
Markov models and dynamical fingerprints: Unraveling the complexity of molecular kinetics

Unraveling the Active Site in Copper-Ceria Systems for the Water-Gas Shift Reaction: InSitu Characterization of an Inverse Powder CeO2-x/CuO-Cu Catalyst
Laura Barrio,† Michael Estrella,† Gong Zhou,† Wen Wen,†,| Jonathan C. Hanson,†
Ana B. Hungrıa,‡ Aitor Hornes,§ Marcos Fernandez-Garcıa,§ Arturo Martınez-Arias,§ andJose A. Rodriguez*,†
Department of Chemistry, BrookhaVen National Laboratory, Upton, New York 11973, Departamento de Cienciade Materiales, Ingenierıa Metalurgica y Quımica Inorganica, Facultad de Ciencias, UniVersidad de Cadiz,11510 Puerto Real, Cadiz, Spain, and Instituto de Catalisis y Petroleoquımica, CSIC, C/ Marie Curie 2,Campus de Cantoblanco, 28049, Madrid, Spain
ReceiVed: October 29, 2009; ReVised Manuscript ReceiVed: December 22, 2009
An inverse powder system composed of CeO2 nanoparticles dispersed over a CuO-Cu matrix is proposed asa novel catalyst for the water-gas shift reaction. This inverse CeO2/CuO-Cu catalyst exhibits a higher activitythan standard Cu/CeO2 catalysts. In situ synchrotron characterization techniques were employed to followthe structural changes of CeO2/CuO-Cu under reaction conditions. Time-resolved X-ray diffraction experimentsshowed the transformation of CuO to metallic Cu via a Cu2O intermediate. Short-order structural changeswere followed by pair distribution function analysis and corroborated the results obtained by diffraction.Moreover, X-ray absorption spectroscopy also revealed oxidation state changes from Cu2+ to Cu0 and thepartial reduction of CeOx nanoparticles. The activity data obtained by mass spectrometry revealed that hydrogenproduction starts once the copper has been fully reduced. The strong interaction of ceria and copper boostedthe catalytic performance of the sample. The inverse catalyst was active at low temperatures, stable to severalreaction runs and to redox cycles. These characteristics are highly valuable for mobile fuel cell applications.The active phases of the inverse CeO2/CuO-Cu catalyst are partially reduced ceria nanoparticles stronglyinteracting with metallic copper. The nature and structure of the ceria nanoparticles are of critical importancebecause they are involved in processes related to water dissociation over the catalyst surface.
Introduction
At present, hydrogen is mainly produced from the reforming ofcrude oil, coal, natural gas, wood, organic waste, and biomass.1,2
The CO (1-10% content) present in the reformed fuel degradesthe performance of the Pt electrode used in fuel cell systems. Thewater-gas shift (WGS) reaction is a critical process for procuringclean hydrogen. As noted in the following chemical equation
for each mole of CO removed, a mole of hydrogen is produced.The WGS reaction then allows not only the removal of CO butalso an upgrade in fuel cell efficiency by increasing the H2
concentration.For mobile fuel cell applications, conventional WGS catalysts
are not suitable and advanced systems are required. CommercialCu-Zn-based catalysts are pyrophoric, require special activationprocedures, and are intolerant to oxidation. Because of theexpected exposure to many start-up/shutdown cycles of mobilefuel cell systems, novel WGS catalysts should be tolerant toredox cycles and steam condensation.1,2 Ceria-based nanocata-
lysts have been investigated extensively in recent years and areexpected to be part of the next generation of WGS catalysts.3-7
To obtain low-temperature WGS activity, cerium oxide isusually loaded with reduction promoter metals, such as Rh, Pt,Cu, or Au.3-7
The design and optimization of new catalysts for the WGSreaction is hindered by the complex reaction mechanism andthe difficulty of identifying active species. The characterizationof the catalysts and reaction intermediates under WGS processconditions is, therefore, important.8,9 The WGS mechanism onCu-based catalysts has been a question of study for manyyears.3,7-16 It has been generally accepted that the active phaseinvolves reduced Cu metal sites supported on metal oxides.Nevertheless, the metal oxide support plays an important rolein the reaction mechanism. CeO2-supported systems have shownenhanced activity in the WGS reaction with various metals, andthis has been ascribed to the excellent oxygen storage capabilityof CeO2. In this sense, although CeO2 alone does not exhibitsignificant WGS activity, it appears typically acting as a support,an essential promoter for achieving enhanced WGS activity.Mechanistic DRIFT studies have suggested that CeO2 is ableto readily dissociate water,17,18 whereas Cu sites adsorb CO,14
and then, the oxygen is transferred from the support in order tooxidize the CO.19 However, it has been demonstrated that Cusurfaces alone are also active in the WGS reaction.10,14,20
Furthermore, Cu nanoparticles have also shown the ability todissociate water and have proved to be far more active than thewell-ordered surfaces of Cu (111).14
Traditionally, precious metal nanoparticles are dispersed overmetal oxide supports, with the conviction that the supports’ main
* To whom correspondence should be addressed. E-mail: [email protected]. Fax: 1-631-344-5815.
† Brookhaven National Laboratory.‡ Universidad de Cadiz.§ Instituto de Catalisis y Petroleoquımica, CSIC.| Current address: Experimental Division, Shanghai Synchrotron Radia-
tion Facility, Shanghai Institute of Applied Physics, Chinese Academy ofSciences, Pudong New Area, Shanghai, People’s Republic of China, 201204.
CO + H2O T CO2 + H2 (1)
J. Phys. Chem. C 2010, 114, 3580–35873580
10.1021/jp910342b 2010 American Chemical SocietyPublished on Web 02/08/2010

role is to stabilize and disperse active sites along its surface.Recently, inverse model catalysts of CeOx nanoparticles sup-ported over Au(111)21 or Cu(111)22 have shown high catalyticperformance in the WGS reaction, up to the point that a CeOx/Cu(111) system is more active than Cu/CeO2(111) and Cu/ZnO(0001j) systems. In the present work, we move away frommodel catalysts and investigate the behavior of inverse powdercatalysts with relatively high specific surface area involvingcerium oxide nanoparticles dispersed over a CuO/Cu support.This approach will allow us to better study the role of CeO2
and Cu in the reaction mechanism. As shown in Scheme 1, theinverse CeO2/CuO-Cu catalyst exposes ceria nanoparticles tothe reactants. Defect sites present in the oxide are not coveredby metal particles, as occurs in the case of a traditional Cu/CeO2 catalyst. On the inverse CeO2/CuO-Cu catalyst, thereactants can interact with defect sites of the oxide nanoparticles,metal sites, and the metal-oxide interface.
In this article, in situ characterization techniques wereemployed to correlate the structure of the inverse CeO2/CuO-Cucatalyst with its activity. Time-resolved X-ray diffraction(TR-XRD)8,23 provided unique information on the crystallinephases present in the sample, and subsequent data refinementrevealed changes in phase composition, unit cell size, andoxygen vacancies. The atomic pair distribution function (PDF)method24-26 gave short- and long-range structural information.X-ray absorption near-edge structure (XANES) was used tomonitor changes in the oxidation state of the catalyst duringthe WGS reaction. With the inverse catalyst proposed in thepresent work, the copper species is the major component, andwe will exploit this feature to clarify details of the copper-to-cerium oxide interactions.
Experimental Section
A. Catalyst Preparation. The inverse CeO2/CuO catalystwas prepared by employing reverse microemulsions containingn-heptane, Triton-X-100, and n-hexanol as organic solvent,surfactant, and cosurfactant, respectively, in amounts similarto those reported previously.27 The required amount of Cu(NO3)2
was dissolved in distilled water and added to the former in orderto form the reverse microemulsion. Simultaneously, anothermicroemulsion of similar characteristics was prepared contain-ing, dissolved in its aqueous phase, the required amount oftetramethyl ammonium hydroxide (TMAH). After 1 h of stirringof the two microemulsions, the TMAH-containing one wasadded to the Cu-containing one and it was left for the period of18-24 h in order to complete the precipitation reaction. Theresulting microemulsion was then heated gently up to 339 Kusing a water bath. This microemulsion containing the precipi-tated copper was then mixed with another one of similarcharacteristics in which Ce had previously been precipitated,following mixing of Ce(NO3)2- and TMAH-containing micro-emulsions. This final microemulsion containing both precipitatedCu and Ce components was kept under agitation for the periodof 18-24 h. The resulting solid was then separated bycentrifugation and decantation, rinsed with methanol, and dried
overnight at 100 °C, and the resulting powder was calcined at773 K for 2 h under air. Nominal Cu/Ce atomic ratios of 7/3was employed for this catalyst, close to 2.57 detected by X-rayfluorescence elemental analysis. A specific area value of SBET
) 66.9 m2 g-1 was obtained from BET analysis of thecorresponding N2 adsorption isotherm.
B. Electron Microscopy. High-resolution electron micros-copy (HREM), high-angle annular dark-field scanning transmis-sion electron microscopy (HAADF-STEM) images, and X-rayenergy-dispersive spectra (XEDS) were recorded on a 200 kVFEI Tecnai F20-G2 TEM/STEM miscroscope equipped withan EDAX r-TEM ultrathin window (UTW) X-ray detector.XEDS analysis was performed in STEM mode, with a probesize of ∼1 nm. Specimens were prepared by depositing particlesof the samples to be investigated onto a molybdenum gridsupporting a perforated carbon film. Deposition was achievedby dipping the grid directly into the powder of the samples toavoid contact with any solvent.
C. In Situ Time-Resolved X-ray Diffraction. In situ time-resolved X-ray diffraction (TR-XRD) experiments were carriedout on beamline X7B of the National Synchrotron Light Source(NSLS) at Brookhaven National Laboratory.8,9 The sample (∼5mg) was loaded into a glass cell with a diameter of 1 mm, whichwas attached to a flow system.28 A small resistance heater waswrapped around the capillary, and the temperature was moni-tored with a 0.1 mm chromel-alumel thermocouple placed inthe capillary near the sample.28 Two-dimensional powderpatterns were collected with a Mar345 image plate detector and
SCHEME 1: Structures of Conventional Cu/CeO2 andInverse CeO2/Cu Catalysts
Figure 1. Rietveld analysis of an XRD pattern (top panel) and PDFanalysis (bottom panel) of fresh inverse CeO2/CuO catalyst.
Inverse CeO2-x/CuO-Cu Catalyst for the WGS Reaction J. Phys. Chem. C, Vol. 114, No. 8, 2010 3581

the powder diffraction rings were integrated using the FIT2Dcode.29 The instrument parameters (Thompson-Cox-Hastingsprofile coefficients) were derived from the fit of a LaB6 referencepattern. Rietveld profile refinements were performed with theaid of GSAS software.30 The series of powder patterns wasrefined by sequential analysis where the starting model is basedon the earlier powder pattern. In addition to Rietveld refine-ments, pair distribution function (PDF) analyses were performedwith the PDFgetX231 program to apply corrections to theintensity data and to calculate the resulting pair distributionfunction. Diffraction patterns were collected over the catalystduring the WGS reaction, reduction in 1% CO/He, and oxidationin 5% O2/He. The WGS reaction was carried out with a steppedtemperature program from room temperature to 500 °C, with3 h soaks at every 100 °C beyond 200 °C amid a 1% CO and99% He gas mixture flow rate of 10 mL/min and a spacevelocity of 4000 h-1 (volumetric feed flow rate/catalytic bedvolume). This gas mixture passed through a water bubbler (roomtemperature) before entering the reactor. The relative ratio ofwater vapor to CO pressures in the gas mixture was 3.
D. In Situ Time-Resolved X-ray Absorption. Cu K-edgeand Ce LIII-edge in situ XANES spectra were collected atbeamline X19A of the NSLS under similar operational condi-tions as those for the TR-XRD experiments. The same cell wasused for the XANES experiments as that for in situ XRD,28
except that the sample was loaded into a Kapton capillary andheated with a hot air blower. The X-ray absorption spectra weretaken repeatedly in the “fluorescence-yield mode” using apassivated implanted planar silicon (PIPS) detector cooled withcirculating water. The XANES data were then analyzed usingthe Athena program.32
The relative product concentrations from both TR-XRD andTR-XANES experiments were measured with a 0-100 amuquadruple mass spectrometer (QMS, Stanford Research Sys-tems). A portion of the exit gas flow passed through a leak valveand into the QMS vacuum chamber. QMS signals at mass-to-charge ratios of 2(H2), 4(He), 17(OH), 18(H2O), 28(CO), 32(O2),and 44(CO2) were monitored and recorded during the experiments.
Results and Discussion
A. Characterization of the Inverse CeO2/CuO Catalyst.We investigated the long- and short-range structure of the CeO2/CuO catalyst using XRD and the PDF method.35-37 In the toppanel of Figure 1, the XRD pattern for the CeO2/CuO catalysthas only diffraction peaks for CeO2
33 and CuO.34 A Rietveldrefinement of this pattern gave a molar composition of 40%CeO2 and 60% CuO. No other phase was detected in the long-range probed with XRD or in the short-range probed by a PDFanalysis. The PDF traces were well-fitted by a sum of CeO2
and CuO features (bottom panel of Figure 1). The CeO2/CuOcatalyst shows peaks at 1.95 and 3.05 Å, which correspond tothe Cu-O and Cu-Cu distances in the CuO phase. The CeO2
phase has a peak at 2.35 Å for the Ce-O distance and a peakat 3.90 Å for the Ce-Ce distance. Using the Scherrer equation35
and the width of the diffraction peaks in Figure 1, we obtainedaverage particle sizes of 8.7 nm for ceria and 29.4 nm for CuO.
The inverse configuration of the CeO2/CuO catalyst wasdemonstrated by means of HAADF-STEM-XEDS and HRTEManalyses. Figure 2 displays a HAADF-STEM image representa-tive for this catalyst. The image illustrates the presence ofrelatively big CuO particles, as evidenced by the XED spectrataken in zone A, onto which small (in the range of 5-10 nm)
Figure 2. HAADF-STEM image of the inverse CeO2/CuO catalyst, showing a CuO particle in (A) and CeO2 nanoparticles in (B).
3582 J. Phys. Chem. C, Vol. 114, No. 8, 2010 Barrio et al.
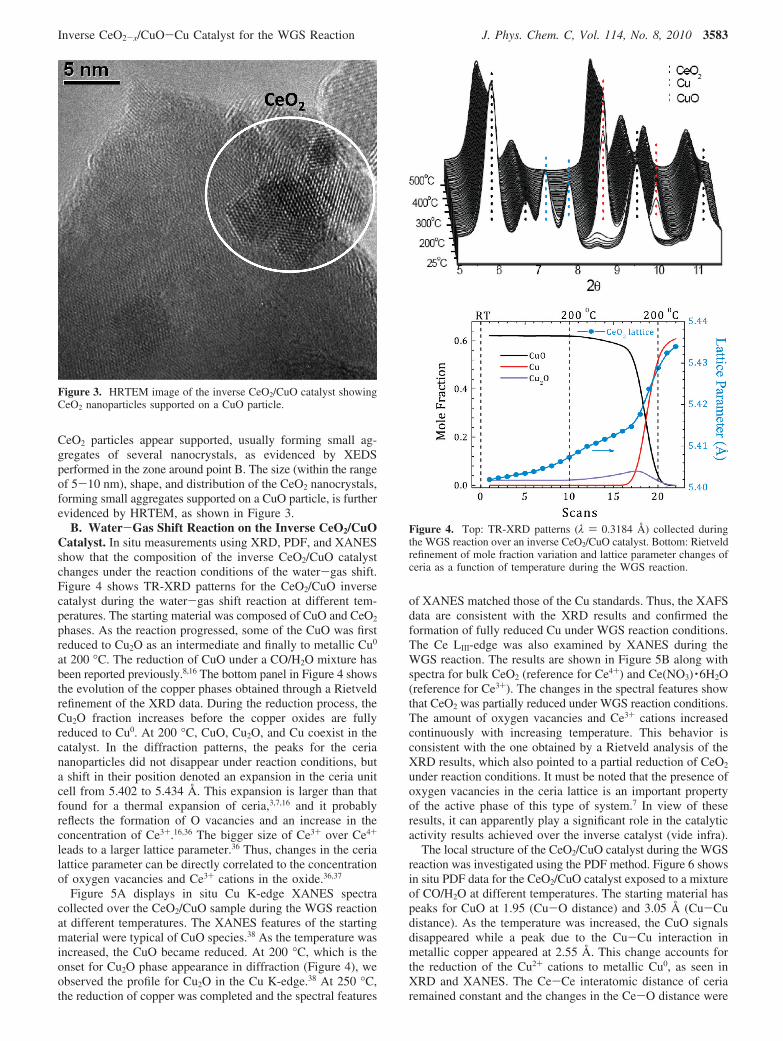
CeO2 particles appear supported, usually forming small ag-gregates of several nanocrystals, as evidenced by XEDSperformed in the zone around point B. The size (within the rangeof 5-10 nm), shape, and distribution of the CeO2 nanocrystals,forming small aggregates supported on a CuO particle, is furtherevidenced by HRTEM, as shown in Figure 3.
B. Water-Gas Shift Reaction on the Inverse CeO2/CuOCatalyst. In situ measurements using XRD, PDF, and XANESshow that the composition of the inverse CeO2/CuO catalystchanges under the reaction conditions of the water-gas shift.Figure 4 shows TR-XRD patterns for the CeO2/CuO inversecatalyst during the water-gas shift reaction at different tem-peratures. The starting material was composed of CuO and CeO2
phases. As the reaction progressed, some of the CuO was firstreduced to Cu2O as an intermediate and finally to metallic Cu0
at 200 °C. The reduction of CuO under a CO/H2O mixture hasbeen reported previously.8,16 The bottom panel in Figure 4 showsthe evolution of the copper phases obtained through a Rietveldrefinement of the XRD data. During the reduction process, theCu2O fraction increases before the copper oxides are fullyreduced to Cu0. At 200 °C, CuO, Cu2O, and Cu coexist in thecatalyst. In the diffraction patterns, the peaks for the ceriananoparticles did not disappear under reaction conditions, buta shift in their position denoted an expansion in the ceria unitcell from 5.402 to 5.434 Å. This expansion is larger than thatfound for a thermal expansion of ceria,3,7,16 and it probablyreflects the formation of O vacancies and an increase in theconcentration of Ce3+.16,36 The bigger size of Ce3+ over Ce4+
leads to a larger lattice parameter.36 Thus, changes in the cerialattice parameter can be directly correlated to the concentrationof oxygen vacancies and Ce3+ cations in the oxide.36,37
Figure 5A displays in situ Cu K-edge XANES spectracollected over the CeO2/CuO sample during the WGS reactionat different temperatures. The XANES features of the startingmaterial were typical of CuO species.38 As the temperature wasincreased, the CuO became reduced. At 200 °C, which is theonset for Cu2O phase appearance in diffraction (Figure 4), weobserved the profile for Cu2O in the Cu K-edge.38 At 250 °C,the reduction of copper was completed and the spectral features
of XANES matched those of the Cu standards. Thus, the XAFSdata are consistent with the XRD results and confirmed theformation of fully reduced Cu under WGS reaction conditions.The Ce LIII-edge was also examined by XANES during theWGS reaction. The results are shown in Figure 5B along withspectra for bulk CeO2 (reference for Ce4+) and Ce(NO3) ·6H2O(reference for Ce3+). The changes in the spectral features showthat CeO2 was partially reduced under WGS reaction conditions.The amount of oxygen vacancies and Ce3+ cations increasedcontinuously with increasing temperature. This behavior isconsistent with the one obtained by a Rietveld analysis of theXRD results, which also pointed to a partial reduction of CeO2
under reaction conditions. It must be noted that the presence ofoxygen vacancies in the ceria lattice is an important propertyof the active phase of this type of system.7 In view of theseresults, it can apparently play a significant role in the catalyticactivity results achieved over the inverse catalyst (vide infra).
The local structure of the CeO2/CuO catalyst during the WGSreaction was investigated using the PDF method. Figure 6 showsin situ PDF data for the CeO2/CuO catalyst exposed to a mixtureof CO/H2O at different temperatures. The starting material haspeaks for CuO at 1.95 (Cu-O distance) and 3.05 Å (Cu-Cudistance). As the temperature was increased, the CuO signalsdisappeared while a peak due to the Cu-Cu interaction inmetallic copper appeared at 2.55 Å. This change accounts forthe reduction of the Cu2+ cations to metallic Cu0, as seen inXRD and XANES. The Ce-Ce interatomic distance of ceriaremained constant and the changes in the Ce-O distance were
Figure 3. HRTEM image of the inverse CeO2/CuO catalyst showingCeO2 nanoparticles supported on a CuO particle.
Figure 4. Top: TR-XRD patterns (λ ) 0.3184 Å) collected duringthe WGS reaction over an inverse CeO2/CuO catalyst. Bottom: Rietveldrefinement of mole fraction variation and lattice parameter changes ofceria as a function of temperature during the WGS reaction.
Inverse CeO2-x/CuO-Cu Catalyst for the WGS Reaction J. Phys. Chem. C, Vol. 114, No. 8, 2010 3583

difficult to follow because it overlapped with the Cu-Cudistance of metallic copper. Above 250 °C, there were noadditional changes in the local and long-range structures of thecatalyst.
Figure 7 shows the product analysis obtained by massspectrometry at the exit of a microreactor28 that contained theinverse CeO2/CuO catalyst. The activity onset (i.e., simultaneousformation of H2 and CO2) was observed at ∼200 °C, when theCeO2/CuO f CeOx/Cu transformation was taking place. As inthe case of CuO/CeO2 and Ce1-xCuxO2 catalysts,16 the generationof reduced states of copper in the inverse catalyst accompaniesthe production of H2 through the water-gas shift reaction. Highcatalytic activity is observed at 300, 400, and 500 °C, temper-atures at which the catalyst consists of CeOx/Cu. Noteworthy,the activity profile shows neither deactivation of the catalystduring long reaction times (∼4 h) nor any appreciable deactiva-tion effect at a reaction temperature as high as 500 °C.
C. Reduction of CeO2/CuO with CO and Reoxidationwith O2. To understand better the redox process occurring inthe CeO2/CuO catalyst during the water-gas shift reaction, weexamined the interaction of CO with the catalyst. For this, theCeO2/CuO sample was exposed to a mixture of 5% CO/95%He and the temperature was ramped from 25 to 500 °C,monitoring changes in the X-ray diffraction pattern. Figure 8summarizes the results obtained through a Rietveld refinementof the XRD data. A direct CuO f Cu transformation occurredat 80-150 °C, without the formation of Cu2O as an intermediate.When the CuO is being reduced, the expansion of the CeO2
lattice is linear due only to thermal effects. At higher temper-atures (∼150 °C), there is a change in the slope of the cerialattice expansion, indicating an increase in the reduction degreeof the ceria species. Under a flow of CO, the reductions of CuOand CeO2 do not appear to correlate (in agreement with previousredox models for catalysts of this type),39 in contrast with thebehavior observed under WGS conditions (bottom panel ofFigure 4). The presence of water in the reaction mixture delaysthe reduction of CuO and makes possible the formation of Cu2Oas a reaction intermediate.
Figure 9 compares the cell dimensions of ceria in CeO2/CuOduring temperature-programmed oxidation, reduction, and duringthe WGS process. The results correspond to a temperature of500 °C, and the dashed line marks the cell dimension of pureceria at that temperature. The ceria cell dimensions increasedin the following sequence: O2 < WGS < CO. Because the cellparameter accounts directly for the ceria reduction degree,16,36,37
Figure 5. (A) Cu K-edge XANES spectra collected under WGSreaction conditions at the indicated temperatures, with a reference Cu0
foil spectrum. (B) Ce LIII-edge XANES collected under WGS conditionsat the indicated temperatures, with reference Ce3+ and Ce4+ spectra.
Figure 6. Time-resolved PDF analysis of the inverse CeO2/CuOcatalyst under WGS reaction conditions. The analysis was performedusing the XRD patterns from Figure 4.
Figure 7. Production of CO2 and H2 during the WGS reaction on theinverse CeO2/CuO catalyst. These data were obtained in the same setof experiments that produced the XRD patterns displayed in Figure 4.
3584 J. Phys. Chem. C, Vol. 114, No. 8, 2010 Barrio et al.

it can be concluded that the concentration of oxygen vacanciesand Ce3+ cations during the WGS is smaller than after reductionin CO and larger than on a fully oxidized CeO2/CuO catalyst.During the WGS, in accordance with previous studies,7 the watermolecules probably adsorb and dissociate on oxygen vacancies(Ovac) created upon interaction with CO
leading to a reduction in the concentration of Ce3+ cations andthe cell dimensions of ceria. Considering the obtained results(Figures 4, 8, and 9), it can be proposed that CO will adsorb
on the Cu sites and then migrate to reduce the ceria nanopar-ticles, where the water will dissociate.
The CeOx/Cu catalyst could also be regenerated to its oxidizedCeO2/CuO state by heating under an oxidizing atmosphere of5% O2 in helium (see Figure 10). The catalyst regeneration cyclewas performed several times, and the process was alwaysreversible. Reaction of O2 with CeOx/Cu led to the removal ofCe3+ centers and a consequent contraction of the ceria celldimensions. From the metallic copper phase, the catalyst evolvedto a CuO phase, passing through the formation of a Cu2Ointermediate. Cu K-edge XANES spectra were recoded duringthe reoxidation and are shown in Figure 10B. Under oxygenflow, the metallic copper is readily oxidized to CuO. Theisosbestic points obtained along the spectra further confirm thephase transition of copper species. The XANES results indicatethat only CuO was present after reoxidation.
Activity data on oxidized and (in situ) reduced samples showno significant changes (see Figure 11), indicating that, underreaction conditions, the activity is governed by reduced speciesthat are already present at 200 °C, as seen in in situ measure-ments with XRD, XANES, and PDF. The insensitivity of thecatalyst performance to redox cycles is a promising characteristicfor mobile fuel cell applications because the catalyst would bestable during multiple start-up/shutdown processes. This appearsto be an interesting characteristic of the inverse CeO2/CuOcatalyst from a practical point of view. Hydrogen productionand hence activity always increase with temperature, and oncethe steady state is reached, the activity remains constant at afixed temperature. This result is similar to that found for theWGS on CeOx/Cu(111)22 but differs from previous work forthe WGS on Cu/CeO2 and Ce1-xCuxO2 systems where adeactivation was observed at high temperatures.7,16 Furthermore,the inverse CeO2/CuO catalyst also exhibits high stability forpreferential oxidation of carbon monoxide (CO-PROX).40
D. WGS Activity of CuO, CuO/CeO2, and CeO2/CuOCatalysts. Nanoparticles or extended surfaces of CeO2 are verypoor catalysts for the water-gas shift reaction.7,14 Figure 12compares the catalytic activity of CuO, a conventional CuO/CeO2 catalyst,7 and the inverse CeO2/CuO catalyst. The use ofthe same experimental conditions allows for a reliable com-parison of reaction rates. Under reaction conditions, the CuOtransforms into metallic copper.7,16 This system exhibits thelowest catalytic activity in Figure 12. Once ceria is added tothe catalyst mixtures, there is a substantial improvement in thecatalytic activity. From experimental studies with singlecrystals14,22,41 and density functional calculations (DF),42-44 itis known that the most difficult step for the water-gas shift onpure copper systems is the dissociation of water. In contrast,the CeOx/Cu system apparently constitutes a highly efficientbifunctional catalyst in which, according to the results obtained,CO adsorbs on copper and water dissociates on the partiallyreduced ceria nanoparticles. The XRD results in Figure 9 pointto a waterT CeOx interaction, as described in eq 3. Furthermore,DF calculations for the dissociation of H2O on a CeOx/Cu(111)surface gave an exothermic ∆E of -0.33 eV and point to anactivation energy smaller than 0.35 eV.22 In contrast, thedissociation of water on pure Cu(111) is endothermic (∆E )0.2-0.6 eV) with a large activation energy (Ea ) 0.9-1.4eV).43,44 This highlights the key role played by the CeOx
nanoparticles supported on Cu.A recent article discusses the advantages of using an inverse
CeOx/Pt system for catalyzing the water-gas shift reaction.45
When optimizing the performance of copper-based WGScatalysts, the major emphasis is usually in controlling the
Figure 8. Rietveld refinement of mole fraction variation and latticeparameter changes of CeO2 as a function of temperature during thereduction of the CeO2/CuO inverse catalyst in a mixture of 5% CO/95% He. For comparison, we also include the variation in the latticeparameter of ceria due to thermal heating.
Figure 9. Comparison of ceria cell dimensions obtained at 500 °Cduring temperature-programmed oxidation, CO reduction, and duringthe WGS reaction. The dashed line denotes the cell dimension of pureceria at 500 °C.
CO(gas) + CeO2(surf) f CO2(gas) + CeO2-Ovac(surf)(2)
CeO2-Ovac(surf) + H2O(gas) f H2(gas) + CeO2(surf)(3)
Inverse CeO2-x/CuO-Cu Catalyst for the WGS Reaction J. Phys. Chem. C, Vol. 114, No. 8, 2010 3585

oxidation state and morphology of the copper within the catalyticsystem.7-15,18 Our results for CeOx/Cu indicate that an optimiza-tion of the physical and chemical properties of the oxidecomponent is as important as the optimization of the propertiesof the metal component. Because ceria is active as a“support”,36,46,47 one must optimize its properties when designingWGS catalysts.
Conclusions
An inverse CeOx/CuO catalyst has been prepared by aninverse microemulsion approach. Characterization of the freshmaterial by XRD and TEM demonstrated the inverse nature ofthe sample with nanoparticles of ceria dispersed on a copperoxide support.
In situ WGS reaction tests showed that the composition ofthe inverse CeOx/CuO catalyst changed under reaction condi-tions. TR-XRD patterns showed the evolution from CuO tometallic copper via a small amount of Cu2O intermediate. Pairdistribution function analysis corroborated the XRD results andruled out the presence of unidentified amorphous phases.Sequential Rietveld refinement showed an expansion of theCeO2 lattice that correlated with the copper reduction. This cellexpansion is mainly ascribed to the partial reduction of Ce4+ toCe3+.
XANES spectra for the Cu K-edge region confirmed thereduction to metallic copper under WGS reaction conditions.Analysis of the Ce LIII-edge verified the partial reduction ofthe ceria nanoparticles to CeO2-x entities. The presence ofoxygen vacancies in the ceria lattice is an important propertyof the active phase because it opens a new reaction pathinvolving the dissociative interaction of water, which enhancesthe WGS activity on the inverse catalyst.
Reduction of the sample under a 5%CO/He flow shows adirect transformation from CuO to Cu0, without formation ofthe Cu1+ intermediate. Reduction of the sample occurs at a muchlower temperature than under WGS reaction conditions. In theabsence of water, the reduction of CeOx is no longer correlatedwith the copper reduction and occurs at higher temperatures.Reoxidation of the sample was achieved by heating up to 500°C in a 5% O2/He flow. Both XRD and XANES data show thereversibility of the system properties with respect to redoxcycles. Furthermore, the sample reduction-oxidation can beconducted several times without any significant loss in activity,proving the excellent redox stability of the system.
WGS activity tests for CuO, Cu/CeOx, and the inverse CeOx/Cu system showed the importance of the presence of CeOx
species on a copper surface. The inverse catalyst apparentlyoperates as a bifunctional system in which the CO is adsorbedon Cu sites while water becomes dissociated over CeOx
nanoparticles. The close interaction between copper and ceriaallows for the oxygen transfer to the CO. Thus, the inverse CeOx/Cu catalyst is a highly active and stable system with promisingcharacteristics for mobile fuel cell applications.
Figure 10. (A) TR-XRD pattern (λ ) 0.3184 Å) and (B) Cu K-edge TR-XANES spectra during reoxidation of the inverse catalyst in a 5% O2/95%He mixture.
Figure 11. Catalyst stability during several WGS reaction runs overthe same sample.
Figure 12. Catalytic performance of the inverse catalyst vs referencesamples (CuO and Cu/CeO2).
3586 J. Phys. Chem. C, Vol. 114, No. 8, 2010 Barrio et al.

Acknowledgment. N. Marinkovic and S. Khalid are grate-fully acknowledged for their help carrying out the XANESexperiments.The work at BNL was financed by the U.S.Department of Energy (DOE), Chemical Sciences Division (DE-AC02-98CH10086). The National Synchrotron Light Source issupported by the Divisions of Materials and Chemical Sciencesof the U.S.-DOE. L.B. acknowledges funding by the FP7 Peopleprogram under the project Marie Curie IOF-219674. A.H. thanksthe MICINN for a FPU grant under which his contribution tothis work was made. Work at ICP-CSIC and U.C. was financedby Comunidad de Madrid (ENERCAM S-0505/ENE/000304)and MICINN (CTQ2006-15600/BQU and CTQ2009-14527/BQU) projects to whom we are grateful.
References and Notes
(1) Spivey, J. J. Catal. Today 2005, 100, 171–180.(2) Ladebeck, J. R.; Wang, J. P. Catalyst development for water-gas
shift In Handbook of Fuel Cells - Fundamentals, Technology, andApplications; Vielstich, W., Lamm, A., Gasteiger, H. A., Eds.; Fuel CellTechnology and Applications, Vol. 3; John Wiley & Sons, Ltd.: Chichester,U.K., 2003.
(3) Wang, X. Q.; Rodriguez, J. A.; Hanson, J. C.; Gamarra, D.;Martinez-Arias, A.; Fernandez-Garcia, M. J. Phys. Chem. B 2005, 109,19595–19603.
(4) (a) Meunier, F. C.; Reid, D.; Goguet, A.; Shekhtman, S.; Hardacre,C.; Burch, R.; Deng, W.; Flytzani-Stephanopoulos, M. J. Catal. 2007, 247,277–287. (b) Fu, Q.; Saltsburg, H.; Flytzani-Stephanopoulos, M. Science2003, 301, 935–938.
(5) Phatak, A. A.; Koryabkina, N.; Rai, S.; Ratts, J. L.; Ruettinger,W.; Farrauto, R. J.; Blau, G. E.; Delgass, W. N.; Ribeiro, F. H. Catal. Today2007, 123, 224–234.
(6) Jacobs, G.; Ricote, S.; Davis, B. H. Appl. Catal., A 2006, 302, 14–21.
(7) Wang, X. Q.; Rodriguez, J. A.; Hanson, J. C.; Gamarra, D.;Martinez-Arias, A.; Fernandez-Garcia, M. J. Phys. Chem. B 2006, 110, 428–434.
(8) Rodriguez, J. A.; Hanson, J. C.; Wen, W.; Wang, X.; Brito, J. L.;Martinez-Arias, A.; Fernandez-Garcia, M. Catal. Today 2009, 145, 188–194.
(9) Clausen, B. S.; Steffensen, G.; Fabius, B.; Villadsen, J.; Feidenhansl,R.; Topsøe, H. J. Catal. 1991, 132, 524.
(10) Jacobs, G.; Chenu, E.; Patterson, P. M.; Williams, L.; Sparks, D.;Thomas, G.; Davis, B. H. Appl. Catal., A 2004, 258, 203–214.
(11) Koryabkina, N. A.; Phatak, A. A.; Ruettinger, W. F.; Farrauto, R. J.;Ribeiro, F. H. J. Catal. 2003, 217, 233–239.
(12) Fukuhara, C.; Ohkura, H.; Gonohe, K.; Igarashi, A. Appl. Catal.,A 2005, 279, 195–203.
(13) Martinez-Arias, A.; Gamarra, D.; Fernandez-Garcia, M.; Wang,X. Q.; Hanson, J. C.; Rodriguez, J. A. J. Catal. 2006, 240, 1–7.
(14) Rodriguez, J. A.; Liu, P.; Hrbek, J.; Evans, J.; Perez, M. Angew.Chem., Int. Ed. 2007, 46, 1329–1332.
(15) Ilinich, O.; Ruettinger, W.; Liu, X. S.; Farrauto, R. J. Catal. 2007,247, 112–118.
(16) Wang, X. Q.; Rodriguez, J. A.; Hanson, J. C.; Gamarra, D.;Martinez-Arias, A.; Fernandez-Garcia, M. Top. Catal. 2008, 49, 7.
(17) Daly, H.; Ni, J.; Thompsett, D.; Meunier, F. C. J. Catal. 2008,254, 238–243.
(18) Meunier, F. C.; Goguet, A.; Hardacre, C.; Burch, R.; Thompsett,D. J. Catal. 2007, 252, 18–22.
(19) Jacobs, G.; Patterson, P. M.; Williams, L.; Chenu, E.; Sparks, D.;Thomas, G.; Davis, B. H. Appl. Catal., A 2004, 262, 177–187.
(20) Nakamura, J.; Campbell, J. M.; Campbell, C. T. J. Chem. Soc.,Faraday Trans. 1990, 86, 2725–2734.
(21) Rodriguez, J. A.; Ma, S.; Liu, P.; Hrbek, J.; Evans, J.; Perez, M.Science 2007, 318, 1757–1760.
(22) Rodriguez, J. A.; Graciani, J.; Evans, J.; Park, J. B.; Yang, F.;Stacchiola, D.; Senanayake, S. D.; Ma, S.; Perez, M.; Liu, P.; Fernandez-Sanz, J.; Hrbek, J. Angew. Chem., Int. Ed. 2009, 48, 8047–8050.
(23) Norby, P.; Hanson, J. C. Catal. Today 1998, 39, 301.(24) Chupas, P. J.; Qiu, X. Y.; Hanson, J. C.; Lee, P. L.; Grey, C. P.;
Billinge, S. J. L. J. Appl. Crystallogr. 2003, 36, 1342–1347.(25) Chupas, P. J.; Chapman, K. W.; Jennings, G.; Lee, P. L.; Grey,
C. P. J. Am. Chem. Soc. 2007, 129, 13822.(26) Egami, T.; Billinge, S. J. L. UNDERNEATH THE BRAGG PEAKS:
Structural Analysis of Complex Materials; Elsevier Ltd.: Oxford, U.K., 2003.(27) Gamarra, D.; Belver, C.; Fernandez-Garcıa, M.; Martınez-Arias,
A. J. Am. Chem. Soc. 2007, 129, 12064.(28) Chupas, P.; Chapman, K.; Kurtz, C.; Hanson, J. C.; Lee, P.; Grey,
C. P. J. Appl. Crystallogr. 2008, 41, 822.(29) Hammersley, A. P.; Svensson, S. O.; Thompson, A. Nucl. Instrum.
Methods Phys. Res., Sect. A 1994, 346, 312.(30) (a) Toby, B. H. J. Appl. Crystallogr. 2001, 34, 210. (b) Larson,
A. C.; von Dreele, R. B. GSAS General Structure Analysis System; ReportLAUR 86-748; Los Alamos National Laboratory: Los Alamos, NM, 1995.
(31) Qiu, X.; Thompson, J. W.; Billinge, S. J. L. J. Appl. Crystallogr.2004, 37, 678–678.
(32) (a) Ravel, B.; Newville, M. J. Synchrotron Radiat. 2005, 12, 537.(b) Newville, M.; Ravel, B.; Haskel, D.; Rehr, J. J.; Stern, E. A.; Yacoby,Y. Physica B 1995, 208-209, 154.
(33) JCPDS Powder Diffraction File; International Centre for DiffractionData: Newtown Square, PA, 1997; PDF No. 04-0593.
(34) JCPDS Powder Diffraction File; International Centre for DiffractionData: Newtown Square, PA, 1997; PDF No. 48-1548.
(35) Scherrer, P. Gottinger Nachrichten 1918, 2, 92.(36) Trovarelli, A. Catal. ReV.-Sci. Eng. 1996, 38, 439.(37) (a) Engler, B.; Koberstein, E.; Schubert, P. Appl. Catal. 1989, 48,
71–92. (b) Yao, H. C. Appl. Surf. Sci. 1984, 19, 398–406.(38) Wang, X.; Hanson, J. C.; Frenkel, A. I.; Kim, J.-W.; Rodriguez,
J. A. J. Phys. Chem. B 2004, 108, 13667–13673.(39) Martınez-Arias, A.; Hungrıa, A. B.; Fernandez-Garcıa, M.; Conesa,
J. C.; Munuera, G. J. Phys. Chem. B 2004, 108, 17983.(40) Hornes, A.; Hungrıa, A. B.; Bera, P.; Lupez Camara, A.; Fernandez-
Garcıa, M.; Martınez-Arias, A.; Barrio, L.; Estrella, M.; Zhou, G.; Fonseca,J. J.; Hanson, J. C.; Rodrıguez, J. A. J. Am. Chem. Soc. 2010, 132, 34.
(41) (a) Campbell, C. T.; Koel, B.; Daube, K. A. J. Vac. Sci. Technol.,A 1987, 5, 810. (b) Nakamura, J.; Campbell, J. M.; Campbell, C. T. J. Chem.Soc., Faraday Trans. 1990, 86, 2725.
(42) Liu, P.; Rodriguez, J. A. J. Chem. Phys. 2007, 126, 164705.(43) Gokhale, A. A.; Dumesic, J. A.; Mavrikakis, M. J. Am. Chem. Soc.
2008, 130, 1402.(44) Fajin, J. L. C.; Illas, F.; Gomes, J. R. B. J. Chem. Phys. 2009, 130,
224702.(45) Yeung, C. M. Y.; Tsang, S. C. J. Phys. Chem. C 2009, 113, 6074.(46) (a) Si, R.; Flytzani-Stephanopoulos, M. Angew. Chem., Int. Ed.
2008, 47, 2884. (b) Li, Y.; Fu, Q.; Flytzani-Stephanopoulos, M. Appl. Catal.,B 2000, 27, 179.
(47) Hilaire, S.; Wang, X.; Luo, T.; Gorte, R. J.; Wagner, J. Appl. Catal.,A 2001, 215, 271.
JP910342B
Inverse CeO2-x/CuO-Cu Catalyst for the WGS Reaction J. Phys. Chem. C, Vol. 114, No. 8, 2010 3587
Copyright © 2022 FDOKUMEN




















