







Bahasa
Halaman
Hukum


PRECLINICAL STUDY
Transactivation of ErbB-2 induced by tumor necrosis factora promotes NF-jB activation and breast cancer cell proliferation
Martın A. Rivas Æ Mercedes Tkach Æ Wendy Beguelin ÆCecilia J. Proietti Æ Cinthia Rosemblit Æ Eduardo H. Charreau ÆPatricia V. Elizalde Æ Roxana Schillaci
Received: 3 June 2009 / Accepted: 3 September 2009
� Springer Science+Business Media, LLC. 2009
Abstract Tumor necrosis factor alpha (TNFa) is a
pleiotropic cytokine which, acting locally, induces tumor
growth. Accumulating evidence, including our findings,
showed that TNFa is mitogenic in breast cancer cells in
vitro and in vivo. In the present study, we explored TNFainvolvement on highly aggressive ErbB-2-overexpressing
breast cancer cells. We found that TNFa induces ErbB-2
phosphorylation in mouse breast cancer C4HD cells and in
the human breast cancer cell lines SK-BR-3 and BT-474.
ErbB-2 phosphorylation at Tyr877 residue was mediated by
TNFa-induced c-Src activation. Moreover, TNFa promoted
ErbB-2/ErbB-3 heterocomplex formation, Akt activation
and NF-jB transcriptional activation. Inhibition of ErbB-2
by addition of AG825, an epidermal growth factor receptor/
ErbB-2-tyrosine kinase inhibitor, or knockdown of ErbB-2
by RNA interference strategy, blocked TNFa-induced NF-
jB activation and proliferation. However, the humanized
monoclonal antibody anti-ErbB-2 Herceptin could not
inhibit TNFa ability to promote breast cancer growth.
Interestingly, our work disclosed that TNFa is able to
transactivate ErbB-2 and use it as an obligatory downstream
signaling molecule in the generation of mitogenic signals.
As TNFa has been shown to be present in the tumor
microenvironment of a significant proportion of human
infiltrating breast cancers, our findings would have clinical
implication in ErbB-2-positive breast cancer treatment.
Keywords ErbB-2 � TNFa � Herceptin � c-Src
Introduction
Tumor necrosis factor alpha (TNFa) is a pleiotropic cyto-
kine originally characterized to cause hemorrhagic necrosis
of tumors at high doses [1]. However, it is now widely
accepted that TNFa, acting locally, induces the growth of
certain tumor types such as ovary and breast [2–5]. In
particular, TNFa has been shown to be produced by
malignant or host cells in the tumor microenvironment of
human infiltrating breast cancer and to be associated with
increasing malignancy [6]. Treatment of cancer cells with
TNFa enhances cell proliferation in vitro [7, 8] and, as we
have recently demonstrated, also supports murine breast
cancer growth in vivo [5]. In addition, we have shown that
TNFa induces in vitro proliferation of breast cancer cells
through a mechanism which requires activation of p42/p44
mitogen-activated protein kinase (MAPK), c-jun NH2-ter-
minal kinase (JNK), Akt and NF-jB transcriptional acti-
vation [5]. However, the exact mechanism by which TNFaenhances tumor growth and its involvement in different
breast cancer subtypes remains elusive.
ErbB-2, a transmembrane tyrosine kinase receptor, is
overexpressed in nearly 30% of human breast cancer and
has been associated with enhanced tumor aggressiveness
and poor clinical outcome [9]. These tumors also display
NF-jB activation [10]. ErbB-2 is an orphan receptor
belonging to the family of type I tyrosine kinase receptors,
which signals by forming heterodimers with epidermal
Electronic supplementary material The online version of thisarticle (doi:10.1007/s10549-009-0546-3) contains supplementarymaterial, which is available to authorized users.
M. A. Rivas � M. Tkach � W. Beguelin �C. J. Proietti � C. Rosemblit � E. H. Charreau �P. V. Elizalde � R. Schillaci (&)
Laboratory of Molecular Mechanisms of Carcinogenesis,
Instituto de Biologıa y Medicina Experimental (IBYME),
CONICET, Vuelta de Obligado 2490,
Buenos Aires C1428ADN, Argentina
e-mail: [email protected]
123
Breast Cancer Res Treat
DOI 10.1007/s10549-009-0546-3

growth factor receptor (EGFR), ErbB-3 and ErbB-4 [11] in
response to ligands including heregulins [12]. After ligand
binding, all ErbB receptors are phosphorylated, serving as
docking sites for the recruitment of cytoplasmic adaptor
proteins, initiating signaling cascades that control multiple
cellular processes. HerceptinTM (Trastuzumab) is a mono-
clonal antibody which binds to the extracellular domain of
the receptor [13] and is administrated to breast cancer
patients whose tumors overexpress ErbB-2. However, the
clinical efficacy of Herceptin is limited to 30% of these
patients. An important reason for this is that other tumor-
cell alterations may influence the response to ErbB-2-tar-
geted inhibitors [14]. Thus, understanding the mechanisms
by which ErbB-2 can be activated through non-classical
receptors and ligands is relevant in order to design a new
therapeutic approach and to predict patients’ response.
Ligands of G protein-coupled receptors which act through
c-Src kinase activation [15], as well as hormones such as
prolactin, acting through Janus kinase 2 activation [16],
and cytokines such as interleukin-6 [17] have all been
demonstrated to transactivate ErbB-2. Several groups have
so far shown transactivation of EGFR by TNFa through the
activation of matrix metalloproteinases (MMPs) which are
able to release EGFR ligands from the cell membrane [18–
20]. In contrast, there are no reports demonstrating TNFaability to transactivate ErbB-2 in breast cancer.
In the present work, we explore the effect of TNFa on
breast cancer cells that overexpress ErbB-2 and found that
TNFa induces not only ErbB-2 autophosphorylation, but
also phosphorylation of its Tyr877 residue through the
activation of the tyrosine kinase c-Src. We further found
that it promotes association between ErbB-2 and ErbB-3.
ErbB-2 transactivation by TNFa is a rapid event that does
not involve either ligand release or MMPs activation, and
leads to cell proliferation even in the presence of Hercep-
tin. Interestingly, we showed that TNFa regulates ErbB-2
phosphorylation as a requisite to activate NF-jB and cell
proliferation in human and murine breast cancer cells. This
is the first demonstration of ErbB-2 transactivation by
TNFa in breast cancer cells, which may be one of the
mechanisms by which ErbB-2-overexpressing tumors show
resistance to anti-ErbB-2 monoclonal antibodies therapy.
Materials and methods
Animals and tumors
Experiments were carried out in virgin female Balb/c mice,
raised at the Instituto de Biologıa y Medicina Experimental
of Buenos Aires. All animal studies were conducted as
described [5]. C4HD mouse mammary tumor expresses
progesterone and estrogen receptors, lacks EGFR
expression, overexpresses ErbB-2 and exhibits high levels
of ErbB-3 and low expression of ErbB-4 [21–23].
Antibodies
Antibodies to the following proteins were used: Neu/ErbB-
2 (C-18), ErbB-3 (C-17), phosphotyrosine (PY99), p85
phosphatidyl inositol 3-kinase (PI3-K), p42/p44 MAPK (C-
14), phospho-p42/p44 MAPK (E-4), JNK (N-18) and
phospho JNK (G-7) all from Santa Cruz Biotechnology
(Santa Cruz, CA, USA), Akt, phospho Akt (Ser 473),
phospho IjBa (Ser32/36), IjBa, phosphotyrosine c-Src
(Tyr416), c-Src (36D10), phospho-ErbB-2 (Tyr1221/
1222), phospho-ErbB-2 (Tyr877) and EGFR from Cell
Signaling (Beverly, MA, USA), v-Src (ab-1) from Cal-
biochem (La Jolla, CA, USA) and actin (Clone ACTN05)
and cyclin D1 from Neomarkers (Fremont, CA, USA).
Cell culture and treatments
Primary cultures of epithelial cells from the mouse mam-
mary tumor C4HD, growing in medroxyprogesterone ace-
tate (MPA)-treated mice, were performed as previously
described [5, 24]. As C4HD cells are sensitive to progestin,
all the experiments were performed in Dulbecco’s Modified
Eagle’s Medium/F12 without phenol red (Sigma, St. Louis,
MO, USA) (DMEM) ? 0.1% charcoal-stripped fetal calf
serum (ChFCS). Human breast cancer cell lines SK-BR-3
and BT-474 were obtained from the American Type Culture
Collection and maintained in Mc Coy’s 5A ? 10% FCS
(Gen SA, Buenos Aires, Argentina) and RPMI 1640 ? 10%
FCS, respectively. Experiments were performed in their
respective medium ? 1% FCS. Cells were treated for
the indicated times with 20 ng/ml of murine or human TNFa(mTNFa, hTNFa, respectively) (Cell Sciences, Canton,
MA, USA) or with 20 ng/ml of recombinant human b1
heregulin (HRG, Upstate, Millipore, Bedford, MA, USA).
The following inhibitors were added to cells 60 min before
incubation with TNFa: AG825, EGFR/ErbB-2 tyrosine
kinase inhibitor from the benzylidene malononitrile family;
PP2, 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,
4-d]pyrimidine, c-Src inhibitor; GF109203X, PKC inhibitor;
GM6001, a broad MMPs inhibitor, and its corresponding
negative control (all from Calbiochem); Dasatinib, c-Src
inhibitor (LC Laboratories, Woburn, MA, USA); GW2974,
EGFR/ErbB-2 tyrosine kinase inhibitor from the indazol-
yamino quinazoline family and Bay 11-7082, inhibitor of
IjB phosphorylation (both from Sigma). To perform certain
experiments, cells were pre-incubated for 16 h with 10 lg/ml
of the anti-ErbB-2 antibody HerceptinTM (Hoffmann-La
Roche Ltd, Basel, Switzerland) for Western blot analysis, and
1 h for proliferation and reporter assays. Cell proliferation was
evaluated by [3H]-thymidine incorporation assay. Cells were
Breast Cancer Res Treat
123

incubated with TNFa for 48 h. 1 lCi [3H]-thymidine (NEN,
Dupont, Boston, MA, USA; specific activity 20 Ci/mmol)
was added at hour 24 of said culture. Cells were then tryp-
sinized and harvested and radioactivity was counted using
standard scintillation procedures [5]. Assays were performed
in octuplicate. In earlier experiments we demonstrated that
[3H]-thymidine uptake correlated with the number of cells/
well [5]. Cell viability was performed by triplicate by Trypan
blue exclusion at 48 h of treatment with the corresponding
inhibitors. For C4HD cells, cultured with 100 lM AG825, it
was of 67.6 ± 4.7%, for BT-474 cells cultured with 100 lM
AG825, 10 lg/ml Herceptin, 1 lM GW2974, 0.5 lM Da-
satinib and 1 lM Bay 11-7082, it was of 62.3 ± 7.2,
70.9 ± 7.6, 73.7 ± 10.3, 77.0 ± 1.3 and 73 ± 8.3%,
respectively, and for SK-BR-3 cells cultured with 100 lM
AG825 and 10 lg/ml Herceptin it was of 55.8 ± 9.3 and
65.6 ± 1.3%, respectively.
Immunofluorescence staining and confocal microscopy
SK-BR-3, BT-474 and C4HD cells grown on glass cover-
slips were treated with TNFa 20 ng/ml for 30 min. Cells
were fixed and permeabilized in ice-cold methanol and
ErbB-2 was localized using ErbB-2 9G-6 (Santa Cruz
Biotechnology) followed by incubation with a rhodamine
conjugated anti-mouse secondary antibody (The Jackson
Laboratory, Bar Harbor, ME, USA). Stained cells were
analyzed using a Nikon C1 confocal laser scanning
microscope.
Flow cytometry analysis
SK-BR-3 and BT-474 cells treated with hTNFa for the
indicated times were harvested with PBS ? EDTA 1% and
incubated with anti-ErbB-2 9G-6 antibody followed by
incubation with anti-mouse phycoerythrin (PE)-conjugated
antibody (Santa Cruz Biotechnology). A total of 104 cells/
sample was analyzed using FACSAria cytometer (Becton–
Dickinson, La Jolla, CA, USA). Background staining was
evaluated in cells incubated with an isotype control IgG
followed by anti-mouse PE-conjugated antibody. Data
analysis was performed using WinMDI software (J. Trot-
ter, Scripps Research Institute, San Diego, CA, USA).
Delta mean fluorescence intensity (MFI) values were
obtained by subtracting the MFI of the cells incubated with
control isotype antibody from the MFI of cells incubated
with ErbB-2 antibody.
For cell cycle analysis, BT-474 cells were subjected to
the different treatments, harvested at 36 and 48 h, and fixed
in 70% ethanol for 24 h at 4�C, as we previously described
[5]. They were washed twice with PBS, followed by RNA
digestion (RNAse A 50 U/ml) and propidium iodide
(20 lg/ml) staining for 30 min at room temperature in the
dark. Cell cycle analysis was performed using a FAC-
Scalibur flow cytometer (Becton–Dickinson) and Modfit
LT software.
Western blot and immunoprecipitation
Lysates were prepared from cells subjected to the different
treatments described in each experiment, as previously
detailed, and proteins were subjected to SDS-PAGE [5].
Association among ErbB-2, ErbB-3 and p85 PI3-K was
studied by performing co-immunoprecipitation experi-
ments as previously described [24]. Briefly, 500 lg of
protein lysates was incubated with 2 lg of rabbit anti-
EGFR, ErbB-2 or ErbB-3 antibody and the immunocom-
plexes were captured by adding protein A-agarose (Santa
Cruz Biotechnology). Beads were washed, boiled in sample
buffer, and proteins were electroblotted as described above.
As negative control, normal rabbit serum was used.
In vitro cold phosphorylation assay
C4HD cells were treated with TNFa for 5 min or prein-
cubated before TNFa stimulation for 60 min with PP2
(10 lM). Cells were lysed in kinase lysis buffer (20 mM
HEPES pH 7.5, 10 mM EGTA, 1% NP-40, 2.5 mM
MgCl2) and Src was immunoprecipitated from 500 lg
protein extracts using an anti-v-Src antibody. ErbB-2 was
immunoprecipitated from 500 lg protein extract from
unstimulated C4HD cells. The immunoprecipitated ErbB-2
was then subjected to an in vitro phosphorylation assay
with Src immunoprecipitated from each treatment as pre-
viously described [25]. Proteins were separated by elec-
trophoresis; gels were transferred onto nitrocellulose and
immunoblotted with anti-Tyr877/927 ErbB-2 and anti-total
c-Src antibodies and ErbB-2.
Transient transfections
jB-Luc vector (jB sites from HIV promoter) and cyclin
D1 promoter-Luc vector were kindly provided by Dr M.
Bell (Mayo Clinic, Rochester, MN, USA) and Dr R. Pestell
(Northwestern University Medical School, Chicago, IL,
USA), respectively. Human ErbB-2 wild type, kinase-
negative (KN) ErbB-2 and ErbB-2-Y877F were kindly
provided by Dr T. Akiyama (Gumma University, Gumma,
Japan), Dr A. Gertler (Protein Laboratories Rehovot,
Israel) and Dr O. Segatto (Centro Recerca Sperimentale,
Rome, Italy). C4HD cells were transfected for 24 h in
DMEM supplemented with 10 nM MPA and 2.5% ChFCS,
and cell lines in the corresponding growth medium without
antibiotics. FuGENE 6 transfection reagent technique
(Roche Biochemicals, Indianapolis, IN, USA) was used in
accordance with the manufacturer’s instructions. Cells
Breast Cancer Res Treat
123

were transiently co-transfected with 1 lg of jB-Luc or
1 lg cyclin D1-luc construct plus 10 ng Renilla luciferase
expression vector CMV-pRL (Promega, Madison, WI,
USA) used to correct variations in transfection efficiency.
As control, cells were transfected with a pGL3-basic
reporter lacking jB. Transfected cells were lysed and
luciferase assays carried out using the Dual-Luciferase
Reporter Assay System (Promega).
siRNAs targeting mouse ErbB-2 mRNAs were synthe-
sized by Dharmacon Inc. (Lafayette, CO, USA) (ErbB-2
siRNA#02: antisense, 50-GAUGUCCUCCGUAAGAAUA-
30, ErbB-2 siRNA#03 antisense 50-GAUGGUGCUUACU
CAUUGA-30, ErbB-2 siRNA#04 antisense 50-GGAAUC
CUAAUCAAACGAA-30). The non-silencing siRNA oli-
gonucleotide from Dharmacon, which does not target any
known mammalian gene, was used as negative control.
Transfection of siRNA plus 1 lg of empty vector
(pcDNA3.1) or 1 lg of human ErbB-2 expression vector
and 1 lg of jB-Luc vector plus 10 ng Renilla luciferase
expression vector was performed using the DharmaFECT
Duo transfection reagent following the manufacturer’s
directions, using 25 nM of siRNA for 3 days. Cell treatment
and reporter activity were measured as described above. In
some experiments, siRNA at a final concentration of 25 nM
was transfected with Dharmafect 1 reagent following the
manufacturer’s directions.
Statistical analysis
The differences between control and experimental groups
were analyzed by ANOVA followed by Tukey t test among
groups. Kolmorgorov–Smirnov test was used for flow
cytometric studies.
Results
TNFa-induced breast cancer cell proliferation
requires ErbB-2 phosphorylation
We have already shown that TNFa induces proliferation of
the murine mammary adenocarcinoma C4HD, both in vitro
and in vivo [5]. As C4HD cells overexpress ErbB-2 and
since ErbB-2 is a key player in C4HD cell proliferation [21,
22, 24], we investigated the potential role of ErbB-2 sig-
naling in the growth stimulatory effects of TNFa. For that
purpose, cell proliferation assays were conducted in the
presence of the selective dual EGFR/ErbB-2 tyrosine kinase
inhibitor tyrphostin AG825. Previous studies have shown
that C4HD cells do not express EGFR [21, 23, 24], therefore
the results obtained with AG825 will be attributable entirely
to ErbB-2 blockage. Interestingly, proliferation of C4HD
cells induced by 20 ng/ml TNFa was inhibited by addition
of 100 lM AG825 (Fig. 1a). As expected, AG825 com-
pletely suppressed HRG-induced growth (Fig. 1a) and
ErbB-2 phosphorylation at Tyr1272, one of the main
C-terminal tail autophosphorylation sites (Supplemental
Fig. 1). In a previous work, we demonstrated that TNFainduced p42/p44 MAPK and JNK activation in breast
cancer cells [5]. We now wanted to explore the involvement
of ErbB-2 on the activation of these signaling pathways.
Addition of AG825 did not affect TNFa-induced p42/p44
MAPK nor JNK phosphorylation in C4HD cells (Supple-
mental Fig. 2). However, addition of AG825 completely
blocked HRG-induced p42/p44 MAPK activation (Sup-
plemental Fig. 2). These contrasting results suggest that
p42/p44MAPK activation has a different up-stream sig-
naling pathway that is dependent on ErbB-2 phosphoryla-
tion when cells are stimulated with HRG, and is ErbB-2
phosphorylation-independent when the mitogen used is
TNFa.
Fig. 1 TNFa requires ErbB-2 phosphorylation for inducing breast
cancer cell proliferation. a, e Cells were preincubated with 100 lM
AG825 for 60 min and then treated or not with 20 ng/ml of TNFa or
20 ng/ml heregulin (HRG) for 48 h. Proliferation was performed by
[3H]-thymidine incorporation assay. Data are presented as
mean ± SE of octuplicate samples (*P \ 0.05, **P \ 0.001 vs.
control). The experiments shown are representative of a total of four.
Controls were performed in order to verify that dimethyl sulfoxide
(DMSO) (1:2,000) did not modify TNFa-induced proliferation.
b C4HD cells were treated with TNFa for the times shown and
subjected to ErbB-2 immunoprecipitation. Immunoprecipitates were
blotted with anti-phosphotyrosine antibody (top panel) and mem-
branes were stripped and blotted with anti-ErbB-2 antibody (bottompanel). IP immunoprecipitation, NRS normal rabbit serum. Bands
were quantified using Image J with untreated cell samples (first lines)
set as 1.0. This is a representative experiment out of a total of three.
c C4HD cells were treated with TNFa for the times shown. Whole cell
lysates were subjected to Western blot analysis for ErbB-2 phos-
phorylation at Tyr1222/1272 residue. ErbB-2 is shown as loading
control and was used for quantification as described in (b). d, f C4HD
cells were preincubated with 50, 80 or 100 lM AG825 for 60 min
and SK-BR-3 and BT-474 cells with 100 lM AG825 and then treated
with TNFa for the times shown. ErbB-2 phosphorylation at Tyr1222/
1272 residue was performed as described in (c). This is a represen-
tative experiment out of a total of three. g ErbB-2 expression by flow
cytometry in cells treated with TNFa. ErbB-2 expression was
revealed by incubation with anti-ErbB-2 antibody followed by a
secondary phycoerythrin (PE)-conjugated antibody (ErbB-2 PE).
Representative histograms of ErbB-2 expression in SK-BR-3 and BT-
474 cells treated or not with TNFa for 30 min are shown. The delta
mean fluorescence intensity of control and TNFa-treated SK-BR3
cells was of 4.4 and 2.9, respectively (P \ 0.01) and of control and
TNFa-treated BT-474 cells it was of 22.2 and 10.0, respectively
(P \ 0.001). Kolmorgorov–Smirnov statistical test was used. hLocalization of ErbB-2 by immunofluorescence and confocal micros-
copy in TNFa-treated cells. Each image is representative of at least 10
(scale bars 10 lm). Control experiments demonstrated no detectable
staining with secondary antibody incubation only or with anti-ErbB-2
antibody preincubated with the specific blocking peptide. Nuclei were
stained with 40,6-diamidino-2-phenylindole (DAPI). hTNFa human
TNFa, mTNFa murine TNFa
c
Breast Cancer Res Treat
123

We examined ErbB-2 phosphorylation levels after TNFatreatment. Figure 1b shows that TNFa augmented total
levels of ErbB-2 tyrosine phosphorylation from 2 to 15 min
of treatment and decline thereafter. Then, we addressed the
specific mouse ErbB-2 tyrosine phosphorylation at Tyr1272,
analogous to human Tyr1222 ErbB-2. There was an increase
in Tyr1272 phosphorylation 2 min after TNFa treatment in
C4HD cells, with maximum phosphorylation at 5 min
(Fig. 1c). Addition of AG825 blocked TNFa-induced
Tyr1272 phosphorylation in a concentration-dependent
manner (Fig. 1d). Since our goal was to determine whether
TNFa-induced ErbB-2 transactivation was taking place also
in human breast cancer, we then used SK-BR-3 [26] and BT-
474 cell lines [27] that are widely used models of ErbB-2-
overexpression. While TNFa induced SK-BR-3 and BT-474
cell proliferation, addition of AG825 blocked TNFa mito-
genic effect (Fig. 1e). Similar effects were obtained with
HRG-treated cells. In addition, TNFa induced phosphory-
lation of the Tyr1222 residue after 2 min stimulation, with a
peak at 5 min (Fig. 1f) which was inhibited by addition of
A
B
0 2 105 15 6030 mTNFα, min
pTyr
ErbB-2
C4HD
IP: ErbB2 NRS
5
pTyr-1272-ErbB-2
ErbB-2
-
- -
+ + + + mTNFαAG825
-- -
C4HD
pTyr-1272-ErbB-2
ErbB-2
C0 2 105 15 6030 mTNFα, min
C4HD
F
0
10
20
- + - - + -mTNFα- - -HRG + - +
AG825 - - ++ +-
[3 H]-
Th
ymid
ine
inco
rpo
rati
on
(C
PM
x 1
03)
*
**C4HD
[3 H]-
Th
ymid
ine
inco
rpo
rati
on
(C
PM
x 1
03)
0
125
250
- + - - + -hTNFα- - -HRG + - +
AG825 - - ++ +-
**
*
SK-BR-3
0
15
30
45
- + - - + -
- - -+ - +
- - ++ +-
* *
BT-474E
ErbB2 DAPI
hTN
Fα
30 m
inC
ontr
ol
SK-BR-3
pTyr-1222-ErbB-2
ErbB-2
SK-BR-3
+ +------
0 2 105 15 30 0 10
AG825
hTNFα, min
BT-474
+ +------
0 2 105 15 30 0 10
AG825
hTNFα, min
pTyr-1222-ErbB-2
ErbB-2
G
BT-474
ErbB2 DAPIErbB2 DAPI
C4HDH
mT
NF
α 30
min
Con
trol
D
1 1.6 1.51.6 1.5 0.90.8
1 1.5 1.71.7 1.6 0.41.8 0.4 1 1.6 2.62.4 2.6 0.62.0 0.6
1 3.3 1.53.7 1.0 0.91.0
Isotype control
Control
hTNFα 30 min
64
0100 101 102
ErbB-2-PE
SK-BR-3 64
100 101 102
ErbB-2-PE
0
BT-474
3232
Breast Cancer Res Treat
123

AG825. Comprehensively, our present findings indicate that
TNFa is able to transactivate ErbB-2 in breast cancer cells.
To examine the effect of TNFa stimulation on ErbB-2
expression on the surface of SK-BR-3 and BT-474 cells,
we performed immunofluorescence and flow cytometry
analysis. ErbB-2 plasma membrane expression was
observed to decrease, reaching its minimum at 30 min
(Fig. 1g) and staying low for at least 1 h of TNFa treatment
in both cell lines. To confirm these data, we performed
confocal microscopy studies. ErbB-2 was localized pri-
marily to the plasma membrane in unstimulated cells.
TNFa treatment for 30 min led to a significant increase in
ErbB-2 localization in the cytoplasm of C4HD, SK-BR-3
and BT-474 cells (Fig. 1h) without affecting ErbB-2 con-
tent in whole cell extracts. Comprehensively, these results
show for the first time that TNFa is able to activate ErbB-2
inducing its phosphorylation and internalization into the
cytoplasm of breast cancer cells.
The finding that TNFa is able to induce ErbB-2 trans-
activation, prompted us to explore the mechanism under-
lying this phosphorylation. It is known that TNFa can
induce EGFR transactivation through MMP-dependent
EGFR ligand release. Treatment of C4HD and SK-BR-3
cells with 10 lM GM6001, a broad MMPs inhibitor, did
not modify TNFa-induced ErbB-2 phosphorylation (Sup-
plemental Fig. 3a). In addition, the use of blocking anti-
bodies to ErbB-3 and ErbB-4 to impede ligand binding did
not modify TNFa-induced cell growth (Supplemental
Fig. 3b) although both antibodies were able to inhibit HRG
proliferative effect in C4HD cells [22]. Thus, these data
suggest that TNFa does not activate ErbB-2 through ligand
release in these breast cancer cells.
TNFa-induced c-Src kinase activation mediates
ErbB-2 phosphorylation
It has been shown recently that ErbB-2 phosphorylation at
the residue Tyr877 (Tyr927 in mice), located in the acti-
vation loop of the kinase domain, is important for its
intrinsic kinase activity [28]. Interestingly, we found that
TNFa induced an increase on Tyr877/927 ErbB-2 phos-
phorylation in C4HD, SK-BR-3 and BT-474 cells (Fig. 2a).
Since tyrosine kinase c-Src directly phosphorylates ErbB-2
at Tyr877 residue [29], we hypothesized that c-Src could be
involved in this event. We observed that TNFa induced
c-Src phosphorylation at Tyr416 in C4HD cells, with a
clear activation at 2–5 min of treatment (Fig. 2b). Addition
of PP2 or Dasatinib, a clinically used c-Src inhibitor, dra-
matically decreased TNFa-induced ErbB-2 phosphoryla-
tion at Tyr877 in SK-BR-3 and BT-474 (Fig. 2b).
To further explore whether activated c-Src can phos-
phorylate Tyr877 ErbB-2 in vitro, we performed a cold
phosphorylation assay. For this purpose, we immunopre-
cipitated c-Src from C4HD cells treated or not with TNFa for
5 min, and from C4HD cells treated with TNFa and PP2. We
also immunoprecipitated ErbB-2 from unstimulated cells
and used it as a source of unphosphorylated ErbB-2 in the
assay. As shown in Fig. 2c, c-Src activated by TNFa was
able to phosphorylate ErbB-2 at Tyr927 residue. Neither
c-Src obtained from control cells nor c-Src inactivated by
PP2 increased ErbB-2 phosphorylation (Fig. 2c). Moreover,
it is known that after TNFa binding to its receptors, protein
kinase C (PKC) is involved in c-Src phosphorylation [30].
Addition of 10 lM GF109203X, a PKC inhibitor, effectively
abolished TNFa ability to induce c-Src phosphorylation and
subsequently ErbB-2 phosphorylation at Tyr927 in C4HD
cells (Fig. 2d). Taken together, these results strongly suggest
that TNFa induces c-Src activation which phosphorylates
ErbB-2 at Tyr877/927 residue.
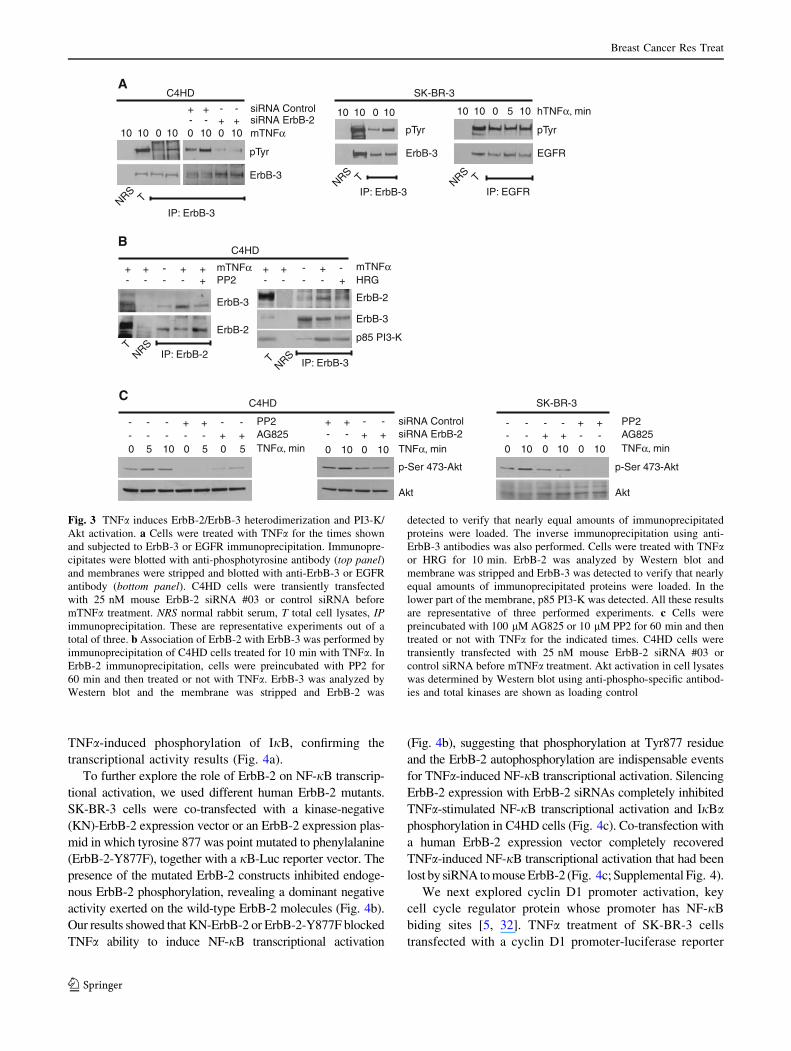
TNFa induces ErbB-2/ErbB-3 heterodimerization
and PI3-K/Akt pathway activation
In the last years, it has been established that ErbB-2 and
ErbB-3 function as an oncogenic unit responsible for
driving breast tumor-cell proliferation by activating PI3-K/
Akt pathway [31]. To gain further insight into the molec-
ular mechanisms triggered by TNFa-induced ErbB-2
transactivation, we monitored ErbB-3 and EGFR phos-
phorylation and ErbB-2 heterodimerization with ErbB-3.
TNFa caused ErbB-3 phosphorylation in C4HD and
SK-BR-3 cells but did not substantially affect EGFR
tyrosine phosphorylation in SK-BR-3 cells (Fig. 3a).
Silencing ErbB-2 expression with 25 nM ErbB-2 siRNAs
(Supplemental Fig. 4) completely inhibited TNFa-induced
ErbB-3 phosphorylation in C4HD cells (Fig. 3a). By co-
immunoprecipitation experiments in C4HD cells, we
showed that TNFa induced ErbB-2 and ErbB-3 heterodi-
merization, which was reduced by addition of PP2
(Fig. 3b). The reverse experiment, in which we immuno-
precipitated ErbB-3 and sought for ErbB-2 presence, is
shown in Fig. 3b with similar results. HRG-induced ErbB-
3/ErbB-2 association was included as control. Among the
members of the ErbB family, ErbB-3 has the unique ability
of binding p85 subunit of PI3-K. TNFa also induced a
rapid p85 PI3-K association with ErbB-3 similar to that
observed upon HRG treatment (Fig. 3b). The activation of
the downstream kinase of PI3-K, Akt, by TNFa was sig-
nificantly inhibited by AG825 in SK-BR-3 and C4HD cells
(Fig. 3c). Blockage of ErbB-2 expression by siRNA also
leads to inhibition of TNFa-induced Akt activation in
C4HD cells (Fig. 3c). Notably, in the absence of c-Src
activation by preincubation with PP2, TNFa capacity to
activate Akt was blocked (Fig. 3c).
Breast Cancer Res Treat
123

ErbB-2 tyrosine kinase inhibitor but not Herceptin
blocks TNFa-induced NF-jB transcriptional activation,
cyclin D1 expression and cell proliferation
Recently we have demonstrated that NF-jB activation is a
key factor in TNFa-induced C4HD cell proliferation [5].
Using the IjB pharmacological inhibitor Bay 11-7082, NF-
jB activation is also shown to be a requisite in TNFa-
induced BT-474 cell proliferation (Supplemental Fig. 5).
Therefore, we examined whether ErbB-2 participates in
NF-jB activation by TNFa. SK-BR-3 and BT-474 cells
were transiently transfected with a jB-luciferase reporter
construct. Treatment with TNFa induced a six- or twofold
increase in jB transcriptional activation, respectively
(Fig. 4a). We next examined the effect of Herceptin, the
humanized anti-ErbB-2 antibody widely used in ErbB-2
positive breast cancer patients [13]. Addition of 10 lg/ml
Herceptin had no effect on TNFa-induced activation of
NF-jB (Fig. 4a). On the other hand, AG825 blocked
TNFa-induced NF-jB transcriptional activation (Fig. 4a).
A similar result was observed using AG825 in C4HD cells
(not shown). HRG induced a threefold induction in jB
transcriptional activation that was blocked by Herceptin
and AG825 treatment (Fig. 4a). Herceptin effectiveness
was proved through its ability to block Akt phosphorylation
in SK-BR-3 and BT-474 cells (Supplemental Fig. 6). To
accomplish NF-jB transcriptional activation through the
canonical pathway, cytoplasmic inhibitor IjBa must be
phosphorylated, ubiquitinated and degraded. Treatment of
SK-BR-3 and BT-474 cell lines with Herceptin did not
modify TNFa-induced phosphorylation of IjB (Fig. 4a).
On the other hand, addition of AG825 completely inhibited
A
B
D
-- -+ + mTNFα
GF109203X- ++
pTyr-927-ErbB-2
ErbB-2
Src
pTyr-416-Src
C4HDC
pTyr-927-ErbB-2
ErbB-2
Src
IP Src Control
IP Src mTNFα + PP2IP Src mTNFα
+-
---
-+---+-
IP ErbB2 Control++++C4HD
C4HD
0 2 105 15 30 60
SK-BR-3
0 2 105 15 30
pTyr-877/927-ErbB-2
ErbB-2
TNFα, min0 2 105 15 30
BT-474
C4HD
0 2 5 mTNFα, min0 5
pTyr-416-Src
Src
pTyr-927-ErbB-2
ErbB-2
- PP2- ++- -- -+ +
SK-BR-3
- ++
hTNFαPP2
pTyr-877-ErbB-2
ErbB-2
-- -+ +
BT-474
- ++
1 1.2 0.82.7 1.0 1.11.0 1 1.3 1.71.6 2.0 1.6 1 1.9 2.82.7 3.6 3.3
1 1.4 1.01.4 0.9
1 1.9 0.41.4 0.5
1 2.0 0.40.41 1.4 0.40.4
1 2.4 1.41.3
1 5.0 1.31.4
hTNFαDasatinibpTyr-877-ErbB-2
ErbB-2
-- -+ +
BT-474
- ++
1 1.7 0.30.3
Src
pTyr-416-Src
1 2.4 0.60.5
Fig. 2 TNFa phosphorylates ErbB-2 through c-Src activation.
a Cells were treated with TNFa for the times shown. Whole cell
lysates were subjected to Western blot analysis for ErbB-2 phos-
phorylation at Tyr877/927 residue. ErbB-2 is shown as loading
control. Bands were quantified using Image J with untreated cell
samples (first lines) set as 1.0. The experiment shown is represen-
tative of a total of five with same results. b C4HD and SK-BR-3 cells
were preincubated with 10 lM PP2 for 60 min and BT-474 cells with
10 lM PP2 or 0.5 lM Dasatinib and then treated or not with TNFa.
c-Src activity in cell lysates was determined by Western blot using
anti-phospho Tyr416 Src antibody and total c-Src is shown as loading
control. ErbB-2 phosphorylation at Tyr877/927 residue was per-
formed as described in (a). These experiments were repeated four
times with same results. c Cold in vitro phosphorylation assay was
performed with C4HD cells preincubated or not with PP2 and then
treated with TNFa for 5 min. c-Src was immunoprecipitated from
each treatment and ErbB-2 immunoprecipitated from unstimulated
C4HD cells was used as substrate. A cold in vitro phosphorylation
assay was performed as described in ‘‘Materials and methods’’. As
specificity control 500 lg protein extract from C4HD cells treated
5 min with TNFa were immunoprecipitated with 2 lg of normal
mouse serum and subjected to the same in vitro phosphorylation
protocol. Western blots of c-Src, anti-phospho Tyr927 ErbB-2 and
ErbB-2 are shown. These experiments were repeated four times with
same results. IP immunoprecipitation. d C4HD cells were preincu-
bated with 10 lM GF109203X for 60 min and then treated with
TNFa. c-Src phosphorylation and ErbB-2 Tyr927 phosphorylation
were performed as described in (b)
Breast Cancer Res Treat
123
TNFa-induced phosphorylation of IjB, confirming the
transcriptional activity results (Fig. 4a).
To further explore the role of ErbB-2 on NF-jB transcrip-
tional activation, we used different human ErbB-2 mutants.
SK-BR-3 cells were co-transfected with a kinase-negative
(KN)-ErbB-2 expression vector or an ErbB-2 expression plas-
mid in which tyrosine 877 was point mutated to phenylalanine
(ErbB-2-Y877F), together with a jB-Luc reporter vector. The
presence of the mutated ErbB-2 constructs inhibited endoge-
nous ErbB-2 phosphorylation, revealing a dominant negative
activity exerted on the wild-type ErbB-2 molecules (Fig. 4b).
Our results showed that KN-ErbB-2 or ErbB-2-Y877F blocked
TNFa ability to induce NF-jB transcriptional activation
(Fig. 4b), suggesting that phosphorylation at Tyr877 residue
and the ErbB-2 autophosphorylation are indispensable events
for TNFa-induced NF-jB transcriptional activation. Silencing
ErbB-2 expression with ErbB-2 siRNAs completely inhibited
TNFa-stimulated NF-jB transcriptional activation and IjBaphosphorylation in C4HD cells (Fig. 4c). Co-transfection with
a human ErbB-2 expression vector completely recovered
TNFa-induced NF-jB transcriptional activation that had been
lost by siRNA to mouse ErbB-2 (Fig. 4c; Supplemental Fig. 4).
We next explored cyclin D1 promoter activation, key
cell cycle regulator protein whose promoter has NF-jB
biding sites [5, 32]. TNFa treatment of SK-BR-3 cells
transfected with a cyclin D1 promoter-luciferase reporter
CC4HD
++-----+ - -+--- ++--
-- + - -+--
SK-BR-3
AG825PP2
0 5 10 TNFα, min0 5 0 5
p-Ser 473-Akt
Akt
0 10 0 10 0 10
A
- + + mTNFαPP2+- -
NRSTIP: ErbB-2
IP: ErbB-3
- + - mTNFαHRG+- -
NRST
ErbB-3
ErbB-2
ErbB-2
ErbB-3
p85 PI3-K
C4HDB
+-
+-
+-
+-
IP: ErbB-3TNRS
0 10
C4HD
10 10 mTNFα
siRNA Control- -++siRNA ErbB-2+ +--
pTyr
ErbB-3
0 100 10
siRNA Control- -++siRNA ErbB-2+ +--
p-Ser 473-Akt
Akt
TNFα, min0 10 0 10
0 5 10 hTNFα, min
pTyr
EGFR
IP: EGFRTNRS
IP: ErbB-3TNRS
0 10
SK-BR-3
10 10 10 10
pTyr
ErbB-3
AG825PP2
TNFα, min
Fig. 3 TNFa induces ErbB-2/ErbB-3 heterodimerization and PI3-K/
Akt activation. a Cells were treated with TNFa for the times shown
and subjected to ErbB-3 or EGFR immunoprecipitation. Immunopre-
cipitates were blotted with anti-phosphotyrosine antibody (top panel)and membranes were stripped and blotted with anti-ErbB-3 or EGFR
antibody (bottom panel). C4HD cells were transiently transfected
with 25 nM mouse ErbB-2 siRNA #03 or control siRNA before
mTNFa treatment. NRS normal rabbit serum, T total cell lysates, IPimmunoprecipitation. These are representative experiments out of a
total of three. b Association of ErbB-2 with ErbB-3 was performed by
immunoprecipitation of C4HD cells treated for 10 min with TNFa. In
ErbB-2 immunoprecipitation, cells were preincubated with PP2 for
60 min and then treated or not with TNFa. ErbB-3 was analyzed by
Western blot and the membrane was stripped and ErbB-2 was
detected to verify that nearly equal amounts of immunoprecipitated
proteins were loaded. The inverse immunoprecipitation using anti-
ErbB-3 antibodies was also performed. Cells were treated with TNFaor HRG for 10 min. ErbB-2 was analyzed by Western blot and
membrane was stripped and ErbB-3 was detected to verify that nearly
equal amounts of immunoprecipitated proteins were loaded. In the
lower part of the membrane, p85 PI3-K was detected. All these results
are representative of three performed experiments. c Cells were
preincubated with 100 lM AG825 or 10 lM PP2 for 60 min and then
treated or not with TNFa for the indicated times. C4HD cells were
transiently transfected with 25 nM mouse ErbB-2 siRNA #03 or
control siRNA before mTNFa treatment. Akt activation in cell lysates
was determined by Western blot using anti-phospho-specific antibod-
ies and total kinases are shown as loading control
Breast Cancer Res Treat
123

vector stimulated luc activity threefold (Fig. 5a). The
presence of Herceptin did not modify TNFa-induced cyclin
D1 promoter expression in SK-BR-3 cells. On the other
hand, AG825 blocked TNFa-induced cyclin D1 promoter
activation. Moreover, in BT-474 cells, Herceptin could not
block TNFa-induced up-regulation of cyclin D1 expres-
sion, while AG825 and GW2974, structurally related to the
ErbB-2 inhibitor clinically used Lapatinib, completely
inhibited TNFa-induced expression of cyclin D1 (Fig. 5b).
Interestingly, addition of Herceptin to BT-474 cells did not
affect TNFa-induced proliferation measured either by cell
count (96 h), propidium iodide staining and flow cytometry
analysis (48 h) or by thymidine incorporation (48 h) as
shown in Fig. 5c. Treatment with Herceptin diminished
BT-474 cell count and induced G0/G1 arrest, although
inhibition of thymidine incorporation was very slight
(Fig. 5c). In line with our previous data on cyclin D1
promoter and protein expression, the presence of AG825
blocked TNFa-induced BT-474 breast cancer cell prolif-
eration. These results show that ErbB-2 transactivation is
essential for TNFa-induced NF-jB transcriptional activa-
tion and proliferation in breast cancer cells.
According to our results in which c-Src acts as an
upstream activator of ErbB-2, we reasoned out that
A
0
3
6
9
HerceptinAG825
κB-l
uc
/ ren
illa
(fo
ld in
du
ctio
n)
- + - - + -- - -+ - +
+- -- - +
++ +- - - - - -++ +- - -- - -
hTNFαHRG
**
*
**
SK-BR-3
0
1,5
3
4,5
- + - +- - - -
+-- -
+ +- - - -+ +- -- -
BT-474
**
**
-
- -
+ +-
+ +
SK-BR-3
-
- -
+ - -
- -
+ -
- -
- +- - +-
- - - + + +
hTNFαAG825
phospho-IκBα
IκBα
HRGHerceptin
-
- -
+ - +
- -
- -- -
- - + +
BT-474
-
- -
+ +-
+ +
1 4.1 1.11.3 1 2.0 1.31.5 1.22.3 1 4.7 0.70.4 1 4.7 101.1
B
0
2
4
6
8
mTNFα
siRNA control
siRNA ErbB-2
- + - +
+ +
- - + +
- -
- +
+ +
- -
κB-l
uc
/ ren
illa
(fo
ld in
du
ctio
n)
pCDNA3.1 hErbB-2wt
*
*
C
0
0,5
1
1,5
2
2,5
κB-l
uc
/ ren
illa
(fo
ld in
du
ctio
n)
- + - + - +
*
pCDNA3.1 ErbB-2-Y877F KN-ErbB-2
hTNFα
SK-BR-3 C4HD
siRNA Control- -++siRNA ErbB-2+ +--TNFα, min0 10 0 10phospho-IκBα
IκBα
- -+ +pCDNA3.1 KN-ErbB-2
- -+ + hTNFαpCDNA3.1 ErbB-2-Y877F
hTNFα
pTyr-877-ErbB-2
pTyr-1222-ErbB-2
ErbB-2
ErbB-2
Fig. 4 ErbB-2 mediates TNFa-induced NF-jB trancriptional activa-
tion. a kB luciferase-transfected cells were preincubated with 100 lM
AG825 or control DMSO or with 10 lg/ml Herceptin for 60 min and
then treated or not with 20 ng/ml hTNFa or with 20 ng/ml HRG for
18 h. Cells were harvested for NF-jB transcriptional activation as
described in ‘‘Materials and methods’’ (*P \ 0.01, **P \ 0.001 vs.
control). Right panel shows Western blot analysis of phospho IjBafrom cells preincubated with AG825 or Herceptin and treated or not
with hTNFa or HRG for 10 min. As loading control, membranes were
stripped and hybridized with an anti-IjBa antibody. Bands were
quantified using Image J with untreated cell samples (first lines) set as
1.0. b SK-BR-3 cells were transiently co-transfected with the
indicated vectors and jB-luciferase construct. Cell treatment and
reporter activity were performed as described in (a). SK-BR-3 cells
transfected with 2 lg ErbB-2-Y877F vector or pCDNA3.1 and treated
with TNFa for 10 min were subjected to Western blot analysis for
ErbB-2 phosphorylation at Tyr877/927 residue as described in Fig. 2a
(right panel). Cells transfected with 2 lg KN-ErbB-2 vector or
pCDNA3.1 were subjected to Western blot analysis for ErbB-2
phosphorylation at Tyr1222 residue as described in Fig. 1f. c C4HD
cells were transiently co-transfected with 25 nM mouse ErbB-2
siRNA #03 or control siRNA, and with jB-luciferase construct before
mTNFa treatment. Similar results were obtained with siRNA #02 and
#04. In reconstitution experiments, 1 lg/well of a human ErbB-2
expression vector was used. Cell treatment and reporter activity were
performed as described in (a) (*P \ 0.001 vs. control). Inset shows
Western Blot analysis of IjBa phosphorylation, as described in (a),
from C4HD cells transiently transfected with 25 nM mouse ErbB-2
siRNA #03 or control siRNA before mTNFa treatment
Breast Cancer Res Treat
123

BT-474
[3 H]-
Thy
mid
ine
inco
rpo
rati
on
(C
PM
x 1
03)
0
50
100
150
200
250
- + - +mTNFα
- - +Dasatinib +
*
* *
D
- + - + - +
AG825 - - ++ - -
Herceptin - - +- +-
Cyc
lin D
1-lu
c / r
enill
a(f
old
ind
uct
ion
)A
SK-BR-3
hTNFα
B
0
1
2
3 * *
0
- -
24 48
+ +- - - -- --+
hTNFα (h)
AG825
GW 2974
Herceptin
0 24 48 0 24 48 0 24 48
- - + +- - - -- -- +- - + +- - - -- -- +
Cyclin D1
Actin
BT-474
1 1.8 0.31.9 0.20.3 0.1 0.2 0.80.1 1.7 1.4
0
25
50
75
100
G0/G1 S G2/M
- + - + - +
- - ++ - -
- - +- +-
BT-474%
Cel
ls
SK-BR-3mTNFα, min
pTyr-416-Src
Src
- Herceptin- + +
0 5 50
- - + +
0 5 50
BT-474
E
- + - -+ +
- - ++ - -
- - +- +-[3
H]-
Thy
mid
ine
inco
rpo
rati
on
(C
PM
x 1
03)
0
5
10
15
20
25
30*
*
0
5
10
15
20
Cel
l nu
mb
er /
wel
l (10
4)
- + - + - +
AG825 - - ++ - -
Herceptin - - +- +-
hTNFα
*
*
†
* *
C
Fig. 5 TNFa-induced proliferation is blocked by pharmacological
inhibitors of ErbB-2 and c-Src but not by Herceptin in human breast
cancer cells. a SK-BR-3 cells were transiently transfected with cyclin
D1-luciferase promoter and then treated as described in Fig. 4a
(*P \ 0.001 vs. control). b BT-474 cells were preincubated with
100 lM AG825, 1 lM GW2974 or 10 lg/ml Herceptin for 60 min
and treated or not with 20 ng/ml hTNFa for 24 or 48 h. Whole cell
lysates were subjected to Western blot analysis for cyclin D1
expression. Actin is shown as loading control and was used for
quantification. c BT-474 cells were preincubated with 100 lM
AG825 or 10 lg/ml Herceptin for 60 min and treated or not with
20 ng/ml hTNFa. Proliferation assay was performed by cell count
with Trypan blue at 96 h of culture (*P \ 0.05 vs. control, �P \ 0.05
vs. Herceptin), left panel, by cell cycle analysis with propidium iodide
staining and flow cytometry analysis at 48 h of culture (G0/G1 phase
P \ 0.02 TNFa vs. control; G2/M phase P \ 0.05 TNFa vs. control;
G0/G1 phase P \ 0.05 TNFa ? Herceptin vs. Herceptin; G2/M phase
P \ 0.05 TNFa ? Herceptin vs. Herceptin), central panel, and by
[3H]-thymidine incorporation assay, as described in Fig. 1a
(*P \ 0.01 vs. control) right panel. d BT-474 and SK-BR-3 cells
were preincubated with 10 lg/ml Herceptin for 6 h and treated or not
with 20 ng/ml hTNFa. c-Src phosphorylation was performed as
described in Fig. 2b. e BT-474 cells were preincubated with 0.5 lM
Dasatinib for 60 min and then treated or not with 20 ng/ml of TNFafor 48 h. Proliferation was performed by [3H]-thymidine incorpora-
tion assay as described in Fig. 1 (*P \ 0.01 vs. control)
Breast Cancer Res Treat
123

Herceptin should not inhibit c-Src activation by TNFa.
Indeed in the presence of Herceptin, TNFa induced c-Src
activity to the same degree as in control cells (Fig. 5d).
These results led us to assess whether c-Src was involved in
the mitogenic effect of TNFa. We performed a proliferation
assay that showed that Dasatinib completely blocked TNFaability to induce BT-474 proliferation (Fig. 5e). These
results disclose that c-Src is a key player on TNFa-induced
signaling cascade leading to breast cancer cell growth.
Discussion
In the present study we disclosed, for the first time, that
TNFa transactivates ErbB-2 in breast cancer cells, thus
becoming another player in the scenario of ErbB-2 onco-
genic activity. Our results provided a time course of events
triggered by TNFa leading to breast cancer prolifera-
tion.We unraveled that in ErbB-2-overexpressing cells,
TNFa initiates ErbB-2 signal transduction by the activation
of c-Src (2–5 min), the kinase involved in Tyr877 ErbB-2
phosphorylation. These induce ErbB-2 autophosphoryla-
tion (5–15 min) and ErbB-2/ErbB-3 heterodimer forma-
tion, leading to the activation of PI3-K/Akt and finally
transcriptional activation of NF-jB. This transcription
factor in turn increases the expression of its target gene,
cyclin D1 which is a key protein involved in breast cancer
proliferation (Fig. 6). Cyclin D1 increase was detectable as
soon as 4 h of TNFa treatment (data not shown) continuing
high 48 h later.
Previous studies have shown that c-Src activation is
dependent on ErbB-2, but it has recently been reported that
c-Src itself plays a role upstream on ErbB-2 phosphoryla-
tion. c-Src interacts directly with the catalytic domain of
ErbB-2 [33, 34] and phosphorylates ErbB-2 at Tyr877/927
residue within the activation loop of the kinase domain,
stabilizing it in an open and extended conformation which
increases its intrinsic kinase activity [28]. Here, we found
that TNFa induces Tyr877/927 ErbB-2 phosphorylation
through c-Src activation, in a PKC-dependent manner, in
murine C4HD and human SK-BR-3 and BT-474 cells. We
observed that TNFa induces Tyr416 phosphorylation of
c-Src, and that this activated kinase phosphorylates ErbB-2
Tyr877/927 residue in vitro. Moreover, we also demon-
strated that blockage of c-Src with the clinically used
inhibitor, Dasatinib, inhibits TNFa-induced BT-474 cell
proliferation. This piece of information would be useful in
future evaluations of breast cancer specimens with the
expression of TNFa and activated c-Src, of patients
undergoing anti-ErbB-2 therapy. It was recently demon-
strated that TNFR1 is constitutively associated with c-Src,
and that TNFa induces recruitment of additional c-Src and
increases its activity in HEK293 and MCF-7 cell lines [35].
During the preparation of this manuscript, Yamaoka et al.
[36] demonstrated that Src kinase activity as well as
transactivation of EGFR and ErbB-2 is required for TNFa-
induced survival of young adult mouse colon (YAMC)
epithelial cells. In the present study, we also confirmed that
c-Src is the kinase responsible for ErbB-2 transactivation,
and we proved that c-Src mediates the phosphorylation on
ErbB-2 Tyr877 residue. Moreover, we demonstrated that
ErbB-2 transactivation is responsible for TNFa-induced
proliferation of breast cancer cells overexpressing ErbB-2,
and that TNFa elicits heterodimerization of ErbB-2 with
ErbB-3. However, we could not detect any participation of
EGFR. These differences might be attributed to tissue-
specific signaling. Another interesting finding is that,
although both TNFa and HRG stimulate p42/p44 MAPK
activation, blockage of ErbB-2 by AG825 did not inhibit the
activation of p42/p44 MAPK induced by TNFa. In contrast,
AG825 blocked HRG-induced p42/p44 MAPK activation.
These results reveal different signaling pathways induced
by ErbB-2 transactivation caused by a classic ErbB-3/ErbB-
TNFR
PKC
c-Src
TNFα
ErbB2ErbB3
Akt
PI3-K
P
P P
IκB
α
> Cyclin D1p65p50
877
1222
Fig. 6 Model of TNFa transactivation of ErbB-2 and proliferation
induction in breast cancer cells. TNFa binds to TNFa receptor and
activates PKC which in turn activates c-Src which is able to
phosphorylate ErbB-2 at Tyr877 residue. ErbB-2 becomes autophos-
phorylated at Tyr1222 residue inducing ErbB-2/ErbB-3 heterodimer-
ization, ErbB-3 phosphorylation and the concomitant recruitment of
p85 PI3-K. This event activates Akt and leads to NF-jB transcrip-
tional activation and the consequent expression of cyclin D1 and
proliferation
Breast Cancer Res Treat
123

4 ligand (HRG) or a ‘‘non-classic’’ activator (TNFa). On the
other hand, transactivation of EGFR by TNFa is a well-
documented mechanism that involves MMPs stimulation;
in particular, of the TNFa converting enzyme, which
releases EGFR ligands [18–20]. However, in this work we
could not detect any evidence supporting the fact that ErbB-
2 transactivation by TNFa was induced by cleavage of
membrane-tethered ErbB ligands, since pretreatment with
GM6001, a broad spectrum inhibitor of MMPs, did not
modify TNFa-induced ErbB-2 phosphorylation either in
C4HD cells or in SK-BR-3 cells.
An exciting novel finding of this study has been the
demonstration that TNFa utilizes ErbB-2 as a downstream
signaling partner in the generation of mitogenic signals.
Our data supporting the fact that TNFa-induced prolifera-
tion of C4HD cells is dependent on ErbB-2 kinase activity
was also confirmed in SK-BR-3 cells, where TNFa has
already been reported as mitogenic [8], and in BT-474
cells. Our findings suggest that NF-jB activation induced
by TNFa requires a functional ErbB-2 because (a) the
inhibition of ErbB-2 by AG825, (b) the protein knockdown
of ErbB-2 by siRNA strategy, (c) the fact that transfection
with KN-ErbB-2 or ErbB-2-Y877F plasmids, impaired
TNFa capacity to activate NF-jB and (d) the fact that co-
transfection with siRNA to mouse ErbB-2 and reconstitu-
tion of ErbB-2 expression with a vector encoding human
ErbB-2, restored TNFa-induced NF-jB activation. How-
ever, in NIH 3T3 cells, which have low levels of ErbB-2,
transiently transfected with ErbB-2 plasmid, we observed
no difference of TNFa effect on NF-jB activation as
compared to the cells transfected with the empty vector
(data not shown). These data suggest that ErbB-2-over-
expressing cells have other/s signaling molecule/s that
allow/s the cross talk between TNFa receptors and ErbB-2.
We observed that either inhibition of ErbB-2 with AG825
or silencing ErbB-2 by siRNA treatment inhibited TNFa-
induced activation of PI3-K/Akt and IjBa phosphoryla-
tion. These results are in line with our previous report
showing that Akt behaves as an upstream molecule in the
NF-jB activation cascade and this activation proceeds
through the canonical pathway [5]. Although it has long
been known that amplified expression of ErbB-2 induces
breast cancer resistance to TNFa-induced cytotoxicity [37],
the mechanisms that promote this effect have scarcely been
characterized. ErbB-2 overexpression has been found to
constitutively activate Akt/NF-jB anti-apoptotic pathway,
conferring resistance to TNFa in cancer cell lines [10].
Yamaoka et al. [36] determined that loss of EGFR
expression suppressed TNFa activation of Akt and induce
apoptosis in YAMC cells, although they did not observe
any modulation of IjB. On the other hand, Biswas et al.
[38] demonstrated that NF-jB activation is predominantly
found in the estrogen receptor (ER)-negative/ErbB-2
positive subclass in comparison with ER-positive human
breast cancer tumors. Recently, this group demonstrated
that NF-jB activation in this subclass of breast cancer is
essential for tumorigenesis and tumor progression [39]. In
the present work, we illustrate that TNFa activates ErbB-2
and uses it as an intermediate to promote NF-jB activation,
adding yet another layer of complexity in ErbB-2 over-
expressing tumor signaling.
An extensive body of evidence has established ErbB-2
as a key mediator of tumor cell growth and survival, since
ErbB-2 overexpression in nearly 30% of human breast
cancers predicts an aggressive course of disease and poor
prognosis [9]. The first anti-ErbB-2 agent used in clinical
practice is the humanized monoclonal antibody Herceptin
[13]. Given alone as first line treatment to metastatic breast
cancer patients, Herceptin shows an overall response rate
of 38% [40]. It has been suggested that one likely mech-
anism of acquired resistance to Herceptin can be the
amplification of ligand-induced activation of the ErbBs
receptor [41]. Consequently, small-molecule inhibitors of
EGFR/ErbB-2 have progressed to clinical trials [42]. Here,
we report TNFa as a new ErbB-2 transactivating factor
unexplored before, and demonstrate that TNFa-induced
ErbB-2 transactivation leading to NF-jB activation and
cell proliferation can be blocked at the level of ErbB-2
phosphorylation through AG825 or GW2974, specific
EGFR/ErbB-2 tyrosine kinase inhibitors, but not at the
level of antibody blockage of the receptor. Interestingly,
Herceptin increased TNFa-induced NF-jB activity and
IjBa phosphorylation in BT-474 but not in SK-BR-3 cells
(Fig. 4a). Since BT-474 cell line expresses estrogen and
progesterone receptors (PR) and as we have demonstrated
that PR is involved in ErbB-2 phosphorylation [43], further
studies have to be carried out to shed light on the above-
mentioned finding.
In this work, we provide a suitable molecular explana-
tion for the resistance to Herceptin as observed in clinical
practice, and hypothesize that TNFa signaling may have
augmented in patients unresponsive to monoclonal anti-
body therapy. Even when TNFa has been shown to be
expressed in a significant proportion of human infiltrating
breast cancers [6], the eventual worth of TNFa signaling as
a prognostic factor in anti-ErbB-2 therapy is yet to be
determined.
Acknowledgments This work was supported by grants IDB 1728/
OC-AR PICT 2006 0211 and PICT 2004 05-25301, both from the
National Agency of Scientific Promotion of Argentina, PIP 5391 from
the Argentine National Council of Scientific Research (CONICET)
and by Oncomed-Reno CONICET 1819/03, from the Henry Moore
Institute of Argentina and by grant KG090250 from the Susan G.
Komen for the Cure. The authors wish to thank Dr Alfredo A. Mo-
linolo (NIH, Bethesda, MD) for his constant help and support. We
thank Dr C. Lanari for providing the MPA-induced mammary tumor
model.
Breast Cancer Res Treat
123

References
1. Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B
(1975) An endotoxin-induced serum factor that causes necrosis of
tumors. Proc Natl Acad Sci USA 72:3666–3670
2. Wu S, Boyer CM, Whitaker RS, Berchuck A, Wiener JR,
Weinberg JB, Bast RC Jr (1993) Tumor necrosis factor alpha as
an autocrine and paracrine growth factor for ovarian cancer:
monokine induction of tumor cell proliferation and tumor
necrosis factor alpha expression. Cancer Res 53:1939–1944
3. Naylor MS, Stamp GW, Foulkes WD, Eccles D, Balkwill FR
(1993) Tumor necrosis factor and its receptors in human ovarian
cancer. Potential role in disease progression. J Clin Invest
91:2194–2206
4. Rubio MF, Werbajh S, Cafferata EG, Quaglino A, Colo GP,
Nojek IM, Kordon EC, Nahmod VE, Costas MA (2006) TNF-
alpha enhances estrogen-induced cell proliferation of estrogen-
dependent breast tumor cells through a complex containing
nuclear factor-kappa B. Oncogene 25:1367–1377
5. Rivas MA, Carnevale RP, Proietti CJ, Rosemblit C, Beguelin W,
Salatino M, Charreau EH, Frahm I, Sapia S, Brouckaert P,
Elizalde PV, Schillaci R (2008) TNFalpha acting on TNFR1
promotes breast cancer growth via p42/P44 MAPK, JNK, Akt
and NF-kappaB-dependent pathways. Exp Cell Res 314:509–529
6. Garcia-Tunon I, Ricote M, Ruiz A, Fraile B, Paniagua R, Royuela
M (2006) Role of tumor necrosis factor-alpha and its receptors in
human benign breast lesions and tumors (in situ and infiltrative).
Cancer Sci 97:1044–1049
7. Pirianov G, Colston KW (2001) Interactions of vitamin D ana-
logue CB1093, TNFalpha and ceramide on breast cancer cell
apoptosis. Mol Cell Endocrinol 172:69–78
8. Lyu MA, Rosenblum MG (2005) The immunocytokine scFv23/
TNF sensitizes HER-2/neu-overexpressing SKBR-3 cells to
tumor necrosis factor (TNF) via up-regulation of TNF receptor-1.
Mol Cancer Ther 4:1205–1213
9. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire
WL (1987) Human breast cancer: correlation of relapse and
survival with amplification of the HER-2/neu oncogene. Science
235:177–182
10. Zhou BP, Hu MC, Miller SA, Yu Z, Xia W, Lin SY, Hung MC
(2000) HER-2/neu blocks tumor necrosis factor-induced apop-
tosis via the Akt/NF-kappaB pathway. J Biol Chem 275:8027–
8031
11. Yarden Y, Sliwkowski MX (2001) Untangling the ErbB signal-
ling network. Nat Rev Mol Cell Biol 2:127–137
12. Tzahar E, Waterman H, Chen X, Levkowitz G, Karunagaran D,
Lavi S, Ratzkin BJ, Yarden Y (1996) A hierarchical network of
interreceptor interactions determines signal transduction by Neu
differentiation factor/neuregulin and epidermal growth factor.
Mol Cell Biol 16:5276–5287
13. Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong
WL, Rowland AM, Kotts C, Carver ME, Shepard HM (1992)
Humanization of an anti-p185HER2 antibody for human cancer
therapy. Proc Natl Acad Sci USA 89:4285–4289
14. Hynes NE, Lane HA (2005) ERBB receptors and cancer: the
complexity of targeted inhibitors. Nat Rev Cancer 5:341–354
15. Cabioglu N, Summy J, Miller C, Parikh NU, Sahin AA, Tuzlali S,
Pumiglia K, Gallick GE, Price JE (2005) CXCL-12/stromal cell-
derived factor-1alpha transactivates HER2-neu in breast cancer
cells by a novel pathway involving Src kinase activation. Cancer
Res 65:6493–6497
16. Yamauchi T, Yamauchi N, Ueki K, Sugiyama T, Waki H, Miki
H, Tobe K, Matsuda S, Tsushima T, Yamamoto T, Fujita T,
Taketani Y, Fukayama M, Kimura S, Yazaki Y, Nagai R,
Kadowaki T (2000) Constitutive tyrosine phosphorylation of
ErbB-2 via Jak2 by autocrine secretion of prolactin in human
breast cancer. J Biol Chem 275:33937–33944
17. Qiu Y, Ravi L, Kung HJ (1998) Requirement of ErbB2 for sig-
nalling by interleukin-6 in prostate carcinoma cells. Nature
393:83–85
18. Lee CW, Lin CC, Lin WN, Liang KC, Luo SF, Wu CB, Wang SW,
Yang CM (2007) TNF-alpha induces MMP-9 expression via
activation of Src/EGFR, PDGFR/PI3K/Akt cascade and promotion
of NF-kappaB/p300 binding in human tracheal smooth muscle
cells. Am J Physiol Lung Cell Mol Physiol 292:L799–L812
19. Chokki M, Mitsuhashi H, Kamimura T (2006) Metalloprotease-
dependent amphiregulin release mediates tumor necrosis factor-
alpha-induced IL-8 secretion in the human airway epithelial cell
line NCI-H292. Life Sci 78:3051–3057
20. Chen WN, Woodbury RL, Kathmann LE, Opresko LK, Zangar
RC, Wiley HS, Thrall BD (2004) Induced autocrine signaling
through the epidermal growth factor receptor contributes to the
response of mammary epithelial cells to tumor necrosis factor
alpha. J Biol Chem 279:18488–18496
21. Balana ME, Labriola L, Salatino M, Movsichoff F, Peters G,
Charreau EH, Elizalde PV (2001) Activation of ErbB-2 via a hier-
archical interaction between ErbB-2 and type I insulin-like growth
factor receptor in mammary tumor cells. Oncogene 20:34–47
22. Labriola L, Salatino M, Proietti CJ, Pecci A, Coso OA, Kornblihtt
AR, Charreau EH, Elizalde PV (2003) Heregulin induces tran-
scriptional activation of the progesterone receptor by a mecha-
nism that requires functional ErbB-2 and mitogen-activated
protein kinase activation in breast cancer cells. Mol Cell Biol
23:1095–1111
23. Lanari C, Kordon E, Molinolo A, Pasqualini CD, Charreau EH
(1989) Mammary adenocarcinomas induced by medroxyproges-
terone acetate: hormone dependence and EGF receptors of
BALB/c in vivo sublines. Int J Cancer 43:845–850
24. Balana ME, Lupu R, Labriola L, Charreau EH, Elizalde PV
(1999) Interactions between progestins and heregulin (HRG)
signaling pathways: HRG acts as mediator of progestins prolif-
erative effects in mouse mammary adenocarcinomas. Oncogene
18:6370–6379
25. Salatino M, Beguelin W, Peters MG, Carnevale R, Proietti CJ,
Galigniana MD, Vedoy CG, Schillaci R, Charreau EH, Sogayar
MC, Elizalde PV (2006) Progestin-induced caveolin-1 expression
mediates breast cancer cell proliferation. Oncogene 25:7723–7739
26. Plowman GD, Culouscou JM, Whitney GS, Green JM, Carlton
GW, Foy L, Neubauer MG, Shoyab M (1993) Ligand-specific
activation of HER4/p180erbB4, a fourth member of the epider-
mal growth factor receptor family. Proc Natl Acad Sci USA
90:1746–1750
27. Brockhoff G, Heiss P, Schlegel J, Hofstaedter F, Knuechel R
(2001) Epidermal growth factor receptor, c-erbB2 and c-erbB3
receptor interaction, and related cell cycle kinetics of SK-BR-3
and BT474 breast carcinoma cells. Cytometry 44:338–348
28. Xu W, Yuan X, Beebe K, Xiang Z, Neckers L (2007) Loss of
Hsp90 association up-regulates Src-dependent ErbB2 activity.
Mol Cell Biol 27:220–228
29. Ishizawar RC, Miyake T, Parsons SJ (2007) c-Src modulates
ErbB2 and ErbB3 heterocomplex formation and function.
Oncogene 26:3503–3510
30. Huang WC, Chen JJ, Inoue H, Chen CC (2003) Tyrosine phos-
phorylation of I-kappa B kinase alpha/beta by protein kinase
C-dependent c-Src activation is involved in TNF-alpha-induced
cyclooxygenase-2 expression. J Immunol 170:4767–4775
31. Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF III,
Hynes NE (2003) The ErbB2/ErbB3 heterodimer functions as an
oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell
proliferation. Proc Natl Acad Sci USA 100:8933–8938
Breast Cancer Res Treat
123

32. Hinz M, Krappmann D, Eichten A, Heder A, Scheidereit C,
Strauss M (1999) NF-kappaB function in growth control: regu-
lation of cyclin D1 expression and G0/G1-to-S-phase transition.
Mol Cell Biol 19:2690–2698
33. Kim H, Chan R, Dankort DL, Zuo D, Najoukas M, Park M,
Muller WJ (2005) The c-Src tyrosine kinase associates with the
catalytic domain of ErbB-2: implications for ErbB-2 mediated
signaling and transformation. Oncogene 24:7599–7607
34. Belsches-Jablonski AP, Biscardi JS, Peavy DR, Tice DA, Romney
DA, Parsons SJ (2001) Src family kinases and HER2 interactions in
human breast cancer cell growth and survival. Oncogene 20:1465–
1475
35. Pincheira R, Castro AF, Ozes ON, Idumalla PS, Donner DB
(2008) Type 1 TNF receptor forms a complex with and uses Jak2
and c-Src to selectively engage signaling pathways that regulate
transcription factor activity. J Immunol 181:1288–1298
36. Yamaoka T, Yan F, Cao H, Hobbs SS, Dise RS, Tong W, Polk
DB (2008) Transactivation of EGF receptor and ErbB2 protects
intestinal epithelial cells from TNF-induced apoptosis. Proc Natl
Acad Sci USA 105:11772–11777
37. Hudziak RM, Lewis GD, Shalaby MR, Eessalu TE, Aggarwal
BB, Ullrich A, Shepard HM (1988) Amplified expression of the
HER2/ERBB2 oncogene induces resistance to tumor necrosis
factor alpha in NIH 3T3 cells. Proc Natl Acad Sci USA 85:5102–
5106
38. Biswas DK, Shi Q, Baily S, Strickland I, Ghosh S, Pardee AB,
Iglehart JD (2004) NF-kappa B activation in human breast cancer
specimens and its role in cell proliferation and apoptosis. Proc
Natl Acad Sci USA 101:10137–10142
39. Singh S, Shi Q, Bailey ST, Palczewski MJ, Pardee AB, Iglehart
JD, Biswas DK (2007) Nuclear factor-kappaB activation: a
molecular therapeutic target for estrogen receptor-negative and
epidermal growth factor receptor family receptor-positive human
breast cancer. Mol Cancer Ther 6:1973–1982
40. Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN,
Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF, Burch-
more M, Shak S, Stewart SJ, Press M (2002) Efficacy and safety
of trastuzumab as a single agent in first-line treatment of HER2-
overexpressing metastatic breast cancer. J Clin Oncol 20:719–
726
41. Ritter CA, Perez-Torres M, Rinehart C, Guix M, Dugger T,
Engelman JA, Arteaga CL (2007) Human breast cancer cells
selected for resistance to trastuzumab in vivo overexpress epi-
dermal growth factor receptor and ErbB ligands and remain
dependent on the ErbB receptor network. Clin Cancer Res
13:4909–4919
42. Johnston S, Trudeau M, Kaufman B, Boussen H, Blackwell K,
LoRusso P, Lombardi DP, Ben Ahmed S, Citrin DL, DeSilvio
ML, Harris J, Westlund RE, Salazar V, Zaks TZ, Spector NL
(2008) Phase II study of predictive biomarker profiles for
response targeting human epidermal growth factor receptor 2
(HER-2) in advanced inflammatory breast cancer with lapatinib
monotherapy. J Clin Oncol 26:1066–1072
43. Proietti CJ, Rosemblit C, Beguelin W, Rivas MA, Diaz Flaque
MC, Charreau EH, Schillaci R, Elizalde PV (2009) Activation of
Stat3 by heregulin/ErbB-2 through the co-option of progesterone
receptor signaling drives breast cancer growth. Mol Cell Biol
29:1249–1265
Breast Cancer Res Treat
123
Top Related
Copyright © 2022 FDOKUMEN

