
YAG wet-chemical synthesis from chlorides and nitrates precursors: effect on phase evolution and...
18
374 Int. J. Materials and Product Technology, Vol. 35, Nos. 3/4, 2009 Copyright © 2009 Inderscience Enterprises Ltd. YAG wet-chemical synthesis from chlorides and nitrates precursors: effect on phase evolution and powder sinterability Paola Palmero*, Serena Di Nunzio and Laura Montanaro R.U. INSTM- PoliTO, LINCE Lab., Department of Materials Science and Chemical Engineering, Politecnico di Torino, Corso Duca degli Abruzzi, 24 – 10129 Torino, Italy Fax: +39 011 5644699 E-mail: [email protected] E-mail: [email protected] E-mail: [email protected] *Corresponding author Abstract: The influence of inorganic precursors on phase evolution and powder sinterability of YAG has been investigated. YAG powders were synthesised by reverse-strike precipitation, from yttrium and aluminium chlorides or nitrates aqueous solution. The powders were characterised by thermal analysis and XRD measurements. Pure-YAG was obtained after calcination at high temperature from both precursors, but the chlorides-derived materials yield mixtures of YAG and metastable YAlO 3 phases from 800°C to 1100°C. On the contrary, nitrates precursors led to the nucleation of pure-YAG at very low temperatures (from 500°C), thus allowing to directly produce well crystallised YAG powders, suitable for enhanced sintered products. Keywords: YAG; wet chemical co-precipitation; precursors; phase evolution; powder sinterability; microstructure. Reference to this paper should be made as follows: Palmero, P., Di Nunzio, S. and Montanaro, L. (2009) ‘YAG wet-chemical synthesis from chlorides and nitrates precursors: effect on phase evolution and powder sinterability’, Int. J. Materials and Product Technology, Vol. 35, Nos. 3/4, pp.374–391. Biographical notes: P. Palmero is an Assistant Professor in Materials Science and Technology at the second Faculty of Architecture of Politecnico di Torino. She received her PhD in Materials Science and Technology in January 2005 in the framework of a project of internationalisation of PhD supported by the Italian Ministry of Education, University and Scientific Research (MIUR). She is author or co-author of 16 scientific publications and two patents. Her current research interest is focused on ceramic materials, and her research activities mostly concern the development of nano-structured ceramic components, both mono-phased and composites, for functional and mechanical applications. She is actually involved in the Integrated Project NanoKer (VI F.P.) in which she acts as Researcher of the WorkPackage concerning nanopowders synthesis and characterisation.
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of YAG wet-chemical synthesis from chlorides and nitrates precursors: effect on phase evolution and...

374 Int. J. Materials and Product Technology, Vol. 35, Nos. 3/4, 2009
Copyright © 2009 Inderscience Enterprises Ltd.
YAG wet-chemical synthesis from chlorides and nitrates precursors: effect on phase evolution and powder sinterability
Paola Palmero*, Serena Di Nunzio and Laura Montanaro R.U. INSTM- PoliTO, LINCE Lab., Department of Materials Science and Chemical Engineering, Politecnico di Torino, Corso Duca degli Abruzzi, 24 – 10129 Torino, Italy Fax: +39 011 5644699 E-mail: [email protected] E-mail: [email protected] E-mail: [email protected] *Corresponding author
Abstract: The influence of inorganic precursors on phase evolution and powder sinterability of YAG has been investigated. YAG powders were synthesised by reverse-strike precipitation, from yttrium and aluminium chlorides or nitrates aqueous solution. The powders were characterised by thermal analysis and XRD measurements. Pure-YAG was obtained after calcination at high temperature from both precursors, but the chlorides-derived materials yield mixtures of YAG and metastable YAlO3 phases from 800°C to 1100°C. On the contrary, nitrates precursors led to the nucleation of pure-YAG at very low temperatures (from 500°C), thus allowing to directly produce well crystallised YAG powders, suitable for enhanced sintered products.
Keywords: YAG; wet chemical co-precipitation; precursors; phase evolution; powder sinterability; microstructure.
Reference to this paper should be made as follows: Palmero, P., Di Nunzio, S. and Montanaro, L. (2009) ‘YAG wet-chemical synthesis from chlorides and nitrates precursors: effect on phase evolution and powder sinterability’, Int. J. Materials and Product Technology, Vol. 35, Nos. 3/4, pp.374–391.
Biographical notes: P. Palmero is an Assistant Professor in Materials Science and Technology at the second Faculty of Architecture of Politecnico di Torino. She received her PhD in Materials Science and Technology in January 2005 in the framework of a project of internationalisation of PhD supported by the Italian Ministry of Education, University and Scientific Research (MIUR). She is author or co-author of 16 scientific publications and two patents. Her current research interest is focused on ceramic materials, and her research activities mostly concern the development of nano-structured ceramic components, both mono-phased and composites, for functional and mechanical applications. She is actually involved in the Integrated Project NanoKer (VI F.P.) in which she acts as Researcher of the WorkPackage concerning nanopowders synthesis and characterisation.

YAG wet-chemical synthesis from chlorides and nitrates precursors 375
S. Di Nunzio is currently Researcher at the LINCE Lab, Research Unit of Politecnico di Torino, for National Consortium in Materials Science and Technology INSTM, Florence, Italy. She carried out 6-months research grant, dealing with the development of ceramic coatings, at FIAT Research Centre. She received her PhD in Biomedical Engineering from Politecnico di Torino in May 2005. She is author or co-author of 14 scientific publications and a patent. Her current research activity focuses on nano-structured ceramic oxides for thermo-mechanical and biomedical applications. She is actually involved in the Integrated Project NanoKer (VI F.P.) in which she acts as Researcher of the WorkPackage concerning nanopowders synthesis and characterisation.
L. Montanaro is full Professor in Materials Science and Technology at the first faculty of Engineering of Politecnico di Torino. She is scientific responsible of the Reference Centre of Ceramic Materials Technology and Engineering LINCE of the National Consortium in Materials Science and Technology INSTM. She is author or co-author of about 100 scientific publications and papers mostly concerning ceramic materials. She is member of the Scientific Council of INSTM Consortium and Vice-President of the Italian Association on Materials Engineering AIMAT. She is scientific responsible of the Research Unit of INSTM involved in the Integrated Project NanoKer (VI F.P.) in which she acts as coordinator of the WorkPackage concerning Nanopowders synthesis and characterisation.
1 Introduction
Y3Al5O12 (YAG) is a compound in the yttria-alumina system that crystallises with a cubic garnet structure. YAG single-crystals and polycrystalline materials are actually applied as laser host materials, having peculiar optical features (Veith et al., 1999; Rabinovitch et al., 2003; Hreniak and Strek, 2002). Polycrystalline YAG also shows interesting thermo-mechanical properties that make it suitable for coatings as well as for ceramic composites as reinforcing phase (French et al., 1994; Torrecillas et al., 2007; Wang et al., 2001; Pullar et al., 1999).
Several different synthesis routes are present in literature: particularly, methods able to yield ultra-fine and pure YAG are actually under investigation since these powders should allow to produce high density materials, almost unaffected by defects and porosity, thus showing high transparency.
Among these methods, which refer to solid-state reactions (Nyman et al., 1997; Lupei et al., 2004), reverse-strike co-precipitations (Apte et al., 1992; Xu et al., 2006; Vrolijk et al., 1990; Su et al., 2005), sol-gel (Gowda et al., 1986; Lo et al., 1998), thermal decomposition and combustion syntheses (Kakade et al., 2003; Ramanathan et al., 2003; Messier and Gazza, 1972), hydrothermal and solvothermal syntheses (Li et al., 2004; Inoue et al., 1991; Cabanas et al., 2007), the reverse-strike co-precipitation method was recently exploited to produce nano-sized YAG particles starting from chlorides precursors (Palmero et al., 2005).
The synthesis of pure YAG powders is sometimes a challenging process since, dependently from the starting raw materials, a series of metastable and secondary stable phases should be produced, such as orthorhombic perovskite (YAP, YAlO3), hexagonal phase (h-YAlO3), cubic YAlO3 having a garnet structure and monoclinic YAM (Y4Al2O9), near the 3 : 5 molar ratio garnet-type YAG.

376 P. Palmero et al.
In some previous papers (Palmero et al., 2005; Li et al., 2000; Wang et al., 2000; Xu et al., 2006) the influence of some process parameters was clearly underlined. Particularly, the effect of the precipitation temperature on the homogeneity of the product and on the phase evolution was clearly stated (Palmero et al., 2005; Palmero and Montanaro, 2007).
In fact, if the synthesis is performed at higher temperatures (such as 60°C instead of 5–25°C), a partial crystallisation of the starting precipitate is induced. A similar phenomenon was already observed in wet-chemical synthesised powders for pure-alumina preparation (Mathieu, 1956; Yoldas, 1982). The traces of bayerite detected in the 60°C precipitate YAG precursor, should be related to a lowering of the homogeneity of cations distribution, respect to the 5–25°C synthesised products, thus justifying the appearance of secondary, undesired phases such as YAM (Y4Al2O9). This stable monoclinic phase was in fact detected in the high-temperature regime (1100–1350°C) needed to develop YAG phase.
At the same time, the precipitation temperature induced different YAG crystallisation paths. The appearance of the metastable h-YAlO3 phase was in fact more or less significant as a function of the preparation temperature: for instance, at 5°C as well as at 25°C, YAG and h-YAlO3 were simultaneously yielded after calcination at about 800–915°C, whereas the metastable phase was prevalent in the 60°C-synthesised product.
This paper represents an improvement in the above knowledge of the influence of process parameters. In particular, it deals with the role of the inorganic precursors on the phase evolution and sintering behaviour of ultra-fine YAG powders. In fact, it should be expected that different precursors could affect the phases evolution and the physico-chemical characteristics of materials, as stated by Montanaro and Saracco (1995) for alumina.
On the ground of those considerations, the aim of this work is to make a direct comparison between the crystallisation paths of two different YAG powders, synthesised by reverse strike co-precipitation. These products only differ on their starting precursors, while all other synthesis parameters (T, pH, precipitant solution, concentration, rate of solutions addition …) were maintained equal for both preparations.
In particular, it was decided to analyse nitrates and chlorides salts as YAG precursors, since literature reports many information about the two different preparation routes (Palmero et al., 2004a; Lu et al., 2002; Wang et al., 2000; Apte et al., 1992), but a direct comparison between the derived products and their phases evolution during heating treatments was not deeply investigated before the present work.
2 Materials and methods
Two powders were prepared starting from yttrium and aluminium chlorides and nitrates salts; they will be labelled Chloride-derived Powder (CP) and Nitrate-derived Powder (NP), respectively.

YAG wet-chemical synthesis from chlorides and nitrates precursors 377
2.1 Chlorides-derived Powders (CP)
CP was prepared starting from an aqueous solution of YCl3 • 6H2O and AlCl3 • 6H2O, in a molar ratio of 3 : 5. This solution was then fed drop-wise in an ammonia dilute solution (pH 9) under continuous stirring and monitoring by a pH-meter; pH was kept constant to the value of 9 ± 0.2 by adding extra ammonia solution. On the ground of previous research the reverse-strike co-precipitation was carried out at 25°C, to avoid any dishomogeneity induced by precipitation temperature.
A jelly and whitish suspension was obtained and submitted to centrifugation to separate the precipitate from mother liquor. In order to remove residual by-products, in particular ammonium chloride, four washing steps of the precipitate were performed by using dilute ammonia (pH 9) and then two washings were carried out in absolute ethanol, prior to drying at 60°C for 48 h. This synthesis was optimised in some previous works thus allowing to obtain very fine and homogeneous crystallite size. In fact, by systematic TEM studies performed on CP pre-treated for various temperatures and times, it was possible to follow the crystallite size evolution from the very beginning of crystallisation (800°C) up to 1350°C. Crystallites showed a limited increase of their size from about 20 nm up to 100–150 nm for the higher-temperature treatment. More details on this investigation are reported elsewhere (Palmero et al., 2004b).
2.2 Nitrates-derived Powders (NP)
NP was prepared from an aqueous solution of Y(NO3)3 • 6H2O and Al(NO3)3 • 9H2O, in a molar ratio of 3 : 5, following the above described procedure. Also in this case, a jelly and whitish suspension was obtained and centrifuged to separate the precipitate from by-products, mainly ammonium nitrate.
The main difference of this preparation, respect to the previous one, is that the jelly material was washed only once in dilute ammonia, and then dispersed in absolute ethanol before drying at 60°C for 48 h. This change in procedure was decided on the ground of previous studies, which demonstrated that the nitrate-based by-products should be easily thermally decomposed in the low-temperature regimes (400–600°C), whereas residual chloride species remain in the material up to higher temperatures (Montanaro and Saracco, 1995). Therefore, nitrate precursors should be effectively exploited for an easier scale-up of the synthesis, by limiting the onerous washing steps necessary when chlorides are used as precursors.
2.3 Phase evolution study
Both CP and NP were firstly subjected to crushing and sieving and then characterised by differential scanning calorimetry and thermal gravimetry (DSC-TG, Netzsch STA 409C thermal analyser) analyses. On the ground of the above results, they were then submitted to various thermal treatments, by varying both temperatures and soaking times, to investigate the phase evolution. Phases development was studied by means of X-Ray Diffraction (XRD, Philips PW 1710, Cu anticathode, angle range 5°–70°2θ, step size of 0.05°2θ, time per step of 5 s). This study was then exploited to select some pre-treatment cycles prior to sintering.
378 P. Palmero et al.
2.4 Sintering study
Pre-treated CP and NP were uniaxially pressed at 300 MPa in bars (15 × 5 × 3 mm) for investigating their sinterability by means of dilatometric analyses (Netzsch 402 E). All samples were sintered in an absolute dilatometer by heating at 10°C/min up to 1500°C, then maintaining the maximum temperature for 3 h, and finally cooling down to room temperature at 20°C/min. Some calcined powders were submitted to a milling procedure, precisely to ball milling by using zirconia spheres (2 mm diameter), for 60 h in absolute ethanol (powder/spheres weight ratio of 1 : 10). In this way, the influence of milling on the sintering behaviour of calcined powders was investigated. The microstructure of the sintered products was observed by scanning electron microscopy (SEM, Hitachi S2300), performed on fracture surfaces.
3 Results and discussion
3.1 Phase evolution
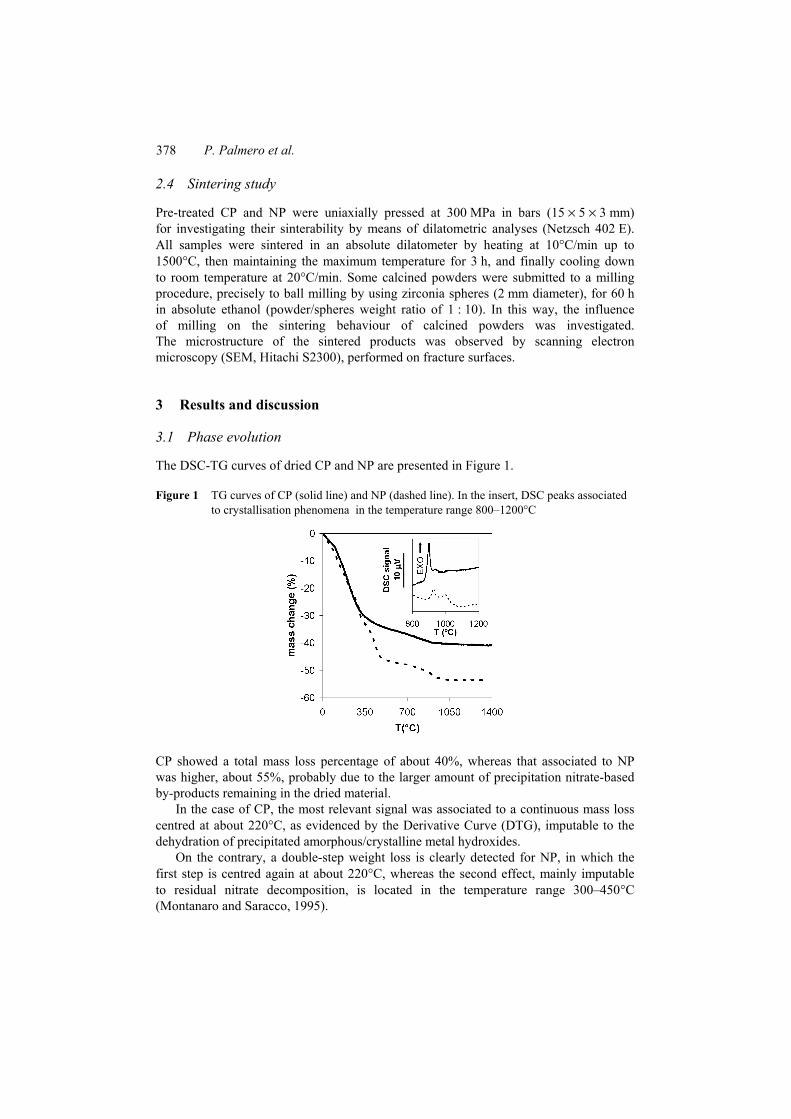
The DSC-TG curves of dried CP and NP are presented in Figure 1.
Figure 1 TG curves of CP (solid line) and NP (dashed line). In the insert, DSC peaks associated to crystallisation phenomena in the temperature range 800–1200°C
CP showed a total mass loss percentage of about 40%, whereas that associated to NP was higher, about 55%, probably due to the larger amount of precipitation nitrate-based by-products remaining in the dried material.
In the case of CP, the most relevant signal was associated to a continuous mass loss centred at about 220°C, as evidenced by the Derivative Curve (DTG), imputable to the dehydration of precipitated amorphous/crystalline metal hydroxides.
On the contrary, a double-step weight loss is clearly detected for NP, in which the first step is centred again at about 220°C, whereas the second effect, mainly imputable to residual nitrate decomposition, is located in the temperature range 300–450°C (Montanaro and Saracco, 1995).

YAG wet-chemical synthesis from chlorides and nitrates precursors 379
A residual mass loss at about 850–900°C was finally detected on both TG curves: it can be related to the first exothermic peak in DSC curves (see the insert of Figure 1).
However, for NP the mass decrease (about 5%) was larger than for CP (less than 2%). Moreover, CP DSC curve is characterised by a sharp, abrupt first exothermic peak,
whereas NP DSC curve shows a weaker and broader signal. The first exotherm is then followed by less intense, broad peak in both cases.
These DSC signals were associated to crystallisation phenomena on the ground of literature data (Yamaguchi et al., 1992) and confirmed in the present work by means of XRD investigations, as shown in the following.
To better evidence the crystallisation peaks and the related maximum temperatures, TG-DSC curves were then recorded on both powders pre-treated at 800°C for a zero-soaking time (Figure 2).
Figure 2 DSC curve of CP (solid line) and DSC-TG curves of NP (dashed lines) pre-treated at 800°C for a zero-soaking time
The maximum of the first exotherm is located at about 890°C and 930°C, whereas the second peak is centred at about 995°C and 1020°C for CP and NP, respectively.
Powder pre-treatment allowed also to put in evidence that each peak is related to a mass loss, since the thermal gravimetry curve in this temperature range is clearly shared in two steps, as shown for NP in Figure 2.
To relate such peaks to phase evolution from the dried precipitates, XRD investigation was performed on powders calcined at various temperatures for different soaking times.
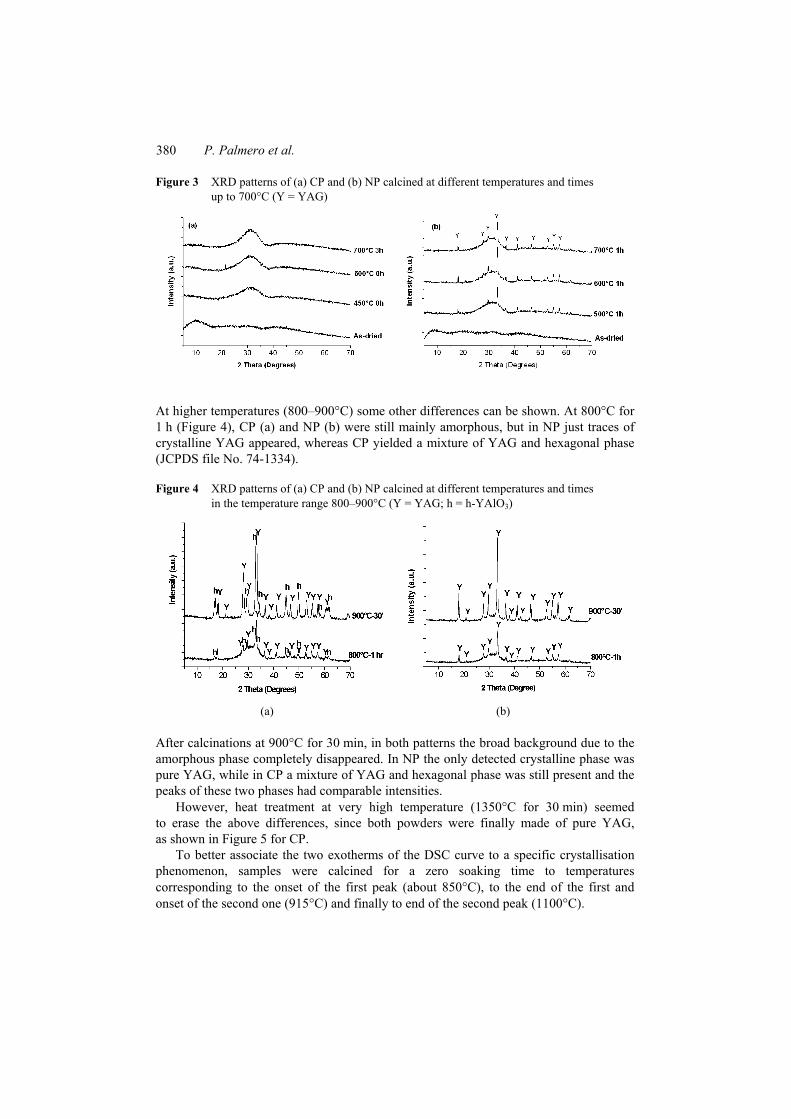
In the low-temperature regimes from 60°C up to 700°C (Figure 3(a) and (b)), the first relevant difference in crystallisation path of these two materials was observed.
In fact, even if both dried powders are completely amorphous, starting from 500°C for 1 h, the more intense peaks of the garnet phase YAG (JCPDS file No. 82-0575) start to appear in NP (Figure 3(b)).
However, the intensity of these peaks is not significantly affected by increasing the treatment temperature up to 700°C.
On the contrary, CP remains amorphous even after 3 h soaking at 700°C (Figure 3(a)).
380 P. Palmero et al.
Figure 3 XRD patterns of (a) CP and (b) NP calcined at different temperatures and times up to 700°C (Y = YAG)
At higher temperatures (800–900°C) some other differences can be shown. At 800°C for 1 h (Figure 4), CP (a) and NP (b) were still mainly amorphous, but in NP just traces of crystalline YAG appeared, whereas CP yielded a mixture of YAG and hexagonal phase (JCPDS file No. 74-1334).
Figure 4 XRD patterns of (a) CP and (b) NP calcined at different temperatures and times in the temperature range 800–900°C (Y = YAG; h = h-YAlO3)
(a) (b)
After calcinations at 900°C for 30 min, in both patterns the broad background due to the amorphous phase completely disappeared. In NP the only detected crystalline phase was pure YAG, while in CP a mixture of YAG and hexagonal phase was still present and the peaks of these two phases had comparable intensities.
However, heat treatment at very high temperature (1350°C for 30 min) seemed to erase the above differences, since both powders were finally made of pure YAG, as shown in Figure 5 for CP.
To better associate the two exotherms of the DSC curve to a specific crystallisation phenomenon, samples were calcined for a zero soaking time to temperatures corresponding to the onset of the first peak (about 850°C), to the end of the first and onset of the second one (915°C) and finally to end of the second peak (1100°C).

YAG wet-chemical synthesis from chlorides and nitrates precursors 381
Figure 5 XRD pattern of CP calcined at 1350°C for 30 min (Y = YAG)
From the XRD analyses of the above samples (Figure 6) it was possible to associate the first peak to the competitive crystallisation of h-YAlO3 and YAG phases and the latter to the conversion of the metastable phase to YAG, in agreement with previous literature data (Yamaguchi et al., 1992). In this case, both materials presented a similar behaviour, independently from the precursors.
Figure 6 XRD patterns of powders calcined in the temperature range of appearance of exothermal DSC peaks: case of CP
For exploiting the differences in crystallisation path put in evidence at low temperature regimes, a systematic study was carried out calcining NP at low temperatures, but for longer soaking times. In fact, if successful, such treatment should allow to produce, at low temperature, pure YAG powder, without forming any intermediate or secondary phase. Moreover, a low calcination temperature should allow to retain the grain growth, to maintain a relevant specific surface area and, consequently, to improve powder sinterability.

382 P. Palmero et al.
The first attempt was performed by calcination at 600°C for 1 up to 24 h -soaking time. XRD patterns of the respective materials are collected in Figure 7.
Even if the heat treatment is prolonged up to 24 h, any significant increase in peaks intensities was achieved and the background due to amorphous phase still remained.
Therefore, the treatment temperature was increased up to 700°C (Figure 8), but also in this case a similar behaviour was observed, so that experimental tests were stopped after 6 h -soaking.
Figure 7 XRD patterns of NP calcined at 600°C for different soaking times (Y = YAG)
Figure 8 XRD patterns of NP calcined at 700°C for different soaking times (Y = YAG)
At 800°C, on the contrary, it was possible to observe a positive evolution in the XRD patterns (Figure 9), since the peaks intensities continuously increased with increasing soaking time, up to a complete crystallisation achieved after 48 h of treatment.
For a sake of comparison, a similar investigation was performed on CP (Figure 10). In this case, at 800°C for a zero-soaking time, the powder was still amorphous; then, prolonging the soaking time, the amorphous phase progressively crystallised, yielding a mixture of both YAG and hexagonal phase, even if from 30 min up to 4 h a progressive increase in percentage of YAG phase was observed. Moreover, if the calcination is

YAG wet-chemical synthesis from chlorides and nitrates precursors 383
prolonged up to 48 h as done for NP, pure YAG was yielded, reaching the same intensities presented by the NP pattern.
Figure 9 XRD patterns of NP calcined at 800°C for different soaking times (Y = YAG)
Figure 10 XRD patterns of CP calcined at 800°C for different soaking times (Y = YAG; h = h-YAlO3)
For better evidencing the differences in the phase evolution of both materials, the intensities of the most important peak of YAG phase (I420) and hexagonal phase (I102) were plotted vs. soaking time, as shown in Figure 11(a) and (b).
At 800°C, NP (Figure 11(a)) was interested by a progressive crystallisation of YAG from the amorphous phase, whereas in the case of CP (Figure 11(b)) the amorphous phase progressively disappeared to yield both YAG and hexagonal phase; during time, the conversion of the metastable phase into the stable one was the predominant effect. In the insert of Figure 11(b), the evolution in CP of the YAG and h-YAlO3 peaks is evidenced for shorter soaking times.
To experimentally confirm the above statements, DSC curves of the powders calcined at 800°C for different times were recorded (Figure 12) and compared to the dried amorphous product and to the fully-crystallised material obtained after treatment at 1500°C. These curves were plotted referring to a normalised mass of the sample, calculated at the onset temperature of the first peak by exploiting the associated TG data.

384 P. Palmero et al.
Figure 11 Evolution of the peak intensity (counts) of the YAG peak I420 vs. soaking times in NP (a) and (b) CP calcined at 800°C. In the insert of (b), the evolution in CP of the YAG peak I420 and h-YAlO3 peak I102 for shorter soaking times is evidenced
(a)
(b)
Figure 12 Evolution of the peak intensities on DSC curves of the NP as-dried, calcined at 800°C for different times and at 1500°C

YAG wet-chemical synthesis from chlorides and nitrates precursors 385
By increasing the soaking time and consequently reducing the residual amount of amorphous phase by YAG crystallisation, both peaks progressively decreased in intensity up to a complete disappearance after 48 h, at which fully crystallisation of YAG phase was achieved.
A complementary information was obtained by the associated enthalpies shown in Table 1.
Phase-transformation related enthalpies were calculated by the integral of each peak, normalised with the mass of the sample.
Table 1 Normalised enthalpy values of the DSC peaks
Normalised enthalpy (J/g) Soaking time at 800°C (h) 1st peak 2nd peak 1 –112.5 –103.3 24 –33.8 –19.3 48 –6.6 /
3.2 Sintering behaviour and fired microstructures
In order to better relate the powders composition to their sintering behaviour, dilatometric investigations were performed on differently calcined samples by applying the same sintering heating rate (10°C/min) employed for XRD and DSC-TG characterisations. In this way all collected data detailed in the previous section were exploited to discuss the present results.
The particular phase evolution described in the Section 3.1 was in fact supposed to cause the abrupt shrinkage phenomenon shown in Figure 13 for a CP sample, previously calcined at 800°C for 30 min. As shown in Figure 10, this material was prevalently amorphous, just having some signals assigned to h-YAlO3 and YAG and the discontinuity observed during the heating step of its sintering curve was so associated to crystallisation and phase conversion steps.
Figure 13 Dilatometric curve (heating step) of the CP pre-treated at 800°C for 30 min and uniaxially pressed at 300 MPa

386 P. Palmero et al.
So, in order to avoid the above phenomenon, the sinterability of NP crystallised at 800°C for 24 h and 48 h, respectively, was investigated by submitting the powders to a dilatometric study up to 1500°C for 3 h -soaking time (Figure 14(a) and (b)). These thermal treatments were selected on the ground of both XRD (Figure 9) and DSC-TG (Figure12) results.
The 24 h treated material was still partially amorphous, so that it is expected the slight contraction observed in the temperature range 800–850°C. In fact, crystallisation to YAG phase was completed at about this temperature during a continuous heating step, as shown in Figure 4. However, such shrinkage effect was significantly slighter than that presented by the powder just pre-treated for 30 min at 800°C, being it a consequence of the lower crystallisation degree, as demonstrated on the ground of XRD and calorimetric data. This shrinkage completely disappeared when a 48 h calcined powder was submitted to the same sintering cycle, strengthening the above statements, since in this case a fully-crystallised powder was submitted to the dilatometric test.
This approach did not allow to achieve a satisfactory result, for two reasons. Firstly, the green density of both NP pre-treated materials were relatively low, reaching about 46% of the theoretical value (4.54 g/cm3); secondly the overall shrinkage recorded during the heating and soaking steps just reached about 8%. As a consequence, both fired materials presented a very poor final density of about 60% of the theoretical value. To improve the densification, NP calcined at 800°C for 48 h was then submitted to ball-milling, performed in absolute ethanol for 60 h. Milling allowed to obtain a significant lowering of the mean agglomerates size, as stated on the ground of particle size distribution analyses (Fritzsch Analysette 22 Compact). In Table 2 the size population values corresponding to 10 (d10), 50 (d50) and 90% (d90) of the volume cumulative distribution for the un-milled and ball milled materials are collected.
In Figure 15, the dilatometric curve of the ball milled powder uniaxially pressed at 300 MPa, is reported.
Figure 14 Dilatometric curves of NP pre-treated at 800°C for (a) 24 h and (b) 48 h and then uniaxially pressed at 300 MPa
(a) (b)
Table 2 Particle size distribution of milled and un-milled NP
d10 (µm) d50 (µm) d90 (µm) Un-milled NP 4.1 28.9 50.3 Ball-milled NP 0.6 1.8 7.2

YAG wet-chemical synthesis from chlorides and nitrates precursors 387
Figure 15 Dilatometric curve of NP pre-treated at 800°C for 48 h, ball milled and uniaxially pressed at 300 MPa prior to sinter at 1500°C for 3 h
The green density was not affected by the milling step, reaching the same above value. On the contrary, ball milling was effective in the improvement of densification. In fact, a significant increase in the total linear shrinkage (from about 8% in the un-milled material to about 18% in the ball-milled one) was observed, leading to a final fired density of about 86% of the theoretical value.
SEM observation (Figure 16) of the final microstructure showed a homogeneous distribution of small, almost equiaxial pores surrounded by spherical particles having a mean size of about 500 nm (see the higher magnification image in the insert of Figure 16). The residual porosity can be easily explained on the ground of the experimental conditions used to perform densification; in fact, as stated in literature, highly dense YAG materials are usually obtained at higher final densification temperatures (about 1700°C) and sometimes in vacuum conditions (Wen et al., 2004; Li et al., 2000; Tachiwaki et al., 2001), not allowed by the adopted apparatus.
Figure 16 SEM micrograph of NP pre-treated at 800°C for 48 h submitted to ball milling prior to uniaxial pressing at 300 MPa and sintering at 1500°C for 3 h (surface fracture). In the insert, a higher magnification image is reported

388 P. Palmero et al.
4 Conclusions
Reverse-strike co-precipitation starting from nitrates or chlorides precursors yields completely amorphous precipitates. However, this study allowed to clearly state the role of the precursor on phases evolution.
In particular, the main differences in crystallisation phenomena among nitrate (NP) – and chloride (CP) -derived powders were drawn.
NP and CP mainly differ on their crystallisation paths, already at low calcination temperatures: CP remains amorphous up to 700°C while NP starts crystallisation at 500°C yielding pure YAG phase. However, even after prolonging the soaking calcination time and/or increasing the calcination temperature up to 700°C, no appreciable increase of the YAG phase amount was detected.
At higher calcination temperatures (800–1100°C) different crystallisation paths were identified. CP evolves from the nucleation of a mixture of YAG and h-YAlO3 phases in the amorphous matrix through a completely crystallised biphasic product up to a final pure-YAG material. On the contrary, NP yields pure YAG in the amorphous matrix, evolving to a well crystallised pure YAG phase, by-passing the formation of any intermediate.
Dilatometric results showed that yttrium aluminates crystallisation from an amorphous matrix led to an abrupt and undesired contraction at about 850°C. So, to minimise this effect, well crystallised product should be selected.
However, even sintering fully crystallised YAG materials, very poor final densities were reached, imputable to the large agglomerates, induced by prolonged calcination, but also to the adopted sintering conditions.
To reduce the first drawback, ball milling was performed leading to a significant gain in powder sinterability. However, to further improve densification, higher performing sintering conditions should be required, at present not allowed by the adopted equipment.
In any case, the study of the sintering behaviour and the obtained microstructure suggests that the material should be successfully used to realise high density components with a tailored grain size, if densified by more effective procedures like SPS (Spark Plasma Sintering) or HP (Hot Pressing).
Finally, the overall results open new research perspective by exploiting the different powder compositions from the two wet-chemical syntheses. In a future investigation, the sintering behaviour of a pure YAG material from NP could be compared to that of a mixture of stable and metastable phases, namely YAG and h-YAlO3, from CP.
Acknowledgement
The authors are greatly indebted to the European Community to have partially supported this research in the frame of the Integrated Project NanoKer.

YAG wet-chemical synthesis from chlorides and nitrates precursors 389
References Apte, P., Burke, H. and Pickup, H. (1992) ‘Synthesis of yttrium aluminium garnet by reverse strike
precipitation’, Journal of Materials Research, Vol. 7, No. 3, March, pp.706–711. Cabanas, A., Li, J., Blood, P. et al. (2007) ‘Synthesis of nanoparticulate yttrium aluminum garnet
in supercritical water-ethanol mixtures’, Journal of Supercritical Fluids, Vol. 40, No. 2, March, pp.284–292.
French, J.D., Zhao, J. et al. (1994) ‘Creep of duplex microstructures’, Journal of the American Ceramic Society, Vol. 77, No. 11, November, pp.2857–2865.
Gowda, G. (1986) ‘Synthesis of yttrium aluminates by the sol-gel process’, Journal of Materials Science Letters, Vol. 5, No. 10, October, pp.1029–1032.
Hreniak, D. and Strek, W. (2002) ‘Synthesis and optical properties of Nd3+ -doped Y3Al5O12 nanoceramics’, Journal of Alloys and Compounds, Vol. 341, 17 July, pp.183–186.
Inoue, M., Otsu, H., Kominami, H. and Inui, T. (1991) ‘Synthesis of yttrium aluminium garnet by the glicothermal method’, Journal of the American Ceramic Society, Vol. 74, No. 6, June, pp.1452–1454.
Kakade, M.B., Ramanathan, S. and Ravindran, P.V. (2003) ‘Yttrium aluminium garnet powders by nitrate decomposition and nitrate-urea combustion reactions – a comparative study’, Journal of Alloys and Compounds, Vol. 350, 17 February, pp.123–129.
Li, J.G., Ikegami, T., Lee, J.H. et al. (2000) ‘Co-precipitation synthesis and sintering of yttrium aluminum garnet (YAG) powders: the effect of precipitant’, Journal of the European Ceramic Society, Vol. 20, Nos. 14–15, pp.2395–2405.
Li, X., Liu, H., Wang, J. et al. (2004) ‘Rapid synthesis of YAG nano-sized powders by a novel method’, Materials Letters, Vol. 58, No. 19, July, pp.2377–2380.
Lo, J.R. and Tseng, T.Y. (1998) ‘Phase development and activation energy of the Y2O3-Al2O3 system by a modified sol-gel process’, Materials Chemistry and Physics, Vol. 56, No. 1, 30 September, pp.56–62.
Lu, J., Ueda, K., Yagi, H., Yanagitani, T., Akiyama, Y. and Kaminskii, A.A. (2002) ‘Neodymium doped yttrium aluminium garnet (Y3Al5O12) nanocrystalline ceramics-a new generation of solid state laser and optical materials’, Journal of Alloys and Compounds, Vol. 341, pp.220–225.
Lupei, V., Lupei, A. and Ikesue, A. (2004) ‘Single crystal and transparent ceramic Nd-doped oxide laser materials: a comparative spectroscopic investigation’, Journal of Alloys and Compounds, Vol. 380, Nos. 1–2, 20 October, pp.61–70.
Mathieu, M.V. (1956) contribution a l’étude des gels d’alumine desorganisées, PhD Thesis, University of Lyon, Lyon, France.
Messier, D.R. and Gazza, G.E. (1972) ‘Synthesis of MgAl2O4 and Y3Al5O12 by thermal decomposition of hydrated nitrate mixtures’, American Ceramic Society Bulletin, Vol. 51, No 9, September, pp.692–694, 697.
Montanaro, L. and Saracco, G. (1995) ‘Influence of some precursors on the physico-chemical characteristics of transition aluminas for the preparation of ceramic catalytic filters’, Ceramics International, Vol. 21, No. 1, p.43.
Nyman, M., Caruso, J. and Hampden-Smith, M.J. (1997) ‘Comparison of solid-state and spray-pyrolysis synthesis of yttrium aluminate powders’, Journal of the American Ceramic Society, Vol. 80, No. 5, May, pp.1231–1238.
Palmero, P. and Montanaro, L. (2007) ‘Thermal and mechanical-induced phase transformation during YAG and Alumina-YAG syntheses’, Journal of Thermal Analysis and Calorimetry, Vol. 88, pp.261–267.

390 P. Palmero et al.
Palmero, P., Esnouf, C., Fantozzi, G. and Montanaro, L. (2004a)‘Sol-gel synthesis of high-purity YAG powders: a kinetic study of crystallisation and growth’, 9th European Interregional Conference on Ceramics (CIEC 9), Bardonecchia (TO), Italy, pp.167–172.
Palmero, P., Esnouf, C., Montanaro, L. and Fantozzi, G. (2004b). ‘Influence of the synthesis temperature on the crystallisation path and kinetics of YAG powders’, Ceramic Engineering and Science Proceedings of the 28th International Conference on Advanced Ceramic, Vol. 25, pp.597–602.
Palmero, P., Esnouf, C., Montanaro, L. and Fantozzi, G. (2005) ‘Influence of the co-precipitation temperature on phase evolution in yttrium-aluminum oxide materials’, Journal of the European Ceramic Society, Vol. 25, No. 9, June, pp.1565–1573.
Pullar, R.C., Taylor, M.D. and Bhattacharya, A.K. (1999) ‘The sintering behaviour, mechanical properties and creep resistance of aligned polycrystalline yttrium aluminium garnet (YAG) fibres, produced from an aqueous sol-gel precursor’, Journal of the European Ceramic Society, Vol. 19, No. 9, July, pp.1747–1758.
Rabinovitch, Y., Tetard, D., Faucher, M.D. and Pham-Thi, M. (2003) ‘Transparent polycrystalline neodymium doped YAG: synthesis parameters, laser efficiency’, Optical Materials, Vol. 24, Nos. 1–2, October/November, pp.345–351.
Ramanathan, S., Kakade, M.B. et al. (2003) ‘Processing and characterization of combustion synthesized YAG powders’, Ceramics International, Vol. 29, No. 5, pp.477–484.
Su, J., Zhang, Q.L., Gu, C.J. et al. (2005) ‘Preparation and characterization of Y3Al5O12 (YAG) nano-powder by co-precipitation method’, Materials Research Bulletin, Vol. 40, No. 8, 11 August, pp.1279–1285.
Tachiwaki, T., Yoshinaka, M., Hirota, K. et al. (2001) ‘Novel synthesis of Y3Al5O12 (YAG) leading to transparent ceramics’, Solid State Communications, Vol. 119, Nos. 10–11, 29 August, pp.603–606.
Torrecillas, R., Schehl, M., Diaz, L.A. et al. (2007) ‘Creep behaviour of alumina/YAG nanocomposites obtained by a colloidal processing route’, Journal of the European Ceramic Society, Vol. 27, No. 1, pp.143–150.
Veith, M., Mathur, S. et al. (1999) ‘Low temperature synthesis of nanocrystalline Y3Al5O12 (YAG) and Ce-doped Y3Al5O12 via different sol-gel methods’, Journal of Materials Chemistry, Vol. 9, p.3069r.
Vrolijk, J.W., Willems, J.W. and Metselaar, R. (1990) ‘Coprecipitation of yttrium and aluminum hydroxides for preparation of yttrium aluminum garnet’, Journal of the European Ceramic Society, Vol. 6, No. 1, pp.47–51.
Wang, H., Gao, L. and Niihara, K. (2000) ‘Synthesis of nanoscaled yttrium aluminum garnet powder by the co-precipitation method’, Materials Science and Engineering A (Structural Materials: Properties, Microstructure and Processing), Vol. A288, No. 1, 31 August, pp.1–4.
Wang, H., Gao, L., Shen, Z. and Nygren, M. (2001) ‘Mechanical properties and microstructures of Al2O3-5vol% YAG composites’, Journal of the European Ceramic Society, Vol. 21, No. 6, pp.779–783.
Wen, L., Sun, X., Xiu, Z. et al. (2004) ‘Synthesis of nanocrystalline yttria powder and fabrication of transparent YAG ceramics’, Journal of the European Ceramic Society, Vol. 24, No. 9, August, pp.2681–2688.
Xu, G., Zhang, X., He, W. et al. (2006) ‘Preparation of highly dispersed YAG nano-sized powder by co-precipitation method’, Materials Letters, Vol. 60, No. 7, April, pp.962–965.
Yamaguchi, O., Takeoka, K., Hirota, K., Takano, H. and Hayashida, A. (1992) ‘Formation of alkoxyde-derived yttrium aluminum oxides’, Journal of Materials Science, Vol. 27, No. 5, 1 March, pp.1261–1264.

YAG wet-chemical synthesis from chlorides and nitrates precursors 391
Yoldas, B.E. (1982) ‘Effect of varations in polymerized oxides on sintering and crystalline transformation’, Journal of the American Ceramic Society, Vol. 65, No. 8, August, pp.387–393.
Website For further information on nano-YAG development and exploitation for advanced optical and
structural applications, see www. nanoker-society.org




















