
A High-Resolution Structure that Provides Insight into Coiled-Coil Thiodepsipeptide Dynamic...
18
Thiodepsipeptides DOI: 10.1002/anie.201303900 A High-Resolution Structure that Provides Insight into Coiled-Coil Thiodepsipeptide Dynamic Chemistry** Zehavit Dadon, Manickasundaram Samiappan, Anat Shahar, Raz Zarivach, and Gonen Ashkenasy* Chemical synthesis and cell-based expression methods can afford incorporation of non-natural entities into proteins. New structures are obtained, and new functions gained, by attachment of amino acids with non-native side-chains, [1, 2] modified backbones such as b- and g-amino acids, [3–5] or peptide bond analogues such as peptoids, esters, and thio- esters. [6–9] Backbone modification with longer and flexible amino acids allows expansion of the conformational space occupied by proteins, while introduction of ester (depsipep- tide) and thioester (thiodepsipeptide) bonds enhances their reactivity towards hydrolysis and other nucleophilic attacks. The durability of thioester bonds in neutral aqueous solutions and their reactivity in thiol–thioester exchange reactions make them a relevant choice for performing dynamic chemistry in water. [10–15] Particularly interesting is the possi- bility of utilizing such transformations for exchanging domains between different protein molecules, owing to sequence mutations or in response to chemical and physical changes. Self-organization of molecular networks has been exten- sively studied by scientists interested in systems chemis- try. [16, 17] When studying protein-based networks, it was demonstrated that the network connectivity and overall topology can be dictated by the sequence-specific information embedded in coiled-coil architectures, [18–20] and that careful design of the interhelical recognition interface can be used to affect the network in a predictable manner. [14, 20, 21] It is suggested here that the adaptive behavior of such networks, namely their rewiring in response to external triggers, can be greatly expanded if the coiled-coil proteins are formed within dynamic networks in which domain exchange readily takes place. Towards this end, we utilize coiled-coil protein analogues that contain thioester bonds within their sequences. We predict that to be mechanistically relevant for domain exchange, the thioester bond should be isostructural with the peptide bonds to maintain the 3D structure, and it should also be kept exposed and reactive towards small-molecule thiols and/or thiol-containing proteins. To highlight these character- istics, we provide here the first high-resolution structure (1.35 ĸ) of a thioester coiled-coil protein, and compare it to the structure of the native and depsipeptide analogue proteins. The integrity of the thioester bonds and their accessibility to other molecules are revealed by analysis of the crystal structure, as well as from complementary thiol- exchange assays. We then show using a set of mutants that the thioester stability can be correlated with the backbone regularity and the coiled-coil unfolding stability. Finally, a small library formed of these thioester mutants is screened for domain exchange in the absence and presence of an external template molecule, revealing significant template effect and exchange-product amplification. The sequence of the key thioester peptide (1 t ) was designed by replacing a glycine residue (G18) in a previously studied (GCN4-based) coiled-coil peptide 1 [21] with a thiogly- colic acid (Figure 1). Peptides 1, 1 t , and their ester analogue 1 e , containing a glycolic acid in position 18, were synthesized and purified (Supporting Information, Figures S1 and S2). Figure 1. a) Peptide-bond analogues and b) peptide sequences. Ar = 4- acetamidobenzoate, Z =-SCH 2 CO, Z b =-SCH 2 CH 2 CO, B =-OCH 2 CO, X = Lys-Ar. 1, 1 t , and 1 e sequences are identical, except for having different peptide-bond analogues at position 18. Minor mutations introduced to non-informal solvent-exposed positions of 1 t b and 1 t E13A , relative to 1 t sequence, to facilitate better HPLC separation in the network experiments (Figure 4). [*] Z. Dadon, Dr. M. Samiappan, Prof. G. Ashkenasy Department of Chemistry, Ben Gurion University of the Negev Beer Sheva, 84105 (Israel) E-mail: [email protected] Prof. G. Ashkenasy Ilse Katz Institute for Nanoscale Science and Technology Ben Gurion University of the Negev Beer Sheva, 84105 (Israel) Dr. R. Zarivach Department of Life Sciences, Ben Gurion University of the Negev Beer Sheva, 84105 (Israel) Dr. A. Shahar, Dr. R. Zarivach Institute of Biotechnology in the Negev Ben Gurion University of the Negev Beer Sheva, 84105 (Israel) [**] This research was supported by the European Research Council (ERC 259204). We thank Vered Zavaro for assistance in early stages of the project, and Dr. Rivka Cohen-Luria for help in the lab. Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201303900. . Angewandte Communications 9944 # 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. Int. Ed. 2013, 52, 9944 –9947
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of A High-Resolution Structure that Provides Insight into Coiled-Coil Thiodepsipeptide Dynamic...

ThiodepsipeptidesDOI: 10.1002/anie.201303900
A High-Resolution Structure that Provides Insight into Coiled-CoilThiodepsipeptide Dynamic Chemistry**Zehavit Dadon, Manickasundaram Samiappan, Anat Shahar, Raz Zarivach, andGonen Ashkenasy*
Chemical synthesis and cell-based expression methods canafford incorporation of non-natural entities into proteins.New structures are obtained, and new functions gained, byattachment of amino acids with non-native side-chains,[1, 2]
modified backbones such as b- and g-amino acids,[3–5] orpeptide bond analogues such as peptoids, esters, and thio-esters.[6–9] Backbone modification with longer and flexibleamino acids allows expansion of the conformational spaceoccupied by proteins, while introduction of ester (depsipep-tide) and thioester (thiodepsipeptide) bonds enhances theirreactivity towards hydrolysis and other nucleophilic attacks.The durability of thioester bonds in neutral aqueous solutionsand their reactivity in thiol–thioester exchange reactionsmake them a relevant choice for performing dynamicchemistry in water.[10–15] Particularly interesting is the possi-bility of utilizing such transformations for exchangingdomains between different protein molecules, owing tosequence mutations or in response to chemical and physicalchanges.
Self-organization of molecular networks has been exten-sively studied by scientists interested in systems chemis-try.[16, 17] When studying protein-based networks, it wasdemonstrated that the network connectivity and overalltopology can be dictated by the sequence-specific informationembedded in coiled-coil architectures,[18–20] and that carefuldesign of the interhelical recognition interface can be used toaffect the network in a predictable manner.[14, 20, 21] It issuggested here that the adaptive behavior of such networks,namely their rewiring in response to external triggers, can be
greatly expanded if the coiled-coil proteins are formed withindynamic networks in which domain exchange readily takesplace. Towards this end, we utilize coiled-coil proteinanalogues that contain thioester bonds within their sequences.We predict that to be mechanistically relevant for domainexchange, the thioester bond should be isostructural with thepeptide bonds to maintain the 3D structure, and it should alsobe kept exposed and reactive towards small-molecule thiolsand/or thiol-containing proteins. To highlight these character-istics, we provide here the first high-resolution structure(1.35 �) of a thioester coiled-coil protein, and compare it tothe structure of the native and depsipeptide analogueproteins. The integrity of the thioester bonds and theiraccessibility to other molecules are revealed by analysis of thecrystal structure, as well as from complementary thiol-exchange assays. We then show using a set of mutants thatthe thioester stability can be correlated with the backboneregularity and the coiled-coil unfolding stability. Finally,a small library formed of these thioester mutants is screenedfor domain exchange in the absence and presence of anexternal template molecule, revealing significant templateeffect and exchange-product amplification.
The sequence of the key thioester peptide (1t) wasdesigned by replacing a glycine residue (G18) in a previouslystudied (GCN4-based) coiled-coil peptide 1[21] with a thiogly-colic acid (Figure 1). Peptides 1, 1t, and their ester analogue1e, containing a glycolic acid in position 18, were synthesizedand purified (Supporting Information, Figures S1 and S2).
Figure 1. a) Peptide-bond analogues and b) peptide sequences. Ar=4-acetamidobenzoate, Z =-SCH2CO, Zb =-SCH2CH2CO, B =-OCH2CO,X = Lys-Ar. 1, 1t, and 1e sequences are identical, except for havingdifferent peptide-bond analogues at position 18. Minor mutationsintroduced to non-informal solvent-exposed positions of 1t
b and 1tE13A,
relative to 1t sequence, to facilitate better HPLC separation in thenetwork experiments (Figure 4).
[*] Z. Dadon, Dr. M. Samiappan, Prof. G. AshkenasyDepartment of Chemistry, Ben Gurion University of the NegevBeer Sheva, 84105 (Israel)E-mail: [email protected]
Prof. G. AshkenasyIlse Katz Institute for Nanoscale Science and TechnologyBen Gurion University of the NegevBeer Sheva, 84105 (Israel)
Dr. R. ZarivachDepartment of Life Sciences, Ben Gurion University of the NegevBeer Sheva, 84105 (Israel)
Dr. A. Shahar, Dr. R. ZarivachInstitute of Biotechnology in the NegevBen Gurion University of the NegevBeer Sheva, 84105 (Israel)
[**] This research was supported by the European Research Council(ERC 259204). We thank Vered Zavaro for assistance in early stagesof the project, and Dr. Rivka Cohen-Luria for help in the lab.
Supporting information for this article is available on the WWWunder http://dx.doi.org/10.1002/anie.201303900.
.AngewandteCommunications
9944 � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. Int. Ed. 2013, 52, 9944 –9947

The coiled-coil fold characteristics and the three-dimen-sional structures of these analogues were analyzed andcompared. CD measurements (Supporting Information, Fig-ure S3) showed the typical coiled-coil spectrum for all three ofthe compounds, albeit with high helix content for 1 (87 %),and somewhat compromised helicity for 1t and 1e (67 % and68% helix, respectively). Peptides 1, 1t, and 1e were thencrystalized from circa 10 mg mL�1 solutions, under similarcrystallization conditions, and into the same space group(P212121). The crystal structures were solved to 2.1, 1.35, and1.40 �, respectively (Supporting Information, Table S1). Allthree peptides assembled into trimer structures of paralleland highly regular a-helices (Figure 2; Supporting Informa-
tion, Figures S4 and S5). Interestingly, we have not observeddimeric coiled-coils, even though a close variant of 1, alsocontaining valine and leucine residues in the hydrophobiccore, was crystalized earlier in both the dimer and trimerforms.[22] The effect of incorporating a thioester into thebackbone of 1t is further highlighted by crystal structurealignment with 1. Only minimal differences were observedalong the entire structure (RMSD = O.37 �), except for somedeformation of 1t structure around the thioester bond (Fig-ure 2b).
Analysis of the high-resolution structure of 1t allowsdetailed characterization of the thioester bond, formedbetween the thioglycolic acid sulfur atom and Ala 17 carbonylcarbon, and its environment, including assignment of bondlengths (Figure 3a; Supporting Information, Figure S6) andidentification of close-by side-chains (Figure 3b; SupportingInformation, Figure S5 b) that are likely to interfere withincoming molecules during the thiol–thioester exchange
reactions. For example, the loss of one hydrogen bond perchain in 1t and 1e (versus 1) can be directly deduced from thecrystal structures. These reveal 0.2–0.4 � longer distancesbetween the thioester S or ester O atoms and the K14carbonyl oxygen atom (Supporting Information, Figure S6).GnHCl-dependent denaturation experiments have shownthat the coiled-coil structure of 1t is less stable than that of1 by 1.1 kcalmol�1,[14] namely implying about 0.4 kcalmol�1
structure destabilization for each thioester bond. The rela-tively minor destabilizing effect is similar to that observedearlier for removing a solvent exposed hydrogen bond,[7] andcan be explained by water molecules solvation of the sulfur(and oxygen) atoms, which reduces repulsion between themand the K14 carbonyl oxygen lone pair.[7]
Figure 3b shows that the thioester bond is solvent-exposed, but it is also engulfed by the side-chains of residuesK15, X21 (X = aryl-modified Lys; Figure 1), and E22 of thesame chain, and E20 from the opposing helix. The regular
Figure 2. a) Side view of the crystal structure of 1t ; the yellow ribbonmarks the thioester thiol position. b) Crystal structure alignment of 1t
(marine blue) and 1 (orange), showing almost identical structures withsome deformation around the thioester site of 1t. The crystal structureof 1e, additional views of 1t structure, and crystallization procedure,conditions, and collected data are given in Supporting Information.
Figure 3. Close-up view of the thioester site of 1t. a) Bond andinteratom distances in the thioester vicinity. The bond and interatomdistances found in the other two chains of 1t and in the structures of1 and 1e are given in the Supporting Information, Figure S6. b) Stickmodel highlighting the side-chains that engulf the thioester and mayinterfere with reacting thiol molecules.
AngewandteChemie
9945Angew. Chem. Int. Ed. 2013, 52, 9944 –9947 � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org

helix structure of the thiodepsipeptide coiled-coils and thisshielding of the thioester bond render 1t stable againsthydrolysis and thiol-dependent decomposition. Indeed, thethioester was only hydrolyzed to a limited extent (ca. 5%)during two days in pH 7 aqueous solution. We have furtherevaluated the decomposition of 1t in the presence of molarexcess of external small-molecule thiols (S), to yield a thio-ester (E) and n-terminal thioglycolic (N) fragments(Scheme 1). Equilibration of 1t (100 mm) in the presence of
the alkyl thiol 2-mercapto ethyl sulfonate (1.0 mm) in pH 7showed some decomposition (Supporting Information, Fig-ure S7), revealing an equilibrium constant Keq = ([E] � [N]/[1t] � [S]) (Scheme 1) of 1.5 � 10�3. The same experiments inunfolding conditions (3m GnHCl) resulted in fast andefficient decomposition into E and N (Keq = 2.1) and moresignificant hydrolysis. Furthermore, shielding of the thioesterwas evidenced from equilibration in the presence of tert-butylthiol (1.0 mm) that resulted in slow decomposition forwhich no equilibrium could be reached after 2 days.
In recent studies, Gellman, Woolfson, and their co-workers have shown that the extent of thioester formationin loop positions of helix–loop–helix structures can becorrelated with the stability of the coiled-coils.[23–26] Toprobe whether such correlation exists for thioesters locatedwithin the helix itself, we have related the unfolding stabilityof 1t and its mutants (1t
E13A, 1tE13K, and 1t
b ; Figure 1) to theirequilibrium in thiol-dependent decomposition (Scheme 1).1t
E13A and 1tE13K mutants consist of Ala and Lys residues,
respectively, in place of Glu13, which constitutes part of thehelix–helix interaction interface. These mutations areexpected to have minor (1t
E13A) and considerable (1tE13K)
destabilizing effects on the formed structures, relative to 1t.Peptide 1t
b consists of 3-mercaptopropanoic acid in posi-tion 18 readily incorporated to form a thioester bond with anextra backbone methylene as a b-amino acid analogue(Figure 1a). The CD measurements (Supporting Information,Figure S3) showed that 1t
E13A forms a stable coiled-coilstructure, with a helix content similar to 1t (63 %); 1t
b canalso form coiled-coils but with lower helix content (51%), and1t
E13K cannot form coiled-coils under the studied conditions.This unfolding stability trend was found indeed to correlatewell with the thiol-dependent decomposition, where we foundKeq values of 3 � 10�3, 4.2 � 10�3, and 1.0 for 1t
E13A, 1tb, and
1tE13K, respectively.
The reversible thiol–thioester exchange reactions candrive domain replacement between proteins in response to
environmental changes, such as changes in folding/denatura-tion conditions and/or introduction of external templatemolecules. Dynamic exchange reactions have been describedbefore[27] for small molecules[27] or libraries of short pep-tides.[15,28–31] We have followed such a process within a smalldynamic library made of the four thiodepsipeptides 1t, 1t
E13A,1t
E13K, and 1tb (95 mm each). These were equilibrated together
in presence of their respective electrophilic fragments, E(200 mm), EE13A (100 mm), and EE13K (100 mm), and excess of 2-mercaptoethyl sulfonate (Figure 4; Supporting Information,
Figure S8). The effect of external template on domainexchange was evaluated when the mixture was seeded withpeptide 2 (Figure 1) that forms stable coiled-coils with themost unstable thioester 1t
E13K.[14] The concentrations of thefour peptides at equilibrium (t� 30 h) were calculated fromHPLC chromatograms of the complex mixture (Figure 4 b;Supporting Information, Figure S8 a). Figure 4c shows that inthe absence of template, the library equilibrated such that1t
E13K concentration decreased significantly (�80%), while1t
E13A and 1tb concentrations stayed almost unchanged and the
concentration of 1t almost doubled. This is explained bydegradation of 1t
E13K, which produces N molecules that then
Scheme 1. Equilibration of a thiodepsipeptide in the presence ofexcess thiol molecule (S) to yield a thioester (E) and thiol-containing(N) fragments.
Figure 4. Network equilibration experiments. a) Network reaction for-mula. b) Representative HPLC chromatogram that was used to followthe network equilibration. c) Measured concentrations (mm) of thio-depsipeptides 1t, 1t
E13A, 1tE13K, and 1t
b within the network mediumbefore equilibration (gray), after equilibration without any externaltemplate (blue), or after equilibration in the presence of peptide 2 asa template (red). d) Calculated amplification factors, obtained bydividing the concentration of a thiodepsipeptide in template-seededreaction by its concentration in the template-free reaction. Experimen-tal details and further characterization of the network experiments aregiven in Supporting Information.
.AngewandteCommunications
9946 www.angewandte.org � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. Int. Ed. 2013, 52, 9944 –9947

react with E to form primarily peptide 1t, leading to formationof the more stable 1t-containing coiled-coil structures. Whenthe reaction was re-equilibrated in the presence of 2, theproduct distribution was significantly different, revealingdegradation of 1t
E13K to a much lower extent and accordinglylower formation of 1t. The template amplification effect onproduct distribution in the library is manifested in Figure 4d,showing exclusively the amplification of 1t
E13K (amplificationfactor = 4.4), no meaningful effect on 1t
E13A and 1tb concen-
trations, and an amplification effect of less than unity as anindirect result of lower degradation of 1t
E13K.In summary, we have shown that detailed characterization
of the thioester bond integrity and its environment can bevaluable for explaining and predicting dynamic behavior ofthioester peptides and proteins. While the coiled-coil motif isvery abundant in nature (appearing in about 5% of allproteins), and as ester and thioester bonds are frequently usedby chemists in research and industry, it was interesting to findout that no high-resolution structure was available for coiled-coil depsipeptides or thiodepsipeptides. We suggest that thethiol–thioester-driven exchange in our model can be relevantfor studying the dynamic chemistry of other technologicallyand pharmaceutically important proteins.
Received: May 7, 2013Revised: June 16, 2013Published online: August 8, 2013
.Keywords: coiled coils · depsipeptides · dynamic chemistry ·peptide networks · thiodepsipeptides
[1] C. C. Liu, P. G. Schultz, Annu. Rev. Biochem. 2010, 79, 413 – 444.[2] Z. Hao, S. Hong, X. Chen, P. R. Chen, Acc. Chem. Res. 2011, 44,
742 – 751.[3] S. H. Gellman, Acc. Chem. Res. 1998, 31, 173 – 180.[4] W. S. Horne, S. H. Gellman, Acc. Chem. Res. 2008, 41, 1399 –
1408.[5] W. S. Horne, Expert Opin. Drug Discovery 2011, 6, 1247 – 1262.[6] E. T. Powers, S. Deechongkit, J. W. Kelly, Adv. Protein Chem.
2006, 59, 39 – 78.[7] J. A. Scheike, C. Baldauf, J. Spengler, F. Albericio, M. T.
Pisabarro, B. Koksch, Angew. Chem. 2007, 119, 7912 – 7916;Angew. Chem. Int. Ed. 2007, 46, 7766 – 7769.
[8] J. Gao, D. A. Bosco, E. T. Powers, J. W. Kelly, Nat. Struct. Mol.Biol. 2009, 16, 684 – 690.
[9] A. Choudhary, R. T. Raines, ChemBioChem 2011, 12, 1801 –1807.
[10] M. G. Woll, S. H. Gellman, J. Am. Chem. Soc. 2004, 126, 11172 –11174.
[11] A. Koglin, C. T. Walsh, Nat. Prod. Rep. 2009, 26, 987 – 1000.[12] Y. Ura, J. M. Beierle, L. J. Leman, L. E. Orgel, M. R. Ghadiri,
Science 2009, 325, 73 – 77.[13] Y. Ura, M. Al-Sayah, J. Montenegro, J. M. Beierle, L. J. Leman,
M. R. Ghadiri, Org. Biomol. Chem. 2009, 7, 2878 – 2884.[14] Z. Dadon, M. Samiappan, N. Wagner, G. Ashkenasy, Chem.
Commun. 2012, 48, 1419 – 1421.[15] S. Ghosh, L. A. Ingerman, A. G. Frye, S. J. Lee, M. R. Gagne,
M. L. Waters, Org. Lett. 2010, 12, 1860 – 1863.[16] R. F. Ludlow, S. Otto, Chem. Soc. Rev. 2008, 37, 101 – 108.[17] Z. Dadon, N. Wagner, G. Ashkenasy, Angew. Chem. 2008, 120,
6221 – 6230; Angew. Chem. Int. Ed. 2008, 47, 6128 – 6136.[18] S. Yao, I. Ghosh, R. Zutshi, J. Chmielewski, Nature 1998, 396,
447 – 450.[19] A. Saghatelian, Y. Yokobayashi, K. Soltani, M. R. Ghadiri,
Nature 2001, 409, 797 – 801.[20] G. Ashkenasy, R. Jagasia, M. Yadav, M. R. Ghadiri, Proc. Natl.
Acad. Sci. USA 2004, 101, 10872 – 10877.[21] Z. Dadon, M. Samiappan, E. Y. Safranchik, G. Ashkenasy,
Chem. Eur. J. 2010, 16, 12096 – 12099.[22] P. B. Harbury, T. Zhang, P. S. Kim, T. Alber, Science 1993, 262,
1401 – 1407.[23] J. L. Price, E. B. Hadley, J. D. Steinkruger, S. H. Gellman,
Angew. Chem. 2010, 122, 378 – 381; Angew. Chem. Int. Ed.2010, 49, 368 – 371.
[24] E. B. Hadley, O. D. Testa, D. N. Woolfson, S. H. Gellman, Proc.Natl. Acad. Sci. USA 2008, 105, 530 – 535.
[25] J. D. Steinkruger, D. N. Woolfson, S. H. Gellman, J. Am. Chem.Soc. 2010, 132, 7586 – 7588.
[26] J. D. Steinkruger, G. J. Bartlett, D. N. Woolfson, S. H. Gellman,J. Am. Chem. Soc. 2012, 134, 15652 – 15655.
[27] P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor,J. K. M. Sanders, S. Otto, Chem. Rev. 2006, 106, 3652 – 3711.
[28] R. L. E. Furlan, Y.-F. Ng, S. Otto, J. K. M. Sanders, J. Am. Chem.Soc. 2001, 123, 8876 – 8877.
[29] M. Rauschenberg, S. Bomke, U. Karst, B. J. Ravoo, Angew.Chem. 2010, 122, 7498 – 7503; Angew. Chem. Int. Ed. 2010, 49,7340 – 7345.
[30] J. M. A. Carnall, C. A. Waudby, A. M. Belenguer, M. C. A.Stuart, J. J. P. Peyralans, S. Otto, Science 2010, 327, 1502 – 1506.
[31] J. Atcher, A. Moure, I. Alfonso, Chem. Commun. 2013, 49, 487 –489.
AngewandteChemie
9947Angew. Chem. Int. Ed. 2013, 52, 9944 –9947 � 2013 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.org

Supporting Information
� Wiley-VCH 2013
69451 Weinheim, Germany
A High-Resolution Structure that Provides Insight into Coiled-CoilThiodepsipeptide Dynamic Chemistry**Zehavit Dadon, Manickasundaram Samiappan, Anat Shahar, Raz Zarivach, andGonen Ashkenasy*
anie_201303900_sm_miscellaneous_information.pdf

High Resolution Structure Provides Insight into Coiled Coil Thiodepsipeptides
Dynamic Chemistry
Zehavit Dadon, Manickasundaram Samiappan, Anat Shahar, Raz Zarivach and Gonen Ashkenasy*
Ben Gurion University of the Negev Beer Sheva, Israel.
*Corresponding author: G. Ashkenasy. Email: [email protected]
s1. Experimental Section 1. Peptides synthesis and characterization
The nucleophilic peptides N and Nβ were synthesized on Rink-Amide MBHA resin using standard Fmoc-
based chemistry, and their thiolated n-terminus residues - thioglycolic acid and 3-mercaptopropionic acid,
respectively - were introduced by attachment of the Trt-protected versions in the last coupling step.
Cleavage and global deprotection were achieved using the standard cocktail for Fmoc chemistry (95%
TFA). Peptides 1 and 2 were synthesized using the same method.
Thioester peptides E, EE13K and EE13A were synthesized on MBHA resin using a modified Boc SPPS
method, in which 3-triphenylmethylthiopropionic acid (3 equivalents relative to resin loading) in
dimethylformamide (DMF) was first coupled to the resin, using HBTU as coupling reagent (3 eq.) and
DIPEA as base (20 eq.). The Trt group was removed using a TFA:TIS mixture (95:5; 2x5 min). The
synthesis continued using the standard Boc-chemistry procedure, and the crude peptide was obtained after
cleavage and global deprotection with a TFMSA/TFA mixture.
Thioester peptides 1t, 1tE13K, 1t
E13A, and 1tβ were synthesized via thiol-thioester exchange reactions. The
respective electrophile peptide (3-5 mM; 1 eq.) and the thiol nucleophilic peptide (1.2 eq.) were dissolved
in 3-(N-morpholino) propanesulfonic acid (MOPS) buffer at pH ~7.5, containing tris(2-
carboxyethyl)phosphine hydrochloride (TCEP) as reducing agent. The reaction was allowed to proceed at
37°C for 4 - 8 hours, until quenched by TFA, and subjected to purification by HPLC (Fig. s1). Ligation
yields ranged between ~60% for producing 1tβ and up to almost quantitative yield (≥ 90%) for 1t and
1tE13A. Note that minor mutations were introduced to solvent-exposed positions of 1t
β and 1tE13A, relative to
1t sequence, in order to facilitate better HPLC separation in the dynamic chemistry experiments (see
Section 4). It has been shown earlier[1,2] that mutations to these non-informative positions imply negligible
effect on the coiled coils stbility.

The ester peptide 1e was synthesized on MBHA resin using standard Boc SPPS method, in which glycolic
acid (10 eq. relative to resin loading) in DMF:DCM (1:1) was coupled to the growing peptide chain, in the
presence of DIC (20 eq.), HOBT (20 eq.) and N-ethylmorpholine (NEM) as base (4 eq.). This reaction
step proceeded to completion in 10 minutes, as judged by standard ninhydrin assay. The coupling of the
following BOC-protected amino acid along the peptide (Ala) was carried out for 60 minutes in
DMF/DCM (1:1) in the presence of DIC (20 eq.), NEM (8 eq.) and catalytic amount of DMAP (0.04 eq.).
The synthesis then continued using the standard Boc-chemistry procedure, and the crude peptide was
obtained after cleavage and global deprotection with HBr/TFA mixture (Fig. s2).
All peptides were purified by preparative HPLC using a C18 reverse phase column (Dionex 1100) with a
step gradient of solvent A (99% water, 1% acetonitrile (ACN), 0.1% TFA) and B (90% ACN, 10% water,
0.07% TFA). The identity and purity of the peptides were analyzed by analytical HPLC (with the same
solvent system for elution) and LCMS. Molecular weights (Mw) observed for all peptides were no more
than ±2 off the calculated Mw. Only peptides of 95% purity or higher were used for further structure and
function analysis.
2. Circular dichroism (CD) measurements
Stock solutions (0.7 - 1.0 mM) were prepared by weighing lyophilized peptides into Eppendorf tubes, and
then further diluting by MOPS buffer (pH 7.4) to result in the desired concentration for analysis (typically
20 µM). The exact concentration of each peptide solution was determined from its UV absorbance at 270
nm, based on the known absorbance of 4-acetamidobenzoic acid (ABA). Measurements were carried out
on a Jasco-815 CD spectropolarimeter, at 25 °C, by using a quartz cell with 1.0 mm path length. CD
spectra were obtained as the average of three scans and collecting data at 1 nm intervals from 260 to 200
nm. The CD spectrum resulting from buffer was subtracted from the spectrum of each peptide solution.
Data was converted to ellipticity (Ө in deg•cm2•dmol-1) according to the equation: [Ө] = Ψ•100 / (nlc),
where Ψ is the CD signal in degrees, n is the number of peptide bonds, l is the path length in centimeters,
and c is the concentration in decimoles per cm3. The helix content in each peptide solution was calculated
using the program provided with the CD instrument.
3. Thiol dependent degradation experiments
Stock solutions of peptides (1t, 1tE13K, 1t
E13A, 1tβ and 1e) were prepared by weighing lyphoilized peptides
into Eppendorf tubes and dissolving in Millipore water to yield 0.5 – 1 mM solutions. Experiments were
performed by mixing aqueous solutions containing each of these peptides separately, TCEP (2 mM) as
reducing agent, ABA (50 µM) as internal standard, and the small-molecule thiol 2-

mercaptoethanesulfonate sodium salt (RSH; 1.0 mM). The mixtures were allowed to equilibrate for 30
minutes under the acidic conditions. After that, the equilibration reactions were initiated by the addition of
MOPS buffer at pH = 7.4, yielding a total volume of 100 µL. Aliquots (10 µL) were removed at various
time points over 48 h, immediately quenched in 5% TFA in water, and stored frozen until subjected to RP-
HPLC analysis. The equilibrium constants were calculated from final concentrations (at t = 28 h; Fig. s7)
of the full length peptide and its respective E and N and fragments. The calculated values were compared
to the equilibrium constants obtained when a higher concentration of 1t (150 µM) allowed to equilibrate,
and to values obtained for equilibration in opposite direction, namely in reactions leading to formation of
1t from E and N fragments. For all cases, the observed equilibrium constants were found to be very
similar (± 10%) to those reported in the manuscript. Equilibration of peptide 1e practically showed no
decomposition over 24 h.
4. Dynamic network equilibration experiments
Experiments were prepared by mixing aqueous solutions of equimolar amounts of the thioester full length
peptides 1t, 1tE13K, 1t
E13A, and 1tβ (95±10 µM each) with E (200 µM), EE13K (100 µM) and EE13A (100±10
µM), TCEP (2 mM), ABA (50 µM) and 2-mercaptoethanesulfonate sodium salt (1 mM). The reactions
were allowed to equilibrate for 30 minutes, and then initiated by the addition of MOPS buffer at pH = 7.4,
yielding a total volume of 100 µL. Aliquots (10 µL) were removed at various time points, immediately
quenched in 5% TFA in water, and stored frozen until subjected to RP-HPLC analysis (see for example
chromatograms in Fig. 4 and Fig. s8a). The system reached equilibrium after 30 h (Fig. s8). The template
induced equilibration experiments were done with the same solution seeded from the beginning with 50
µM of peptide 2. When the same experiment was repeated by seeding with peptide 2 three hours after
starting equilibration, the system converged to the same products distribution.
5. Crystallization and structure determination of 1, 1t and 1e
Crystallization of peptides 1 and 1t was performed using a 24-well plate using the sitting drop vapor
diffusion method at 293K. Crystals were obtained after 12 hours by mixing 1/0.5 µl of 6 mg/ml 1/1t
sample in 20mM MOPS pH 7.0 with an equal crystallization solution volume containing 50% Tacsimate
pH 7.0 /2.2M Sodium Acetate respectively. For data collection, crystals were incubated briefly in a
solution containing 60% of the crystallization condition, 25% Ethylene Glycol and 15% ultrapure water
prior to flash freezing in liquid nitrogen.

Crystallization of peptide 1e was performed on a 96-well plate using the sitting drop vapor diffusion
method at 293K. Crystals were obtained after 12 hours by mixing 0.3 µl of 5 mg/ml sample in 20 mM
MOPS pH 7.0 with an equal volume of crystallization solution containing 54% Tacsimate pH 7.0. For data
collection, crystals were incubated briefly in a solution containing 60% of the crystallization condition,
25% Ethylene Glycol and 15% ultrapure water prior to flash freezing in liquid nitrogen.
The data were indexed, integrated and scaled using the HKL2000 program suite. The structures were
determined by molecular replacement using Phaser[3] (CCP4 suite) with the coordinates of PDB entries
1IJ2 & 1IJ3. Refinement was performed using REFMAC program[4] (CCP4 suite).
The research leading to these results has received funding from the European Community's Seventh
Framework Programme (FP7/2007-2013) under BioStruct-X (grant agreement No. 283570). In addition,
we thank the European Synchrotron Radiation Facility (ESRF, Grenoble, France) beamline ID 23-1 and
Diamond Light Source (DLS) beamline I04 for their assistance during data collection.

s2. Additional tables and figures
Table s1: Data collection and refinement statistic
PDB code 3W8V (1)
3W92 (1t)
3W93 (1e)
Data collection X-ray source Rigaku RU-H3RHB ID23-1 (ESRF) I04 (Diamond) Space group P212121 P212121 P212121 Cell dimensions a,b,c (Å) 31.168, 34.162, 95.067 31.760, 33.552, 94.998 31.722, 33.881, 95.179 α,β,γ (°) 90.000, 90.000, 90.000 90.000, 90.000, 90.000 90.000, 90.000, 90.000 Wavelength (Å) 1.5418 0.9724 0.9795 Resolution (Å) 2.1 1.35 1.40 Completeness (%) 99.1 (90.7) 99.8 (99.0) 97.4 (96.8) Total observations 190,154 1,070,719 121,981 Rmerge 10.6 (41.6) 8.2 (87.6) 5.3 (25.6) I/σI 34.34 (4.5) 74.3 (3.8) 31.68 (4.92) Redundancy 10.2 (6.9) 24.8 (20.1) 3 (2.9) Refinment Resolution range(Å) 47.53-2.1 47.5-1.35 47.59-1.5 Rwork/Rfree 19.2/28.8 16.4/19 21.28/24.35 No. atoms Protein 821 900 789 Waters 72 114 77 Average B factors (Å2)
Protein 15.515 16.849 13.49 Waters 17.712 25.393 19.385 R.m.s deviation Bond lengths (Å) 0.02 0.025 0.029 Bond angles (°) 2.093 2.406 2.415
* Values in parentheses refer to data in the highest resolution shell.
* All residues are in the allowed regions of Ramachandran plot.

Figure S1: Characterization of the thiodepsipeptides syntheses, from their electrophile (E) and nucleophile (N) fragments, by HPLC analysis of the ligation crude mixtures (left panels) and LCMS of the purified products (right).

Figure s2: Characterization of depsipeptide 1e synthesis. a) HPLC analysis of the crude mixture. b) HPLC and LCMS analysis of purified 1e product.
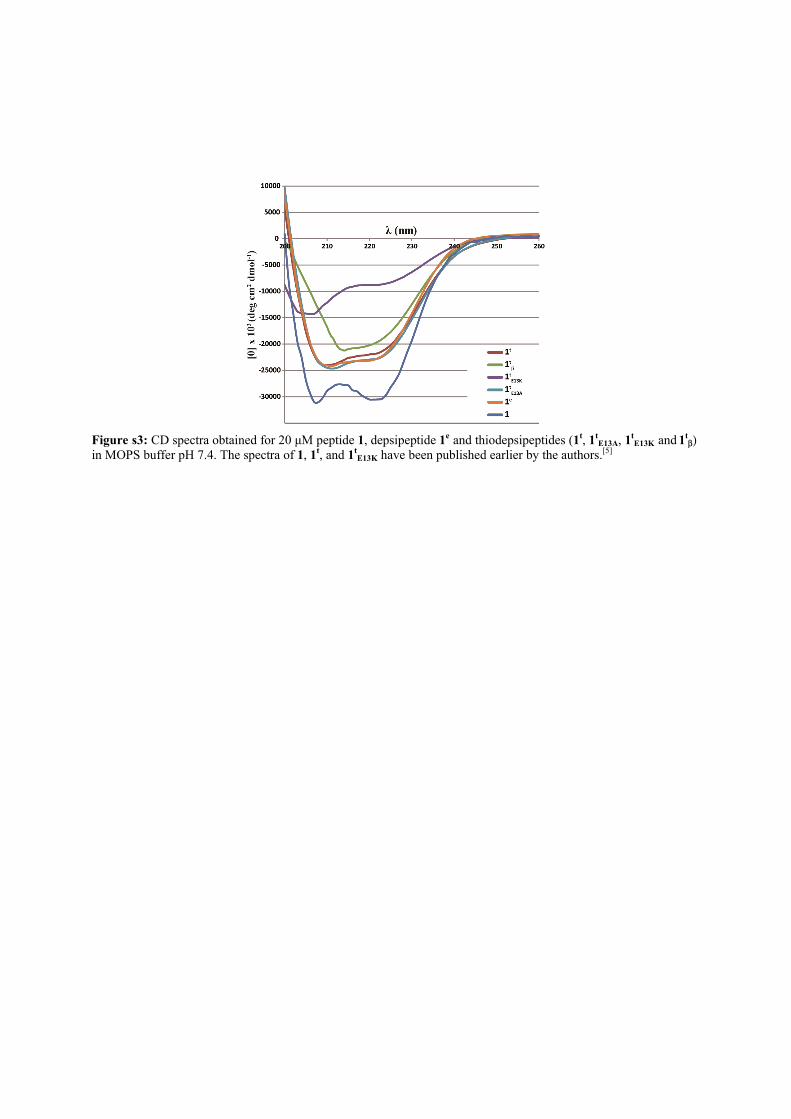
Figure s3: CD spectra obtained for 20 µM peptide 1, depsipeptide 1e and thiodepsipeptides (1t, 1t
E13A, 1tE13K and 1t
β) in MOPS buffer pH 7.4. The spectra of 1, 1t, and 1t
E13K have been published earlier by the authors.[5]

Figure s4: Side view crystal structures of peptides 1 and depsipeptide 1e.

Figure s5: Top view crystal structure of 1t (a), and side view stick model highlighting the peptide side chains (b).

Figure s6: Close-up views highlighting bond and inter-atom lengths, for each individual helix chain, in vicinity of position 18 occupied by a thioglycolic acid (1t; top row), glycine (1; middle) and glycolic acid (1e; bottom). The lengths assignment follows the a,b,c markers at the top left panel (1t chain a).

Figure s7: Analysis of the thiol dependent decomposition of 1t. a) Example of HPLC chromatogram (270 nm), showing 1t and its decomposition products (E and N). * Denotes disulfide dimer of N. b) Kinetic profile of 1t decomposition. Reaction conditions are described in detail in the Experimental Section.

Figure s8: Analysis of the equilibration of a library made of the four thiodepsipeptides 1t, 1t
E13A, 1tE13K and 1t
β. These thiodepsipeptides were mixed in equal amounts (95±10 µM each), and equilibrated in presence of their respective electrophilic fragments, E (200 µM), EE13A (100 µM), and EE13K (100 µM), and excess of 2-mercapto ethyl sulfonate (1.0 mM). Reaction conditions are described in detail in the Experimental Section. a) Representative HPLC chromatograms obtained when the mixture was equilibrated without any external templae (blue), or in the presence of peptide 2 as a template (red). Very similar final products distribution was observed if the mixture was seeded with 2 at the beginning, or three hours after equilibration without a template. b) Kinetic profile of the library equilibration without an external template. c) Kinetic profile of the library equilibration in the presence of peptide 2. References
[1] G. Ashkenasy, R. Jagasia, M. Yadav, M. R. Ghadiri, Proc. Natl. Acad. Sci. USA 2004, 101, 11872-10877. [2] Z. Dadon, M. Samiappan, E. Y. Safranchik, G. Ashkenasy, Chem. Eur. J. 2010, 16, 12096-12099. [3] A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni, R. J. Read, J. Appl. Cryst. 2007, 40, 658-674. [4] G. N. Murshudov, P. Skubák, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long, A. A. Vagin, Acta Crystallogr. D Biol. Crystallogr. 2011 67, 355-367. [5] Z. Dadon, M. Samiappan, N. Wagner, G. Ashkenasy, Chem. Commun. 2012, 48, 1419-1421




















