
A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures
Upload
independentCategory
view
1download
0
Q4
Q1
Q2
lable at ScienceDirect
European Journal of Medical Genetics xxx (2015) 1e8
123456789101112131415161718192021222324252627282930313233343536373839404142434445464748495051525354
55
EJMG3011_proof ■ 17 January 2015 ■ 1/8
Contents lists avai
European Journal of Medical Genetics
journal homepage: http: / /www.elsevier .com/locate/ejmg
565758596061626364
Clinical research 65666768697071727374757677787980818283848586878889909192939495969715q11.2 microdeletion (BP1eBP2) and developmental delay,behaviour issues, epilepsy and congenital heart disease: A seriesof 52 patients
Clémence Vanlerberghe a,*, Florence Petit b, Valérie Malan c, Catherine Vincent-Delorme b,Sonia Bouquillon a, Odile Boute b, Muriel Holder-Espinasse b,d, Bruno Delobel e,Bénédicte Duban e, Louis Vallee f, Jean-Marie Cuisset f, Marie-Pierre Lemaitre f,Marie-Christine Vantyghemg, Marie Pigeyre g, Sandrine Lanco-Dosen h, Ghislaine Plessis i,Marion Gerard i, Matthieu Decamp i, Michèle Mathieu j, Gilles Morin j,Guillaume Jedraszak j, Frédéric Bilan k, Brigitte Gilbert-Dussardier l, Delphine Fauvert m,Joëlle Roume n, Valérie Cormier-Daire o, Roseline Caumes o, Jacques Puechberty p,David Genevieve p, Pierre Sarda p, Lucie Pinsonp, Patricia Blanchet p, Nathalie Lemeur q,Frenny Sheth r, Sylvie Manouvrier-Hanu b, Joris Andrieux a
a Institut de génétique médicale, Hôpital Jeanne de Flandre, CHRU Lille, Franceb Service de génétique clinique, Hôpital Jeanne de Flandre, CHRU Lille, Francec Service de cytogénétique et d’embryologie, Hôpital Necker-Enfants Malades, AP-HP, Paris, FrancedDepartment of Clinical Genetics, Guy’s Hospital, London, UKeCentre de cytogénétique, Hôpital Saint Vincent de Paul, Lille, Francef Service de neuropédiatrie, Hôpital Jeanne de Flandre, CHRU Lille, Franceg Service d’endocrinologie, Hôpital Claude Huriez, CHRU Lille, Franceh Service de neuropédiatrie, Hôpital de Sambre e Avesnois, Maubeuge, Francei Service de génétique clinique, CHU Caen, Francej Service de génétique clinique, CHU Amiens, Francek Laboratoire de Génétique cellulaire et moléculaire, Pôle Biologie Santé, CHU Poitiers, Francel Service de génétique clinique, La Milétrie, CHU Poitiers, Francem Service de cytogénétique, CHI Poissy, FrancenUnité de génétique médicale, CHI Poissy, FranceoUnité de génétique médicale, Hôpital Necker-Enfants malades, AP-HP, Paris, Francep Service de génétique médicale, CHRU Montpellier, Franceq Service de cytogénétique, CHU Rouen, Francer Institute of Human Genetics, Jodhpur Village Road, Gujarat, India
9899
100101102103104105106107108109110111112
a r t i c l e i n f o
Article history:Received 2 March 2014Accepted 4 January 2015Available online xxx
Keywords:BP1eBP215q11.2 microdeletionCongenital heart diseaseCYFIP1NIPA1NIPA2TUBGCP5
* Corresponding author. Institut de génétique médic20 44 40 18; fax: þ33 3 20 44 68 04.
E-mail address: clemence.vanlerberghe@chru-lille
http://dx.doi.org/10.1016/j.ejmg.2015.01.0021769-7212/� 2015 Published by Elsevier Masson SAS
Please cite this article in press as: Vanlerbergand congenital heart disease: A series of 52 p
a b s t r a c t
Proximal region of chromosome 15 long arm is rich in duplicons that, define five breakpoints (BP) for 15qrearrangements. 15q11.2 microdeletion between BP1 and BP2 has been previously associated withdevelopmental delay and atypical psychological patterns. This region contains four highly-conserved andnon-imprinted genes: NIPA1, NIPA2, CYFIP1, TUBGCP5. Our goal was to investigate the phenotypesassociated with this microdeletion in a cohort of 52 patients.
This copy number variation (CNV) was prevalent in 0.8% patients presenting with developmentaldelay, psychological pattern issues and/or multiple congenital malformations. This was studied by array-CGH at six different French Genetic laboratories. We collected data from 52 unrelated patients (including3 foetuses) after excluding patients with an associated genetic alteration (known CNV, aneuploidy orknown monogenic disease).
Out of 52 patients, mild or moderate developmental delay was observed in 68.3%, 85.4% had speechimpairment and 63.4% had psychological issues such as Attention Deficit and Hyperactivity Disorder,
ale, Hôpital Jeanne de Flandre, Barre Nord 2ème étage, 2, avenue Oscar Lambret, 59037 LILLE CEDEX, France. Tel.: þ33 3
.fr (C. Vanlerberghe).
.
113114115116117118119
he C, et al., 15q11.2 microdeletion (BP1eBP2) and developmental delay, behaviour issues, epilepsyatients, European Journal of Medical Genetics (2015), http://dx.doi.org/10.1016/j.ejmg.2015.01.002

C. Vanlerberghe et al. / European Journal of Medical Genetics xxx (2015) 1e82
1234567891011121314151617181920212223242526272829303132333435363738394041424344454647484950515253545556575859606162636465
66
EJMG3011_proof ■ 17 January 2015 ■ 2/8
676869707172737475
Please cite this article in press as: Vanlerbergand congenital heart disease: A series of 52 p
Autistic Spectrum Disorder or Obsessive-Compulsive Disorder. Seizures were noted in 18.7% patients andassociated congenital heart disease in 17.3%. Parents were analysed for abnormalities in the region in65.4% families. Amongst these families, ‘de novo’ microdeletions were observed in 18.8% and 81.2% wereinherited from one of the parents. Incomplete penetrance and variable expressivity were observedamongst the patients.
Our results support the hypothesis that 15q11.2 (BP1eBP2) microdeletion is associated with devel-opmental delay, abnormal behaviour, generalized epilepsy and congenital heart disease. The later featurehas been rarely described. Incomplete penetrance and variability of expression demands furtherassessment and studies.
� 2015 Published by Elsevier Masson SAS.
7677
78 798081828384858687888990919293949596979899100101102103104105106107108109110111112
1. Introduction
Array-CGH in human genetics has led to the detection of newsubmicroscopic genomic disorders, associated with an increasedrisk of developmental delay, psychological issues and/or epilepsy.Some CNVs are well known and classified as pathogenic or poly-morphic. But, the CNVs for which an association to phenotype isunclear are known as Variant Of Unknown Significance (VOUS). Theproximal long arm of chromosome 15 is a region rich in segmentalduplications, that house five breakpoints (BP) for recurrent 15qCNVs as defined by non-allelic homologous recombination. Themicrodeletion, extending from BP1 to BP3 (type 1) or fromimprinted region BP2 to BP3 (type 2), cause PradereWilli/Angel-man syndrome, depending on the parental origin of the deletedallele (Fig. 1) [Christian et al., 1999]. 15q13.3 microdeletion,between BP4 and BP5; comprising CHRNA7 gene causes mild tomoderate intellectual disabilities associated to epilepsy with avariable phenotype [Masurel-Paulet et al., 2010; Shinawi et al.,2009].
Previous publications have reported patients with 15q11.2microdeletion between BP1 and BP2 presenting with develop-mental delay, speech impairment, learning disabilities and/orbehavioural issues [Abdelmoity et al., 2012; Burnside et al., 2011;Doornbos et al., 2009; Madrigal et al., 2012; Murthy et al., 2007;Sempere Pérez et al., 2011; von der Lippe et al., 2011].Nevertheless, several patients harboured another associatedgenetic alteration and added confusion to genotypeephenotypecorrelations. In addition, this CNV is frequently inherited fromunaffected parent; thus the pathogenicity of 15q11.2microdeletion seem unclear, leading to its interpretation as VOUS.
The goal of the present study therefore was to refine thephenotype associated with this 15q11.2 microdeletion by descrip-tion of a large and multicentre series of patients without a knownassociated pathogenic genetic alteration.
113114115116117118119120121122123124125126127128129130
2. Material and methods
2.1. Patients
A total of 9852 patients were referred for array-CGH duringJanuary 2008 to February 2013 because of intellectual deficiency,behaviour issues and/or multiple congenital abnormalities, inseveral genetics departments in France (Lille, Poitiers, Montpellier,Poissy, Rouen and Paris). Informed consent was obtained from allthe patients and/or their parents. A 15q11.2 microdeletion wasidentified in 80 patients out of referrals. Twenty eight patients wereexcluded from the study because of an associated chromosomalalteration (VOUS or pathogenic CNV), aneuploidy (Klinefelter syn-drome), a monogenic associated diagnosis (GallowayeMowatsyndrome, CASH syndrome) or lack of clinical data. The clinical dataof the remaining 52 patients was retrospectively collected by aquestionnaire sent to each referring clinician to obtain information
he C, et al., 15q11.2 microdeleatients, European Journal of
on prenatal abnormalities, birth parameters, psychomotor devel-opment, neurological examination, behavioural pattern, growth,dysmorphism, associated malformations, paraclinical in-vestigations (audiophonologic examination, cerebral MRI, cardiacultrasound, molecular investigations) and familial history (SeeSupplementary data). Intellectual deficiency was defined as mild(IQ range: 55 and 70), moderate (IQ range: 40e55) and severe(IQ < 40). Speech impairment was defined by an age of firstwords above 20 months or dysphasia. Delayed motor milestonescorresponded to an age of walking above 18 months. All datawere reported on a chart and analysed.
2.2. Array comparative genomic hybridization (aCGH)
Genomic DNA was extracted from the patients’ peripherallymphocytes using the QIAamp DNA Blood Midi kit� (Qiagen�,Valencia, CA). DNA concentration was determined with Nano-Drop� ND-1000 spectrophotometer and software (NanoDrop�Technologies, Berlin, Germany). Detection of copy number varia-tions was performed by array-Comparative Genomic Hybridization(array-CGH) experiments using 44.000 or 60.000 oligo probes(Human Genome CGH microarray 44B or 60Bkit, Agilent� orAffymetrix 6.0) according to the manufacturer’s protocols. Male orfemale genomic DNA (Promega�) was used as reference in sex-matched hybridizations that were analysed with the CGH-analytics software v3.4 and ADM2 algorithm application. Analysiswas performed at filter settings: 3-point filter and 0.2 of variation.
Validation of the microdeletion detected by array CGH wasvalidated by quantitative PCR or Fluorescence In Situ Hybridization.
2.3. Quantitative PCR (qPCR)
Quantitative real time PCR (qPCR; using primers amplifying aunique sequence within exon 6 of CYFIP1) was performed in ma-jority of the probands to confirm 15q11.2 microdeletion and studythe parental inheritance following the manufacturer’s recommen-dations [QuantiTect SYBR Green PCR Kit (Qiagen, CA, USA)] on LightCycler (Roche Diagnostics, Mannheim, Germany). Relative quanti-fication was performed with ddct method and RPPH1 as referencegene (primer sequences available upon request).
2.4. Fluorescence In Situ Hybridization (FISH) analysis
Fluorescence In Situ Hybridization was performed to confirmthe 15q11.2 microdeletion and for study of parental inheritancein other probands using- Bacterial Artificial Chromosome(BAC) clones as follows: onewithin themicrodeletion region [RP11-307C10 located at chr15:22,973,229e23,141,039(hg19) or RP11-289D12 located at chr15:22,812,353e22,990,924(hg19) andone on 15qter (RP11-90E5 located at chr15:100,570,581e100,756,964(hg19)].
tion (BP1eBP2) and developmental delay, behaviour issues, epilepsyMedical Genetics (2015), http://dx.doi.org/10.1016/j.ejmg.2015.01.002

Fig. 1. Chromosome 15 ideogramwith proximal long arm expanded showing the 3 recurrent breakpoints (BP), the critical region for 15q11.2 (BP1eBP2) CNV and PradereWilli(PW)/Angelman syndromes.
C. Vanlerberghe et al. / European Journal of Medical Genetics xxx (2015) 1e8 3
1234567891011121314151617181920212223242526272829303132333435363738394041424344454647484950515253545556575859606162636465
66676869707172737475767778798081828384858687888990919293949596979899
100101102103104105106107108109110111112113114115116117118119120121122123124125126127128129130
EJMG3011_proof ■ 17 January 2015 ■ 3/8
3. Results
The prevalence of 15q11.2 microdeletion (BP1eBP2) was 0.8%(80/9852) in the population under study by array-CGH. Unclearlydefined segmental duplications and a centromeric polymorphicregion (BP1 and BP2) extend respectively betweenchr15:20408283e22784523 and chr15:23047696e23487533 dueto different microarray platforms.
We collected clinical data of 52 unrelated patients. The sex ratiowas 1.5 (31 males versus 21 females). In three prenatal cases, array-CGHwas performedbecause no non-genetic causewas found linkedto ultrasonography findings. 15q11.2 microdeletion was antenatallydiagnosed in these cases and the observed malformations by ul-trasonography led in favour of termination of pregnancy. An over-view of the patients’ features carrying a 15q11.2 (BP1eBP2)microdeletion is presented inTable 1. Phenotypes of the foetuses arespecified in Table 2. The mean age of patients at diagnosis was 8.6years (Range: new borne54 years) and the median age was 6years. Karyotypes did not reveal any cytogenetic rearrangementsassociated with microdeletion at 550-band resolution. Retrospec-tive analysis did not allow us to collect complete information for all52 patients. Amongst the patients that were older enough to getevaluated, 68.3% (28/41) presented intellectual deficiency (ID):39.3% of them (11/28) underwent neuropsychological evaluation,showed mild ID in 6/10 (60%) and moderate ID in 5/10 (50%).
Please cite this article in press as: Vanlerberghe C, et al., 15q11.2 microdeleand congenital heart disease: A series of 52 patients, European Journal of M
Cerebral MRI was performed in 59.6% (31/52) patients; andabnormalities were detected in 38.7%. The postnatal abnormalitieswere: non-specific white matter involvement, cerebral atrophy,ventriculomegaly, cerebellar hypoplasia, thin corpus callosum andparietal and occipital ischaemia.
Echocardiography was performed in 50% (26/52); variablecongenital heart defects were seen in 34.6% (9/26; Table 3).
Both parents were analysed in 34/52 (65.3%) families. ‘De novo’microdeletions were seen in 18.8% (6/34) of these families, 29.4%(10/34) were inherited from a mildly affected parent and 52.9% (18/34) were inherited from an apparently healthy parent. Clinical in-formation about the parent who transmitted was usually incom-plete. In the 10 mildly affected children who got transmission fromparents, 2 (20%) had mild or moderate developmental delay, 5(50%) had speech impairment, 2 (20%) had behavioural issues (ASD,ADHD, Obsessive compulsive behaviour), 3 (30%) presented sei-zures and none had congenital heart disease, but none of themwere investigated with echocardiography.
4. Discussion
An interest in BP1eBP2 regionwas generated due to observationof significant phenotypic differences in patients harbouring BP1eBP3 (type 1) deletion and BP2eBP3 (type 2) deletion in PradereWilli and Angelman syndromes. In PradereWilli syndrome, some
tion (BP1eBP2) and developmental delay, behaviour issues, epilepsyedical Genetics (2015), http://dx.doi.org/10.1016/j.ejmg.2015.01.002

Table 1Overview of the clinical features of patients carrying a 15q11.2 (BP1eBP2) microdeletion. Cases of the present study are compared to previously described cases [Abdelmoityet al., 2012; Burnside et al., 2011; Doornbos et al., 2009; Madrigal et al., 2012; Murthy et al., 2007; Sempere Pérez et al., 2011; von der Lippe et al., 2011].
Sexe Present study [Murthyet al., 2007]
[Doornboset al., 2009]
[von der Lippeet al., 2011]
[Burnsideet al., 2011]
[Sempere Pérezet al., 2011]
[Madrigalet al., 2012]
[Abdelmoityet al., 2012]
% Total (presentstudy and literature)
31M/21F 1M 7M/2F 5M/2F 44M/25F 1M 1M/1F 9M/7F M/F ¼ 1.69
AuxologyIUGR 11/49 (22.4%) 0/1 3/9 1/7 ni 0/1 ni ni 15/67 (22.4%)Height< �2SD 9/45 (20%) ni ni ni ni ni ni ni 9/45 (20%)Obesity 6/43 (14%) ni ni ni ni ni ni ni 6/43 (14%)Failure to thrive 14/44 (31.8%) 0/1 1/9 2/7 ni 0/1 ni ni 17/62 (27.4%)Microcephaly 10/42 (23.8%) ni ni ni ni ni 2/2 4/16 16/60 (26.7%)Psychomotor developmentHypotonia 9/41 (22%) ni ni ni ni ni ni 2/15 11/56 (19.6%)Delayed motor milestones 18/41 (43.9%) 1/1 8/9 3/7 20/56 1/1 2/2 ni 53/117 (45.3%)Speech impairment 35/41 (85.4%) 1/1 8/8 7/7 44/49 1/1 2/2 ni 98/109 (90%)Learning difficulties 27/41 (65.9%) ni ni 6/6 ni 1/1 2/2 ni 36/50 (72%)Neurology-psychiatryIntellectual disability 28/41 (68.3%) 1/1 ni ni 11/49 1/1 2/2 13/16 56/109 (51.4%)Ataxia 5/41 (12.2%) 1/1 ni 0/7 15/49 0/1 0/2 ni 21/101 (20.8%)Seizures 9/48 (18.7%) 0/1 2/8 1/7 14/56 0/1 0/2 2/15 28/138 (20.3%)Psychiatric troubles 0/40 (0%) 0/1 0/9 1/7 ni 0/1 ni ni 1/58 (1.7%)ASD 4/41 (9.8%) 0/1 4/8 2/7 14/49 0/1 2/2 2/16 28/125 (22.4%)ADHD 20/41 (48.8%) 1/1 2/8 1/7 16/49 1/1 0/2 7/12 48/121 (39.7%)OCD 2/41 (4.9%) 0/1 2/8 0/7 16/49 0/1 0/2 ni 20/109 (18.3%)MalformationsHeart defect 9/26 (34.6%) 0/1 2/9 ni ni ni ni ni 11/36 (30.6%)Cerebral malformation 12/31 (38.7%) 0/1 1/4 1/2 ni ni ni ni 14/37 (37.8%)Visual impairment 18/33 (54.5%) ni 3/4 1/2 ni 0/1 ni ni 22/40 (55%)Deafness 0/27 (0%) ni 2/4 ni ni 0/1 ni ni 2/32 (6.2%)Cleft palate/lip 3/42 (7.1%) 1/1 2/9 ni ni 0/1 ni 1/16 7/69 (10.1%)Facial dysmorphismGeneral 30/42 (71.4%) ni 6/9 ni 27/56 1/1 ni ni 64/108 (59.3%)High forehead 10/42 (23.8%) ni 5/9 ni ni 1/1 2/2 1/16 19/77 (24.7%)Hypertelorism 8/42 (19%) ni 5/9 ni ni 1/1 ni 1/16 16/75 (21.3%)Dysplastic ears 13/42 (31%) ni 6/9 ni ni 1/1 2/2 2/16 24/77 (31.2%)Long philtrum 5/42 (12%) ni ni ni ni ni ni 0/16 5/58 (8.6%)High arched palate 5/42 (12%) ni 1/9 ni ni 1/1 2/2 2/15 11/69 (16%)Micrognathia 8/42 (19%) ni ni ni ni ni ni 0/16 8/58 (13.8%)InheritanceDe novo 6/34 (18.8%) 0/1 2/9 0/6 1/44 0/1 0/2 0/7 9/104 (8.7%)Inherited from an
healthy parent18/34 (52.9%) 0/1 6/9 1/6 ni 1/1 0/2 5/7 31/60 (51.7%)
Inherited from a mildlyaffected parent
10/34 (29.4%) 1/1 1/9 5/6 ni 0/1 2/2 2/7 21/60 (35%)
Sex ratio of thetransmitted parent
13M/15F (0.86) ni ni ni ni ni ni ni 13M/15F (0.86)
M: male; F: female; SD: Standard Deviation; ni: no information; IUGR: intrauterinegrowth retardation; ASD: autismspectrumdisorder; ADHD: Attention Deficit-HyperactivityDisorder; OCD: obsessive compulsive disorder.
C. Vanlerberghe et al. / European Journal of Medical Genetics xxx (2015) 1e84
1234567891011121314151617181920212223242526272829303132333435363738394041424344454647484950515253545556575859606162636465
66676869707172737475767778798081828384858687888990919293949596979899
100101102103104105106107108109110111112113114115116117118119120121122123124125126127128129130
EJMG3011_proof ■ 17 January 2015 ■ 4/8
authors observed a severe phenotype in patients with type 1deletion than type 2 deletion considering the behavioural pattern,reading impairment, mathematical skills and visual-motor inte-gration [Bittel et al., 2006; Butler et al., 2004; Hartley et al., 2005].However, other studies did not find any phenotypic differencesbetween the two groups [Milner et al., 2005]. Type 1 deletionpatients have less performance in vocalization and motordevelopment in Angelman syndrome, and greater severity inAutistic Spectrum Disorder (ASD) features than type 2 deletedsubjects [Peters et al., 2012; Sahoo et al., 2006; Varela et al., 2004].
The BP1eBP2 region of 300 kb encompasses four genes: NIPA1,NIPA2, CYFIP1, TUBGCP5 [Chai et al., 2003]. Functional analysisshowed a bi-allelic expression of those four genes, proving theirnon-imprinted expression [Chai et al., 2003]. Studies have alreadydemonstrated haploinsufficiency of these four genes inindividuals carrying BP1eBP2 microdeletion [Chai et al., 2003].Three of these genes are widely expressed in the central nervoussystem (CYFIP1, NIPA1, NIPA2), while TUBGCP5 is specificallyexpressed in the subthalamic nuclei [Nagase et al., 2001]. NIPA1gene mutations are known to cause autosomal dominanthereditary spastic paraplegia (SPG6) by a dominant negativeeffect [Rainier et al., 2003].
Please cite this article in press as: Vanlerberghe C, et al., 15q11.2 microdeleand congenital heart disease: A series of 52 patients, European Journal of
4.1. Prevalence
Prevalence of 15q11.2 microdeletion (BP1eBP2) was 0.8% in ourstudy population. In Database of Genomic Variant (DGV), this CNVis reported in 0.09% (2/2026) controls subjects [Shaikh et al., 2009].Pooled frequency of this CNV in control populations was 0.2% (30/15382) in association studies [Cooper et al., 2011; Itsara et al., 2009;Krawczak et al., 2006; Soemedi et al., 2012;Wichmann et al., 2005].These results are consistent with Rosenfeld et al. who reported afrequency of 0.81% (203/25113) in postnatal aCGH cases and of0.38% (84/22246) in controls [Rosenfeld et al., 2013].
Even if 15q11.2 (BP1eBP2) microdeletion seems to be morefrequent in the population studied by array-CGH for intellectualdeficiency, behaviour issues and/or multiple congenital abnor-malities, this was not statistically significant in the populationstudied.
4.2. Neurodevelopmental disorders
Mild to moderate Intellectual Deficiency was present in 68.3%patients in the current study who underwent IQ evaluation. This iscoherent with previous literature data [Abdelmoity et al., 2012;
tion (BP1eBP2) and developmental delay, behaviour issues, epilepsyMedical Genetics (2015), http://dx.doi.org/10.1016/j.ejmg.2015.01.002
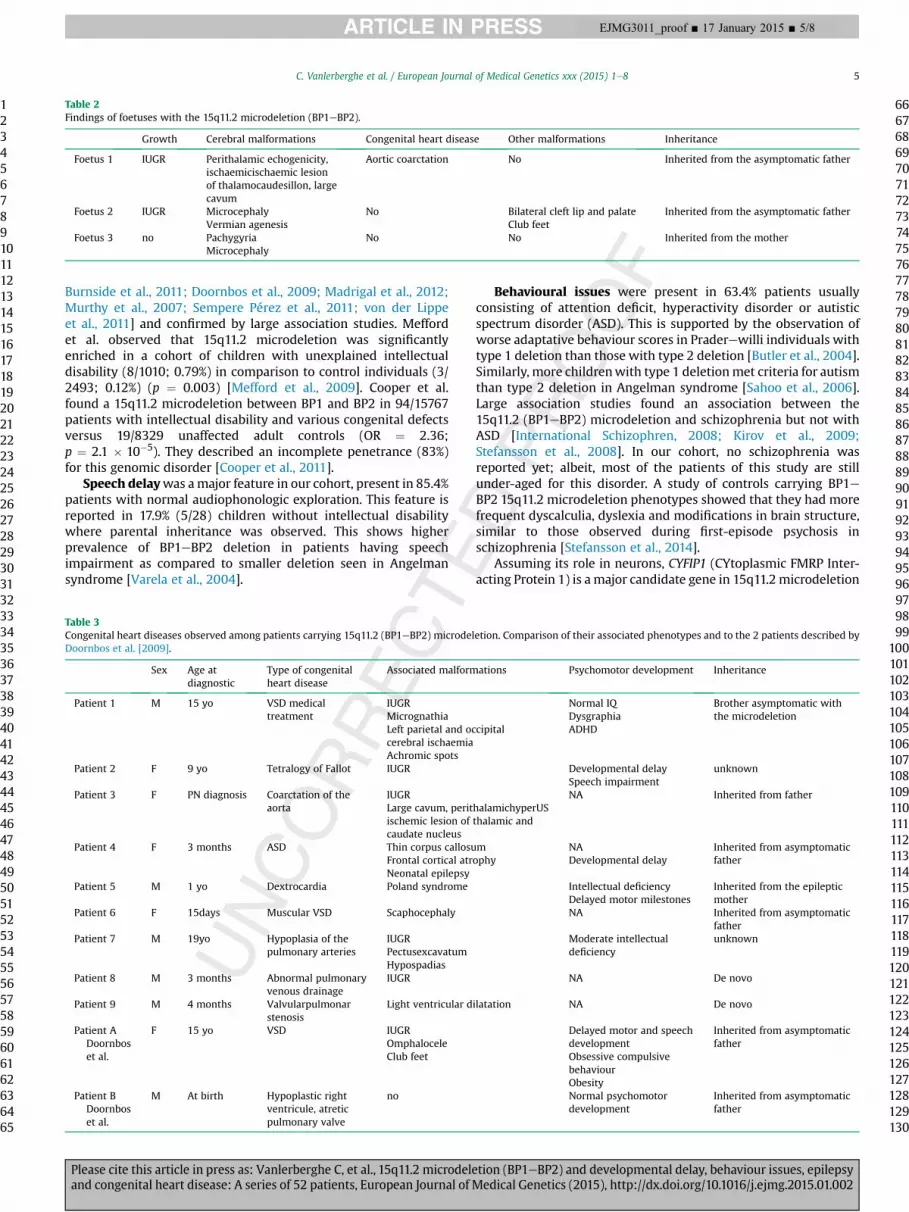
Table 2Findings of foetuses with the 15q11.2 microdeletion (BP1eBP2).
Growth Cerebral malformations Congenital heart disease Other malformations Inheritance
Foetus 1 IUGR Perithalamic echogenicity,ischaemicischaemic lesionof thalamocaudesillon, largecavum
Aortic coarctation No Inherited from the asymptomatic father
Foetus 2 IUGR MicrocephalyVermian agenesis
No Bilateral cleft lip and palateClub feet
Inherited from the asymptomatic father
Foetus 3 no PachygyriaMicrocephaly
No No Inherited from the mother
C. Vanlerberghe et al. / European Journal of Medical Genetics xxx (2015) 1e8 5
1234567891011121314151617181920212223242526272829303132333435363738394041424344454647484950515253545556575859606162636465
66676869707172737475767778798081828384858687888990919293949596
EJMG3011_proof ■ 17 January 2015 ■ 5/8
Burnside et al., 2011; Doornbos et al., 2009; Madrigal et al., 2012;Murthy et al., 2007; Sempere Pérez et al., 2011; von der Lippeet al., 2011] and confirmed by large association studies. Meffordet al. observed that 15q11.2 microdeletion was significantlyenriched in a cohort of children with unexplained intellectualdisability (8/1010; 0.79%) in comparison to control individuals (3/2493; 0.12%) (p ¼ 0.003) [Mefford et al., 2009]. Cooper et al.found a 15q11.2 microdeletion between BP1 and BP2 in 94/15767patients with intellectual disability and various congenital defectsversus 19/8329 unaffected adult controls (OR ¼ 2.36;p ¼ 2.1 � 10�5). They described an incomplete penetrance (83%)for this genomic disorder [Cooper et al., 2011].
Speech delaywas amajor feature in our cohort, present in 85.4%patients with normal audiophonologic exploration. This feature isreported in 17.9% (5/28) children without intellectual disabilitywhere parental inheritance was observed. This shows higherprevalence of BP1eBP2 deletion in patients having speechimpairment as compared to smaller deletion seen in Angelmansyndrome [Varela et al., 2004].
Table 3Congenital heart diseases observed among patients carrying 15q11.2 (BP1eBP2) microdelDoornbos et al. [2009].
Sex Age atdiagnostic
Type of congenitalheart disease
Associated malform
Patient 1 M 15 yo VSD medicaltreatment
IUGRMicrognathiaLeft parietal and occerebral ischaemiaAchromic spots
Patient 2 F 9 yo Tetralogy of Fallot IUGR
Patient 3 F PN diagnosis Coarctation of theaorta
IUGRLarge cavum, peritischemic lesion ofcaudate nucleus
Patient 4 F 3 months ASD Thin corpus callosuFrontal cortical atrNeonatal epilepsy
Patient 5 M 1 yo Dextrocardia Poland syndrome
Patient 6 F 15days Muscular VSD Scaphocephaly
Patient 7 M 19yo Hypoplasia of thepulmonary arteries
IUGRPectusexcavatumHypospadias
Patient 8 M 3 months Abnormal pulmonaryvenous drainage
IUGR
Patient 9 M 4 months Valvularpulmonarstenosis
Light ventricular d
Patient ADoornboset al.
F 15 yo VSD IUGROmphaloceleClub feet
Patient BDoornboset al.
M At birth Hypoplastic rightventricule, atreticpulmonary valve
no
Please cite this article in press as: Vanlerberghe C, et al., 15q11.2 microdeleand congenital heart disease: A series of 52 patients, European Journal of M
Behavioural issues were present in 63.4% patients usuallyconsisting of attention deficit, hyperactivity disorder or autisticspectrum disorder (ASD). This is supported by the observation ofworse adaptative behaviour scores in Praderewilli individuals withtype 1 deletion than those with type 2 deletion [Butler et al., 2004].Similarly, more childrenwith type 1 deletionmet criteria for autismthan type 2 deletion in Angelman syndrome [Sahoo et al., 2006].Large association studies found an association between the15q11.2 (BP1eBP2) microdeletion and schizophrenia but not withASD [International Schizophren, 2008; Kirov et al., 2009;Stefansson et al., 2008]. In our cohort, no schizophrenia wasreported yet; albeit, most of the patients of this study are stillunder-aged for this disorder. A study of controls carrying BP1eBP2 15q11.2 microdeletion phenotypes showed that they had morefrequent dyscalculia, dyslexia and modifications in brain structure,similar to those observed during first-episode psychosis inschizophrenia [Stefansson et al., 2014].
Assuming its role in neurons, CYFIP1 (CYtoplasmic FMRP Inter-acting Protein 1) is a major candidate gene in 15q11.2 microdeletion
etion. Comparison of their associated phenotypes and to the 2 patients described by
ations Psychomotor development Inheritance
cipital
Normal IQDysgraphiaADHD
Brother asymptomatic withthe microdeletion
Developmental delaySpeech impairment
unknown
halamichyperUSthalamic and
NA Inherited from father
mophy
NADevelopmental delay
Inherited from asymptomaticfather
Intellectual deficiencyDelayed motor milestones
Inherited from the epilepticmother
NA Inherited from asymptomaticfather
Moderate intellectualdeficiency
unknown
NA De novo
ilatation NA De novo
Delayed motor and speechdevelopmentObsessive compulsivebehaviourObesity
Inherited from asymptomaticfather
Normal psychomotordevelopment
Inherited from asymptomaticfather
979899
100101102103104105106107108109110111112113114115116117118119120121122123124125126127128129130
tion (BP1eBP2) and developmental delay, behaviour issues, epilepsyedical Genetics (2015), http://dx.doi.org/10.1016/j.ejmg.2015.01.002

C. Vanlerberghe et al. / European Journal of Medical Genetics xxx (2015) 1e86
1234567891011121314151617181920212223242526272829303132333435363738394041424344454647484950515253545556575859606162636465
66676869707172737475767778798081828384858687888990919293949596979899
100101102103104105106107108109110111112113114115116117118119120121122123124125126127128129130
EJMG3011_proof ■ 17 January 2015 ■ 6/8
for neurodevelopmental disorders. CYFIP1is a partner of FMRP, theprotein impaired in Fragile X syndrome. FMRP belongs to the familyof RNA binding proteins and has a role in mRNA stability, transportand translation. CYFIP1 acts as an eIF4E-binding protein (4E-BP),repressing mRNA translation initiation by sequestration of the cap-binding protein eIF4E [Napoli et al., 2008]. Control of the synthesisof these proteins is crucial in synaptic plasticity and long-lastingchanges, for consolidation and storage of long-term memories,through mTOR pathway. Absence of one component of theeIF4E-CYFIP1-FMRP complex leads to its dislocation and to the alleviationof translation repression. Thus, haploinsufficiency of CYFIP1 canreproduce the effect of FMR inactivation in Fragile X syndrome [DeRubeis and Bagni, 2011]. This hypothesis is supported by micemodels in which haploinsufficiency of Cyfip1 mimics chemicalaspects of Fmr1 knockout [Bozdagi et al., 2012]. The whole codingregion of CYFIP1was sequenced in three patients with a 15q11.2deletion, presenting with ASD and no mutation was found[Depienne et al., 2009]. Reduced CYFIP1 mRNA expression, butnormal FMR1 mRNA levels were observed in four 15q11.2 (BP1eBP2) microdeletion carriers [Madrigal et al., 2012].
In addition, TUBGCP5 could be another candidate gene forbehavioural issues in these patients, since this gene is expressed insubthalamic nuclei; an area implicated in attention deficit hyper-activity disorder and obsessive-compulsive disorder [Grabli et al.,2004; Nagase et al., 2001].
4.3. Seizures
Seizures were reported in 18.7% patients in our cohort, versus5% in the general population [OMS, n.d.]. No specific type ofepilepsy was noticed. Nine patients had epilepticencephalopathy. Significant association has been describedbetween Idiopathic Generalized Epilepsy (IGE) and the 15q11.2(BP1eBP2) microdeletion [de Kovel et al., 2010]. This CNV wasobserved in 1% (12/1234) patients with IGE, and 0.2% (6/3022)controls [de Kovel et al., 2010]. In addition, Mefford et al.reported 5 carriers of the 15q11.2 microdeletion in a populationof 517 individuals with various idiopathic cryptogenic epilepsies[Mefford et al., 2010]. However, no 15q11.2 CNV has beenidentified in a group of 315 patients with epilepticencephalopathies [Mefford et al., 2011].
Assuming that most of the previously described genes for hu-man idiopathic epilepsies encode ions channels, NIPA1 and NIPA2are strong candidate genes for epilepsy associated with 15q11.2BP1eBP2 region. These genes encode selective Mg2þ transporters[Goytain et al., 2008; Rainier et al., 2003]. Three different NIPA2heterozygous mutations (2 missenses, one in-frame insertion)were identified in 380 Chinese patients presenting with childhoodabsence epilepsy. All mutations were inherited from an asymp-tomatic father, however no mutation was found in 700 controls(p¼ 0.043) [Jiang et al., 2012]. These data are controversial since theresults could not be confirmed in Caucasian patients withchildhood absence epilepsy or other generalized epilepsy. Thesame in-frame insertion was present in 2.6% controls rather than1.4% in patients [Hildebrand et al., 2014; Jiang et al., 2014].
In the current cohort, we observed a high prevalence of severalcerebral abnormalities (38.7% patients in whom MRI was per-formed). Previous clinical descriptions did not mention cerebralmalformations. Systematic MRI examination of forth coming pa-tients could result in to more precise information of this condition.
4.4. Congenital heart disease (CHD)
Amongst the patients investigated by echocardiography, 34.6%presented with CHD. Two patients with a 15q11.2 encompassing
Please cite this article in press as: Vanlerberghe C, et al., 15q11.2 microdeleand congenital heart disease: A series of 52 patients, European Journal of
BP1eBP2 microdeletion an heart defect have already beendescribed [Doornbos et al., 2009]. In addition, significantassociation between congenital heart disease (CHD) and 15q11.2microdeletion has also been observed earlier: 12 microdeletionswere identified in a cohort of 2256 patients with CHD, while onlyone was found in 1538 controls (OR ¼ 8.2; p ¼ 0.02). Phenotypeswere complex; left or right-sided malformations, coarctation ofaorta, atrial septal defect, ventricular septal defect, total anomalouspulmonary venous drainage and Tetralogy of Fallot [Soemedi et al.,2012]. Among the genes in the deletion, TUBGCP5 and NIPA1 arereported to be expressed in the foetal heart; however their rolein the developing heart has not yet been fully elucidated[Soemedi et al., 2012]. TUBGCP5 encodes human g-globulincomplex required for microtubule nucleation at the core of thecentrosome; this gene therefore may interfere in development[Xiong and Oakley, 2009]. However, as occurrence of CHD may beunderestimated in patients with 15q11.2 BP1eBP2 deletion,echocardiographic examination should be performedsystematically.
4.5. Minor features
Association of the BP1eBP2 15q11.2 microdeletion with otherminor signs is also reported. Some dysmorphic features have beendescribed [Abdelmoity et al., 2012; Burnside et al., 2011; Doornboset al., 2009; Madrigal et al., 2012; Sempere Pérez et al., 2011]. Thesefindings however are inconstant and no recognizable facial featureshave been delineated in the current larger series. In our cohort, 14%of patients had obesity. One patient was previously described witha central obesity in the context of obsessive compulsive behaviour[Doornbos et al., 2009]. Obesity may be linked to behaviouraldisorders as obsessive compulsive behaviour and intellectualdisability in these patients.
4.6. Prenatal
We have reported 3 foetal cases of 15q11.2 microdeletion. Allpregnancies were terminated because of the severity of cerebralmalformations. 15q11.2 microdeletion in these three cases wasinherited from apparently healthy parents.
4.7. Inheritance
In most cases, 15q11.2 microdeletion was inherited fromapparently healthy parents (52.9%). We did not observe anyparental sex bias consistent with the absence of parentalimprinting in this region [Bittel et al., 2006]. The parents of affectedpatients in this study might have been underestimated since wecould not obtain complete information on parents’ developmentalmilestones. De novo microdeletion was observed in 18.8%patients. No differences in the phenotypes was noticed between‘de novo’ or inherited cases.
4.8. Incomplete penetrance and variability of expression
We observed an incomplete penetrance of 15q11.2 micro-deletion and a wide variability of expression ranging from severelyaffected patients (epileptic encephalopathy, cerebral malforma-tions, cardiac malformations) to patients suffering from milddevelopmental delay. In the first study on developmental delay, apenetrance rate of 83% was reported [Cooper et al., 2011]. Rosenfeldet al. conducted a Bayesian analysis on an increased population sizeand penetrance estimates for this CNV at 10.4% (95% CI; 8.45e12.7)[Rosenfeld et al., 2013]. Bittel et al. studied expression level of BP1eBP2 region’s four genes in individuals with PradereWilli syndrome
tion (BP1eBP2) and developmental delay, behaviour issues, epilepsyMedical Genetics (2015), http://dx.doi.org/10.1016/j.ejmg.2015.01.002

C. Vanlerberghe et al. / European Journal of Medical Genetics xxx (2015) 1e8 7
1234567891011121314151617181920212223242526272829303132333435363738394041424344454647484950515253545556575859606162636465
6667686970717273747576777879808182838485868788899091929394959697
EJMG3011_proof ■ 17 January 2015 ■ 7/8
with type 1 or type 2 deletion by quantification of mRNA isolatedfrom lymphoblastoid cell cultures. They found reduced butdetectable quantities of mRNA in individuals with type 1 deletioncompared with subjects to type 2 deletion supporting a biallelicexpression of the four genes. Furthermore, there was acorrelation between mRNA quantity and cognitive andbehavioural parameters; the highest of correlation for NIPA2 gene[Bittel et al., 2006].
Several hypotheses can be considered to explain the incompletepenetrance and the interfamilial variability of expression. Asdescribed previously for other recurrent CNVs [Girirajan et al., 2010,2012], a “two-hit model” in which a second event occur for theremaining allele causing bi-allelic haploinsufficiency of one of thegenes included in microdeletion can be suggested. A strongcandidate gene could be CYFIP1 because of the interaction betweenCYFIP1 and FMRP proteins. However, sequencing of these fourgenes between BP1 and BP2 has been performed in several patientssuffering from autism and harbouring 15q11.2 microdeletionwithout any mutations [Depienne et al., 2009]. Otherwise, apotential second event could occur in non-coding regions or atanother locus, affecting a modifier gene. By replication-timingstudies, Chai et al. determined the replication of mouse genomicregion spanning Nipa1-Nipa2-Cyfip1 that showed a pattern ofasynchrony and was unrelated to parent-of-origin influences [Chaiet al., 2003]. Finally, epigenetic modifications, mi RNA action and/orenvironmental implication may be considered. Further molecularand functional studies are warranted to understand theincomplete penetrance and the variability of expression of thisCNV.
Thus, genetic counselling is very difficult regarding this locus.This CNV may be interpreted differently depending on patient-specific and family-specific informations [De Wolf et al., 2013].
9899
100101102103104105106107108109110111112113114115116117118119120
5. Conclusion
We confirmed the association of 15q11.2 (BP1eBP2) micro-deletion with mild/moderate intellectual deficiency, behaviouralissues, speech impairment and epilepsy, and with non-specificcongenital heart disease; the later has been described rarely. Thecore phenotype of this deletion seems to be mild intellectualdeficiency with speech impairment, attention deficit and hyper-activity disorder.
Cardiac or cerebral malformations are described less frequently.An ascertainment bias has to be considered as patients with asso-ciated malformations are more likely to be analysed by array CGHthan patients with isolated intellectual deficiency. The brain func-tion and cardiac malformations remain under diagnosed since thepatients with isolated intellectual deficiency are not thoroughlysubjected to evaluation by MRI and echocardiography.
Furthermore, incomplete penetrance and high variability ofexpression amongst patients imply that associated genetic and/orenvironmental events could cause the more severe phenotypes,notably the prenatal ones. Thus further explorations and geneticstudiesmay be advised for severe and/ormalformative phenotypes.
121122123124
Conflict of interest
We declare no conflict of interest.
125126127128129130Acknowledgements
We thank the patients and their parents for their participationto this study.
Please cite this article in press as: Vanlerberghe C, et al., 15q11.2 microdeleand congenital heart disease: A series of 52 patients, European Journal of M
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejmg.2015.01.002.
References
Abdelmoity AT, LePichon J-B, Nyp SS, Soden SE, Daniel CA, Yu S. 15q11.2 proximalimbalances associated with a diverse array of neuropsychiatric disorders andmild dysmorphic features. J. Dev. Behav. Pediatr. JDBP 2012;33:570e6. http://dx.doi.org/10.1097/DBP.0b013e31826052ae.
Bittel DC, Kibiryeva N, Butler MG. Expression of 4 genes between chromosome 15breakpoints 1 and 2 and behavioral outcomes in Prader-Willi syndrome. Pe-diatrics 2006;118:e1276e1283. http://dx.doi.org/10.1542/peds.2006-0424.
Bozdagi O, Sakurai T, Dorr N, Pilorge M, Takahashi N, Buxbaum JD. Hap-loinsufficiency of Cyfip1 produces fragile X-like phenotypes in mice. PLoS One2012;7:e42422. http://dx.doi.org/10.1371/journal.pone.0042422.
Burnside RD, Pasion R, Mikhail FM, Carroll AJ, Robin NH, Youngs EL, et al. Micro-deletion/microduplication of proximal 15q11.2 between BP1 and BP2: a sus-ceptibility region for neurological dysfunction including developmental andlanguage delay. Hum. Genet. 2011;130:517e28. http://dx.doi.org/10.1007/s00439-011-0970-4.
Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T. Behavioral differ-ences among subjects with Prader-Willi syndrome and type I or type II deletionand maternal disomy. Pediatrics 2004;113:565e73.
Chai J-H, Locke DP, Greally JM, Knoll JHM, Ohta T, Dunai J, et al. Identification of fourhighly conserved genes between breakpoint hotspots BP1 and BP2 of thePrader-Willi/Angelman syndromes deletion region that have undergoneevolutionary transposition mediated by flanking duplicons. Am. J. Hum. Genet.2003;73:898e925. http://dx.doi.org/10.1086/378816.
Christian SL, Fantes JA, Mewborn SK, Huang B, Ledbetter DH. Large genomicduplicons map to sites of instability in the Prader-Willi/Angelman syndromechromosome region (15q11-q13). Hum. Mol. Genet. 1999;8:1025e37.
Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, et al. A copy numbervariation morbidity map of developmental delay. Nat. Genet. 2011;43:838e46.http://dx.doi.org/10.1038/ng.909.
Depienne C, Moreno-De-Luca D, Heron D, Bouteiller D, Gennetier A, Delorme R,et al. Screening for genomic rearrangements and methylation abnormalities ofthe 15q11-q13 region in autism spectrum disorders. Biol. Psychiatry 2009;66:349e59. http://dx.doi.org/10.1016/j.biopsych.2009.01.025.
von der Lippe C, Rustad C, Heimdal K, Rødningen OK. 15q11.2 microdeletion e sevennew patients with delayed development and/or behavioural problems. Eur. J.Med. Genet. 2011;54:357e60. http://dx.doi.org/10.1016/j.ejmg.2010.12.008.
Doornbos M, Sikkema-Raddatz B, Ruijvenkamp CAL, Dijkhuizen T, Bijlsma EK,Gijsbers ACJ, et al. Nine patients with a microdeletion 15q11.2 betweenbreakpoints 1 and 2 of the Prader-Willi critical region, possibly associated withbehavioural disturbances. Eur. J. Med. Genet. 2009;52:108e15. http://dx.doi.org/10.1016/j.ejmg.2009.03.010.
Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, Itsara A, et al.A recurrent 16p12.1 microdeletion supports a two-hit model for severe devel-opmental delay. Nat. Genet. 2010;42:203e9. http://dx.doi.org/10.1038/ng.534.
Girirajan S, Rosenfeld JA, Coe BP, Parikh S, Friedman N, Goldstein A, et al. Phenotypicheterogeneity of genomic disorders and rare copy-number variants. N. Engl. J.Med. 2012;367:1321e31. http://dx.doi.org/10.1056/NEJMoa1200395.
Goytain A, Hines RM, Quamme GA. Functional characterization of NIPA2, a selectiveMg2þ transporter. Am. J. Physiol. Cell. Physiol. 2008;295:C944e53. http://dx.doi.org/10.1152/ajpcell.00091.2008.
Grabli D, McCairn K, Hirsch EC, Agid Y, Féger J, François C, et al. Behavioural dis-orders induced by external globus pallidus dysfunction in primates: I. Behav-ioural study. Brain J. Neurol. 2004;127:2039e54. http://dx.doi.org/10.1093/brain/awh220.
Hartley SL, Maclean Jr WE, Butler MG, Zarcone J, Thompson T. Maladaptive be-haviors and risk factors among the genetic subtypes of Prader-Willi syndrome.Am. J. Med. Genet. A 2005;136:140e5. http://dx.doi.org/10.1002/ajmg.a.30771.
Hildebrand MS, Damiano JA, Mullen SA, Bellows ST, Scheffer IE, Berkovic SF. Doesvariation in NIPA2 contribute to genetic generalized epilepsy? Hum. Genet.2014. http://dx.doi.org/10.1007/s00439-013-1414-0.
International Schizophrenia Consortium. Rare chromosomal deletions and dupli-cations increase risk of schizophrenia. Nature 2008;455:237e41. http://dx.doi.org/10.1038/nature07239.
Itsara A, Cooper GM, Baker C, Girirajan S, Li J, Absher D, et al. Population analysis oflarge copy number variants and hotspots of human genetic disease. Am. J. Hum.Genet. 2009;84:148e61. http://dx.doi.org/10.1016/j.ajhg.2008.12.014.
Jiang Y, Zhang Y, Zhang P, Sang T, Zhang F, Ji T, et al. NIPA2 located in 15q11.2 ismutated in patients with childhood absence epilepsy. Hum. Genet. 2012;131:1217e24. http://dx.doi.org/10.1007/s00439-012-1149-3.
Jiang Y, Zhang Y, Zhang P, Zhang F, Xie H, Chan P, et al. NIPA2 mutations arecorrelative with childhood absence epilepsy in the Han Chinese population.Hum. Genet. 2014. http://dx.doi.org/10.1007/s00439-014-1428-2.
Kirov G, Grozeva D, Norton N, Ivanov D, Mantripragada KK, Holmans P, et al. Sup-port for the involvement of large copy number variants in the pathogenesis ofschizophrenia. Hum. Mol. Genet. 2009;18:1497e503. http://dx.doi.org/10.1093/hmg/ddp043.
tion (BP1eBP2) and developmental delay, behaviour issues, epilepsyedical Genetics (2015), http://dx.doi.org/10.1016/j.ejmg.2015.01.002

3
C. Vanlerberghe et al. / European Journal of Medical Genetics xxx (2015) 1e88
123456789101112131415161718192021222324252627282930313233343536373839
404142434445464748495051525354555657585960616263646566676869707172737475
EJMG3011_proof ■ 17 January 2015 ■ 8/8
de Kovel CGF, Trucks H, Helbig I, Mefford HC, Baker C, Leu C, et al. Recurrentmicrodeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalizedepilepsies. Brain J. Neurol. 2010;133:23e32. http://dx.doi.org/10.1093/brain/awp262.
Krawczak M, Nikolaus S, von Eberstein H, Croucher PJP, El Mokhtari NE, Schreiber S.PopGen: population-based recruitment of patients and controls for the analysisof complex genotype-phenotype relationships. Community Genet. 2006;9:55e61. http://dx.doi.org/10.1159/000090694.
Madrigal I, Rodríguez-Revenga L, Xunclà M, Milà M. 15q11.2 microdeletion andFMR1 premutation in a family with intellectual disabilities and autism. Gene2012;508:92e5. http://dx.doi.org/10.1016/j.gene.2012.07.023.
Masurel-Paulet A, Andrieux J, Callier P, Cuisset JM, Le Caignec C, Holder M, et al.Delineation of 15q13.3 microdeletions. Clin. Genet. 2010;78:149e61. http://dx.doi.org/10.1111/j.1399-0004.2010.01374.x.
Mefford HC, Cooper GM, Zerr T, Smith JD, Baker C, Shafer N, et al. A method forrapid, targeted CNV genotyping identifies rare variants associated with neu-rocognitive disease. Genome Res. 2009;19:1579e85. http://dx.doi.org/10.1101/gr.094987.109.
Mefford HC, Muhle H, Ostertag P, von Spiczak S, Buysse K, Baker C, et al. Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathicgeneralized and focal epilepsies. PLoS Genet. 2010;6:e1000962. http://dx.doi.org/10.1371/journal.pgen.1000962.
Mefford HC, Yendle SC, Hsu C, Cook J, Geraghty E, McMahon JM, et al. Rare copynumber variants are an important cause of epileptic encephalopathies. Ann.Neurol. 2011;70:974e85. http://dx.doi.org/10.1002/ana.22645.
Milner KM, Craig EE, Thompson RJ, Veltman MWM, Thomas NS, Roberts S, et al.Prader-Willi syndrome: intellectual abilities and behavioural features by ge-netic subtype. J. Child. Psychol. Psychiatry 2005;46:1089e96. http://dx.doi.org/10.1111/j.1469-7610.2005.01520.x.
Murthy SK, Nygren AOH, El Shakankiry HM, Schouten JP, Al Khayat AI, Ridha A, et al.Detection of a novel familial deletion of four genes between BP1 and BP2 of thePrader-Willi/Angelman syndrome critical region by oligo-array CGH in a childwith neurological disorder and speech impairment. Cytogenet. Genome Res.2007;116:135e40. http://dx.doi.org/10.1159/000097433.
Nagase T, Kikuno R, Ohara O. Prediction of the coding sequences of unidentifiedhuman genes. XXI. The complete sequences of 60 new cDNA clones from brainwhich code for large proteins. DNA Res. Int. J. Rapid Publ. Rep. Genes. Genomes2001;8:179e87.
Napoli I, Mercaldo V, Boyl PP, Eleuteri B, Zalfa F, De Rubeis S, et al. The fragileX syndrome protein represses activity-dependent translation through CYFIP1,a new 4E-BP. Cell 2008;134:1042e54. http://dx.doi.org/10.1016/j.cell.2008.07.031.
OMS j Epilepsie: étiologie, épidémiologie et prognostic, WHO. (n.d.).Peters SU, Horowitz L, Barbieri-Welge R, Taylor JL, Hundley RJ. Longitudinal follow-
up of autism spectrum features and sensory behaviors in Angelman syndromeby deletion class. J. Child. Psychol. Psychiatry 2012;53:152e9. http://dx.doi.org/10.1111/j.1469-7610.2011.02455.x.
Please cite this article in press as: Vanlerberghe C, et al., 15q11.2 microdeleand congenital heart disease: A series of 52 patients, European Journal of
Rainier S, Chai J-H, Tokarz D, Nicholls RD, Fink JK. NIPA1 gene mutations causeautosomal dominant hereditary spastic paraplegia (SPG6). Am. J. Hum. Genet.2003;73:967e71. http://dx.doi.org/10.1086/378817.
Rosenfeld JA, Coe BP, Eichler EE, Cuckle H, Shaffer LG. Estimates of penetrance forrecurrent pathogenic copy-number variations. Genet. Med. Off. J. Am. Coll. Med.Genet. 2013;15:478e81. http://dx.doi.org/10.1038/gim.2012.164.
De Rubeis S, Bagni C. Regulation of molecular pathways in the Fragile X Syndrome:insights into Autism Spectrum Disorders. J. Neurodev. Disord. 2011;3:257e69.http://dx.doi.org/10.1007/s11689-011-9087-2.
Sahoo T, Peters SU, Madduri NS, Glaze DG, German JR, Bird LM, et al. Microarraybased comparative genomic hybridization testing in deletion bearing patientswith Angelman syndrome: genotype-phenotype correlations. J. Med. Genet.2006;43:512e6. http://dx.doi.org/10.1136/jmg.2005.036913.
Sempere Pérez A, Manchón Trives I, Palazón Azorín I, Alcaraz Más L, Pérez Lledó E,Galán Sánchez F. 15Q11.2 (BP1-BP2) microdeletion, a new syndrome with var-iable expressivity. An. Pediatría Barc 2011;75:58e62. http://dx.doi.org/10.1016/j.anpedi.2011.01.033. Spain 2003.
Shaikh TH, Gai X, Perin JC, Glessner JT, Xie H, Murphy K, et al. High-resolutionmapping and analysis of copy number variations in the human genome: a dataresource for clinical and research applications. Genome Res. 2009;19:1682e90.http://dx.doi.org/10.1101/gr.083501.108.
Shinawi M, Schaaf CP, Bhatt SS, Xia Z, Patel A, Cheung SW, et al. A small recurrentdeletion within 15q13.3 is associated with a range of neurodevelopmentalphenotypes. Nat. Genet. 2009;41:1269e71. http://dx.doi.org/10.1038/ng.481.
SoemediR,Wilson IJ, BenthamJ,DarlayR,TöpfA,ZelenikaD,etal. Contributionofglobalrare copy-number variants to the risk of sporadic congenital heart disease. Am. J.Hum. Genet. 2012;91:489e501. http://dx.doi.org/10.1016/j.ajhg.2012.08.003.
Stefansson H, Rujescu D, Cichon S, Pietiläinen OPH, Ingason A, Steinberg S, et al.Large recurrent microdeletions associated with schizophrenia. Nature2008;455:232e6. http://dx.doi.org/10.1038/nature07229.
Stefansson H, Meyer-Lindenberg A, Steinberg S, Magnusdottir B, Morgen K,Arnarsdottir S, et al. CNVs conferring risk of autism or schizophrenia affectcognition in controls. Nature 2014;505:361e6. http://dx.doi.org/10.1038/nature12818.
Varela MC, Kok F, Otto PA, Koiffmann CP. Phenotypic variability in Angelman syn-drome: comparison among different deletion classes and between deletion andUPD subjects. Eur. J. Hum. Genet. EJHG 2004;12:987e92. http://dx.doi.org/10.1038/sj.ejhg.5201264.
Wichmann H-E, Gieger C, Illig T, MONICA/KORA Study Group. KORA-generesourcefor population genetics, controls and a broad spectrum of disease phenotypes.Gesundheitswesen Bundesverb. Ärzte Öffentl. Gesundheitsdienstes Ger2005;67(Suppl. 1):S26e30. http://dx.doi.org/10.1055/s-2005-858226. Q
De Wolf V, Brison N, Devriendt K, Peeters H. Genetic counseling for susceptibilityloci and neurodevelopmental disorders: the del15q11.2 as an example. Am. J.Med. Genet. A 2013;161A:2846e54. http://dx.doi.org/10.1002/ajmg.a.36209.
Xiong Y, Oakley BR. In vivo analysis of the functions of gamma-tubulin-complexproteins. J. Cell. Sci. 2009;122:4218e27. http://dx.doi.org/10.1242/jcs.059196.
767778
tion (BP1eBP2) and developmental delay, behaviour issues, epilepsyMedical Genetics (2015), http://dx.doi.org/10.1016/j.ejmg.2015.01.002
Copyright © 2022 FDOKUMEN




















