
Vasopressin up-regulates the expression of growth-related immediate-early genes via two distinct EGF...
9
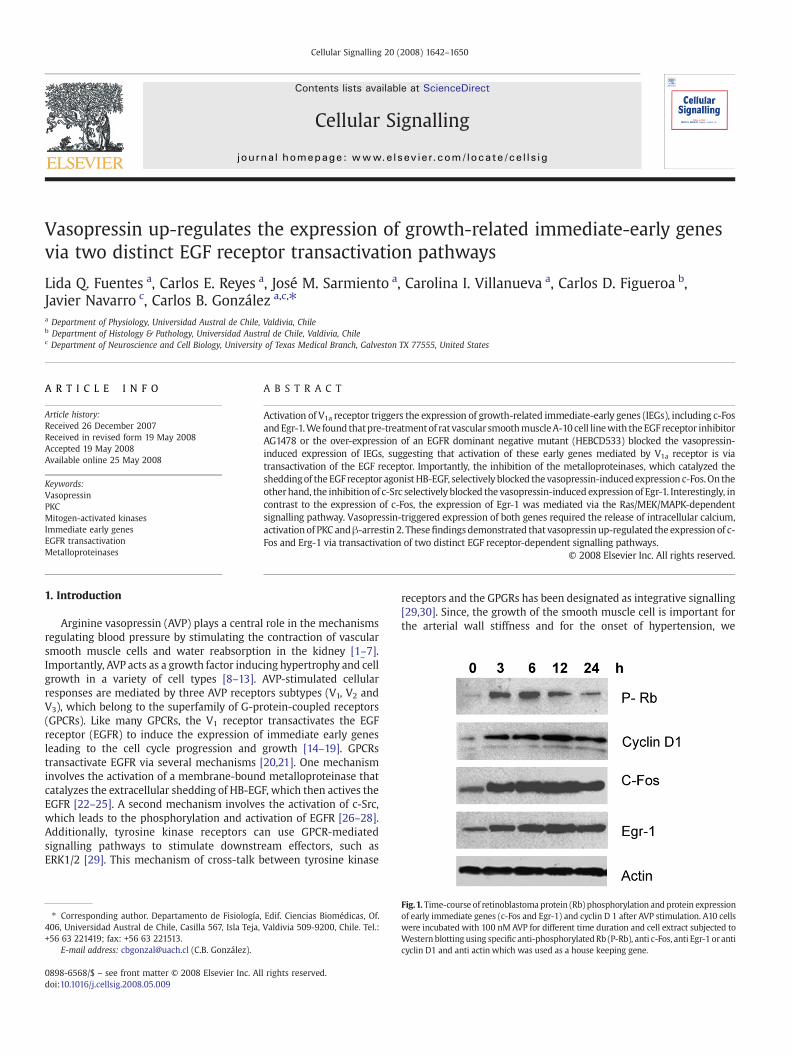
Vasopressin up-regulates the expression of growth-related immediate-early genes via two distinct EGF receptor transactivation pathways Lida Q. Fuentes a , Carlos E. Reyes a , José M. Sarmiento a , Carolina I. Villanueva a , Carlos D. Figueroa b , Javier Navarro c , Carlos B. González a,c, ⁎ a Department of Physiology, Universidad Austral de Chile, Valdivia, Chile b Department of Histology & Pathology, Universidad Austral de Chile, Valdivia, Chile c Department of Neuroscience and Cell Biology, University of Texas Medical Branch, Galveston TX 77555, United States ABSTRACT ARTICLE INFO Article history: Received 26 December 2007 Received in revised form 19 May 2008 Accepted 19 May 2008 Available online 25 May 2008 Keywords: Vasopressin PKC Mitogen-activated kinases Immediate early genes EGFR transactivation Metalloproteinases Activation of V 1a receptor triggers the expression of growth-related immediate-early genes (IEGs), including c-Fos and Egr-1. We found that pre-treatment of rat vascular smooth muscle A-10 cell line with the EGF receptor inhibitor AG1478 or the over-expression of an EGFR dominant negative mutant (HEBCD533) blocked the vasopressin- induced expression of IEGs, suggesting that activation of these early genes mediated by V 1a receptor is via transactivation of the EGF receptor. Importantly, the inhibition of the metalloproteinases, which catalyzed the shedding of the EGF receptor agonist HB-EGF, selectively blocked the vasopressin-induced expression c-Fos. On the other hand, the inhibition of c-Src selectively blocked the vasopressin-induced expression of Egr-1. Interestingly, in contrast to the expression of c-Fos, the expression of Egr-1 was mediated via the Ras/MEK/MAPK-dependent signalling pathway. Vasopressin-triggered expression of both genes required the release of intracellular calcium, activation of PKC and β-arrestin 2. These findings demonstrated that vasopressin up-regulated the expression of c- Fos and Erg-1 via transactivation of two distinct EGF receptor-dependent signalling pathways. © 2008 Elsevier Inc. All rights reserved. 1. Introduction Arginine vasopressin (AVP) plays a central role in the mechanisms regulating blood pressure by stimulating the contraction of vascular smooth muscle cells and water reabsorption in the kidney [1–7]. Importantly, AVP acts as a growth factor inducing hypertrophy and cell growth in a variety of cell types [8–13]. AVP-stimulated cellular responses are mediated by three AVP receptors subtypes (V 1 ,V 2 and V 3 ), which belong to the superfamily of G-protein-coupled receptors (GPCRs). Like many GPCRs, the V 1 receptor transactivates the EGF receptor (EGFR) to induce the expression of immediate early genes leading to the cell cycle progression and growth [14–19]. GPCRs transactivate EGFR via several mechanisms [20,21]. One mechanism involves the activation of a membrane-bound metalloproteinase that catalyzes the extracellular shedding of HB-EGF, which then actives the EGFR [22–25]. A second mechanism involves the activation of c-Src, which leads to the phosphorylation and activation of EGFR [26–28]. Additionally, tyrosine kinase receptors can use GPCR-mediated signalling pathways to stimulate downstream effectors, such as ERK1/2 [29]. This mechanism of cross-talk between tyrosine kinase receptors and the GPGRs has been designated as integrative signalling [29,30]. Since, the growth of the smooth muscle cell is important for the arterial wall stiffness and for the onset of hypertension, we Cellular Signalling 20 (2008) 1642–1650 ⁎ Corresponding author. Departamento de Fisiología, Edif. Ciencias Biomédicas, Of. 406, Universidad Austral de Chile, Casilla 567, Isla Teja, Valdivia 509-9200, Chile. Tel.: +56 63221419; fax: +56 63 221513. E-mail address: [email protected] (C.B. González). Fig. 1. Time-course of retinoblastoma protein (Rb) phosphorylation and protein expression of early immediate genes (c-Fos and Egr-1) and cyclin D 1 after AVP stimulation. A10 cells were incubated with 100 nM AVP for different time duration and cell extract subjected to Western blotting using specific anti-phosphorylated Rb (P-Rb), anti c-Fos, anti Egr-1 or anti cyclin D1 and anti actin which was used as a house keeping gene. 0898-6568/$ – see front matter © 2008 Elsevier Inc. All rights reserved. doi:10.1016/j.cellsig.2008.05.009 Contents lists available at ScienceDirect Cellular Signalling journal homepage: www.elsevier.com/locate/cellsig
Transcript of Vasopressin up-regulates the expression of growth-related immediate-early genes via two distinct EGF...
Cellular Signalling 20 (2008) 1642–1650
Contents lists available at ScienceDirect
Cellular Signalling
j ourna l homepage: www.e lsev ie r.com/ locate /ce l l s ig
Vasopressin up-regulates the expression of growth-related immediate-early genesvia two distinct EGF receptor transactivation pathways
Lida Q. Fuentes a, Carlos E. Reyes a, José M. Sarmiento a, Carolina I. Villanueva a, Carlos D. Figueroa b,Javier Navarro c, Carlos B. González a,c,⁎a Department of Physiology, Universidad Austral de Chile, Valdivia, Chileb Department of Histology & Pathology, Universidad Austral de Chile, Valdivia, Chilec Department of Neuroscience and Cell Biology, University of Texas Medical Branch, Galveston TX 77555, United States
⁎ Corresponding author. Departamento de Fisiología406, Universidad Austral de Chile, Casilla 567, Isla Teja,+56 63 221419; fax: +56 63 221513.
E-mail address: [email protected] (C.B. González).
0898-6568/$ – see front matter © 2008 Elsevier Inc. Aldoi:10.1016/j.cellsig.2008.05.009
A B S T R A C T
A R T I C L E I N F OArticle history:
Activation of V1a receptor trig Received 26 December 2007Received in revised form 19 May 2008Accepted 19 May 2008Available online 25 May 2008Keywords:VasopressinPKCMitogen-activated kinasesImmediate early genesEGFR transactivationMetalloproteinases
gers the expression of growth-related immediate-early genes (IEGs), including c-FosandEgr-1.We found thatpre-treatmentof rat vascular smoothmuscleA-10 cell linewith theEGFreceptor inhibitorAG1478 or the over-expression of an EGFR dominant negative mutant (HEBCD533) blocked the vasopressin-induced expression of IEGs, suggesting that activation of these early genes mediated by V1a receptor is viatransactivation of the EGF receptor. Importantly, the inhibition of the metalloproteinases, which catalyzed thesheddingof the EGFreceptor agonistHB-EGF, selectively blocked the vasopressin-inducedexpression c-Fos.On theotherhand, the inhibitionof c-Src selectively blocked the vasopressin-inducedexpression of Egr-1. Interestingly, incontrast to the expression of c-Fos, the expression of Egr-1 was mediated via the Ras/MEK/MAPK-dependentsignalling pathway. Vasopressin-triggered expression of both genes required the release of intracellular calcium,activationof PKC andβ-arrestin 2. Thesefindingsdemonstrated that vasopressin up-regulated the expression of c-Fos and Erg-1 via transactivation of two distinct EGF receptor-dependent signalling pathways.
© 2008 Elsevier Inc. All rights reserved.
1. Introduction
Arginine vasopressin (AVP) plays a central role in the mechanismsregulating blood pressure by stimulating the contraction of vascularsmooth muscle cells and water reabsorption in the kidney [1–7].Importantly, AVP acts as a growth factor inducing hypertrophy and cellgrowth in a variety of cell types [8–13]. AVP-stimulated cellularresponses are mediated by three AVP receptors subtypes (V1, V2 andV3), which belong to the superfamily of G-protein-coupled receptors(GPCRs). Like many GPCRs, the V1 receptor transactivates the EGFreceptor (EGFR) to induce the expression of immediate early genesleading to the cell cycle progression and growth [14–19]. GPCRstransactivate EGFR via several mechanisms [20,21]. One mechanisminvolves the activation of a membrane-bound metalloproteinase thatcatalyzes the extracellular shedding of HB-EGF, which then actives theEGFR [22–25]. A second mechanism involves the activation of c-Src,which leads to the phosphorylation and activation of EGFR [26–28].Additionally, tyrosine kinase receptors can use GPCR-mediatedsignalling pathways to stimulate downstream effectors, such asERK1/2 [29]. This mechanism of cross-talk between tyrosine kinase
, Edif. Ciencias Biomédicas, Of.Valdivia 509-9200, Chile. Tel.:
l rights reserved.
receptors and the GPGRs has been designated as integrative signalling[29,30]. Since, the growth of the smooth muscle cell is important forthe arterial wall stiffness and for the onset of hypertension, we
Fig.1. Time-course of retinoblastoma protein (Rb) phosphorylation and protein expressionof early immediate genes (c-Fos and Egr-1) and cyclin D 1 after AVP stimulation. A10 cellswere incubated with 100 nM AVP for different time duration and cell extract subjected toWestern blotting using specific anti-phosphorylatedRb (P-Rb), anti c-Fos, anti Egr-1 or anticyclin D1 and anti actin which was used as a house keeping gene.
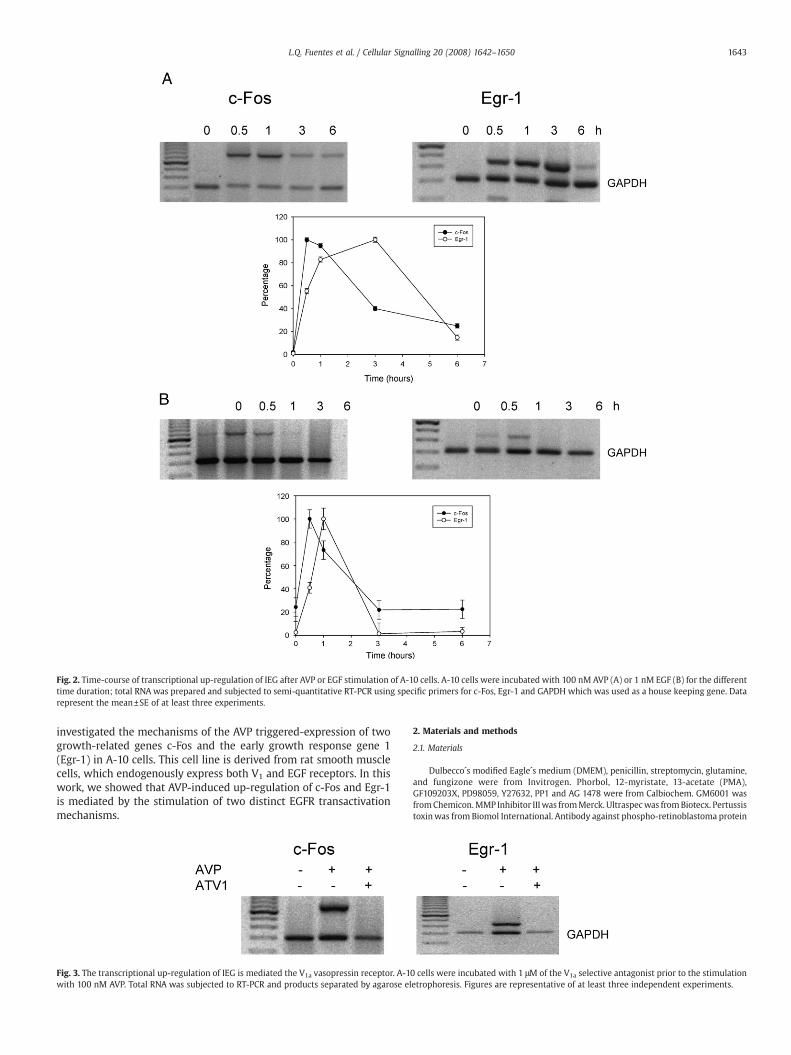
Fig. 2. Time-course of transcriptional up-regulation of IEG after AVP or EGF stimulation of A-10 cells. A-10 cells were incubated with 100 nM AVP (A) or 1 nM EGF (B) for the differenttime duration; total RNA was prepared and subjected to semi-quantitative RT-PCR using specific primers for c-Fos, Egr-1 and GAPDH which was used as a house keeping gene. Datarepresent the mean±SE of at least three experiments.
1643L.Q. Fuentes et al. / Cellular Signalling 20 (2008) 1642–1650
investigated the mechanisms of the AVP triggered-expression of twogrowth-related genes c-Fos and the early growth response gene 1(Egr-1) in A-10 cells. This cell line is derived from rat smooth musclecells, which endogenously express both V1 and EGF receptors. In thiswork, we showed that AVP-induced up-regulation of c-Fos and Egr-1is mediated by the stimulation of two distinct EGFR transactivationmechanisms.
Fig. 3. The transcriptional up-regulation of IEG is mediated the V1a vasopressin receptor. A-1with 100 nM AVP. Total RNA was subjected to RT-PCR and products separated by agarose el
2. Materials and methods
2.1. Materials
Dulbecco´s modified Eagle´s medium (DMEM), penicillin, streptomycin, glutamine,and fungizone were from Invitrogen. Phorbol, 12-myristate, 13-acetate (PMA),GF109203X, PD98059, Y27632, PP1 and AG 1478 were from Calbiochem. GM6001 wasfromChemicon.MMP Inhibitor IIIwas fromMerck. Ultraspecwas fromBiotecx. Pertussistoxinwas from Biomol International. Antibody against phospho-retinoblastoma protein
0 cells were incubated with 1 μM of the V1a selective antagonist prior to the stimulationetrophoresis. Figures are representative of at least three independent experiments.
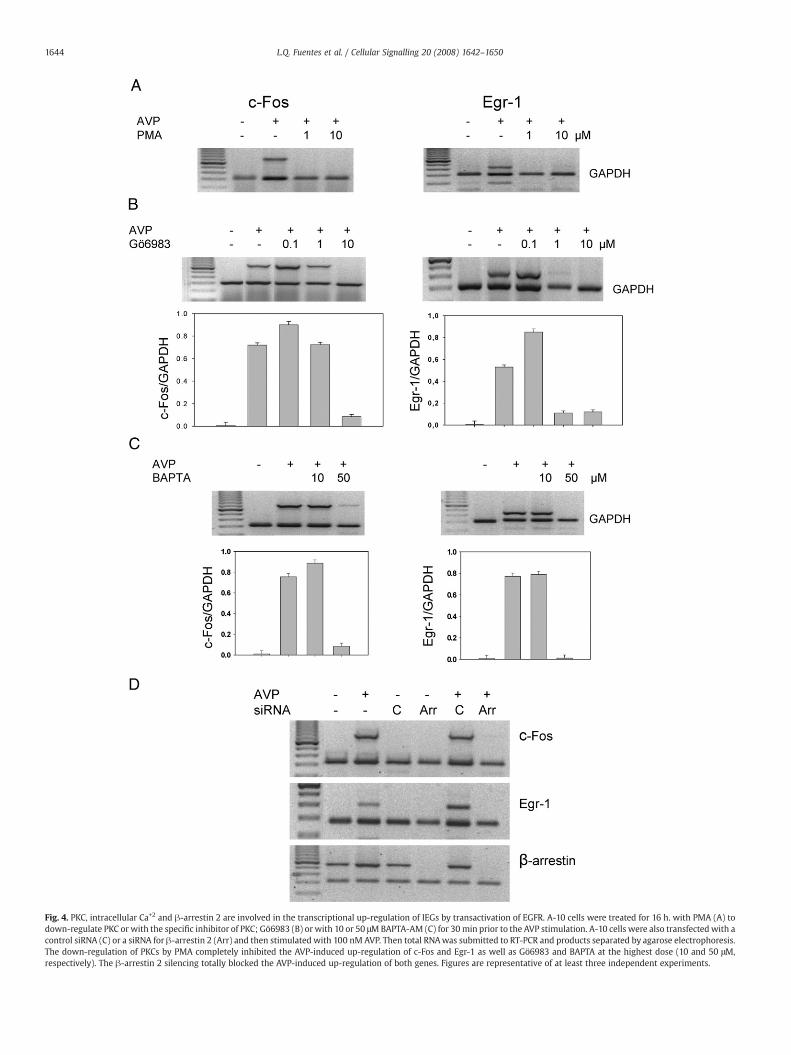
Fig. 4. PKC, intracellular Ca+2 and β-arrestin 2 are involved in the transcriptional up-regulation of IEGs by transactivation of EGFR. A-10 cells were treated for 16 h. with PMA (A) todown-regulate PKC or with the specific inhibitor of PKC; Gö6983 (B) or with 10 or 50 μMBAPTA-AM (C) for 30min prior to the AVP stimulation. A-10 cells were also transfected with acontrol siRNA (C) or a siRNA for β-arrestin 2 (Arr) and then stimulated with 100 nM AVP. Then total RNAwas submitted to RT-PCR and products separated by agarose electrophoresis.The down-regulation of PKCs by PMA completely inhibited the AVP-induced up-regulation of c-Fos and Egr-1 as well as Gö6983 and BAPTA at the highest dose (10 and 50 μM,respectively). The β-arrestin 2 silencing totally blocked the AVP-induced up-regulation of both genes. Figures are representative of at least three independent experiments.
1644 L.Q. Fuentes et al. / Cellular Signalling 20 (2008) 1642–1650

1645L.Q. Fuentes et al. / Cellular Signalling 20 (2008) 1642–1650
was from Cell Signaling Technology, anti Egr-1 and anti c-Fos were from Santa CruzBiotechnology. The V1 antagonist d(CH2)5[Tyr2(Me)Tyr9(NH2)]AVP was kindly providedby Prof. M. Manning (Medical College of Ohio, Toledo, USA). The siRNA for β-arrestin 2was purchased from Invitrogen.
2.2. Expression vectors
Plasmids encoding wild type c-Src and c-SrcK295R/Y527F were a generous giftfrom Dr. Joan S. Brugge (Harvard Medical School, USA). The plasmid encodingL61S186Ras was generously provided by Dr. Kun-Liang Guan (University of MichiganMedical School, USA). The EGFR dominant negative mutant HERCD533 was generouslyprovided by Dr. Sylvain Meloche (University of Montreal, Quebec, Canada). The plasmidencoding the C-terminus of β-adrenergic receptor kinase (CT-βGRK2) was a generousgift from Dr. Juan Olate (Universidad de Concepción, Chile). The plasmid encoding theS1 catalytic subunit of Pertussis toxin was kindly provided by Dr. Halvard Bonig(University of Washington, USA).
2.3. Cell culture and transfections
The smooth muscle cell line A-10 (ATCC CRL 1476) was cultured to subconfluencyon 35 mm dishes in DMEM containing 10% FBS. Serum starved cells were treated withvasopressin in the absence and presence of inhibitors. The reaction was stopped byaddition of 100 μl of ice-cold RIPA buffer (150 mM NaCl, Tris/HCl pH 8.0, 1% deoxycholicacid, 1% Nonidet P40, 0.1% SDS, 4 mM EDTA, 1 mM Na3VO4, 250 μg/ml p-nitrophenylphosphate, 1 mM phenylmethane-sulphonyl fluoride, 1 μg/ml each of leupeptin,pepstatin A and aprotinin). Cells were lysed and proteins were precipitated by additionof trichloroacetic acid and resuspended in electrophoresis sample buffer containing1 mM Na3VO4. In some experiments, cells were incubated with the V1 antagonist d(CH2)5[Tyr2(Me)Tyr9(NH2)]AVP, GF109203X or with PD98059 or with AG 1478 or withMMP or GM6001 inhibitors prior the stimulation with AVP. Transient transfectionswere carried out using FuGENE 6 Transfection Reagent (Roche Diagnostics). The siRNAfor β arrestin 2 was transfected using Block-iT Transfection Kit (Invitrogen).
2.4. Western blotting
Cell extracts were fractionated using SDS-PAGE, and the proteins were electro-transferred ontonitrocellulosefilters using 0.05% SDS in the transfer buffer (20mMTris–glicine pH 8.3 and 20% methanol). Blots were incubated with anti c-Fos or anti Egr-1 oranti phospho-retinoblastoma protein antibodies at a dilution of 1:1000. The blots werethen incubated with peroxidase-labeled secondary antibody at a dilution of 1:50,000followed by chemiluminescence.
2.5. Reverse transcription and polymerase chain reaction
Total RNA from A-10 cells was prepared using Ultraspec (Biotecx). cDNAs weresynthesized using oligo dt primers and SuperScript II (Gibco BRL). PCR were carried outusing the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene as an internalnon-competitive standard to normalize the expression of the gene to be studied. Thepair of primers for the c-Fos were the sense 5´GCCAACTTTATCCCCACG3´ and the anti-sense 5´TAGAAGGAACCAGACAGGTCC3´ which should generate a fragment of 725 bp,for the Egr-1 were the sense 5´-CTTCAGTCGTAGTGACCACCT-3´ and the anti-sense
Fig. 5. The AVP-induced up-regulation of the IEGs is due to the transactivation of the EGFR. Awith the dominant negative mutant HERCD533 (2, 4 and 6 μg of DNA) (B) prior to the stimuagarose eletrophoresis. Figures are representative of at least three independent experiment
5´-ATGTCTGAAAGACCAGTTGAG-3´which should generate a fragment of 441 bp,and for the GAPDH were the sense 5´TCCCTCAAGATTGTCAGCAA-3 and the anti-sense5´-AGATCCACAACGGATACATT-3´´ which should generate a fragment of 309 bp. Theratio of the co-amplification of products was estimated at the exponential phase of thePCR. The PCR reaction mixture contained 50 pMol of each primer for gene to bestudied and for GAPDH, 1 mM deoxynucleotides 1x Taq polymerase reaction buffer,1.5 mM MgCl2 and 2.5 U Taq polymerase. PCR products were fractionated byelectrophoresis on 1.6% agarose gel, stained with ethidium bromide and the bandsvisualized with a UV transilluminator.
3. Results
3.1. The V1a receptor mediated the expression of IEGs
Because AVP triggers cell proliferation in several cell types, weinitially tested the AVP-induced expression of genes regulating cellgrowth in the aorta smooth muscle derived A-10 cell line. Western blotanalysis showed that AVP up-regulated the expression of genesregulating cell proliferation, including cyclin D1 and the immediateearly genes c-Fos and Egr-1 (Fig. 1). In addition, we showed the AVP-dependent phosphorylation of the retinoblastomaprotein (Rb),which isessential in the cell cycle progression. RT-PCR assays demonstrated thatthe AVP-dependent expression of c-Fos was transient, reaching amaximum level at 30 min; whereas the expression of Egr-1 wassustained for up to 3 h (Fig. 2A). Interestingly, EGF–triggered expressionof both c-Fos and Egr-1 was fast and transient (Fig. 2B). To identify theAVP receptor subtype responsible for the up-regulation of these earlyimmediate genes we employed AVP receptor-specific antagonists. Wefound that the V1a antagonist d(CH2)5[Tyr2(Me)Tyr9(NH2)]AVP blockedtheAVP-inducedup-regulationof both IEGs (Fig. 3), suggesting that theirexpression is mediated via the V1a receptor subtype. The V2 specificagonist, desmopressin, did not affect the expression of IEGs in A-10 cells(Data not shown).
3.2. AVP-induced up-regulation of IEGs required activation of PKC,intracellular calcium and β-arrestin 2
A-10 cells depleted of PKC by the long-treatmentwith phorbol estersfailed to elicit AVP-dependent expression of both c-Fos and Erg-1(Fig. 4A). Similarly, the PKC inhibitor Gö6983 blocked the AVP-inducedup-regulation of both genes (Fig. 4B). In contrast, the PKC inhibitorGF109203X (GFX) was a weak inhibitor of the AVP-dependent upregulation of these genes (Data not shown). Further, cells treated withthe intracellular calcium chelator BAPTA failed to respond to AVP-
-10 cells were treated with 1 μM of the EGFR tyrosine kinase inhibitor (A) or transfectedlation with 100 nM AVP. Total RNA was subjected to RT-PCR and products separated bys.
1646 L.Q. Fuentes et al. / Cellular Signalling 20 (2008) 1642–1650
induced up-regulation of IEGs (Fig. 4C), suggesting that the release ofintracellular calcium is required for the AVP effect.
Since the activated and phosphorylated V1 receptor binds β-arrestin2, desensitizing the responses mediated by the receptor andstimulating non-G protein signalling pathways [31,32], we examinedthe role of β-arrestin 2 in the AVP-induced expression of IEG. Weshowed that A-10 cells depleted of β-arrestin 2 by specific siRNAfailed to elicit AVP-induced up-regulation of both IEGs (Fig. 4D). Thisfinding indicates that β-arrestin 2 is required in the AVP-induced up-regulation of IEGs.
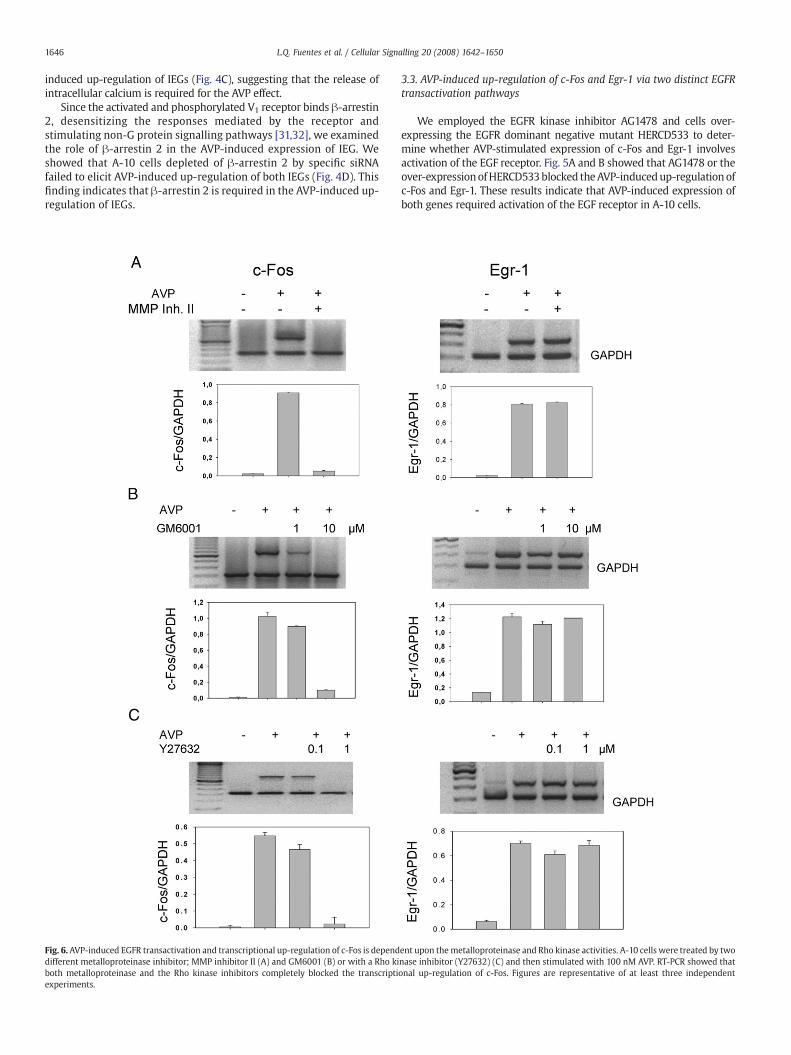
Fig. 6. AVP-induced EGFR transactivation and transcriptional up-regulation of c-Fos is dependdifferent metalloproteinase inhibitor; MMP inhibitor II (A) and GM6001 (B) or with a Rho kiboth metalloproteinase and the Rho kinase inhibitors completely blocked the transcriptiexperiments.
3.3. AVP-induced up-regulation of c-Fos and Egr-1 via two distinct EGFRtransactivation pathways
We employed the EGFR kinase inhibitor AG1478 and cells over-expressing the EGFR dominant negative mutant HERCD533 to deter-mine whether AVP-stimulated expression of c-Fos and Egr-1 involvesactivation of the EGF receptor. Fig. 5A and B showed that AG1478 or theover-expressionofHERCD533blocked theAVP-inducedup-regulation ofc-Fos and Egr-1. These results indicate that AVP-induced expression ofboth genes required activation of the EGF receptor in A-10 cells.
ent upon themetalloproteinase and Rho kinase activities. A-10 cells were treated by twonase inhibitor (Y27632) (C) and then stimulated with 100 nM AVP. RT-PCR showed thatonal up-regulation of c-Fos. Figures are representative of at least three independent
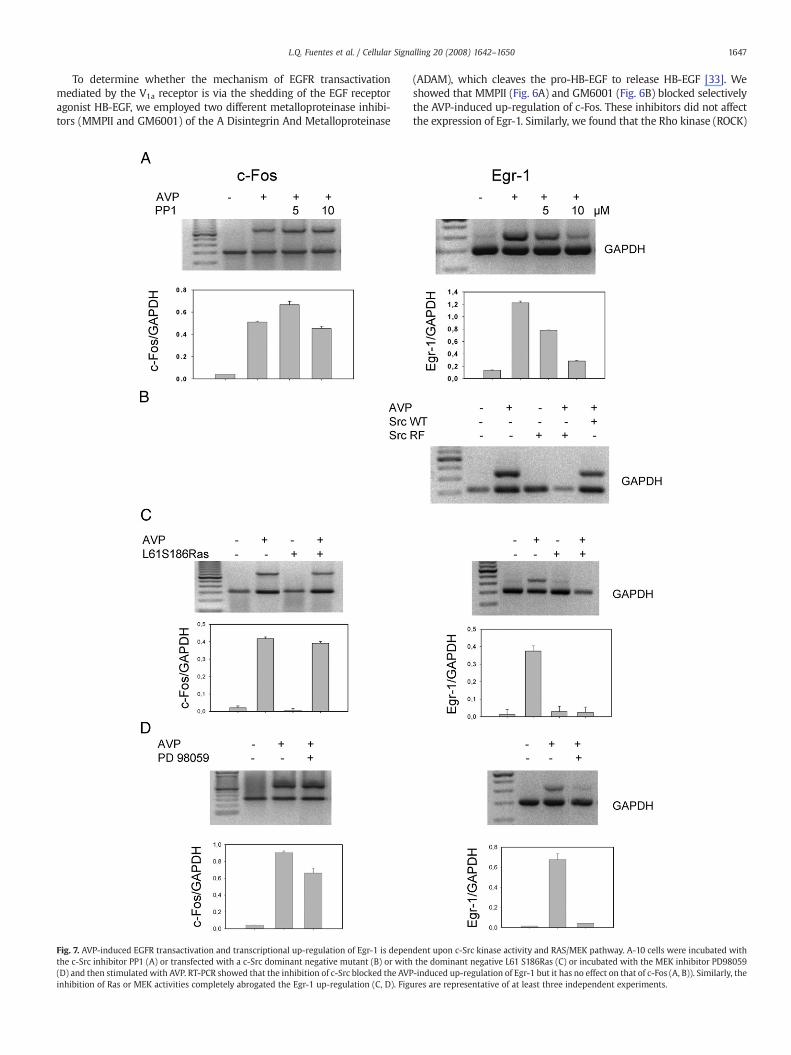
1647L.Q. Fuentes et al. / Cellular Signalling 20 (2008) 1642–1650
To determine whether the mechanism of EGFR transactivationmediated by the V1a receptor is via the shedding of the EGF receptoragonist HB-EGF, we employed two different metalloproteinase inhibi-tors (MMPII and GM6001) of the A Disintegrin And Metalloproteinase
Fig. 7. AVP-induced EGFR transactivation and transcriptional up-regulation of Egr-1 is depenthe c-Src inhibitor PP1 (A) or transfected with a c-Src dominant negative mutant (B) or with(D) and then stimulated with AVP. RT-PCR showed that the inhibition of c-Src blocked the AVPinhibition of Ras or MEK activities completely abrogated the Egr-1 up-regulation (C, D). Fig
(ADAM), which cleaves the pro-HB-EGF to release HB-EGF [33]. Weshowed that MMPII (Fig. 6A) and GM6001 (Fig. 6B) blocked selectivelythe AVP-induced up-regulation of c-Fos. These inhibitors did not affectthe expression of Egr-1. Similarly, we found that the Rho kinase (ROCK)
dent upon c-Src kinase activity and RAS/MEK pathway. A-10 cells were incubated withthe dominant negative L61 S186Ras (C) or incubated with the MEK inhibitor PD98059-induced up-regulation of Egr-1 but it has no effect on that of c-Fos (A, B)). Similarly, theures are representative of at least three independent experiments.
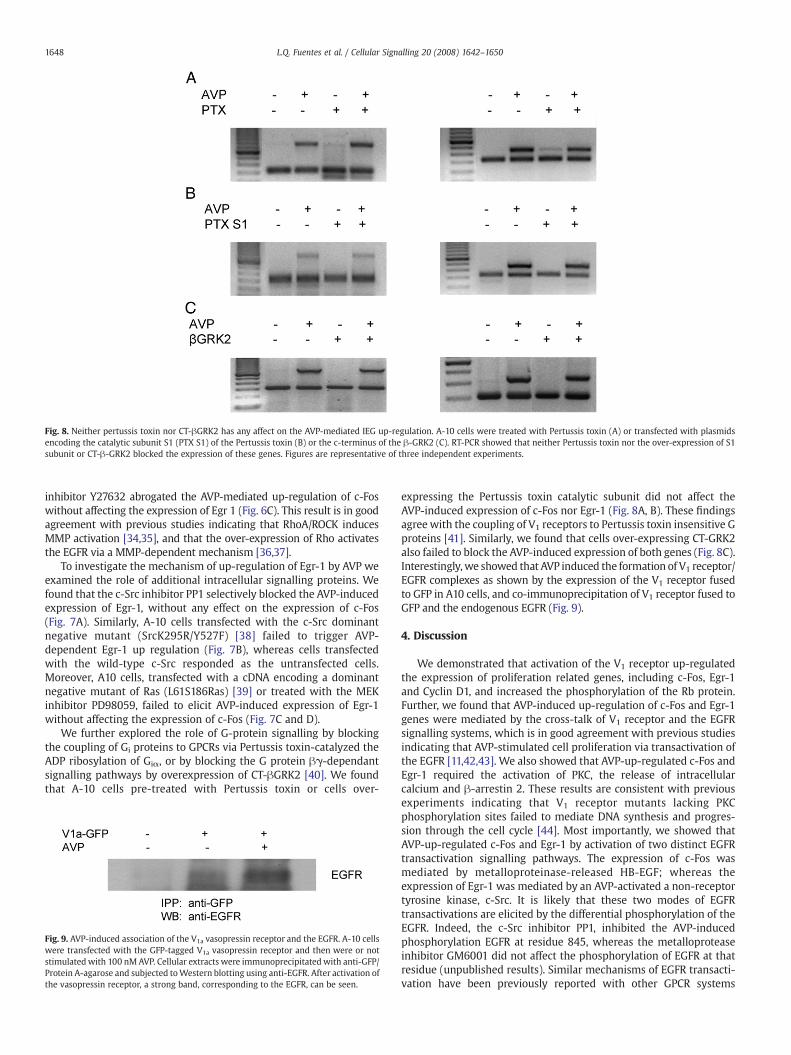
Fig. 8. Neither pertussis toxin nor CT-βGRK2 has any affect on the AVP-mediated IEG up-regulation. A-10 cells were treated with Pertussis toxin (A) or transfected with plasmidsencoding the catalytic subunit S1 (PTX S1) of the Pertussis toxin (B) or the c-terminus of the β-GRK2 (C). RT-PCR showed that neither Pertussis toxin nor the over-expression of S1subunit or CT-β-GRK2 blocked the expression of these genes. Figures are representative of three independent experiments.
1648 L.Q. Fuentes et al. / Cellular Signalling 20 (2008) 1642–1650
inhibitor Y27632 abrogated the AVP-mediated up-regulation of c-Foswithout affecting the expression of Egr 1 (Fig. 6C). This result is in goodagreement with previous studies indicating that RhoA/ROCK inducesMMP activation [34,35], and that the over-expression of Rho activatesthe EGFR via a MMP-dependent mechanism [36,37].
To investigate the mechanism of up-regulation of Egr-1 by AVP weexamined the role of additional intracellular signalling proteins. Wefound that the c-Src inhibitor PP1 selectively blocked the AVP-inducedexpression of Egr-1, without any effect on the expression of c-Fos(Fig. 7A). Similarly, A-10 cells transfected with the c-Src dominantnegative mutant (SrcK295R/Y527F) [38] failed to trigger AVP-dependent Egr-1 up regulation (Fig. 7B), whereas cells transfectedwith the wild-type c-Src responded as the untransfected cells.Moreover, A10 cells, transfected with a cDNA encoding a dominantnegative mutant of Ras (L61S186Ras) [39] or treated with the MEKinhibitor PD98059, failed to elicit AVP-induced expression of Egr-1without affecting the expression of c-Fos (Fig. 7C and D).
We further explored the role of G-protein signalling by blockingthe coupling of Gi proteins to GPCRs via Pertussis toxin-catalyzed theADP ribosylation of Giα, or by blocking the G protein βγ-dependantsignalling pathways by overexpression of CT-βGRK2 [40]. We foundthat A-10 cells pre-treated with Pertussis toxin or cells over-
Fig. 9. AVP-induced association of the V1a vasopressin receptor and the EGFR. A-10 cellswere transfected with the GFP-tagged V1a vasopressin receptor and then were or notstimulatedwith 100 nM AVP. Cellular extracts were immunoprecipitated with anti-GFP/Protein A-agarose and subjected toWestern blotting using anti-EGFR. After activation ofthe vasopressin receptor, a strong band, corresponding to the EGFR, can be seen.
expressing the Pertussis toxin catalytic subunit did not affect theAVP-induced expression of c-Fos nor Egr-1 (Fig. 8A, B). These findingsagree with the coupling of V1 receptors to Pertussis toxin insensitive Gproteins [41]. Similarly, we found that cells over-expressing CT-GRK2also failed to block the AVP-induced expression of both genes (Fig. 8C).Interestingly,we showed that AVP induced the formation of V1 receptor/EGFR complexes as shown by the expression of the V1 receptor fusedto GFP in A10 cells, and co-immunoprecipitation of V1 receptor fused toGFP and the endogenous EGFR (Fig. 9).
4. Discussion
We demonstrated that activation of the V1 receptor up-regulatedthe expression of proliferation related genes, including c-Fos, Egr-1and Cyclin D1, and increased the phosphorylation of the Rb protein.Further, we found that AVP-induced up-regulation of c-Fos and Egr-1genes were mediated by the cross-talk of V1 receptor and the EGFRsignalling systems, which is in good agreement with previous studiesindicating that AVP-stimulated cell proliferation via transactivation ofthe EGFR [11,42,43]. We also showed that AVP-up-regulated c-Fos andEgr-1 required the activation of PKC, the release of intracellularcalcium and β-arrestin 2. These results are consistent with previousexperiments indicating that V1 receptor mutants lacking PKCphosphorylation sites failed to mediate DNA synthesis and progres-sion through the cell cycle [44]. Most importantly, we showed thatAVP-up-regulated c-Fos and Egr-1 by activation of two distinct EGFRtransactivation signalling pathways. The expression of c-Fos wasmediated by metalloproteinase-released HB-EGF; whereas theexpression of Egr-1 was mediated by an AVP-activated a non-receptortyrosine kinase, c-Src. It is likely that these two modes of EGFRtransactivations are elicited by the differential phosphorylation of theEGFR. Indeed, the c-Src inhibitor PP1, inhibited the AVP-inducedphosphorylation EGFR at residue 845, whereas the metalloproteaseinhibitor GM6001 did not affect the phosphorylation of EGFR at thatresidue (unpublished results). Similar mechanisms of EGFR transacti-vation have been previously reported with other GPCR systems

Fig. 10. Diagrammatic representation of the pathways involved in the IEG transcriptional up-regulation by the AVP-mediated EGFR transactivation.
1649L.Q. Fuentes et al. / Cellular Signalling 20 (2008) 1642–1650
[27,45–49]. On the basis of our co-immunoprecipitation studies weargue that V1a mediated EGFR transactivation involves the formationof V1a/EGFR complex assembled with β-arrestin 2 and intracellularproteins of the trafficking mechanisms [50].
Interestingly, AVP-induced up-regulation of Egr-1, but not the c-Fosup-regulation, was mediated by a Ras/MEK-dependent signallingpathway. These results are in agreement with previous studiesindicating that the GPCR-dependent expression of Egr-1 is via theactivation of MEK/ERK signalling pathway [51–55]. Although ERKactivation enhances transcription of c-Fos and Egr-1 [53,56–59], wefound that AVP-induced expression of c-Fos is via an ERK-independentsignalling pathway, indicating that c-Fos transcription is activated viamultiple signalling pathways. Our data are consistent with a signallingmodel inwhich the activated and phosphorylated V1 receptor binds β-arrestin to trigger the activation of Rho/ROCK- and c-Src-dependentsignalling pathways. The Rho/ROCK pathway activates MMP to releaseHB-EGF, which then triggers the expression of c-Fos via the activationof the EGF receptor. On the other hand, the c-Src pathway catalyzesthe phosphorylation of the EGFR, which elicit the activation of the Ras/MEK/ERK signalling mechanism to induce the expression of Erg-1(Fig. 10). The novel mechanism of the AVP-triggered transactivation ofEGFR provides the basis for regulating the selective expression of EIGs,as Egr-1 is a major vascular transcription factor involved in athero-sclerosis and restenosis [60,61].
Acknowledgments
Wewould like to thank the expert technical assistance of E. Oyarzunand to G. Perdomo for preliminary experiments. This work wassupported by grants 1030261 and 1060158 from FONDECYT (CBG) and2005-19 (LQF) and (CER) from DIUACH; andWelch Foundation and NIHgrants (R01 EY014218 and GM064855) to JN.
References
[1] A.T. Hirsch, V.J. Dzau, J.A. Majzoub, M.A. Creager, J. Clin Invest 84 (1989) 418.[2] C.B. Gonzalez, C.D. Figueroa, Biol. Res. 32 (1999) 63.[3] J.M. Sarmiento, P. Ehrenfeld, C.C. Anazco, C.E. Reyes, S. Troncoso, C.D. Figueroa,
W. Muller-Esterl, C.B. Gonzalez, Kidney Int. 68 (2005) 487.[4] J.I. Kreisberg, M. Venkatachalam, D. Troyer, Am. J. Physiol. 249 (1985) F457.[5] G. Segarra, P.Medina, J.M.Vila, P. Chuan,C.Domenech, S. Lluch, J.Hypertens. 20 (2002)
1373.[6] P. Medina, A. Acuna, J.B. Martinez-Leon, E. Otero, J.M. Vila, M. Aldasoro, S. Lluch,
Circulation 97 (1998) 865.[7] B.M. Altura, B.T. Altura, Fed. Proc. 36 (1977) 1853.[8] F.Y. Bhora, P.C. Kothary, H. Imanishi, F.E. Eckhauser, S.E. Raper, J. Surg. Res. 57 (1994)
706.[9] C. Serradeil-Le Gal, B. Bourrie, D. Raufaste, P. Carayon, C. Garcia, J.P. Maffrand, G. Le
Fur, P. Casellas, Biochem. Pharmacol. 47 (1994) 633.[10] C. Serradeil-Le Gal, J.M. Herbert, C. Delisee, P. Schaeffer, D. Raufaste, C. Garcia, F. Dol,
E. Marty, J.P. Maffrand, G. Le Fur, Am. J. Physiol. 268 (1995) H404.[11] T. Chiu, S.S. Wu, C. Santiskulvong, P. Tangkijvanich, H.F. Yee Jr., E. Rozengurt, Am. J.
Physiol Cell Physiol. 282 (2002) C434.
[12] E. Rozengurt, A. Rodriguez-Pena, J. Sinnett-Smith, Ciba Found. Symp.116 (1985) 66.[13] C. Santiskulvong, E. Rozengurt, Exp. Cell Res. 290 (2003) 437.[14] R.D. Brinton, R. Yamazaki, C.M. Gonzalez, K. O'Neill, S.S. Schreiber, Brain Res. Mol.
Brain Res. 57 (1998) 73.[15] J. Cortner, P.J. Farnham, Cell Growth Differ. 2 (1991) 465.[16] A. Dey, H. She, L. Kim, A. Boruch, D.L. Guris, K. Carlberg, S.M. Sebti, D.T. Woodley,
A. Imamoto, W. Li, Mol. Biol. Cell 11 (2000) 3835.[17] M.J. Gomez-Lechon, I. Guillen, X. Ponsoda, R. Fabra, R. Trullenque, T. Nakamura,
J.V. Castell, Hepatology 23 (1996) 1012.[18] D.E. Hallahan, E. Dunphy, S. Virudachalam, V.P. Sukhatme, D.W. Kufe, R.R.
Weichselbaum, J. Biol. Chem. 270 (1995) 30303.[19] M.F. Roussel, Mol. Reprod. Dev. 46 (1997) 11.[20] A. Gschwind, E. Zwick, N. Prenzel, M. Leserer, A. Ullrich, Oncogene 20 (2001) 1594.[21] R. Wetzker, F.D. Bohmer, Nat. Rev. Mol. Cell Biol. 4 (2003) 651.[22] B. Schafer, B. Marg, A. Gschwind, A. Ullrich, J. Biol. Chem. 279 (2004) 47929.[23] K.L. Pierce, A. Tohgo, S. Ahn, M.E. Field, L.M. Luttrell, R.J. Lefkowitz, J. Biol. Chem.
276 (2001) 23155.[24] C. Gallet, S. Blaie, S. Levy-Toledano, A. Habib, Biochem. J. 371 (2003) 733.[25] Y. Itoh, T. Joh, S. Tanida, M. Sasaki, H. Kataoka, K. Itoh, T. Oshima, N. Ogasawara,
S. Togawa, T. Wada, H. Kubota, Y. Mori, H. Ohara, T. Nomura, S. Higashiyama, M. Itoh,Cytokine 29 (2005) 275.
[26] L.M. Luttrell, G.J. Della Rocca, T. van Biesen, D.K. Luttrell, R.J. Lefkowitz, J. Biol.Chem. 272 (1997) 4637.
[27] W. Wu, L.M. Graves, G.N. Gill, S.J. Parsons, J.M. Samet, J. Biol. Chem. 277 (2002)24252.
[28] J.M. Samet, B.J. Dewar,W.Wu, L.M. Graves, Toxicol. Appl. Pharmacol. 191 (2003) 86.[29] N.J. Pyne, C.Waters, N.A.Moughal, B. Sambi, M. Connell, S. Pyne, Methods Enzymol.
390 (2004) 451.[30] N.J. Pyne, C.Waters, N.A.Moughal, B.S. Sambi, S. Pyne, Biochem. Soc. Trans. 31 (2003)
1220.[31] S. Terrillon, M. Bouvier, EMBO J. 23 (2004) 3950.[32] S. Terrillon, C. Barberis, M. Bouvier, Proc. Natl. Acad. Sci. U. S. A. 101 (2004) 1548.[33] N. Prenzel, E. Zwick, H. Daub, M. Leserer, R. Abraham, C. Wallasch, A. Ullrich,
Nature 402 (1999) 884.[34] I. Abecassis, B.Olofsson,M. Schmid,G. Zalcman,A.Karniguian, Exp. CellRes. 291 (2003)
363.[35] B. Desai, M.J. Rogers, M.A. Chellaiah, Mol. Cancer 6 (2007) 18.[36] M. Caceres, J. Guerrero, J. Martinez, Exp. Cell Res. 309 (2005) 229.[37] J. Guerrero, J.F. Santibanez, A. Gonzalez, J. Martinez, Exp. Cell Res. 292 (2004) 201.[38] D. Mukhopadhyay, L. Tsiokas, X.M. Zhou, D. Foster, J.S. Brugge, V.P. Sukhatme,
Nature 375 (1995) 577.[39] S. Stewart, K.L. Guan, J. Biol. Chem. 275 (2000) 8854.[40] Y. Daaka, J.A. Pitcher, M. Richardson, R.H. Stoffel, J.D. Robishaw, R.J. Lefkowitz, Proc.
Natl. Acad. Sci. U.S.A. 94 (1997) 2180.[41] L.R. Grillone, M.A. Clark, R.W. Godfrey, F. Stassen, S.T. Crooke, J. Biol. Chem. 263 (1988)
2658.[42] P.M. Ghosh, R. Bedolla, C.A. Thomas, J.I. Kreisberg, J. Cell Biochem. 91 (2004) 1109.[43] P.M. Ghosh, M. Mikhailova, R. Bedolla, J.I. Kreisberg, Am. J. Physiol. Renal. Physiol.
280 (2001) F972.[44] M. Thibonnier, C.L. Plesnicher, K. Berrada, L. Berti-Mattera, Am. J. Physiol.
Endocrinol. Metab. 281 (2001) E81.[45] J. Andreev, M.L. Galisteo, O. Kranenburg, S.K. Logan, E.S. Chiu, M. Okigaki, L.A. Cary,
W.H. Moolenaar, J. Schlessinger, J. Biol. Chem. 276 (2001) 20130.[46] C. Cheng-Hsien, H. Yung-Ho, S. Yuh-Mou, H. Chun-Cheng, L. Horng-Mo, H. Huei-
Mei, C. Tso-Hsiao, Pflugers Arch. 452 (2006) 16.[47] L.D. Alexander, Y. Ding, S. Alagarsamy, X.L. Cui, J.G.Douglas, Kidney Int. 69 (2006) 1823.[48] S. Drube, J. Stirnweiss, C. Valkova, C. Liebmann, Cell Signal 18 (2006) 1633.[49] B.L. Slomiany, A. Slomiany, Inflammopharmacology 12 (2004) 233.[50] A.C. Hanyaloglu, M. von Zastrow, Annu. Rev. Pharmacol. Toxicol. 48 (2008) 537.[51] A. Al-Sarraj, G. Thiel, Neurosci. Lett. 332 (2002) 111.[52] M.T. Wiiger, H. Prydz, Thromb. Haemost. 92 (2004) 13.[53] D. Zhao, J. Letterman, B.M. Schreiber, J. Biol. Chem. 276 (2001) 30579.[54] D. Zhao, Y. Zhan, H. Zeng, H.W. Koon, M.P. Moyer, C. Pothoulakis, Int. J. Cancer
120 (2007) 1652.

1650 L.Q. Fuentes et al. / Cellular Signalling 20 (2008) 1642–1650
[55] H. Zhao, W. Tian, H. Xu, D.M. Cohen, Biochem J. 370 (2003) 479.[56] B.K. Dieckgraefe, D.M. Weems, Am. J. Physiol. 276 (1999) G322.[57] C.Hodge, J. Liao,M. Stofega, K.Guan, C. Carter-Su, J. Schwartz, J. Biol. Chem. 273 (1998)
31327.[58] L.P. De Sousa, B.S. Brasil, B.M. Silva, M.H. Freitas, S.V. Nogueira, P.C. Ferreira, E.G.
Kroon, C.A. Bonjardim, Biochem. Biophys. Res. Commun. 329 (2005) 237.
[59] S. Goetze, U. Kintscher, K. Kaneshiro, W.P. Meehan, A. Collins, E. Fleck, W.A. Hsueh,R.E. Law, Atherosclerosis 159 (2001) 93.
[60] F. Blaschke, D. Bruemmer, R.E. Law, Rev. Endocr. Metab. Disord. 5 (2004) 249.[61] L.M. Khachigian, Circ. Res. 98 (2006) 186.




















