ELP4 in rolandic epilepsy and BRD2 in juvenile myoclonic epilepsy
15
Major Susceptibility Genes for Common Idiopathic Epilepsies: ELP4 in Rolandic Epilepsy and BRD2 in Juvenile Myoclonic Epilepsy Deb K Pal, MD PhD David A Greenberg, PhD INTRODUCTION Clinical observations confirm that the idiopathic epilepsies are neurodevelopmental disorders strongly influenced by genetic factors. Genetic epidemiological studies prove major genetic influences on two common forms of idiopathic epilepsy to be discussed here: rolandic epilepsy (RE) and juvenile myoclonic epilepsy (JME). The common forms of these idiopathic syndromes have a complex genetic inheritance and this fact complicates finding and elucidating the susceptibility genes as well as proving their pathogenic role. Genetic heterogeneity and phenotype definition are much more serious factors in studying common complex epilepsies than they are in the study of densely affected epilepsy pedigrees showing Mendelian inheritance. In complex disorders, several genes and sometimes environmental factors are believed to contribute to disease etiology; as a result, proving a causative role for any one gene, in a genetic model involving multiple genes, can present a challenge. In this chapter, we first review the methodological issues associated with analyzing complex genetic disorders, then explain the strategy that we have used in investigating these two particular syndromes. Genomewide linkage analyses and fine-mapping have resulted in the identification of susceptibility genes ELP4 in Rolandic epilepsy and BRD2 in juvenile myoclonic epilepsy. We discuss the evidence in favor of a pathogenic role for each gene and the further steps that are required to establish causality. THE IDIOPATHIC EPILEPSIES AS NEURODEVELOPMENTAL DISORDERS Determining the etiology of the common idiopathic epilepsies is a difficult challenge. Although there have been several discoveries of genes involved in rare Mendelian variants of idiopathic epilepsies, for example in autosomal dominant juvenile myoclonic epilepsy 1 and benign familial infantile convulsions (BFIC) 2 , mutations in these genes have not been demonstrated in the common forms of epilepsy that show complex, rather than simple Mendelian, inheritance. Further, the phenomenon of heterogeneity, where similar phenotypes have different genetic etiologies, means that data thought to reflect a single disease may actually contain diseases of different etiologies. In contrast to the rare Mendelian forms of epilepsy, the common forms have a complex inheritance in which susceptibility likely results from the combination of the interaction of a number of genes, more likely oligogenic (few genes of major effect) rather than polygenic (many genes each of equal small effect) and there is no evidence for an environmental influence 3 . Many of the gene mutations identified in rare epilepsies are located in ion channel or neurotransmitter subunits, giving rise to the term “channelopathy”, but there is evidence that susceptibility genes influencing common idiopathic epilepsies may be involved in pathways related to neurodevelopment. The hypothesis for a neurodevelopmental origin to common epilepsies comes from three lines of evidence: natural history, neuropathology and neuroimaging, and is further supported by Jasper's Basic Mechanisms of the Epilepsies Jasper's Basic Mechanisms of the Epilepsies
Transcript of ELP4 in rolandic epilepsy and BRD2 in juvenile myoclonic epilepsy

Major Susceptibility Genes for Common Idiopathic Epilepsies:ELP4 in Rolandic Epilepsy and BRD2 in Juvenile MyoclonicEpilepsyDeb K Pal, MD PhD
David A Greenberg, PhD
INTRODUCTIONClinical observations confirm that the idiopathic epilepsies are neurodevelopmental disordersstrongly influenced by genetic factors. Genetic epidemiological studies prove major geneticinfluences on two common forms of idiopathic epilepsy to be discussed here: rolandic epilepsy(RE) and juvenile myoclonic epilepsy (JME). The common forms of these idiopathicsyndromes have a complex genetic inheritance and this fact complicates finding and elucidatingthe susceptibility genes as well as proving their pathogenic role. Genetic heterogeneity andphenotype definition are much more serious factors in studying common complex epilepsiesthan they are in the study of densely affected epilepsy pedigrees showing Mendelianinheritance. In complex disorders, several genes and sometimes environmental factors arebelieved to contribute to disease etiology; as a result, proving a causative role for any one gene,in a genetic model involving multiple genes, can present a challenge.
In this chapter, we first review the methodological issues associated with analyzing complexgenetic disorders, then explain the strategy that we have used in investigating these twoparticular syndromes. Genomewide linkage analyses and fine-mapping have resulted in theidentification of susceptibility genes ELP4 in Rolandic epilepsy and BRD2 in juvenilemyoclonic epilepsy. We discuss the evidence in favor of a pathogenic role for each gene andthe further steps that are required to establish causality.
THE IDIOPATHIC EPILEPSIES AS NEURODEVELOPMENTAL DISORDERSDetermining the etiology of the common idiopathic epilepsies is a difficult challenge. Althoughthere have been several discoveries of genes involved in rare Mendelian variants of idiopathicepilepsies, for example in autosomal dominant juvenile myoclonic epilepsy1 and benignfamilial infantile convulsions (BFIC)2, mutations in these genes have not been demonstratedin the common forms of epilepsy that show complex, rather than simple Mendelian, inheritance.Further, the phenomenon of heterogeneity, where similar phenotypes have different geneticetiologies, means that data thought to reflect a single disease may actually contain diseases ofdifferent etiologies. In contrast to the rare Mendelian forms of epilepsy, the common formshave a complex inheritance in which susceptibility likely results from the combination of theinteraction of a number of genes, more likely oligogenic (few genes of major effect) ratherthan polygenic (many genes each of equal small effect) and there is no evidence for anenvironmental influence 3. Many of the gene mutations identified in rare epilepsies are locatedin ion channel or neurotransmitter subunits, giving rise to the term “channelopathy”, but thereis evidence that susceptibility genes influencing common idiopathic epilepsies may be involvedin pathways related to neurodevelopment.
The hypothesis for a neurodevelopmental origin to common epilepsies comes from three linesof evidence: natural history, neuropathology and neuroimaging, and is further supported by
Jasper's Basic Mechanism
s of the EpilepsiesJasper's Basic M
echanisms of the Epilepsies

experimental data outlined later in this chapter. There is increasing recognition of pre-morbidsymptoms in rolandic epilepsy (RE). Children with RE are at 2.5 times higher odds of speechsound disorder than the general population, and 5.8 times higher odds of dyslexia 4. Speechsound disorder presents in the first two years of life, long before the median onset age forseizures, and dyslexia is also recognized at, or soon after, school entry, usually prior to, oraround the time of, the first seizure. In RE families, these symptoms were reported as epilepsy-associated, but it is evident that their occurrence precedes the onset of seizures and thereforetheir sequential onset may indicate a neurodevelopmental pathogenesis of rolandic epilepsy.Furthermore, the presence of these same symptoms in family members of RE patients (whothemselves do not have epilepsy) suggests an underlying genetic etiology for these traits relatedto, but partly independent from, RE itself4.
Similar claims for a premorbid or comorbid diathesis have been made for a distinct frontal lobepersonality in juvenile myoclonic epilepsy 5, although this has been debated 6, 7. However, agrowing body of neuroimaging studies suggest that juvenile myoclonic epilepsy shows focalbrain structural abnormalities8 associated with specific neuropsychological impairments, andsimilar findings are emerging from studies in childhood absence epilepsy 9. Detailedmorphometric neuropathological studies in IGE suggest an abnormally increased number ofneurons in the molecular layer of the cortex 10, also described as microdysgenesis. Thesefindings have proven hard to replicate, owing to the scarcity of biologic material. Nonetheless,the pertinence of brain structural abnormalities is further supported by the JME mouse model,discussed below. Taken together, these various lines of evidence suggest that the pathogenesisof idiopathic epilepsies is related to subtle deviations in the normal postnatal development ofthe brain.
METHODOLOGICAL ISSUES AND EXPERIMENTAL STRATEGIES INCOMMON EPILEPSIES
The experimental approaches available in common epilepsies are somewhat different thanthose in Mendelian disorders because, in most common epilepsies, there are usually not manyaffected people in a single kindred. Thus, a single family of a person with a common type ofepilepsy does not contain sufficient information for a genetic study. One way to overcome thislimitation is to collect many families with the same phenotype; each family then contributessome genetic information. However, using this strategy, genetic heterogeneity becomes acritical confounder in both genetic association and linkage studies 11, because signals fromdifferent genetic forms of disease may “cancel each other out”. An obvious solution would beto separate out possibly different genetic forms on the basis of phenotypic differences.Sometimes though, there are no obvious phenotypic features to distinguish different geneticforms. Nonetheless, what are often considered minor differences can be exploited to limitpotential genetic heterogeneity. For example, generalized tonic–clonic seizures (GTCS) thatoccur on awakening may be inherited differently from GTCS that occur at random times ofthe day 12 13. These studies show that some apparently minor distinguishing features can befeasibly used to stratify datasets to guide gene searches.
Genetic heterogeneity may occur within or between different sub-populations. For example,evidence for linkage to EJM1 in juvenile myoclonic epilepsy is limited to European originfamilies and not found in Hispanic origin or African-American families 14–17. Another JMElocus at 6p12-p11 was found in families from Belize and Mexico 18. In a third example, GTCSare linked to markers on chromosome 10 in families from India, but no such linkage peak existsin corresponding European datasets19. This diversity of findings in genomic geography isincreasingly recognized as the norm in international studies of complex disease, and likelyreflects differing genetic selection pressures on subpopulations in different environments. In
Page 2
Major Susceptibility Genes for Common Idiopathic Epilepsies: ELP4 in Rolandic Epilepsy and BRD2 in Juvenile
Myoclonic Epilepsy
Jasper's Basic Mechanism
s of the EpilepsiesJasper's Basic M
echanisms of the Epilepsies

epilepsy, the origins of genetic heterogeneity also reflect the multiple genes that contribute tothe neural circuits that regulate cortical excitability.
A second complementary approach to overcome genetic heterogeneity is to dissect the overallphenotype into simpler traits and then genetically analyze those traits. The success of thisapproach depends on stringent phenotyping. For example, in idiopathic generalized epilepsies,not only do probands have different types of seizures (eg absence, generalized tonic-clonic,myoclonic) but also clinically affected family members often have different seizure types thanare seen in the proband 20. By parsing the epilepsy syndrome into separate seizure types,different loci have been identified that lead to shared or distinct influences on the differentseizure types in IGE 21. Thus, malic enyme 2 (ME2) has been identified as a major susceptibilitygene common to all forms of IGE22, while myoclonic seizures have been mapped tobromodomain 2 (BRD2)23. Similarly, in Rolandic epilepsy, probands have combinations ofneurodevelopmental traits including speech dyspraxia and dyslexia while family memberswithout seizures may be affected by the same disorders and/or exhibit centrotemporal sharpwaves (CTS) on their EEGs. Thus, instead of conducting genetic analysis on a family in whichonly one person has epilepsy, it is possible to increase the power of genetic analysis byanalyzing traits that segregate in more than one family member. Using this strategy, 4–6Hzspike-and-wave EEG trait seen in unaffected family members of JME proband families wasfound, like JME itself, to be influenced by the chromosome 6p21 locus 15, 24, and possiblyphotosensitivity as well 25, 26. We discovered linkage to the 11p13 region for CTS27, andshowed that speech dyspraxia and CTS share linkage at the same locus28. Loci for dyslexia inRE appear to be distinct from those for CTS or speech dyspraxia in RE29, adding complexityto the genetic model of the overall phenotype.
Below we review the evidence for the role of ELP4 in Rolandic epilepsy and BRD2 in JME.
ELP4 AND ROLANDIC EPILEPSYSegregation analysis
Segregation analysis is a method that allows one to test hypotheses about different modes ofinheritance. It only has the power to exclude tested modes of inheritance rather thanconclusively prove one mode is correct, and it is often complicated by methodological errorsin ascertainment and analysis. Although some attempts have been made to estimate theinheritance of CTS and of rolandic epilepsy in the past, they have been affected by suchmethodological errors. We designed our study to avoid such errors: we collected 23 probandswith rolandic epilepsy through unambiguous single ascertainment. Thirty siblings of REprobands in the age range 4–16 years underwent sleep-deprived EEG; observations from thosewho remained awake were omitted. CTS were rated as present or absent by two independentobservers blinded to the study hypothesis and subject identities. Twenty-three siblings showedevidence of sleep in their EEG recordings. Eleven of 23 recordings demonstrated CTS, yieldinga corrected segregation ratio of 0.48 (95% CI: 0.27–0.69). The male to female ratio of CTSaffectedness was approximately equal. The segregation ratio of CTS in rolandic epilepsyfamilies is therefore consistent with a highly penetrant autosomal dominant inheritance, withequal sex ratio. Autosomal recessive and X-linked inheritance can be rejected.
Linkage analysisLinkage analysis is a method for localizing major effect genes to within several million basepairs. Linkage can be used either to test if a chromosomal region containing a suspected geneis related to disease expression, or to do a genome-wide search for disease-related loci. Weconducted genome-wide linkage analysis of the CTS trait in 38 US families singly ascertainedthrough a RE proband. In 11 of the families, one additional sibling was known to carry the
Page 3
Major Susceptibility Genes for Common Idiopathic Epilepsies: ELP4 in Rolandic Epilepsy and BRD2 in Juvenile
Myoclonic Epilepsy
Jasper's Basic Mechanism
s of the EpilepsiesJasper's Basic M
echanisms of the Epilepsies

CTS trait, but the CTS status of individuals younger than 4 years, or older than 16 years, wasunknown because of its age-limited expression. Only markers on chromosome 11 yielded two-point LOD scores exceeding 3.0. Markers in the region of chromosomal band 11p13 providedstrong and compelling evidence for linkage to CTS, with a maximum two-point LOD score of4.01, and multipoint LOD score of 4.30. Both European and non-European ancestry familiescontributed proportionally to the LOD score. LOD score maximization in this region of 11pmost often occurred at 95% penetrance, consistent with predictions from segregation analysis.We did not observe significant evidence of linkage at markers previously reported for CTS at15q1430, nor for a rare recessive variant of RE at 16p12 –11.231, nor for X-linked rolandicseizures and cognitive deficit32. Similarly, we did not find evidence of linkage to 11p13 in anautosomal dominant variant of RE with speech dyspraxia and cognitive impairment 33.
Association analysisAssociation analysis can show that having a specific allele (variant) at a locus correlates withincreased risk for disease. We designated a 13-cM linkage region at 11p13, encompassing thearea in which LOD scores >2.0, as our region of interest for fine mapping. We then tested forassociation of CTS with SNP markers distributed across genes in this region. We initially useda ‘discovery’ data set that included 68 cases and 187 controls group matched for ancestry andgender – 38 of these cases were included in the original linkage screen. In addition to case–control analysis, we used family-based analysis to guard against the potential for positiveconfounding due to population stratification. We took a pure likelihood approach to thestatistical analysis of linkage and association 34. We then typed additional SNPs around genesthat showed compelling evidence of association in the preliminary analysis. In a second,independent, ‘replication’ case-control dataset, we typed a subset of the SNPs in our region ofinterest. The replication set included 40 RE cases and 120 controls from Western Canada; thetwo data sets were then jointly analyzed.
We discovered significant association with SNPs in ELP4 introns 9, 6 and 5, with estimatedodds ratios 1.80–2.04 at these markers (Figure 1). These associations were demonstrated inboth family-based analysis and case-control analysis. Joint analysis of US and Canadiandatasets provided confirmation of these associations, the maximum likelihood ratio forassociation at intron 9 was 589:1 (Figure 2). All associated markers are in linkagedisequilibrium with each other (Figure 3), so it is unlikely that they are exerting independenteffects. However, none of the associated SNPs are predicted to have deleterious functionalconsequences, and we therefore resequenced this genomic region to search for a causativevariant.
Page 4
Major Susceptibility Genes for Common Idiopathic Epilepsies: ELP4 in Rolandic Epilepsy and BRD2 in Juvenile
Myoclonic Epilepsy
Jasper's Basic Mechanism
s of the EpilepsiesJasper's Basic M
echanisms of the Epilepsies

Figure 1. Pure likelihood plot of association evidence in discovery set and in joint analysis of datasets (Figure 2)
This pure likelihood analysis plots odds ratio (OR) on the y axis and base-pair position on the x axis. Each vertical line representsa likelihood interval (LI) for the OR at a given SNP. The OR 1/4 1 line is plotted as a solid black horizontal line, for reference.LIs in color are denoted as SNPs of interest, whereas a gray line indicates that the SNP is not of interest because the 1/32 LI forthat SNP covers the OR 1/4 1 line. The small horizontal tick on each LI is the maximum likelihood estimator for the OR. Theportion of the colored LI that covers the OR 1/4 1 horizontal line indicates the strength of the association information at that SNP.In particular, if the navy blue portion is above the OR 1/4 1 line while the yellow portion of the LI covers the OR 1/4 1 line, thenthe LOD evidence at that SNP is between 1.5 and 2 (ie, the 1/32 LI does not include the OR 1/4 1 value, but the 1/100 LI does);similarly, if both the yellow and navy blue portions are above the OR 1/4 1 line but the turquoise portion covers the line, then theLOD evidence is between 2 and 3 (ie, the 1/100 LI does not include OR 1/4 1 as a plausible value but the 1/1000 LI does). Thefurther the colored line is above the OR 1/4 1 line, the stronger the association evidence. The max LR for each SNP in color isalso provided as text in the plot, providing evidence not only of whether the LOD evidence is between 2 and 3, but also the exactvalue of the max LR.
Figure 2. Joint Analysis of Discovery (US) and Replication (Canadian) datasets
Fig 3. Linkage Disequilibrium map in 11p13 region from SNP association dataBlock 4 spans the DPH4, IMMP1L, ELP4, PAX6 genes
Genomic resequencingWe resequenced the coding portions, exon–intron boundaries and 50kb upstream region of theELP4 gene in 40 RE probands from the discovery set. The 274 kb ELP4 gene is transcribedinto a 1584 bp mRNA consisting of 12 exons, a 35 b 5′-UTR and a 257 bp 30 -UTR. Alternativetranscripts have been reported that include or exclude the last two exons. Three previouslyreported SNP variants were found in these 40 individuals but the distribution of these SNPswas not different to that found in controls or reference databases.
Page 5
Major Susceptibility Genes for Common Idiopathic Epilepsies: ELP4 in Rolandic Epilepsy and BRD2 in Juvenile
Myoclonic Epilepsy
Jasper's Basic Mechanism
s of the EpilepsiesJasper's Basic M
echanisms of the Epilepsies

Summary of evidence for ELP4 as a susceptibility gene for CTSCTS is obviously a mandatory component of the overall rolandic epilepsy phenotype. The sumof our analyses point to CTS as a highly penetrant, autosomal dominant trait. We have verystrong evidence of a single genomewide locus at 11p13 influencing the CTS trait. Associationevidence unambiguously points to association with markers in the ELP4 gene, but not withmarkers in the DCDC1, DPH4, IMMP1L or PAX6 genes under the 11p13 linkage peak. Thisassociation evidence is present in family-based analysis, making population stratification anunlikely explanation for the finding, and was also replicated in an independent dataset.However, no obvious coding mutation has been discovered in the ELP4 gene. There are severalpossible explanations for this that are yet to be fully explored. First, coding changes are notcommon in complex diseases, in contrast to the simple loss or gain of function coding mutationsfound in Mendelian diseases. Hence the sought after variant may reside in a regulatory orconserved region of the gene; second, some regulatory regions may be distant from the genethat they regulate, and so the causative variant may not be in ELP4; third, it is possible that thecausative variant lies in another part of the 11p13 linkage region and that the association withELP4 represents long-range linkage disequilibrium with the “true” variant. The search for thecausative variant will continue, meanwhile a coherent evaluation of the evidence should takeaccount of the biological function of potential candidate genes. The relevant information aboutthe role of Elongator in the nervous system is presented below.
Elongator biologyThere is increasing evidence that impairment of the Elongator complex may be involved inseveral different neurological disorders and this topic has recently been extensivelyreviewed35 and excerpted below. Elongator is composed of six subunits (ELP1–ELP6).ELP1 is the main scaffold protein whereas ELP3 is the main enzymatic subunit that has histoneacetyltransferase activity36. Elongator is believed to play an important role in thetranscriptional elongation of some genes37, 38; an hypothesis further supported by theobservation of transcriptional defects in multiple genes in ELP1-depleted cells and mouseembryos 37, 39.
As well as a nuclear function associated with RNA polymerase II and histone H3 acetylation,there is evidence for the Elongator complex having a role in tRNA modification in thecytoplasm. Cytoplasmic ELP1 co-localizes with filamin A in membrane ruffles, and a defectiveactin cytoskeleton has been reported in ELP1-depleted cells, possibly explaining cell motilitydefects 40. Thus is appears that Elongator critically regulates the migration of multiple celltypes.
Page 6
Major Susceptibility Genes for Common Idiopathic Epilepsies: ELP4 in Rolandic Epilepsy and BRD2 in Juvenile
Myoclonic Epilepsy
Jasper's Basic Mechanism
s of the EpilepsiesJasper's Basic M
echanisms of the Epilepsies
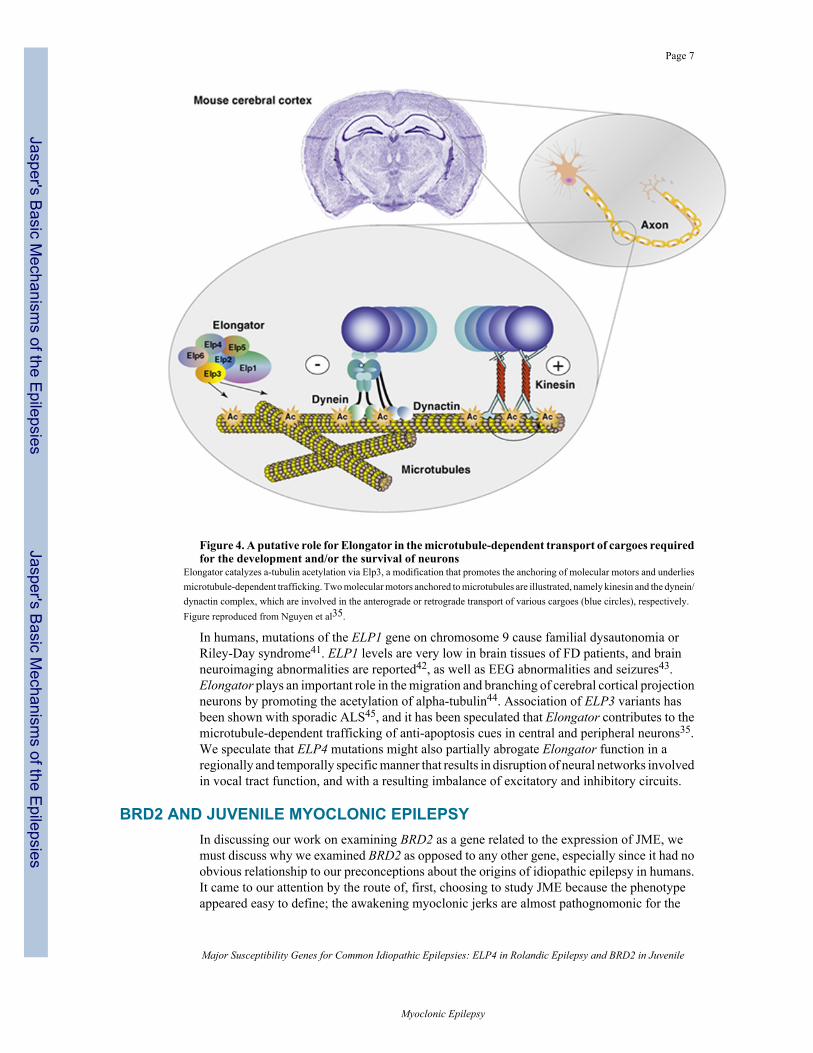
Figure 4. A putative role for Elongator in the microtubule-dependent transport of cargoes requiredfor the development and/or the survival of neurons
Elongator catalyzes a-tubulin acetylation via Elp3, a modification that promotes the anchoring of molecular motors and underliesmicrotubule-dependent trafficking. Two molecular motors anchored to microtubules are illustrated, namely kinesin and the dynein/dynactin complex, which are involved in the anterograde or retrograde transport of various cargoes (blue circles), respectively.Figure reproduced from Nguyen et al35.
In humans, mutations of the ELP1 gene on chromosome 9 cause familial dysautonomia orRiley-Day syndrome41. ELP1 levels are very low in brain tissues of FD patients, and brainneuroimaging abnormalities are reported42, as well as EEG abnormalities and seizures43.Elongator plays an important role in the migration and branching of cerebral cortical projectionneurons by promoting the acetylation of alpha-tubulin44. Association of ELP3 variants hasbeen shown with sporadic ALS45, and it has been speculated that Elongator contributes to themicrotubule-dependent trafficking of anti-apoptosis cues in central and peripheral neurons35.We speculate that ELP4 mutations might also partially abrogate Elongator function in aregionally and temporally specific manner that results in disruption of neural networks involvedin vocal tract function, and with a resulting imbalance of excitatory and inhibitory circuits.
BRD2 AND JUVENILE MYOCLONIC EPILEPSYIn discussing our work on examining BRD2 as a gene related to the expression of JME, wemust discuss why we examined BRD2 as opposed to any other gene, especially since it had noobvious relationship to our preconceptions about the origins of idiopathic epilepsy in humans.It came to our attention by the route of, first, choosing to study JME because the phenotypeappeared easy to define; the awakening myoclonic jerks are almost pathognomonic for the
Page 7
Major Susceptibility Genes for Common Idiopathic Epilepsies: ELP4 in Rolandic Epilepsy and BRD2 in Juvenile
Myoclonic Epilepsy
Jasper's Basic Mechanism
s of the EpilepsiesJasper's Basic M
echanisms of the Epilepsies

condition (when there are no exclusionary symptoms). Second, we could make use of theinterictal 4–6Hz spike-and-wave seen in almost all JME patients as the phenotype because thistrait is also seen with increased frequency in clinically unaffected family members 46, 47. Thismeant we might be able to use the presence of the EEG trait as the phenotype for the linkageanalysis instead of the epilepsy itself, which proved to be a useful approach 15, 47, an approachlater confirmed when it led to the discovery that ELP4 is related to the centrotemporal spikesof RE 48.
Segregation analysisA segregation analysis of JME was able to exclude simple Mendelian modes of inheritance,even when using the 4–6Hz spike and wave trait as the phenotype of interest (clinicallyunaffected family members with the trait were included as “affected”) 49. However, thesegregation analysis showed that the best fit to the data was a model of inheritance involvingtwo genes, one showing dominant and one showing recessive inheritance. Models in whichboth loci were dominant or both recessive did not fit the data as well. Interestingly, two locilater identified as contributing to JME supported that mode of inheritance: linkage analysisidentified a dominantly inherited locus at chromosome 6p21, which was eventually identifiedas BRD2 and one on chromosome 18, ME2, that indicated recessive inheritance.
Linkage and Association AnalysisOur initial linkage scan revealed the presence of a JME-related locus on chromosome 6p21, afinding confirmed in the data set of Janz 14, 15. Later, after an entirely new and larger data setwas collected, we were able not only to confirm the linkage again, but the discovery of twofamilies with recombinants allowed us to narrow down the region containing the gene tobetween the HLA-DQ and HLA-DP loci. Furthermore, the advance in genetic technology hadallowed us to discover an association of JME, first with a microsatellite marker 17 and laterwith SNP haplotypes23. Both of these results supported BRD2 as the gene influencing JMEexpression. Subsequently, we demonstrated strong association of BRD2 variants with JME,with odds ratio 6.4523.
There are two important points to emphasize. First, these results started with a highly specificphenotype, then used linkage data from entire families to find a locus containing a disease-related gene, and then used association analysis to identify the gene within that locus. No geneswere excluded from examination because they did not fit a preconceived epilepsy mechanism.Second, in the absence of any idea of what the mechanism might be, and confronted with acandidate gene of unknown function and containing no epilepsy-related exonic mutations aftersequencing, the problem remained (and remains) of proving that the gene we found is the genewe are looking for. Until this point, all the evidence is statistical. In the absence of any idea ofhow a so-called transcription factor element could lead to a subtle form of epilepsy, we neededsome biologic evidence that BRD2 was somehow involved in expression. This led to ourdevelopment of the Brd2 knockout mouse.
Brd2 knockout mouseOur Brd2 knockout mouse was created using the gene-trap technique.50 and embryos wereraised on a B57Bl/CJ background. We made several observations about mice homozygous forthe knockout (Brd2−/−):
i. Brd2−/− embryos do not survive past the 13th gestational day (E13).ii. Brd2−/− embryos are considerably smaller than their wild-type brd2+/+ or brd2+/−
littermates. Figure 5 shows a homozygote embryo and wild type embryo at E11,showing the much smaller size of the homozygote.
iii. The neural tube of the homozygote is not closed, as the arrow in figure 5 shows.
Page 8
Major Susceptibility Genes for Common Idiopathic Epilepsies: ELP4 in Rolandic Epilepsy and BRD2 in Juvenile
Myoclonic Epilepsy
Jasper's Basic Mechanism
s of the EpilepsiesJasper's Basic M
echanisms of the Epilepsies

iv. The most obvious effect of a lack of brd2 during development is on the mouse nervoussystem. Figure 6 shows a wild type and brd2−/− mouse in cross section at E11. Notethat the brain and spinal cord for the homozygote are severely dysmorphic, with braindevelopment apparently spatially disorganized. Note also how other organs, whilesmaller than in the wild type, appear structurally intact.
v. Confirming the apparent major effect of brd2 mostly on nervous system development,a brd2 probe showed that brd2 is expressed mostly in developing nervous tissue. Figure7 shows a wild type mouse (left) with the densest areas of stain, indicating the greatestexpression of Brd2, being the nervous system. The Brd2−/− mouse on the right showsno Brd2 expression.
Figure 5. Photographs of wild-type (Brd2−/−, left) and Brd2−/− (right) embryos at embryonic day(E) 9.5 of gestation
The mutant embryo is smaller and the developing neural tube is unfused (arrow). (Figure reproduced from Shang et al.50)
Figure 6. Sagittal sections of E11.5 embryos of wild-type and mutant embryosGrossly normal appearing heart, lung, and so on, are seen in the mutant, but the brain of the mutant is aberrant. FB, forebrain;MB, midbrain; HB, hindbrain. (Figure reproduced from Shang et al.50)
Page 9
Major Susceptibility Genes for Common Idiopathic Epilepsies: ELP4 in Rolandic Epilepsy and BRD2 in Juvenile
Myoclonic Epilepsy
Jasper's Basic Mechanism
s of the EpilepsiesJasper's Basic M
echanisms of the Epilepsies
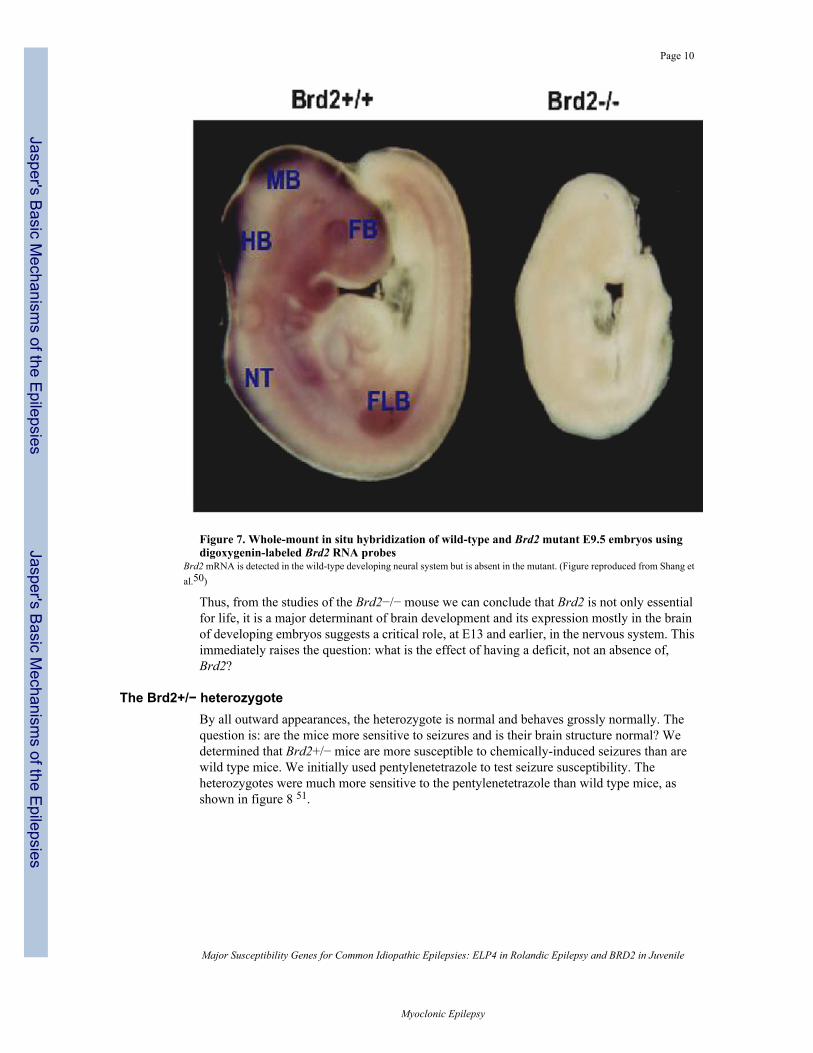
Figure 7. Whole-mount in situ hybridization of wild-type and Brd2 mutant E9.5 embryos usingdigoxygenin-labeled Brd2 RNA probes
Brd2 mRNA is detected in the wild-type developing neural system but is absent in the mutant. (Figure reproduced from Shang etal.50)
Thus, from the studies of the Brd2−/− mouse we can conclude that Brd2 is not only essentialfor life, it is a major determinant of brain development and its expression mostly in the brainof developing embryos suggests a critical role, at E13 and earlier, in the nervous system. Thisimmediately raises the question: what is the effect of having a deficit, not an absence of,Brd2?
The Brd2+/− heterozygoteBy all outward appearances, the heterozygote is normal and behaves grossly normally. Thequestion is: are the mice more sensitive to seizures and is their brain structure normal? Wedetermined that Brd2+/− mice are more susceptible to chemically-induced seizures than arewild type mice. We initially used pentylenetetrazole to test seizure susceptibility. Theheterozygotes were much more sensitive to the pentylenetetrazole than wild type mice, asshown in figure 8 51.
Page 10
Major Susceptibility Genes for Common Idiopathic Epilepsies: ELP4 in Rolandic Epilepsy and BRD2 in Juvenile
Myoclonic Epilepsy
Jasper's Basic Mechanism
s of the EpilepsiesJasper's Basic M
echanisms of the Epilepsies

Figure 8. Seizure sensitivity to pentylenetetrazol in Wild-type and Brd2+/− miceSeizure intensity was scored blind to genotype by four independent observers on a scale of 0–3; where 0=no change in muscletone or activity, 1= mild tremor, 2=moderate seizure, 3=severe seizure. 3 of 5 heterozygotes died following severe convulsion butall five wild type animals completely recovered.
These results were confirmed using the seizure-inducing ether, flurothyl (Velisek et al,submitted) in which we also showed that female heterozygote mice are much more sensitiveto tonic-clonic seizures than male mice, a finding perhaps echoing a result found by Pal etal 20 in which human females had up to a 12 fold increased risk for seizures in JME familiesthan do males.
We also found that the brain structures of the Brd2+/− mice were subtly abnormal. In the frontallobes there was up to a 50% deficit of parvalbumin staining interneurons than in wild type mice(figure 9).
Page 11
Major Susceptibility Genes for Common Idiopathic Epilepsies: ELP4 in Rolandic Epilepsy and BRD2 in Juvenile
Myoclonic Epilepsy
Jasper's Basic Mechanism
s of the EpilepsiesJasper's Basic M
echanisms of the Epilepsies

Figure 9. Differences in density of parvalbumin-stained neurons in Brd2+/− vs WT in four brainregions: Cg1 – cingulated cortex, area 1; Prl – prelimbic cortex; IL – infralimbic cortex; DP – dorsalpeduncular cortex
Densities represent counts from over 100 slices from two Brd2+/− and two WT.
These results were also later confirmed and expanded (Velisek et al, submitted). Almost allbrain areas examined showed varying levels of GABAergic neuron deficit. Taken togetherwith early ultrastructural studies in post-mortem human IGE brains10, as well as with recentvolumetric studies in JME8, the corpus of evidence supports a neurodevelopmental origin forcommon idiopathic generalized epilepsy.
The implications of these findings could have a profound effect on our view of the origins ofIGE. They imply that, for JME if not for other IGEs, part of the neural substrate for the epilepsyis a deficit of inhibitory neurons caused by a subtle insufficiency of, for JME, BRD2. Thisdeficit leads to a under production or abnormal migrations or early apoptosis of such neurons,a deficit that later will lead to epilepsy in the presence of some other, probably subtle,abnormality caused by a second gene or genes.
These results are compelling evidence that BRD2 is, in fact, the EJM1 locus on chromosome6p21. However, mice are not the same as humans. In the absence of human biologic materialto test the hypothesis that there are fewer GABAergic inhibitory neurons in the brains of JMEpatients, we must continue to investigate the effects of Brd2 in mice to discover a definitiveconnection that will allow us to predict the risk for JME. Identifying the BRD2 JME-relatedalleles will enable us to do that.
Numerous genes have been identified in densely-affected families showing Mendelianinheritance or, like the gene SCN1A’s role in severe myoclonic epilepsy of infancy, have beenshown to play a role in severe epilepsy. However, none of these genes has yet been linked,associated, or by mutation analysis, been shown to play a role in common, genetically complexforms of epilepsy. The results of the RE and JME work on ELP4 and BRD2 suggest thatcommon epilepsies are not disorders of a single gene but require several genes for theirexpression. More important is the suggestion that these are disorders that result from changesin gene expression that result in subtle changes during development. Investigators studyingepilepsy will need to adopt more subtle observational approaches in order to understand thesechanges.
References1. Cossette P, Liu L, Brisebois K, et al. Mutation of GABRA1 in an autosomal dominant form of juvenile
myoclonic epilepsy. Nat Genet 2002;31:184–9. [PubMed: 11992121]2. Malacarne M, Gennaro E, Madia F, et al. Benign familial infantile convulsions: mapping of a novel
locus on chromosome 2q24 and evidence for genetic heterogeneity. Am J Hum Genet 2001;68:1521–6. [PubMed: 11326335]
3. Greenberg DA, Durner M, Delgado-Escueta AV. Evidence for multiple gene loci in the expression ofthe common generalized epilepsies. Neurology 1992;42:56–62. [PubMed: 1574177]
4. Clarke T, Strug LJ, Murphy PL, et al. High risk of reading disability and speech sound disorder inrolandic epilepsy families: case-control study. Epilepsia 2007;48:2258–65. [PubMed: 17850323]
5. Janz D. The idiopathic generalised epilepsies of adolescence with childhood and juvenile age of onset.Epilepsia 1997;38:4–11. [PubMed: 9024180]
6. Gelisse P, Genton P, Samuelian JC, Thomas P, Bureau M. [Psychiatric disorders in juvenile myoclonicepilepsy]. Rev Neurol (Paris) 2001;157:297–302. [PubMed: 11319492]
7. Trinka E, Kienpointner G, Unterberger I, et al. Psychiatric comorbidity in juvenile myoclonic epilepsy.Epilepsia 2006;47:2086–91. [PubMed: 17201708]
Page 12
Major Susceptibility Genes for Common Idiopathic Epilepsies: ELP4 in Rolandic Epilepsy and BRD2 in Juvenile
Myoclonic Epilepsy
Jasper's Basic Mechanism
s of the EpilepsiesJasper's Basic M
echanisms of the Epilepsies

8. Roebling R, Scheerer N, Uttner I, Gruber O, Kraft E, Lerche H. Evaluation of cognition, structural,and functional MRI in juvenile myoclonic epilepsy. Epilepsia 2009;50:2456–65. [PubMed:19490045]
9. Caplan R, Levitt J, Siddarth P, et al. Frontal and temporal volumes in Childhood Absence Epilepsy.Epilepsia 2009;50:2466–72. [PubMed: 19624714]
10. Meencke HJ, Janz D. Neuropathological findings in primary generalized epilepsy: a study of eightcases. Epilepsia 1984;25:8–21. [PubMed: 6692795]
11. Vieland VJ. The replication requirement. Nat Genet 2001;29:244–5. [PubMed: 11687787]12. Greenberg DA, Durner M, Resor S, Rosenbaum D, Shinnar S. The genetics of idiopathic generalized
epilepsies of adolescent onset: differences between juvenile myoclonic epilepsy and epilepsy withrandom grand mal and with awakening grand mal. Neurology 1995;45:942–6. [PubMed: 7746411]
13. Durner M, Zhou G, Fu D, et al. Evidence for linkage of adolescent-onset idiopathic generalizedepilepsies to chromosome 8-and genetic heterogeneity. Am J Hum Genet 1999;64:1411–9. [PubMed:10205274]
14. Weissbecker KA, Durner M, Janz D, Scaramelli A, Sparkes RS, Spence MA. Confirmation of linkagebetween juvenile myoclonic epilepsy locus and the HLA region of chromosome 6. Am J Med Genet1991;38:32–6. [PubMed: 1901452]
15. Durner M, Sander T, Greenberg DA, Johnson K, Beck-Mannagetta G, Janz D. Localization ofidiopathic generalized epilepsy on chromosome 6p in families of juvenile myoclonic epilepsypatients. Neurology 1991;41:1651–5. [PubMed: 1922810]
16. Sander T, Bockenkamp B, Hildmann T, et al. Refined mapping of the epilepsy susceptibility locusEJM1 on chromosome 6. Neurology 1997;49:842–7. [PubMed: 9305351]
17. Greenberg DA, Durner M, Keddache M, et al. Reproducibility and complications in gene searches:linkage on chromosome 6, heterogeneity, association and maternal inheritance in juvenile myoclonicepilepsy. American Journal of Human Genetics 2000;66:508–16. [PubMed: 10677311]
18. Liu AW, Delgado-Escueta AV, Serratosa JM, et al. Juvenile myoclonic epilepsy locus in chromosome6p21.2-p11: linkage to convulsions and electroencephalography trait. Am J Hum Genet1995;57:368–81. [PubMed: 7668263]
19. Puranam RS, Jain S, Kleindienst AM, et al. A locus for generalized tonic-clonic seizure susceptibilitymaps to chromosome 10q25-q26. Ann Neurol 2005;58:449–58. [PubMed: 16130088]
20. Pal DK, Durner M, Klotz I, et al. Complex inheritance and parent-of-origin effect in juvenilemyoclonic epilepsy. Brain Dev 2006;28:92–8. [PubMed: 16414227]
21. Durner M, Keddache MA, Tomasini L, et al. Genome scan of idiopathic generalised epilepsy:evidence for major susceptibility gene and modifying genes influencing the seizure type. Annals ofNeurology 2001;49:328–35. [PubMed: 11261507]
22. Greenberg DA, Cayanis E, Strug L, et al. Malic enzyme 2 may underlie susceptibility to adolescent-onset idiopathic generalized epilepsy. Am J Hum Genet 2005;76:139–46. [PubMed: 15532013]
23. Pal DK, Evgrafov OV, Tabares P, Zhang F, Durner M, Greenberg DA. BRD2 (RING3) is a probablemajor susceptibility gene for common juvenile myoclonic epilepsy. Am J Hum Genet 2003;73:261–70. [PubMed: 12830434]Epub 2003 Jun 25
24. Greenberg DA, Delgado-Escueta AV, Widelitz H, et al. Juvenile myoclonic epilepsy may be linkedto the BF and HLA loci on human chromosome 6. American Journal of Medical Genetics 1988;31
25. Lorenz S, Taylor KP, Gehrmann A, et al. Association of BRD2 polymorphisms with photoparoxysmalresponse. Neurosci Lett 2006;400:135–9. [PubMed: 16516380]
26. Tauer U, Lorenz S, Lenzen KP, et al. Genetic dissection of photosensitivity and its relation toidiopathic generalized epilepsy. Ann Neurol 2005;57:866–73. [PubMed: 15929039]
27. Strug LJ, Clarke T, Chiang T, et al. Centrotemporal sharp wave EEG trait in rolandic epilepsy mapsto Elongator Protein Complex 4 (ELP4). Eur J Hum Genet 2009;17:1171–81. [PubMed: 19172991]
28. Pal DK, Strug LJ, Bali B, Clarke T, Lieberman P. Pleiotropic Effects of the 11p13 Locus onDevelopmental Verbal Dyspraxia and EEG Centrotemporal Sharp Waves Genes. Brain and Behavior.2010in press
29. Pal DK, Strug LJ, Clarke T, Murphy PL. Major genetic loci for reading disability in rolandic epilepsyfamilies. Epilepsia 2007;48:376.
Page 13
Major Susceptibility Genes for Common Idiopathic Epilepsies: ELP4 in Rolandic Epilepsy and BRD2 in Juvenile
Myoclonic Epilepsy
Jasper's Basic Mechanism
s of the EpilepsiesJasper's Basic M
echanisms of the Epilepsies

30. Neubauer BA, Fiedler B, Himmelein B, et al. Centrotemporal spikes in families with rolandic epilepsy:linkage to chromosome 15q14. Neurology 1998;51:1608–12. [PubMed: 9855510]
31. Guerrini R, Bonanni P, Nardocci N, et al. Autosomal recessive Rolandic epilepsy with paroxysmalexercise-induced dystonia and writer’s cramp: delineation of the syndrome and gene mapping tochromosome 16p12-11.2. Ann Neurol 1999;45:344–52. [PubMed: 10072049]
32. Roll P, Rudolf G, Pereira S, et al. SRPX2 mutations in disorders of language cortex and cognition.Hum Mol Genet 2006;15:1195–207. [PubMed: 16497722]
33. Kugler SL, Bali B, Lieberman P, et al. An autosomal dominant genetically heterogeneous variant ofrolandic epilepsy and speech disorder. Epilepsia 2008;49:1086–90. [PubMed: 18248446]
34. Strug LJ, Hodge SE, Chiang T, Pal DK, Corey P, Rohde C. A pure likelihood approach to the analysisof genetic association data: an alternative to Bayesian and Frequentist analysis. Eur J Hum Genet.2010in press
35. Nguyen L, Humbert S, Saudou F, Chariot A. Elongator - an emerging role in neurological disorders.Trends Mol Med 2010;16:1–6. [PubMed: 20036197]
36. Wittschieben BO, Otero G, de Bizemont T, et al. A novel histone acetyltransferase is an integralsubunit of elongating RNA polymerase II holoenzyme. Mol Cell 1999;4:123–8. [PubMed:10445034]
37. Close P, Hawkes N, Cornez I, et al. Transcription impairment and cell migration defects in elongator-depleted cells: implication for familial dysautonomia. Mol Cell 2006;22:521–31. [PubMed:16713582]
38. Kouskouti A, Talianidis I. Histone modifications defining active genes persist after transcriptionaland mitotic inactivation. EMBO J 2005;24:347–57. [PubMed: 15616580]
39. Chen YT, Hims MM, Shetty RS, et al. Loss of mouse Ikbkap, a subunit of elongator, leads totranscriptional deficits and embryonic lethality that can be rescued by human IKBKAP. Mol CellBiol 2009;29:736–44. [PubMed: 19015235]
40. Johansen LD, Naumanen T, Knudsen A, et al. IKAP localizes to membrane ruffles with filamin Aand regulates actin cytoskeleton organization and cell migration. J Cell Sci 2008;121:854–64.[PubMed: 18303054]
41. Slaugenhaupt SA, Blumenfeld A, Gill SP, et al. Tissue-specific expression of a splicing mutation inthe IKBKAP gene causes familial dysautonomia. Am J Hum Genet 2001;68:598–605. [PubMed:11179008]
42. Axelrod FB, Hilz MJ, Berlin D, et al. Neuroimaging supports central pathology in familialdysautonomia. J Neurol 2010;257:198–206. [PubMed: 19705052]
43. Niedermeyer E, McKusick VA, Brunt P, Mahloudji M. The EEG in familial dysautonomia (Riley-Day syndrome). Electroencephalogr Clin Neurophysiol 1967;22:473–5. [PubMed: 4164636]
44. Creppe C, Malinouskaya L, Volvert ML, et al. Elongator controls the migration and differentiationof cortical neurons through acetylation of alpha-tubulin. Cell 2009;136:551–64. [PubMed:19185337]
45. Simpson CL, Lemmens R, Miskiewicz K, et al. Variants of the elongator protein 3 (ELP3) gene areassociated with motor neuron degeneration. Hum Mol Genet 2009;18:472–81. [PubMed:18996918]
46. Tsuboi T, Christian W. On the genetics of primary generalized epilepsy with sporadic myoclonias ofimpulsive petit mal. A clinical and electroencephalographic study of 399 probands. Humangenetik1973;19:155–82. [PubMed: 4200688]
47. Greenberg DA, Delgado-Escueta AV, Widelitz H, et al. Juvenile myoclonic epilepsy (JME) may belinked to the BF and HLA loci on human chromosome 6. Am J Med Genet 1988;31:185–92.[PubMed: 3146924]
48. Strug LJ, Clarke T, Chiang T, et al. Centrotemporal sharp wave EEG trait in rolandic epilepsy mapsto Elongator Protein Complex 4 (ELP4). Eur J Hum Genet 2009;17:1171–81. [PubMed: 19172991]
49. Greenberg DA, Delgado-Escueta AV, Maldonado HM, Widelitz H. Segregation analysis of juvenilemyoclonic epilepsy. Genet Epidemiol 1988;5:81–94. [PubMed: 3136050]
50. Shang E, Wang X, Wen D, Greenberg DA, Wolgemuth DJ. Double bromodomain-containing geneBrd2 is essential for embryonic development in mouse. Dev Dyn 2009;238:908–17. [PubMed:19301389]
Page 14
Major Susceptibility Genes for Common Idiopathic Epilepsies: ELP4 in Rolandic Epilepsy and BRD2 in Juvenile
Myoclonic Epilepsy
Jasper's Basic Mechanism
s of the EpilepsiesJasper's Basic M
echanisms of the Epilepsies

51. Greenberg DA, Shang E, Luo J, et al. Knockout mouse data support BRD2 as a gene for JuvenileMyoclonic Epilepsy. Epilepsia. 2007;(Suppl)
Page 15
Major Susceptibility Genes for Common Idiopathic Epilepsies: ELP4 in Rolandic Epilepsy and BRD2 in Juvenile
Myoclonic Epilepsy
Jasper's Basic Mechanism
s of the EpilepsiesJasper's Basic M
echanisms of the Epilepsies