
Nanomaterials in the Environment: From Materials to High-Throughput Screening to Organisms

Research Article
A constitutive expression system for high-throughput screening
To reduce the amount of consumables and number of pipetting steps in high-throughput screening, a constitutive expression system was developed thatcomprises four different promoters of varying strength. The system was validated bythe expression of different sucrose phosphorylase enzymes from Leuconostocmesenteroides, Lactobacillus acidophilus and Bifidobacterium adolescentis in 96-deep-and low-well plates at three temperatures. Drastically improved soluble expressionin mini-cultures was observed for the enzymes from L. mesenteroides strains byreducing the promoter strength from strong to intermediate and by expressing theproteins at lower temperatures. In contrast, the enzymes from B. adolescentis andL. acidophilus were expressed most efficiently with a strong promoter. The consti-tutive expression of sucrose phosphorylases in low-well plates resulted in a level ofactivity that is equal or even better than what was achieved by inducible expression.Therefore, our plasmid set with varying constitutive promoters will be an indis-pensable tool to optimize enzyme expression for high-throughput screening.
Keywords: Constitutive expression / Directed evolution / High-throughput screening /Promoter / Sucrose phosphorylase
Received: March 29, 2010; revised: August 3, 2010; accepted: August 11, 2010
DOI: 10.1002/elsc.201000065
1 Introduction
Enzymes are increasingly used as powerful biocatalysts for thesynthesis of compounds traditionally produced by chemicalsynthesis. Natural enzymes are, however, often not well suitedfor industrial applications, and might need improvements intheir substrate specificity, selectivity or stability [1, 2]. Theseproperties can be optimized by protein engineering, of whichdirected evolution has been shown to be highly effective [3].The high-throughput screening of mutant libraries is typicallyperceived as the bottleneck in directed evolution [4]. To thatend, assays are usually performed in multiwell plates [5–8].
Before a library can be screened, soluble expression of theenzymes has to be achieved. Typically, libraries for directedevolution are produced in Escherichia coli in 24-, 48- or 96-wellplates. Individual colonies are then transferred to microtiter
plates, grown until stationary phase and induced for a fewhours with IPTG, arabinose or rhamnose [9, 10]. A majorconcern of this small-scale expression is that the expressionlevel and the solubility of the recombinant protein are influ-enced by the culture conditions and the degree of aeration.Recent advances in microtiter plate design has allowedsimilar growth conditions to be reached either in 24-, 48- or96-square-deep-well plates, in shake flasks or at bioreactorscale [11–14]. In contrast, the oxygen transfer ratio is oftenimpaired in 96-low-well plates when conventional shakingdevices are used [11, 15], resulting in reduced expression ofsoluble protein [16–18]. However, expression kinetics similarto that of a bioreactor have been achieved when 96-low-wellmicrotiter plates were shaken at 995 rpm [19]. To improvesoluble expression in low-well plates, lowering the promoterstrength and/or induction temperature can be a major strategy[20, 21].
As an alternative to inducible expression, a constitutiveexpression system can be used that would be more convenientfor high-throughput screening. Several experimenters havealready used a constitutive-like-expression system to circum-vent the induction step by adding inducer from the start of thefermentation [22–24]. Recently, tyrosine phenol-lyase wasconstitutively expressed using the HCE promoter fromGeobacillus toebii in high-throughput screening [25]. A fewreports are also available that describe the constitutiveproduction of different recombinant proteins in E. coli
Dirk Aerts
Tom Verhaeghe
Marjan De Mey
Tom Desmet
Wim Soetaert
Centre of Expertise for
Industrial Biotechnology and
Biocatalysis, Department of
Biochemical and Microbial
Technology, Ghent
University, Ghent, Belgium
Supporting informationavailable online
Abbreviations: BaSP, Bifidobacterium adolescentis SP; GFP, green
fluorescent protein; LaSP, Lactobacillus acidophilus SP; LmSP 12291,
Leuconstoc mesenteroides ATTC 12291 SP; LmSP B1355, Leuconstoc
mesenteroides NRRL B1355 SP; LmSP B1149, Leuconstoc mesenteroides
NRRL B1149 SP; rpoS, Deletion of sigma factor S; SP, Sucrose phos-
phorylase
Correspondence: Dirk Aerts ([email protected]), Centre of
Expertise for Industrial Biotechnology and Biocatalysis, Department of
Biochemical and Microbial Technology, Ghent University, Coupure
Links 653, 9000 Ghent, Belgium
& 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim http://www.els-journal.com
10 Eng. Life Sci. 2011, 11, No. 1, 10–19

[25–32]. However, these expression systems were limited to theuse of strong promoters. Although a strong promoter wouldbe the first choice for large-scale expression, for small-scaleexpression it can be advisable to lower the promoter strengthin order to increase expression in the soluble fraction [20, 21].Therefore, a plasmid set comprising of a range of constitutivepromoters with varying promoter strength would be anindispensible tool for the successful constitutive small-scaleexpression of proteins in the soluble fraction.
There are only a few studies conducted where differentconstitutive promoters are compared according to theirpromoter strength in E. coli [33–36]. In the study of De Meyand colleagues [27], 57 constitutive promoters with varyingpromoter strength were developed. These promoters wereobtained from a library of a degenerated oligonucleotidesequence that encodes consensus sequences for E. colipromoters separated by spacers of random sequences. Thestrength of the synthetic promoters was determined byexpressing green fluorescent protein in E. coli MA8 cells andmeasuring the relative fluorescence units. From this promotercollection, four constitutive promoters with varying strengthswere chosen to use in the current work: one very strongpromoter P14, two intermediate promoters P34 and P22 and aweak one P78 [27].
In this study, the design of a set of constitutive expressionplasmids with varying promoter strengths is described. The useof this expression system for high-throughput small-scaleexpression was demonstrated by expression of sucrose phos-phorylase (SP) enzymes from a range of microorganisms inE. coli. SP (E.C. 2.4.1.7) is classified into the a-amylase family(GH-13) and catalyzes the reversible phosphorolysis of sucroseto a-D-glucose-1-phosphate (a-Glc-1-P) and D-fructose.Moreover, this enzyme can also transfer a glucosyl moiety to avariety of monosacharides, sugar alcohols and even phenoliccompounds and is therefore a very interesting biocatalyst forthe production of a-D-glucosides as industrial fine chemicals[37]. The optimization of the enzyme’s specificity will,however, require the application of engineering strategies andthe availability of a high-throughput screening methodology.The recombinant proteins were expressed at three differenttemperatures in 96-square-deep-well plates. For two enzymes,the constitutive expression system was also tested in 96-round-low-well plates and compared with an IPTG inducibleexpression system driven by the trc promoter.
2 Materials and methods
2.1 Materials and bacterial strains
Restriction endonucleases and T4 DNA ligase were obtainedfrom New England Biolabs, except for PfoI, which wasobtained from Fermentas. DNA fragments and genes wereamplified with high-fidelity polymerase from Roche. Oligo-nucleotides were synthesized by Sigma-genosys. Vectorelements for the plasmid set were obtained from plasmidspTrcHis Topo (Invitrogen) and pGEMT (Promega) andpTRC99A. Constitutive promoters were amplified from plas-mids called pVIK165 PXX [27]. For the isolation of genomic
DNA the GenElute Bacterial Genomic DNA isolation kit fromSigma was used. Kits for gel extraction, PCR purification andplasmid isolation were purchased from Qiagen. Unless notedotherwise, all chemicals were obtained from Sigma or Merckand were of the highest purity.
Escherichia coli XL10-Gold (Stratagene) was used as cloningstrain. In contrast, E. coli Rosetta 2 (Novagen) was used for theexpression of the recombinant SP enzymes. Lactobacillusacidophilus LMG 9433, Leuconostoc mesenteroides LMG 18967(or B1149) and Bifidobacterium adolescentis LMG 10502T wereobtained from BCCMTM. Other strains from L. mesenteroideswere ATCC 12291 and NRRL B1355. The former waspurchased from the ATCC and the latter was obtained from theARS culture collection.
2.2 Culture conditions
E. coli cells were grown in LB-medium (10% Trypton (Oxoid),5% Yeast Extract (Oxoid) and 5% NaCl) supplemented withappropriate antibiotics at 371C and 200 rpm, unless notedotherwise. Lactic acid bacteria were grown at 301C withoutshaking in M.R.S. broth (Oxoid) supplemented with 10% v/vof a solution containing 0.1% FeSO4.7 H2O, 2% MnSO4.H2O,0.1% thiamine.HCl, 0.5% ascorbate and 0.3% citrate. Bifido-bacterium adolescentis LMG 10502T was grown anaerobically at371C in a medium containing 2.3% special peptone (Oxoid),0.5% NaCl, 0.03% cysteine hydrochloride, 0.1% soluble starchand 0.5% glucose.
2.3 Construction of a constitutive expressionplasmid set
The expression plasmid pCXP22h was created first, after whichpromoter P22 was replaced by other constitutive promoters toobtain a set of plasmids (Fig. 1). Promoter P22 was amplifiedfrom pVIK165 P22 by high-fidelity PCR using primersConstpromFw and ConstpromRv (Table 1) to add restrictionsites PfoI and PciI. This fragment was cut with these restrictionenzymes and ligated in vector pTRC99A predigested with PfoIand NcoI (whose overhang sequences are compatible withPciI), thereby replacing the LacIq gene and the trc promoter inthe pTRC99A vector to create pCXP22. Next, a fragmentcontaining a His-tag and an enterokinase recognition site wasamplified from plasmid pTrcHis TOPO with high-fidelityDNA polymerase by PCR using the following pair of oligo-nucleotide primers: FwpTRChis frag and RvpTRChis fragcontaining an EcoRI and an AflII restriction site, respectively(Table 1). The resulting PCR-product was purified and ligatedinto the pGEMT vector. Then, the constructed plasmidpGHisfrag was transformed in E. coli XL10-Gold and checkedfor correct orientation of the fragment in the vector by colonyPCR. Following digestion of this construct with EcoRI andNsiI, a fragment containing the His-tag, enterokinase recog-nition site and the multiple cloning site from the pGEMTvector was obtained. This fragment was subsequently ligated inpCXP22, predigested with EcoRI and PstI (whose overhangsequences are compatible with NsiI), finally forming the
Eng. Life Sci. 2011, 11, No. 1, 10–19 A constitutive expression system for high-throughput screening 11
& 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim http://www.els-journal.com

expression plasmid pCXP22h, which was confirmed bysequencing at the AGOWA sequence service (Berlin).
Starting from expression plasmid pCXP22h, three otherexpression plasmids with varying promoter strength werecreated. First, the three different promoters (PXX: P14, P34 orP78) were amplified with primers ConstpromFw andConstpromRv (Table 1) from a plasmid pVIK165-PXX. Thecreated fragment was digested with PfoI and EcoRI and ligatedinto the predigested pCXP22h vector to replace promoter P22with promoter P14, P34 or P78. The resulting plasmids werecalled pCXPXXh and confirmed by sequencing.
2.4 Molecular cloning of SP genes into expressionplasmids
The SP genes were amplified from the genomic DNA by high-fidelity PCR using the primer pairs listed in Table 1. Eachforward primer includes a NheI or SpeI restriction site andeach reverse primer includes a PstI restriction site. Because ofthe high GC-content of the genomic DNA from B. adolescentis,5% DMSO was added to the PCR reaction mixture. The fivegenes were purified and digested with the proper restrictionendonucleases and ligated in the predigested pCXPXXh vector
Figure 1. Cloning scheme for theconstruction of plasmid set pCXPXXh.RD: restriction digest, PXX: constitu-tive promoter, 6�His: His-tag, ECS:enterokinase cleavage site, MCS:multiple cloning site, rrnB T1 andrrnB T2: transcription terminator,b-lac: b-lactamase gene, Ori: Origin ofreplication.
Table 1. Primers for the amplification of vector elements and genes.
Fragment Primer pair Sequence (50-30)
PXX ConstpromFw GAATTTCCGGGAGAGCTCGATATCC
ConstpromRv TACACATGTTTGTTTCCTCCGAATTCG
His-Tag1ECS FwpTRChis frag GAATTCGGAGGAAACAAAGATGGGGGGTTCTCATCATC
RvpTRChis frag ACTTAAGGGTTGGATCCTTATC
LmSP 12291 12291 Fcl02Sh TAGCTAGCATGGAAATTCAAAACAAAGC
12291 Rvcl01 TCACTGCAGTTAGTTCTGAGTCAAATTATCACTGC
LmSP B1149 B1149 Fw02 TAGCTAGCATGGAAATTCAAAACAAAGCAATG
B1149 Rvcl02 TCACTGCAGTTACAATTGTTGCTTATCAGC
LmSP B1355 B1355 Fwcl04 TAGCTAGCATGGAAATTCAAAATAAAGC
B1355 Rvcl04 TCACTGCAGTTATTTGTTTTGTAAGACTG
LaSP LaSP Fwcl02 TAGCTAGCATGCCAATTGAAAATAAAGTAATG
LaSP rvcl02 TCACTGCAGTTAAAAGTTAATAACCTC
BaSP BaSP Fcl02 TAGCTAGCATGAAAAACAAGGTGCAGCTCATC
BaSP Rcl02 TCACTGCAGTCAGGCGACGACAGGCGGATT
12 D. Aerts et al. Eng. Life Sci. 2011, 11, No. 1, 10–19
& 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim http://www.els-journal.com

with NheI and PstI. The resulting constructs are namedpCXPXXh ‘enzyme name’, for example pCXP22h BaSP.
The genes coding for Leuconstoc mesenteroides ATTC 12291SP (LmSP 12291) and Bifidobacterium adolescentis SP (BaSP)were also cloned into the inducible pTRC99A expressionplasmid using NcoI and PstI as restriction sites. The resultingplasmids were named pIXLmSP 12291 and pIXBaSP.
2.5 Expression and enzyme extraction usingmini-cultures
2.5.1 96-square-deep-well plates
E. coli Rosetta 2 transformed with the expression plasmidwas grown overnight at 371C in 3 mL LB medium containing100mg/mL ampicillin and 25mg/mL chloramphenicol in24-square-deep-well blocks enclosed with a sandwich cover(CR1424 and CR1224, Enzyscreen, Leiden, Netherlands). In eachwell, inoculum was build up for a plasmid consisting of a differentpromoter–enzyme combination. The deep-well plate wascontinuously shaken at 220 rpm with shaking amplitude of 50 mm.From each preculture, 20mL was inoculated into four wells of a 96-square-deep-well block with round bottoms covered with a sand-wich closure system (CR1496 and CR1296, Enzyscreen, Leiden,Netherlands) containing 1 mL fresh medium and grown at 371Cwith continuous shaking at 300 rpm on the same shaker. Once thecultures reached the exponential phase (OD600 5 0.5–0.6),recombinant proteins were further constitutively expressed at either37, 30 or 251C. For the inducible system, the trc promoter wasinduced at the same moment with 1 mM IPTG. At regular timeintervals, 150mL sample was withdrawn from the cultures, trans-ferred to microtiter plates and centrifuged at 41C and 5000� g for20 min. The obtained cell pellets were stored at �201C.
2.5.2 96-low-well flat-bottomed plates
Individual colonies were picked with an automated colony-picker(QPix2, Genetix) and inoculated into sterile 96-well flat-bottomedmicrotiter plates (260860, Nunc, Roskilde, Denmark) enclosed bya sandwich cover (CR1597, Enzyscreen, Leiden, Netherlands)containing 175mL LB medium per well, supplemented withantibiotics. The microtiter plates were incubated for 16 h at 371Cand 250 rpm (50 mm shaking amplitude). Recombinant enzymeswere then expressed by inoculation of 25mL of the grown mini-cultures into 150mL fresh medium in new microtiter plates.Following constitutive expression for 16h at different tempera-tures and 250 rpm, the microtiter plates were centrifuged at5000� g for 20 min, and the formed pellets were frozen at�201C.For the inducible expression of BaSP and LmSP 12291 from thetrc promoter, cultures were induced after the cultures reached thestationary phase (16h) with 15mL IPTG to a final concentrationof 1 mM and the expression was prolonged for another 4 h.
2.5.3 Extraction from mini-cultures
After thawing at room temperature, the pellets were lysed with200 mL of lysis buffer composed of 50 mM Tris–HCl (pH 7.5),
1 mM EDTA, 4 mM MgCl2, 50 mM NaCl, 0.1 mM PMSF and1 mg/mL lysozyme to extract the intracellular enzyme from thecells [38]. Lysis was carried out for 30 min at 371C on aFreedom EVO 200 liquid handling robot (Tecan) followed bycentrifugation at 5000� g for 20 min. The supernatants wereused for screening optimal expression conditions.
2.6 Enzyme assay
SP activity was determined at 371C using a continuous assay, inwhich production of a-glucose-1-phosphate from sucrose andinorganic phosphate is coupled to the reduction of NAD1 in thepresence of phosphoglucomutase (PGM) and glucose-6-phos-phate dehydrogenase (G6P-DH) [39, 40]. The standard assaysolution contained 50 mM Tris-HCL, pH 7.0, 1 mM EDTA,5 mM MgSO4, 1 mM b-NAD, 5mM Glc-1,6-PP, 0.6 U PGM and0.6 U G6P-DH. As substrate, 50 mM sucrose in 50 mM sodiumphosphate buffer, pH 7.0 was used. The increase in absorbanceat 340 nm was recorded in a spectrophotometer equilibrated at371C. One unit of the SP activity was defined as the amount ofenzyme that released 1mmol a-Glc-1-P min�1.
2.7 Analysis of soluble and insoluble fractions of96-low-well cultures
For the analysis of the soluble and insoluble fractions of the celllysate, a cell pellet was obtained from 7 mL of the combinedculture of 48 wells from a 96-low-well plate normalized for anOD600 of 3 by centrifugation for 20 min at 5000� g. Theobtained cell pellet was lysed with 1.5 mL lysis buffer for 30 minat 371C in a water bath followed by centrifugation at 16000� gfor 20 min. The insoluble fraction was then washed twice withlysis buffer and resolved in 750mL loading buffer composed of62.5 mM Tris-HCl, pH 6.8, 25% v/v glycerol, 2% w/v SDS,0.01% w/v bromophenol blue and 5% v/v b-mercaptoethanoland incubated for 30 min at 951C.
Prior to Western blot analysis, 15% SDS-PAGE wasperformed as described in [41]. The loading volume ofColorPlus Prestained Protein marker (New England Biolabs)was 5 mL and of both soluble and insoluble fraction were 15 mL.Following SDS-PAGE, the protein bands on the gel weretransferred to nitrocellulose membranes (Hybond-ECLTM,Amersham Biosciences, USA) as described in [41]. Mousemonoclonal Anti-poly his antibody (H1029, Sigma) as the firstantibody and anti-mouse IgG alkaline phosphatase conjugate(A3562, Sigma) as the second anti-body were used at a 1:2000dilution. The nitrocellulose membranes were then stained withBCIP/NBT phosphatase substrate (Roche).
3 Results and discussion
3.1 Construction of a constitutive expressionplasmid set
A prerequisite for high-throughput screening is that therecombinant enzymes are expressed in a soluble form and do
Eng. Life Sci. 2011, 11, No. 1, 10–19 A constitutive expression system for high-throughput screening 13
& 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim http://www.els-journal.com

not form inclusion bodies. As the strength of the promotercan have a very big influence on the amount of solubleenzyme, a plasmid set with four different constitutivepromoters was constructed. Plasmids pCXPXXh were designedby combining elements from pTRC99A, pTrcHis Topo,pGEMT and pVIK165 PXX. The strength of the constitutivepromoters was previously determined using green fluorescentprotein as reporter by De Mey and colleagues [27].Promoter P14 was found to be 2, 2.4 and 4 times strongerthan P34, P22 and P78, respectively. Further theexpression plasmids allow constitutive expression ofproteins with or without an N-terminal fusion peptidecontaining either only a 6� His-tag or a 6� His-tag and anenterokinase recognition site to remove the fusion peptideafter purification. In order to facilitate cloning, a multiplecloning site originating from pGEMT was introduced. All theseelements were combined with the transcription terminatorsrrnB T1 and rrnB T2, the b-lactamase resistance gene and thepBR322 origin of replication originating from pTRC99A toform the expression plasmids pCXPXXh as illustrated inFigure 1.
3.2 Optimization of expression conditions in96-square-deep-well plates
Enzyme expression in 96-square-deep-well plates is not onlyconvenient for high-throughput screening, but can also beused for the optimization of expression conditions beforescale-up [42]. Here, five different SP enzymes were constitu-tively expressed at three different temperatures to evaluate theeffectiveness of our approach. E. coli Rosetta 2, whichsupplements tRNAs for rare codons, was chosen as host strainto prevent difficulties in expression due to the presence of rarecodons in the sp genes.
Cultures expressing the SP enzymes constitutivelyshowed no significant difference in growth rates between deep-well plates and shake flasks as illustrated in SupportingInformation Fig. S1 A. This result was expected sincerecent developments in well design and closure systemsensure similar growth conditions in these mini-cultures asoccur in Erlenmeyer flasks [11, 13]. Furthermore, nodifferences in growth were observed between the differentpromoters or between the different enzymes (SupportingInformation Figs. S1B and S2). However, induction of thePtrc promoter with IPTG at an optical density of 0.5–0.6reduced the growth of the expressing cultures (SupportingInformation Fig. S2A, B and C). Not surprisingly, growthslowed down with 10–35% when proteins were expressed at 30and 251C, respectively, instead of at 371C. As can be seen fromFigure 2, constitutive expression was achieved for all five SPenzymes and all of them were active. To the best of ourknowledge, the recombinant expression of the SP fromL. acidophilus in E. coli is reported here for the first time. Theother SP enzymes were previously expressed in E. coli using aninducible expression system in a shake flask or in a bioreactor[43–46].
In this work, specific activity is defined as SP activity from1 mL culture with an optical density at 600 nm of 1, which
allows a comparison of the amount of active enzyme present ineach cell (Fig. 2). In general, the highest specific activity isfound after 2 h expression. This observation holds not only forthe constitutive expression, but it is also true for the inducibleexpression of BaSP with the lacIq trc operator–promotersystem (Fig. 2A–C). The decreasing specific activity during thecourse of the fermentation is most likely caused by growth ratedependence of cellular parameters like concentration of freeRNA polymerase and ribosomes and the physiological state ofthe cell during the fermentation [36, 47]. In addition, theproduced recombinant protein could also be degraded by hostproteases, although E. coli Rosetta 2 that was used in this studyis deficient in the intracellular Lon protease and the periplas-matic OmpT protease.
A second important observation is that the specific activityis not necessarily correlated with the promoter strength(Fig. 2). For some enzymes, the strength of the promoter haslittle influence (Lactobacillus acidophilus SP (LaSP)), whereasfor others either the strongest (BaSP) or an intermediatepromoter (LmSP) gives the best results. The low specificactivity of LmSP enzymes expressed with the strongestpromoter P14 is most likely attributable to the formation ofmore inclusion bodies. Even at lower temperatures, the use ofthe strongest promoter P14 did not result in the most activeenzyme extract.
Although the growth rate of E. coli is maximal at 371C,expression at suboptimal temperature (30–251C) can reduceunwanted metabolic responses to the synthesis of a recombi-nant protein. As a consequence the yield and solubility of thetarget protein can be improved [48]. Therefore, the fourdifferent promoters have been tested at 37, 30 and 251C. Thelevels of soluble enzyme at different temperatures arecompared using the average activity from the four constitutivepromoters. This averaged activity is shown by a horizontaldashed line in Figures 2, 3. As can be seen from these graphs(A–C), the expression temperature for BaSP is not of signifi-cant importance. In contrast, the activity is drasticallyimproved when expressed at lower temperatures for LmSPenzymes (Figs. 2, 3D–L). This observation is in accordancewith the observation that these enzymes are better expressedwith a promoter of intermediate strength implying reducedformation of inclusion bodies. The expression of LaSP wasoptimal at 301C.
A remarkable observation for promoter P22 is that forBaSP, this promoter produces more active enzyme at 301C ifcompared with 37 and 251C. Possibly the combination of thispromoter with the BaSP gene is temperature dependent. Giladiet al. [49] observed an increased activity of phage l PL
promoter when decreasing the expression temperature. It wassuggested that this increased activity was caused by a betterbinding of E. coli’s integration host factor to the promoter atlower temperatures.
As can be seen from Figure 3, the maximal total activity(activity for 1 mL culture with the actual sampling OD600) isgenerally reached at the end of the exponential phase, i.e.around 3, 5 and 7 h at 37, 30 and 251C, respectively, and isindependent from the promoter strength. For BaSP, themaximum activity was obtained with the strongest promoterP14 at 371C after 6 h expression (Fig. 3A). This maximal total
14 D. Aerts et al. Eng. Life Sci. 2011, 11, No. 1, 10–19
& 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim http://www.els-journal.com

activity obtained with the constitutive expression system wasonly 8.2% less than the maximal activity obtained using theinducible LaqIq Ptrc expression system at 371C. LaSP was bestexpressed with P34 at 301C for 4 h (Fig. 3N), although nosignificant difference was obtained using P14 or P22. ForLmSP enzymes the highest yields were obtained at 251C withpromoter P34 after 8 h of expression (Fig. 3F, I, L).
3.3 Application of the expression system in 96-low-well flat-bottomed plates
To further increase the throughput in screenings, microbialgrowth and constitutive expression in 96-low-well flat-bottomedplates were investigated for the SP enzymes fromL. mesenteroides ATTC 12291 and B. adolescentis. In order to
Figure 2. Specific activity for thedifferent SP enzymes at differentexpression times expressed withdifferent promoters and at threetemperatures. BaSP: B. adolescentisSP, LmSP 12291: L. mesenteroidesATTC 12291 SP, LmSP B1149:L. mesenteroides NRRL B1149 SP,LmSP B1355: L. mesenteroides NRRLB1355 SP, LaSP: L. acidophilus SP.
Eng. Life Sci. 2011, 11, No. 1, 10–19 A constitutive expression system for high-throughput screening 15
& 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim http://www.els-journal.com
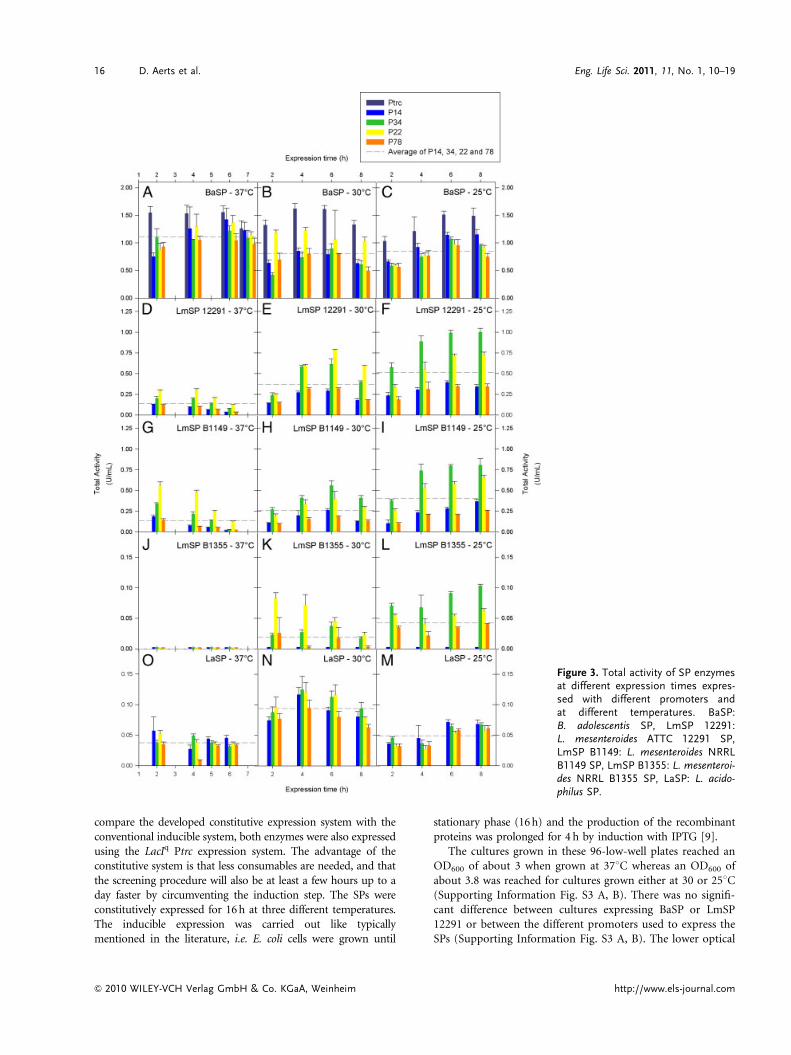
compare the developed constitutive expression system with theconventional inducible system, both enzymes were also expressedusing the LacIq Ptrc expression system. The advantage of theconstitutive system is that less consumables are needed, and thatthe screening procedure will also be at least a few hours up to aday faster by circumventing the induction step. The SPs wereconstitutively expressed for 16 h at three different temperatures.The inducible expression was carried out like typicallymentioned in the literature, i.e. E. coli cells were grown until
stationary phase (16 h) and the production of the recombinantproteins was prolonged for 4 h by induction with IPTG [9].
The cultures grown in these 96-low-well plates reached anOD600 of about 3 when grown at 371C whereas an OD600 ofabout 3.8 was reached for cultures grown either at 30 or 251C(Supporting Information Fig. S3 A, B). There was no signifi-cant difference between cultures expressing BaSP or LmSP12291 or between the different promoters used to express theSPs (Supporting Information Fig. S3 A, B). The lower optical
Figure 3. Total activity of SP enzymesat different expression times expres-sed with different promoters andat different temperatures. BaSP:B. adolescentis SP, LmSP 12291:L. mesenteroides ATTC 12291 SP,LmSP B1149: L. mesenteroides NRRLB1149 SP, LmSP B1355: L. mesenteroi-des NRRL B1355 SP, LaSP: L. acido-philus SP.
16 D. Aerts et al. Eng. Life Sci. 2011, 11, No. 1, 10–19
& 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim http://www.els-journal.com

densities obtained in these 96-low-well plates are a result of alower oxygen transfer rate as compared with 96-square-deep-well plates [11]. The total activity obtained by expressing BaSPand LmSP in 96-low-well plates at different temperatures isshown in Fig. 4. The maximal total activities obtained for BaSPand LmSP were 23 and 5.5%, respectively, if compared withthose obtained in deep-well plates. For the constitutiveexpression of BaSP in these low-well plates the promoterstrength is respected, although there are no significant differ-ences in the total activity obtained with the strongest promoterP14 and the two intermediate ones. The maximal total activityfor BaSP was reached at 371C and decreased at lowertemperatures. If the constitutive expression at 371C iscompared with the inducible system, the total activity is about3 times higher. However, the total activity of the inducibleexpression can be increased by prolonging expression for a fewhours, but this operation will drastically lower the throughput.
As already found for the expression of LmSP 12291 in deep-well plates, promoter P14 is too strong and the resulting totalactivity is comparable to the weakest promoter P78 (Fig. 4B).Expression driven by the intermediate promoters resulted in nosignificant difference in total activity. In contrast to the low-wellexpression of BaSP, the inducible expression of LmSP for 4 hreached a similar level of activity as obtained with the constitutiveexpression system. As can be seen in Figure 4B the growth andexpression temperature is of striking importance for LmSP 12291expression. At 371C almost no activity could be measured foreither the constitutive or inducible expression and no clear bandswere observed on a Western blot (Supporting InformationFig. S4 C). As observed for the deep-well expression, decreasinggrowth and expression temperature significantly improves thesoluble expression of LmSP 12291 (Supporting InformationFig. S4 B). A decrease to 301C was necessary for low-well expres-sion but no significant difference was obtained when expressed at251C. A similar trend was also observed by Goedl et al. [50] for thesame enzyme using an inducible expression system.
Overall the improved yields at lower temperatures and withweaker promoters obtained using the deep-well or low-wellexpression indicate a decreased formation of inclusion bodies,although these still are the dominant fraction (SupportingInformation Fig. S4). As is widely known, unwanted hydro-phobic interactions are more prevalent at higher temperatures,which favor protein aggregation. Together with a reducedexpression of heat shock protease, the activity and expression
of a number of E. coli chaperones are increased at temperaturesaround 301C. These factors increase the chance of properfolding, and the decreased expression rate reduces unwantedinteraction between unfolded intermediates [21, 48].
4 Concluding remarks
In this study, we have demonstrated the successful constitutiveexpression of different SPs in E. coli Rosetta 2 in deep- andlow-well plates. It would also be interesting to see if expressionlevels can be further improved when expressed in a rpoS�
strain as was previously reported for the constitutive produc-tion of human leptin by Jeong et al. [28].
For enzymes that are not toxic and not prone to forminclusion bodies when expressed in deep-well plates or inshake flasks, a strong constitutive promoter may be advisablefor small-scale expression. In contrast, intermediate or weakconstitutive promoters are advisable for enzymes that have atendency to form inclusion bodies. Therefore, the plasmid setconstructed here will be an inevitable tool to optimize theconstitutive expression of enzymes for high-throughputscreening. As observed for LmSP enzymes, the expressiontemperatures are of striking importance for constitutive as wellas inducible expression in mini-cultures, especially whenscaling down to 96-low-well plates. It is therefore advisable totest your recombinant expression at lower temperatures ifexpression at 371C fails in mini-cultures.
The use of the developed expression system circumvents notonly the labor-intensive and time-consuming induction step,but also saves expensive consumables. Bypassing the inductionstep in low-well expression expedite high-throughput screen-ing with a few hours up to a day. Cai et al. [51] describe amethod for the reagentless extraction of intracellular proteins.If both methods are combined, a ‘minimal pipetting high-throughput screening’ can be further developed. Such a systemwill be ideal for high-throughput screening in directedevolution to alter enzymes’ activity, specificity or stability.
Acknowledgements
The authors wish to thank the Institute for the Promotionof Innovation through Science and Technology in Flanders
Figure 4. Total activity for eitherconstitutive or inducible expressionof A. BaSP and B. LmSP 12291 in 96-low-well flat-bottomed plates. BaSP:B. adolescentis SP, LmSP 12291:L. mesenteroides ATTC 12291 SP.
Eng. Life Sci. 2011, 11, No. 1, 10–19 A constitutive expression system for high-throughput screening 17
& 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim http://www.els-journal.com

(IWT-Vlaanderen) for financial support through a Ph.D.-grant(81323) and an SBO-project (50191).
Conflict of interest
The authors have declared no conflict of interest.
References
[1] U. T. Bornscheuer, Directed evolution of enzymes, Angew.
Chem. Int. Ed. 1998, 37, 3105–3108.
[2] K. A. Powell, S. W. Ramer, S. B. del Cardayre, W. P. C.
Stemmer, M. B. Tobin, P. F. Longchamp, G. W. Huisman,
Directed evolution and biocatalysis, Angew. Chem. Int. Ed.
2001, 40, 3948–3959.
[3] C. A. Tracewell, F. H. Arnold, Directed enzyme evolution:
climbing fitness peaks one amino acid at a time, Curr. Opin.
Chem. Biol. 2009, 13, 3–9.
[4] H. M. Zhao, F. H. Arnold, Combinatorial protein design:
strategies for screening protein libraries, Curr. Opin. Struct.
Biol. 1997, 7, 480–485.
[5] M. T. Reetz, D. Kahakeaw, R. Lohmer, Addressing the
numbers problem in directed evolution, ChemBioChem
2008, 9, 1797–1804.
[6] M. T. Reetz, D. Kahakeaw, J. Sanchis, Shedding light on the
efficacy of laboratory evolution based on iterative saturation
mutagenesis, Mol. BioSyst. 2009, 5, 115–122.
[7] A. V. Shivange, J. Marienhagen, H. Mundhada, A. Schenk,
U. Schwaneberg, Advances in generating functional diversity
for directed protein evolution, Curr. Opin. Chem. Biol. 2009,
13, 19–25.
[8] N. J. Turner, Directed evolution drives the next generation of
biocatalysts, Nat. Chem. Biol. 2009, 5, 568–574.
[9] D. Hoffmeister, J. Yang, L. Liu, J. S. Thorson, Creation of the
first anomeric D/L-Sugarlinase by means of directed evolu-
tion, Proc. Natl. Acad. Sci. USA 2003, 100, 13184–13189.
[10] F. H. Arnold, G. Georgiou, Directed enzyme evolution:
screening and selection methods. Methods in Molecular
Biology, vol. 230. Humana Press Inc, Totowa, NJ 2003.
[11] W. A. Duetz, Microtiter plates as mini-bioreactors: minia-
turization of fermentation methods, Trends Microbiol. 2007,
15, 469–475.
[12] W. A. Duetz, L. Ruedi, R. Hermann, K. O’Connor, J. Buchs,
B. Witholt, Methods for intense aeration, growth, storage,
and replication of bacterial strains in microtiter plates, Appl.
Environ. Microbiol. 2000, 66, 2641–2646.
[13] H. Waegeman, J. Beauprez, J. Maertens, M. De Mey,
L. Demolder, M. R. Foulqui e-Moreno, N. Boon, D. Charlier,
W. Soetaert, Validation study of 24 deepwell microtiterplates
to screen libraries of strains in metabolic engineering,
J. Biosci. Bioeng. 2010, doi: 10.1016/j.jbiosc.2010.07.008.
[14] T. A. Barrett, A. Wu, H. Zhang, M. S. Levy, G. J. Lye, Microwell
engineering characterization for mammalian cell culture
process development, Biotechnol. Bioeng. 2010, 105, 260–275.
[15] R. Hermann, M. Lehmann, J. Buchs, Characterization of
gas–liquid mass transfer phenomena in microtiter plates,
Biotechnol. Bioeng. 2003, 81, 178–186.
[16] S. Graslund, P. Nordlund, J. Weigelt, J. Bray, B. Struct
Genomics Consortium; Architecture Fonction Macromol, C.
Struct Genomics Ctr; China Struct Genomics Consortium;
Integrated, S. Struct Function; Israel Struct Proteomics Ctr;
Joint Ctr, X. R. C. Genomics; Midwest Ctr Struct Genomics;
New York Struct Genomi, N. E. S. G. C. O. P. P. F. P. Sample,
R. S. G. Prod Facility; Max Delbruck Ctr Mol Med, S. C.
Proteomics, Protein production and purification, Nat.
Methods 2008, 5, 135–146.
[17] J. O. Konz, J. King, C. L. Cooney, Effects of oxygen on
recombinant protein expression, Biotechnol. Prog. 1998, 14,
393–409.
[18] K. H. Jung, Y. J. Lee, J. H. Yeon, S. K. Yoo, B. C. Chung,
Improvement of soluble recombinant interferon-alpha
expression by methyl alpha-D-glucopyranoside in araBAD
promoter system of Escherichia coli, Biotechnol. Bioprocess
Eng. 2009, 14, 274–280.
[19] F. Kensy, C. Engelbrecht, J. Buchs, Scale-up from microtiter
plate to laboratory fermenter: evaluation by online moni-
toring techniques of growth and protein expression in
Escherichia coli and Hansenula polymorpha fermentations,
Microb. Cell Fact. 2009, 8, 64.
[20] S. C. Makrides, Strategies for achieving high-level expression
of genes in Escherichia coli, Microbiol. Rev. 1996, 60 (3), 512.
[21] H. P. Sorensen, K. K. Mortensen, Soluble expression of
recombinant proteins in the cytoplasm of Escherichia coli,
Microb. Cell Fact. 2005, 4, 1.
[22] M. R. M. De Groeve, M. De Baere, L. Hoflack, T. Desmet,
E. J. Vandamme, W. Soetaert, Creating lactose phosphorylase
enzymes by directed evolution of cellobiose phosphorylase,
Protein Eng. Des. Sel. 2009, 22, 393–399.
[23] B. A. van der Veen, G. Potocki-Veronese, C. Albenne,
G. Joucla, P. Monsan, M. Remaud-Simeon, Combinatorial
engineering to enhance amylosucrase performance:
construction, selection, and screening of variant libraries for
increased activity, FEBS Lett. 2004, 560, 91–97.
[24] B. A. van der Veen, L. K. Skov, G. Potocki-Veronese,
M. Gajhede, P. Monsan, M. Remaud-Simeon, Increased
amylosucrase activity and specificity, and identification of
regions important for activity, specificity and stability
through molecular evolution. FEBS J. 2006, 273, 673–681.
[25] E. Rha, S. Kim, S. L. Choi, S. P. Hong, M. H. Sung, J. J. Song,
S. G. Lee, Simultaneous improvement of catalytic activity
and thermal stability of tyrosine phenol-lyase by directed
evolution. FEBS Journal 2009, 276, 6187–6194.
[26] V. Chauhan, A. Singh, S. M. Waheed, S. Singh, R. Bhatnagar,
Constitutive expression of protective antigen gene of Bacillus
anthracis in Escherichia coli. Biochem. Biophys. Res. Commun.
2001, 283, 308–315.
[27] M. De Mey, J. Maertens, G. J. Lequeux, W. K. Soetaert, E. J.
Vandamme, Construction and model-based analysis of a
promoter library for E. coli: an indispensable tool for
metabolic engineering, BMC Biotechnol. 2007, 7, 14.
[28] K. J. Jeong, J. H. Choi, W. M. Yoo, K. C. Keum, N. C. Yoo,
S. Y. Lee, M. H. Sung, Constitutive production of human
leptin by fed-batch culture of recombinant rpoS(�) Escher-
ichia coli. Prot. Expr. Purif. 2004, 36, 150–156.
[29] D. C. Lee, G. J. Kim, Y. K. Cha, C. Y. Lee, H. S. Kim, Mass
production of thermostable D-hydantoinase by batch culture
18 D. Aerts et al. Eng. Life Sci. 2011, 11, No. 1, 10–19
& 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim http://www.els-journal.com

of recombinant Escherichia coli with a constitutive expression
system. Biotechnol. Bioeng. 1997, 56, 449–455.
[30] Y. Q. Liu, Q. Li, X. J. Hu, J. C. Yang, Using native
hydantoinase promoter to induce D-carbamoylase soluble
expression in Escherichia coli. Biochem. Eng. J. 2008, 41,
12–16.
[31] V. Menart, S. Jevsevar, M. Vilar, A. Trobis, A. Pavko,
Constitutive versus thermoinducible expression of hetero-
logous proteins in Escherichia coli based on strong P-R,P-L
promoters from phage lambda. Biotechnol. Bioeng. 2003, 83,
181–190.
[32] H. Poo, J. J. Song, S. P. Hong, Y. H. Choi, S. W. Yun, J. H.
Kim, S. C. Lee, S. G. Lee, M. H. Sung, Novel high-level
constitutive expression system, pHCE vector, for a
convenient and cost-effective soluble production of human
tumor necrosis factor-alpha. Biotechnol. Lett. 2002, 24,
1185–1189.
[33] H. Alper, C. Fischer, E. Nevoigt, G. Stephanopoulos, Tuning
genetic control through promoter engineering. Proc. Natl.
Acad. Sci. U. S. A. 2005, 102, 12678–12683.
[34] M. De Mey, J. Maertens, G. J. Lequeux, W. K. Soetaert, E. J.
Vandamme, Construction and model-based analysis of a
promoter library for E-coli: an indispensable tool for meta-
bolic engineering. BMC Biotechnol. 2007, 7, 14.
[35] P. R. Jensen, K. Hammer, Artificial promoters for metabolic
optimization. Biotechnol. Bioeng. 1998, 58, 191–195.
[36] S. T. Liang, M. Bipatnath, Y. C. Xu, S. L. Chen, P. Dennis,
M. Ehrenberg, H. Bremer, Activities of constitutive promo-
ters in Escherichia coli. J. Mol. Biol. 1999, 292, 19–37.
[37] C. Goedl, T. Sawangwan, P. Wildberger, B. Nidetzky, Sucrose
phosphorylase: a powerful transglucosylation catalyst for
synthesis of I7-D-glucosides as industrial fine chemicals.
Biocatal. Biotransform. 2010, 28, 10–21.
[38] M. R. M. De Groeve, H. G. Tran, A. Van Hoorebeke, J. Stout,
T. Desmet, S. N. Savvides, W. Soetaert, Development
and application of a screening assay for glycoside phos-
phorylases. Anal. Biochem. 2010, 401, 162–167.
[39] R. Silverstein, J. Voet, D. Reed, R. H. Abeles, purification and
mechanism of action of sucrose phosphorylase. J. Biol. Chem.
1967, 242, 1338–1346.
[40] T. Koga, K. Nakamura, Y. Shirokane, K. Mizusawa, S. Kitao,
M. Kikuchi, Purification and some properties of sucrose
phosphorylase from Leuconostoc mesenteroides. Agric. Biol.
Chem. 1991, 55, 1805–1810.
[41] J. Sambrook, D. Russell, Molecular cloning: a laboratory
manual. 2001, Cold spring harbor laboratory press, New
York.
[42] W. Peti, R. Page, Strategies to maximize heterologous protein
expression in Escherichia coli with minimal cost. Prot. Expr.
Purif. 2007, 51, 1–10.
[43] L. A. M. van den Broek, E. L. van Boxtel, R. P. Kievit,
R. Verhoef, G. Beldman, A. G. J. Voragen, Physico-chemical
and transglucosylation properties of recombinant sucrose
phosphorylase from Bifidobacterium adolescentis DSM20083.
Appl. Microbiol. Biotechnol. 2004, 65, 219–227.
[44] S. Kitao, E. Nakano, Cloning of the sucrose phosphorylase
gene from Leuconostoc mesenteroides and its overexpression
using a sleeper bacteriophage vector. J. Ferment. Bioeng.
1992, 73, 179–184.
[45] J. H. Lee, Y. H. Moon, N. Kim, Y. M. Kim, H. K. Kang, J. Y.
Jung, E. Abada, S. S. Kang, D. Kim, Cloning and expression of
the sucrose phosphorylase gene from Leuconostoc mesenter-
oides in Escherichia coli. Biotechnol. Lett. 2008, 30, 749–754.
[46] J. H. Lee, S. H. Yoon, S. H. Nam, Y. H. Moon, Y. Y. Moon,
D. Kim, Molecular cloning of a gene encoding the sucrose
phosphorylase from Leuconostoc mesenteroides B-1149 and
the expression in Escherichia coli. Enzyme Microb. Technol.
2006, 39, 612–620.
[47] S. Klumpp, Z. G. Zhang, T. Hwa, Growth rate-dependent
global effects on gene expression in bacteria. Cell 2009, 139,
1366–1375.
[48] R. S. Donovan, C. W. Robinson, B. R. Glick, Review: opti-
mizing inducer and culture conditions for expression of
foreign proteins under the control of the lac promoter. J. Ind.
Microbiol. 1996, 16, 145–154.
[49] H. Giladi, D. Goldenberg, S. Koby, A. B. Oppenheim,
Enhanced activity of the bacteriophage lambda PL promoter
at low temperature. Proc. Natl. Acad. Sci. USA 1995, 92,
2184–2188.
[50] C. Goedl, A. Schwarz, A. Minani, B. Nidetzky, Recombinant
sucrose phosphorylase from Leuconostoc mesenteroides:
Characterization, kinetic studies of transglucosylation, and
application of immobilised enzyme for production of alpha-
D-glucose 1-phosphate. J. Biotechnol. 2007, 129, 77–86.
[51] Z. Cai, W. H. Xu, R. Xue, Z. L. Lin, Facile, Reagentless and in
situ release of Escherichia coli intracellular enzymes by heat-
inducible autolytic vector for high-throughput screening.
Protein Eng. Des. Sel. 2008, 21, 681–687.
Eng. Life Sci. 2011, 11, No. 1, 10–19 A constitutive expression system for high-throughput screening 19
& 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim http://www.els-journal.com
Copyright © 2022 FDOKUMEN




















