dissol�ed-phase and pure-phaseTCE by KMnO4
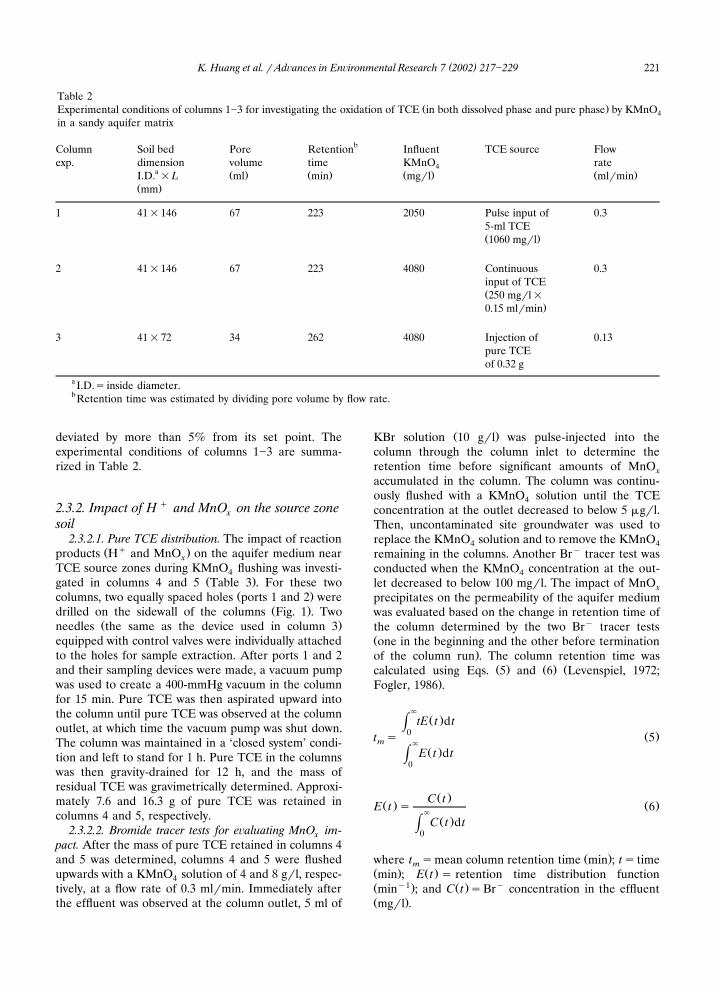
Columns 1�3 were conducted to evaluate the extentof oxidation of dissolved-phase and pure-phase TCE ina sandy aquifer matrix by KMnO flushing. The4
Žobserved chloride recovery i.e. 110, 106 and 99.7% in.columns 1, 2 and 3, respectively indicated that TCE in
either dissolved-phase or pure-phase in the aquiferŽmatrix was completely dechlorinated by KMnO Fig.4
.2 . This conclusion was supported by the GC-MS analy-sis, which showed no significant levels of other VOCsin the effluents of these columns. In addition, the ionchromatograph analysis demonstrated that the reac-tions did not produce chloroacetic acids. During theexperiments, TCE concentration in the effluents wasless than 100 �g�l in column 1 and below 5 �g�l incolumn 2, revealing that KMnO rapidly oxidized dis-4solved-phase TCE in the aquifer matrix. However, in
Ž .column 3, the destruction of pure TCE 0.32 g by
Fig. 2. Extent of TCE dechlorination by KMnO vs. time in columns 1�3. Data are shown with 5% error bar.4

( )K. Huang et al. � Ad�ances in En�ironmental Research 7 2002 217�229 223
KMnO flushing appeared to be relatively slow as4Ž .evidenced by a long-tailed chloride recovery Fig. 2 .
In general, the oxidation of TCE by KMnO in4�columns 1�3 caused a slight decrease in pH e.g. from
ŽpH 7.7 to 6.6 in column 2, and from pH 8.5 at the. Ž .beginning of the run to 6.8 the lowest pH observed in
�column 3 . The aquifer matrix in columns 1�3 ap-peared to be able to buffer�react with the H� gener-ated from TCE oxidation. The results are indeed ex-pected because the pH buffering capacity of most natu-ral soils is generally sufficient to resist significant changein pH in the regions away from TCE source zonesduring KMnO flushing. However, it is suspected that a4substantial decrease in the pH value may occur nearTCE source zones because large quantities of H� maybe released in local regions where source TCE is incontact with KMnO .4
( �3.2. Impact of the reaction products H and)MnOx
Columns 4 and 5 were conducted to investigate theimpact of H� and MnO generated from thexTCE�KMnO reactions on pH, metal leaching and the4permeability of the aquifer medium near TCE sourcezones, where the highest impact may occur duringKMnO flushing. The results indicated that KMnO4 4was consumed rapidly once it was in contact with TCEin the columns. For instance, �14, 26 and 32 pore
Ž .volumes PVs of KMnO solutions were used before4KMnO became detectable at port 1, port 2 and the4
Ž .outlet of column 5, respectively data not shown here .It is also evident from Fig. 3 that substantial amountsof H� and Cl� were generated while the TCE in
columns 4 and 5 was oxidized. The pH at the outlets ofŽcolumns 4 and 5 decreased from �7.5 at the begin-
. Žning of the run to �2 the lowest pH observed during. �the run ; Cl concentration increased from �30 mg�l
Ž .at the beginning of the run to a maximum of �1500and 3000 mg�l in columns 4 and 5, respectively. Theresults imply that the conditions of low pH and highCl� concentration might occur near TCE source zonesŽespecially for the aquifer with low pH buffering capac-
.ity when KMnO solutions contact source zone TCE.4
Indeed, the condition was observed in a pilot-scale testinvolving source zone TCE destruction by flushing with
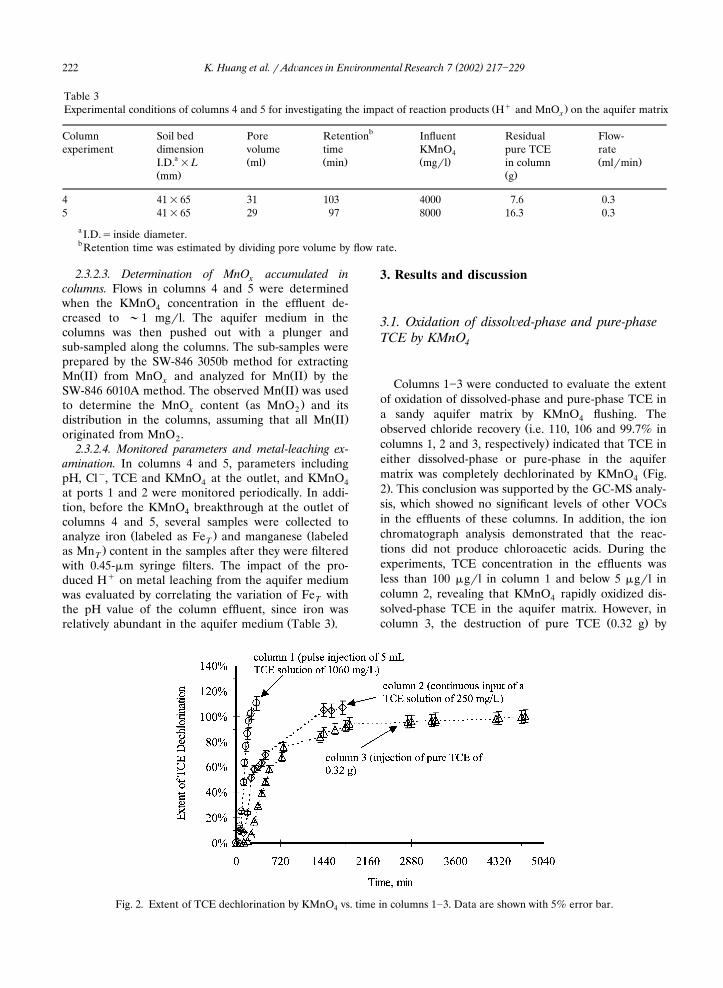
Ž .an NaMnO solution of 10 g�l Huang et al., 2000 .4� Ž � .The quantity of H observed H in the columnobs
effluent was compared with that of H� generatedŽ � . Ž . �H Fig. 4 , which was estimated on the basis of Clgen
production and the assumption that 3 moles of Cl�� � Ž .�production yield 1 mole of H Eq. 1 .
The reaction of H� with the aquifer substancesgen
was evident from the noticeable difference betweenH� and H� in columns 4 and 5. The graph in Fig. 4obs gen
is divided into three reaction stages. Stage I shows acondition of nearly complete reaction of H� withgen
aquifer substances, which occurred in the early stage ofthe experiments, as evidenced by ‘H� �H� ’ in thisobs gen
period. The aquifer matrix in stage I was sufficient tobuffer�react H� and resist change in pH. As thegen
experiments proceeded, the H� increased and reachedobs
�46 and 23% of the H� in columns 4 and 5, respec-genŽ .tively stage II , implying that the reactions of the
aquifer matrix with H� were limited and that thegen
aquifer matrix was not able to buffer�react most of theH� . In column 5, more TCE was present and a highergen
� Ž . Ž . Ž .Fig. 3. Variation in Cl and pH in the effluent of columns 4 and 5: KMnO ��4 g�l column 4 and 8 g�l column 5 ; flow4 inŽ . Ž .rate �0.3 ml�min; pure TCE in columns�7.6 g column 4 and 16.3 g column 5 ; and temperature ��21 �C.
( )K. Huang et al. � Ad�ances in En�ironmental Research 7 2002 217�229224
� � Ž . Ž . Ž .Fig. 4. Comparison of H observed with H generated in columns 4 and 5: KMnO ��4 g�l column 4 and 8 g�l column 5 ;4 inflow rate �0.3 ml�min; and temperature ��21 �C.
KMnO dose was used as compared with column 4.4Thus, a greater H� in column 5 was expected becauseobsthis column produced more Cl�. However, the resultsindicated that the quantity of H� was similar in theseobs
� Ž .two columns, and neither approached H Fig. 4 . Itgenappeared that part of the H� was consumed by sidegenreactions that occurred in columns 4 and 5. For exam-
Žple, the oxidation of formic acid Taylor and Halpern,. Ž .1959 and oxalic acid Pimienta et al., 1994 by perman-
Ž . Ž .ganate as presented in Eqs. 7 and 8 , respectively,probably consumed protons in the reactions.
2MnO��3HCO H�2H��2MnO �3CO4 2 2Žs . 2Žg .
Ž .�4H O 72
2MnO��3H CO �2H��2MnO �6CO4 2 4 2Žs . 2Žg .
Ž .�4H O 82
In stage III, the H� decreased with the decrease inobsH� , reflecting the limited amount of TCE remaininggenin the columns. Gradually, the H� decreased to nearlygenthe H� at the column outlet in both columns.obs
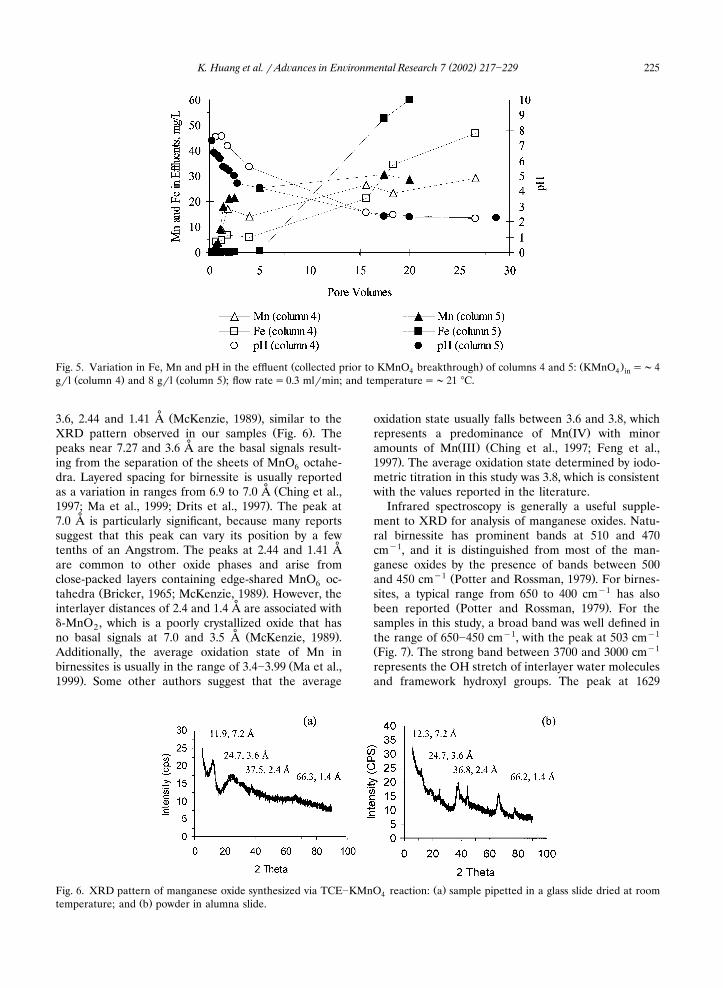
The pH and redox potential are master variablescontrolling metal solubility and dissolution of soil min-
Ž .erals Hem, 1972; Satawathananont et al., 1991 . Re-ducing the soil pH generally increases the leaching ofmetal ions from the soil, as most metals have highersolubility at low pH conditions. In columns 4 and 5, itwas observed that the Fe in the effluent increased asT
Ž .pH decreased Fig. 5 . This indicated that theTCE�KMnO reactions facilitated the leaching of iron4oxides from the aquifer medium, most likely by lower-
ing the pH values in the columns. The results are inagreement with other studies that have indicated thatmetal oxides, including chromium and selenium, insoils were oxidized and mobilized by KMnO oxidation4Ž .Chambers et al., 2000a,b; Li and Schwartz, 2000 .
Data on Mn observed in the effluents were used toTŽ .evaluate the potential increase in Mn II in ground-
water and to examine the predominantly reduced Mnspecies in columns 4 and 5. When the pH in columns 4
�and 5 with an influent KMnO solution of 4 g�l4Ž .experimentally determined pH�6.5; E �670 mVh
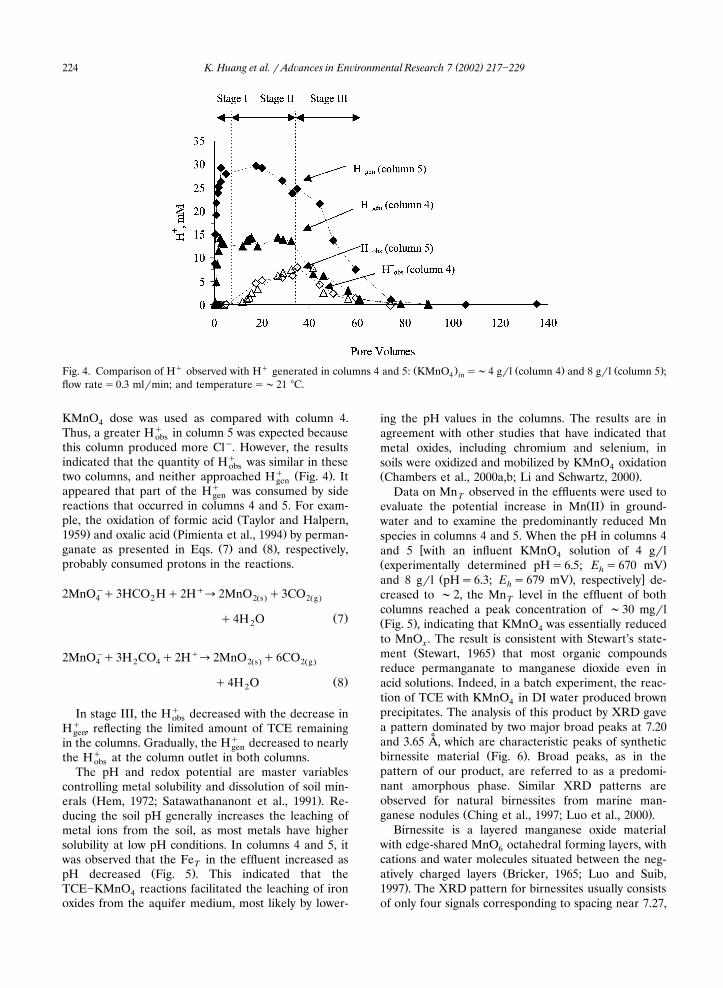
Ž . �and 8 g�l pH�6.3; E �679 mV , respectively de-hcreased to �2, the Mn level in the effluent of bothTcolumns reached a peak concentration of �30 mg�lŽ .Fig. 5 , indicating that KMnO was essentially reduced4to MnO . The result is consistent with Stewart’s state-x
Ž .ment Stewart, 1965 that most organic compoundsreduce permanganate to manganese dioxide even inacid solutions. Indeed, in a batch experiment, the reac-tion of TCE with KMnO in DI water produced brown4precipitates. The analysis of this product by XRD gavea pattern dominated by two major broad peaks at 7.20
˚and 3.65 A, which are characteristic peaks of syntheticŽ .birnessite material Fig. 6 . Broad peaks, as in the
pattern of our product, are referred to as a predomi-nant amorphous phase. Similar XRD patterns areobserved for natural birnessites from marine man-
Ž .ganese nodules Ching et al., 1997; Luo et al., 2000 .Birnessite is a layered manganese oxide material
with edge-shared MnO octahedral forming layers, with6cations and water molecules situated between the neg-
Žatively charged layers Bricker, 1965; Luo and Suib,.1997 . The XRD pattern for birnessites usually consists
of only four signals corresponding to spacing near 7.27,
( )K. Huang et al. � Ad�ances in En�ironmental Research 7 2002 217�229 225
Ž . Ž .Fig. 5. Variation in Fe, Mn and pH in the effluent collected prior to KMnO breakthrough of columns 4 and 5: KMnO ��44 4 inŽ . Ž .g�l column 4 and 8 g�l column 5 ; flow rate �0.3 ml�min; and temperature ��21 �C.
˚ Ž .3.6, 2.44 and 1.41 A McKenzie, 1989 , similar to theŽ .XRD pattern observed in our samples Fig. 6 . The
˚peaks near 7.27 and 3.6 A are the basal signals result-ing from the separation of the sheets of MnO octahe-6dra. Layered spacing for birnessite is usually reported
˚ Žas a variation in ranges from 6.9 to 7.0 A Ching et al.,.1997; Ma et al., 1999; Drits et al., 1997 . The peak at
˚7.0 A is particularly significant, because many reportssuggest that this peak can vary its position by a few
˚tenths of an Angstrom. The peaks at 2.44 and 1.41 Aare common to other oxide phases and arise fromclose-packed layers containing edge-shared MnO oc-6
Ž .tahedra Bricker, 1965; McKenzie, 1989 . However, the˚interlayer distances of 2.4 and 1.4 A are associated with
�-MnO , which is a poorly crystallized oxide that has2˚ Ž .no basal signals at 7.0 and 3.5 A McKenzie, 1989 .
Additionally, the average oxidation state of Mn inŽbirnessites is usually in the range of 3.4�3.99 Ma et al.,
.1999 . Some other authors suggest that the average
oxidation state usually falls between 3.6 and 3.8, whichŽ .represents a predominance of Mn IV with minor
Ž . Žamounts of Mn III Ching et al., 1997; Feng et al.,.1997 . The average oxidation state determined by iodo-
metric titration in this study was 3.8, which is consistentwith the values reported in the literature.
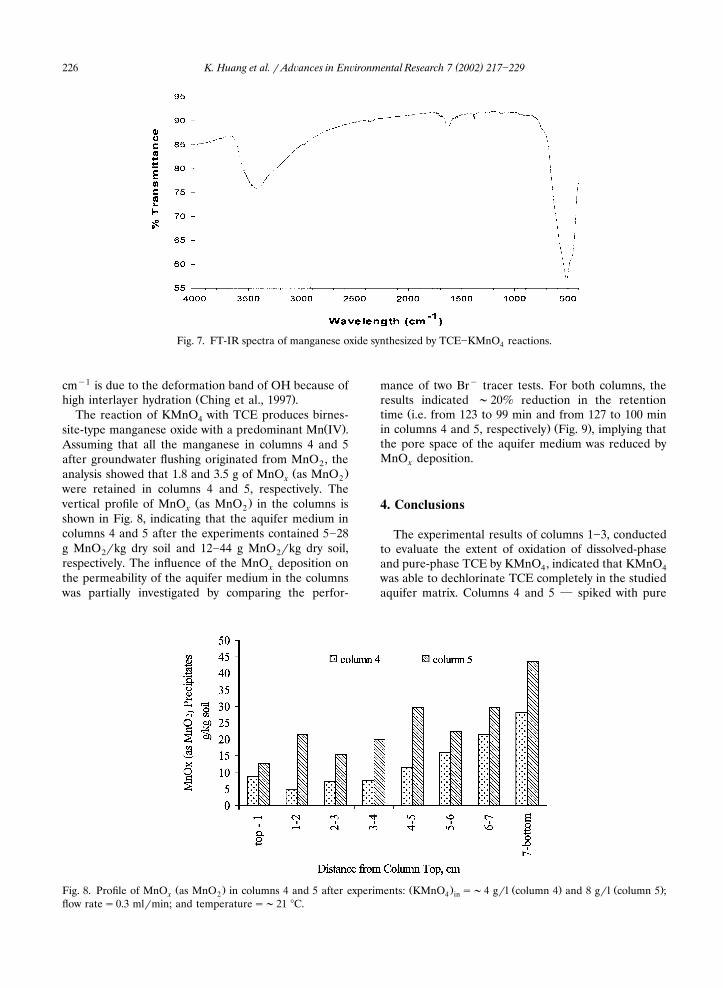
Infrared spectroscopy is generally a useful supple-ment to XRD for analysis of manganese oxides. Natu-ral birnessite has prominent bands at 510 and 470cm�1, and it is distinguished from most of the man-ganese oxides by the presence of bands between 500
�1 Ž .and 450 cm Potter and Rossman, 1979 . For birnes-sites, a typical range from 650 to 400 cm�1 has also
Ž .been reported Potter and Rossman, 1979 . For thesamples in this study, a broad band was well defined inthe range of 650�450 cm�1, with the peak at 503 cm�1
Ž . �1Fig. 7 . The strong band between 3700 and 3000 cmrepresents the OH stretch of interlayer water moleculesand framework hydroxyl groups. The peak at 1629
Ž .Fig. 6. XRD pattern of manganese oxide synthesized via TCE�KMnO reaction: a sample pipetted in a glass slide dried at room4Ž .temperature; and b powder in alumna slide.
( )K. Huang et al. � Ad�ances in En�ironmental Research 7 2002 217�229226
Fig. 7. FT-IR spectra of manganese oxide synthesized by TCE�KMnO reactions.4
cm�1 is due to the deformation band of OH because ofŽ .high interlayer hydration Ching et al., 1997 .
The reaction of KMnO with TCE produces birnes-4Ž .site-type manganese oxide with a predominant Mn IV .
Assuming that all the manganese in columns 4 and 5after groundwater flushing originated from MnO , the2
Ž .analysis showed that 1.8 and 3.5 g of MnO as MnOx 2were retained in columns 4 and 5, respectively. The
Ž .vertical profile of MnO as MnO in the columns isx 2shown in Fig. 8, indicating that the aquifer medium incolumns 4 and 5 after the experiments contained 5�28g MnO �kg dry soil and 12�44 g MnO �kg dry soil,2 2respectively. The influence of the MnO deposition onxthe permeability of the aquifer medium in the columnswas partially investigated by comparing the perfor-
mance of two Br� tracer tests. For both columns, theresults indicated �20% reduction in the retention
Žtime i.e. from 123 to 99 min and from 127 to 100 min. Ž .in columns 4 and 5, respectively Fig. 9 , implying that
the pore space of the aquifer medium was reduced byMnO deposition.x
4. Conclusions
The experimental results of columns 1�3, conductedto evaluate the extent of oxidation of dissolved-phaseand pure-phase TCE by KMnO , indicated that KMnO4 4was able to dechlorinate TCE completely in the studiedaquifer matrix. Columns 4 and 5 � spiked with pure
Ž . Ž . Ž . Ž .Fig. 8. Profile of MnO as MnO in columns 4 and 5 after experiments: KMnO ��4 g�l column 4 and 8 g�l column 5 ;x 2 4 inflow rate �0.3 ml�min; and temperature ��21 �C.

( )K. Huang et al. � Ad�ances in En�ironmental Research 7 2002 217�229 227
Ž .Fig. 9. Bromide breakthrough curves one conducted in the beginning and the other at the end of the run in columns 4 and 5:Ž �. Ž .Br �10 g�l 5-ml pulse injection ; and flow rate �0.3 ml�min.in
TCE and used to investigate the impact of H� andMnO on pH, metal leaching and permeability of thexaquifer medium near TCE source zones during KMnO4flushing � showed that significant amounts of H�, Cl�
and MnO were generated from the reactions. Largexquantities of MnO precipitates accumulated in thexcolumns. Two bromide tracer tests indicated that theMnO precipitates retained in columns 4 and 5, re-xduced approximately 20% of the pore space in thecolumns. The chemical and physical impact of MnOxon the effectiveness of KMnO flushing appeared to be4a critical issue. Additionally, the decrease in the pHlevel in columns 4 and 5 led to an increase in the ironleaching from the studied aquifer medium.
Acknowledgements
United Technologies Corporation provided the fund-ing for this research. MnO characterization was per-xformed by Sinue Gomez of the Chemistry Department
Ž .of the University of Connecticut UConn . The authorsthank Farhad Nadim and Gary Ulatowski of the Envi-ronmental Research Institute, UConn for their techni-cal assistance.
References
Bowles, J.E., 1984. Physical and Geotechnical Properties ofSoils. McGraw-Hill Inc, New York.
Bricker, O., 1965. Some stability relations in the systemMn�O �H O at 25 �C and one atmosphere total pressure.2 2Am. Mineral. 50, 1296�1354.
Chambers, J., Leavitt, A., Walti, C., Schreier, C.G., Melby, J.,Goldstein, L., 2000a. In-situ destruction of chlorinated sol-vents with KMnO oxidizes chromium. Proceedings of the4
Second International Conference on Remediation of Chlo-rinated and Recalcitrant Compounds. Monterey, CA, USA,pp. 49�56.
Chambers, J., Leavitt, A., Walti, C., Schreier, C.G., Melby, J.,2000b. Treatability study � fate of chromium during oxi-dation of chlorinated solvents. Proceedings of the SecondInternational Conference on Remediation of Chlorinatedand Recalcitrant Compounds. Monterey, CA, USA, pp.57�66.
Ching, S., Petrovay, D.J., Jorgensen, M.L., Suib, S.L., 1997.Gel synthesis of layered birnesseite-type manganese oxides.Inorg. Chem. 36, 883�890.
Cline, S.R., West, O.R., Siegrist, R.L., Holden, W.L., 1997.Performance of in situ chemical oxidation field demonstra-tion at DOS sites. Proceedings of In Situ Remediation ofthe Geoenvironment Conference. Minneapolis, MI, USA.
Drits, V.A., Silvester, A., Gorshkov, A.I., Manceau, A., 1997.Structure of synthetic monoclinic Na-rich birnessite andhexagonal birnessite: 1. Results from X-ray diffraction andselected-area electron diffraction. Am. Mineral. 82,946�961.
Feng, Q., Yanagisawa, K., Yamasaki, N., 1997. Synthesis ofbirnessite-type potassium manganese oxide. J. Mater. Sci.Lett. 16, 110�112.
Fogler, H.S., 1986. Elements of Chemical Reaction Engineer-ing. Prentice-Hall Inc, New Jersey.
Gardner, K.A., Mayer, J.M., 1995. Understanding C�H BondOxidation: H � and H� transfer in the oxidation of tolueneby permanganate. Science 269, 1849�1851.
Gates, D.D., Siegrist, R.L., Cline, S.R., 1995. Chemical oxida-tion of contaminants in clay or sandy soil. J. Environ. Eng.121, 582�588.
Glover, D., Schumm Jr., B., Kazowa, A., 1989. In: Glover, D.,Ž .Schumm Jr., B., Kazowa, A. Eds. , Handbook of Man-
ganese Dioxides Battery Grade. International Battery Ma-Ž .terial Association IBA , Cleveland, OH, USA.
Hedges, J.I., Stern, J.H., 1984. Carbon and nitrogen determi-nations of carbonate-containing solids. Limnol. Oceanogr.29, 657�663.

( )K. Huang et al. � Ad�ances in En�ironmental Research 7 2002 217�229228
Hem, J.D., 1972. Chemical factors that influence the availabil-ity of iron and manganese in aqueous systems. Geol. Soc.Am. Bull. 83, 443�450.
Huang, K.C., Hoag, G.E., 2000. A study of oxidation ofchlorinated ethenes with permanganate in aqueous andporous media. Ph.D. Dissertation. The University of Con-necticut, Storrs, CT, USA.
Huang, K.C., Hoag, G.E., Chheda, P., Woody, B.A., Dobbs,G.M., 2000. A pilot scale study of oxidation of trichloroeth-ylene by sodium permanganate. Proceedings of the SecondInternational Conference on Remediation of Chlorinatedand Recalcitrant Compounds. Monterey, CA, USA, pp.145�152.
Huang, K.C., Hoag, G.E., Chheda, P., Woody, B.A., Dobbs,G.M., 1999. Kinetic study of oxidation of trichloroethyleneby potassium permanganate. Environ. Eng. Sci. 1116,265�274.
Levenspiel, O., 1972. Chemical Reaction Engineering. JohnWiley and Sons Inc, New York.
Li, X.D., Schwartz, F.W., 2000. Efficiency problems related topermanganate oxidation schemes. Proceedings of the Sec-ond International Conference on Remediation of Chlori-nated and Recalcitrant Compounds. Monterey, CA, USA,pp. 41�48.
Luo, J., Suib, S., 1997. Preparative parameters, magnesiumeffects, and anion effects in the crystallization of birnes-sites. J Phys. Chem. B. 101, 10403.
Luo, J., Zhang, Q., Suib, S.L., 2000. Mechanistic and kineticstudies of crystallization of birnessite. Inorg. Chem. 39,741�747.
Ma, J., Li, G., Graham, N.J.D., 1994. Efficiency and mecha-nism of acrylamide removal by permanganate oxidation. JWater SRT-Aqua 43, 287�293.
Ma, Y., Luo, J., Suib, S.L., 1999. Synthesis of birnessites usingalcohols as reducing reagents: effects of synthesis parame-ters on the formation of birnessites. Chem. Mater. 11,1972�1979.
Matsubara, H., Nakayama, S., 1992. Stability of premethylatedaromatic model compounds of constituents of humic subs-tances toward KMnO oxidation. Water Res. 26, 1471�1478.4
McKay, D.J., Stark, J.A,. Young, B.L., Govoni, J.W., Berini,C.M, Cronan, T.J., Hewitt, T.J., 2000. A field demonstra-tion of trichloroethylene oxidation using potassium per-manganate. Proceedings of the Second International Con-ference on Remediation of Chlorinated and RecalcitrantCompounds. Monterey, CA, USA, pp. 109�116.
McKenzie, R.M., 1989. Manganese oxides and hydroxides. In:Ž .Dixon, J.B., Weed, S.B. Eds. , Minerals in Soil Environ-
ments, 2nd ed. Soil Science Society of America, Madison,WI, USA, pp. 439�465.
Perkin-Elmer, 1988. 2400 CHN Elemental Analyzer, Instruc-Ž .tion Manual �0990-7147 . Perkin-Elmer Inc, CT, USA.
Pimienta, V., Lavabre, G., Levy, G., Micheau, J.C., 1994.Ž . Ž .Reactivity of the Mn III and Mn IV intermediates in the
permanganate�oxalic acid�sulfuric acid reaction: kineticdetermination of the reducing species. J. Phys. Chem. 98,13294�13299.
Potter, R.M., Rossman, G.R., 1979. The tetravalent man-ganese oxides: identification, hydration, and structural rela-
tionships by infrared spectroscopy. Am. Mineral. 64,1199�1218.
Reitsma, S., Marshall, M., 2000. Experimental study of oxida-tion of pooled NAPL. Proceedings of the Second Interna-tional Conference on Remediation of Chlorinated and Re-calcitrant Compounds. Monterey, CA, USA, pp. 25�32.
Satawathananont, A., Patrick Jr., W.H., Moore Jr., P.A., 1991.Effect of controlled redox conditions on metal solubility inacid sulfate soils. Plant and Soil 133, 281�290.
Schnarr, M., Truax, C., Farquhar, G., Hood, E., Gonulla, T.,Stickney, B., 1998. Laboratory and controlled field experi-ments using potassium permanganate to remediatetrichloroethylene and tetrachloroethylene DNAPLs inporous media. J. Contam. Hydrol. 29, 205�224.
Siegrist, R.L., Lowe, K.S., Murdoch, L.C., Case, T.L., Picker-ing, D.A., 1999. In situ oxidation by fracture emplacedreactive solids. J. Environ. Eng. 125, 429�440.
Stewart, R., 1964. Oxidation Mechanisms. W.A. Benjamin Inc,New York.
Ž .Stewart, R., 1965. In: Wiberg, K.B. Ed. , Oxidation in OrganicChemistry. Academic Press.
Taylor, S.M., Halpern, J., 1959. Kinetics of the permanganateoxidation of formic acid and formate ion in aqueous solu-tion. J Am. Chem. Soc. 81, 2933�2937.
Tratnyek, P.G., Johnson, T.M., Warner, S.D., Clarke, H.S.,and Baker, J.A., 1998. In situ treatment of organics bysequential reduction and oxidation. Proceedings of theFirst International Conference on Remediation of Chlori-nated and Recalcitrant Compounds. Cl-5, pp. 371�376.
Truax, C.T., 1993. Investigation of the in-situ potassium per-manganate oxidation of residual DNAPLs located belowthe groundwater table. M.S. Thesis. University of Waterloo,Ontario, Canada.
United States Environmental Protection Agency, 1980. SW-846Test Methods for Evaluating Solid Waste Physical�Chem-ical Methods. Office of Solid Waste.
Urynowicz, M.A., Siegrist, R.L., 2000. Chemical degradationof TCE DNAPL by Permanganate. Proceedings of theSecond International Conference on Remediation of Chlo-rinated and Recalcitrant Compounds. Monterey, CA, USA,pp. 75�82.
Vella, P.A., Veronda, B., 1992. Oxidation of trichloroethylene:a comparison of potassium permanganate and Fenton’sreagent. Chemical Oxidation: Technology for the Ninetiesin Proceedings of the Third International Symposium. PA,USA, pp. 62�73.
West, O.R., Cline, S.R., Holden, W.L. et al., 1997. A Full-ScaleDemonstration of In Situ Chemical Oxidation through Re-circulation at X-701b Site. Oak Ridge National Laboratory,Oak Ridge, TN.
Wiberg, K.B., Saegebarth, K.A., 1957. The mechanisms ofpermanganate oxidation. IV. Hydroxylation of olefins andrelated reactions. J Am. Chem. Soc. 79, 2822�2824.
Yan, Y.E., Schwartz, F.W., 1998. Oxidative degradation ofchlorinated ethylenes by potassium permanganate. J. Con-tam. Hydrol. 37, 343�365.
Yan, Y.E., Schwartz, F.W., 2000. Kinetics and mechanisms forTCE oxidation by permanganate. Environ. Sci. Technol. 34,2535�2541.

( )K. Huang et al. � Ad�ances in En�ironmental Research 7 2002 217�229 229
Dr Kun-Chang Huang, Engineering Laboratory Manager ofthe Environmental Research Institute of the University ofConnecticut, has been studying the destruction of toxic chemi-cals such as TCE, PCE, MTBE and PCBs using abioticoxidation methods for many years. He is also interested in sitecharacterization, risk assessment and remediation schemeplanning.
Dr George E. Hoag, Professor in the Environmental Engineer-ing Program and Director of the Environmental ResearchInstitute of the University of Connecticut, has focused on thefate and transport of organic chemicals in soil and groundwa-ter. His research interests also include developing innovativetechnologies for soil and groundwater clean-up.
Dr Pradeep Chheda is Project Manager at the EnvironmentalResearch Institute of the University of Connecticut. His re-
search interests include the investigation and development ofinnovative technology for remediating soil and groundwatercontaminated with chlorinated solvents and polychlorinatedbiphenyls using physical and chemical processes.
Mr Bernard A. Woody, Senior Scientist at the United Tech-nologies Research Center, holds an M.S. degree from theUniversity of Illinois and currently leads a group of scientistsand engineers developing and employing new techniques toclean contaminated soil and groundwater.
Dr Gregory M. Dobbs, Senior Consulting Scientist at the UnitedTechnologies Research Center, received his Ph.D. in physicalchemistry at Princeton University. He leads a research groupon the application of new technologies for protecting environ-mental resources.




















