
Variabilidade genética de isolados do Grapevine leafroll-associated virus-3 originários de...
88
1 RESUMO A videira é afetada por diversas viroses e o enrolamento da folha se destaca pela importância econômica. Essa doença é causada por um complexo formado por até oito vírus (GLRaV-1 ao 8). O objetivo desse trabalho foi verificar a variabilidade da extremidade 3´ do gene da polimerase e do gene do capsídeo de três isolados de Grapevine leafroll associated virus-3 (GLRaV – 3), provenientes do Submédio do Vale do Rio São Francisco (Petrolina, PE) e relatar a ocorrência do mesmo. O RNA viral foi extraído de amostras infectadas, reagentes em teste de ELISA para GLRaV-3, e fragmentos de 340 pb e 942 pb foram amplificados por RT-PCR, utilizando-se oligonucleotídeos para a região do gene da polimerase viral, compreendida entre os nucleotídeos 8267 a 8606 e para o gene do capsídeo, compreendido entre os nucleotídeos 13269 a 14210. As seis seqüências geradas foram alinhadas com as seqüências disponíveis no GenBank. Na comparação da região 3´da polimerase, observou-se que os três isolados da região do São Francisco apresentaram entre 94% e 98% de similaridade com o isolado norte-americano (NC_004667). A principal variação observada foi a troca de um aminoácido da posição 2766 de fenilalanina para tirosina. Na comparação do gene do capsídeo, observou-se similaridade de 99% entre os nucleotídeos, mas com seis substituições de aminoácidos não-conservativas. Esses dados preliminares indicam a existência de variabilidade entre isolados de GLRaVs de diferentes regiões geográficas.
Transcript of Variabilidade genética de isolados do Grapevine leafroll-associated virus-3 originários de...

1
RESUMO
A videira é afetada por diversas viroses e o enrolamento da folha se
destaca pela importância econômica. Essa doença é causada por um complexo
formado por até oito vírus (GLRaV-1 ao 8). O objetivo desse trabalho foi verificar a
variabilidade da extremidade 3´ do gene da polimerase e do gene do capsídeo de
três isolados de Grapevine leafroll associated virus-3 (GLRaV – 3), provenientes
do Submédio do Vale do Rio São Francisco (Petrolina, PE) e relatar a ocorrência
do mesmo. O RNA viral foi extraído de amostras infectadas, reagentes em teste
de ELISA para GLRaV-3, e fragmentos de 340 pb e 942 pb foram amplificados por
RT-PCR, utilizando-se oligonucleotídeos para a região do gene da polimerase
viral, compreendida entre os nucleotídeos 8267 a 8606 e para o gene do
capsídeo, compreendido entre os nucleotídeos 13269 a 14210. As seis
seqüências geradas foram alinhadas com as seqüências disponíveis no GenBank.
Na comparação da região 3´da polimerase, observou-se que os três isolados da
região do São Francisco apresentaram entre 94% e 98% de similaridade com o
isolado norte-americano (NC_004667). A principal variação observada foi a troca
de um aminoácido da posição 2766 de fenilalanina para tirosina. Na comparação
do gene do capsídeo, observou-se similaridade de 99% entre os nucleotídeos,
mas com seis substituições de aminoácidos não-conservativas. Esses dados
preliminares indicam a existência de variabilidade entre isolados de GLRaVs de
diferentes regiões geográficas.

2
ABSTRACT
Grapevines are affected by many viral diseases and leafroll has great
economical importance. A complex of eight viruses (Grapevine leafroll-associated
virus-1 to 8) is associated to this disease. The objective of this study was to
analyze the variability of the 3´ terminal region of the polymerase gene and the
capsid protein gene of three isolates of GLRaV–3 (Grapevine leafrol- associated
virus-3), from Submédio do Vale do Rio São Francisco (Petrolina-PE) and its
occurrence. The viral RNA was extracted from infected ELISA reactive plants for
GLRaV-3 and two fragments, one of 340 bp for the polymerase gene and one of
942 bp for the capsid gene, were amplified, by RT-PCR, using primers that
recognize the portion of the polymerase gene between nucleotides 8267 and 8606
and the capsid gene between nucleotides 13269 and 14210. The six sequences
obtained were aligned and compared to other sequences of GLRaV–3, available at
GenBank. The analysis of the polymerase gene showed that the three isolates
from Vale do Rio São Francisco had between 94% to 98% of nucleotide similarity
to the North American isolate, NC_004667. The main variation found was an amino
acid change at position 2766 from phenylalanine to tyrosine. The analysis of the
capsid gene showed 99% of nucleotide similarity with the same isolate, but with six
non-conservative aminoacid changes. These preliminary data indicate the
existence of variation between different GLRaV–3 isolates from distinct geographic
regions.

3
ÍNDICE DE TABELAS
Tabela 1. Espécies do gênero Closterovirus (adaptado de Martelli et al., 2002) .. 37
Tabela 2. Espécies do gênero Crinivirus (adaptado de Martelli et al., 2002). ....... 39
Tabela 3. Espécies do gênero Ampelovirus (adaptado de Martelli et al., 2002). .. 42
Tabela 4. Seqüências dos oligonucleotídeos (primers) utilizados para amplificação
do gene da RNA polimerase dependente de RNA e da Proteína do Capsídeo.
....................................................................................................................... 50
Tabela 5. Absorbância obtida em leitora de ELISA, em comprimento de onda de
405 nm, para amostras consideradas reagentes em ELISA e originárias do
Vale do São Francisco. .................................................................................. 58

4
ÍNDICE DE FIGURAS
Figura 1. Vitis Sezannensis e Vitis olrikii, principais precursoras das videiras
cultivadas (fonte: Souza, 1969). ..................................................................... 13
Figura 2. Região mais provável como centro de origem da viticultura (fonte:
Souza, 1969). ................................................................................................. 13
Figura 3. Rota da introdução de Vitis vinifera no Brasil, desde o ponto de origem
até o primeiro plantio em 1532. 1: Ásia Menor; 2: Portugal; 3: Ilha da Madeira;
4: Capitania de São Vicente (fonte: Souza, 1969). ........................................ 14
Figura 4. Sintomas do enrolamento da folha em variedades tintas. A folha
localizada a esquerda apresenta sintomas da doença descrita como
enrolamento, a saber: enrolamento dos bordos para baixo e avermelhamento.
A folha da direita foi extraída de uma planta sadia (fonte:
http://winegrapes.tamu.edu/grow/diseases/ leafroll.shtml). ............................ 31
Figura 5. Sintomas do enrolamento das folhas nos cachos. Os frutos localizados a
direita foram extraídos de uma planta contaminada pelo vírus, apresentando
pouco desenvolvimento quando comparada com frutos extraídos de uma
planta sadia (à esquerda) (fonte: Pearson & Goheen, 1998). ........................ 32
Figura 6. Partículas de Citrus tristeza virus (CTV) decoradas com antisoro
homólogo (fonte: van Regenmortel et al., 2000). ........................................... 34
Figura 7. Estrutura genômica do BYV, mostrando posições relativas das ORFs e
seus produtos: PRO - domínio de protease; MTR – domínio de
metiltransferase; HEL – domínio de helicase; RdRp - RNA polimerase
dependente de RNA; HSP70 - proteína análoga a HSP70 descrita em células
animais; CPd - proteína do capsídeo divergente; CP - proteína do capsídeo.
As funções das demais ORFs ainda não foram descritas (adaptado de Martelli
et al., 2002). ................................................................................................... 37
Figura 8. Estrutura genômica de LIYV, mostrando a posição relativa das ORFs e
seus produtos: PRO – domínio de protease; MTR – domínio de
metiltransferase; HEL – domínio de helicase; RdRp - RNA polimerase
dependente de RNA; HSP70 - proteína análoga à HSP70 descrita em células
animais; CP - proteína do capsídeo; CPd - proteína do capsídeo divergente.

5
As funções das demais ORFs ainda não foram descritas (adaptado de Martelli
et al., 2002). ................................................................................................... 38
Figura 9. Micrografias apresentando partículas de GLRaV-1 e GLRaV-3 (a, c), e
as mesmas decoradas por antisoro homólogo (b, d). Barras = 100 nm (fonte:
Credi & Giunchedi, 1996). .............................................................................. 41
Figura 10. Estrutura genômica do GLRaV-3, mostrando a posição relativa das
ORFs e seus produtos: PRO – domínio de protease; MTR – domínio de
metiltransferase; HEL – domínio de helicase; RdRp - RNA polimerase
dependente de RNA; HSP70 - proteína análoga a HSP70 descrita em células
animais; CP - proteína do capsídeo; CPd - proteína do capsídeo divergente
As funções das demais ORFs ainda não foram descritas (adaptado de Martelli
et al., 2002). ................................................................................................... 42
Figura 11. Sintomas em plantas reagentes no teste DAS-ELISA em relação à
infecção por GLRaV-3 em casas de vegetação da Estação Experimental da
Biologia - UnB. Observa-se avermelhamento entre as nervuras e enrolamento
dos bordos para baixo. ................................................................................... 57
Figura 12. Gel de agarose a 1,5%. Os poços 3 e 4 mostram a amplificação do
gene do capsídeo viral com 942 pb, o poço 2 mostra o controle negativo, o
poço 1 não possui amostras. M: marcador 100 pb ladder. ............................ 59
Figura 13. Gel de agarose a 1,5%. Os poços de 2 a 5 mostram a amplificação de
parte do gene da polimerase viral com 340 pb, o poço 1 mostra o controle
negativo. M: marcador 100 pb ladder. ............................................................ 60
Figura 14. PCR das minipreparações plasmidiais com o inserto da RNA
polimerase. Poços 1, 3, 5, 7, 9, 11, 13, 15, 17 e 19 mostram o resultado dos
PCRs; M = marcador 100 pb ladder; Primers H229 e C547 (Ling et al., 1998).
....................................................................................................................... 61
Figura 15 . PCR das minipreparações dos plasmídeos com o inserto do capsídeo.
Poços 1 e 5 contém os resultados do PCR. Poços 2 e 4 são vazios. M =
marcador 100 pb ladder. Primers LR3-9445C e LR3-8504V.......................... 61
Figura 16. Alinhamento das seqüências de nucleotídeos da região 3´ da
polimerase de isolados de GLRaV-3 do Vale do São Francisco (Pet-1, Pet-2 e

6
Pet-3) com isolados disponíveis no Genbank (NY1: isolado descrito por Ling
et al., 1998; AF438411: isolado descrito por Fajardo et al., 2002). Os pontos
mostram equivalência de nucleotídeos na referida posição. Nucleotídeos em
negrito representam o códon de terminação do gene da polimerase. O
asterisco mostra a posição onde um número igual de seqüências apontou a
presença tanto de A quanto de G. ................................................................. 63
Figura 17. Alinhamento das seqüências de aminoácidos deduzidas da região 3´
do gene da polimerase de isolados de GLRaV-3. Nucleotídeos em negrito
representam trocas de bases com relação a dois isolados estudados.
Mudanças na seqüência de aminoácidos dos isolados Pet-1, Pet-2 e Pet-3
comparados com NY1 e AF438411 são marcados com uma barra cinza. A
substituição ocorrida somente em um isolado foi marcada com uma barra
escura. ........................................................................................................... 64
Figura 18. Alinhamento das seqüências de nucleotídeos da proteína do capsídeo
(CP) de isolados de GLRaV-3 do Vale do São Francisco (Pet-1, Pet-2 e Pet-3)
com o único isolado disponível no GenBank (NY1: isolado descrito por Ling et
al., 1998). Os pontos mostram equivalência de nucleotídeos na referida
posição. .......................................................................................................... 67
Figura 19. Alinhamento das seqüências de aminoácidos deduzidas do gene do
capsídeo de isolados de GLRaV-3. Nucleotídeos em negrito representam
trocas de bases em relação a um isolado estudado. Mudanças na seqüência
de aminoácidos dos isolados Pet-1, Pet-2 e Pet-3 comparados com NY1 são
marcados com uma barra cinza. .................................................................... 70
Figura 20. Alinhamento da seqüência de nucleotídeos e aminoácidos mostrando
as variações nos isolados Pet-1, Pet-2 e Pet-3 com relação às seqüências da
polimerase dos isolados NY1 e AF438411. Posições dos nucleotídeos são
indicadas na parte de cima da figura. O número se refere ao primeiro
nucleotídeo do genoma de GLRaV-3. A seqüência de referência é indicada
como Ref. Cada linha indica, da esquerda para a direita, a identificação do
isolado e o alinhamento da seqüência de nucleotídeos comparada com a

7
referência. A substituição de aminoácido e sua posição são indicados abaixo
da linha, em comparação com o aminoácido original. ................................... 71
Figura 21. Alinhamento da seqüência de nucleotídeos e aminoácidos mostrando
as variações nos isolados Pet-1, Pet-2 e Pet-3 com relação à seqüência do
capsídeo do isolado NY1. Posições dos nucleotídeos são indicadas na parte
de cima da figura. O número se refere ao primeiro nucleotídeo do genoma de
GLRaV-3. A seqüência de referência é indicada como Ref. Cada linha indica,
da esquerda para a direita, a identificação do isolado e o alinhamento da
seqüência de nucleotídeos comparada com a referência. As substituições de
aminoácidos e suas posições são indicadas abaixo da linha, em comparação
com o aminoácido original. ............................................................................. 72
Figura 22. Análise fiologenética, realizada pelo método de máxima
verossimilhança das seqüências nucleotídicas da polimerase viral e do
capsídeo de Pet-1, Pet-2 e Pet-3 comparadas à seqüência de referência NY1
retirada do GenBank. Os números mostrados nos pontos de ramificação
foram obtidos pelo teste dos ramos internos e representam valores de
consenso para 1000 réplicas. ........................................................................ 73

8
LISTA DE SIGLAS E ABREVIATURAS
bp – base pairs
BYSV – Beet yellow stunt virus
BYV – Beet yellows virus
cDNA – DNA complementar
CP – proteína do capsídeo
CPd – análogo da proteína do capsídeo
CTV – Citrus tristeza virus
DAS-ELISA – Double antibody sandwich enzyme linked imunnosorbent assay
ELISA – Enzyme linked Imunnosorbent assay
g – gramas
GFkV – Grapevine fleck virus
GFLV – Grapevine fanleaf virus
GLRaV – Grapevine leafroll-associated virus
GVA – Grapevine virus A
GVB – Grapevine virus B
GVC – Grapevine virus C
GVD – Grapevine virus D
HSP70 – proteína de choque térmico 70
kb – mil pares de bases
kDa – kilodalton
kg/cm 2 – quilogramas por centímetro quadrado
LChV-2 – Little cherry virus-2
LIYV – Lettuce infectious yellows virus
M – molar
mg/ml – miligramas por mililitro
ml – mililitros
mm – milimetros
mM – milimolar
nm – nanômetros

9
ORF – região codificadora
pb – pares de bases
PCR – Polymerase chain reaction
PE – Pernambuco
Ref. - referência
rpm – rotações por minuto
RT-PCR – Reverse transcription – Polymerase chain reaction
µg/ml – microgramas por mililitro
µl - microlitros

10
1. REVISÃO BIBLIOGRÁFICA
1.1. Introdução
A videira (Vitis spp., família Vitaceae) é uma das frutíferas mais cultivadas
no mundo. A área utilizada para plantio da videira em 1994 já era de
aproximadamente 10 milhões de hectares (Pearson & Goheen, 1998).
O cultivo da videira é afetado por inúmeras viroses que podem diminuir a
produtividade, comprometer a qualidade das uvas e, em algumas cultivares,
provocar o definhamento da planta. No Brasil, o enrolamento da folha da videira é
uma das viroses de destaque pela sua importância econômica crescente, já que
pode chegar a infectar até 98% de um cultivo (Kuhn & Nickel, 1998; Tavares et
al.,2000; Fajardo et al., 2002).
O estudo do complexo viral responsável pelo enrolamento da folha da
videira é dificultado por algumas características dos membros da família
Closteroviridae. Dentre estas está a restrição do vírus ao tecido floemático,
resultando em baixa concentração de partículas virais nos processos de
purificação. Dois fatores que também contribuem para a grande dificuldade no
estudo molecular destes vírus são: o grande tamanho de seu RNA genômico e a
difícil transmissão mecânica (Karasev, 2000), possível somente com o Grapevine
leafroll-associated vírus-2 e, mesmo assim, restrita a Nicotiana sp. (Fajardo et al.,
2003).
O ensaio imunoenzimático do tipo ELISA é amplamente utilizado para a
detecção dos GLRaVs, principalmente do GLRaV-3 (Hu et al., 1990; Zimmermann

11
et al., 1990; Ling et al., 2001) e várias técnicas relacionadas à detecção molecular
destes vírus já foram desenvolvidas (Rowhani et al., 1993; Rowhani et al., 1995;
MacKenzie et al., 1997; Dovas & Katis, 2003). Diversos testes mostraram que a
sensibilidade do PCR para a detecção desses vírus é superior ao ELISA
(Chevalier et al., 1995; Acheche et al., 1999; Ling et al., 2001; Dovas & Katis,
2003).
O Grapevine leafroll-associated vírus-3 é a espécie de vírus do enrolamento
mais estudada e o genoma de um isolado de Nova York (acesso NC_004667) já
foi completamente seqüenciado (Ling et al., 1998). Entretanto, substituições de
nucleotídeos e aminoácidos em regiões específicas das seqüências podem indicar
a existência de variantes virais, cuja infectividade, patogenicidade e etc. pode ser
diferente daquele caracterizado anteriormente.
1.2. Histórico
Os prováveis centros de origem da videira são a Groenlândia e outras
regiões hiperbóreas. Nesses locais, foram encontrados os fósseis mais antigos de
plantas ancestrais das atuais espécies cultivadas. Essas espécies ancestrais são
de ocorrência anterior ao surgimento do homem, no período quaternário. A partir
desse centro, as videiras ganharam as terras meridionais, seguindo duas rotas
principais: uma américo-asiática e outra euro-asiática. Deste último percurso se
originou Vitis sezannensis, precursora de Vitis vinifera (Figura 1). Antes da
glaciação ocorrida neste período, existiam dois subgêneros diferenciados: Euvitis
e Muscadinia, que se localizavam desde a Groenlândia, Islândia e Alasca até as
orlas do Mediterrâneo. Após este período, as espécies que suportaram o inverno

12
extremamente prolongado sobreviveram em centros menos atingidos pelo gelo:
um americano, um europeu e um asiático-ocidental. No centro americano,
localizado na costa leste dos Estados Unidos e México, expandindo-se até Costa
Rica, originaram-se as espécies americanas: Vitis labrusca, V. vulpina, V.
rupestris, V. aestivalis, V. rotundifolia, V. tiliaefolia, V. smalliana, V. lincecumii, V.
cordifolia, V. berlandieri, etc. No centro europeu, que abrangia áreas
mediterrâneas francesas e italianas, seguindo até a península balcânica,
sobreviveu apenas uma espécie: Vitis vinifera silvestris. No centro asiático-
ocidental, localizado ao sul do Mar Cáspio, entre o Ararat, o Cáucaso e o Taurus,
populações de Vitis vinifera caucasica se estabeleceram. Após o estabelecimento
de espécies consideradas produtivas, a cultura da videira finalmente começa a
surgir, provavelmente, no final da era do bronze, quando o homem começa a se
fixar, abandonando o estilo de vida nômade. A origem da viticultura se deu
provavelmente no Oriente Médio, mais precisamente entre a Armênia e a Pérsia,
região delimitada pelo mar Negro, Cáspio e Mediterrâneo (Figura 2). Os primeiros
plantios foram de Vitis vinifera caucasica originária do Oeste da Ásia, por produzir
bagas de melhor qualidade do que as produzidas por V. vinifera silvestris.
Achados arqueológicos encontrados na região indicam a ocorrência de viticultura
há cerca de 3.500 anos A.C. (Souza, 1969).

13
Figura 1. Vitis Sezannensis e Vitis olrikii, principais precursoras das videiras cultivadas (fonte: Souza, 1969).
Figura 2. Região mais provável como centro de origem da viticultura (fonte: Souza, 1969).
Do ponto de origem ao Leste do mar Mediterrâneo, o cultivo se espalhou
por toda a Ásia Menor e para a Trácia e Península Balcânica. Em direção ao sul,
se expandiu para Síria e Egito. Foi introduzido na Grécia e a partir daí,

14
aproximadamente 20 séculos a.C., foi levado a Roma, de onde se disseminou pela
Europa, juntamente com as conquistas do Império Romano que fixaram seus
vinhedos no Vale do Ródano, na Ermitage, Borgonha e Champanha, indo até a
Suíça, Alemanha, Grã-Bretanha, Espanha e Portugal. Variedades mais resistentes
ao frio tornaram possível o transporte e o estabelecimento de cultivos mais ao
norte do continente europeu. Aproximadamente no século I a.C. se deu o
estabelecimento de plantios em terras portuguesas. A descoberta da Ilha da
Madeira em 1418-1420 e a introdução da viticultura nessa nova região prepararam
o transporte e a introdução da videira em terras que seriam brevemente
descobertas (Figura 3) (Souza, 1969).
Figura 3. Rota da introdução de Vitis vinifera no Brasil, desde o ponto de origem até o primeiro plantio em 1532. 1: Ásia Menor; 2: Portugal; 3: Ilha da Madeira; 4: Capitania de São Vicente (fonte: Souza, 1969).

15
No Brasil, a introdução da viticultura ocorreu a partir da expedição do
Almirante Martim Afonso de Souza, que veio para o novo continente 32 anos após
o descobrimento, efetuar o estabelecimento das capitanias que iniciariam a
formação do Brasil–Colônia. Na primeira capitania formada, a de São Vicente,
atual Estado de São Paulo, Brás Cubas estabeleceu o primeiro vinhedo,
caracterizando então essa região como o berço da viticultura brasileira. Do final do
século XVII até a primeira metade do século XIX, a viticultura nacional foi colocada
de lado, graças à exploração do ouro, do cultivo da cana e do café. Com a
introdução de variedades americanas em São Paulo, entre 1830-1840, a produção
vitivinicultora se inicia e se fortalece com a chegada dos imigrantes italianos a
partir de 1888. Desde então, a introdução de novas variedades e híbridos resultou
na distribuição dos plantios de uva desde a região Sul até o extremo norte da
região Nordeste (Souza, 1969; Leão & Possídio, 2000).
1.3. Situação Atual da Viticultura no Brasil e no S emi-árido
O cultivo da videira (Vitis spp.) é responsável por boa parte do comércio de
frutíferas do Brasil, tanto para uvas de mesa quanto para produção de vinho. Em
2001, a produção foi estimada em 998.857 toneladas em uma área plantada de
61.382 hectares. Já em 2002, esse cultivo foi responsável por uma área plantada
de aproximadamente 65.381 hectares, resultando em uma produção de 1.120.570
toneladas, com um aumento de mais de 120.000 toneladas (FAO, 2003).
Os estados que se destacam na produção de uvas no Brasil são: Rio
Grande do Sul, Santa Catarina, algumas regiões de São Paulo, Bahia e
Pernambuco. No Rio Grande do Sul, os municípios de Flores da Cunha, Bento

16
Gonçalves, Garibaldi, Santana do Livramento e Caxias do Sul são os grandes
produtores de vinho. Outras áreas se dedicam a produção de uvas finas de mesa
tais como, Maringá e Marialva, no Norte do Paraná, produzindo variedades Itália e
Rubi. Em São Paulo, destacam-se os municípios de Jales, São Miguel do Arcanjo,
Vinhedo e Porto Feliz. O município de Pirapora, no Norte de Minas Gerais, no Vale
do São Francisco, também começa a surgir como importante pólo produtor de
uvas finas de mesa (Robbs & Neto, 1999).
A Região Sul é responsável pela maior produção de uvas no Brasil, com
uma área plantada de aproximadamente 46.600 hectares e uma produção em
torno de 710.392 toneladas. O Rio Grande do Sul é maior produtor, com uma
colheita no ano de 2002 de 570.181 toneladas (IBGE, 2003). Entretanto, a
produção dessa região está voltada principalmente para a produção de vinho. A
região do Submédio do Vale do São Francisco é considerada a principal produtora
e exportadora de uvas de mesa do Brasil, podendo ser citado como exemplo o
ano de 1996, quando foi responsável por 27% da produção nacional. As condições
de clima e solo, vinculadas a um manejo eficiente proporcionam 2,5 safras ao ano,
o que é considerado acima da média em outras regiões produtoras (Silva &
Correia, 2000). As características que favorecem a produção são: o baixo índice
de precipitação; alta luminosidade e alta temperatura. Essas características
facilitam o controle de doenças, além do controle da irrigação e tratos culturais
adequados que proporcionam a diminuição da incidência principalmente de míldio
e oídio. Por outro lado, tais áreas estão sujeitas à degradação ambiental. Sem o
manejo adequado da adubação mineral e irrigação, pode ocorrer a salinização do
solo, que resulta em queda de desempenho da cultura e até abandono de áreas

17
de cultivo. O uso de porta-enxertos resistentes à salinidade amenizam estes
problemas (Viana et al., 2001).
A importância dessa cultura na região semi-árida é cada vez maior,
considerando o grande aumento da área plantada e também a sua importância
social, pois é uma cultura que depende de um potencial de mão-de-obra elevado
para sua manutenção. Foram verificados casos onde 6,4 empregos anuais por
hectare foram criados, sendo que a área cultivada na região em 1996 era de 4.800
hectares e que para cada 100 empregos diretos correspondem entre 40 e 55
empregos indiretos (Silva & Correia, 2000). No período entre 1991-1998 houve um
aumento de 303% na área colhida, enquanto que nas demais regiões produtoras
verificou-se uma diminuição de aproximadamente 9,1% (Prognóstico Agrícola,
1998).
1.4. Doenças que Afetam a Videira
A videira passa por vários problemas fitossanitários, com uma sensibilidade
explorada por uma ampla gama de patógenos. Dentre alguns de importância
econômica destacam-se o cancro bacteriano causado por Xantomonas campestris
pv. viticola, o Mal de Pierce causado por Xylella fastidiosa, várias doenças
fúngicas, dentre elas a antracnose causada pelo fungo Elsinoe ampelina, o míldio
causado por Plasmopara viticola e a ferrugem recentemente identificada
(Tessmann et al., 2004). Também ocorrem prejuízos desencadeados por
nematóides, principalmente os do gênero Meloidogyne. Além destes patógenos, a
videira é afetada por cerca de 50 viroses, que resultam em baixa na produtividade
e comprometem a qualidade das uvas produzidas, podendo levar à perda do

18
plantio se atitudes corretas e voltadas para um melhor conhecimento destas
viroses não forem adotadas (Pearson & Goheen, 1998; Trindade, 2002 ;Fajardo et
al., 2003).
A seguir passaremos a relatar as principais doenças em videiras.
1.4.1. Doenças bacterianas
- Cancro Bacteriano
Detectado no início de 1998, o cancro-da-videira, causado por
Xanthomonas campestris pv. viticola Nayudu é a primeira bacteriose com
incidência expressiva e causadora de danos econômicos em videira. Ataca
principalmente variedades Red Globe e sem sementes, sendo Thompson
Seedless a mais afetada com danos de 10% a 100%. Foi constatada nos Estados
de Pernambuco, Bahia e Piauí, em mudas originárias da região de ocorrência da
doença (Trindade, 2002; Lima, 2003).
Os sintomas nas folhas são caracterizados por manchas angulares escuras
com 1 a 2 mm de diâmetro, circundadas ou não por um halo amarelado,
distribuídas nas folhas entre as nervuras. Em estágios avançados de infecção, as
folhas tornam-se amareladas e caem. Em ramos verdes e maduros há o
surgimento de cancros que gradualmente alargam-se, expondo os tecidos
internos. Na inflorescência ocorre necrose, surgindo a partir da extremidade em
direção à base. A bactéria é disseminada por meio de material propagativo de
copa e de porta-enxertos infectados, caracterizando-se como um dos principais
problemas fitossanitários da cultura da videira em áreas irrigadas no Submédio do
Vale do São Francisco (Lima & Moreira, 2002; Trindade, 2002).

19
-Galhas de Agrobacterium vitis (Agrobacterium tumefasciens biovar 3)
Foi descoberta na França, em 1853, e é problema principalmente em
variedades de V. vinifera em clima frio, pois as plantas ficam propensas a
ferimentos a baixas temperaturas, que podem servir como porta de entrada para a
bactéria. Foi relatada em 1889 nos Estados Unidos e é um sério problema para os
plantios na Califórnia (Burr, 1998). Mesmo não possuindo expressão na cultura da
videira no Brasil, a bactéria já foi detectada afetando parreirais em Minas Gerais,
Rio Grande do Norte, São Paulo e no Submédio do Vale do São Francisco (Lima
& Moreira, 2002).
- Mal de Pierce
Esta doença é muito importante nos Estados Unidos, onde ocorre desde
1892, tendo sido detectada décadas mais tarde no México, Costa Rica e
Venezuela, possivelmente ocorrendo na maioria das regiões da América Central
(Goheen & Hopkins, 1998). É fator limitante da produção de Vitis labrusca e V.
vinifera no Estado da Califórnia. A bactéria Xylella fastidiosa, responsável pela
doença, ainda não foi detectada infectando videiras no Brasil, mas é de
importância quarentenária (Lima, 2003).
1.4.2. Doenças Fúngicas
- Ferrugem da Videira
Causada pelo fungo Phakopsora euvitis Ono, a ferrugem foi encontrada
pela primeira vez em 2001 em parreirais do sul do país, mais especificamente na
região Norte do Paraná, se espalhando rapidamente e chegando, em 2003, aos
plantios de São Paulo (Tessmann et al., 2003).

20
São observadas pústulas de tamanho pequeno e coloração amarela na face
inferior das folhas, que podem cobrir grande extensão do limbo. Na face superior,
são verificadas áreas necrosadas opostas às posições das pústulas. As folhas
severamente atacadas amarelam e secam, causando desfolha precoce das
plantas. Os urediniósporos são o inóculo primário e são disseminados
principalmente pelo vento. Entre as safras, o patógeno sobrevive em material
verde das plantas, que é constante ao longo do ano em várias regiões produtoras.
Medidas fitossanitárias e um monitoramento constante foram implantados para se
evitar que esse fungo chegue até os vinhedos do Submédio do Vale do São
Francisco, causando danos que em conjunto com os causados pelo Cancro
Bacteriano podem comprometer seriamente a produção dessa região (Tessmann
et al., 2003).
- Podridão Seca
Causada pelo fungo Lasiodiplodia theobromae (sin. Botryodiplodia
theobromae Pat.), é um dos maiores problemas fitossanitários da região. O
primeiro relato da doença foi feito em 1991 e por ser relativamente recente, é de
difícil detecção por parte dos produtores. Em 2001, causou declínio generalizado,
chegando a causar comprometimento de cerca de 50 mil plantas com 5 anos de
idade em um plantio da região (Tavares et al., 2000; Tavares & Cruz, 2002).
- Míldio
É considerada a principal doença da videira no Brasil. Foi introduzida em
São Paulo quando se iniciou o cultivo de videiras americanas. É causada por
Plasmopara viticola (Berk & Curtis) Berl & Toni (Oomycota, Chromista) e é
considerada como a primeira doença a ser controlada por um fungicida. Causa

21
destruição parcial ou total dos frutos, podendo também produzir efeitos negativos
sobre a produção futura, graças à desfolha que causa em plantas infectadas.
Cultivares européias são muito mais sensíveis do que as americanas, explicando
a falha ao se tentar introduzir estas variedades em solo americano (Sônego et al.,
2003).
- Oídio
Em 1854 o oídio quase comprometeu por completo a produção vitícola da
França. No Brasil, foi introduzido em 1888, através de cultivares americanas.
Ocorre em todas as regiões produtoras do país, com efeitos mais acentuados na
região do Submédio do Vale do São Francisco graças ao clima favorável. As
manchas nos frutos causadas por Uncinula necator (Schw.) Burr são irreversíveis,
tornando-os impróprios para a comercialização. Em uvas para vinho, aumenta a
acidez do mosto, que mostra odor de mofo e problemas na fermentação. Causa
perdas na região Nordeste, no noroeste de São Paulo e nos países vitícolas da
Europa (Tavares & Cruz, 2002; Sônego et al., 2003).
- Podridão-cinzenta-da-uva
Causada por Botrytis cinérea (De Bary) Whetzel, ocorre em todos os países
vitícolas do mundo. Reduz qualitativa e quantitativamente a produção, sendo
considerada a mais importante das podridões de cacho. Perdas significativas
podem ocorrer em cultivares viníferas, principalmente as de cacho compacto. Em
locais de baixa umidade, ocorre o fenômeno da podridão-nobre. Esta podridão
ocorre no final do ciclo de maturação da uva e proporciona grande concentração
de açúcares nas uvas, o que é apreciado na produção de vinhos doces altamente
aromáticos (Sônego et al. 2003). No Submédio essa doença só ocorre em

22
pomares mais densos, com pouca aeração e muito sombreados. O apodrecimento
pode evoluir, causando perda generalizada (Tavares et al., 2000).
- Outras doenças fúngicas
Também ocorrem a verrugose causada por Elsinoe ampelina (De Bary)
Shear, escoriose causada por Phomopsis viticola Sacc., fusariose causada por
Fusarium oxysporum f. sp. herbemontis Tocchetto, podridão-de-armillaria causada
por Armillaria mellea (Vahl.) Quetet, podridão-ácida causada por leveduras,
roseliniose causada por Rosellinia necatrix Prill , podridão-amarga causada por
Melanconium fuligineum (Scrib & Viala) Cav , podridão-da-uva-madura causada
por Glomerella cingulata (Ston.) Spauld & Schrenk, mancha-da-folha causada por
Pseudocercospora vitis (Lév.) Speg e, mais recentemente a podridão-de-raízes
causada por Cylindrocarpon destructans (Zinnsm.) Scholten (Sônego et al., 2003).
1.4.3. Doenças causadas por Nematóides
Todos os nematóides identificados atacando videiras até o momento são
parasitas de raízes. A detecção do ataque é bastante complicada, pois as plantas
afetadas não apresentam sintomas evidentes. Mesmo assim, causam prejuízos,
provocando o declínio das plantas. No Submédio do Vale do São Francisco, além
de nematóides das galhas como Meloydogine incognita, M. javanica e M. arenaria,
outros também são observados, por exemplo: Xiphinema spp., Longidorus spp,
Trichodorus spp, Paratrichodorus spp, Pratylenchus spp, Tylenchulus
semipenetrans, Paratrichodorus christiei e etc (Campos et al., 2003).

23
1.4.4. Principais viroses da videira
- Intumescimento dos ramos da videira
Conhecida como “Grapevine corky bark”, ocorre na maioria dos países
produtores, afetando muitas cultivares comerciais e porta-enxertos sem exibição
de sintomas aparentes. O vírus causa redução de vigor, queda na produção e
definhamento de ramos, podendo causar a morte de plantas altamente suscetíveis
(Kuhn & Nickel, 1998). Já foi detectada no Rio Grande do Sul e em São Paulo com
uma incidência que varia de 2,3% a 20%. Em algumas áreas excepcionalmente
infectadas a incidência pode chegar a 50% (Kuniyuki, 1973; Pearson & Goheen,
1998; Fajardo et al., 2003).
O agente causal desta doença é o Grapevine vírus B (GVB) que pertence
ao gênero Vitivirus. Seu genoma é composto por 7.600 nucleotídeos constituído
de RNA fita simples, positivo, com cerca de 800 nanômetros de comprimento e
proteína do capsídeo com 23 kDa de peso molecular (Fajardo et al., 2003).
Em cultivares americanas (Vitis labrusca), a infecção é facilmente
identificada graças ao surgimento característico do intumescimento dos entrenós
dos ramos mais novos, com fendilhamento longitudinal do tecido. Quando os
ramos amadurecem o tecido dessa região morre, ficando com aspecto de cortiça.
Os ramos afetados se curvam para baixo destacando-se com facilidade. Em
plantas severamente afetadas a brotação é retardada e fraca, com os bordos das
folhas se enrolando para baixo. A uva não completa a maturação e sofre um
definhamento gradual, terminando em seca parcial ou total dos ramos afetados
seguida de morte (Fajardo et al., 2003). Em cultivares viníferas e híbridas verifica-
se o avermelhamento ou amarelamento das folhas abrangendo toda a área foliar,

24
inclusive os tecidos ao longo das nervuras (Pearson & Goheen, 1998). Outro
sintoma relacionado à presença do vírus é o engrossamento na região da enxertia,
que quando é removido apresenta caneluras avançando na direção do lenho da
produtora (Fajardo et al., 2003).
O vírus é transmitido através de material vegetativo, por multiplicação por
estacas ou gemas e enxertia e a indexação é feita através do uso de indicadoras
da variedade LN33. A transmissão via inseto vetor é possível, com transmissão
experimental confirmada através de cochonilhas da família Pseudococcidae:
Planococcus fícus, Planococcus citri, Pseudococcus longispinus e Pseudococcus
affinis. Não há constatação de contaminação de plantas por ferramentas e
tesouras de poda (Kuhn, 1992; Kuhn & Nickel, 1998; Pearson & Goheen, 1998;
Fajardo et al. 2003.)
- Caneluras do tronco da videira
Conhecida na maior parte das regiões produtoras mundiais, foi descrita pela
primeira vez na Itália com o nome de “legno riccio”, sendo posteriormente
denominada “stem-pitting” e “wood-pitting”. Foi constatada em São Paulo afetando
cultivares Itália e Rupestris du Lot (Kuniyuki & Costa, 1972) e também com
sintomas menos intensos em porta enxertos como Golia e Kober 5BB (Kuniyuki &
Muller, 1987), no Paraná (Kuniyuki, 1981) e Rio Grande do Sul (Kuhn, 1992). Um
isolado severo em Kober 5BB mas mais brando em Rupestris du Lot foi
encontrado em São Paulo (Kuniyuki & Costa, 1992). Os níveis de incidência desta
doença variam, podendo se manter entre 3 e 10%. Mas em cultivares suscetíveis
com mais de 12 anos a incidência pode chegar a mais de 50% (Kuniyuki & Costa,
1987).

25
A severidade varia dependendo de fatores como a combinação
produtora/porta-enxerto, suscetibilidade das plantas e nível de virulência da estirpe
viral. O declínio sempre é acompanhado de redução na colheita que piora
gradativamente levando à improdutividade total da planta. Em cultivares
suscetíveis a morte pode ocorrer 7 a 8 anos após a infecção (Kuhn & Nickel,
1998).
Etiologicamente o agente causal ainda não foi esclarecido. O que se sabe é
que o responsável pelas Caneluras do Tronco é um complexo viral, onde estão
incluídos os Vitivirus Grapevine virus A (GVA), Grapevine virus B (GVB),
Grapevine virus C (GVC) e Grapevine vírus D (GVD) (Chevalier et al., 1995). Este
complexo viral é conhecido como o Complexo Rugoso da Videira (Rugose wood
complex). Os componentes deste complexo (Rupestris stem pitting, Corky bark,
Kober stem grooving e LN33 Stem grooving) podem ser separados por testes de
indexação usando-se as seguintes indicadoras: Rupestris du Lot, LN33 e Kober
5BB (Kuhn & Nickel, 1998).
GVA, GVC e GVD possuem partículas de aproximadamente 700 a 825 nm,
composto por RNA fita simples de cerca de 7349 a 7600 nucleotídeos, com
subunidades da capa protéica de 20,45 até 22,5 kDa. Podem ser transmitidos
mecanicamente para Chenopodium quinoa, C. amaranticolor, Gomphrena globosa
e várias espécies de Nicotiana (Agran et al., 1990; Galiakparov et al., 2003).
Cultivares sensíveis à doença apresentam as caneluras penetrando até o
lenho, dificultando a formação dos vasos condutores. O número dessas caneluras,
o comprimento e a largura variam de acordo com a sensibilidade da cultivar e da
estirpe do vírus. As plantas afetadas sofrem retardamento no desenvolvimento,

26
diminuindo o vigor e atrasando o brotamento das gemas em uma ou duas
semanas. Os porta-enxertos normalmente mostram sintomas nítidos da doença. O
mesmo se aplica a várias produtoras européias e americanas (Fajardo et al.,
2003).
Os sintomas podem ser verificados nas raízes de cultivares muito
suscetíveis, como em Rupestris du Lot, podendo ocorrer também na região de
enxertia. As folhas em cultivares tintas apresentam avermelhamento quando muito
afetadas e pode ocorrer a morte entre 6 e 10 anos após o plantio (Kuhn & Nickel,
1998).
O principal modo de transmissão dos vírus do complexo ocorre através de
material vegetativo contaminado, e de maneira natural por meio das cochonilhas
Pseudococcus longispinus, P. affinis, Planococcus citri e P. fícus. Ainda não se
obteve registros de transmissão dos vírus por tesoura de poda ou ferramentas de
trabalho (Agran et al., 1990).
- Degenerescência da videira
Conhecida como “Court noué”, “Dégénérescence infectieuse” ou
“Grapevine fanleaf degeneration”, ocorre em todos os países víticolas. Apresenta
pouca expressão no Brasil, com incidência de 2 a 3% (Fajardo et al., 2003).
Nos Estados Unidos e Europa é uma doença de grande importância,
causando danos que podem levar a quedas na produção de até 80%, com perda
na qualidade da uva, diminuição na pega da enxertia e no enraizamento das
mudas (Martelli & Savino, 1998).
O agente causal é um Nepovirus da família Comoviridae, o Grapevine
fanleaf vírus (Vírus da folha em leque). Possui partículas isométricas de 30 nm de

27
diâmetro, triparticulado, com proteínas do capsídeo de aproximadamente 55 a 60
kDa. Possui dois RNAs fita simples, positivos, e um RNA linear ou circular de
baixo peso molecular, denominado satélite. Pode ser facilmente transmitido
mecanicamente para uma ampla gama de hospedeiras herbáceas, entre elas:
Chenopodium quinoa, C. amaranticolor, Cucumis sativus e Gomphrena globosa
(Martelli & Savino, 1998).
As plantas afetadas apresentam folhas com deformações como: assimetria
e dentes pontiagudos e distribuições anormais das nervuras (fanleaf). Também
ocorre redução no tamanho das folhas e aparecimento de manchas translúcidas
de formas variadas. Nos ramos ocorrem entrenós curtos, bifurcações,
achatamentos e nós duplos, com proliferação de gemas e brotações fracas e
atrasadas. Nos cachos notam-se bagas menores e em menor número, com atraso
ou não ocorrência de maturação (Kuhn & Nickel, 1998). Foi verificada ocorrendo
em plantas do porta enxerto 106-8 “Traviú” com sintomas de mosaico, manchas
cloróticas irregulares e esparsas; faixas cloróticas em zig-zag; folhas menores e
deformadas (Kuniyuki, 1972b).
A disseminação natural ocorre através dos nematóides Xiphinema index e
X. italiae, também ocorrendo a transmissão por longa distância através de material
propagativo e por enxertia (Kuniyuki, 1972b; Martelli & Savino, 1998).
- Necrose das nervuras da videira
Ocorre nas principais regiões vitícolas do mundo. No Brasil, Kuniyuki &
Costa (1987) verificaram incidência de 70,8% nas cultivares de uvas viníferas,
46% nos porta-enxertos e 34,4% em uvas comuns em produtoras do Estado de
São Paulo. Foi verificada causando anomalias no porta-enxerto R110, constatada

28
no Rio Grande do Sul. Aparenta estar amplamente difundido, sem causar
sintomas (Kuhn, 1994).
Os efeitos em cultivares afetadas parecem não ter relevância econômica.
Mas, pelo estado latente que apresenta em todas as cultivares comerciais e por
ocorrer em um grande percentual de plantas, ela tem sido incluída em programas
de seleção sanitária (Kuhn & Nickel, 1998).
O agente causal da necrose das nervuras da videira ainda não é conhecido,
tudo que se sabe é que pode ser perpetuado através de material vegetativo, é
transmitido por enxertia e pode ser eliminado por termoterapia (Fajardo et al.,
2003).
Como sintoma principal, verifica-se necrose das nervuras, bem visível na
parte abaxial das folhas inferiores, que se transmite para o restante das folhas ao
longo do crescimento do ramo. Estas marcas também ocorrem na superfície de
ramos verdes e nos pecíolos. Em plantas muito afetadas os sintomas podem
evoluir para manchas necróticas abrangendo grande parte da área foliar, com
maior intensidade nas folhas basais (Tavares et al., 2000).
É transmitido por enxertia através de material propagativo, e não há
constatação de contaminação de plantas através de tesoura de poda ou outras
ferramentas. Não foi reportado nenhum vetor para o patógeno e nenhuma
confirmação de outro hospedeiro que não seja Vitis spp. (Kuhn & Nickel, 1998).
- Manchas das nervuras da videira
Esta doença ocorre em todas as regiões produtoras do mundo. No Brasil é
também conhecida como mosaico das nervuras e é encontrada em todos os
estados produtores (Kuniyuki & Costa, 1987). De acordo com Kuniyuki & Costa

29
(1994) o vírus ocorre com uma incidência média de 58% em cultivares de copa e
em porta-enxertos com incidência média de 18,1%.
Por ter uma alta ocorrência e por ser latente em praticamente todas as
cultivares viníferas e porta-enxertos, Kuhn e Nickel (1998) justificam a inclusão
desta virose nos programas de seleção sanitária.
O agente causal é o vírus-das-manchas-das-nervuras-das-videiras
(Grapevine fleck vírus, GFkV), pertencente ao gênero Moculavirus com partículas
isométricas de 30 nm de diâmetro e RNA fita simples de aproximadamente 7,5 kb,
e subunidades da proteína da capa com cerca de 28 kDa (Fajardo et al., 2003).
Plantas da variedade George apresentam a maior sensibilidade ao vírus,
mostrando manchas translúcidas e sem forma definida acompanhando as
nervuras terciárias e quarternárias em folhas novas e de meia idade, aparecendo
em parte ou em toda a lâmina foliar. Quando essas manchas surgem em grande
intensidade, as folhas apresentam-se torcidas e enrugadas (Stellmach & Goheen,
1998). Também ocorrem deformações no seio peciolar, verificando-se uma
abertura excessiva dos mesmos com conseqüente assimetria e distorção das
folhas (Kuhn & Nickel, 1998). Plantas infectadas tem seu desenvolvimento
comprometido e apresentam folhas com bordos virados para cima (Tavares et al.,
2000). Induz manchas translúcidas nas nervuras da indicadora Rupestris du Lot
(Kuniyuki, 1972c).
O vírus é transmitido através de material propagativo infectado, transmitido
para outras plantas através de enxertia. Já foi confirmada a transmissão do vírus
por meios naturais, mas o vetor ainda não foi definido (Fajardo et al., 2003).

30
- Enrolamento das folhas da videira
Conhecida também como vermelhão ou amarelo, é a doença de maior
ocorrência nos plantios de videira e que se destaca por sua importância
econômica. O primeiro relato da doença no Brasil foi feita no estado de São Paulo
(Kuniyuki, 1972a), com incidência de até 78%. Foi relatada no Rio Grande do Sul,
atingindo de 15,6 a 98% dos vinhedos e ocorre também nos estados de Goiás,
Minas Gerais, Paraná, Santa Catarina e no Submédio do Vale do São Francisco
(Kuniyuki, 1981; Kuhn, 1989b; Tavares et al., 2000). No Brasil já foram detectados
o Grapevine leafroll-associated vírus-1 (GLRaV-1), Grapevine leafroll-associated
virus-2 (GLRaV-2), Grapevine leafroll-associated vírus-3 (GLRaV-3) e Grapevine
leafroll-associated vírus-6 (GLRaV-6). Em plantas severamente afetadas,
verificou-se redução de 42,4% no número de cachos, 62,8% na produção e 65,2%
no vigor, além de decréscimo no teor de açúcares redutores da uva (Kuhn, 1989a;
Kuhn & Nickel, 1998; Fajardo et al., 2002).
O complexo viral responsável pela doença do enrolamento da folha da
videira é formado, até o momento, por oito espécies de vírus denominados GLRaV
1-8 (Grapevine leafroll associated vírus 1 to 8), que podem ocorrer de maneira
isolada (Fazeli & Rezaian, 2000; Alkowni et al., 2002).
No campo, a confirmação visual da infecção em Vitis vinifera L. é
relativamente fácil. Durante a primavera, as folhas de plantas infectadas e
saudáveis são bem similares. Ao longo das outras estações, até o final do outono,
as folhas infectadas se tornam amareladas em variedades brancas, ou vermelhas
em variedades tintas, e os bordos se enrolam para baixo (Goheen & Hewitt, 1964)
(Figura 1). Este sintoma surge a partir da base dos ramos e se distribui até a

31
extremidade (Fajardo et al., 2002). Cultivares americanas (V. labrusca) e híbridos
mostram pouco ou nenhum sintoma de enrolamento (Kuhn & Nickel, 1998).
Figura 4. Sintomas do enrolamento da folha em variedades tintas. A folha localizada a esquerda apresenta sintomas da doença descrita como enrolamento, a saber: enrolamento dos bordos para baixo e avermelhamento. A folha da direita foi extraída de uma planta sadia (fonte: http://winegrapes.tamu.edu/grow/diseases/ leafroll.shtml).
O sintoma mais comum nos cachos, principalmente em cultivares tintas, é a
maturação retardada ou irregular dos frutos. Os cachos são menores em tamanho
e número, as bagas apresentam casca menos pigmentada, permanecendo
esverdeadas e esbranquiçadas. Em alguns casos, quando a planta é severamente
afetada, o processo de maturação chega a ser interrompido (Goheen & Hewitt,
1964).

32
Figura 5. Sintomas do enrolamento das folhas nos cachos. Os frutos localizados a
direita foram extraídos de uma planta contaminada pelo vírus, apresentando pouco desenvolvimento quando comparada com frutos extraídos de uma planta sadia (à esquerda) (fonte: Pearson & Goheen, 1998).
De acordo com Lima (2002), os principais prejuízos causados por esse
vírus em variedades suscetíveis são a redução na produção dos frutos, menor
enraizamento de estacas e porcentagem de pegamento de enxertias, bem como
aumento na suscetibilidade das plantas às injúrias causadas por geadas.
A doença é disseminada principalmente através de material propagativo
infectado (Habili & Nutter, 1997), podendo também ser transmitida por vetores,
que são as cochonilhas pertencentes aos gêneros Heliococcus bohemicus,
Phenacoccus aceris, Pseudococcus longispinus, P. calceolariae, P. viburni,
Planococcus fícus, P. citri, Pulvinaria vitis, Neopulvinaria sp. e Parthenolecanium
sp. A transmissão por vetor só foi confirmada em duas espécies do vírus, GLRaV
1 e 3, ambas com transmissão natural confirmada (Habili et al., 1995; Petersen &
Charles, 1997; Sforza et al., 2003). A transmissão mecânica para hospedeiras
herbáceas ocorre somente para GLRaV-2, e se restringe a Nicotiana spp. (Fajardo
et al., 2003).

33
O controle se dá através do uso de plantas certificadas, livres de vírus, e
também através do uso de termoterapia (Valero et al., 2003).
A diagnose do vírus do enrolamento das folhas pode ser feita através de
testes de indexação nas cultivares tintas indicadoras Pinot Noir, Cabernet Franc,
Cabernet Sauvignon, Barbera, Mission e LN33 (Kuhn, 1989a; Tavares et al.,
2000). A detecção também pode ser feita por ELISA, RT-PCR (Zimmermann et al.,
1990; Karazev et al., 1994; Minafra & Hadidi, 1994; Rowhani et al., 1995;
MacKenzie et al., 1997; Ling et al., 2001) e hibridização com uso de sondas
(Saldarelli et al., 1994).
Como o GLRaV-3, assunto dessa dissertação, pertence à família
Closteroviridae, a seguir passaremos à descrição geral dessa família.
1.5. A Família Closteroviridae
Todas as espécies de vírus da família possuem uma gama restrita de
hospedeiras. Os sintomas causados são amarelecimento, enrolamento de folhas,
atraso no desenvolvimento de frutos e rompimentos ou surgimento de caneluras
nos troncos. A infecção é sistêmica, mas normalmente limitada ao floema.
Algumas espécies são dificilmente transmitidas por inoculação mecânica e a
transmissão à longa distância se dá através de material propagativo, sendo que a
transmissão por sementes é extremamente rara (van Regenmortel et al., 2000).
Estão entre os vetores dos membros da família Closteroviridae os afídeos
(Nasonovia, Myzus, Aphis, Toxoptera, Rhopalosiphum), moscas-brancas (Bemisia,
Trialeurodes) e cochonilhas (Heliococcus, Pseudococcus, Planococcus;
Phenacoccus, Saccharicoccus e Dysmicoccus; Pulvinaria, Neopulvinaria e

34
Parthenolecanium). A transmissão é semi-persistente (van Regenmortel et al.,
2000; Little et al., 2001).
De acordo com Martelli et al. (2002) os vírions que fazem parte desta
família são flexuosos, com simetria helicoidal. As partículas de todos os membros
da família possuem cerca de 12 nm de diâmetro, mas seu comprimento varia de
acordo com o gênero e/ou a espécie (Figura 6). As Partículas virais dos gêneros
desta família são encapsidadas em uma das extremidades por um análogo da
proteína do capsídeo, expresso por uma duplicata desse gene (CPd).
Figura 6. Partículas de Citrus tristeza virus (CTV) decoradas com antisoro homólogo (fonte: van Regenmortel et al., 2000).
Mesmo possuindo genoma bipartido ou monopartido, todos os vírus da
família Closteroviridae tem em sua composição uma única fita linear de RNA,
sentido positivo, que constitui 5 a 6% do seu peso total. O tamanho do genoma
varia com o comprimento da partícula. Os closterovírus possuem o maior genoma
entre os vírus de plantas com genoma de RNA fita simples positivo. O genoma
dos três gêneros que compõem a família é caracterizado pela presença de genes
que codificam uma proteína homóloga à HSP70 e uma análoga à proteína do
capsídeo. A organização genômica, o número e a posição relativa das regiões

35
codificadoras (open reading frames – ORFs) variam de acordo com o gênero e a
espécie. As ORFs que codificam uma pequena proteína hidrofóbica com massa
molar de aproximadamente 6 x 103, uma proteína homóloga à HSP70, um produto
de 55-64 pares de bases, a proteína do capsídeo e a CPd formam um módulo de
cinco genes que se mantém conservado entre os membros da família (Martelli et
al., 2000).
Excluindo-se GLRaV-2, que pertence ao gênero Closterovirus, e GLRaV-7,
ainda não classificado, todas as outras espécies de GLRaV pertencem à família
Closteroviridae, gênero Ampelovirus, e a espécie tipo é o GLRaV-3 (van
Regenmortel et al., 2002).
1.5.1. Classificação dos membros da família Closteroviridae
Quando foi estabelecida, em 1998, a divisão das espécies de vírus da
família Closteroviridae em gêneros era baseada no tipo de RNA genômico: se
monoparticulado então classificava-se como do gênero Closterovirus, se
biparticulado, o vírus era classificado como pertencente ao gênero Crinivirus (van
Regenmortel et al., 2000). Martelli et al. (2002), com base em dados moleculares e
biológicos mais acurados revisaram a família, modificando a classificação das
espécies e criando um novo gênero, Ampelovirus (Ampelos=uva), onde foram
agrupados os vírus do enrolamento da folha da videira, com exceção do GLRaV-2.
Com base nessas informações, a família Closteroviridae foi classificada
pelo tipo de inseto vetor, abandonando-se a idéia de classificação baseada nos
RNAs genômicos. A estrutura taxonômica ficou da seguinte forma: Família:
Closteroviridae; Gênero Closterovirus, espécie tipo: Beet yellows virus (BYV);

36
Gênero Ampelovirus, espécie tipo: Grapevine leafroll-associated virus 3 (GLRaV-
3); Gênero Crinivirus, espécie tipo: Lettuce infectious yellows virus (LYIV) (Martelli
et al., 2002).
1.5.2. Gênero Closterovirus
Os vírions tem aproximadamente 1250 – 2200 nm de comprimento e
possuem uma única molécula de RNA fita simples positivo, com 15.5 – 19.3 kb. As
subunidades da proteína do capsídeo, com 22-25 kDa, cobrem a maior parte do
vírus e a CPd, com 24-27 kDa, cobre uma extremidade. Existem três tipos de
estrutra gênomica no gênero, representadas por três espécies: Beet yellows virus
(BYV), Citrus tristeza virus (CTV) e Beet yellow stunt virus (BYSV). A organização
do genoma do BYSV é intermediária entre BYV e CTV, o que sugere que esses
três vírus representam três estágios distintos na evolução dos closterovírus.
Diferenciando-se dos dois outros gêneros da família, Crinivirus e Ampelovirus, o
gene que codifica a CPd dos closterovirus se localiza à juzante com relação ao
gene da CP. O gênero possui vírus que são transmitidos por afídeos, de maneira
semi-persistente (Tabela 1), infectando primariamente hospedeiras dicotiledôneas.
Alguns são transmissíveis por inoculação mecânica mas com dificuldade (Martelli
et al., 2002).

37
ORF 1a
Figura 7. Estrutura genômica do BYV, mostrando posições relativas das ORFs e seus produtos: PRO - domínio de protease; MTR – domínio de metiltransferase; HEL – domínio de helicase; RdRp - RNA polimerase dependente de RNA; HSP70 - proteína análoga a HSP70 descrita em células animais; CPd - proteína análoga a proteína do capsídeo; CP - proteína do capsídeo. As funções das demais ORFs ainda não foram descritas (adaptado de Martelli et al., 2002).
Tabela 1. Espécies do gênero Closterovirus (adaptado de Martelli et al., 2002) Transmi ssíveis por afídeos:
Beet yellows virus (BYV)
Beet yellow stunt virus (BYSV)
Burlock yellows virus (BuYV)
Carnation necrotic fleck virus (CNFV)
Carrot yellow leaf virus (CYLV)
Citrus tristeza virus (CTV)
Wheat yellow leaf virus (WYLV)
Vetor descon hecido:
Grapevine leafroll-associated virus 2 (GLRaV-2)
Prováveis espécies do gênero:
Clover yellows virus (CYV)
Dendrobium vein necrosis virus (DVNV)
Heracleum virus 6 (HV-6)
Festuca necrosis virus (FNV)

38
1.5.3. Gênero Crinivirus
Os vírions são menores que 1000 nm e possuem dois comprimentos, de
aproximadamente 650 – 850 nm e 700 – 900 nm. O genoma é composto por RNA
fita simples positiva, com 15.3 – 19 kb divididos em duas moléculas que são
necessárias para a infecção e são separadamente encapsidadas. As subunidades
do capsídeo pesam entre 28 e 33 kDa, mas a CPd pode chegar até 80 kDa. O
RNA-1 de LIYV é uma molécula bicistronica que codifica proteínas relacionadas
com a replicação (ORF1). O RNA-2 possui sete ORFs, que contém o módulo de
cinco genes típico da família mas difere um pouco pela presença de um gene
pequeno (ORF4) que se localiza a montante do gene da CP (Figura 5). Em todos
os membros do gênero (Tabela 2) o gene da CPd se localiza a juzante do gene da
CP. Nenhum dos membros pode ser transmitido por inoculação mecânica. Os
vetores são moscas-brancas (Bemisia e Trialeurodes) que transmitem de maneira
semi-persistente.
ORF 1a
Figura 8. Estrutura genômica de LIYV, mostrando a posição relativa das ORFs e seus produtos: PRO – domínio de protease; MTR – domínio de metiltransferase; HEL – domínio de helicase; RdRp - RNA polimerase dependente de RNA; HSP70 - proteína análoga à HSP70 descrita em células animais; CP - proteína do capsídeo; CPd - proteína análoga a proteína do capsídeo. As funções das demais ORFs ainda não foram descritas (adaptado de Martelli et al., 2002).

39
Tabela 2. Espécies do gênero Crinivirus (adaptado de Martelli et al., 2002). Abutilon yellows virus (AbYV)
Cucurbit yellow stunting disorder virus (CYSDV)
Lettuce chlorosis virus (LCV)
Lettuce infectious yellows virus (LIYV)
Sweet potato chlorotic stunt virus (SPCSV)
Tomato chlorosis virus (ToCV)
Tomato infectious chlorosis virus (TICV)
Prováveis espécies do gênero:
Potato yellow vein virus (PYVV)
Beet pseudoyellows virus (BPYV)
Diodea vein chlorosis virus (DVCV)
1.5.4. Gênero Ampelovirus
Os vírions tem aproximadamente 1400 – 2200 nm de comprimento e
possuem uma única molécula de RNA fita simples positivo, com 16,9 – 19,5 kb
(Figura 9). As subunidades da proteína do capsídeo possuem grande massa
molecular de 35 – 43 kDa (Zimmermann et al., 1990). Existem dois tipos de
estrutura de genoma no gênero, cujas espécies tipo são GLRaV-3 e Little cherry
virus 2 (LChV-2) (Rott & Jelkmann, 2001). Com relação à posição da CPd, no
GLRaV-3 e em vários outros membros do gênero ela ocorre a juzante em relação
ao gene da CP, enquanto que em LChV-2 esta ocorre cinco ORFs a montante do
gene da CP (Martelli et al., 2002). Grapevine leafroll-associated vírus-1 (GLRaV-1)
apresenta algo peculiar com relação aos outros membros do gênero: seu gene da

40
CPd é duplicado (Little et al., 2001). Os vírus deste gênero (Tabela 3) infectam
somente hospedeiras dicotiledôneas e são transmissíveis por cochonilhas
coccídeas ou pseudococcídeas. Nenhum deles é transmissível mecanicamente
(Martelli et al., 2002).
Little et al. (2001), verificaram que as ORFs 3, 6 e 7 do GLRaV-1, que
correspondem respectivamente ao gene homólogo à HSP70, e duas cópias da CP
(CPd1 e CPd2), apresentam um alto grau de variação. Após verificar 75 clones
que correspondiam a cada uma das ORFs, foi detectada uma variação de até 60%
nas posições dos nucleotídeos dessas regiões em um ou mais dos clones
seqüenciados. Estas variações sugeriram que o GLRaV-1 pode existir na forma de
uma população heterogênea, resultado da falta de pressão de seleção ou da
mistura de variantes virais devido às práticas culturais de enxertia ou propagação
vegetativa utilizadas através dos séculos.
O GLRaV-3 já foi seqüenciado e possui 17919 nucleotídeos. A análise
dessa seqüência revelou a presença de 13 regiões de leitura (ORFs), definidas
como ORFs 1a, 1b e 2 a 12. As ORFs 1a e 1b estão envolvidas na replicação
viral, a primeira codifica uma proteína semelhante as helicases dos vírus de RNA
fita simples positivos, enquanto a segunda codifica uma RNA polimerase
dependente de RNA. A ORF 4 codifica uma proteína similar a HSP70,
possivelmente a proteína de movimento desse vírus. A ORF 6 codifica a proteína
do capsídeo viral. A ORF 7 é responsável pela síntese de uma cópia divergente do
capsídeo viral (CPd), essa proteína interage com as extremidades dos vírions,
gerando estruturas complexas possivelmente relacionadas à replicação e/ou

41
movimento das partículas entre células. As ORFs restantes não possuem função
definida (Ling et al., 1998).
A expressão dessas ORFs é realizada através de, pelo menos, três
mecanismos: processamento proteolítico, mudança de fase ribossomal e síntese
de RNAs subgenômicos. Os dois primeiros processos estão envolvidos na
expressão das ORFs 1a e 1b. Já o último mecanismo está relacionado às ORFs
restantes (Karasev, 2000).
Figura 9. Micrografias apresentando partículas de GLRaV-1 e GLRaV-3 (a, c), e as mesmas decoradas por antisoro homólogo (b, d). Barras = 100 nm (fonte: Credi & Giunchedi, 1996).

42
ORF 1a
Figura 10. Estrutura genômica do GLRaV-3, mostrando a posição relativa das ORFs e seus produtos: PRO – domínio de protease; MTR – domínio de metiltransferase; HEL – domínio de helicase; RdRp - RNA polimerase dependente de RNA; HSP70 - proteína análoga a HSP70 descrita em células animais; CP - proteína do capsídeo; CPd - proteína do capsídeo divergente As funções das demais ORFs ainda não foram descritas (adaptado de Martelli et al., 2002).
Tabela 3. Espécies do gênero Ampelovirus (adaptado de Martelli et al., 2002). Espécies do gênero:
Grapevine leafroll-associated virus 1 (GLRaV-1)
Grapevine leafroll-associated virus 3 (GLRaV-3)
Grapevine leafroll-associated virus 5 (GLRaV-5)
Pineapple mealybug wilt-associated virus 1 (PMWaV-1)
Pineapple mealybug wilt-associated virus 2 (PMWaV-2)
Little cherry virus 2 (LChV-2)
Prováveis espécies do gênero:
Transmissível por cochonilhas:
Sugarcane mild mosaic virus (SMMV)
Vetor desconhecido:
Grapevine leafroll-associated virus 4 (GLRaV-4)
Grapevine leafroll-associated virus 6 (GLRaV-6)
Grapevine leafroll-associated virus 8 (GLRaV-8)
Plum bark necrosis and stem pitting-associated virus (PBNSPaV)

43
2. OBJETIVOS
O presente trabalho teve como objetivos:
1) Detectar, por meio de ensaio imunológico e amplificação de ácidos nucléicos
(polymerase chain reaction – PCR), a ocorrência do GLRaV-3 em videiras
provenientes do Vale do São Francisco;
2) Comparar e verificar se a detecção por métodos imunológicos e moleculares
apresentaram alguma diferença;
3) Iniciar a caracterização da variabilidade genética do GLRaV-3 por meio da
amplificação de ácidos nucléicos (PCR) e do seqüenciamento.

44
3. MATERIAIS E MÉTODOS
O primeiro passo para o estudo do GLRaV-3 foi a detecção por meio do
teste imunoenzimático ELISA em plantas originárias do Vale do São Francisco
(Petrolina) seguido da realização de um procedimento de extração de RNA
baseado em uma técnica já descrita (MacKenzie et al., 1997), utilização do RNA
extraído em RT-PCR e seqüenciamento dessas regiões do genoma viral que
foram amplificadas.
Todas as amostras recebidas de Petrolina foram testadas tanto para ELISA
quanto em RT-PCR.
3.1. Local e período de realização do trabalho
O presente trabalho foi realizado no Laboratório de Fitopatologia, no
Instituto de Ciências Biológicas da Universidade de Brasília e na Estação
Experimental da Biologia, no período de Fevereiro de 2002 a Maio de 2004.
3.2. Obtenção e manutenção das amostras
Nove estacas de videira (Vitis vinifera) da variedade Alicante Bouchet,
provenientes de Petrolina/PE, município de Lagoa Grande, Fazenda Vitivinícola,
foram mantidas em casas de vegetação na Estação Experimental da Biologia na
Universidade de Brasília. Estas foram testadas através do método ELISA para
comprovação da suspeita de infecção por GLRaV, através do uso de um kit
comercial (Sanofi-Pasteur). Após cerca de seis meses, sete estacas das
variedades Caner, Liberti, CG40016, 10-6, CG351, CG28467 e CG39915 foram

45
recebidas de Petrolina, Estação Experimental de Bebedouro/PE. Estas foram
mantidas no mesmo local que as outras e também foram utilizadas nos testes.
3.3. ELISA
O procedimento de ELISA adotado foi o do ensaio direto de duplo-
sanduiche (DAS-ELISA) (Clark & Adams, 1977). Folhas e pecíolos apresentando
sintomas de infecção viral como avermelhamento e enrolamento dos bordos para
baixo, foram trituradas com nitrogênio líquido em um almofariz, com posterior
adição de um tampão de extração na proporção 1/5 (peso/volume) contendo: TRIS
3 M pH 8,0; NaCl 0,5 M; PVP-40 0,8 M; 0,001% de merthiolate; 0,5 ml de Tween-
20 em 1 litro de água destilada.
A suspensão obtida foi transferida para tubos Falcon (Corning Incorporated)
e centrifugada por 10 minutos a 3000 rpm para clarificação (IEC Centra CL2).
Todo o sobrenadante obtido foi transferido para tubos tipo Eppendorf e a parte
sólida foi descartada. Um ciclo de centrifugação por aproximadamente 5 segundos
à 4000 rpm foi realizado em cada uma das amostras contidas nos tubos e a
suspensão foi transferida para novos tubos para que esta ficasse quase que
completamente livre de partículas sólidas. Toda a suspensão obtida nos
processos de extração do vírus foi depositada em congeladores a –80o C.
Previamente à realização do teste, foi preparado o tampão de lavagem
PBS-Tween contendo: NaCl 0,5 M; KH2PO4 25 mM; Na2HPO4.2H2O 0,5 M; KCl 10
mM e 0,5 mililitros de Tween 20 em 1 litro de água destilada.
Foi utilizado um kit comercial da Sanofi Pasteur contendo: anticorpos anti-
GLRaV 1 + 3 para sensibilização da placa; conjugado de anticorpos policlonais

46
adicionados à enzima fosfatase alcalina e controles positivos e negativos (N° do
catálogo: P51343, lote: 92219).
Inicialmente, a microplaca de 96 poços (Dynatech) foi sensibilizada através
da adição de 100 µl/poço da solução de sensibilização contendo anticorpos e
tampão de sensibilização na proporção 1:50. Todos os poços onde iriam ser
aplicadas as amostras foram preenchidos com este tampão, com exceção
daqueles onde seriam aplicados os controles do substrato.
Vedou-se a microplaca e a mesma foi colocada em uma câmara úmida,
sendo posteriormente acondicionada em estufa a 37o C.
Após um período de duas horas, descartou-se o conteúdo dos poços e
lavou-se a placa com a solução de PBS-Tween, adicionando-se 200 µl em cada
poço em 3 repetições de aproximadamente 30 segundos, descartando-se o
conteúdo em um recipiente adequado. Aplicou-se 100 µl dos extratos
descongelados em cada poço sensibilizado e a microplaca foi então levada à
estufa a 37o C por aproximadamente 12 horas.
Após este período, o conteúdo da placa foi descartado e o tampão de
lavagem PBS-Tween foi novamente aplicado (100 µl), agora em 4 repetições com
duas pausas de aproximadamente 3 minutos entre as duas últimas aplicações.
Após este ciclo de lavagem, aplicou-se 100 µl da solução de conjugado (diluição
1:50), depositando-se a microplaca em estufa a 37o C por 2 horas.
Novamente o conteúdo da placa foi descartado e o mesmo ciclo de
lavagem utilizando-se a solução de PBS-Tween foi repetido. Como último passo
do processo, foi adicionada aos poços a solução de substrato à temperatura

47
ambiente com posterior leitura em espectrofotômetro a 405 nm, após um período
de aproximadamente 1 hora. Foram definidas como infectadas as que
apresentaram valores médios de absorbância, pelo menos, duas vezes superiores
aos verificados nos controles negativos (Almeida & Lima, 2001).
As amostras e os tampões foram aplicados utilizando-se uma pipeta de 8
canais (Finnpipette Digital Multichannel 50-300 µl – Sigma chemical co.).
As amostras que apresentaram resultado positivo no teste de ELISA foram
processadas para a extração de RNA, PCR e posterior seqüenciamento.
3.4. Extração de RNA e Síntese de cDNA
3.4.1. Extração de RNA
Alguns métodos de extração de RNA viral foram testados e comparados.
Os procedimentos de extração foram baseados em algumas publicações
científicas nas quais os processos de extração do RNA viral de GLRaV (Minafra &
Hadidi, 1994; Mackenzie et al., 1997) e Grapevine fanleaf vírus (GFLV) (Rowhani
et al., 1993, Rowhani et al., 1995) foram bem sucedidos. Também foi testado um
método de extração de RNA a partir do procedimento de Sambrook, modificado
por Câmara et al. (2001).
Para a extração do RNA foram utilizadas aproximadamente 0,10 gramas de
tecido das folhas, incluindo-se partes dos pecíolos e nervuras. Este tecido foi
triturado em tubos tipo Eppendorf utilizando-se nitrogênio líquido e um bastão de
vidro, que era devidamente lavado e esterilizado com álcool quando utilizado em
amostras diferentes.

48
Aos tubos contendo o tecido triturado, foi adicionado aproximadamente 1
mililitro do tampão de extração contendo: acetato de sódio 0,2 M, pH 5.0; EDTA 25
mM; 2,5% (peso/volume) de PVP-40 e 1% (volume/volume) de 2-mercaptoetanol
(adicionado pouco antes do uso). Os tubos contendo o extrato foram centrifugados
a 4000 rpm por 5 minutos (Jouan, ST. Herblain-France, Type A 14/V1). O
sobrenadante foi transferido para outros tubos e 140 µl foram utilizados no kit
QIAamp de purificação de RNA viral (Qiagen).
Primeiramente, os 140 µl de extrato e 560 µl do tampão AVL contendo
carrier RNA preparado anteriormente (fornecido pelo kit) foram misturados através
do uso de vortex (Vortex Genie-2 – Scientific Industries) e incubados por 10
minutos.
Adicionou-se 560 µl de etanol (96-100%) à mistura, com mais um ciclo de
utilização do vortex até ocorrer uma homogeneização completa. Seiscentos e
trinta microlitros da solução foram aplicados à uma coluna de rotação, com
elemento filtrante composto por sílica gel previamente posicionada sobre um tubo
de 2 ml (ambos fornecidos pelo kit). Os tubos foram então centrifugados à 8000
rpm por 1 minuto.
Após a centrifugação, a coluna foi transferida para outro tubo de coleta de 2
ml. O tubo contendo o filtrado foi descartado. Aplicou-se novamente 630 µl da
solução na coluna e um novo ciclo de centrifugação à mesma velocidade e tempo
foi realizado. Quinhentos microlitros do tampão denominado AW (fornecido pelo
kit) foi adicionado à coluna. Um novo ciclo de centrifugação foi realizado.

49
Adicionou-se à coluna mais 500 µl do mesmo tampão e um ciclo de centrifugação
à velocidade máxima por 3 minutos foi realizado.
O tubo de 2 ml contendo o filtrado foi descartado e a coluna foi transferida
para um tubo eppendorf de 1,5 ml. O RNA contido na coluna foi eluído através da
adição de 50 µl de água livre de RNAses pré aquecida a 80o C. As amostras foram
acondicionadas em congeladores a –80o C.
3.4.2. Síntese de cDNA
Antes de se realizar o PCR é necessário sintetizar, a partir das fitas de RNA
fita simples viral, o DNA complementar (cDNA), para tal é necessário se fazer a
reação de transcrição reversa (Minafra & Hadidi, 1994; Fazeli et al., 1998; Zhang &
Rowhani, 2000).
A reação foi realizada em um volume final de 25 µl: 4 µl de primers
aleatórios, 6 µl de RNA das amostras e 5 µl de H2O livre de RNAses foram
aquecidos a 70o C por 5 minutos e colocados imediatamente no gelo,
proporcionando o anelamento do RNA aos primers randômicos. A essa reação
foram adicionados: 5 µl de tampão de reação, 5 µl de dNTPs, 1 µl de RNAsin, 1 µl
da enzima M-MLV transcriptase reversa (Promega) e 13 µl de H2O livre de
RNAses. A reação foi então aquecida a 37o C por 60 minutos em banho-maria
para a síntese do cDNA.
3.5. Amplificação do gene do capsídeo viral e da re gião 3´ da RNA
polimerase
Para realizar o processo de caracterização molecular do GLRaV-3 presente
nas amostras, dois genes foram escolhidos para amplificação e seqüenciamento:

50
o gene da RNA polimerase dependente de RNA (RdRp) e o gene da proteína do
capsídeo (Cp). Primers específicos selecionados a partir da literatura (Minafra &
Hadidi, 1994; Ling et al., 1997) foram utilizados para essa finalidade (Tabela 4).
Para a amlificação dos 340 pares de base do gene da polimerase viral
realizou-se o PCR em um volume final de 50 µl com 5 µl de cDNA, 0,2 mM de
dNTPs (Gibco BRL), 2 U da enzima Taq polimerase (Gibco BRL), utilizando-se os
primers C547 e H229 propostos por Minafra & Hadidi (1994) e o programa do
termociclador proposto por MacKenzie et al. (1997) com modificações: 95o C por 2
minutos seguido de 35 ciclos de desnaturação a 95o C por 1 minuto, anelamento a
53o C por 1 minuto e extensão a 72o C por 1 minuto, terminando em uma extensão
final a 72o C por 7 minutos.
Tabela 4. Seqüências dos oligonucleotídeos (primers) utilizados para amplificação do gene da RNA polimerase dependente de RNA e da Proteína do Capsídeo. RNA polimerase Proteína do Capsídeo
Primers
H229 (nucleotídeos 8286 a 8267)
5´ATAAGCATTCGGGATGGACC3´
C547 (nucleotídeos 8585 a 8606)
5´ATTAACTTGACGGATGGCACGC3´
LR3-8504V (nucleotídeos 13269 a 13288)
5´ATGGCATTTGAACTGAAATT3´
LR3-9445C (nucleotídeos 14191 a 14210)
5´CTACTTCTTTTGCAATAGTT3´
Para a amplificação de 942 pares de base do gene do capsídeo viral,
realizou-se o PCR em um volume final de 50 µl com 5 µl de cDNA, 0,2 mM de
dNTPs (Gibco BRL), 2 U da enzima Taq polimerase (Gibco BRL), utilizando-se os
primers LR3-9445C e LR3-8504V propostos por Ling et al. (1997) e o seguinte

51
programa do termociclador: 95o C por 2 minutos seguido de 35 ciclos de
desnaturação a 95o C por 1 minuto, anelamento a 48o C por 1 minuto e extensão a
72o C por 1 minuto terminando em uma extensão final a 72o C por 7 minutos
(MacKenzie et al., 1997).
Os resultados foram visualizados em gel de agarose a 1,5%, preparado em
tampão 0,5X TBE (5,4 g de Tris-base, 2,75 g de ácido bórico e 0,375 g de EDTA
para 1000 ml), onde 10 µl de cada amostra foram misturados em 2 µl de tampão
de carregamento (0,25% de azul de bromofenol em 87% de glicerol) e aplicados
no gel. As condições de corrida se deram a 90 V durante 1 hora. Os marcadores
utilizados foram 1 Kb DNA Ladder e 100 pb Ladder (Gibco-BRL). Após a corrida, o
gel foi corado em solução de brometo de etídeo e visualizado sobre luz ultravioleta
e fotografado utilizando-se fotodocumentador Eagle EYE – II (Strategene).
3.6. Clonagem dos segmentos de DNA amplificados
A clonagem foi realizada usando-se o vetor pGEM T Easy Vector
(Promega) e bactérias Escherichia colli da linhagem TH5α.
Para a reação de ligação foram utilizados 5 µl do tampão fornecido pelo kit,
1 µl do vetor, 5 µl da amostra a ser clonada e 1 µl da enzima DNA ligase que
foram pipetados em tubos tipo Eppendorf de 1,5 ml. Esses tubos foram vedados
com Parafilm® e colocados em banho-maria à 4o C por uma noite.
O meio de cultura para o crescimento das bactérias foi preparado de acordo
com a seguinte formulação: 16 g de bacto peptona; 10 g de extrato de levedura; 5
g de cloreto de sódio e água destilada para 1 litro. Em seguida o meio foi

52
autoclavado por 20 minutos à 120o C e pressão de 1 kg/cm2 (Sterilmatic - Market
Forge Industries). Cerca de 200 ml deste meio foi separado. No restante
adicionou-se ainda: 15 g de ágar e 10 ml de ampicilina (adicionada pouco antes do
processo de plaqueamento, com a temperatura do meio de cultura chegando
quase à temperatura ambiente). O meio de cultura ao qual foi adicionado o ágar e
a ampicilina foi colocado em placas de Petri para que ocorresse sua solidificação.
O meio de cultura líquido foi mantido em câmara fria.
A transformação da E. colli da linhagem TH5α� foi realizada a partir de
células competentes congeladas a –80o C. Trezentos microlitros destas células
foram adicionadas a cada tubo tipo Eppendorf contendo as ligações e em seguida
os tubos foram colocados em gelo por 40 minutos. Após este período, os tubos
foram tranferidos para banho-maria à 42o C por 1 minuto e meio e depositados
novamente em gelo por, no mínimo, 5 minutos.
Após o choque térmico, 700 µl do meio de cultura líquido foram adicionados
à cada tubo contendo as células, e os tubos foram colocados em estufa à 37o C
por 1 hora, com homogeneizações de 15 em 15 minutos. Às placas de Petri
contendo os meios de cultura solidificados foram adicionados 40 µl de IPTG 100
mM (GibcoBRL) e 70 µl de X-GAL 50 mg/ml (Invitrogen).
Após o período de 1 hora, os tubos foram retirados da estufa, centrifugados
à 3000 rpm por 5 minutos e 700 µl do sobrenadante foram descartados.
De cada tubo duas repetições foram realizadas no processo de
plaqueamento das células bacterianas: em uma placa colocou-se 270 µl e em

53
outra colocou-se os 30 µl restantes. As placas foram depositadas por uma noite
em estufa à 37o C.
As colônias bacterianas que adquiriram o inserto apresentaram coloração
branca, enquanto que as colônias que não o adquiriram apresentaram coloração
azul. As colônias brancas foram selecionadas através do uso de palitos
previamente autoclavados e posteriormente colocados em tubos de vidro
contendo 2 ml de meio de cultura líquido e 20 µl de ampicilina 100 µg/ml. Estes
tubos de vidro foram acondicionados em um agitador (New Brunswick Industries) à
50 rpm e 37o C de temperatura por uma noite.
A seguir os plasmídeos foram purificados pela técnica de minipreparação
de DNA através do uso de um kit comercial (FlexiPrep Kit – Pharmacia).
Um mililitro e meio de cada meio de cultura contido nos tubos foi transferido
para tubos tipo Eppendorf e centrifugados a velocidade máxima por 30 segundos.
O subrenadante foi descartado, deixando o precipitado o mais seco possível.
Duzentos microlitros da solução 1 (fornecida pelo kit) foram adicionados aos tubos
que foram subseqüentemente agitados em vortex. Duzentos microlitros da solução
2 (fornecida pelo kit) foram adicionados e misturados invertendo-se os tubos
diversas vezes. Finalmente, 200 µl da solução 3 (fornecida pelo kit) foram
adicionados e misturados através da inversão repetitiva dos tubos. Os tubos foram
então centrifugados à velocidade máxima por 5 minutos a temperatura ambiente.
A adição dessas soluções proporcionou a lise celular e conseqüente liberação do
plasmídeo.

54
O sobrenadante foi transferido para novos tubos. Quatrocentos e vinte
microlitros de isopropanol, em temperatura ambiente, foram adicionados, agitados
através do vortex e incubados em temperatura ambiente por 10 minutos. Após o
período de incubação, um novo ciclo de centrifugação foi realizado à velocidade
máxima por 10 minutos para ocorrer a precipitação do DNA plasmidial. O
sobrenadante foi descartado e os tubos foram invertidos sobre papel toalha para
secagem do precipitado.
Aos tubos contendo o precipitado já seco adicionou-se 150 µl da solução
Sephaglastm FP e misturou-se os mesmos em vórtex por 1 minuto para dissolução.
Um ciclo de centrifugação por 15 segundos foi realizado e o sobrenadante foi
descartado sem perturbar o precipitado.
Duzentos microlitros do tampão de lavagem (fornecido pelo kit) foram
utilizados para ressuspensão através do uso do vórtex. Um ciclo de centrifugação
a velocidade máxima por 15 segundos foi realizado removendo-se o sobrenadante
em seguida. Trezentos microlitros de etanol 70% foi adicionado, seguido de
centrifugação a velocidade máxima por 15 segundos e subseqüente remoção do
sobrenadante. Os tubos foram abertos por cerca de 10 minutos para ocorrer a
secagem do precipitado.
Cinqüenta microlitros de tampão TE (Tris 50 mM, EDTA 10 mM) foram
adicionados e o precipitado foi ressuspendido através do uso do vórtex. Um último
ciclo de centrifugação a velocidade máxima por 1 minuto foi realizado. Transferiu-
se os sobrenadantes contendo o DNA para novos tubos. Os que continham o
precipitado foram descartados.

55
Para a confirmação da presença dos insertos nos plasmídeos, foram
realizados PCRs, seguindo as mesmas condições utilizadas no item anterior,
utilizando-se os primers para amplificação da RNA polimerase dependente de
RNA e do capsídeo viral. Esses PCRs foram feitos a partir das minipreparações de
plasmídeos.
3.7. Seqüenciamento
Os produtos de PCR e os clones foram seqüenciados automaticamente, em
ambas as direções através do uso dos primers sense e anti-sense para a região 3´
da RNA polimerase dependente de RNA H229 e C547 (Minafra & Hadidi, 1994) e
para o gene do capsídeo viral LR3-8504V e LR3-9445C (Ling et al., 1997). As
reações de seqüenciamento nucleotídico foram realizadas pelo método “Taq Dye-
terminator”, no Sistema Megabace (Amersham, Pharmacia).
3.8. Análise das Seqüências
A qualidade das seqüências obtidas nas reações de seqüenciamento
automático foi verificada por PHRED (Base-Calling Software with Quality
Information) (Erwing et al., 1998). As homologias das seqüências nucleotídicas
obtidas foram determinadas pelo programa BLAST (Basic Local Alignment Search
Tool) (Altschul et al., 1997), e foram alinhadas com seqüências de GLRaV
disponíveis no banco genômico (www.ncbi.nlm.nih.gov). O alinhamento das
seqüências em ambas as direções foi realizado pelo programa CLUSTAL W
(Thompson et al., 1994) e otimizado por inspeção visual. Após a correção das
divergências nas duas direções, as seqüências alinhadas foram traduzidas e

56
alinhadas com as seqüências de nucleotídeos e de aminoácidos de consenso.
Para o gene da RNA polimerase dependente de RNA as seqüências foram
alinhadas com a seqüência completa denominada NY1 e a seqüencia de um
isolado brasileiro cujo código é AF438411. Para o gene do capsídeo viral, o
alinhamento foi feito somente com a seqüência NY1.
3.9. Análise Filogenética
As análises filogenéticas foram conduzidas utilizando-se o programa
PHYLIP (Retief, 2000). A construção da árvore filogenética foi realizada pelo
método de “maximal likehood”, com taxa de transição/transversão de 2,0. O teste
dos ramos internos (1000 réplicas) foi usado para verificação da confiabilidade da
árvore gerada. Na geração das árvores foram utilizadas as seqüências da
polimerase e do capsídeo do isolado de referência NY1 (Ling et al., 1997) e das
amostras Pet-1, Pet-2 e Pet-3.

57
4. RESULTADOS
4.1. ELISA
Os testes foram realizados em amostras provenientes das estacas
recebidas de Petrolina (Fazenda Vitivinícola e Estação Experimental de
Bebedouro) e mantidas em casas de vegetação da Estação Experimental da
Biologia. Das nove estacas testadas inicialmente, duas apresentaram resultados
reagentes em ELISA e foram denominadas Pet-1 e Pet-2. Das sete estacas
recebidas seis meses depois, uma apresentou resultado reagente em ELISA,
sendo da variedade CG351 e foi denominada Pet-3. Os sintomas observados
nessas três hospedeiras foram bem característicos de enrolamento, a saber:
enrolamento dos bordos das folhas para baixo; coloração avermelhada em Pet-1 e
Pet-2 e amarelada em Pet-3 (Figura 11); redução no tamanho quando
comparadas com estacas de mesma idade sadias.
Figura 11. Sintomas em plantas reagentes no teste DAS-ELISA em relação à
infecção por GLRaV-3 em casas de vegetação da Estação Experimental da Biologia - UnB. Observa-se avermelhamento entre as nervuras e enrolamento dos bordos para baixo.

58
A tabela 5 mostra os valores de absorbância obtidos para as três plantas
em ELISA. Todas as reações foram realizadas em triplicata para o cálculo da
média das absorbâncias obtidas.
Os controles positivos e negativos foram fornecidos pelo kit e eram
compostos, respectivamente, por extratos de plantas de videira contaminadas e
livres de vírus.
Tabela 5. Absorbância obtida em leitora de ELISA, em comprimento de onda de 405 nm, para amostras consideradas reagentes em ELISA e originárias do Vale do São Francisco.
Absorbância (405nm) Média da Absorbância
Controle Positivo
1 – 0.386
2 – 0.430
3 – 0.407
0.407
Controle Negativo
1 – 0.125
2 – 0.123
3 – 0.121
0.123
Pet-1
1 – 0.389
2 – 0.373
3 – 0.341
0.367
Pet-2
1 – 0.286
2 – 0.283
3 – 0.284
0.284
Pet-3
1 – 0.273
2 – 0.264
3 – 0.347
0.295
Controle do Substrato
1 – 0.130
2 – 0.131
3 – 0.133
0.131

59
4.2. Métodos de extração de RNA
Dentre todos os métodos testados, o desenvolvido por MacKenzie et al.
(1997) foi o que mostrou melhor resultado durante os testes preliminares, por
fornecer RNA de boa qualidade para as sínteses de cDNA. Os métodos de
Minafra & Hadidi (1994), Rowhani et al. (1993), Rowhani et al. (1995) e o
procedimento de Sambrook, modificado por Câmara et al. (2001) não
apresentaram bons resultados na síntese de cDNA.
4.3. Amplificação do gene do capsídeo viral e da re gião 3´ da RNA
polimerase
As amostras foram amplificadas para o gene do capsídeo viral (CP) e da
região 3´ do gene da RNA polimerase dependente de RNA (RdRp), obtendo-se
fragmentos de 942 e 340 pares de bases, respectivamente (Figuras 12 e 13).
Figura 12. Gel de agarose a 1,5%. Os poços 3 e 4 mostram a amplificação do gene do capsídeo viral com 942 pb, o poço 2 mostra o controle negativo, o poço 1 não possui amostras. M: marcador 100 pb ladder.

60
1 2 3 4 5 M
Figura 13. Gel de agarose a 1,5%. Os poços de 2 a 5 mostram a amplificação de parte do gene da polimerase viral com 340 pb, o poço 1 mostra o controle negativo. M: marcador 100 pb ladder.
4.4. Clonagem dos segmentos de DNA.
As amostras Pet-1, Pet-2 e Pet-3 tiveram dois produtos de PCR distintos
clonados em vetor pGEM T easy (Promega). Após a clonagem, feitas as
minipreparações de plasmídeos, foi possível observar a presença das bandas
esperadas obtidas por meio de reações de PCR. As figuras 14 e 15 ilustram o
PCR das minipreparações de DNA com os segmentos da polimerase e do
capsídeo respectivamente.

61
M 1 3 5 7 9 11 13 15 17 19
Figura 14. PCR das minipreparações plasmidiais com o inserto da RNA
polimerase. Poços 1, 3, 5, 7, 9, 11, 13, 15, 17 e 19 mostram o resultado dos PCRs; M = marcador 100 pb ladder; Primers H229 e C547 (Ling et al., 1998).
M 1 2 3 4 5
Figura 15 . PCR das minipreparações dos plasmídeos com o inserto do capsídeo. Poços 1 e 5 contém os resultados do PCR. Poços 2 e 4 são vazios. M = marcador 100 pb ladder. Primers LR3-9445C e LR3-8504V.
4.5. Análise do seqüenciamento
4.5.1. Seqüenciamento da região 3´ do gene da polim erase viral
Após o seqüenciamento e a análise das seqüências obtidas por meio do
programa BLAST (http://www.ncbi.nlm.nih.gov/BLAST), as três amostras
apresentaram substituições de nucleotídeos quando comparadas entre si e com
isolados já descritos (Figura 16). A seqüência de nucleotídeos do primeiro e
340 pb

62
segundo isolados (Pet-1 e Pet-2) apresentaram 98% e 94% de identidade com os
isolados NY1 (NC_004667, norte-americano) e AF438411 (Isolado do sul do
Brasil) (Fajardo et al., 2002), respectivamente. Pet-3 apresentou um polimorfismo
na posição 8418. Alguns clones apresentaram uma adenina e alguns
apresentaram uma guanina nesta posição. Quando a adenina estava presente,
Pet-3 apresentava identidade de 98% e 96% com os isolados NY1 e AF438411
respectivamente. Quando a guanina estava presente, demonstrava identidade de
99% e 96%.
A seqüência de nucleotídeos dos três isolados foi traduzida e os
aminoácidos deduzidos foram comparados. Pet-1 e Pet-3 apresentaram
identidade de 100% com NY1 e 95% com AF438411. Pet-2 apresentou identidade
de 98% e 94% respectivamente (Figura 17). A troca de aminoácidos nas posições
2718 (de alanina para valina), 2736 (de alanina para valina) e 2758 (de leucina
para isoleucina) ocorrida no isolado AF438411 não foi observada em nenhum dos
isolados. O isolado Pet-2 apresentou uma troca conservativa de aminoácido na
posição 2766 (de fenilalanina para tirosina).

63
....|....| ....|....| ....|....| ....|....| ....|....| ....|....| 8271 8281 8291 8301 8311 8321 NY1 ATAAGCATTC GGGATGGACC TACTCGGCTT TGTGTGTCTT GCACGTTTTA AGTGCAAATT Pet-1 .......... .......... .......... .A........ .......... .......... Pet-2 .......... .......... .......... .......... .......... .......... Pet-3 .......... .......... .......... .......... .......... .......... AF438411 .......... .......... .......... .......... .....C.... .....T.... ....|....| ....|....| ....|....| ....|....| ....|....| ....|....| 8331 8341 8351 8361 8371 8381 NY1 TTTCGCAGTT CTGTAGGTTA TATTACCACA ATAGCGTGAA TCTCGATGTG CGCCCTATTC Pet-1 .......... .......... .......... .......... ...T...... .......... Pet-2 .......... .......... .......... .......... ...T..C... .......... Pet-3 .......... .......... .......... .......... ...T...... .......... AF438411 .......... T......... .......... ......C... .......... .......... ....|....| ....|....| ....|....| ....|....| ....|....| ....|....| 8391 8401 8411 8421 8431 8441 NY1 AGAGGACCGA GTCGCTTTCC TTGCTGGCCT TGAAGGCAAG AATTTTAAGG TGGAAAGCTT Pet-1 .......... .......... .......... .......... .......... .......... Pet-2 .......... .......... .......... .......C.. .......C.. .......... Pet-3 .......... .......... .......... .*........ .......... .......... AF438411 .......... .......... .......... .A........ .C........ .......... ....|....| ....|....| ....|....| ....|....| ....|....| ....|....| 8451 8461 8471 8481 8491 8501 NY1 CTCGTTTTGC CTTTTCGATA AAGAGGGGTT AATCGCGTTG GCCACGCTAT AGTGTTTCTG Pet-1 .......... .......... .......... .......... .T........ .......... Pet-2 ......A... .......... .......... .......... ...G...... .......... Pet-3 .......... .......... .......... .......... .......... .C........ AF438411 .......... .........T .......... .......... C......... .C........ ....|....| ....|....| ....|....| ....|....| ....|....| ....|....| 8511 8521 8531 8541 8551 8561 NY1 TGCCTCGGTT CTTCGTGAGG TTAATACCGA AGGGTCGTCG TACTTATCTC AGTTATTTAT Pet-1 .......... .......... .......... .......... .......... ..C...C... Pet-2 .......... .......... .......... .......... .......... .......... Pet-3 .......... .......... .......... .......... .......... .......... AF438411 ......T... .......... ......T... .......... .......T.. .......... ....|....| ....|....| ....|....| ....|....| 8571 8581 8591 8601 NY1 TTTTTCGTCT TCTCTTAGGC GTGCCATCCG TGAAGTTAAT Pet-1 ...C...... .......... .......... .......... Pet-2 .......... .......... .......... .......... Pet-3 .......... ...G...... .......... .......... AF438411 .......... ...G...... .......... ..........
Figura 16. Alinhamento das seqüências de nucleotídeos da região 3´ da polimerase de isolados de GLRaV-3 do Vale do São Francisco (Pet-1, Pet-2 e Pet-3) com isolados disponíveis no Genbank (NY1: isolado descrito por Ling et al., 1998; AF438411: isolado descrito por Fajardo et al., 2002). Os pontos mostram equivalência de nucleotídeos na referida posição. Nucleotídeos em negrito representam o códon de terminação do gene da polimerase. O asterisco mostra a posição onde um número igual de seqüências apontou a presença tanto de A quanto de G.

64
Pet-1 8269 AAG CAT TCG GGA TGG ACC TAC TCG GCT TTA TGT GTC TTG CAC GTT 8313 Pet-2 8269 AAG CAT TCG GGA TGG ACC TAC TCG GCT TTG TGT GTC TTG CAC GTT 8313 Pet-3 8269 AAG CAT TCG GGA TGG ACC TAC TCG GCT TTG TGT GTC TTG CAC GTT 8313 Pet-1 2704 K H S G W T Y S A L C V L H V 2718 Pet-2 2704 K H S G W T Y S A L C V L H V 2718 Pet-3 2704 K H S G W T Y S A L C V L H V 2718 NY1 2704 K H S G W T Y S A L C V L H V 2718 AF438411 2704 K H S G W T Y S A L C V L H A 2718 Pet-1 8314 TTA AGT GCA AAT TTT TCG CAG TTC TGT AGG TTA TAT TAC CAC AAT 8358 Pet-2 8314 TTA AGT GCA AAT TTT TCG CAG TTC TGT AGG TTA TAT TAC CAC AAT 8358 Pet-3 8314 TTA AGT GCA AAT TTT TCG CAG TTC TGT AGG TTA TAT TAC CAC AAT 8358 Pet-1 2719 L S A N F S Q F C R L Y Y H N 2734 Pet-2 2719 L S A N F S Q F C R L Y Y H N 2734 Pet-3 2719 L S A N F S Q F C R L Y Y H N 2734 NY1 2719 L S A N F S Q F C R L Y Y H N 2734 AF438411 2719 L S A N F S Q F C R L Y Y H N 2734 Pet-1 8359 AGC GTG AAT CTT GAT GTG CGC CCT ATT CAG AGG ACC GAG TCG CTT 8403 Pet-2 8359 AGC GTG AAT CTT GAC GTG CGC CCT ATT CAG AGG ACC GAG TCG CTT 8403 Pet-3 8359 AGC GTG AAT CTT GAT GTG CGC CCT ATT CAG AGG ACC GAG TCG CTT 8403 Pet-1 2735 S V N L D V R P I Q R T E S L 2749 Pet-2 2735 S V N L D V R P I Q R T E S L 2749 Pet-3 2735 S V N L D V R P I Q R T E S L 2749 NY1 2735 S V N L D V R P I Q R T E S L 2749 AF438411 2735 S A N L D V R P I Q R T E S L 2749 Pet-1 8404 TCC TTG CTG GCC TTG AAG GCA AGA ATT TTA AGG TGG AAA GCT TCT 8448 Pet-2 8404 TCC TTG CTG GCC TTG AAG GCC AGA ATT TTA CGG TGG AAA GCT TCT 8448 Pet-3 8404 TCC TTG CTG GCC TTA AAG GCA AGA ATT TTA AGG TGG AAA GCT TCT 8448 Pet-1 2750 S L L A L K A R I L R W K A S 2764 Pet-2 2750 S L L A L K A R I L R W K A S 2764 Pet-3 2750 S L L A L K A R I L R W K A S 2764 NY1 2750 S L L A L K A R I L R W K A S 2764 AF438411 2750 S L L A L K A R L L R W K A S 2764 Pet-1 8449 CGT TTT GCC TTT TCG ATA AAG AGG GGT TAA Pet-2 8449 CGT TAT GCC TTT TCG ATA AAG AGG GGT TAA Pet-3 8449 CGT TTT GCC TTT TCG ATA AAG AGG GGT TAA Pet-1 2765 R F A F S I K R G * Pet-2 2765 R Y A F S I K R G * Pet-3 2765 R F A F S I K R G * NY1 2765 R F A F S I K R G * AF438411 2765 R F A F S I K R G *
Figura 17. Alinhamento das seqüências de aminoácidos deduzidas da região 3´
do gene da polimerase de isolados de GLRaV-3. Nucleotídeos em negrito representam trocas de bases com relação a dois isolados estudados. Mudanças na seqüência de aminoácidos dos isolados Pet-1, Pet-2 e Pet-3 comparados com NY1 e AF438411 são marcados com uma barra cinza. A substituição ocorrida somente em um isolado foi marcada com uma barra escura.

65
4.5.2. Seqüenciamento do gene do capsídeo
Foi possível seqüenciar um segmento de tamanho variável do gene do
capsídeo para cada isolado do GLRaV-3: Para Pet-1 foi seqüenciado um
segmento de 801 pb; para Pet-2 um segmento de 768 pb e para Pet-3 um
segmento de 708 pb.
As seqüências geradas foram alinhadas e comparadas à única seqüência
de GLRaV-3 disponível (NC_004667). Observou-se que os isolados da região do
São Francisco apresentaram grande homologia de nucleotídeos com o isolado
norte-americano NY1 (NC_004667), mantendo uma similaridade de 99% na região
seqüenciada (Figura 18). Foram observadas treze substituições de nucleotídeos.
Desse total, duas foram comuns aos três isolados: posição 13511 (de citosina
para timina) e posição 14113 (de timina para citosina). A substituição na posição
13511 implicou em uma troca de aminoácido na posição 3982, de alanina para
valina, considerada conservativa. Essa foi a substituição encontrada nos 3
isolados seqüenciados em relação a seqüência de aminoácidos do isolado NY1
(Figura 19). Com relação ao isolado Pet-1, foram verificadas substituições não
conservativas nas posições 4025 (de alanina para treonina), 4033 (de valina para
serina) e 4070 (de glutamato para lisina). Na seqüência do isolado Pet-2 foi
detectada uma substituição não conservativa na posição 3962 (de treonina para
alanina). O isolado Pet-3 apresentou substituições também não conservativas nas
posições 4025 (de alanina para treonina) e 4070 (de glutamato para lisina).
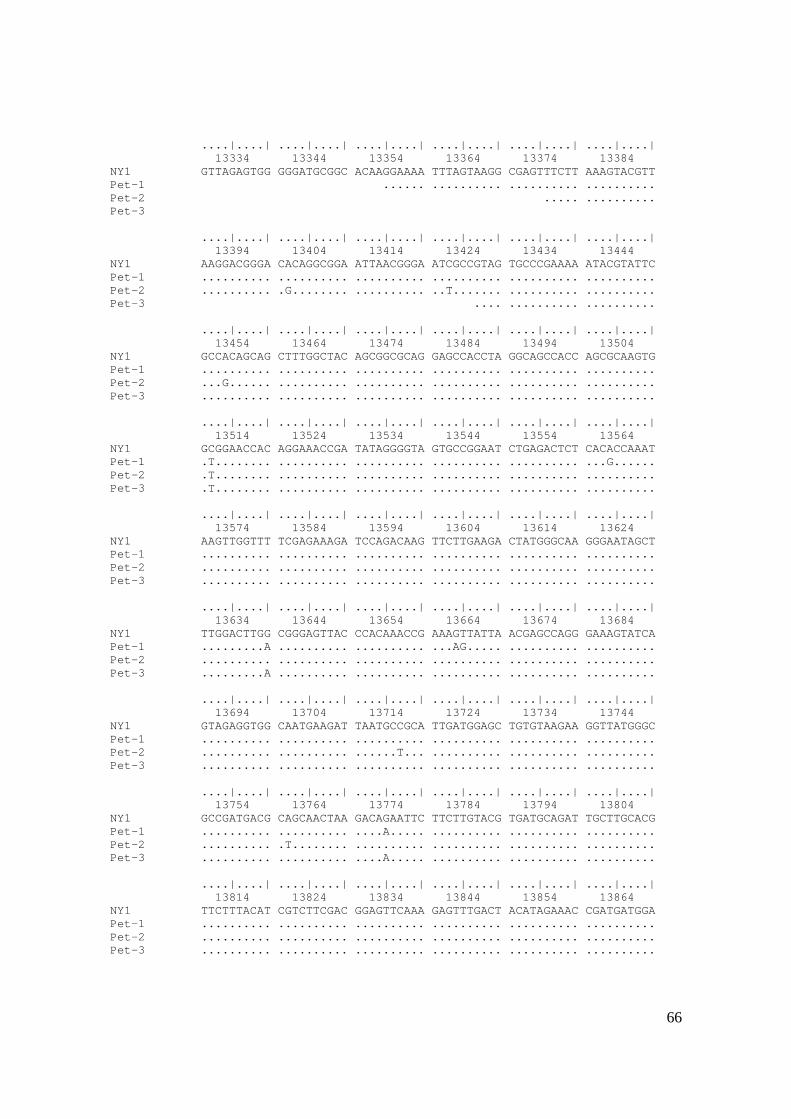
66
....|....| ....|....| ....|....| ....|....| ....|....| ....|....| 13334 13344 13354 13364 13374 13384 NY1 GTTAGAGTGG GGGATGCGGC ACAAGGAAAA TTTAGTAAGG CGAGTTTCTT AAAGTACGTT Pet-1 ...... .......... .......... .......... Pet-2 ..... .......... Pet-3 ....|....| ....|....| ....|....| ....|....| ....|....| ....|....| 13394 13404 13414 13424 13434 13444 NY1 AAGGACGGGA CACAGGCGGA ATTAACGGGA ATCGCCGTAG TGCCCGAAAA ATACGTATTC Pet-1 .......... .......... .......... .......... .......... .......... Pet-2 .......... .G........ .......... ..T....... .......... .......... Pet-3 .... .......... .......... ....|....| ....|....| ....|....| ....|....| ....|....| ....|....| 13454 13464 13474 13484 13494 13504 NY1 GCCACAGCAG CTTTGGCTAC AGCGGCGCAG GAGCCACCTA GGCAGCCACC AGCGCAAGTG Pet-1 .......... .......... .......... .......... .......... .......... Pet-2 ...G...... .......... .......... .......... .......... .......... Pet-3 .......... .......... .......... .......... .......... .......... ....|....| ....|....| ....|....| ....|....| ....|....| ....|....| 13514 13524 13534 13544 13554 13564 NY1 GCGGAACCAC AGGAAACCGA TATAGGGGTA GTGCCGGAAT CTGAGACTCT CACACCAAAT Pet-1 .T........ .......... .......... .......... .......... ...G...... Pet-2 .T........ .......... .......... .......... .......... .......... Pet-3 .T........ .......... .......... .......... .......... .......... ....|....| ....|....| ....|....| ....|....| ....|....| ....|....| 13574 13584 13594 13604 13614 13624 NY1 AAGTTGGTTT TCGAGAAAGA TCCAGACAAG TTCTTGAAGA CTATGGGCAA GGGAATAGCT Pet-1 .......... .......... .......... .......... .......... .......... Pet-2 .......... .......... .......... .......... .......... .......... Pet-3 .......... .......... .......... .......... .......... .......... ....|....| ....|....| ....|....| ....|....| ....|....| ....|....| 13634 13644 13654 13664 13674 13684 NY1 TTGGACTTGG CGGGAGTTAC CCACAAACCG AAAGTTATTA ACGAGCCAGG GAAAGTATCA Pet-1 .........A .......... .......... ...AG..... .......... .......... Pet-2 .......... .......... .......... .......... .......... .......... Pet-3 .........A .......... .......... .......... .......... .......... ....|....| ....|....| ....|....| ....|....| ....|....| ....|....| 13694 13704 13714 13724 13734 13744 NY1 GTAGAGGTGG CAATGAAGAT TAATGCCGCA TTGATGGAGC TGTGTAAGAA GGTTATGGGC Pet-1 .......... .......... .......... .......... .......... .......... Pet-2 .......... .......... ......T... .......... .......... .......... Pet-3 .......... .......... .......... .......... .......... .......... ....|....| ....|....| ....|....| ....|....| ....|....| ....|....| 13754 13764 13774 13784 13794 13804 NY1 GCCGATGACG CAGCAACTAA GACAGAATTC TTCTTGTACG TGATGCAGAT TGCTTGCACG Pet-1 .......... .......... ....A..... .......... .......... .......... Pet-2 .......... .T........ .......... .......... .......... .......... Pet-3 .......... .......... ....A..... .......... .......... .......... ....|....| ....|....| ....|....| ....|....| ....|....| ....|....| 13814 13824 13834 13844 13854 13864 NY1 TTCTTTACAT CGTCTTCGAC GGAGTTCAAA GAGTTTGACT ACATAGAAAC CGATGATGGA Pet-1 .......... .......... .......... .......... .......... .......... Pet-2 .......... .......... .......... .......... .......... .......... Pet-3 .......... .......... .......... .......... .......... ..........

67
....|....| ....|....| ....|....| ....|....| ....|....| ....|....| 13874 13884 13894 13904 13914 13924 NY1 AAGAAGATAT ATGCGGTGTG GGTATATGAT TGCATTAAAC AAGCTGCTGC TTCGACGGGT Pet-1 .......... .......... .......... .......... .......... .......... Pet-2 .......... .......... .......... .......... .......... .......... Pet-3 .......... .......... .......... .......... .......... .......... ....|....| ....|....| ....|....| ....|....| ....|....| ....|....| 13934 13944 13954 13964 13974 13984 NY1 TATGAAAACC CGGTAAGGCA GTATCTAGCG TACTTCACAC CAACCTTCAT CACGGCGACC Pet-1 .......... .......... .......... .......... .......... T......... Pet-2 .......... .......... .......... .......... .......... .......... Pet-3 .......... .......... .......... .......... .......... .......... ....|....| ....|....| ....|....| ....|....| ....|....| ....|....| 13994 14004 14014 14024 14034 14044 NY1 CTGAATGGTA AACTAGTGAT GAACGAGAAG GTTATGGCAC AGCATGGAGT ACCACCGAAA Pet-1 .......... .......... .......... .......... .......... .......... Pet-2 .......... .......... .......... .......... .......... .......... Pet-3 .......... .......... .......... .......... .......... .......... ....|....| ....|....| ....|....| ....|....| ....|....| ....|....| 14054 14064 14074 14084 14094 14104 NY1 TTCTTTCCGT ACACGATAGA CTGCGTTCGT CCGACGTACG ATCTGTTCAA CAACGACGCA Pet-1 .......... .......... .......... .......... .......... .......... Pet-2 .......... .......... .......... .......... .......... .......... Pet-3 .......... .......... .......... .......... .......... .......... ....|....| ....|....| ....|....| ....|....| ....|....| ....|....| 14114 14124 14134 14144 14154 14164 NY1 ATATTAGCAT GGAATTTAGC TAGACAGCAG GCGTTTAGAA ACAAGACGGT AACGGCCGAT Pet-1 ...C...... .......... .......... .......... ..... Pet-2 ...C...... .......... .......... ... Pet-3 ...C...... .......... ....
Figura 18. Alinhamento das seqüências de nucleotídeos da proteína do capsídeo (CP) de isolados de GLRaV-3 do Vale do São Francisco (Pet-1, Pet-2 e Pet-3) com o único isolado disponível no GenBank (NY1: isolado descrito por Ling et al., 1998). Os pontos mostram equivalência de nucleotídeos na referida posição.

68
Pet-1 13360 TTT AGT AAG GCG AGT TTC TTA AAG TAC GTT AAG GAC GGG ACA CAG 13404 Pet-2 13360 TTC TTA AAG TAC GTT AAG GAC GGG ACG CAG 13404 Pet-3 13360 13404 Pet-1 3931 F S K A S F L K Y V K D G T Q 3945 Pet-2 3931 F L K Y V K D G T Q 3945 Pet-3 3931 3945 NY1 3931 F S K A S F L K Y V K D G T Q 3945 Pet-1 13405 GCG GAA TTA ACG GGA ATC GCC GTA GTG CCC GAA AAA TAC GTA TTC 13449 Pet-2 13405 GCG GAA TTA ACG GGA ATT GCC GTA GTG CCC GAA AAA TAC GTA TTC 13449 Pet-3 13405 GTA GTG CCC GAA AAA TAC GTA TTC 13449 Pet-1 3946 A E L T G I A V V P E K Y V F 3960 Pet-2 3946 A E L T G I A V V P E K Y V F 3960 Pet-3 3946 V V P E K Y V F 3960 NY1 3946 A E L T G I A V V P E K Y V F 3960 Pet-1 13450 GCC ACA GCA GCT TTG GCT ACA GCG GCG CAG GAG CCA CCT AGG CAG 13494 Pet-2 13450 GCC GCA GCA GCT TTG GCT ACA GCG GCG CAG GAG CCA CCT AGG CAG 13494 Pet-3 13450 GCC ACA GCA GCT TTG GCT ACA GCG GCG CAG GAG CCA CCT AGG CAG 13494 Pet-1 3961 A T A A L A T A A Q E P P R Q 3975 Pet-2 3961 A A A A L A T A A Q E P P R Q 3975 Pet-3 3961 A T A A L A T A A Q E P P R Q 3975 NY1 3961 A T A A L A T A A Q E P P R Q 3975 Pet-1 13495 CCA CCA GCG CAA GTG GTG GAA CCA CAG GAA ACC GAT ATA GGG GTA 13539 Pet-2 13495 CCA CCA GCG CAA GTG GTG GAA CCA CAG GAA ACC GAT ATA GGG GTA 13539 Pet-3 13495 CCA CCA GCG CAA GTG GTG GAA CCA CAG GAA ACC GAT ATA GGG GTA 13539 Pet-1 3976 P P A Q V V E P Q E T D I G V 3991 Pet-2 3976 P P A Q V V E P Q E T D I G V 3991 Pet-3 3976 P P A Q V V E P Q E T D I G V 3991 NY1 3976 P P A Q V A E P Q E T D I G V 3991 Pet-1 13540 GTG CCG GAA TCT GAG ACT CTC ACG CCA AAT AAG TTG GTT TTC GAG 13584 Pet-2 13540 GTG CCG GAA TCT GAG ACT CTC ACA CCA AAT AAG TTG GTT TTC GAG 13584 Pet-3 13540 GTG CCG GAA TCT GAG ACT CTC ACA CCA AAT AAG TTG GTT TTC GAG 13584 Pet-1 3992 V P E S E T L T P N K L V F E 4006 Pet-2 3992 V P E S E T L T P N K L V F E 4006 Pet-3 3992 V P E S E T L T P N K L V F E 4006 NY1 3992 V P E S E T L T P N K L V F E 4006 Pet-1 13585 AAA GAT CCA GAC AAG TTC TTG AAG ACT ATG GGC AAG GGA ATA GCT 13629 Pet-2 13585 AAA GAT CCA GAC AAG TTC TTG AAG ACT ATG GGC AAG GGA ATA GCT 13629 Pet-3 13585 AAA GAT CCA GAC AAG TTC TTG AAG ACT ATG GGC AAG GGA ATA GCT 13629 Pet-1 4007 K D P D K F L K T M G K G I A 4021 Pet-2 4007 K D P D K F L K T M G K G I A 4021 Pet-3 4007 K D P D K F L K T M G K G I A 4021 NY1 4007 K D P D K F L K T M G K G I A 4021 Pet-1 13630 TTG GAC TTG ACG GGA GTT ACC CAC AAA CCG AAA AGT ATT AAC GAG 13674 Pet-2 13630 TTG GAC TTG GCG GGA GTT ACC CAC AAA CCG AAA GTT ATT AAC GAG 13674 Pet-3 13630 TTG GAC TTG ACG GGA GTT ACC CAC AAA CCG AAA GTT ATT AAC GAG 13674 Pet-1 4022 L D L T G V T H K P K S I N E 4036 Pet-2 4022 L D L A G V T H K P K V I N E 4036 Pet-3 4022 L D L T G V T H K P K V I N E 4036 NY1 4022 L D L A G V T H K P K V I N E 4036 Pet-1 13675 CCA GGG AAA GTA TCA GTA GAG GTG GCA ATG AAG ATT AAT GCC GCA 13719 Pet-2 13675 CCA GGG AAA GTA TCA GTA GAG GTG GCA ATG AAG ATT AAT GCT GCA 13719 Pet-3 13675 CCA GGG AAA GTA TCA GTA GAG GTG GCA ATG AAG ATT AAT GCC GCA 13719 Pet-1 4037 P G K V S V E V A M K I N A A 4051 Pet-2 4037 P G K V S V E V A M K I N A A 4051 Pet-3 4037 P G K V S V E V A M K I N A A 4051 NY1 4037 P G K V S V E V A M K I N A A 4051

69
Pet-1 13720 TTG ATG GAG CTG TGT AAG AAG GTT ATG GGC GCC GAT GAC GCA GCA 13764 Pet-2 13720 TTG ATG GAG CTG TGT AAG AAG GTT ATG GGC GCC GAT GAC GCT GCA 13764 Pet-3 13720 TTG ATG GAG CTG TGT AAG AAG GTT ATG GGC GCC GAT GAC GCA GCA 13764 Pet-1 4052 L M E L C K K V M G A D D A A 4066 Pet-2 4052 L M E L C K K V M G A D D A A 4066 Pet-3 4052 L M E L C K K V M G A D D A A 4066 NY1 4052 L M E L C K K V M G A D D A A 4066 Pet-1 13765 ACT AAG ACA AAA TTC TTC TTG TAC GTG ATG CAG ATT GCT TGC ACG 13809 Pet-2 13765 ACT AAG ACA GAA TTC TTC TTG TAC GTG ATG CAG ATT GCT TGC ACG 13809 Pet-3 13765 ACT AAG ACA AAA TTC TTC TTG TAC GTG ATG CAG ATT GCT TGC ACG 13809 Pet-1 4067 T K T K F F L Y V M Q I A C T 4081 Pet-2 4067 T K T E F F L Y V M Q I A C T 4081 Pet-3 4067 T K T K F F L Y V M Q I A C T 4081 NY1 4067 T K T E F F L Y V M Q I A C T 4081 Pet-1 13810 TTC TTT ACA TCG TCT TCG ACG GAG TTC AAA GAG TTT GAC TAC ATA 13854 Pet-2 13810 TTC TTT ACA TCG TCT TCG ACG GAG TTC AAA GAG TTT GAC TAC ATA 13854 Pet-3 13810 TTC TTT ACA TCG TCT TCG ACG GAG TTC AAA GAG TTT GAC TAC ATA 13854 Pet-1 4082 F F T S S S T E F K E F D Y I 4096 Pet-2 4082 F F T S S S T E F K E F D Y I 4096 Pet-3 4082 F F T S S S T E F K E F D Y I 4096 NY1 4082 F F T S S S T E F K E F D Y I 4096 Pet-1 13855 GAA ACC GAT GAT GGA AAG AAG ATA TAT GCG GTG TGG GTA TAT GAT 13899 Pet-2 13855 GAA ACC GAT GAT GGA AAG AAG ATA TAT GCG GTG TGG GTA TAT GAT 13899 Pet-3 13855 GAA ACC GAT GAT GGA AAG AAG ATA TAT GCG GTG TGG GTA TAT GAT 13899 Pet-1 4097 E T D D G K K I Y A V W V Y D 4111 Pet-2 4097 E T D D G K K I Y A V W V Y D 4111 Pet-3 4097 E T D D G K K I Y A V W V Y D 4111 NY1 4097 E T D D G K K I Y A V W V Y D 4111 Pet-1 13900 TGC ATT AAA CAA GCT GCT GCT TCG ACG GGT TAT GAA AAC CCG GTA 13944 Pet-2 13900 TGC ATT AAA CAA GCT GCT GCT TCG ACG GGT TAT GAA AAC CCG GTA 13944 Pet-3 13900 TGC ATT AAA CAA GCT GCT GCT TCG ACG GGT TAT GAA AAC CCG GTA 13944 Pet-1 4112 C I K Q A A A S T G Y E N P V 4126 Pet-2 4112 C I K Q A A A S T G Y E N P V 4126 Pet-3 4112 C I K Q A A A S T G Y E N P V 4126 NY1 4112 C I K Q A A A S T G Y E N P V 4126 Pet-1 13945 AGG CAG TAT CTA GCG TAC TTC ACA CCA ACC TTC ATT ACG GCG ACC 13989 Pet-2 13945 AGG CAG TAT CTA GCG TAC TTC ACA CCA ACC TTC ATC ACG GCG ACC 13989 Pet-3 13945 AGG CAG TAT CTA GCG TAC TTC ACA CCA ACC TTC ATC ACG GCG ACC 13989 Pet-1 4127 R Q Y L A Y F T P T F I T A T 4141 Pet-2 4127 R Q Y L A Y F T P T F I T A T 4141 Pet-3 4127 R Q Y L A Y F T P T F I T A T 4141 NY1 4127 R Q Y L A Y F T P T F I T A T 4141 Pet-1 13990 CTG AAT GGT AAA CTA GTG ATG AAC GAG AAG GTT ATG GCA CAG CAT 14034 Pet-2 13990 CTG AAT GGT AAA CTA GTG ATG AAC GAG AAG GTT ATG GCA CAG CAT 14034 Pet-3 13990 CTG AAT GGT AAA CTA GTG ATG AAC GAG AAG GTT ATG GCA CAG CAT 14034 Pet-1 4142 L N G K L V M N E K V M A Q H 4156 Pet-2 4142 L N G K L V M N E K V M A Q H 4156 Pet-3 4142 L N G K L V M N E K V M A Q H 4156 NY1 4142 L N G K L V M N E K V M A Q H 4156

70
Pet-1 14035 GGA GTA CCA CCG AAA TTC TTT CCG TAC ACG ATA GAC TGC GTT CGT 14079 Pet-2 14035 GGA GTA CCA CCG AAA TTC TTT CCG TAC ACG ATA GAC TGC GTT CGT 14079 Pet-3 14035 GGA GTA CCA CCG AAA TTC TTT CCG TAC ACG ATA GAC TGC GTT CGG 14079 Pet-1 4157 G V P P K F F P Y T I D C V R 4171 Pet-2 4157 G V P P K F F P Y T I D C V R 4171 Pet-3 4157 G V P P K F F P Y T I D C V R 4171 NY1 4157 G V P P K F F P Y T I D C V R 4171 Pet-1 14080 CCG ACG TAC GAT CTG TTC AAC AAC GAC GCA ATA CTA GCA TGG AAT 14124 Pet-2 14080 CCG ACG TAC GAT CTG TTC AAC AAC GAC GCA ATA CTA GCA TGG AAT 14124 Pet-3 14080 CCG ACG TAC GAT CTG TTC AAC AAC GAC GCA ATA CTA GCA TGG AAT 14124 Pet-1 4172 P T Y D L F N N D A I L A W N 4186 Pet-2 4172 P T Y D L F N N D A I L A W N 4186 Pet-3 4172 P T Y D L F N N D A I L A W N 4186 NY1 4172 P T Y D L F N N D A I L A W N 4186 Pet-1 14125 TTA GCT AGA CAG CAG GCG TTT AGA AAC AAG 14154 Pet-2 14125 TTA GCT AGA CAG CAG GCG 14142 Pet-3 14125 TTA GCT AGA 14133 Pet-1 4187 L A R Q Q A F R N K 4196 Pet-2 4187 L A R Q Q A 4192 Pet-3 4187 L A R 4189 NY1 4187 L A R Q Q A F R N K T V T A D 4215
Figura 19. Alinhamento das seqüências de aminoácidos deduzidas do gene do
capsídeo de isolados de GLRaV-3. Nucleotídeos em negrito representam trocas de bases em relação a um isolado estudado. Mudanças na seqüência de aminoácidos dos isolados Pet-1, Pet-2 e Pet-3 comparados com NY1 são marcados com uma barra cinza.
As figuras 20 e 21 resumem as susbstituições observadas na região 3´ do
gene da RNA polimerase dependente de RNA e do gene do capsídeo
respectivamente.

71
8 2 9 8
8 3 7 0
8 3 7 3
8 4 1 8
8 4 2 4
8 4 3 4
8 4 5 3
8 4 8 8
8 4 9 0
8 4 9 8
8 5 5 9
8 5 6 3
8 5 7 0
8 5 8 0
NY1 G C T G A A T C A G T T T C Ref. AF43 - - - A - - - - - - - - - - Brasil Pet-1 A T - - - - - T - - C C C - Pet-2 - T C - C C A - G - - - - - Pet-3 - T - A - - - - - C - - - G - - - - - - F - - - - - - -
Y 2766
Figura 20. Alinhamento da seqüência de nucleotídeos e aminoácidos mostrando
as variações nos isolados Pet-1, Pet-2 e Pet-3 com relação às seqüências da polimerase dos isolados NY1 e AF438411. Posições dos nucleotídeos são indicadas na parte de cima da figura. O número se refere ao primeiro nucleotídeo do genoma de GLRaV-3. A seqüência de referência é indicada como Ref. Cada linha indica, da esquerda para a direita, a identificação do isolado e o alinhamento da seqüência de nucleotídeos comparada com a referência. A substituição de aminoácido e sua posição são indicados abaixo da linha, em comparação com o aminoácido original.

72
1 3 4 0 1
1 3 4 2 2
1 3 4 5 3
1 3 5 1 1
1 3 5 6 3
1 3 6 3 9
1 3 6 6 3
1 3 6 6 4
1 3 7 1 6
1 3 7 6 1
1 3 7 7 4
1 3 9 8 0
1 4 1 1 3
NY1 A C A C A G G T C A G C T Ref. Pet-1 - - - T G A A G - - A T C Pet-2 G T G T - - - - T T - - C Pet-3 - - - T - A - - - - A - C - - T A - A V V - - E - -
A 3962
V 3981
T 4025
S 4033
S 4033
K 4070
Figura 21. Alinhamento da seqüência de nucleotídeos e aminoácidos mostrando
as variações nos isolados Pet-1, Pet-2 e Pet-3 com relação à seqüência do capsídeo do isolado NY1. Posições dos nucleotídeos são indicadas na parte de cima da figura. O número se refere ao primeiro nucleotídeo do genoma de GLRaV-3. A seqüência de referência é indicada como Ref. Cada linha indica, da esquerda para a direita, a identificação do isolado e o alinhamento da seqüência de nucleotídeos comparada com a referência. As substituições de aminoácidos e suas posições são indicadas abaixo da linha, em comparação com o aminoácido original.
4.6. Análise Filogenética
A análise filogenética foi realizada utilizando-se o programa PHYLIP para
todas as seqüências obtidas. Foi construída uma árvore utilizando-se o método de
máxima verossimilhança com taxa de transição/transversão de 2,0. Foram
utilizadas as seqüências dos isolados Pet-1, Pet-2, Pet-3 e a seqüência de
referência NY1.

73
Figura 22. Análise fiologenética, realizada pelo método de máxima verossimilhança das seqüências nucleotídicas da polimerase viral e do capsídeo de Pet-1, Pet-2 e Pet-3 comparadas à seqüência de referência NY1 retirada do GenBank. Os números mostrados nos pontos de ramificação foram obtidos pelo teste dos ramos internos e representam valores de consenso para 1000 réplicas.
A análise filogenética das seqüências revelou uma maior proximidade dos
isolados Pet-1 e Pet-2 com relação ao isolado americano NY1. Pet-3 se localizou
em um ramo distinto.

74
5. DISCUSSÃO
As técnicas moleculares usadas atualmente na identificação de
fitopatógenos possuem grande especificidade, eficiência e precisão no
diagnóstico. O uso da técnica de PCR (Polymerase Chain Reacion) envolve o
desenvolvimento de primers (iniciadores da reação) específicos para detecção e
identificação de patógenos (Minafra & Hadidi, 1994; Karasev et al., 1994). Vários
trabalhos já demonstraram a eficiência do PCR na detecção dos GLRaVs, apesar
da existência de compostos presentes na planta com possível ação inibitória da
reação (Mackenzie et al., 1997; Routh et al., 1998; Acheche et al., 1999; Martin et
al., 2000; Ling et al., 2001; Fajardo et al., 2002; Dovas & Katis, 2003). Essa
limitação requer a utilização de técnicas de extração de RNA viral que além de
gerarem uma quantidade satisfatória de ácidos nucléicos, devem também coibir ou
diminuir a ação dos compostos inibitórios (Mackenzie et al., 1997).
Para o GLRaV-3 o uso de transcrição reversa seguido de PCR (RT-PCR –
Reverse Transcription Polymerase Chain Reaction) é recomendado (Rowhani et
al., 1997). O RT-PCR baseia-se na transcrição reversa do DNA a partir do RNA
viral previamente à reação de PCR. Nesse caso, primers randômicos são
utilizados para se complementar o RNA extraído.
Várias cultivares de videira não evidenciam sintomas quando infectadas por
GLRaV, devido à característica latente da infecção ou por fatores como a
variedade ou a idade da planta (Rowhani et al., 1997). O uso de técnicas de RT-
PCR, como os aplicados neste trabalho, são justificados. Outro fator que promove

75
o uso dessas técnicas é a ocorrência de infecções múltiplas, o que torna a
identificação no campo virtualmente impossível (Fajardo et al., 2002).
Neste trabalho verificou-se que todas as amostras tidas como não
reagentes ou reagentes para ELISA tiveram os resultados confirmados no PCR. A
concentração mais elevada de anticorpos e conjugado pode ter contribuído para
uma maior sensibilidade da detecção por ELISA, não revelando diferença, quando
comparados com a detecção por PCR, como verificado por Chevalier et al. (1995),
Acheche et al. (1999) e Dovas & Katis (2003). Esses autores observaram maior
sensibilidade no PCR, detectando o vírus em amostras não reagentes em ELISA.
A diluição dos anticorpos e do conjugado recomendada pelo kit era de 1:100.
Nesse trabalho foi utilizada a diluição 1:50, elevando para o dobro da
concentração de anticorpos recomendada no procedimento, resultando em
aumento da sensibilidade sem comprometimento da especificidade.
Este foi o primeiro estudo molecular de GLRaV-3 afetando videiras no Vale
do São Francisco. Estudos preliminares descreveram o GLRaV-3 nessa região
apenas pela detecção por ELISA (Tavares et al., 2000; Fajardo et al., 2002).
Mesmo não tendo sido verificada a infecção por GLRaV-1, a ocorrência
deste vírus não deve ser descartada já que foi anteriormente verificado que ocorre
a coexistência das duas espécies em uma mesma planta (Monis & Bestwick,
1996), sendo que a ocorrência de GLRaV-3 é mais abrangente (Zimmermann et
al., 1990). Fajardo et al. (2001), trabalhando com plantas da coleção de cultivares
da EMBRAPA – Uva e Vinho e algumas amostras do Submédio do Vale do São
Francisco, detectaram, através de DAS-ELISA, somente GLRaV-3 em amostras
da coleção de cultivares e GLRaV-1 e -3 em amostras do Submédio. O que

76
justifica a presença de GLRaV-1 somente nas amostras do Vale do São Francisco
seria a procedência do material: na região Petrolina/Juazeiro predominam
cultivares que produzem uvas finas de mesa, já na Serra Gaúcha predominam
cultivares voltadas à elaboração de vinho. Como conseqüência desse fato, a
origem dos cultivares ou porta-enxertos utilizados nos plantios de ambas as
regiões é diferente, demonstrando diferenças também em seu estado
fitossanitário.
A utilização do protocolo de MacKenzie et al. (1997) possibilitou a
realização da RT-PCR, resultando em RNA total sem propriedades inibitórias da
reação. Em conjunto com o ELISA, o PCR é útil para indexar matrizes ou material
básico de videira, não tendo limitações que são comuns aos testes sorológicos,
como por exemplo a época do ano, o tipo da amostra e o estado fisiológico da
planta (MacKenzie et al., 1997).
5.1. Análise da região 3´ da polimerase
Foram observadas substituições diferentes em uma mesma posição (8419)
na amostra Pet-3. Essas substituições podem corresponder a subpopulações
virais infectando a mesma planta, o que resultaria em pequenas diferenças na
seqüência. Isso pode ser explicado pois a RNA polimerase dependente de RNA,
exclusiva dos vírus de RNA, não possui um mecanismo de reparo das
substituições de nucleotídeos que ocorrem na fita que está sendo sintetizada.
Conseqüentemente, as substituições são incorporadas às novas fitas, fazendo
com que essas novas mutações façam parte do genoma viral.

77
Os resultados obtidos através da comparação das três seqüências (Pet-1,
Pet-2 e Pet-3) com o isolado AF438411 (também brasileiro) apresentaram uma
diferença de 4 a 6%. Isso indica uma variabilidade que pode ser explicada pelas
diferenças nas regiões geográficas onde essas amostras foram coletadas: Pet-1,
Pet-2 e Pet-3 são do Vale do São Francisco, enquanto que AF438411 foi isolada
na Serra Gaúcha (Fajardo et al., 2002).
5.2. Análise do gene do capsídeo
Foi verificado, ao longo do trabalho, que não foi possível o seqüenciamento
completo do gene do capsídeo utilizando-se os primers recomendados, gerando-
se seqüências cujas extremidades 5´ e 3´ não puderam ser completadas. Esse
problema está sendo abordado por meio do sequenciamento do gene do capsídeo
a partir dos clones, usando primers universais presentes nas extremidades do
vetor de clonagem.
A análise da seqüência de aminoácidos demonstrou que a maior parte das
substituições não são conservativas, o que indica a possibilidade da existência de
variantes, já que a conformação da proteína pode ser afetada por essas
substituições. A ocorrência dessas substituições na proteína do capsídeo podem
influenciar na capacidade de detecção do vírus através de ELISA, já que a
funcionalidade desse método se baseia na especificidade entre os anticorpos e a
proteína capsidial do vírus. Por esse motivo, a utilização de anticorpos policlonais
para a detecção do GLRaV-3, como os utilizados nesse trabalho, é recomendada.

78
5.3. Considerações Finais
A alta homologia de nucleotídeos verificada para o capsídeo (99% para os
três isolados) e para a polimerase viral (média de 96,6% para os três isolados)
confirma a detecção específica deste vírus. As diferenças encontradas podem ser
explicadas pela variabilidade natural que ocorre entre diferentes isolados de um
mesmo vírus.
Proporcionalmente, o gene da polimerase revelou um maior número de
substituições de nucleotídeos quando comparado com as verificadas no gene do
capsídeo. Essas substituições do gene da polimerase resultaram em apenas uma
troca de aminoácido caracterizada como conservativa. Já na investigação do gene
do capsídeo, várias trocas não conservativas foram verificadas. A variação
encontrada no gene da polimerase deve ser necessariamente menor, por ser um
gene que codifica uma proteína que faz parte de um processo vital da replicação
viral, que é a cópia do genoma. Qualquer mudança significativa na funcionalidade
deste gene pode resultar na inativação do processo de infecção e conseqüente
inviabilidade da partícula. Já a proteína do capsídeo pode mostrar maior
variabilidade, sem comprometer a viabilidade da partícula viral, mas dificultando
sensivelmente as técnicas de detecção sorológica. Uma variabilidade maior da
proteína do capsídeo pode comprometer o diagnóstico através desses métodos, o
que mostra a importância do monitoramento constante das seqüências virais.

79
6. CONCLUSÕES
• O GLRaV-3 pode ser detectado tanto por ELISA quanto por RT-PCR.
• Não foi relatada a ocorrência de falsos positivos ou falsos negativos na
detecção por ELISA em relação ao diagnóstico por RT-PCR. Conclui-se
que a detecção por ELISA apresentou sensibilidade e especificidade
elevadas quando comparada com os resultados obtidos por RT-PCR.
• As amostras Pet-1 e Pet-2, isoladas de plantas da variedade Alicante
Bouchet se mostraram mais semelhantes ao isolado de referência NY1 do
que Pet-3, isolado a partir do porta-enxerto CG351.
• A ocorrência de substituições não-conservativas na seqüência da proteína
do capsídeo revelou a importância do monitoramento das seqüências virais,
sendo recomendado o uso de anticorpos policlonais para a detecção do
GLRaV-3.
• O seqüenciamento dos genes da RNA polimerase e do capsídeo
apresentou variações com relação à seqüência de referência, revelando a
possibilidade da existência de variantes de GLRaV-3 de diferentes regiões
geográficas.

80
7. REFERÊNCIAS BIBLIOGRÁFICAS
ACHECHE, H., FATTOUCH, S., M´HIRSI, S., NÉJIB, M. & MARRAKCHI, M. Use
of optimized PCR methods for the detection of GLRaV3: a closterovirus associated with grapevine leafroll in Tunisian grapevine plants. Plant Molecular Biology Reporter. 17: 31-42. 1999.
AGRAN, M.K., TERLIZZI, B.D., MINAFRA, A., SAVINO, V., MARTELLI G.P. &
ASKRI, F. Occurrence of grapevine virus A (GVA) and other closteroviruses in Tunisian grapevine affected by leafroll disease. Vitis 92(1):43-48. 1990.
ALKOWNI, R., ROWHANI, A. & GOLINO, D.A. Partial nucleotide sequence and
molecular detection of a putative new grapevine leafroll associated virus. Phytopathology 92:S3. 2002. (resumo)
ALTSCHUL, S.F., MADDEN, T.L., SCHÄFFER, A.A., ZHANG, J., ZHAND, Z.,
MILLER, W. & LIPMAN, D.J.K. Gapped BLAST and PSI-Blast: a new generation of protein database search programs. Nucleic Acids Research. 325:3389-3402.1997.
ALMEIDA, A.M.R. & LIMA, J.A.A. (Eds.) Princípios e técnicas de diagnose
aplicados em fitovirologia. Publicações SBF. 2001.
BURR, T.J. Crown Gall. In: PEARSON, C. & GOHEEN, R. (Eds) Compendium of grape diseases. APS Press. 41-42. 1998.
CÂMARA, G.N.N.L., CERQUEIRA, D.M., NEVES, D., Silva, E.D., Carvalho, L.G.S., Martins, C.R.F. Caracterização Molecular de Papilomavírus Humano em Mulheres com Lesões no Colo Uterino, em Brasília, DF. Revista de Saúde do Distrito Federal 12. 2001.
CAMPOS, V.P., MAXIMINIANO, C. & FERREIRA, E.A. Doenças causadas por
nematódes. In: FAJARDO, T.V.M. (Ed). Uva para processamento. Fitossanidade. Embrapa informações tecnológicas 72-81. 2003.
CHEVALIER, S., GREIF, C., CLAUZEL, J.M., WALTER, B. & FRITSCH, C. Use of
immunocapture-polymerase chain reaction procedure for the detection of grapevine virus A in Kober Stem Grooving-infected grapevines. Journal of Phytopathology 143:369-373. 1995.
CLARK, M.F. & ADAMS, A.N. Characteristics of the microplate method of enzyme-
linked immunosorbent assay for the detection of plant viruses. Journal of General Virology 34: 475-483. 1977.

81
CREDI, R. & GIUNCHEDI L. Grapevine leafroll-associated viruses and grapevine virus A in selected Vitis vinifera cultivars in northern Italy. Plant Pathology 45:1110-1116. 1996.
DOVAS, C.I. & KATIS, N.I. A spot multiplex nested RT-PCR for the simultaneous
and generic detection of viruses involved in the aetiology of grapevine leafroll and rugose wood of grapevine. Journal of Virological Methods 109: 217-226. 2003.
ERWING, B., HILLIER, L.D., WENDL M.C. & GREEN, P. Base-calling of
automated sequencer traces using phred. I. Accuracy assessment. Genome Research 8:175-185. 1998.
FAJARDO, T.V.M., KUHN, G.B., EIRAS, M. & NICKEL, O. Detecção de
Closterovirus em videira e caracterização parcial de um isolado do grapevine leafroll-associated virus 3. Fitopatologia Brasileira 27:58-64. 2002.
FAJARDO, T.V.M., KUHN, G.B., NICKEL, O. Doenças virais. In: FAJARDO,
T.V.M. (Ed). Uva para processamento. Fitossanidade. Embrapa informações tecnológicas 45-62. 2003.
FAO, Statistical Databases – FAOSTAT Agriculture Data. 2003.
http://apps.fao.org/page/collections?subset=agriculture FAZELI, C.F. & REZAIAN, M.A. Nucleotide sequence and organization of ten open
reading frames in the genome of grapevine leafroll-associated virus 1 and identification of three subgenomic RNAs. Journal of General Virology 81:605-615. 2000.
FAZELI, C.F., HABILI, N. & REZAIAN, M.A. Efficient cloning of cDNA from
grapevine leafroll-associated virus 4 and demonstration of probe specificity by viral antibody. Journal of Virological Methods 70:201-211. 1998.
GALIAKPAROV, N., TANNE, E., SELA, I. & GAFNY, R. Functional analysis of the
grapevine virus A genome. Virology 306:42-50. 2003. GOHEEN, A.C. & HEWITT, W.B. Diagnosis of leafroll of grapevine. Rivista di
Patologia Vegetale 4(8):427-442. 1964. HABILI, N. & NUTTER, F.W. Temporal and spatial analysis of grapevine leafroll-
associated virus 3 in Pinot Noir grapevines in Australia. Plant Disease 81:625-628. 1997.
HABILI, N., FAZELI, C.F., EWART, A., HAMILTON, R., CIRAMI, R., SALDARELLI,
P., MINAFRA, A. & REZAIAN, M.A. Natural spread and molecular analysis of grapevine leafroll-associated virus 3 in Australia. Phytopathology 85:1418-1422.1995.

82
HU, J.S., GONSALVES, D., BOSCIA, D. & NAMBA, S. Use of monoclonal
antibodies to characterize grapevine leafroll associated closteroviruses. Phytopathology 80: 920-925. 1990.
IBGE, Instituto Brasileiro de Geografia e Estatística – Indicadores de Produção
Agrícola. 2003. http://www.ibge.gov.br KARASEV, A. Genetic diversity and evolution of closteroviruses. Annual Review of
Phytopathology 38:293-324. 2000. KARASEV, A.V., NIKOLAEVA, O.V., KOONIN, E.V., GUMPF, D.J. & GARNSEY,
S. Screening of the closterovirus genome by degenerate primer-mediated polymerase chain reaction. Journal of General Virology 75:1415-1422. 1994.
KUHN, G.B. Efeitos causados pelo vírus do enrolamento da folha da videira na
cultivar Cabernet Franc. Fitopatologia Brasileira 14:280-283. 1989a. KUHN, G.B. Identificação, incidência e controle do vírus do enrolamento da folha
da videira no estado do Rio Grande do Sul. Fitopatologia Brasileira 14:220-226. 1989b.
KUHN, G.B. Intumescimento dos ramos da videira (corky bark) doença constatada
no Rio Grande do Sul. Fitopatologia Brasileira 17(4):339-406. 1992. KUHN, G.B. Necrose das nervuras, doença que ocorre de forma latente na maioria
das cultivares de videira no Rio Grande do Sul. Fitopatologia Brasileira 19(1):79-83. 1994.
KUHN, G.B. & NICKEL, O. Viroses e sua importância na viticultura brasileira.
Informe Agropecuário 19(194):85-91. 1998. KUNIYUKI, H. Intumescência dos entre-nós dos ramos de videira, provavelmente
causada por vírus, em São Paulo. Revista da Sociedade Brasileira de Fitopatologia 8:10-11. 1973.
KUNIYUKI, H. Mosaico do Traviú, uma moléstia de vírus da videira. Revista da
Sociedade Brasileira de Fitopatologia 5:123. 1972b. KUNIYUKI, H. Ocorrência de um vírus latente na videira em São Paulo. Revista da
Sociedade Brasileira de Fitopatologia 5:189-190. 1972c. KUNIYUKI, H. Viroses da videira no Brasil. Fitopatologia Brasileira 6(2):300-301.
1981. KUNIYUKI, H. & COSTA, A.S. Incidência de vírus da videira em São Paulo.
Fitopatologia Brasileira 12:240-245. 1987.

83
KUNIYUKI, H. & COSTA, A.S. Evidência preliminar de transmissão do
enrolamento da folha (vermelhão ou amarelo) da videira em São Paulo. Revista da Sociedade Brasileira de Fitopatologia 5:165-166. 1972a.
KUNIYUKI, H. & COSTA, A.S. Nota sobre a ocorrência do cascudo, moléstia
semelhante ao legno riccio da videira. Revista da Sociedade Brasileira de Fitopatologia 5:137. 1972.
KUNIYUKI, H. & COSTA, A.S. Caneluras no porta-enxerto Kober 5BB são
provavelmente causadas por um novo isolado do vírus do cascudo da videira. Summa Phytopathogica 18:13. 1992.
KUNIYUKI, H. & COSTA, A.S. Mosaico das nervuras, uma virose da videira em
São Paulo. Summa Phytopathologica 20(3):152-157. 1994. KUNIYUKI, H. & MULLER, G.W. Diagnose do cascudo da videira em São
Paulo. Summa Phytopathologica 13:26. 1987. LEÃO, P.C.S. & POSSÍDIO, E.L. Histórico da videira. In: LEÃO, P.C.S. &
SOARES, J.M. (Eds). A viticultura no semi-árido brasileiro. Embrapa Semi-Árido 15-17. 2000.
LIMA, M.F. Principais viroses. In: LIMA, M.F. & MOREIRA, W.A. Uva de mesa
fitossanidade. Embrapa Informações Tecnológicas 35-44. 2002. LIMA, M.F. Doenças Bacterianas. In: FAJARDO, T.V.M. (Ed). Uva para
processamento. Fitossanidade. Embrapa Informações Tecnológicas 63-71. 2003.
LIMA, M.F. & MOREIRA, W.A. Doenças causadas por bactérias. In: LIMA, M.F. &
MOREIRA, W.A. Uva de mesa: Fitossanidade. Embrapa Informação Tecnológica 27-34. 2002.
LING, K.S., ZHU, H.Y., ALVIZO, H., HU, J.S., DRONG, R.F., SLIGHTOM, J.L. &
GONSALVES, D. The coat protein gene of grapevine leafroll associated closterovirus-3: cloning, nucleotide sequencing and expression in transgenic plants. 142:1101-1116. 1997.
LING, K.S., ZHU, H.Y., DRONG, R.F., SLIGHTOM, J.L., McFERSON, J.R. &
GONSALVES, D. Nucleotide sequence of the 3’-terminal two-thirds of the grapevine leafroll-associated virus-3 genome reveals a typical monopartite closterovirus. Journal of General Virology 79:1299-1307. 1998.
LING, K.S., ZHU, H.Y., PETROVIC, N. & GONSALVES, D. Comparative
Effectiveness of ELISA and RT-PCR for detecting grapevine leafroll-

84
associated closterovirs-3 in field samples. American Journal of Enology and Viticulture 52(1):21-27. 2001. (resumo)
LITTLE, A., FAZELLI, C.F. & REZAIAN, M.A. Hypervariable genes in grapevine
leafroll associated virus 1. Virus Research 80:109-116. 2001. MACKENZIE, D.J., MCLEAN, M.A., MUKERJI, S. & GREEN, M. Improved RNA
extraction from woody plants for the detection of viral pathogens by reverse transcription-polymerase chain reaction. Plant Disease 81:222-226. 1997.
MARTELLI, G.P., AGRANOVSKY, A.A., BAR-JOSEPH, M., BOSCIA, D.
CANDRESSE, T. et al. The family Closteroviridae revised. Archives of Virology 147:2039-2044. 2002.
MARTELLI, G.P. & SAVINO, V. Fanleaf degeneration. In: PEARSON, C. &
GOHEEN, R. (Eds) Compendium of grape diseases. APS Press. 48-49. 1998. MARTIN, R.R., JAMES, D. & LÉVESQUE, C.A. Impacts of Molecular Diagnostic
Technologies on Plant Disease Management. Annual Review of Phytopathology 38:207-239. 2000.
MONIS, J. & BESTWICK, R.K. Detection and localization of grapevine leafroll
associated closterovirus in greenhouse and tissue culture grown plants. Amercan Journal of Enology and Viticulture 47:199-205. 1996. (resumo)
MINAFRA, A. & HADIDI, A. Sensitive detection of grapevine virus A, B, or leafroll-
associated III from viruliferous mealybugs and infected tissue by cDNA amplification. Journal of Virological Methods 47:175-188. 1994.
PEARSON, C. & GOHEEN, R. (Eds) Compendium of grape diseases. APS Press.
4th ed. 1998. PETERSEN, C.L. & CHARLES, J.G. Transmission of grapevine leafroll-associated
closteroviruses by Pseudococcus longispinus and P. calceolariae. Plant Pathology 46:509-515. 1997.
PROGNÓSTICO AGRÍCOLA. IEA 2:210. 1998. RETIEF, J.D. Phylogenetic analysis using PHYLIP. Methods of Molecular Biology
132: 243-258. 2000. ROBBS, C.F. & NETO, J.R. Enfermidades causadas por bactérias em frutíferas
tropicais no Brasil. Summa Phytopathologica 25(1):47-52. 1999. ROTT, M.E. & JELKMANN, W. Detection and partial characterization of a second
closterovirus associated with little cherry disease, little cherry virus-2. Phytopathology 91:261-267. 2001.

85
ROUTH, G., ZHANG, Y., SALDARELLI, P. & ROWHANI, A. Use of degenerate
primers for partial sequencing and RT-PCR-based assays of grapevine leafroll-associated viruses 4 and 5. Phytopathology 88:1238-1243. 1998.
ROWHANI, A., CHAY, C., GOLINO, A. & FALK, B.W. Development of a
polymerase chain reaction technique for the detection of grapevine fanleaf virus in grapevine tissue. Phytopathology 83:749-753. 1993.
ROWHANI, A., MANINGAS, M.A., LILE, L.S., DAUBERT, S.D. & GOLINO, D.A.
Development of a detection system for viruses of woody plants based on PCR analysis of immobilized virions. Phytopathology 85:347-352. 1995.
SALDARELLI, P., MINAFRA, A., MARTELLI, G.P. & WALTER, B. Detection of
grapevine leafroll-associated closterovirus III by molecular hybridization. Plant Pathology 43:91-96. 1994.
SFORZA, R., BOUDON-PADIEU, E. & GREIF, C. New mealybug species
vectoring Grapevine leafroll-associated viruses-1and -3 (GLRaV-1 and –3). European Journal of Plant Pathology 109:975-981. 2003. (resumo)
SILVA, P.C.G. & CORREIA, R.C. Caracterização social e econômica da videira. In:
LEÃO, P.C.S. & SOARES, J.M. (Eds). A viticultura no semi-árido brasileiro. Embrapa Semi-Árido 21-32. 2000.
SÔNEGO, O.R., GARRIDO, L.R. & GRIGOLETTI JÚNIOR, A. Doenças Fúngicas.
In: FAJARDO, T.V.M. (Ed). Uva para processamento. Fitossanidade. Embrapa informações tecnológicas 11-44. 2003.
SOUZA, J.S.I. (Ed) Uvas para o Brasil. Edições Melhoramentos.1969. STELLMACH, G. & GOHEEN, A.C. Other virus and viruslike diseases. In:
PEARSON R.C. & GOHEEN, A.C. Compendium of grape diseases. APS Press 54. 1998.
TAVARES, S.C.C.H. & CRUZ, S.C. Doenças causadas por fungos. In: LIMA, M.F.
& MOREIRA, W.A. Uva de mesa: Fitossanidade. Embrapa Informação Tecnológica 9-26. 2002.
TAVARES, S.C.C.H., LIMA, M.F. & MELO, N.F. Principais doenças da videira e
alternativas de controle. In: LEÃO, S. & SOARES, J.M. (Eds) A viticultura no semi-árido brasileiro. Embrapa Semi-Árido 294-356. 2000.
TESSMANN, D.J.; DIANESE, J.C.; GENTA, W.; VIDA, J.B. & MAY-DE MIO, L.L.
Grape rust (Phakopsora euvitis): a new disease for Brazil. Fitopatologia Brasileira 29(3): 435. 2004.

86
TESSMANN, D.J., VIDA, J.B. & LOPES, D.B. Um novo problema. Cultivar HF 22-25. 2003.
THOMPSON, J., HIGGINS, D., GIBSON, T. CLUSTAW W: improving the
sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acid Research 22: 4673-4680. 1994.
TRINDADE, L.C. Caracterização molecular de Xanthomonas campestris pv.
viticola, agente causal do cancro bacteriano da videira (dissertação de mestrado). Universidade de Brasília. 2002.
VALERO, M., IBAÑES, A. & MORTE, A. Effects of high vineyard temperatures on
the grapevine leafroll associated virus elimination from Vitis vinifera L. cv. Napoleon tissue cultures. Scientia Horticulturae 97:289-296. 2003.
van REGENMORTEL, M.H.V., FAUQUET, C.M., BISHOP, D.H.L., CARSTENS,
E.B., ESTES, M.K. et al. (Eds). Virus Taxonomy: Classification and Nomenclature of Viruses. Academic Press. 2000.
VIANA, A.P., BRUCKNER, C.H., MARTINEZ, H.E.P., HUAMAN, C.A.M. &
MOSQUIM, P.R. Características fisiológicas de porta-enxertos de videira em solução salina. Scientia Agrícola 58(1):139-143. 2001.
ZHANG, Y. & ROWHANI, A. A strategy for rapid cDNA cloning from double-
stranded RNA templates isolated from plants infected with RNA viruses by using taq DNA polymerase. Journal of Virological Methods 84:59-63. 2000.
ZIMMERMANN, D., BASS, P., LEGIN, R. & WALTER B. Characterization and
serological detection of four closterovirus-like particles associated with leafroll disease of grapevine. Journal of Phytopathology 130: 205-218. 1990.

87
SUMÁRIO
RESUMO................................................................................................................. 1
ABSTRACT .......................................... ................................................................... 2
ÍNDICE DE TABELAS ................................. ........................................................... 3
ÍNDICE DE FIGURAS ............................................................................................. 4
LISTA DE SIGLAS E ABREVIATURAS .................... ............................................. 8
1. REVISÃO BIBLIOGRÁFICA .......................... ................................................... 10
1.1. INTRODUÇÃO ................................................................................................. 10
1.2. HISTÓRICO .................................................................................................... 11
1.3. SITUAÇÃO ATUAL DA VITICULTURA NO BRASIL E NO SEMI-ÁRIDO ........................ 15
1.4. DOENÇAS QUE AFETAM A VIDEIRA .................................................................. 17
1.4.1. Doenças bacterianas ............................................................................ 18
1.4.2. Doenças Fúngicas................................................................................ 19
1.4.3. Doenças causadas por Nematóides ..................................................... 22
1.4.4. Principais viroses da videira ................................................................. 23
1.5. A FAMÍLIA CLOSTEROVIRIDAE ......................................................................... 33
1.5.1. Classificação dos membros da família Closteroviridae ........................ 35
1.5.2. Gênero Closterovirus ........................................................................... 36
1.5.3. Gênero Crinivirus ................................................................................. 38
1.5.4. Gênero Ampelovirus ............................................................................. 39
2. OBJETIVOS ...................................... ................................................................ 43
3. MATERIAIS E MÉTODOS ............................ .................................................... 44
3.1. LOCAL E PERÍODO DE REALIZAÇÃO DO TRABALHO ............................................. 44
3.2. OBTENÇÃO E MANUTENÇÃO DAS AMOSTRAS ..................................................... 44
3.3. ELISA .......................................................................................................... 45
3.4. EXTRAÇÃO DE RNA E SÍNTESE DE CDNA ........................................................ 47
3.4.1. Extração de RNA.................................................................................. 47

88
3.4.2. Síntese de cDNA .................................................................................. 49
3.5. AMPLIFICAÇÃO DO GENE DO CAPSÍDEO VIRAL E DA REGIÃO 3´ DA RNA POLIMERASE
........................................................................................................................... 49
3.6. CLONAGEM DOS SEGMENTOS DE DNA AMPLIFICADOS....................................... 51
3.7. SEQÜENCIAMENTO......................................................................................... 55
3.8. ANÁLISE DAS SEQÜÊNCIAS ............................................................................. 55
3.9. ANÁLISE FILOGENÉTICA ................................................................................. 56
4. RESULTADOS ..................................... ............................................................. 57
4.1. ELISA .......................................................................................................... 57
4.2. MÉTODOS DE EXTRAÇÃO DE RNA ................................................................... 59
4.3. AMPLIFICAÇÃO DO GENE DO CAPSÍDEO VIRAL E DA REGIÃO 3´ DA RNA POLIMERASE
........................................................................................................................... 59
4.4. CLONAGEM DOS SEGMENTOS DE DNA. ........................................................... 60
4.5. ANÁLISE DO SEQÜENCIAMENTO ....................................................................... 61
4.5.1. Seqüenciamento da região 3´ do gene da polimerase viral ................. 61
4.5.2. Seqüenciamento do gene do capsídeo ................................................ 65
4.6. ANÁLISE FILOGENÉTICA ................................................................................. 72
5. DISCUSSÃO ..................................................................................................... 74
5.1. ANÁLISE DA REGIÃO 3´ DA POLIMERASE ........................................................... 76
5.2. ANÁLISE DO GENE DO CAPSÍDEO ..................................................................... 77
5.3. CONSIDERAÇÕES FINAIS ................................................................................ 78
6. CONCLUSÕES ................................................................................................. 79
7. REFERÊNCIAS BIBLIOGRÁFICAS ..................... ............................................ 80




















