
Microfacies and diagenesis of the reefal limestone, Callovian ...

W A T E R - S E D I M E N T EXCHANGE OF NUTRIENTS DURING EARLY DIAGENESIS AND RESUSPENSION OF ANOXIC SEDIMENTS FROM THE
N O R T H E R N ADRIATIC SEA SHELF.
F e d e r i c o Spagno l i and M a r i a Cr i s t ina B e r g a m i n i . Istituto di Geologia Marina. Consi~,,lio Nazionale delle Ricerche. Bologna. Italy
,'tbstract. This paper presents the results of a study on nutrient exchange at the sediment-water interface which ~s caused by early diagencsis and resuspension of bottom sediments. The research was carried out on anoxic ,ihy-clay sediment cores collected south of the Po river delta (Northern Adriatic Sea, Italy) in late summer.
The early diagenetic processes were investigated by means of the integrated study of pore-water chemistry md solid phase composition. Exchange at the sediment-water interface was studied by comparing the fluxes measured in incubated cores with the fluxes calculated by modelling pore-water profiles. Nutrient exchange Juring resuspension was analysed by simulating a storm event in the laboratory.
The high production of nutrients near the sediment-water interface is mainly caused by the anoxic legradation of organic matter and the successive reductions of Mn and Fe-oxyhydroxides and, to a lesser extent, of sulphate. The oxic degradation of organic maner occurs only at the sediment-water interface.
In the incubation expemnent the increases of phosphate, ammonia, nitrate, silica, and Fe in bottom waters ~ere measured. The comparison between calculated and measured fluxes showed that: a) the fluxes are mainly controlled by molecular diffusion: b) phosphate and Fe sink because of the Fe-oxyhydroxide precipitation and c) nitrification process influences the ammonia and nitrate fluxes.
Resuspension caused the release of: a) phosphate through surficial desorption and authigenic apatite lissolution; b) ammonia by means of the oxic degradation of organic nmner; and c) dissolved silica generated t~y biogenic silica dissolution. Resuspension also caused a weak removal of Fe. The more oxic conditions following resuspension favourcd the formation of a Ee-oxyhydroxide fihn at the sediment-water interface which inhibited the phosphate fluxes from sediments to the water column.
Keywords: Diagenesis, resuspension, sediment, pore-water, marine
1. Introduction
In coas ta l mar ine e n v i r o n m e n t s the chemica l t r an s fo rma t ions that take place at the
s e d i m e n t - w a t e r in te r face d e t e r m i n e the cyc l ing of nu t r ien ts be tween s e d i m e n t s and
waters. The fl~rmer can cons t i tu t e a source or a sink for nutr ients , so that in areas with
sha l low waters s e d i m e n t s can be one of the ma jo r factors which cont ro l the t rophic level
of the aquat ic sys tem. M a n y of these chemica l r eac t ions are b io log ica l ly med ia ted ; the i r
re la t ive i m p o r t a n c e d e p e n d s on several factors, such as s e d i m e n t c o m p o s i t i o n ,
s e d i m e n t a t i o n rate, hyd rodynan l i c s , b io tu rba t ion and i r r igat ion, as well as the phys ica l
and c h e m i c a l cha rac te r i s t i c s of bo t tom waters .
Ear ly d i agenes i s can p roduce solute c o n c e n t r a t i o n d i f f e rences be tween pore waters and
sea waters , ma in ly becaus e of organic mat te r d e g r a d a t i o n and var ia t ions of pH and redox
potent ia l . Such d i f f e rences cause f luxes t h rough d i f fus ion , b io tu rba t ion and i r r iga t ion
( H a m m o n d et al., 1985) at the s e d i m e n t - w a t e r interface, where a lso a d s o r p t i o n / d e s o r p t i o n
p rocesses can take place (Santsch i et al., 1990).
M a n y au thors inves t iga ted the re lease of nu t r ien ts f rom s e d i m e n t s by means of in si tu
m e a s u r e m e n t s ( H a m m o n d et al., 1985; W e s t e r l u n d et al., 1986; B e n d e r et al., 1989;
Be re l son et al., 1987b, 1990, 1994), micro and m e s o c o s m s (Sundby et al., 1986; Pe te r sen
et al., 1995), water d y n a m i c s mode l l i ng (Be re l son et al., 1987a) or pore water prof i le
Water. Air and Soil Pollution 99: 541-556, 1997. �9 1997 Kluwer Academic Publishers. Printed in the Netherlands.

542 F. SPAGNOLI AND M. C. BERGAMINI
modelling (Aller, 1980; Berner, 1980; Klump and Martens, 1981; Berelson et al., 1987a; Hammond et al., in press)
Resuspension may influence the chemical composition of the water column because of pore water and bottom water mixing, sorption-desorption on solid surfaces, precipitation-dissolution processes, and an enhanced organic matter degradation. Solid- water exchanges during resuspension have been relatively neglected because of sampling difficulties during storms (Oviatt et al., 1981; Fanning et al., 1982)
The Northwestern Adriatic Sea is an economically important region undergoing heavy eutrophication and pollution pressure (Marchetti, 1992) and where nutrient release from sediments plays a major role (Giordani et al., 1992). Fluxes at the sediment-water interface have been measured in situ (Giordani and Hammond, 1985; Giordani et al.,
1992), by means of incubation or pore water modelling (Barbanti et al., 1995). The chemical effects of resuspension were investigated only by Frignani and Turci (1981) and, in lagoonal areas, by Sfriso et al. (1990) and Calvo et al. ( 1991 ).
The aim of this work was to investigate the role played by anoxic sediments as nutrient sinks or sources, as well as the changes in speciation on the particulate phase of some elements caused by early sediment-water interface in low interactions during resuspension intact cores.
diagenesis and resuspension processes. Fluxes at the hydrodynamic conditions, and the water-solid matter were studied by means of a laboratory experiment on
2. Sampl ing area
The cores were collected from the 22.5 m deep .... seafloor in the southern prodelta area of the Po ........ ~Ee~ ~ ' ' :\ River, North Adriatic Sea (Fig. I). This area has a l ~" sedimentation rate as high as 0.88 cmly (Frignani and Langone, 1991) and silty clay sediments which are mainly supplied by the Po River, the largest Italian river (mean water discharge 1500 m 3 s -1, ~.0~. , ...........
mean solid discharge 14 million tonnes year-l; Dal k . , , Cin, 1983) which drains very industrialised and intensively cultivated areas. The sediments have high concentrations of continental and marine organic matter and host the typical populations of muddy seabeds. Polychaetes, such as Aricidea ...... " ~ " 7 " W " assimilis, A. claudiae. Prionospio malmgreni, P. / \ cirrifera, Maldane sarsi, and Nephtys incisa .......
dominate the macrofauna (Crema et al., 1991 ). ~ . . . . . . . . . . . . .
2
Figure 1 Study Area
The water circulation of the area follows the general circulation of the Northern Adriatic Sea, with seasonal dynamics which are strongly conditioned by thermohaline and barocline factors and, to a lesser extent, by winds (Franco et al., 1982; Orlich et al., 1992; Artegiani et al., in press). In summer the circulation of waters is rather slow, with

WATER-SEDIMENT EXCHANGE OF NUTRIENTS 543
horizontal stratification in the water column mainly caused by the outflow of the Po River (Nelson, 1970; Orlich et al., 1992). In winter a cyclonic water circulation prevails, with a southward fast-moving current and a more homogenous water column which mostly consists of marine waters, even if the Po River waters may be occasionally discernable.
This area has frequent resuspension events which are caused chiefly by the bora and scirocco winds and secondarily by tidal currents (Drake et al., 1992). The dissolved oxygen in bottom waters varies considerably: it is maximum in winter and very low in summer when waters often reach an anoxic state.
3. Materials and methods
3. l . SAMPLING OPERATIONS
Field operations were performed on September 1st, 1992. Continuous temperature, salinity, dissolved oxygen, chlorophyll a and pH profiles were performed by CTD probe. Dissolved oxygen in the bottom waters indicate anoxic conditions. The water column was sampled by means of a Niskin bottle, while bottom water was collected by a diver. Using Plexiglas tubes of 10.4 cm in diameter, divers also collected the cores close to one another, to minimize disparity in sediment composition. The cores were immediately stored at 4~ and kept refrigerated for about five hours, the time required to take them to the laboratory.
One core which was about 30 cm long, was used to characterise the sediments. Core extrusion and pore water extraction were performed in a nitrogen atmosphere within +2~ of the in situ temperature. The core was cut into 0.5 to 3 cm thick slices, using the higher resolution near the sediment-water interface. Each slice was placed in a polypropylene tube and centrifuged at 50(/0 rpm at the in situ temperature for 15 mins. The supernatant was then filtered through a 0.45 bun Durapore membrane and split into two aliquots: one was left unacidified lor nutrient, alkalinity, sulphate, sulphide and salinity determinations, while the other was acidified to pH 1-2 with Suprapure HNO3 for Ca, Mg, Fe, Mn and trace metal analysis (Spagnoli, 1993). The pH and redox potential were determined through precalibrated punch-in electrodes. Visual descriptions of the core were recorded. The entire procedure was completed in 6 hours. After centrifugation, sediments were freeze-dried and ground by means of an agate pestle and mortar for subsequent chemical analyses: organic and inorganic C, total content of Ca, Mg, Fe, Mn, Ti, AI, Cu, Zn. Cr, Cd, Pb and P, as well as Fe, Mn, Cu, Zn, Cr, Cd, Pb and P sequential extraction (Spagnoli, 1993). Grain sizes were determined with the wet samples.
3.2. LABOP, ATORY EXPERIMENT PROCEI)UP, ES
Seven cores were used for laboratory experiments. All preparatory operations were performed at 20+2~ in a nitrogen atmosphere. Cores A and B were used for simulating resuspension events and core C for measuring the fluxes at the sediment-water interface in low hydrodynamic conditions (stirring experiment). The remaining four cores were used for slice "shaking experiments".

544 F. SPAGNOLI AND M. C. BERGAMINI
The supernatants of the A, B and C cores were removed carefully and replaced with 4 1 of natural, oligotrophic and filtered seawater. The seawater was added very slowly through Teflon capillary tubing so that it trickled gently down the tube wall above the sediment; the replacement took 3 to 4 h. Subsequently, a 8.5 cm wide Teflon paddle was placed about 18.5 cm above the sediment-water interface, in order to create resuspension (high angular speed) or to keep the diffusive boundary layer thickness at the sediment-water interface unchanged during incubation (low angular speed: 2-3 rpm). After setting up these apparatuses, all cores were kept at 20+2~ with their tops exposed. Supernatant samples were collected by means of a syringe. At the end of the stirring experiment, the surficial sediments (0.5 cm) were collected.
Fig. 2 shows the different steps of the stirring experiment for the A and B cores�9 The paddle rotated for the first 45 rains at low speed in order to bring the added seawater and sediments to a balance. During this time the suspended solids ranged from 1 to 23 mg/I. In the second and third steps the speed of the paddle was increased further for 3.5 h and 8.25 h, respectively. The maximum stirring speed for the A core was faster than that for the B core. The resuspended solids of the A core increased at the beginning and at the end of the maximum stirring step, while those of the B core increased continuously throughout the same step. The maximum resuspended solid value for A was 0.89 g/l, while it was 0.11 g/l for B. The distance between paddle and sediment interface, the fastest angular speed and the maximum stirring time were set to simulate the duration (about 7-8 h) and magnitude (about 600 rag/l) of an average resuspension event as recorded by the Geoprobe deployed in the sampling area (Drake et al., 1992)�9
A 9O
lO
o
rpm + suspended solid
90
0.9
0.8
0.7
0.5
0.4 ~L~ o.3_~ o 0.2 m
0.1
0 120
B 0.9
7O
E ~
2O 10
O r , I k i i L
40 ~ 80 100
hours
0.8 0.7
04 XN 0 3 ~ , o 0.2 m
0.1 0
Figure 2. The RPM of t.he stirring paddles and suspended solids in cores A and B.

WATER-SEDIMENT EXCHANGE OF NUTRIENTS 545
In contrast with the literature, at the end of the maximum stirring step no resuspension steady state was reached (it took about 10 minutes according to Tsai and Lick, 1986). We presume that this was caused by a degradation process of the highly reactive organic matter (Westrich and Berner, 1984) at the interface following the more oxic conditions created by our resuspension. The measurement of maximum solid resuspension made it possible to determine the maximum thickness of sediment erosion. With a dry density of 2.7 and a porosity of 0.83, the erosion was I mm in core A and 0.12 mm in core B.
After resuspension the paddle was slowed down to intermediate and low speeds in two steps. The last slow-speed step (incubation) lasted tour days. The same slow speed was used throughout the experiment with the C core (low hydrodynamic conditions). During such incubation the suspended solids ranged from 5 to 35 mg/l.
In the shaking experiment, one slice per core (the first 0.1, 0.5, 1 and 2 cm, respectively) was collected and split into four subsamples. The subsamples then were put in to sealed vessels with 100 ml of natural, oligotrophic, filtered seawater and shaken for 1,2, 10 and 21 h, respectively. In order to assess the solid-water fluxes for each chemical, the pore water content of each species was subtracted from its final amount in the 100 ml after shaking. The final content/solid matter ratios were also determined.
After pH, Eh, salinity, O2 and chlorophyll a determinations, samples of the stirring and shaking experiments were filtered through pre-weighed, 0.45 gm Nucleopore filters. Then filters were washed with Milli-Q water, freeze-dried, and weighed again. The resulting weights were used to determine the resuspended particle concentrations. The filtered solutions and solid samples were analyzed for the same sediment parameters as above. No algal growth was found during either the stirring or shaking experiments.
3.3. CHEMICAL ANALYSIS
3.3.1 Water anah'sis Dissolved oxygen and salinity were measured by suitable electrodes; chlorophyll a was determined by means of a fluorimeter. Dissolved phosphate, ammonia, nitrite and nitrate were determined using the colorimetric technique of Strickland and Parsons (1972). Alkalinity was determined by titration with HC1 (Gieskes and Rogers, 1973); Total carbonate was calculated from alkalinity, pH, salinity and temperature. Sulphides were removed by adding ZnCI2 first to prevent oxidation to sulphates and then determined with
tile method of Cline (1969). Sulphates were measured by the indirect titration of precipitated barium sulphates (Howarth, 1978). Fe, Mn, Ca, Mg and other trace metals (Spagnoli, 1993) were determined on the acidified aliquot by means of ICP-AES.
3.3.2 Solid phase anah'sis Grain size analysis was performed after the complete destruction of aggregates by hydrogen peroxide and sonication. The > 63 ~tm traction was separated from the mud by wet sieving, and the silt-clay fractions were subsequently analysed by means of an X-ray Sedigraph. Porosity was calculated from the sediment weight loss after drying at 60~ (Berner, 1971). Mineralogical composition was determined using X-ray diffraction analysis (Cook et al., 1975). Organic carbon and total nitrogen were determined using a CHN elemental analyser after removal of the inorganic carbon with HC1 (Froelich, 1980).

546 F. SPAGNOLI AND M. C. BERGAMINI
Iron speciation was determined using the procedure of Forstner and Salomons (1980) which was integrated with the procedure of Tessier et al. (1979). This method (E&T) (Barbanti and Sighinolfi; 1988) separates and quantifies the exchangeable fraction (exc), the carbonate fraction (carb), the easily and moderately reducible fraction (red), the organic and sulphide fraction (oxid) and the residual mineral fraction (res) of metals associated with the solid phase. The total iron content was determined by total digestion of the sediment samples with concentrated mineral acid mixtures (Guerzoni et al., 1984). Fe concentrations were determined in extracts by means of ICP-AES.
P speciation was obtained using two different selective, sequential chemical extraction techniques: the Golterman and Booman method (1988) as modified by Barbanti et al.
(1994) and the F&T procedure satisfactorily tested by Barbanti and Sighinolfi (1988) for P. The modified Golterman and Booman sequence (G&B) identifies exchangeable P (P- exc), P bound to Fe (P-Fe) and P bound to Ca (P-Ca). Total P was determined with the same method used for total iron. Organic P (P-org) in G&B was determined as the difference between total P and inorganic P. Reactive phosphates in all extracts were determined using the colorimetric technique of Strickland and Parsons (1972). The total P extracts were pre-treated by digestion with fuming HNO~HC104.
4. Results and discussion
4.1 . DIAGENETIC PROCESSES
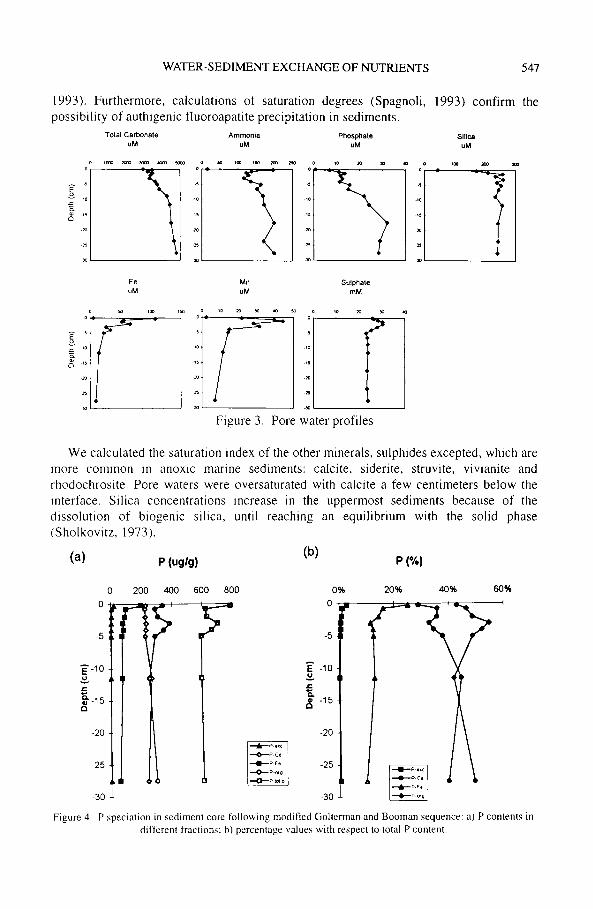
The distribution of organic matter degradation products in pore waters (total carbonate, ammonia and phosphate) (Fig. 3) shows a near-surface peak and a regular increase at deeper sediments levels. The anoxic degradation of organic matter by the successive reduction of Mn, Ee-oxyhydroxides and sulphates takes place just below the sediment- water interface; this is supported by the negative Ell values and by the blackish grey colour of the sediments, the typical shade of anoxic conditions. The slight and gradual increase of total carbonate, ammonia and phosphate at deeper levels can be accounted for only by a small sulphate depletion, indicating that the reactivity of the buried organic carbon is probably too low to drive significant sulphate reduction. The oxic degradation of organic matter is confined to tile sediment-water interface.
The diagenesis of phosphorus deserves special consideration, since its concentration in pore waters is controlled not only by tile organic matter degradation, but also by the dissolution-precipitation of Fe-oxyhydroxides with which phosphate tends to associate under oxic conditions (Berner, 1980; Krom and Berner, 1981). The influence of the reduction process of Fe-oxyhydroxides on phosphate regeneration m the uppermost sediments is confirmed by the profile of the P-Fe fraction (Fig. 4): ttle P-Fe concentration decreases rapidly in the solid phase and a rapid rise in pore water phosphate concentrations occurs at the same time. Below the 4 cm level phosphate the pore water phosphate increases gradually, while the P-Fe remains constant; this suggests that the contribution from tile organic matter degradation process prevails. The precipitation of authigenic apatite is another important process controlling the phosphate concentration in pore waters. The slight increase of P-Ca fraction with depth (Fig. 4) indicates a diagenetic removal of P for precipitation with the authigenic solid phase (Ruttenberg and Berner,
WATER-SEDIMENT EXCHANGE OF NUTRIENTS 547
1993). Furthermore, calculations of saturation degrees (Spagnoli, 1993) confirm the possibility of authigenic fluoroapatite precipitation in sediments.
o
v ,o s
Tolal Carbonate Ammonia Phosphate Silica uM uM uM uM
, o s
Fe Mn S u l p h a l e
uM uM m M
Figure 3. Pore water profiles
We calculated the saturation index of the other minerals, sulphides excepted, which are more common in anoxic marine sediments: calcite, siderite, struvite, vivianite and rhodochrosite. Pore waters were oversaturated with calcite a few centimeters below the interface. Silica concentrations increase in the uppermost sediments because of the dissolution of biogenic silica, until reaching an equilibrium with the solid phase (Sholkovitz, 1973).
(a) P {uglg} (b) P (%}
0 200 400
- 5 I ,
~ ' - 1 0 -
u �9
~-15 - a
600 800 0% 20% 40% 60%
E" -10 &
' - 1 5
-20 - -20
-25 L ~ -25 \ ,
-~o i -3o I
Figure 4. P speciation in sediment core following modified Golterman and Booman sequence: a) P contents in different fractions: b) percentage values with respect to total P content.

548 F. SPAGNOLI AND M. C. BERGAMINI
4.2. LABORATORY EXPERIMENT
At the beginning of the stirring experiment the concentrations of all chemicals were irregular, because the replacement of the supernatant with fresh seawater caused a slight resuspension of bottom sediments and the appearance of more oxic conditions.
During resuspension (Fig. 5) the phosphate release was proportional to the resuspended solid matter. In the few days following resuspension the phosphate concentration increases in the supernatant from the C core (low hydrodynamic conditions) exceeding that for the A and B cores.
F* (uM)
~3
N
II] 1! ~'L _ _ . _ ~
NNrWa - N ~ luW
N { ~a
8
g~ ~e
. . . . . . . . . . I ~ '
*l
g,
~ L _ ]
l-If b
8
Figure 5. Dissolved chemical concentrations versus time in A,B, and C cores of the stirring experiment.

WATER-SEDIMENT EXCHANGE OF NUTRIENTS 549
In the shaking experiment, the net phosphate concentration increased (Fig. 6). This indicates that phosphate was released from the solid phase: such release was greater in the three thinner slices. The relative phosphate release per gram of shaken sediment was much higher in the thinnest slice and proportional to the shaking time in the 0.1 and 0.5 cm slices.
0 ~ rNd PO4 (umol) 0 3 ~ l t NH4 (umol)
OO2 ~ 0 2
015 h ~ - 01 o , o I I _ _ o~
1 2 10 2~ 1 2 10 21
03 05 o,!
0= oo~ 01 -01
0 1 2 10 21
002 ~ 04 02
015 01 0
OO5 ~ 0 Ol
2 I0 21
05
I ~ ! 0 2 0 2
I~ | NO3+NO2 i tttt SI~OH)4 ~ ~ (umol) n41( r-e (um,o4) 1 (umol) I e , (umc4) 03
10 !2 O~ 02
0 - - 0 1 2 10 21 i 1 2 10 21
06 02 �9
05 0 1 .
, , I o~ - 0 , - 0 0 L - O 2 -
1 2 10 21 / , 2 10 21 1 2 10 21
" J ; t o, ! o: '~ 21 o2 l
_ _ ! o21U -02 , 2, L . . . . . . j 20 16 T ' 0 8 ~
,o 3 ; i I 5 12 ot= _mia;a a _ l l
1 2 10 21 1 2 10 21 " 2 0 2
hour~ h o u ~ h o u ~
Figure 6. Concentrations of dissolved chemicals in the supernatants versus shaking time in the shaking experiment. Net amounts of chemicals are obtained by subtracting the pore water and initial contents from
final quantities in the supematant.
The results of the two sequential extraction procedures (Fig. 7) used in the shaking experiment show that the P-exc fraction decreased in time in the 0.5 cm slice, suggesting that the phosphate release during resuspension was caused partly by desorption of the P weakly bound to the solid surfaces. Furthermore, the P-Ca fraction decreased in the solid phase from the 0.5 and 1 cm slices and in the surface sediments taken at the end of the stirring experiment. This indicates that P was released because of the dissolution of the fresh authigenic apatite which precipitated near the interface. At the same time, the P- Fe traction increase in the solid phase of the shaken samples, indicates that P precipitated together with Fe-oxyhydroxides. The high concentrations of the P-red (associated with the Fe-oxyhydroxide) fraction in the suspended solids of the F&T sequences indicates that P probably precipitated with this mineral phase. However, the extent of this P sinking process was lower than the release processes of the other P fractions. Undoubtedly, the high P-red concentrations were caused partly by the diagenetic enrichment process near the interface. The existence of such a process is confirmed also by the fact that the high P- total concentrations found in the resuspended solids and in the samples from the thinnest slice are in contrast to the general phosphate releases recorded at the end of the shaking of the O. 1 cm slice samples. The P-exc concentrations increased in the shaken samples of the thicker slices and were higher than in the samples of the thinnest slice. This was due to the

550 F. SPAGNOLI AND M. C. BERGAMINI
stronger adsorption processes determined by the presence of partially dissolved Fe- oxyhydroxides at deeper levels.
This observation together with the P-Fe fraction increase and the reduced phosphate release in the samples of the thickest slice suggest that the phosphate release is low when resuspension involves anoxic sediments and is greater when resuspension involves oxic sediments. The oxidizing conditions in the water column determine a considerable removal of phosphate, because there are larger amounts of dissolved Fe which precipitates in oxyhydroxide forms, as well as numerous free sites for adsorption on the surface of partially dissolved oxyhydroxides.
The increase of P-Fe and P-red fractions in the surface sediments of the A and B core indicate that after resuspension the oxidizing conditions at the sediment-water interface of both cores lead to the formation of a Fe-oxyhydroxide film acting as a chemical trap for the P which ditfuses into the water. The same indication comes from the after- resuspension phosphate fluxes detected for the A and B cores which were lower than the sediment-water interface fluxes of the incubated C core. The decrease of the P-Fe, P-red, and P-exc fractions in the surface sediments from the C core indicates reducing conditions at the sediment-water interlace which cause the dissolution of Fe oxyhydroxides. These in turn release the P bound to them.
, ,co
o
(b) (c}
~ p p ~ Q~IB ~ ~ P I ~mqm~ P IT N~mmc~
I I I
/// i i ,, =1= i J'!ll ill::
. . . . ; " i : , , , o
Figure 7. Total phosphorus concentrations (a) and percentages of (a) obtained with the G&B (b) and F&T (c) sequences. D 0.5 and D I indicate solid phase samples before the stirring and shaking experiment (number is
sample thic "kness). A,B, and C = stirring experiment samples, max = suspended solid sample at end of nlaximum stirring, end = surficial sediment sample at end of stirring, S= solid phase samples from shaking
experiment where first number is thickness and second number is shaking time.
During resuspension the ammonia concentrations (Fig. 5) increased slightly while in the shaking experiment the ammonia release (Fig. 6) was relevant in the thinnest slice. The lower ammonia increase in the cores A and B with respect to the shaken experiment was due to a partial nitrification of ammonia. This was confirmed by the nitrite+nitrate increase in the stirring and in the shaking experiments (Fig. 5 and 6). The ammonia

WATER-SEDIMENT EXCHANGE OF NUTRIENTS 551
release in the both experiments is caused by organic matter degradation which is stronger near the interface where the content of reactive organic matter is greater.
After resuspension in cores A and B, the ammonia concentration gradient was less than in the incubated C core. In fact, the nitrificaticn process was stronger for the A and B cores, because more oxidizing conditions were created by stirring the supernatant. Also in this situation the nitrification process is confirmed by the trends of nitrite+nitrate: their gradients are proportional to the degree of stirring (greater for A, medium for B and lower for C) and inverse to the ammonia gradient (greater for C, medium for B and lower A). The resuspension in the water column causes the release of nitrogen as ammonia which then tends to be transformed successively into nitrite and nitrate.
During resuspension tile dissolved silica concentration (Fig. 5) shows a slight increase which continues also during the final incubation step. Indeed, for the C core the dissolved silica gradient increased less than for the A and B cores.
In the shaking experiment samples (Fig. 6) the dissolved silica concentration increased with shaking time. The release of silica was caused by its desorption from the solid phase (Sholkovitz, 1973) and by the dissolution of fresh biogenic silica, the content of which was greater in the first millimeters of sediments. In fact, the released silica amount was lower for the thicker slices. The silica increase observed for the C core and, after the end of resuspension, also for the A and B cores was mainly caused by diffusive fluxes from pore waters.
During resuspension, the sulphate concentrations (Fig. 5) increased slightly and then decreased to steady values similar to those determined in the initial stirring step and for the C core. In the two thinner slices of the shaking experiment (Fig. 6) a slight sulphate release was noticed. This was caused by the dissolution of the unstable sulphides which were poorly crystallised and present in larger amounts near the interface (Canfield, 1989) because of their recent precipitation.
With respect to alkalinity and calculated total carbonate, it must be said that the stirring experiment was set up as an open system; therefore, the dissolved oxygen and CO2 reached an equilibrium with the atmosphere and it was impossible to observe the
processes occurring at the sediment-water interface. In fact, these two parameters had only minor variations. However, during resuspension a slight increase in alkalinity concentrations could be identified, which was attributable mainly to organic matter oxidation and ammonium nitrification. In the shaking experiment alkalinity concentrations decreased in all sc:mple slices, perhaps because the Fe-oxyhydroxide reduction processes prevailed over the oxidation of sulphides and ammonium (Berelson e t
al., 1987a). During resuspension iron concentrations (Fig. 5) were variable. The correlation
between sulphate and iron concentrations for the A and B cores was nearly perfect. This suggests that any sudden Fe concentration increase produced by the dissolution of particularly unstable Fe-sulphides was compensated by the successive re-precipitation of Fe as oxyhydroxides.
After resuspension the Fe concentration decreased in the A and B cores. In the incubated C core the Fe concentration had a rapid, initial increase because of the fast dissolution of the fresh Fe-oxyhydroxides formed immediately after the bottom water replacement. In the following days the dissolution of more crystalline Fe-oxyhydroxides and the diffusive fluxes from the sediment-water interface caused a weak increase in the Fe concentration.

552 F. SPAGNOLI AND M. C. BERGAM1NI
In the shaking experiment, Fe concentrations (Fig. 6) increased slightly in the two thinner slices. The reason for this is that the oxidation of newly formed, poorly cristallized sulphides (present in larger amounts in the first millimeters of sediment) dominated over the Fe-oxyhydroxide precipitation. On the contrary, this precipitation is prevalent at deeper levels because of the larger quantities of dissolved Fe and adsorption sites on partially dissolved Fe-oxyhydroxides.
The total Fe content in the solid phase (Fig. 8) indicates that considerable amounts of this element was present in both resuspended solids of A and B cores and the O. 1 cm slice samples because of Fe reprecipitation and recycling at the sediment-water interface. The results of sequential extractions do not help in understanding these Fe-related processes, since resuspension created Fe concentration variations in the solid phase which were lower than analytical error. Only the Fe-carb and Fe-oxid fractions have recognizable trends. The Fe-carb fraction decreased in the surficial sediments collected from the cores after the stirring experiment and in all shaked subsamples. This decrease indicates that Fe is desorbed from this mineral phase. The Fe-oxid fraction decreased in the 0.5 cm slice, whereas it was almost constant in the remaining slices; this confirms that the oxidation processes of sulphides and organic matter prevail at surficial levels. The Fe-res fractions and the Fe-red fractions from the thicker slices were higher and lower, respectively, than the corresponding fractions detected from the thinnest slice. The reason for this is the ageing process of Fe-oxyhydroxides which, with increasing depth and time, acquire a more stable'crystal form.
(.)
, s
~o
3o
, s
I o
s
o
~,,
(b)
~, | -
Ft
/
m | ~ 1 1 : , ~,
- i | |
Figure 8. Total iron concentrations (a) and percentages of (a) obtained with the F&T sequence (b). A,B,C, max, end and S ,are the same as indicated in Figure 7.
In general, Fe tended to precipitate as oxyhydroxides during resuspension in oxidizing environments. Such phenomena would reverse following Fe-sulphide oxidation and, to a lesser extent, Fe desorption from the carbonate traction. Fe precipitation was fast but

WATER-SEDIMENT EXCHANGE OF NUTRIENTS 553
limited, and this perhaps was caused by the complexation of Fe with organic and inorganic colloids which were released during resuspension and prevented Fe precipitation (Hunter and Liss, 1982). After resuspension (cores A and B), the oxidizing conditions at the water-sediment interface promoted the precipitation of Fe- oxyhydroxides. On the contrary, a reducing environment determined by stable hydrodynamic conditions (core C) promoted the dissolution of Fe-oxyhydroxides.
4.3. CALCULATED FLUXES
The fluxes at the sediment-water interface were calculated applying Fick's First Law
(Berner, 1980): Ji = --q)Di/~edl(--~-)z=o where Ji is the diffusive flux of the solute i
(mmol m -2 day-l), ~0 is the porosity, Dieted) is the bulk sediment diffusion coefficient of
the solute i (cm 2 s- 1) and ~ i ] ~ is the concentration gradient of the solute i at depth
z--0. The bulk sediment diffusion coefficient was calculated as proposed by Ullman and Aller (1982) for nearshore sediment when porosity is > 0.7. The free solution diffusion
coefficient of solute i ( D i ) (Wollast and Garrel, 1971: Li and Gregory, 1974) was
corrected for temperature and salinity with the function Di=D0(I+aT)(1-bS), where D O is
the free solution diffusion coefficient at temperature 0 ~ T is the temperature, S is the salinity, a and b are two interpolation constants. To calculate the concentration gradients at the sediment-water interface we applied an exponential function describing the pore water profile of dissolved silica and a quadratic function for phosphate, nitrate and Fe (Berelson et al., 1987a). Then we derived the appropriate function for depth z=0. For
dissolved silica we have: C = C ( 1 - - e - b Z ) + C 0 ( e - b Z ) , where C is the silica
concentration at depth z, C a is the silica concentration at the solid-liquid phase
equilibrium and b is a regression constant. The concentration gradient for depth z=0 is:
b( C - C0), where C O is the silica concentration at z=0. For phosphate, nitrate and Fe we
have: Ci= C0+alz+a2z where C i is the concentration of solute i, and a 1 and a 2 are two
regression constants. The concentration gradient for depth z=0 is a 2. If only one useful
sample is available near the water-sediment interface (such as for ammonia), we calculate a linear concentration gradient (Klump and Martens, 1981). The fluxes are then corrected for the benthic boundary layer effect by successive approximations. The concentration gradients were calculated using the in situ bottom water concentrations.
Table 1. Calculated and measured fluxes at the sediment-water interface (positive values indicate upward fluxes).
Fluxes phosphate (retool m -2 d- 1 )
Calculated 0.14 Measured 0.19 Measured after 0.05 resuspension
ammonia nitrate silica Fe
5.66 0.09 3.16 0.27 2.69 0.71 2.40 0.03 1.27 1.27 3.07 A -0.14
B -0.03

554 F. SPAGNOLI AND M. C. BERGAM1NI
The calculated phosphate fluxes are comparable to the measured ones. This means that diffusion is the major transport mechanism However, there is a flux component due to the degradation of organic matter near the sediment-water interface. This component can balance phosphate precipitation with Fe-oxyhydroxides. The phosphate fluxes measured after resuspension are smaller than the calculated ones by one order of magnitude, because of the precipitation of Fe-oxyhydroxides which act as sinks for phosphate.
The difference between calculated and measured ammonia fluxes is attributable to the nitrification which takes place in the supernatant where oxidizing conditions occurs In the A and B cores the nitrification process was stronger and fluxes were lower accordingly. The nitrification process is evidenced by the nitrate fluxes determined during incubation and after resuspension which are higher than the calculated nitrate fluxes. In low hydrodynamic conditions the ammonia produced in the sediment by organic matter degradation flows out of sediments mainly by molecular diffusion.
Silica fluxes support the prevalence of diffusive process. Calculated iron fluxes are higher than tile measured ones by one order of magnitude. This difference is attributed to a process which took place near the sediment-water interface but was not recorded in the core sampled, i.e. the precipitating Fe-oxyhydroxide induced sinking of the iron flowing out of sediments. This same process does not influence tile measured and calculated phosphate fluxes, because it was balanced by the production of organic matter degradation. The more oxic conditions following resuspension favour a considerable Fe- oxyhydroxide precipitation which determines the Fe negative flux.
5. Conclusions
During resuspension, phosphate release occurs mainly by desorption from solid surfaces and dissolution of authigenic apatite. If the resuspension event involves the oxic layer of sediments, the phosphate release is proportional to the resuspended matter. If tile resuspension event involves tile anoxic layer too, the phosphate release is more limited. Following resuspension, tile oxic environment favours the precipitation of Fe- oxyhydroxides at tile sediment-water interface, which act as sinks for phosphate. This process creates lower phosphate fluxes than those measured in low hydrodynamic conditions. The phosphate fluxes are mainly diffusive in low hydrodynamic conditions, although the degradation of organic matter at tile sediment-water interface gives some contribution.
During resuspension, ammonia is released because of the degradation of organic matter, but the more oxic conditions filvour the fast transformation of ammonia into nitrate. After resuspension, ammonia diffusive fluxes prevail, as in a low hydrodynamic environment, but the oxic conditions of waters cause a strong nitrification process.
During resuspension, silica is released mainly because of biogenic silica dissolution. In low hydrodynamic conditions tile silica fluxes are mainly diffusive. During resuspension, iron sulphides with low cristallinity dissolve and iron associated with the carbonate traction desorbs. Only a portion of the free iron precipitates as Fe-oxyhydroxides, because some iron becomes bound with organic colloids. After resuspension, the removal of Fe from the water colunm prevails because of the precipitation of Fe-oxyhydroxides at the

WATER-SEDIMENT EXCHANGE OF NUTRIENTS 555
sediment-water interface. In low hydrodynamic conditions low diffusive Fe fluxes prevail over the precipitation of Fe-oxyhydroxides at the sediment-water interface.
Acknowledgements
We wish to thank Dott.ssa Franca Frascari of the Istituto per la Geologia Marina-CNR for her technical, spiritual and financial support during this research; Prof. Antonio Brambati of the Trieste University for funding the grant which made this work possible; Dott. Stefano Miserocchi, Mr. Angelo Magagnoli, Mr. Maurizio Mengoli and Ms. Gabriella Rovatti of the CNR Institute of Marine Geology, as well as Dott.ssa Carla Ferrari of the U.O. Daphne of the Emilia Romagna Regional Administration for their assistance with the laboratory analyses and technical support. This is contribution n. 1070 of Istituto di Geologia Marina-CNR, Bologna, Italy.
References
Aller, RC.: 1980, Advances in Geophysics 22, 237-350. Artegiani, A., Bregant, D., Paschini, E., Pinardi. N., Raichic, F. and Russo. A., J.Physical Oceanogr., in press. Barbanti, A. and Sighinolfi, G.P.: 1988, Environ. Technol. Lett. 9, 127-134. Barbanti, A., Bergamini, M.C., Frascari, F., Miserocchi, S. and Rosso, G.: 1994, J. Environ. Qual. 23, 1093-
1102. Barbanti, A , Bergamini, M.C., Frascari. F,, Miserocchi, S., Ratta, M and Rosso, G.: 1995, Mar. Freshwater
Res. 46, 55-67. Bender. M., Jahnke, R., Weiss, R , Martin, W., Heggie. D.T., Orchardo, J. and Sowers, T,: 1989, Geochim.
Cosmochim. Acta 53, 685-697. Berelson. W.M., Hammond. DE. and Johnson, K.S.: 1987a, Geochim. Cosmochin~. Acta 51, 1345-1363, Berelson, W.M., Hammond. DE., Smith, K.L., Jahnke, RA.. Devol, A.H., Hinga, K.R, Rowe, G.T. and
Sayles, F.: 1987b, Mar. Technol. Soc. J. 21, 26-32. Berelson, WM. , Hammond, D.E., O'Neill, D., Zu, .M., Chin, C. and Zukin, J.: 199(I, Geochim. Cosmochim.
Acta 54, 3001-3012. Berelson, W.M., Hammond, D.E., McManus, J. and Kilgore, TE.: 1994. Global Biogeochem. Cycles 8, 219-
235. Bemer, R.A.: 1971, Priciples O/chemical sedimentolrJ~,,y, McGraw-Hill (eds.), New York. p. 240. Berner, RA.: 1980, Early diagenesis: a theoretical approach, Princeton Univ. Press (eds.), N.J., p. 241. Calvo, C., Donazzolo, R., Guidi, F. and Orio, A.: 1991, WaterRes. 25, 1295-1302. Canfield, D.E.: I989, Geochinl. C(~smochim. Acta 53, 619-632. Cline, J.D.: 1969, Limnol. Oceanogr. 14, 454-458. Cook, H.E.. Johnson, PD., Matti, C. and Zemmels. I.: 1975, Methods of sanlple preparation and X ray
di[J-raction data analysis. X ray mineralogy laboratot3', deep-sex drilling project, University of California, Riverside, lnit. Rep. DS[)P 28, 999-1007.
Crema, R., Castelli, A. and Prevedelli, D.: 1991, Mar. Pollut. Bull. 22, 503-508. Dal Cin, R.: 1983, Boll. Soc. Geol. 102, 9-56. Drake, E.D., Cacchione, DA., Frascari, F., Bortoluzzzi, G., Tate, G., Miserocchi, S., Thede, J. and Viall, R.:
1992. Rapp. Comm. hit. Met Medit. 33. Fanning, K.A.. Carder, KL. and Betzer, P R : 1982, Deep-Sea Res. 29, 953-996. Forstner. U. and Salomons, W.: 1980, Environ. Technol. Lett. 11,494-517. Franco, P., Jeftic, L., Malanotte Rizzoli, P,. Michelato, A. and Orlic. M.: 1982, Oceanologiea Acta 5, 379-389. Frignani, M. and Turci, C.: 1981, CNR. AQ/2/12 101. Frignani, M, and Langone, L.: 1991, Continental Shell Res. 11,525-542. Froelich, PN.: 1980.1,imnol. Oce~mo~,,r. 25, 242-248. Gieskes, J.M. and Rogers, W.C: 1973, J. Sedim. Petr. 43, 272-277.

556 F. SPAGNOLI A N D M. C. B E R G A M I N I
Giordani, P. and Hammond, D.E.: 1985, Technical Report No. 20, Consiglio Nazionale delle Ricerche, Istituto per la Geologia Marina (Bologna), p. 33.
Giordani, P., Hammond, D.E., Berelson, WM., Montanari, G.. Poletti, R., Milandri, A., Frignani, M., Langone, L., Ravaioli, M., Rovatti, G. and Rabbi, E.: 1992, Sci. Total Environ. Supplement , 251-275.
Golterman, H.L. and Booman. A.: 1988, Verh. hzt. Ver. Limnol. 23, 904-909. Guerzoni, S., Frignani, M., Giordani, P. and Frascari, F.: 1984, Environ. Geol. 6, 111-119, Hammond, D.E., Fuller, C., Harmon, D., Hartman, B., Korosec, M., Miller, L.G., Rea, R., Warren, S., Berelson, W. and Hager, S.W.: 1985, Hydrobiologia 129, 69-90. Hammond, D.E., McManus, J., Berelson, W., Kilgore. T. and Pope, R.. Deep-Sea Res., in press. Howarth, R.W.: 1978, Limnol. Oceanogr. 23, 1066-1069. Hunter, K.A. and Liss, P.S.: 1982. Limnol. Oceam~gr. 27, 322-335. Klump, V.J. and Martens, C.S.: 1981, Geochim. Cosmochim. Acta 45, 101-121. Krom, M, and Berner, R.A.: 1981, Geochim. Cosmochim. Acta 45, 207-216. Li, Y. and Gregory, S.: 1974, Geochim. Cosmochim. A cta 38, 703-714. Marchetti, R.: 1992, Sci. Total Environ. Supplement, 21-33. Nelson, B.W.: 1970, Deltaic sedimentation - modern and ancient, Morgan T. Ed., Soc. Ec. Paleon.
Sp. Pubbl. 15, 152-184. Orlich, M., Gacic, M. and La Violette, PE.: 1992, Oceanoh;gica Acta 15, 109-124. Oviatt, C.A., Hunt, C.D., Vargo, G.A. and Kopchynski, K.W.: 1981, J. Mar. Res. 34, 605-626. Petersen, W., Wallmann, K., Pinglin, Li, Schroeder, F. and Knauth, H.D.: 1995, Mar. Freshwater Res. 46, 19- 26. Ruttenberg, K .C and Berner, R.A.: 1993, Geochim. Cosmochim. Acta 57, 991 - 1007. Santschi, P., Hohener, P., Benoit, G. and Buchohz-ten Brink, M.: 1990, Mar. Chem. 30, 269-315. Sfriso, A., Donazzolo, R., Calvo, C. and Orio, A.A.: 1990, Environ. Technol. 12, 371-379. Sholkovitz. E.: 1973. Geochim. Cosmochim. Acta 37, 2043-2073. Spagnoli, F.: 1993, Diagenesi precoce e processi di scambio di sostanze nutrienti e metalli in tracce
Ira acqua e sedimento in condizioni di quiete e di risospensione di un 'area marina a sud del delta del P,, PhD Dissertation. Univ. Trieste, Italy.
Strickland, J.D.H and Parsons, T.R.: 1972, A practical Handbook of seawater anakysis, Fish. Res. Board Canada, Ottawa, p. 310.
Sundby, B., Anderson, LG., Hall, P.O.J., Iverfeldt, A., Rutgers Van der Loeff, M.M. and Westerlund, S.F.G.: 1986, Geochim, Cosmochim. Acta 50, 1281 - 1288.
Tessier, A., Campbell, P.C.G, and Bisson, M.: 1979, Anal. Chem. 51,844-850. Tsai, C.H. and Lick, W.: 1986, J. Great l~lkes Res. 12, 314-321. U[Iman, W.J. and Aller, R.C.: 1982, Limn,I. Oceanogr. 27, 552-556. Westerlund, S.F.G., Anderson, L.G., Hall, P.O.J., lverfeldt, A., Rutgers Van der Loft, M.M. and Sundby, B.:
1986, Geochim. Cosmochim Acta 50, 1289-1296. Westrich, J.T. and Berner, A.: 1984, Limnol. Oceanogr. 29, 236-249. Wollast, R. and Garrels, R.: 1971, Nature l~mdon Phys. Sci. 229, 94.
Copyright © 2022 FDOKUMEN



















