
THE PERSISTENCE OF UNION REPRESSION IN AN ERA OF RECOGNITION

1
A conformational switch between transcriptional repression and replication initiation in RepA dimerization domain Rafael Giraldo1, Carlos Fernández-Tornero2, Philip R. Evans3, Ramón Díaz-Orejas1 and Antonio Romero2 Departments of 1Molecular Microbiology and 2Protein Structure and Function, Centro de Investigaciones Biológicas – CSIC, C/ Velázquez 144, Madrid, 28006, Spain. 3MRC Laboratory of Molecular Biology, Hills Road, Cambridge, CB2 2QH, UK. Correspondence should be addressed to R.G. e-mail: [email protected] Plasmids are natural vectors for gene transfer. In Gram-negative bacteria, plasmid DNA replication is triggered upon binding of monomers of an initiator protein (Rep) to direct repeats at the origin sequence. Rep dimers, inactive as initiators, bind to an inverse repeat operator repressing rep transcription. Rep proteins are composed of N-terminal dimerization plus C-terminal DNA binding domains. Activation is coupled to dimer dissociation, converting the dimerization domain into a second origin binding module. The structure of the monomeric F plasmid initiator (mRepE) was known, but the molecular nature of Rep activation remained elusive. Here we report the crystal structure of the dimeric N-terminal domain of the pPS10 plasmid initiator (dRepA). dRepA has a winged-helix fold, as its homologous domain in mRepE. However, dimerization transforms an interdomain loop plus β-strand (mRepE) into an α-helix (dRepA). dRepA resemble the C terminus of eukaryotic/archaeal Cdc6, giving clues on the phylogeny of DNA replication initiators. Plasmids are autonomously replicating DNA molecules1 that are common vectors in the horizontal transfer of genes between bacteria. In Gram-negative bacteria, most plasmids encode two elements for replication1,2: a cis-acting DNA sequence (the origin of replication, ori) and a trans-acting protein (Rep), that binds to ori establishing a nucleoprotein pre-initiation assembly. Rep binding contributes to melt ori and acts as a landing pad for bacterial host factors (helicases, primases, DNA polymerases) required to proceed with plasmid replication. In solution, Rep proteins are mainly found as dimers. These can act as autorepressors by binding to an inversely-repeated operator sequence found at the rep promoter3–5. Chaperones (either DnaK-J-GrpE or ClpA/X)6–11, or the allosteric effect of specific directly repeated ori sequences (iterons)12, can cause the dissociation of Rep dimers into monomers. These assemble a functional replication initiator complex4,13,14. A number of biochemical and biophysical studies have suggested that Rep proteins are composed of two winged helix (WH) domains14. In the dimers, the C-terminal domain (WH2) binds to each operator DNA repeat (5´ GGACAGGG in pPS10 plasmid15) through the major groove, whereas the N-terminal domain (WH1) acts as the dimerization interface14. In the monomeric form, WH2 binds to the end of each iteron that includes the core of the operator repeat sequence5 (underlined in the above sequence), while WH1 is structurally remodelled, enabling it to bind to the opposite iteron end (5´ TTTAAAGG in pPS10)14. The structure of the monomeric species of Escherichia coli F plasmid initiator (mRepE) has been previously solved in complex with an iteron repeat, revealing the details of origin DNA recognition16. However, the molecular basis for the transformation of the replication-inert Rep dimers into the initiator monomers remains essentially unknown. This switch constitutes an attractive,

2
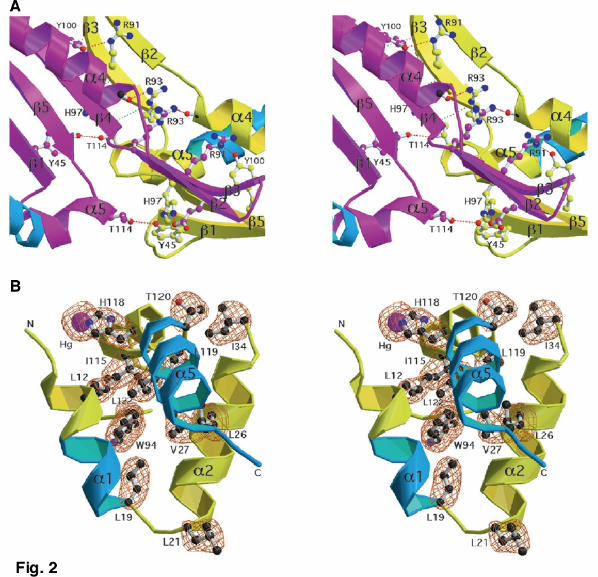
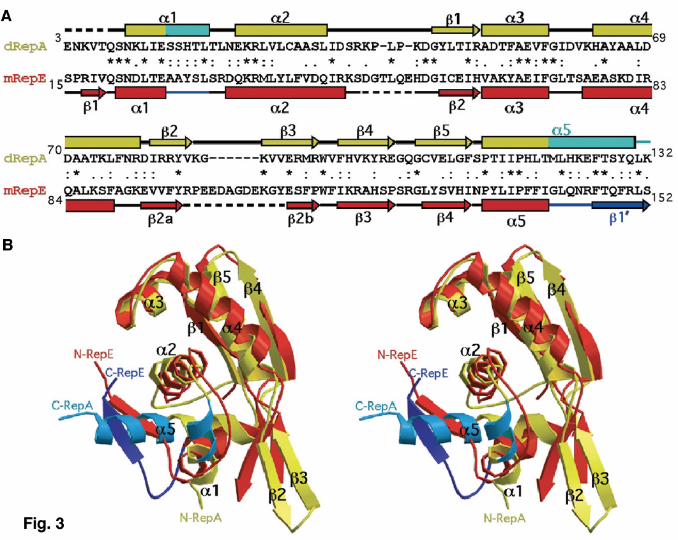
yet unexplored, target for the design of new drugs to combat antibiotic resistance in Gram-negative bacteria. We have solved the crystal structure of the dimeric N-terminal domain of the Pseudomonas pPS10 plasmid Rep protein15,14 (dRepA) to 2.75 Å resolution using the MAD method on a Hg derivative. The comparison of the structures of dRepA dimer with the previously reported mRepE monomer16 provides a solid ground for understanding the molecular basis of plasmid replication activation1,2 and a unique and elegant example of a functionally relevant large structural transformation in a DNA binding domain (DBD). Results Architecture of dRepA The overall shape of dRepA resembles a montera, the hat used by bullfighters, with dimensions about 33 Å in height and 55-75 Å wide (Fig. 1). The refined dRepA model includes residues 8–132 (the C terminus of cloned WH114), but clear continuous density is not evident for residues 36–42 in one of the protomers. Both protomers are nearly identical, r.m.s. deviation = 0.84 Å for 124 superimposed Cα atoms, and are composed of five α-helices and five β-strands. The helices are arranged in two contiguous three-helix bundles (α1-α2-α5 and α2-α3-α4, respectively) with α2 in common. The C-terminal helix (α5) projects between the N-terminal helices α1 and α2, resulting in an almost closed domain. The β-strands are clustered in two oppositely oriented, twisted antiparallel β-sheets (β1-β5-β4 and β2-β3, respectively). Helices α2-α4 plus the first β-sheet constitute a WH fold17,2. In a Dali18 search for structurally similar proteins, all significant hits (Z-score >3.5) belong to the WH family of Helix-Turn-Helix (HTH) DBDs. Dimerization interface and hydrophobic core In dRepA the dimerization interface mainly comprises strands β3 and β4 from different protomers, thus forming a central five-stranded pleated (β1-β5-β4 plus β3-β2) antiparallel β-sheet (Figs. 1a, 2a). Interactions between strands β3 and β4 are made through main chain hydrogen bonding. However, on both sides of the sheet several side chains establish hydrogen bonds linking not only the β-sheets, but also linking the β-sheets to the C terminus of α4 and the N terminus of α5 (Fig. 2a). Formation of RepA dimers through these regions buries 1,804 Å2 (22%) of solvent accessible (1.4 Å probe radius) surface in the monomers, constituting the largest protein contact area in the whole unit cell. There is no direct contribution of the α-helical elements to dimerization, although it had been expected that residues Leu12, Leu19 and Leu26 would form a hydrophobic heptad resembling a leucine zipper (LZ) motif19. However, those leucines, found in helices α1 and α2, contribute significantly to stabilize the conformation of each protomer in dRepA by means of: i) direct hydrophobic interactions with α5, and ii) together with the central Trp94 residue12,14, forming part of an extensive hydrophobic network that holds α5 by fixing the angle between the V-shaped structure formed by helices α1 and α2 (Fig. 2a). Structural transformation of Rep dimers into monomers The structural basis of Rep activation can be inferred by comparing dRepA and mRepE16 because of their significant similarities, both in sequence (25% identities plus 42% conservative changes) and in secondary structure elements (Fig. 3a). dRepA (protomer) and mRepE structures can be superimposed with a r.m.s. deviation of 1.56 Å for 82 Cα atoms, being nearly identical in their WH moiety (according to dRepA labelling, helices α2-α4 plus strands β1, β5 and β4). However, large differences are evident between the two structures outside of the WH domain (Fig. 3b). The antiparallel sheet β2a-β2b (in mRepE16) is bent ~30o inwards relative to its position at the equivalent

3
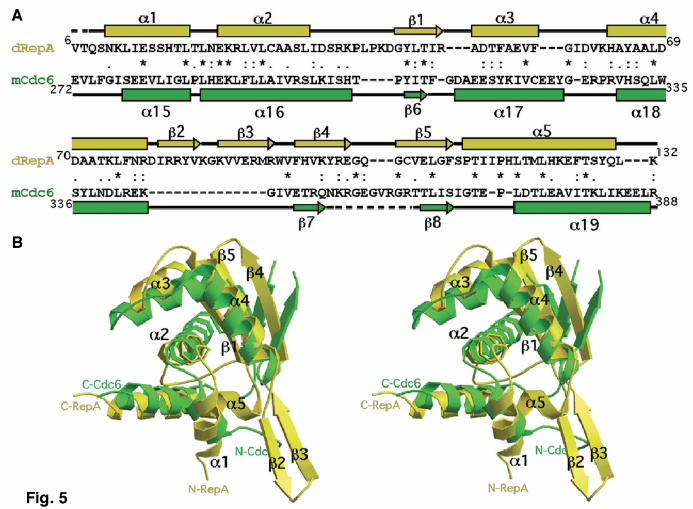
dimerization interface in dRepA (β2-β3) (Fig. 1a). This movement in mRepE seems sufficient to destabilize the extended antiparallel β-sheet, and thus precludes dimerization. The most drastic conformational changes affect both the N- and C-terminal ends of WH1 (Fig. 3b). In dRepA the seven N-terminal residues are disordered and the C terminus forms helix α5, about five and a half turns long and kinked at Pro117. α5 folds between the V-shaped α1 and α2 (interaxial angle ~65o). In contrast, in mRepE the two ends form a short antiparallel β-sheet (β1-β1´). Strand β1 is built from the disordered residues in dRepA N terminus, whereas strand β1´ and its preceding loop are made at the expense of three helical turns from the C terminus of α5. β1´ is rotated ~60o counterclockwise relative to a virtual axis normal to α5. In mRepE, α5 is much shorter (about two turns) so it is not long enough to be buried between α1 and α2, as it is in dRepA. The loop connecting these two helices becomes longer in mRepE through the loss of one helical turn from α1. This allows α1 and α2 to align roughly antiparallel. Overall, upon dimer dissociation, the conversion of 16 residues from two α-helices (α5 and α1) into two loops and a β-strand (β1´) involves 7% of the residues in the whole RepA. This value is close to the estimate derived from CD spectroscopy measurements for the conformational change coupled to RepA activation12. This confirms that the structural transformation proposed from comparing dRepA and mRepE is functionally relevant, and not an artifact due to having solved the structure of the isolated WH1 domain, rather than the whole RepA dimer. It also indicates that the observed structural changes are not due to intrinsic differences between RepA and RepE sequences or crystal packing. Modeling operator DNA binding by dRepA In dRepA, α3–α4 constitute a near orthogonal HTH motif20 in which α4 would correspond to the DNA binding α-helix (Fig. 1a). However, the closest distance between the antiparallel axes of both α4 helices in the dimer is ~11 Å, much shorter than the value for contiguous major grooves in B-DNA (~36 Å) but similar to the separation between the phosphates flanking a minor groove. Thus, dRepA can be modeled on operator DNA straddling across the minor groove, centered at the middle of the 11 bp sequence linking the two 8 bp arms of the inverted repeat14,5 (Fig. 4, top panel). Six basic residues distributed along α4 (Lys74), β2 (Arg81), β3 (Arg91 and Arg93) and their adjacent loops (Lys62 and Arg78) (Figs. 1b, 4) follow the DNA backbone. Furthermore, this model is in agreement with the footprints reported for RepA dimers5. The WH2 domains from both protomers (absent in the dRepA crystal structure) would bind to the operator arms through the major groove, on alternate sides of the DNA double helix14. Clues to the activation of eukaryotic/archaeal initiators Besides mRepE (see above), the most similar fold to dRepA found in Dali18 corresponds to the C-terminal domain of the chromosomal replication initiator Cdc6/Orc1 from the archaea Pyrobaculum aerophilum21 (Z-score = 5.9). Cdc6 is a monomer (mCdc6) and, thus, lacks one of the two antiparallel β-sheets (β2-β3) forming the dimerization interface in dRepA (Fig. 5). On the basis of their reduced sequence similarities (16% identical residues; Fig. 5a) and based on biochemical and spectroscopic observations, it had been proposed22 that: i) RepA, the C-terminal domain of Cdc6 and some subunits of ORC initiator (Orc1/4/5) share a common WH fold, and ii) Saccharomyces cerevisiae Orc4-WH is a dimer in solution. This is compatible with showing Orc4-WH a partial conservation of dRepA dimerization interface22. mRepE and mCdc6-WH were previously superimposed with a r.m.s. deviation of 2.14 Å for 41 Cα atoms22. dRepA and mCdc6-WH superposition improves that value (r.m.s. deviation = 1.86 Å, for 59 Cα atoms; Fig. 5b), now including the three-helix bundle that undergoes structural

4
transformation (α1, α2 and α5). In addition, the secondary structure elements in both proteins are connected with the same topology. Therefore, with dRepA structure in hand, the proposed similarity between Rep and archaeal/eukaryal initiators2,22 can be confirmed and extended further. Discussion Implications of dRepA structure in initiator activation The structure of RepA N-terminal dimerization domain (dRepA), described in this paper, provides insights on the molecular mechanisms responsible for the initiation of DNA replication in Gram-negative bacteria plasmids. This is fulfilled when dRepA is compared with the monomeric form of the same domain in a homologous initiator (mRepE16), and when the available biophysical, biochemical and genetic evidences14,12 are also considered. Two coupled phenomena occur in the activation of Rep initiators: i) dissociation of dimers, and ii) a conformational change, both affecting precisely the N-terminal dimerization domain. Regarding to dimer dissociation, in pPS1023 and in other related plasmids24-26 a number of mutations in their rep genes have been characterized to result in high plasmid copy number, and thus in more efficient replication initiation. These mutations affect residues (such as Arg91, Arg93 and Thr114 in dRepA) found at the β-sheet dimerization interface (β3 and β4, Fig. 2a). In particular, the mutation R93C in pPS10 RepA23, that results in enhanced protein monomerization (data not shown), maps to the C terminus of β3, at the center of the dimerization β-sheet and close to the pseudo-two fold axis in dRepA (Fig. 2a). Thus plasmid copy-up mutants confirm that Rep dimerization relies on that β-sheet rather than in the LZ-like sequence motif, as previously though19,13,16. However, it had been shown that mutations in the first two leucines of the putative LZ (Leu12 and Leu19) increase dissociation of RepA dimers13,12. This paradox can be readily explained after inspecting dRepA model (Fig. 2b). When Leu12 and Leu19 are mutated12, the three-helix bundle constituted by α1, α2 and α5 cannot be formed and dRepA becomes unstable. A significant proportion of RepA thus shifts into monomers, together with larger aggregates due to the exposure of hydrophobic residues to the solvent12. The hydrogen bond between Thr114 (N terminus of α5) and Tyr45 (β1) from different protomers (Fig 2a) could be used to sense dRepA dissociation and so to trigger the transformation of part of α5 into a loop plus a β-strand. It is noteworthy that the α1-α2-α5 bundle extends the hydrophobic core of WH1 in dRepA (Fig. 2b) further than in mRepE16. This explains both the extraordinary thermal stability of Rep dimers compared with monomers14 and the requirement for chaperones to dissociate Rep6–11. Alternatively, chaperones could mask hydrophobic residues transiently exposed to the solvent during allosteric activation of Rep by iteron DNA. Iteron binding stabilizes the monomeric conformation of RepA (ΔTm = 4.8 oC)12. Interdomain geometry and DNA recognition Although the structural details of the allosteric activation of Rep proteins by iteron DNA remain to be determined, comparison of dRepA and mRepE structures suggests some clues on how the conformational change in WH1 affects DNA recognition. The transformation (Fig. 3) of part of α5 (in dRepA) into β1´ (in mRepE) must affect the interface between WH1 and WH216, having consequences in iteron versus operator recognition14,16,12 (Fig. 4). In mRepE, the β1-β1´ sheet brings both domains in close contact each to the other16, locating WH1 at the precise distance to bind to its site in the iteron repeat14. However, in dRepA α5 would push WH1 apart from the operator-bound WH2. Each WH1 domain in the dimer would be thus forced to sit over the phosphate backbone of the DNA at the sequence spacing both operator arms. Binding to DNA through Arg91 and Arg93 (Figs. 1b, 2a, 4) could reinforce RepA dimerization: in fact, operator binding stabilizes RepA dimers against thermal denaturation (ΔTm = 12.4 oC)12. Multiple regulatory surfaces in Rep proteins

5
When mRepE molecules are bound to contiguous iteron repeats, arranged head-to-tail, the β-sheet formed by β2-β4-β3 (β1-β5-β4 in dRepA) would face the equivalent β-sheet of the WH2 domain in the next monomer16. This is feasible because, in mRepE, domains WH1 and WH2 are related by pseudo-twofold symmetry16. Contacts between those sheets would be responsible for the cooperative binding of RepA to the four adjacent iterons in pPS1012. In addition, once bound to Rep monomers, iteron repeats located in either the same or different plasmid molecules can be brought together by protein–protein interactions, forming a complex which cannot function in initiation. This is the basis for the regulatory mechanism termed ‘handcuffing’27–31. Comparison of dRepA and mRepE structures (Fig. 3) reveals that a partially disordered β-sheet in the monomers (β2a-β2b)16, overlapping with the dimerization element (β3-β4 in dRepA), is potentially available for handcuffing interactions. This would thus be the third protein–protein contact interface in Rep proteins, apart from those involved in dimerization and in cooperative binding to the iterons. Concluding remarks The conformational change that the RepA dimerization domain (WH1) undergoes upon dissociation, enabling it to bind to origin DNA, is one of the largest found so far in any DBD. Conformational changes in DBDs are usually linked to DNA binding32,33, implying either subtle side chain re-orientations or discrete interconversions between secondary structure elements, rather than extensive remodeling in their folds, as described here for RepA. The initial (dRepA) and final (mRepE) states in the activation of initiator proteins in Gram-negative bacteria plasmids2,12,14 are now defined with molecular detail. This leaves the way open to the design of new drugs that would block the dimeric conformation of Rep proteins, thus disabling plasmid DNA replication, to leave pathogenic bacteria helpless against current antibiotics. This would be of clinical relevance since increasing spread of antibiotic-resistant bacteria is a world threat to human health. Furthermore, the astonishing similarity between dRepA and the C-terminal WH domain in Cdc6 confirms the proposal2,22 for a common phylogenetic origin of initiator proteins in Gram-negative bacteria plasmids (Rep) and archaea/eukaryotes (ORC, Cdc6), that would be traceable back to the universal ancestor of all cellular life. Their structural homology also suggests that some ORC initiator subunits might undergo the same kind of activation process characterized for Rep, either upon assembling or binding to origin DNA or to other replication factors (Cdc6). Methods Protein crystallization. dRepA WH1 domain (RepA-ΔC133) was purified as described14. Crystallization was achieved by vapor diffusion in hanging drops. Well reservoirs contained 500 µl of 14% (w/v) PEG4000 plus 6% (v/v) MPD. 10 µl drops included equal volumes of protein (5 mg ml–1 in 50 mM K2HPO4/KH2PO4, pH = 6.2) and well solutions, plus 1 µl of p-Cl-mercuribenzoate (~1 mM in water). Thin large crystal prisms grew at 295 K over a period of one month. Data collection and processing. Crystals were harvested in 20% (w/v) PEG4000, 6% (v/v) MPD, 0.1 M Mes-KOH, pH 6.2 and immediately supplemented with PEG400 to 15% (v/v) for flash-cooling under liquid N2. Diffraction was collected at the DESY-X31 tuneable synchrotron beamline. After measuring the fluorescence emission from Hg, two complete datasets, showing weak anisotropic diffraction to 2.6 Å, were acquired (1.5 o oscillations) at peak and inflection wavelengths (Table 1). Data were processed with MOSFLM34 and reduced with the CCP4 suite programs35 to a maximum resolution of 2.75 Å, based on a I/σI > 2.0 cut-off. Crystals belong to the space group P212121 (a = 51.87 Å, b = 55.53 Å, c = 96.07 Å) and include one dRepA dimer per asymmetric unit, with a Matthews solvent content of 55%. Phasing, model building and refinement. Three main Hg sites were found, both manually and with CNS36, and confirmed in anomalous difference Patterson maps. Three additional Hg sites were added during MAD phasing with SHARP37. The resulting, solvent-flattened, electron density map was readily interpretable and was

6
used, after bone skeletonization, to build an initial model with O38. Two more Hg sites, with lower occupancy, and two benzoate rings were included. Restrained maximum likelihood refinement with phases was carried out with REFMAC39, yielding Rwork 24.3% and Rfree 28.7%. Then simulated annealing (2500 K) plus B-factor refinement, using CNS36, further improved the map to model the two C-terminal Lys132 residues and resulted in dropping Rwork to 23.8% and Rfree to 27.2% (Table 1). In the current model, only Lys85 in chain A falls slightly outside of the favored or allowed regions of the Ramachandran plot40, due to interaction with a phosphate anion involved in crystal packing. Coordinates. dRepA coordinates have been deposited in the Protein Data Bank (accession code 1HKQ). Acknowledgments We thank the staff at beamline X31 in EMBL-DESY for help with data collection. We are indebted to D. Rhodes for the critical reading of the manuscript and, together with V. Ramakrishnan, for encouragement. This work has been financed with grants of Spanish MCyT and a FIS network for research on infectious pathology. Competing interests statement The authors declare that they have no competing financial interests. References 1. del Solar, G., Giraldo, R., Ruíz-Echevarría, M.J., Espinosa, M. & Díaz, R. Replication and control of circular bacterial plasmids. Microbiol. Mol. Biol. Rev. 62, 434-464 (1998). 2. Giraldo, R. Common domains in the initiators of DNA replication in Bacteria, Archaea and Eukarya: Combined structural, functional and phylogenetic perspectives. FEMS Microbiol. Rev. 26, 533-554 (2003). 3. Manen, D., Upegui, G.L. & Caro, L. Monomers and dimers of the RepA protein in plasmid pSC101 replication: Domains in RepA. Proc. Natl. Acad. Sci. USA, 89, 8923-8927 (1992). 4. Ishiai, M., Wada, C., Kawasaki, Y. & Yura, T. Replication initiator protein RepE of mini-F plasmid: Functional differentiation between monomers (initiator) and dimers (autogenous repressor). Proc. Natl. Acad. Sci. USA 91, 3839-3843 (1994). 5. García de Viedma, D., Giraldo, R., Ruíz-Echevarría, M.J., Lurz, R. & Díaz-Orejas, R. Transcription of repA, the gene of the initiation protein of the Pseudomonas plasmid pPS10, is autoregulated by sequential interactions of the RepA protein at a symmetrical operator. J. Mol. Biol. 247, 211-223 (1995). 6. Wickner, S., Skowyra, D., Hoskins, J. & McKenney, K. DnaJ, DnaK, and GrpE heat shock proteins are required in oriP1 DNA replication solely at the RepA monomerization step. Proc. Natl. Acad. Sci. USA 89, 10345-10349 (1992). 7. Sozhamannan, S. & Chattoraj, D.K. Heat shock proteins DnaJ, DnaK and GrpE stimulate P1 plasmid replication by promoting initiator binding to the origin. J. Bacteriol. 175, 3546-3555 (1993). 8. DasGupta, S., Mukhopadhyay, G., Papp, P.P., Lewis, M.S. & Chattoraj, D.K. Activation of DNA binding by the monomeric form of the P1 replication initiator RepA by heat shock proteins DnaJ and DnaK. J. Mol. Biol. 232, 23-34 (1993). 9. Wickner, S., Gottesman, S., Skowyra, D.F., Hoskins, J., McKenney, K. & Maurizi, M.R. A molecular chaperone, ClpA, functions like DnaK and DnaJ. Proc. Natl. Acad. Sci. USA 91, 12218-12222 (1994). 10. Dibbens, J.A., Muraiso, K. & Chattoraj, D.K. Chaperone-mediated reduction of RepA dimerization is associated with RepA conformational change. Mol. Microbiol. 26, 185-195 (1997). 11. Konieczny, I. & Helinski, D.R. The replication initiator protein of the broad-host-range plasmid RK2 is activated by the ClpX chaperone. Proc. Natl. Acad. Sci. USA 94, 14378-14382 (1997).

7
12. Díaz-López, T., Lages-Gonzalo, M., Serrano-López, A., Alfonso, C., Rivas, G., Díaz-Orejas, R. & Giraldo, R. Structural changes in RepA, a plasmid replication initiator, upon binding to origin DNA. J. Biol. Chem. 278, in press (2003). 13. García de Viedma, D., Giraldo, R., Rivas, G., Fernández-Tresguerres, E. & Díaz-Orejas, R. A Leucine-Zipper motif determines different functions in a DNA replication protein. EMBO J. 15, 925-934 (1996). 14. Giraldo, R., Andreu, J.M. & Díaz-Orejas, R. Protein domains and conformational changes in the activation of RepA, a DNA replication initiator. EMBO J. 17, 4511-4526 (1998). 15. Nieto, C., Giraldo, R., Fernández-Tresguerres, E. & Díaz, R. Genetic and functional analysis of the basic replicon of pPS10, a plasmid specific of Pseudomonas isolated from Pseudomonas syringae pv. savastanoi. J. Mol. Biol. 223, 415-426 (1992). 16. Komori, H., Matsunaga, F., Higuchi, Y., Ishiai, M., Wada, C. & Miki, K. Crystal structure of a prokaryotic replication initiator protein bound to DNA at 2.6Å resolution. EMBO J. 18, 4597-4607 (1999). 17. Gajiwala, K.S. & Burley, S.K. Winged helix proteins. Curr. Op. Struct. Biol. 10, 110-116 (2000). 18. Holm, L. & Sander, C. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233, 123-138 (1993). 19. Giraldo, R., Nieto, C., Fernández-Tresguerres, M.E. & Díaz, R. Bacterial zipper. Nature 342, 866 (1989). 20. García de Viedma, D., Serrano-López, A. & Díaz-Orejas, R. Specific binding of the replication protein of plasmid pPS10 to direct and inverted repeats is mediated by an HTH motif. Nucl. Acids Res. 23, 5048-5054 (1995). 21. Liu, J., Smith, C.L., DeRyckere, D., DeAngelis, K., Martin, G.S. & Berger, J.M. Structure and function of Cdc6/Cdc18: Implications for origin recognition and checkpoint control. Mol. Cell 6, 637-648 (2000). 22. Giraldo, R. & Díaz-Orejas, R. Similarities between the DNA replication initiators of Gram-negative bacteria plasmids (RepA) and eukaryotes (Orc4p) / archaea (Cdc6p). Proc. Natl. Acad. Sci. USA 98, 4938-4943 (2001). 23. Maestro, B., Sanz, J.M., Díaz-Orejas, R. & Fernández-Tresguerres, E. Modulation of pPS10 host range by plasmid-encoded RepA initiator protein. J. Bacteriol. 185, 1367-1375 (2003). 24. Matsunaga, F., Ishiai, M., Kobayashi, G., Uga, H., Yura, T. & Wada, C. The central region of RepE initiator protein of mini-F plasmid plays a crucial role in dimerization required for negative replication control. J. Mol. Biol. 274, 27-38 (1997). 25. Xia, G., Manen, D., Yu, Y. & Caro, L. In vivo and in vitro studies of a copy number mutation of the RepA replication protein of plasmid pSC101. J. Bacteriol. 175, 4165-4175 (1993). 26. Miron, A., Patel, I. & Bastia, D. Multiple pathways of copy control of γ replicon of R6K: Mechanisms both dependent on and independent of cooperativity of interaction of π protein with DNA affect the copy number. Proc. Natl. Acad. Sci. USA 91, 6438-6442 (1994). 27. Chattoraj, D.K. Control of plasmid DNA replication by iterons: No longer paradoxical. Mol. Microbiol. 37, 467-476 (2000). 28. Blasina, A., Kittell, B.L., Toukdarian, A.E. & Helinski, D.R. Copy-up mutants of the plasmid RK2 replication initiation protein are defective in coupling RK2 replication origins. Proc. Natl. Acad. Sci. USA 93, 3559-3564 (1996). 29. Urh, M., Wu, J., Wu, J., Forest, K., Inman, R.B. & Filutowicz, M. Assemblies of replication initiation protein on symmetric and asymmetric DNA sequences depend on multiple protein oligomerization surfaces. J. Mol. Biol. 283, 619-631 (1998). 30. Uga, H., Matsunaga, F. & Wada, C. Regulation of DNA replication by iterons: An interaction between the ori2 and incC regions mediated by RepE-bound iterons inhibits DNA replication of mini-F plasmid in Escherichia coli. EMBO J. 18, 3856-3867 (1999). 31. Park, K., Han, E., Paulsson, J. & Chattoraj, D.K. Origin pairing (´handcuffing´) as a mode of negative control of P1 plasmid copy number. EMBO J. 20, 7323-7332 (2001).

8
32. Spolar, R.S. & Record, T.M.Jr. Coupling of local folding to site-specific binding of proteins to DNA. Science 263, 777-784 (1994). 33. Jen-Jacobson, L., Engler, L.E. & Jacobson, L.A. Structural and thermodynamic strategies for site-specific DNA binding proteins. Structure 8, 1015-1023 (2000). 34. Leslie, A.G.W. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallogr. 26 (1992). 35. Collaborative Computational Project-4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D 50, 760-763 (1994). 36. Brünger, A.T. et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D 54, 905-921 (1998). 37. de La Fortelle, E. & Bricogne, G. Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol. 276, 472-494 (1997). 38. Jones, T.A., Zou, J.-Y., Cowan, S.W. & Kjeldgaard, M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47, 110-119 (1991). 39. Murshudov, G.N., Vagin, A.A. & Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 53, 240-255 (1997). 40. Laskowski, R.A, MacArthur, M.W., Moss, D.S. & Thornton, J.M. PROCHECK: a program to check the stereochemical quality of protein structure. J. Appl. Crystallogr. 26, 283-291 (1993). 41. Kraulis, P.J. MOLSCRIPT - a program to produce both detailed and schematic plots of protein structures. Appl. Crystallogr. 24, 946-950 (1996). 42. Merritt, E.A.& Bacon, D.J. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277, 505-524 (1997). 43. Nicholls, A., Sharp. K.A. & Honig, B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11, 281-296 (1991).

9
Table 1. Statistics on data collection, phasing and refinement1 ---------------------------------------------------------------------------------- Data collection Peak Inflection Wavelength (Å) 1.0090 1.0120 Resolution (Å) 48.2 - 2.75 48.2 - 2.75 Reflections Measured 44839 43605 Unique 8343 8288 Rsym (%) 8.1 (32.0) 8.7 (35.8) I/σ (I) 8.0 (2.4) 7.3 (2.1) Completeness (%) 97.7 (97.8) 97.5 (97.6) Phasing (MAD on Hg, 6 sites) Rcullis Isomorph. Reference 0.833 Anomal. 0.758 0.879 Ph. P. Isomorph. Reference 1.331 Anomal. 0.576 0.827 FOM 0.42 initial / 0.85 density mod. Model refinement Resolution (Å) 27.89 - 2.75 Reflections Working set 7421 Test set (10.9%) 806 Rwork / Rfree (%) 23.8 / 27.2 Contents Protein atoms 2026 Hg atoms 8 Benzoate groups 2 Phosphate ion 1 Solvent (O) 30 Bfact Wilson / Average (Å2) 45.9 / 48.1 Stereochemistry: r.m.s. deviation Bond lengths (Å) 0.008 Bond angles (o) 1.400 Ramachandran Chain A Chain B Favoured 92.0 84.0 Allowed 7.1 16.0 Gen. All. 0.9 0.0 Disallow 0.0 0.0 GFact (Aver.) 0.32 0.25 ---------------------------------------------------------------------------------- 1Values from the highest resolution bin (2.90-2.75 Å) are bracketed

10
Figure legends Fig. 1. Overall architecture of dRepA. a, Two orthogonal views of RepA-WH1 dimer, with protomers coloured in yellow and magenta. Model was sketched with MOLSCRIPT41 and rendered with RASTER3D42. b, GRASP43 representation of the electrostatic potential (red: –11 kT.e-1; blue: +11 kT.e-1) on the accessible molecular surface (probe radius 1.4 Å) of dRepA, in the same orientations shown above. Grey oval marks the electropositive region modelled (see Fig. 4) to straddle minor groove phosphates at operator; dotted line represents the DNA helix axis. Fig. 2. Stereo close-ups into details of dRepA structure. a, dRepA dimerization interface. Hydrogen bonds between main chain β-sheet atoms are in green. Side chains involved in dimerization contacts (dotted red lines) are displayed as balls and sticks, with the former colored as their corresponding protomer. Mutations in these side chains result in enhanced Rep monomerization and thus more efficient replication in vivo23-26. b, The three-helix bundle α1-α2-α5. Side chains that contribute to helix package are displayed and fitted into the final 2Fo–Fc electron density map (contoured at 1.0 σ, orange). One of the Hg atoms co-crystallized (linked to His118) is shown as a purple CPK sphere. Figure was created with MOLSCRIPT41 and RASTER3D42. Cyan colouring in part of the ribbons is according to the criteria discussed in Fig. 3. Fig. 3. Comparison of dRepA and mRepE reveals structural differences linked to Rep activation. a, Sequence alignment of dRepA (yellow) and mRepE (red) WH1 domains based on the secondary structure elements found in their crystal structures. Regions showing differences between the two structures are highlighted in cyan (dRepA) and blue (mRepE). Disordered loops are dashed. Asterisks mark identical residues, whereas dots identify conservative (colons) and compatible (single points) sequence substitutions. b, A stereo view of a least squares superposition of dRepA and mRepE (PDB entry 1REP)16 WH1 Cα backbones. Secondary structure elements are labeled according to dRepA. Structural changes in the transformation of dimers (cyan) into monomers (blue) extensively remodel the N and C termini of the domain but do not alter its overall WH fold. Superposition was performed with O38 and then displayed with MOLSCRIPT41 and RASTER3D42. Fig. 4. Scheme showing the conformational activation of Rep proteins, based on dRepA and mRepE structures and on available biochemical and biophysical data12,14. Full-length RepA dimer was modeled by building a WH2 domain from RepE16 (orange) at the C terminus of each dRepA protomer (WH1, yellow). Regions in the dimer undergoing structural changes upon monomerization are in cyan. RepA dimer was manually docked to a 29 bp DNA duplex (light green, for clarity just its sugar-phosphate backbone is displayed), placing the WH2 domains at operator repeats and optimizing potential interactions between protein basic side chains (magenta, Fig. 1b) and the DNA phosphates. In this complex, RepA suffers no significant conformational change12,14 and represses repA transcription5. Rep proteins can dissociate three ways2: i) spontaneously (not favored, because equilibrium is displaced towards dimers12); ii) by chaperones6–11 (dark green) targeting dRepA; and iii) by the allosteric effect of iteron origin DNA12 (pink). Chaperones could also stabilize transient folding intermediates (as RepA-2L2A mutant12). Upon binding to iteron DNA, the structure of WH2 does not change12,14 (orange), but WH1 is extensively remodelled (blue) to become WH1* (in red). This helps WH2 in iteron binding, either for initiation14 or negative control of replication (handcuffing)27–31. The monomer and its complex with iteron DNA correspond to RepE16. Fig. 5. Eukaryotic/archaeal replication initiator mCdc6 is structurally related to bacterial dRepA. a, Sequence alignment of a protomer of dRepA (yellow) and the C-terminal WH domain in the archaea P. aerophilum mCdc621 (green), based on their crystal structures. Secondary structure elements and identical and conserved residues are

11
labeled as in Fig. 3a. b, Least squares superposition of dRepA and mCdc6 (PDB entry 1FNN)21 WH domains, with labeling according to the former. Their nearly identical WH and three-helix bundle moieties are evident. Stereo view was created as in Fig. 3b.


Copyright © 2022 FDOKUMEN




















