
Università degli Studi di Firenze Dottorato di Ricerca in ...
341
Università degli Studi di Firenze Dottorato di Ricerca in Scienze Cliniche Indirizzo Fisiopatologia Clinica e dell'invecchiamento CICLO XXVI Confronting the archipelago of primary myocardial diseases: from the comprehension of genetic basis, molecular mechanisms and clinical correlates to the development of novel therapeutic approaches. Dottorando Tutore Dott.ssa Tomberli Benedetta Prof.ssa Abbate Rosanna Coordinatore Prof. Laffi Giacomo Anni 2011/2013
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of Università degli Studi di Firenze Dottorato di Ricerca in ...

Università degli Studi di Firenze
Dottorato di Ricerca in Scienze Cliniche
Indirizzo Fisiopatologia Clinica e dell'invecchiamento
CICLO XXVI
Confronting the archipelago of primary myocardial diseases:
from the comprehension of genetic basis, molecular mechanisms and clinical correlates
to the development of novel therapeutic approaches.
Dottorando Tutore Dott.ssa Tomberli Benedetta Prof.ssa Abbate Rosanna
Coordinatore Prof. Laffi Giacomo
Anni 2011/2013

To Bianca, my little lovely niece.
She lights up my heart every time I see her playing and smiling.
She has been a true blessing to our family.


Index
1. Introduction: Tales of the Unexpected
1.1. Brief history and contemporary classification of cardiomyopathies. Pag. 5
1.2. A thousand conditions with a lot in common. Translational overview supporting a comprehensive approach to primary diseases of the myocardium. Pag. 22
1.3. Making sense of diversity: finding common answers in clinical practice. Pag. 35
1.4. Aim of the thesis and project design Pag. 41
2. Genotype-‐Phenotype Correlations
2.1. Into the myocardium: the distinct phenotypic expression of thin filament mutations in hypertrophic cardiomyopathy. Pag. 47
2.2. Early cardiac phenotype in patients with Anderson-‐Fabry disease. Pag. 60
2.3. Next Generation Sequencing: new horizons in the field of cardiomyopathies. Pag. 76
3. Environmental Modifiers
3.1. The role of environmental modifiers on the phenotypic expression and outcome of Hypertrophic cardiomyopathy: the “weight” of obesity. Pag. 109

4. Natural History and Predictors of Outcome
4.1. Predicting the risk of sudden cardiac death using cardiac magnetic resonance. Pag. 121
4.2. A hundred tales the blood can tell: NT-‐pro BNP as a clinical barometer of disease stability in HCM. Pag. 134
4.3. Long-‐term outcome of idiopathic dilated cardiomyopathy: the real world. Pag. 141
4.4. Myocardial damage due to infectious disease: Chagas cardiomyopathy. Pag. 149
5. Translational Routes from Altered Molecular
Homeostasis to Treatment Opportunities
5.1. Electrophysiological cardiomyocyte remodeling as a new therapeutic target in HCM Pag. 164
5.2. The real story of life-‐saving devices: a backstage tour. Pag. 170
5.3. Changing the destiny of unfolded proteins: pharmacological chaperone as a new therapy for Anderson-‐Fabry disease. Pag. 178
6. Conclusive Remarks Pag. 191
7. References Pag. 193
8. Publications
8.1. List of papers. Pag. 227
8.2. Full-‐text papers and abstracts Pag. 230
9. Acknowledgments Pag. 337

1. Introduction: Tales of the
Unexpected

1. Introduction: tales of the unexpected
4
“We hear only those questions for which
we are in a position to find answers”
Friedrich Nietzsche

1. Introduction: tales of the unexpected
5
1.1 Brief history and contemporary classification of
cardiomyopathies.
It is somewhat of a paradox that cardiologists invest most of their
time treating conditions that only secondarily affect the heart muscle,
due to coronary disease, hypertension or the valvular abnormalities.
Conversely, substantially less time and resources are devoted to the large
and heterogeneous family of diseases originated primarily from the
myocardium – the cardiomyopathies. Although less prevalent than the
previously quoted conditions, cardiomyopathies have a considerable
impact on the community, with a 3% estimated prevalence worldwide,
generating mortality and morbidity preferentially in the young.
Furthermore, they represent valuable paradigms allowing translational
investigation of the normal and abnormal functions of the myocardium,
creating opportunities for the development of novel treatment with
broad implications for all kinds of cardiac patients. Therefore, increased
awareness, investments and research efforts in this field are highly
desirable.
In its present definition, the term “cardiomyopathy” refers to a
myocardial disorder, often genetic in nature, in which the heart muscle is
structurally and functionally abnormal in the absence of coronary artery
disease, hypertension, valvular or congenital heart disease sufficient to
cause the observed myocardial abnormality. This term was first used in
1957 by Brigden, who described a group of uncommon, non-‐coronary
myocardial diseases [1]. In 1961 Goodwin defined cardiomyopathies as
“myocardial diseases of unknown cause” [2]. He described three different
entities, namely “dilated, hypertrophic and restrictive”, terms which are

1. Introduction: tales of the unexpected
6
still in use today. However, only in the 70s and 80s, following the advent
of non-‐invasive imaging, such as M-‐mode and 2D echocardiography, the
true frequency and spectrum and of cardiomyopathies began to be
recognized. In an attempt to provide a practical framework for clinicians
involved in the care of these patients, the first classification of
cardiomyopathies was published in 1980, by the World Health
Organization (WHO) and International Society and Federation of
Cardiology (ISFC), and included the three entities proposed by Goodwin
[3]. The definition of “myocardial diseases of unknown cause” was
maintained to define cardiomyopathies, as opposed to “specific heart
muscle diseases”, comprising heart diseases with similar phenotypes, but
due to an identifiable cause.
In the last 30 years, progress in imaging techniques and intensive
genetic investigation have produced major advancements in our
understanding of the causes and manifestations of cardiomyopathies
[4,5], and new nosologic entities have been described. This led to the
new revision of the classification, carried out in 1996 by the WHO and
ISFC [6]. Representing a major advancement, both “arrhythmogenic right
ventricular dysplasia” (with the inappropriate term “dysplasia” later
changed to “cardiomyopathy”) and a group of “unclassified
cardiomyopathies'', defined as “those that do not fit in any group”, were
added to the three original subgroups. The definition of cardiomyopathy
was changed to “diseases of the myocardium associated with myocardial
dysfunction”. Moreover, three additional subgroups termed
“hypertensive”, “valvular” and “ischemic” cardiomyopathies were -‐
confusingly -‐ added to the group of “specific heart muscle diseases” in
order to resolve a terminology controversy between US and European
experts [6]. These were defined as cardiac conditions characterized by the

1. Introduction: tales of the unexpected
7
presence of hypertension, coronary or valvular disease, in a degree that
would not explain the magnitude of LV dysfunction observed.
Nevertheless, a substantial difference in terminology persisted on the two
sides of the Atlantic, reflecting the refusal of these fine distinctions by US
experts [7].
In 2006, an American Historical Association (AHA) panel of experts
published a scientific statement on the “Contemporary classification and
definitions of Cardiomyopathies” [7]. They proposed a novel approach, by
which the etiology, rather than the phenotype, was used as the main
criterion. “Primary” cardiomyopathies were defined as those “involving
only the heart”, as opposed to the “secondary”. Primary
cardiomyopathies for the first time also included “ion channel diseases”
and were differentiated in three subgroups based on their etiology as
“genetic, mixed and acquired” [Figure 1.1-‐1].
Figure 1.1-‐1: American Heart Association classification of the cardiomyopathies (2006)

1. Introduction: tales of the unexpected
8
Of note, the term “primary” was used to describe diseases in which
the heart is the sole or predominantly involved organ, while “secondary”
described diseases in which myocardial dysfunction is part of a systemic
disorder [5]. However, the challenge of distinguishing primary and
secondary disorders is illustrated by the fact that many of the diseases
classified as primary cardiomyopathies can be associated with major
extra-‐cardiac manifestations; conversely, pathology in many of the
diseases classed as secondary cardiomyopathies can predominantly (or
exclusively) involve the heart.
The radical shift from a phenotypic to an etiological classification, as
well as the inclusion of ion channel diseases among cardiomyopathies,
although proposed for research rather than clinical purposes, sparked a
passionate transatlantic debate, culminating in a thorough re-‐visitation of
the original 1995 classification by the European Society of Cardiology
(ESC) Working Group on Myocardial and Pericardial diseases, in 2008 [8].
Intrinsically faithful to the concept of classifying cardiomyopathies based
on phenotype, the 2008 European classification maintained each of the
time-‐honoured categories including dilated, hypertrophic, restrictive and
arrhythmogenic right ventricular cardiomyopathy. The confusing
“hypertensive'', “valvular'' and “ischemic'' categories were removed. In
the “unclassified” subgroup, “Left Ventricular Non-‐compaction” and
“Tako-‐Tsubo” cardiomyopathy made their official debut [Figure 1.1-‐2].
Conversely, ion channel diseases were excluded, despite their genetic
nature, in view of their lack of a structural cardiac phenotype. Each
cardiomyopathy subtype was subdivided in a familial and non-‐familial
subset, and, to replace the pre-‐genetic era concept of “unknown
etiology”, a list of potential genetic and non-‐genetic causes was provided
[Tables 1 to 5].

1. Introduction: tales of the unexpected
9
Figure 1.1-‐2: ESC classification of the cardiomyopathies (2008)
Table 1 to table 5: Genetic and non-‐genetic causes of cardiomyopathies

1. Introduction: tales of the unexpected
10

1. Introduction: tales of the unexpected
11
The precise identification of the disease etiology has obvious clinical
implications, by virtue of its direct impact to totally different
management. For example, amyloidosis, Anderson Fabry diseases and
glycogen storage diseases may be diagnosed as hypertrophic
cardiomyopathy (HCM); yet their treatment varies widely. Of note, the
inclusion of amyloidosis in this classification was widely debated [9].
Substantial doubts also regarded Takotsubo, a disease that is generally
transient, has no proven inherited cause, and appear related to regional
myocardial hypoperfusion rather than to heart muscle abnormalities.
Ultimately, both were included as this was felt to be conceptually useful
in clinical practice.
To follow is a brief overlook of the major cardiomyopathies subtypes,
based on contemporary definitions.
1.1.1 Hypertrophic Cardiomyopathy
HCM is a genetic disease characterized by unexplained LV
hypertrophy, associated with non-‐dilated ventricular chambers, in the
absence of another cardiac or systemic disease capable of producing that
degree of hypertrophy [Figure 1.1-‐3]. HCM is diagnosed by a maximal LV
wall thickness greater than 15 mm, based on echocardiography (Echo) or
cardiac magnetic resonance (CMR) [10]. This value is lowered to 13-‐14
mm, when family members are screened. In children, a wall thickness
greater than 2 standard deviations (SD) for age, sex or body size is
considered diagnostic.

1. Introduction: tales of the unexpected
12
Figure 1.1-‐3: Figure 3. Hypertrophic cardiomyopathy.
Echocardiographic and cardiac magnetic resonance images from a 17-‐ year old female patient with HCM. Parasternal long and short axis views show severe LV thickness values (max LV wall thickness 31 mm), with redundant mitral leaflets (panels A, B and D) and small cavity size. Apical 4 chambers views show massive hypertrophy of the septum and the antero-‐lateral wall (panels C and E). Images of late gadolinium enhancement showing limited and nontransmural area of fibrosis of the IVS (panel F: black arrow).
The distribution of hypertrophy is usually asymmetric and
sometimes confined to one or two LV segments. As a consequence, LV
mass (measured by CMR) can be within the normal range. LV outflow
tract obstruction is an important feature of HCM, and may be
demonstrated in up to 70% patients [11]. Overall, the clinical course of
patients with HCM is relatively benign, with an annual mortality rate of
about 1%. Contrary to prior perceptions, the risk of sudden cardiac death
is relatively low [12], although still a major concern in young individuals
and athletes. Furthermore, about half of patients show some degree of
disease progression and functional limitation, with a small but significant
subset of about 5% developing the so-‐called end-‐stage HCM. Family

1. Introduction: tales of the unexpected
13
screenings, following the introduction of genetic testing, has led to the
identification of genotype-‐positive/phenotype-‐negative individuals, a
novel category within the HCM spectrum, characterized by absence of LV
hypertrophy, assessed by ECG and ECHO [10].
Sarcomeric gene mutations, often private, are the most frequent
cause of HCM, accounting for approximately 30-‐65% of probands in
different cohorts [13]. In the remaining subset the genetic substrate is
unknown. Furthermore, a small proportion of patients with the HCM
phenotype are affected by cardiofacial syndromes (e.g. Noonan, LEOPARD,
Costello), neuromuscular diseases (e.g. Frederich's ataxia), mitochondrial
diseases [14], metabolic disorders of lysosomal storage diseases (i.e.
Fabry, Pompe, Danon) [15]. These rare conditions sometimes exhibit an X-‐
linked rather than the autosomal pattern of inheritance, usually observed
in HCM [Table 1].
1.1.2 Dilated cardiomyopathy
DCM is characterized by LV dilatation and global systolic dysfunction
(EF < 50%), in the absence of coronary artery disease or other identifiable
causes (such as systemic hypertension, valve disease, drugs, inflammatory
heart diseases) capable of causing that magnitude of impairment [Table
2]. In familial DCM, screening of first-‐degree relatives will identify the
disease in up to 50% [16]. As for many other cardiomyopathies, the
prevalence of DCM is underestimated, because many patients may have a
subclinical form of the disease, which may be difficult to diagnose for the
lack of symptoms. Familial and sporadic forms of DCM have similar
morphological manifestation and clinical course [Figure 1.1-‐4].

1. Introduction: tales of the unexpected
14
However some gene mutations, such as Lamin A/C seem to carry a
more adverse outcome, in particular for sudden death [17-‐19]. DCM is a
progressive disease, with a prognosis that, although improved in the last
decades, is usually poor due to heart failure, atrial and ventricular
arrhythmias, stroke and sudden death [20]. In patients with refractory
heart failure, heart transplant represent the final option. The low yield of
genetic testing for DCM (i.e. 30%) limits its clinical use and it is related to
the large number of potentially disease-‐causing genes. Furthermore,
genetic mutations are usually private and the interpretation of the
analysis results may be difficult [16]. However, the advent of new
sequencing technologies is probably going to change this paradox in the
near future (see chapter 2).

1. Introduction: tales of the unexpected
15
Figure 1.1-‐4: Dilated cardiomyopathy
Echocardiographic and cardiac magnetic resonance images from a 57-‐year old female patient with DCM and normal coronary angiogram. Parasternal long axis view and CMR images show dilated LV (panels A-‐B and E-‐F), with severe systolic dysfunction – EF = 21%; (panel C = diastole, panel D = systole). Abbreviations: LV = left ventricle, RV = right ventricle IVS = inter-‐ventricular septum.

1. Introduction: tales of the unexpected
16
1.1.3 Restrictive cardiomyopathy
RCM is defined by the presence of a restrictive LV physiology, with
normal or more often reduced diastolic/systolic volumes, normal wall
thickness and systolic function, marked diastolic flow impairment and
biatrial dilatation. RCMs are rather uncommon, although their prevalence
is still unknown. Either Amyloid Light-‐chain (AL) amyloidosis or
amyloidosis due to transthyretin gene mutations with heart involvement,
often cause RCM [Table 3] [9]. A striking subtype of disease with
restrictive physiology, endomyocardial fibrosis, endemic in areas of the
African continent, has an unknown etiology and very poor prognosis [21].
Moreover a “restrictive phenotype” may be part of the clinical spectrum
of end-‐stage HCM [22], and may occasionally originate as a primary, non
HCM-‐related phenotype from sarcomere gene mutations (generally in the
thin filament protein coding genes – see chapter 2). RCM is usually
associated with severe functional limitation, mainly related to the
extreme diastolic dysfunction, with reduced diastolic filling and stroke
volume, and a poor prognosis [23].
1.1.4 Arrhythmogenic right ventricular cardiomyopathy
ARVC is characterized by fibro-‐fatty replacement of the right
ventricular myocardium and ventricular arrhythmias [24]. In the most
common right-‐dominant form, structural changes may be absent or
confined to a localized region of the right ventricle (inflow and outflow
tract, right ventricular apex, known as the `triangle of dysplasia') at an
early stage [Figure 1.1-‐5]. Progression to more diffuse right ventricular
disease and LV involvement (typically affecting the posterior lateral wall),
associated with ventricular systolic dysfunction, is common at later stages
[25]. Ventricular arrhythmias are the clinical hallmark of the disease, but
1. Introduction: tales of the unexpected
17
atrial fibrillation may also occur. The diagnosis of ARVC is often
challenging for the cardiologist, in particular during the early “concealed
phase”, when individuals are still asymptomatic and structural
abnormalities are subtle. Predominant LV disease has also been
recognized in a small subset of patients of about 5%. New diagnostic
criteria with higher sensitivity and specificity have recently been
published [24,26]. ARVC is generally a familial disease with autosomal
dominant inheritance but it may be recessive when associated with
woolly hair and palmopalmar hyperkeratosis (eg, Naxos disease, Carvajal
syndrome). Mutations in desmosomal and non-‐desmosomal genes have
been identified, but interpretation of their pathogenicity is often
challenging in the affected individual [Table 4].
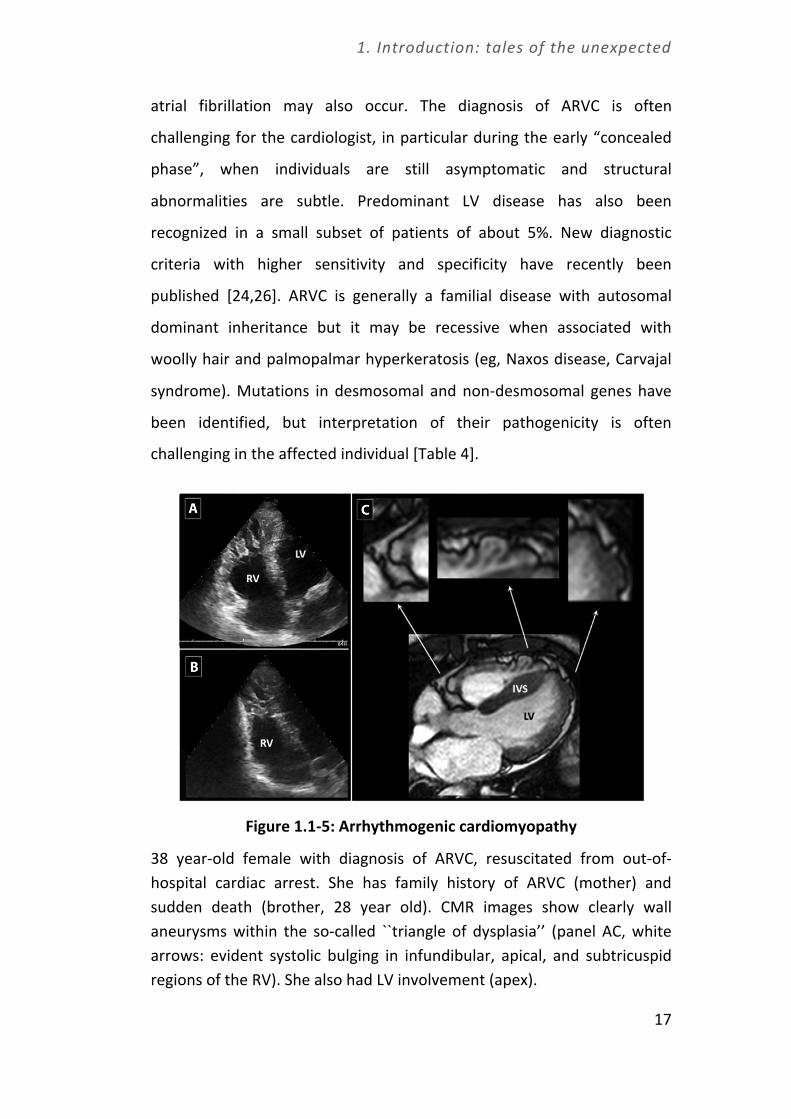
Figure 1.1-‐5: Arrhythmogenic cardiomyopathy
38 year-‐old female with diagnosis of ARVC, resuscitated from out-‐of-‐hospital cardiac arrest. She has family history of ARVC (mother) and sudden death (brother, 28 year old). CMR images show clearly wall aneurysms within the so-‐called ``triangle of dysplasia’’ (panel AC, white arrows: evident systolic bulging in infundibular, apical, and subtricuspid regions of the RV). She also had LV involvement (apex).

1. Introduction: tales of the unexpected
18
Abbreviations: LV = left ventricle, RV = right ventricle, IVS= interventricular septum.
1.1.5 Unclassified cardiomyopathies
Isolated LV non-‐compaction (LVNC) is characterized by prominent LV
trabeculae and deep inter-‐trabecular recesses, that can be associated
with LV dilatation and systolic dysfunction [Figure 1.1-‐6].
Figure 1.1-‐6: Left ventricular non compaction
Isolated left ventricular non-‐compaction in a 45 year-‐old male, with mild systolic dysfunction (EF 48%), ventricular arrhythmias and normal LV diameters. Multiple trabeculations and recesses are evident, particularly

1. Introduction: tales of the unexpected
19
in the apex and the free wall of the LV (panels A and C: apical 4 chambers view; panels B and D: apical 3 chambers view). CMR confirmed the diagnosis (panels E-‐F). Abbreviations: LV= left ventricle, RV = right ventricle IVS = inter-‐ventricular septum.
LVNC is familial, with 25% of asymptomatic first-‐degree relatives
having some echocardiographic abnormalities [Table 5]. Of note, this
rather mysterious disease shows substantial phenotypic overlap with
other cardiomyopathies (in particular HCM and DCM, which often exhibit
limited areas of non-‐compaction in the left ventricle), as well as a
common genetic substrate [8]. Furthermore, LVNC may be associated
with congenital cardiac disorders (such as Ebstein's anomaly or complex
cyanotic heart disease) and some neuromuscular diseases. Therefore, it is
still debated whether isolated LVNC should be considered a separate
clinical and genetic entity, or a morphological trait shared by many
distinct cardiomyopathies. As a result of the difficult comprehension of
this clinical entity, the real prevalence of LVNC and its outcome remain
largely unknown.
Takotsubo cardiomyopathy, also known as LV apical ballooning or
stress-‐induced cardiomyopathy, is characterized by transient regional
systolic dysfunction involving the apex and/or mid-‐ventricle in the
absence of obstructive coronary artery disease on angiogram [8]. The
condition is reported all over the world, and most reported cases occur in
post-‐menopausal women following physical or psychological stress, but it
has been described also in patients with intracranial haemorrhage or
other acute cerebral accidents (so-‐called “neurogenic myocardial
stunning”). Typically, takotsubo cardiomyopathy has a sudden onset, with
chest pain, diffuse T-‐wave inversion and mild cardiac enzyme elevation.
Symptoms are often preceded by emotional or physical stress. If the
patient survives the acute phase of disease, the prognosis is almost

1. Introduction: tales of the unexpected
20
invariably favourable, with a normalization of LV function over a period of
days to weeks; recurrence is possible, but rare.
1.1.6 A never-‐ending story
As more families with cardiomyopathies are genotyped, and new
diseases are being described, the paradigm “one gene, one disease”
appears no longer sustainable. The same mutation can be expressed at a
different age and give rise to hugely different phenotypes within the
same family. Different phenotype patterns may originate from the same
genetic substrates, in a spectrum encompassing HCM, DCM, RCM and
LVNC (all associated with sarcomere genes), or ARVC/DCM (associated
with desmosomal genes). Such heterogeneity is thought to derive from
the interaction between one or more genetic mutations, modifier genes
and environmental factors [10]. When genetic analysis is performed in
candidate genes, the probability of identifying the pathogenic gene
mutation is in the range of 40 60%, for patients with HCM, with
approximately 5% of complex mutations [11,12]. Results for dilated
cardiomyopathy (DCM), restrictive cardiomyopathy (RCM) and isolated LV
non-‐compaction are considerably less rewarding [10], although the
advent of next generation, genome-‐wide techniques may increase the
yield substantially, as recent data on titin in DCM suggests [27]. The
recent introduction of next generation sequencing has started what
promises to be a revolution in molecular diagnostics, allowing rapid and
affordable testing of hundreds of genes, or even whole genomes. As an
example, a wide range of truncating gene mutations encoding Titin, a
very large cytoskeleton gene which could not be assessed by traditional
sequencing techniques, has recently been discovered to represent a
prevalent cause of familial DCM, up to 25% [27]. In the near future, the

1. Introduction: tales of the unexpected
21
list of causative genes will therefore likely require an update. The focus
for researchers will necessarily shift from analyzing single mutations in
candidate genes, to interpreting the hundreds of variants of unknown
significance in putative causative as well as modifier genes, requiring
entirely new skills and significant interaction with biophysicists and
computer scientists.
At present, and in the foreseeable future, however, clinical
classifications of cardiomyopathies based on clinical presentation and
morphological criteria represent an important tool for clinicians involved
with these complex diseases. While calling for constant improvement and
update in the light of advances provided by imaging, genetics and basic
science, individual patient phenotypes continue to represent the core of
any classification in clinical medicine, something that has not changed
with time.

1. Introduction: tales of the unexpected
22
1.2 A thousand conditions with a lot in common.
Translational overview supporting a comprehensive
approach to primary diseases of the myocardium.
The pathophysiology of cardiomyopathies is extraordinarily complex
and encompasses a constellation of different mechanisms, most of which
at present unresolved. Even when the disease model is narrowly defined
and recognizes a well-‐defined etiology, such as a gene mutation, the
processes leading to the phenotypic manifestations are largely beyond
our current understanding. For example, early hopes of establishing strict
genotype-‐phenotype correlations in HCM, probably the most extensively
studied disease in this group, have definitely been abandoned. Even when
the link between mutations and myocardial damage is relatively
understood, as in Anderson Fabry disease, the reasons of clinical
heterogeneity observed even among individuals from the same family
remain obscure. It is now well established that the same genes can give
rise to radically different phenotypic and clinical manifestations and –
conversely – that the same phenotype can be caused by mutations in
different genes. In other words, gene mutations are necessary but not
sufficient to cause clinical phenotypes, and what comes in between is at
least as important as the etiology itself. In what may seem like an
inextricable labyrinth, however, a number of general mechanisms seem
to represent common pathophysiologic determinants of cardiomyopathy
phenotypes. While not specifically related to the disease etiology, these
features represent shared pathways by which the disease becomes overt
and leads to its clinical consequences; in some cases they represent
features are common with heart failure due to any cause. Any attempt to
comprehensively cover this field goes, is would be too ambitious.

1. Introduction: tales of the unexpected
23
What follows is an overview of some of the most intriguing elements
characterizing cardiomyopathy pathophysiology, representing both keys
to further understanding of the “core” of heart muscle disease, and
potential targets for treatment.
1.2.1 Genetic and post-‐transcriptional mechanisms
The variable, often age related, penetrance and variable disease
expression suggest that the effects of cardiomyopathy-‐causing mutations
are modifiable both by modifier genes and environmental factors [28].
Mutations generally cause single amino-‐acid substitutions in proteins that
become incorporated into the sarcomere and exert their pathological
effects by acting as poison peptides that alter normal sarcomere function
in a concentration dependent manner. Thus, homozygous mutations,
multiple mutations, and compound genotypes (2 or more mutations in
multiple genes) often result into earlier presentation and rapid disease
progression [29-‐31]: this has been shown for HCM, ARVC and ion
channel disease. An exception to this rule are most MYBPC3 mutations in
HCM, that result in insufficient protein production for normal sarcomere
function (haploinsufficiency) [32-‐33]. Haploinsufficiency can be
attributed to cell surveillance mechanisms, including nonsense-‐mediated
decay of mRNA transcripts that contain premature termination codons
and/or ubiquitin-‐mediated proteasomal (UPS) degradation of misfolded
proteins. It remains to be demonstrated whether impairment of UPS due
to excess degradation of mutant proteins may trigger phenotypic onset or
contribute to disease progression [34]. In addition, differential levels of
activity in these cell surveillance mechanisms may explain individual
heterogeneity in phenotype, eg within families with the same genetic
mutation. Finally, MicroRNAs (miRNAs) are small conserved RNA

1. Introduction: tales of the unexpected
24
molecules nucleotides which negatively modulate gene expression in
animals and plants. MiRNAs are involved in a variety of basic biological
processes, for example, cell proliferation and apoptosis and stress
responses. A subset of miRNAs are either specifically or highly expressed
in cardiac muscle, providing an opportunity to understand how gene
expression is controlled by miRNAs at the post-‐transcriptional level in this
muscle type. miR-‐1, miR-‐133, miR-‐206, and miR-‐208 have been found to
be muscle-‐specific, and thus have been called myomiRs; among their
functions, they have been shown to regulate cardiac development and
differentiation. MiRNAs are thought to play an important role in
modulating the development of phenotype in patients with
cardiomyopathies. For example, in vivo miR-‐133 levels are down-‐
modulated in patients with HCM, and other MiRNAs have been implicated
in the regulation of cardiac hypertrophy [35]. Therefore, both the initial
genetic burden and post-‐transcriptional mechanisms seem to significantly
impact the development of cardiomyopathies.
1.2.2 Abnormal calcium homeostasis
Most models of heart failure, including human disease, are
characterized by decreased SR Ca2+-‐ ATPase (SERCA) expression and
upregulation of Na+/Ca2+ exchanger (NCX) expression and function.
Decreased SERCA activity, by slowing down Ca2+ reuptake to the
sarcoplasmic reticulum, allows more Ca2+ to be extruded via the NCX;
coupled with the increased NCX expression [36] this results in a net loss
of cell Ca2+ and contributes to the reduction of sarcoplasmic reticulum
Ca2+ load [37]. Another contributor to the lower Ca2+ content in heart
failure is the increased diastolic leakage of Ca2+ from the sarcoplasmic
reticulum, which is determined by the hyperphosphorylation of ryanodine

1. Introduction: tales of the unexpected
25
receptors by protein-‐kinase A and/or Ca2+-‐Calmodulin dependent protein
kinase-‐II (CaMKII) [38]. CaMKII activity is increased in HF, and CaMKII-‐
dependent phosphorylation of RyR enhances Ca2+ spark frequency and
thus spontaneous diastolic sarcoplasmic reticulum Ca2+ leak [39], making
it a leading pathway causing contractile dysfunction and
arrhythmogenesis in HF. Enhanced NCX function, combined with the
higher probability of spontaneous Ca2+ release from the SR, contribute
directly to arrhythmogenesis via delayed-‐afterdepolarizations (DADs).
When a large spontaneous Ca2+ release event occurs during diastole
giving rise to a generalized Ca2+ wave, part of the released Ca2+ is
extruded through the NCX, which generates an inward current that
depolarizes the membrane (i.e. a DAD). If large enough, a DAD may reach
the threshold for a premature AP, giving rise to a premature activation
that can propagate through the myocardium, triggering sustained
arrhythmias. Thus, abnormal calcium homeostasis is a main determinant
of several manifestations that are common to several cardiomyopathies,
including diastolic dysfunction (due to the excess residual cytoplasmic
Ca2+ at the end of systole), impaired energetics, myocardial ischemia and
arrhythmgenesis. This has been specifically shown in models and human
tissue with HCM, for example.
1.2.3 Lack of energetic sustainability
Primary myocardial disease is often characterized by abnormal
energy generation and/or consumption. In rare diseases, such as
cardiomyopathies associated with mitochondrial disease, this feature is
taken to the extreme. However, varying levels of energetic impairment
can be found in virtually all models. Notably, this is not always due to
insufficient energy generation; in HCM for example, disease causing

1. Introduction: tales of the unexpected
26
mutations are often gain-‐of-‐function, and lead to sarcomere energetic
inefficiency, due to the excess ATP utilization required to generate
isometric tension [40, 41], which may ultimately compromise overall
cardiomyocyte energetic balance [42]. Energy deficiency would be
expected to reduce the activity of membrane-‐bound energy-‐requiring ion
transporters, potentially triggering arrhythmias and contributing to
diastolic dysfunction, and to decrease contractile reserve. In addition,
cardiomyocyte energetic compromise might contribute to generation of
pathologic hypertrophy (and potentially adverse remodeling) as a
consequence of intracellular energy sensor activation [4]. In addition,
residual, force-‐generating, ATP-‐consuming acto-‐myosin interactions
during diastole lead to incomplete relaxation and directly contribute to
diastolic dysfunction while increasing the energetic compromise of HCM
myocytes. Sarcomeres and their Z-‐disk components are now recognized
centers of mechano-‐sensation, -‐transmission and -‐transduction [43, 44].
Cardiac stress leads to mechanical and chemical signals which remodel
sarcomeres and either offset or exacerbate the stress. In HCM patients,
altered sarcomere mechanics due to faster kinetics of force generation,
hypercontractility or incomplete relaxation may trigger hypertrophy and
adverse remodeling. That this may represent an important primary
mechanism is supported by the HCM-‐causing role of mutations in genes
encoding Z-‐disk proteins [45]. In the long term, reduced energy
production or inefficient utilization may become non-‐sustainable, and
directly contribute to a vicious cycle of worsening cardiomyocyte
dysfunction, increased LV wall stress, increased oxygen demand and
further energetic mismatch, leading to disease progression and heart
failure.

1. Introduction: tales of the unexpected
27
1.2.4 Electrophysiological remodeling
The fact that cardiomyopathies may express an electrophysiological,
as well as structural, cellular phenotype has only recently been
appreciated. While clinicians have always known the surface ECG
manifestations associated with each disease, these have classically been
attributed to macroscopic abnormalities of the heart such as disarray,
strain, hypertrophy and necrosis. However, it is now evident that some of
these manifestations (such as QT prolongation) and even the
arrhythmogenic potential associated with primary heart muscle disease
may originate at the level of the cardiomyocyte sarcolemma. Among the
most prominent abnormalities are alterations of late Na+ current in HF.
Increased [Na+]i in HF may be due to increased Na+ influx or decreased
Na+ efflux. Early studies suggested that an excess of Na+ influx is the
major contributor to Na+ overload and the main source of the increased
Na+ entry is the enhanced “late” or “persistent” Na+ current [46, 47]. As
explained before, a component of Na+ current with slow or incomplete
inactivation can be measured in normal human and animal cardiac
myocytes [48]. However, acquired and primary cardiac diseases are
commonly characterized by abnormally large INaL. Enhancement of late
Na+ current has been identified in HCM, and may contribute to its
pathogenesis. In addition, besides LQT3 syndrome, which is directly due
to Na+ channel mutations [49], increase INaL was found as a consequence
of ankyrin B mutations (LQT4 syndrome) and caveolin-‐3 mutations (LQT-‐
CAV3) [50, 51], as well as end stage heart failure [52, 53] and following
myocardial infarction [54]. Increased Ca2+ -‐Calmodulin and calmodulin
kinase II (CaMKII) activity, both common features of myocardial
remodelling in HF, have been shown to increase INaL. Recent evidence
identified CaMKII-‐mediated phosphorylation of cardiac Na+ channels at
multiple sites is the main regulator of Na+ channel inactivation. INaL

1. Introduction: tales of the unexpected
28
enhancement is associated with prolonged repolarisation causing a
remarkable increase in action potential duration [55, 56]. Atrial potential
prolongation leads to reduced repolarisation reserve and therefore
increased incidence of early afterdepolarizations (EADs, i.e. premature
depolarizations occurring during the plateau phase) and potentially fatal
arrhythmias [57]. All these conditions are all characterized by increased
susceptibility to perturbations of repolarization (e.g. drugs blocking K+
currents or electrolyte imbalances) and overall increased risk of
arrhythmias.
1.2.5 Coronary microvascular dysfunction
By definition, cardiomyopathies are characterized by the absence of
acquired or congenital coronary artery disease at the epicardial level. Yet,
a quota of myocardial ischemia is virtually always present. This is due to
impairment of flow reserve at the microvascular level – a crucial
determinant of myocardial perfusion. Coronary microvascular dysfunction
has been demonstrated in diseases ranging from HCM, DCM, ARVC to
Anderson Fabry disease [58-‐61]. In most studies, impairment of
coronary reserve at this level has been shown to have profound
prognostic implications [62]. Therefore, whatever the mechanism(s)
leading to microvascular dysfunction, the latter tends to take center stage
and determine disease progression and outcome when severe,
representing an important (and unfortunately yet unattainable)
therapeutic target. Mechanisms may be several: the most common is
non-‐specific, represented by extravascular compression of the small
coronary vessels due to increased LV wall tension and fibrosis [63].
However, disease-‐specific mechanisms exist, such as endothelial

1. Introduction: tales of the unexpected
29
infiltration in storage disease (most notably Anderson-‐Fabry disease) and
microvascular remodeling in HCM.
In HCM patients, microvascular remodeling is a striking and
consistent feature which occurs independent of hypertrophy, appears to
be genetically regulated [64], and may initiate as early as during
development [65]. Abnormalities include marked thickening of the
intramural coronary arteriole wall, due to smooth muscle hyperplasia and
an abundance of disorganized elastic fibres, causing deformation and
irregular narrowing of the vessel lumen: as a consequence, their capacity
to vasodilate in response to physiological stimuli is markedly impaired.
Other factors contributing to microvascular dysfunction, such as disarray,
reduced capillary density and increased extravascular compressive forces,
are probably active in the most hypertrophied regions of the myocardium.
1.2.6 Fibrosis
Marked increase in the extracellular matrix is another common
theme in cardiomyopathy. Collagen synthesis may be primarily triggered
by altered fibroblast function (eg due to paracrine or neuroendocrine
influences) or as a reparative phenomenon following myocyte loss [66].
Primary (i.e. non ischemia-‐mediated) activation of profibrotic
pathways is a consistent finding in patients with cardiomyopathies, and
has been demonstrated even in the pre-‐clinical phase [67]. These
pathways are associated with variable levels of interstitial fibrosis, the
clinical relevance of which remains uncertain. Of note, biomarkers of
collagen synthesis and degradation reflect collagen metabolism and
correlate with adverse outcomes in hypertension, heart failure, and
myocardial infarction. In HCM, studies of the links relating sarcomere

1. Introduction: tales of the unexpected
30
protein mutations to the mechanisms that regulate vascular remodeling
and fibrosis are likely a key to our understanding of disease progression.
Recent studies have suggested that some phenotypic expressions
correlate more with altered function of fibroblasts than with myocyte-‐
related pathology. Animal models of HCM that recapitulate human
disease have recently shed light on the earliest cellular and molecular
responses to sarcomere-‐gene mutations [68]. Cardiac transcriptional
profiling in young mice in which hypertrophy has not yet developed
shows activation of pathways involved in fibrosis and collagen deposition.
These studies indicate that a profibrotic milieu is present early in hearts
with HCM even when cardiac histologic findings are normal.
Replacement fibrosis is discrete, rather than interstitial, may be
transmural and occupy significant proportions of the left and/or right
ventricle, can be visualized in vivo as late gadolinium enhancement (LGE)
by CMR, and is generally associated with systolic dysfunction [69-‐76].
LGE is believed to largely reflect a reparative process following
microvascular ischemia-‐mediated damage – i.e. a scar [77].
The deposition of LGE varies according to the specific disease. In
HCM, areas of LGE have typical mid-‐wall localization sparing the
subendocardial region [70-‐76]. CMR late gadolinium enhancement is
present in about two-‐thirds of HCM patients, varying from very limited to
large, confluent, infarct-‐like patches occupying significant proportions of
the LV [70, 74]. LGE localizes preferentially to the most hypertrophied
regions of the ventricle, often represented by the basal and mid-‐septum,
and are more often found in patients with diffuse and severe hypertrophy.
Conversely, LGE is often absent in patients with mild HCM phenotype and
limited extension of hypertrophy [70,71]. LGE is inversely related to LV
systolic function in HCM patients, and that those individuals who have

1. Introduction: tales of the unexpected
31
reached the end-‐stage phase of the disease, characterized by overt
systolic dysfunction and LV remodelling with progressive wall thinning,
constantly exhibit large and often transmural areas of LGE, which may
occupy as much as 50% of the whole ventricular mass [78]. Furthermore,
preliminary evidence points to LGE areas as a potential substrate of
ventricular arrhythmias [69, 72].
In ARVC patients, phenotype, although genetically determined,
develops postnatally and is progressive with age, initiated by necrotic cell
death, which subsequently triggers an inflammatory response and
massive calcification within the myocardium, followed later by injury
repair with fibrous tissue replacement. In a Desmoglein 2 mutant mouse
model, necrosis always preceded other pathological signs of disease, such
as inflammation and calcification, and later on, fibrosis, ventricular
dilation, and aneurysm formation. Furthermore, myocyte necrosis
originated in the subepicardial myocardium, followed by a wave-‐front
extension toward the endocardium [79]. This wave-‐front phenomenon of
myocardial atrophy is well documented in human ARVC, both by
histopathologic examinations and cardiac magnetic resonance studies
[80, 81].
Both reactive (interstitial and perivascular) and reparative
(replacement) myocardial fibrosis (MF) are hallmarks of DCM [82-‐84]. In
vivo assessment of myocardial fibrosis by CMR in DCM patients have
shown a prevalence of significant LGE in almost 50%, with mid-‐wall
distribution and typical sparing of the subendocardium – a useful marker
to rule out ischemic dilated cardiomyopathy. Fibrosis is a predictor of
outcome and reverse remodeling following HF therapy; of note, it is less
prevalent in DCM caused by myocarditis, in which LV dysfunction may be
reversible, compared to genetic DCM [85].

1. Introduction: tales of the unexpected
32
1.2.7 Developmental aspects
At birth, patients with genetic cardiomyopathies generally exhibit
normal hearts, and the full-‐fledged phenotype may develop at
adolescence or during adult life; pediatric onset is rare but possible.
When the phenotype develops, a number of tissues and cell types that do
not express the mutated gene are found to actively participate in the
disease process, in ways that are hard to explain simply on the basis of
secondary, bystander, involvement. To date, the link between the genetic
defect such “extended” phenotype remains elusive. Among the possible
explanation, the hypothesis of cardiomyopathies as cell lineage diseases
has emerged, by which pre-‐natal mechanisms directly linked to the causal
gene defect acts upon multipotent progenitors to influence their
commitment and ultimate development [65].
For example, theories explaining the development of hypertrophy in
HCM patients fail to address aspects of HCM as diverse as interstitial
fibrosis, microvascular remodeling and mitral valve. Specific features,
such as direct papillary insertion into the mitral leaflet or myocardial
bridging, clearly suggest a developmental defect [86]. Our group has
proposed [65] that a common lineage ancestry for these extramyocardial
phenotypes can be traced back to the proepicardial organ, originating
from the posterior component of the secondary heart field [87]. Early
during development, the migration of proepicardial cells over the naked
heart tube originates the epicardium. Following a process called
epithelial-‐mesenchymal transformation, apparently pluripotent
epicardium-‐derived cells (EPDCs) subsequently migrate diffusely into the
myocardium and differentiate into diverse cell types which give rise to
cardiac scaffolding as interstitial fibroblasts, to the coronary vasculature

1. Introduction: tales of the unexpected
33
as smooth muscle cells and adventitial fibroblasts, and to the atrio-‐
ventricular cushion tissue as mesenchymal cells [87]. At the time when
EPDC migration occurs from the epicardium into the myocardium, the
heart has already begun to contract, and most known HCM-‐causing
mutations, such as those involving the beta-‐myosin heavy chain and
myosin-‐binding protein C genes, are already expressed in the embryonic
heart [87, 88]. Therefore, it is tempting to speculate that an interference
with the EPDC migration and differentiation processes, by a putative
mechanism of mechanotransduction [89], may account for features as
diverse as myocardial disarray, interstitial fibrosis, mitral valve
abnormalities and microvascular remodeling [65]. Another explanation
for why a mutation in sarcomeric proteins can affect valve development is
that during development, EPDCs—rather than becoming hypertrophic like
other cardiomyocytes—differentiate or revert into fibroblastic-‐like cells. If
this is true, one would expect increased levels of periostin production in
hypertrophic hearts since the hallmark of fibroblastic differentiation is
expression of periostin. Consistent with this hypothesis, markedly
elevated levels of periostin are indeed expressed in HCM mice [90].
Similar hypotheses may explain the cellular origin of adipocytes in
ARVC, which represents an enigma [91]. In the heart, the only cell type
known to express desmosomal proteins is the cardiomyocyte, which are
terminally differentiated in the adult and hence not candidates to
dedifferentiate to adipocytes. However, lineage tracing experiments have
shown showed that adipocytes in ARVC originate from second heart field
progenitor cells that preferentially differentiate into adipocytes because
of suppressed canonical Wnt signaling. Indeed, Wnt/β-‐catenin signaling is
an important switch regulator of myogenesis versus adipogenesis and a
differentiation of cardiac progenitor cells. These findings may also explain

1. Introduction: tales of the unexpected
34
the predominant involvement of the right ventricle in ARVC, as that the
second heart field progenitors give rise to the right ventricle and its
outflow tracts through mechanisms governed by canonical Wnt signaling.
Further understanding of the effects of cardiomyopathy-‐causing
mutations during development, and their protean implication on clinical
phenotype, may provide important clues on the essence of genetic
cardiac disease. These are difficult studies to perform, and require hard
multidisciplinary work. However, the rewards may include a completely
new way of conceiving the spectrum of these conditions, as well as novel
therapeutic targets.

1. Introduction: tales of the unexpected
35
1.3 Making sense of diversity: finding common answers
in clinical practice.
Imagining a disease as a fruit or a planet, there are several levels at
which one can intervene with any therapeutic approach. The first and
most obvious is to simply scratch the surface and control symptoms. This
objective can be achieved in most cardiac patients: however, it is the very
least we can do. The second step is to interfere and possibly halt disease
progression, thus preventing its consequences on outcome: this can be
done in several cardiac conditions, but is definitely harder to achieve
[92-‐93]. Third, we can try to prevent the development of full-‐blown
disease in patients who are predisposed due to acquired risk factors
and/or genetic substrate [94]. And fourth, we can address the core of the
problem by acting directly on the etiology, removing the actual cause and
ultimately cure the patient [95]. Despite extraordinary progress over the
last decades, these last two steps have hardly ever been achieved in
cardiovascular medicine. As a consequence, it is important to realize that
our practice is based on highly sophisticated palliation. What this
approach usually does is change a disease into a milder one. For example
septal myectomy turns obstructive into non-‐obstructive HCM, and the ICD
can prevent malignant arrhythmias in ARVC or DCM, all highly significant
benefits for the patients [96, 97]. Therefore, this kind of very effective
palliation is something we should definitely keep on doing, and improving,
until a cure becomes available. However, all efforts should be directed at
improving the state of things by accumulating new evidence and
knowledge [98].

1. Introduction: tales of the unexpected
36
Despite decades of increasing attention and research efforts by the
scientific community, treatment strategies for cardiomyopathies remain
largely based on a small number of clinical studies, or empirically based
on personal experience or extrapolation from other cardiac conditions. As
stated in the recent Report of the Working Group of the National Heart,
Lung, and Blood Institute on Research Priorities in Hypertrophic
Cardiomyopathy [98], “nearly 50 years after the identification of HCM as
an autosomal dominant disease, and 20 years after its linkage to
sarcomeric protein mutations, we still do not understand the most
proximal mechanism(s) that initiates the disorder”; and “treatment
recommendations in HCM are based on observational series without
prospective randomized controls. While clinical usage provides support
that various pharmacologic agents reduce HCM symptoms, no evidence
has demonstrated that they alter disease progression or outcomes.” In a
recent review of original articles, reviews and editorials addressing any
pharmacological agent ever used in HCM cohorts, only 45 studies were
identified over the last sixty years (i.e. less than 1 per year), enrolling a
total of 2,121 HCM patients [95]. Of these, only 5 were randomized,
double blind placebo-‐controlled trials. Remarkably, a comparison of the
period 1991-‐2011 vs. 1971-‐1990 demonstrated no increase in the number
of studies, and only a modest increase in the number of patients enrolled
(627 vs. 1,473, patients respectively).
Several reasons -‐ some obvious, other less so -‐ stand behind this
state of things. The first lies with the practical challenges inherent in
designing trials with cardiomyopathy patients. The epidemiology of these
conditions is complex and only partially known, due to issues such as
scarce awareness, incomplete penetrance and prevalence of subclinical
disease [67]. Despite not being rare, cardiomyopathies are uncommonly

1. Introduction: tales of the unexpected
37
encountered and possibly neglected at community-‐based cardiac centers
and outpatient clinics. Furthermore, even when overt and correctly
diagnosed, their clinical spectrum is highly heterogeneous, encompassing
different stages that may not be directly comparable. A preventive trial in
genotype-‐positive / phenotype-‐negative individuals will necessarily enroll
subsets that are different from patients with end-‐stage disease. Each
research question should be addressed by targeting the appropriate
patient subgroups, with imaginable problems in achieving the desired
yield in any given cohort [98].
As highlighted in the previous section, several targets for treatment
have been identified, some of which overlap with other cardiac diseases
and with heart failure at large.
Figure 1.3-‐1: Therapeutic targets and goals in cardiomyopathies

1. Introduction: tales of the unexpected
38
For example, progressive interstitial cardiac fibrosis, resulting from
non–myocyte (e.g., fibroblast)-‐mediated activation of transforming
growth factorβ signaling, is a common feature of most cardiomyopathies
[4]. The finding that preemptive angiotensin II type 1–receptor inhibition
prevented myocardial fibrosis in a mouse model of cardiomyopathy, as
well as encouraging results from a small clinical study, supports further
investigation of this approach [28].
Furthermore, interventions aimed at normalizing energy
homeostasis represent a viable approach, as shown by a recent study on
perhexiline, a metabolic modulator which inhibits the metabolism of free
fatty acids and enhances carbohydrate utilization by the cardiomyocyte.
In a randomized, double-‐blind placebo-‐controlled trial, perhexiline has
recently shown the capacity to improve the energetic profile of the LV,
resulting in improved diastolic function and exercise capacity in HCM
patients [99]. HCM cardiomyocytes exhibit well-‐established abnormalities
in intracellular calcium handling, contributing to excessive energy
expenditure and enhanced arrhythmogenesis, that are largelydue to
enhanced membrane late sodium current [100]. Such defect may be
selectively and dramatically reversed in vitro by ranolazine. Following the
demonstration of its beneficial effects on HCM cardiomyocytes, a
multicenter, double blind, placebo-‐controlled pilot study is currently
underway in Europe, to test the efficacy of ranolazine on exercise
tolerance and diastolic function in symptomatic HCM patients (RESTYLE-‐
HCM; EUDRA-‐CT 2011-‐004507-‐20). Besides the specific merits of
ranolazine, similar examples of translational approach identify a
fundamental pre-‐requisite for the identification of novel, potentially
effective agents, based on thorough investigation of the molecular basis
of these diseases.

1. Introduction: tales of the unexpected
39
In the future, a more specific approach may be tailored to specific
mutations or groups of mutations associated with cardiomyopathies, by
screening large panels of candidate molecules in assays based on induced
pluripotent stem cells isolated from human fibroblasts [101]. As shown
recently, the possibility of modulating the activity of sarcomere
contractile proteins, such as beta-‐myosin, is beginning to surface, with
huge potential implications for treatment of DCM, HCM and heart failure
in general [102]. Finally, during heart development, immature
cardiomyocytes proliferate actively to accommodate increasing heart size
and function [103]; however, this proliferation is abruptly abrogated
shortly after birth, leaving the heart with a limited regenerative capacity
insufficient to replace substantial amounts of tissue lost after injury. For
conditions characterized by loss of viable myocardial tissue and
dysfunction, such as DCM and ARVC, an promising approach is
constituted by the possibility of reactivating the dormant proliferative
capacity of adult cardiomyocytes as a direct effect of miRNA delivery. In a
recent study, selected miRNAs showed the ability of selectively induce
cardiomyocyte proliferation in vitro and in vivo. miRNA delivery to the
infarcted heart resulted in structural and functional recovery. Therefore,
the broader action of miRNAs impacting multiple pathways opens up a
new translational perspective for the treatment of complex cardiac
disease as a stand-‐alone therapy or in combination with other
regenerative resources. Likewise, control of gene transcription by specific
interaction with histone acetylation and deacetylation (by the
antagonistic families of histone acetyltransferases and histone
deacetylases) may represent a viable therapeutic pathway in genetic
heart disease, with targets ranging from modulation of hypertrophy to
cardiac regeneration [104].

1. Introduction: tales of the unexpected
40
Overall, these broad concepts provide a broad intellectual
framework supporting the idea of a common pathophysiologic “core” of
primary heart muscle disease, representing a unitary target for research
efforts in the field. Although the final application of future therapies will
be necessarily individualized (i.e. both disease-‐ and patient-‐specific), the
beginning of this therapeutic revolution will be based on the
comprehensive understanding of cardiomyopathies as a whole. Similar to
what is happening in cancer or autoimmune diseases, it will be impossible
to cure a single entity without being near to curing the rest. Rather than
subdividing the field in water-‐proof niches with little communication, the
approach to cardiomyopathies should constantly strive to pursue the
inter-‐disciplinary cross-‐fertilization. It is hoped that the present work may
contribute to this novel perspective.

1. Introduction: tales of the unexpected
41
1.4 Aim of the thesis and project design.
The present thesis reflects several aspects of my work at the
Florence Referral Center for Cardiomyopathy and Careggi University
Hospital during the last 3 years. The leading theme is the effort to
embrace the complexities of cardiomyopathies using a translational
approach ranging from clinical to imaging, to genetics, to basic science.
Section 2 focuses on genotype-‐phenotype correlations in two
specific disease models (thin filament-‐associated HCM and Anderson
Fabry disease) and the impact of next generation sequencing on the
diagnosis of clinically challenging cardiomyopathies. Section 3 deals with
the hypothesis that environmental modifiers may exert a significant
impact on phenotype and clinical course, by addressing one of the most
prevalent cardiovascular risk factor in the western world, i.e. obesity, in
HCM patients undergoing cardiac magnetic resonance imaging. Section 4
addresses clinical markers of risk and predictors of outcome in patients
with HCM (by evaluating the value of NT-‐pro BNP and late gadolinium
enhancement as a clinical barometers of disease progression and
arrhythmic risk), DCM (assessing the impact of advances in management
on outcome), and an acquired model of myocardial disease involving cell-‐
mediated immunity – Chagas cardiomyopathy. Section 5 illustrates novel
therapeutic approaches that are being developed for patients with HCM
and Anderson-‐Fabry disease, and a critical reappraisal of a well-‐
established – but not perfect – preventive option such as the implantable
cardioverter defibrillator. Finally, in my conclusive remarks, I will outline
work that is presently ongoing and future directions for research to be
pursued in Florence.

1. Introduction: tales of the unexpected
42

2. Genotype-‐Phenotype Correlations

2. Genotype – phenotype correlations
44
“The important thing in science
is not so much to obtain new facts,
as to discover new ways
of thinking about them”
Sir William Bragg

2. Genotype – phenotype correlations
45
Genetics is a new science. It is little more than a century since
Mendel's laws were rediscovered in 1900, and less than 60 years since the
structure of DNA was discovered in 1953. Human and medical genetics
were late developers: they started to slowly develop during the first half
of the 20th century, then saw an increasingly rapid rise and continues in
the 21st century. Thanks to the outstanding efforts of Victor Almon
McKusick, now regarded as the “father of medical genetics”, in the
second half of the 20th century genetics met medicine. Since then, the
Holy Grail in medical genetic has been the ability to deduce the clinical
phenotype of an individual from his genotype. It was once naively
assumed that, at least for ‘‘monogenic’’ disorders, genotype–phenotype
relationships would be simple and straightforward to understand. This era
of substantial optimism conquered the whole word of medicine:
physicians and geneticists shared the unrealistic expectation that
molecular genetics would lead to a new paradigm in predicting the
outcome of patients.
What was initially thought to be one-‐to-‐one gene-‐disease has
turned out to display important variability, dependent in large part on the
genetic and environmental backgrounds into which the genes express.
Therefore, the reality is that we cannot readily draw straight lines of
causation from known genotypes to specific clinical phenotypes [Fig 2-‐1].
Although the original goal to link genotype to phenotype remains, it is
now clear that the overall complexity of this relationship will require a far
more subtle understanding of both molecular mechanisms and clinical
correlates. Furthermore, there is an additional and perhaps central issue
that compromise our ability to match genotype to phenotype: the
progressive nature of the cardiac pathogenic process. Longitudinal studies
of patients with cardiomyopathies have documented the dynamic nature

2. Genotype – phenotype correlations
46
of the ventricular remodeling. Thus, it is apparent that focusing only on
the end phenotype as the supposed “link” to the molecular mechanism is
not only limiting but also likely to be misleading.
Figure 2-‐1: Genotype-‐phenotype correlations
The connections between phenotype and genotype are complex and quite hard to understand. Even in monogenic disorders, the same genetic variant can lead to different clinical pictures. The expression of phenotype may be modulated by a variety of genetic and non-‐genetic factors. Among the former, the type of mutation, the phenomenon of allele dosage, the presence of modifiers gene and epigenetics modifications are those better known. Environmental factors are those who are modifiable and can be have a vigorous influence on phenotypic expression (see chapter 3).

2. Genotype – phenotype correlations
47
2.1 Into the myocardium: the distinct phenotypic
expression of thin filament mutations in hypertrophic
cardiomyopathy
It was in a cold and rainy day of November that we first have the
idea of designing this study. At the end of a very busy morning in the
outpatient clinic, we were reviewing the most interesting cases of the day.
That morning we evaluated, totally by chance, three patients with HCM
due to mutations in the thin filament genes. We were discussing the
patients, their clinical history and disease progression. We were also
analyzing echo images, one after another, and in that moment we
realized how they were part of a distinct subgroup, if compared to other
patients with mutations in thick filament genes.
HCM is a disease of the sarcomere: mutations are very often found
in one of the two genes encoding for thick filament proteins (myosin
binding protein C -‐MYPC3 and myosin heavy chain -‐ MYH7) [Fig. 2.1-‐1] [1].
Mutations in the thin filament regulatory protein genes accounts for a
minority of molecular defect involved in HCM and includes cardiac
troponin T (TNNT2) and I (TNNI3), alpha-‐tropomyosin (TPM1), cardiac
actin (ACTC) and, very rarely, troponin C [2]. These mutations, although
rare, have always seduced physicians and geneticists involved in the field,
because of them ominous outcome and high prevalence of malignant
arrhythmias especially in the young.
Prior reports of patients with thin filament mutations described a
severe form of HCM characterized by early onset, mild degrees of
hypertrophy and high prevalence of juvenile sudden cardiac death [3-‐6].
Despite the low statistic power of these studies, limited by sample size

2. Genotype – phenotype correlations
48
and a cross-‐sectional design, such rumor spread quickly among
cardiologists, and these mutations are now known as “malignant
mutations”. However, recent reports have shown that the overall
spectrum of thin filament-‐HCM is far more heterogeneous than
previously thought, extensively overlapping the more prevalent forms of
HCM associated with thick filament mutations [7].
Figure 2.1-‐1: The sarcomere
The upper panel shows a transmission electron micrograph of a human cardiac sarcomere. The lower portion of the figure shows a simplified illustration of the sarcomere.
Two important questions arise from these considerations: is thin
filament HCM a distinct disease? How and why is thin filament HCM
different from the more common thick-‐filament disease?

2. Genotype – phenotype correlations
49
Given the international network in which our center was involved,
we decided to design a multicenter study, in order to describe the clinical
features and outcome of a large cohort of patients with thin filament
HCM, compared to matched patients with pathogenic thick filament
mutations. The study cohort comprised 84 patients with a clinical
diagnosis of HCM associated with one or more thin filament gene
mutations (including cardiac troponin T (TNNT2) and I (TNNI3),
tropomyosin (TPM1) and cardiac actin (ACTC), while no troponin C
mutations were found), while 157 HCM patients with pathogenic
mutations in the thick filament genes (MYBPC3 and MYH7) were used as
controls [Fig 2.1-‐1]. Four referral centers for cardiomyopathies were
involved in the study: Careggi University Hospital in Florence, Italy;
Brigham and Women's Hospital in Boston; Stanford Medical Center and
University of Michigan Medical Center in Ann Arbor.
Patients underwent a thorough clinical and instrumental
evaluation, including clinical history, echocardiography, 12-‐lead basal and
24-‐hours ambulatory ECG and cardiac magnetic resonance. They were
followed-‐up at yearly intervals with review of history and symptoms,
physical examination, and echocardiographic examination, and
electrocardiography. 24-‐ to 48-‐hour ambulatory ECG monitoring and CMR
were performed if clinically indicated.
The phenotype of patients with thin-‐filament mutations slowly
started to reveal itself. Compared with patients with thick filament
disease, thin filament HCM seemed to represent a distinct and well-‐
defined subset, in terms of cardiac morphology, systolic and diastolic
myocardial function and patterns of disease progression.

2. Genotype – phenotype correlations
50
2.1.1 Cardiac morphology and arrhythmic burden
The “thin filament” heart appeared to be less prone to develop
severe hypertrophy. Mean maximal LV wall thickness values were on
average milder than that observed in the thick filament group.
Furthermore, the distribution of hypertrophy within the left ventricle was
atypical, with high prevalence of concentric and apical patterns whereas
thick filament HCM was almost uniformly (94%) manifest as classic
asymmetric LVH involving the basal septum and anterior wall [Figure 2.1-‐
2]. The atypical distribution of hypertrophy, together with the mild
degree of hypertrophy, also accounted for the low prevalence of resting
LV outflow tract obstruction.
The arrhythmic risk of HCM has been known since the first reports
of the disease in the fifties. With time, due to the increasing knowledge of
the complexity of this cardiomyopathy, many other features of the
disease have been described and ventricular arrhythmias are now
considered only part of a heterogeneous spectrum of characteristics, with
an incidence of sudden cardiac death of 1%/year in non selected
populations. The arrhythmogenic burden and the risk of sudden cardiac
death of thin filament mutations have always gained most of the
attention. Indeed, our results are consistent with previous reports of
enhanced arrhythmic propensity associated with thin filament HCM. In
our cohort, the annual rate of major arrhythmic events, including sudden
cardiac death, resuscitated cardiac arrest and appropriate ICD
intervention, was almost double that of the thick filament group
(2%/year).

2. Genotype – phenotype correlations
51
Figure 2.1-‐2: Phenotypic variability in thin filament HCM.
(A-‐H) Echocardiographic apical 4-‐chamber views showing different patterns of distribution of hypertrophy (ID number and mutation for each patient are reported). (A-‐B) Mild-‐to-‐moderate hypertrophy, with “classical” septal localization of maximal LV thickness. (C) Moderate septal hypertrophy with apical involvement; (D) mid-‐apical septal hypertrophy; (E) moderate mid-‐septal hypertrophy with severe left atrial dilatation; (F) mild concentric hypertrophy (G) apical hypertrophy with severe left atrial dilatation; (H), moderate concentric hypertrophy with severe apical involvement and marked bi-‐atrial enlargement. (I) Echocardiographic and cardiac magnetic resonance (CMR) images from a patient with the TNNT2-‐R92W mutation. From left: apical four-‐chamber and parasternal long

2. Genotype – phenotype correlations
52
axis view showing marked asymmetric LV hypertrophy involving the septum and anterior wall; parasternal short axis and 2-‐chamber CMR section confirming the marked degree of hypertrophy involving multiple LV segments.
(L) CMR images from a patient with the TNNT2-‐F110L mutation. From left: 4-‐chamber and 3-‐chamber sections showing extensive regions of non-‐compaction (black arrowheads) in the apical and apico-‐lateral wall of the LV; 2-‐chamber section and 4-‐chamber section showing large areas of late gadolinium enhancement (black arrowheads) in the anterior free wall and in the septum, respectively.
In this specific subset of patients, the rate of lethal or potentially
lethal arrhythmic events was significantly higher, suggesting the presence
of a severe arrhythmogenic substrate probably linked to this specific
molecular alteration. In mutant mouse hearts, the increased Ca2+
sensitivity of myofilament induced by thin filament mutations is directly
associated with a susceptibility to arrhythmias, which can be observed
even in the absence of any detectable cardiac hypertrophy or fibrosis [8].
However, the absolute magnitude of such risk remains low and,
consequently, the mere identification of thin filament mutations does not
justify aggressive management strategies.
2.1.2 Thin filament HCM as a progressive disease
The analysis of our data depicted a novel and unexpected profile of
thin filament disease, which emerged in our cohort as a progressive
disease, more than as a purely arrhythmogenic disease, a feature that
appears to have been largely underappreciated in the past. The different

2. Genotype – phenotype correlations
53
architecture of the thin filament HCM was accompanied by substantial
functional alterations, regarding both systolic and diastolic function.
The high prevalence of severe diastolic dysfunction, represented
by abnormal filling patterns such as restrictive MV inflow or triphasic LV
filling, reflected a profound impairment in the compliance of the left
ventricle. In particular, the presence of a triphasic pattern, characterized
by prominent mid-‐diastolic flow velocity (L-‐wave), reveals the reduced LV
compliance and elevated filling pressures [Figure 2.1-‐3]. Moving from
bedside to bench, preliminary data of isolated myofibrils with thin
filament mutation showed that the “extra” wave in the diastolic pattern
could probably be explained by their incapacity to achieve a fast and fully
relaxed state during the diastolic phase.
Thin filament HCM were associated with a higher likelihood of
major clinical outcomes, as reflected by composite CV death, life-‐
threatening arrhythmia, and progression to NYHA class III-‐IV. This
difference was largely driven by the higher rate of symptoms progression
to NYHA Class III/IV, which was related to the development of LV systolic
dysfunction [Figure 2.1-‐4 and 2.1-‐5].
The prevalence of “end-‐stage” disease, a morpho-‐functional state
represented by overt systolic dysfunction and/or restrictive LV filling
pattern, is around 1% in unselected HCM populations [9, 10]. In our
population, adverse LV remodeling and disease progression was found in
more than one quarter, with an incidence of end-‐stage disease close to
3% per year. Furthermore, the considerable prevalence of heart failure-‐
related symptoms and complications are only rarely associated by
dynamic LV outflow tract obstruction in thin filament HCM and rather
reflect progressive systolic and diastolic LV dysfunction.

2. Genotype – phenotype correlations
54
Figure 2.1-‐3 Morphologic aspects and in-‐vitro correlates of triphasic LV filling.
(A) Representative examples of transmitral blood flow velocity pattern at Doppler echocardiography from different patients of the thin-‐filament cohort, showing examples mid-‐diastolic flow velocity (L-‐wave).
(B) Comparison of full relaxation from maximal Ca2+ activation in isolated myofibrils from an HCM patient with the MYH7 mutation (red tracing) and a patient with the TNNT2 mutation (black tracing). In spite of a similar initial rate of tension decay, the early linear isometric phase of relaxation is prolonged and the final exponential phase of relaxation is decelerated in the thin filament mutant myofibril, suggesting impairment of the molecular mechanisms that switch contraction off.

2. Genotype – phenotype correlations
55
Fig. 2.1-‐4 Evidence of disease progression in thin-‐filament HCM.
Echo and CMR images from patient ID#5 carrying the TNNT2-‐F110L mutation.
(A) Echocardiographic images from an early clinical assessment in 1992. Left: parasternal short axis view showing severe asymmetric septal hypertrophy. Right: color-‐Doppler image displaying turbulent flow in the LV outflow tract (white arrowhead) due to severe dynamic obstruction.
(B) Same views from last evaluation in 2013, showing marked reduction in septal thickness and spontaneous loss of dynamic obstruction, with laminar flow in the LV outflow (white arrowhead); the left atrium is markedly enlarged.
(C) Left: CMR scan from 2013 showing mild LV hypertrophy and areas of non-‐compaction in the lateral wall (arrow). Right: progressive septal thinning observed over the years is associated with extensive transmural fibrous substitution, as indicated by the large areas of late gadolinium enhancement (white arrowhead).

2. Genotype – phenotype correlations
56
Figure 2.1-‐5. Outcomes of thin-‐ versus thick-‐filament HCM
Kaplan Meier survival curves showing: (A) cumulative incidence of the primary end-‐point (combination of cardiovascular death, resuscitated cardiac arrest or appropriate ICD shock, non-‐fatal stroke and progression to NYHA class III or IV) during follow-‐up; (B) lifelong occurrence of the secondary end-‐point, assessing functional changes in LV function due to adverse remodeling, based on the echocardiographic detection of LV systolic dysfunction (ejection fraction <50%) and/or restrictive LV filling (mitral ratio of peak early to late diastolic filling velocity ≥2 in conjunction with deceleration time of 150 ms or less or, in patients with atrial fibrillation, a transmitral deceleration time <120 ms).

2. Genotype – phenotype correlations
57
Notably, a purely restrictive evolution with preserved LV systolic
function was observed in 11% of patients with thin-‐filament HCM,
consistent with prior reports emphasizing the prevalence of isolated,
severe diastolic dysfunction in patients with troponin mutations, with
particular regard to troponin I.
Both the development of LV systolic dysfunction and restrictive
filling, bear major adverse prognostic implications in HCM patients [9, 11].
Life-‐threatening arrhythmic events remain an important feature of thin
filament HCM, as previously emphasized, but their absolute prevalence is
low. Preclinical studies on animal models support the view that HCM due
to thin filament mutations is a progressive disease, and suggest that the
same molecular mechanisms may underlie both arrhythmogenesis and LV
dysfunction [2]. Skinned tissue from patient samples and animal models
have consistently shown marked increases in myofilament Ca2+ sensitivity
in the presence of thin filament mutations [12, 13], closely related with
abnormalities of cardiac relaxation [14, 15], diastolic dysfunction and an
early increase in susceptibility to arrhythmias.
These elements, consistent with a recent report based on a large
group of HCM patients with TNNT2 mutations, depict a novel and
unexpected profile of thin filament disease [7], potentially relevant to risk
stratification and management. Specifically, follow-‐up strategies should
not be limited to risk stratification for potentially lethal ventricular
arrhythmias, but also focus on the early detection of adverse LV
remodeling, significant increases in myocardial fibrosis and decline in LV
function. Present options and future advances in antiremodeling therapy
may prove useful, if introduced in a timely fashion, to prevent disease
progression in this challenging HCM subset.

2. Genotype – phenotype correlations
58
Furthermore, thin filament mutations can alter cardiac function by
increasing the energy cost of contraction [15, 16]. While several of these
abnormalities are in common with other HCM-‐related genes [17], their
extent is generally greater in thin filament mutant samples. The
constellation of early impairment in excitation-‐contraction coupling,
myocardial energetic derangement and intrinsic abnormalities of
sarcomeric relaxation constituting direct consequences of thin filament
mutation, all drive an aggressive remodeling process at the level of the
single cardiomyocyte [18]. The ultimate result of these abnormalities is a
reduction in contractile capacity both at the molecular and cellular level,
which precedes structural changes and clinically evident LV dysfunction.
Of note, thin filament-‐HCM transgenic mouse lines display a
remarkable tendency towards disease progression by developing
restrictive diastolic patterns and systolic dysfunction over time [19],
consistent with the severe diastolic impairment observed in our patients.
One of the most intriguing findings in the present cohort was the elevated
prevalence of triphasic LV filling pattern in the thin-‐filament subset
compared to the thick-‐filament subgroup. Trans-‐mitral diastolic blood
flow assessed by pulsed wave Doppler classically comprises two velocity
peaks, the E and A waves, occurring during early rapid filling and atrial
contraction. However, a triphasic LV filling pattern may be seen when an
additional velocity peak of at least 0.2m/s, the L wave, is present during
diastasis [20]. The presence of the L-‐wave has been associated with
severe diastolic dysfunction and extensive septal scarring in HCM patients.
Interestingly, the presence of the L wave in thin filament-‐HCM patients
was independent of the overall pattern of LV filling, suggesting that
patients with various degrees of diastolic dysfunction, and not only those
with most severe impairment, may display such abnormality [21, 22].

2. Genotype – phenotype correlations
59
These data therefore support the view that triphasic filling may
represent the clinical counterpart of intrinsic molecular abnormalities
associated with thin-‐filament mutations, although features such as severe
microvascular ischemia and myocardial fibrosis are likely to concur, as
shown in transgenic TNNT2 mutant mice [23]. When relaxation kinetics
were comparable in myofibrils from HCM patients carrying mutations in
TNNT2 and MYH7, overall relaxation upon Ca2+ removal was clearly
prolonged in the former, but not in the latter. In agreement with
available evidence from the literature, these findings suggest important
differences in the mechanisms leading to diastolic dysfunction for the two
forms of HCM. Indeed, while diastolic abnormalities in MYH7-‐mutant
HCM hearts are likely related to secondary changes in energetics and E-‐C
coupling of HCM [24, 25], thin filament mutations appear to directly
impair cardiac relaxation. This idea is supported by the finding that, at
variance with thick filament mutant myofibrils, resting tension is higher in
thin filament mutant myofibrils compared to controls. If confirmed by
ongoing studies, these preliminary findings may help identify novel, gene-‐
specific therapeutic targets in thin filament HCM.
In conclusions, HCM associated with thin filament mutations is
characterized by significant differences in phenotype and clinical course
compared to the more prevalent thick-‐filament HCM, including increased
likelihood of LV dysfunction and progressive heart failure. Management
strategies, classically focusing on arrhythmic prophylaxis in this subset,
should be reconsidered to include adequate surveillance for adverse LV
remodeling and worsening systolic and diastolic function, pointing to
specific molecular mechanisms leading to diastolic impairment in this
challenging HCM subgroup.

2. Genotype – phenotype correlations
60
2.2 Early cardiac phenotype in patients with Anderson-‐
Fabry disease.
Those who are familiar with Anderson Fabry disease could find this
simple title very intriguing. For those who are not, we should take a step
back.
Anderson-‐Fabry disease (AFD) (OMIM#301500) was named after
two dermatologists, W.Anderson and J. Fabry, who separately described
two cases of patients with angiocheratoma corporis diffusum in 1898 [26,
27]. Almost a century later AFD was showed to be a rare X-‐linked
multisystemic lysosomal storage disorder of glycosphingolipids
catabolism, due to the deficiency of the lysosomal enzyme a galactosidase
A (a-‐Gal A) [28, 29]. As a consequence of α-‐gal A deficiency, progressive
accumulation of globotriaosylceramide (Gb3) and its deacylated form, the
globotriaosylsphingosine (lyso-‐Gb3), occurs in various tissues throughout
the body, within the lysosomes and in the plasma [30].
AFD is considered a rare disease, with a reported prevalence in the
general population of about 1:117.000 [31]. Recently however, newborn
screening initiatives have found an unexpectedly high prevalence, as high
as 1 in ~3900 newborns [32], suggesting that prior figures may represent
a substantial underestimate. While the classic phenotype of AFD is
actually rare, these data show that milder variant with late-‐onset
phenotype may be much more frequent than expected [33, 34]. The real
strength of these newborn-‐screening initiatives goes well beyond the
simple data itself. They shifted a historic paradigm of a rare disease, with
multisystemic involvement in young males, to a more common disease,
with a wide phenotype, sometimes with disease manifestation confined

2. Genotype – phenotype correlations
61
to one organ system [Fig 2.2-‐1]. Furthermore, screening for AFD among
selected groups of patients, i.e. patients with juvenile cryptogenic stroke,
renal failure or left ventricular hypertrophy, suggests that many patients
are misdiagnosed.
Fig 2.2-‐1: Symptoms onset and disease progression in classic Anderson-‐Fabry disease (AFD).
Signs and symptoms of AFD progressively increase with age. In infancy and adolescence neuropathic pain, gastrointestinal manifestations and angiokeratomas are the most frequent manifestations. Proteinuria, the first sign of kidney involvement, is also present in young affected males. Major organ involvement usually develops slowly with age.
Although having an X-‐linked pattern of inheritance, the disease
may affect males and females, the latter with an average 10 years delay
[36]. Metabolites storage leads to progressive cellular and multiorgan
dysfunction, with either early and late onset variable clinical
manifestations that usually reduce quality of life and life expectancy [37].

2. Genotype – phenotype correlations
62
Heart and kidney failure, stroke and sudden death are the most
devastating complications.
A specific therapy for AFD, enzyme replacement therapy (ERT), is
available since 2001[38]. New treatment approaches, such as chemical
chaperone therapy, alone or in combination with ERT, are currently under
investigation [39, 40]. While long-‐term efficacy of ERT in advanced stage
is still debated, increasing evidence shows greater efficacy of early
treatment initiation [41].
2.2.1 Hypertrophic cardiomyopathy and Anderson Fabry disease: the
linking point
The referral center for cardiomyopathies in Florence has been
historically known as primary committed to the management of patients
with hypertrophic cardiomyopathy. However, in 2004 genetic analysis of
one patient with an established diagnosis of HCM revealed a mutation in
the GLA gene. Later, he was proved to have a late-‐onset variant of AFD,
with manifestation confined to the heart. The fact that he was
misdiagnosed for almost 4 years, spurred our curiosity and we started to
study AFD in its genetic and clinical aspects. To present, almost 70
patients with AFD have been evaluated in our center and more than 50
are in regular follow-‐up.
Given the expertise our center had with the issue of microvascular
dysfunction in hypertrophic cardiomyopathy, in 2006 we decided to study
the presence, prevalence and clinical significance of cardiac microvascular
dysfunction (CMD) in our cohort of AFD patients.

2. Genotype – phenotype correlations
63
The linking point between AFD cardiomyopathy and HCM is the
myocardium: both these conditions affect primary the heart muscle.
In AFD patients, cardiomyopathy represents one of the features of
a multisystemic condition, which affects not only the heart, but also the
kidney and the peripheral and central nervous system [29]. Cardiac
involvement has been described in both genders and is mainly
characterized by variable degrees of hypertrophy and interstitial fibrosis.
Although commonly classified as a storage disease of the heart, AFD
cardiomyopathy is characterized by true hypertrophy, as Gb3
accumulation only accounts for a very limited proportion of cardiac mass
increase. In hypertrophic cardiomyopathy, a genetic mutation in one of
the gene encoding for sarcomeric proteins leads to variable degrees of
hypertrophy [45]. Tissue architecture in HCM appears overturned, with
cardiomyocytes derangement (the so-‐called “disarray”) and interstitial
fibrosis [46].
The structure and function of coronary microcirculation are also
altered in both these conditions [Fig 2.2-‐2]. Narrowing of the arterioles
lumen is caused by endothelial Gb3 storage in AFD [47] and hypertrophy
of the media wall in HCM [46], and it is accompanied by functional
alterations. Therefore, blood supply of the myocardium may be normal at
rest but it is usually impaired during exercise and may lead to myocardial
ischemia, cardiomyocytes death and cardiac dysfunction.
Our interest in the issue of microvascular function arises from the
fact that an impaired response of myocardial blood flow to dipyridamole
has been proved to represent a strong and independent predictor of
clinical deterioration and death and is associated with poor prognosis in
patients with HCM and idiopathic dilated cardiomyopathy [48]. Thus,

2. Genotype – phenotype correlations
64
since microvascular dysfunction may represent a common pathway
leading to disease progression in different cardiomyopathies, we thought
to investigate the presence, characteristics and clinical severity of
microvascular function also in patients with AFD.
Fig 2.2-‐2: Gb3 inclusions in the endothelium and cardiomyocytes.
Panel A shows an intramural coronary artery in a patient with hypertrophic cardiomyopathy. Wall thickening is due primarily to severe thickening of medial wall, while the intima is mildly thickened.
Panel B shows interstitial capillaries in the myocardium of a patient with Anderson-‐Fabry disease. Glycosphingolipid inclusions are present in the endothelium (arrow) and cardiomyocytes (arrowhead)
At that time, the knowledge of coronary microcirculation in Fabry
disease was very limited [47]. CMD had been described only in male
patients with hypertrophy and advanced stage of disease [49, 50]. Despite
the absence of available data about this issue, we believed that CMD

2. Genotype – phenotype correlations
65
could be a feature of AFD cardiomyopathy, independently of the presence
of LV hypertrophy and other instrumental signs of cardiac involvement.
What made our hypothesis plausible was the fact that in patients with
sarcomeric hypertrophic cardiomyopathy, CMD was known to be a diffuse
phenomenon within the LV, involving even non-‐hypertrophied LV walls,
and it could possibly precede LVH development.
This data spurred our curiosity: was CMD also present in AFD
female patients, generally exhibiting more subtle cardiac manifestation
than men? Was CMD present in patients without any instrumental sign of
cardiac involvement? Thus, the present study was undertaken to
investigate coronary microvascular function in a cohort of male and
female AFD patients at different stages of disease, with and without
evidence of LVH, in order to assess whether CMD may occur independent
of LVH and in the absence of any other sign of cardiomyopathy.
2.2.2 Coronary microvascular dysfunction as a universal manifestation
of Fabry cardiomyopathy.
Thirty patients with an established diagnosis of AFD were selected
and underwent a thorough cardiac evaluation by clinical examination,
resting ECG, 24-‐hours Holter monitoring and echocardiography. The same
study protocol was employed in 24 healthy controls, who were
investigated for exclusion of heart disease, and were comparable to
patients in terms of gender and age. Two-‐third of our patients showed
increased LV wall thickness values, ranging from 13 to 27 mm. In contrast
to patients with HCM, the distribution of LV hypertrophy was mostly
concentric, with milder degree of hypertrophy [Fig 2.2-‐3 and 2.2-‐4]. The
remaining 10 patients had no evidence of cardiac involvement, with
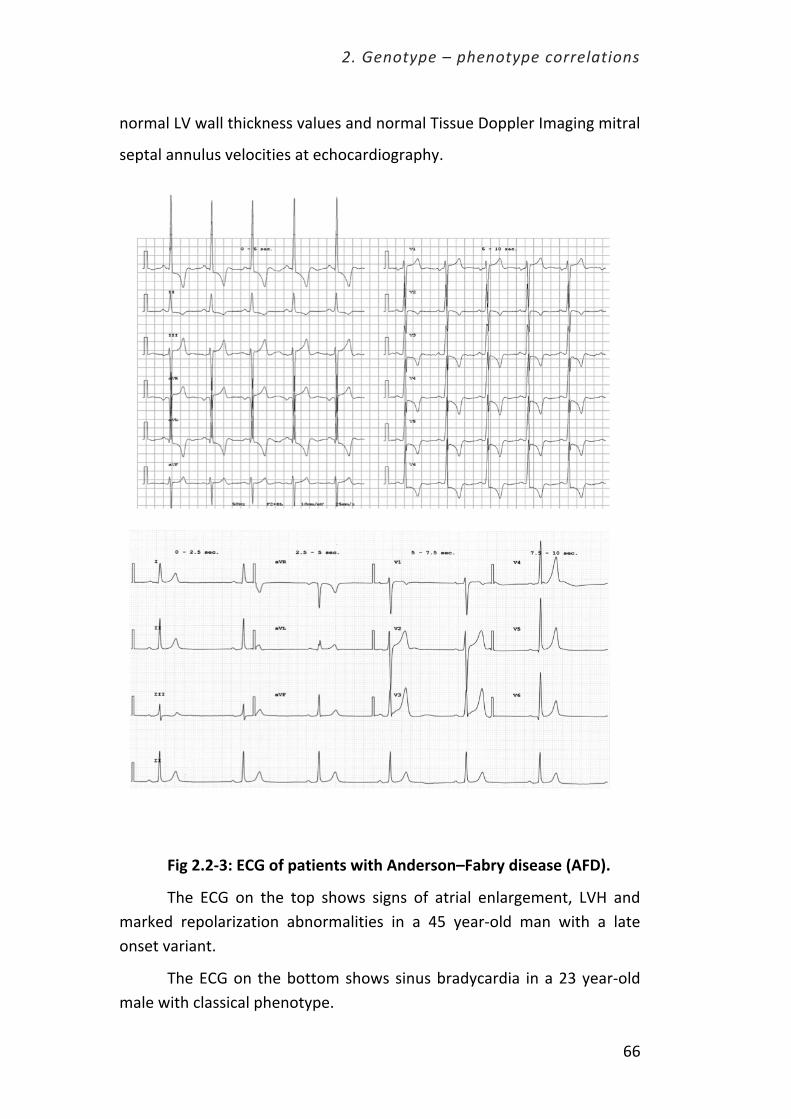
2. Genotype – phenotype correlations
66
normal LV wall thickness values and normal Tissue Doppler Imaging mitral
septal annulus velocities at echocardiography.
Fig 2.2-‐3: ECG of patients with Anderson–Fabry disease (AFD).
The ECG on the top shows signs of atrial enlargement, LVH and marked repolarization abnormalities in a 45 year-‐old man with a late onset variant.
The ECG on the bottom shows sinus bradycardia in a 23 year-‐old male with classical phenotype.

2. Genotype – phenotype correlations
67
Fig 2.2-‐4: Echocardiography images of a patient with Anderson–Fabry disease (AFD), carrying the N215S mutation.
Upper and middle panel show different echocardiographic images with LV hypertrophy, which is concentric. Right ventricle and papillary muscles are also hypertrophied. The lower panel shows diastolic dysfunction (from left to right: pulse-‐waved Doppler of mitral inflow showing mild diastolic dysfunction; TDI images with reduced velocities and pulmonary vein flow with prominent A reverse wave).
Coronary microvascular function was assessed by Positron
emission tomography (PET), a technique that allows a direct and precise
quantification of microvascular function, through the measurement of
maximal global and regional myocardial blood flow after dipyridamole
(Dip-‐MBF) [51]. The measurement of myocardial blood flow in conditions

2. Genotype – phenotype correlations
68
of near-‐maximal vasodilatation, obtained by intravenous infusion of
dipyridamole, permits calculation of maximum MBF. In the absence of
obstructive epicardial coronary disease, flow resistance is primarily
determined by the microvasculature (>90% of coronary resistance resides
in arterioles <300 um in diameter), and therefore a reduced MBF is a
marker of coronary microvascular dysfunction. In normal subjects,
maximal myocardial blood flow values >3 ml/min/g after dipyridamole
infusion are considered normal. Such values reflect the ability of increase
myocardial perfusion to match increased demand.
All AFD patients enrolled in this study exhibited a blunted coronary
microvascular response to dipyridamole, as compared to control subjects,
with a mean Dip-‐MBF of only 2.0±0.4 mL/min/g, compared to 3.2±0.5 in
normal controls, i.e. a 60% reduction in coronary flow [Fig. 2.2-‐5].
Fig 2.2-‐5: Coronary microvascular dysfunction in patients with Anderson–Fabry disease.
Myocardial blood flow following dipyridamole infusion is markedly impaired in AFD patients compared with control subjects.

2. Genotype – phenotype correlations
69
As expected, patients with LVH invariably showed marked degrees
of CMD, suggesting that the profound remodeling of the heart structure
was invariably accompanied by changes in the architecture of small
vessels. However, even patients without LVH showed a blunted response
to dipyridamole, with mean myocardial blood values ranging from 2.4 to
as low as 1.2 mL/min/g. As a result, CMD in patients without LVH was on
average milder, but not statistically different, from those observed in
patients with LVH.
Altogether, these findings point to CMD as a constant
manifestation of AFD, which may be present independent of other
instrumental evidence of cardiac involvement [Fig. 2.2-‐6]. Moreover, AFD
patients without LVH and CMD generally had none of the other features
that are considered very early disease manifestations, such as reduced
diastolic mitral annulus velocities or presence of LGE [52, 53]. Therefore,
this observation implies that myocardial flow abnormalities can indeed be
considered to represent initial cardiac involvement, possibly followed by
overt cardiomyopathy over time. This novel concept is suggested by the
young age of patients with CMD in the absence of LVH.
Although myocardial blood flow was globally impaired in AFD
hearts, there was a marked regional heterogeneity of myocardial flow,
with prevalent hypoperfusion of the apical region. Such regional nature of
cardiac involvement may appear counterintuitive in a systemic storage
disease. However, the novel idea of a non-‐homogeneous cardiac
involvement in AFD is in line with prior observations in a number of
cardiomyopathies, and particularly to those related to storage or
infiltrative disorders, such as for example amyloidosis [54]. The
mechanisms accounting for such regionally heterogeneous manifestations
remain unresolved.

2. Genotype – phenotype correlations
70
Fig 2.2-‐6: Relationship of coronary microvasculature dysfunction(CMD) to phenotype in individual patients with Anderson–Fabry disease (AFD).
(A) A 28-‐year-‐old male with extreme sinus bradycardia (36 b.p.m.) and normal interventricular septal thickness values (*echocardiographic parasternal long axis and cardiac magnetic resonance four-‐chamber views). Colour-‐coded polar map obtained by positron emission tomography (PET) after dipyridamole infusion showing regional CMD, with relatively preserved flow in the antero-‐lateral region.
(B) A 60-‐year-‐old female with normal ECG, without normal LV wall thickness values. The polar map shows severe CMD, despite normal LV wall thickness values.
(C) A 52-‐year-‐old male with marked ECG abnormalities, severe LV hypertrophy (LVH) (*echocardiographic apical four-‐chamber view and

2. Genotype – phenotype correlations
71
cardiac magnetic resonance four-‐chamber view). The polar map shows severe CMD.
(D) A 47-‐year-‐old female with markedly abnormal ECG, and severe LVH. The polar map shows relatively preserved microvascular function, despite the LVH. Dip-‐MBF, myocardial blood flow following dipyridamole infusion,
2.2.3. Cardiac involvement in females with AFD and influence of
genotype
Although AFD is an X-‐linked disease, female patients are well
known to develop the clinical manifestations of the disease, which can be
severe and determine outcome [55]. Cardiomyopathy is not an exception
to this rule, and has been described in women; however, cardiac
manifestations generally have a later onset and are less marked than
those occurring in men [56, 57]. Thus, we were expecting milder degree
of microvascular dysfunction in females. However, women with and
without LVH had considerable degrees of CMD, although to a less sever
degree than males. Of note, in females without LVH, a reduced MBF
represented the only detectable evidence of cardiac involvement. As a
result, heightened awareness should be considered for cardiac
involvement in female patients with AFD, even in the absence of an
echocardiographic phenotype.
With regard to the genotype of patients, we had the opportunity
to study a recurrent mutation in Fabry disease. The c.644A>G GLA-‐gene
mutation, leading to the p.Asn215Ser amino acid substitution was present
in three index patients and seven family members. This mutation is
known to be related to the so-‐called “cardiac variant” of the disease [58].
Patients harboring this mutation usually have a predominant cardiac
phenotype, characterized by moderate to severe degree of hypertrophy,

2. Genotype – phenotype correlations
72
with an asymmetrical distribution, which is rather uncommon in AFD.
Disease onset is usually late, with males developing signs of cardiac
involvement during their thirty. Extracardiac involvement, such as renal
or cerebrovascular, is uncommon although not impossible.
In our cohort, the p.Asn215Ser group showed similar degree of
hypertrophy compared to patients with other genotype. Nevertheless,
myocardial blood flow was markedly lower when compared to other
genotypes [Fig 2.2-‐7], suggesting a severe impairment in the
microvascular function. Furthermore, at logistic regression analysis, only
the mutation p.Asn215Ser proved to be an independent determinant of
severe CMD (hazard ratio for Dip-‐MBF<1.25 ml/g/min 9.0; 95%
confidence intervals: 1.3-‐61.1; p=0.03).
Figure 2.2-‐7: Genotype influence on microvascular function
Despite similar degree of hypertrophy, patients with the p.Ans215Ser mutation showed a more severe impairment of the myocardial perfusion when compared to other patients.

2. Genotype – phenotype correlations
73
The consistent and severe blunting of MBF in patients with the p.
Ans215Ser GLA gene mutation may suggest a role of genetic status in
determining the severity of MBF. Furthermore, the concept of the
possible influence of genetic status to microvascular function has been
proved in other more common genetic cardiomyopathy (i.e. hypertrophic
cardiomyopathy) [59]. Such variability suggests selective microvascular
involvement activated by specific gene mutations, and is consistent with
the phenotypic variability of clinical manifestations in AFD. In the light of
these findings, the molecular links between GLA gene mutations and
coronary microvascular remodeling need to be further investigated.
2.2.4. Potential mechanisms of coronary microvascular dysfunction
The potential pathophysiological mechanisms underlying coronary
blood flow impairment in AFD are several, and include those mediated by
LVH (reduced capillary density, extravascular compression forces) as well
as those directly affecting the microvasculature (endothelial dysfunction
due to GB3 storage, nitric oxide pathway dysregulation, microvascular
remodeling) [60]. However, in patients without LVH, only the latter are
present, suggesting that LVH per se is indeed likely a contributor to CMD,
rather than a cause.
Despite considerable advances in our understanding of AFD, the
mechanisms leading to LVH are not well understood. Storage of Gb3
within cardiac myocytes accounts for a small amount of the whole LV
mass, which is mainly represented by true myocardial hypertrophy [44].
One hypothesis is that intracellular accumulation of Gb3 may disturb
cardiac energetic metabolism, representing an early trigger for the
activation of the intracellular signaling pathways leading to hypertrophy,

2. Genotype – phenotype correlations
74
fibrosis, apoptosis and necrosis [61]. Furthermore, microvascular
dysfunction and chronic hypoperfusion, due to Gb3 accumulation in
endothelial cells, may also play a crucial role in the activation and
perpetuation of these pathophysiological processes, even at early stages.
An unavoidable limitation of the present study lies in the small
sample size and heterogeneity of our study population. Selecting a more
homogeneous study cohort, by excluding patients with comorbidities or
cardiovascular risk-‐factors, would not have been feasible in a rare and
multisystemic disorder such as AFD. Thus, myocardial blood flow may
have been affected in our cohort by the interplay of comorbidities such as
hypertension, smoking or dyslipidaemia, as well as ERT itself. This may
have potentially led to overestimation of CMD in our patients with regard
to the healthy controls, in whom none of the conventional cardiovascular
risk factors were present. While we acknowledge this unavoidable bias,
however, MBF values in AFD with no history of smoking, hypertension
and dyslipidaemia, ideally representing “pure” AFD patients, were
comparable to the rest of the cohort, showing similar degree of MBF
impairment. Thus, our data are consistent with a disproportionate
microvascular involvement in AFD, overruling the effect of these potential
environmental confounders.
2.2.5. Early detection of heart involvement in Fabry disease: a crucial
point for patient’s management
The issue of early organ damage identification has become critical
following the introduction in clinical practice of ERT and novel therapeutic
molecules, including pharmacological chaperone therapy [39, 40]. To date,
evidence of benefit of ERT in AFD patients with overt signs of

2. Genotype – phenotype correlations
75
cardiomyopathy and LVH is disappointing, and earlier timing of treatment
initiation has been advocated in order to improve efficacy [41].
Our findings imply that a normal echocardiogram cannot exclude
deterioration of microvascular function related to the disease, raising
important issues regarding risk stratification and management. Thus,
early detection of subclinical cardiac involvement, as allowed by PET
studies of microvascular function, may become a critical element in
clinical decision making especially in young AFD patients. In particular,
CMD may represent a viable treatment target, potentially relevant to
prevention of disease progression and outcome. In the future, the advent
of cardiac magnetic resonance (CMR), by virtue of its larger availability
and more favourable safety profile compared to PET [62], may allow
large-‐scale investigation of CMD in AFD patients. To date, however,
quantitative assessment of microvascular flow by CMR is time-‐consuming
and largely limited to research purposes.
Furthermore, is it possible to hypothesize that the severity of CMD
may be an important predictor of adverse outcome, as for other
genetically determined cardiomyopathies [48].
These findings represent a picture of our AFD population at this
time. However, since everything we know, especially in medicine, is
destined to change or modify, our present goal is to continue to follow
this patients in order to be able to answer many more questions arisen
from this work. We are in particular interested in determining whether
CMD may predict the development of LVH or may be a predictor of
outcome.
Full text of the publication may be found in chapter 7.

2. Genotype – phenotype correlations
76
2.3 Next Generation Sequencing: new horizons in the
field of cardiomyopathies
Cardiomyopathies are a clinically heterogeneous group of heart
muscle disorders, largely genetic in nature, defined by the presence of
abnormal myocardial structure and/or function in the absence of
ischemic heart disease or abnormal loading conditions. Cardiomyopathies
have been variously classified over the years based on their phenotypes.
The current classifications continue to be based on phenotype defined by
clinical evaluation of affected individuals and incorporating genotype
when possible (see chapter 1)[63]. Contemporary classifications
distinguish the dilated (DCM), hypertrophic (HCM) and restrictive forms
(RCM), as well as arrhythmogenic right ventricular cardiomyopathy
(ARVC) and an unclassified group including isolated left ventricular non-‐
compaction (LVNC).
While the familial nature of cardiomyopathies has been known
since their earliest clinical description, the first cardiac disease-‐causing
mutation was identified in a family with hypertrophic cardiomyopathy in
1990 [64]. During the past two decades, numerous genes and genetic
variants associated with different cardiomyopathies have been identified.
However, the great complexity of genotype is paralleled by huge
phenotype variability, thus making a genotype-‐phenotype correlation
almost impossible to achieve.
Many attempts at providing distinct boundaries between these
clinical entities have been hindered by the increasing evidence of bi-‐
directional overlap: a) different cardiomyopathies share common genetic
background; for example, genes encoding sarcomere protein genes may

2. Genotype – phenotype correlations
77
be associated with HCM, DCM, RCM and LVNC, and Z-‐disc or calcium
handling protein genes may cause both HCM and DCM; b) the same
clinical phenotype may be produced by defects in genes that are
profoundly diverse in structure and function, such as the lamin A-‐C gene,
titin or beta-‐myosin heavy chain in the case of DCM. As a consequence,
genetic investigations of patients with cardiomyopathies based on
traditional sequencing techniques have grown at a relatively slow pace, as
each phenotype could only be tested for a limited amount of candidate
genes in clinical practice.
Sanger sequencing, even when performed by automated high-‐
throughput machines, analyzes ~2 million bases per day, a very low
performance compared to the collective size of the genes involved in
familial heart disease. This has led different institutions and companies
over the years to design small panels of 8-‐20 genes for each condition,
with yields ranging from ~60% in HCM to ~20% in DCM. The various
panels were often not all available in single institutions, because of costs
and time constraints, forcing patients to travel and perform repeated
analyses before reaching a diagnosis.
The recent advent of next generation sequencing (NGS)
technologies have led to massive increase in throughput, up to ~50 billion
sequenced bases per day, and reduction in cost. Therefore, NGS, also
described as “second generation” or “massively parallel”, has replaced
Sanger sequencing as the primary methodology employed by researchers
and clinicians to identify disease variant. The ability to simultaneously
analyze multiple or very large genes at a cheaper cost per base makes
next generation sequencing an attractive solution for clinical diagnostic
testing to identify the disease-‐causing mutation (or mutations) in patients
with genetically heterogeneous disorders. Three different approaches

2. Genotype – phenotype correlations
78
have become feasible in clinical practice (in increasing order of
complexity): a targeted approach (with panels possibly including over 100
genes), whole exome and whole genome. While the last two remain
expensive, labor-‐intensive and mainly used for research purpose, a
targeted approach is becoming increasingly popular in referral centers for
inherited heart diseases.
2.3.1. NGS in the clinical setting: clinical pictures of familial phenotype
and genetic study
At our Institution, genetic screening has been offered to HCM
patients since 2000. At that time, denaturing high pressure liquid
chromatography (DHPLC) was used as an initial approach, and only the 3
most prevalent genes (MYH7, MYBPC3 and TNNT2) were analyzed, as
more comprehensive testing would not have been sustainable in a
national health system environment. Furthermore, turn-‐around times
were very long (close to 2 years) because of the complex and lengthy
procedures employed. By 2006, only 88 index cases had been completed,
with an overall yield of mutations approaching 60%. In 2008, robotic
automation allowed an expansion of the standard screening set of genes
from 3 to 8 (by including TNNI3, MYL2, MYL3, ACTC1, TPM1), and DHPLC
was abandoned in favour of direct Sanger sequencing. Costs and
turnaround time decreased significantly (to less than 6 months). At the
end of 2010, 672 index cases had been completed, with an overall yield of
64%. Thus, even considering most novel variants as pathogenic (an issue
that remains controversial to this day), over one-‐third of our probands
proved negative for the most prevalent sarcomere genes and thus the
molecular basis of their disease remains unresolved – a problem shared

2. Genotype – phenotype correlations
79
by all laboratories involved in HCM screening. However, NGS is rapidly
changing the landscape of genetic testing because of its power and
sensitivity.
In the present section I would like to illustrate the first experience
of our referral center, historically committed to genetic studies of familial
cardiomyopathies, with these new genetic technologies. Thirteen families
with different form of cardiomyopathy, negative after standard genetic
analysis, were selected and analyzed using whole exome analysis and a
targeted approach [Table 2.3-‐1].
For the purpose of this thesis, only the most informative families
have been selected and will be described in the next sections.
Table 2.3-‐1: familial cardiomyopathies studied using NGS
Whole exome analysis Targeted approach
1 family with unexplained juvenile sudden cardiac death and ventricular fibrillation
1 heterogeneous phenotype * (HCM / LVNC cardiomyopathy and supraventricular arrhythmias)
8 families with HCM 1 family with DCM * 1 family with ARVC *
1 family with LV aneurism *
[* = families described in the text].

2. Genotype – phenotype correlations
80
2.3.2 Whole exome analysis
A family with idiopathic dilated cardiomyopathy
The proband was 31 years old when he was first seen at our center.
He was totally asymptomatic and came to our center because of familiar
screening. His 39-‐year-‐old brother had died suddenly a couple of months
earlier and the post-‐mortem examination revealed dilated
cardiomyopathy. His 63-‐year-‐old mother had died 3 years earlier because
of refractory heart failure. She had had a diagnosis of dilated
cardiomyopathy when she was 45 and required a pacemaker at the age of
53 [Figure 2.3-‐2]. Despite the early onset of the disease in this lady, who
had no major cardiovascular risk factors, the possible familial nature of
the disease was missed and none of the doctors advise a family screening.
In our patient, echocardiography revealed a dilatation of the left
ventricle, with a mild impairment of the systolic function (ejection
fraction 45%). Furthermore, the ECG showed I degree atrio-‐ventricular
block (PR interval 420 msec), while dynamic 24-‐hours holter showed
many episodes of type 2 II degree AV block. He also had elevated CPK
level, without any sings of skeletal myopathy. These elements prompted
us to consider a diagnosis of cardiolaminopathy as the most plausible
[Figure 2.3-‐2].

2. Genotype – phenotype correlations
81
Figure 2.3-‐2: Clinical features of the proband with DCM
Top right: family pedigree showing a dominant inheritance
Bottom right: echocardiogram and cardiac magnetic resonance images showing dilated LV (EF 45%).
ECG performed during the first examination (top left) and during a follow-‐up visit (bottom left): marked first degree atrio-‐ventricular block was present.
Therefore, after a detailed pre-‐test counseling with our geneticist,
a blood sample was drawn. Since the clinical picture was highly suggestive
of a genetic cardiomyopathy related to mutation in the LMNA A/C gene,
we choose standard genetic with Sanger sequencing as a first-‐line
diagnostic technique. Surprisingly, genetic analysis came back negative.
Filled with curiosity to detect the mutation responsible for such
cardiomyopathy, we decided to analyze the whole exome of this subject.

2. Genotype – phenotype correlations
82
Initial genetic analysis identified almost 55000 variants. The
standard bioinformatics analysis (as described in methods) did not allow
the identification of the causative mutation.
Sanger sequencing, as well as whole-‐exome sequencing, have been
successfully used to discover the great majority of genomic variants, (i.e
common and rare single nucleotide variants (SNVs), small
insertions/deletions (indels) and breakpoints of structural variations).
However, they are unsuitable for detecting large deletions or duplications
because they span one or both primer binding sites. This can be achieved
through the analysis of copy number variations (CNVs), a comparative
depth of coverage analysis between normal controls and patient samples.
In order to do that, the EXCAVATOR software was applied and a huge
deletion of 163±22 kb was found. The deleted region, located on the long
arm of the 1 chromosome (1q22), comprised the entire LMNA gene.
A family with arrhythmogenic cardiomyopathy
The proband was a 37-‐year-‐old woman who was admitted to our
hospital after resuscitated cardiac arrest. She had negative cardiological
clinical history, without syncope, dyspnea or palpitations. She had been a
competitive runner until her thirties and at the age of 35 she gave birth to
spontaneous conceived triplets. At the time of the cardiologic evaluation
she presented clinical features consistent with a diagnosis of
arrhythmogenic cardiomyopathy with biventricular involvement,
associated with palmar keratoderma [Figure 2.3-‐3]. Echocardiography and
cardiac magnetic resonance showed a mildly dilated right ventricle with
reduced systolic function and multiple areas of bulging of the free wall,
associated with reduced left systolic function (EF 45%).

2. Genotype – phenotype correlations
83
A therapy with ACE-‐inhibitors, amiodarone and beta-‐blockers was
initiated and an implantable cardioverter-‐defibrillator was implanted in
secondary prevention. She also had family history of juvenile sudden
cardiac death: her brother, with a history of syncope, died suddenly at the
age of 28 years while playing soccer. He also had scalp dermatitis and
woolly hair. A post-‐mortem evaluation of echocardiograms and
electrocardiograms, performed a couple of months before his death,
showed clinical findings suggestive of arrhythmogenic right ventricular
cardiomyopathy. The grandmother’s sister had dilated cardiomyopathy
and she also died suddenly at the age of forty. A mild form of ARVC and
scalp dermatitis was observed both in the mother and in the uncle of the
proband. Clinical features observed were consistent with the diagnosis of
Carvajal syndrome [66], although the family’s pedigree strongly suggested
a dominant model of inheritance [Figure 2.3-‐4].
Traditional Sanger sequencing failed to identify any mutation in
those genes usually associated with ARVC (transforming growth factor
beta3, ryanodine receptor 2, desmoplakin, plakophilin2, desmoglein2,
desmocollin2 transmembrane protein 43, junction plakoglobin protein).
A whole exome sequencing study was therefore performed in the
proband. Data analysis identified 51337 variants, 7517 of which located in
coding regions and intron-‐exon junctions. After frequency filtering
(variants with minor allele frequency < 5% in 1000 Genomes Project), 834
variants were selected. Among these, 101 variants were not present in
dbSNP Release [Figure 2.3-‐4].

2. Genotype – phenotype correlations
84
Figure 2.3-‐3: clinical features of the proband with ARVC
A-‐C: echocardiogram and cardiac MRI images show wall aneurysms within the so-‐called ``triangle of dysplasia'' (white arrows: evident systolic bulging in infundibular, apical, and subtricuspid regions of the RV). The LV apex is also involved.
D: ECG of the proband
E: palmar keratosis
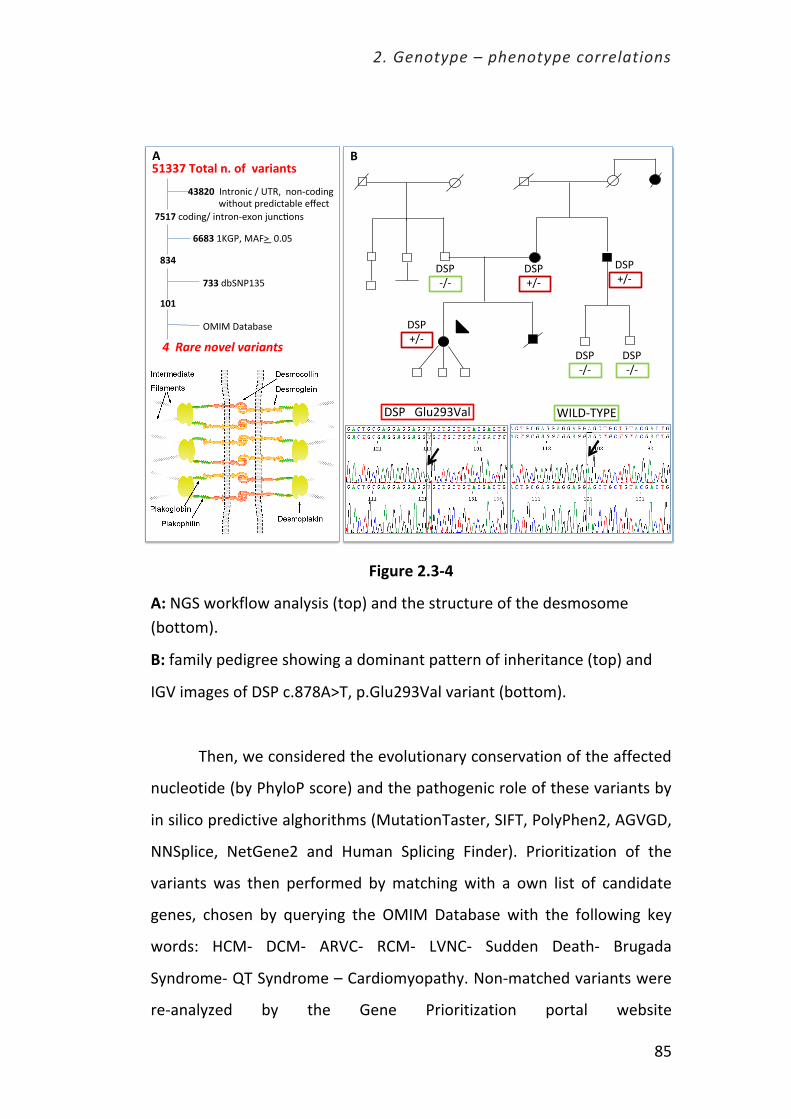
2. Genotype – phenotype correlations
85
Figure 2.3-‐4
A: NGS workflow analysis (top) and the structure of the desmosome (bottom).
B: family pedigree showing a dominant pattern of inheritance (top) and
IGV images of DSP c.878A>T, p.Glu293Val variant (bottom).
Then, we considered the evolutionary conservation of the affected
nucleotide (by PhyloP score) and the pathogenic role of these variants by
in silico predictive alghorithms (MutationTaster, SIFT, PolyPhen2, AGVGD,
NNSplice, NetGene2 and Human Splicing Finder). Prioritization of the
variants was then performed by matching with a own list of candidate
genes, chosen by querying the OMIM Database with the following key
words: HCM-‐ DCM-‐ ARVC-‐ RCM-‐ LVNC-‐ Sudden Death-‐ Brugada
Syndrome-‐ QT Syndrome – Cardiomyopathy. Non-‐matched variants were
re-‐analyzed by the Gene Prioritization portal website
!"#$%&'$
!"#$%&'$
!"#$%&'$
!"#$'&'$
!"#$'&'$
!"#$'&'$
()*!'+,#-$!"#$$$./012345/$
!"#!$6789:;&$9:<=7:'>?7:$@0:6A7:B$$
"#$$!%&'()*%+,%'-%%.)/0)+(1%
2$3%%
3$245$$):<=7:96$&$C+DE$$:7:'6789:;$$$$$$$$$$$$$$F9<G70<$H=>896<5I/>$>J>6<$$
$
662$$KL.#E$MNOP$$QRQS$$
#5#%!$$$8I"T#K3S$
!""#$%&"'()&*")$%+$',-"
UM)M$!5<5I5B>$$
7% 8%

2. Genotype – phenotype correlations
86
(http://homes.esat.kuleuven.be/~bioiuser/gpp/tools.php) by using the
three tools ToppGene, Endeavour and Suspects, allowing a prioritization
of candidate genes based on functional similarity with a training gene list.
Prioritization identified a candidate variant in DST gene (OMIM#614653),
which encodes dystonin, a large protein member of the plakin protein
family, which bridges the cytoskeletal filament networks. Different DST
transcripts are expressed in the central nervous system, muscle, and skin,
although mutations in this gene are not usually associated with
cardiomyopathies. We therefore excluded the present gene from our
analysis.
After data filtering and integration of filtered results with patient’s
clinical features, 4 potential mutations were selected. Only two of them
co-‐segregated in the family: c.878A>T, p.Glu293Val variant in the
Desmoplakin gene and c.626A>C, p.Tyr209Ser variant in the cytochrome c
oxidase assembly gene (SCO2). Since the cardiomyopathy related to
SCO2 gene mutation is usually associated with encephalopathy and is
fatal in children, we excluded the causative role of such gene in our family.
Therefore, we considered the DSPGlu293Val variant as disease causing
and we hypothesized a modifier role for the SCO2 variant.
Naxos disease and Carvajal syndrome
In 1986, an association of cardiomyopathy with wooly hair and
palmoplantar keratoderma was first reported in families from the Greek
island of Naxos [67]. This autosomal recessive cardiocutaneous syndrome
(‘‘Naxos disease’’)[68] presented clinical and histopathological
characteristics of arrhythmogenic right ventricular cardiomyopathy [69].
In 1998 Carvajal [70] reported a variant of Naxos disease in families from
India and Ecuador, respectively, in which the heart disorder presented

2. Genotype – phenotype correlations
87
with a more pronounced left ventricular involvement and early morbidity.
Naxos disease and Carvajal syndrome are now considered as one entity
with clinical and genetic heterogeneity.
Molecular genetic investigations in families with this
cardiocutaneous phenotype have identified mutations in genes encoding
the cell adhesion proteins plakoglobin and desmoplakin. The structural
and functional integrity of cardiac tissue is supported by desmosomes,
adherens junctions and gap junctions located at intercalated disks [71]. In
desmosomes, plakoglobin, plakophilin2 and desmoplakin anchor desmin
intermediate filaments to desmosomal cadherins, securing the
mechanical cell–cell adhesion. Desmosomal cadherins (desmoglein and
desmocollin) are transmembrane proteins forming an extracellular
zipper‐like dimer with the corresponding part of desmosomal cadherins
of the adjacent myocardial cell. Plakoglobin and plakophilin2 are
armadillo proteins located at the outer dense plaque of desmosomes
which bind with the N‐terminal of desmoplakin and with the C‐
terminal of desmosomal cadherins. Desmoplakin is a larger dumbbell‐
shaped molecule which makes up the inner dense plaque with its middle
coiled‐coiled rod domain and binds via its C‐terminal with desmin
intermediate filaments. Plakoglobin is the only desmosomal protein
which is also found at adherens junctions where it is involved in linking
with the actin cytoskeleton of adjacent myocardial cells.
Normal functioning and integrity of desmosomes is of utmost
importance in myocardium and skin particularly of palms and soles,
tissues that experience constant mechanical stress. Intercellular junctions
not only maintain tissue integrity but also integrate mechanical and
signaling pathways in the regulation of cell growth, differentiation and
development.

2. Genotype – phenotype correlations
88
A family with early onset of LV aneurysm and ventricular
arrhythmias
The proband was a 28 year-‐old man who experienced repeated
episodes of sustained ventricular tachycardia, treated in the ER with DC
shock. The ECG showed pathological Q waves in the infero-‐lateral leads
[Figure 2.3-‐5], while the echocardiogram revealed a small aneurysm of
the basal segment of the infero-‐lateral wall. The coronary angiogram
performed showed normal coronary artery. The cardiac magnetic
resonance, performed after a few months, confirmed the presence of the
LV aneurysm [Figure 2.3-‐6]. The patient had no relevant clinical history
and a normal cardiovascular risk profile. Therefore a diagnosis of
myocarditis was made.
A couple of months later, his father underwent cardiological
evaluation as a part of pre-‐operative assessment. His ECG was
indistinguishable from that of his son and the echocardiogram showed an
infero-‐lateral aneurism. He was asymptomatic, with no history of angina,
dyspnea or palpitations. Therefore, we supposed a familial form of
cardiomyopathy. The brother of the proband was also examined and he
presented the same clinical phenotype.
Figure 2.3-‐5: ECG of the proband (next page)
ECG showing pathological Q waves in the inferior (II-‐III-‐ avF) and lateral leads (V5-‐V6).
Figure 2.3-‐6: Pedigree and clinical phenotype
Top left panel: family pedigree showing a dominant inheritance Top right and bottom panels: cardiac magnetic resonance images showing a small aneurysm of the basal segment of the infero-‐lateral wall, with transmural fibrosis (LGE).

2. Genotype – phenotype correlations
89
Figure 2.3-‐5: ECG of the proband
Figure 2.3-‐6: Pedigree and clinical phenotype

2. Genotype – phenotype correlations
90
While the familial nature of the cardiac disease was clear, the
clinical phenotype was atypical and undetermined. Therefore, since
standard genetic analysis could not be performed, we employed NGS to
perform a whole exome sequencing study.
Data analysis identified 64115 variants, 19209 of which located in
coding regions and intron-‐exon junctions. After frequency filtering
(variants with minor allele frequency < 5% in 1000 Genomes Project),
1343 variants were selected. Among these, 955 variants were not present
in dbSNP Release. Prioritization identified a candidate variant in DSG2
gene (OMIM#610193), desmoglein 2, c.1912G>A p.Gly638Arg, that co-‐
segregated in the family. Desmoglein is an essential component of
desmosomes and it is known to be associated with arrhythmogenic
cardiomyopathy, as described in the previous paragraph.
Genetic analysis allowed the definition (re-‐definition) of the clinical
phenotype as an atypical, non-‐syndromic form of ARVC with localized
involvement of the LV. This atypical presentation, initially suspected to be
novel phenotypes, has been reassigned as atypical presentations of a
well-‐known genetic disorder, expanding its phenotypic spectrum.
2.3.3 Targeted approach
Family with HCM, noncompaction and supraventricular
arrhythmias
We assessed 19 subjects from a large four-‐generation Italian family
living in central and northern Italy, who remained undiagnosed following
conventional Sanger sequencing testing. Affected individuals of this family
had evidence of cardiomyopathic involvement, transmitted with an

2. Genotype – phenotype correlations
91
autosomal dominant inheritance pattern, comprising variable
combinations of 3 distinctive features: asymmetric LV hypertrophy (LVH)
consistent with HCM, early onset of supraventricular arrhythmias and
atrioventricular (AV) block, and regional LV noncompaction [Figure 2.3-‐7:
family pedigree and figure legend].
Figure 2.3-‐7: pedigree (next page) and figure legend.
Pedigree including the results of ACTN2 Met 228Thr cosegregation in 18 family members. Arrow: proband. +/-‐ presence of the heterozygous Met228Thr; -‐/-‐ absence of the heterozygous Met228Thr .

2. Genotype – phenotype correlations
92

2. Genotype – phenotype correlations
93
The proband (II-‐15) was an 82 year-‐old man with mild, asymmetric
LV hypertrophy localized to the basal and mid-‐septum, marked bi-‐atrial
dilatation and a restrictive LV filling pattern with preserved systolic
function. He had been diagnosed with nonobstructive HCM almost three
decades earlier, and followed at our Institution since 2005. Remarkably,
he had a history of paroxysmal atrial fibrillation (AF) which presented at
the age of 30, which subsequently evolved into permanent AF with
advanced AV block, requiring VVI pacing at the age of 68 [Figure 2.3-‐8]. In
2008 he proved to be negative for mutations in the coding regions and
splice sites of the 8 most prevalent sarcomere genes. Despite his early
onset of disease manifestations and adverse cardiac remodelling,
consistent with restrictive evolution of HCM, he remained fully active
with only mild functional limitation (functional class NYHA class II).
Furthermore, he remained free from cardioembolic complications
although he repeatedly refused treatment with oral anticoagulants. He
currently remains on diltiazem, loop diuretics, kanrenone and low-‐dose
aspirin. His pacemaker had been replaced 4 times, and has never been
upgraded to an implantable defibrillator; no significant ventricular
arrhythmias have been noted on repeated 24-‐hour ECG Holter monitoring.
His Pro-‐BNP in 2009 was 1013 pg/ml.

2. Genotype – phenotype correlations
94
Figure 2.3-‐8: Clinical features of the proband (II-‐15)
A-‐B: Parasternal long axis (2d and M-‐mode) of the proband, showing marked left atrial dilatation (52 mm), with mild hypertrophy of the septum (16 mm) and small LV cavity (telediastolic diameter 40 mm).
C-‐D: Apical 4 chamber view (diastole and systole). Asymmetric LV hypertrophy, with maximal LV wall thickness of the basal and mid-‐septum, small LV cavity (telediastolic volume 63 ml) and marked bi-‐atrial dilatation (LA volume 220 ml). Ejection fraction was normal (70%).
E: Restrictive mitral inflow filling pattern, with deceleration time of 144 ms(rhythm: AF with ventricular pacing).

2. Genotype – phenotype correlations
95
F: Tricuspid regurgitation jet velocity showing pulmonary hypertension, with an RV/RA gradient of 44 mmHg. Inferior vena cava diameter (not showed) was 3.1 cm, with respiratory variation of diameter <50%.
G: ECG (DI, DII and DIII, V1-‐3 leads) showed AF with complete AV block and VVI pacing.
Three additional family members had similar echocardiographic
and clinical features, characterized by nonobstructive HCM with
restrictive evolution, AV block and biatrial dilatation: the proband’s
brother (II-‐5), aged 80, who developed permanent AF at the age of 64
years and required VVI pacing for advanced AV block at 68 years; and two
of his nephews (III-‐6 and III-‐7). Patient III-‐6 was a 50-‐year old man with a
long-‐standing history of frequent ventricular ectopic beats and sustained
supraventricular arrhythmias associated with apical HCM and regional LV
noncompaction; he recently developed paroxysmal AF and advanced AV
block requiring permanent pacing. His brother (III-‐7) required surgery at
the age of 15 due to a large atrial septal defect (ostium secundum)
associated with severe right-‐sided heart failure. At that age, he already
showed first degree AV block and frequent supraventricular ectopic
beats; his echocardiogram showed apical HCM with marked bi-‐atrial
dilatation [Figure 2.3-‐9]. He unfortunately died of heart failure-‐related
complications a few years later.

2. Genotype – phenotype correlations
96
Figure 2.3-‐9: Marked clinical heterogeneity in family members (next page).
Panel A-‐F: morphological features of some family members.
A: PatientIII-‐17 showedmild left atrial dilatation (index volume 44 ml/m2).
B: The 15 year-‐old proband’s nephew (III-‐7) had apical HCM with marked bi-‐atrial dilatation; he required surgery at the age of 15 due to a large atrial septal defect (ostiumsecundum). At that age, he already showed 1st degree AV block and frequent supraventricular ectopic beats. He died a few years later due to right-‐sided heart failure complications.
C: Patient III-‐6 was a 50-‐year old man had HCM with restrictive evolution, marked biatrial dilatation and regional LV noncompaction. He had a long-‐standing history of frequent ventricular ectopic beats and sustained supraventricular arrhythmias, including paroxysmal AF. He also required permanent pacing due to advanced AV block (see also panel B6).
D: The proband’s brother (II-‐5), aged 80, had nonobstructive HCM with restrictive evolution, and biatrial dilatation: he developed permanent AF at the age of 64 years and required VVI pacing for advanced AV block at 68 (see also panel B5).
E-‐F: Echocardiogram and cardiac MRI of two female patients (patient III-‐13 and IV-‐8respectively), showing LV non compaction of the apex, distal septum and lateral wall.
Panel G-‐N: broad spectrum of electrocardiographic abnormalities and supraventricular arrhythmias.
G: Isolated and short run of supraventricular ectopic beats on Holter ECG monitoring (individual III-‐13).
H: Second Degree sinoatrial block, Type II, of individual III-‐6, at the age of 9. (Arrow indicates the dropped P wave).
I: Supraventricular tachycardia (individual III-‐17). Arrows indicate P waves.
L: First degreeatrio-‐ventricular block (individual III-‐19).
M: Atrial fibrillation with a slow ventricular response (individual III-‐24).
N: Junctional rhythm (P waves are absent), of individual III-‐6, who required permanent pacing at the age of 50.

2. Genotype – phenotype correlations
97

2. Genotype – phenotype correlations
98
The 4-‐year old son of patient III-‐6 (IV-‐2) presented at birth with
esophageal atresia and tracheal fistula associated with ostrium secundum
atrial septal defect, requiring multiple surgery. He had multiple runs of
supraventricular tachycardia, but no evidence of LVH or noncompaction.
In addition cardiomyocytes show hypertrophy by intraoperative surgical
biopsy of the ventricular septum [Figure 2.3-‐10].
Figure 2.3-‐10: Intraoperative surgical biopsy of the ventricular septum
A) panoramic view of the surgical biopsy sample with endocardial fibrous thickening and mild disarray (trichrome stain).
B,C) cardiomyocytes show hypertrophy (mean diameter 20 micron), diffuse cytoplasmic vacuolization with perinuclear halo and dysmetric and dysmorphic nuclei, associated with focal fibrosis” (haematoxylin eosin stain, scale bar =100 micron).
The remaining patients all showed milder forms of cardiac disease,
including mild apical LVH (patient III-‐26); regional LV non-‐compaction
(patient III-‐13 and IV-‐8, associated with first degree AV block in the
former); left atrial dilatation and supraventricular arrhythmias (patients

2. Genotype – phenotype correlations
99
III-‐17, III-‐21 and III-‐24, associated with first degree AV block in the latter);
and left atrial dilatation with 1st degree AV block (patient III-‐19). None of
the other family members assessed had evidence of heart disease
suggesting genetic transmission of the cardiomyopathic trait. Individual
III-‐18 died suddenly at the age of 49: autopsy showed evidence of
ischemic heart disease associated with coronary atherosclerosis, with no
evidence of LVH, disarray or interstitial fibrosis.
Genetic analysis
Following genetic counseling and informed consent, proband’s
DNA was screened by targeted massively parallel sequencing of 36
cardiomyopathy-‐associated genes [Table 2.3-‐2].
Data analysis identified 1467 variants, 50 of which are located in
coding regions and intron-‐exon junctions. In order to identify clinically
relevant variants, data obtained from the analysis were filtered based on
frequency using dbSNP Release 137, Exome Sequencing Project (ESP),
1000 Genomes Project (1KGP) and HGMDTM Professional. Then they
were filtered according to the function (coding, 5’ or 3’ junctions), the
evolutionary conservation of the affected nucleotide (PhyloP score) and
pathogenic potential by in silico predictive algorithms (MutationTaster),
SIFT, PolyPhen2. Following bioinformatics analysis, four likely pathogenic
variants were identified: TTN NM_003319.4:c.21977G>A (p.Arg7326Gln);
TTN NM_003319.4:c.8749A>C (p.Thr2917Pro); ACTN2
NM_001103.2:c.683T>C (p.Met228Thr) and OBSCN
NM_052843.2:c.13475T>G (p.Leu4492Arg). The novel variant ACTN2
p.Met228Thr, located in the Actin–binding domain, proved to be the only
mutation fully co-‐segregating with the cardiomyopathic trait in 18
additional family members (of whom 11 clinically affected).

2. Genotype – phenotype correlations
100
ACTN2_Met228Thr was absent in 570 alleles of healthy controls and in
1000 Genomes Project, and was labelled as “Causative” by in silico
analysis using PolyPhen-‐2, as “Deleterious” by SIFT and as “Disease-‐
Causing” by MutationTaster.
Table 2.3-‐2: List f 36 Hypertrophic Cardiomyopathy-‐associated genes
Gene Symbol NM Gene name
ACTA1 NM_001100.3 Actin, alpha 1 , skeletal muscle ACTC1 NM_005159.4 Actin alpha, cardiac muscle1 ACTN2 NM_001103.2 Actin,alpha 2 CALM3 NM_005184.2 Calmodulin 3 (phosphorylase kinase, delta) CALR3 NM_145046.3 Calreticulin 3 CASQ2 NM_001232.3 Calsequestrin 2 (cardiac muscle) CAV3 NM_033337.2 Caveolin 3 COX15 NM_078470.4 Cytochrome c oxidase subunit 15
ANKRD1 NM_014391.2 Ankyrin repeat domain 1 (cardiac muscle) CSRP3 NM_003476.2 Cysteine and glycine-rich protein 3
DES NM_001927.3 Desmin FXN NM_000144.4 Frataxin GLA NM_000169.2 Galactosidase-alpha JPH2 NM_020433.4 Junctophilin 2
LAMP2 NM_002294.2 Lysosomal-associated membrane protein 2 MYBPC3 NM_000256.3 Myosin binding protein C, cardiac
MYH6 NM_002471.3 Myosin heavy chain 6, cardiac muscle,alpha MYH3 NM_002470.2 Myosin heavy chain 6, skeletal muscle MYH7 NM_000257.2 Myosin heavy chain 7, cardiac muscle,beta MYL2 NM_000432.3 Myosin light chain 2, regulatory, cardiac MYL3 NM_000258.2 Myosin light chain 3
MYLK2 NM_033118.3 Myosin light chain kinase 2, skeletal muscle MYO6 NM_004999.3 Myosin VI
MYOZ2 NM_021245.2 Myozenin 2 NDUFV2 NM_021074.4 NADH dehydrogenase (ubiquinone) Flavoprotein 2
OBSCN NM_001098623.1
Obscurin, cytoskeletal calmodulin and titin-interacting RhoGEF
PLN NM_002667.3 Phospholamban PRKAG2 NM_016203.3 Protein kinase, AMP-activated, gamma 2
RAF1 NM_002880.3 V-raf-1 murine leukemia viral oncogene homolog SRI NM_003130.2 Sorcin
TCAP NM_003673.3 Titin-cap or Telethonin TNNI3 NM_000363.4 Troponin I type3 (cardiac) TNNT2 NM_000364.2 Troponin T type2 (cardiac) TPM1 NM_000366.5 Tropomyosin 1 (alpha)
TTN NM_001256850 Titin

2. Genotype – phenotype correlations
101
ACTN2 and Z-‐disc protein genes as a cause of HCM
Over the last decade, many genes that do not strictly belong to the
sarcomere have been associated with rare forms of HCM [72]. These
include genes coding for proteins involved in intracellular calcium
handling or constituting the Z-‐disc -‐ the interface between the
cardiomyocyte contractile apparatus and the cytoskeleton [73].
These results support the role of Z-‐disc protein as a rare cause of -‐
LVH, and suggest a potential involvement of ACTN2 mutations in the
genesis of supraventricular arrhythmias. Notably, our family displayed
several atypical features compared to classic HCM. First, HCM patients
presented with mild to moderate mid-‐apical LVH, with slow but
distinctive progression towards a restrictive pathophysiology; none of our
patients had LVH preferentially localized to the basal septum or anterior
wall, and none had dynamic LV outflow obstruction. These features are
consistent with ACTN2-‐related HCM recently described in an Australian
family [74]. Furthermore, our family displayed a spectrum of clinical
manifestations beyond LVH itself, including supraventricular arrhythmias
and regional LV non-‐compaction, which were associated with HCM in the
most affected family members including the proband, but were present in
isolation (i.e. in the absence of LVH) in subjects with milder disease. In
addition, ostium secundum atrial septal defect was present in two
brothers and one of their sons: the latter finding, however, was confined
to a single branch of the family, suggesting fortuitous association with a
distinct genetic defect superimposed to the ACTN2 mutation. Conversely,
we found little propensity to malignant ventricular arrhythmias in our
patients, in that none of the affected individuals died suddenly or had
sustained VT or cardiac arrest, even when marked progression of disease
was noted. The most striking example is represented by the proband,

2. Genotype – phenotype correlations
102
who remains active and with only mild functional limitation at 90 years of
age. The only patient who died prematurely had undergone repair of an
extensive atrial septal defect during adolescence, and his demise largely
resulted from right heart failure secondary to the congenital defect on a
cardiomyopathic substrate. The relatively favourable long-‐term clinical
course in our family is in contrast with that of the ACTN2 pedigree
described by Chiu et al, characterized by elevated prevalence of
progressive heart failure and sudden cardiac death. Such discrepancy is
evident despite the fact that both ACTN2 mutations occur within the
actin-‐binding domain, in two adjacent calponin homology domains,
suggesting that different mutations in this gene may dictate radically
diverse clinical evolution and arrhythmic profile. This is in line with the
heterogeneity of phenotypes associated with ACTN2, which include HCM
with sigmoidal type hypertrophy and even dilated cardiomyopathy [74].
In a larger perspective, such diversity highlights the broad
pathophysiological implications of mutations affecting the molecular
apparatus connecting the cardiomyocyte contractile apparatus to the
cytoskeleton and muscle membrane, involving other HCM –associated
genes such as titin, telethonin, muscle LIM, and myozenin.
2.3.4. Translation of NGS into a diagnostic tool for patients with genetic
cardiovascular disease: the targeted approach
Unlike traditional approaches, where genes are generally studied
one at a time, high throughput approaches can provide a global view of
all the genes in a genome in a relatively fast and cost‐effective manner.
Among various high throughput methods, a targeted NGS approach

2. Genotype – phenotype correlations
103
describes a strategy where a specific set of genes related to the patient’s
phenotype are analyzed within the context of a genetic “test”.
By including virtually all known cardiomyopathy-‐associated genes,
these panels can serve as a first line, one-‐fits-‐all approach in the clinic for
any given patient, irrespective of his/her phenotype. This approach has
brought several advantages: a) most diagnoses may find molecular
confirmation at any center implementing NGS, avoiding the need for
traveling and repeated blood sampling; b) global costs are reduced; c)
even rare forms and phenocopies, such as Anderson-‐Fabry disease for
HCM, generally requiring several years for correct identification, may be
diagnosed at first attempt (and even when not clinically suspected); d)
very large genes, such as titin causing DCM, technically off-‐limits by
Sanger techniques, may be explored.
A very important implication of these enhanced possibilities is that
unclassified, atypical phenotypes, for which a candidate gene approach
could not be proposed, are being serendipitously diagnosed by using the
powerful targeted NGS approach. For example, systematic screening
exploring desmosomal genes have led to the recognition of forms of
ARVC with prevalent or even isolated LV involvement (thus the proposal
to rename ARVC simply as “arrhythmogenic cardiomyopathy”). Thus, NGS
is helping clinicians redefine the phenotypic spectrum of single
cardiomyopathies by expanding the gold standard of genetic diagnosis
beyond the limits suggested by clinical investigation.
The implementation of other NGS-‐based approaches such as
whole-‐exome sequencing (WES) and whole-‐genome sequencing (WGS) is
well underway. At the present time, these approaches remain expensive,
time-‐consuming and fraught with interpretational difficulties, largely

2. Genotype – phenotype correlations
104
limiting their use to research purpose aimed at identifying novel disease-‐
causing genes. Ultimately, WES or WGS will become part of a standard
assessment for most patients. The pace of this transition will depend on
the presence of the requisite infrastructure, regulatory standards, training
and best practice guidelines for reporting.
2.3.5. Challenges and opportunities: the evolving role of the medical
geneticist.
The many assets of NGS come at the cost of increasing complexity.
The multidisciplinary team required to interpret the biostatistical, genetic
and clinical implication of NGS findings, even when a focused targeted
approach is employed, are extremely demanding. Two are the main
challenges: the first is to confidently discriminate disease-‐causing variants
from rare, benign mutations with no clinical relevance. The wealth of
information derived from NGS is not yet paralleled by the clinical
knowledge needed to gauge the value of each identified mutation. For
novel mutations (i.e. the majority), this mandates clinical studies of
familial co-‐segregation involving dedicated physicians. Unfortunately,
large, informative families are not always available. The second challenge
(pertaining more to the whole-‐exome and –genome studies) is
represented by the ethical implications of unexpected findings that may
not be directly relevant to the original question (for example a mutation
in a cancer-‐causing gene during investigations for familial
cardiomyopathies). Clinical implications, often uncertain, need to be
balanced against the certainty of onerous psychological consequences
and a substantial risk of overdiagnosis diseases that may never occur.

2. Genotype – phenotype correlations
105
The NGS era of gene discovery and, ultimately, molecular
diagnoses heralds a fundamental change for medical genetics and
medical geneticists. In addition to continuing to define phenotypes, the
medical geneticist of the NGS era will have an unprecedented opportunity
to identify human disease-‐causing genes, enable the translation of NGS
into diagnostic tools and understand the role of disease modifiers to
advance the care of patients with rare diseases. These geneticists will also
become vital members of collaborative teams along with providers from
other specialties as they use genomic results to guide patient care. As
such, medical geneticists will need to complement their clinical skills with
expertise in the clinical interpretation of NGS data.
In the future, medical geneticists will instead phenotype patients
to facilitate the interpretation of the large data set generated by WES or
WGS. Perhaps most importantly, as increasing molecular insight inevitably
suggests novel therapeutic avenues, some medical geneticists will also
become interventionists, configuring, trialing and instituting treatments
for genetic disorders.
In conclusion, NGS represents the (ultimate) revolution for
mutational screening, and is bound to change our practice in the near
future. However, the wealth of information derived from NGS is not yet
paralleled by the clinical knowledge needed to gauge the value of each
identified mutation. As for each prior revolution in genetic diagnosis, the
only reliable key to understanding is represented by our patients and
their pedigrees. The psychological and ethical challenges associated with
the massive potential of NGS, as well as the issue of over-‐diagnosis,
require specific investigation in patients with inherited heart disease.

2. Genotype – phenotype correlations
106

3. Environmental Modifiers

3. Environmental Modifiers
108
“In nature we never see anything isolated,
but everything in connection with something else
which is before it, beside it, under it and over it.”
Johann Wolfgang von Goethe

3. Environmental Modifiers
109
3.1 The role of environmental modifiers on the
phenotypic expression and outcome of hypertrophic
cardiomyopathy: the “weight” of obesity
Say you are studying three individuals who carry the same disease-‐
causing mutation; two of these individuals suffer from the disease but
exhibit different symptoms, while the third person is completely
unaffected. For many inherited diseases, the same mutation is not always
expressed in all individuals who carry it; moreover, when the mutation is
expressed, it is not always expressed in the same way, even among
different individuals of the same family. How would you explain this
phenomenon? How is it possible that one family member carrying a
sarcomere mutation has HCM, while another carrying the same mutation
does not? What accounts for such a difference in disease expression?
Answering these questions is not an easy task. Nonetheless, research has
shown that variable phenotypes can be caused by a number of factors,
including complex interactions between genetic and environmental
factors.
This is the case of our research, conducted in a cohort of patients
with hypertrophic cardiomyopathy (HCM). HCM is the most common
genetic heart disease, characterized by heterogeneous phenotypic
expression with extreme diversity in the pattern and extent of left
ventricular hypertrophy (LVH) [1-‐5]. The reasons of such diversity are
related to molecular pathways and triggers that remain largely
unexplained. In the vast majority of genotype-‐positive patients, HCM is
associated with mutations in genes encoding proteins of the cardiac
sarcomere, most commonly beta-‐myosin heavy chain and myosin-‐binding
protein C. While these molecular defects are considered responsible for

3. Environmental Modifiers
110
the development of LVH, there is currently no conclusive evidence to
explain the variability in phenotypic expression of HCM, ranging from
massive degrees to absence of LVH even within the same family [6].
Among several hypotheses, the interplay of modifier genes and
environmental factors has been commonly offered as a potential
explanation for phenotypic diversity [7,8]. However, the extreme
heterogeneity of phenotypic expression among HCM patients, even in
family members sharing the same mutation, implies that other
determinants of cardiac morphology must be operative. The increased
cardiac mass in patients with obstructive HCM is a striking example
proving that extrinsic factors can play a primary role in the modulation of
the phenotype [9, 10]. In order to address this issue, we hypothesized
that obesity, an established cardiovascular risk factor known to promote
LVH in the general population [11-‐14], could be associated with an
increased left ventricular mass and cavity remodeling in this genetically
determined condition.
3.1 Obesity as a modulator of HCM phenotype
Obesity is a leading preventable cause of death worldwide, and it is
considered one of the most serious public health problems of the 21st
century. It is s classified as a disease, though it was widely perceived as a
symbol of wealth and fertility at other times in history [15, 16]. As of 2008
the WHO estimates that at least 500 million adults (greater than 10%) are
obese [Figure 3-‐1]. Obesity is known to promote LVH in the general
population due to numerous pathophysiological mechanisms, such as
increased sympathetic tone and leptin levels, myocardial fatty infiltration,
insulin resistance, and enhanced renin-‐angiotensin activity. Obesity may

3. Environmental Modifiers
111
also cause heart failure, and has been shown to result in worsening
diastolic function independent of other comorbid conditions [17].
Figure 3-‐1: A world map showing the prevalence and distribution of obesity
The hypothesis that obesity could influence the magnitude of LV
mass and prognosis in patients with HCM, although intuitive, had never
been studied, and in 2009 it was considered a mere speculation. We
therefore decided to design an observational multicenter study in order
to prove this hypothesis.
Three major referral HCM centers were involved: Minneapolis
Heart Institute Foundation (Minneapolis, Minnesota), Tufts Medical
Center (Boston, Massachusetts) and Careggi University Hospital (Florence,
Italy). The study cohort comprised 275 adult patients with HCM

3. Environmental Modifiers
112
consecutively referred at the 3 participating referral centers. All patients
enrolled underwent a thorough clinical and instrumental evaluation,
including ECG, echocardiography, cardiac magnetic resonance. The
population was divided into three groups, based on body mass index
(BMI): normal weight (BMI <25Kg/m2), pre-‐obese (BMI 25-‐30 Kg/m2) and
obese (BMI <30Kg/m2). Only 25% of patients were classified as normal,
while the majority of individuals were pre-‐obese or obese (respectively 38
and 37%).
The analysis of cardiac morphology and function showed that LV
mass index progressively increased in pre-‐obese and obese patients,
reflecting a direct relationship between BMI and LV mass measured by
CMR [Figure 3-‐2]. Not only the heart of obese and pre-‐obese patients was
more hypertrophied, but was also characterized by grater LV cavity size
and volume, indicating profound remodeling of cardiac morphology
[Figure 3-‐3]. Conversely, maximum LV wall thickness and systolic function
were identical in the three subgroups.
Figure 3-‐2 (left) and 3-‐3 (right): Impact of BMI on LV Mass and volume
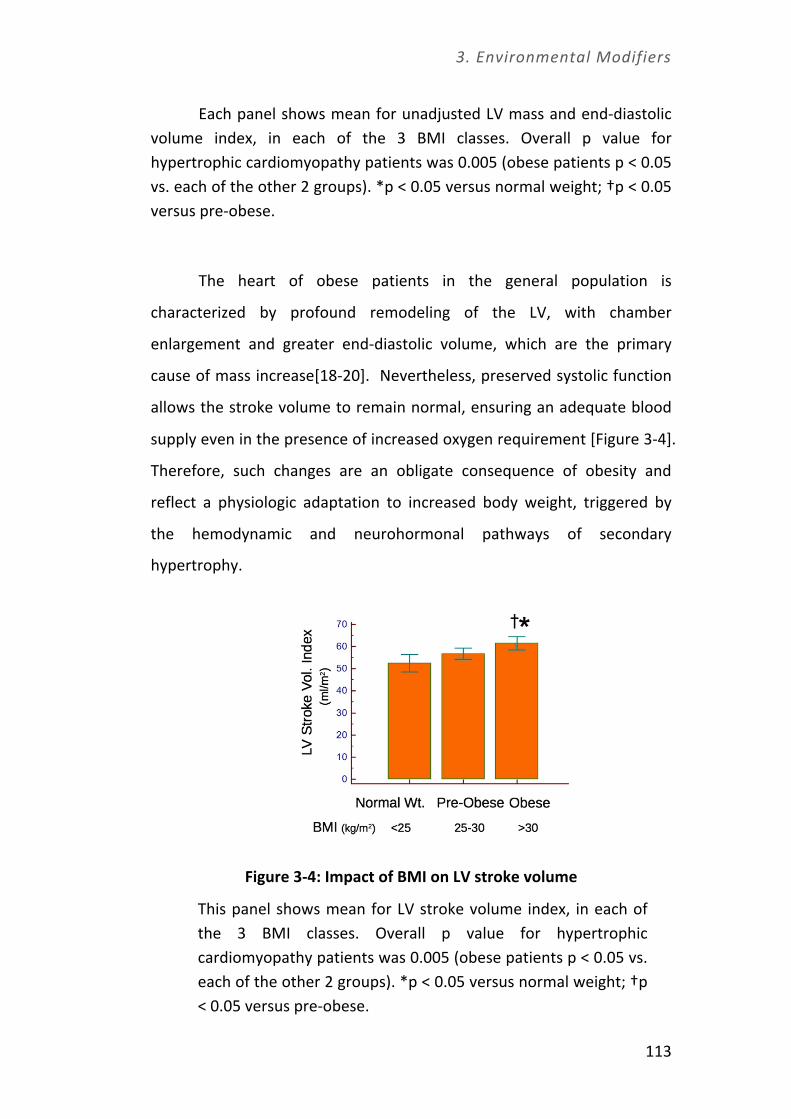
3. Environmental Modifiers
113
Each panel shows mean for unadjusted LV mass and end-‐diastolic volume index, in each of the 3 BMI classes. Overall p value for hypertrophic cardiomyopathy patients was 0.005 (obese patients p < 0.05 vs. each of the other 2 groups). *p < 0.05 versus normal weight; †p < 0.05 versus pre-‐obese.
The heart of obese patients in the general population is
characterized by profound remodeling of the LV, with chamber
enlargement and greater end-‐diastolic volume, which are the primary
cause of mass increase[18-‐20]. Nevertheless, preserved systolic function
allows the stroke volume to remain normal, ensuring an adequate blood
supply even in the presence of increased oxygen requirement [Figure 3-‐4].
Therefore, such changes are an obligate consequence of obesity and
reflect a physiologic adaptation to increased body weight, triggered by
the hemodynamic and neurohormonal pathways of secondary
hypertrophy.
Figure 3-‐4: Impact of BMI on LV stroke volume
This panel shows mean for LV stroke volume index, in each of the 3 BMI classes. Overall p value for hypertrophic cardiomyopathy patients was 0.005 (obese patients p < 0.05 vs. each of the other 2 groups). *p < 0.05 versus normal weight; †p < 0.05 versus pre-‐obese.

3. Environmental Modifiers
114
Notably, in our cohort the absolute LV wall thickness was
unaffected by body weight, suggesting that the degree of regional and
asymmetrical hypertrophy, distinct feature of HCM, is primary driven by
genetic factors and remain largely unaffected by environmental
modulation. Therefore, neither the current clinical diagnostic criteria for
HCM [3] nor decision-‐making for primary prevention of sudden death
with implantable cardioverter-‐defibrillators (both based on maximum
absolute LV wall thickness) require adjustment with respect to BMI in
adult.
Myocardial fibrosis, represented by areas of late gadolinium
enhancement (LGE) at CMR, was more prevalent in obese and pre-‐obese
patients. However, average percentage of fibrosis, expressed as % of LV
mass occupied by LGE, was comparable among the subgroups. The
multivariate model identified four variables as independently associated
with an increased LV mass: showed BMI, male sex, hypertension and
outflow obstruction. In particular pre-‐obese and obese HCM patients
showed respectively a 65% and 300% increased likelihood of assignment
to the highest LV mass index quartile.
Not only the cardiac morphology and function, but also the
outcome was related to the BMI class. Although all-‐cause mortality did
not differ among the three different subgroups, obese patients who were
alive at the end of follow-‐up showed an increased risk of developing
severe functional limitation (NYHA functional class III to IV -‐ HR 3.6)
[Figures 3-‐5 and 3-‐6]. However, the presence of severe symptoms was not
due to progressive systolic dysfunction (the so-‐called “end-‐stage HCM”),
since mean ejection fraction of the obese patients remains in the normal
range. Therefore, it is challenging to ascertain whether the functional

3. Environmental Modifiers
115
limitation was directly related to obesity or to the presence of HCM. Both
these factors may be closely related and embraced in a negative cycle of
event in which obesity leads to a sedentary life-‐style, further increases in
BMI, and, ultimately, worsening of heart failure symptoms.
Despite the impact on symptomatic status, the presence of obesity
itself did not confer a survival disadvantage in patients with HCM. While
this is partially counterintuitive, it is in line with the well know
phenomenon of “obesity paradox”, which states that, in patients with
chronic heart disease, the presence of obesity is a strong predictor of
favorable outcome [21]. Conversely, in the general population, obesity is
associated with increased mortality and morbidity [11, 17, 22].
Figure 3-‐5: Prevalence of HCM-‐related mortality and severe heart failure symptoms (NYHA functional classes III or IV)
among survivors at the end of follow-‐up period
HCM: hypertrophic cardiomyopathy; NYHA: New York Heart Association; Wt: weight. CMR. cardiac magnetic resonance

3. Environmental Modifiers
116
Figure 3-‐6: Cumulative risk of all-‐cause mortality in normal weight, pre-‐obese, and obese patients during follow-‐up
(Abbreviations as in figure 3.5)
HCM is a genetic-‐determined cardiac disease, whose phenotype
was thought to be entirely the expression of sarcomeric mutations. This
study proved, for the first time, that phenotype and outcome of patients
with a genetically determined cardiac hypertrophy can be modulated and
influenced by environmental and modifiable factors such as obesity.
These data also raise the possibility that control of modifiable risk factors
may be beneficial to cardiomyopathy, although further study are
mandatory in order to determine whether reduction of BMI could lead to
an improvement in clinical course.
Beyond the understanding of the physiopathological mechanisms
leading to a different phenotypic expression of the disease, this data may
be useful in our everyday clinical practice. Nowadays the primary target in
the treatment of HCM is symptoms relief, since neither drugs nor surgery
capable of treating the disease itself are currently available [23]. Therapy
usually employed to relief symptoms in HCM might be effective when
targeting hemodynamic alterations. In obese HCM patients, symptoms
may be caused not only by the hemodynamic changes, but mostly by

3. Environmental Modifiers
117
deconditioning and excess oxygen requirements, which are primarily
related to the obesity. In that case, both medical and even invasive
therapies risk to be ineffective and could have a limited impact on
symptoms.
So, when treating an obese patient with HCM and deciding
whether to start, increase, or decrease therapy, it is extremely important
taking into account that his symptoms are the result of the balance
between hemodynamics and excess body weight. Yet, treating the
hemodynamic abnormalities will lead to less than satisfying results unless
weight loss and healthy habits are taught to and adopted by the patients.
Readers with sharp eyes would have noticed that only a minority
of patients enrolled was in the normal weight range, like HCM and obesity
were somehow related. However, the challenging question is: which one
comes first? Is it a more severe expression of HCM that leads to obesity,
or is it the general trend of increasing body weight in Western society
that complicates HCM? The cross-‐sectional design of the study didn’t
allow the understanding of this association. However, both patients and
physicians should be aware of the negative implications of obesity in
outcome of HCM and should therefore prevent it. The inappropriate
suggestion to avoid physical exercise arises from the fact that the
incidence of sudden cardiac death in HCM patients involved in
competitive sports is much higher than in those who are not. However,
the extension of such recommendation to regular, non-‐competitive
exercise might lead to an unhealthy lifestyle, including weight gain and
increased cardiovascular risk.
Full text of the publication may be found in chapter 7.


4. Natural History and Predictors of Outcome
119
4. Natural History and Predictors
of Outcome

4. Natural History and Predictors of Outcome
120
“Risk stratification for any disease is, in many ways,
medicine’s attempt to predict the future”.
Rod Passman and Jeffrey J. Goldberger

4. Natural History and Predictors of Outcome
121
4.1 Predicting the Risk of Sudden Cardiac Death using
Cardiac Magnetic Resonance
Risk stratification for sudden cardiac death (SCD) has always been
painful for cardiologists, especially for those involved in the filed of
cardiomyopathies. Cardiomyopathies are disease of the heart muscle
with high arrhythmogenic propensity, usually affecting young subjects
and entire families. SCD is the most visible and devastating complication
of cardiomyopathies and the attempt to stratify patients, identifying
those who are at higher risk for malignant arrhythmias, is difficult.
Although the huge amount of risk factors individuated and the effort to
assemble risk score system, the issue of risk stratification and prevention
of SCD remains incomplete and very far from perfect [1-‐10]. The need to
better stratify patients, with the aspiration to prevent SCD, has been
realized with the systematic use of implantable cardioverter defibrillators
(ICDs) only in the past 10 years.
Estimates of the SCD rate in HCM emerged from hospital-‐based
cohorts were as high as 6%/year. This incredibly high incidence of SCD in
now recognized as an overestimate, was based on data collected in
tertiary center contaminated by the preferential referral of high-‐risk
patients. Recent reports, originated from less selected regional or
community-‐based cohorts placed annual HCM mortality rates at a much
more realistic ≤1%. Nevertheless, the traditional profile of SCD in HCM
remains unchanged; that is, it usually occurs without warning in
asymptomatic or mildly symptomatic young patients (predominantly <25
years of age).
While is now universally accepted the need for secondary
prevention with ICDs for patients who have survived a cardiac arrest with

4. Natural History and Predictors of Outcome
122
documented VF or an episode of sustained VT [12, 13], there has been a
substantial effort in reliably identifying the relatively small subset of
patients within the wide HCM spectrum who are at unacceptably high risk
for SCD and could benefit for ICD in primary prevention.
The conventional primary prevention risk factors in HCM, are [14]:
(1) family history of ≥1 HCM-‐related SCDs;
(2) ≥1 episode of unexplained, recent syncope;
(3) massive LV hypertrophy (thickness ≥30 mm);
(4) frequent nonsustained VT on serial ambulatory 24-‐h Holter;
(5) hypotensive or attenuated blood pressure response to exercise
Furthermore, certain morphological and functional features of
HCM can contribute to risk stratification, since they are known to be
associated with higher risk of arrhythmic event:
(1) LV Apical Aneurysms: the fibrotic thin-‐walled aneurysm, which
is usually present in patients with mid-‐ventricular obstruction, creates the
arrhythmogenic substrate triggering VT and VF [15].
(2) End-‐stage disease: this small subset of patients (around 5% of
HCM population) develops a systolic dysfunction characterized by wall
thinning and cavity dilatation, due to a widespread and often transmural
LV scarring. Inevitably, an adverse clinical course with progressive heart
failure and atrial and ventricular tachyarrhythmias occurs [16].
(3) LV Outflow Obstruction: a gradient ≥30 mmHg at rest is a
quantitative measure of elevated intraventricular pressure and wall stress.
Some studies showed that obstruction had a very modest, though
statistically significant, relationship to SCD risk [17, 18].

4. Natural History and Predictors of Outcome
123
(4) Percutaneous Alcohol Septal Ablation: this is a technique used
as an alternative therapeutic strategy for selected obstructive HCM
patients to the preferred option of surgical myectomy. It has the
capability of reducing the LV outflow gradient by creating a transmural
scar by alcohol injection in a septal coronary artery. There is accumulating
evidence and concern that the potential arrhythmogenicity of the scar
created by alcohol septal ablation can augment risk in the HCM
population [19, 20].
(5) Genotype: Although now commercially available, genetic
testing has not achieved the initial expectation that it would become a
reliable strategy for predicting prognosis or selecting patients for primary
prevention ICDs. However, the presence of multiple mutations, with the
hypothesis of “gene dosage effect”, has been proved to be associated
with higher risk of negative outcome, especially development of systolic
dysfunction [21].
Because current risk stratification cannot precisely guide SCD
prevention for each HCM patient, and SCD is known to occur in occasional
patients without any conventional risk factors, there is the ongoing
aspiration to identify more sensitive and specific clinical markers. Ideally,
this would lead to a single, noninvasive, repeatable and accurate
quantitative test.
Ventricular tachyarrhythmias, emanating from regions of
structurally abnormal myocardium (including areas of disorganized
architecture and myocardial fibrosis), represent the likely mechanism of
SD in HCM [22, 23]. Contrast-‐enhanced cardiovascular magnetic
resonance (CMR) imaging with late gadolinium enhancement (LGE) is
capable of noninvasive identification of myocardial fibrosis. While recent
investigations in HCM have demonstrated strong association between the

4. Natural History and Predictors of Outcome
124
presence of LGE and ventricular tachyarrhythmias, the utility of LGE as a
predictor of SD events remains unresolved [24-‐26].
Therefore, we have assembled a large multicenter HCM cohort to
investigate the independent prognostic value of LGE with respect to SD
and other adverse disease consequences. We prospectively evaluated
1300 HCM patients presenting to 7 HCM centers. HCM patients at each
institution consecutively eligible for CMR imaging comprised the initial
study cohort.
4.1.1 Extension of LGE as a powerful predictor of SCD
Most of the contrast-‐enhanced CMR studies in HCM have focused
almost entirely on the association between presence of LGE and SD.
However, the mere presence of any amount of LGE cannot be regarded as
a risk marker, since this designation gives equal weight to LGE across a
broad spectrum of amounts, from minimal to extensive [25]. Furthermore,
attributing increased risk to the presence of LGE per se conveys an
impractical and also imprudent message, since in some studies up to 70%
of all HCM patients have some LGE [26], all of whom could be regarded as
potential candidates for primary prevention ICDs, including a very large
proportion who would not benefit from this therapy and could be
exposed only to potential device complications.
Indeed, our data showed that SD risk increased in direct relation to
extent of LGE, with 10% increase in LGE associated with 40% increase in
relative SD risk. Notably, LGE ≥20% conferred a 2-‐fold greater risk
compared to the absence of LGE [Figure 4.1-‐1]. Multivariable analysis
identified extent of LGE as a strong independent predictor of SD,
independent of patient age, and of all cause mortality (p=0.007).

4. Natural History and Predictors of Outcome
125
Figure 4.1-‐1. Relation between extent of late gadolinium enhancement (LGE) and sudden death (SD) in 1293 patients with HCM.
Top panel: Incidence of SD events increased progressively and in direct relation to extent of LGE (p<0.001 for chi-‐square trend test).
Bottom panel: Hazard plot based on multivariable Cox regression analysis (p=0.02).
More than half patients with SD events had none of the
conventional risk markers. Among these patients, SD risk increased in
direct proportion to extent of LGE, with 20% LGE conferring nearly a 3-‐
fold greater risk of SCD [Figure 4.1-‐2]. Therefore, extensive LGE provided

4. Natural History and Predictors of Outcome
126
the only opportunity to identify some young asymptomatic patients
otherwise not regarded at increased risk, given absence of conventional
risk markers, and who would have remained unprotected against SD
without an ICD. Since approximately one-‐half of all clinically identified
HCM patients do not have conventional risk factors [27], CMR could
potentially identify a number of otherwise under-‐recognized patients at
increased risk who would potentially benefit from this enhanced risk
stratification model.
Figure 4.1-‐2: Relation between extent of LGE, conventional risk factors, and sudden death risk in 1293 patients with HCM.
Incidence of sudden death events increased progressively with increasing amounts of LGE in patients without conventional risk factors, while in HCM patients with ≥1 risk factor the incidence of sudden death events was highest in patients with >10% LGE compared to those with less LGE.
Comparably, amongst all patients with ≥1 risk factors, sudden
death risk increased in direct relation to the extent of LGE, with at least
moderate amounts of LGE (≥10%), conferring a 30% increase in SD risk
compared to those with no LGE. For patients in this “grey area” of HCM
risk stratification, we found that LGE can act as a potential arbitrator for

4. Natural History and Predictors of Outcome
127
resolving ambiguous ICD decisions [Figure 4.1-‐2]. These data support a
role for contrast enhanced CMR in more precisely defining patients at risk
for SD, even in the presence of conventional risk factors, with important
implications for ICD decision-‐making.
On the other hand, the absence of LGE was associated with a low
risk of SD and a potential source of reassurance to patients. However, this
finding did not absolutely immunize our patients against SD risk,
suggesting that susceptibility to potentially lethal ventricular
tachyarrhythmias can be influenced by factors other than myocardial
fibrosis [28].
4.1.2 Identification of progressive disease
The present data also identified an association between marked
and diffuse LGE and systolic dysfunction with progressive heart failure
[Figure 4.1-‐3 A and 4.1-‐4].
The amount of LGE was greater in the 26 patients who developed
end-‐stage during the follow-‐up compared to the 1206 HCM patients in
whom systolic function remained normal. Therefore, such extensive LGE
can be predictive of subsequent evolution into the end-‐stage, even when
initially identified in patients with normal ejection fraction and without
severe symptoms, with each 10% increase in LGE associated with 80%
increase in relative risk of developing end-‐stage HCM. Indeed, there was a
3-‐fold increase in risk for end-‐stage progression when LGE was ≥20%.
Notably, this same degree of fibrosis was also strongly associated with the
much different risk for SD in HCM (usually in the absence of progressive
heart failure), highlighting the power of extensive LGE as a noninvasive
marker with dual value in predicting sudden death and end-‐stage HCM.

4. Natural History and Predictors of Outcome
128
The ability to prospectively identify patients who will progress to
end-‐stage has clinical relevance. Indeed, this patient subset experiences
an unpredictable clinical course associated with unfavorable (albeit
treatable) disease consequences, including rapidly progressive heart
failure, that require alteration in management strategies including
tailored drugs and early consideration for heart transplant and
prophylactic defibrillators [16].
On the other hand, advanced heart failure with normal EF (both
with and without obstruction) was associated with no or small amounts
of LGE, suggesting that myocardial fibrosis is not an important
determinant of heart failure in the presence of preserved systolic function
[Figure 4.1-‐3 B]. As expected, patients with progressive heart failure due
to LV outflow obstruction showed a very low amount of LGE, of only 2%.
From a physiopatological standpoint, this result is quite intuitive, since
the heart muscle needs to be very efficient in order to create a significant
outflow gradient.

4. Natural History and Predictors of Outcome
129
Figure 4.1-‐3 A-‐C: relationship between LGE and progression to End-‐Stage.

4. Natural History and Predictors of Outcome
130
Figure 4.1-‐3 A-‐C: relationship between LGE and progression to End-‐Stage.
A. Hazard plot based on multivariable Cox regression showing the risk of developing the end-‐stage among 1232 HCM patients with normal systolic function at study entry. Incidence of end-‐stage development increased progressively with greater amounts of LGE (p<0.001)
B. Graph illustrating the strong relationship between the extent of LGE and the risk of progression to overt systolic dysfunction (end-‐stage), of all-‐cause death and sudden cardiac death.
C. Extent of LGE among 26 patients who developed end-‐stage HCM, 903 patients with normal EF without outflow obstruction, and 303 patients with normal EF and outflow obstruction (p<0.01).
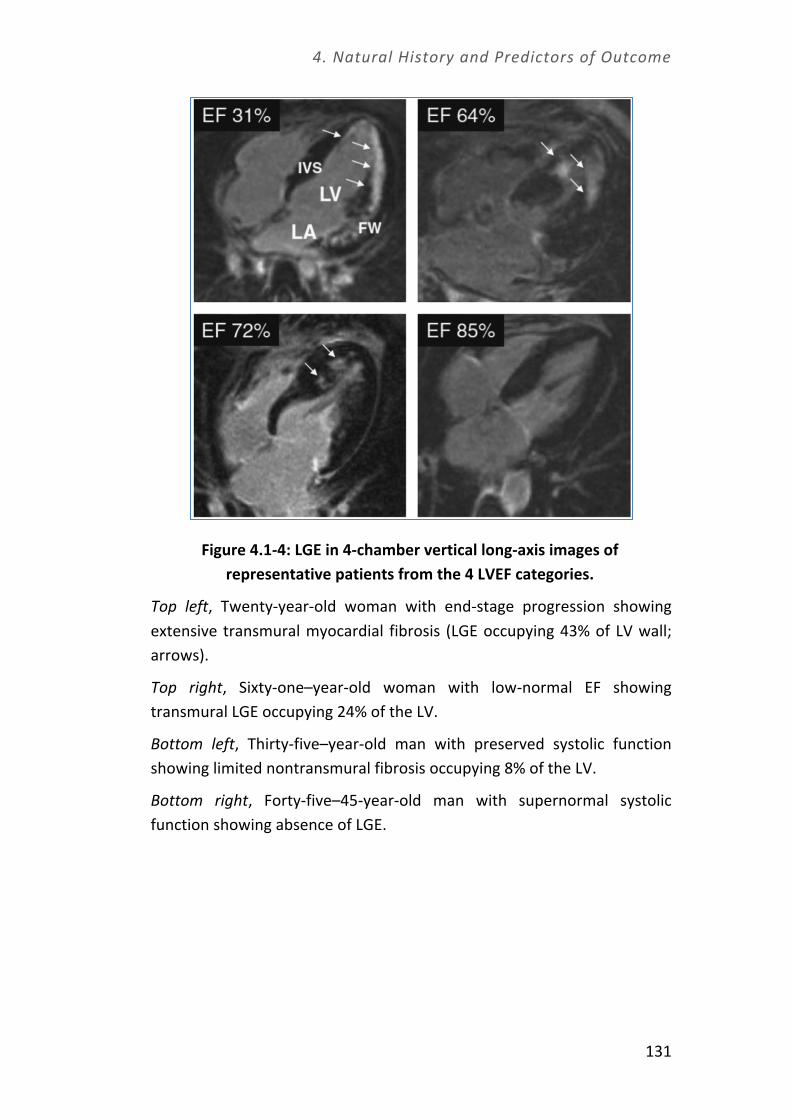
4. Natural History and Predictors of Outcome
131
Figure 4.1-‐4: LGE in 4-‐chamber vertical long-‐axis images of representative patients from the 4 LVEF categories.
Top left, Twenty-‐year-‐old woman with end-‐stage progression showing extensive transmural myocardial fibrosis (LGE occupying 43% of LV wall; arrows).
Top right, Sixty-‐one–year-‐old woman with low-‐normal EF showing transmural LGE occupying 24% of the LV.
Bottom left, Thirty-‐five–year-‐old man with preserved systolic function showing limited nontransmural fibrosis occupying 8% of the LV.
Bottom right, Forty-‐five–45-‐year-‐old man with supernormal systolic function showing absence of LGE.

4. Natural History and Predictors of Outcome
132
4.1.3 Utility of risk assessment in clinical decision-‐making
ICDs have partially changed the natural history of HCM for many
patients and provided an opportunity to longevity [29]. Indeed,
prevention of SCD has now become a new paradigm in the management
of HCM.
Targeting candidates for prophylactic ICD therapy can be complex,
due to the unpredictability of the arrhythmogenic substrate, the absence
of a dominant risk factor, and difficulty in assembling randomized trials.
Clinical dilemmas inevitably arise concerning ICD recommendations,
because many HCM patients fall into ambiguous gray zones in which the
level of risk cannot be assessed with precision using conventional risk
factors, and a measure of individual clinical judgment and experience of
the managing physician with direct knowledge of the patient’s clinical
profile and desires is necessary.
Implanting an ICD in primary prevention is not an easy call, nor for
patients nor for physicians. Despite the advances in ICD technology, ICD-‐
related complications are well documented and include infection, pocket
hematoma, pneumothorax and venous thrombosis [11]. Furthermore,
25% of HCM patients experience inappropriate shocks (5%/year),
resulting from lead fracture or dislodgement, oversensing, double
counting and programming malfunctions, or when triggered improperly
by sinus tachycardia or AF [11]. Unfortunately, the highest rate of
complications is in younger HCM patients, largely because their activity
level and body growth places a continual strain on the leads, which are
regarded as the weakest point in the system.
The reputation of the ICD as a “shock box” may even push the
patient to delay or refuse the device. Because of fear of present and/or
future discharges, some patients increasingly limit their range of activities

4. Natural History and Predictors of Outcome
133
and inadvertently diminish the benefits of the ICD in terms of quality of
life [30]. In such patients, especially those under the age of 50, the
benefits of a life-‐saving device may be attenuated by symptoms of anxiety
and depression due to significant difficulties in emotional adjustment
after ICD implantation [30].
In conclusion, while the present data do not resolve all remaining
questions in the arena of risk stratification in HCM, the imaging capability
of contrast-‐enhanced CMR with extensive LGE advances the risk
stratification strategy in HCM by providing the opportunity to identify
additional patients at increased SCD risk in whom prophylactic ICD
therapy would otherwise not be considered. Conversely, the absence of
LGE was associated with lower SCD risk. In addition, extensive LGE was
also predictive of adverse LV remodeling with systolic dysfunction
(endstage) and therefore is associated with 2 diverse consequences of
HCM. These novel findings support greater application of CMR in the
routine clinical evaluation and risk stratification of patients with HCM.

4. Natural History and Predictors of Outcome
134
4.2 A hundred tales the blood can tell: NT-‐pro BNP as a
clinical barometer of disease stability in HCM
There is an embarrassing gap between the availability of
information derived from serum biomarkers and our ability to understand
their true significance in order to create appropriate personalized
remedies.
Natriuretic peptides have been validated as markers of severity
and decompensation in various cardiac disorders and represent powerful
predictors of outcome in heart failure [31, 32]. The value of BNP or NT-‐
proBNP to identify those at highest risk for adverse outcome, has led to
the concept of using measurement of these biomarkers to ‘monitor’ and
‘guide’ adequacy of therapy. They have also been useful as screening
tools in large populations at risk of developing cardiovascular events,
including diabetic and elderly patients [32].
NT-‐proBNP, the N-‐terminal part of the BNP pro-‐hormone, is
released during hemodynamic stress by stretched cardiac myocytes and
split by proteolytic enzymes into 2 peptides; the C-‐terminal fragment
(BNP) represents the biologically active hormone [31, 33]. Because of a
different clearance mechanism (renal for NT-‐proBNP and receptor
mediated for BNP), NT-‐proBNP represents a more reliable predictor of
outcome than BNP [33].
To date, the prognostic accuracy of natriuretic peptides in patients
with hypertrophic cardiomyopathy (HCM), the most common genetic
heart disease, is unknown [34, 35]. Therefore, the serum NT-‐proBNP
levels were determined in 218 consecutive outpatients with HCM who

4. Natural History and Predictors of Outcome
135
had been referred to the Referral Center for Cardiomyopathies in
Florence (Italy) before January 2011.
4.2.1 Enrollment of patients and clinical follow-‐up
Of the 218 patients, 35 with known coronary artery disease (n=15)
or evidence of systolic dysfunction at the first evaluation (defined as a LV
ejection fraction <50%; n=20) were excluded. The remaining 183 patients
with HC constituted the study group enrolled in the present project. All
had had a stable clinical course, without episodes of acute HF or
worsening of symptoms, in the previous 3 months.
After enrollment, the patients were evaluated annually or more
often if dictated by the clinical status, using 12-‐lead electrocardiography,
echocardiography, and 24-‐hour electrocardiographic Holter monitoring.
Therapy was prescribed as appropriate, according to the current HCM
guidelines.
The primary end point (PE) was defined as a composite of
cardiovascular death (including sudden cardiac death), heart
transplantation, and resuscitated cardiac arrest.
The secondary end point (SE) was a composite of HF-‐related events,
including HF death, pulmonary edema requiring hospitalization,
progression to advanced symptomatic status (New York Heart Association
class III or IV), end-‐stage disease, onset of atrial fibrillation, and stroke.

4. Natural History and Predictors of Outcome
136
4.2.2 Results
The median NT-‐ProBNP value for the study group was 615 pg/ml
(range 22 to 16,013). Patients in the upper tertile, with higher NT-‐proBNP
values, had a larger left atrial diameter and maximum LV wall thickness
and were more likely to have advanced symptoms (New York Heart
Association class III), atrial fibrillation, and dynamic LV outflow tract
obstruction. The women had greater NT-‐proBNP values than the men and
represented most patients in the upper tertile. These data are in
agreement with those derived from previous studies, confirming that NT-‐
proBNP values correlate with the degree of symptomatic impairment and
functional capacity, LV mass index, left atrial volume, and LV outflow tract
obstruction [36-‐38].
In our HC cohort, followed up for an average of almost 4 years, NT-‐
proBNP proved to be an independent predictor of cardiovascular
mortality (our primary end point), with a six-‐fold increase in the likelihood
for each increment in the log NT-‐proBNP [Figure 4.2-‐1]. The cumulative
rate of the primary end point at the end of the follow-‐up period was 5%
(1.3%/yr) and 8% (2.1%/yr) in the middle and upper tertile, respectively.
Furthermore, the presence of a restrictive LV filling pattern conferred
additional prognostic stratification for patients in the middle NT-‐proBNP
tertile, although it had no additional value for patients in the upper tertile.
In the present analysis, however, sudden death represented a
potential confounder with regard to the predictive values of NT-‐proBNP,
because life-‐threatening ventricular arrhythmias can occur in stable and
often asymptomatic or only mildly symptomatic patients, lacking LV
overload and hemodynamic decompensation at death and, thus, might
not be predictable using the biomarkers of HF.

4. Natural History and Predictors of Outcome
137
Figure 4.2-‐1.
(A) Survival free from the primary end point (PE; a composite of cardiovascular death, heart transplantation, and resuscitated cardiac arrest) stratified by NT-‐proBNP tertiles (overall log-‐rank p ¼ 0.01). After pairwise comparison with Bonferroni correction, only the lower to upper tertile comparison reached statistical significance (p ¼ 0.01; HR 9.26, 95% CI 1.58 to 53.9).
(B) Freedom from PE stratified by NT-‐proBNP tertiles and presence or absence of mitral pulsed wave restrictive filling pattern (RFP).
The secondary end point, specifically focused on HF-‐related events,
was more accurately predicted by the NT-‐proBNP levels, with favorable
rate of survival free from the SE in the lower NT-‐proBNP tertile compared
with the middle and upper tertiles [Figure 4.2-‐2]. The cumulative rates of
SE were 18% (4.6%/yr) in the lower, 47% (12.0%/yr) in the middle and
44% (11.2%/yr) in the upper tertile. With regard to the SE, the presence
of congestive symptoms at enrollment (expressed as New York Heart
Association functional class) significantly contributed to the risk
stratification of patients within the middle and upper NT-‐proBNP tertiles
[Figure 4.2-‐2].

4. Natural History and Predictors of Outcome
138
Figure 4.4-‐2.
(A) Survival free from the SE (a composite of HF death, acute decompensation, progression to end-‐stage disease, and stroke) stratified by NT-‐proBNP tertiles (overall log-‐rank p ¼ 0.002). After Bonferroni correction, the lower to middle and lower to upper tertile comparisons reached statistical significance (p ¼ 0.001 and p ¼ 0.002, respectively).
(B) In a Cox regression model including other independent predictors of SE, only the initial New York Heart Association (NYHA) functional class helped to further stratify high-‐risk patients.
Receiver operating characteristic curve analysis demonstrated
moderate power for NT-‐proBNP in the prediction of the primary and
secondary end point [Figure 4.2-‐3].
The relatively high NT-‐proBNP value of 810 pg/ml proved the best
cutoff for the identification of patients at risk of cardiovascular death (PE),
although with limited specificity (61%). Comparably, an NT-‐proBNP level
<310 pg/ml was associated with a 75% reduction in risk of HF-‐related
events compared with values of >310 pg/ml [Figure 4.2-‐3], with high
sensitivity (85%) but low specificity (45%).

4. Natural History and Predictors of Outcome
139
Figure 4.2-‐3.
Receiver operating characteristic curves evaluating accuracy of NT-‐proBNP levels for prediction of (A) primary end point and (B) SE. *95% CI 0.61 to 0.86; †95% CI 0.58 to 0.74.
The threshold of 310 pg/ml, representing the cutoff value for the
lower tertile, had very high negative predictive accuracy (85%) and
retained important prognostic value even after adjustment for other
powerful adverse clinical features, such as age, New York Heart
Association functional class, and LV outflow tract obstruction. Similarly,
the positive predictive accuracy for end-‐stage progression was low (25%),
despite an excellent negative predictive value (88%).
Therefore, NT-‐proBNP appeared most useful for its high negative
predictive value, as a screening test to identify low-‐risk patients, rather
than to single out those with a greater risk of events.
4.2.3 Understanding the language of serum biomarkers
In stable outpatients with HC, plasma NT-‐proBNP proved a
powerful independent predictor of death and HF-‐related events. While

4. Natural History and Predictors of Outcome
140
the positive predictive accuracy of the elevated NT-‐proBNP values was
modest, low values reflected true clinical stability,
Low NT-‐proBNP values can identify truly stable disease and allow
cautious reassurance of patients with HC regarding the risk of disease
progression and HF, even in the presence of potentially adverse clinical
features, suggesting the possibility of avoiding or postponing aggressive
treatment options. For patients with HC and dynamic LV outflow tract
obstruction, for example, low NT-‐proBNP values might represent a useful
arbitrator, supporting the decision to avoid or postpone invasive septal
reduction therapies, particularly when the symptoms are sufficiently well-‐
controlled by pharmacologic treatment.
In contrast, greater NT-‐proBNP values represent a rather
nonspecific “red flag,” lacking sufficient specificity for the identification of
the truly high-‐risk subset, and needs to be complemented by additional
clinical and instrumental workup.

4. Natural History and Predictors of Outcome
141
4.3 Long term outcome of idiopathic dilated
cardiomyopathy: the real world
Idiopathic dilated cardiomyopathy (IDCM) is a primary myocardial
disease of unknown cause characterized by cardiac enlargement and
impaired systolic function of one or both ventricles, in the absence of
abnormal load conditions (such as hypertension, valve disease, or
congenital heart disease) or coronary artery disease of sufficient severity
to cause global impairment of ventricular function [39-‐41]. Different
causes can lead to DCM, including inherited, infectious, and inflammatory
diseases. However, the majority of cases remain unexplained after a
thorough review for secondary cause. IDCM is an important cause of
sudden cardiac death (SCD) and heart failure (HF) and is one of the
leading indications for cardiac transplantation in children and adults
worldwide [41].
Over the last three decades, the outcome of IDCM has radically
changed due to major advances in pharmacological and device-‐based
therapeutic strategies. However, studies addressing the outcome of HF
have been characterized by patients with DCM, with a relatively small
representation of individuals with IDCM, so that limited data exists with
specific regard to this condition [42-‐45]. Furthermore, the everyday
practice can be very different from that described in the trials, with rates
of patient’s compliance usually considerably lower than that reported in
clinical trials [46-‐48].
The present study is based on a real clinical experience, whereby
603 patients with angiographically proven idiopathic dilated
cardiomyopathy have been followed during the past 30 years in the
Regional referral center for cardiomyopathies in Florence. This cohort of

4. Natural History and Predictors of Outcome
142
unselected, consecutively enrolled and angiographically negative IDCM
patients was evaluated in a systematic fashion by the same team,
providing substantial continuity of care during an extended time period.
4.3.1. Enrollment of patients and follow-‐up
All study patients were evaluated and followed up at our
institution by clinical and family history, physical examination, 12-‐lead
ECG, standard chest radiograph, routine laboratory tests, 24-‐hour Holter
ECG monitoring (since early 1980s), M-‐mode and 2D echocardiography
(since 1970s), and Doppler echocardiography (since mid-‐1980s). Patients
were regularly followed up by outpatient visits every 6 months (or more
frequently when clinically indicated) and, when necessary, by interviews
with referring physicians or by telephone contacts.
HF treatment has radically changed during the past decades,
following major randomized clinical trials and their subsequent impact on
international HF guidelines[44, 49]. The use of neurohormonal inhibitors
has become established during the past 2 decades. While in the 1980s HF
treatment was virtually confined to diuretics and digoxin, the 1990s have
seen an explosion in the use of neurohormonal blockers.
Therefore, to assess long-‐term changes in outcome in relation to
treatment options, we subdivided our patients with IDCM into 4 periods,
coinciding with different therapeutic eras of HF treatment:
• period 1: 66 patients (11%) enrolled from 1977 to 1984, defined as the pre-‐ACE inhibition era, when standard therapy consisted of diuretic agents, digoxin, and early vasodilators;
• period 2: 102 patients (17%) enrolled from 1985 to 1990, marking the beginning of the ACEI era;

4. Natural History and Predictors of Outcome
143
• period 3: 197 patients (33%) enrolled from 1991 to 2000, characterized by increasing use of ACEI and ARBs and the introduction of β-‐blockers;
• period 4: 238 patients (39%) enrolled from 2001 to 2011, the device-‐era, characterized by the introduction of ICD and CRT on top of extensive neurohormonal blockade
4.3.2 Evolving treatment opportunities and outcome improvement
At initial evaluation, the 4 groups of patients were comparable
with regard to sex and NYHA class. However, there was a slight trend
toward enrollment of older patients, with less severe LV dilatation and
dysfunction during the years, with permanent atrial fibrillation at initial
diagnosis less prevalent over time.
At the end of follow-‐up, patients enrolled in periods 3 and 4
showed a better clinical profile, both in terms of cardiac function and
symptoms. The improvement in systolic LV function, together with a
reduction in end-‐diastolic diameter index, both expression of LV reverse
remodeling, was paralleled by a clear improvement of functional status
over time. Conversely, patients enrolled in periods 1 and 2 showed
progression of symptoms and limited improvement in systolic function
[Figure 4.3-‐1].
Figure 4.3-‐1 (following page):
Changes in New York Heart Association (NYHA) class [top panel], left ventricular ejection fraction (LVEF) [middle panel], and end-‐diastolic diameter index (iEDD) [bottom panel], from initial evaluation to end of follow-‐up based on enrollment period. Each bar indicates the change from enrollment (horizontal line) to final evaluation (blunt bar extremity).

4. Natural History and Predictors of Outcome
144

4. Natural History and Predictors of Outcome
145
The presence of such favourable clinical profile resulted in a better
outcome, so that each enrollment period was associated with a 42%
reduction in risk of all-‐cause mortality and transplantation, compared
with the previous one [Figure 4.3-‐2 A]. Likewise, an earlier period of
enrollment proved a potent independent predictor of refractory HF death
and SD [Figure 4.3-‐2 B,C].
HF death rates declined at a relatively constant rate after the
introduction of ACE inhibition, in the transition from periods 1 to 2.
Conversely, marked and progressive impact on SD was evident in the
transition to periods 3 and 4, after the introduction of the ICD/CRT and
the complete penetration of β-‐blocker treatment for HF in real-‐world
practice.
Figure 4.3-‐2 (following page)
Cox multivariate regression survival plot (adjusted for age, New York Heart Association [NYHA] class, sex, left ventricular ejection fraction and indexed left atrium diameter) indicating differences in outcome based on enrollment period for the 3 study end points: A, All-‐cause mortality/heart transplantation; (B) death for refractory heart failure; and (C) sudden death.
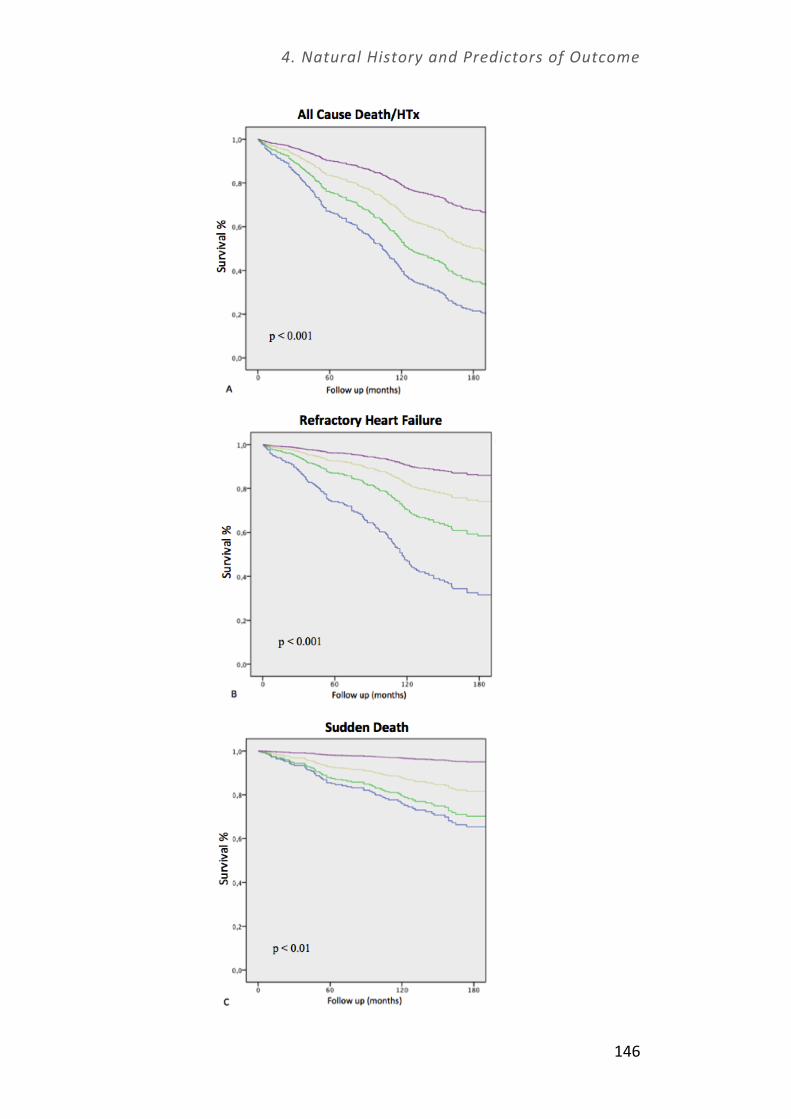
4. Natural History and Predictors of Outcome
146

4. Natural History and Predictors of Outcome
147
It is important to notice that in our cohort, patients enrolled later
had a less severe profile in terms of symptoms, LV size and function. This
trend presumably reflects increasing awareness of the disease in the
cardiology community and availability of more sensitive diagnostic tools
that allowed an early diagnosis of the disease. This may have contributed
to the reduction in mortality observed during the years, resulting in an
overestimation of benefits related to treatment. However, this trend is
counterbalanced by the more advanced age at enrollment of the most
recent groups. In our multivariate models, the enrolment period proved
to be a very potent predictor of outcome independent of the most
relevant baseline clinical and demographic features including well-‐
recognized prognostic factors (ie, age, sex, NYHA functional class, LV and
left atrial diameters, LVEF, and degree of functional mitral regurgitation)
[50-‐54]. Although it is virtually impossible to quantify the net result of
these trends on outcome, it is plausible to attribute most of the survival
benefit observed to improvements in management rather than to the
evolving patient demographics.
4.3.3 Conclusions
The present study demonstrates that the long-‐term prognosis of
patients with idiopathic dilated cardiomyopathy has radically improved
during the past 30 years, in terms of overall mortality, refractory heart
failure death, and sudden cardiac death, reflecting continuing progress in
pharmacological and device-‐based management.
The power of our data goes well beyond the huge number of highly
selected patients, the extend period of follow-‐up and the stability of the

4. Natural History and Predictors of Outcome
148
team-‐care over time. It indeed resides in the possibility to replicate the
setting of a clinical trial in the real world, overcoming those limitations
usually associated with purely retrospective and observational
community studies with long follow-‐up where data are not usually
collected in a systematic fashion.

4. Natural History and Predictors of Outcome
149
4.4 Myocardial damage due to infectious disease: Chagas
cardiomyopathy
Patients suffering from spatial neglect may eat from only one side
of a plate, write on only one side of a page, or shave only one side of the
face. The neglect may be so strong so as to deny ownership of the other
side, stating that it belongs to someone else, or to avoid or withdrawn
from the contralateral side of space.
There is a form of special neglect affecting the medical community:
more than one billion of people worldwide suffer from a group of
infections caused by a variety of pathogens, responsible for a significant
numbers of death in tropical and sub-‐tropical areas. However, the
majority of these tropical diseases afflict the poorest people, who live on
less than $2 per day [55]. Therefore, despite their significance in terms of
burden of ill carried by more than one-‐sixth of the world's population,
little financial support has been provided to address these diseases, like
they don't even exist. That is the reason why the World Health
Organization decided to unify them under the name of “neglected
tropical diseases” and prioritize 17 of them in an official list, in order to
increase their awareness among physicians of the eastern countries [55-‐
57]. Moreover, due to the increasing immigration, international travel
and adoption, many of these diseases now represent a matter of
international concern even for non-‐endemic countries, such as Europe or
US [58-‐60]. They are a ‘neglect’ too important to ignore.
We started to explore this “new” branch of cardiology in 2011, as a
result of our collaboration with the Infectious Disease Unit of our Hospital.
Two of young women with the chronic form of Chagas disease were
referred to our center for cardiologic evaluation. They showed heart

4. Natural History and Predictors of Outcome
150
failure due to dilated cardiomyopathy and isolated ventricular
arrhythmias as dominant features of cardiac involvement, which was
substantially comparable to other more common form of heart disease.
Therefore, we decided to share our experience describing the story
of two patients with CD, in order to emphasizes the role of Chagas
disease as an important cause of cardiac disease in patients from endemic
regions, even when long removed from their native countries,
highlighting the need for increased awareness in non-‐endemic settings.
4.4.1 Clinical pictures
Patient # 1:
A 46-‐year old Brazilian woman, who has resided in Italy for 24
years, was admitted to our hospital following the onset of worsening
dyspnea and cough over the previous 3 months. The patient had a
longstanding history of smoking (1 pack/day), but no other known
cardiovascular risk factors. Two months previously, she had been seen by
a pneumologist, and had a chest CT and pulmonary function tests, upon
which a tentative diagnosis of interstitial pulmonary disease was made.
Treatment with oral steroids had been started over the ensuing 4 weeks,
with little benefit. On admission, she complained of shortness of breath
during daily activities and was unable to climb stairs (NYHA functional
class III), but had no history of chest pain or palpitations. On examination,
she had a 3rd heart sound but no murmurs; JVP was slightly elevated, and
she had fine bilateral basal crackles on chest auscultation. A repeat CT of
her chest showed evidence of pulmonary congestion, with mild interstitial
thickening; pulmonary function tests were consistent with moderate
restrictive respiratory impairment. Her electrocardiogram (ECG) showed

4. Natural History and Predictors of Outcome
151
sinus rhythm with occasional ventricular ectopic beats, but was otherwise
unremarkable. A transthoracic echocardiogram, however, showed a
mildly dilated, diffusely hypokinetic left ventricle (LV), with an ejection
fraction of 38% and severe functional mitral regurgitation. Routine
laboratory tests and standard virology, including cardiotropic and
neurologic agents, were inconclusive. A normal coronary angiogram
excluded ischemic heart disease as a cause of LV dysfunction.
When other potential etiologies were explored, a detailed family
history revealed that the patient’s sister had died suddenly years before
in Brazil with a prior diagnosis of Chagas, prompting specific
investigations for T. Cruzi. Serology tests resulted positive, leading to the
diagnosis of chronic CD complicated by cardiomyopathy.
Standard treatment for heart failure was begun with carvedilol,
ramipril and loop diuretics. Following multidisciplinary consultation,
antitrypanosmomal therapy was felt not indicated at this stage and the
patient was discharged in stable conditions.
After one month, her dyspnea had progressively improved and she
remained in NYHA functional class II. However, she complained of
recurrent palpitations and a 24-‐hour ambulatory Holter ECG showed
extremely frequent polymorphic premature ventricular beats, with
several couples and triplets. Amiodarone was added to her therapeutic
regimen at a dose of 200 mg/day.
At 3 months, she reported further improvement, with only mild
exercise limitation and no further episodes of palpitations. A repeat
Holter ECG showed dramatic reduction in the frequency of ventricular
and supraventricular beats. Her echocardiogram had also improved, with
significant increment in LV ejection fraction (to 52%) and only mild
residual mitral regurgitation [Figure 4.4-‐1]. Cardiac MRI showed discrete

4. Natural History and Predictors of Outcome
152
LV akinesia localized to the apical region, apical septum and infero-‐lateral
wall; following gadolinium injection, there was evident delayed
enhancement of the apex with prevalent sub-‐epicardial distribution,
consistent with Chagas cardiomyopathy [Figure 4.4-‐2].
Figure 4.4-‐1: Transthoracic echocardiogram from patient #1.
Parasternal long axis and short axis views (panel A1 and panel A2) and apical four chamber view (panel B1 and panel B2), showed mild left ventricular dilatation and dysfunction, with functional mitral regurgitation (panel A3) and moderate tricuspid regurgitation (panel B3). A triphasic pattern of left ventricular diastolic filling recorded with pulsed Doppler (panel C1) associated with reduced tissue Doppler lateral (panel C2) and septal E’ (panel C3), consistent with moderate diastolic dysfunction.

4. Natural History and Predictors of Outcome
153
Figure 4.4-‐2: Cardiac MRI from patient #1.
Four-‐chamber enlargement is accompanied by a-‐dyskinesia of apical region, distal septum and infero-‐lateral wall (panel A-‐C), associated with transmurallate gadolinium enhancement of the apical region (arrow) (panel B), with sub-‐epicardial distribution evident in two-‐chamber view (arrowhead) (panel D).
After one year of optimal medical therapy, the patient was in
stable clinical conditions, with no symptoms except for occasional fatigue.
An implantable cardioverter defibrillator (ICD) was discussed with the
patient, who declined based on her excellent response to medical
treatment. At this stage, antitrypanosomal treatment with benznidazole
was introduced but had to be withdrawn on day 12 due to a diffuse
itching maculopapular skin rash, which worsened despite oral treatment
with hydroxyzine dihydrochloride and prednisone. Benznidazole was

4. Natural History and Predictors of Outcome
154
therefore discontinued and no further antytripanosomal treatment has
been considered so far. She’s currently been considered for nifurtimox.
One year later, during a scheduled follow-‐up visit, she complained
of palpitations, insomnia and anxiety, started a couple of weeks earlier.
The ECG showed frequent isolated ventricular ectopic beats, while LV
function was stable. Blood tests revealed a severe hyperthyroidism
probably related to amiodarone, with T3 level about 100 times higher
than the normal range. Therefore, methimazole was started with
progressive reduction of thyroids hormones level and clinical
improvement. Amiodarone was immediately stopped, but no other
antiarrhythmic drugs could be considered because of the underlying
cardiac condition. Thus, after a careful reevaluation of risk factor for
sudden cardiac death, the ICD was reconsidered and then implanted.
Patient #2
A 48-‐year old Bolivian woman, resident in Italy for 32 years, was
referred for cardiac evaluation by the infectious disease unit after a
recent diagnosis of CD, following screening of the local Bolivian
Community in central Italy. Her father and sister had both died at a young
age, possibly due to complications of CD. She complained of recurrent
exertional chest discomfort, but no dyspnoea or palpitations. On
examination she was found to have an irregular pulse, while the rest of
her examination was otherwise unremarkable. A transthoracic
echocardiogram showed normal LV function with trivial mitral
regurgitation and normal pulmonary pressures. However, her 24-‐hours
Holter ECG showed numerous monomorphic premature ventricular beats
with frequent periods of bigeminy and trigeminy. A symptom-‐limited,
maximal exercise test ruled out inducible ischemia. Treatment with

4. Natural History and Predictors of Outcome
155
amiodarone was started at the dose of 200 mg/day followed after one
month by antiparasitic treatment with benznidazole 150 mg twice daily
for 66 days. The latter was well tolerated. At 2 months, a repeat Holter
ECG showed marked reduction of the arrhythmic burden, an
improvement paralleled by relief of her exertional chest discomfort. At
one year, she was asymptomatic on amiodarone, with no evidence of
structural heart disease on her echocardiogram.
4.4.2 The kissing bug
Chagas disease (CD), also known as American Trypanosomiasis, is a
parasitic disease caused by the protozoan Trypanosoma cruzi, endemic to
Central and South America, where 16-‐18 million people are infected and
about 100-‐120 million people are at risk of contracting the disease [61,
62].
T. Cruzi is commonly transmitted to humans by the blood-‐sucking
triatomine bug (also known as the kissing bug) [62]. The disease is
characterized by two phases: the acute infection, which occurs a few days
to some weeks after the transmission, which is usually asymptomatic [62,
63]. Sometimes patients may manifest acute symptoms such as fever,
inflammation at the inoculation site, unilateral palpebral edema (Romaña
sign), enlarged lymph nodes, and splenomegaly that resolve
spontaneously within 2-‐4 months. Rarely acute cardiac and neurological
involvement, such as severe myocarditis and meningo-‐encephalitis, may
also develop [64, 65].
In the absence of successful treatment, individual remains infected
for life: this is the beginning of the chronic phase. Individuals may remain
asymptomatic for a long period of time, usually decades, or even for their

4. Natural History and Predictors of Outcome
156
entire life, although representing infected reservoir. Following a period of
20-‐30 years, approximately 30-‐40% of subjects develop evidence of
cardiac involvement and 5-‐10% develop gastrointestinal signs associated
with megaesophagus and/or megacolon [62, 65].
4.2.3 Chagas cardiomyopathy
Clinical involvement in Chagas disease is characterized by
myocardial damage, with variable combination of LV dilatation and
dysfunction, potentially leading to heart failure [62, 66-‐68 ]. The
patological mechanism responsible for the adverse cardiac remodeling
and chamber dilatation is represented by chronic fibrosing myocarditis,
leading to progressive loss of cardiomyocites, muscle fibers and fascicles
breakdown, and increasing extent of replacement myocardial fibrosis.
Such profound morphological overturn is accompanied by functional
alteration with decreased systolic function over time and a high likelihood
of HF development [62, 67].
Furthermore, malfunctioning of the electrophysiological syncytia
are responsible for potentially lethal ventricular and supraventricular
tachy-‐ and bradyarrhythmias, which can explain the high risk of sudden
death in patients with CD cardiomyopathy [66].
From a physiopathological standpoint, the pathogenesis of Chagas
cardiomyopathy may result from a direct tissue damage by the parasite or
an indirect damage mediated by inflammatory and immune system
responses [69]. Although obvious triggers of reactivation may be absent,
conditions such as immunosuppression or chronic steroid treatment may
predispose to chronic CD complications.

4. Natural History and Predictors of Outcome
157
The clinical spectrum of CD cardiomyopathy may be very
heterogeneous, encompassing adverse LV remodeling with chamber
enlargement and systolic dysfunction or sub-‐epicardial fibrosis localized
to the apical region.
Characteristic electrocardiographic abnormalities include right
bundle-‐branch block (with or without associated left anterior hemiblock),
premature ventricular beats, second or third degree atrioventricular block,
sinus bradycardia and atrial fibrillation [68, 70]. Arrhythmias are also
frequent features of CD cardiomyopathy, and may be present in the
absence of detectable structural heart disease. Of note, both patients
underscore the considerable arrhythmic potential of Chagas
cardiomyopathy, emphasizing that CD should be considered in the
differential diagnosis of “idiopathic” ventricular arrhythmias. Sudden
cardiac death is common and may occur even in patients with normal
ECGs [70]. A risk score for death related to CD has been developed based
on 6 independent predictors, of which 3 relate to cardiovascular status,
including an abnormal echocardiogram, a small QRS on the ECG and runs
nonsustained VT on ambulatory Holter monitoring [68].
4.2.4 Management of Chagas cardiomyopathy
Two anti-‐trypansomal drugs have been introduced in the 1970s,
benznidazole and nifurtimox and, although evidence from randomized
control trials are lacking, benznidazole is currently considered first line
therapy [71, 72]. However, while the drug is considered highly effective
for eradication during the acute and congenital infection, the efficacy of
treatment during the chronic phase is highly debated, where only 40-‐60%
of patients achieving complete eradication [72]. Furthermore, treatment
is associated with considerable side effects, that more frequent and

4. Natural History and Predictors of Outcome
158
severe in adult than in children [74]. The most frequent side effects are
gastrointestinal disorder, skin rash and paresthesia.
Nevertheless, because persistence of Trypanosoma Cruzi in human
tissue is considered crucial for organ damage progression, antiparasitic
therapy is recommended for chronic CD, especially in patients with early
stages of cardiomyopathy [75].
Despite the lacking of evidence-‐based studies on Chagas
cardiomyopathy, the specific management relies on the management of
heart failure, arrhythmias and thromboembolism. Standard heart failure
therapy is employed for treatment of LV dysfunction and related
symptoms, including ACE-‐inhibitors or angiotensin receptor blockers,
betablockers, anti-‐aldosterone agents and loop diuretics [76, 77].
Considering the high risk of thromboembolism and the elevated incidence
of ischemic stroke, oral anticoagulation represent a possible important
therapeutic strategy, who should guided by standard clinical
recommendations.
Amiodarone is considered the agent of for treatment of ventricular
arrhythmias in these patients, due to its efficacy in reducing the
arrhythmic frequency, safety profile, and a reported direct antiparasitic
effect [78]. Indeed, preliminary data suggest that amiodarone treatment
may reduce mortality in CD patients. Selected high-‐risk patients with
Chagas cardiomyopathy may require an implantable cardioverter-‐
defibrillator, particularly in the presence of sustained ventricular
tachycardia, syncopal episodes or persistent LV dysfunction [79].

4. Natural History and Predictors of Outcome
159
4.2.5. Chagas disease: why should European cardiologists care?
Due to the increasing immigration, international travel and
adoption, Chagas disease now represent a matter of international
concern even for non-‐endemic countries, such as Europe or US.
According to the World Health Organization, 59.000 to 108.000
individuals in Europe are affected with CD. A recent serological survey for
T. cruzi infection offered to all migrants from Latin American living in the
North of Italy, has shown that 4% are infected. Bolivian and Brazil
migrants are at particularly high risk, with a prevalence of T. cruzi
infection as high as 19.8% [58, 59].
In non-‐endemic countries, potential mechanism of infection
include blood transfusions, transplacenteal transmission, solid organ
transplantation and ingestion food or liquid contamined with feces of
triatomines [62].
While screening programs of blood donors and pregnant women
coming from or exposed in endemic countries are being increasingly
promoted in non-‐endemic areas, the majority of cases are still
undiagnosed or probably misdiagnosed [60]. Of note, in Europe the index
of underdiagnosis of T. cruzi infection has been estimated between 94%
and 96%. The long latency period from infection to the clinical
manifestations, often ranging from 10 to 30 years, makes CD an
extremely challenging diagnosis in non-‐endemic countries, where lack of
awareness is a critical cause of delay or misdiagnosis. Furthermore,
because of easier access to health-‐care compared to endemic countries,
cardiac involvement in Europe and the US may be detected at the initial
stages, when clinical manifestations of CD are non-‐specific and more
difficult to interpret.

4. Natural History and Predictors of Outcome
160
4.2.6 Conclusions
Despite its growing epidemiological relevance in the western world,
and considerable emphasis by the World Health Organization, awareness
of CD among physicians in Europe remains low, promoting neglect and
misdiagnosis. It is hoped that CD may increasingly be considered as a
potential diagnosis in European residents from endemic Latin American
regions presenting with “idiopathic” cardiac manifestations, even when
long removed from their country of origin, with potential implications for
implementation of early treatment and control of CD transmission.

5. Translational Routes from
Altered Molecular Homeostasis
to Treatment Opportunities

5. Treatment Opportunities
162
“The art of medicine consists
in amusing the patient
while nature cures the disease”
Voltaire

5. Treatment Opportunities
163
Despite the modern recognition cardiomyopathies as distinct
clinical entities for over five decades, and in spite of considerable
advances in its phenotyping and molecular genetics, current
pharmacological treatment of these patients has remained largely
unchanged and empiric. In conjunction with the implementation of a
DNA-‐based diagnosis and the comprehension of molecular phenotype,
pharmacological treatment of cardiomyopathies is also likely to evolve
and become largely individualized.
The overview of molecular, genetic and clinical features of
cardiomyopathy reviewed in the previous sections gives the opportunity
to describe the possibility to translate the acquired knowledge into
treatment opportunities.
The present section illustrates novel therapeutic approaches that
are being developed for patients with hypertrophic cardiomyopathy and
Anderson-‐Fabry disease, and a critical reappraisal of a well-‐established –
but not perfect – preventive option such as the implantable cardioverter
defibrillator.

5. Treatment Opportunities
164
5.1 Electrophysiological cardiomyocyte remodeling as a
new therapeutic target in HCM
Hypertrophic cardiomyopathy (HCM) is the most prevalent
monogenic cardiac disorder, with a reported prevalence of 1 in 500
worldwide [1]. Despite its epidemiological relevance, HCM is largely an
orphan condition because it lacks a disease-‐specific pharmacological
treatment. HCM is frequently associated with reduced exercise capacity
and symptoms such as angina and syncope [1].
The pathophysiology of symptoms and disease progression in HCM
is complex and only incompletely understood [2-‐4]. However, several
important elements have been identified which are closely interwoven,
and impact decisively on the genesis of symptoms and long-‐term clinical
course: A) diastolic dysfunction, multifactorial in origin, with causes
ranging from intracellular calcium overload to increased interstitial
fibrosis [1-‐4]; B) dynamic LV outflow obstruction, whether in resting
conditions or during exercise [5]; C) microvascular dysfunction and
blunted coronary flow reserve, secondary to marked anatomic
remodelling of the arterioles [6, 7]; D) arrhythmias, with particular regard
to atrial fibrillation and its consequences [8]. Moreover, HCM represents
the most common cause of arrhythmic sudden cardiac death in young
athletes, because of the increased propensity for potentially life-‐
threatening ventricular arrhythmias [1]. HCM is associated with a complex
electrophysiological cardiomyocyte remodelling involving multiple
changes in transmembrane currents ultimately leading to susceptibility to
ventricular arrhythmias; particularly, in animal models of ventricular
hypertrophy, ventricular myocytes show functional down regulation of
potassium currents and up-‐regulation of inward currents (Calcium

5. Treatment Opportunities
165
currents, Chloride currents, others), all leading to action potential
prolongation. Prolonged action potential can lead to increased duration of
QT interval at the surface electrocardiogram, a feature that has been
found associated with HCM and secondary hypertrophy in patients [9-‐
12].Marked prolongation of the QT interval is a pro-‐arrhythmic risk factor
in both congenital and drug-‐induced long QT syndrome (LQTS) [11-‐12].
Developed in the 90s as a cardiac specific partial fatty acid
oxidation inhibitor, Ranolazine was first experimented as a possible
antianginal compound due to its ability to shift cardiac metabolism from
fatty acid to glucose consumption, that requires less O2 to produce ATP
[13]: this metabolic modulatory effect of ranolazine was thought to be
responsible for its clear protective effect on myocardial tissue subjected
to ischemia/reperfusion damage. It becomes clear that the antianginal
and antiischemic effect of ranolazione were not due to the supposed
metabolic effects (which are negligible at therapeutic concentrations), but
rather to its selective blockade of the late sodium current. Its
electrophysiological effects were in fact described in 2004 [14].
Ranolazine exerts an inhibitory effect on IKr and a slight ICaL block but the
main effect was a potent inhibition of INaL ,with small or no effect on
peak INa, within the therapeutic concentration range. Inhibition of IKr is
generally considered pro-‐arrhythmic: however, in a large scale trial on non
ST-‐elevation myocardial infarction patients, Ranolazine significantly
reduced the incidence of ventricular tachycardia and atrial arrhythmias
after the first event [15]. The late sodium current is enhanced under
several pathological conditions such as ischemia, heart failure, cardiac
hypertrophy and various conditions associated with oxidative stress. An
increase in INaL causes Na+ overload, which in turn, promotes an
increased exchange of intracellular Na+ for extracellular Ca2+ through the
reverse mode of the NCX. In its reverse mode, the NCX increases Ca2+

5. Treatment Opportunities
166
entry into the cell in exchange to the extrusion of 3 Na+ per cycle,
reducing the overall cellular capacity to eliminate calcium outside of the
cytosol, and thus causing Ca 2+ overload. The antianginal effect observed
at therapeutic concentrations of ranolazine (≤ 10 µM) has been related to
its ability to inhibit the late INa and, as a consequence, the increase in
intracellular Na+ and Ca2+ overload in myocardial cells [16].
Recently, a paper published by our group described the
electrophysiological profile, Ca2+i handling properties, and contractile
function of isolated cardiomyocytes and trabeculae from HCM patients
undergoing surgical myectomy []. Among the several ion channel and
Ca2+i handling proteins changes identified, an enhanced INaL seems to be
a major contributor to the electrophysiological and Ca2+i dynamic
abnormalities of ventricular myocytes and trabeculae from patients with
HCM. These data suggest a favourable electrophysiological effect of
ranolazine on HCM cardiomyocytes, by reverting the abnormal
prolongation of action potential observed [Figure 5.1-‐1].
Figure 5.1-‐1: Effects of INaL inhibition by ranolazine on action potentials. Action potentials at 0.2 Hz from a hypertrophic cardiomyopathy (HCM) cardiomyocyte before (basal) and after exposure to 10 μmol/L ranolazine (Ran).

5. Treatment Opportunities
167
In HCM patients, ranolazine represents a promising option,
because of its pharmacological profile, comprising several different effects
of potential benefit to their condition. By reducing intracellular Na+
overload, and thus diastolic Ca2+ ranolazine has the potential to improve
diastolic function, a cause of symptoms in most HCM patients, thus
impacting on functional limitation and quality of life. In the long term, the
effect on calcium homeostasis is expected to down regulate a number of
remodelling pathways that strictly depend on myocyte driven signaling:
this may in turn bring benefit to extracellular matrix remodelling, reducing
fibroblasts growth and collagen production in hypertrophied myocardium.
Second, ranolazine administration has been shown to reduce the degree
of myocyte hypertrophy and interstitial fibrosis, and to ultimately lower
the propensity to arrhythmias [18]. Third, ranolazine has been shown
improve myocardial perfusion indirectly, by normalizing capillary density
in the ventricles, as well as, in patients with stable CAD, via a direct action
on the microvasculature leading to improved endothelial function [19].
Thus, treatment with ranolazine may be capable of alleviating angina in
HCM patients, and, most importantly, to prevent disease progression
towards the end-‐stage phase, by acting upon one of its most critical
determinants -‐ microvascular ischemia. In principle, ranolazine has an
ideal profile for clinical use in HCM patients, suggesting a wide range of
positive actions which may critically impact on acute symptoms as well as
on the natural history of the disease. Such a strong rationale calls for a
concerted action aimed at investigated the efficacy of ranolazine in this
not-‐so-‐uncommon disease, which is still orphan of appropriate
pharmacological treatment.
In the light of this view, a pilot study has been designed in order to
test the effectiveness of ranolazine on exercise tolerance and symptoms
in HCM patients. The present study is intended to represent the first

5. Treatment Opportunities
168
meaningful step in such direction, by assessing the clinical and
instrumental effects of a 5-‐month treatment with ranolazine in a selected
cohort of HCM patients.
The protocol of this randomized, Double blind, Placebo controlled,
study is here presented: a total of 120 patients with symptomatic HCM
(SHCM) will be recruited in 11 European referral centers and randomized
to receive ranolazine or placebo for 5 months. During the 5 months
treatment phase, the randomized patients will perform 3 visits, for safety
and efficacy assessments [Figure 5.1-‐2].
Figure 5.1-‐2: study design.
Visit 1 to visit 2: up-‐titration of drug dose.
Visit 4 to Visit 6: treatment period
Study name and code:
RESTYLE-‐HCM: RANOLAZINE IN PATIENTS WITH SYMPTOMATIC HYPERTROPHIC CARDIOMYOPATHY: A PILOT STUDY ASSESSING THE EFFECTS ON EXERCISE CAPACITY, DIASTOLIC FUNCTION AND SYMPTOMATIC STATUS
Code: MEIN/11/RAN-‐HCM/001 (EUDRA-‐CT number: 2011-‐004507-‐20)

5. Treatment Opportunities
169
Improvement in exercise capacity, as assessed by cardiopulmonary
test with the Peak Oxygen Consumption (V02 peak) technique, will be the
primary endpoint.
Secondary objectives will be to demonstrate the efficacy of
ranolazine on diastolic function, symptomatic status and natriuretic
peptide biomarker proBNP in patients affected by SHCM. The assessment
of safety by evaluation of adverse events, laboratory findings, the rest
standard 12-‐lead ECG, detection of 24-‐hour arrhythmic burden by Holter
ECG monitoring and physical examination will also be considered a
secondary study objectives.
The ongoing study is still recruiting patients in Europe, with the
leading center of Florence with the highest number of enrolled individuals.
Despite the idea of using established drug therapies in an “old” disease
such HCM, the real purpose of the study is to take a step toward the
world of evidence-‐based medicine.
The development of new experimental drugs, such new sodium
channel blocker, more specific than ranolazine, is ongoing. Interest of
pharmaceutical company in this fascinating group of disease is growing
faster and the possibility to intervene on the complex pathophysiology of
the disease in order to alter its natural course is close at hand.

5. Treatment Opportunities
170
5.2 The real story of life-‐saving devices: a backstage tour
Hypertrophic cardiomyopathy (HCM) is the most common genetic
cardiac disease, with mutations in genes coding for myofilament
contractile proteins of the cardiac sarcomere representing the most
common genetic substrate. In most cohort studies the prevalence of
genotype positive patients is as high as 65%, representing over two third
of the total [20]. To date, large clinical studies on HCM have assessed
outcome irrespective of genetic background. Although clinically
indistinguishable, this genotype-‐negative subset is likely to include a
number of rare etiologies, including phenocopies, like infiltrative disease,
whose natural history might differ significantly from sarcomere-‐related
HCM [20, 21].
Therefore, the large proportion of patients with no detectable
sarcomere myofilament gene mutations may confound the natural history
of HCM with proven sarcomere etiology. Furthermore, such concept is
supported by the finding that genotype-‐positive patients have more
severe outcome compared to those who are genotype-‐negative [20]. Thus,
data deriving from non-‐genotyped populations may significantly confound
our perception of the clinical correlates and outcome associated with a
specific genetic background, and large studies on genotyped HCM
populations appear desirable [22].
In order to address this important issue, we sought to investigate
the clinical features and outcome of a large cohort consisting solely of
patients with proven myofilament-‐positive HCM, followed prospectively
at our Institution after systematic genetic screening. In addition, the
impact of the implantable cardioverter-‐defibrillator (ICD), both in terms of

5. Treatment Opportunities
171
appropriate intervention rates and adverse effects, were specifically
assessed.
5.2.1 Clinical picture of the study cohort
We followed for 6±3 years 250 HCM patients (age 46±17 years,
36% female) consecutively found positive at mutational screening in the
protein-‐coding exons and splice sites of 8 sarcomere gene, including
myosin binding protein C (MYBPC3), beta-‐myosin heavy chain [MYH7],
regulatory and essential light chains [MYL2 and MYL3], troponin-‐T
[TNNT2], troponin-‐I [TNNI3], alpha-‐tropomyosin [TPM1], and alpha-‐actin
[ACTC]).
The distribution of affected genes in this population was consistent
with most reported cohorts internationally, with a large majority carrying
single mutations in either of MYBPC3 and MHY7 gene [Figure 5.2-‐1]. Only
20 patients (8%) had complex genotypes characterized by two or more
mutations in the same or in different sarcomere genes.
Figure 5.2-‐21: Distribution of sarcomere mutation in the study cohort
MYBPC3 (48%), MHY7 (28%), TNNT2 (4%), or the other genes (12%);
complex genotype (8%).

5. Treatment Opportunities
172
Overall, 64 patients (26%) received an ICD, for secondary (n=10) or
primary prevention of SCD (n=54). Among the latter, the ICD was
implanted because of the high-‐risk profile (n=37) or progression to overt
LV systolic dysfunction (LVEF<50%; n=17, including 10 with CRT
capabilities) [Figure 5.2-‐2]. Three clinical scenarios led to the
recommendation of ICD implantation in individual our HCM patients: 1)
prior cardiac arrest or sustained ventricular tachycardia (secondary
prevention); 2) primary prophylaxis of SCD in patients with ≥2 of the
established risk factors indicated by current guidelines, i.e. malignant
family history, extreme LV wall thickness, unexplained syncope, abnormal
blood pressure response to exercise and repeated runs on nonsustained
ventricular tachycardia (NSVT) on 24-‐hour ECG monitoring; 3) progression
towards systolic dysfunction, the so-‐called “end-‐stage” phase, per se
associated with high SCD rated, in whom cardiac resynchronization was
routinely considered at the time of ICD implantation.
Figure 5.2-‐2: Patients with ICD
64 patients had an ICD implanted for primary or secondary prevention
(respectively 54 and 10).

5. Treatment Opportunities
173
5.2.2. Outcome
At final evaluation, the incidence of death from cardiovascular
causes was quite low, with an overall 6% and an annual incidence of 1%.
Indeed, the most frequent cause of death was related to progressive heart
failure, while SCD appeared to be less frequent.
The comparison of subgroups of patients, based on affected gene
was not statistically significant, although complex genotypes showed
more severe clinical course and adverse outcome. However, while the
incidence of SCD and appropriate ICD discharge were comparable to
previously reported unselected HCM cohorts, progression to overt systolic
dysfunction was more common in our cohort of genotype-‐positive
patients. Thus, the significant propensity towards LV systolic dysfunction
and HF, with a prevalence of 10 and 14% respectively, is consistent with
the hypothesis of long-‐term energy depletion due to inefficient energetic
handling by the mutated sarcomere. Indeed, this concept is accentuated
in patients with multiple genetic defects, representing extreme paradigms
of deranged sarcomere function.
Compared to patients without ICD, those who received a device
were younger at HCM diagnosis, more symptomatic, had greater
maximum LV wall thickness and left atrial dimensions, and, as expected, a
greater number of risk factors for SCD. Thus, patients with ICD had an
unfavorable baseline risk profile, with a greater prevalence of end-‐stage
disease, which is known to be associated with a particularly ominous
outcome. Despite that, the incidence of cardiovascular mortality was only
slightly higher, but not statistically significant, from that observed in
patients without ICD [Figure 5.2-‐3]. Of note, two of the deaths in the ICD
group were sudden, occurring despite the device: unfortunately, neither

5. Treatment Opportunities
174
an autopsy nor an ICD interrogation was performed in these patients, and
the exact causes of death could not be ascertained.
Figure 5.2-‐3: Survival benefit attributable to the ICD
ICD allowed favorable (or normalize) survival rates in a subset of HCM patients.
5.2.2. ICD: the good, the bad and the ugly
Implantable cardioverter defibrillators have now been in clinical
practice for 25 years. Despite great skepticism and opposition in the early
years, the weight of data from multiple prospective randomized clinical
trials has proved beyond any doubt that ICD therapy is highly effective in
reducing sudden cardiac death in patients with a great variety of cardiac
pathologies. Faced with ‘evidence-‐based medicine’, HCM guidelines have
become increasingly clear, and we all now recognize that there are
patients for whom an ICD is highly recommended. Furthermore, multiple
trials of ICD treatment compared with drugs showed that in specific
clinical setting, ICD therapy might be superior to drugs.

5. Treatment Opportunities
175
Overall, 11% of patients experienced an appropriate ICD
intervention (shocks for VT of VF), with an annual incidence of 2%.
Therefore, ICD allowed favorable survival rates in a subset of high-‐risk
HCM patients [Figure 5.2-‐3].
However, 16 patients (25% of the ICD subset, including 2 with
appropriate shocks) experienced device-‐related complications such as
inappropriate ICD interventions (n=10; including 4 with electric storms
due to lead fracture), infections (n=4) and lead dislocation (n=6) [Figure
5.2-‐4]. As a consequence of multiple inappropriate shock and
psychological distress, two young patients required their ICD to be
explanted, 5 and 7 years after implantation; both are still alive. Finally, 31
patients required one or more substitutions of the device at end of
battery life.
Figure 5.2-‐4: ICD-‐related complications
Overall, 16% of patients with ICD experienced device-‐related complication, while only 11% had appropriate device intervention. The rate of
complication increase over time and become significant after 3 year from implantation.

5. Treatment Opportunities
176
Therefore, the decision to implant an ICD prophylactically for SD
prevention in HCM patients involve consideration of the potential
complications and inconvenience incurred by a permanent device versus
obvious lifesaving benefit [23, 24]. Particularly, these considerations
should arise in pediatrics patients when physicians are confronted by the
clinical paradox in which active and healthy-‐appearing HCM patients
(exposed to greatest SD risk by age) have the highest device complication
rates over long time periods [25]. Device-‐related complications, including
infection, pocket hematoma, pneumothorax, and venous thrombosis, are
well documented and occur most commonly in younger patients,
primarily because their activity level and body growth place continual
strain on leads, considered the weakest link in this system [25-‐27].
Although controversial, there is evidence that defibrillator shocks
can cause myocardial damage [28, 29], and the shocks have been
associated with increased mortality [30]. Recent findings from one large
randomized trial showed that different programming approach may
reduce potentially dangerous inappropriate therapies and increased
survival among patients with ICDs [31]. Furthermore, device technologies
move on at a fast rate and new alternative to the classic intravenous ICD
are currently available, although with limited clinical experience especially
in patients with cardiomyopathy [32].

5. Treatment Opportunities
177
5.2.3 Conclusions
In our large cohort of HCM patients with proven sarcomere
myofilament disease, risk of sudden cardiac death or appropriate ICD
shock was low even in the presence of multiple risk factors. Conversely,
end-‐stage progression proved relatively common, supporting the
hypothesis of long-‐term myocyte energy depletion related to mutations in
sarcomeric genes.
The ICD allowed favorable survival rates in a subset of HCM
patients at higher risk, at the cost of considerable complication rates, and
sudden cardiac death occasionally occurred despite the device.

5. Treatment Opportunities
178
5.4 Changing the destiny of unfolded proteins:
pharmacological chaperone as a new therapy for
Anderson-‐Fabry disease
Fabry disease is a hereditary, X-‐linked lysosomal storage disorder
caused by a deficiency of the lysosomal enzyme α-‐galactosidase A, which
results in the accumulation of the glycosphingolipid globotriaosylceramide
(Gb3), in different cells and organs, especially in endothelial cells and
smooth muscle cells of blood vessel [33, 34].
To date, more than 500 mutations have been reported for the GLA
gene, 57% of which are missense mutations. Patients with missense
mutations often have some residual enzyme activity, ranging from 2% to
25% [33]. Studies of the residual GLA activity of mutant forms of the
enzyme revealed that many had kinetic properties similar to the wildtype
enzyme, but were significantly less stable [35-‐37]. These results suggest
that the compromised enzyme activity in many Fabry patients is due to
protein misfolding and the inability to traffic the enzyme to the lysosomes,
where they are needed to break down substrate [36]. Misfolded enzymes
may be stopped by the ER quality control system and the mutant protein
is retained in the ER and degraded prematurely [Figure 5.4-‐1]. Since most
misfolded enzymes are never sent to the lysosome, substrate may
accumulate within the lysosome, damage the cell, and cause the signs and
symptoms of a lysosomal storage disorder [36]. Therefore, the discovery
of small molecules that assist mutant enzymes to fold correctly may
rescue them from premature degradation and increase the amount of
active enzyme in the lysosomes.

5. Treatment Opportunities
179
Figure 5.4-‐1: A simplified schematics of the molecular chaperones rescue
mechanism, sorting proteins for folding or degradation
ERT is presently considered the standard of care for LSDs [38]. It
has considerable success in the treatment or in ameliorating the quality of
life of patients with several of these diseases. However, ERT has
significant limitations in terms of tissue distribution of therapeutic
enzymes, impact on patients’ quality of life and high costs []. ERT is based
on the intravenous infusion of recombinant or gene-‐activated human
enzyme. The enzyme is delivered into the blood in order to reach specific
receptor cell surfaces, to be taken up by cells and then transported to the
lysosome. Upon entering the lysosome, this enzyme can perform the
function of the unstable endogenous enzyme. However, the pH in the
infusion bag and in blood is higher than the enzyme’s natural acidic

5. Treatment Opportunities
180
environment in the lysosome. In result, the infused enzyme rapidly
denatures and may be misdirected to non-‐target tissues or delivered in
inactive form to the lysosome. Exposure to these infused enzymes may
also mount an immune response or neutralizing antibodies than can
impact efficacy or cause adverse effects. Possible problems related to the
unfolding of infused enzyme include: rapid denaturation and misfolding in
blood, leading to short circulating half-‐life, immunogenicity, poor delivery
and uptake of active enzyme into key tissues of disease and reduced
activity.
Pharmacological chaperone therapy (PC) has recently emerged as a
potential therapeutic alternative for Fabry disease and appears
advantageous when compared to ERT, as the chaperones are better
distributed in tissues, including the brain, and because therapy may be
administered orally [36, 37, 40]. PCT, however, also has limitations, since
only patients with responsive mutations will be amenable to this therapy
[40]. Pharmacological chaperones are small molecules that are designed
to selectively bind to a misfolded protein thereby increase the protein’s
stability, assisting their correct folding, maturation, and trafficking to their
functional site, such as the lysosome [Figure 5.4-‐1 and 5.4-‐2]. Once in the
lysosome, the pharmacological chaperone disassociates and the enzyme is
free to break down substrate []. By stabilizing a misfolded protein,
pharmacological chaperones may be able to restore the intended
biological function of the protein. Because each chaperone is designed to
bind to only one particular lysosomal enzyme, a specific chaperone is
developed for each targeted lysosomal disease []. PC are reversible
enzyme inhibitors and bind to the active enzyme site, thus stabilizing, but
also inhibiting, the enzyme. To be effective, PC must be able to dissociate
from the enzyme once they have reached the lysosome, a process which
is promoted by the acidic environment of lysosomes and excess of natural

5. Treatment Opportunities
181
substrate, with a higher affinity to the catalytic sites []. Therefore, the
concentration of chaperone in the ER and in the lysosome is a critical
factor. If the amount of chaperone in the ER is low, they are ineffective in
assisting the enzyme folding, thus resulting in a reduced enzyme
concentration (and activity) in the lysosome. However, the dissociation of
the PC from the active site of the enzyme is impossible if the
concentration of chaperone in the lysosome is too high. In that case, the
PC occupies the active site of the enzyme and the enzyme activity is
therefore reduced. In theory, an enzyme activator or a pure enzyme
binder with chaperone activity would be a better choice as a candidate for
drug development. If an enzyme activator is used as a therapy, both the
chaperone and enzyme stimulatory actions of the activator would
synergistically increase enzyme activity in the lysosomes.
Figure 5.4-‐2: Proposed Mechanism of Enzyme Stabilization by Chemical Chaperones.

5. Treatment Opportunities
182
Panel A shows the processing of normal a-‐galactosidase A. Newly synthesized a-‐galactosidase A is translocated into the endoplasmic reticulum, where molecular chaperones facilitate its proper folding and dimerization by specialized processing enzymes. The molecular chaperones then dissociate from the folded, dimerized enzyme, which moves to the Golgi apparatus and then to lysosomes, where the enzyme is stable and active in the acidic environment of these organelles. Panel B shows the processing of mutant a-‐galactosidase A. Most mutations in the α-‐galactosidase A gene encode α-‐galactosidase A molecules that are misfolded, misassembled, or aggregated in the endoplasmic reticulum, where they are degraded, presumably by the ubiquitin–proteasome pathway. However, certain missense mutations decrease the stability of the enzyme, but the conformation of the active site is retained. Most of this type of mutant enzyme is degraded in the endoplasmic reticulum. However, these mutant forms of α-‐galactosidase A may be stabilized by chemical chaperones, such as galactose, that bind to the active site of the enzyme, promote folding, and stabilize the mutant enzyme. Some of the enzyme then reaches the lysosomes, where it retains low levels of activity. In the lysosomes, the accumulated glycosphingolipid substrates displace the chemical chaperones and are hydrolyzed by the enzyme [Modified from 44].
Recent studies suggest that new solutions to improve the efficacy
of treatments for LSDs may lie on the combination of distinct therapies
[45]. The combination of ERT and PCT resulted in a synergistic effect in
Pompe and Fabry disease models, and may be helpful in patients
responding poorly to ERT and in tissues where corrective levels of
recombinant enzymes are difficult to obtain. These studies greatly expand
the use of pharmacological chaperones and suggest a change in the use of
these drugs. One GLA inhibitor, 1-‐ deoxygalactonojirimycin (DGJ,
marketed as AmigalTM by Amicus Therapeutics, Inc. [46]) is currently
being studied, as a chaperone therapeutic agent for Fabry disease, in
phase 2 and phase 3 clinical trials:

5. Treatment Opportunities
183
1) Migalastat HCl Monotherapy: Phase 3 Study 011 (The FACETS, or FAB-‐
AT1001-‐011 Study): Study of the Effects of Oral AT1001 (Migalastat
Hydrochloride) in Patients with Fabry Disease. Design: Placebo-‐controlled,
double-‐blind Phase 3 study of migalastat HCl
2) Migalastat HCl Monotherapy: Phase 3 Study 012 (The ATTRACT, or
FAB-‐AT1001-‐012 Study): Study to Compare the Efficacy and Safety of Oral
AT1001 and Enzyme Replacement Therapy in Patients with Fabry Disease.
Design: Randomized, open-‐label, 18-‐month Phase 3 study investigating
the safety and efficacy of migalastat HCI compared to current standard-‐of-‐
care ERTs Fabrazyme® (agalsidase beta) or Replagal® (agalsidase alfa) for
Fabry disease
3) Migalastat HCl Monotherapy: Phase 2 Extension Study 205 (FAB-‐CL-‐
205 Study): Open Label Long-‐term Safety Study of AT1001 in Patients with
Fabry Disease Who Have Completed a Previous AT1001 Study. Design:
Open-‐label Phase 2 extension study to evaluate the long-‐term safety and
tolerability and to explore the efficacy of migalastat HCl in patients who
have previously completed a Phase 2 clinical study of migalastat HCl for
Fabry disease.
4) Migalastat HCl Co-‐Administered with ERT: Phase 2 Study 013 (AT1001-‐
013 Study): Drug-‐Drug Interaction Study Between AT1001 and Agalsidase
in Subjects With Fabry Disease. Design: Open-‐label Phase 2 study to
compare a single administration of oral migalastat HCl co-‐administered
with infused ERT (Fabrazyme or Replagal) versus ERT alone. Each patient
receives their current dose and regimen of ERT alone at one infusion and
oral migalastat HCl (150 mg or 450 mg) administered prior to ERT at their
next infusion

5. Treatment Opportunities
184
5.2.1 The ATTRACT study
ATTRACT is an acronyms for “AT1001 therapy compared to enzyme
replacement therapy in Fabry patients with AT1001-‐responsive mutations:
a global clinical trial”. The ATTRACT study is a phase 3 clinical trial that
aims to measure the effectiveness and safety of an investigational
pharmacological chaperone migalastat HCl (AT1001), when compared to
enzyme replacement therapy (ERT) alone, for the treatment of patients
with Fabry disease.
The study enrolled approximately 50 participants currently
receiving agalsidase (ERT), who have been randomized to receive either
migalastat HCl or agalsidase (ERT). Approximately 30 participants stopped
their agalsidase (ERT) regimen and started with migalastat HCl and 20
participants remained on their prescribed agalsidase (ERT) regimen. The
ATTRACT study was fully enrolled on December 4th, 2012.
Study participation will last approximately 21 months, including a
screening/baseline period (2 months), an open-‐label treatment period (18
months), and a follow-‐up period (1 month). After completing the 18
month treatment period, participants will have the option to continue in a
12-‐month treatment extension, where all participants will be treated with
Migalastat.
The primary end-‐point of the study is the assessment of renal
function by measuring the glomerular filtration rate (GFR) as assessed by
plasma clearance of iohexol (“iohexol GFR”). Iohexol is a non-‐ionic x-‐ray
contrast medium of low osmolality, extensively used in clinical radiology
and considered essentially free from side effects. Like other iodine-‐
containing contrast media, it is eliminated from the body by excretion in
the urine. These substances are therefore potential markers for renal
function. Iohexol is a suitable marker for glomerular filtration rate (GFR):

5. Treatment Opportunities
185
after intravenous injection, it is quantitatively recovered in the urine; the
elimination occurs by glomerular filtration, with no signs of tubular
secretion or reabsorbtion.
The secondary end-‐points are more specifically focused on those
signs expression of major organ involvement, such as 24-‐hour urine
protein for kidney disease, cardiac morphology and function assessed by
echocardiography, burden of pain and quality of life reported by the
patients (standard questionnaire). Furthermore, a composite clinical
outcome is also assessed by the occurrence of renal, cardiac,
cerebrovascular events or death.
Our center is actively involved in the ATTRACT study. Two female
patients were enrolled on July 2012 and both randomized to receive
Migalastat and stop ERT. So far, no serious adverse events have been
reported in our patients.
5.2.2 Next-‐Generation Approach for Lysosomal Storage Diseases (LSDs)
A substantial amount of novel and promising data support the
potential of pharmacological chaperones as therapeutic agents in Fabry
disease [36]. Future efforts should be directed towards the identification
of new chaperones for additional LSDs and the identification of ‘second-‐
generation’ molecules with a good safety profile and better enhancing
properties and intracellular distribution [42]. An ideal compound should
have weaker inhibitory effects and higher enhancing activity. Novel
chaperone molecules should not only be advantageous in terms of
improved enhancing profile, but may also increase the rate of responsive
mutations by assisting the folding of enzymes with mutations in domains
other than the catalytic site.

5. Treatment Opportunities
186
Given the limitations of all therapeutic approaches so far, looking
for new strategies to optimize therapies and to target the diverse cellular
and phenotypic aspects of these diseases is becoming an important goal
of current research. In this respect, a leap forward was made, thanks to
recent studies that indicated new solutions to improve the efficacy of
treatments for LSDs and opened new avenues to the use of PCT. Although
the concept of PCT is based on the enhancing effect of chaperones on
mutated misfolded proteins, some evidence points to a possible effect of
chaperones on wild-‐type enzymes [45, 40]. Chaperones induce
conformational stabilization and increase thermal stability of normal
recombinant enzymes used for ERT, which may be prone to mistrafficking
and degradation like the mutant enzymes. Some studies suggested that
protection of recombinant enzymes from degradation can be achieved by
chaperones [47].
The CHART™ (Chaperone-‐Advanced Replacement Therapy)
platform combines unique pharmacological chaperones with enzyme
replacement therapies (ERTs) for LSDs. Amicus is leveraging the CHART
platform to improve currently marketed ERTs through co-‐administration
of a pharmacological chaperone prior to ERT infusion, and to develop
next-‐generation ERTs that consist of a proprietary lysosomal enzyme
therapy co-‐formulated with a pharmacological chaperone. This platform
has demonstrated proof-‐of-‐concept in the clinic and in multiple preclinical
studies. In the clinic, consistent increases in active enzyme in plasma and
enzyme uptake into tissue have been observed following chaperone-‐ERT
co-‐administration compared to ERT alone. In preclinical studies, this
increased activity and tissue uptake has lead to greater substrate
reduction than ERT alone. In each CHART program, a unique
pharmacological chaperone is designed to bind to a specific therapeutic
(exogenous) enzyme, stabilizing the enzyme in its properly folded and

5. Treatment Opportunities
187
active form. This may allow for enhanced tissue uptake, greater lysosomal
activity, more reduction of substrate, and lower immunogenicity
compared to standard of care ERTs. This combination approach may
benefit patients with lysosomal storage diseases, including patients with
inactive endogenous proteins who are not amenable to chaperone
monotherapy. Therefore, such coadministration may provide an improved
therapeutic strategy that is broadly applicable to a diverse population of
Fabry patients.

5. Treatment Opportunities
188

6. Conclusive Remarks

6. Conclusive remarks
190
"An investment in knowledge
pays the best interest."
Benjamin Franklin

6. Conclusive remarks
191
Conclusive remarks
New hopes
After more than fifty years of passionate clinical and molecular
description of cardiomyopathy, the enthusiasm for these fascinating
heart diseases has reached a record high level. Now, the time is ripe to
consider what we know and reflect on what remains to be discovered in
order to fill the existent gaps.
The interest of the scientific world and pharmaceutical companies
is slowly emerging, giving to physicians and researchers involved in the
fields a remarkable opportunity to translate basic insights into new
clinical models for diagnosis, prevention, and therapy. Novel initiatives,
supported by private companies and international medical societies, are
paradigmatic examples of the increased awareness and subsequent
interest in this group of diseases.
The EURObservational Research Programme (EORP) was launched
by the European Society of Cardiology in 2009, with the aim to provide a
better understanding of medical practice, based on observational data
collected with more robust methodological procedures. Three years later,
a special EORP Registry on Cardiomyopathies was added to the list, with
the intent to collect previously unobtainable data on the epidemiology
and outcomes of patients seen across referral centers in Europe. The ESC
Cardiomyopathy Registry is a prospective, multicenter, observational
study that describes the demographic, clinical and genetic characteristics
of patients with cardiomyopathies. The study was conducted as a pilot for
the first year in a small number of expert centers, with 1149 patients
enrolled in the first twelve months, and the long-‐term registry will start in
2014.

6. Conclusive remarks
192
The call for action, emphasized also in an official report of
international working group [1], lighted up the enthusiasm for
cardiomyopathies, making them a group of diseases particularly
appealing for investments of private and public companies, and the
number of clinical trials, as well as the development of new specific
therapeutic strategies, will hopefully increase in the immediate and near
future.
In conclusion, cardiomyopathies are not uncommon, but remain
orphan diseases with regard to treatment. In the era of translational
research, physicians and researchers have been very good at asking the
right questions, but they need to direct more effort into finding the best
clinical answers for these patients. The changing epidemiology of these
diseases now allows what was previously impossible: it is high time to
move cardiomyopathies into the era of evidence-‐based management.
In a broader perspective, cardiomyopathies should be seen as
paradigms providing invaluable insights on disease mechanisms that may
be of general relevance to patients with more prevalent cardiac
conditions. What has long been considered impossible to achieve is now
closer at hand: the time is now ripe to promote robust clinical research in
these complex conditions, in order to advance and standardize
management of patients.

7. References

7. References
194
Chapter 1: Introduction
1) Brigden W. Uncommon myocardial diseases: the non-‐coronary cardiomyopathies. Lancet. 1957 Dec. 21; 273:7008, 1243-‐1249.
2) Goodwin JF, Gordon H, Hollman A and Bishop MB. Clinical aspects of cardiomyopathy. Br Med J. 1961 Jan.14; 1:5219, 69-‐79.
3) [No authors listed]. Report of the WHO/ISFC Task Force on the definition and classification of cardiomyopathies. Br Heart J. 1980; 44, 672-‐673.
4) Watkins H, Ashrafian H and Redwood C. Inherited cardiomyopathies. N Engl J Med. 2011 Apr. 28; 364:17, 1643-‐1656.
5) Bos JM, Towbin JA and Ackerman MJ. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009 July 14; 54:3, 201-‐211.
6) Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O'Connell J, Olsen E, Thiene G and Goodwin J. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation. 1996; 93:841-‐842.
7) Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman C and Young JB. Contemporary definitions and classification of the cardiomyopathies. An American Heart Association scientific statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006; 113:1807-‐1816.
8) Elliott P, Anderson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kuhl U, Maisch B, McKenna WJ, Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L and Keren A. Classification of cardiomyopathies: a position statement from the European working group on myocardial and pericardial diseases. Eur Heart J. 2008; 29:270-‐276.
9) Rapezzi C, Quarta CC, Riva L, Longhi S, Gallelli I, Lorenzini M, Ciliberti P, Biagini E, Salvi F and Branzi A. Transthyretin-‐related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol. 2010 July; 7:7, 398-‐408.
10) Gersh BJ, Maron BJ, Bonow RO, Dearani A, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE and Yancy CW. ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011; 124:2761-‐2796.

7. References
195
11) Maron MS, Olivotto I and Zenovich AG et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation. 2006; 114:21, 2232-‐2239.
12) Maron BJ. Contemporary insights and strategies for risk stratification and prevention of sudden death in hypertrophic cardiomyopathy. Circulation. 2010; 121:445-‐456.
13) Girolami F, Ho CY, Semsarian C, Baldi M, Will ML, Baldini K, Torricelli F, Yeates L, Cecchi F, Ackerman MJ and Olivotto I. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J Am Coll Cardiol. 2010 Apr 6; 55:14, 1444-‐1453.
14) Schapira AH. Mitochondrial diseases. Lancet. 2012 May 12; 379(9828):1825-‐1834.
15) Keren A, Syrris P and McKenna W. Hypertrophic cardiomyopathy: the genetic determinants of clinical disease expression. Nat Clin Pract Cardiovasc Med. Mar. 2008; 5:3, 158-‐168.
16) Jacoby D and McKenna WJ. Genetics of inherited cardiomyopathy. Eur Heart J. 2012 Feb.; 33:3, 296-‐304.
17) Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Peterson A, Li D, Jakobs P, Litt M, Porter CB, Rahko PS and Hershberger RE. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J. 2008; 156:161-‐169.
18) Van Berlo JH, de Voogt WG and van der Kooi AJ et al. Meta-‐analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death?. J Mol Med. 2005; 83:79-‐83.
19) Meune C, Van Berlo JH, Anselme F, Bonne G, Pinto YM and Duboc D. Primary prevention of sudden death in patients with lamin A/C gene mutations. N Engl J Med. 2006; 354:209-‐210.
20) Merlo M, Pyxaras SA, Pinamonti B, Barbati G, Di Lenarda A and Sinagra G. Prevalence and prognostic significance of left ventricular reverse remodeling in dilated cardiomyopathy receiving tailored medical treatment. J Am Coll Cardiol. 2011; 57:13, 1468-‐1476.
21) Mocumbi AO, Yacoub S and Yacoub MH. Neglected tropical cardiomyopathies: II. Endomyocardial fibrosis: myocardial disease. Heart. 2008 Mar.; 94:3, 384-‐390.
22) Kubo T, Gimeno JR and Bahl A et al. Prevalence, clinical significance, and genetic basis of hypertrophic cardiomyopathy with restrictive phenotype. Journal of the American College of Cardiology. June 2007; 49:25, 2419-‐2426.
23) Zangwill SD, Naftel D, L'Ecuyer T, Rosenthal D, Robinson B, Kirklin JK, Stendahl G and Dipchand AI. Pediatric Heart Transplant Study Investigators. Outcomes of children with restrictive cardiomyopathy

7. References
196
listed for heart transplant: a multi-‐institutional study. J Heart Lung Transplant. 2009 Dec; 28:12, 1335-‐1340.
24) Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffitz JE, Sanborn DM, Steinberg JS, Tandri H, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T and Zareba W. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J. 2010; 31:806-‐814.
25) Basso C, Bauce B, Corrado D and Thiene G. Pathophysiology of arrhythmogenic cardiomyopathy. Nat Rev Cardiol. 2011 Nov 29; 9:4, 223-‐233
26) Ackerman MJ, Priori SG and Willems S et al. HRS/EHRA Expert Consensus Statement on the State of Genetic Testing for Channelopathies and Cardiomyopathies. Europace. 2011 Aug; 13:8, 1077-‐1109.
27) Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG and Seidman CE. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012 Feb. 16; 366:7, 619-‐628.
28) Ashrafian H, McKenna WJ, Watkins H. Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circ Res. 2011 Jun 24; 109(1):86-‐96.
29) Ho CY, Lever HM, DeSanctis R, Farver CF, Seidman JG, Seidman CE. Homozygous mutation in cardiac troponin T: implications for hypertrophic cardiomyopathy. Circulation. 2000 Oct 17; 102(16):1950-‐1955.
30) Ingles J, Doolan A, Chiu C, Seidman J, Seidman C, Semsarian C. Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. J Med Genet. 2005 Oct; 42(10):e59.
31) Girolami F, Ho CY, Semsarian C, Baldi M, Will ML, Baldini K, Torricelli F, Yeates L, Cecchi F, Ackerman MJ, Olivotto I. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J Am Coll Cardiol. 2010 Apr 6; 55(14):1444-‐1453.
32) Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, Jalilzadeh S, Carballo S, Redwood C, Watkins H. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res. 2009 Jul 31; 105(3):219-‐222.

7. References
197
33) Vignier N, Schlossarek S, Fraysse B, Mearini G, Krämer E, Pointu H, Mougenot N, Guiard J, Reimer R, Hohenberg H, Schwartz K, Vernet M, Eschenhagen T, Carrier L. Nonsense-‐mediated mRNA decay and ubiquitin-‐proteasome system regulate cardiac myosin-‐binding protein C mutant levels in cardiomyopathic mice. Circ Res. 2009 Jul 31; 105(3):239-‐248.
34) Schlossarek S, Frey N, Carrier L. Ubiquitin-‐proteasome system and hereditary cardiomyopathies. J Mol Cell Cardiol. 2013 Dec 28
35) Carè A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Høydal M, Autore C, Russo MA, Dorn GW 2nd, Ellingsen O, Ruiz-‐Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G. MicroRNA-‐133 controls cardiac hypertrophy. Nat Med. 2007 May; 13(5):613-‐618. Epub 2007 Apr 29.
36) Pogwizd, S.M.; Schlotthauer, K.; Li, L.; Yuan, W.; Bers, D.M. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-‐calcium exchange, inward rectifier potassium current, and residual beta-‐adrenergic responsiveness. Circulation research 88 2001, 1159-‐1167
37) Hasenfuss, G.; Schillinger, W.; Lehnart, S.E.; Preuss, M.; Pieske, B.; Maier, L.S.; Prestle, J.; Minami, K.; Just, H. Relationship between Na+-‐Ca2+-‐exchanger protein levels and diastolic function of failing human myocardium. Circulation 99 1999, 641-‐648
38) Shannon, T.R.; Pogwizd, S.M.; Bers, D.M. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circulation research 93 2003, 592-‐594
39) Maier, L.S.; Zhang, T.; Chen, L.; DeSantiago, J.; Brown, J.H.; Bers, D.M. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circulation research 92 2003, 904-‐911
40) Belus A, Piroddi N, Scellini B, Tesi C, D'Amati G, Girolami F, Yacoub M, Cecchi F, Olivotto I, Poggesi C. The familial hypertrophic cardiomyopathy-‐associated myosin mutation R403Q accelerates tension generation and relaxation of human cardiac myofibrils. J Physiol. 2008 Aug 1; 586(Pt 15):3639-‐3644
41) Ferrantini C, Belus A, Piroddi N, Scellini B, Tesi C, Poggesi C. Mechanical and energetic consequences of HCM-‐causing mutations. J Cardiovasc Transl Res. 2009 Dec; 2(4):441-‐451
42) Ashrafian H, Redwood C, Blair E, Watkins H. Hypertrophic cardiomyopathy:a paradigm for myocardial energy depletion. Trends Genet. 2003 May; 19(5):263-‐268.
43) Pyle WG, Solaro RJ. At the crossroads of myocardial signaling: the role of Z-‐discs in intracellular signaling and cardiac function. Circ Res. 2004 Feb 20; 94(3):296-‐305.

7. References
198
44) Linke WA. Sense and stretchability: the role of titin and titin-‐associated proteins in myocardial stress-‐sensing and mechanical dysfunction. Cardiovasc Res. 2008 Mar 1; 77(4):637-‐648.
45) Arimura T, Bos JM, Sato A, Kubo T, Okamoto H, Nishi H, Harada H, Koga Y, Moulik M, Doi YL, Towbin JA, Ackerman MJ, Kimura A. Cardiac ankyrin repeat protein gene (ANKRD1) mutations in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009 Jul 21; 54(4):334-‐342. doi: 10.1016/j.jacc.2008.12.082.
46) Pieske, B.; Houser, S.R. [Na+]i handling in the failing human heart. Cardiovascular research 57 2003; 874-‐886
47) Pogwizd, S.M.; Sipido, K.R.; Verdonck, F.; Bers, D.M. Intracellular Na in animal models of hypertrophy and heart failure: contractile function and arrhythmogenesis. Cardiovascular research 57 2003; 887-‐896
48) Maltsev, V.A.; Sabbah, H.N.; Higgins, R.S.; Silverman, N.; Lesch, M.; Undrovinas, A.I. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation 98 1998; 2545-‐2552
49) Bennett, P.B.; Yazawa, K.; Makita, N.; George, A.L., Jr. Molecular mechanism for an inherited cardiac arrhythmia. Nature 376 1995; 683-‐685
50) Mohler, P.J.; Schott, J.J.; Gramolini, A.O.; Dilly, K.W.; Guatimosim, S.; duBell, W.H.; Song, L.S.; Haurogne, K.; Kyndt, F.; Ali, M.E.; Rogers, T.B.; Lederer, W.J.; Escande, D.; Le Marec, H.; Bennett, V. Ankyrin-‐B mutation causes type 4 long-‐QT cardiac arrhythmia and sudden cardiac death. Nature 421 2003; 634-‐639
51) Vatta, M.; Ackerman, M.J.; Ye, B.; Makielski, J.C.; Ughanze, E.E.; Taylor, E.W.; Tester, D.J.; Balijepalli, R.C.; Foell, J.D.; Li, Z.; Kamp, T.J.; Towbin, J.A. Mutant caveolin-‐3 induces persistent late sodium current and is associated with long-‐QT syndrome. Circulation 114 2006; 2104-‐2112
52) Maltsev, V.A.; Sabbah, H.N.; Higgins, R.S.; Silverman, N.; Lesch, M.; Undrovinas, A.I. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation 98 1998; 2545-‐2552
53) Maltsev, V.A.; Silverman, N.; Sabbah, H.N.; Undrovinas, A.I. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: implications for repolarization variability. European journal of heart failure 9 2007; 219-‐227
54) Huang, B.; El-‐Sherif, T.; Gidh-‐Jain, M.; Qin, D.; El-‐Sherif, N. Alterations of sodium channel kinetics and gene expression in the postinfarction remodeled myocardium. Journal of cardiovascular electrophysiology 12 2001; 218-‐225
55) Undrovinas, A.I.; Maltsev, V.A.; Sabbah, H.N. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cellular and molecular life sciences: CMLS 55 1999; 494-‐505

7. References
199
56) Belardinelli, L.; Shryock, J.C.; Fraser, H. Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart 92 Suppl 4 2006, iv6-‐iv14
57) Antzelevitch, C.; Belardinelli, L. The role of sodium channel current in modulating transmural dispersion of repolarization and arrhythmogenesis. Journal of cardiovascular electrophysiology 17 Suppl 1 2006; S79-‐S85
58) Maron MS, Olivotto I, Maron BJ, Prasad SK, Cecchi F, Udelson JE, Camici PG. The case for myocardial ischemia in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009 Aug 25; 54(9):866-‐875.
59) Neglia D, L'Abbate A. Coronary microvascular dysfunction and idiopathic dilated cardiomyopathy. Pharmacol Rep. 2005; 57 Suppl:151-‐155
60) Paul M, Rahbar K, Gerss J, Kies P, Schober O, Schäfers K, Breithardt G, Schulze-‐Bahr E, Wichter T, Schäfers M. Microvascular dysfunction in nonfailing arrhythmogenic right ventricular cardiomyopathy. Eur J Nucl Med Mol Imaging. 2012 Mar; 39(3):416-‐420.
61) Tomberli B, Cecchi F, Sciagrà R, Berti V, Lisi F, Torricelli F, Morrone A, Castelli G, Yacoub MH, Olivotto I. Coronary microvascular dysfunction is an early feature of cardiac involvement in patients with Anderson-‐Fabry disease. Eur J Heart Fail. 2013 Dec; 15(12):1363-‐1373. doi: 10.1093/eurjhf/hft104. Epub 2013 Jun 30.
62) Cecchi F, Olivotto I, Gistri R, Lorenzoni R, Chiriatti G, Camici PG. Coronary microvascular dysfunction and prognosis in hypertrophic cardiomyopathy. N Engl J Med. 2003 Sep 11; 349(11):1027-‐1035.
63) Olivotto I, Cecchi F, Camici PG. Coronary microvascular dysfunction and ischemia in hypertrophic cardiomyopathy. Mechanisms and clinical consequences. Ital Heart J. 2004 Aug; 5(8):572-‐80.
64) Olivotto I, Girolami F, Sciagrà R, Ackerman MJ, Sotgia B, Bos JM, Nistri S, Sgalambro A, Grifoni C, Torricelli F, Camici PG, Cecchi F. Microvascular function is selectively impaired in patients with hypertrophic cardiomyopathy and sarcomere myofilament gene mutations. J Am Coll Cardiol. 2011; 58:839–848.
65) Olivotto I, Girolami F, Nistri S, Rossi A, Rega L, Garbini F, Grifoni C, Cecchi F, Yacoub MH. The many faces of hypertrophic cardiomyopathy: from developmental biology to clinical practice. J Cardiovasc Transl Res. 2009; 2:349–367.
66) Lombardi, R., Betocchi, S., Losi, M. A., Tocchetti, C. G., Aversa, M., Miranda, M., et al. (2003). Myocardial collagen turnover in hypertrophic cardiomyopathy. Circulation, 108, 1455–1460.
67) Ho CY, López B, Coelho-‐Filho OR, Lakdawala NK, Cirino AL, Jarolim P, Kwong R, González A, Colan SD, Seidman JG, Díez J, Seidman CE. Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N Engl J Med. 2010 Aug 5; 363(6):552-‐563.

7. References
200
68) Geisterfer-‐Lowrance AA, Christe M, Conner DA, et al. A mouse model of familial hypertrophic cardiomyopathy. Science 1996; 272:731-‐734.
69) Adabag AS, Maron BJ, Appelbaum E, Harrigan CJ, Buros JL, Gibson CM, Lesser JR, Hanna CA, Udelson JE, Manning WJ, Maron MS. Occurrence and frequency of arrhythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular magnetic resonance. J Am Coll Cardiol. 2008 Apr 8; 51(14):1369-‐1374.
70) Maron MS, Appelbaum E, Harrigan CJ, Buros J, Gibson CM, Hanna C, Lesser JR, Udelson JE, Manning WJ, Maron BJ. Clinical profile and significance of delayed enhancement in hypertrophic cardiomyopathy. Circ Heart Fail. 2008 Sep; 1(3):184-‐191
71) Maron MS. The current and emerging role of cardiovascular magnetic resonance imaging in hypertrophic cardiomyopathy. J Cardiovasc Transl Res. 2009 Dec; 2(4):415-‐425.
72) Moon JC, Mogensen J, Elliott PM, Smith GC, Elkington AG, Prasad SK, Pennell DJ, McKenna WJ. Myocardial late gadolinium enhancement cardiovascular magnetic resonance in hypertrophic cardiomyopathy caused by mutations in troponin I. Heart. 2005 Aug; 91(8):1036-‐1040.
73) Moon JC, Prasad SK. Cardiovascular magnetic resonance and the evaluation of heart failure. Curr Cardiol Rep. 2005 Jan; 7(1):39-‐44.
74) Olivotto I, Maron MS, Autore C, Lesser JR, Rega L, Casolo G, De Santis M, Quarta G, Nistri S, Cecchi F, Salton CJ, Udelson JE, Manning WJ, Maron BJ. Assessment and significance of left ventricular mass by cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2008 Aug 12; 52(7):559-‐566.
75) Schulz-‐Menger J, Abdel-‐Aty H, Rudolph A, Elgeti T, Messroghli D, Utz W, Boyé P, Bohl S, Busjahn A, Hamm B, Dietz R. Gender-‐specific differences in left ventricular remodelling and fibrosis in hypertrophic cardiomyopathy: insights from cardiovascular magnetic resonance. Eur J Heart Fail. 2008 Sep; 10(9):850-‐854.
76) Choudhury L, Mahrholdt H, Wagner A, Choi KM, Elliott MD, Klocke FJ, Bonow RO, Judd RM, Kim RJ. Myocardial scarring in asymptomatic or mildly symptomatic patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2002 Dec 18; 40(12):2156-‐2164.
77) Basso C, Thiene G, Corrado D, Buja G, Melacini P, Nava A. Hypertrophic cardiomyopathy and sudden death in the young: pathologic evidence of myocardial ischemia. Hum Pathol. 2000 Aug; 31(8):988-‐998.
78) Yacoub MH, Olivotto I, Cecchi F. 'End-‐stage' hypertrophic cardiomyopathy: from mystery to model. Nat Clin Pract Cardiovasc Med. 2007 May; 4(5):232-‐233
79) Krusche CA, Holthöfer B, Hofe V, van de Sandt AM, Eshkind L, Bockamp E, Merx MW, Kant S, Windoffer R, Leube RE. Desmoglein 2 mutant mice develop cardiac fibrosis and dilation. Basic Res Cardiol. 2011 Jun; 106(4):617-‐633. doi: 10.1007/s00395-‐011-‐0175-‐y. Epub 2011 Apr 1.

7. References
201
80) Sen-‐Chowdhry, S., P. Syrris, S.K. Prasad, S.E. Hughes, R. Merrifield, D. Ward, D.J. Pennell, W.J. McKenna. 2008. Left-‐dominant arrhythmogenic cardiomyopathy: an under-‐recognized clinical entity. J. Am. Coll. Cardiol. 52:2175–2187
81) Basso, C., D. Corrado, F.I. Marcus, A. Nava, G. Thiene. 2009. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 373:1289–1300.
82) de Leeuw N, Ruiter DJ, Balk AH, de Jonge N, Melchers WJ, Galama JM. Histopathologic findings in explanted heart tissue from patients with end-‐stage idiopathic dilated cardiomyopathy. Transpl Int. 2001; 14:299–306.
83) Roberts WC, Siegel RJ, McManus BM. Idiopathic dilated cardiomyopathy: analysis of 152 necropsy patients. Am J Cardiol. 1987; 60:1340–1355.
84) Unverferth DV, Baker PB, Swift SE, Chaffee R, Fetters JK, Uretsky BF, Thompson ME, Leier CV. Extent of myocardial fibrosis and cellular hypertrophy in dilated cardiomyopathy. Am J Cardiol. 1986; 57:816–820
85) Masci PG, Schuurman R, Andrea B, Ripoli A, Coceani M, Chiappino S, Todiere G, Srebot V, Passino C, Aquaro GD, Emdin M, Lombardi M. Myocardial fibrosis as a key determinant of left ventricular remodeling in idiopathic dilated cardiomyopathy: a contrast-‐enhanced cardiovascular magnetic study. Circ Cardiovasc Imaging. 2013; 6:790-‐799.
86) Klues HG, Proschan MA, Dollar AL, Spirito P, Roberts WC, Maron BJ. Echocardiographic assessment of mitral valve size in obstructive hypertrophic cardiomyopathy. Anatomic validation from mitral valve specimen. Circulation. 1993 Aug; 88(2):548-‐555.
87) Lie-‐Venema H, van den Akker NM, Bax NA, Winter EM, Maas S, Kekarainen T, Hoeben RC, deRuiter MC, Poelmann RE, Gittenberger-‐de Groot AC. Origin, fate, and function of epicardium-‐derived cells (EPDCs) in normal and abnormal cardiac development. ScientificWorldJournal. 2007 Nov 12; 7:1777-‐1798.
88) Kattman SJ, Adler ED, Keller GM. Specification of multipotential cardiovascular progenitor cells during embryonic stem cell differentiation and embryonic development. Trends Cardiovasc Med. 2007 Oct; 17(7):240-‐246.
89) Dahl KN, et al. Nuclear shape, mechanics and mechanotransduction. Circulation Respiratory. 2008; 108, 173–189.
90) Markwald RR, Norris RA, Moreno-‐Rodriguez R, Levine RA. Developmental basis of adult cardiovascular diseases: valvular heart diseases. Ann N Y Acad Sci. 2010; 1188:177-‐183
91) Lombardi R, Marian AJ. Arrhythmogenic right ventricular cardiomyopathy is a disease of cardiac stem cells. Curr Opin Cardiol. 2010; 25:222-‐228.

7. References
202
92) Braunwald E. Cardiovascular science: opportunities for translating research into improved care. J Clin Invest. 2013; 123:6–10.
93) Maron BJ, Braunwald E. Evolution of hypertrophic cardiomyopathy to a contemporary treatable disease. Circulation. 2012; 126:1640–1644.
94) Marian AJ. Experimental therapies in hypertrophic cardiomyopathy. J CardiovascTransl Res. 2009; 2:483–492.
95) Spoladore R, Maron MS, D’Amato R, Camici PG, Olivotto I. Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur Heart J. 2012; 33:1724–1733.
96) Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW. ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy; executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011; 124:2761–2796.
97) Ommen SR, Maron BJ, Olivotto I, Maron MS, Cecchi F, Betocchi S, Gersh BJ, Ackerman MJ, McCully RB, Dearani JA, Schaff HV, Danielson GK, Tajik AJ, Nishimura RA. Long-‐term effects of surgical septalmyectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am CollCardiol. 2005; 46:470–476.
98) Force T, Bonow RO, Houser SR, Solaro RJ, Hershberger RE, Adhikari B, Anderson ME, Boineau R, Byrne BJ, Cappola TP, Kalluri R, LeWinter MM, Maron MS, Molkentin JD, Ommen SR, Regnier M, Tang WH, Tian R, Konstam MA, Maron BJ, Seidman CE. Research priorities in hypertrophic cardiomyopathy: report of a Working Group of the National Heart, Lung, and Blood Institute. Circulation. 2010; 122(11):1130–1133.
99) Abozguia K, Elliott P, McKenna W, Phan TT, Nallur-‐Shivu G, Ahmed I, Maher AR, Kaur K, Taylor J, Henning A, Ashrafian H, Watkins H, Frenneaux M. Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation. 2010; 122:1562–1569.
100) Coppini R, Ferrantini C, Yao L, Fan P, Del Lungo M, Stillitano F, Sartiani L, Tosi B, Suffredini S, Tesi C, Yacoub M, Olivotto I, Belardinelli L, Poggesi C, Cerbai E, Mugelli A. Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation. 2013; 127:575–584.
101) Lan F, Lee AS, Liang P, Sanchez-‐Freire V, Nguyen PK, Wang L, Han L, Yen M, Wang Y, Sun N, Abilez OJ, Hu S, Ebert AD, Navarrete EG, Simmons CS, Wheeler M, Pruitt B, Lewis R, Yamaguchi Y, Ashley EA, Bers DM, Robbins RC, Longaker MT, Wu JC. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-‐specific induced pluripotent stem cells. Cell Stem Cell. 2013; 12:101–113.
102) Malik FI, Hartman JJ, Elias KA, Morgan BP, Rodriguez H, Brejc K, Anderson RL, Sueoka SH, Lee KH, Finer JT, Sakowicz R, Baliga R, Cox DR,

7. References
203
Garard M, Godinez G, Kawas R, Kraynack E, Lenzi D, Lu PP, Muci A, Niu C, Qian X, Pierce DW, Pokrovskii M, Suehiro I, Sylvester S, Tochimoto T, Valdez C, Wang W, Katori T, Kass DA, Shen YT, Vatner SF, Morgans DJ. Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science. 2011; 331:1439–1443.
103) Eulalio A, Mano M, Dal Ferro M, Zentilin L, Sinagra G, Zacchigna S, Giacca M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012 Dec 20; 492(7429):376-‐81. doi: 10.1038/nature11739. Epub 2012 Dec 5.
104) Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009 Jan; 10(1):32-‐42. doi: 10.1038/nrg2485.

7. References
204
Chapter 2: Genotype-‐Phenotype Correlations
1) Olivotto I, Girolami F, Ackerman MJ, Nistri S, Bos JM, Zachara E, et al. Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin. Proc. 2008 Jun; 83(6):630–638.
2) Tardiff JC. Thin filament mutations: developing an integrative approach to a complex disorder. Circulation research 2011; 108:765-‐782.
3) Moolman JC, Corfield VA, Posen B et al. Sudden death due to troponin T mutations. Journal of the American College of Cardiology 1997; 29:549-‐555.
4) Varnava AM, Elliott PM, Baboonian C, Davison F, Davies MJ, McKenna WJ. Hypertrophic cardiomyopathy: histopathological features of sudden death in cardiac troponin T disease. Circulation. 2001
5) Anan R, Greve G, Thierfelder L, Watkins H, McKenna WJ, Solomon S, Vecchio C, Shono H, Nakao S, Tanaka H, Mares AJ, Towbin JA, Spirito P, Roberts R, Seidman JG, Seidman CE. Prognostic implications of novel beta cardiac myosin heavy chain gene mutations that cause familial hypertrophic cardiomyopathy. J Clin Invest. 1994
6) Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O'Donoghue A, Spirito P, Matsumori A, Moravec CS, Seidman JG, et al. Mutations in the genes for cardiac troponin T and alpha-‐tropomyosin in hypertrophic cardiomyopathy. N Engl J Med. 1995 Apr 20; 332(16):1058-‐1064.
7) Pasquale F, Syrris P, Kaski JP, Mogensen J, McKenna WJ, Elliott P. Long-‐term outcomes in hypertrophic cardiomyopathy caused by mutations in the cardiac troponin T gene. Circ Cardiovasc Genet. 2012 Feb 1; 5(1):10-‐17.
8) Baudenbacher F, Schober T, Pinto JR et al. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. The Journal of clinical investigation 2008; 118:3893-‐3903.
9) Harris KM, Spirito P, Maron MS et al. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-‐stage phase of hypertrophic cardiomyopathy. Circulation 2006; 114:216-‐225.
10) Olivotto I, Cecchi F, Gistri R et al. Relevance of coronary microvascular flow impairment to long-‐term remodeling and systolic dysfunction in hypertrophic cardiomyopathy. Journal of the American College of Cardiology 2006; 47:1043-‐1048.
11) Pinamonti B, Di Lenarda A, Nucifora G, Gregori D, Perkan A, Sinagra G. Incremental prognostic value of restrictive filling pattern in hypertrophic cardiomyopathy: a Doppler echocardiographic study. Eur J Echocardiogr. 2008 Jul; 9(4):466-‐471.
12) Lin D, Bobkova A, Homsher E, Tobacman LS. Altered cardiac troponin T in vitro function in the presence of a mutation implicated in familial

7. References
205
hypertrophic cardiomyopathy. The Journal of clinical investigation 1996; 97:2842-‐2848.
13) Redwood C, Lohmann K, Bing W et al. Investigation of a truncated cardiac troponin T that causes familial hypertrophic cardiomyopathy: Ca(2+) regulatory properties of reconstituted thin filaments depend on the ratio of mutant to wild-‐type protein. Circulation research 2000; 86:1146-‐1152.
14) Miller T, Szczesna D, Housmans PR et al. Abnormal contractile function in transgenic mice expressing a familial hypertrophic cardiomyopathy-‐linked troponin T (I79N) mutation. The Journal of biological chemistry 2001; 276:3743-‐3755.
15) Chandra M, Tschirgi ML, Tardiff JC. Increase in tension-‐dependent ATP consumption induced by cardiac troponin T mutation. American journal of physiology Heart and circulatory physiology 2005; 289:H2112-‐2119.
16) Javadpour MM, Tardiff JC, Pinz I, Ingwall JS. Decreased energetics in murine hearts bearing the R92Q mutation in cardiac troponin T. The Journal of clinical investigation 2003; 112:768-‐75.
17) Ashrafian H, Redwood C, Blair E, Watkins H. Hypertrophic cardiomyopathy:a paradigm for myocardial energy depletion. Trends in genetics: TIG 2003; 19:263-‐268.
18) Tardiff JC. Sarcomeric proteins and familial hypertrophic cardiomyopathy: linking mutations in structural proteins to complex cardiovascular phenotypes. Heart failure reviews 2005; 10:237-‐248.
19) He H, Hoyer K, Tao H et al. Myosin-‐driven rescue of contractile reserve and energetics in mouse hearts bearing familial hypertrophic cardiomyopathy-‐associated mutant troponin T is mutation-‐specific. The Journal of physiology 2012; 590:5371-‐5388.
20) Keren G, Meisner JS, Sherez J, Yellin EL, Laniado S. Interrelationship of mid-‐diastolic mitral valve motion, pulmonary venous flow, and transmitral flow. Circulation 1986; 74:36-‐44.
21) Debl K, Djavidani B, Buchner S et al. Triphasic mitral inflow pattern and regional triphasic mitral annulus velocity in hypertrophic cardiomyopathy. Journal of the American Society of Echocardiography: official publication of the American Society of Echocardiography 2008; 21:407 e5-‐6.
22) Ha JW, Ahn JA, Moon JY et al. Triphasic mitral inflow velocity with mid-‐diastolic flow: the presence of mid-‐diastolic mitral annular velocity indicates advanced diastolic dysfunction. European journal of echocardiography: the journal of the Working Group on Echocardiography of the European Society of Cardiology 2006; 7:16-‐21.
23) Ertz-‐Berger BR, He H, Dowell C et al. Changes in the chemical and dynamic properties of cardiac troponin T cause discrete cardiomyopathies in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America 2005; 102:18219-‐18224.

7. References
206
24) Belus A, Piroddi N, Scellini B, Tesi C, D'Amati G, Girolami F, Yacoub M, Cecchi F, Olivotto I, Poggesi C. The familial hypertrophic cardiomyopathy-‐associated myosin mutation R403Q accelerates tension generation and relaxation of human cardiac myofibrils. J Physiol. 2008 Aug 1; 586(Pt 15):3639-‐3644.
25) Ferrantini C, Belus A, Piroddi N, Scellini B, Tesi C, Poggesi C. Mechanical and energetic consequences of HCM-‐causing mutations. J Cardiovasc Transl Res. 2009 Dec; 2(4):441-‐451.
26) Anderson W. A case of angio-‐keratoma. Br J Dermatol 1898; 10:113-‐117.
27) Fabry J. Purpura papulosa haemorrhagica Hebrae. Arch Dermatol Syphilol 1898; 43:187-‐201. [In German].
28) Desnick RJ, Ioannou YA, Eng CM. a-‐ Galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS and Valle D, eds. The metabolic and molecular bases of inherited disease, 8th Ed. New York: McGraw-‐Hill; 2001. pp 3733-‐3774.
29) Germain DP. Fabry disease. Orphanet J Rare Dis 2010; 5:30-‐79. 30) Aerts JMFG, Kallemeijn WW, Wegdam W, et al. Biomarkers in the
diagnosis of lysosomal storage disorders: proteins, lipids, and inhibodies. J Inherit Metab Dis 2011; 1-‐15.
31) Meikle PJ, Hopwood JJ, Clague AE, Carrey WF. Prevalence of lysosomal storage disorders. JAMA 1999; 281:249–254.
32) Mechtler TP, Stary S, Metz TF, De Jesús VR, Greber-‐Platzer S, Pollak A, Herkner KR, Streubel B, Kasper DC. Neonatal screening for lysosomal storage disorders: feasibility and incidence from a nationwide study in Austria. Lancet 2012; 379: 335-‐341.
33) Spada M, Pagliardini S, Yasuda M, et al. High incidence of later-‐onset fabry disease revealed by newborn screening. Am J Hum Genet 2006; 79:31-‐40.
34) 19. Hwu W-‐L, Chien Y-‐H, Lee N-‐C, et al. Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-‐onset GLA mutation c.936+919G>A (IVS4+919G>A). Hum Mutat 2009; 30: 1397-‐1405.
35) Linthorst GE, Bouwman MG, Wijburg F a, et al. Screening for Fabry disease in highrisk populations: a systematic review. J Med Genet 2010; 47:217-‐222.
36) Wilcox WR, Oliveira JP, Hopkin RJ, Ortiz A, Banikazemi M, Feldt-‐Rasmussen U, et al. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol. Genet. Metab. 2008 Feb; 93(2):112–128
37) Waldek S, Patel MR, Banikazemi M, Lemay R, Lee P. Life expectancy and cause of death in males and females with Fabry disease: findings from the Fabry Registry. Genet. Med 2009 Nov; 11(11):790–796.

7. References
207
38) Lidove O, West ML, Pintos-‐Morell G, Reisin R, Nicholls K, Figuera LE, et al. Effects of enzyme replacement therapy in Fabry disease-‐A comprehensive review of the medical literature. Genet. Med. 2010 Nov; 12(11):668–679.
39) IShii BS. Pharmacological chaperone therapy for Fabry disease. In Vitro. 2012; 88:18–30.
40) Porto C, Pisani A, Rosa M, Acampora E, Avolio V, Tuzzi MR, Visciano B, Gagliardo C, Materazzi S, la Marca G, Andria G, Parenti G. Synergy between the pharmacological chaperone 1-‐deoxygalactonojirimycin and the human recombinant alphagalactosidase A in cultured fibroblasts from patients with Fabry disease. J Inherit Metab Dis 2012; 35:513–520.
41) Weidemann F, Niemann M, Breunig F, Herrmann S, Beer M, Sto¨rk S, Voelker W, Ertl G,Wanner C, Strotmann J. Long-‐term effects of enzyme replacement therapy on Fabry cardiomyopathy: evidence for a better outcome with early treatment. Circulation 2009; 119:524–529.
42) Cecchi F, Olivotto I, Gistri R, Lorenzoni R, Chiriatti G, Camici PG. Coronary microvascular dysfunction and prognosis in hypertrophic cardiomyopathy. N Engl J Med. 2003; 349:1027–1035
43) Eng CM, Fletcher J, Wilcox WR, Waldek S, Scott CR, Sillence DO, Breunig F, Charrow J, Germain DP, Nicholls K, Banikazemi M. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis. 2007; 30:184-‐192.
44) O'Mahony C, Elliott P. Anderson-‐Fabry disease and the heart. Prog Cardiovasc Dis. 2010 Jan-‐Feb; 52(4):326-‐335.
45) Maron BJ, Niimura H, Casey S a, Soper MK, Wright GB, Seidman JG, et al. Development of left ventricular hypertrophy in adults in hypertrophic cardiomyopathy caused by cardiac myosin-‐binding protein C gene mutations. J. Am. Coll. Cardiol. 2001 Aug; 38(2):315–321.
46) Maron MS, Olivotto I, Maron BJ, Prasad SK, Cecchi F, Udelson JE, et al. The case for myocardial ischemia in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2009 Aug; 54(9):866–875
47) Elliott PM, Kindler H, Shah JS, Sachdev B, Rimoldi OE, Thaman R, Tome MT, McKenna WJ, Lee P, Camici PG. Coronary microvascular dysfunction in male patients with Anderson-‐Fabry disease and the effect of treatment with alpha galactosidase A. Heart. 2006; 92:357-‐360.
48) Cecchi F, Olivotto I, Gistri R, Lorenzoni R, Chiriatti G, Camici PG. Coronary microvascular dysfunction and prognosis in hypertrophic cardiomyopathy. N. Engl. J. Med. 2003 Sep; 349(11):1027–1035.
49) Kalliokoski RJ, Kalliokoski KK, Sundell J, Engblom E, Penttinen M, Kantola I, Raitakari OT, Knuuti J, Nuutila P. Impaired myocardial perfusion reserve but preserved peripheral endothelial function in patients with Fabry disease. J Inherit Metab Dis. 2005; 28:563-‐573

7. References
208
50) Kalliokoski RJ, Kantola I, Kalliokoski KK, Engblom E, Sundell J, Hannukainen JC, Janatuinen T, Raitakari OT, Knuuti J, Penttinen M, Viikari J, Nuutila P. The effect of 12-‐month enzyme replacement therapy on myocardial perfusion in patients with Fabry disease. J Inherit Metab Dis. 2006 ; 29:112-‐118.
51) Camici PG, Crea F. Coronary microvascular dysfunction. N. Engl. J. Med. 2007 Feb; 356(8):830–840.
52) Pieroni M, Chimenti C, Ricci R, Sale P, Russo MA, Frustaci A. Early detection of Fabry cardiomyopathy by tissue Doppler imaging. Circulation 2003; 107:1978–1984.
53) Niemann M, Herrmann S, Hu K, Breunig F, Strotmann J, Beer M, Machann W, VoelkerW, Ert F,Wanner C,Weidemann F. Differences in Fabry cardiomyopathy between female and male patients consequences for diagnostic assessment. JACC Cardiovasc Imaging 2011; 4:592–601.
54) Perugini E, Rapezzi C, Piva T, Leone O, Bacchi-‐Reggiani L, Riva L, Salvi F, Lovato L, Branzi A, Fattori R. Non-‐invasive evaluation of the myocardial substrate of cardiac amyloidosis by gadolinium cardiac magnetic resonance. Heart 2006; 92:343–349.
55) Wilcox WR, Oliveira JP, Hopkin RJ, Ortiz A, Banikazemi M, Feldt-‐Rasmussen U, et al. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol Genet Metab 2008; 93:112–128.
56) Pinto LL, VieiraTA, Giugliani R, Schwartz IV. Expression of the disease on female carriers of X-‐linked lysosomal disorders: a brief review. Orphanet J Rare Dis 2010; 5:14.
57) Kampmann C, Baehner F, Whybra C, Martin C, Wiethoff CM, Ries M, Gal A, Beck M. Cardiac manifestations of Anderson–Fabry disease in heterozygous females. J Am Coll Cardiol 2002; 40:1668–1674.
58) Nakao S, Takenaka T, Maeda M, Kodama C, Tanaka A, Tahara M, et al. An atypical variant of Fabry’s disease in men with left ventricular hypertrophy. N. Engl. J. Med. 1995 Aug 3; 333(5):288–293.
59) Olivotto I, Girolami F, Sciagrà R, Ackerman MJ, Sotgia B, Bos JM, et al. Microvascular function is selectively impaired in patients with hypertrophic cardiomyopathy and sarcomere myofilament gene mutations. J. Am. Coll. Cardiol 2011 Aug 16; 58(8):839–848.
60) Camici PG, Olivotto I, Rimoldi OE. The coronary circulation and blood flow in left ventricular hypertrophy. J Mol Cell Cardiol 2012; 52:857–864.
61) MachannW, Breunig F,Weidemann F, Sandstede J, Hahn D, Ko¨ stler H, Neubauer S, Wanner C, Beer M. Cardiac energy metabolism is disturbed in Fabry disease and improves with enzyme replacement therapy using recombinant human galactosidase A. Eur J Heart Fail 2011; 13:278–283.
62) Petersen SE, Jerosch-‐Herold M, Hudsmith LE, Robson MD, Francis JM, Doll HA, Selvanayagam JB, Neubauer S,Watkins H. Evidence for

7. References
209
microvascular dysfunction in hypertrophic cardiomyopathy: new insights from multiparametric magnetic resonance imaging. Circulation 2007; 115:2418–2425.
63) Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kühl U, Maisch B, McKenna WJ, Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L, Keren A. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008 Jan; 29(2):270-‐276
64) Geisterfer-‐Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 1990 Sep 7; 62(5):999-‐1006.
65) Alberto Magi, Lorenzo Tattini, Ingrid Cifola, Romina D’Aurizio, Matteo Benelli, Eleonora Mangano, Cristina Battaglia, Elena Bonora, Ants Kurg, Marco Seri, Pamela Magini, Betti Giusti, Giovanni Romeo, Tommaso Pippucci, Gianluca De Bellis, Rosanna Abbate and Gian Franco Gensini. EXCAVATOR: detecting copy number variants from whole-‐exome sequencing data. Genome Biology 2013, 14:R120.
66) Boulé S, Fressart V, Laux D, Mallet A, Simon F, de Groote P, Bonnet D, Klug D, Charron P. Expanding the phenotype associated with a desmoplakin dominant mutation: Carvajal/Naxos syndrome associated with leukonychia and oligodontia. Int J Cardiol. 2012 Nov 1; 161(1):50-‐52.
67) Protonotarios N, Tsatsopoulou A, Patsourakos P, Alexopoulos D, Gezerlis P, Simitsis S, Scampardonis G. Cardiac abnormalities in familial palmoplantar keratosis. Br Heart J 1986; 56:321–326.
68) Luderitz B. Naxos disease. J Interv Card Electrophysiol 2003; 9:405–406. 69) Fontaine G, Fontaliran F, Frank R. Arrhythmogenic right ventricular
cardiomyopathies. Clinical forms and main differential diagnoses. Circulation 1998; 97:1532–1535.
70) Carvajal-‐Huerta L. Epidermolytic palmoplantar keratoderma with wooly hair and dilated cardiomyopathy. J Am Acad Dermatol 1998; 39:418– 421.
71) Perriard J C, Hirschy A, Ehler E. Dilated cardiomyopathy: a disease of the intercalated disk?. Trends Cardiovasc Med 2003; 1330–1338.38.
72) Bos JM, Towbin JA, Ackerman MJ. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009; 54:201-‐211.
73) Wang H, Li Z, Wang J, Sun K, et al. Mutations in NEXN, a Z-‐disc gene, are associated with hypertrophic cardiomyopathy. Am J Hum Genet. 2010; 87:687-‐893.

7. References
210
74) Chiu C, Bagnall RD, Ingles J, et al. Mutations in alpha-‐actinin-‐2 cause hypertrophic cardiomyopathy. A genome-‐wide analysis. J Am Coll Cardiol. 2010; 55:1127-‐1135.

7. References
211
Chapter 3: Environmental Modifiers
1) Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet 2012; 381:242–255.
2) Watkins H, Ashrafian H, Redwood C. Inherited cardiomyopathies. N Engl J Med 2011; 364:1643–1656.
3) Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2011; 58:2703–2738.
4) Olivotto I, Girolami F, Nistri S, et al. The many faces of hypertrophic cardiomyopathy: from developmental biology to clinical practice. J Cardiovasc Transl Res 2009; 2:349–367.
5) Olivotto I, Cecchi F, Poggesi C, Yacoub MH. Patterns of disease progression in hypertrophic cardiomyopathy: an individualized approach to clinical staging. Circ Heart Fail 2012; 5:535–546.
6) Ciró E, Nichols PF 3rd, Maron BJ. Heterogeneous morphologic expression of genetically transmitted hypertrophic cardiomyopathy. Two-‐dimensional echocardiographic analysis. Circulation 1983; 67:1227–1233.
7) Wang L, Seidman JG, Seidman CE. Narrative review: harnessing molecular genetics for the diagnosis and management of hypertrophic cardiomyopathy. Ann Intern Med 2010; 152:513–520, W181.
8) Daw EW, Chen SN, Czernuszewicz G, et al. Genome-‐wide mapping of modifier chromosomal loci for human hypertrophic cardiomyopathy. Hum Mol Genet 2007; 16:2463–2471.
9) Olivotto I, Maron MS, Autore C, et al. Assessment and significance of left ventricular mass by cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Am Coll Cardiol 2008; 52:559–566.
10) Piran S, Liu P, Morales A, Hershberger RE. Where genome meets phenome: rationale for integrating genetic and protein biomarkers in the diagnosis and management of dilated cardiomyopathy and heart failure. J Am Coll Cardiol 2012; 60:283–289.
11) Flegal KM, Kit BK, Orpana H, Graubard BI. Association of all-‐cause mortality with overweight and obesity using standard body mass index categories: a systematic review andmeta-‐analysis. JAMA2013; 309:71–82.
12) Heckbert SR, Post W, Pearson GD, et al. Traditional cardiovascular risk factors in relation to left ventricular mass, volume, and systolic function by cardiac magnetic resonance imaging: the Multiethnic Study of Atherosclerosis. J Am Coll Cardiol 2006; 48:2285–2292.

7. References
212
13) Rider OJ, Francis JM, Ali MK, et al. Determinants of left ventricular mass in obesity; a cardiovascular magnetic resonance study. J Cardiovasc Magn Reson 2009;11:9.
14) Turkbey EB, McClelland RL, Kronmal RA, et al. The impact of obesity on the left ventricle: the Multi-‐Ethnic Study. JACC Cardiovasc Imaging. 2010 Mar; 3(3):266-‐274.
15) Kahn R, Robertson RM, Smith R, Eddy D. The impact of prevention on reducing the burden of cardiovascular disease. Circulation 2008; 118:576–585.
16) Parikh NI, Pencina MJ, Wang TJ, et al. Increasing trends in incidence of overweight and obesity over 5 decades. Am J Med 2007; 120:242–250.
17) Kushner RF. Clinical assessment and management of adult obesity. Circulation 2012; 126:2870–2877.
18) Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiol Rev 2008; 88:389–419.
19) Danias PG, Tritos NA, Stuber M, Kissinger KV, Salton CJ, Manning WJ. Cardiac structure and function in the obese: A cardiovascular magnetic resonance imaging study. J Cardiovasc Magn Reson 2003; 5:431–438.
20) Wong CY, O’Moore-‐Sullivan T, Leano R, Byrne N, Beller E, Marwick TH. Alterations of left ventricular myocardial characteristics associated with obesity. Circulation 2004; 110:3081–3087.
21) Oreopoulos A, Padwal R, Kalantar-‐Zadeh K, Fonarow GC, Norris CM, McAlister FA. Body mass index and mortality in heart failure: a meta-‐analysis. Am Heart J 2008; 156:13–22.
22) Kenchaiah S, Evans JC, Levy D, et al. Obesity and the risk of heart failure. N Engl J Med 2002; 347:305–313.
23) Olivotto I, Tomberli B, Spoladore R, Mugelli A, Cecchi F, Camici PG. Hypertrophic cardiomyopathy: The need for randomized trials. Global Cardiology Science and Practice 2013:31

7. References
213
Chapter 4: Natural History and Predictors of Outcome
1) Jacoby DL, DePasquale EC, McKenna WJ. Hypertrophic cardiomyopathy: diagnosis, risk stratification and treatment. CMAJ. 2013 Feb 5; 185(2):127-‐134
2) O'Mahony C, Jichi F, Pavlou M, Monserrat L, Anastasakis A, Rapezzi C, Biagini E, Gimeno JR, Limongelli G, McKenna WJ, Omar RZ, Elliott PM. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM Risk-‐SCD). Eur Heart J. 2013 Oct 14
3) O'Mahony C, Elliott P, McKenna W. Sudden cardiac death in hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol. 2013 Apr; 6(2):443-‐451
4) Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013 Jan 19; 381(9862):242-‐255.
5) Maron BJ. Risk stratification and role of implantable defibrillators for prevention of sudden death in patients with hypertrophic cardiomyopathy. Circ J. 2010 Nov; 74(11):2271-‐2282
6) Okutucu S, Oto A. Risk stratification in nonischemic dilated cardiomyopathy: Current perspectives. Cardiol J. 2010; 17(3):219-‐229
7) Klein GJ, Krahn AD, Skanes AC, Yee R, Gula LJ. Primary prophylaxis of sudden death in hypertrophic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, and dilated cardiomyopathy. J Cardiovasc Electrophysiol. 2005
8) Meune C, Van Berlo JH, Anselme F, Bonne G, Pinto YM, Duboc D. Primary prevention of sudden death in patients with lamin A/C gene mutations. N Engl J Med. 2006 Jan 12; 354(2):209-‐210
9) Silvano M, Corrado D, Köbe J, Mönnig G, Basso C, Thiene G, Eckardt L. Risk stratification in arrhythmogenic right ventricular cardiomyopathy. Herzschrittmacherther Elektrophysiol. 2013 Dec; 24(4):202-‐208
10) Oechslin E, Jenni R. Left ventricular non-‐compaction revisited: a distinct phenotype with genetic heterogeneity?. Eur Heart J. 2011 Jun; 32(12):1446-‐1456
11) Maron BJ. Contemporary insights and strategies for risk stratification and prevention of sudden death in hypertrophic cardiomyopathy. Circulation. 2010 Jan 26; 121(3):445-‐456.
12) Massimo Santini, Carlo Lavalle, and Renato Pietro Ricci. Primary and secondary prevention of sudden cardiac death: who should get an ICD?. Heart. 2007 November; 93(11): 1478–1483.
13) N.A. Mark Estes. Predicting and Preventing Sudden Cardiac Death. Circulation. 2011; 124: 651-‐656
14) Maron BJ, Spirito P, Shen W-‐K, Haas TS, Formisano F, Link MS, Epstein AE, Almquist AK, Daubert JP, Lawrenz T, Boriani G, Estes NA III, Favale S, Piccininno M, Winters SL, Santini M, Betocchi S, Arribas F, Sherrid MV,

7. References
214
Buja G, Semsarian C, Bruzzi P. Implantable cardioverter-‐defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA. 2007; 298: 405–412.
15) Maron MS, Finley JJ, Bos JM, Hauser RH, Manning WJ, Haas TS, Lesser JR, Udelson JE, Ackerman MJ, Maron BJ. Prevalence, clinical significance and natural history of left ventricular apical aneurysms in hypertrophic cardiomyopathy. Circulation. 2008; 118: 1541–1549
16) Harris KM, Spirito P, Maron MS, Zenovich AG, Formisano F, Lesser JR, Mackey-‐Bojack S, Manning WJ, Udelson JE, Maron BJ. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-‐stage phase of hypertrophic cardiomyopathy. Circulation. 2006; 114: 216–225
17) Maron MS, Olivotto I, Betocchi S, Casey SA, Lesser JR, Losi MA, Cecchi F, Maron BJ. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med. 2003; 348: 295–303.
18) Elliott PM, Gimeno JR, Tomé MT, Shah J, Ward D, Thaman R, Mogensen J, McKenna WJ. Left ventricular outflow tract obstruction and sudden risk in patients with hypertrophic cardiomyopathy. Eur Heart J. 2006; 27: 1933–1941.
19) Sorajja P, Valeti U, Nishimura R, Ommen SR, Rihal CS, Gersh BJ, Hodge DO, Schaff HV, Holmes DR. Outcome of alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Circulation. 2008; 118: 131–139
20) Boltwood CM Jr, Chien W, Ports T. Ventricular tachycardia complicating alcohol septal ablation. N Engl J Med. 2004; 351: 1914–1915.
21) Girolami F, Ho CY, Semsarian C, Baldi M, Will ML, Baldini K, et al. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J. Am. Coll. Cardiol. 2010 Apr; 55(14):1444–1453
22) Maron BJ, Spirito P. Implantable defibrillators and prevention of suddendeath in hypertrophic cardiomyopathy. J Cardiovasc Electrophysiol. 2008; 19:1118 –1126.
23) Elliott PM, Sharma S, Varnava A, Poloniecki J, Rowland E, McKenna WJ. Survival after cardiac arrest or sustained ventricular tachycardia in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 1999; 33: 1596–1601.
24) Moon JC, McKenna WJ, McCrohon JA, Elliott PM, Smith GC, Pennell DJ. Toward clinical risk assessment in hypertrophic cardiomyopathy with gadolinium cardiovascular magnetic resonance. J Am Coll Cardiol. 2003; 41:1561–1567.
25) Maron MS, Appelbaum E, Harrigan C, Buros J, Gibson M, Hanna CA, Lesser JR, Udelson JE, Manning WJ, Maron BJ. Clinical profile and significance of delayed enhancement in hypertrophic cardiomyopathy. Circ Heart Fail. 2008; 1:184 –191.

7. References
215
26) Adabag AS, Maron BJ, Appelbaum E, Harrigan CJ, Buros JL, Gibson CM, Lesser JR, Hanna CA, Udelson JE, Manning WJ, Maron MS. Occurrence and frequency of arrhythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular magnetic resonance. J Am Coll Cardiol. 2008; 51:1369 –1374.
27) Maron BJ, Maron MS, Lesser JR, Hauser RG, Haas TS, Harrigan CJ, Appelbaum E, Main ML, Roberts WC. Sudden cardiac arrest in hypertrophic cardiomyopathy in the absence of conventional criteria for high risk status. Am J Cardiol. 2008; 101:544 –547.
28) Adabag AS, Casey SA, Kuskowski MA, Zenovich AG, Maron BJ. Spectrum and prognostic significance of arrhythmias on ambulatory Holter electrocardiogram in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2005; 45:697-‐704.
29) Maron BJ, Shen W-‐K, Link MS, Epstein AE, Almquist AK, Daubert JP, Bardy GH, Favale S, Rea RF, Boriani G, Estes NA III, Spirito P. Efficacy of implantable cardioverter-‐defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med. 2000; 342:365–373.
30) Magyar-‐Russell G, Thombs BD, Cai JX, Baveja T, Kuhl EA, Singh PP, Montenegro Braga Barroso M, Arthurs E, Roseman M, Amin N, Marine JE, Ziegelstein RC. The prevalence of anxiety and depression in adults with implantable cardioverter defibrillators: a systematic review. J Psychosom Res. 2011 Oct; 71(4):223-‐231.
31) Daniels LB, Maisel AS. Natriuretic peptides. J Am Coll Cardiol 2007; 50:e2357-‐2368.
32) Wang TJ, Larson MG, Levy D, Benjamin EJ, Leip EP, Omland T, Wolf PA, Vasan RS. Plasma natriuretic peptide levels and the risk of cardiovascular events and death. N Engl J Med 2004; 350:e655-‐663.
33) Collinson PO, Barnes SC, Gaze DC, Galasko G, Lahiri A, Senior R. Analytical performance of the N terminal pro B type natriuretic peptide (NT-‐proBNP) assay on the Elecsys 1010 and 2010 analysers. Eur J Heart Fail 2004; 6:e365-‐368.
34) Mutlu B, Bayrak F, Kahveci G, Degertekin M, Eroglu E, Basaran Y. Usefulness of N-‐terminal pro-‐B-‐type natriuretic peptide to predict clinical course in patients with hypertrophic cardiomyopathy. Am J Cardiol 2006; 98:e1504-‐1506.
35) Kitaoka H, Kubo T, Okawa M, Takenaka N, Sakamoto C, Baba Y, Hayashi K, Yamasaki N, Matsumura Y, Doi YL. Tissue Doppler imaging and plasma BNP levels to assess the prognosis in patients with hypertrophic cardiomyopathy. J Am Soc Echocardiogr 2011; 24:e1020-‐1025.
36) Maron BJ, Tholakanahalli VN, Zenovich AG, Casey SA, Duprez D, Aeppli DM, Cohn JN. Usefulness of B-‐type natriuretic peptide assay in the assessment of symptomatic state in hypertrophic cardiomyopathy. Circulation 2004; 109:e984-‐989.

7. References
216
37) Park JR, Choi JO, Han HJ, Chang SA, Park SJ, Lee SC, Choe YH, Park SW, Oh JK. Degree and distribution of left ventricular hypertrophy as a determining factor for elevated natriuretic peptide levels in patients with hypertrophic cardiomyopathy: insights from cardiac magnetic resonance imaging. Int J Cardiovasc Imaging 2012; 28:e763-‐772.
38) Vanderheyden M, Goethals M, Verstreken S, De Bruyne B, Muller K, Van Schuerbeeck E, Bartunek JJ. Wall stress modulates brain natriuretic peptide production in pressure overload cardiomyopathy. Am Coll Cardiol 2004; 44:e2349-‐2354.
39) Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kühl U, Maisch B, McKenna WJ, Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L, Keren A. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008; 29:270–276.
40) Codd MB, Sugrue DD, Gersh BJ, Melton LJ III. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A populationbased study in Olmsted County, Minnesota, 1975-‐1984. Circulation. 1989; 80:564–572.
41) Manolio TA, Baughman KL, Rodeheffer R, Pearson TA, Bristow JD, Michels VV, Abelmann WH, Harlan WR. Prevalence and etiology of idiopathic dilated cardiomyopathy (summary of a National Heart, Lung, and Blood Institute workshop. Am J Cardiol. 1992; 69:1458–1466.
42) Fuster V, Gersh BJ, Giuliani ER, Tajik AJ, Brandenburg RO, Frye RL. The natural history of idiopathic dilated cardiomyopathy. Am J Cardiol. 1981; 47:525–531.
43) Gavazzi A, Lanzarini L, Cornalba C, Desperati M, Raisaro A, Angoli L, De Servi S, Specchia G. Dilated (congestive) cardiomyopathy. Followup study of 137 patients. G Ital Cardiol. 1984; 14:492–498.
44) Kubo T, Matsumura Y, Kitaoka H, Okawa M, Hirota T, Hamada T, Hitomi N, Hoshikawa E, Hayato K, Shimizu Y, Yamasaki N, Yabe T, Nishinaga M, Takata J, Doi Y. Improvement in prognosis of dilated cardiomyopathy in the elderly over the past 20 years. J Cardiol. 2008; 52:111–117.
45) Aleksova A, Sabbadini G, Merlo M, Pinamonti B, Barbati G, Zecchin M, Bussani R, Silvestri F, Iorio AM, Stolfo D, Dal Ferro M, Dragos AM, Meringolo G, Pyxaras S, Lo Giudice F, Perkan A, di Lenarda A, Sinagra G. Natural history of dilated cardiomyopathy: from asymptomatic left ventricular dysfunction to heart failure–a subgroup analysis from the Trieste Cardiomyopathy Registry. J Cardiovasc Med (Hagerstown). 2009; 10:699–705.
46) Lenzen MJ, Boersma E, Reimer WJ, Balk AH, Komajda M, Swedberg K, Follath F, Jimenez-‐Navarro M, Simoons ML, Cleland JG. Underutilization of evidence-‐based drug treatment in patients with heart failure is only partially explained by dissimilarity to patients enrolled in landmark trials: a report from the Euro Heart Survey on Heart Failure. Eur Heart J. 2005; 26:2706–2713.

7. References
217
47) Bursi F, Weston SA, Redfield MM, Jacobsen SJ, Pakhomov S, Nkomo VT, Meverden RA, Roger VL. Systolic and diastolic heart failure in the community. JAMA. 2006; 296:2209–2216.
48) Patel P, White DL, Deswal A. Translation of clinical trial results into practice: temporal patterns of beta-‐blocker utilization for heart failure at hospital discharge and during ambulatory follow-‐up. Am Heart J. 2007; 153:515–522.
49) McMurray JJV, Adamopoulos S, Anker SD, Auricchio A, Böhm M, Dickstein K, Falk V, Filippatos G, Fonseca C, Gomez-‐Sanchez MA, Jaarsma T, Køber L, Lip GY, Maggioni AP, Parkhomenko A, Pieske BM, Popescu BA, Rønnevik PK, Rutten FH, Schwitter J, Seferovic P, Stepinska J, Trindade PT, Voors AA, Zannad F, Zeiher A; ESC Committee for Practice Guidelines. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012. Eur Heart J. 2012; 33:1787–1847.
50) Bahler RC. Assessment of prognosis in idiopathic dilated cardiomyopathy. Chest. 2002; 121:1016–1019.
51) Miura K, Matsumori A, Nasermoaddeli A, Soyama Y, Morikawa Y, Sakurai M, Kitabatake A, Nagai M, Inaba Y, Nakagawa H. Prognosis and prognostic factors in patients with idiopathic dilated cardiomyopathy in Japan—results from a nationwide study. Circulation. 2008; 72:343–348.
52) Karaca O, Avci A, Guler GB, Alizade E, Guler E, Gecmen C, Emiroglu Y, Esen O, Esen AM. Tenting area reflects disease severity and prognosis in patients with non-‐ischaemic dilated cardiomyopathy and functional mitral regurgitation. Eur J Heart Fail. 2011; 13:284–291.
53) Pecini R, Thune JJ, Torp-‐Pedersen C, Hassager C, Køber L. The relationship between mitral regurgitation and ejection fraction as predictors for the prognosis of patients with heart failure. Eur J Heart Fail. 2011; 13:1121–1125.
54) Tamura H, Watanabe T, Nishiyama S, Sasaki S, Arimoto T, Takahashi H, Shishido T, Miyashita T, Miyamoto T, Nitobe J, Hirono O, Kubota I. Increased left atrial volume index predicts a poor prognosis in patients with heart failure. J Card Fail. 2011; 17:210–216.
55) Dye C, Mertens T, Hirnschall G, Mpanju-‐Shumbusho W, Newman RD, Raviglione MC, Savioli L, Nakatani H. WHO and the future of disease control programmes. Lancet. 2013 Feb 2; 381(9864):413-‐418.
56) World Health Organization (WHO). Working to overcome the global impact of neglected tropical diseases: first WHO report on neglected tropical diseases. Geneva: WHO; 2010. Available from: http://www.who.int/neglected_diseases/2010report/en/9
57) WHO. Working to overcome the global impact of neglected tropical diseases. First WHO report on neglected tropical diseases [online], (2010).
58) Repetto E.C., Egidi A.M., Angheben A., Anselmi M., Al Rousan A., Ledezma G., Ruiz R., Torrico C., Buoninsegna M., Andreoni F., Maccagno B., De Maio G., Garelli S. Addressing the challenge of Chagas Disease in

7. References
218
a nonendemic country: The collaboration between Mèdicines Sans Frontieres (MSF), the NGO OikosCenter and the Center of Tropical Diseases of Sacro Cuore Hospital (Negrar) in Bergamo Province, Italy. Rev EspSaludPublica 2013; 87:61-‐62.
59) Angheben A, Anselmi M, Gobbi F, Marocco S, Monteiro G, Buonfrate D, Tais S, Talamo M, Zavarise G, Strohmeyer M, Bartalesi F, Mantella A, Di Tommaso M, Aiello Kh, Veneruso G, Graziani G, Ferrari M, Spreafico I, Bonifacio E, Gaiera G, Lanzafame M, Mascarello M, Cancrini G, Albajar-‐Vinas P, Bisoffi Z, Bartoloni A. Chagas disease in Italy: breaking an epidemiological silence. Euro Surveill. 2011; 16(37)
60) Guerri-‐Guttenberg RA., Grana D.R., Ambrosio G., Milei J. Chagas cardiomyopathy: Europe is not spared. Eur Heart J. 2008; 29(21):2587-‐2591.
61) Yacoub S., Mocumbi AO.,Yacoub MH. Neglected tropical disease: I. Chagas disease: myocardial disease. Heart 2008; 94:244-‐248
62) Rassi A. Jr., Rassi A. Marin-‐Neto JA. Chagas disease. Lancet 2010; 375:1388-‐1402
63) Munoz-‐Saravia S.G., Haberland A., Wallukat G., Schimke I. Chronic Chagas’ heart disease: a disease on its way to becoming a worldwide health problem: epidemiology, etiopathology, treatment, pathogenesis and laboratory medicine. Heart Fail Rev. 2012; 17(1):45-‐64
64) Shikanai-‐Yasuda MA, Carvalho NB. Oral transmission of Chagas disease. Clin Infect Dis 2012; 54:845-‐852.
65) Perez-‐Molina JA, Norman F, Lopez-‐Velez R. Chagas disease in non-‐endemic countries: epidemiology, clinical presentation and treatment. Curr Infect Dis Rep 2012; 14:263-‐274.
66) Biolo A., Ribeiro AL., Clausell N. Chagas cardiomyopathy-‐Where do we stand after a hundred years? Progress in Cardiovascular Disease. 2010; 52:300-‐316
67) Marin-‐Neto JA., Cunha-‐Neto E., Maciel BC., Simoes MV. Pathogenesis of chronic Chagas heart disease. Circulation 2007; 115:1109-‐1123.
68) Rassi AJr, Rassi A, Little WC, Xavier SS, Rassi SG, Rassi AG, Rassi GG, Hasslocher-‐Moreno A, Sousa AS, Scanavacca MI. Development and Validation of a Risk Score for Predicting Death in Chagas’ Heart Disease. N. Eng J Med 2006; 355:799-‐808
69) Pinazo MJ, Espinosa G, Cortes-‐Lletget C, Posada Ede J, Aldasoro E, Oliveira I, et al. Immunosuppression and chagas disease: a management challenge. PLoS Negl Trop Dis 2013; 7:e1965.
70) Rocha MOC., Teixeira MM., Ribeiro AL. An update on the management of Chagas cardiomyopathy. Expert Rev. Anti Infect. 2007; 5(4):727-‐743
71) Marin-‐Neto JA , Rassi A Jr, Morillo CA, Avezum A, Connolly SJ, Sosa-‐Estani S, Rosas F, Yusuf S. Rationale and design of a randomized placebo-‐controlled trial assessing the effects of etiologic treatment in

7. References
219
Chagas' cardiomyopathy: The BENznidazole Evaluation For Interrupting Trypanosomiasis (BENEFIT). Am Heart J. 2008; 156(1):37-‐43
72) Bern C., Montgomery SP., Herwaldt BL, Rassi A Jr, Marin-‐Neto JA., Dantas RO., Maguire JH., Acquatella H., Morillo C., Kirchhoff LV., Gilman RH., Reyes PA., Salvatella R., Moore AC. Evaluation and treatment of chagas disease in the United States: a systematic review. JAMA. 2007 Nov 14; 298(18):2171-‐2181.
73) Perez-‐Molina JA, Perez-‐Ayala A, Moreno S, Fernandez-‐Gonzalez MC, Zamora J, Lopez-‐Velez R. Use of benznidazole to treat chronic Chagas' disease: a systematic review with a meta-‐analysis. J AntimicrobChemother 2009; 64:1139-‐1147.
74) SosaEstani, S., E. L. Segura, A. M. Ruiz, E. Velazquez, B. M. Porcel, and C. Yampotis. Efficacy of chemotherapy with benznidazole in children in the indeterminate phase of Chagas’ disease. Am. J. Trop. Med. Hyg. 1998; 59:526–529
75) De Andrade, A. L., F. Zicker, R. M. de Oliveira, S. Almeida Silva, A. Luquetti, L. R. Travassos, I. C. Almeida, S. S. de Andrade, J. G. de Andrade, and C. M. Martelli. Randomized trial of efficacy of benznidazole in treatment of early Trypanosomacruzi infection. Lancet. 1998; 348:1407–1413.
76) Roberti RR, Martinez EE, Andrade JL, et al. Chagas cardiomyopathy and captopril. Eur Heart J 1992; 13:966-‐970.
77) Botoni FA, Poole-‐Wilson PA, Ribeiro AL, et al. A randomized trial of carvedilol after renin-‐angiotensin system inhibition in chronic Chagas cardiomyopathy. Am Heart J 2007; 153:544-‐548
78) Singh SN., Fletcher RD., Fisher SG., Singh BN, Lewis HD, Deedwania PC, Massie BM, Colling C, Lazzeri D. Amiodarone in patients with congestive heart failure and asymptomatic ventricular arrhythmia. Survival Trial of Antiarrhythmic Therapy in Congestive Heart Failure. N. Engl. J. Med 1995; 333(2):77-‐82
79) Rassi A. Jr. Implantable cardioverter-‐defibrillators in patients with chagas heart disease: misperceptions, many questions and the urgent need for a randomized clinical trial. J. Cardiovasc. Electrophysiol. 2007; 18:1241-‐1243

7. References
220
Chapter 5: Translational Routes from Altered Molecular Homeostasis to Treatment Opportunities
1) Maron, B.J., Hypertrophic cardiomyopathy: a systematic review. JAMA, 2002; 287(10): p. 1308-‐1320.
2) Bos, J.M., lA. Towbin, and M.J. Ackerman. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coli Cardiol, 2009; 54(3): p. 201-‐211.
3) Maron, B.J., et al. Implantable cardioverter-‐defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. Jama, 2007; 298(4): p. 405-‐412.
4) Olivotto l, Girolami F, Nistri S, Rossi A, Rega L, Garbini F, Grifoni C, Cecchi F, Yacoub MH. The Many Faces of Hypertrophic Cardiomyopathy:From Developmental Biology to Clinical Practice. J Cardiovasc Trans Res, 2009; 2:349-‐367
5) Maron MS, Olivotto I, Betocchi S, Casey SA, Lesser JR, Losi MA, Cecchi F, Maron BJ. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med. 2003; 348:295-‐303.
6) Cecchi F, Olivotto I, et al. Coronary microvascular dysfunction and prognosis in hypertrophic cardiomyopathy. N Engl J Med. 2003; 349:1027-‐1035.
7) Olivotto I, Cecchi F, Gistri R, Lorenzoni R, Chiriatti G, Girolami F, Torricelli F, Camici PG. Relevance of coronary microvascular flow impairment to long-‐term remodeling andsystolic dysfunction in hypertrophic cardiomyopathy. J Am Coli Cardiol. 2006; 47:1043-‐1048.
8) Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation. 2001; 104:2517-‐2524
9) Goldenberg I., et al. Long-‐QT syndrome after age 40. Circulation, 2008; 117(17): p. 2192-‐2201.
10) Hobbs JB, et al. Risk of aborted cardiac arrest or sudden cardiac death during adolescence in the long-‐QT syndrome. Jama, 2006; 296(10):1249-‐1254.
11) Jouven X., et al. Relation between QT duration and maximal wall thickness in familial hypertrophic cardiomyopathy. Heart, 2002; 88(2):153-‐157.
12) Martin A.B., A. Garson, Jr., and J.c. Perry. Prolonged QT interval in hypertrophic and dilated cardiomyopathy in children. Am Heart J, 1994; 127(1):64-‐70.
13) James G. McCormack, Rick L. Barr, Andrew A. Wolff, Gary D. Lopaschuk. Ranolazine Stimulates Glucose Oxidation in Normoxic, Ischemic, and Reperfused Ischemic Rat Hearts. Circulation, 1996; 93: 135-‐142.

7. References
221
14) Antzelevitch C, Belardinelli L, Zygmunt AC, Burashnikov A, Di Diego JM, Fish JM, Cordeiro JM, Thomas G. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation, 2004; 110(8): 904-‐910.
15) Scirica BM, Morrow DA, Hod H, Murphy SA, Belardinelli L, Hedgepeth CM, Molhoek P, Verheugt FW, Gersh BJ, McCabe CH, Braunwald E. Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non ST-‐segment elevation acute coronary syndrome: results from the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST-‐Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-‐TIMI 36) randomized controlled trial. Circulation, 2007; 116(15): 1647-‐1652.
16) Sossalla S, Wagner S, Rasenack EC, Ruff H, Weber SL, Schöndube FA, Tirilomis T, Tenderich G, Hasenfuss G, Belardinelli L, Maier LS. Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts. Role of late sodium current and intracellular ion accumulation. J Mol Cell Cardiol, 2008; 45(1): 32-‐43
17) Coppini R, Ferrantini C, Yao L, Fan P, Del Lungo M, Stillitano F, Sartiani L, Tosi B, Suffredini S, Tesi C, Yacoub M, Olivotto I, Belardinelli L, Poggesi C, Cerbai E, Mugelli A. Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation. 2013 Feb 5; 127(5):575-‐584.
18) Rastogi S, Sharov VG, Mishra S, Gupta RC, Blackburn B, Belardinelli L, Stanley WC, Sabbah HN. Ranolazine combined with enalapril or metoprolol prevents progressive LV dysfunction and remodeling in dogs with moderate heart failure. Am J Physiol Heart Circ Physiol. 2008; 295:H2149-‐2155.
19) Deshmukh SH, Patel SR, Pinassi E, Mindrescu C, Hermance EV, Infantino MN, Coppola JT, Staniloae CS. Ranolazine improves endothelial function in patients with stable coronary artery disease. Coron Artery Dis. 2009; 20:343-‐347.
20) Olivotto I, Girolami F, Ackerman MJ, Nistri S, Bos JM, Zachara E, et al. Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin. Proc. 2008 Jun; 83(6):630–638.
21) Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J. Am. Coll. Cardiol. [Internet]. American College of Cardiology Foundation; 2012 Aug 21 [cited 2012 Oct 7]; 60(8):705–715.
22) Ali J. Marian. Experimental Therapies in Hypertrophic Cardiomyopathy. J. of Cardiovasc. Trans. Res. (2009) 2:483–492.
23) Lin G, Nishimura RA, Gersh BJ, Ommen S, Ackerman M, Brady PA. Device complications and inappropriate implantable

7. References
222
cardioverterdefibrillator shocks in patients with hypertrophic cardiomyopathy. Heart. 2009; 95:709 –714.
24) Maron BJ, Spirito P. Implantable defibrillators and prevention of sudden death in hypertrophic cardiomyopathy. J Cardiovasc Electrophysiol. 2008; 19:1118 –1126.
25) Berul CI, Van Hare GF, Kertesz NJ, Dubin AM, Cecchin F, Collins KK, Cannon BC, Alexander ME, Triedman JK, Walsh EP, Friedman RA. Results of a multicenter retrospective implantable cardioverterdefibrillator registry of pediatric and congenital heart disease patients. J Am Coll Cardiol. 2008; 51:1685–1691.
26) Maisel WH. Transvenous implantable cardioverter-‐defibrillator leads: the weakest link. Circulation. 2007; 115:2461–2463.
27) Jayatilleke I, Doolan A, Ingles J, McGuire M, Booth V, Richmond DR, Semsarian C. Long-‐term follow-‐up of implantable cardioverter defibrillator therapy for hypertrophic cardiomyopathy. Am J Cardiol. 2004; 93:1192–1194.
28) Tereshchenko LG, Faddis MN, Fetics BJ, Zelik KE, Efimov IR, Berger RD. Transient local injury current in right ventricular electrogram after implantable cardioverter-‐defibrillator shock predicts heart failure progression. J Am Coll Cardiol 2009; 54:822-‐828.
29) Xie J, Weil MH, Sun S, et al. Highenergy defibrillation increases the severity of postresuscitation myocardial dysfunction. Circulation 1997; 96:683-‐688.
30) Poole JE, Johnson GW, Hellkamp AS, et al. Prognostic importance of defibrillator shocks in patients with heart failure. N Engl J Med 2008; 359:1009-‐1017.
31) Moss AJ, Schuger C, Beck CA, Brown MW, Cannom DS, Daubert JP, Estes NA 3rd, Greenberg H, Hall WJ, Huang DT, Kautzner J, Klein H, McNitt S, Olshansky B, Shoda M, Wilber D, Zareba W, MADIT-‐RIT Trial Investigators. Reduction in inappropriate therapy and mortality through ICD programming. N Engl J Med. 2012 Dec 13; 367(24):2275-‐2283
32) Bardy GH, Smith WM, Hood MA, Crozier IG, Melton IC, Jordaens L, Theuns D, Park RE, Wright DJ, Connelly DT, Fynn SP, Murgatroyd FD, Sperzel J, Neuzner J, Spitzer SG, Ardashev AV, Oduro A, Boersma L, Maass AH, Van Gelder IC, Wilde AA, van Dessel PF, Knops RE, Barr CS, Lupo P, Cappato R, Grace AA. An entirely subcutaneous implantable cardioverter-‐defibrillator. N Engl J Med. 2010 Jul 1; 363(1):36-‐44.
33) Germain DP. Fabry disease. Orphanet J Rare Dis 2010; 5:30-‐79. 34) Cecchi F, Tomberli B, Morrone A. Anderson Fabry disease, the histrionic
disease: from genetic to clinical management. Cardiogenetics, 2013; 3 (1s), e3.
35) Porto C, Pisani A, Rosa M, Acampora E, Avolio V, Tuzzi MR, Visciano B, Gagliardo C, Materazzi S, la Marca G, Andria G, Parenti G. Synergy between the pharmacological chaperone 1-‐deoxygalactonojirimycin

7. References
223
and the human recombinant alphagalactosidase A in cultured fibroblasts from patients with Fabry disease. J Inherit Metab Dis 2012; 35:513–520.
36) IShii BS. Pharmacological chaperone therapy for Fabry disease. In Vitro. 2012; 88:18–30.
37) Sugawara K, Tajima Y, Kawashima I, Tsukimura T, Saito S, Ohno K. Molecular interaction of imino sugars with human a -‐galactosidase : Insight into the mechanism of complex formation and pharmacological chaperone action in Fabry disease. Mol. Genet. Metab. 2009; 96(4):233–238.
38) Waldek S, Germain DP, Wanner C, Warnock DG, Deegan P. Enzyme replacement therapy for Fabry’s disease. Lancet [Internet]. Elsevier Ltd; 2010 May [cited 2010 Aug 24]; 375(9725):1523–1523
39) Brady R. Enzyme replacement therapy: conception, chaos and culmination. Philos. Trans. R. Soc. Lond. B. Biol. Sci. [Internet]. 2003 May [cited 2010 Aug 28]; 358(1433):915–919
40) Wu X, Katz E, Valle C Della, Mascioli K, Flanagan J, Castelli JP, et al. A pharmacogenetic approach to identify mutant forms of α-‐galactosidase a that respond to a pharmacological chaperone for Fabry disease. Hum. Mutat.. 2011 May [cited 2011 Jun 4];
41) Yam GH-‐F, Bosshard N, Zuber C, Steinmann B, Roth J. Pharmacological chaperone corrects lysosomal storage in Fabry disease caused by trafficking-‐incompetent variants. Am. J. Physiol. Cell Physiol. [Internet]. 2006 Apr [cited 2010 Aug 28]; 290(4):C1076–1082.
42) Shin S, Murray G, Kluepfel-‐Stahl S, AM. Screening for pharmacological chaperones in fabry disease. Biochem.. 2007 [cited 2010 Aug 27]; 359(1):168–173.
43) Kolter T, Wendeler M. Chemical chaperones-‐-‐a new concept in drug research. Chembiochem . 2003 Apr; 4(4):260–264.
44) Galina Kuznetsov, Sanjay K. Nigam. Folding of Secretory and Membrane Proteins. N Engl J Med 1998; 339:1688-‐1695.
45) Benjamin ER, Khanna R, Schilling A, Flanagan JJ, Pellegrino LJ, Brignol N, et al. Co-‐administration With the Pharmacological Chaperone AT1001 Increases Recombinant Human α-‐Galactosidase A Tissue Uptake and Improves Substrate Reduction in Fabry Mice. Mol. Ther. [Internet]. Nature Publishing Group; 2009.
46) Khanna R, Soska R, Lun Y, Feng J, Frascella M, Young B, et al. The pharmacological chaperone 1-‐deoxygalactonojirimycin reduces tissue globotriaosylceramide levels in a mouse model of Fabry disease. Mol. Ther. [Internet]. Nature Publishing Group; 2010 Jan [cited 2010 Aug 26]; 18(1):23–33.
47) Shen JS, Edwards NJ, Hong YB, Murray GJ. Isofagomine increases lysosomal delivery of exogenous glucocerebrosidase. Biochem Biophys Res Commun 2008; 369: 1071-‐1075.

7. References
224
Chapter 6: Conclusive remarks
1) Force T, Bonow RO, Houser SR, Solaro RJ, Hershberger RE, Adhikari B, Anderson ME, Boineau R, Byrne BJ, Cappola TP, Kalluri R, LeWinter MM, Maron MS, Molkentin JD, Ommen SR, Regnier M, Tang WH, Tian R, Konstam MA, Maron BJ, Seidman CE. Research Priorities in Hypertrophic Cardiomyopathy. Report of a Working Group of the National Heart, Lung, and Blood Institute. Circulation. 2010 Sep 14;122(11):1130-‐3.

8. Publications

8. Publications
226
“If the facts don`t fit the theory,
change the facts”
Albert Einstein d

8. Publications
227
8.1 List of publications
1. Tomberli B, Cecchi F, Sciagrà R, Berti V, Lisi F, Torricelli F, Morrone A, Castelli G, Yacoub MH, Olivotto I. “Coronary microvascular dysfunction is an early feature of cardiac involvement in patients with Anderson-‐Fabry disease.” Eur J Heart Fail 2013 Dec; 15(12):1363-‐73. [Full text in section 8.2]
2. Olivotto I, Maron BJ, Tomberli B, Appelbaum E, Salton C, Haas TS,
Gibson M, Nistri S, Servettini E, Chan RH, Udelson JE, Lesser JR,
Cecchi F, Manning WJ, Maron MS. “Obesity and its Association to Phenotype and Clinical Course in Hypertrophic Cardiomyopathy. “ J Am Coll Cardiol 2013 Jul 30;62(5):449-‐57. [Full text in section 8.2]
3. Castelli G, Fornaro A, Ciaccheri M, Dolara A, Troiani V, Tomberli B,
Olivotto I, Gensini GF. “Improving survival rates of patients with idiopathic dilated cardiomyopathy in Tuscany over three decades: impact of evidence based medicine”. Circulation Heart Failure 2013
Sep 1;6 (5):913-‐21. [Full text in section 8.2]
4. D'Amato R, Tomberli B, Castelli G, Spoladore R, Girolami F, Fornaro
A, Caldini A, Torricelli F, Camici P, Gensini GF, Cecchi F, Olivotto I.
“Prognostic Value of N-‐Terminal Pro-‐Brain Natriuretic Peptide in Outpatients With Hypertrophic Cardiomyopathy”. Am J Cardiol. 2013
Oct 15;112 (8):1190-‐1196. [Full text in section 8.2]
5. Cecchi F, Tomberli B, Morrone A. “Anderson Fabry disease, the histrionic disease: from genetic to clinical management.”
Cardiogenetics, 2013, 3 (1s), e3. [Full text in section 8.2]
6. Olivotto I, Tomberli B, Spoladore R, Mugelli A, Cecchi F, Camici PG.
“Hypertrophic cardiomyopathy: the need for randomized trials”. Global Cardiology Science and Practice 2013:31. [Full text in section
8.2]
7. Tomberli B, Girolami F, Coppini R, Ferrantini C, Rossi A, Cecchi F,
Olivotto I. “Management of refractory symptoms in hypertrophic

8. Publications
228
cardiomyopathy with restrictive pathophysiology: novel perspectives for ranolazine”. [Article in Italian] G Ital Cardiol (Rome) 2012 Apr;13
(4):297-‐303. [Full text in section 8.2]
8. Cecchi F, Tomberli B, Olivotto I. “Clinical and molecular classification of cardiomyopathies.” Review article. Global Cardiology Science and
Practice 2012. Vol. 2012 1, 4. [Full text in section 8.2]
9. Girolami F, Bardi S, Berti L, Cecchi F, Servettini E, Tomberli B.,
Torricelli F, Olivotto I. ”Rilevanza clinica del test genetico nella
cardiomiopatia ipertrofica” [Clinical relevance of genetic testing in hypertrophic cardiomyopathy]. Recenti progressi in medicina, 2011
vol. 102; p. 486-‐493. [Full text in section 8.2]
10. Chan RH, Maron BJ, Olivotto I, Assenza EG, Lesser JR, Haas T, Gruner
C, Crean AM, Rakowski H, Udelson JE, Rowin E, Lombardi M,
Tomberli B, et al. “Prognostic Utility of Quantitative Contrast-‐Enhanced Cardiovascular Magnetic Resonance in Hypertrophic
Cardiomyopathy.” Sumbitted. [Abstract in section 8.2]
11. Vannucchi V, Tomberli B, Zammarchi L, Fornaro A, Castelli G, Pieralli
F, Berni B, Yacoub S, Bartoloni A, Olivotto I. “Chagas disease as a cause of heart failure and ventricular arrhythmias in patients long removed from endemic areas: an emerging problem in Europe”. Journal of Cardiovascular Medicine, in press. [Abstract in section
8.2]
12. Girolami F, Iascone M, Tomberli B, Bardi S. Benelli M, Marseglia G,
Pescucci C, Pezzoli L, Sana ME, Basso C, Marziliano N, Merlini P.A,
Fornaro A, Cecchi F, Torricelli F, Olivotto I. “Novel alpha-‐actinin-‐2 variant associated with familial hypertrophic cardiomyopathy and juvenile atrial arrhythmias: a massively parallel sequencing study”.
Submitted. [Abstract in section 8.2]
13. Coppini R, Ho CY, Ashley E, Day S, Ferrantini C, Girolami F, Tomberli
B, Bardi S, Torricelli F, Cecchi F, Poggesi C, Tardiff J, Olivotto I.”

8. Publications
229
Clinical phenotype and outcome of Hypertrophic Cardiomyopathy associated with sarcomere thin filament gene mutations”.
Submitted. [Abstract in section 8.2]
14. Tomberli B, Olivotto I, Ferrantini C, Coppini R, Fornaro A, Girolami F,
Castelli G, Pieragnoli P, Padeletti L, Cecchi F. “Outcome of Hypertrophic Cardiomyopathy associated with sarcomere protein gene mutations: impact of the Implantable Cardioverter-‐
Defibrillator”. [Abstract in section 8.2]
15. B. Tomberli, A. Fornaro, S. Bardi, F. Torricelli, M. Benelli, C. Pescucci,
F. Cecchi, F. Girolami, I. Olivotto. “A novel desmoplakin dominant mutation responsible for Carvajal/Naxos syndrome identified by
exome sequencing”. [Abstract in section 8.2]
16. E.J. Rowin, B. Tomberli, A. Arretini, B.J. Maron, S.A. Casey, R.
Garberich, M.S. Link, R.H.M. Chan, E. Appelbaum, J.R. Lesser, J.E.
Udelson, M.S. Maron, F. Cecchi, I. Olivotto. “Prevalence and clinical significance of heart failure symptoms in patients with nonobstructive hypertrophic cardiomyopathy”. [Abstract in section
8.2]
17. Girolami F, Tomberli B, Bardi S, Berti L, Pippucci T, , Cecchi F,
Torricelli F, Olivotto I. “Outcome of hypertrophic cardiomyopathy associated to MYBPC3 mutations: a founder effect in Tuscany”.
[Abstract available in section 8.2].
18. Fornaro A, Castelli G, Ciaccheri M, Tomberli B, Olivotto I, Gensini GF.
“Survival of chemotherapy-‐induced cardiomyopathy in the last two
decades”. [Abstract available in section 8.2]

8. Publications
230
8.2 Full-‐text papers and abstracts

. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Coronary microvascular dysfunction is an earlyfeature of cardiac involvement in patients withAnderson–Fabry diseaseBenedetta Tomberli1,2*, Franco Cecchi1, Roberto Sciagra3, Valentina Berti3,Francesca Lisi4, Francesca Torricelli5, Amelia Morrone6, Gabriele Castelli2,Magdi H. Yacoub7, and Iacopo Olivotto1,2
1Department of Clinical and Experimental Medicine, University of Florence, Italy; 2Referral Center for Myocardial Diseases, Department of Cardiology, Careggi University Hospital,Florence, Italy; 3Nuclear Medicine Unit, Department of Clinical Physiopathology, Careggi University Hospital, Florence, Italy; 4Department of Radiology, Careggi University Hospital,Florence, Italy; 5Unit for Genetic Diagnosis, Careggi University Hospital, Florence, Italy; 6Metabolic and Muscular Unit, Clinic of Pediatric Neurology, Meyer University Hospital, Florence,Italy; and 7Heart Science Centre, Imperial College London, Harefield, UK
Received 27 February 2013; revised 31 May 2013; accepted 7 June 2013
Aims Male patients with Anderson–Fabry disease (AFD) often exhibit cardiac involvement, characterized by LV hypertrophy(LVH), associated with severe coronary microvascular dysfunction (CMD). Whether CMD is present in patients withoutLVH, particularly when female, remains unresolved. The aim of the study was to investigate the presence of CMD by posi-tron emission tomography (PET) in AFD patients of both genders, with and without evidence of LVH.
Methodsand results
We assessed myocardial blood flow following dipyridamole infusion (Dip-MBF) with 13N-labelled ammonia by PET in 30AFD patients (age 51+ 13 years; 18 females) and in 24 healthy controls. LVH was defined as echocardiographic maximalLV wall thickness ≥13 mm. LVH was present in 67% of patients (n ¼ 20; 10 males and 10 females). Dip-MBF was reducedin all patients compared with controls (1.8+ 0.5 and 3.2+0.5 mL/min/g, respectively, P , 0.001). For both genders,flow impairment was most severe in patients with LVH (1.4+0.5 mL/min/g in males and 1.9+0.5 mL/min/g infemales), but was also evident in those without LVH (1.8+ 0.3 mL/min/g in males and 2.1+ 0.4 mL/min/g in females;overall P ¼ 0.064 vs. patients with LVH). Analysis of variance (ANOVA) for the 17 LV segments showed marked regionalheterogeneity of MBF in AFD (F ¼ 4.46, P , 0.01), with prevalent hypoperfusion of the apical region. Conversely, con-trols showed homogeneous LV perfusion (F ¼ 1.25, P ¼ 0.23).
Conclusions Coronary microvascular function is markedly impaired in AFD patients irrespective of LVH and gender. CMD mayrepresent the only sign of cardiac involvement in AFD patients, with potentially important implications for clinicalmanagement.
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -Keywords Fabry disease † Coronary microvascular dysfunction † PET † Early phenotype
Introduction
Anderson–Fabry disease (AFD) is an X-linked disorder of glyco-sphingolipid catabolism caused by deficient or absent activity of thelysosomal enzyme a-galactosidase A (a-gal A). AFD is considereda rare disease, with a reported prevalence in the general populationof � 1:117 000.1,2 Recently, however, newborn screening initiatives
have found an unexpectedly high prevalence of AFD, as high as 1 in�3900 newborns,3,4 suggesting that previous figures may representa substantial underestimate.
As a consequence of a-gal A deficiency, progressive accumulationof globotriaosylceramide (Gb3) occurs in various tissues throughoutthe body, including the endothelium, and is associated with proteanclinical manifestations, including renal failure, juvenile stroke, dys-pnoea, angina, and sudden cardiac death.5,6 Cardiac involvement
* Corresponding author. Dipartimento Cuore e vasi, San Luca Vecchio, Centro di Riferimento Cardiomiopatie, Azienda Ospedaliero Universitaria Careggi, Largo Brambilla 3, 50134,Firenze, Italy. Tel +39 055 7945138, Fax +39 055 7949335, Email: [email protected]
Published on behalf of the European Society of Cardiology. All rights reserved. & The Author 2013. For permissions please email: [email protected].
European Journal of Heart Failuredoi:10.1093/eurjhf/hft104
European Journal of Heart Failure Advance Access published June 30, 2013 by guest on July 1, 2013
http://eurjhf.oxfordjournals.org/D
ownloaded from

has been described in both genders and is mainly characterized byvariable degrees of LV hypertrophy (LVH) and interstitial fibrosis.7
Although commonly classified as a storage disease of the heart,AFD cardiomyopathy is characterized by true LVH, as Gb3 accumu-lation only accounts for a very limited proportion of cardiac mass in-crease.8
Coronary microvascular dysfunction (CMD) is an importantfeature of AFD cardiomyopathy, accounting for the considerableprevalence of angina, in the absence of epicardial coronarydisease.8,9 CMD has mostly been described in male AFD patientswith LVH.9– 11 However, in patients with sarcomeric hypertrophiccardiomyopathy, a different model of genetically determined LVH,CMD is a diffuse phenomenon within the LV, may involve non-hypertrophied LV walls, and may possibly precede LVH develop-ment.12–15 Whether CMD may be present in AFD patientswithout LVH remains unresolved. Furthermore, the prevalenceand severity of CMD in female AFD patients, generally exhibitingmore subtle cardiac manifestation than men, are undefined. Bothissues are potentially relevant to early diagnosis of AFD cardiomyop-athy and long-term risk stratification.12–15
Thus, the present study was undertaken to investigate coronarymicrovascular function in a cohort of male and female AFD patientsat different stages of disease, with and without evidence of LVH, inorder to assess whether CMD may occur independently of LVHand in the absence of any other sign of cardiomyopathy. We there-fore assessed maximal global and regional myocardial blood flowafter dipyridamole (Dip-MBF), as an expression of coronary micro-vascular function, using positron emission tomography (PET).
Methods
Study populationThirty patients from 13 families (12 males, 18 females; mean age 51+13years) with AFD were included in a cross-sectional study. Diagnosis wasbased on measurement ofa-gal A enzyme activity in leucocytes and con-firmed by direct sequencing of the a-gal A gene (GLA gene) on genomicDNA isolated from whole blood. Enzyme activity and genetic analysiswere assessed as previously described.16 In two patients (individual II-1from family 3 and individual III-7 from family 7), the final diagnosis ofAFD was achieved by means of endomyocardial biopsy from the rightinterventricular septum. Between 2006 and 2010, all patients underwentthorough cardiac evaluation by clinical examination, resting ECG, Holtermonitoring, and two-dimensional (2D) echocardiography includingtissue Doppler analysis.
Coronary microvascular function was assessed by PET as describedbelow. CAD was excluded in AFD patients at the time of enrolment bymaximal, symptom-limited treadmill or cycloergometer exercise test,in asymptomatic patients with low pre-test probability (i.e. young andwithout cardiovascular risk factors), followed by coronary angiographyor computed tomography (CT)-angiography in the presence of a positiveor dubious test result; patients with a high risk profile and/or angina weredirectly referred for coronary angiography or CT-angiography. Further-more, patients with diabetes were excluded. The same study protocolwas employed in 24 healthy controls, who were investigated for exclu-sion of heart disease, and were comparable with the patients in termsof gender and age (13 males and 11 females, P ¼ 0.55 vs. patients; age46+16 years, P ¼ 0.23 vs. patients). Healthy controls had no evidenceof cardiomyopathy, with normal ECG, echocardiographic examination,
and without common cardiovascular risk factors. The study protocolwas approved by the local research ethics committee, and writteninformed consent in lay Italian language was obtained from eachsubject included in the study
EchocardiographyStandard echocardiographic studies were performed with commerciallyavailable instruments. Clinical diagnosis of LVH was based on the demon-stration by 2D echocardiogram of a hypertrophied and non-dilated LV(wall thickness ≥ 13 mm) in the absence of another cardiac or systemicdisease capable of producing a similar degree of hypertrophy. The LVend-diastolic and end-systolic diameter, left atrial diameter, and magni-tude and distribution of LVH were assessed as previously described.17
In all patients, maximum LV wall thickness values were measured at thetime of PET. The LVEF was measured in the standard four-chamberviewby thearea–lengthmethod. Obstruction of theLVoutflowwascon-sidered present when a peak outflow gradient of ≥ 30 mmHg waspresent under basal conditions. Diastolic function was assessed accord-ingly to current guidelines.18
Positron emission tomographyAll cardiac PET scans were performed in the Nuclear Medicine Labora-tory (Nuclear Medicine Unit, Department of Clinical Physiopathology,Careggi University Hospital) in Florence between 2006 and 2011, afteran appropriate period of pharmacological wash-out for patients receivingpharmacological treatment. Patients were positioned on the couch of thePET scanner (General Electrics Advance PET, Milwaukee, WI, USA) and a5 min transmission scan was recorded for subsequent attenuation correc-tion of emission data, according to a previously described procedure.19
Then, near maximal hyperaemia was induced by i.v. administration ofdipyridamole (0.56 mg/kg of body weight over 4 min). Three minutes fol-lowing theendofdipyridamole infusion, abolusof370MBqofnitrogen-13ammonia (13N-labelled ammonia) diluted in 10 mL of saline solution wasinjected i.v. over a period of 15–20 s and followed by a 10 mL saline solu-tion flush at a rate of 2 mL/s. A dynamic scan with 15 frames of increasingduration was acquired for4 min, followed bya prolonged static acquisitionof 15min. Particular carewas taken toavoid anypatient motion in order tominimize possible misalignment problems between transmission andemission scans. Data were analysed with an operator interactive com-puter program [PMOD Cardiac Modelling (PCARD), version 3.3,PMOD Technologies, Zurich, Switzerland]. Briefly, anatomic imageswere reconstructed using the static acquisition, and reoriented accordingto the heart axis. After reconstruction, the dynamic images were reor-iented as well. On the short axis slices, regions of interest were manuallydrawn including: (i) therightventricularcavity; (ii) theLVcavity; and(iii) theLV wall from the apex through the base (identified by the appearance ofthe membranous septum). The regions of interest were edited by thestandard PMOD volume of interest (VOI) tool to derive the relatedVOIs. The three VOIs were then copied on all 13N-labelled ammoniadynamic images to extract the corresponding time–activity curves. Thearterial input function was obtained from the LV cavity time–activitycurve. The myocardial uptake was derived from the LV wall VOI. Myocar-dial perfusion was calculated from model fitting of the arterial input func-tion and tissue time–activity curves.20 The LV wall was divided into 17segments: septal (apical septal, midinferoseptal, midanteroseptal, basalinferoseptal, and basal anteroseptal), anterior (apical anterior, midanter-ior, and basal anterior), lateral (apical lateral, midlateral, and basal lateral),inferior (apical inferior, midinferolateral, midinferior, basal inferolateral,and basal inferior), and apical.21 Mean hyperaemic MBF for the entireleft ventricle was obtained by volume-weighted averaging of the 17 LVsegment territories (apex, septum, anterior, lateral, and inferior).
B. Tomberli et al.Page 2 of 11
by guest on July 1, 2013http://eurjhf.oxfordjournals.org/
Dow
nloaded from

All images studies were analysed by one expert observer (R.S.), blinded topatients’ genetic, clinical, and echocardiographic data.
Statistical analysisData are expressed as mean+ standard deviation. Two-tailed unpairedStudent’s t-testwasemployed for the comparison of normally distributeddata. x2 or Fisher’s exact test, as appropriate, were utilized to comparenon-continuous variables expressed as proportions. Independent deter-minants of CMD were evaluated by logistic regression analysis. P-valuesare two-sided and considered significant when ,0.05. Calculationswere performed using the SPSS 12.0 software (Chicago, IL, USA).
Results
Demographic features and mutationalstatusThe mean age of the 30 AFD patients, at the time of PET, was 51+13years (range 23–75); 12 patients (40%) were male (Table 1). Geneticanalysis identified 11 distinct mutations in the GLA gene (Table 2,Figure 1); three index patients and seven family members sharedthe c.644A . G GLA gene mutation leading to the p.Asn215Seramino acid substitution. As expected, mean a-gal A activity in leuco-cytes was lower in males than in females (2.1+ 1.4 vs. 21+12 nmol/mg/h, respectively; P , 0.001); conversely, mean levels were com-parable in patients with and without echocardiographic evidence ofLVH (13+13 and 14+14 nmol/mg/h, respectively; P ¼ 0.75).The two patients with endomyocardial biopsy, both with echocar-diographic evidence of LVH, had typical AFD histological findings.
Overall, 8 hadahistoryof smoking, 5werecurrent smokers, and11had hypercholesterolaemia (Table 1). Three patients were obese[body mass index (BMI) .30; 10%], while hypertension, well con-trolled by medical therapy, was present in 12 patients (40%)without differences between the two subgroups. Evidence of extra-cardiac involvement was present in 19 patients (63%) (detailed inTable 2): 12 patients (40%, 9 males) had proteinuria, 5 males hadmild to moderate renal impairment (17%), and 2 patients (6%, 1male) underwent renal transplantation. Five patients had priorstroke (17%, 2 males), while neuropathic pain was present in 11patients (37%, 5 males). Nine patients with angina and dubiousstress test underwent coronary angiogram or coronaryCT-angiogram (as reported in Table 2), in order to exclude epicardialCAD. In the remaining patients, coronary disease was excluded bysymptom-limited exercise test.
Eleven patients (37%) were receiving enzyme replacementtherapy (ERT) at the time of the study, eight patients with LVH andthree without LVH. Most patients were on ACE inhibitors/ARBs(57%) and antiplatelet agents (43%), whereas only a minority wereon beta-blockers or calcium channel antagonists (17% for each class).
Evidence of Anderson–Fabry diseasecardiomyopathyTwentyof the 30 AFD patients (67%, 10 males, 10 females) had echo-cardiographic evidence of LVH, with a maximum LV wall thicknessvalue of 20+4 mm (range 13–27 mm). The remaining 10 patients(33%, 2 males, 8 females) had normal LV wall thickness values.Most patients with LVH (76%) had ECG abnormalities compatible
with LV hypertrophy/strain. Conversely, only one of the patientswithout LVH (10%) had an abnormal ECG morphology (Table 1).
Compared with patients without LVH, those with LVH were moreoften males (10/20 or 50% vs. 2/10 or 20% respectively,P ¼ 0.23) andmore often symptomatic for chest pain or dyspnoea (P , 0.05 forboth; Table 1). Patients with LVH had larger atrial volumes andmore frequent evidenceofdiastolic dysfunction.Noneof thepatientshad LV outflow tract obstruction at rest. Of note, patients with LVHwere older (56+9 years vs. 42+ 16 in those without LVH, P ¼0.005), with a strong direct relationship between age and degree ofhypertrophy, particularly in males, suggesting age-related develop-ment of AFD cardiomyopathy (Figure 2A). Finally, 8 of the 10 patientswithout LVH (i.e. 7 females and 1 males) showed normal tissueDoppler imaging mitral septal annulus velocities (adjusted per age)and absence of late gadolinium enhancement (LGE) at cardiac mag-netic resonance imaging (MRI).
Evidence of coronary microvasculardysfunction by positron emissiontomographyCoronary microvascular response to dipyridamole was blunted in allAFD patients, as compared with control subjects (1.8+0.5 and3.2+ 0.5 mL/min/g, respectively; P , 0.001) (Figure 3). Dip-MBF inAFD patients ranged from 0.8 to 2.6 mL/min/g, and was ,1.25 mL/min/g, reflecting severe impairment of coronary microvascular func-tion, in 7 patients (23%). Patients with LVH invariably showed markeddegrees of CMD (Figure 2B). However, patients without LVH alsoshowed blunted Dip-MBF values, ranging from 2.4 to as low as1.2 mL/min/g. As a result, CMD in patients without LVH was onaverage milder, but not statistically different, from that of thosewith LVH (mean 2.0+ 0.4 vs. 1.6+ 0.5 mL/min/g, respectively;P ¼ 0.064) (Figure 3).
Analysis of variance (ANOVA) for the 17 LV segments showedmarked regional heterogeneity of MBF in AFD (P , 0.01), withprevalent hypoperfusion of the apical region (Figure 4). Conversely,controls showed homogeneous perfusion of the LV (patients, F ¼4.46, P , 0.01; controls, F ¼ 1.25, P ¼ 0.23).
To exclude the possible influence of common CMD risk factors,other than AFD, we compared MBF values between patients withno history of smoking, hypertension, and dyslipidaemia with therest of the cohort. Patients without risk factors (n ¼ 12) showed asimilar degree of MBF impairment, as compared with other patients(n ¼ 18; MBF ¼ 1.6+ 0.5 mL/min/g vs. MBF ¼ 1.9+0.4 mL/min/g,respectively; P ¼ 0.12). Furthermore, there was no difference inMBF between patients on ERT vs. naıve patients (1.7+0.5 mL/min/g vs. 1.7+ 05 mL/min/g, respectively; P ¼ 0.9).
Impact of gender on microvascularfunctionCoronary microvascular dysfunctionwas more severe in males, com-pared with female AFD patients (1.5+0.4 and 1.9+0.5 mL/min/g,respectively, P ¼ 0.01) (Figure 3). Unexpectedly, however, ablunted Dip-MBF was also present in the 18 female patients, irre-spective of the presence of LVH (1.9+0.5 mL/min/g in femaleswith LVH and 2.1+ 0.4 mL/min/g in females without LVH; P ¼0.38). Of note, a 61-year-old female patient without LVH had
Microvascular dysfunction in Fabry disease Page 3 of 11
by guest on July 1, 2013http://eurjhf.oxfordjournals.org/
Dow
nloaded from

. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Table 1 Baseline and cardiac characteristics of 30 patients with Anderson–Fabry disease
All patients (n 5 30) LVH – (n 5 10) LVH 1 (n 5 20) P-value
Age, years 51+13 42+16 56+9 0.005
Age ,40 years, n (%) 6 (10) 5 (50) 1 (5) 0.009
Gender 0.23
Male, n (%) 12 (40) 2 (20) 10 (50)
Female, n (%) 18 (60) 8 (80) 10 (50)
Height, cm 167+8 165+9 168+7 0.42
Weight, kg 69+12 65+10 72+13 0.15
BMI 26+7 27+11 26+4 0.63
Genotype 0.98
p.Asn215Ser (c.644A . G), n (%) 10 (33) 3 (30) 7 (35)
Other mutations, n (%) 20 (67) 7 (70) 13 (65)
Cardiovascular risk factors
Smoking
Smokers, n (%) 5 (17) 1 (10) 4 (20) 0.64
Ex-smokers, n (%) 8 (27) 3 (30) 5 (25) 0.93
Hypercholesterolaemia, n. (%) 11 (37) 2 (20) 9 (45) 0.25
Hypertension, n (%) 12 (40) 2 (20) 10 (50) 0.23
BMI .30, n (%) 3 (10) 1 (10) 2 (10) 0.96
Therapy
Beta-blockers, n (%) 5 (17) 0 5 (25) 0.14
Calcium channel antagonists, n (%) 5 (17) 1 (10) 4 (20) 0.64
Amiodarone, n (%) 2 (7) 0 2 (10) 0.54
ACE inhibitors, n (%) 8 (27) 1 (10) 7 (35) 0.21
ARBs, n (%) 9 (30) 1 (10) 8 (40) 0.20
Antiplatelets, n (%) 13 (43) 1 (10) 12 (60) 0.017
ERT, n (%) 11 (37) 3 (30) 8 (40) 0.70
NYHA class 0.02
I, n (%) 17 (57) 9 (90) 8 (40)
II, n (%) 10 (33) 1 (10) 9 (45)
III/IV, n (%) 3 (10) 0 3 (15)
Dyspnoea, n (%) 13 (43) 1 (10) 12 (60) 0.017
Chest pain, n (%)
At rest 2 (7) 1 (10) 1 (5) 0.95
On effort 11 (37) 1 (10) 10 (50) 0.049
Palpitations, n (%) 16 (53) 5 (50) 11 (55) 0.99
Syncope, n (%) 5 (17) 2 (20) 3 (15) 0.97
NSVT, n (%) 3 (10) 0 3 (15) 0.53
PAF, n (%) 4 (13) 1 (10) 3 (15) 0.97
Pacemaker, n (%) 3 (10) 0 3 (15) 0.53
ECG
LVH, n (%) 14 (52) 1 (10) 13 (76) 0.001
Bradycardia, n (%) 11 (37) 4 (40) 7 (35) 0.94
Heart rate, b.p.m. 61+8 61+12 62+7 0.79
Short PR, n (%) 3 (10) 1 (10) 2 (10) 0.97
Mean flow ,1.25 mL/min/g, n (%) 7 (23) 1 (10) 6 (30) 0.37
Echocardiography
LV max WT, mm 16+6 9+1 20+4 ,0.01
IV septum, mm 15+6 9+1 18+5 ,0.01
LV posterior wall, mm 12+3 9+1 13+2 ,0.01
Left atrium, mm 37+7 31+5 40+6 ,0.01
Left atrium volume, mm/m2 39+13 29+9 45+11 ,0.01
Continued
B. Tomberli et al.Page 4 of 11
by guest on July 1, 2013http://eurjhf.oxfordjournals.org/
Dow
nloaded from

extreme degrees of coronary flow impairment (Dip-MBF ¼ 1.2 mL/min/g) (family 6, subject II-1 in Table 2). An inverse relationshipbetween mean Dip-MBF and degree of LVH, suggesting moresevere CMD in the presence of increasing LVH, was present only inmales (Figure2B). The10patientswith the p.Ans215SerGLA genemu-tation showed significant blunting of MBF as compared with patientswith other mutations (1.5+0.5 vs. 1.9+0.5 mL/min/g, respectively;P ¼ 0.019), despite a similar degree of hypertrophy (maximum LVwall thickness 18+8 mm vs. 15+ 5 mm, respectively, P ¼ 0.28).
Discussion
Coronary microvascular dysfunction isindependent of left ventricularhypertrophy in Anderson–Fabry diseaseThe present study demonstrates that microvascular function is mark-edly impaired in AFD patients, irrespective of any other evidence ofcardiac involvement. In our cohort, all AFD patients with normalechocardiographic findings exhibited blunting of the vasodilator re-sponse to dipyridamole, reflecting impaired microvascular function,with a mean Dip-MBF of only 2.0+ 0.4 mL/min/g, compared with3.2+ 0.5 mL/min/g in normal controls, i.e. a 60% reduction in coron-ary flow. Such a degree of CMD was only slightly milder than thatobserved in patients with clear evidence of AFD cardiomyopathyand LVH. Indeed the most severe impairment in microvascular func-tion was observed in a 61-year-old female patient, without evidenceof LVH (Dip-MBF 1.2 mL/min/g).
Of note, while MBF was globally impaired in AFD hearts, there wasmarked heterogeneity in flow, with prevalent hypoperfusion of theapical region of the left ventricle, suggesting a regional nature ofcardiac involvement. While counterintuitive in a systemic storagedisease, the novel idea of a non-homogeneous cardiac involvement
in AFD is in line with prior observations in a number of cardiomyop-athies, and particularly those related to storage or infiltrativedisorders, such as, for example, amyloidosis.22 The mechanismsaccounting for such regionally heterogeneous manifestationsremain unresolved.
Altogether, these findings point to CMD as a constant manifest-ationof AFD, which may be present independent of other instrumen-tal evidence of cardiac involvement. In males, the correlationbetween LV wall thickness and maximal MBF, and the direct relation-ship of LV wall thickness to age, both suggest that a reduced coronarymaximal blood flow may precede development of LVH (Figure 2).This concept is novel and strongly supported by the young age ofpatients with CMD in the absence of LVH. Furthermore, AFDpatients without LVH and CMD generally had none of the other fea-tures that are considered very early disease manifestations, such asreduced diastolic mitral annulus velocities or presence of LGE.23,24
Nevertheless, longitudinal studies are required to understandwhether flow abnormalities can indeed be considered to representinitial cardiac involvement followed by overt cardiomyopathy inAFD patients.
The consistent and severe blunting of MBF in patients with thep.Ans215Ser GLA gene mutation may suggest a role for geneticstatus in determining the severity of MBF. Furthermore, theconcept of the possible influence of genetic status on microvascularfunction has been proved in another more common genetic cardio-myopathy (i.e. hypertrophic cardiomyopathy).25 However, in thepresent study, the relatively small number of patients did not allowa meaningful comparison between individual mutations.
Coronary microvascular dysfunction infemale Anderson–Fabry disease patientsIn our cohort, females with LVH showed blunted Dip-MBF, althoughto a less severedegree thanmales (1.9+0.5 and 1.5+0.4 mL/min/g,
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Table 1 Continued
All patients (n 5 30) LVH – (n 5 10) LVH 1 (n 5 20) P-value
LV telediastolic diameter, mm 48+6 48+5 49+6 0.75
LV telesystolic diameter, mm 27+8 27+5 27+7 0.86
LV telediastolic volume, mL 99+32 91+19 102+37 0.40
LV telesystolic volume, mL 37+20 33+13 39+23 0.42
EF, % 63+10 65+9 63+10 0.62
Diastolic pattern 0.03
Normal, n (%) 6 (20) 6 (60) 0
Delayed relaxation, n (%) 12 (40) 4 (40) 8 (40)
Pseudonormal, n (%) 11 (37) 0 11 (50)
Restrictive, n (%) 1 (3) 0 1 (5)
LVOT obstruction, n (%) 0 0 0 N/A
Mitral regurgitation (mild or moderate) 16 (53) 4 (40) 12 (60) 0.44
Aortic regurgitation (mild or moderate) 5 (17) 2 (20) 3 (15) 0.92
Data are shown as mean+ SD or n (%).BMI, body mass index; ERT, enzyme replacement therapy; IV, interventricular; LVH –,patientswithoutLVhypertrophy; LVH+, patientswithLVhypertrophy; LVmaxWT,LVmaximalwall thickness; LVOT, LV outflow tract obstruction; N/A, not applicable; NSVT, non-sustained ventricular tachycardia; PAF, paroxysmal atrial fibrillation.
Microvascular dysfunction in Fabry disease Page 5 of 11
by guest on July 1, 2013http://eurjhf.oxfordjournals.org/
Dow
nloaded from

. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Table 2 Clinical and genetic features of the study population at the time of positron emission tomography
Family Genotype(effect onprotein)
Subject Sex Age a-Gal ERT Symptoms(NYHA class)
LVH MaxLVWT
EF Dip-MBF CAD exclusion Major organinvolvement
At clinicalonset
Atdiagnosis
AtPET
1 p.Met1Val (c.1A . G)
III-1 M 4 28 29 0.9 Yes Palpitations (1) Yes 16 67 2.4 Stress test Proteinuria, acroparaesthesia,abdominal pain
II-1 F 16 47 48 46 Yes Dyspneoa, palpitations,syncope, angina (2)
Yes 16 62 2.3 Coronaryangiogram
Acroparaesthesia
2 p.Trp44X (c.131G . A)
III-1 M 12 52 54 0.01 Yes Dyspnoea, palpitations (2) Yes 23 70 1.1 CoronaryCT-angiogram
Acroparaesthesia, RI,proteinuria
III-2 F - 39 41 11.8 No Palpitations (1) No 9 60 1.8 Stress test None
3 p.Arg112Cys (c.334C . T)
II-1 M 11 34 52 1.9 Yes Angina (1) Yes 23 66 0.8 Coronaryangiogram
Acroparaesthesia stroke,renal transplant
4 p.Ala143Thr (c.427G . A)
III-2 F - 40 42 5.8 No Palpitations (1) No 8 64 2.4 Stress test None
5 p.Asn215Ser (c.644A . G)
II-1 M 44 56 60 1.82 No None (1) Yes 25 59 1.7 CoronaryCT-angiogram
None
6 p.Asn215Ser (c.644A . G)
II-1 F - 56 60 13 No Dyspnoea, palpitations (2) No 11 67 1.2 Stress test NoneII-2 M 48 55 55 3.4 No Dyspnoea, angina (2) Yes 28 58 1.2 Coronary
angiogramRI, proteinuria
II-3 F N/A 53 56 16 No Dyspnoea (2) Yes 13 70 0.9 Stress test AcroparaesthesiaII-4 M N/A 46 53 4.2 No Dyspnoea, palpitations (2) Yes 27 62 1.0 Stress test RI, proteinuriaII-5 M – 51 48 3.6 No Dyspnoea, angina (2) Yes 22 70 1.3 Stress test NoneIII-3 F - 24 27 44 No None (1) No 7 76 2.2 Stress test NoneIII-5 F 20 25 31 5.3 No Syncope, palpitations (1) No 9 67 2.3 Stress test Acroparaesthesia
7 p.Asn215Ser (c.644A . G)
III-7 M 59 69 69 3.2 No Palpitations, dyspnea,angina (3)
Yes 23 33 1.2 Coronaryangiogram
Proteinuria
III-12 M 50 68 69 2.8 No Dyspnea, angina (2) Yes 23 71 1.5 Stress test Proteinuria, RI, stroke
8 p.Arg220X (c.658C . T)
II-2 F 22 37 47 26.6 Yes None (1) Yes 21 82 1.6 Coronaryangiogram
Renal transplant
9 p.Leu243Ser (c.728T . C)
III-1 F N/A 63 62 26 No Palpitations, dyspnoea (2) Yes 16 62 2.3 Stress test NoneIII-2 F 36 56 57 22 No Dyspnoea, palpitations,
angina (3)Yes 23 47 2.2 Stress test Proteinuria
10 p Asp299Gly (c.896A . C)
II-1 F N/A 66 68 21 No None (1) Yes 17 58 1.9 Stress test NoneIII-1 F - 49 49 31 No Palpitations (1) No 11 59 1.9 Stress test NoneIII-2 M 18 46 49 0.01 Yes None (1) Yes 15 61 2.0 Stress test Proteinuria, RI,
acroparaesthesia,abdominal pain
11 p.Arg301Pro (c.902G . C)
III-2 F 16 49 63 13.5 Yes Palpitations, dyspnoea,syncope, angina (2)
Yes 21 58 1.6 Stress test Proteinuria, acroparaesthesia,strokes
III-4 F N/A 46 56 31.6 No None (1) Yes 14 63 2.6 Stress test NoneIII-5 F N/A 55 62 18.2 No Dyspnoea (2) Yes 15 63 1.4 Stress test StrokeIV-1 M 25 30 40 0.9 Yes None (1) No 10 59 2.0 Stress test Proteinuria
12 p.Arg301X (c.901C . T)
I-1 F – 70 75 No Palpitations (1) No 9 77 2.2 Coronaryangiogram
None
Continued
B.Tom
berlietal.P
age6
of11
by guest on July 1, 2013 http://eurjhf.oxfordjournals.org/ Downloaded from

respectively; P ¼ 0.01). In addition, and rather unexpectedly, womenwithout LVH also had considerable degrees of CMD, representingthe only evidence of AFD in some of these individuals. As a result,there should be heightened awareness of the consideration ofcardiac involvement in female patients with AFD, even in theabsence of an echocardiographic phenotype. Although AFD is anX-linked disease, female patients are well known to develop the clin-ical manifestations of the disease, which can be severe and determineoutcome.26 Cardiomyopathy is not an exception to this rule, and hasbeen described in women; however, cardiac manifestations generallyhave a later onset and are less marked than those occurring in men(Figure 4).27,28
Potential mechanisms of coronarymicrovascular dysfunction and implicationfor managementThere are several potential pathophysiological mechanismsunderlying coronary blood flow impairment in AFD, and theseinclude those mediated by LVH (reduced capillary density, extravas-cular compression forces) as well as those directly affecting themicrovasculature (endothelial dysfunction due to Gb3 storage,nitric oxide pathway dysregulation, or microvascular remodelling).13
However, in patients without LVH, only the latter are present, sug-gesting that LVH per se is indeed a likely contributor to CMD,rather than a cause.
Despite considerable advances in our understanding of AFD, themechanisms leading to LVH are not well understood. Storage ofGb3 within cardiac myocytes accounts for a small amount of thewhole LV mass, which is mainly represented by true myocardialhypertrophy.8 One hypothesis is that intracellular accumulation ofGb3 may disturb cardiac energetic metabolism, representing anearly trigger for the activation of the intracellular signalling pathwaysleading to hypertrophy, fibrosis, apoptosis, and necrosis.29 Further-more, microvascular dysfunction and chronic hypoperfusion, dueto Gb3 accumulation in endothelial cells, may also play a crucialrole in the activation and perpetuation of these pathophysiologicalprocesses, even at early stages.13
The issue of identification of early organ damage has become crit-ical following the introduction in clinical practice of ERT and noveltherapeutic molecules, including pharmacological chaperonetherapy.30–33 To date, evidence of benefit of ERT in AFD patientswith overt signs of cardiomyopathy and LVH is disappointing, andearlier timing of treatment initiation has been advocated in orderto improve efficacy.11,30
Thus, early detection of subclinical cardiac involvement, as allowedby PET studies of microvascular function, may become a criticalelement in clinical decision-making especially in young AFD patients.In particular, CMD may represent a viable treatment target, poten-tially relevant to prevention of disease progression and outcome.30
In the future, the advent of cardiac magnetic resonance (CMR), byvirtue of its wider availability and more favourable safety profile com-pared with PET, may allow large-scale investigation of CMD in AFDpatients. To date, however, quantitative assessment of microvascularflow by CMR is time-consuming and largely limited to research pur-poses.15
13c.
1235
_123
6de
lCT
II-1
F15
4054
6.6
Yes
Dys
pnoe
a,pa
lpita
tions
(2)
Yes
1376
1.8
Cor
onar
yC
T-a
ngio
gram
Acr
opar
aest
hesi
a,st
roke
s,ab
dom
inal
pain
III-1
M4
1528
2.4
Yes
Non
e(1
)N
o10
531.
5St
ress
test
Acr
opar
aest
hesi
a,ab
dom
inal
pain
,pro
tein
uria
III-3
F14
1023
13.8
Yes
Non
e(1
)N
o8
632.
3St
ress
test
Acr
opar
aest
hesi
a,ab
dom
inal
pain
a-G
al¼
a-g
alac
tosi
dase
Aac
tivity
assa
yed
onle
ucoc
ytes
(nor
mal
valu
es20
–60
nmol
/mg/
h);C
T-a
ngio
gram
,com
pute
dto
mog
raph
yan
giog
ram
;Dip
-MBF
,mea
nm
yoca
rdia
lblo
odflo
wfo
llow
ing
dipy
rida
mol
ein
fusi
on;E
RT
,enz
yme
repl
acem
ent
ther
apy;
LVH
,left
vent
ricu
larh
yper
trop
hy;M
ajor
orga
nin
volv
emen
t,on
lyki
dney
(RI,
rena
lim
pair
men
tdia
gnos
edas
glom
erul
arfil
trat
ion
rate
,60
mL/
min
),pe
riph
eral
.and
cent
raln
ervo
ussy
stem
invo
lvem
enta
rere
port
edin
this
tabl
e;M
axLV
WT,
max
imal
left
vent
ricu
lar
wal
lthi
ckne
ss;N
/A,a
geat
onse
tun
know
n(d
iagn
osis
mad
eby
fam
ilysc
reen
ing)
.
Microvascular dysfunction in Fabry disease Page 7 of 11
by guest on July 1, 2013http://eurjhf.oxfordjournals.org/
Dow
nloaded from

Furthermore, is it possible tohypothesize that the severityofCMDmay be an important predictor of adverse outcome, as for other gen-etically determined cardiomyopathies.12,34
Limitations of the studyAn unavoidable limitation of the present study lies in the smallsample size and heterogeneity of our study population. Selectinga more homogeneous study cohort, by excluding patients withco-morbidities or cardiovascular risk factors, would not havebeen feasible in a rare and multisystemic disorder such as AFD.Thus, myocardial blood flow may have been affected in ourcohort by the interplay of co-morbidities such as hypertension,
smoking, or dyslipidaemia, as well as ERT itself. This may have po-tentially led to overestimation of CMD in our patients with regardto the healthy controls, in whom none of the conventional car-diovascular risk factors was present. The presence of epicardialcoronary disease was not systematically excluded by coronaryangiogram or coronary CT-angiogram in all patients, based onradioprotection and safety considerations. Nevertheless, none ofour enrolled AFD patients, at .3 years average from the execu-tion of PET studies, has been found to have significant CADduring follow-up.
Whileweacknowledge this unavoidablebias, however,MBFvaluesin AFD with no history of smoking, hypertension, and dyslipidaemia,
Figure 1 Pedigree of the 13 index patients.
B. Tomberli et al.Page 8 of 11
by guest on July 1, 2013http://eurjhf.oxfordjournals.org/
Dow
nloaded from

ideally representing ‘pure’ AFD patients, were comparable with therest of the cohort. Likewise, MBF in patients who were on ERT atthe time of the study was comparable with that of those who wereuntreated at that time, consistent with previous studies which
confirmed the questionable efficacy of ERT in patients with clear-cutcardiomyopathy.9,11,30 Thus, our data are consistent with a dispro-portionate microvascular involvement in AFD, overruling the effectof these potential environmental confounders.
Figure 2 Relationship between LV wall thickness and myocardial flow. Linear regression analysis showed a direct relationship between age anddegree of LV hypertrophy (LVH) (A) and between degree of LVH and myocardial blood flow following dipyridamole infusion. (B). Both relationshipswere stronger in males, suggesting that the development of Anderson–Fabry cardiomyopathy is age related and that coronary microvasculaturedysfunction may precede LVH.
Figure 3 Comparison of microvascular dysfunction in patients with Anderson–Fabry disease (AFD), based on the presence of LV hypertrophy(LVH) and gender. Myocardial blood flow following dipyridamole infusion is markedly impaired in AFD patients compared with control subjects.Values are shown as mean+ SD.
Microvascular dysfunction in Fabry disease Page 9 of 11
by guest on July 1, 2013http://eurjhf.oxfordjournals.org/
Dow
nloaded from

Comparably, due to the small and heterogeneous patientcohort and the cross-sectional design of the study, it is not pos-sible to extrapolate whether CMD represents an early phase ofcardiac involvement in AFD, invariably followed by onset of aclear-cut cardiomyopathy over time. Likewise, it is not possibleat this stage to envisage whether these abnormalities are
susceptible to regression with specific therapy; both such hypoth-eses need to be specifically addressed in longitudinal studies.However, our findings imply that a normal echocardiogramcannot exclude deterioration of microvascular function relatedto the disease, raising important issues regarding risk stratificationand management.
Figure4 Relationship of coronary microvasculaturedysfunction (CMD) to phenotype in individual patients with Anderson–Fabrydisease (AFD).(A) A 28-year-old male with extreme sinus bradycardia (36 b.p.m.) and normal interventricular septal thickness values (*echocardiographic para-sternal long axis and cardiac magnetic resonance four-chamber views). Colour-coded polar map obtained by positron emission tomography(PET) after dipyridamole infusion showing regional CMD, with relatively preserved flow in the antero-lateral region (family 13, subject III-1; Table2). (B) A 60-year-old female with normal ECG, without normal LV wall thickness values. The polar map shows severe CMD, despite normal LVwall thickness values (family 6, subject II-1; Table 2). (C) A 52-year-old male with marked ECG abnormalities, severe LV hypertrophy (LVH) (*echo-cardiographic apical four-chamber view and cardiac magnetic resonance four-chamber view). The polar map shows severe CMD (family 3, subjectII-1; Table 2). (D) A 47-year-old female with markedly abnormal ECG, and severe LVH. The polar map shows relatively preserved microvascularfunction, despite the LVH (family 8, subject II-2; Table 2). Dip-MBF, myocardial blood flow following dipyridamole infusion,
B. Tomberli et al.Page 10 of 11
by guest on July 1, 2013http://eurjhf.oxfordjournals.org/
Dow
nloaded from

ConclusionsCoronary microvascular function is markedly impaired in AFDpatients irrespective of LVH and gender. CMD may represent thefirst sign of cardiac involvement in patients who would be otherwiseconsidered unaffected. These findings suggest that coronary bloodflow impairment may precede the development of cardiac hyper-trophy in AFD, representing an important element for patient clinicalmanagement and decision-making in this challenging disease.
AcknowledgementsThe authors would like to thank Katia Baldini, professional nurse (Re-ferral Center for Myocardial Diseases, Department of Cardiology,Careggi University Hospital, Florence, Italy).
Conflict of interest: F.C., B.T., and A.M. received speaker’s fees andresearch support from Shire HGT, Genzyme Corporation. All otherauthors report no potential conflict of interest.
References1. Meikle PJ, Hopwood JJ, Clague AE, Carrey WF. Prevalence of lysosomal storage dis-
orders. JAMA 1999;281:249–254.2. Poorthuis BJ, Wevers RA, Kleijer WJ, Groener JE, de Jong JG, van Weely S,
Niezen-Koning KE, van Diggelen OP. The frequency of lysosomal storage diseasesin The Netherlands. Hum Genet 1999;105:151–156.
3. Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Sakuraba H, Ponzone A,Desnick RJ. High incidence of later-onset fabry disease revealed by newborn screen-ing. Am J Hum Genet 2006;79:31–40.
4. Mechtler TP, Stary S, Metz TF, De Jesus VR, Greber-Platzer S, Pollak A, Herkner KR,Streubel B, Kasper DC. Neonatal screening for lysosomal storage disorders: feasibil-ity and incidence from a nationwide study in Austria. Lancet 2012;379:335–341.
5. Germain DP. Fabry disease. Orphanet J Rare Dis 2010;5:306. Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C,
Linhart A, Sunder-Plassmann G, Ries M, Beck M. Fabry disease defined: baseline clin-ical manifestations of 366 patients in the FabryOutcome Survey. Eur J Clin Invest 2004;34:236–242.
7. Eng CM, Fletcher J, Wilcox WR, Waldek S, Scott CR, Sillence DO, Breunig F,Charrow J, Germain DP, Nicholls K, Banikazemi M. Fabry disease: baseline medicalcharacteristics of a cohort of 1765 males and females in the Fabry Registry. J InheritMetab Dis 2007;30:184–192.
8. O’Mahony C, Elliott P. Anderson–Fabry disease and the heart. Prog Cardiovasc Dis2010;52:326–335.
9. Elliott PM, Kindler H, Shah JS, Sachdev B, Rimoldi OE, Thaman R, Tome MT,McKenna WJ, Lee P, Camici PG. Coronary microvascular dysfunction in malepatients with Anderson–Fabry disease and the effect of treatment with alpha galac-tosidase A. Heart 2006;92:357–360.
10. Kalliokoski RJ, Kalliokoski KK, Sundell J, Engblom E, Penttinen M, Kantola I,Raitakari OT, Knuuti J, Nuutila P. Impaired myocardial perfusion reserve but pre-served peripheral endothelial function in patients with Fabry disease. J InheritMetab Dis 2005;28:563–573.
11. Kalliokoski RJ, Kantola I, Kalliokoski KK, Engblom E, Sundell J, Hannukainen JC,Janatuinen T, Raitakari OT, Knuuti J, Penttinen M, Viikari J, Nuutila P. The effect of12-month enzyme replacement therapy on myocardial perfusion in patients withFabry disease. J Inherit Metab Dis 2006;29:112–118.
12. Cecchi F, Olivotto I, Gistri R, Lorenzoni R, Chiriatti G, Camici PG. Coronary micro-vascular dysfunction and prognosis in hypertrophic cardiomyopathy. N Engl J Med2003;349:1027–1035.
13. Camici PG, Olivotto I, Rimoldi OE. The coronary circulation and blood flow in leftventricular hypertrophy. J Mol Cell Cardiol 2012;52:857–864.
14. MaronMS,Olivotto I, MaronBJ, PrasadSK,Cecchi F,Udelson JE,Camici PG.The casefor myocardial ischemia in hypertrophic cardiomyopathy. J Am Coll Cardiol 2009;54:866–875.
15. Petersen SE, Jerosch-Herold M, Hudsmith LE, Robson MD, Francis JM, Doll HA,Selvanayagam JB, Neubauer S, Watkins H. Evidence for microvascular dysfunction
in hypertrophic cardiomyopathy: new insights from multiparametric magnetic res-onance imaging. Circulation 2007;115:2418–2425.
16. Ferri L, Guido C, la Marca G, Malvagia S, Cavicchi C, Fiumara A, Barone R, Parini R,Antuzzi D, Feliciani C, Zampetti A, Manna R, Giglio S, Della Valle CM, Wu X,Valenzano KJ, Benjamin R, Donati MA, Guerrini R, Genuardi M, Morrone A. Fabrydisease: polymorphic haplotypes and a novel missense mutation in the GLA gene.Clin Genet 2012;81:224–233.
17. Maron MS, Olivotto I, Betocchi S, Casey SA, Lesser JR, Losi MA, Cecchi F, Maron BJ.Effect of left ventricular outflow tract obstruction on clinical outcome in hyper-trophic cardiomyopathy. N Engl J Med 2003;348:295–303.
18. Nagueh SF, Appleton CP, Gillebert TC, Marino PN, Oh JK, Smiseth OA,Waggoner AD, Flachskampf FA, Pellikka PA, Evangelisa A. Recommendations forthe evaluation of left ventricular diastolic function by echocardiography. J Am SocEchocardiogr 2009;22:107–133.
19. Sotgia B, Sciagra R, Olivotto I, Casolo G, Rega L, Betti I, Pupi A, Camici PG, Cecchi F.Spatial relationship between coronary microvascular dysfunction and delayed con-trast enhancement in patients with hypertrophic cardiomyopathy. J Nucl Med 2008;49:1090–1096.
20. DeGrado TR, Hanson MW, Turkington TG, Delong DM, Brezinski DA, Vallee JP,Hedlund LW, Zhang J, Cobb F, Sullivan MJ, Coleman RE. Estimation of myocardialblood flow for longitudinal studies with 13N-labeled ammonia and positron emissiontomography. J Nucl Cardiol 1996;3:494–507.
21. Cerqueira MD, Weissman NJ, Dilsizian V, Jacobs AK, Kaul S, Laskey WK, Pennell DJ,Rumberger JA, Ryan T, Verani MS; American Heart Association Writing Group onMyocardial Segmentation and Registration for Cardiac Imaging. Standardized myo-cardial segmentation and nomenclature for tomographic imaging of the heart. Circu-lation 2002;105:539–542.
22. Perugini E, Rapezzi C, Piva T, Leone O, Bacchi-Reggiani L, Riva L, Salvi F, Lovato L,Branzi A, Fattori R. Non-invasive evaluation of the myocardial substrate of cardiacamyloidosis by gadolinium cardiac magnetic resonance. Heart 2006;92:343–349.
23. Pieroni M, Chimenti C, Ricci R, Sale P, Russo MA, Frustaci A. Early detection of Fabrycardiomyopathy by tissue Doppler imaging. Circulation 2003;107:1978–1984.
24. Niemann M, Herrmann S, Hu K, Breunig F, Strotmann J, Beer M, Machann W,Voelker W, Ert F, Wanner C, Weidemann F. Differences in Fabry cardiomyopathybetween female and male patients consequences for diagnostic assessment. JACCCardiovasc Imaging 2011;4:592–601.
25. Olivotto I, Girolami F, Sciagra R, Ackerman MJ, Sotgia B, Bos JM, Nistri S,Sgalambro A, Grifoni C, Torricelli F, Camici PG, Cecchi F. Microvascular functionis selectively impaired in hypertrophic cardiomyopathy patients with sarcomeremyofilament gene mutation. J Am Coll Cardiol 2011;58:839–848.
26. Wilcox WR, Oliveira JP, Hopkin RJ, Ortiz A, Banikazemi M, Feldt-Rasmussen U,Sims K, Waldek S, Pastores GM, Lee P, Eng CM, Marodi L, Stanford KE, Breunig F,Wanner C, Warnock DG, Lemay RM, Germain DP, Fabry Registry. Females withFabry disease frequently have major organ involvement: lessons from the FabryRegistry. Mol Genet Metab 2008;93:112–128.
27. Pinto LL, Vieira TA, Giugliani R, Schwartz IV. Expression of the disease on female car-riers of X-linked lysosomal disorders: a brief review. Orphanet J Rare Dis 2010;5:14.
28. Kampmann C, Baehner F, Whybra C, Martin C, Wiethoff CM, Ries M, Gal A, Beck M.Cardiac manifestations of Anderson–Fabry disease in heterozygous females. J AmColl Cardiol 2002;40:1668–1674.
29. Machann W, Breunig F, Weidemann F, Sandstede J, Hahn D, Kostler H, Neubauer S,Wanner C, Beer M. Cardiac energy metabolism is disturbed in Fabry disease andimproves with enzyme replacement therapy using recombinant human galactosi-dase A. Eur J Heart Fail 2011;13:278–283.
30. Weidemann F, Niemann M, Breunig F, Herrmann S, Beer M, Stork S, Voelker W,Ertl G, Wanner C, Strotmann J. Long-term effects of enzyme replacement therapyon Fabry cardiomyopathy: evidence for a better outcome with early treatment. Cir-culation 2009;119:524–529.
31. Ishii S. Pharmacological chaperone therapy for Fabry disease. Proc Jpn Acad Ser B PhysBiol Sci 2012;88:18–30.
32. Porto C, Pisani A, Rosa M, Acampora E, Avolio V, Tuzzi MR, Visciano B, Gagliardo C,Materazzi S, la Marca G, Andria G, Parenti G. Synergy between the pharmacologicalchaperone 1-deoxygalactonojirimycin and the human recombinant alpha-galactosidase A in cultured fibroblasts from patients with Fabry disease. J InheritMetab Dis 2012;35:513–520.
33. Motabar O, Sidransky E, Goldin E, Zheng W. Fabry disease—current treatment andnew drug development. Curr Chem Genomics 2010;4:50–56.
34. Olivotto I, Cecchi F, Gistri R, Lorenzoni R, Chiriatti G, Girolami F, Torricelli F,Camici PG. Relevance of coronary microvascular flow impairment to long-term re-modeling and systolic dysfunction in hypertrophic cardiomyopathy. J Am Coll Cardiol2006;47:1043–1048.
Microvascular dysfunction in Fabry disease Page 11 of 11
by guest on July 1, 2013http://eurjhf.oxfordjournals.org/
Dow
nloaded from

Cardiomyopathy
Obesity and its Association to Phenotype andClinical Course in Hypertrophic Cardiomyopathy
Iacopo Olivotto, MD,* Barry J. Maron, MD,y Benedetta Tomberli, MD,* Evan Appelbaum, MD,zxCarol Salton, AB,zx Tammy S. Haas, RN,y C. Michael Gibson, MD,zx Stefano Nistri, MD,*
Eleonora Servettini, MD,* Raymond H. Chan, MD,x James E. Udelson, MD,k John R. Lesser, MD,yFranco Cecchi, MD,* Warren J. Manning, MD,zx Martin S. Maron, MDkFlorence, Italy; Minneapolis, Minnesota; and Boston, Massachusetts
Objectives This study sought to assess the impact of body mass index (BMI) on cardiac phenotypic and clinical course ina multicenter hypertrophic cardiomyopathy (HCM) cohort.
Background It is unresolved whether clinical variables promoting left ventricular (LV) hypertrophy in the general population, suchas obesity, may influence cardiac phenotypic and clinical course in patients with HCM.
Methods In 275 adult HCM patients (age 48 � 14 years; 70% male), we assessed the relation of BMI to LV mass, determinedby cardiovascular magnetic resonance (CMR) and heart failure progression.
Results At multivariate analysis, BMI proved independently associated with the magnitude of hypertrophy: pre-obeseand obese HCM patients (BMI 25 to 30 kg/m2 and >30 kg/m2, respectively) showed a 65% and 310%increased likelihood of an LV mass in the highest quartile (>120 g/m2), compared with normal weight patients(BMI<25 kg/m2; hazard ratio [HR]: 1.65; 95% confidence interval [CI]: 0.73 to 3.74, p¼ 0.22 and 3.1; 95% CI: 1.42to 6.86, p ¼ 0.004, respectively). Other features associated with LV mass >120 g/m2 were LV outflow obstruction(HR: 4.9; 95% CI: 2.4 to 9.8; p < 0.001), systemic hypertension (HR: 2.2; 95% CI: 1.1 to 4.5; p ¼ 0.026), and malesex (HR: 2.1; 95% CI: 0.9 to 4.7; p ¼ 0.083). During a median follow-up of 3.7 years (interquartile range: 2.5 to 5.3),obese patients showed an HR of 3.6 (95% CI: 1.2 to 10.7, p ¼ 0.02) for developing New York Heart Association(NYHA) functional class III to IV symptoms compared to nonobese patients, independent of outflow obstruction.Noticeably, the proportion of patients in NYHA functional class III at the end of follow-up was 13% among obesepatients, compared with 6% among those of normal weight (p ¼ 0.03).
Conclusions In HCM patients, extrinsic factors such as obesity are independently associated with increase in LV mass and maydictate progression of heart failure symptoms. (J Am Coll Cardiol 2013;62:449–57) ª 2013 by the AmericanCollege of Cardiology Foundation
Hypertrophic cardiomyopathy (HCM) is the most commongenetic heart disease, characterized by heterogeneousphenotypic expression with extreme diversity in the patternand extent of left ventricular hypertrophy (LVH), dueto molecular pathways and triggers that remain largely
unexplained (1–5). In the vast majority of genotype-positivepatients, HCM is associated with mutations in genesencoding proteins of the cardiac sarcomere, most commonlybeta-myosin heavy chain and myosin-binding protein C(1–3). While these molecular defects are consideredresponsible for the development of LVH, there is currentlyno conclusive evidence to explain the variability in pheno-typic expression of HCM, ranging from massive degrees toabsence of LVH even within the same family (1,4–6).
Among several hypotheses, the interplay of modifier genesand environmental factors has been commonly offered asa potential explanation for phenotypic diversity (7,8). To date,however, the possibility of an environmental modulation ofthe HCM phenotype remains speculative, and even the impact
See page 458
From the *Referral Center for Myocardial Diseases, Azienda Ospedaliera Universitaria
Careggi, Florence, Italy; yHypertrophic Cardiomyopathy Center, Minneapolis Heart
Institute Foundation, Minneapolis, Minnesota; zDepartment of Medicine,
Cardiovascular Division, Beth Israel Deaconess Hospital and Harvard Medical
School, Boston, Massachusetts; xPERFUSE Core Laboratory and Data Coordinating
Center, Harvard Medical School, Boston, Massachusetts; and the kDivision of
Cardiology, Tufts Medical Center, Boston, Massachusetts. This work was supported
by the Italian Ministry for University and Research (PRIN), and the European Union
(STREP Project 241577 “BIG HEART,” 7th European Framework Program).
Dr. B. J. Maron is grantee for GeneDx and Medtronic. All other authors have re-
ported that they have no relationships relevant to the contents of this paper to disclose.
Manuscript received October 3, 2012; revised manuscript received March 3, 2013,
accepted March 5, 2013.
Journal of the American College of Cardiology Vol. 62, No. 5, 2013� 2013 by the American College of Cardiology Foundation ISSN 0735-1097/$36.00Published by Elsevier Inc. http://dx.doi.org/10.1016/j.jacc.2013.03.062

of an obvious candidate variablesuch as obesity, known to pro-mote LVH in the general pop-ulation, is unresolved (9–15). Inaddition, it is unknown whetherthe adverse metabolic and hemo-dynamic effects of obesity, towhich HCM patients may beexposed during the long-termcourse of their disease, ultimatelyaffect symptomatic status andprognosis (11,12,16–18). There-fore, the present study wasdesigned, in a consecutive multi-center cohort studied with cardiacmagnetic resonance (CMR), to
assess the impact of body mass index (BMI) on the phenotype,as well as clinical course, of HCM.
Methods
Study population. The study cohort comprised 275 adultpatients with HCM (age >18 years, mean 48 � 14 years atstudy entry; 70% male, maximum left ventricular (LV) wallthickness 21 � 5 mm) consecutively referred for CMRstudies between January 2005 and June 2008 at 3 partici-pating referral centers in the United States and Italy: Min-neapolis Heart Institute Foundation (Minneapolis,Minnesota; n ¼ 168); Tufts Medical Center (Boston,Massachusetts; n ¼ 45); and Careggi University Hospital(Florence, Italy; n ¼ 62). Diagnosis of HCM was based on2-dimensional echocardiographic evidence of a hypertro-phied, nondilated LV (maximal wall thickness �15 mm), inthe absence of another cardiac or systemic disease that couldproduce the magnitude of hypertrophy evident (1,3). Weexcluded significant atherosclerotic coronary artery disease(>50% stenosis in a major artery) by virtue of 2 specificclinical or CMR criteria: 1) no study patient experienced anacute coronary event associated with increased cardiacenzymes or Q waves on electrocardiogram; and 2) in allpatients with late gadolinium enhancement (LGE) distrib-uted in a single coronary vascular territory, hemodynamicallysignificant coronary artery disease was excluded by arteriog-raphy or computed tomography angiogram. Furthermore,patients with prior cardiac surgery (including septal myec-tomy), alcohol septal ablation, and chronic renal failure wereexcluded (3). The study protocol was approved by therespective Internal Review Boards or research ethicscommittees of each institution, and written inform consentwas obtained from each subject.Definitions. Body mass index was calculated as weight/(height $ height) and expressed in kg/m2. Patients were clas-sified as normal weight (BMI range <25 kg/m2), pre-obese(25 to 30 kg/m2), and obese (>30 kg/m2), according toexisting guidelines (14). Type 2 diabetes was defined (andtreated) according to standard guidelines (12,18).
Systemic hypertension was diagnosed based on restingblood pressure values >140/90 mm Hg on �3 differentexaminations and treated medically to optimize bloodpressure control, as per standard international guidelines(18). All patients with hypertension had a diagnosisof HCM based on 1 or more of the following criteria: 1)HCM-causing sarcomere gene mutation or family historyof HCM; 2) onset of hypertension occurring years after thediagnosis of HCM; 3) maximum LV wall thickness ex-ceeding that expected by hypertension alone (i.e., >20 mm);4) presence of marked mitral leaflet elongation (19); 5)dynamic LV outflow obstruction (�30 mm Hg) underresting conditions (20); and 6) distribution of LGE bycontrast CMR consistent with HCM (i.e., preferentiallymid-wall or transmural, and not confined to a singlecoronary vascular territory) (3,5,21).Echocardiography. Echocardiographic studies were per-formed with commercially available instruments. Leftventricular hypertrophy was assessed with 2-dimensionalechocardiography, and the site and extent of maximal wallthickness were identified. Maximal end-diastolic LV wallthickness was taken as the dimension of greatest magnitudeat any site within the chamber. Left ventricular outflowobstruction, due to mitral valve systolic anterior motion andmitral-septal contact, was identified by a peak instantaneousoutflow gradient �30 mm Hg occurring under basal con-ditions (n ¼ 57) (20). Two hundred and eighteen patientswere nonobstructive at rest (basal gradient <30 mm Hg), ofwhom 105 (age 43 � 13 years, 72% males) underwentmaximal symptom-limited exercise echocardiography, aspreviously described (18); 50 developed dynamicgradients �30 mm Hg during effort or recovery (range 48 to155 mm Hg), and were considered to have provokableoutflow obstruction (20).CMR. All CMR examinations were performed usingcommercially available scanners (Philips ACS-NT 1.5-TGyroscan-Intera, Best, the Netherlands) and a commercialcardiac coil. Electrocardiographic gated, steady-state, freeprecession breath-hold cines in sequential 10-mm short-axisslices (no gap) were acquired starting parallel to the atrio-ventricular ring and covering the entire ventricle. Leftventricular end-diastolic and end-systolic volumes, leftventricular mass and wall thickness were calculated withcommercially available work stations (View Forum, PhilipsMedical System, Best, the Netherlands) (19,21).
For calculation of LV mass, the endocardial and epicardialborders of the left ventricle were manually planimetered onsuccessive short-axis cine images at end-diastole. The mostbasal slice at end-diastole was visually inspected and, ifventricular myocardium was present, it was planimeteredand included in the mass calculation. If myocardium but nointracavitary blood pool was present on the most apical slice,it was included in the mass calculation by planimetering onlythe epicardial border. Particular care was taken to avoidincluding papillary muscles in the LV mass calculation.Left ventricular mass was derived by the summation of
Abbreviationsand Acronyms
BMI = body mass index
CMR = cardiovascular
magnetic resonance
HCM = hypertrophic
cardiomyopathy
HR = hazard ratio
LGE = late gadolinium
enhancement
LV = left ventricular
LVH = left ventricular
hypertrophy
NYHA = New York Heart
Association
Olivotto et al. JACC Vol. 62, No. 5, 2013Obesity and LV Mass in Hypertrophic Cardiomyopathy July 30, 2013:449–57
450

discs method and multiplying myocardial muscle volumeby 1.05 g/cm3 (21). Left ventricular mass was indexed tobody surface area. Maximum end-diastolic LV wall thick-ness was taken as the dimension of greatest magnitude atany site within the LV wall. CMR measurements wereperformed by an experienced investigator at each center,blinded to the results of echocardiography. The presence ofLGE was assessed by visual inspection 15 min after intra-venous administration of 0.2 mmol/kg gadolinium-diethylenetriaminepentaacetic acid (Magnevist, Schering,Berlin, Germany) with breath-held segmented inversion-recovery sequence (inversion time 240 to 300 ms), whichwas acquired in the same views as the cine images (21).Statistical methods. Continuous variables were expressed asmean� SD, or median or interquartile range, as appropriate.For the comparison of normally distributed variables, weemployed Student t test and 1-way analysis of variancefollowed by Bonferroni post-hoc test, as appropriate. Chi-square test (not adjusted for multiple comparisons) wasutilized to compare noncontinuous variables expressed asproportions; Fisher exact test was employed when 1 or morecells in the comparison table had an expected frequency of<5.
When comparing noncontinuous variables among the 3 BMIclasses, an overall p value was obtained using theMonte Carlomethod; when the p value was<0.05, individual comparisonsbetween the subgroups were then assessed by cross-tabulationanalysis.
Clinical features independently associated with increasedLV mass index were assessed by multivariate logisticregression analysis using the stepwise (forward conditional)method. Therefore, the multivariate model was ultimatelyconstructed only with the variables that proved significant atunivariate analysis. Survival was assessed by Cox propor-tional hazards regression. The survival curve was constructedaccording to the Kaplan-Meier method, and comparisonswere performed using the log-rank test; p values are 2-sidedand considered significant when <0.05. Calculations wereperformed with SPSS 12.0 software (Chicago, Illinois).
Results
Prevalence of obesity. The 275 HCM patients had anaverage BMI of 29.1 � 6.1 kg/m2, ranging from 16.2 to49.3 kg/m2. Sixty-nine patients (25%) were in the normal
Table 1 Clinical and Echocardiographic Features of the 275 HCM Patients in Relation to BMI
OverallNormal
BMI <25 kg/m2Pre-Obese
BMI 25–30 kg/m2Obese
BMI >30 kg/m2 p Value
n 275 69 (25%) 105 (38%) 101 (37%)
Male 192 (70%) 32 (46%) 81 (77%) 79 (78%) <0.001
Age at diagnosis, yrs 43 � 14 41 � 15 45 � 13 44 � 13 0.257
Age at CMR, yrs 48 � 14 46 � 14 50 � 14 49 � 13 0.123
Body surface area, m2 1.97 � 0.25 1.73 � 0.18 1.99 � 0.2 2.11 � 0.23y <0.001
BMI, kg/m2 29.1 � 6.1 22.3 � 2.1 27.4 � 1.4 35.4 � 4.8y <0.001
Height, m 1.71 � 0.12 1.68 � 0.09 1.75 � 0.12y 1.68 � 0.14 <0.001
Weight, kg 85 � 19 63 � 9 84 � 11 101 � 16y <0.001
NYHA functional class at first evaluation
I 149 (54%) 37 (54%) 64 (61%) 48 (47%) overall
II 67 (24%) 19 (27%) 22 (21%) 26 (26%) 0.183
III 15 (5%) 3 (4%) 3 (3%) 9 (9%)
Syncope 57 (20%) 10 (14%) 26 (25%) 21 (21%) 0.263
Atrial fibrillation 30 (11%) 4 (6%) 12 (11%) 14 (14%) 0.248
Hypertension 75 (27%) 8 (12%) 29 (28%) 38 (38%) 0.001
Type II diabetes 14 (5%) 4 (4%) 10 (10%) 0.012
Hypercholesterolemia 84 (31%) 9 (13%) 40 (38%) 35 (35%) 0.001
Echocardiography
Left atrial diameter, mm 44 � 8 42 � 7 44 � 8 46 � 7* 0.032
LV end-diastolic diameter, mm 45 � 6 43 � 6 45 � 6 46 � 6* 0.025
Maximum LV wall thickness, mm 21 � 5 22 � 6 21 � 5 21 � 5 0.272
With LV outflow obstruction 107 (39%) 15 (22%) 41 (39%) 51 (50%)* 0.001
Medical treatment
Beta-blockers 151 (55%) 28 (40%) 62 (59%) 61 (60%) 0.427
Verapamil 52 (19%) 9 (13%) 22 (21%) 21 (21%) 0.578
Amiodarone 15 (5%) 4 (6%) 6 (6%) 5 (5%) 0.850
Disopyramide 10 (4%) 2 (3%) 4 (4%) 4 (4%) 0.613
Diuretics 29 (10%) 2 (3%) 9 (8%) 18 (18%)* <0.001
ACE inhibitors/sartans 40 (14%) 4 (6%) 14 (13%) 22 (21%)* <0.01
Warfarin 14 (5%) 0 5 (5%) 9 (9%) 0.155
Values are n (%) or mean � SD. *p < 0.05 versus the other 2 groups; yp < 0.05 versus normal.ACE ¼ angiotensin-converting enzyme; BMI ¼ body mass index; CMR ¼ cardiac magnetic resonance; HCM ¼ hypertrophic cardiomyopathy; LV ¼ left ventricular; NYHA ¼ New York Heart Association.
JACC Vol. 62, No. 5, 2013 Olivotto et al.July 30, 2013:449–57 Obesity and LV Mass in Hypertrophic Cardiomyopathy
451
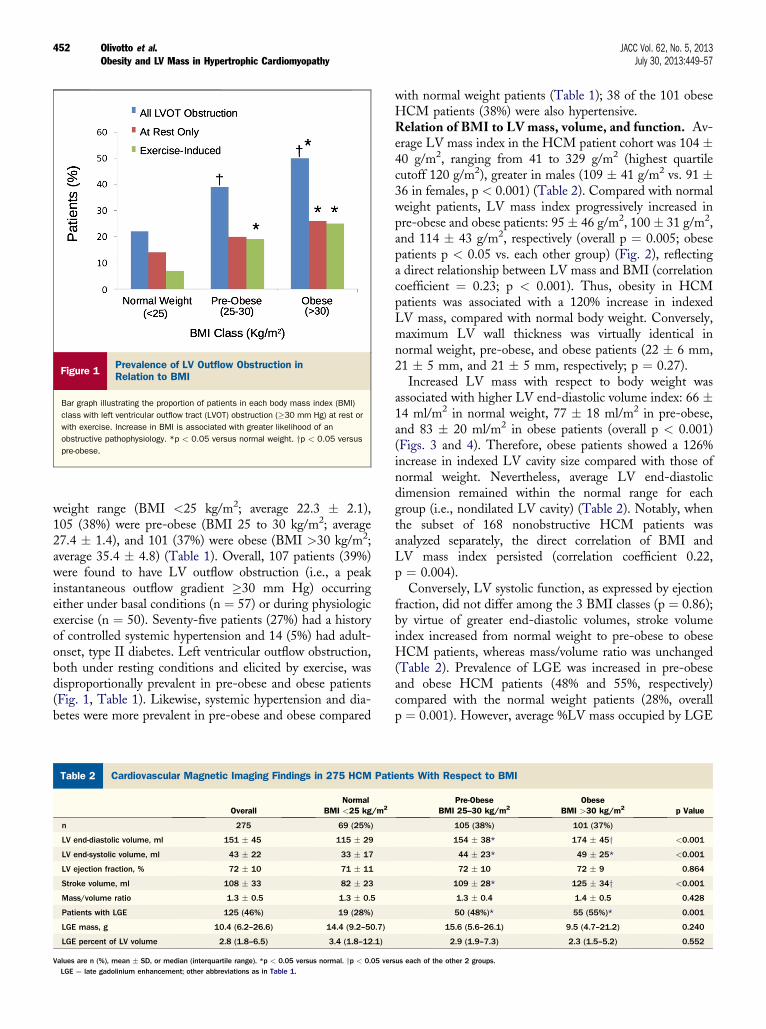
weight range (BMI <25 kg/m2; average 22.3 � 2.1),105 (38%) were pre-obese (BMI 25 to 30 kg/m2; average27.4 � 1.4), and 101 (37%) were obese (BMI >30 kg/m2;average 35.4 � 4.8) (Table 1). Overall, 107 patients (39%)were found to have LV outflow obstruction (i.e., a peakinstantaneous outflow gradient �30 mm Hg) occurringeither under basal conditions (n ¼ 57) or during physiologicexercise (n ¼ 50). Seventy-five patients (27%) had a historyof controlled systemic hypertension and 14 (5%) had adult-onset, type II diabetes. Left ventricular outflow obstruction,both under resting conditions and elicited by exercise, wasdisproportionally prevalent in pre-obese and obese patients(Fig. 1, Table 1). Likewise, systemic hypertension and dia-betes were more prevalent in pre-obese and obese compared
with normal weight patients (Table 1); 38 of the 101 obeseHCM patients (38%) were also hypertensive.Relation of BMI to LV mass, volume, and function. Av-erage LV mass index in the HCM patient cohort was 104 �40 g/m2, ranging from 41 to 329 g/m2 (highest quartilecutoff 120 g/m2), greater in males (109 � 41 g/m2 vs. 91 �36 in females, p < 0.001) (Table 2). Compared with normalweight patients, LV mass index progressively increased inpre-obese and obese patients: 95 � 46 g/m2, 100 � 31 g/m2,and 114 � 43 g/m2, respectively (overall p ¼ 0.005; obesepatients p < 0.05 vs. each other group) (Fig. 2), reflectinga direct relationship between LV mass and BMI (correlationcoefficient ¼ 0.23; p < 0.001). Thus, obesity in HCMpatients was associated with a 120% increase in indexedLV mass, compared with normal body weight. Conversely,maximum LV wall thickness was virtually identical innormal weight, pre-obese, and obese patients (22 � 6 mm,21 � 5 mm, and 21 � 5 mm, respectively; p ¼ 0.27).
Increased LV mass with respect to body weight wasassociated with higher LV end-diastolic volume index: 66 �14 ml/m2 in normal weight, 77 � 18 ml/m2 in pre-obese,and 83 � 20 ml/m2 in obese patients (overall p < 0.001)(Figs. 3 and 4). Therefore, obese patients showed a 126%increase in indexed LV cavity size compared with those ofnormal weight. Nevertheless, average LV end-diastolicdimension remained within the normal range for eachgroup (i.e., nondilated LV cavity) (Table 2). Notably, whenthe subset of 168 nonobstructive HCM patients wasanalyzed separately, the direct correlation of BMI andLV mass index persisted (correlation coefficient 0.22,p ¼ 0.004).
Conversely, LV systolic function, as expressed by ejectionfraction, did not differ among the 3 BMI classes (p ¼ 0.86);by virtue of greater end-diastolic volumes, stroke volumeindex increased from normal weight to pre-obese to obeseHCM patients, whereas mass/volume ratio was unchanged(Table 2). Prevalence of LGE was increased in pre-obeseand obese HCM patients (48% and 55%, respectively)compared with the normal weight patients (28%, overallp ¼ 0.001). However, average %LV mass occupied by LGE
Figure 1Prevalence of LV Outflow Obstruction inRelation to BMI
Bar graph illustrating the proportion of patients in each body mass index (BMI)
class with left ventricular outflow tract (LVOT) obstruction (�30 mm Hg) at rest or
with exercise. Increase in BMI is associated with greater likelihood of an
obstructive pathophysiology. *p < 0.05 versus normal weight. yp < 0.05 versus
pre-obese.
Table 2 Cardiovascular Magnetic Imaging Findings in 275 HCM Patients With Respect to BMI
OverallNormal
BMI <25 kg/m2Pre-Obese
BMI 25–30 kg/m2Obese
BMI >30 kg/m2 p Value
n 275 69 (25%) 105 (38%) 101 (37%)
LV end-diastolic volume, ml 151 � 45 115 � 29 154 � 38* 174 � 45y <0.001
LV end-systolic volume, ml 43 � 22 33 � 17 44 � 23* 49 � 25* <0.001
LV ejection fraction, % 72 � 10 71 � 11 72 � 10 72 � 9 0.864
Stroke volume, ml 108 � 33 82 � 23 109 � 28* 125 � 34y <0.001
Mass/volume ratio 1.3 � 0.5 1.3 � 0.5 1.3 � 0.4 1.4 � 0.5 0.428
Patients with LGE 125 (46%) 19 (28%) 50 (48%)* 55 (55%)* 0.001
LGE mass, g 10.4 (6.2–26.6) 14.4 (9.2–50.7) 15.6 (5.6–26.1) 9.5 (4.7–21.2) 0.240
LGE percent of LV volume 2.8 (1.8–6.5) 3.4 (1.8–12.1) 2.9 (1.9–7.3) 2.3 (1.5–5.2) 0.552
Values are n (%), mean � SD, or median (interquartile range). *p < 0.05 versus normal. yp < 0.05 versus each of the other 2 groups.LGE ¼ late gadolinium enhancement; other abbreviations as in Table 1.
Olivotto et al. JACC Vol. 62, No. 5, 2013Obesity and LV Mass in Hypertrophic Cardiomyopathy July 30, 2013:449–57
452

in individual patients did not differ between the subgroups(overall p ¼ 0.55) (Table 2).
Systemic hypertension was associated with increased LVmass index in our HCM cohort (118 � 44 g/m2 vs. 98 �36 g/m2 in normotensive; p < 0.001), although type 2 dia-betes was not (LV mass index 104 � 41 g/m2 vs. 104 �21 g/m2 in nondiabetic patients; p ¼ 0.98). Patients whowere both obese and hypertensive hadLVmass index values of126� 44 g/m2, comparedwith 93� 42 g/m2 in those patientswho were neither obese nor hypertensive (p < 0.001).Correlates of LV mass. A multivariate regression modelwas constructed to identify variables independently associ-ated with greater magnitude of LVH, defined by an LV massin the highest quartile for the overall cohort, or >120 g/m2.Variables assessed included BMI, age, sex, resting, or pro-vokable LV outflow obstruction, systemic hypertension, and
type 2 diabetes. The model was ultimately constructed withthe 3 variables that proved significant at univariate analysis(i.e., obesity, sex, and LV outflow obstruction). In thismodel, BMI proved an independent correlate of LV mass>120 g/m2, with a hazard ratio (HR) per unit increase of1.07 (95% CI: 1.01 to 1.13; p ¼ 0.019).
Pre-obese HCM patients showed a 65% increased like-lihood of assignment to the highest LV mass index quartile,compared with normal weight patients (HR: 1.65; 95% CI:0.73 to 3.74; p ¼ 0.22), while in obese patients this likeli-hood increased >300% (HR: 3.1; 95% CI: 1.42 to 6.86;p ¼ 0.004). Other variables associated with LV mass >120g/m2 were resting or provokable outflow obstruction (HR:4.9; 95% CI: 2.4 to 9.8; p < 0.001), systemic hypertension(HR: 2.2; 95% CI: 1.1 to 4.5; p ¼ 0.026), and male sex(HR: 2.1; 95% CI: 0.9 to 4.7; p ¼ 0.08).Symptomatic status and outcome. During a medianfollow-up of 3.7 years (interquartile range: 2.5 to 5.3 years)following CMR, there were 25 deaths (or equivalents), ofwhich 6 were noncardiac and 19 were HCM-related. Of thelatter 19 death events, 12 were sudden (including 7 deaths,2 patients resuscitated from cardiac arrest, and 3 appropriateimplantable cardioverter-defibrillator discharges for ventric-ular tachycardia/fibrillation). In addition, there were 6 heartfailure–related events (3 deaths and 3 heart transplants), and1 post-operative death (surgical septal myectomy). Therewas no difference in all-cause mortality among the 3 BMIclasses (Fig. 5).
In the 256 patients who were alive at the end of follow-up, those with obesity were however more likely to havedeveloped progressive New York Heart Association(NYHA) functional class III to IV symptoms at most recentevaluation, compared with normal weight patients (overallp ¼ 0.027). Noticeably, the proportion of patients in NYHAfunctional class III at the end of follow-up was 13% amongobese patients, compared with 6% among those of normalweight (p ¼ 0.03) (Fig. 5). Independent predictors ofNYHA functional class III to IV symptoms at end offollow-up were obesity (HR: 3.6; 95% CI: 1.2 to 10.7;p ¼ 0.02), female sex (HR: 4.3; 95% CI: 1.5 to 12.4;p ¼ 0.007), and LV outflow obstruction (HR: 2.7; 95% CI:0.9 to 7.8; p ¼ 0.07), whereas age, history of atrial fibrilla-tion, and hypertension were not.
Discussion
Obesity and the HCM phenotype. In HCM, the primarymorphologic expression of LVH has historically beenconsidered solely a consequence of the gene mutation, withno evidence to date that environmental variables can influ-ence phenotypic expression (2,5,7,8). However, the extremeheterogeneity of phenotypic expression among HCMpatients, even in family members sharing the same mutation(6), implies that other determinants of cardiac morphologymust be operative (2,3–5). For example, greater LV mass hasbeen observed in male patients and those with dynamic LV
Figure 2 Impact of BMI on LV Mass
Each panel shows mean (�95% confidence interval for mean) for unadjusted LV
mass and LV mass index in each of the 3 BMI classes. Overall p value for
hypertrophic cardiomyopathy patients was 0.005 (obese patients p < 0.05 vs.
each of the other 2 groups). *p < 0.05 versus normal weight; yp < 0.05 versus
Pre-Obese. Wt ¼ weight; other abbreviations as in Figure 1.
JACC Vol. 62, No. 5, 2013 Olivotto et al.July 30, 2013:449–57 Obesity and LV Mass in Hypertrophic Cardiomyopathy
453

outflow obstruction (21), suggesting the disease phenotypemay be sensitive to environmental modulation (22). In orderto address this issue, we have considered whether obesity, anestablished cardiovascular risk factor known to promoteLVH in the general population, may influence the magni-tude of LV mass and prognosis in a multicenter HCMcohort.
Our data demonstrate that obesity is independentlyassociated with increased LV mass, establishing a novelprinciple that environmental variables can influence diseaseexpression in a primary genetic cardiomyopathy such asHCM, a concept also relevant to other cardiomyopathies(22). Indeed, BMI was powerfully associated with severe LVmass increase in our HCM patients, independent of otherimportant determinants such as sex and dynamic LVoutflow obstruction (21), as well as systemic hypertension(18). In our HCM patients, the relationship between BMI
and LV mass became particularly evident for BMI values>30. Obese patients were >3 times as likely to havea marked increase in LV mass exceeding 120 g/m2,compared with those of normal weight. This increase in LVmass was driven primarily by greater end-diastolic volume(which nevertheless remained within normal limits whenindexed to body size). In the general population, LVremodeling associated with chamber enlargement is anestablished consequence of obesity, which normalizes strokevolume index in the presence of increased oxygen require-ment, thereby reflecting a physiologic adaptation to bodyweight (14,15,17,23,24). This principle was also supportedby our observation that greater LV cavity volume in obeseHCM patients was accompanied by preserved systolicfunction, resulting in an increased stroke volume index(14,15,23,24). Such impact of obesity on the HCMphenotype is likely mediated by the same triggers producing
Figure 3 Cardiac Remodeling in an Obese Patient With Hypertrophic Cardiomyopathy
Images from a 35-year-old male patient with a body mass index of 28 kg/m2. Left ventricular mass was 367 g (indexed 153 g/m2), with a maximal wall thickness of 29 mm.
Left ventricular end-diastolic volume was 235 ml (indexed 97 ml/m2) and left ventricular ejection fraction was 80%. (A) Cardiac magnetic resonance steady state free
precession 4-chamber showing diffuse thickening with sparing of the apex. (B) Corresponding late gadolinium enhancement imaging shows lack of fibrosis in LV.
*Subcutaneous fat, Dintrathoracic visceral fat. LA ¼ left atrium; LV ¼ left ventricle; RA ¼ right atrium; RV ¼ right ventricle.
Figure 4 Impact of BMI on LV Volume and Function
Panels show mean (�95% confidence interval for mean) LV end-diastolic volume index, LV ejection fraction, and stroke volume index for the 3 BMI classes. Overall p values for
each variable are provided in Table 1. *p < 0.05 versus normal weight; yp < 0.05 versus pre-obese. Wt ¼ weight; other abbreviations as in Figure 1.
Olivotto et al. JACC Vol. 62, No. 5, 2013Obesity and LV Mass in Hypertrophic Cardiomyopathy July 30, 2013:449–57
454
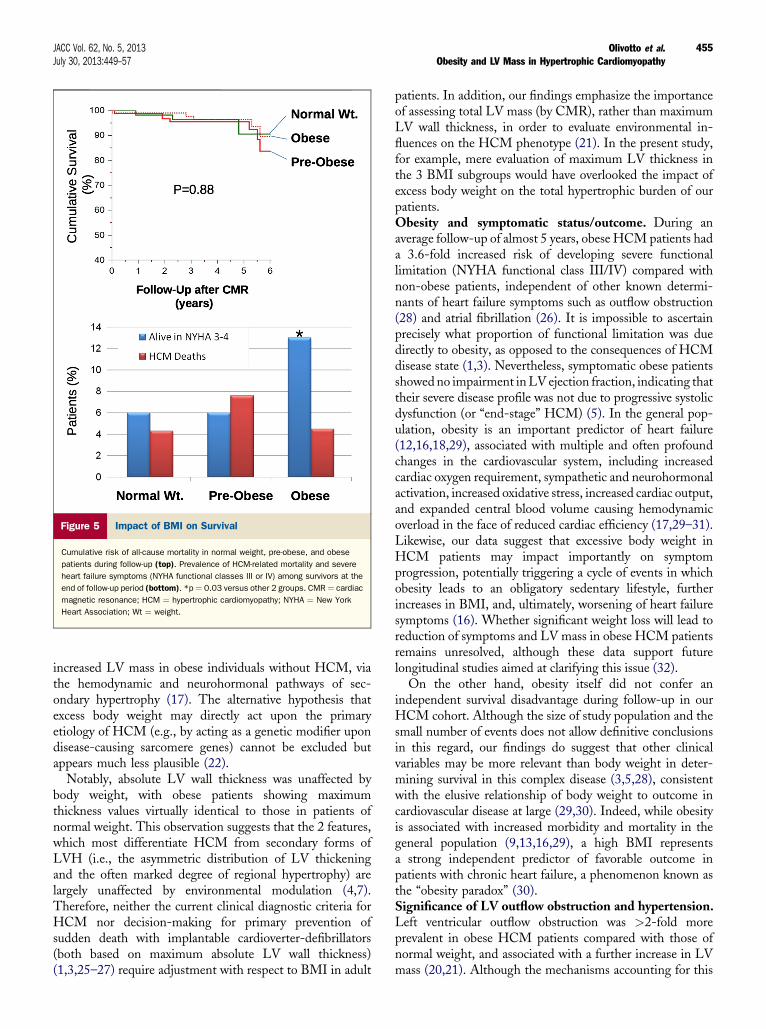
increased LV mass in obese individuals without HCM, viathe hemodynamic and neurohormonal pathways of sec-ondary hypertrophy (17). The alternative hypothesis thatexcess body weight may directly act upon the primaryetiology of HCM (e.g., by acting as a genetic modifier upondisease-causing sarcomere genes) cannot be excluded butappears much less plausible (22).
Notably, absolute LV wall thickness was unaffected bybody weight, with obese patients showing maximumthickness values virtually identical to those in patients ofnormal weight. This observation suggests that the 2 features,which most differentiate HCM from secondary forms ofLVH (i.e., the asymmetric distribution of LV thickeningand the often marked degree of regional hypertrophy) arelargely unaffected by environmental modulation (4,7).Therefore, neither the current clinical diagnostic criteria forHCM nor decision-making for primary prevention ofsudden death with implantable cardioverter-defibrillators(both based on maximum absolute LV wall thickness)(1,3,25–27) require adjustment with respect to BMI in adult
patients. In addition, our findings emphasize the importanceof assessing total LV mass (by CMR), rather than maximumLV wall thickness, in order to evaluate environmental in-fluences on the HCM phenotype (21). In the present study,for example, mere evaluation of maximum LV thickness inthe 3 BMI subgroups would have overlooked the impact ofexcess body weight on the total hypertrophic burden of ourpatients.Obesity and symptomatic status/outcome. During anaverage follow-up of almost 5 years, obese HCM patients hada 3.6-fold increased risk of developing severe functionallimitation (NYHA functional class III/IV) compared withnon-obese patients, independent of other known determi-nants of heart failure symptoms such as outflow obstruction(28) and atrial fibrillation (26). It is impossible to ascertainprecisely what proportion of functional limitation was duedirectly to obesity, as opposed to the consequences of HCMdisease state (1,3). Nevertheless, symptomatic obese patientsshowedno impairment inLV ejection fraction, indicating thattheir severe disease profile was not due to progressive systolicdysfunction (or “end-stage” HCM) (5). In the general pop-ulation, obesity is an important predictor of heart failure(12,16,18,29), associated with multiple and often profoundchanges in the cardiovascular system, including increasedcardiac oxygen requirement, sympathetic and neurohormonalactivation, increased oxidative stress, increased cardiac output,and expanded central blood volume causing hemodynamicoverload in the face of reduced cardiac efficiency (17,29–31).Likewise, our data suggest that excessive body weight inHCM patients may impact importantly on symptomprogression, potentially triggering a cycle of events in whichobesity leads to an obligatory sedentary lifestyle, furtherincreases in BMI, and, ultimately, worsening of heart failuresymptoms (16). Whether significant weight loss will lead toreduction of symptoms and LV mass in obese HCM patientsremains unresolved, although these data support futurelongitudinal studies aimed at clarifying this issue (32).
On the other hand, obesity itself did not confer anindependent survival disadvantage during follow-up in ourHCM cohort. Although the size of study population and thesmall number of events does not allow definitive conclusionsin this regard, our findings do suggest that other clinicalvariables may be more relevant than body weight in deter-mining survival in this complex disease (3,5,28), consistentwith the elusive relationship of body weight to outcome incardiovascular disease at large (29,30). Indeed, while obesityis associated with increased morbidity and mortality in thegeneral population (9,13,16,29), a high BMI representsa strong independent predictor of favorable outcome inpatients with chronic heart failure, a phenomenon known asthe “obesity paradox” (30).Significance of LV outflow obstruction and hypertension.Left ventricular outflow obstruction was >2-fold moreprevalent in obese HCM patients compared with those ofnormal weight, and associated with a further increase in LVmass (20,21). Although the mechanisms accounting for this
Figure 5 Impact of BMI on Survival
Cumulative risk of all-cause mortality in normal weight, pre-obese, and obese
patients during follow-up (top). Prevalence of HCM-related mortality and severe
heart failure symptoms (NYHA functional classes III or IV) among survivors at the
end of follow-up period (bottom). *p ¼ 0.03 versus other 2 groups. CMR ¼ cardiac
magnetic resonance; HCM ¼ hypertrophic cardiomyopathy; NYHA ¼ New York
Heart Association; Wt ¼ weight.
JACC Vol. 62, No. 5, 2013 Olivotto et al.July 30, 2013:449–57 Obesity and LV Mass in Hypertrophic Cardiomyopathy
455

relationship are uncertain, the abnormally increased adren-ergic drive associated with obesity may predispose todevelopment of intraventricular gradients (31). Such obser-vations may suggest that excess LVH in obese patients isprincipally mediated by outflow obstruction and afterloadmismatch (21). Nevertheless, when the present analysis wasrestricted to nonobstructive HCM patients, the associationbetween BMI and LV mass persisted, consistent with theconcept that cardiac remodeling due to excess body weight islargely independent of (although synergistic to) outflowobstruction (14,21). We acknowledge that the overallprevalence of LV outflow obstruction in the present cohort(39%) was significantly lower than that reported in a pre-vious study by our group (i.e., 70%) (20). This is largely dueto the fact that, in the quoted study, all patients withoutresting obstruction at baseline were systematically exercisedin order to assess provokable obstruction (20), whereas only105 of 218 such patients were assessed during exercise in thepresent cohort. Therefore, the true prevalence of obstructionelicited by physiological exercise may have been under-estimated in our study.
Systemic hypertension was another modifier of the HCMphenotype (9,13). As expected, the prevalence of elevatedblood pressure increased with body weight, and was presentin almost 40% of obese patients in our HCM cohort. Eventhough pharmacologically treated according to existingguidelines (18), hypertension doubled the likelihood ofsevere LVH in these patients, independent of other deter-minants of LV mass. Furthermore, the combination ofobesity and hypertension was associated with the highest LVmass values observed for any subset within the cohort. Thus,the present findings support the concept that the neuro-hormonal abnormalities associated with hypertension mayimpact LV mass in HCM patients (16,23,33), and therebyrepresent a relevant therapeutic target (16,18). To thisregard, it is important to acknowledge that environmentalinfluences of single cardiovascular risk factors on the HCMphenotype are unavoidably linked to a number of coexistingfactors (22). For example, obesity is per se associated with anincreased likelihood of hypertension, diabetes, and dynamicLV outflow obstruction (11,16). Therefore, interpretation ofthe clinical role of each of these factors, in isolation from theoverall pathophysiological picture, remains challenging.
Conclusions
The present study provides evidence that obesity is inde-pendently associated with adverse cardiac remodeling andincrease in LV mass in HCM patients. These observationsunderscore the novel principle that the primary phenotypicexpression in this complex, heterogenous heart disease is alsosubject to environmental variables and not solely the productof disease-causing sarcomere mutations. In addition, obesityappears to play a role in the development and progression ofheart failure symptoms in HCM, supporting the need for
follow-up studies clarifying whether modulating obesity canimprove clinical course.
Reprint requests and correspondence: Dr. Iacopo Olivotto,Dipartimento Cuore e Vasi, Azienda Ospedaliera UniversitariaCareggi, Viale Pieraccini 19, 50134 Firenze, Italy. E-mail:[email protected].
REFERENCES
1. Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet 2012;381:242–55.
2. Watkins H, Ashrafian H, Redwood C. Inherited cardiomyopathies.N Engl J Med 2011;364:1643–56.
3. Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guidelinefor the diagnosis and treatment of hypertrophic cardiomyopathy:executive summary: a report of the American College of CardiologyFoundation/American Heart Association Task Force on PracticeGuidelines. J Am Coll Cardiol 2011;58:2703–38.
4. Olivotto I, Girolami F, Nistri S, et al. The many faces of hypertrophiccardiomyopathy: from developmental biology to clinical practice.J Cardiovasc Transl Res 2009;2:349–67.
5. Olivotto I, Cecchi F, Poggesi C, Yacoub MH. Patterns of diseaseprogression in hypertrophic cardiomyopathy: an individualizedapproach to clinical staging. Circ Heart Fail 2012;5:535–46.
6. Ciró E, Nichols PF 3rd, Maron BJ. Heterogeneous morphologicexpression of genetically transmitted hypertrophic cardiomyopathy.Two-dimensional echocardiographic analysis. Circulation 1983;67:1227–33.
7. Wang L, Seidman JG, Seidman CE. Narrative review: harnessingmolecular genetics for the diagnosis and management of hypertrophiccardiomyopathy. Ann Intern Med 2010;152:513–20, W181.
8. Daw EW, Chen SN, Czernuszewicz G, et al. Genome-wide mappingof modifier chromosomal loci for human hypertrophic cardiomyopathy.Hum Mol Genet 2007;16:2463–71.
9. Kahn R, Robertson RM, Smith R, Eddy D. The impact of preventionon reducing the burden of cardiovascular disease. Circulation 2008;118:576–85.
10. Parikh NI, Pencina MJ, Wang TJ, et al. Increasing trends in incidenceof overweight and obesity over 5 decades. Am J Med 2007;120:242–50.
11. Kushner RF. Clinical assessment and management of adult obesity.Circulation 2012;126:2870–7.
12. Flegal KM, Kit BK, Orpana H, Graubard BI. Association of all-causemortality with overweight and obesity using standard body mass indexcategories: a systematic review andmeta-analysis. JAMA2013;309:71–82.
13. Heckbert SR, Post W, Pearson GD, et al. Traditional cardiovascularrisk factors in relation to left ventricular mass, volume, and systolicfunction by cardiac magnetic resonance imaging: the Multiethnic Studyof Atherosclerosis. J Am Coll Cardiol 2006;48:2285–92.
14. Rider OJ, Francis JM, Ali MK, et al. Determinants of left ventricularmass in obesity; a cardiovascular magnetic resonance study. J Car-diovasc Magn Reson 2009;11:9.
15. Turkbey EB, McClelland RL, Kronmal RA, et al. The impact ofobesity on the left ventricle: the Multi-Ethnic Study of Atherosclerosis(MESA). J Am Coll Cardiol Img 2010;3:266–74.
16. National Institutes of Health. Clinical guidelines on the identification,evaluation, and treatment of overweight and obesity in adultsdtheevidence report. Obes Res 1998;6 Suppl 2:51S–209S.
17. Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity.Physiol Rev 2008;88:389–419.
18. Smith SC Jr., Benjamin EJ, Bonow RO, et al. AHA/ACCF secondaryprevention and risk reduction therapy for patients with coronary andother atherosclerotic vascular disease: 2011 update: a guideline from theAmerican Heart Association and American College of CardiologyFoundation. J Am Coll Cardiol 2011;58:2432–46.
19. Maron MS, Olivotto I, Harrigan C, et al. Mitral valve abnormalitiesidentified by cardiovascular magnetic resonance represent a primaryphenotypic expression of hypertrophic cardiomyopathy. Circulation2011;124:40–7.
Olivotto et al. JACC Vol. 62, No. 5, 2013Obesity and LV Mass in Hypertrophic Cardiomyopathy July 30, 2013:449–57
456

20. Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomy-opathy is predominantly a disease of left ventricular outflow tractobstruction. Circulation 2006;114:2232–9.
21. Olivotto I, Maron MS, Autore C, et al. Assessment and significance ofleft ventricular mass by cardiovascular magnetic resonance in hyper-trophic cardiomyopathy. J Am Coll Cardiol 2008;52:559–66.
22. Piran S, Liu P, Morales A, Hershberger RE. Where genome meetsphenome: rationale for integrating genetic and protein biomarkers inthe diagnosis and management of dilated cardiomyopathy and heartfailure. J Am Coll Cardiol 2012;60:283–9.
23. Danias PG, Tritos NA, Stuber M, Kissinger KV, Salton CJ,Manning WJ. Cardiac structure and function in the obese: A cardio-vascular magnetic resonance imaging study. J Cardiovasc Magn Reson2003;5:431–8.
24. Wong CY, O’Moore-Sullivan T, Leano R, Byrne N, Beller E,Marwick TH. Alterations of left ventricular myocardial characteristicsassociated with obesity. Circulation 2004;110:3081–7.
25. Maron BJ. Sudden death in hypertrophic cardiomyopathy. J CardiovascTransl Res 2009;2:368–80.
26. Spirito P, Bellone P, Harris KM, Bernabo P, Bruzzi P, Maron BJ.Magnitude of left ventricular hypertrophy and risk of sudden death inhypertrophic cardiomyopathy. N Engl J Med 2000;342:1778–85.
27. Olivotto I, Gistri R, Petrone P, Pedemonte E, Vargiu D, Cecchi F.Maximum left ventricular thickness and risk of sudden death in
patients with hypertrophic cardiomyopathy. J AmColl Cardiol 2003;41:315–21.
28. Maron MS, Olivotto I, Betocchi S, et al. Effect of left ventricularoutflow tract obstruction on clinical outcome in hypertrophic cardio-myopathy. N Engl J Med 2003;348:295–303.
29. Kenchaiah S, Evans JC, Levy D, et al. Obesity and the risk of heartfailure. N Engl J Med 2002;347:305–13.
30. Oreopoulos A, Padwal R, Kalantar-Zadeh K, Fonarow GC,Norris CM, McAlister FA. Body mass index and mortality in heartfailure: a meta-analysis. Am Heart J 2008;156:13–22.
31. Grassi G, Seravalle G, Quarti-Trevano F, et al. Excessive sympatheticactivation in heart failure with obesity and metabolic syndrome:characteristics and mechanisms. Hypertension 2007;49:535–41.
32. Clarke R. Long-term weight loss and prevention of cardiovasculardisease. Circulation 2011;124:2801–2.
33. Tsybouleva N, Zhang L, Chen S, et al. Aldosterone, through novelsignaling proteins, is a fundamental molecular bridge between thegenetic defect and the cardiac phenotype of hypertrophic cardiomy-opathy. Circulation 2004;109:1284–91.
Key Words: cardiac magnetic resonance - hypertrophiccardiomyopathy - hypertrophy - obesity - outcome.
JACC Vol. 62, No. 5, 2013 Olivotto et al.July 30, 2013:449–57 Obesity and LV Mass in Hypertrophic Cardiomyopathy
457

EDITORIAL COMMENT
Obesity and HypertrophicCardiomyopathyChickens, Eggs, and Causality:Clinical Skills Remain the Key toCaring for Patients*
Steve R. Ommen, MD,
Francisco Lopez-Jimenez, MD, MSC
Rochester, Minnesota
The impact of obesity on the cardiovascular system con-tinues to reveal itself. In this issue, Olivotto et al. (1) sharethe results of a 3-center collaboration on body mass indexand its relation to left ventricular morphology in patientswith hypertrophic cardiomyopathy (HCM). The investiga-tors found that obese HCM patients had significantly largerleft ventricular mass, left ventricular mass index, and highersubsequent rates of symptom development. These resultssuggest that environmental/situational factors can influenceventricular morphology in a condition that is geneticallyinherited. Notably, only 25% of the patients in this study fell
See page 449
into the normal weight classification. So, like the age-oldquestion, can we tell which came first? Is it a more severeexpression of HCM that leads to obesity, or is it the generaltrend of increasing body weight in Western society thatcomplicates HCM?
Because this was an observational study, we cannot tell thedirection of the association. There are numerous potentialpathophysiologic mechanisms by which obesity could resultin more hypertrophy. These include increased sympathetictone, increased leptin levels, myocardial fatty infiltration,insulin resistance, and enhanced renin-angiotensin activity(2). Likewise, a more severe phenotypic expression of HCMcould readily lead to a less active life with all the attendantnegative impacts, including increasing body mass. While atbaseline, the study subjects had statistically similar symptomstatus; there is no indication of daily activity levels, nor
objective measures of exercise capacity. In the end, treatingpatients with HCM and obesity will necessarily involvetreating both aspects.
The impact of obesity on symptom status is a troublingclinical conundrum. Obesity is a well-known cause of heartfailure, and has been shown to result in worsening diastolicfunction independent of other comorbid conditions (3,4).Patients with obesity have higher overall oxygen require-ments as well as abnormal myocardial function as evidencedby strain and strain rate imaging (5). In the Olivotto et al.study, the patients with obesity were more likely to have leftventricular outflow tract obstruction, and to be receivingdiuretics, and/or pure vasodilators as part of their therapy.Those medications are well known to exacerbate leftventricular outflow tract obstruction.
Why is this a problem in patients with HCM? Other thandefibrillators for prevention of sudden cardiac death, alltherapies in HCM have the sole indication of symptomrelief (6). The therapies employed, including medicationsand invasive procedures, can be quite effective when com-bating symptoms due to hemodynamic derangements, butnot likely to be effective at when directed at problems such asdeconditioning and excess oxygen requirements. Similarly,the need to consider surgical or invasive therapy is based onpersistent symptoms that are unresponsive to pharmacologictherapy. However, if the symptoms are persistent becausethey are primarily related to the obesity, then the medica-tions (and the subsequent procedures) have little chance ofmaking an impact on symptoms.
So, when facing an obese patient with HCM and decidingwhether to start, increase, or decrease therapy one has to makeimportant considerations of the balance between hemody-namics and excess body weight. By the time such patientshave become highly symptomatic, the combination of sed-entary status, deconditioning, and increased tendency tooutflow tract obstruction may make therapeutic lifestylechange untenable. Yet, treating the hemodynamic abnor-malities will lead to less than satisfying results unless weightloss and healthy habits are taught to and adopted by thepatients.
How does one then approach this situation? With car-diopulmonary exercise testing we can determine whetherpatients exercise capacity is limited by cardiac output limi-tations based on a plateau in the peak oxygen consumption(VO2). Conversely, if a patient is purely limited due todeconditioning and obesity, then the peak oxygen con-sumption (VO2) will continue to rise through the point atwhich the patient has to stop exercising. Armed with thisinformation, appropriate therapeutic decisions and focusedcounseling can help our patients understand the expectedimpact, and their role in achieving a better quality oflife. Even with completely successful treatment of hemody-namic considerations, the obese patient needs to understandthis is the first step in their path to improved functionalcapacity. Long-term exercise and dietary modifications willneed to continue. Importantly, therapeutic lifestyle change,
*Editorials published in the Journal of the American College of Cardiology reflect the
views of the authors and do not necessarily represent the views of JACC or the
American College of Cardiology.
From the Division of Cardiovascular Diseases and Internal Medicine, Mayo Clinic,
Rochester, Minnesota. Both authors have reported that they have no relationships
relevant to the contents of this paper to disclose.
Journal of the American College of Cardiology Vol. 62, No. 5, 2013� 2013 by the American College of Cardiology Foundation ISSN 0735-1097/$36.00Published by Elsevier Inc. http://dx.doi.org/10.1016/j.jacc.2013.03.063

particularly if the cardiac hemodynamic abnormalities aremild, may represent the most important step to recovery.Whether purposeful weight loss in symptomatic patientswith HCM improves left ventricular morphology andhemodynamics is yet to be determined.
The study by Olivotto et al. (1) suggested that some ofthese findings were independent of comorbid conditions.However, as clinicians we must not discount the increasedrates of hypertension, diabetes, coronary artery disease, andsleep apnea have important roles not only in the cardiovas-cular system but in general well-being. Furthermore, eachof these conditions has been shown to be associated withadverse outcomes in patients with HCM. Treating andcaring for patients with HCM means not just understandingthe pathophysiology of diastole and outflow tract obstruc-tion, but must incorporate treating comorbid conditionsaggressively.
Another of the management considerations for patientswith HCM deserves mention. Many patients perceive, or arein fact inappropriately counseled by their physicians, thatthey should not engage in regular exercise. This stems fromthe fact that sudden cardiac death among competitiveathletes is most often related to HCM such that competitiveathletics are discouraged for these patients. To extend thatdiscouragement to regular, noncompetitive exercise is a trav-esty. Counseling and encouraging healthy habits, includingmild to moderate intensity exercise, is an important aspect ofcaring for patients.
Hypertrophic cardiomyopathy is generally well tolerated,with a good prognosis, and completely compatible with anormal lifestyle (often with no therapeutic interventions).That obesity modifies the ventricular morphology, and theoverall well-being of our patients, provides further evidence
that we need to actively teach our patients about healthychoices. Which comes first? Maybe we will find out one day,but for now it is our responsibility to treat both. Clinically,we treat people, not just pathophysiology.
Reprint requests and correspondence: Dr. Steve R. Ommen,Mayo Clinic, College of Medicine, Division of CardiovascularDiseases and Internal Medicine, 200 First Street SW, Rochester,Minnesota 55905. E-mail: [email protected].
REFERENCES
1. Olivotto I, Maron BJ, Tomberli B, et al. Obesity and its association tophenotype and clinical course in hypertrophic cardiomyopathy. J AmColl Cardiol 2013;62:449–57.
2. Poirier P, Giles TD, Bray GA, et al. Obesity and cardiovascular disease:pathophysiology, evaluation, and effect of weight loss: an update ofthe 1997 American Heart Association Scientific Statement on Obesityand Heart Disease from the Obesity Committee of the Council onNutrition, Physical Activity, and Metabolism. Circulation 2006;113:898–918.
3. Kenchaiah S, Sesso HD, Gaziano JM. Body mass index and vigorousphysical activity and the risk of heart failure among men. Circulation2009;119:44–52.
4. Cil H, Bulur S, Turker Y, et al., for the MELEN Investigators. Impactof body mass index on left ventricular diastolic dysfunction. Echocar-diography 2012;29:647–51.
5. Orhan AL, Uslu N, Dayi SU, et al. Effects of isolated obesity on leftand right ventricular function: a tissue Doppler and strain rate imagingstudy. Echocardiography 2010;27:236–43.
6. Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline forthe diagnosis and treatment of hypertrophic cardiomyopathy: executivesummary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J AmColl Cardiol 2011;58:2703–38.
Key Words: body mass index - hypertrophic cardiomyopathy -
left ventricular hypertrophy - obesity - outcome.
JACC Vol. 62, No. 5, 2013 Ommen and Lopez-JimenezJuly 30, 2013:458–9 Obesity and HCM
459

Benedetta Tomberli, Iacopo Olivotto and Gian Franco GensiniGabriele Castelli, Alessandra Fornaro, Mauro Ciaccheri, Alberto Dolara, Vito Troiani,
Over 3 Decades: Impact of Evidence-Based ManagementImproving Survival Rates of Patients With Idiopathic Dilated Cardiomyopathy in Tuscany
Print ISSN: 1941-3289. Online ISSN: 1941-3297 Copyright © 2013 American Heart Association, Inc. All rights reserved.
75231is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TXCirculation: Heart Failure
doi: 10.1161/CIRCHEARTFAILURE.112.0001202013;6:913-921; originally published online July 25, 2013;Circ Heart Fail.
http://circheartfailure.ahajournals.org/content/6/5/913World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circheartfailure.ahajournals.org//subscriptions/
is online at: Circulation: Heart Failure Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further information isthe Editorial Office. Once the online version of the published article for which permission is being requested
can be obtained via RightsLink, a service of the Copyright Clearance Center, notCirculation: Heart Failurein Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
at UNIV DI FIRENZE on October 25, 2013http://circheartfailure.ahajournals.org/Downloaded from at UNIV DI FIRENZE on October 25, 2013http://circheartfailure.ahajournals.org/Downloaded from at UNIV DI FIRENZE on October 25, 2013http://circheartfailure.ahajournals.org/Downloaded from at UNIV DI FIRENZE on October 25, 2013http://circheartfailure.ahajournals.org/Downloaded from at UNIV DI FIRENZE on October 25, 2013http://circheartfailure.ahajournals.org/Downloaded from at UNIV DI FIRENZE on October 25, 2013http://circheartfailure.ahajournals.org/Downloaded from at UNIV DI FIRENZE on October 25, 2013http://circheartfailure.ahajournals.org/Downloaded from at UNIV DI FIRENZE on October 25, 2013http://circheartfailure.ahajournals.org/Downloaded from at UNIV DI FIRENZE on October 25, 2013http://circheartfailure.ahajournals.org/Downloaded from at UNIV DI FIRENZE on October 25, 2013http://circheartfailure.ahajournals.org/Downloaded from

913
Idiopathic dilated cardiomyopathy (IDCM) is a severe myo-cardial disease characterized by dilatation and impaired
function of the left or both ventricles,1 affecting >36.5 indi-viduals per 100 000.2 IDCM accounts for ≈50 000 hospital-izations and 10 000 deaths each year and is responsible for ≈25% of all cases of heart failure (HF) in the United States.3 During the past 3 decades, the outcome of IDCM is presumed to have radically changed after major advances in pharma-cological and device-based therapeutic strategies for HF; however, studies addressing the outcome of HF have been characterized by relatively small representation of individu-als with IDCM, so that limited data exist with specific regard to this condition.4–11
Clinical Perspective on 921
In addition, real-world implementation of standard HF guidelines is challenging, with rates of compliance that can be considerably lower than those reported in clinical trials documenting survival benefits.12–17 As a result, the issue of long-term outcome of IDCM in the community remains largely unresolved. In the present study, we there-fore chose to examine the clinical features and prognosis of a sizeable cohort of unselected, consecutively enrolled and angiographically negative patients with IDCM from a well-defined regional population in Tuscany, evaluated dur-ing the past 30 years in a systematic fashion and by the same team, in relation to the evolving treatment strategies includ-ing evidence-based pharmacological and device-based interventions.
Original Article
© 2013 American Heart Association, Inc.
Circ Heart Fail is available at http://circheartfailure.ahajournals.org DOI: 10.1161/CIRCHEARTFAILURE.112.000120
Background—Contemporary therapeutic options have led to substantial improvement in survival of patients with heart failure. However, limited evidence is available specifically on idiopathic dilated cardiomyopathy. We thus examined changes in prognosis of a large idiopathic dilated cardiomyopathy cohort systematically followed during the past 30 years.
Methods and Results—From 1977 to 2011, 603 consecutive patients (age, 53±12 years; 73% men; left ventricular ejection fraction, 32±10%) fulfilling World Health Organization criteria for idiopathic dilated cardiomyopathy, including negative coronary angiography, were followed up for 8.8±6.3 years. Patients were subdivided in 4 enrollment periods on the basis of heart failure treatment eras: (1) 1977–1984 (n=66); (2) 1985–1990 (n=102); (3) 1991–2000 (n=197); (4) 2001–2011 (n=238). Rates of patients receiving angiotensin-converting enzyme inhibitors/angiotensin receptors blockers, β-blockers, and devices at final evaluation increased from 56%, 12%, 8% (period 1) to 97%, 86%, 17% (period 4), respectively (P<0.05). There was a trend toward enrollment of older patients with less severe left ventricular dilatation and dysfunction during the years. During follow-up, 271 patients (45%) reached a combined end point including death (heart failure related, n=142; sudden death, n=71; and noncardiac, n=22) or cardiac transplant (n=36). A more recent enrollment period represented the most powerful independent predictor of favorable outcome {period 2 versus 1 (hazard ratio [HR], 0.64; P=0.04), period 3 versus 1 (HR, 0.35; P<0.001), period 4 versus 1 (HR, 0.14; P<001)}. Each period was associated with a 42% risk reduction versus the previous one (HR, 0.58; 95% confidence interval, 0.50–0.67; P<0.001), reflecting marked decreases in heart failure–related mortality and sudden death (period 4 versus 1: HR, 0.10; P<001 and HR, 0.13; P<0.0001, respectively).
Conclusions—Evidence-based treatment has led to dramatic improvement in the prognosis of idiopathic dilated cardiomyopathy during the past 3 decades. The benefits of controlled randomized trials can be replicated in the real world, emphasizing the importance of tailored follow-up and long-term continuity of care. (Circ Heart Fail. 2013;6:913-921.)
Key Words: cardiac resynchronization therapy ■ cardiomyopathy, dilated ■ drug therapy ■ heart failure ■ outcomes assessment
Received December 29, 2012; accepted July 15, 2013.From the Heart and Vessel Department, Referral Center for Cardiomyopathies, Careggi University Hospital, Florence, Italy.Correspondence to Gabriele Castelli, MD, Cardiologia Generale 1–San Luca, Dipartimento del Cuore e dei Vasi, Azienda Ospedaliero Universitaria
Careggi, Largo Brambilla 1, 50134 Firenze, Italy. E-mail [email protected]
Improving Survival Rates of Patients With Idiopathic Dilated Cardiomyopathy in Tuscany Over 3 Decades
Impact of Evidence-Based Management
Gabriele Castelli, MD; Alessandra Fornaro, MD; Mauro Ciaccheri, MD; Alberto Dolara, MD; Vito Troiani, MD; Benedetta Tomberli, MD; Iacopo Olivotto, MD; Gian Franco Gensini, MD

914 Circ Heart Fail September 2013
MethodsSetting and Study PopulationThe Referral Center for Cardiomyopathies has been established in the mid-1970s in Careggi University Hospital, a large community-based multispecialty hospital in Florence, Italy. Careggi is a tertiary center with >1500 beds, serving the Florence metropolitan area (population ≈1 million) and the surrounding geographic region of Tuscany (total population ≈3 700 000 within ≈23 000 km2).18 In this setting, between January 1977 and September 2011, we consecutively enrolled 1085 pa-tients with a diagnosis of dilated cardiomyopathy. In 271 patients, the condition was judged to be secondary to ischemic heart disease, sys-temic hypertension, chemotherapy, alcoholic abuse, diabetes mellitus, cor pulmonalis, valve disease, or other cardiac or systemic diseases. In addition, 211 patients in whom a coronary angiogram was not available were excluded. The remaining 603 were classified as IDCM, accord-ing to the World Health Organization criteria and in the presence of a negative coronary angiogram: these comprise the present study cohort.
Evaluation and Follow-UpAll study patients were evaluated and followed up at our institution by clinical and family history, physical examination, 12-lead ECG, standard chest radiograph, routine laboratory tests, 24-hour Holter ECG monitoring (since early 1980s), M-mode and 2D echocardiogra-phy (since 1970s), and Doppler echocardiography (since mid-1980s). Exercise stress test and cardiac catheterization were performed as dictated by clinical requirements. Endomyocardial biopsy has been performed routinely until the early 1990s and thereafter only on se-lected patients with suspect active myocarditis. Although the family history of each patient was investigated, systematic family screenings have not been performed. In 51 patients (8%) a genetic transmission of IDCM was evident or suspected by virtue of clinical and echocar-diographic documentation of the disease in a relative or by a family history of HF-related death or premature sudden death (SD).
Patients were regularly followed up by outpatient visits every 6 months (or more frequently when clinically indicated) and, when necessary, by interviews with referring physicians or by tele-phone contacts. For the purposes of this study, follow-up ended on September 30th, 2011. In patients who died or were transplanted, end of follow-up was considered as time of death or heart transplanta-tion. In the minority of patients lost to follow-up (ie, not traceable by September 30th, 2011), the last clinical evaluation of telephone contact was considered. In the past 5 years of the study (2006–2011), the proportion of patients who were actively followed up, had died/transplanted, or were lost in period was 44%, 45%, and 11%, respec-tively. For the entire study period, patients were seen by the same cardiologists who assumed primary responsibility for management. The use of pharmacological agents, implantable cardioverter defibril-lator (ICD), and biventricular pacing for cardiac resynchronization therapy (CRT) was carefully considered as these became available and implemented when felt appropriate, according to existing guide-lines.19 Pharmacological treatment was carefully titered to achieve maximum tolerated doses of angiotensin-converting enzyme inhibi-tors/angiotensin receptors blockers (ACEI/ARBs) and β-blockers. Candidates for heart transplantation (HTx) were assessed jointly with the Regional Heart Transplant Referral Center in Siena, where the op-erations were performed. The study was approved by the institutional review committee and patients gave an informed consent.
Enrollment PeriodsTo assess long-term changes in outcome in relation to treatment op-tions, we subdivided our patients with IDCM into 4 periods, coin-ciding with different therapeutic eras of HF treatment: period 1: 66 patients (11%) enrolled from 1977 to 1984, defined as the pre-ACE inhibition era, when standard therapy consisted of diuretic agents, di-goxin, and early vasodilators; period 2: 102 patients (17%) enrolled from 1985 to 1990, marking the beginning of the ACEI era; period 3: 197 patients (33%) enrolled from 1991 to 2000, characterized by increasing use of ACEI and ARBs and the introduction of β-blockers;
and period 4: 238 patients (39%) enrolled from 2001 to 2011, the device-era, characterized by the introduction of ICD and CRT on top of extensive neurohormonal blockade.20
Statistical AnalysisStudent t test and 1-way ANOVA were used to compare continu-ous variables, whereas categorical variables were compared by χ2 test or Fisher method, as appropriate. Univariate survival estimates were obtained using Kaplan–Meier method. Forward conditional Cox proportional hazards regression analysis was used to evaluate the relationship between periods of enrollment, clinical and instru-mental baseline data of patients, and long-term outcome for the fol-lowing end points: (1) death from any cause including appropriate ICD interventions and HTx; (2) SD; (3) death because of refractory HF. The following variables were included in the models: age, New York Heart Association (NYHA) class, sex, left ventricular ejection fraction (LVEF), baseline LV end-diastolic diameter index, left atrial diameter index, and the presence of moderate to severe mitral regur-gitation at enrollment. A P value <0.05 was considered statistically significant. Statistical analysis was performed with the SPSS pack-age, version 20 (SPSS Inc, Chicago, IL).
ResultsBaseline Clinical FeaturesMean age of the 603 patients at first evaluation was 53±12 (range, 16–75) years; 442 (73%) were men. In 51 patients (8%) there was a family history of IDCM. Average NYHA functional class was 2.3±0.8; 82 patients (14%) were in class I, 265 (44%) in class II, 198 (33%), and 58 (10%) in class III or IV. Mean end-diastolic diameter index was 36±6 mm/m2, LVEF was 31±10%, and left atrial diameter index was 24±4 mm/m2; moderate to severe mitral regurgitation was diagnosed in 106 patients (18%). One hundred seventeen patients (19%) had ECG evidence of left bundle-branch block, whereas a history of paroxysmal or permanent atrial fibrillation was recorded in 123 (21%). Of the 531 patients with 24-hour ambulatory Holter monitoring, 183 (30%) showed non–sustained ventricular tachycardias ≥3 beats and sustained ventricular tachycardias (Table 1).
Comparison of Enrollment PeriodsAt initial evaluation, the 4 groups of patients identified based on the period of enrollment were comparable with regard to sex and NYHA class (Table 1). However, there was a slight trend toward enrollment of older patients with less severe LV dilatation and dysfunction during the years: in period 4 versus period 1, age was 55±12 versus 50±11 years, respec-tively (P<0.0001); end-diastolic diameter index was 34±5 versus 39±6 mm/m2 (P<0.0001), and LVEF was 33±9 versus 29±11% (P=0.016). The overall prevalence of atrial fibrilla-tion was similar in the 4 periods; however, permanent atrial fibrillation at initial diagnosis became less prevalent over time (overall P<0.001; Table 1).
Evolution in ManagementExpectedly, medical treatment at enrollment differed signifi-cantly between the 4 patient periods (Table 2; Figure 1). ACEI and ARBs were extensively used at our institution since early 1990s, reaching a 96% rate at enrollment after 2001 versus 4% before 1985 (P<0.0001). The use of β-blockers increased significantly after 2001: 79% of patients were already treated with these agents at enrollment in period 4 compared with 0%

Castelli et al Long-Term Outcome of IDCM in Tuscany 915
in period 1, 1% in period 2, and 21% in period 3 (P<0.0001). These differences were still evident at the end of follow-up, although the rate of treated patients considerably increased in all patient subgroups, reflecting changes in management over time (Table 2). Of note, 97% and 86% of patients in period 4 were on ACEI/ARBs and β-blockers, respectively, at final evaluation. The use of mineralocorticoid receptor antagonists
also increased progressively in periods 1 to 4, whereas that of digoxin decreased (at final evaluation, only 37% of patients in period 4 were on digoxin versus 92% in period 1; P<0.0001). The ICD and CRT were introduced in 1998 and 2000, respec-tively, and their use increased steadily over time (Table 2). Overall, 79 patients (13%) were implanted with an ICD, whereas 69 (11%) received combined ICD/CRT (Figure 1).
Table 1. Baseline Clinical and Instrumental Characteristics of Patients at Enrollment
TotalPeriod 1 (1977–1984)
n=66Period 2 (1985–1990)
n=102Period 3 (1991–2000)
n=197Period 4 (2001–2011)
n=238 P Value
Men, % 442 (73%) 50 (76%) 75 (73%) 149 (76%) 168 (71%) NS
BSA, kg/m2 1.9±0.2 1.8±0.2 1.8±0.2 1.8±0.2 1.9±0.2 <0.001
Age, y 53±12 50±11 48±13 54±12 55±12 <0.001
Follow-up, mo 106±75 113±108 135±99 122±64 79±47 <0.001
Familial IDCM, % 51 (8%) 1 (1%) 2 (2%) 22 (11%) 26 (8%) 0.003
LBBB, % 117 (19%) 19 (29%) 32 (31%) 35 (18%) 31 (13%) NS
Paroxysmal AF 29 (5%) 0 (0%) 2 (2%) 8 (4%) 19 (8%) <0.001
Chronic AF 94 (16%) 18 (27%) 21 (21%) 29 (15%) 26 (11%) <0.001
Complex VA 183 (30%) 27 (41%) 46 (45%) 65 (33%) 45 (19%) <0.001
NYHA class I 82 (14%) 6 (9%) 10 (10%) 31 (16%) 35 (15%) NS
NYHA class II 265 (44%) 33 (50%) 50 (49%) 89 (45%) 93 (39%)
NYHA class III 198 (33%) 21 (32%) 31 (30%) 63 (32%) 83 (35%)
NYHA class IV 58 (10%) 6 (9%) 11 (11%) 14 (7%) 27 (11%)
EDD, mm 67±8 69±8 69±9 67±9 65±8 <0.001
iEDD, mm/m2 36±6 39±6 38±6 36±5 34±5 <0.001
LVEF, % 31±10 29±11 30±10 31±9 33±9 0.016
LVFS, % 18±6 16±5 17±6 19±6 19±6 0.015
ARD, mm 33±4 33±4 32±4 32±4 33±4 0.022
iARD, mm/m2 17±3 17±5 18±3 17±2 17±3 NS
LAD, mm 44±7 45±7 46±8 44±7 44±7 NS
iLAD, mm/m2 24±4 25±4 25±5 24±4 23±4 <0.001
Moderate to severe MR 106 (18%) 3 (4%) 12 (12%) 29 (15%) 62 (26%) <0.001
Data are presented as mean±SD or percentages. AF indicates atrial fibrillation; ARD, aortic root diameter; BSA, body surface index; EDD, end-diastolic diameter; iARD, aortic root diameter index; IDCM, idiopathic dilated cardiomyopathy; iEDD end-diastolic diameter index; iLAD, left atrial diameter index; LAD, left atrial diameter; LBBB, left bundle-branch block; LVEF, left ventricular ejection fraction; LVFS, left ventricular fractional shortening; MR, mitral regurgitation; NS, not significant; NYHA, New York Heart Association; and VA, ventricular arrhythmias.
Table 2. Treatment at Enrollment and at the End of Follow-Up
TotalPeriod 1 (1977–1984)
n=66Period 2 (1985–1990)
n=102Period 3 (1991–2000)
n=197Period 4 (2001–2011)
n=238 P Value
Enrollment End F-Up Enrollment End F-Up Enrollment End F-Up Enrollment End F-Up Enrollment End F-Up Enrollment End F-Up
ACEI/ARBs 467 (77%) 552 (91%) 3 (4%) 37 (56%) 53 (52%) 93 (91%) 182 (92%) 192 (97%) 229 (96%) 230 (97%) <0.001 <0.001
β-Blockers 232 (38%) 344 (57%) 0 (0%) 8 (12%) 1 (1%) 22 (22%) 42 (21%) 110 (56%) 189 (79%) 204 (86%) <0.001 <0.001
Digoxin 338 (56%) 359 (59%) 60 (91%) 61 (92%) 80 (78%) 83 (81%) 117 (59%) 127 (64%) 81 (34%) 88 (37%) <0.001 <0.001
Diuretics 483 (80%) 502 (83%) 61 (92%) 63 (95%) 87 (85%) 91 (89%) 153 (78%) 164 (83%) 182 (76%) 184 (77%) 0.01 0.001
MRA 155 (26%) 243 (40%) 7 (11%) 24 (36%) 12 (12%) 39 (38%) 41 (21%) 77 (39%) 95 (40%) 103 (43%) <0.001 <0.001
Vasodilators 58 (10%) 96 (16%) 8 (12%) 20 (30%) 5 (5%) 13 (13%) 22 (11%) 37 (19%) 23 (10%) 26 (11%) NS 0.001
Warfarin 131 (22%) 203 (34%) 6 (9%) 27 (41%) 20 (20%) 44 (43%) 43 (22%) 56 (28%) 62 (26%) 76 (32%) 0.03 0.04
Amiodarone 103 (17%) 155 (26%) 10 (15%) 26 (39%) 20 (20%) 36 (35%) 33 (17%) 44 (22%) 40 (17%) 49 (21%) NS 0.001
ICD … 79 (13%) … 5 (8%) … 9 (9%) … 24 (12%) … 41 (17%) … NS
CRT … 69 (11%) … 3 (4%) … 7 (7%) … 18 (9%) … 41 (17%) … 0.003
ACEI indicates angiotensin-converting enzyme inhibitors, ARBs, angiotensin receptor blockers; CRT, cardiac resynchronization therapy; ICD, implantable cardioverter defibrillator; MRA, mineralocorticoid receptor antagonists; and NS, not significant.

916 Circ Heart Fail September 2013
Notably, 52% of patients that received an ICD and 59% of those with CRT were enrolled in period 4; the remaining were implanted in patients enrolled in previous periods, who were still actively followed up when the devices became available.
Changes in Functional Status and Reverse RemodelingOn average, patients enrolled in periods 3 and 4 improved their functional status during follow-up, whereas those enrolled earlier showed symptomatic progression (Figure 2). Improvement in systolic LV function compared with baseline was observed in all groups, although most evident in periods 3 and 4. Likewise, a reduction in end-diastolic diameter index occurred to a greater extent in those patients enrolled most
recently (Figure 2). This was paralleled by a less common occurrence of worsening mitral regurgitation after 1990 (33% in period 1; 37% in period 2; 28% in period 3; and 11% in period 4; overall P<0.0001).
Long-Term Outcome and Predictors of RiskDuring an average follow-up of 8.8±6.3 years, 271 patients (45%) reached the combined end point including all-cause mortality and HTx. Of these, 142 patients (23%) died because of refractory HF, 71 (12%) because of SD, 22 (4%) died of noncardiac causes, and 36 patients (6%) underwent HTx. Overall survival was 79% and 63% at 5 and 10 years, respec-tively. There was marked and progressive improvement in outcome from periods 1 to 4 for all end points (Figure 3).
A B
C D
E
Figure 1. Evolution in pharmacological and device therapy from initial evaluation to end of follow-up based on enrollment period. A to D, The dotted line indicates treatment rates at enrollment for each period, whereas the straight line indicates treatment rates at final evalua-tion. E, The dotted line indicates implant rates at enrollment for each period, whereas the straight line indicates implant rates at final eval-uation. ACEI/ARB indicates angiotensin-converting enzyme inhibitor/angiotensin receptor blockers; and MRA, mineralocorticoid receptor antagonists.

Castelli et al Long-Term Outcome of IDCM in Tuscany 917
When adjusted for age, sex, NYHA class, LVEF, and left atrial diameter index, event-free rates for the combined end point (all-cause mortality and HTx) at 5 years were 62%, for patients enrolled in period 1; 74% for period 2; 84% for period 3; and 93% for period 4 (Figure 3A). At multivari-ate analysis, an earlier enrollment period proved the most powerful predictor of the combined end point, independent of age, NYHA functional class, LVEF, left atrial diameter index, and sex (Table 3). Of note, each enrollment period was associated with a 42% reduction in risk compared with the previous one (hazard ratio, 0.58; 95% confidence inter-val, 0.50–0.67; P<0.001).
Likewise, an earlier period of enrollment proved a potent independent predictor of refractory HF death and SD. Risk
Figure 2. Changes in New York Heart Association (NYHA) class, left ventricular ejection fraction (LVEF), and end-diastolic diameter index (iEDD) from initial evaluation to end of follow-up based on enrollment period. Each bar indicates the change from enrollment (horizontal line) to final evaluation (blunt bar extrem-ity). Overall P values (ANOVA) for average differences based on enrollment period are shown.
Figure 3. Cox multivariate regression survival plot (adjusted for age, New York Heart Association [NYHA] class, sex, left ventricu-lar ejection fraction and indexed left atrium diameter) indicating differences in outcome based on enrollment period for the 3 study end points: A, All-cause mortality/heart transplantation; (B) death for refractory heart failure; and (C) sudden death.
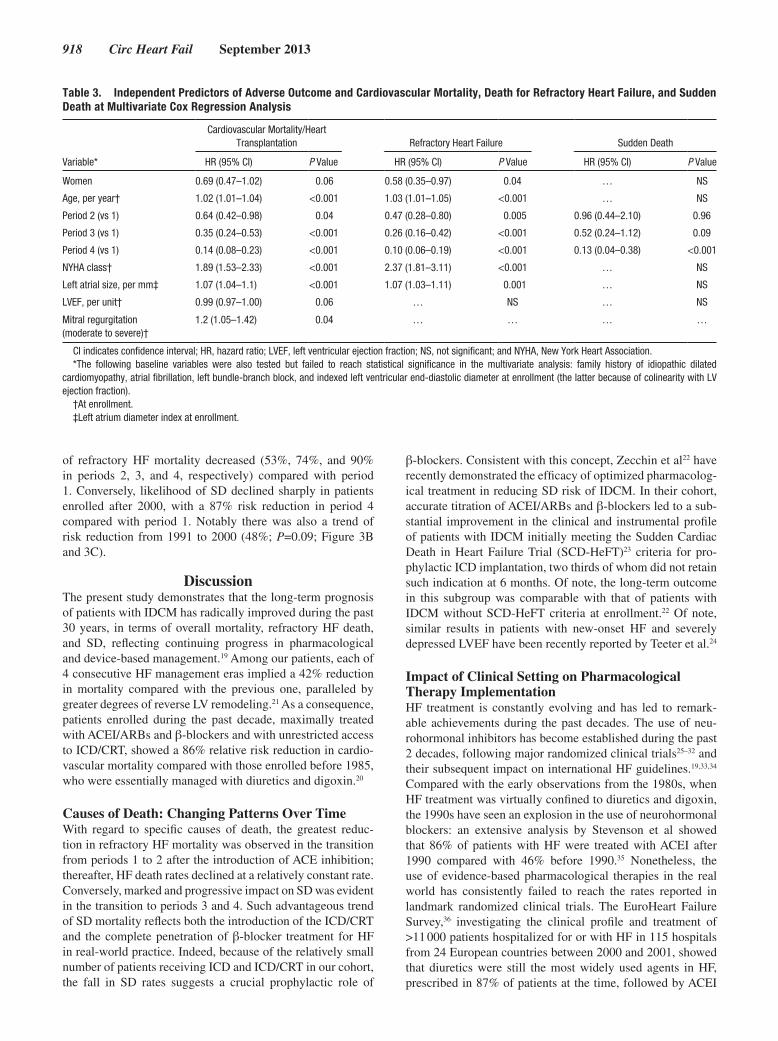
918 Circ Heart Fail September 2013
of refractory HF mortality decreased (53%, 74%, and 90% in periods 2, 3, and 4, respectively) compared with period 1. Conversely, likelihood of SD declined sharply in patients enrolled after 2000, with a 87% risk reduction in period 4 compared with period 1. Notably there was also a trend of risk reduction from 1991 to 2000 (48%; P=0.09; Figure 3B and 3C).
DiscussionThe present study demonstrates that the long-term prognosis of patients with IDCM has radically improved during the past 30 years, in terms of overall mortality, refractory HF death, and SD, reflecting continuing progress in pharmacological and device-based management.19 Among our patients, each of 4 consecutive HF management eras implied a 42% reduction in mortality compared with the previous one, paralleled by greater degrees of reverse LV remodeling.21 As a consequence, patients enrolled during the past decade, maximally treated with ACEI/ARBs and β-blockers and with unrestricted access to ICD/CRT, showed a 86% relative risk reduction in cardio-vascular mortality compared with those enrolled before 1985, who were essentially managed with diuretics and digoxin.20
Causes of Death: Changing Patterns Over TimeWith regard to specific causes of death, the greatest reduc-tion in refractory HF mortality was observed in the transition from periods 1 to 2 after the introduction of ACE inhibition; thereafter, HF death rates declined at a relatively constant rate. Conversely, marked and progressive impact on SD was evident in the transition to periods 3 and 4. Such advantageous trend of SD mortality reflects both the introduction of the ICD/CRT and the complete penetration of β-blocker treatment for HF in real-world practice. Indeed, because of the relatively small number of patients receiving ICD and ICD/CRT in our cohort, the fall in SD rates suggests a crucial prophylactic role of
β-blockers. Consistent with this concept, Zecchin et al22 have recently demonstrated the efficacy of optimized pharmacolog-ical treatment in reducing SD risk of IDCM. In their cohort, accurate titration of ACEI/ARBs and β-blockers led to a sub-stantial improvement in the clinical and instrumental profile of patients with IDCM initially meeting the Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT)23 criteria for pro-phylactic ICD implantation, two thirds of whom did not retain such indication at 6 months. Of note, the long-term outcome in this subgroup was comparable with that of patients with IDCM without SCD-HeFT criteria at enrollment.22 Of note, similar results in patients with new-onset HF and severely depressed LVEF have been recently reported by Teeter et al.24
Impact of Clinical Setting on Pharmacological Therapy ImplementationHF treatment is constantly evolving and has led to remark-able achievements during the past decades. The use of neu-rohormonal inhibitors has become established during the past 2 decades, following major randomized clinical trials25–32 and their subsequent impact on international HF guidelines.19,33,34 Compared with the early observations from the 1980s, when HF treatment was virtually confined to diuretics and digoxin, the 1990s have seen an explosion in the use of neurohormonal blockers: an extensive analysis by Stevenson et al showed that 86% of patients with HF were treated with ACEI after 1990 compared with 46% before 1990.35 Nonetheless, the use of evidence-based pharmacological therapies in the real world has consistently failed to reach the rates reported in landmark randomized clinical trials. The EuroHeart Failure Survey,36 investigating the clinical profile and treatment of >11 000 patients hospitalized for or with HF in 115 hospitals from 24 European countries between 2000 and 2001, showed that diuretics were still the most widely used agents in HF, prescribed in 87% of patients at the time, followed by ACEI
Table 3. Independent Predictors of Adverse Outcome and Cardiovascular Mortality, Death for Refractory Heart Failure, and Sudden Death at Multivariate Cox Regression Analysis
Variable*
Cardiovascular Mortality/Heart Transplantation Refractory Heart Failure Sudden Death
HR (95% CI) P Value HR (95% CI) P Value HR (95% CI) P Value
Women 0.69 (0.47–1.02) 0.06 0.58 (0.35–0.97) 0.04 … NS
Age, per year† 1.02 (1.01–1.04) <0.001 1.03 (1.01–1.05) <0.001 … NS
Period 2 (vs 1) 0.64 (0.42–0.98) 0.04 0.47 (0.28–0.80) 0.005 0.96 (0.44–2.10) 0.96
Period 3 (vs 1) 0.35 (0.24–0.53) <0.001 0.26 (0.16–0.42) <0.001 0.52 (0.24–1.12) 0.09
Period 4 (vs 1) 0.14 (0.08–0.23) <0.001 0.10 (0.06–0.19) <0.001 0.13 (0.04–0.38) <0.001
NYHA class† 1.89 (1.53–2.33) <0.001 2.37 (1.81–3.11) <0.001 … NS
Left atrial size, per mm‡ 1.07 (1.04–1.1) <0.001 1.07 (1.03–1.11) 0.001 … NS
LVEF, per unit† 0.99 (0.97–1.00) 0.06 … NS … NS
Mitral regurgitation (moderate to severe)†
1.2 (1.05–1.42) 0.04 … … … …
CI indicates confidence interval; HR, hazard ratio; LVEF, left ventricular ejection fraction; NS, not significant; and NYHA, New York Heart Association. *The following baseline variables were also tested but failed to reach statistical significance in the multivariate analysis: family history of idiopathic dilated
cardiomyopathy, atrial fibrillation, left bundle-branch block, and indexed left ventricular end-diastolic diameter at enrollment (the latter because of colinearity with LV ejection fraction).
†At enrollment.‡Left atrium diameter index at enrollment.

Castelli et al Long-Term Outcome of IDCM in Tuscany 919
(62%), β-blockers (37%), cardiac glycosides (36%), nitrates (32%), calcium channel blockers (21%), and spironolactone (20%). Notably only 17.2% of patients received the combi-nation of diuretic, ACEI, and β-blockers. These rates have significantly improved in more recent HF registries, such as the S-HFR registry37 or the EURObservational Research Pro-gramme: the Heart Failure Pilot.38 Yet, in the S-HFR registry, as many as 18% of patients with HF did not receive ACEI/ARBs, whereas 11% did not receive a β-blocker in the absence of any reported contraindication or side effect. Similar results originated from other extensive studies of HF cohorts show-ing that, in spite of the widely available evidence-based inter-national treatment guidelines, recommended therapies are slowly adopted and inconstantly applied in clinical practice, with considerable proportions of eligible patients failing to receive optimal treatment.39,40
Conversely, our observations support the view that, in appro-priate settings, the treatment rates and benefits of controlled randomized HF trials can be replicated in the real world.41 The present IDCM cohort is representative of those seen at other com-munity hospitals, in that ≈95% of our patients were from a well-defined geographic region in Central Italy, most of them having lived virtually their entire life in Tuscany. All patients were sys-tematically followed up on a 6-month basis by the same team >3 decades, providing a high level of continuity of care during this extended time period and allowing continuous reassessment of risk profile and management of the primary condition as well as cardiovascular risk factors and comorbidities. This provided the opportunity to gradually optimize pharmacological therapy, including uptitration of ACEI/ARBs and β-blockers to the maxi-mum tolerated dosage.19 It is noteworthy that 92% and 57% of the overall population received ACEI/ARBs and β-blockers during the whole study period, reflecting stringent implementation of international guidelines even in the earliest cohorts, as soon as new treatments became available. In the past decades, these rates were as high as 97% and 86%, respectively, resembling those of land-mark HF randomized control trials, rather than most HF surveys and registries.36 However, our data are consistent with selected reports from dedicated tertiary hospital settings based on patients with chronic systolic HF because of ischemic heart disease.42 Overall, these findings considerably expand previous observa-tions showing favorable temporal trends in the outcome of IDCM and HF, based on smaller cohorts and less extended follow-up. Matsumura and Kubo9,10 have shown significant improvement in the prognosis of a relatively small group of Japanese patients with IDCM, partly explained through an increased use of ACEI/ARBs and β-blockers and a declining use of class Ia/Ib antiar-rhythmic drugs after the 1990s, and a similar trend was observed by Aleksova et al11 in a study focusing mainly on asymptomatic patients with IDCM without prior history of HF.
Changes in Demographics Among Enrollment PeriodsIt is important to acknowledge that, in our IDCM cohort, patients enrolled later had a less severe profile in terms of symptoms, LV size and function. This trend presumably reflects increasing awareness of the disease and availability of more sensitive diagnostic tools and may have contributed to the reduction in mortality observed during the years, resulting
in an overestimation of benefits related to treatment. However, this trend is counterbalanced by the more advanced age at enrollment of the most recent groups. Although it is virtually impossible to quantify the net result of these trends on out-come, their opposite prognostic weight is likely to limit their combined effect substantially. In our multivariate models, the enrolment period proved to be a very potent predictor of out-come independent of the most relevant baseline clinical and demographic features including well-recognized prognostic factors (ie, age, sex, NYHA functional class, LV and left atrial diameters, LVEF, and degree of functional mitral regurgita-tion).43–47 Thus, it is plausible to attribute most of the survival benefit observed to improvements in management rather than to the evolving patient demographics.
ConclusionsThe evolution of evidence-based treatment has led to progres-sive improvement in the prognosis of IDCM, with dramatic reduction in heart failure–related mortality and SD during the past 3 decades. In the appropriate setting, the benefits of controlled randomized trials can be replicated in the real world, emphasizing the importance of tailored and systematic follow-up providing long-term continuity of care.
Sources of FundingThis work was supported by the Italian Ministry for University and Research (Programmi di Ricerca di rilevante Interesse Nazionale) and the European Union (Specific Targeted Research Projects, STREP Project 241577 “BIG HEART,” 7th European Framework Program).
DisclosuresNone.
References 1. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P,
Dubourg O, Kühl U, Maisch B, McKenna WJ, Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L, Keren A. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29:270–276.
2. Codd MB, Sugrue DD, Gersh BJ, Melton LJ III. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975-1984. Circulation. 1989;80:564–572.
3. Manolio TA, Baughman KL, Rodeheffer R, Pearson TA, Bristow JD, Michels VV, Abelmann WH, Harlan WR. Prevalence and etiology of id-iopathic dilated cardiomyopathy (summary of a National Heart, Lung, and Blood Institute workshop. Am J Cardiol. 1992;69:1458–1466.
4. Fuster V, Gersh BJ, Giuliani ER, Tajik AJ, Brandenburg RO, Frye RL. The natural history of idiopathic dilated cardiomyopathy. Am J Cardiol. 1981;47:525–531.
5. Gavazzi A, Lanzarini L, Cornalba C, Desperati M, Raisaro A, Angoli L, De Servi S, Specchia G. Dilated (congestive) cardiomyopathy. Follow-up study of 137 patients. G Ital Cardiol. 1984;14:492–498.
6. Ciaccheri M, Castelli G, Nannini M, Santoro G, Troiani V, Di Lenarda A, Miani D, Sinagra G, Mestroni L, Risoli A. Valutazione prognostica della cardiomiopatia dilatativa: follow up di 138 pazienti. G Ital Cardiol. 1990;20:645–650.
7. Di Lenarda A, Secoli G, Perkan A, Gregori D, Lardieri G, Pinamonti B, Sinagra G, Zecchin M, Camerini F. Changing mortality in dilated cardiomyopathy. The Heart Muscle Disease Study Group. Br Heart J. 1994;72(6 Suppl):S46–S51.
8. Gavazzi A, De Maria R, Porcu M, Beretta L, Casazza F, Castelli G, Luvini M, Parodi O, Recalcati F, Renosto G; on behalf of the SPIC. Dilated cardiomyopathy: a new natural history? The experience of

920 Circ Heart Fail September 2013
the Italian Multicenter Cardiomyopathy Study (SPIC). G Ital Cardiol. 1995;25:1109–1125.
9. Matsumura Y, Takata J, Kitaoka H, Kubo T, Baba Y, Hoshikawa E, Hamada T, Okawa M, Hitomi N, Sato K, Yamasaki N, Yabe T, Furuno T, Nishinaga M, Doi Y. Long-term prognosis of dilated cardiomyopa-thy revisited: an improvement in survival over the past 20 years. Circ J. 2006;70:376–383.
10. Kubo T, Matsumura Y, Kitaoka H, Okawa M, Hirota T, Hamada T, Hitomi N, Hoshikawa E, Hayato K, Shimizu Y, Yamasaki N, Yabe T, Nishinaga M, Takata J, Doi Y. Improvement in prognosis of dilated cardiomyopathy in the elderly over the past 20 years. J Cardiol. 2008;52:111–117.
11. Aleksova A, Sabbadini G, Merlo M, Pinamonti B, Barbati G, Zecchin M, Bussani R, Silvestri F, Iorio AM, Stolfo D, Dal Ferro M, Dragos AM, Meringolo G, Pyxaras S, Lo Giudice F, Perkan A, di Lenarda A, Sinagra G. Natural history of dilated cardiomyopathy: from asymptomatic left ventricular dysfunction to heart failure–a subgroup analysis from the Trieste Cardiomyopathy Registry. J Cardiovasc Med (Hagerstown). 2009;10:699–705.
12. Cleland JG, Cohen-Solal A, Aguilar JC, Dietz R, Eastaugh J, Follath F, Freemantle N, Gavazzi A, van Gilst WH, Hobbs FD, Korewicki J, Madeira HC, Preda I, Swedberg K, Widimsky J; IMPROVEMENT of Heart Failure Programme Committees and Investigators. Improvement programme in evaluation and management; Study Group on Diagnosis of the Working Group on Heart Failure of The European Society of Cardiology. Management of heart failure in primary care (the IMPROVEMENT of Heart Failure Programme): an international survey. Lancet. 2002;360:1631–1639.
13. Lee DS, Mamdani MM, Austin PC, Gong Y, Liu PP, Rouleau JL, Tu JV. Trends in heart failure outcomes and pharmacotherapy: 1992 to 2000. Am J Med. 2004;116:581–589.
14. Lee DS, Tu JV, Juurlink DN, Alter DA, Ko DT, Austin PC, Chong A, Stukel TA, Levy D, Laupacis A. Risk-treatment mismatch in the pharma-cotherapy of heart failure. JAMA. 2005;294:1240–1247.
15. Lenzen MJ, Boersma E, Reimer WJ, Balk AH, Komajda M, Swedberg K, Follath F, Jimenez-Navarro M, Simoons ML, Cleland JG. Under-utilization of evidence-based drug treatment in patients with heart failure is only partially explained by dissimilarity to patients enrolled in land-mark trials: a report from the Euro Heart Survey on Heart Failure. Eur Heart J. 2005;26:2706–2713.
16. Bursi F, Weston SA, Redfield MM, Jacobsen SJ, Pakhomov S, Nkomo VT, Meverden RA, Roger VL. Systolic and diastolic heart failure in the community. JAMA. 2006;296:2209–2216.
17. Patel P, White DL, Deswal A. Translation of clinical trial results into practice: temporal patterns of beta-blocker utilization for heart failure at hospital discharge and during ambulatory follow-up. Am Heart J. 2007;153:515–522.
18. Cecchi F, Olivotto I, Montereggi A, Santoro G, Dolara A, Maron BJ. Hypertrophic cardiomyopathy in Tuscany: clinical course and outcome in an unselected regional population. J Am Coll Cardiol. 1995;26:1529–1536.
19. McMurray JJV, Adamopoulos S, Anker SD, Auricchio A, Böhm M, Dickstein K, Falk V, Filippatos G, Fonseca C, Gomez-Sanchez MA, Jaarsma T, Køber L, Lip GY, Maggioni AP, Parkhomenko A, Pieske BM, Popescu BA, Rønnevik PK, Rutten FH, Schwitter J, Seferovic P, Stepinska J, Trindade PT, Voors AA, Zannad F, Zeiher A; ESC Committee for Practice Guidelines. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012. Eur Heart J. 2012;33:1787–847.
20. Staylor A. Heart Failure Therapy: Past, Present and Future posted 05/07/2002 on Medscape Cardiology. http://www.medscape.org/view-article/433746. Accessed June 20, 2012.
21. Merlo M, Pyxaras SA, Pinamonti B, Barbati G, Di Lenarda A, Sinagra G. Prevalence and prognostic significance of left ventricular reverse remod-eling in dilated cardiomyopathy receiving tailored medical treatment. J Am Coll Cardiol. 2011;57:1468–1476.
22. Zecchin M, Merlo M, Pivetta A, Barbati G, Lutman C, Gregori D, Serdoz LV, Bardari S, Magnani S, Di Lenarda A, Proclemer A, Sinagra G. How can optimization of medical treatment avoid unnecessary implantable cardioverter-defibrillator implantations in patients with idiopathic dilated cardiomyopathy presenting with “SCD-HeFT criteria?”. Am J Cardiol. 2012;109:729–735.
23. Bardy GH, Lee KL, Mark DB, Poole JE, Packer DL, Boineau R, Domanski M, Troutman C, Anderson J, Johnson G, McNulty SE, Clapp-Channing N, Davidson-Ray LD, Fraulo ES, Fishbein DP, Luceri RM, Ip JH; Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT)
Investigators. Amiodarone or an implantable cardioverter-defibrillator for congestive heart failure. N Engl J Med. 2005;352:225–237.
24. Teeter WA, Thibodeau JT, Rao K, Brickner ME, Toto KH, Nelson LL, Mishkin JD, Ayers CR, Miller JG, Mammen PP, Patel PC, Markham DW, Drazner MH. The natural history of new-onset heart failure with a severely depressed left ventricular ejection fraction: implications for timing of implantable cardioverter-defibrillator implantation. Am Heart J. 2012;164:358–364.
25. The CONSENSUS Trial Study Group. Effects of enalapril on mortal-ity in severe congestive heart failure: Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N Engl J Med. 1987; 316:1429–1435.
26. The SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med. 1991;325:293–302.
27. Cohn JN, Tognoni G; Valsartan Heart Failure Trial Investigators. A ran-domized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N Engl J Med. 2001;345:1667–1675.
28. Granger CB, McMurray JJ, Yusuf S, Held P, Michelson EL, Olofsson B, Ostergren J, Pfeffer MA, Swedberg K; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function intolerant to angiotensin-converting-enzyme inhibitors: the CHARM-Alternative trial. Lancet. 2003;362:772–776.
29. Waagstein F, Bristow MR, Swedberg K, Camerini F, Fowler MB, Silver MA, Gilbert EM, Johnson MR, Goss FG, Hjalmarson A. Beneficial effects of metoprolol in idiopathic dilated cardiomyopathy. Lancet. 1993;342:1441–1446.
30. CIBIS-II Investigators. The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): A randomized trial. Lancet. 1999;353:9–13.
31. Metoprolol CR/XL. Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF) Study Group. Effect of metoprolol CR/XL in chronic heart failure. Lancet. 1999;353:2001–2007.
32. Packer M, Fowler MB, Roecker EB, Coats AJ, Katus HA, Krum H, Mohacsi P, Rouleau JL, Tendera M, Staiger C, Holcslaw TL, Amann-Zalan I, DeMets DL; Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) Study Group. Effect of carvedilol on the morbidity of patients with severe chronic heart failure: results of the carvedilol prospective randomized cumulative survival (COPERNICUS) study. Circulation. 2002;106:2194–2199.
33. Jessup M, Abraham WT, Casey DE, Feldman AM, Francis GS, Ganiats TG, Konstam MA, Mancini DM, Rahko PS, Silver MA, Stevenson LW, Yancy CW. 2009 focused update: ACCF/AHA Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collabora-tion with the International Society for Heart and Lung Transplantation. Circulation. 2009;119:1977–2016.
34. Heart Failure Society of America, Lindenfeld J, Albert NM, Boehmer JP, Collins SP, Ezekowitz JA, Givertz MM, Katz SD, Klapholz M, Moser DK, Rogers JG, Starling RC, Stevenson WG, Tang WH, Teerlink JR, Walsh MN. Executive Summary: HFSA 2010 Comprehensive Heart Failure Practice Guideline. J Cardiac Fail. 2010;16:475–539.
35. Stevenson WG, Stevenson LW, Middlekauff HR, Fonarow GC, Hamilton MA, Woo MA, Saxon LA, Natterson PD, Steimle A, Walden JA. Improving survival for patients with advanced heart failure: a study of 737 consecutive patients. J Am Coll Cardiol. 1995;26:1417–1423.
36. Komajda M, Follath F, Swedberg K, Cleland J, Aguilar JC, Cohen-Solal A, Dietz R, Gavazzi A, Van Gilst WH, Hobbs R, Korewicki J, Madeira HC, Moiseyev VS, Preda I, Widimsky J, Freemantle N, Eastaugh J, Mason J; Study Group on Diagnosis of the Working Group on Heart Failure of the European Society of Cardiology. The EuroHeart Failure Survey programme–a survey on the quality of care among patients with heart failure in Europe. Part 2: treatment. Eur Heart J. 2003;24:464–474.
37. Jonsson A, Edner M, Alehagen U, Dahlström U. Heart failure registry: a valuable tool for improving the management of patients with heart fail-ure. Eur J Heart Fail. 2010;12:25–31.
38. Maggioni AP, Dahlström U, Filippatos G, Chioncel O, Leiro MC, Drozdz J, Fruhwald F, Gullestad L, Logeart D, Metra M, Parissis J, Persson H, Ponikowski P, Rauchhaus M, Voors A, Nielsen OW, Zannad F, Tavazzi L; Heart Failure Association of ESC (HFA). EURObservational Research Programme: the Heart Failure Pilot Survey (ESC-HF Pilot). Eur J Heart Fail. 2010;12:1076–1084.
39. Fonarow GC, Albert NM, Curtis AB, Stough WG, Gheorghiade M, Heywood JT, McBride ML, Inge PJ, Mehra MR, O’Connor CM, Reynolds

Castelli et al Long-Term Outcome of IDCM in Tuscany 921
D, Walsh MN, Yancy CW. Improving evidence-based care for heart fail-ure in outpatient cardiology practices: primary results of the Registry to Improve the Use of Evidence-Based Heart Failure Therapies in the Outpatient Setting (IMPROVE HF). Circulation. 2010;122:585–596.
40. Komajda M, Lapuerta P, Hermans N, Gonzalez-Juanatey JR, van Veldhuisen DJ, Erdmann E, Tavazzi L, Poole-Wilson P, Le Pen C. Adherence to guidelines is a predictor of outcome in chronic heart fail-ure: the MAHLER survey. Eur Heart J. 2005;26:1653–1659.
41. Albert NM, Fonarow GC, Yancy CW, Curtis AB, Stough WG, Gheorghiade M, Heywood JT, McBride M, Mehra MR, O’Connor CM, Reynolds D, Walsh MN. Influence of dedicated heart failure clinics on delivery of rec-ommended therapies in outpatient cardiology practices: findings from the Registry to Improve the Use of Evidence-Based Heart Failure Therapies in the Outpatient Setting (IMPROVE HF). Am Heart J. 2010;159:238–244.
42. Franke J, Zugck C, Wolter JS, Frankenstein L, Hochadel M, Ehlermann P, Winkler R, Nelles M, Zahn R, Katus HA, Senges J. A decade of devel-opments in chronic heart failure treatment: a comparison of therapy and outcome in a secondary and tertiary hospital setting. Clin Res Cardiol. 2012;101:1–10.
43. Bahler RC. Assessment of prognosis in idiopathic dilated cardiomyopa-thy. Chest. 2002;121:1016–1019.
44. Miura K, Matsumori A, Nasermoaddeli A, Soyama Y, Morikawa Y, Sakurai M, Kitabatake A, Nagai M, Inaba Y, Nakagawa H. Prognosis and prognostic factors in patients with idiopathic dilated cardio-myopathy in Japan—results from a nationwide study. Circulation. 2008;72:343–348.
45. Karaca O, Avci A, Guler GB, Alizade E, Guler E, Gecmen C, Emiroglu Y, Esen O, Esen AM. Tenting area reflects disease severity and prognosis in patients with non-ischaemic dilated cardiomyopathy and functional mitral regurgitation. Eur J Heart Fail. 2011;13:284–291.
46. Pecini R, Thune JJ, Torp-Pedersen C, Hassager C, Køber L. The rela-tionship between mitral regurgitation and ejection fraction as predic-tors for the prognosis of patients with heart failure. Eur J Heart Fail. 2011;13:1121–1125.
47. Tamura H, Watanabe T, Nishiyama S, Sasaki S, Arimoto T, Takahashi H, Shishido T, Miyashita T, Miyamoto T, Nitobe J, Hirono O, Kubota I. Increased left atrial volume index predicts a poor prognosis in patients with heart failure. J Card Fail. 2011;17:210–216.
CLINICAL PERSPECTIVEThe present study is based on a clinical experience whereby 603 patients with angiographically proven idiopathic dilated cardiomyopathy have been followed by the same team during the past 30 years in a Regional Hospital in Florence, providing substantial continuity of care during an extended time period. Outcome in our cohort was associated with the treatment era in which each patient was enrolled (1977–1984; 1985–1990; 1991–2000; and 2001–2011), reflecting progress in management and changing demographics of patients with idiopathic dilated cardiomyopathy. Of note, each period was associated with a 40% reduction in mortality compared with the previous one, with patients enrolled during the most recent decade showing a 75% relative risk reduction in cardiovascular mortality compared with those enrolled before 1985. With regard to specific causes of death, the greatest reduction in mortality related to refractory heart failure was observed in the transition from periods 1 to 2 after the introduction of angiotensin-converting enzyme inhibition. Conversely, progressive impact on sudden death was evident in the past 2 periods, reflecting the introduction of device-based therapies and full penetration of β-blocker treatment. These results quantify the impact of evolving treatment options for idiopathic dilated cardiomyopathy in the real world, emphasizing the importance of tailored follow-up and long-term continuity of care.

Prognostic Value of N-Terminal Pro-Brain Natriuretic Peptidein Outpatients With Hypertrophic Cardiomyopathy
Rossella D’Amato, MDa, Benedetta Tomberli, MDb,c,*, Gabriele Castelli, MDb, Roberto Spoladore, MDa,Francesca Girolami, BSce, Alessandra Fornaro, MDb, Anna Caldini, BScd, Francesca Torricelli, BSce,Paolo Camici, MDa, Gian Franco Gensini, MDb, Franco Cecchi, MDc, and Iacopo Olivotto, MDb
In hypertrophic cardiomyopathy, the plasma levels of N-terminal pro-brain natriureticpeptide (NT-proBNP) correlate with functional capacity. However, their prognostic rele-vance remains unresolved. We followed up 183 stable outpatients with hypertrophiccardiomyopathy (age 50 – 17 years, 64% men) for 3.9 – 2.8 years after NT-proBNPmeasurement. The primary end point included cardiovascular death, heart transplantation,resuscitated cardiac arrest, and appropriate implantable cardioverter-defibrillator inter-vention. The secondary end point (SE) included heart failure-related death or hospitaliza-tion, progression to end-stage disease, and stroke. The median NT-proBNP level was 615 pg/ml (intertertile range 310 to 1,025). The incidence of the primary end point in the lower,middle, and upper tertiles was 0%, 1.3%, and 2.1% annually, respectively (overall p[ 0.01).On multivariate analysis, the only independent predictors of the primary end point were NT-proBNP (hazard ratio for log-transformed values 5.8, 95% confidence interval 1.07 to 31.6;p [ 0.04) and a restrictive left ventricular filling pattern (hazard ratio 5.19, 95% confidenceinterval 1.3 to 21.9; p [ 0.02). The NT-proBNP cutoff value of 810 pg/ml had the bestsensitivity for the primary end point (88%), but the specificity was low (61%). The incidenceof the SE in the lower, middle, and upper NT-proBNP tertiles was 4.6%, 12.0%, and 11.2%annually, respectively (overall p [ 0.001). An NT-proBNP level of <310 pg/ml was asso-ciated with a 75% reduction in the rate of SE compared with a level of ‡310 pg/ml (hazardratio 0.25, 95% confidence interval 0.11 to 0.57; p [ 0.001), independent of age, leftventricular outflow tract obstruction, or atrial fibrillation. In conclusion, in stable outpatientswith hypertrophic cardiomyopathy, plasma NT-proBNP proved a powerful independentpredictor of death and heart failure-related events. Although the positive predictive accuracyof an elevated NT-proBNP level was modest, low values reflected true clinical stability,suggesting the possibility of avoiding or postponing aggressive treatment options. � 2013Elsevier Inc. All rights reserved. (Am J Cardiol 2013;112:1190e1196)
Natriuretic peptides have been validated as markers ofseverity and decompensation in various cardiac disorders andrepresent powerful predictors of outcome inheart failure (HF).1
They have also been useful as screening tools in large pop-ulations at risk of developing cardiovascular events, includingdiabetic and elderly patients.2 To date, the prognostic accuracyof natriuretic peptides in patients with hypertrophic cardio-myopathy (HC), the most common genetic heart disease, is
unknown.3e9 In the present study,we assessed the outcomes ofa consecutive outpatient cohort of patients with HC, who werelargely asymptomatic ormildly symptomatic at enrollment andwere followed up for almost 4 years after a comprehensiveclinical evaluation that had included serum N-terminal pro-brain natriuretic peptide (NT-proBNP) titration. NT-proBNP,theN-terminal part of the BNPpro-hormone, is released duringhemodynamic stress by stretched cardiacmyocytes and split byproteolytic enzymes into 2 peptides; the C-terminal fragment(BNP) represents the biologically active hormone.1,10 Becauseof a different clearance mechanism (renal for NT-proBNP andreceptor mediated for BNP), NT-proBNP represents a morereliable predictor of outcome than BNP10 and was, therefore,chosen for our analysis.
Methods
The serum NT-proBNP levels were determined in 218consecutive outpatients with HC who had been referred tothe Referral Center for Cardiomyopathies in Florence (Italy)before January 2011 as a part of a comprehensive nonin-vasive cardiovascular evaluation, including clinical history,physical examination, 12-lead electrocardiography, andechocardiography. The diagnosis of HC was determined by
aVita Salute University and San Raffaele Scientific Institute, Milan,Italy; bReferral Centre for Cardiomyopathies, dGeneral Laboratory, andeCytogenetics Unit, Careggi University Hospital, Florence, Italy; andcDepartment of Clinical and Experimental Medicine, University Florence,Italy. Manuscript received March 18, 2013; revised manuscript receivedand accepted June 11, 2013.
Drs. D’Amato and Tomberli contributed equally to the report.This work was supported by the Italian Ministry for University and
Research (Research Project of National Interest, Florence, Italy) and theEuropean Union (STREP Project 241577 “BIG HEART,” Seventh Euro-pean Framework Program, Florence, Italy).
See page 1195 for disclosure information.*Corresponding author: Tel: (þ39) 055-794-5138; fax: (þ39) 055-794-
9335.E-mail address: [email protected] (B. Tomberli).
0002-9149/13/$ - see front matter � 2013 Elsevier Inc. All rights reserved. www.ajconline.orghttp://dx.doi.org/10.1016/j.amjcard.2013.06.018

2-dimensional echocardiographic identification of leftventricular (LV) hypertrophy associated with nondilatedventricular chambers in the absence of any other cardiac orsystemic disease capable of producing the magnitude ofhypertrophy evident.11 Of the 218 patients, 35 with knowncoronary artery disease (n ¼ 15) or evidence of systolicdysfunction at the first evaluation (defined as a LV ejectionfraction <50%; n ¼ 20) were excluded. The remaining 183patients with HC constituted the study group enrolled in thepresent project. All had had a stable clinical course, withoutepisodes of acute HF or worsening of symptoms, in theprevious 3 months.
The local ethics committee approved the study, and allpatients gave informed consent to participate. Peripheral bloodsamples, collected after 30 minutes of complete rest, weremeasured using an electrochemiluminescent immunoassay(Elecsys proBNP II assay, Roche Diagnostics, Indianapolis,Indiana) on a Cobas E601 analyzer (Roche Diagnostics).12
Transthoracic echocardiography was performed usingcommercially available equipment. The standard evaluationincluded M-mode, 2-dimensional, and Doppler study,according to the recommendations of the American Society ofEchocardiography.13 LV systolic dysfunction, characterizingend-stage HC, was defined as an LV ejection fraction <50%.A restrictive LV filling pattern was defined as a ratio of early tolate velocity ofmitral inflow�2, associatedwith a decelerationtime of�150 ms (or a deceleration time<120 ms for patients
with atrial fibrillation).14 LV outflow tract obstruction wasconsidered presentwhen a peak instantaneous outflowgradientof�30mmHgwas measured using continuous wave Dopplerechocardiography under basal (at rest) conditions or withphysiologic provocation (Valsalva maneuver or exercise).
After enrollment, the patients were evaluated annually ormore often if dictated by the clinical status, using 12-leadelectrocardiography, echocardiography, and 24-hour electro-cardiographic Holter monitoring. Pharmacologic therapy wasprescribed as appropriate, according to the current HC guide-lines.3 Patientswith refractory symptoms related toLVoutflowtract obstruction despite optimal medical therapywere referredfor surgical myectomy; alcohol septal ablation was reservedfor patients at high operative risk owing to advanced age andco-morbidities.3 An implantable cardioverter-defibrillator wasoffered to survivors of cardiac arrest or sustained ventriculararrhythmias, for primary prevention in high-risk patientsevaluated using established risk markers for sudden cardiacdeath, and in patients with end-stage disease.3
Genetic counseling and mutational analysis were offeredto all patients as a part of the standard policy at our center.After providing informed consent, 133 of the study patients(73%) were screened for mutations in the protein-codingexons and splice sites of 8 myofilament genes, includingmyosin binding protein C, b-myosin heavy chain, theregulatory and essential light chains (MYL2 and MYL3),troponin-T, troponin-I, a-tropomyosin, and a-actin. Direct
Table 1Baseline patient characteristics within each N-terminal pro-brain natriuretic peptide (NT-proBNP) tertile
Variable Overall Population(n ¼ 183)
Lower Tertile(<310 pg/ml;
n ¼ 61)
Middle Tertile(310e1,025 pg/ml;
n ¼ 61)
Upper Tertile(>1,025 pg/ml;
n ¼ 61)
p Value
Men 118 (64%) 53 (86%)*,† 39 (64%)† 26 (42%)* <0.01z
Age (yrs) 50 � 17 47 � 17 52 � 16 50 � 19 0.256NYHA class III-IV at first evaluation 33 (18%) 4 (6.5%)*,† 13 (21%) 16 (26%) 0.01z
Atrial fibrillation 48 (26%) 7 (11%)*,† 19 (31%) 22 (36%) <0.01z
LA diameter (mm) 44 � 7 42 � 6*,† 45 � 8 46 � 7 <0.01z
Restrictive LV filling pattern 12 (6.5%) 1 (1.6%) 6 (10%) 5 (8%) 0.26Peak LVOT gradient �30 mm Hg 65 (36%) 14 (23%)*,† 25 (41%) 27 (44%) 0.03z
Positive genotype 98 (53%) 30 (49%) 32 (52%) 36 (59%) 0.14Maximum LV wall thickness (mm) 22 � 6 20 � 3*,† 24 � 5 23 � 8 <0.01z
b-Blocker use 122 (67%) 34 (55%)* 45 (74%) 43 (70%) 0.07Amiodarone use 23 (12%) 5 (8%) 8 (13%) 10 (16%) 0.38With ICD 21 (17%) 7 (11%) 11 (18%) 14 (23%) 0.24Primary end point 8 (4%) 0*,† 3 (4.9%) 5 (8.1%) 0.01z
Sudden death 4 (2%) 0 2 (3.3%) 2 (3.3%) 0.18HF-related death 2 (1%) 0 1 (1.6%) 1 (1.6%) 0.44Cardiac surgery-related death 1 (0.5%) 0 0 1 (1.6%) 0.20Heart transplantation 1 (0.5%) 0 0 1 (1.6%) 0.20Appropriate ICD discharge 0 0 0 0 —
Secondary end point 67 (37%) 11 (18%)*,† 29 (47%) 27 (44%) <0.01z
Hospitalization for HF 38 (21%) 4 (6.5%)*,† 15 (24.5%) 19 (31%) <0.01z
Progression to NYHA III-IV (during follow-up) 20 (11%) 4 (6.5%) 9 (15%) 7 (11.5%) 0.17Progression to end-stage disease 21 (11.5%) 2 (3%)*,† 9 (15%) 10 (16%) <0.01z
New-onset AF 14 (8%) 4 (6.5%) 6 (10%) 4 (6.5%) 0.59Cerebrovascular event 8 (4%) 0† 2 (3%) 6 (10%) <0.01z
ICD ¼ implantable cardioverter-defibrillator; LA ¼ left atrial; LVOT ¼ LV outflow tract; NYHA ¼ New York Heart Association.* p <0.05 versus middle tertile.† p <0.05 versus upper tertile.z statistically significant.
Cardiomyopathy/NT-ProBNP and Outcome in HC 1191
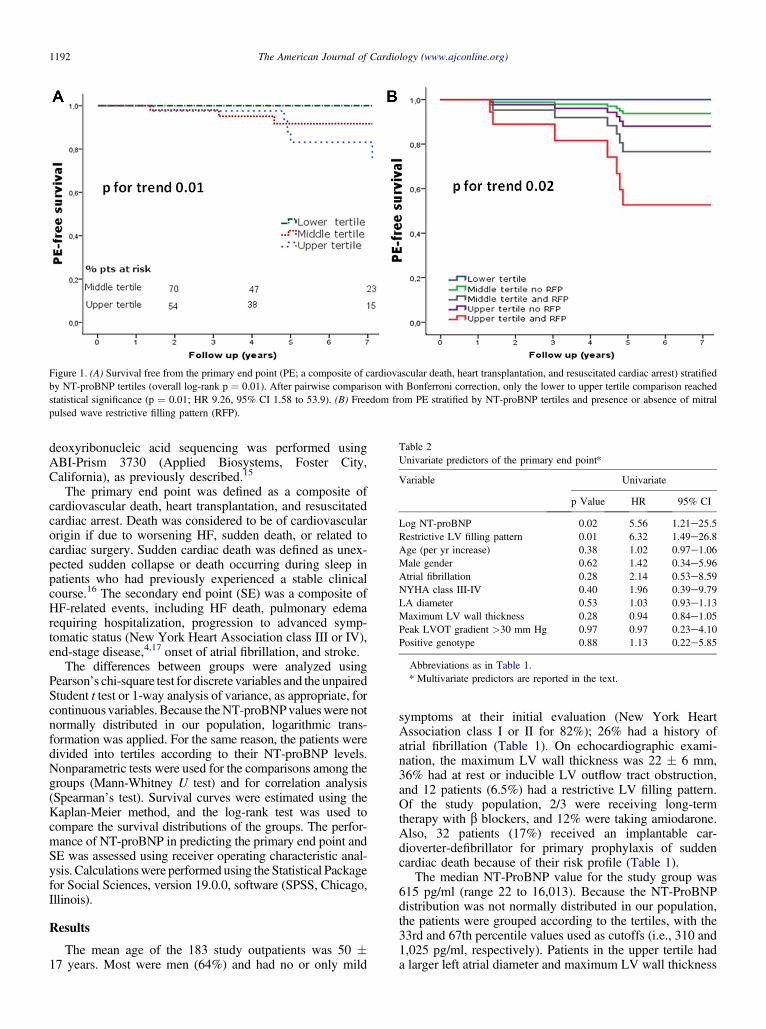
deoxyribonucleic acid sequencing was performed usingABI-Prism 3730 (Applied Biosystems, Foster City,California), as previously described.15
The primary end point was defined as a composite ofcardiovascular death, heart transplantation, and resuscitatedcardiac arrest. Death was considered to be of cardiovascularorigin if due to worsening HF, sudden death, or related tocardiac surgery. Sudden cardiac death was defined as unex-pected sudden collapse or death occurring during sleep inpatients who had previously experienced a stable clinicalcourse.16 The secondary end point (SE) was a composite ofHF-related events, including HF death, pulmonary edemarequiring hospitalization, progression to advanced symp-tomatic status (New York Heart Association class III or IV),end-stage disease,4,17 onset of atrial fibrillation, and stroke.
The differences between groups were analyzed usingPearson’s chi-square test for discrete variables and the unpairedStudent t test or 1-way analysis of variance, as appropriate, forcontinuous variables.Because theNT-proBNPvalueswere notnormally distributed in our population, logarithmic trans-formation was applied. For the same reason, the patients weredivided into tertiles according to their NT-proBNP levels.Nonparametric tests were used for the comparisons among thegroups (Mann-Whitney U test) and for correlation analysis(Spearman’s test). Survival curves were estimated using theKaplan-Meier method, and the log-rank test was used tocompare the survival distributions of the groups. The perfor-mance of NT-proBNP in predicting the primary end point andSE was assessed using receiver operating characteristic anal-ysis. Calculationswere performed using the Statistical Packagefor Social Sciences, version 19.0.0, software (SPSS, Chicago,Illinois).
Results
The mean age of the 183 study outpatients was 50 �17 years. Most were men (64%) and had no or only mild
symptoms at their initial evaluation (New York HeartAssociation class I or II for 82%); 26% had a history ofatrial fibrillation (Table 1). On echocardiographic exami-nation, the maximum LV wall thickness was 22 � 6 mm,36% had at rest or inducible LV outflow tract obstruction,and 12 patients (6.5%) had a restrictive LV filling pattern.Of the study population, 2/3 were receiving long-termtherapy with b blockers, and 12% were taking amiodarone.Also, 32 patients (17%) received an implantable car-dioverter-defibrillator for primary prophylaxis of suddencardiac death because of their risk profile (Table 1).
The median NT-ProBNP value for the study group was615 pg/ml (range 22 to 16,013). Because the NT-ProBNPdistribution was not normally distributed in our population,the patients were grouped according to the tertiles, with the33rd and 67th percentile values used as cutoffs (i.e., 310 and1,025 pg/ml, respectively). Patients in the upper tertile hada larger left atrial diameter and maximum LV wall thickness
Figure 1. (A) Survival free from the primary end point (PE; a composite of cardiovascular death, heart transplantation, and resuscitated cardiac arrest) stratifiedby NT-proBNP tertiles (overall log-rank p ¼ 0.01). After pairwise comparison with Bonferroni correction, only the lower to upper tertile comparison reachedstatistical significance (p ¼ 0.01; HR 9.26, 95% CI 1.58 to 53.9). (B) Freedom from PE stratified by NT-proBNP tertiles and presence or absence of mitralpulsed wave restrictive filling pattern (RFP).
Table 2Univariate predictors of the primary end point*
Variable Univariate
p Value HR 95% CI
Log NT-proBNP 0.02 5.56 1.21e25.5Restrictive LV filling pattern 0.01 6.32 1.49e26.8Age (per yr increase) 0.38 1.02 0.97e1.06Male gender 0.62 1.42 0.34e5.96Atrial fibrillation 0.28 2.14 0.53e8.59NYHA class III-IV 0.40 1.96 0.39e9.79LA diameter 0.53 1.03 0.93e1.13Maximum LV wall thickness 0.28 0.94 0.84e1.05Peak LVOT gradient >30 mm Hg 0.97 0.97 0.23e4.10Positive genotype 0.88 1.13 0.22e5.85
Abbreviations as in Table 1.* Multivariate predictors are reported in the text.
1192 The American Journal of Cardiology (www.ajconline.org)
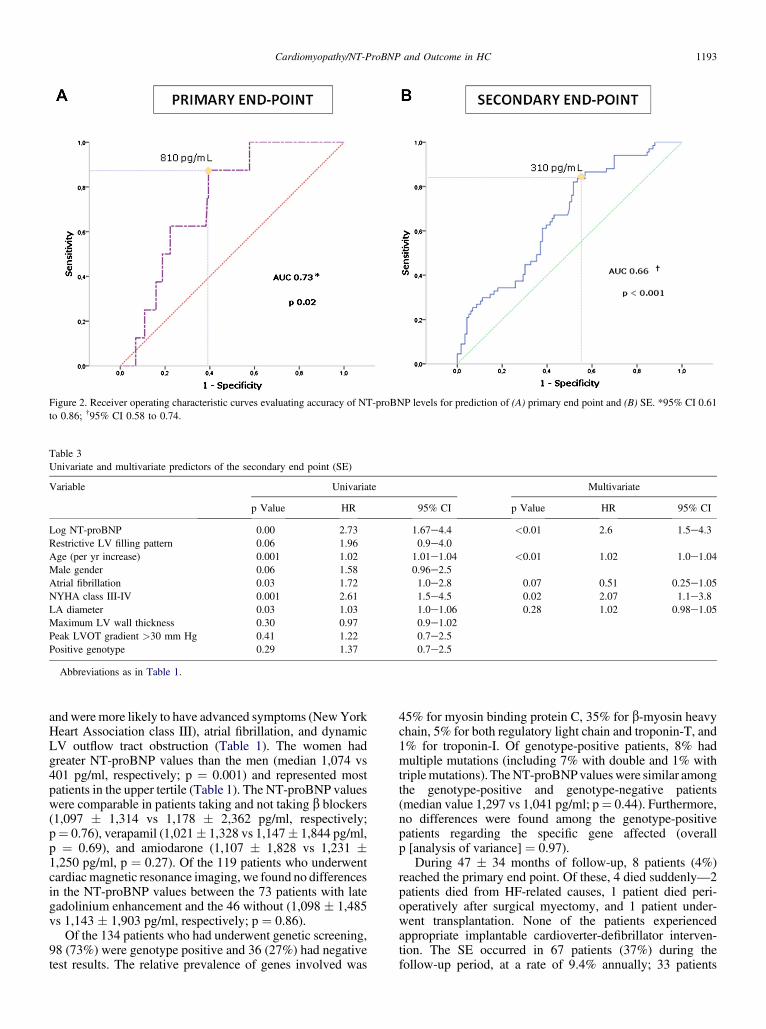
andwere more likely to have advanced symptoms (NewYorkHeart Association class III), atrial fibrillation, and dynamicLV outflow tract obstruction (Table 1). The women hadgreater NT-proBNP values than the men (median 1,074 vs401 pg/ml, respectively; p ¼ 0.001) and represented mostpatients in the upper tertile (Table 1). The NT-proBNP valueswere comparable in patients taking and not taking b blockers(1,097 � 1,314 vs 1,178 � 2,362 pg/ml, respectively;p¼ 0.76), verapamil (1,021� 1,328 vs 1,147� 1,844 pg/ml,p ¼ 0.69), and amiodarone (1,107 � 1,828 vs 1,231 �1,250 pg/ml, p ¼ 0.27). Of the 119 patients who underwentcardiacmagnetic resonance imaging, we found no differencesin the NT-proBNP values between the 73 patients with lategadolinium enhancement and the 46 without (1,098 � 1,485vs 1,143 � 1,903 pg/ml, respectively; p ¼ 0.86).
Of the 134 patients who had underwent genetic screening,98 (73%) were genotype positive and 36 (27%) had negativetest results. The relative prevalence of genes involved was
45% for myosin binding protein C, 35% for b-myosin heavychain, 5% for both regulatory light chain and troponin-T, and1% for troponin-I. Of genotype-positive patients, 8% hadmultiple mutations (including 7% with double and 1% withtriplemutations). TheNT-proBNPvalueswere similar amongthe genotype-positive and genotype-negative patients(median value 1,297 vs 1,041 pg/ml; p¼ 0.44). Furthermore,no differences were found among the genotype-positivepatients regarding the specific gene affected (overallp [analysis of variance] ¼ 0.97).
During 47 � 34 months of follow-up, 8 patients (4%)reached the primary end point. Of these, 4 died suddenly—2patients died from HF-related causes, 1 patient died peri-operatively after surgical myectomy, and 1 patient under-went transplantation. None of the patients experiencedappropriate implantable cardioverter-defibrillator interven-tion. The SE occurred in 67 patients (37%) during thefollow-up period, at a rate of 9.4% annually; 33 patients
Figure 2. Receiver operating characteristic curves evaluating accuracy of NT-proBNP levels for prediction of (A) primary end point and (B) SE. *95% CI 0.61to 0.86; †95% CI 0.58 to 0.74.
Table 3Univariate and multivariate predictors of the secondary end point (SE)
Variable Univariate Multivariate
p Value HR 95% CI p Value HR 95% CI
Log NT-proBNP 0.00 2.73 1.67e4.4 <0.01 2.6 1.5e4.3Restrictive LV filling pattern 0.06 1.96 0.9e4.0Age (per yr increase) 0.001 1.02 1.01e1.04 <0.01 1.02 1.0e1.04Male gender 0.06 1.58 0.96e2.5Atrial fibrillation 0.03 1.72 1.0e2.8 0.07 0.51 0.25e1.05NYHA class III-IV 0.001 2.61 1.5e4.5 0.02 2.07 1.1e3.8LA diameter 0.03 1.03 1.0e1.06 0.28 1.02 0.98e1.05Maximum LV wall thickness 0.30 0.97 0.9e1.02Peak LVOT gradient >30 mm Hg 0.41 1.22 0.7e2.5Positive genotype 0.29 1.37 0.7e2.5
Abbreviations as in Table 1.
Cardiomyopathy/NT-ProBNP and Outcome in HC 1193

(18%) experienced >1 event. The cumulative rates ofHF-related events were 21% (5.3%/yr) for acute HF requiringhospitalization, 11% (2.8%/yr) for worsening of symptomaticstatus to New York Heart Association class III or IV; and 8%(1.9%/yr) for new-onset atrial fibrillation. Eight patients (4%;1.1%/yr) experienced cerebrovascular events judged to becardioembolic. The global rate of progression to end-stagedisease was 11.5% (n ¼ 21, 2.8%/yr).
When the events were analyzed with regard to the NT-proBNP distribution, the cumulative rate of the primary endpoint at the end of the follow-up period was 5% (1.3%/yr)and 8% (2.1%/yr) in the middle and upper tertile, respec-tively. No event occurred in the lower tertile. As shown inFigure 1, the patients in the lower tertile (<310 pg/ml) hada favorable outcome compared with those in the other ter-tiles (overall p ¼ 0.01). The patients in the middle and uppertertiles, however, had comparable survival rates. On multi-variate Cox regression analysis, only NT-proBNP anda restrictive LV filling pattern were independently associ-ated with the primary end point (hazard ratio [HR] for log-transformed NT-proBNP 5.8, 95% confidence interval [CI]1.07 to 31.6, p ¼ 0.04; HR for a restrictive LV filling pattern5.19, 95% CI 1.23 to 21.9, p ¼ 0.02; Table 2). Of thepatients in the middle NT-proBNP tertile, the presence ofa restrictive LV filling pattern conferred additional prog-nostic stratification, although it had no additional value forpatients in the upper tertile (Figure 1). Receiver operatingcharacteristic curve analysis demonstrated moderate powerfor NT-proBNP in the prediction of the primary end point(area under the curve 0.73, p ¼ 0.02). The NT-proBNPvalue of 810 pg/ml proved the best threshold for the iden-tification of patients at risk of cardiovascular death, withhigh sensitivity (88%) but low specificity (61%; Figure 2).
On multivariate analysis, NT-proBNP proved a powerfulpredictor of the SE, independent of other relevant clinicalvariables such as age, atrial fibrillation, and left atrial
dimensions (HR for log-transformed NT-ProBNP 2.6,95% CI 1.5 to 4.3, p <0.01; Table 3). Specifically, anNT-proBNP level <310 pg/ml was associated with a 75%reduction in risk of HF-related events compared with valuesof �310 pg/ml (HR for HF-related events 0.25, 95% CI 0.11to 0.57; p ¼ 0.001). On receiver operating characteristicanalysis, the sensitivity for the cutoff value of NT-proBNPof 310 pg/ml was 85%; the specificity, however, was low(45%; Figure 2). Overall, the negative predictive accuracyfor the SE associated with an NT-proBNP level <310 pg/mlwas 85%. Similarly, the positive predictive accuracy forend-stage progression was low (25%), despite an excellentnegative predictive value (88%). The combination of an NT-proBNP level in the upper tertile with left atrial dilation of�50 mm was associated with a 2.8 HR for the SE (95% CI1.39 to 5.69; p ¼ 0.004).
The cumulative rates of SE were 18% (4.6%/yr) in thelower, 47% (12.0%/yr) in the middle and 44% (11.2%/yr) inthe upper tertile. The rate of the SE among the patients in thelower tertile was similar for the patients with and without LVoutflow tract obstruction (p¼ 0.73). Survival free from the SEwas favorable in the lower NT-proBNP tertile compared withthe middle and upper tertiles (overall p <0.01; Figure 3). Asobserved for the primary end point, however, the middle andupper tertiles had comparable event-free SE survival rates.The presence of congestive symptoms at enrollment(expressed as New York Heart Association functional class)significantly contributed to the risk stratification of patientswithin the middle and upper NT-proBNP tertiles (Figure 3).
Although serial NT-proBNP testing was not systemati-cally performed, repeat values were available at variableintervals from the original titration (2.6� 1.2 years) for 48 ofthe 183 study patients (26%). Of these 48 patients, 24 showedan increase inNT-proBNP during the follow-up period (range30 to 5,958 pg/ml; average 1,207� 1,412) and 24 a decrease(range �5 to �3,558 pg/ml; average �663 � 740). The
Figure 3. (A) Survival free from the SE (a composite of HF death, acute decompensation, progression to end-stage disease, and stroke) stratified by NT-proBNPtertiles (overall log-rank p ¼ 0.002). After Bonferroni correction, the lower to middle and lower to upper tertile comparisons reached statistical significance(p ¼ 0.001 and p ¼ 0.002, respectively). (B) In a Cox regression model including other independent predictors of SE, only the initial New York HeartAssociation (NYHA) functional class helped to further stratify high-risk patients.
1194 The American Journal of Cardiology (www.ajconline.org)

prevalence of the SE was almost 3 times more common in thegroup with an NT-BNP increase (n ¼ 8; 33%) than in thepatients with an NT-BNP decrease (n ¼ 3; 12%; p ¼ 0.03,univariate survival analysis).
Discussion
In patients with HC, the natriuretic peptides are oftenelevated and have been shown to correlate with the degree ofsymptomatic impairment and functional capacity,18 LV massindex, left atrial volume, and LV outflow tract obstruc-tion.19,20 To date, however, the predictive accuracy of natri-uretic peptides with regard to outcomes in this complexdisease has received limited attention.21,22 In our HC cohort,followed up for an average of almost 4 years, NT-proBNPproved to be an independent predictor of cardiovascularmortality (our primary end point), with a sixfold increase inthe likelihood for each increment in the log NT-proBNP. Therelatively high NT-proBNP value of 810 pg/ml proved thebest cutoff for the identification of patients at risk of cardio-vascular death, althoughwith limited specificity (61%) owingto the expected paucity of events during the follow-up period.In the present analysis, however, sudden death representeda potential confounder with regard to the predictive values ofNT-proBNP, because life-threatening ventricular arrhythmiascan occur in stable and often asymptomatic or only mildlysymptomatic patients, lacking LV overload and hemody-namic decompensation at death and, thus, might not bepredictable using the biomarkers of HF.
Our SE was more specifically focused on HF-relatedevents and, as expected, was more accurately predicted bythe NT-proBNP levels. However, NT-proBNP appearedmost useful for its high negative predictive value (i.e., asa screening test to identify low-risk patients, rather than tosingle out those with a greater risk of events). The threshold of310 pg/ml, representing the cutoff value for the lower tertile,had very high negative predictive accuracy (85%) andretained important prognostic value even after adjustment forother powerful adverse clinical features, such as age, NewYorkHeart Association functional class, and LVoutflow tractobstruction. However, neither this nor other cutoffs were ableto further stratify the risk in the 2 upper tertiles, despite a widespectrum of values ranging to >16,000 pg/ml. These datahave confirmed and expanded those of previous studies21e23
showing a significant association between the NT-proBNPlevel and HF-related prognosis in patients with HC. Veryrecently, Coats et al23 reported a sevenfold increased risk ofdeath and transplantation in patients with HC and abnormalNT-proBNP values. That study enrolled patients with overtsystolic dysfunction at baseline, a subgroup with particularlyominous short-term outcomes9; however, that subgroup wasexcluded from our cohort to avoid a potential confounder.23
The most important clinical implication of our findingswas that low NT-proBNP values can identify truly stabledisease and allow cautious reassurance of patients with HCregarding the risk of disease progression and HF, even in thepresence of potentially adverse clinical features. For patientswith HC and dynamic LV outflow tract obstruction, forexample, low NT-proBNP values might represent a usefularbitrator, supporting the decision to avoid or postponeinvasive septal reduction therapies, particularly when the
symptoms are sufficiently well-controlled by pharmacologictreatment.24 This hypothesis deserves to be tested in specifi-cally designed, longitudinal studies. In contrast, greaterNT-proBNP values represent a rather nonspecific “red flag,”lacking sufficient specificity for the identification of the trulyhigh-risk subset, and needs to be complemented by additionalclinical and instrumental workup.4 For example, the presenceof a restrictive LV filling pattern significantly increased therisk of the primary end point in our patients with HCbelonging to the intermediate NT-proBNP tertile.14 Further-more, in a subgroup of 48 of the 183 study patients for whomNT-BNP repeat values were available during follow-up, theprevalence of the SE was almost 3 times more common in thegroupwith anNT-BNP increase than in those with decreasingtiters. Thus, it is plausible that longitudinal, serial testing ofNT-proBNP during follow-up could provide incrementalclinical benefit comparedwith a single baseline evaluation, byrevealing the trend of over time and the response totreatment.25
Our results have suggested that the prognostic value ofNT-proBNP is only partly explained by other covariablestraditionally regarded as determinants of pressure overload,such as diastolic dysfunction and LV outflow tract obstruction,indicating that someadditionalmechanism, other thanmyocytestretch,must be involved. InnonobstructiveHC, the ventricularexpression of BNP has been suggested to partly reflectmyocardial disarray, hypertrophy, and fibrosis, rather thansolely a response to the hemodynamic load.26 For example,recent evidence that a quota ofBNP can beproducedby cardiacfibroblasts, involved in the regulation of extracellular matrixturnover,27 might explain the correlation observed among theNT-proBNP levels, the extent of late gadolinium enhancementat cardiac magnetic resonance imaging,19 and plasmabiochemical markers of collagen turnover.28 Finally, micro-vascular dysfunction, a key pathophysiologic feature ofHC, might represent an additional stimulus to NT-proBNPproduction in asymptomatic patients.29 This concept isconsistent with the work by Nakamura et al,30 demonstratingthat myocardial ischemia might stimulate peptide release inpatients with HC and angiographically normal coronaryarteries.
In conclusion, in stable outpatients with HC, plasmaNT-proBNP proved a powerful independent predictor ofdeath andHF-related events. Although the positive predictiveaccuracy of the elevated NT-proBNP values was modest, lowvalues reflected true clinical stability, suggesting the possi-bility of avoiding or postponing aggressive treatment options.
Disclosures
The authors have no conflicts of interest to disclose.
1. Daniels LB, Maisel AS. Natriuretic peptides. J Am Coll Cardiol2007;50:2357e2368.
2. Wang TJ, Larson MG, Levy D, Benjamin EJ, Leip EP, Omland T, WolfPA, Vasan RS. Plasma natriuretic peptide levels and the risk ofcardiovascular events and death. N Engl J Med 2004;350:655e663.
3. GershBJ,MaronBJ, BonowRO,Dearani JA, FiferMA, LinkMS,NaiduSS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA,Udelson JE, Yancy CW; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; Amer-ican Association for Thoracic Surgery; American Society of Echocar-diography; American Society of Nuclear Cardiology; Heart Failure
Cardiomyopathy/NT-ProBNP and Outcome in HC 1195

Society of America; Heart Rhythm Society; Society for CardiovascularAngiography and Interventions; Society of Thoracic Surgeons. 2011ACCF/AHA guideline for the diagnosis and treatment of hypertrophiccardiomyopathy: executive summary: a report of the American Collegeof Cardiology Foundation/American Heart Association Task Force onPractice Guidelines. Circulation 2011;124:2761e2796.
4. Olivotto I, Cecchi F, Poggesi C, Yacoub MH. Patterns of diseaseprogression in hypertrophic cardiomyopathy: an individualized approachto clinical staging. Circ Heart Fail 2012;5:535e546.
5. Maron BJ. Sudden death in hypertrophic cardiomyopathy. J CardiovascTransl Res 2009;2:368e380.
6. Olivotto I, Girolami F, Nistri S, Rossi A, Rega L, Garbini F, Grifoni C,Cecchi F, Yacoub MH. The many faces of hypertrophic cardiomyop-athy: from developmental biology to clinical practice. J CardiovascTransl Res 2009;2:349e367.
7. Ommen SR, Maron BJ, Olivotto I, Maron MS, Cecchi F, Betocchi S,Gersh BJ, Ackerman MJ, McCully RB, Dearani JA, Schaff HV,Danielson GK, Tajik AJ, Nishimura RA. Long-term effects of surgicalseptal myectomy on survival in patients with obstructive hypertrophiccardiomyopathy. J Am Coll Cardiol 2005;46:470e476.
8. Maron BJ. Controversies in cardiovascular medicine: surgical myec-tomy remains the primary treatment option for severely symptomaticpatients with obstructive hypertrophic cardiomyopathy. Circulation2007;116:196e206.
9. Yacoub MH, Olivotto I, Cecchi F. “End-stage” hypertrophic cardio-myopathy: from mystery to model. Nat Clin Pract Cardiovasc Med2007;4:232e233.
10. Collinson PO, Barnes SC, Gaze DC, Galasko G, Lahiri A, Senior R.Analytical performance of the N terminal pro B type natriuretic peptide(NT-proBNP) assay on the Elecsys 1010 and 2010 analysers. Eur JHeart Fail 2004;6:365e368.
11. Maron BJ,MaronMS. Hypertrophic cardiomyopathy. Lancet 2012;381:242e255.
12. Yeo KT, Wu AH, Apple FS, Kroll MH, Christenson RH, LewandrowskiKB, Sedor FA, Butch AW. Multicenter evaluation of the RocheNT-proBNP assay and comparison to the Biosite Triage BNP assay. ClinChim Acta 2003;338:107e115.
13. Nagueh SF, Bierig SM, Budoff MJ, Desai M, Dilsizian V, Eidem B,Goldstein SA, Hung J, Maron MS, Ommen SR, Woo A. AmericanSociety of Echocardiography clinical recommendations for multi-modality cardiovascular imaging of patients with hypertrophic cardio-myopathy: endorsed by the American Society of Nuclear Cardiology,Society for Cardiovascular Magnetic Resonance, and Society ofCardiovascular Computed Tomography. J Am Soc Echocardiogr2011;24:473e498.
14. Pinamonti B, Di Lenarda A, Nucifora G, Gregori D, Perkan A, SinagraG. Incremental prognostic value of restrictive filling pattern in hyper-trophic cardiomyopathy: a Doppler echocardiographic study. Eur JEchocardiogr 2008;9:466e471.
15. Olivotto I, Girolami F, Ackerman MJ, Nistri S, Bos JM, Zachara E,Ommen SR, Theis JL, Vaubel RA, Re F, Armentano C, Poggesi C,Torricelli F, Cecchi F. Myofilament protein gene mutation screeningand outcome of patients with hypertrophic cardiomyopathy. Mayo ClinProc 2008;83:630e638.
16. Maron BJ, Olivotto I, Spirito P, Casey SA, Bellone P, Gohman TE,Graham KJ, Burton DA, Cecchi F. Epidemiology of hypertrophiccardiomyopathy-related death: revisited in a large non-referral-basedpatient population. Circulation 2000;102:858e864.
17. Harris KM, Spirito P,MaronMS, ZenovichAG, Formisano F, Lesser JR,Mackey-Bojack S, Manning WJ, Udelson JE, Maron BJ. Prevalence,
clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation 2006;114:216e225.
18. Maron BJ, Tholakanahalli VN, Zenovich AG, Casey SA, Duprez D,Aeppli DM, Cohn JN. Usefulness of B-type natriuretic peptide assay inthe assessment of symptomatic state in hypertrophic cardiomyopathy.Circulation 2004;109:984e989.
19. Park JR, Choi JO, Han HJ, Chang SA, Park SJ, Lee SC, Choe YH,Park SW, Oh JK. Degree and distribution of left ventricular hyper-trophy as a determining factor for elevated natriuretic peptide levels inpatients with hypertrophic cardiomyopathy: insights from cardiacmagnetic resonance imaging. Int J Cardiovasc Imaging 2012;28:763e772.
20. Vanderheyden M, Goethals M, Verstreken S, De Bruyne B, Muller K,Van Schuerbeeck E, Bartunek JJ. Wall stress modulates brain natri-uretic peptide production in pressure overload cardiomyopathy. AmColl Cardiol 2004;44:2349e2354.
21. Mutlu B, Bayrak F, Kahveci G, Degertekin M, Eroglu E, Basaran Y.Usefulness of N-terminal pro-B-type natriuretic peptide to predictclinical course in patients with hypertrophic cardiomyopathy. Am JCardiol 2006;98:1504e1506.
22. Kitaoka H, Kubo T, Okawa M, Takenaka N, Sakamoto C, Baba Y,Hayashi K, Yamasaki N, Matsumura Y, Doi YL. Tissue Dopplerimaging and plasma BNP levels to assess the prognosis in patients withhypertrophic cardiomyopathy. J Am Soc Echocardiogr 2011;24:1020e1025.
23. Coats CJ, Gallagher MJ, Foley M, O’Mahony C, Critoph C, Gimeno J,Dawnay A, McKenna WJ, Elliott PM. Relation between serum N-terminal pro-brain natriuretic peptide and prognosis in patients withhypertrophic cardiomyopathy. Eur Heart J 2013 Mar 8.
24. Maron MS, Olivotto I, Betocchi S, Casey SA, Lesser JR, Losi MA,Cecchi F, Maron BJ. Effect of left ventricular outflow tract obstructionon clinical outcome in hypertrophic cardiomyopathy. N Eng J Med2003;348:295e303.
25. Jourdain P, Jondeau G, Funck F, Gueffet P, Le Helloco A, Donal E,Aupetit JF, Aumont MC, Galinier M, Eicher JC, Cohen-Solal A,Juillière Y. Plasma brain natriuretic peptide-guided therapy to improveoutcome in heart failure: the STARS-BNP Multicenter Study. J AmColl Cardiol 2007;49:1733e1739.
26. Hasegawa K, Fujiwara H, Doyama K, Miyamae M, Fujiwara T, SugaS, Mukoyama M, Nakao K, Imura H, Sasayama S. Ventricularexpression of brain natriuretic peptide in hypertrophic cardiomyopathy.Circulation 1993;88:372e380.
27. Tsuruda T, Boerrigter G, Huntley BK, Noser JA, Cataliotti A, Costello-Boerrigter LC, Chen HH, Burnett JC Jr. Brain natriuretic peptide isproduced in cardiac fibroblasts and induces matrix metalloproteinases.Circ Res 2002;91:1127e1134.
28. Lombardi R, Betocchi S, Losi MA, Tocchetti CG, Aversa M, MirandaM, D’Alessandro G, Cacace A, Ciampi Q, Chiariello M. Myocardialcollagen turnover in hypertrophic cardiomyopathy. Circulation2003;108:1455e1460.
29. Olivotto I, Cecchi F, Gistri R, Lorenzoni R, Chiriatti G, Girolami F,Torricelli F, Camici PG. Relevance of coronary microvascular flowimpairment to long-term remodeling and systolic dysfunction inhypertrophic cardiomyopathy. J Am Coll Cardiol 2006;47:1043e1048.
30. Nakamura T, Sakamoto K, Yamano T, Kikkawa M, Zen K, HikosakaT, Kubota T, Azuma A, Nishimura T. Increased plasma brain natri-uretic peptide level as a guide for silent myocardial ischemia in patientswith non-obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol2002;39:1657e1663.
1196 The American Journal of Cardiology (www.ajconline.org)

[Cardiogenetics 2013; 3(s1):e3] [page 13]
Anderson-Fabry, the histrionicdisease: from genetics toclinical managementFranco Cecchi,1 Benedetta Tomberli,1,2Amelia Morrone3,41Department of Clinical and ExperimentalMedicine; 2Department of Heart andVessels, Referral Center forCardiomyopathies, Careggi Hospital;3Department of Neurosciences,Psychology, Pharmacology and ChildHealth, University of Florence;4Molecular and Cell Biology Laboratory,Paediatric Neurology Unit, NeuroscienceDepartment, Meyer Children’s Hospital,Florence, Italy
Abstract
Anderson-Fabry disease (AFD) is an X-linked lysosomal storage disorder of glycosph-ingolipid catabolism, due to deficiency orabsence of a galactosidase A (a-gal A) enzyme.The disease may affect males and females, thelatter with an average 10 years delay.Metabolites storage (mostly Gb3 and lyso-Gb3)leads to progressive cellular and multiorgandysfunction, with either early and late onsetvariable clinical manifestations that usuallyreduce quality of life and life expectancy. Heartand kidney failure, stroke and sudden deathare the most devastating complications. AFD isalways been considered a very rare disease,although new epidemiologic data, based onnewborn screening, showed that AFD preva-lence is probably underestimated and muchhigher than previously reported, especially forlate-onset atypical phenotypes. Currently, thediagnosis may be easier and simpler by evalu-ating a-gal A enzyme activity and geneticanalysis for GLA gene mutations on dried bloodspot. While a marked a-gal A deficiency leadsto diagnosis of AFD in hemizygous males, themolecular analysis is mandatory in heterozy-gous females. However, referral to a centerwith an expert multidisciplinary team is high-ly advisable, in order to ensure careful man-agement and treatment of patients, based alsoon accurate molecular and biochemical datainterpretation. While long-term efficacy ofenzyme replacement therapy (ERT) inadvanced stage is still debated, increasing evi-dence shows greater efficacy of early treat-ment initiation. Concomitant, organ-specifictherapy is also needed. New treatmentapproaches, such as chemical chaperone ther-apy, alone or in combination with ERT, are cur-rently under investigation. The present reviewillustrates the major features of the disease,
focusing also on biochemical and geneticaspects.
Introduction
Anderson-Fabry disease (AFD) (OMIM#301500) is named after two dermatologists, W.Anderson and J. Fabry, who separately describedtwo cases of patients with angiocheratoma cor-poris diffusum in 1898.1,2 Almost a century laterAFD was showed to be a rare X linked multisys-temic lysosomal storage disorder of glycosphin-golipid catabolism, due to the deficiency of thelysosomal enzyme a galactosidase A (a-Gal A;EC 3.2.1.22).3,5 This deficiency leads to systemicaccumulation of globotriaosylceramide (Gb3)and its deacylated Gb3, the globotriaosylsphin-gosine (lysoCTH or lyso-Gb3), throughout thebody within the lysosomes and in the plasma. Insymptomatic AFD male patients Gb3 and lyso-Gb3 are both increased, in plasma as well as inurine.6 However, symptomatic female patientsoften have Gb3 values within the normal range.Contrary to Gb3, lyso-Gb3 is absent in healthycontrols and it is also markedly increased insymptomatic female patients. Furthermore, theabsence of manifestations in male AFD childrenwith no residual enzyme activity, and Gb3 accu-mulation within the cells, suggests that Gb3may not be directly responsible for diseasesigns and symptoms, while lyso-Gb3 is likely toplay a direct role in the pathogenesis of AFD.7
A specific therapy for AFD, enzyme replace-ment therapy (ERT), is available since 2001.The clinical and scientific interest in this raredisease is growing all over the world, withmore than 2600 papers published up to date(Figure 1).
Lysosomes anda-galactosidase A
Glycosphingolipids are an essential part ofthe lipid bilayer in the intra and extracellularhuman membranes. The lysosomal enzyme a-Gal A is involved in the catabolism of glycosph-ingolipids within the lysosomes whereas it cat-alyzes the hydrolysis of a-galactosidic linkagesof glycosphingolipids, glycoproteins and poly-saccharides.3 This complex catabolic pathwayallows a reutilization of the membrane compo-nents. Mutations in the GLA gene encoding thea-Gal A may affect the synthesis, processing,and stability of the enzyme, leading to theimpairment of glycosphingolipids catabolismand to their progressive accumulation withinthe lysosomes. The storage of Gb3 and othertoxic metabolites involves every cell of thehuman body and starts during the fetal life.8
However, cellular dysfunction and organ dam-age usually become evident with early signsand symptoms in infancy (classic phenotype),or decades later, in the late-onset variant.5
Genetic bases
The lysosomal enzyme a-GAL A is encodedby the GLA gene (MIM 300644) mapping on theX chromosome at locus Xq22.1 and organizedin seven exons encompassing over 12 Kb.9 TheGLA cDNA of 1290 bp encodes for a precursorprotein of ~50kDa (429 amino acids), which isproteolytically cleaved into the lysosomalmature protein.10,11
The mature a-GAL A enzyme is a homod-imeric glycoprotein of about 46 kDa (398amino acids). Each monomer is composed oftwo domains. The domain 1 contains the activesite (encompassing amino acids 32-330), atthe center of the b strands in the (b/a)8 barrel,while the domain 2 contains antiparallel bstrands.12
At present 665 GLA gene mutations, most of
Cardiogenetics 2013; 3(s1):e3
Correspondence: Benedetta Tomberli, Diparti-mento Cuore e Vasi, San Luca Vecchio, Centro diRiferimento Cardiomiopatie, Azienda OspedalieroUniversitaria Careggi, Largo Brambilla 3, 50134,Firenze, Italy.Tel. +39.055.7945138 - Fax: +39.055.7949335.E-mail: [email protected]
Key words: Anderson-Fabry, heart failure, renalfailure, stroke, genetics, enzyme replacementtherapy.
Acknowledgments: the authors would like tothank, for their constant support in research andeducation, Genzyme Corporation and Shire HGT,which had no direct role in the developmentand/or content of this manuscript.
Contributions: all authors contributed equally.
Conflict of interests: FC, BT, received speaker’sfees and research support from Shire HGT,Genzyme Corporation; AM, received financialsupport for research from Shire HGT andGenzyme Corporation.
Received for publication: 14 November 2012.Revision received: 21 January 2013.Accepted for publication: 4 February 2013.
This work is licensed under a Creative CommonsAttribution NonCommercial 3.0 License (CC BY-NC 3.0).
©Copyright F. Cecchi et al., 2013Licensee PAGEPress, ItalyCardiogenetics 2013; 3(s1):e3doi:10.4081/cardiogenetics.2013.s1.e3
Non-co
mmercial
use o
nly

[page 14] [Cardiogenetics 2013; 3(s1):e3]
them private and spread throughout all exons,have been reported (HGMD professional2012.2), confirming a high genetic hetero-geneity. Over 65% of the AFD causing geneticvariants are missense or nonsense mutations,while 5% are splicing mutations, 22% smalldeletions/small insertions or regulatory muta-tions, and only 6% are gross deletion/insertion.While missense mutations not affecting theactive site may lead to partial reduction of a-GAL A activity, no residual enzyme activity isexpected for the remaining mutations.AFD is transmitted as an X-linked disorder.
Sons of affected males are always disease free,while their daughters are obligate heterozy-gotes. The heterozygous females can transmittheir mutated allele to their male and femaleoffspring. Thus in each pregnancy 50% of theirsons can be hemizygotes and 50% of theirdaughters can be heterozygotes. Although X-linked, penetrance and expres-
sivity rate are high in both genders (100% inmales and 70% in females).13 Heterozygousfemales are usually affected with a milder andlate-onset disease (about 10 years later thanmales), but in a minority, symptoms and organdamage can be as severe and early as in males.Therefore it would be preferable to discontinuefemale carrier and X-linked recessive terms, asmisleading.13 This huge variability of femalephenotype can be partially explained by therandom X inactivation process.A genotype-phenotype correlation is often
difficult, as most of the AFD mutations are pri-vate and unique within single families.However some common missense mutations,leading to residual a-GAL activity, have beenlinked mainly to specific organ involvement inAFD patients (e.g. N215S [c.644A>G➔p.Asn215Ser] to hypertrophic cardiomyopathy).14,15
Epidemiology
AFD is rare disease, with a reported preva-lence in the general population ranging from1:117,000 to 1:467,000.16,17 However, recent bio-chemical and genetic screening data in new-borns report an unexpectedly much higher
incidence (Table 1).18-21 While the classic phe-notype of AFD is actually rare, these data showthat milder variant with late-onset phenotypemay be much more frequent than expected.The late-onset subgroup comprises patientswith disease onset much later in life thanpatients with classic AFD, usually in the fourthto six decades, and manifestations confinedmainly to one organ system. Patients withatypical late-onset variants have residualenzyme activity, which ranges from 2 to 20% ofnormal values and results in a milder pheno-type. However the correlation between resid-ual enzyme activity and disease phenotype isnot strong.22 The real strength of these new-born-screening initiatives goes well beyondthe simple data itself. They shifted a historicparadigm of a rare disease, with multisystemicinvolvement in young males, to a more com-mon disease, with a wide phenotype, some-times with disease manifestation confined toone organ system.Furthermore, screening for AFD among
selected groups of patients, i.e. patients with
juvenile cryptogenic stroke, renal failure or leftventricular hypertrophy, suggests that manypatients are misdiagnosed.23 Indeed, the preva-lence of AFD among patients with hypertrophiccardiomyopathy (HCM) ranges from 0.5% to6%, depending on different selection criteriafor age and gender, and diagnostic meth-ods.24,25 It is about 1% if only males older than35 years at diagnosis of HCM are selected. AFD is a pan-ethnic disease, with few areas
of high prevalence (i.e. Canada and WestVirginia, USA), probably due to a foundereffect.4
Clinical manifestations, clinicalcourse and prognosis
AFD is a progressive disease, characterizedby a wide variability of signs, symptoms andpatterns, which, for the classical phenotype,are clearly different in three periods of life(Figure 2):
Review
Figure 1. The number of papers on Anderson-Fabry disease is significantly increased sincethe advent of enzyme replacement therapy, in 2001.
Table 1. Epidemiology of Anderson-Fabry disease. Data from newborn screening based on a-Gal A activity and GLA gene analysis ondried blood spot.
Gender CountryRef Total incidence Male incidence Classic phenotype Late-onset phenotype
M Italy18 - ∼1:3100 ∼1:37,000 ∼1:3400M+F Taiwan19 1:2300* ∼1:1250* ∼1:22,600 (M)* ∼1:1400 (M)*M+F Taiwan20 1:2500* ∼1:1400* ∼1:57,000 (M)* ∼1:1500 (M)*M+F Austria21 ∼1:3900 - ∼1:17,000 ∼1:4100M, male; F, female. *Newborn screening in Taiwan reveald a high incidence of the IVS4+919G>A cryptic splicing mutation, probably due to a founder effect. This mutation has been described in late-onset cardiacphenotype.19,20
Non-co
mmercial
use o
nly

[Cardiogenetics 2013; 3(s1):e3] [page 15]
- Childhood: although the pathologic accumu-lation of sphingolipids in various tissuesthroughout the body usually starts in thefetal life,8 early symptoms, mostly neuro-pathic pain, usually emerge in infancy.26
- Age 20 to 30: progressive cellular dysfunc-tion, related to the slow but continuousintracellular storage, leads to organ damageand failure. Symptoms tend to progress, andthe first, subclinical signs of organ involve-ment may be detected [e.g. proteinuria,electrocardiogram (ECG) changes and silentcerebral abnormalities].
- Over age 30: major organ involvement, suchas renal, cerebrovascular and cardiovascu-lar, may lead to disease progression andeventually to an end-stage phase.27 Averagereduction of life expectancy is about 20years in males and 10 in females.28
Anderson-Fabry disease in thechildhood: first signs andsymptoms
The first clinical symptom is usually neuro-pathic pain, which occurs in 70% of patients andusually emerges between age 4 and 12, earlier inmales than in females.29 The pain is located atthe extremities and can either be chronic orcharacterized by crisis. It may be triggered byexercise, fever, sun exposure, changes in tem-peratures or stress.30 The burning pain may be sointense to interfere with every-day life and sub-stantially reduce quality of life.31 Neurologicexamination is normal, as well as electromyogra-phy (EMG) and electroneurography.32 Bothexaminations only assess large nerve fibersfunction, while in AFD small nerve fibers areinvolved, and their reduced density on skin biop-sy is sometimes required to diagnose AFD neu-ropathy.33 The neuropathic pain is difficult tomanage with standard analgesic and mayrespond to narcotic analgesic (such as codeine ormorphine) or anticonvulsant drugs (such as car-bamazepine).34 Anhidrosis and hypohidrosis, areduced or absent ability to sweat, can cause heatand exercise intolerance, often reported as recur-rent fever after exercise by the patients and theirparents. They may also trigger AFD pain crisis.35
Gastrointestinal involvement, characterizedby abdominal pain, diarrhea, nausea, can alsobe reported during childhood as well as in theadult life.36
Retinal vessel tortuosity and cornea verticil-lata, a corneal lesion caused by Gb3 deposition,can also be detected in childhood.37 Vertigo,tinnitus and sudden hearing loss have beenreported at an early stage and are thought to berelated to small vessel involvement.38
Angiokeratoma is another early feature ofAFD.39 Small purple skin lesions may be present
in children and tend to increase in size andnumber with age. They can be found typically onthe lower back, thighs, genital areas, umbilicusand sometimes on mucosae (Figure 3). Histo-logically they are characterized by vessel dilata-tion (angiomas) and hyperkeratosis. Major organ involvement, such as renal,
cerebrovascular or cardiovascular is uncom-
mon in children.40 Microalbuminuria and pro-teinuria, signs of early renal damage, may bedetected in the second decade of life.41 Smallwhite matter lesion on magnetic resonanceimaging (MRI) may reveal an initial centralnervous system involvement, as well as ECGchanges of the heart. Although the early clinical picture is sugges-
Review
Figure 2. Symptoms onset and disease progression in classic Anderson-Fabry disease(AFD). Signs and symptoms of AFD progressively increase with age. In infancy and ado-lescence neuropathic pain, gastrointestinal manifestations and angiokeratomas are themost frequent manifestations. Proteinuria, the first sign of kidney involvement, is alsopresent in young affected males (Modified from Zarate and Hopkin4).
Figure 3. Various aspects of angiokeratoma in Anderson-Fabry disease patients: angioker-atoma of the periungual and subungual area (panel A), of the palms, lips and umbilicus(panel B-D respectively) (Reprinted with permission of the publisher from Zampetti et al.39).
Non-co
mmercial
use o
nly

[page 16] [Cardiogenetics 2013; 3(s1):e3]
tive of AFD, the diagnosis is rarely made inchildhood and adolescence, with an averagediagnostic delay of 10 years for both genders.Recognition of early and advanced clinicalmanifestations of AFD represents a real chal-lenge for clinicians, due to variable clinicalmanifestations and symptoms that may resem-ble other common diseases.42
Anderson-Fabry disease in theadult life The full spectrum of AFD phenotype slowly
develops with age.
Cardiac involvementGb3 and related sphingolipids storage may
occur in cardiomyocites, cardiac valves,endothelial cells and conduction system.43
While ECG changes may be found in youngpatients, an overt cardiomyopathy with leftventricular hypertrophy (LVH), with diastolicdysfunction, preserved systolic function, oftenwith mild valvular regurgitation emerges inthe third or fourth decade of life in affectedmales, more rarely in a few females.44
Conduction abnormalities andarrhythmiasECG changes are often present in AFD
(Figure 4). A short PQ interval (<120 ms),described as a feature of AFD, is a non-specif-ic and rather uncommon finding with a preva-lence of 14%.45 Signs of LVH on ECG, alongwith repolarization changes (including deep Twaves inversion) are frequent. They may bedetected even in patients without a markedincrease of left ventricular wall thickness atechocardiography.45,46
Bradycardia, sinus node disease, prolongedPQ intervals with variable degree of AV-block,and QRS broadening (particularly with a rightbundle branch block pattern) are ECG alter-ations often seen later in life.47 PQ interval andQRS duration are directly proportional to age,suggesting a relationship to progressive stor-age of sphingolipids and development of fibro-sis. Apoptosis and autonomic nervous systeminvolvement might also play a role. Recently AVand sinus node disease were showed to becommon in AFD patients.47 The prevalence ofatrial fibrillation and non-sustained ventricu-lar-tachycardia (VT) are higher in AFDpatients when compared to the general popula-tion.48 Furthermore the risk of arrhythmiasincreases with age, while there are no gender-related differences. Sudden cardiac death mayoccur, usually in the end stage phase, althoughits prevalence is unknown.48 It may be trig-gered by episodes of sustained VT.
Anderson-Fabry diseasecardiomyopathyAFD cardiomyopathy is characterized by
diastolic dysfunction and LVH, usually con-centric and non-obstructive. Basal LV outflowtract obstruction (LVOTO) is rare, while exer-cise provokable LVOTO has been detected in43% of patients,49 most of them with LV smallcavity size. LVH is usually mild, although male patients
may also show severe and asymmetric septal orapical hypertrophy, both in classical and late-onset disease (Figure 5). Systolic function isusually preserved in most patients until
advanced stage, but a few patients may gradu-ally develop a systolic dysfunction, often lead-ing to progressive refractory heart failure.50
The use of some echocardiographic technique,such as tissue doppler imaging or strain rateimaging, may detect early cardiac involvementbefore LVH develops.51,52 Replacement fibrosis,detected by cardiac MRI usually in the inferioror posterolateral wall, is considered a sign ofdisease progression (Figure 6).53 Myocardialfibrosis may also be present in female patients
Review
Figure 4. Electrocardiogram in a male patient with Anderson-Fabry disease showing leftventricular hypertrophy with T-waves inversion and a relatively short PR (133 ms)(Reprinted with permission of the publisher from O’Mahony and Elliott46).
Figure 5. Typical echocardiographic aspect of Anderson-Fabry disease-related cardiomy-opathy. The 2-D images in panel A and B show mild, concentric left ventricular hypertro-phy. There is also mild diastolic dysfunction (panel C and D) (Reprinted with permissionof the publisher from O’Mahony and Elliott46).
Non-co
mmercial
use o
nly

[Cardiogenetics 2013; 3(s1):e3] [page 17]
without LVH, suggesting that hypertrophy andfibrosis are not necessarily associated.54 Theendocardial binary appearance (binary sign)was suggested as an echocardiographic hall-mark of AFD cardiomyopathy;55 however, recentstudies showed that these echo features can bedetected also in patients with sarcomericHCM.56 The binary sign is of limited utility indistinguishing the cause of LVH. Of note, rightventricular hypertrophy, is detected by MRI inabout 2/3 of patients with LVH.57
Angina and coronary artery diseaseChest discomfort and angina may be pres-
ent, despite the absence of atheroscleroticcoronary artery disease. Both are likely to berelated to microvascular dysfunction (Figures7 and 8).58-60 Coronary microvascular blood flowimpairment can be detected in both genders,even without LVH. It can lead to chronicischemia and myocardial replacement fibrosis.
Other cardiac abnormalitiesThe prevalence of mild aortic root enlarge-
ment is higher in male AFD patients,61 whileexercise capacity is usually reduced, whenmeasured by a cardiorespiratory test.62
Kidney involvementMicroalbuminuria and proteinuria, due to
kidney impairment related to Gb3 accumula-tion and glomerular fibrosis, are usually pres-ent by the third decade of life, sometimes ear-lier when a-GAL enzyme activity is <1%(Figure 9).63,64 Proteinuria levels may varywidely, but tend to increase in severity withage and are inversely correlated to kidneyfunction, based on glomerular filtration rate,which is often normal or even higher in youngpatients.65 After renal dysfunction has pro-gressed to end-stage, dialysis and renal trans-plant may be necessary.66-67
Cerebrovascular involvementTransient ischemic attack and strokes are
the most relevant and disabling complicationof AFD and may significantly contribute to dis-ease-related morbidity and mortality.68
Multifocal leukoencephalopathy is the typicallesions of AFD and results from small vesselsinvolvement and microvascular dysfunction(Figure 10).69 The prevalence of strokes in AFDis about 7% in males and 5% in females, usual-ly in the third and the fourth decade of life.However, the prevalence of cerebrovascularinvolvement rises up to 60-70% if minor signsor symptoms and abnormal brain MR imageare considered.68 Psychiatric disorders, depres-sion and dementia may be present and theycan only be partially attributed to the cerebralmicrovascular disease.70 The severity of cere-brovascular disease may vary widely.71
Combining neurologic symptoms and instru-
Review
Figure 6. Myocardial fibrosis as a sign of disease progression. Panel A) cardiovascularmagnetic resonance image showing an area of myocardial fibrosis involving the postero-lateral wall (arrows in four-chamber horizontal long-axis view). Panel B) the extension offibrosis is significantly increased, despite 6 years of enzyme replacement therapy(Reprinted with permission of the publisher from Imbriaco et al.101).
Figure 7. Gb3 inclusions in the endothelium and cardiomyocytes. Panel A shows an intra-mural coronary artery in a patient with hypertrophic cardiomyopathy. Wall thickening isdue primarily to severe thickening of medial wall, while the intima is mildly thickened.Panel B shows interstitial capillaries in the myocardium of a patient with Anderson-Fabrydisease. Glycosphingolipid inclusions are present in the endothelium (arrow) and cardiomy-ocytes (arrowhead) (Reprinted with permission of the publisher from Camici e Crea58).
Figure 8. Microvascular dysfunction detected by myocardial positron emission tomogra-phy. Male patients with Anderson-Fabry disease (AFD) show a marked reduction inmyocardial blood flow (MBF) during adenosine-induced hyperaemia (ADO), comparedto controls (Reprinted with permission of the publisher from Elliott et al.59).
Non-co
mmercial
use o
nly

[page 18] [Cardiogenetics 2013; 3(s1):e3]
mental findings, CNS disease can be brieflyclassified into 3 categories: i) symptomaticpatients with repeated cerebrovascular events,ii) patients with minor neurological manifes-tation such as vertigo, dizziness, hearing loss,headache and migraine; iii) asymptomaticpatients with abnormal brain MRI findings.68
Other findings Symptoms compatible with autonomic dys-
function, such as arterial hypotension, sinusbradycardia or gastrointestinal symptoms,have always been attributed to autonomicnervous system neuropathy,72 which is oftenneglected in the single patient. Obstructive airway disease and osteopenia/
osteoporosis in AFD patients have also beenreported.73,74
Diagnosis
Once the clinical pictures rises a suspicionof AFD, biochemical and genetic tests arerequired to confirm the diagnosis.
Biochemical diagnosisThe detection of markedly reduced or absent
a-gal A activity in leukocytes is sufficient toconfirm AFD diagnosis,3 although it should becarefully evaluated with clinical and geneticmolecular findings. While patients with amilder AFD phenotype usually have residual a-gal A activity, in classically affected youngmales enzyme activity it is very low or unde-tectable.75,76
a-gal A activity can be assessed in plasma.However, when reduced, its value needs to beconfirmed in leukocytes or fibroblasts, in orderto avoid diagnostic pitfalls, due to enzymepseudodeficiency in plasma.77 The GLA genesequence analysis should always be performedand correctly interpreted, in order to revealpolymorphisms, that potentially lead to anenzymatic pseudodeficiency.78 Biochemicaldiagnosis may be unreliable in heterozygousfemales, as they can have a normal or onlymildly reduced a-gal A activity, due to randomX-chromosome inactivation. Therefore, it iscrucial to combine biochemical assays withappropriate molecular analyses of the GLAgene in order to attribute or exclude the AFDheterozygous status in females.79
More recently, screening tests on driedblood spot (DBS), using fluorescent methods,were introduced,80 together with multiplexassays of lysosomal enzymes by tandem massspectrometry.81,82 The availability of multiplextechnology using DBS on filter paper madepossible newborn screening programs of treat-able lysosomal storage diseases.83 DBS is sim-ply transportable and represent an easier,
faster and less expensive method for enzymeactivity evaluation in selected at risk popula-tion. As false positive or negative results mayoccur, it is necessary to confirm a reducedresidual enzyme activity by standard laborato-ry diagnostic procedures.84,85
Genetic analysisMolecular analysis is usually performed in
probands by direct sequencing analysis of theGLA gene coding regions, of its exons/intronsboundaries and of the intronic region encom-passing the known deep intronic muta-tions.86,87 Rarely, in males with low or noenzyme activity, and females with AFD clinicalmanifestations and normal or low enzymeactivity, exon-based sequence analysis may failto identify the disease-causing mutation. Suchin these cases, further molecular and cellularlaboratory investigations are needed to evalu-ate the GLA gene expression profile at mRNAand protein level. Both Ishii and Filoni identi-fied, by mRNA analysis, deep intronic patho-genic mutations, which alter the GLA splicingregulation.86,87 In females, due to the presenceof the wild-type allele, gross GLA generearrangements, such as deletion or insertionof entire exons, may be missed, unless specif-ic laboratory technique, such as multiplex liga-tion-dependent probe amplification analysisand/or quantitative polymerase chain reactionamplification, are used.88,89
Additional investigations are also necessaryin order to assess the pathogenicity of new vari-ants. In silico and mRNA analysis, computation-al modeling, and functional studies are neededto prove or exclude the effect of the new identi-fied variant as a disease-causing mutation.78
Co-segregation in male family members, tissuebiopsies and determination of specific metabo-lites (i.e. Gb3 or lyso-Gb3) may also be neces-sary. Recently, several authors suggest to con-sider GLA gene mutations on the bases of theirpathogenic significance, with variants predict-
ed to have high (Class 1) or low probability to bedisease-causing (Class 2, polymorphisms).90
Single nucleotide polymorphisms (SNPs) areexonic or intronic nucleotide variants detectedwith incidence higher than 1% in a healthy con-trol population. They are usually interpreted asnot disease-causing mutation. Several GLAgene polymorphisms, leading to molecular het-erogeneity have been described (SNP, http://www.ncbi.nlm.nih.gov/SNP).79 Sometime exon-ic polymorphisms, including rare variants lead-ing to aminoacid change, may be found in maleswith normal or decreased enzyme activity, butstill well above the pathologic range of values.They are almost certainly non-pathogenic vari-ants or Class 2 mutations and they include poly-morphisms or sequence changes described aspseudodeficiency alleles, e.g. c.937 G>T(p.D313Y).89,90 Pseudodeficiency is a conditionin which individuals show a reduced a-Gal Aactivity in vitro, but remain clinically healthy. However, in a few patients, when the combi-
nation of clinical, histological, biochemical andfunctional data are still not clear-cut, the AFDdiagnosis may remain uncertain91 (Figure 11).
Role and significance of Gb3and lyso-Gb3
Biomarkers are chemical molecules that canbe clinically useful to assess disease progres-sion, therapeutic efficacy, as well as diagnosticconfirmation. Although many attempts havebeen made in order to identify a biomarker forAFD, a valid and reliable molecule is still miss-ing. Gb3 is the primary storage molecule andcan be find inside cells and in biologic fluids,such as urine and plasma. For many years Gb3has been considered a reliable marker for AFD,so that its reduction in tissue biopsies hasserved has a major criteria to prove the effica-cy of the enzyme replacement therapy. With
Review
Figure 9. Evidence of Gb3 inclusions in the kidney. Panel A) sphingolipid inclusions inpodocytes (arrow) and distal tubule epithelial cells (double arrow). Panel B) lysosomalinclusions in podocytes (arrow), endothelial (open arrow) and mesangial cells (openarrowhead) (Reprinted with permission of the publisher from Tøndel et al.64).
Non-co
mmercial
use o
nly

[Cardiogenetics 2013; 3(s1):e3] [page 19]
time, new research investigations proved thatGb3 do not correlate with disease manifesta-tions. Plasma Gb3 levels are abnormally high ata very young age (sometimes detectable evenin utero), while clinical manifestations devel-op much later in life.92 Furthermore, Gb3 mightbe within the normal range in symptomaticfemale. The diagnostic value of this parameterhas been questioned as well, since it can bedetected in normal healthy subjects.7
Globotriaosylsphingosine, also known aslyso-Gb3, is the deacylated form of Gb3. In con-trast to Gb3, this relatively new biomarker isabnormally high in male and female patientswith AFD while this is absent in control healthysubjects.7,93 lyso-Gb3 may be a useful additionalelement for confirmation of AFD diagnosis inpatients with GLA variant of unknown signifi-cance (Figure 11). Furthermore, lifetime expo-sure to lyso-Gb3 correlates with disease severi-ty, even after adjustments for age, gender and
cardiovascular risk factors, and suggests itspathophysiological role. However, the useful-ness of lyso-Gb3 as a biomarker for assessingthe response to ERT is still under investiga-tion.94,95
Genetic counseling andmulti-disciplinary approach
Once biochemical and molecular geneticdata are ascertained and/or available, theirinterpretation should be performed by a multi-disciplinary team with expertise in AFD, usual-ly available in referral centers, in order evalu-ate the appropriate treatment strategy. Geneticcounseling should then be offered to theproband, and his family members, who shouldundergo clinical screening.
Review
Figure 10. Brain magnetic resonance imag-ing of an asymptomatic 54-year old femaleshowing multifocal leukoencephalopathy(Reprinted with permission of the publish-er from Buechner et al.68).
Figure 11. Diagnostic flow chart of Anderson-Fabry disease (AFD) in males and females: step-by-step approach. Panel A) males. 1)Clinical picture indicative of AFD, family history compatible with an X-linked transmission. 2) In males, normal values of a-gal A activ-ity exclude the diagnosis of AFD. Reduced (<20%) or absent enzyme activity values confirm the suspicion of AFD and require genetictesting. Biochemical analysis on dried blood spot (DBS) must be confirmed by standard laboratory procedures (i.e. whole blood analy-sis). 3) If no mutations are identified by genetic analysis, and the a-gal A activity is low or undetectable, further laboratory analyses arerequired to confirm the diagnosis (e.g. mRNA and protein analysis, tissue biopsies). Comparably, the identification of new unknownvariant requires laboratory analysis, along with co-segregation studies within the family. Panel B) females. 1) Clinical picture indicativeof AFD, family history compatible with an X-linked transmission. 2) In female the a-gal A activity can be within the normal range.Therefore genetic analysis is mandatory to confirm the diagnosis; if no mutations are identified by genetic analysis, and the clinical sus-picion is high, further laboratory analyses are needed (e.g.multiplex ligation-dependent probe amplification to detect deletions or inser-tions, mRNA and protein analysis, tissue biopsies). Comparably, the identification of new unknown variant requires laboratory analy-sis, along with co-segregation studies within the family. 3) Enzyme activity may be helpful in diagnosis and management of female AFDpatients and should be measured after the genetic diagnosis is made. Biochemical analysis on DBS must be confirmed by standard lab-oratory procedures (i.e. whole blood analysis).
Non-co
mmercial
use o
nly

[page 20] [Cardiogenetics 2013; 3(s1):e3]
Management and follow-up
AFD is a multi-systemic disorder and a thor-ough evaluation of organ involvement is high-ly recommended. Neurologic assessment comprises diagnosis
and staging of neuropathic pain, through EMGand skin biopsy, if needed.33 Since leukoen-cephalopathy may be present even in youngpatients without symptoms and with a normalneurologic exam, brain MRI is required at initialassessment.29 It should be repeated at a 2-3years interval, depending on clinical picture. Nephrologic assessment is based on microal-
buminuria, 24 h proteinuria, renal function(creatinine level and estimated glomerular fil-tration rate), which should be routinely andrepeatedly assessed.5 Ultrasound abdominalexamination is also required for kidney mor-phology, as well as, in selected cases, renalbiopsy.
Cardiologic evaluationPatients should undergo ECG, colordoppler
echocardiography, 24 h ambulatory ECG moni-toring and exercise (possibly cardiorespirato-ry) test, in order to define cardiac involvementand future risk.96 Since arterial hypertension,although rare in these patients, may increasethe risk of cerebrovascular and cardiovasculardisease, blood pressure should be carefullycontrolled even with 24 h blood pressure mon-itoring. Pulmonary function tests are some-
times required in patients whose main symp-tom is dyspnea. A rigorous control of conventional atheroscle-
rotic risk factors is mandatory. Indeed, meas-urement of blood cholesterol, homocysteinelevel, and fasting glucose, should be performedonce or twice a year, although it was suggestedthat statin treatment might be deleterious inAFD patients. Lifestyle change, especially smok-ing cessation, dietary modifications and mildphysical activity, are also recommended.
Therapy
Since 2001 ERT is available and it has beenvalidated as standard treatment for AFD patientsin more than 45 countries. In Europe ERT isavailable in two commercial formulations: agal-sidase a (Replagal®; Shire, Cambridge, MA,USA) and agalsidase b (Fabrazyme®; GenzymeCorp., Cambridge, MA, USA), but the former isnot yet approved by Food and DrugsAdministration in USA. Randomized, placebo-controlled clinical trials assessed safety and effi-cacy of both enzyme formulations. However,these trials enrolled small cohort of patients and,despite over 10 years of clinical practice, manyquestions about ERT long-term efficacy remainunanswered. Both preparations are infusedintravenously once every other week, at differ-ent dose regimens (0.2 mg/kg for Replagal®; 1mg/kg for Fabrazyme®). Patients should be
treated with licensed recommended doses ofagalsidase a and agalsidase b. Currently thereis no level 1 scientific evidence showing superi-ority of one enzyme preparation over theother.97,98 Treatment failures may occur withboth drugs and are probably related to age andadvanced disease stage, because severe organdamage is often irreversible (Figure 6).99,100
Moreover patients can develop complicationsdespite receiving optimal treatment, with con-comitant therapy and ERT at licensed recom-mended dose.97
There is increasing evidence that ERT ismore effective when started in the early stageof disease, and early diagnosis is highly prefer-able.100,101 However, timing of ERT, especially inchildren and females, is still a matter of debateand may vary in different countries. Currentexpert opinions about timing of ERT are pre-sented in Table 2,98 but real guidelines havenever been written. Concomitant treatment: appropriate organ-
specific and adjuvant therapy must always beconsidered for all patients, along with ERTinfusion, for symptom control and disease sta-bilization (Table 3).
Conclusions and futureperspectives
AFD is a lysosomal storage disease withmultiorgan involvement and reduced life
Review
Table 2. Priority stages for treatment in naïve male and female patients with Anderson-Fabry disease.
High priorityChildren Adults
Neuropathic pain unresponsive to optimal medical therapy Neuropathic pain unresponsive to optimal medical therapyPersistent microalbuminuria Normal or mild to moderate reduction of renal function
(GFR 30-90 mL/min)Proteinuria >250 mg/day Proteinuria >300 mg/day
Left ventricular mass index >90th percentile (age adjusted) LVH without extensive fibrosis (on MRI)TIA/stroke or white matter lesion on MRI Cerebrovascular disease
- Disease onset <50 yearsIntermediate priority
Age onset >50 years ➔ Less severe diseaseLVH with fibrosis
Severe renal dysfunction (GFR<30 mL/min) ➔ Less reversible disease
Low priority
Severe cardiac or CNS disease (end stage)Multiple organ failure
Other comorbidities with reduction of life expectancy <1 yearVery mild multiorgan disease (e.g. normal renal function, no LVH, neuropathic pain well controlled by conventional medical therapy)
GFR, glomerular filtration rate; LVH, left ventricular hypertrophy; MRI, magnetic resonance imaging; TIA, transient ischemic attack; CNS, central nervous system.A consensus of expert wrote this document, on October 2010, in order to support clinicians’ decisions during the period of enzyme replacement therapy (ERT) shortage. Although the present recommendations arenot supposed to represent real treatment guidelines, they underline some important issues about the efficacy of ERT in advanced stage of Anderson-Fabry disease. Priority to receive ERT was based on diseaseseverity, potential reversibility and rate of disease progression, while age and gender did not represent valid criteria (Modified from G.E. Linthorst et al. 201293).
Non-co
mmercial
use o
nly

[Cardiogenetics 2013; 3(s1):e3] [page 21]
expectancy in many affected patients. AFDhas always been considered a very rare dis-ease. However, new screening data reporteda much higher incidence. Currently, AFDdiagnosis may be easier and simpler by eval-uating a-gal A enzyme activity and geneticanalysis for GLA gene mutations on driedblood spot. A marked a-gal A deficiency leadsto diagnosis of AFD in hemizygous males,while molecular analysis is mandatory in het-erozygous females. However, referral to acenter with an expert multidisciplinary teamis highly advisable, in order to ensure carefulmanagement and treatment of patients,based also on accurate molecular and bio-chemical data interpretation. Enzyme replacement therapy in patients
with early symptoms and initial organ disease,such as microalbuminuria and neuropathicpain, seem to be effective. Furthermore, newtreatment approaches are currently underinvestigation, such as chemical chaperonetherapy alone (phase III clinical studies;http://www.clinicaltrials.com) or in combina-tion with ERT (preclinical studies).102-104
While many aspects of this disease havebeen elucidated, further clinical and laborato-ries studies are still needed to clarify residualareas of doubt.
References
1. Anderson W. A case of angio-keratoma. BrJ Dermatol 1898;10:113-7.
2. Fabry J. [Purpura papulosa haemorrhagi-ca Hebrae]. Arch Dermatol Syphilol1898;43:187-201. [In German].
3. Desnick RJ, Ioannou YA, Eng CM. a-Galactosidase A deficiency: Fabry disease.In: Scriver CR, Beaudet AL, Sly WS andValle D, eds. The metabolic and molecular
bases of inherited disease, 8th Ed. NewYork: McGraw-Hill; 2001. pp 3733-74.
4. Zarate Y, Hopkin R. Fabry’s disease. Lancet2008;372:1427-35.
5. Germain DP. Fabry disease. Orphanet JRare Dis 2010;5:30-79.
6. Aerts JMFG, Kallemeijn WW, Wegdam W, etal. Biomarkers in the diagnosis of lysoso-mal storage disorders: proteins, lipids, andinhibodies. J Inherit Metab Dis 2011;1-15.
7. Rombach SM, Dekker N, Bouwman MG, etal. Plasma globotriaosylsphingosine: diag-nostic value and relation to clinical mani-festations of Fabry disease. BiochimBiophys Acta 2010;1802:741-8.
8. Bouwman MG, Hollak CEM, van den BerghWeerman M, et al. Analysis of placental tis-sue in Fabry disease with and withoutenzyme replacement therapy. Placenta2010;31:344-6.
9. Kornreich R, Desnick R, Bishop D.Nucleotide sequence of the human a-galactosidase A gene. Nucleic Acids Res1989;17:3301-2.
10. Bishop DF, Calhoun DH, Bernstein HS, etal. Human a-galactosidase A: nucleotidesequence of a cDNA clone encoding themature enzyme. Proc Natl Acad Sci U S A1986:83:4859-63.
11. Lemansky P, Bishop DF, Desnick RJ, et al.Synthesis and processing of a-galactosi-dase A in human fibroblasts. Evidence fordifferent mutations in Fabry disease. JBiol Chem 1987:262:2062-65.
12. Garman SC, Garboczi DN. The moleculardefect leading to Fabry disease: structureof human a-galactosidase. J Mol Biol2004;337:319-35.
13. Pinto LL, Vieira TA, Giugliani R, SchwartzIV. Expression of the disease on femalecarriers of X-linked lysosomal disorders: abrief review. Orphanet J Rare Dis 2010;5:14-23.
14. Nakao S, Takenaka T, Maeda M, et al. An
atypical variant of Fabry’s disease in menwith left ventricular hypertrophy. N Engl JMed 1995;333:288-93.
15. Sachdev B, Takenaka T, Teraguchi H, et al.Prevalence of Anderson-Fabry disease inmale patients with late onset hypertrophiccardiomyopathy. Circulation 2002;105:1407-11.
16. Meikle PJ, Hopwood JJ, Clague AE, CarreyWF. Prevalence of lisosomal storage disor-ders. J Am Med Assoc 1999;281:249-54.
17. Poorthuis BJ, Wevers R, Kleijer WJ, et al.The frequency of lysosomal storage dis-eases in The Netherlands. Hum Genet1999;105:151-6.
18. Spada M, Pagliardini S, Yasuda M, et al.High incidence of later-onset fabry diseaserevealed by newborn screening. Am J HumGenet 2006;79:31-40.
19. Hwu W-L, Chien Y-H, Lee N-C, et al.Newborn screening for Fabry disease inTaiwan reveals a high incidence of thelater-onset GLA mutation c.936+919G>A(IVS4+919G>A). Hum Mutat 2009;30:1397-405.
20. Lin H-Y, Chong K-W, Hsu J-H, et al. Highincidence of the cardiac variant of Fabrydisease revealed by newborn screening inthe Taiwan Chinese population. CircCardiovasc Genet 2009;2:450-6.
21. Mechtler TP, Stary S, Metz TF, et al.Neonatal screening for lysosomal storagedisorders: feasibility and incidence from anationwide study in Austria. Lancet2012;379:335-41.
22. Schaefer E, Mehta A, and Gal A. Genotypeand phenotype in Fabry disease: analysisof the Fabry Outcome Survey. ActaPaediatr 2005;94:87-92.
23. Linthorst GE, Bouwman MG, Wijburg F a,et al. Screening for Fabry disease in high-risk populations: a systematic review. JMed Genet 2010;47:217-22.
24. Monserrat L, Gimeno-Blanes JR, Marín F,
Review
Table 3. Conventional medical treatment in Anderson-Fabry disease.
Symptomatic management of Anderson-Fabry disease
Analgesics to control neuropathic painAvoidance of triggers of acute pain crisis
Anti-arrhythmic drugsRigorous control of any coronary risk factor (smoke, dyslipidemia, hypertension, hyperhomocysteinemia)
Long-term therapy to prevent organ damage
ACEi/ARB for proteinuria and kidney dysfunctionAnti-aggregant (aspirin, clopidogrel) or anticoagulant (warfarin) to prevent TIA and strokes
End-stage disease management
Dialysis or kidney transplantation for renal failureHeart transplantation for patient with refractory heart failure
ACEi, angiotensin-converting-enzyme inhibitors; ARB, angiotensin receptor blockers; TIA, transient ischemic attack.
Non-co
mmercial
use o
nly

[page 22] [Cardiogenetics 2013; 3(s1):e3]
et al. Prevalence of Fabry disease in acohort of 508 unrelated patients withhypertrophic cardiomyopathy. J Am CollCardiol 2007;50:2399-403.
25. Elliott P, Baker R, Pasquale F, et al.Prevalence of Anderson-Fabry disease inpatients with hypertrophic cardiomyopa-thy: the European Anderson-Fabry DiseaseSurvey. Heart 2011;1957-61.
26. Hopkin RJ, Bissler J, Banikazemi M, et al.Characterization of Fabry disease in 352pediatric patients in the Fabry Registry.Pediatr Res 2008;64:550-5.
27. Eng C, Fletcher J, Wilcox WR, et al. Fabrydisease: baseline medical characteristicsof a cohort of 1765 males and females inthe Fabry Registry. J Inherit Metab Dis2007;30:184-92.
28. Waldek S, Patel MR, Banikazemi M, et al.Life expectancy and cause of death inmales and females with Fabry disease:findings from the Fabry Registry. GenetMed 2009;11:790-6.
29. Salviati A, Burlina AP, Borsini W. Nervoussystem and Fabry disease, from symptomsto diagnosis: damage evaluation and fol-low-up in adult patients, enzyme replace-ment, and support therapy. Neurol Sci2010;31:299-306.
30. Pagnini I, Borsini W, Cecchi F, et al. Distallimb pain as presenting feature of fabrydisease. Arthrit Care Res 2010;63:390-5.
31. Gibas AL, Klatt R, Johnson J, et al. A surveyof the pain experienced by males andfemales with Fabry disease. Pain Res ClinMan 2006;11:185.
32. Lacomis D, Roeske-Anderson L, Mathie L.Neuropathy and Fabry’s disease. MuscleNerve 2005;31:102-7.
33. Liguori R, Di Stasi V, Bugiardini E, et al.Small fiber neuropathy in female patientswith Fabry disease. Muscle Nerve2010;41:409-12.
34. Burlina AP, Sims KB, Politei JM, et al. Earlydiagnosis of peripheral nervous systeminvolvement in Fabry disease and treat-ment of neuropathic pain: the report of anexpert panel. BMC Neurol. 2011;11:61.
35. Schiffmann R, Scott LJC. Pathophysiolo-gy and assessment of neuropathic painin Fabry disease. Acta Paediatr 2002;91:48-52.
36. Hoffmann B, Schwarz M, Mehta A, KeshavS. Gastrointestinal symptoms in 342patients with Fabry disease: prevalenceand response to enzyme replacement ther-apy. Clin Gastroenterol 2007;5:1447-53.
37. Sodi A, Ioannidis AS, Mehta A, et al. Ocularmanifestations of Fabry’s disease: datafrom the Fabry Outcome Survey. Br JOphthalmol 2007;91:210-4.
38. Palla A, Hegemann S, Widmer U,Straumann D. Vestibular and auditorydeficits in Fabry disease and their
response to enzyme replacement therapy.J Neurol 2007;254:1433-42.
39. Zampetti A, Orteu CH, Antuzzi D, et al.Angiokeratoma: decision-making aid forthe diagnosis of Fabry disease. Br JDermatol 2012;166:712-20.
40. Ramaswami U, Parini R, Pintos-Morell G,et al. Fabry disease in children andresponse to enzyme replacement therapy:results from the Fabry Outcome Survey.Clin Genet 2011:1-6.
41. Ries M, Gupta S, Moore DF, et al. PediatricFabry disease. Pediatrics 2005;115:e344-55.
42. Marchesoni CL, Roa N, Pardal AM, et al.Misdiagnosis in Fabry disease. J Pediatr2010;156:828-31.
43. Sheppard MN, Cane P, Florio R, et al. Adetailed pathologic examination of hearttissue from three older patients withAnderson-Fabry disease on enzymereplacement therapy. Cardiovasc Pathol2009;19:293-301.
44. Linhart A, Kampmann C, Zamorano JL, etal. Cardiac manifestations of Anderson-Fabry disease: results from the interna-tional Fabry outcome survey. Eur Heart J2007; 28:1228-35.
45. Namdar M, Steffel J, Vidovic M, et al.Electrocardiographic changes in earlyrecognition of Fabry disease. Heart 2011;97:485-90.
46. O’Mahony C, Elliott PM. Anderson-Fabrydisease and the heart. Progr CardiovascDis 2010;52:326-35.
47. O’Mahony C, Coats C, Cardona M, et al.Incidence and predictors of anti-bradycar-dia pacing in patients with Anderson-Fabry disease. Europace 2011;13:1781-8.
48. Shah JS, Hughes DA, Sachdev B, et al.Prevalence and clinical significance of car-diac arrhythmia in Anderson-Fabry dis-ease. Am J Cardiol 2005;96:842-6.
49. Calcagnino M, O’Mahony C, Coats C, et al.Exercise-induced left ventricular outflowtract obstruction in symptomatic patientswith Anderson-Fabry disease. J Am CollCardiol 2011;58:88-9.
50. Shah JS, Lee P, Hughes D, et al. The natu-ral history of left ventricular systolic func-tion in Anderson-Fabry disease. Heart2005;91:533-4.
51. Pieroni M, Chimenti C, Ricci R, et al. Earlydetection of Fabry cardiomyopathy by tis-sue Doppler imaging. Circulation 2003;107:1978-84.
52. Weidemann F, Breunig F, Beer M, et al. Thevariation of morphological and functionalcardiac manifestation in Fabry disease:potential implications for the time courseof the disease. Eur Heart J 2005;26:1221-7.
53. Koeppe S, Neubauer H, Breunig F, et al.MR-based analysis of regional cardiacfunction in relation to cellular integrity inFabry disease. Int J Cardiol 2012;160:53-8.
54. Niemann M, Herrmann S, Hu K, et al.Differences in fabry cardiomyopathybetween female and male patients conse-quences for diagnostic assessment. JACCCardiovasc Imaging 2011;4:592-601.
55. Pieroni M, Chimenti C, De Cobelli F, et al.Fabry’s disease cardiomyopathy: echocar-diographic detection of endomyocardialglycosphingolipid compartmentalization. JAm Coll Cardiol 2006;47:1663-71.
56. Mundigler G, Gaggl M, Heinze G, et al. Theendocardial binary appearance (‘binarysign’) is an unreliable marker for echocar-diographic detection of Fabry disease inpatients with left ventricular hypertrophy.Eur J Echocardiogr 2011;12:744-9.
57. Palecek T, Dostalova G, Kuchynka P, et al.Right ventricular involvement in Fabrydisease. J Am Soc Echocardiogr 2008;21:1265-8.
58. Camici PG, Crea F. Coronary microvasculardysfunction. N Engl J Med 2007;356: 830-40.
59. Elliott PM, Kindler H, Shah JS, et al.Coronary microvascular dysfunction inmale patients with Anderson-Fabry dis-ease and the effect of treatment with agalactosidase A. Heart 2006;92:357-60.
60. Kalliokoski RJ, Kantola I, Kalliokoski KK,et al. The effect of 12-month enzymereplacement therapy on myocardial perfu-sion in patients with Fabry disease. JInherit Metab Dis 2006; 29:112-8.
61. Barbey F, Qanadli SD, Juli C, et al. Aorticremodelling in Fabry disease. Eur Heart J2010; 31:347.53.
62. Bierer G, Kamangar N, Balfe D, et al.Cardiopulmonary exercise testing in Fabrydisease. Respiration 2005;72:504-11.
63. Breunig F, Wanner C. Update on Fabry dis-ease: kidney involvement, renal progres-sion and enzyme replacement therapy. JNephrol 2008;21:32-7.
64. Tøndel C, Bostad L, Hirth A, Svarstad E.Renal biopsy findings in children and ado-lescents with Fabry disease and minimalalbuminuria. Am J Kidney Dis 2008;51:767-76.
65. Ortiz A, Oliveira JP, Waldek S, et al.Nephropathy in males and females withFabry disease: cross-sectional descriptionof patients before treatment with enzymereplacement therapy. Nephrol Dial Trans-plant 2008;23:1600-7.
66. Cybulla M, Walter KN, Schwarting A, et al.Kidney transplantation in patients withFabry disease. Transpl Int 2009;22:475-81.
67. Shah T, Gill J, Malhotra N, et al. Kidneytransplant outcomes in patients with Fabrydisease. Transplantation 2009;87:280-5.
68. Buechner S, Moretti M, Burlina AP, et al.Central nervous system involvement inAnderson-Fabry disease: a clinical andMRI retrospective study. J NeurolNeurosurg Psychiatry 2008;1249-54.
Review
Non-co
mmercial
use o
nly

[Cardiogenetics 2013; 3(s1):e3] [page 23]
69. Fellgiebel A, Müller MJ, Ginsberg L. CNSmanifestations of Fabry’s disease. LancetNeurol 2006;5:791-5.
70. Schermuly I, Müller MJ, Müller K-M, et al.Neuropsychiatric symptoms and brainstructural alterations in Fabry disease. EurJ Neurol 2011;18:347-53.
71. Møller AT, Jensen TS. Neurological mani-festations in Fabry’s disease. Nat ClinPract Neurol 2007;3:95-106.
72. Biegstraaten M, van Schaik IN, Wieling W,et al. Autonomic neuropathy in Fabry dis-ease: a prospective study using the auto-nomic symptom profile and cardiovascularautonomic function tests. BMC Neurol2010;10:38.
73. Magage S, Lubanda JC, Susa Z, et al.Natural history of the respiratory involve-ment in Anderson-Fabry disease. J InheritMetab Dis 2007;30:790-9.
74. Germain DP, Benistan K, Boutouyrie P,Mutschler C. Osteopenia and osteoporosis:previously unrecognized symptoms ofFabry disease. Clin Genet 2005;68:93-5.
75. Nakao S, Kodama C, Takenaka T, et al.Fabry disease: detection of undiagnosedhemodialysis patients and identificationof a “renal variant” phenotype. Kidney Int2003;64:801-7.
76. Nakao S, Takenaka T, Maeda M, et al. Anatypical variant of Fabry’s disease in menwith left ventricular hypertrophy. N Engl JMed 1995;333:288-93.
77. Hoffmann B, Georg Koch H, Schweitzer-Krantz S, et al. Deficient a-galactosidase Aactivity in plasma but no Fabry disease-apitfall in diagnosis. Clin Chem Lab Med2005;43:1276-7.
78. Filocamo M, Morrone A. Lysosomal storagedisorders: molecular basis and laboratorytesting. Hum Genomics 2011;5:156-69.
79. Ferri L, Guido C, La Marca G, et al. Fabrydisease: polymorphic haplotypes and anovel missense mutation in the GLA gene.Clin Genet 2012;81:224-33.
80. Gelb MH, Turecek F, Scott CR, ChamolesNA. Direct multiplex assay of enzymes indried blood spots by tandem mass spec-trometry for the newborn screening oflysosomal storage disorders. J InheritMetab Dis 2006;29:397-404.
81. Li Y, Scott CR, Chamoles NA, et al. Directmultiplex assay of lysosomal enzymes indried blood spots for newborn screening.Clin Chem 2004;50:1785-96.
82. La Marca G, Casetta B, Malvagia S, et al.New strategy for the screening of lysoso-mal storage disorders: the use of theonline
trapping-and-cleanup liquid chromatogra-phy/mass spectrometry. Anal Chem 2009;1:6113-21.
83. Lin H-Y, Chong K-W, Hsu J-H, et al. Highincidence of the cardiac variant of Fabrydisease revealed by newborn screening inthe Taiwan Chinese population. CircCardiovasc Genet 2009;2:450-6.
84. Reuser AJ, Verheijen FW, Bali D, et al. Theuse of dried blood spot samples in thediagnosis of lysosomal storage disorders-current status and perspectives. Mol GenetMetab 2011;104:144-8.
85. Linthorst GE, Vedder AC, Aerts JMFG,Hollak CEM. Screening for Fabry diseaseusing whole blood spots fails to identifyone-third of female carriers. Clin ChimActa 2005;353:201-3.
86. Ishii S, Nakao S, Minamikawa-Tachino R,et al. Alternative splicing in the a-galac-tosidase A gene: increased exon inclusionresults in the Fabry cardiac phenotype. AmJ Hum Genet 2002;70:994-1002.
87. Filoni C, Caciotti A, Carraresi L, et al.Unbalanced GLA mRNAs ratio quantifiedby real time PCR in Fabry patients’ fibrob-lasts results in Fabry disease. Eur J HumGenet 2008;16:1311-7.
88. Schirinzi A, Centra M, Prattichizzo C, et al.Identification of GLA gene deletions inFabry patients by multiplex ligation-dependent probe amplification (MLPA).Mol Genet Metab 2008;94:382-5.
89. Yoshimitsu M, Higuchi K, Miyata M, et al.Identification of novel mutations in the a-galactosidase A gene in patients withFabry disease: pitfalls of mutation analy-ses in patients with low a-galactosidase Aactivity. J Cardiol 2011;57:345-53.
90. Gal A, Hughes DA, Winchester B. Toward aconsensus in the laboratory diagnostics ofFabry disease recommendations of aEuropean expert group. J Inherit MetabDis 2011;34:509-14.
91. Al-Thihli K, Ebrahim H, Hughes DA, et al.A variant of unknown significance in theGLA gene causing diagnostic uncertaintyin a young female with isolated hyper-trophic cardiomyopathy. Gene 2012;497:320-2.
92. Vedder AC, Strijland A, Bergh WeermanMA, et al. Manifestations of Fabry diseasein placental tissue. J Inherit Metab Dis2006;29:106-11.
93. Togawa T, Kodama T, Suzuki T, et al.Plasma globotriaosylsphingosine as a bio-marker of Fabry disease. Molecular genet-ics and metabolism. 2010;100:257-61.
94. Togawa T, Kawashima I, Kodama T, et al.Tissue and plasma globotriaosylsphingo-sine could be a biomarker for assessingenzyme replacement therapy for Fabry dis-ease. Biochem Biophys Res Commun2010;399:716-20.
95. van Breemen MJ, Rombach SM, Dekker N,et al. Reduction of elevated plasma globo-triaosylsphingosine in patients with clas-sic Fabry disease following enzymereplacement therapy. Biochim BiophysActa 2011;1812:70-6.
96. Pieruzzi F, Pieroni M, Chimenti C, et al.[Cardiological follow-up in patients withFabry disease]. G Ital Cardiol 2010;11:566-72. [In Italian].
97. Linthorst G, Burlina A, Cecchi F, et al.Recommendations on reintroduction ofAgalsidase Beta for patients with Fabrydisease in Europe, following a period ofshortage. J Inherit Metab Dis Rep2013;8:51-6.
98. Linthorst GE, Germain DP, Hollak CEM, etal. Expert opinion on temporary treatmentrecommendations for Fabry disease dur-ing the shortage of enzyme replacementtherapy (ERT). Mol Genet Metab 2011;102:99-102.
99. Pisani A, Visciano B, Roux GD, et al.Enzyme replacement therapy in patientswith Fabry disease: State of the art andreview of the literature. Mol Genet Metab2012;107:267-75.
100.Weidemann F, Niemann M, Breunig F, etal. Long-term effects of enzyme replace-ment therapy on fabry cardiomyopathy:evidence for a better outcome with earlytreatment. Circulation 2009;119:524-9.
101.Imbriaco M, Messalli G, Avitabile G, et al.Cardiac magnetic resonance imagingillustrating Anderson-Fabry disease pro-gression. Br J Radiol 2010;83:e249-51.
102.Porto C, Pisani A, Rosa M, Acampora E.Synergy between the pharmacologicalchaperone 1-deoxygalactonojirimycin andthe human recombinant a-galactosidase Ain cultured fibroblasts from patients. JInherit Metab Dis 2012;35:513-20.
103.Motabar O, Sidransky E, Goldin E, ZhengW. Fabry disease - current treatment andnew drug development. Curr Chem Genom2010;4:50-6.
104.Benjamin ER, Khanna R, Schilling A, et al.Co-administration with the pharmacologi-cal chaperone AT1001 increases recombi-nant human a-galactosidase A tissueuptake and improves substrate reductionin Fabry mice. Mol Ther 2012;20:717-26.
Review
Non-co
mmercial
use o
nly

Non-co
mmercial
use o
nly
Non-co
mmercial
use o
nly

OPEN ACCESS Review
Hypertrophic cardiomyopathy:The need for randomized trialsIacopo Olivotto1,2,3,*, Benedetta Tomberli1,2,3, Roberto Spoladore1,2,3,
Alessandro Mugelli1,2,3, Franco Cecchi1,2,3, Paolo G Camici1,2,3
ABSTRACT
Hypertrophic cardiomyopathy (HCM) is a complex cardiac condition characterized by variable degrees
of asymmetric left ventricular (LV) hypertrophy, generally associated with mutations in sarcomere
protein genes. While generally perceived as rare, HCM is the most common genetic heart disease with
over one million affected individuals in Europe alone and represents a prevalent cause of sudden
cardiac death in the young. To date, HCM remains an orphan disease, as recommended treatment
strategies are based on the empirical use of old drugs with little evidence supporting their clinical
benefit in this context. In the six decades since the original description of the disease, less than fifty
pharmacological studies have been performed in HCM patients, enrolling little over 2,000 HCM
patients, mostly comprising small non-randomized cohorts. No specific agent has been convincingly
shown to affect outcome, and critical issues such as prevention of myocardial energy depletion,
microvascular ischemia, progressive myocardial fibrosis and the peculiar mechanisms of
arrhythmogenesis in HCM still need to be addressed in a systematic fashion. However, there is
increasing evidence that a variety of drugs may counter the effects of sarcomere protein mutations and
the resulting pathophysiological abnormalities at the molecular, cellular and organ level. Following
major advances in our understanding of HCM and increasing opportunities for networking among large
international referral centres, the opportunity now exists to identify potentially effective treatments and
implement adequately designed pharmacological trials, with the ultimate aim to impact the natural
course of the disease, alleviate symptoms and improve quality of life in our patients.
Keywords: hypertrophic cardiomyopathy, translational research, pharmacological therapy, clinical trials,outcome
Cite this article as: Olivotto I, Tomberli B, Spoladore R, Mugelli A, Cecchi F, Camici PG.Hypertrophic cardiomyopathy: The need for randomized trials, Global Cardiology Science andPractice 2013:31 http://dx.doi.org/10.5339/gcsp.2013.31
http://dx.doi.org/10.5339/gcsp.2013.31
Submitted: 1 September 2013Accepted: 20 September 2013ª 2013 Olivotto, Tomberli,Spoladore, Mugelli, Cecchi, Camici,licensee Bloomsbury QatarFoundation Journals. This is an openaccess article distributed under theterms of the Creative CommonsAttribution license CC BY 3.0, whichpermits unrestricted use,distribution and reproduction in anymedium, provided the original workis properly cited.
1Referral Center for Cardiomyopathies
(IO, BT, FC), Careggi University Hospital,
Florence, Italy2Vita-Salute University and San Raffaele
Scientific Institute, Milan, Italy (RS, PGC)3Section of Pharmacology, Neurofarba
Department, University of Florence, Italy
(AM)
*Email: olivottoi@aou-careggi.
toscana.it

Imagining a disease as a fruit or a planet, there are several levels at which one can intervene with any
therapeutic approach (Figure 1). The first and most obvious is to simply scratch the surface and control
symptoms. This objective can be achieved in most cardiac patients: however, it is the very least that we
can do. The second step is to interfere and possibly halt disease progression, thus preventing its
consequences on outcome: this can be done in several cardiac conditions, but is definitely harder to
achieve.1,2 Third, we can try to prevent the development of full-blown disease in patients who are
predisposed due to acquired risk factors and/or genetic substrate.3 And fourth, we can address the
core of the problem by acting directly on the etiology, removing the actual cause and ultimately cure
the patient.1,4 Despite extraordinary progress over the last decades, these last two steps have hardly if
ever been achieved in cardiovascular medicine.
As a consequence, it is important to realize that our practice is based on highly sophisticated
palliation. What this approach usually does is change a disease into a milder one. For example, septal
myectomy turns obstructive into non-obstructive hypertrophic cardiomyopathy (HCM), a highly
significant change for the patient.5,6 Therefore, this kind of very effective palliation is something we
should definitely keep on doing and improving until a cure becomes available.2 However, all efforts
should be directed at improving the state of things by accumulating new evidence and knowledge.4,7
HYPERTROPHIC CARDIOMYOPATHY AS AN ORPHAN DISEASE
HCM is a complex cardiac condition characterized by variable degrees of left ventricular (LV)
hypertrophy occurring in the absence of secondary triggers, generally associated with mutations in
sarcomeric protein genes.2,5 Although perceived as rare, HCM is the most common genetic heart
disease, as well as a prevalent cause of sudden cardiac death in the young. Based on the reported
prevalence of 1:500 in the general population, a total estimate exceeding one million individuals with
HCM are expected in Europe alone. HCM is characterized by a very complex pathophysiological
background that accounts for the heterogeneity of its clinical manifestations and natural history.5
Established features of the disease, besides left ventricular (LV) hypertrophy, include a constellation of
deranged cardiomyocyte energetics, diastolic dysfunction, microvascular ischemia, enhanced
myocardial fibrosis, autonomic dysfunction and enhanced arrhythmogenesis.8,9 In addition, most
patients exhibit dynamic LV outflow tract obstruction either at rest or with physiological provocation.10
Outflow obstruction is a major determinant of symptoms, such as dyspnea, chest pain or presyncope
and, together with sudden death prevention, has represented the most visible and consistent target of
therapeutic efforts in HCM.2,5–7
Despite decades of increasing attention and research efforts by the scientific community, treatment
strategies for HCM remain largely based on a small number of clinical studies, or empirically based on
personal experience or extrapolation from other cardiac conditions. As stated in the recent Report of
the Working Group of the National Heart, Lung, and Blood Institute on Research Priorities in
Hypertrophic Cardiomyopathy,7 “nearly 50 years after the identification of HCM as an autosomal
dominant disease, and 20 years after its linkage to sarcomeric protein mutations, we still do not
understand the most proximal mechanism(s) that initiates the disorder”; and “treatment
recommendations in HCM are based on observational series without prospective randomized controls.
While clinical usage provides support that various pharmacologic agents reduce HCM symptoms,
no evidence has demonstrated that they alter disease progression or outcomes.”
In a recent review of original articles, reviews and editorials addressing any pharmacological agent
ever used in HCM cohorts, only forty-five studies were identifiedover the last sixty years (i.e. less than
1 per year), enrolling a total of 2,121 HCM patients.4 Of these, only 5 were randomized double blind
SYMPTOMS
PROGRESSION
SYMPTOMS
ETIOLOGY
DEVELOPMENT
ETIOLOGY
Figure 1. Levels of therapeutic intervention: towards the core.
Page 2 of 6
Olivotto et al. Global Cardiology Science and Practice 2013:31

placebo-controlled trials. Remarkably, a comparison of the period 1991–2011 vs. 1971–1990
demonstrated no increase in the number of studies, and only a modest increase in the number of
patients enrolled (627 vs. 1,473, patients respectively). With regard to sample size, only 7 studies (15%)
enrolled more than 50 patients, whereas 22 (49%) had less than 20 patients.4 The maximum number of
patients in a single prospective study was 118, in a multicenter registry evaluating the efficacy and
safety of disopyramide.11 Overall, these data eloquently demonstrate how poorly pharmacological
research in HCM compares with that performed in other, more prevalent conditions such as coronary
artery disease and heart failure.1
Notably, when randomized trials have been performed in HCM, results have been far less intuitive
than expected. The story of dual-chamber pacing for control of LV outflow obstruction is certainly the
best case in point. Based on anecdotal observations showing an effect of LV pacing on obstruction,
several and often intriguing pathophysiological explanations were provided, leading to three
randomized trials. All three showed that pacing had in fact little if any effect on the gradient or exercise
capacity in most patients, and that any improvement in symptoms was largely related to a placebo
effect of the device.5,12 Thus, we should be aware that several widely accepted recommendations for
management of HCM based on expert opinion or case series still need to stand the test of properly
designed trials.
INHERENT CHALLENGES OF CLINICAL TRIALS IN HCM
Several reasons – some obvious, other less so – stand behind this state of things. The first lies with
the practical challenges inherent in designing trials with HCM patients (Table 1). The epidemiology of
HCM is complex and as yet only partially resolved, due to issues such as incomplete penetrance and
prevalence of subclinical disease.3,13 Despite not being rare, HCM is uncommonly encountered and
possibly neglected at many community-based cardiac centers and outpatient clinics. Furthermore,
even when overt and correctly diagnosed, its clinical spectrum is highly heterogeneous, encompassing
different stages that may not be directly comparable.9 A preventive trial in genotype-positive/
phenotype-negative individuals will necessarily enroll subsets that are different from HCM patients with
classic phenotype and dynamic outflow obstruction, or in the end-stage phase. Each research question
should be addressed by targeting the appropriate patient subgroups, with imaginable problems in
achieving the desired yield in any given cohort.7,9
Another very complex issue concerns the assessment of outcome in HCM, and the effects of
treatment on survival. Even at tertiary referral centers, cardiovascular mortality rates in contemporary
HCM cohorts are very low, i.e. 1–2%/year, although the selected subset with systolic dysfunction and
end-stage progression may exceed 10%/year.2,5,9,14 Rates of sudden cardiac death, formerly believed
to be very common among HCM patients, are even lower, i.e. ,1% in most cohorts and only 3%/year
even in patients carrying implantable defibrillators because judged to have a “high-risk” status.15
Based on these estimates, large patient cohorts and extended follow-up duration are required to detect
differences in survival, even when a highly effective drug or procedure is tested. Thus, the relatively
favorable long-term prognosis of most HCM patients may represent a formidable obstacle to clinical
trials targeting outcome, including a hypothetical study comparing surgical myectomy and
percutaneous alcohol septal ablation.16
In order to overcome the limitations associated with the low hard event rates, surrogate end-points
have often been employed in HCM trials, including indexes of LV performance, functional capacity and
oxygen consumption, progression of symptoms, prevalence of arrhythmias and appropriate
Table 1. Challenges for clinical trials in HCM.
Relatively low prevalence (1:500) Need for adequate networking, hard to perform single-center trialsHeterogeneous disease spectrum Need to tailor trials to specific subsets, hard to include all patients
in a single studyLow event rates Difficult to power studies to assess differences in survival/hard
outcome end-pointsSurrogate end-points End-points such as oxygen consumption, variation in symptoms,
regression of LV hypertrophy or arrhythmias may be difficult todefine and/or have un uncertain relation to outcome
Complex, unresolved pathophysiology Need to identify appropriate targets for treatmentPerceived as economically irrelevantto the pharmaceutical industry
However, this is a wrong perception based on absolute number ofpatients and need for very extended treatment
Page 3 of 6
Olivotto et al. Global Cardiology Science and Practice 2013:31

defibrillator shocks, circulating markers of collagen synthesis and natriuretic peptides.4,13,17–19
Advanced imaging techniques such as positron emission tomography (PET) and cardiac magnetic
resonance (CMR) can be exploited today to quantify the effect of novel treatments on relevant features
including coronary microvascular dysfunction and tissue fibrosis, respectively.13,20–23 Each of these
may represent reasonable end-points in certain contexts, particularly in proof-of-concept and pilot
studies. Furthermore, pilot studies based on these surrogate end-points might provide the rationale
and justify the effort of large, multicenter, prospective studies enrolling the number of patients
necessary to determine the benefit of novel treatments on more robust measures of outcome.4,7 At the
same time, the favourable long-term prognosis associated with HCM identifies control of symptoms
and improvement in quality of life as major objectives for pharmacological treatment, to be pursued
and investigated independent of outcome.4
Last but not least, HCM has never been the focus of strong economic interest by pharmaceutical
companies, because of the perceived rarity of the disease as well as the lack of novel and expensive
therapeutic agents such as have been approved, for example, in rare conditions such as Fabry disease
or pulmonary arterial hypertension.24 This aspect, however, may be expected to change soon. In recent
years, the widespread use of imaging and genetic testing for familial screening, as well as an increasing
awareness in the medical community, have led to the identification of large HCM populations, regularly
followed at international referral centers.2,4 For the first time in the history of this disease, it has
become feasible to address HCM management issues in an evidence-based fashion, by means of
adequately designed clinical trials. The full exploitation of this potential may lead to a paradigm shift in
research methodology involving the whole field of genetic heart diseases. Such favorable conjunction
is beginning to attract the interest of private companies, promoting investments that are likely to reach
a critical mass for effective clinical research in a very near future.4,25 When this time comes, the
powerful combination of basic science, clinical knowledge and adequate trial methodology may finally
come to fruition.
POTENTIAL TARGETS OF PHARMACOLOGIC TREATMENT
Progress in HCM treatment is hindered by the incomplete knowledge of its pathophysiologic
mechanisms and lack of specific agents capable of interfering with disease-causing pathways.7–9
However, several targets for treatment have been identified (Figure 2), some of which overlap with
other cardiac diseases and with heart failure at large.8,26 For example, interventions aimed at
normalizing energy homeostasis represent a viable approach, as shown by a recent study on
perhexiline, a metabolic modulator which inhibits the metabolism of free fatty acids and enhances
carbohydrate utilization by the cardiomyocyte. In a recent randomized double-blind placebo-controlled
trial, perhexilinehas have shown the capacity to improve the energetic profile of the LV, resulting in
improved diastolic function and exercise capacity in HCM patients.18
Furthermore, HCM cardiomyocytes exhibit with well-established abnormalities in intracellular
calcium handling, contributing to excessive energy expenditure and enhanced arrhythmogenesis that
are largely due to enhanced membrane late sodium current.26 Such defect may be selectively and
dramatically reversed in vitro by ranolazine.26 Following the demonstration of its beneficial effects on
HCM cardiomyocytes, a multi-center, double blind, placebo-controlled pilot study is currently underway
in Europe to test the efficacy of ranolazine on exercise tolerance and diastolic function in symptomatic
HCM patients (RESTYLE-HCM; EUDRA-CT 2011-004507-20). Besides the specific merits of ranolazine,
similar examples of translational approach to HCM identifies a fundamental pre-requisite for the
identification of novel, potentially effective agents, based on thorough investigation of the molecular
basis of the disease.1 In the future, a more specific approach may be tailored to specific mutations or
groups of mutations, by screening large panels of candidate molecules in assays based on induced
pluripotent stem cells isolated from human fibroblasts.27 As shown recently, the possibility of
modulating the activity of sarcomere contractile proteins, such as beta-myosin, is beginning to surface,
with huge potential implications for HCM treatment.28
Finally, an option that is less seductive but should not be underestimated, is to investigate potential
role in HCM patients of drugs that represent the standard of care for other cardiac patients. Agents such
as modulators of the renin-angiotensin system, statins and calcium antagonists have all shown
promise in preventing development of LV hypertrophy in genotype positive individuals,3 and may
potentially normalize cellular homeostasis, prevent microvascular remodeling and ischemia, reduce
fibrosis and prevent end-stage progression.4,8,9,17,21,22 However, the indications, timing and doses of
Page 4 of 6
Olivotto et al. Global Cardiology Science and Practice 2013:31

these treatments remain unresolved in HCM, and their efficacy in impacting its natural history remains
untested. Likewise, the effects of betablockers and antiarrhythmics, such as amiodarone, in preventing
appropriate defibrillator shocks and sudden cardiac death has only been addressed retrospectively
and inconclusively.29 As noted above, many recommendations present in current HCM guidelines still
require adequate evidence to their support, including several that few would dare question. And while
it is unrealistic to expect that each of these indications will become evidence-based in the near future,
it is the change of attitude that really matters, from passive acceptance of the empirical to
uncompromising quest for the methodologically appropriate and convincing.2,4,7
CONCLUSIONS
HCM is not uncommon, but remains an orphan disease with regard to pharmacological treatment.
In the era of translational research, physicians and researchers have been very good at asking the
right questions, but need to direct more effort into finding the best clinical answers for HCM patients.
In a broader perspective, cardiomyopathies should be seen as paradigms providing invaluable insights
on disease mechanisms that may be of general relevance to patients with more prevalent cardiac
conditions. What has long been considered impossible to achieve is now closer at hand: the time is
now ripe to promote robust clinical research in this complex condition in order to advance and
standardize management of HCM patients.
AcknowledgementsThis work was supported by the European Union (STREP Project 241577 “BIG HEART,” 7th EuropeanFramework Program).
REFERENCES
[1] Braunwald E. Cardiovascular science: opportunities for translating research into improved care. J Clin Invest.2013;123:6–10.
[2] Maron BJ, Braunwald E. Evolution of hypertrophic cardiomyopathy to a contemporary treatable disease. Circulation.2012;126:1640–1644.
[3] Marian AJ. Experimental therapies in hypertrophic cardiomyopathy. J CardiovascTransl Res. 2009;2:483–492.[4] Spoladore R, Maron MS, D’Amato R, Camici PG, Olivotto I. Pharmacological treatment options for hypertrophic
cardiomyopathy: high time for evidence. Eur Heart J. 2012;33:1724–1733.
LEVEL OFINTERVENTION
TARGET GOAL
MOLECULAR
Abnormal Sarcomere Function
Enhanced Ca++ Sensitivity
Unfavourable EP Remodeling
Improve Contractile Efficiency
Inhibit Triggers for LVH
Reduce Arrhythmogesis
CELLULARDeranged Energy Homeostasis
Disturbed Biomechanical Stress Sensing
Abnormal Ca++ Handling
Prevent Energy Depletion
Prevent Hypertrophy/Apoptosis
Control Collagen Synthesis
ORGANMicrovascular remodelling
Myocardial Fibrosis
Diastolic Dysfunction
Reentry Arrhythmias
Reduce Myocardial Ischemia
Preserve Systolic Function
Improve Diastole
Prevent Arrhythmias
Figure 2. Therapeutic targets and goals in HCM.
Page 5 of 6
Olivotto et al. Global Cardiology Science and Practice 2013:31

[5] Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H,Seidman CE, Towbin JA, Udelson JE, Yancy CW. ACCF/AHA guideline for the diagnosis and treatment of hypertrophiccardiomyopathy; executive summary: a report of the American College of Cardiology Foundation/American HeartAssociation Task Force on Practice Guidelines. Circulation. 2011;124:2761–2796.
[6] Ommen SR, Maron BJ, Olivotto I, Maron MS, Cecchi F, Betocchi S, Gersh BJ, Ackerman MJ, McCully RB, Dearani JA,Schaff HV, Danielson GK, Tajik AJ, Nishimura RA. Long-term effects of surgical septalmyectomy on survival in patientswith obstructive hypertrophic cardiomyopathy. J Am CollCardiol. 2005;46:470–476.
[7] Force T, Bonow RO, Houser SR, Solaro RJ, Hershberger RE, Adhikari B, Anderson ME, Boineau R, Byrne BJ, Cappola TP,Kalluri R, LeWinter MM, Maron MS, Molkentin JD, Ommen SR, Regnier M, Tang WH, Tian R, Konstam MA, Maron BJ,Seidman CE. Research priorities in hypertrophic cardiomyopathy: report of a Working Group of the National Heart,Lung, and Blood Institute. Circulation. 2010;122(11):1130–1133.
[8] Frey N, Luedde M, Katus HA. Mechanisms of disease: hypertrophic cardiomyopathy. Nat Rev Cardiol. 2012;9:91–100.[9] Olivotto I, Cecchi F, Poggesi C, Yacoub MH. Patterns of disease progression in hypertrophic cardiomyopathy: an
individualized approach to clinical staging. Circ Heart Fail. 2012;5:535–546.[10] Maron MS, Olivotto I, Zenovich AG, Link MS, Pandian NG, Kuvin JT, Nistri S, Cecchi F, Udelson JE, Maron BJ.
Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation.2006;114:2232–2239.
[11] Sherrid MV, Barac I, McKenna WJ, Elliott PM, Dickie S, Chojnowska L, Casey S, Maron BJ. Multicenter study of theefficacy and safety of disopyramide in obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol.2005;45:1251–1258.
[12] Binder J, Ommen SR, Sorajja P, Nishimura RA, Tajik AJ. Clinical and echocardiographic variables fail to predict responseto dual-chamber pacing for hypertrophic cardiomyopathy. J Am Soc Echocardiogr. 2008;21:796–800.
[13] Ho CY, Lopez B, Coelho-Filho OR, Lakdawala NK, Cirino AL, Jarolim P, Kwong R, Gonzalez A, Coan SD, Seidman JG,Diez J, Seidman CE. Myocardialfibrosisas an early manifestation of hypertrophic cardiomyopathy. N Engl J Med.2010;363:552–563.
[14] Harris KM, Spirito P, Maron MS, Zenovich AG, Formisano F, Lesser JR, Mackey-Bojack S, ManningWJ, Udelson JE, Maron BJ.Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophiccardiomyopathy. Circulation. 2006;114:216–225.
[15] Schinkel AF, Vriesendorp PA, Sijbrands EJ, Jordaens LJ, ten Cate FJ, Michels M. Outcome and complications afterimplantable cardioverter defibrillator therapy in hypertrophic cardiomyopathy: systematic review and meta-analysis.Circ Heart Fail. 2012;5:552–559.
[16] Olivotto I, Ommen SR, Maron MS, Cecchi F, Maron BJ. Surgical myectomy versus alcohol septal ablation for obstructivehypertrophic cardiomyopathy. Will there ever be a randomized trial? J Am Coll Cardiol. 2007;50:831–834.
[17] Penicka M, Gregor P, Kerekes R, Marek D, Curila K, Krupicka J, Candesartan use in Hypertrophic And Non-obstructiveCardiomyopathy Estate (CHANCE) Study Investigators. The effects of candesartan on left ventricular hypertrophy andfunction in nonobstructive hypertrophic cardiomyopathy: a pilot, randomized study. J Mol Diagn. 2009;11:35–41.
[18] Abozguia K, Elliott P, McKenna W, Phan TT, Nallur-Shivu G, Ahmed I, Maher AR, Kaur K, Taylor J, Henning A, Ashrafian H,Watkins H, Frenneaux M. Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacityin symptomatic hypertrophic cardiomyopathy. Circulation. 2010;122:1562–1569.
[19] Coats CJ, Gallagher MJ, Foley M, O’Mahony C, Critoph C, Gimeno J, Dawnay A, McKenna WJ, Elliott PM. Relation betweenserum N-terminal pro-brain natriuretic peptide and prognosis in patients with hypertrophic cardiomyopathy. Eur HeartJ. 2013;34(32):2529–2537. doi:10.1093/eurheartj/eht070
[20] Camici PG, Olivotto I, Rimoldi OE. The coronary circulation and blood flow in left ventricular hypertrophy. J Mol CellCardiol. 2012;52:857–864.
[21] Mourad J, Hanon O, Deverre JR, Camici PG, Sellier P, Duboc D, Safar ME. Improvement of impaired coronary vasodilatorreserve in hypertensive patients by low dose ace-inhibitor/diuretic therapy: a pilot PET study. J Renin AngiotensinAldosterone Syst. 2003;4:94–95.
[22] Neglia D, Fommei E, Varela-Carver A, ManciniM, Ghione S, LombardiM, Pisani P, Parker H, D’Amati G, Donato L, Camici PG.Perindopril and indapamide reverse coronary microvascular remodelling and improve flow in arterial hypertension. JHypertens. 2011;29:364–372.
[23] Maron MS. Clinical utility of cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Cardiovasc MagnReson. 2012;14:13. doi: 10.1186/1532-429X-14-13
[24] Mehta A, Beck M, Elliott P, Giugliani R, Linhart A, Sunder-Plassmann G, Schiffmann R, Barbey F, Ries M, Clarke JT, FabryOutcome Survey investigators. Enzyme replacement therapy with agalsidase alfa in patients with Fabry’s disease: ananalysis of registry data. Lancet. 2009;374:1986–1996.
[25] http://www.myokardia.com/approach-programs.php accessed on 31/04/2013.[26] Coppini R, Ferrantini C, Yao L, Fan P, Del LungoM, Stillitano F, Sartiani L, Tosi B, Suffredini S, Tesi C, YacoubM, Olivotto I,
Belardinelli L, Poggesi C, Cerbai E, Mugelli A. Late sodium current inhibition reverses electromechanical dysfunction inhuman hypertrophic cardiomyopathy. Circulation. 2013;127:575–584.
[27] Lan F, Lee AS, Liang P, Sanchez-Freire V, Nguyen PK, Wang L, Han L, Yen M, Wang Y, Sun N, Abilez OJ, Hu S, Ebert AD,Navarrete EG, Simmons CS, Wheeler M, Pruitt B, Lewis R, Yamaguchi Y, Ashley EA, Bers DM, Robbins RC, Longaker MT,Wu JC. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell. 2013;12:101–113.
[28] Malik FI, Hartman JJ, Elias KA, Morgan BP, Rodriguez H, Brejc K, Anderson RL, Sueoka SH, Lee KH, Finer JT, Sakowicz R,Baliga R, Cox DR, Garard M, Godinez G, Kawas R, Kraynack E, Lenzi D, Lu PP, Muci A, Niu C, Qian X, Pierce DW,Pokrovskii M, Suehiro I, Sylvester S, Tochimoto T, Valdez C, Wang W, Katori T, Kass DA, Shen YT, Vatner SF, Morgans DJ.Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science. 2011;331:1439–1443.
[29] Melacini P, Maron BJ, Bobbo F, Basso C, Tokajuk B, Zucchetto M, Thiene G, Iliceto S. Evidence that pharmacologicalstrategies lack efficacy for the prevention of sudden death in hypertrophic cardiomyopathy. Heart. 2007;93:708–710.
Page 6 of 6
Olivotto et al. Global Cardiology Science and Practice 2013:31

297G ITAL CARDIOL | VOL 13 | APRILE 2012
Trattamento dei sintomi refrattari nella cardiomiopatiaipertrofica con fisiopatologia restrittiva:
nuove prospettive per la ranolazinaBenedetta Tomberli1, Francesca Girolami2, Raffaele Coppini3, Cecilia Ferrantini4, Alessandra Rossi1,
Franco Cecchi1, Iacopo Olivotto1
1Centro di Riferimento per le Cardiomiopatie, Azienda Ospedaliero-Universitaria Careggi, Firenze2SOD Diagnostica Genetica, Azienda Ospedaliero-Universitaria Careggi, Firenze
3Dipartimento di Farmacologia Clinica e Preclinica, Università degli Studi, Firenze4Dipartimento di Scienze Fisiologiche, Università degli Studi, Firenze
The management of patients with hypertrophic cardiomyopathy (HCM) and refractory symptoms due to mas-sive hypertrophy and severe diastolic dysfunction represents a real challenge for the clinical cardiologist. Suchpatients often require novel therapeutic approaches, both invasive and pharmacological, involving multidis-ciplinary teamwork; however, the implementation of potentially viable treatment options is hindered by lackof disease-specific evidence.
We report the case of a young woman with severe HCM and restrictive physiology, who underwent exten-sive myectomy via the transaortic and transapical approach, followed by biventricular pacing for cardiac re-synchronization, with significant but incomplete symptomatic improvement. The subsequent introduction ofranolazine, based on promising preclinical data, has led to an excellent final result. An ongoing randomizedclinical trial is currently testing the efficacy of ranolazine in symptomatic HCM.
Key words. Angina; Hypertrophic cardiomyopathy; Massive hypertrophy; Ranolazine; Restrictive cardiomy-opathy.
G Ital Cardiol 2012;13(4):297-303
La cardiomiopatia ipertrofica (CMI) è la più frequente malattiagenetica del muscolo cardiaco, con una prevalenza di 1:500nella popolazione generale1 ed è caratterizzata da espressionemorfologica e decorso clinico estremamente eterogenei2. In unsottogruppo di pazienti con CMI, ad espressione fenotipicaestrema, si riscontrano gradi molto marcati di ipertrofia asso-ciata a ridotte dimensioni cavitarie del ventricolo sinistro (VS) efisiopatologia restrittiva. Tali pazienti presentano quasi sempresintomi limitanti, quali dispnea o angina per sforzi anche lievi,e sono di difficile gestione clinica3,4. Le strategie terapeutichevengono rese ancor più complesse dalla mancanza di evidenzescientifiche specifiche per la malattia5.
Viene qui presentato un caso clinico, la cui gestione è ri-sultata particolarmente impegnativa ed ha richiesto nel tempoil ricorso ad approcci innovativi, sia da un punto di vista chirur-gico che farmacologico. Il caso rappresenta un buon esempiodi come, per una corretta gestione del paziente con cardio-miopatia, sia necessario un team davvero multidisciplinare.
CASO CLINICO
La paziente è una giovane donna giunta alla nostra osservazio-ne all’età di 30 anni, in seguito alla diagnosi di CMI, sulla basedi accertamenti per limitazione funzionale. Alla prima valuta-zione presso il nostro centro riferiva palpitazioni, astenia, dolo-re toracico e dispnea da sforzo inquadrabile in una classe NYHAIII, di intensità molto variabile a seconda dei giorni, ma abba-stanza marcati da impedire lo svolgimento delle normali attivi-tà lavorative nel suo negozio (Figura 1). L’esame obiettivo si ca-ratterizzava per bassi valori pressori a riposo (100/55 mmHg) edun terzo tono, in assenza di soffi patologici o segni di scom-penso. L’ECG a 12 derivazioni mostrava ritmo sinusale con iper-trofia e sovraccarico del VS (Figura 2, pannello 1C). All’ecocar-diografia era presente un’ipertrofia diffusa del VS di grado mar-cato, a massima espressione medio-apicale, con spessore mas-simo 30 mm a livello del setto basale anteriore (Figura 3, pan-nelli A-C). Il ventricolo sinistro appariva ipercinetico, con una ca-vità ridotta, a “piede di ballerina” e obliterazione completa del-l’apice in sistole. Era presente una grave disfunzione diastolicadel VS, con pattern del flusso transmitralico restrittivo (Figura 2,pannello 1B; Figura 3, pannelli D). I lembi mitralici erano ridon-danti, con accenno a movimento sistolico anteriore, ma non era-no misurabili gradienti sistolici al tratto di efflusso né in sedemedio-ventricolare. Si riscontravano inoltre una insufficienza mi-tralica centrale, di grado lieve, ed un’ipertensione polmonaremoderata (Figura 2, pannello A1; Figura 3, pannelli D1-D2). Intale occasione venne iniziata una terapia farmacologica con na-
© 2012 Il Pensiero Scientifico EditoreRicevuto 23.02.2012; accettato 27.02.2012.Il dr. Iacopo Olivotto è investigator per lo studio pilota “Ranolazina inpazienti con cardiomiopatia ipertrofica sintomatica: studio pilota”,sponsorizzato da Menarini. Gli altri autori dichiarano nessun conflittodi interessi.Per la corrispondenza:Dr.ssa Benedetta Tomberli Dipartimento Cuore e Vasi, Centro diRiferimento per le Cardiomiopatie, Cardiologia c/o San Luca Vecchio,AOU Careggi, Largo Brambilla 3, 50134 Firenzee-mail: [email protected]
CASO CLINICO
© Il Pensiero Scientifico Editore downloaded by BENEDETTA TOMBERLI IP 87.12.247.5 Sat, 01 Sep 2012, 17:32:02

G ITAL CARDIOL | VOL 13 | APRILE 2012298
B TOMBERLI ET AL
Figura 1. Decorso clinico ed interventi terapeutici. Dall’alto in basso: classe funzionale NYHA per la dispnea,angina, terapie (farmacologica e chirurgica), episodi di TVNS (registrati all’ECG dinamico o al controllo ICD),livelli di NT-proBNP ed esami strumentali. ICD, defibrillatore impiantabile; NT-proBNP, porzione N-terminale del propeptide natriuretico di tipo B; PET, to-mografia ad emissione di positroni; PM, pacemaker; RMN, risonanza magnetica nucleare; TVNS, tachicardiaventricolare non sostenuta.
Figura 2. Quadro elettrocardiografico ed ecocardiografico alla valutazione preoperatoria, postoperatoria e dopoterapia di resincronizzazione cardiaca (CRT). Pannello 1: alla prima valutazione è presente un’insufficienza mitralicadi grado lieve, una disfunzione diastolica severa ed un quadro elettrocardiografico di ipertrofia e sovraccarico delventricolo sinistro. Pannello 2: a 6 mesi dall’intervento di miectomia si riscontra un’insufficienza mitralica di gra-do marcato con dilatazione della cavità ventricolare sinistra, pattern diastolico restrittivo sostanzialmente invariato.L’ECG presenta ritmo sinusale con blocco di branca sinistra. Pannello 3: 3 mesi dopo impianto di pacemaker bi-ventricolare si osserva una netta riduzione del grado di insufficienza mitralica, con miglioramento del patterndiastolico. L’ECG conferma il buon grado di resincronizzazione.AS, atrio sinistro; SIV, setto interventricolare; VS, ventricolo sinistro.
© Il Pensiero Scientifico Editore downloaded by BENEDETTA TOMBERLI IP 87.12.247.5 Sat, 01 Sep 2012, 17:32:02

299G ITAL CARDIOL | VOL 13 | APRILE 2012
CMI SINTOMATICA E RANOLAZINA
dololo, titolato fino alla massima dose tollerata di 80 mg/die,ottenendo una parziale remissione dei sintomi.
Nel corso di controlli successivi, una tomografia ad emissio-ne di positroni mostrava una disfunzione microvascolare diffu-sa, complessivamente di grado lieve (flusso medio per l’intero VSdopo dipiridamolo 2.6 ml/min/g), ma con aree settali marcata-mente ipoperfuse; alla risonanza magnetica cardiaca veniva con-fermata l’ipertrofia massiva con obliterazione medio-apicale (Fi-gura 4); erano inoltre presenti estese aree di impregnazione tar-diva con gadolinio, suggestive per fibrosi miocardica. Un eco-color-Doppler cardiaco da sforzo, in terapia, non evidenziavagradienti significativi in sede subaortica o medio-ventricolare;venivano evidenziati una ridotta capacità funzionale (test inter-rotto per dispnea al termine 100 W), anche se con adeguato in-cremento pressorio (90/60 mmHg al basale, 160/80 mmHg alpicco). La paziente presentava inoltre frequenti salve di tachi-cardia ventricolare non sostenuta all’ECG dinamico e valori del-la porzione N-terminale del propeptide natriuretico di tipo B (NT-proBNP) ripetutamente elevati (Figura 1). Infine, all’esame ge-netico veniva documentata una doppia mutazione in eterozi-gosi sul gene della proteina C legante la miosina (Figura 5).
In seguito a peggioramento della dispnea e soprattutto del-l’angina, la terapia farmacologica è stata progressivamente po-tenziata; nel corso di un follow-up di circa 5 anni sono stati ag-
giunti al nadololo sia calcioantagonisti che nitrati transdermici (Fi-gura 1), con modesti risultati. All’età di 35 anni, in seguito ad ag-gravamento dei sintomi, la paziente veniva ricoverata per ulterioriaccertamenti. Una coronarografia mostrava vasi epicardici inden-ni da lesioni, con modesto bridging miocardico al tratto medio del-l’arteria interventricolare anteriore, di dubbio significato clinico; al-la ventricolografia veniva confermata la grave riduzione di cavitàdel VS, con obliterazione sistolica dell’apice in toto (Figura 6).
Data la gravità del quadro clinico e la refrattarietà dei sinto-mi alla terapia medica, venne proposta alla paziente la possibi-lità di un intervento di miectomia estesa, allo scopo di aumen-tare le dimensioni del VS e la gittata sistolica. In assenza di indi-cazioni “classiche” all’intervento, tale proposta era basata suuna positiva esperienza, maturata presso questo ed altri centri,nel trattamento delle forme di CMI con ostruzione medio-ven-tricolare, che presentano una fisiopatologia per molti aspetti si-mili al caso in questione6. La paziente è stata quindi operata nel2009 dal prof. Magdi Yacoub e dall’equipe del dr. Pierluigi Ste-fano, mediante un approccio combinato per via transaortica etransapicale. Quest’ultimo, mediante incisione a “bocca di pe-sce” dell’apice ventricolare, ha permesso di rimuovere porzionidi miocardio a livello del VS medio-apicale, con significativo am-pliamento della cavità (Figura 4). Il decorso postoperatorio è sta-to complicato da fibrillazione atriale parossistica, per cui è stata
Figura 3. Ecocardiogramma preoperatorio. Proiezione parasternale asse lungo(A), asse corto (B) e apicale 4 camere (C). È presente ipertrofia severa e diffusa, conspessore massimo a livello del setto medio. Si noti la completa obliterazione del-la cavità in sistole nelle varie proiezioni e l’iper-riflettenza irregolare del setto in-terventricolare. L’analisi Doppler del flusso mitralico mostra una disfunzione dia-stolica severa, con pattern restrittivo ad andamento trifasico (D1) e notevole ri-duzione della velocità dell’anulus mitralico al Doppler tissutale (D3-D4). Il flussodelle vene polmonari è alterato, con una A reverse prominente (D2).AS, atrio sinistro; SIV, setto interventricolare; VS, ventricolo sinistro.
© Il Pensiero Scientifico Editore downloaded by BENEDETTA TOMBERLI IP 87.12.247.5 Sat, 01 Sep 2012, 17:32:02

G ITAL CARDIOL | VOL 13 | APRILE 2012300
B TOMBERLI ET AL
iniziata terapia con amiodarone. L’ecocardiogramma alla dimis-sione confermava l’ottimo esito dell’intervento, con un volumeventricolare residuo ora ai limiti della norma per dimensioni cor-poree, una funzione sistolica globale conservata ed una insuffi-
cienza mitralica lieve. A poche settimane dall’intervento, la pa-ziente riferiva netto miglioramento della dispnea (classe funzio-nale NYHA II stabile), associato a riduzione dei valori di NT-proBNP (Figura 1).
Figura 4. Risonanza magnetica nucleare pre- e postoperatoria. Immagini di risonanza magnetica nucleare ottenu-te con sequenze cine in telediastole e telesistole, 2 mesi prima (A, B) e 8 mesi dopo la miectomia (C, D). A e C: leimmagini in 4 camere mostrano l’importante ispessimento del setto interventricolare e della parete laterale medio-apicale. La rimozione di miocardio a livello del setto e della parete postero-laterale (*) mediante miectomia ha per-messo la creazione di una cavità ventricolare. Evidente in C il rimodellamento post-chirurgico della parete laterale(frecce). B e D: sezione coronale asse corto a livello della valvola mitrale e dei muscoli papillari: è evidente l’oblite-razione sistolica a livello medio-apicale prima dell’intervento (B).AS, atrio sinistro; SIV, setto interventricolare; VS, ventricolo sinistro.
Figura 5. Elettroferogramma della sequenza del DNA che mostra la presenza di un genotipo complesso, caratte-rizzato da due mutazioni nello stesso gene (MYBPC3, proteina C legante la miosina). I rettangoli indicano la posi-zione della mutazione nella sequenza. A: mutazione di frameshift caratterizzata dalla duplicazione di una singolabase che ha come conseguenza la creazione di un codone di stop a valle. La sintesi della corrispondente proteinasi arresta prematuramente. B: mutazione missense caratterizzata dalla sostituzione di una singola base (citosina)con un’altra (timina) che comporta a livello della proteina il cambiamento dell’aminoacido prolina con leucina.
© Il Pensiero Scientifico Editore downloaded by BENEDETTA TOMBERLI IP 87.12.247.5 Sat, 01 Sep 2012, 17:32:02

301G ITAL CARDIOL | VOL 13 | APRILE 2012
CMI SINTOMATICA E RANOLAZINA
Dopo circa 6 mesi veniva effettuato un nuovo controllo cli-nico per significativa riaccentuazione della dispnea nelle ultimesettimane. Veniva evidenziato un aumento spiccato del grado diinsufficienza mitralica, attribuibile a rimodellamento della pare-te laterale, che si associava a dilatazione dell’anulus, ipomobilitàe tethering del lembo mitralico posteriore (Figura 2, pannelli 2A-C). Veniva inoltre ipotizzato che la presenza di un’asincronia mar-cata del VS, secondaria al blocco di branca sinistra postoperato-rio, costituisse un fattore aggravante della valvulopatia. Negli an-ni era stata più volte discussa con la paziente l’indicazione ad undefibrillatore impiantabile in prevenzione primaria, dati i nume-rosi fattori di rischio presenti. Venne pertanto proposto di pro-cedere all’impianto di un dispositivo biventricolare, con il razio-nale che la terapia di resincronizzazione potesse migliorare il gra-do di insufficienza mitralica7, come confermato dal successivodecorso. A 3 mesi dall’impianto del dispositivo, l’insufficienza mi-tralica era solo di poco superiore a quella pre-miectomia, stima-bile di grado moderato (Figura 2, pannelli 3A-C), e si rifletteva inun notevole miglioramento della dispnea. Restava invece inva-riata la severità dell’angina, sia come intensità che come fre-quenza degli episodi.
Nel tentativo di potenziare in modo massimale la terapiamedica, data la scarsa tolleranza ai betabloccanti ed un’intol-leranza assoluta agli inibitori dell’enzima di conversione del-l’angiotensina, veniva aggiunto al trattamento in atto (nadolo-lo a basse dosi ed amiodarone), una dose scalare di ranolazina.Tale raccomandazione si basava sulla letteratura disponibile perla cardiopatia ischemica8,9 e sugli ottimi risultati preliminari ot-tenuti in vitro su campioni di miectomia di pazienti con CMI,compreso quello della paziente (Figura 7). Il farmaco è stato ti-tolato fino ad un dosaggio di 1000 mg 2 volte/die, ben tolle-rato eccetto che per disturbi gastrointestinali lievi, che si sonoattenutati dopo le prime somministrazioni. A pochi giorni dal-l’assunzione della dose piena di ranolazina la paziente riferivaun netto miglioramento dei sintomi, con scomparsa degli epi-sodi anginosi durante le normali attività quotidiane e gli sforzimoderati. Non si sono osservate variazioni significative del QTc.Nei 6 mesi successivi la paziente è rimasta stabilmente in clas-se funzionale NYHA II, con buona qualità di vita, mantenendoinvariata una terapia a base di nadololo e ranolazina.
COMMENTI E IMPLICAZIONI CLINICHE
Il caso descritto, fortunatamente raro nella sua complessità,presenta numerosi spunti di interesse. Anzitutto, un grado mas-sivo di ipertrofia nel giovane con CMI si associa molto spesso avari altri marcatori di gravità della malattia, quali un genotipocomplesso10, disfunzione microvascolare e fibrosi del VS, aritmie
ventricolari all’ECG dinamico, elevati livelli di biomarcatori e dis -funzione diastolica di grado severo con pattern restrittivo, tut-ti indicatori di prognosi avversa. Spesso si osserva anche un ri-dotto incremento pressorio allo sforzo, non presente in questo
Figura 6. Ventricolografia preoperatoria. La ventricolografia mostra la riduzione della cavità ventricolare sinistra durante lasistole, che appare molto ridotta (frecce).
Figura 7. Misure del potenziale d’azione e del transiente di calciointracellulare sui cardiomiociti isolati al momento della miectomia. A:pezzo chirurgico di miectomia settale della paziente. Il pezzo è mo-strato sul lato endocardico (A1), dove è possibile apprezzare ampiezone di fibrosi, e nella sua porzione profonda (A2). La barra corri-sponde a 3 mm. B: tramite digestione enzimatica del pezzo chirur-gico sono stati ottenuti cardiomiociti ventricolari isolati, mostrati nel-l’immagine fotomicroscopica in figura. La barra corrisponde a 20µm. C: le cellule sono state utilizzate per simultanee misure di patch-clamp e calcio intracellulare (previo caricamento con il colorante fluo-4). Il pannello C1 mostra un potenziale d’azione ottenuto da unacellula della paziente, nettamente prolungato rispetto a un norma-le potenziale d’azione ventricolare; il pannello C2 mostra un tran-siente di calcio ottenuto dalla stessa cellula, anch’esso di durata au-mentata. L’esposizione al farmaco accorcia la durata del potenzialed’azione e del transiente di calcio, con potenziali benefici sulla fun-zione miocardica.
© Il Pensiero Scientifico Editore downloaded by BENEDETTA TOMBERLI IP 87.12.247.5 Sat, 01 Sep 2012, 17:32:02

G ITAL CARDIOL | VOL 13 | APRILE 2012302
B TOMBERLI ET AL
caso, che esprime il precario equilibrio emodinamico di questipazienti durante l’esercizio fisico. Sul piano clinico, l’esaspera-ta espressione fenotipica e fisiopatologica della malattia si tra-duce nella comparsa di una sintomatologia severa, in partico-lare angina e dispnea da sforzo, causati dal notevole aumentodelle pressioni di riempimento, dall’assenza di cavità con ridu-zione della gittata sistolica e dalla dis funzione microvascolare.I sintomi, limitanti e difficili da trattare, si collocano nel conte-sto di un progressivo rimodellamento del VS, caratterizzato dagradi crescenti di fibrosi alla risonanza magnetica cardiaca3. Al-cuni studi italiani hanno dimostrato che un pattern di riempi-mento restrittivo del VS nella CMI è un fattore predittivo fortee indipendente per l’evoluzione verso la fase terminale e loscompenso refrattario4,11.
In casi di questa gravità una terapia farmacologica standard,basata su betabloccanti e calcioantagonisti, è raramente in gra-do di raggiungere un adeguato controllo dei sintomi. Inoltre, ipazienti con fisiopatologia restrittiva tendono a non tolleraredosi massimali di questi farmaci e sono notoriamente intolle-ranti ai diuretici. D’altra parte il trattamento chirurgico di que-ste forme è un territorio ancora inesplorato. Appaiono invecepromettenti i risultati ottenuti nella risoluzione chirurgica del-l’ostruzione medio-ventricolare nella CMI12,13, un intervento tec-nicamente complesso, che, se eseguito in centri altamente spe-cializzati, può portare ad ottimi risultati a distanza. Nel nostrocaso era di fatto presente la fisiopatologia dell’ostruzione me-dio-ventricolare (e cioè il contatto sistolico di papillari anomalicon il setto interventricolare), anche se la totale obliterazionedell’apice del VS non consentiva lo sviluppo di gradienti a que-sto livello. Il razionale per l’impiego di una miectomia nella pa-ziente presentata appariva quindi promettente, purché asso-ciato ad un gesto specificamente rivolto alla “creazione” di vo-lume intraventricolare. La difficoltà di ottenere tale risultatosenza interferire con la meccanica del VS è ben esemplificatadalla complicanza osservata nel breve termine: un eccessivo ri-modellamento della parete laterale, secondario alla miectomia,ha infatti prodotto una perdita di coaptazione dei lembi mitra-lici, dalle conseguenze potenzialmente gravi. Fortunatamente,il ricorso ad una terapia di resincronizzazione, anch’essa sug-gerita dalla letteratura7, ha permesso di correggere in buonaparte il problema. Il decorso postoperatorio della nostra pa-ziente pone tuttavia l’accento sulla necessità di approfondire lenostre conoscenze sulla fisiopatologia di questa condizione, perstandardizzare un intervento chirurgico risolutivo e privo di dan-ni collaterali. Le nuove possibilità offerte dall’imaging contem-poraneo14 possono consentirci oggi un tale risultato.
Razionale dell’utilizzo della ranolazina in pazienti con cardiomiopatia ipertroficaLe terapie farmacologiche impiegate nella CMI si basano su far-maci antichi, utilizzati in modo empirico, e privi di azioni speci-fiche sui meccanismi fisiopatologici peculiari della malattia2.
Terapie sperimentali, testate su modelli animali, hanno for-nito risultati promettenti soprattutto per quanto riguarda la pre-venzione dello sviluppo di ipertrofia nei soggetti con genotipopositivo, ma non esistono dati riguardo ad interventi poten-zialmente efficaci sui meccanismi molecolari della disfunzioneventricolare sinistra e della progressione di malattia. Un recen-te studio del gruppo di Oxford ha mostrato dei risultati inco-raggianti per quanto riguarda l’efficacia della perexilina sui sin-tomi e sulla limitazione funzionale nei pazienti con CMI. La pe-rexilina è un farmaco noto da molti anni che svolge un’azione
favorevole dal punto di vista metabolico, spostando il metabo-lismo cardiomiocitario dalla catena ossidativa alla glicolisi15-17.Tale farmaco risulta però poco maneggevole per una tossicitàepatica ed una neuropatia dose-dipendente, che rendono ne-cessaria la misurazione della concentrazione ematica del far-maco. Ne appare pertanto improbabile l’uso clinico su vastascala nel prossimo futuro. Lo studio rappresenta tuttavia un uti-le proof of concept, in quanto ha evidenziato il potenziale ef-fetto favorevole di farmaci metabolici nella CMI.
La ranolazina è un inibitore selettivo della corrente lenta delsodio, che ha dimostrato una notevole efficacia antischemicanei pazienti con malattia coronarica, senza interferire in ma-niera significativa sui parametri emodinamici, in quanto legataad una normalizzazione delle elevate concentrazioni di calciointracitoplasmatico tipica del cardiomiocita ischemico. Oltre al-l’effetto antischemico la ranolazina ha mostrato un promet-tente effetto antiaritmico e lusitropo, entrambi oggetto di stu-di clinici in corso8,9. Questi dati fanno ipotizzare un effetto fa-vorevole della ranolazina in una patologia come la CMI, carat-terizzata da elevata calcio-sensibilità miocardica ed elevati va-lori di calcio intracellulare. A conferma di ciò, dati preliminari ot-tenuti presso il laboratorio di farmacologia dell’Università di Fi-renze hanno dimostrato che la corrente lenta del sodio è iper -attivata nei cardiomiociti umani di pazienti con CMI, provo-cando un marcato allungamento del potenziale d’azione e lacomparsa di post-potenziali tardivi18. In tali cellule la ranolazi-na, a concentrazioni terapeutiche, è in grado di accorciare si-gnificativamente il potenziale d’azione e di produrre un nettomiglioramento del profilo elettrofisiologico, con scomparsa deipost-potenziali e riduzione del calcio intracitoplasmatico. A li-vello clinico tali effetti hanno prodotto nella nostra paziente unbuon controllo dei sintomi legati ad ischemia microvascolare.Tale riscontro, sebbene aneddotico, rappresenta un risultatopromettente per l’impiego del farmaco su più ampia scala inpazienti con CMI. Oltre all’azione antischemica, un possibile ef-fetto sulla disfunzione diastolica e sul rischio aritmico, suggeri-to dai dati in vitro, completano il profilo di un razionale estre-mamente forte per un impiego clinico della ranolazina in que-sto contesto.
Seguendo un approccio traslazionale fortemente invocatodalla comunità scientifica internazionale5, i dati preclinici otte-nuti a Firenze sono sfociati in uno studio multicentrico rando-mizzato europeo, volto a valutare l’effetto della ranolazina sucapacità funzionale, biomarcatori e parametri diastolici in pa-zienti con CMI sintomatica, attualmente in corso. Questo stu-dio rappresenta una delle primissime iniziative metodologica-mente solide in questo ambito. Dopo cinque decadi di empiri-smo appassionato la speranza è che stia iniziando anche per ipazienti con cardiomiopatie genetiche l’era di una medicina ba-sata sull’evidenza.
RIASSUNTO
La gestione dei sintomi nei pazienti con cardiomiopatia ipertrofica(CMI) associata a gradi marcati di ipertrofia, ridotte dimensioni ca-vitarie del ventricolo sinistro e fisiopatologia restrittiva, rappresen-ta una vera sfida per il cardiologo clinico. Le strategie terapeutichevengono rese ancor più complesse dalla mancanza di dati relativialle opzioni mediche e chirurgiche potenzialmente più idonee peri pazienti con CMI; soprattutto sul piano farmacologico, la malat-tia è a tutt’oggi da considerarsi orfana di opzioni specifiche e ba-sate sulle evidenze.
© Il Pensiero Scientifico Editore downloaded by BENEDETTA TOMBERLI IP 87.12.247.5 Sat, 01 Sep 2012, 17:32:02

303G ITAL CARDIOL | VOL 13 | APRILE 2012
CMI SINTOMATICA E RANOLAZINA
Presentiamo il caso di una giovane paziente affetta da CMI confisiopatologia restrittiva, sottoposta ad un intervento chirurgicodi miectomia estesa, con approccio transaortico e transapicale,ed a terapia di resincronizzazione cardiaca, con miglioramentosignificativo ma incompleto dei sintomi. Il successivo potenzia-mento della terapia con ranolazina, introdotta sulla base di da-
ti preclinici promettenti, ha portato ad un netto miglioramen-to. È in corso uno studio clinico multicentrico randomizzato pervalutare l’efficacia della ranolazina in pazienti sintomatici conCMI.
Parole chiave. Angina; Cardiomiopatia ipertrofica restrittiva; Iper-trofia massiva; Ranolazina.
1. Gersh BJ, Maron BJ, Bonow RO, et al.2011 ACCF/AHA guideline for the diagno-sis and treatment of hypertrophic car-diomyopathy: executive summary: a reportof the American College of CardiologyFoundation/American Heart AssociationTask Force on Practice Guidelines. Circula-tion 2011;124:2761-96.2. Maron BJ. Hypertrophic cardiomyopathy:a systematic review. JAMA 2002;287:1308-20.3. Biagini E, Spirito P, Rocchi G, et al. Prog-nostic implications of the Doppler restrictivefilling pattern in hypertrophic cardiomyopathy.Am J Cardiol 2009;104:1727-31.4. Pinamonti B, Di Lenarda A, Nucifora G,Gregori D, Perkan A, Sinagra G. Incrementalprognostic value of restrictive filling pattern inhypertrophic cardiomyopathy: a Dopplerechocardiographic study. Eur J Echocardiogr2008;9:466-71.5. Force T, Bonow RO, Houser SR, et al. Re-search priorities in hypertrophic cardiomyopa-thy: report of a Working Group of the Na-tional Heart, Lung, and Blood Institute. Circu-lation 2010;122:1130-3.6. Cecchi F, Olivotto I, Nistri S, Antoniucci D,Yacoub MH. Midventricular obstruction andclinical decision-making in obstructive hyper-trophic cardiomyopathy. Herz 2006;31:871-6.7. Kanzaki H, Bazaz R, Schwartzman D, DohiK, Sade LE, Gorcsan J 3rd. A mechanism for
immediate reduction in mitral regurgitationafter cardiac resynchronization therapy: in-sights from mechanical activation strain map-ping. J Am Coll Cardiol 2004;44:1619-25.8. Morrow DA, Scirica BM, Karwatowska-Prokopczuk E, Skene A, McCabe CH, Braun-wald E; MERLIN-TIMI 36 Investigators. Evalu-ation of a novel anti-ischemic agent in acutecoronary syndromes: design and rationale forthe Metabolic Efficiency with Ranolazine forLess Ischemia in Non-ST-elevation acute coro-nary syndromes (MERLIN)-TIMI 36 trial. AmHeart J 2006;151:1186.e1-9.9. Wilson SR, Scirica BM, Braunwald E, et al.Efficacy of ranolazine in patients with chron-ic angina: observations from the randomized,double-blind, placebo-controlled MERLIN-TIMI (Metabolic Efficiency With Ranolazinefor Less Ischemia in Non-ST-Segment Eleva-tion Acute Coronary Syndromes) 36 Trial. JAm Coll Cardiol 2009;53:1510-6.10. Girolami F, Ho CY, Semsarian C, et al.Clinical features and outcome of hypertrophiccardiomyopathy associated with triple sar-comere protein gene mutations. J Am CollCardiol 2010;55:1444-53.11. Melacini P, Basso C, Angelini A, et al.Clinicopathological profiles of progressiveheart failure in hypertrophic cardiomyopathy.Eur Heart J 2010;31:2111-23.12. Schaff HV, Brown ML, Dearani JA, et al.Apical myectomy: a new surgical technique
for management of severely symptomatic pa-tients with apical hypertrophic cardiomyopa-thy. J Thorac Cardiovasc Surg 2010;139:634-40.13. Dearani JA, Ommen SR, Gersh BJ, SchaffHV, Danielson GK. Surgery insight: Septalmyectomy for obstructive hypertrophic car-diomyopathy - the Mayo Clinic experience. NatClin Pract Cardiovasc Med 2007;4:503-12.14. Maron MS. Clinical utility of cardiovascu-lar magnetic resonance in hypertrophic car-diomyopathy. J Cardiovasc Magn Reson 2012;14:13.15. Marian AJ. Experimental therapies in hy-pertrophic cardiomyopathy. J CardiovascTransl Res 2009;2:483-92.16. Abozguia K, Elliott P, McKenna W, et al.Metabolic modulator perhexiline correctsenergy deficiency and improves exercise ca-pacity in symptomatic hypertrophic car-diomyopathy. Circulation 2010;122:1562-9.17. Horowitz JD, Chirkov YY. Perhexiline andhypertrophic cardiomyopathy: a new horizonfor metabolic modulation. Circulation 2010;122:1547-9.18. Olivotto I, Coppini R, Ferrantini C, et al. Atranslational approach to treatment of hyper-trophic cardiomyopathy: pre-clinical rationaleand design of a prospective randomized pilottrial with ranolazine [abstract]. G Ital Cardiol2011;12(12 Suppl 3):e75.
BIBLIOGRAFIA
© Il Pensiero Scientifico Editore downloaded by BENEDETTA TOMBERLI IP 87.12.247.5 Sat, 01 Sep 2012, 17:32:02

OPEN ACCESS
http://dx.doi.org/10.5339/gcsp.2012.4
Published: 3 July 2012c© 2012 Cecchi, Tomberli &Olivotto, licensee BloomsburyQatar Foundation Journals. This isan open access article distributedunder the terms of the CreativeCommonsAttribution-NonCommercial licenseCC BY-NC 3.0 which permitsunrestricted non-commercial use,distribution and reproduction inany medium, provided the originalwork is properly cited.
Review article
Clinical and molecular classificationof cardiomyopathiesFranco Cecchi*, Benedetta Tomberli, Iacopo Olivotto
Referral Center for Myocardial Diseases,Careggi University Hospital, Florence,Italy*Email: [email protected]
ABSTRACTThe term ‘‘cardiomyopathies’’ was used for the first time 55 years ago, in 1957. Since thenawareness and knowledge of this important and complex group of heart muscle diseases haveimproved substantially. Over these past five decades a large number of definitions, nomenclature andschemes, have been advanced by experts and consensus panel, which reflect the fast and continuedadvance of the scientific understanding in the field.Cardiomyopathies are a heterogeneous group of inherited myocardial diseases, which represent animportant cause of disability and adverse outcome. Although considered rare diseases, the overallestimated prevalence of all cardiomyopathies is at least 3% in the general population worldwide.Furthermore, their recognition is increasing due to advances in imaging techniques and greaterawareness in both the public and medical community.Cardiomyopathies represent an ideal translational model of integration between basic and clinicalsciences. A multidisciplinary approach is therefore essential in order to ensure their correct diagnosisand management.In the present work, we aim to provide a concise overview of the historical background, genetic andphenotypic spectrum and evolving concepts leading to the various attempts of cardiomyopathyclassifications produced over the decades.
Keywords: classification, cardiomyopathies, myocardial disease
Cite this article as: Cecchi F, Tomberli B, Olivotto I. Clinical and molecular classification ofcardiomyopathies, Global Cardiology Science and Practice 2012:4http://dx.doi.org/10.5339/gcsp.2012.4

Page 2 of 11Cecchi, Tomberli & Olivotto. Global Cardiology Science and Practice 2012:4
INTRODUCTIONCardiomyopathies (CM) are a fascinating group of myocardial diseases, which constitute an importantcause of disability and adverse outcome due to heart failure or sudden and unexpected death. Theirrecognition is increasing due to advances in imaging techniques and greater awareness in themedical community, although the majority of patients are still likely to be undiagnosed ormisdiagnosed with more prevalent cardiac conditions. Population cross-sectional studies show thatthe overall estimated prevalence of all cardiomyopathies is at least 3% in the general populationworldwide. They are often inherited heart muscle diseases, generally with an autosomic dominant,more rarely recessive or X-linked transmission. As a variety of gene abnormalities are identified as thecause of cardiomyopathies, the need for a close cooperation among clinicians, geneticists andmolecular biologists, in addition to imaging experts, pathologists, neurologists, nephrologists andpaediatricians is well recognized: a multidisciplinary approach is essential in order to ensure theircorrect diagnosis and management. Furthermore, cardiomyopathies represent an ideal translationalmodel of integration between basic and clinical sciences. In the present work, we aim to provide aconcise overview of the historical background, genetic and phenotypic spectrum and evolvingconcepts leading to the various attempts of cardiomyopathies classifications produced by expertsover the decades.
HISTORYThe term ‘cardiomyopathy ’ was first used in 1957 by Brigden, who described a group of uncommon,non-coronary myocardial diseases [1]. In 1961 Goodwin defined cardiomyopathies as ‘‘myocardialdiseases of unknown cause’’ [2]. He described three different entities, namely ‘‘dilated, hypertrophicand restrictive’’, terms which are still in use today. In the 70s, the expanding clinical use ofnon-invasive imaging, such as m-mode and 2D echocardiography, allowed cardiologists andinternists to easily measure left ventricular (LV) wall thickness, cavity dimension and systolic function.Cardiomyopathies began to be recognized with increasing frequency in different populations. In anattempt to provide a useful intellectual framework for clinicians involved in the care of these patients,the first classification of cardiomyopathies was published in 1980, by the World Health Organization(WHO) and International Society and Federation of Cardiology (ISFC), and included the threesubgroups proposed by Goodwin [3]. The definition of ‘‘myocardial diseases of unknown cause’’ wasmaintained to define cardiomyopathies, which were distinguished from ‘‘specific heart musclediseases’’, the latter comprising heart diseases with similar phenotypes, but due to an identifiablecause.In the last 30 years, intensive genetic investigation carried out with linkage analysis on large
affected families lead to major breakthroughs in the identification of genes associated with familialcardiomyopathies [4,5]. Meanwhile, new nosologic entities were described and the new revision ofthe classification was carried out in 1996 by the WHO and ISFC [6].Representing a major advancement, both ‘‘arrhythmogenic right ventricular dysplasia’’ (with the
inappropriate term ‘‘dysplasia’’ later changed to ‘‘cardiomyopathy’’) and a group of ‘‘unclassifiedcardiomyopathies’’, defined as ‘‘those that do not fit in any group’’, were added to the three originalsubgroups. The definition of cardiomyopathy was changed to ‘‘diseases of the myocardiumassociated with myocardial dysfunction’’. Moreover, three additional subgroups termed‘‘hypertensive’’, ‘‘valvular’’ and ‘‘ischemic’’ cardiomyopathies were – somewhat confusingly – addedto the group of ‘‘specific heart muscle diseases’’ in order to resolve a terminology controversybetween US and European experts [6]. These were defined as cardiac conditions characterized by thepresence of hypertension, coronary or valvular disease, in a degree that would not explain themagnitude of LV dysfunction observed. Nevertheless, a substantial difference in terminology persistedon the two sides of the Atlantic, reflecting the refusal of these fine distinctions by US experts [7].In 2006, an American Historical Association (AHA) panel of experts published a scientific
statement on the ‘‘Contemporary classification and definitions of Cardiomyopathies’’ [7]. Theyproposed a novel approach, by which ‘‘Primary ’’ cardiomyopathies were defined as those ‘‘involvingonly the heart ’’, as opposed to the ‘‘secondary ’’, characterized by a ‘‘generalized multiorganinvolvement ’’. Primary cardiomyopathies for the first time also included ‘‘ion channel diseases’’ andwere differentiated in three subgroups based on their etiology as ‘‘genetic, mixed and acquired ’’ [Fig. 1]. The radical shift from a phenotypic to an etiological classification, as well as the inclusion ofion channel diseases among cardiomyopathies, although proposed to guide future research rather

Page 3 of 11Cecchi, Tomberli & Olivotto. Global Cardiology Science and Practice 2012:4
PRIMARY CARDIOMYOPATHIES(Predominantly involving the heart)
Genetic Mixed Acquired
HCM
ARVC/D
LVNC
PRKAG2
Danon
Glycogenstorage
Conduction defects
Mitochondrial myopathies
Ion channel disorders
LQTS Brugada
AsianSUNDS
SQTS CVPT
DCM Restrictive(non-hypertrophied
and non-dilated)
Inflammatory (myocarditis)
Stress provoked("takotsubo")
Peripartum
Tachycardia - induced
Infants of insulin-dependentdiabetic mothers
Figure 1. Classification of the cardiomyopathies – American Heart Association.Modified from [7].
than to be employed in the clinical arena, sparked a passionate transatlantic debate, culminating in athorough reworking of the original 1995 classification by the European Society of Cardiology (ESC)Working Group on Myocardial and Pericardial diseases, in 2008 [8]. Intrinsically faithful to theconcept of classifying cardiomyopathies based on phenotype, the 2008 classification aimed toprovide a simple operational framework for the medical community, which might have a direct impactin diagnosing and managing these complex diseases. Each of the time-honoured categories dilated,hypertrophic, restrictive and arrhythmogenic right ventricular were maintained, divided into familialand non-familial to replace the pre-genetic era concept of ‘‘unknown etiology’’. Furthermore, only twonew entities were included into the unclassified group, while the confusing ‘‘hypertensive’’, ‘‘valvular’’and ‘‘ischemic’’ categories were removed.
CURRENT CLASSIFICATION OF CARDIOMYOPATHIES (ESC WORKING GROUP ON MYOCARDIALAND PERICARDIAL DISEASES)The panel felt the proposed classification should be useful for everyday clinical practice. The verydefinition of cardiomyopathies was changed, from ‘‘myocardial diseases of unknown cause’’, to‘‘myocardial disorders in which the heart muscle is structurally and functionally abnormal in theabsence of coronary artery disease, hypertension, valvular or congenital heart disease sufficient tocause the observed myocardial abnormality’’ [8]. Ion channel diseases were excluded, despite theirgenetic nature, in view of their lack of a structural cardiac phenotype affecting the heart muscle.Because cardiomyopathies are diagnosed based on clinical examination and imaging, the four
classical morphological subgroups of ‘‘hypertrophic, dilated, restrictive, arrhytmogenic ’’ weremaintained, with a fifth subgroup of ‘‘unclassified ’’, comprising the recently described ‘‘LVnon-compaction’’ and ‘‘Takotsubo cardiomyopathy’’ [Fig. 2].Each category was subdivided in a familial and non-familial subset, with the latter including all
known causes that might be responsible for that phenotype. A list of potential genetic andnon-genetic causes is provided for each subgroup of cardiomyopathies [Tables 1–5]. The precise

Page 4 of 11Cecchi, Tomberli & Olivotto. Global Cardiology Science and Practice 2012:4
Figure 2. Classification of the cardiomyopathies – European Society of Cardiology.Modified from [8].
Table 1. Hypertrophic cardiomyopathy.FAMILIAL Unknown gene
Sarcomeric protein disease ß myosin heavy chain, Cardiac myosin binding protein C Cardiac troponin I,T and C, a-tropomyosin, Essential myosin light chain, Regulatory myosin light chain,Cardiac actin, a- myosin heavy chain, Titin
Glycogen storage diseases (e.g. GSD II (Pompe’s disease); GSD III (Forbes’ disease),AMP kinase (WPW, HCM, conduction disease)
Lysosomal storage diseases (e.g. Anderson-Fabry disease, Hurler’s syndrome)Disorders of Fatty Acid Metabolism Carnitine, Phosphorylase B kinase deficiencyMitochondrial cytopathies (e.g. MELAS, MERFF, LHON)Syndromic HCM Noonan’s syndrome, LEOPARD syndrome, Friedreich’s ataxia,
Beckwith-Wiedermann syndrome; Swyer’s syndrome (pure gonadal dysgenesis)Other: Muscle LIM protein Phospholamban promoter Familial Amyloid
NON-FAMILIALObesity; Infants of diabetic mothers; Athletic training; Amyloid (AL / prealbumin)
Table 2. Dilated cardiomyopathy.FAMILIAL, unknown gene
Sarcomeric protein mutations (see HCM)Z band : Cypher/Zasp, Muscle LIM protein, TCAPCytoskeletal genes: Dystrophin, Desmin, Metavinculin, Sarcoglycan complex, CRYAB, EpicardinNuclear membrane: Lamin A/C, EmerinMildly dilated CMIntercalated disc protein mutations (see ARVC)Mitochondrial cytopathy
NON FAMILIALMyocarditis (infective/toxic/immune)Kawasaki diseaseEosinophilic (Churg Strauss syndrome)Viral persistenceDrugs, Pregnancy, EndocrineNutritional: thiamine, carnitine, selenium, hypophosphataemia, hypocalcemia.AlcoholTachycardiomyopathy
identification of the disease etiology has obvious clinical implications, by virtue of its direct impact tototally different management. For example, amyloidosis, Anderson Fabry diseases and glycogenstorage diseases may be diagnosed as hypertrophic cardiomyopathy (HCM); yet their treatment varieswidely. Of note, the inclusion of amyloidosis in this classification was widely debated [9]. Substantialdoubts also regarded takotsubo, a disease that is generally transient, has no proven inherited cause,and appear related to regional myocardial hypoperfusion rather than to heart muscle abnormalities.Ultimately, both were included as this was felt to be conceptually useful in clinical practice.Finally, a stepwise approach was proposed for diagnostic work-up. Step one is the identification of
cardiomyopathies on the basis of the presenting morphologic features. Following diagnosis in theproband, a comprehensive family screening by ECG and echocardiography should be offered tofirst-degree relatives, in order to assess whether there is a familiar transmission. The third step

Page 5 of 11Cecchi, Tomberli & Olivotto. Global Cardiology Science and Practice 2012:4
Table 3. Restrictive cardiomyopathy.FAMILIAL, unknown gene
Sarcomeric protein mutations: Troponin I (RCM+/− HCM), Essential myosin light chainFamilial Amyloidosis Transthyretin (RCM+ neuropathy)
Apolipoprotein (RCM+ nephropathy)DesminopathyPseuxanthoma elasticumHaemochromatosisAnderson-Fabry diseaseGlycogen storage diseaseEndomyocardial fibrosis (Familial) (Fusion FIP1-like-1 / PDGFRA genes)
NON FAMILIALAmyloid (AL/prealbumin)SclerodermaEndomyocardial fibrosis
Hypereosinophilic syndrome, Idiopathic chromosomal causeDrugs: serotonin, methysergide, ergotamine, mercurial agents, busulfan, anthracyclinesCarcinoid heart disease, Metastatic cancers, Radiation
Table 4. Arrhythmogenic right ventricular cardiomyopathy.FAMILIAL, unknown gene
Intercalated disc protein mutations: Plakoglobin,DesmoplakinPlakophilin 2Desmoglein 2Desmocollin 2
Cardiac ryanodine receptor (RyR2)Transforming growth factor-β3 (TGFβ3)
NON FAMILIALInflammation?
Table 5. Unclassified cardiomyopathies.FAMILIAL unknown gene
Left ventricular non-compaction:Barth SyndromeLamin A/CZASPa-dystrobrevin
NON FAMILIALTakotsubo cardiomyopathy
consists in the search for the specific cause of the disease, with the help of genetic analysis, metabolicand biochemical laboratory tests, additional imaging and, in selected instances, myocardial biopsy.
ROLE OF GENETIC TESTINGMany cardiomyopathies are believed to derive from the interaction between one or more geneticmutations, often unidentified modifier genes and environmental factors [10]. When genetic analysisis performed in candidate genes, the probability of identifying the pathogenic gene mutation is in therange of 40–60%, for patients with HCM, with approximately 5% of complex mutations [11,12].Results for dilated cardiomyopathy (DCM), restrictive cardiomyopathy (RCM) and isolated LVnon-compaction are considerably less rewarding [10], although the advent of next generation,genome-wide techniques may increase the yield substantially, as recent data on titin in DCMsuggests [13].In the meantime, however, cross-talk between geneticists and clinicians has developed slowly, with
modalities of interaction and degree of mutual comprehension that vary wildly in various settings. Inmany institutions, particularly in the US, a geneticist is not available on-site, and genetic testing isoften performed remotely via private companies [14]. In addition, clinicians often question theclinical utility of genetic testing in cardiomyopathy patients and their families. The apparent lack ofpractical benefit, in the face of considerable costs, has long hindered large-scale diffusion of genetictesting, and still accounts for understandable (but not always justifiable) resistance by the clinician.An indisputable benefit of systematic genetic testing lies in the cross-fertilization between

Page 6 of 11Cecchi, Tomberli & Olivotto. Global Cardiology Science and Practice 2012:4
cardiologists and geneticists. The former, generally show limited expertise and propensity atinvestigating the hereditary nature of cardiac diseases, and at identifying complex, syndromicphenotypes associated with cardiomyopathies (e.g. Noonan’s, Leopard’s, mitochondrial disease andAnderson Fabry) [15–18]. Standard protocols for genetic testing routinely include pre-testcounselling by a multidisciplinary team involving clinical geneticists [15]. This is a valuable momentfor reciprocal education among professionals, ultimately benefitting a wide spectrum of patients withrare conditions.
HYPERTROPHIC CARDIOMYOPATHYHCM is a genetic disease characterized by unexplained LV hypertrophy, associated with non-dilatedventricular chambers, in the absence of another cardiac or systemic disease capable of producingthat degree of hypertrophy [Fig. 3]. HCM is diagnosed by a maximal LV wall thickness greater than15 mm, based on echocardiography (ECHO) or cardiac magnetic resonance (CMR) [19]. This value islowered to 13–14 mm, when family members are screened. In children, a wall thickness greater than2 standard deviations (SD) for age, sex or body size is considered diagnostic.
A B C
D E F
Figure 3. Hypertrophic cardiomyopathy. Echocardiographic and cardiac magnetic resonance images from a 17-year old female patient with HCM. Parasternal long and short axis views show severe LV thickness values (maxLV wall thickness 31 mm), with redundant mitral leaflets (panels A, B and D) and small cavity size. Apical 4chambers view shows massive hypertrophy of the septum and the antero-lateral wall (panels C and E). Imageof late gadolinium enhancement showing limited and nontransmural area of fibrosis of the IVS (panel F: blackarrow). Abbreviations: LV= left ventricle, RV= right ventricle IVS= inter-ventricular septum.
The distribution of hypertrophy is usually asymmetric and sometimes confined to one or two LVsegments. As a consequence, LV mass (measured by CMR) can be within the normal range. LV outflowtract obstruction is an important feature of HCM, and may be demonstrated in up to 70%patients [20]. Overall, the clinical course of patients with HCM is relatively benign, with an annualmortality rate of about 1%. Contrary to prior perceptions, the risk of sudden cardiac death is relativelylow [21], although still a major concern in young individuals and athletes. Furthermore, about half ofpatients show some degree of disease progression and functional limitation, with a small subset ofabout 5% developing the so-called end-stage HCM. Family screenings, following the introduction ofgenetic testing has led to the identification of genotype-positive phenotype-negative individuals, anovel category within the HCM spectrum, characterized by absence of LV hypertrophy, assessed byECG and ECHO [19].Sarcomeric gene mutations, often private, are the most frequent cause of HCM, accounting for
approximately 30–65% of probands in different cohorts [22]. In the remaining subset the geneticsubstrate is unknown. Furthermore, a small proportion of patients with the HCM phenotype areaffected by cardiofacial syndromes (e.g. Noonan, LEOPARD, Costello), neuromuscular diseases(e.g. Frederich’s ataxia), mitochondrial diseases [23], metabolic disorders of lysosomal storage

Page 7 of 11Cecchi, Tomberli & Olivotto. Global Cardiology Science and Practice 2012:4
diseases (i.e. Fabry, Pompe, Danon) [24]. These rare conditions sometimes exhibit an X-linked ratherthan the autosomal pattern of inheritance, usually observed in HCM [Table 1].
DILATED CARDIOMYOPATHYDCM is characterized by LV dilatation and global systolic dysfunction (EF < 50%), in the absence ofcoronary artery disease or other identifiable causes (such as systemic hypertension, valve disease,drugs, inflammatory heart diseases) capable of causing that magnitude of impairment [Table 2]. Infamilial DCM, screening of first-degree relatives will identify the disease in up to 50% [10]. As formany other cardiomyopathies, the prevalence of DCM is underestimated, because many patients mayhave a subclinical form of the disease which may be difficult to diagnose for the lack of symptoms.Familial and sporadic forms of DCM have similar morphological manifestation and clinical course[Fig. 4]. They are progressive diseases, with a prognosis that, although improved in the last decades,is usually poor due to heart failure, atrial and ventricular arrhythmias, stroke and sudden death [25].In patients with refractory heart failure, heart transplant represent the final option.
A B
C
E F
D
Figure 4. Dilated cardiomyopathy. Echocardiographic and cardiac magnetic resonance images from a 57-yearold female patient with DCM and normal coronary angiogram. Parasternal long axis view and CMR imagesshow dilated LV (panels A–B and E–F), with severe systolic dysfunction - EF = 21%; (panel C = diastole, panelD= systole). Abbreviations: LV= left ventricle, RV= right ventricle IVS= inter-ventricular septum.
The low yield of genetic testing for DCM (i.e. 30%) limits its clinical use. This is related to the largenumber of potentially disease-causing genes. Furthermore, genetic mutations are usually private andthe interpretation of the analysis results may be difficult [10]. As noted above, the advent of nextgeneration sequencing may radically change this scenario [13]. However some gene mutations, suchas Lamin A/C seem to carry a more adverse outcome, in particular for sudden death [26–28].
RESTRICTIVE CARDIOMYOPATHYRCM is defined by the presence of a restrictive LV physiology, with normal or more often reduceddiastolic/systolic volumes, normal wall thickness and systolic function, marked diastolic flow

Page 8 of 11Cecchi, Tomberli & Olivotto. Global Cardiology Science and Practice 2012:4
impairment and biatrial dilatation. RCM are rather uncommon, although their prevalence is stillunknown. Either Amyloid Light-chain (AL) amyloidosis or amyloidosis due to transthyretin genemutations with heart involvement, often cause RCM [Table 3] [9]. A striking subtype of disease withrestrictive physiology, endomyocardial fibrosis, endemic in areas of the African continent, has anunknown etiology and very poor prognosis [29]. Moreover a ‘‘restrictive phenotype’’ may be part ofthe clinical spectrum of end-stage HCM [30], and may occasionally originate as a primary, nonHCM-related phenotype from sarcomere gene mutations (generally in the thin filament protein codinggenes). RCM is usually associated with severe functional limitation, mainly related to the extremediastolic dysfunction, with reduced diastolic filling and stroke volume, and a poor prognosis [31].
ARRHYTHMOGENIC RIGHT VENTRICULAR CARDIOMYOPATHYARVC is characterized by fibrofatty replacement of the right ventricular myocardium and ventriculararrhythmias [32]. In the most common right-dominant form, structural changes may be absent orconfined to a localized region of the right ventricle (inflow and outflow tract, right ventricular apex,known as the ‘triangle of dysplasia’) at an early stage [Fig. 5]. Progression to more diffuse rightventricular disease and LV involvement (typically affecting the posterior lateral wall), associated withventricular systolic dysfunction, is common at later stages [33]. Ventricular arrhythmias are theclinical hallmark of the disease, but atrial fibrillation may also occur. The diagnosis of ARVC is oftenchallenging for the cardiologist, in particular during the early ‘concealed phase’, when individuals arestill asymptomatic. Predominant LV disease has also been recognized. New diagnostic criteria withhigher sensitivity and specificity have recently been published [32,34]. ARVC is generally a familialdisease with autosomal dominant inheritance but it may be recessive when associated with woollyhair and palmopalmar hyperkeratosis (eg, Naxos disease, Carvajal syndrome). Mutations indesmosomal and non-desmosomal genes have been identified, but interpretation of theirpathogenicity is often challenging in the affected individual [Table 4].
A
B
C
Figure 5. Arrhythmogenic right ventricular cardiomyopathy. 38 year old female with diagnosis of ARVC,resuscitated from out-of-hospital cardiac arrest. She has family history of ARVC (mother) and sudden death(brother, 28 year old). CMR images show clearly wall aneurysms within the so-called ‘‘triangle of dysplasia’’(panel A–C, white arrows: evident systolic bulging in infundibular, apical, and subtricuspid regions of the RV).Abbreviations: LV= left ventricle, RV= right ventricle.
UNCLASSIFIED CARDIOMYOPATHIESIsolated LV non-compaction (LVNC) is characterized by prominent LV trabeculae and deepinter-trabecular recesses, that can be associated with LV dilatation and systolic dysfunction [Fig. 6].LVNC is familial, with 25% of asymptomatic first-degree relatives having some echocardiographicabnormalities [Table 5]. Of note, this rather mysterious disease shows substantial phenotypic overlapwith other cardiomyopathies (in particular HCM and DCM, which often exhibit limited areas ofnon-compaction in the left ventricle), as well as a common genetic substrate [8]. Furthermore, LVNCmay be associated with congenital cardiac disorders (such as Ebstein’s anomaly or complex cyanotic

Page 9 of 11Cecchi, Tomberli & Olivotto. Global Cardiology Science and Practice 2012:4
A B
C D
E F
Figure 6. Unclassified cardiomyopathy. Isolated left ventricular non-compaction in a 45 year-old male, with mildsystolic dysfunction (EF 48%), ventricular arrhythmias and normal LV diameters. Multiple trabeculations andrecesses are evident, particularly in the apex and the free wall of the LV (panels A and C: apical 4 chambers view;panels B and D: apical 3 chambers view). CMR confirmed the diagnosis (panels E–F). Abbreviations: LV = leftventricle, RV= right ventricle IVS= inter-ventricular septum.
heart disease) and some neuromuscular diseases. Therefore, it is still debated whether isolated LVNCshould be considered a separate clinical and genetic entity, or a morphological trait shared by manydistinct cardiomyopathies. As a result of the difficult comprehension of this clinical entity, the realprevalence of LVNC and its outcome remain largely unknown.Takotsubo cardiomyopathy, also known as LV apical ballooning or stress-induced cardiomyopathy,
is characterized by transient regional systolic dysfunction involving the apex and/or mid-ventricle inthe absence of obstructive coronary artery disease on angiogram [8]. The condition is reported allover the world, and most reported cases occur in post-menopausal women following physical orpsychological stress, but it has been described also in patients with intracranial haemorrhage orother acute cerebral accidents (so-called ‘‘neurogenic myocardial stunning’’). Typically, takotsubocardiomyopathy has a sudden onset, with chest pain, diffuse T-wave inversion and mild cardiacenzyme elevation. Symptoms are often preceded by emotional or physical stress. If the patientsurvives the acute phase of disease, the prognosis is almost invariably favourable, with anormalization of LV function over a period of days to weeks; recurrence is possible, but rare.
LIMITATIONS OF CURRENT CLASSIFICATIONS AND NOVEL PERSPECTIVESAs more families with cardiomyopathies are genotyped, and new diseases are being described, theparadigm ‘‘one gene, one disease’’ appears no longer sustainable. The same mutation can beexpressed at a different age and give rise to hugely different phenotypes within the same family, dueto environmental factors and modifier genes. Different phenotype patterns may originate from thesame genetic substrates, in a spectrum encompassing HCM, DCM, RCM and LVNC (all associated withsarcomere genes), or ARVC/DCM (associated with desmosomal genes).
benedetta
Sticky Note

Page 10 of 11Cecchi, Tomberli & Olivotto. Global Cardiology Science and Practice 2012:4
Furthermore, the recent introduction of next generation sequencing has started what promises tobe a revolution in molecular diagnostics, allowing rapid and affordable testing of hundreds of genes,or even whole genomes. As an example, a wide range of truncating gene mutations encoding Titin, avery large cytoskeleton gene which could not be assessed by traditional sequencing techniques, hasrecently been discovered to represent a prevalent cause of familial DCM, up to 25% [13].In the near future, the list of causative genes will therefore likely require an update. This should
ideally become an ongoing process, under the auspices of the ESC Working Group on Myocardial andPericardial Diseases and AHA experts. The focus for researchers will necessarily shift from analyzingsingle mutations in candidate genes, to interpreting the hundreds of variants of unknown significancein putative causative as well as modifier genes, requiring entirely new skills and significant interactionwith biophysicists and computer scientists. At present, and in the foreseeable future, however, clinicalclassifications of cardiomyopathies based on clinical presentation and morphological criteriarepresent an important tool for clinicians involved with these complex diseases. While calling forconstant improvement and update in the light of advances provided by imaging genetics and basicscience, individual patient phenotypes continue to represent the core of any classification in clinicalmedicine, something that has not changed with time.
LIST OF ABBREVIATIONS:ESC: European Society of CardiologyAHA: American Heart AssociationECHO: echocardiographyCMR: Cardiac magnetic resonanceLV: left ventricularEF: ejection fractionHCM: hypertrophic cardiomyopathyDCM: dilated cardiomyopathyRCM: restrictive cardiomyopathyARVC: arrhythmogenic right ventricular cardiomyopathyLVNC: left ventricular non compaction
COMPETING INTERESTSThe authors have no competing interests.
ACKNOWLEDGMENTS:This work was supported by Ministero Istruzione Università e Ricerca (PRIN), and European Union(STREP Project 241577 ‘‘BIG HEART,’’ 7th European Framework Program).
References[1] Brigden W. Uncommon myocardial diseases: the non-coronary cardiomyopathies. Lancet. 1957 Dec.
21;273:7008, 1243–1249.[2] Goodwin JF, Gordon H, Hollman A and Bishop MB. Clinical aspects of cardiomyopathy. Br Med J. 1961 Jan.
14;1:5219, 69–79.[3] Report of the WHO/ISFC Task Force on the definition and classification of cardiomyopathies. Br Heart J. 1980;44,
672–673.[4] Watkins H, Ashrafian H and Redwood C. Inherited cardiomyopathies. N Engl J Med. 2011 Apr. 28;364:17,
1643–1656.[5] Bos JM, Towbin JA and Ackerman MJ. Diagnostic, prognostic, and therapeutic implications of genetic testing for
hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009 July 14;54:3, 201–211.[6] Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O’Connell J, Olsen E, Thiene G and Goodwin J. Report
of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on thedefinition and classification of cardiomyopathies. Circulation. 1996;93:841–842.
[7] Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman C andYoung JB. Contemporary definitions and classification of the cardiomyopathies. An American Heart Associationscientific statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Qualityof Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary WorkingGroups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816.
[8] Elliott P, Anderson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kuhl U, Maisch B, McKenna WJ,Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L and Keren A. Classification of cardiomyopathies: aposition statement from the European working group on myocardial and pericardial diseases. Eur Heart J.2008;29:270–276.

Page 11 of 11Cecchi, Tomberli & Olivotto. Global Cardiology Science and Practice 2012:4
[9] Rapezzi C, Quarta CC, Riva L, Longhi S, Gallelli I, Lorenzini M, Ciliberti P, Biagini E, Salvi F andBranzi A. Transthyretin-related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol. 2010 July;7:7,398–408.
[10] Jacoby D and McKenna WJ. Genetics of inherited cardiomyopathy. Eur Heart J. 2012 Feb.;33:3, 296–304.[11] Olivotto I, Girolami F and Ackerman MJ et al. Myofilament protein gene mutation screening and outcome of
patients with hypertrophic cardiomyopathy. Mayo Clin Proc. 2008 June;83:6, 630–638.[12] Friedrich FW and Carrier L. Genetics of hypertrophic and dilated cardiomyopathy. Curr Pharm Biotechnol. 2012
Jan. 20;[Epub ahead of print].[13] Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B,
Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM,Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG andSeidman CE. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012 Feb. 16;366:7, 619–628.
[14] Olivotto I, Kassem HS and Girolami F. Genetic testing for hypertrophic cardiomyopathy: ongoing voyage fromexploration to clinical exploitation. DOI: 10.4081. Cardiogenetics. 2011.e3 Published: 2011-07-05 10:15:10.
[15] Charron P, Arad M, Arbustini E, Basso C, Bilinska Z, Elliott P, Helio T, Keren A, McKenna WJ, Monserrat L,Pankuweit S, Perrot A, Rapezzi C, Ristic A, Seggewiss H, van Langen I and Tavazzi L. Genetic counselling andtesting in cardiomyopathies: a position statement of the European Society of Cardiology Working Group onMyocardial and Pericardial Diseases. Eur Heart J. 2010;31:2715–2726.
[16] Wang L, Seidman JG and Seidman CE. Narrative review: harnessing molecular genetics for the diagnosis andmanagement of hypertrophic cardiomyopathy. Ann Intern Med. 2010;152:513–520.
[17] Hershberger RE, Lindenfeld J and Mestroni L et al. Genetic evaluation of cardiomyopathy- a Heart Failure Societyof America practice guideline. J Card Fail. 2009;15:83–97.
[18] van Langen IM, Birnie E and Leschot NJ et al. Genetic knowledge and counselling skills of Dutch cardiologists:sufficient for thegenomics era?. Eur Heart J. 2003;24:560–566.
[19] Gersh BJ, Maron BJ, Bonow RO, Dearani A, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H,Seidman CE, Towbin JA, Udelson JE and Yancy CW. ACCF/AHA guideline for the diagnosis and treatment ofhypertrophic cardiomyopathy: executive summary: a report of the American College of CardiologyFoundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124:2761–2796.
[20] Maron MS, Olivotto I and Zenovich AG et al. Hypertrophic cardiomyopathy is predominantly a disease of leftventricular outflow tract obstruction. Circulation. 2006;114:21, 2232–2239.
[21] Maron BJ. Contemporary insights and strategies for risk stratification and prevention of sudden death inhypertrophic cardiomyopathy. Circulation. 2010;121:445–456.
[22] Girolami F, Ho CY, Semsarian C, Baldi M, Will ML, Baldini K, Torricelli F, Yeates L, Cecchi F, Ackerman MJ andOlivotto I. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere proteingene mutations. J Am Coll Cardiol. 2010 Apr 6;55:14, 1444–1453.
[23] Schapira AH. Mitochondrial diseases. Lancet. 2012 Apr 4;Epub ahead of print.[24] Keren A, Syrris P and McKenna W. Hypertrophic cardiomyopathy: the genetic determinants of clinical disease
expression. Nat Clin Pract Cardiovasc Med. Mar. 2008;5:3, 158–168.[25] Merlo M, Pyxaras SA, Pinamonti B, Barbati G, Di Lenarda A and Sinagra G. Prevalence and prognostic significance
of left ventricular reverse remodeling in dilated cardiomyopathy receiving tailored medical treatment. J Am CollCardiol. 2011;57:13, 1468–1476.
[26] Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Peterson A, Li D, Jakobs P, Litt M, Porter CB, Rahko PSand Hershberger RE. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familialdilated cardiomyopathy. Am Heart J. 2008;156:161–169.
[27] van Berlo JH, de Voogt WG and van der Kooi AJ et al. Meta-analysis of clinical characteristics of 299 carriers ofLMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death?. J Mol Med. 2005;83:79–83.
[28] Meune C, Van Berlo JH, Anselme F, Bonne G, Pinto YM and Duboc D. Primary prevention of sudden death inpatients with lamin A/C gene mutations. N Engl J Med. 2006;354:209–210.
[29] Mocumbi AO, Yacoub S and Yacoub MH. Neglected tropical cardiomyopathies: II. Endomyocardial fibrosis:myocardial disease. Heart. 2008 Mar.;94:3, 384–390.
[30] Kubo T, Gimeno JR and Bahl A et al. Prevalence, clinical significance, and genetic basis of hypertrophiccardiomyopathy with restrictive phenotype. Journal of the American College of Cardiology. June 2007;49:25,2419–2426.
[31] Zangwill SD, Naftel D, L’Ecuyer T, Rosenthal D, Robinson B, Kirklin JK, Stendahl G and Dipchand AI. Pediatric HeartTransplant Study Investigators. Outcomes of children with restrictive cardiomyopathy listed for heart transplant: amulti-institutional study. J Heart Lung Transplant. 2009 Dec;28:12, 1335–1340.
[32] Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP,Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffitz JE, Sanborn DM, Steinberg JS, Tandri H,Thiene G, Towbin JA, Tsatsopoulou A, Wichter T and Zareba W. Diagnosis of arrhythmogenic right ventricularcardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J. 2010;31:806–814.
[33] Basso C, Bauce B, Corrado D and Thiene G. Pathophysiology of arrhythmogenic cardiomyopathy. Nat Rev Cardiol.2011 Nov 29;9:4, 223–233.
[34] Ackerman MJ, Priori SG and Willems S et al. HRS/EHRA Expert Consensus Statement on the State of GeneticTesting for Channelopathies and Cardiomyopathies. Europace. 2011 Aug;13:8, 1077–109.

486
Rilevanza clinica del test genetico nella cardiomiopatia ipertrofica
Francesca Girolami1, Sara Bardi1, Laura Berti1, Franco Cecchi2, Eleonora Servettini2, Benedetta Tomberli2,Francesca Torricelli1, Iacopo Olivotto2
1Struttura Organizzativa Dipartimentale Diagnostica Genetica, Azienda Ospedaliero-Universitaria Careggi, Firenze;2Centro di Riferimento per le Cardiomiopatie, Firenze.
Pervenuto il 15 luglio 2011.
Riassunto. Sono trascorsi più di 20 anni dalla scoperta delprimo gene sarcomerico associato all’insorgenza della car-diomiopatia Ipertrofica (CMI). Durante tale periodo lo svi-lupparsi di moderne tecnologie per il sequenziamento delgenoma umano, unitamente alla crescita delle conoscenzesulla genetica, hanno reso disponibile un test genetico spe-cifico per la CMI effettuabile a scopo diagnostico e diffusoin molti paesi. Nel frattempo, per l’intensificarsi dello scam-bio reciproco di conoscenze tra cardiologi e genetisti, èemersa la necessità di discutere sulla vera utilità clinica deltest genetico. La prescrizione del test ha incontrato spessodifficoltà da parte del cardiologo perché ritenuto di scarsautilità nella pratica clinica anche a fronte del costo elevato.In realtà tale resistenza è in contrasto con una serie di evi-denze che supportano la necessità del test genetico nellagestione del paziente con CMI. Tali ragioni sono passate inrassegna nel presente lavoro e spaziano dall’importanzadell’identificazione dei genotipi complessi perché progno-stici di un decorso più sfavorevole della malattia, alla possi-bilità di effettuare diagnosi nei casi dubbi mediante l’iden-tificazione della mutazione, fino a porre diagnosi differen-ziale con le forme non sarcomeriche. In ultimo, ma non perimportanza, vale la pena sottolineare che il test geneticopermette di stabilire la natura ereditaria della malattia, ren-dendo possibile la diagnosi talvolta presintomatica nei fa-miliari del probando.
Parole chiave. Cardiomiopatia ipertrofica, consulenza ge-netica, mutazioni sarcomeriche, test genetico specifico perla cardiomiopatia ipertrofica.
Clinical relevance of genetic testing in hypertrophic cardiomy-opathy.
Summary. More than two decades have elapsed since thediscovery that sarcomere gene defects cause familial hy-pertrophic cardiomyopathy (HCM). Since then, genetic test-ing in HCM has developed, and become an important tool inclinical practice for diagnosis and prognosis overall in theWestern countries. However its practical benefits are still un-derstimated and clinicians often question about cost-effec-tiveness of genic testing in HCM patients and their families.This resistance is in contrast with considerable evidence sup-porting the role of genetics in tailoring management forHCM patients. Several current clinical uses of genetic test-ing in HCM, ranging from diagnosis in ambiguous situations,identification of disease phenocopies and HCM complexgenotypes and confirmation of inherited disease in familymembers are reviewed.In the near future it is hoped that next generation sequenc-ing will provide further diffusion of genetic testing in HCMand improvement in care.
Key words. Genetic counselling, genetic test in hyper-trophic cardiomyopathy, sarcomeric mutation.
venne descritta come entità clinica a sé stante,denominata «ostruzione funzionale del ventrico-lo sinistro» e «ipertrofia settale asimmetrica»grazie alle osservazioni pubblicate dal patologoTeare. Nel 1957, in seguito ad esami autoptici ef-fettuati su nove pazienti (di cui otto morti dimorte improvvisa), Teare individuò una spiccataipertrofia del muscolo cardiaco e ne intuì la na-tura familiare2. Poco dopo, nel 1959, venne effet-tuata per la prima volta una diagnosi di CMI invivo, presso il Clinical Center of National Insti-tutes of Health, negli USA3. Negli anni ’70, l’in-troduzione della metodica ecocardiografica e isuoi continui progressi hanno consentito di ca-ratterizzare con precisione il grado e la sede del-
Introduzione
La cardiomiopatia ipertrofica (CMI) è la piùfrequente malattia genetica del muscolo cardia-co, con una prevalenza di 1:500 nella popolazio-ne generale, pari a oltre 100.000 pazienti stima-ti in Italia. La patologia è caratterizzata daespressione morfologica e decorso clinico etero-genei1, e si identifica per la presenza di ipertro-fia ventricolare sinistra, generalmente asimme-trica, in assenza di malattie cardiache o sistemi-che in grado di produrre tale ipertrofia. Nel 1869,Liouville e Hallopeau descrissero per la primavolta quadri di ipertrofia asimmetrica del settointerventricolare, ma solo negli anni ’50 la CMI
Rassegna Recenti Prog Med 2011; 102: 486-493

F. Girolami et al.: Rilevanza clinica del test genetico nella cardiomiopatia ipertrofica 487
l’ipertrofia, di visualizzare in tempo reale il con-tatto sistolico del lembo anteriore della mitrale(SAM) con il setto interventricolare, e di misura-re i gradienti sistolici intraventricolari e i flussiintracavitari4-7. L’ecocardiografia ha inoltre per-messo di osservare l’evoluzione nel tempo dei pa-zienti con CMI, portando alla luce i diversi profilievolutivi della malattia8,9 e di eseguire screeningfamiliari su ampia scala. Recenti contributi nel-la diagnosi di CMI sono stati portati dall’intro-duzione della RMN cardiaca che permette unamigliore definizione anatomica rispetto all’eco-cardiografia, ma soprattutto la valutazione dellapresenza ed estensione della fibrosi intramiocar-dica10 (figura 1).
Le manifestazioni morfofunzionali e clinichedella malattia sono molto eterogenee. I pazienticon CMI possono rimanere asintomatici per tut-ta la vita; nella maggioranza dei casi, tuttavia,sono presenti sintomi che possono comparire aqualsiasi età e che comprendono la dispnea,l’astenia, il dolore toracico tipico o atipico per an-gina pectoris, le palpitazioni e la sincope. Il qua-dro sintomatologico è in genere di entità lieve omoderata, e risulta compatibile con uno stile divita normale: nei bambini e negli adolescenti lamalattia viene spesso riconosciuta solo nel corsodi studi di screening familiari. In circa un terzodei pazienti, tuttavia, la CMI progredisce verso
lo scompenso cronico, chepuò divenire refrattario, fi-no ad una fase ipocinetico-dilatativa definita “end-stage”. Sfortunatamente,predire il decorso clinico el’esito della malattia neisingoli pazienti con CMI siè rivelato molto difficile. Leprime grandi casistiche dipazienti con CMI, raccoltepresso i Centri di riferi-mento internazionali neglianni ’70, presentavano laCMI come una condizionead alto rischio, soprattuttonei giovani, con una morta-lità annuale del 3-6%11,12.Studi più recenti, in popo-lazioni con minor grado diselezione, hanno mostratoche la CMI presenta in re-altà un decorso relativa-mente stabile, una progno-si complessivamente beni-gna ed una mortalità glo-bale assai minore di quan-to precedentemente ripor-tato (intorno all’1% all’an-no)1,13,14. La complicanzapiù temibile, la morte im-provvisa aritmica, si osser-va in una minoranza di pa-zienti; purtroppo, l’identifi-
cazione dei soggetti a rischio resta molto difficile,in assenza di marker specifici per la prevenzioneprimaria.
La cardiomiopatia ipertroficacome malattia del sarcomero
La scoperta delle basi genetiche della CMI èscaturita dalla collaborazione tra clinici, genetisti,ricercatori e famiglie di pazienti. La natura fami-liare della malattia era nota fin dagli anni ’50; tut-tavia l’identificazione della prima mutazione ge-netica associata a CMI, nel gene codificante la ca-tena pesante della β-miosina (MYH7), risale al1987, in seguito a studi di linkage su famiglieestremamente ampie15. Oggi la CMI è universal-mente conosciuta come una malattia autosomicadominante del sarcomero, causata da varianti inalmeno 13 geni codificanti proteine dell’apparatocontrattile del cardiomiocita. (figura 2 e tabella 1).Tra i geni sarcomerici, 8 sono quelli comunementeanalizzati a scopo diagnostico: il gene della protei-na C legante la miosina, la cui sigla internaziona-le è MYBPC3; il gene della catena pesante della β-miosina, o MYH7; il gene della catena essenzialeleggera 1 della miosina, o MYL3; il gene della ca-tena regolatrice leggera 2 della miosina, o MYL2;il gene della troponina T cardiaca, o TNNT2;
Figura 1. Paziente di 32 anni affetta da cardiomiopatia Ipertrofica con ipertrofia di grado marcatoprevalente a livello medio-apicale. Immagini di risonanza magnetica nucleare ottenute con se-quenze cine in telediastole (A,B,C) ed in telesistole (D,E,F). A,D: Le immagini in 4 camere mostranol’importante ispessimento del setto interventricolare e della parete anteriore medio-apicale, e di-latazione biatriale. B,E: Sezione coronale asse corto a livello medio-ventricolare: è evidente l’oblite-razione sistolica di cavità a questo livello. C,F: Immagini ottenute rispettivamente in 4 camere ed as-se corto, dopo somministrazione di mezzo di contrasto: sono evidenti le aree di impregnazione tar-diva del setto interventricolare (frecce), espressione di fibrosi intramiocardica. AD atrio destro; ASatrio sinistro; SIV setto interventricolare; VD ventricolo destro; VS ventricolo sinistro

Recenti Progressi in Medicina, 102 (12), dicembre 2011488
il gene della troponina I, o TNNI3; il gene dell’al-fa-actina, o ACTC; ed il gene dell’alfa-tropomiosi-na, o TPM1. Una mutazione in questi geni vieneidentificata in circa i due terzi dei pazienti sotto-posti a screening genetico. I difetti di MYH7 eMYBPC3 sono i più frequenti, caratterizzando cir-ca il 50% delle diagnosi genetiche; gli altri 6 genicoprono una percentuale che va da un 3% ad un15% del totale a seconda dei Centri16.
Inoltre, dal 3% al 6% dei soggetti con CMI pre-senta un genotipo complesso, caratterizzato cioèda più mutazioni coesistenti, sia nello stesso gene(eterozigote composto) sia in geni diversi (eterozi-gote doppio); nell’1% dei pazienti sono addiritturadescritte triple mutazioni17.
Al contrario di altre malattie genetiche qualila fibrosi cistica o l’emofilia, in cui le mutazionigenetiche sono ricorrenti, la CMI è caratterizza-ta da estrema variabilità nelle mutazioni riscon-trate. Nei geni sarcomerici sono state infatti de-scritte oltre 1000 varianti diverse, la maggiorparte delle quali identificate in una sola famigliae perciò definite “private” Eccezionalmente sonodescritte mutazioni con “effetto founder” e perciòtipiche di una precisa area geografica18,19. Le va-rianti identificate, soprattutto nel gene MYH7,sono di tipo missense, ossia caratterizzate dallasostituzione di una singola base di DNA che dàluogo a sua volta alla sostituzione di un singoloaminoacido nella struttura proteica. Nel geneMYBPC3, invece, circa la metà delle varianti so-no di tipo frameshift, dette anche mutazioni dascivolamento.
Figura 2. Il Sarcomero cardiaco. (Immagine modificata da Watkins,2003).
Tabella 1. Geni coinvolti nella cardiomiopatia Ipertrofica.
Gene Simbolo Locus Prevalenza (%)
• Proteine sarcomeriche
Catena pesante della β-miosina MYH7 14q11.2-q12 25-30
Proteina C legante la miosina MYBPC3 11p11.2 25-30
Troponina T TNNT2 1q32 5
Troponina I TNNI3 19p13.4 ~5
α-tropomiosina TPM1 15q22.1 <5
Catena leggera della miosina
Essenziale MYL3 3p21.2-p21.3 <1
Regolatrice MYL2 12q23-q24.3 <1
Catena pesante dell’α-miosina MYH6 14q11.2-q12 rara
Troponina C TNNC 15q14 rara
Actina ACTC 3p21 rara
• Altre proteine
Proteina LIM CSRP3 10q22.2-q23.3 <2
α-actina 2 ACTN2 1q42-q43 rara
Titina TTN 2q13-33 rara
Fosfolambano PLN 6q22.1 rara
Caveolina-3 CAV3 3p25 rara
Teletonina TCAP 17q12-q21.1 rara
Giuntofilina JPH2 20q12 rara
LIM binding domain 3 (ZASP) LBD3 10q22.2-q23.3 rara

Queste sono caratterizzate dall’aggiunta (in-serzione) o eliminazione (delezione) di uno o po-chi nucleotidi alterando tutta la struttura dellaproteina a valle (in inglese: frame). Molto fre-quentemente tale meccanismo porta ad un arre-sto della sintesi proteica e conseguentemente al-la produzione di una proteina tronca. Per le for-me di CMI associate a mutazioni frameshift diMYBPC3, è stato ipotizzato un meccanismo pa-togenetico di aploinsufficienza, per cui l’allelenon mutato (wild-type) non riesce a compensarela carenza di proteina determinata da quello mu-tato. Per le mutazioni missense, la maggior par-te dei modelli transgenici indica che la CMI deri-va da effetti di dominanza negativa (cioè da uneffetto “tossico” della proteina mutata), più cheda aploinsufficienza1. Ad oggi, tuttavia, la pato-genesi della CMI a partire dalla mutazione sar-comerica resta largamente ignota, e verosimil-mente sono coinvolti più meccanismi, come sug-gerito dalla marcata eterogeneità di espressioneinterindividuale della malattia20.
Nell’ambito delle casistiche cliniche di CMI, èstata inoltre accertata in tempi recenti la pre-senza delle cosiddette “fenocopie”, cioè di patolo-gie con espressione morfologica molto simile o in-distinguibile dalla CMI sarcomerica, ma causateda meccanismi molecolari diversi e associate a si-gnificative differenze sul piano prognostico e te-rapeutico. Sono state, ad esempio, identificatecardiomiopatie a fenotipo ipertrofico associate amutazioni nei geni del metabolismo energetico edella produzione di ATP. Rientrano in questogruppo le cardiomiopatie secondarie a deficit del-la catena respiratoria mitocondriale, le cardio-miopatie da alterazioni del metabolismo lipidicoe un gruppo geneticamente eterogeneo rappre-sentato dalle cardiomiopatie da accumulo. Traqueste ultime troviamo due malattie X-linkedquali la sindrome di Anderson Fabry (da difettodel gene per l’alfa-galattosidasi A) in cui le ma-nifestazioni cardiologiche si associano spesso aun quadro clinico con interessamento multiorga-no, e la malattia di Danon (da difetto del geneLAMP2)21,22, caratterizzata da aumento estremodella massa cardiaca in età pediatrica. Infine,una cardiomiopatia di incerta classificazione èquella secondaria a mutazioni nella subunità ca-talitica �2 della proteina chinasi AMP-attivata(PRKAG2), in cui il fenotipo ipertrofico si associaa frequente e precoce comparsa di pre-eccitazioneventricolare23.
Ipotesi patogenetiche
L’incorporazione di proteine mutate nel sar-comero porta alla compromissione del normalefunzionamento delle proteine wild-type: è laprincipale causa di sviluppo dell’ipertrofia nellaCMI24. Inizialmente si pensava che mutazioninei geni sarcomerici fossero la causa di una de-pressione della funzione contrattile, con conse-
guente deplezione energetica del cardiomiocitadeterminando così un’ipertrofia di tipo compen-satorio25. Tuttavia, studi successivi hanno dimo-strato che mutazioni a carico della miosina pos-sono addirittura portare ad un incremento del-l’attività contrattile e ad un aumento della sen-sibilità al Ca2+ delle proteine regolatorie dei fi-lamenti sottili26,27. Per questo, alcuni autori han-no ipotizzato che la CMI rappresenti non tantoil risultato di un deficit contrattile, quanto delladeplezione energetica del cardiomiocita, a causadi un inefficiente impiego dei depositi di energiacellulare. Ashrafian, nel 2003, ha dimostrato chela presenza di proteine mutate nel sarcomeroporta ad un incremento della richiesta energeti-ca causata da un inefficiente utilizzo dell’ATP.Questa alterazione comporta una ridotta capaci-tà del cardiomiocita di mantenere i livelli ener-getici nel compartimento responsabile della con-trazione e compromette importanti funzioniomeostatiche come il re-uptake del calcio intra-cellulare. Il conseguente, prolungato transientedi calcio stimola i processi trascrizionali, attra-verso l’attivazione di messaggeri cellulari (pro-tein chinasi calcio-calmodulina-dipendente; viadelle MAP-chinasi; fattori di trascrizione): l’in-sieme di tutti questi segnali può indurre lo svi-luppo dell’ipertrofia, come meccanismo di com-penso.
Nuove ipotesi sullo sviluppo della CMI riguar-dano la possibilità che le mutazioni sarcomericheinterferiscano con lo sviluppo embriologico delcuore, in particolare con i meccanismi di migra-zione e differenziazione delle cellule pluripotentiche derivano dal pro-epicardio. Durante le fasi ini-ziali dello sviluppo embrionale, cellule pro-epicar-diche migrano a costituire l’epicardio, e da qui mi-grano diffusamente all’interno del miocardio e sidifferenziano in varie linee cellulari, che dannoluogo ai fibroblasti interstiziali, alla muscolaturaliscia vascolare, alle cellule avventizie, alle valvo-le atrioventricolari. Le mutazioni del sarcomero el’alterata contrattilità del cuore primordiale al-l’epoca di questa colonizzazione, potrebbero in-fluenzare l’espressione genica delle cellule epicar-diche, mediante un meccanismo di meccano-tra-sduzione. Infatti, al momento della migrazione diqueste cellule dall’epicardio al miocardio, il cuoreha già iniziato a contrarsi e le mutazioni causa diCMI sono già espresse28. Tale meccanismo potreb-be spiegare le molte espressioni fenotipiche dellaCMI che riguardano tessuti diversi dal miocardio,in cui le mutazioni non sono espresse: come, adesempio, le alterazioni della valvola mitralica, ilrimodellamento del microcircolo e la fibrosi inter-stiziale.
Correlazioni genotipo-fenotipo
La CMI è caratterizzata da una grande eteroge-neità genetica, fenotipica e clinica, con scarsa cor-relazione tra i vari aspetti nel singolo individuo.
F. Girolami et al.: Rilevanza clinica del test genetico nella cardiomiopatia ipertrofica 489

Per tale motivo, una volta identificata la mutazionecausante CMI in un paziente, non è in genere possi-bile trarne informazioni prognostiche o implicazioniriguardo allo sviluppo di un fenotipo più o menomarcato nei familiari. La stessa mutazione può de-terminare un ampio spettro di fenotipi perfino nel-l’ambito della stessa famiglia, e suggerisce l’impor-tanza di geni modificatori (varianti in cis o in trans,polimorfismi, varianti rare), fattori epigenetici (gra-do di metilazione del DNA, di acetilazione degli isto-ni, e modificazioni post trascrizionali), e fattori am-bientali (età, attività sportiva, fumo di sigaretta,ecc.) nel determinare le manifestazioni morfologichee cliniche della malattia. Sebbene labili, correlazio-ni genotipo-fenotipo sono state ricercate attivamen-te in passato, soprattutto nei primi anni dopo la sco-perta dei geni coinvolti nella CMI. Ad esempio, in al-cuni studi le mutazioni nel gene MYH7 venivano as-sociate ad insorgenza precoce della malattia, eleva-ta penetranza ed ipertrofia di grado marcato, men-tre le mutazioni nel gene TNNT2 sembravano com-portare una ipertrofia di grado modesto ma un altorischio di morte improvvisa, e mutazioni del geneMYBPC3 si correlavano ad insorgenza tardiva dimalattia, penetranza incompleta e prognosi favore-vole1. Inoltre, specifiche mutazioni, identificate neigeni MYH7 e TNNT2, fortunatamente rare, sonostate correlate con un alto rischio di morte improv-visa in giovane età e perciò definite “maligne”29. Tut-tavia, tali associazioni genotipo-fenotipo si basavanosu piccoli numeri di pazienti, appartenenti a fami-glie molto ampie, altamente selezionate per rischioaritmico. In realtà, studi successivi su grandi casi-stiche hanno spesso contraddetto tali assunti. Unesempio eclatante è quello delle mutazioni diTNNT2, che in alcuni Centri venivano consideratedi per sé un fattore di rischio maggiore per morte im-provvisa, indipendentemente dal profilo clinico.L’esperienza del nostro e di altri Centri ha dimo-strato che la CMI da TNNT2 può associarsi a profi-li estremamente diversi di malattia, che spaziano daforme lievi e asintomatiche fino a forme con ipertro-fia marcata e sintomi invalidanti, con un rischio dimorte improvvisa non diverso dagli altri geni30.
Ruolo clinico del test genetico
Fino a poco tempo fa considerata un mero stru-mento di ricerca, la diagnostica genetica è oggi unmomento estremamente importante dell’inquadra-mento di pazienti con CMI e delle loro famiglie, e ri-veste un ruolo pratico che è ancora poco apprezza-to dai cardiologici, anche per le limitate opportuni-tà di interazione con i genetisti in ambito cardiolo-gico. Il contributo del test genetico si riflette su va-ri momenti dell’iter clinico dei pazienti con CMI.
CONFERMA DELLA DIAGNOSI NEI FENOTIPI DUBBI
Nell’ambito della CMI, sono frequenti i co-siddetti casi “grigi”: il grado di ipertrofia è tal-mente lieve da non consentire una diagnosi si-
cura, e i restanti reperti strumentali (soprattut-to l’ECG) sono nella norma. Questa situazione èricorrente ad esempio nel caso degli atleti. Inquesti individui, l’identificazione di una muta-zione in uno dei geni sarcomerici consente diconfermare diagnosi di CMI31. Tale possibilitàha consentito di svelare gli aspetti più lievi del-la malattia e di caratterizzare meglio lo spettrofenotipico, soprattutto in ambito di screening fa-miliare; questo ha permesso di validare le ma-nifestazioni ancillari della malattia (dilatazioneatriale sinistra, aspetti di non compattazionedel VS, anomalie della mitrale) che vengono de-scritte frequentemente con le tecniche di ima-ging oggi disponibili.
DIAGNOSI DIFFERENZIALE
Il test genetico può essere utile per distinguereuna CMI primitiva, cioè causata da mutazioni ingeni sarcomerici, dalle forme cosiddette pseudo-ipertrofiche (fenocopie). Particolarmente impor-tante è la possibilità di distinguere forme autoso-miche dominanti da forme X-linked e perciò la pos-sibilità di stabilire un preciso rischio di ricorrenzae di trasmissibilità familiare32.
IDENTIFICAZIONE DEI GENOTIPI COMPLESSIE RISCHIO DI PROGRESSIONE DI MALATTIA
La possibilità di identificare genotipi comples-si rappresenta un elemento prognostico impor-tante. La presenza di due o più mutazioni in unostesso paziente è solitamente associata ad insor-genza precoce della malattia, decorso sfavorevolee rischio aumentato di morte improvvisa o perscompenso16. Questi dati sono importanti nel-l’identificazione del paziente a rischio. In un re-cente lavoro del nostro gruppo17, si descrivonoquattro famiglie con CMI, in cui il soggetto indi-ce presenta un genotipo complesso con tre muta-zioni in vari geni. Anche se la presenza di tre mu-tazioni si riscontra raramente (0,8%), essa costi-tuisce un rischio aumentato di progressione versola fase end-stage e di aritmie ventricolari, sugge-rendo un’associazione tra genotipo complesso eprogressione sfavorevole di malattia. Escludendoquesti casi estremi, è comunque regola generaleche il gruppo di pazienti con test genetico positi-vo presenti un aumentato rischio di decorso sfa-vorevole della malattia rispetto al gruppo dei pa-zienti negativi al test16.
RIVALUTAZIONE CRITICA DELLA DIAGNOSINEI PAZIENTI GENOTIPO-NEGATIVI
I pazienti negativi al test genetico rappresen-tano un gruppo composito, in cui la malattia è ve-rosimilmente causata da geni non ancora identifi-cati, oppure è ad eziologia non genetica1,16.
Recenti Progressi in Medicina, 102 (12), dicembre 2011490

Nella pratica clinica, ciascun paziente con fe-notipo CMI e test genetico negativo deve essere ri-valutato, sottoponendolo ad ulteriori esami. In par-ticolare, è importante escludere una CMI associa-ta a malattie neuromuscolari, o mutazione nei ge-ni che possono produrre fenocopie. Soprattutto seil soggetto è un giovane, è fondamentale andare al-la ricerca di manifestazioni extra-cardiache qualidimorfismi facciali, deficit neurologico, coinvolgi-mento renale etc. Queste evidenze, che spesso sfug-gono al cardiologo, possono invece essere utili perorientare la diagnosi nella giusta direzione. Unaconsulenza genetica clinica di II livello può per-mettere di distinguere le forme sindromiche asso-ciate a cardiomiopatia, come ad esempio le sindro-mi di Noonan e di Leopard, la malattia di Ander-son-Fabry, ecc.16,21,22,30. Perciò nei protocolli stan-dard per l’esecuzione del test genetico deve essereinclusa la consulenza pre test e post test eseguitain team multidisciplinare.
DIAGNOSI PRE-NATALE
In situazioni in cui la CMI familiare è associa-ta ad un’insorgenza precoce ed in forma partico-larmente severa, con più soggetti morti improvvi-samente in giovane età, la diagnosi prenatale po-trebbe essere effettuata allo scopo di identificarela presenza della mutazione nel feto33.
Tuttavia a causa delle incerte correlazioni ge-notipo-fenotipo, dovute alla variabile espressioneclinica, non si ha indicazione ad effettuare la dia-gnosi prenatale nel caso della CMI primitiva.
PROCEDURA SEGUITAPER EFFETTUARE IL TEST GENETICO
Il rapporto del paziente (edel cardiologo) con la geneti-ca va ben oltre il semplicetest, e comporta un vero eproprio iter con diverse tappedi significato diverso (figura3). La procedura seguitapresso il Centro di Riferi-mento per le Cardiomiopatiein Firenze negli ultimi 10 an-ni prevede una consulenzapre-test (fase pre-analitica),la fase analitica durante laquale viene eseguito il test inlaboratorio ed una consulen-za post-test (fase post-anali-tica). Le procedura è la stessaper probandi e familiari. Du-rante la fase pre-analitica, ilcardiologo propone al pazien-te di concordare una consu-lenza genetica con il geneti-sta. Per tale colloquio si im-piegano circa 30-45 minuti.
F. Girolami et al.: Rilevanza clinica del test genetico nella cardiomiopatia ipertrofica 491
La consulenza genetica è stata definita come“Processo di comunicazione che concerne i problemipsicologici, etici e sociali correlati all’occorrenza o alrischio di ricorrenza di una patologia genetica inuna famiglia”32. Durante la consulenza geneticapre-test vengono raccolte informazioni e quesiti po-sti dal consultando, viene costruito l’albero genea-logico della famiglia, si effettua una prima valuta-zione del rischio di ricorrenza, si discute con l’inte-ressato di ciò che è possibile fare per meglio preci-sare l’entità del rischio, chiarendo il significato e i li-miti del test genetico; infine viene raccolto uno spe-cifico consenso informato.
La consulenza genetica ed il consenso informa-to sono parti integranti di un test genetico poichépermettono la discussione preliminare di tutte lepossibili implicazioni dei diversi risultati ottenutidal test e devono fornire gli strumenti per la com-prensione della malattia genetica; per questo im-plicano l’utilizzo di particolari procedure atte adaffrontare le problematiche psicologiche, etiche esociali inerenti all’utilizzo dei test genetici32.
Segue una fase analitica durante la quale vieneeffettuata la ricerca di mutazioni nei geni implica-ti nella CMI mediante sequenziamento diretto, apartire da DNA estratto da un prelievo di sangueperiferico.
Nella fase post-analitica vengono effettuate laverifica dei risultati, la preparazione del referto ela consulenza genetica post-test. La consegna delreferto al paziente viene fatta dal cardiologo insie-me al genetista, secondo una procedura multidi-sciplinare che prevede in particolare la spiegazio-ne delle ricadute pratiche del test.
Paziente con Cardiomiopatia
Ipertro�ca (probando)
Consenso InformatoDNA
Consulenza genetica pre-test Cardiologo-Genetista
Screening Molecolaredei geni sarcomerici
Consulenza genetica post-test Comunicazione del risultato
Cardiologo-Genetista-Psicologo
Mutazione presente
No Mutazione Rivalutazioneclinica
Diagnoside�nitiva su
base genetica
Conferma della diagnosi nei fenotipi dubbiDiagnosi di!erenziale
Identi�cazione dei genotipi complessi
Screening genetico peri familiari del probando
Figura 3. Procedura per la diagnosi genetica di cardiomiopatia ipertrofica.(Modificata da Torricelli F. et al 2003)30.

Talvolta è presente anche uno psicologo. È inquesta fase che si apre la possibilità di far accede-re al test genetico i familiari del probando.
Il test genetico effettuato in un paziente conCMI può dare tre risultati diversi:
1. Identificazione di una mutazione sicura-mente patogenetica già descritta in letteratura; inquesto caso la diagnosi genetica è certa, per cui ilgenotipo osservato è compatibile con la diagnosiclinica e viene sempre proposto il test ai familiari.
2. Non identificare alcuna mutazione nei genistudiati a scopo diagnostico; in questo caso vieneprecisato che l’eventuale causa genetica della CMIpotrebbe trovarsi o in geni non analizzati (più ra-ri) oppure in geni al momento ancora non identifi-cati. Ovviamente non può essere esclusa la dia-gnosi clinica sulla base di un risultato negativo deltest.
3. Identificazione di una variante non classifi-cabile (UVs), ossia di una alterazione precedente-mente non descritta, per la quale è necessario de-finire una probabilità di associazione con la ma-lattia tramite ulteriori approfondimenti34.
Test presintomatico
In questa situazione il probando è clinicamen-te sano, con un ecocardiogramma ed un elettro-cardiogramma normali, appartenente ad una fa-miglia che presenta uno o più casi di CMI. Il pa-ziente richiede il test genetico perché vuole sape-re se ha ereditato o meno la mutazione responsa-bile della malattia nella sua famiglia. Il rischioteorico di presentare la mutazione è del 50% (pro-babilità a priori), nel caso in cui il soggetto sia fa-miliare di primo grado del paziente con CMI. Tut-tavia la penetranza è incompleta (70% nell’adul-to), incrementa con l’età ed è legata al sesso, oltreche a fattori ambientali (abitudini di vita quali fu-mo, alcol, attività sportiva). La ragione per cui unsoggetto richiede il test è frequentemente duplice:il rischio di essere “portatore” della mutazionecausa della malattia e di sviluppare la malattiastessa, e il rischio di trasmettere la malattia alladiscendenza.
Tuttavia non è noto nessun trattamento medi-co in grado di prevenire o ritardare l’insorgenzadella malattia. Il solo trattamento efficace nel pre-venire la morte improvvisa è un defibrillatore im-piantabile, ma esso viene proposto solo per i pa-zienti con CMI ad alto rischio di morte cardiaca im-provvisa1. Nei pazienti sani, portatori della muta-zione, può essere raccomandata la restrizione del-l’attività fisica, tuttavia l’effetto di tale provvedi-mento nel prevenire la morte improvvisa è pura-mente speculativo32.
Nel caso in cui il soggetto risulti positivo, sa-rà necessario un follow-up medico, volto alla dia-gnosi precoce dell’espressione fenotipica dellamalattia e alla valutazione – se possibile – del ri-schio di morte improvvisa. Questo soggetto sarà,inoltre a rischio di trasmettere la mutazione.
Nel caso, invece, di negatività al test, il pa-ziente avrà un notevole beneficio a livello psico-logico, e con l’interruzione dei controlli clinici pe-riodici, si avrà un vantaggio anche per il sistemasanitario30,32.
Il test genetico nei minori a scopo presintomatico
L’applicazione del test genetico a scopo presin-tomatico nei bambini è un argomento controverso:alcuni studi hanno enfatizzato i potenziali beneficî,specialmente perché la morte improvvisa può esse-re il primo sintomo della malattia; altri, per man-canza di efficaci trattamenti per prevenire l’insor-genza della malattia e la morte improvvisa, consi-derano deleterio il test presintomatico nei bambi-ni, per i possibili risvolti psicologici negativi32. At-tualmente molti gruppi hanno riserve nell’effet-tuare il test genetico presintomatico prima dellamaggiore età, riferendosi al decreto del Garantedella Privacy, dove si dice che: «I trattamenti di da-ti connessi all’esecuzione di test genetici presinto-matici possono essere effettuati sui minori non af-fetti, ma a rischio per patologie genetiche, solo nelcaso in cui esistano concrete possibilità di terapie odi trattamenti preventivi prima del raggiungimen-to della maggiore età» (Autorizzazione generale deltrattamento dei dati genetici, pubblicato nella Gaz-zetta Ufficiale della Repubblica Italiana n. 159dell’11 luglio 201130.
Conclusioni
Il test genetico rappresenta un punto di forzaper il clinico allo scopo di inquadrare correttamen-te i pazienti con cardiomiopatia ipertrofica. Risul-ta fondamentale al fine di confermare la diagnosinei fenotipi dubbi, riveste un ruolo importante nel-la diagnosi differenziale ed è utile nell’identifica-zione dei genotipi complessi. Inoltre, stabilendo lanatura ereditaria della malattia, interessa anche ifamiliari del probando, nei quali può essere impie-gato a scopo presintomatico.
Lo sviluppo di nuove tecnologie per l’analisi si-multanea di molti geni, quali le piattaforme diNext-Generation Sequencing, unitamente allasempre più stretta collaborazione tra clinici e ge-netisti, permetterà di ampliare sempre più le co-noscenze sulla genetica di questa malattia.
Bibliografia
1. Maron BJ. Hypertrophic Cardiomyopathy. A syste-matic review. JAMA 2002; 287: 1308-20.
2. Teare D. Asymmetrical hypertrophy of the heart inyoung adults. British Heart Journal 1958; 20: 1-8.
3. Maron BJ, MCKenna WJ, Danielson GK, et al.ACC/ESC EXPERT CONSENSUS DOCUMENTAmerican College of Cardiology/European Society ofCardiology Clinical Expert Consensus Document onHypertrophic Cardiomyopathy. J Am Coll Cardiol2003; 42: 1687-713.
Recenti Progressi in Medicina, 102 (12), dicembre 2011492

F. Girolami et al.: Rilevanza clinica del test genetico nella cardiomiopatia ipertrofica 493
4. Morrow AG, Braunwald E. Functional aortic steno-sis; a malformation characterized by resistance toleft ventricular outflow without anatomic obstruc-tion. Circulation 1959; 20: 181-9.
5. Shah PM, Gramiak R, Adelman AG, et al. Role ofechocardiography in diagnostic and hemodynamicassessment of hypertrophic subaortic stenosis. Cir-culation 1971; 44: 891-8.
6. Epstein SE, Morrow AG, Henry WL, et al., Editorial:The role of operative treatment in patients with idio-pathic hypertrophic subaortic stenosis. Circulation1973; 48: 677-80.
7. McKenna W, Deanfield J, Faruqui, et al. Prognosisin hypertrophic cardiomyopathy: role of age and cli-nical, electrocardiographic and hemodynamic featu-res. Am J Cardiol 1981; 47: 532-8.
8. Marian AJ, Roberts R. The molecular genetic basisfor hypertrophic cardiomyopathy. J Mol Cell Cardiol2001; 33: 655-70.
9. Olivotto I, Cecchi F, Gistri R, et al. Relevance of co-ronary microvascular flow impairment to long-termremodeling and systolic dysfunction in hypertro-phic cardiomyopathy. J Am Coll Cardiol 2006; 47:1043-8.
10. Nagueh SF, Bierig SM, Budoff MJ, et al. AmericanSociety of Echocardiography clinical recommenda-tions for multimodality cardiovascular imaging ofpatients with hypertrophic cardiomyopathy: Endor-sed by the American Society of Nuclear Cardiology,Society for Cardiovascular Magnetic Resonance, andSociety of Cardiovascular Computed Tomography.American Society of Echocardiography; AmericanSociety of Nuclear Cardiology; Society for Cardiova-scular Magnetic Resonance; Society of Cardiovascu-lar Computed Tomography. J Am Soc Echocardiogr2011; 24: 473-98.
11. Watkins H, McKenna WJ, Thierfelder L, et al. Mu-tations in the genes for cardiac troponin T and al-pha-tropomyosin in hypertrophic cardiomyopathy. NEngl J Med 1995; 332: 1058-64.
12. Marian AJ, Roberts R. The molecular genetic basisfor hypertrophic cardiomyopathy. J Mol Cell Cardiol2001; 33: 655-70.
13. Niimura H, Patton KK, McKenna WJ, et al. Sarco-mere protein gene mutations in hypertrophic car-diomyopathy of the elderly. Circulation 2002; 105:446-51.
14. Nistri S, Olivotto I, Girolami F, et al. Looking for hy-pertrophic cardiomyopathy in the community: whyis it important? J Cardiovasc Transl Res 2009; 2:392-7.
15. Seidman CE, Seidman JG. Identifying sarcomere ge-ne mutations in hypertrophic cardiomyopathy: a per-sonal history. Circ Res 2011; 108: 743-50.
16. Olivotto I, Girolami F, Ackerman MJ, et al. Myofila-ment protein gene mutation screening and outcomeof patients with hypertrophic cardiomyopathy. MayoClin Proc 2008; 83: 630-8.
17. Girolami F, Ho CY, Semsarian C, et al. Clinical fea-tures and outcome of hypertrophic cardiomyopathyassociated with triple sarcomere protein gene muta-tions. J Am Coll Cardiol 2010; 55: 1444-53.
18. Girolami F, Olivotto I, Passerini I, et al. A molecularscreening strategy based on beta-myosin heavychain, cardiac myosin binding protein C and tropo-nin T genes in Italian patients with hypertrophiccardiomyopathy. J Cardiovasc Med 2006; 7: 601-7.
19. www.angis.org.au/Databases/Heart/heartbreak.htmlcardiogenomics.med.harvard.edu; www.gdb.org;www.hgmd.cf.ac.uk
20. McKenna WJ, Elliott PM. Cardiomiopatia Ipertrofica.Textbook of Cardiovascular Medicine 2009; 28: 433-47.
21. Patel MR, Cecchi F, Cizmarik M, et al Cardiovascu-lar events in patients with fabry disease natural hi-story data from the fabry registry. J Am Coll Cardiol2011; 57: 1093-9.
22. Maron BJ, Roberts WC, Arad M, et al. Clinical Out-come and Phenotypic Expression in LAMP2 Cardio-myopathy. JAMA 2009; 301: 1253-9.
23. Giordano C, D’Amati G. Cardiomyopathies due todefective energy metabolism: morphological and fun-ctional features]. Pathologica 2005; 97: 361-8.
24. Ashrafian H, Redwood C, Blair E, et al. Hypertro-phic cardiomyopathy: a paradigm for myocardialenergy depletion. Trends Genet 2003; 19: 263-8.
25. Watkins H, McKenna WJ, Thierfelder L, et al. Mu-tations in the genes for cardiac troponin T and al-pha-tropomyosin in hypertrophic cardiomyopathy. NEngl J Med 1995; 332: 1058-64.
26. Lowey S. Functional consequences of mutations inthe myosin heavy chain at sites implicated in fami-lial hypertrophic cardiomyopathy. Trends Cardio-vasc Med 2002; 12: 348-54.
27. Elliott P, Andersson B, Arbustini E, et al. Classifica-tion of the cardiomyopathies: a position statementfrom the european society of cardiology workinggroup on myocardial and pericardial diseases. EurHeart J 2008; 29: 270-276.
28. Olivotto I, Girolami F, Nistri S, et al. The many fa-ces of hypertrophic cardiomyopathy: from develop-mental biology to clinical practice. J CardiovascTrans Res 2009; 2: 349-67.
29. Ackerman MJ, VanDriest SL, Ommen SR, et al. Pre-valence and age-Dependance of maliganant muta-tions in the Beta-myosin heavy chain and troponin Tgenes in Hypertrophic Cardiomyopathy. J Am CollCardiol 2002; 39: 2042-8.
30. Torricelli F, Girolami F, Olivotto I, et al. Prevalenceand clinical profile of troponin T mutations amongpatients with hypertrophic cardiomyopathy in tu-scany. Am J Cardiol 2003; 92: 1358-62.
31. Baldi M, Girolami F. A new type of “tracing” for car-diologists? G Ital Cardiol 2009; 10: 266.
32. Charron P, Heron D, Gargiulo M, et al. Genetic te-sting and genetic counselling in hypertrophic car-diomyopathy: the French experience. J Med Genet2002; 39: 741-6.
33. Charron P, Héron D, Gargiulo M, et al. Prenatal mo-lecular diagnosis in hypertrophic cardiomyopathy: re-port of the first case. Prenat Diagn 2004; 24(9): 701-3.
34. Clinical Molecular Genetics Society (CMGS). Prati-ce guidelines for the interpretation and reporting ofunclassified variants (UVs) in clinical molecular ge-netics. Avaible at: http://www.cmgs.org/.
Indirizzo per la corrispondenza:Dott. Francesca GirolamiStruttura Organizzativa Dipartimentale Diagnostica GeneticaAzienda Ospedaliero Universitaria CareggiLargo Brambilla, 350134 FirenzeE-mail: [email protected]

Disc
laim
er: T
he man
uscrip
t and it
s conte
nts ar
e
confid
entia
l, inte
nded fo
r journ
al re
view p
urpose
s
only,
and n
ot to b
e furth
er d
isclo
sed.
1
Quantitative Contrast-Enhanced Cardiovascular Magnetic
Resonance Predicts Sudden Death in Patients with
Hypertrophic Cardiomyopathy
1,2Raymond H. Chan, MD, MPH, 3Barry J. Maron, MD, 4Iacopo Olivotto, MD, 5Michael J. Pencina, PhD,
6Gabriele Egidy Assenza, MD, 3Tammy Haas, RN, 3John R. Lesser, MD, 7Christiane Gruner, MD,
7Andrew M. Crean, MD, 7Harry Rakowski, MD, 8James E. Udelson, MD, 8Ethan Rowin, MD, 9Massimo
Lombardi, MD, 4Franco Cecchi, MD 4Benedetta Tomberli, MD, 10Paolo Spirito, MD, 10Francesco
Formisano, MD, 11Elena Biagini, MD,11Claudio Rapezzi, MD, 6Carlo Nicola De Cecco, MD, 6Camillo
Autore, MD, 12E. Francis Cook, PhD, 1,2Susie N. Hong, MD,1,2 C. Michael Gibson, MD, MS, 1,2Warren J.
Manning, MD,1,2Evan Appelbaum, MD and 8Martin S. Maron, MD
1PERFUSE Study Group, Harvard Medical School, Boston, Massachusetts; 2Department of Medicine, Cardiovascular Division,
Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Massachusetts; 3The Hypertrophic Cardiomyopathy
Center, Minneapolis Heart Institute Foundation, Minneapolis MN; 4Referral Center for Myocardial Diseases, Azienda
Ospedaliera Universitaria Careggi, Florence Italy; 5 Harvard Clinical Research Institute and Boston University Biostatistics;
6Ospedale Sant’Andrea Universita “La Sapienza”, Rome, Italy; 7Division of Cardiology, Toronto General Hospital, University
Health Network, University of Toronto, Toronto, Ontario, Canada; 8Hypertrophic Cardiomyopathy Center, Division of
Cardiology, Tufts Medical Center, Boston, Massachusetts; 9Fondazione C.N.R./Regione Toscana G. Monasterio, Pisa, Italy;
10Ente Ospedaliero Ospedali Galliera, Genoa, Italy; 11Policlinico S. Orsola-Malpighi, Bologna, Italy; 12Department of
Epidemiology, Harvard School of Public Health, Boston, MA,
Running title: LGE and sudden death in HCM Address for Correspondence: Martin S. Maron, MD 800 Washington St, #70 Boston, MA 02111 Email: [email protected] Phone/Fax: 617 636-8066/617 636-7175

Disc
laim
er: T
he man
uscrip
t and it
s conte
nts ar
e
confid
entia
l, inte
nded fo
r journ
al re
view p
urpose
s
only,
and n
ot to b
e furth
er d
isclo
sed.
3
ABSTRACT
Background. Hypertrophic cardiomyopathy (HCM) is the most common cause of sudden
cardiac death (SCD) in the young, although not all patients eligible for SCD prevention with the
implantable cardioverter-defibrillator (ICD) are identified. Contrast-enhanced cardiovascular
magnetic resonance (CMR) with late gadolinium enhancement (LGE) has emerged as an in vivo
marker of myocardial fibrosis, although its role in stratifying SCD risk in subgroups of HCM
patients remains unresolved.
Methods. We prospectively assessed the relation between LGE (as % of left ventricular mass)
and cardiovascular outcomes in 1293 consecutive HCM patients followed for a median of 3.3
years.
Results. Sudden cardiac death events occurred in 37 (3%) patients. A continuous relationship
was evident between %LGE (p=0.001) and SCD risk in HCM patients. Extent of LGE was
independently associated with increased risk of SCD (HRadj 1.4/10% increase in LGE; p=0.002),
even after adjustment for other relevant disease variables. LGE of ≥15% of LV mass
demonstrated a 2-fold increase for SCD in those patients otherwise considered to be at low risk,
with an estimated likelihood for SCD of 6% at 5 years. Performance of the SD risk model was
enhanced by LGE (net reclassification index of 12.9%; 95% CI: 0.3-38.3% with a relative
integrated discrimination improvement of 0.565; 95% CI: 0.019-3.564). Absence of LGE was
associated with lower risk for SD (HRadj 0.39; p=0.02). Extent of LGE also predicted
development of the end-stage of HCM with systolic dysfunction (HRadj 1.8/10% increase in
LGE; p<0.03).

Disc
laim
er: T
he man
uscrip
t and it
s conte
nts ar
e
confid
entia
l, inte
nded fo
r journ
al re
view p
urpose
s
only,
and n
ot to b
e furth
er d
isclo
sed.
4
Conclusions. Extensive LGE measured by quantitative contrast-enhanced CMR is an
independent predictor of SCD in HCM and a new marker for judging high-risk status, identifying
patients otherwise considered to be at low risk and who may benefit from ICD therapy.

CHAGAS DISEASE AS A CAUSE OF HEART FAILURE AND VENTRICULAR
ARRHYTHMIAS IN PATIENTS LONG REMOVED FROM ENDEMIC AREAS: AN
EMERGING PROBLEM IN EUROPE
Vieri Vannucchi MD, Benedetta Tomberli MD, Lorenzo Zammarchi* MD,
Alessandra Fornaro MD, Gabriele Castelli MD, Filippo Pieralli** MD, Andrea Berni*** MD, Sophie
Yacoub**** MD, Alessandro Bartoloni* MD, Iacopo Olivotto MD
Referral Center for Myocardial Diseases, Internal Medicine and Infectious Disease Unit, CareggiUniversity Hospital, Florence, Italy
* Dipartimento di Medicina Sperimentale e Clinica, Sezione Medicina Critica e Medicine Specialistiche, Clinica Malattie Infettive, Università degli Studi di Firenze, Firenze, Italia** Internal and Emergency Medicine Unit, Careggi Uninersity Hospital, Florence, Italy*** Medicina Interna all'Orientamento all'alta complessità aziendale 2, Università degli Studi di Firenze, Italia**** Department of Medicine, Imperial College, Hammersmith Campus, Du Cane Rd, London, UK
Running Head: Chagas in Europe
Total word count: 2052
Address for correspondence: Dr. Vieri Vannucchi, MDUnità Medicina Interna e di UrgenzaAzienda Ospedaliera Universitaria CareggiViale Pieraccini 19, 50134 Firenze, ItalyTel/Fax: 39 055 7949335Email: [email protected]
Keywords: Chagas’ cardiomyopathy, Trypanosomiasis cardiovascular, Heart failure.
Conflict of Interest Disclosures: None.
1

ABSTRACT
Chagas disease (CD) is a parasitic disease caused by the protozoan Trypanosoma cruzi. In endemic
areas (South and Central America), CD represents a relevant public health issue, and is the most frequent
cause of cardiomyopathy. In non-endemic areas such as Europe, CD represents an emerging problem
following the establishment of sizeable communities from Brazil and Bolivia. Chagas cardiomyopathy
represents the most frequent and serious complication of chronic CD, affecting about 20-30% of patients,
potentially leading to heart failure, arrhythmias, thromboembolism, stroke and sudden death. Because late
complications of CD may develop several years or even decades after the acute infection, it may be
extremely challenging to reach the correct diagnosis in patients long removed from the countries of origin.
We report two examples of Chagas cardiomyopathy in South American women permanently residing in Italy
for >20 years, presenting with cardiac manifestations ranging from left ventricular dysfunction and heart
failure to isolated ventricular arrhythmias. The present review emphasizes that CD should be considered as a
potential diagnosis in patients from endemic areas presenting with “idiopathic” cardiac manifestations, even
when long removed from their country of origin, with potential implications for treatment and control of CD
transmission.
Word count = 195
2

1
NOVEL ALPHA-ACTININ-2 VARIANT ASSOCIATED WITH
FAMILIAL HYPERTROPHIC CARDIOMYOPATHY AND JUVENILE
ATRIAL ARRHYTHMIAS:
A MASSIVELY PARALLEL SEQUENCING STUDY
1Girolami F. (BSc), 2Iascone M. (BSc), 3Tomberli B. (MD), 1Bardi S. (BSc), 1Benelli
M.(PhD), 1Marseglia G. (BSc), 1Pescucci C. (BSc), 2Pezzoli L. (BSc), 2Sana ME.(PhD), 4Basso C. (MD), 5-6Marziliano N.(BSc), 6Merlini P.A. (MD), 3Fornaro A. (MD), 7Cecchi
F.(MD), 1Torricelli F.(BSc), 3Olivotto I.(MD).
1Genetic Diagnostic Unit, Careggi University Hospital, Florence, Italy; 2USSD Laboratorio Genetica Medica, Ospedali Riuniti, Bergamo, Italy
3Referral Centre for Myocardial Diseases, Careggi University Hospital, Florence, Italy. 4Division of Cardiology, Department of Cardiological Thoracic and Vascular Sciences, University of
Padua, Padua, Italy. 5Azienda Ospedaliera Ospedale Niguarda Cà Granda, IV Division of Cardiology, Milan, Italy
6 Azienda Ospedaliera Universitaria di Parma, Division of Cardiology, Parma, Italy 7University of Florence, Department of Clinical and Experimental Medicine, Florence, Italy
(Short title: ACTN2-related HCM identified by deep sequencing)
Address for correspondence: Dr. Francesca Girolami SOD Diagnostica Genetica Azienda Ospedaliera Universitaria Careggi Largo Brambilla 3, 50134 Firenze Tel. ##39-0557949364 - Fax. 0557949686 e-mail: [email protected]
Acknowledgments: This work was supported by Ministero Istruzione Università e Ricerca (PRIN), European Union (STREP Project 241577 “BIG HEART,” 7th European Framework Program), and RF2010-2313451”Hypertrophic Cardiomyopathy:new insights from deep sequencing and psycosocial evaluation”.

2
ABSTRACT
Aims. Next generation sequencing (NGS) analysis might be particularly advantageous in genetically heterogeneous conditions, such as familial hypertrophic cardiomyopathy (HCM), in which a considerable proportion of patients remain undiagnosed following conventional Sanger sequencing testing. In the present study we present an Italian family with atypical HCM in which a novel disease-causing variant in alpha-actinin2 (ACTN2) was identified by NGS.
Methods and Results. A large family spanning four generations was examined, exhibiting an autosomal dominant cardiomyopathic trait comprising a variable spectrum of a) mid-apical HCM with restrictive evolution with marked bi-atrial dilatation, b) early onset atrial fibrillation and atrioventricular block, and c) left ventricular (LV) noncompaction. In the proband, 36 candidate genes for HCM (based on published reports) were studied by targeted resequencing with a customized enrichment system. Following bioinformatics analysis, four likely pathogenic variants were identified: TTN NM_003319.4:c.21977G>A (p.Arg7326Gln); TTN NM_003319.4:c.8749A>C (p.Thr2917Pro); ACTN2 NM_001103.2:c.683T>C (p.Met228Thr) and OBSCN NM_052843.2:c.13475T>G (p.Leu4492Arg). The novel variant ACTN2 p.Met228Thr, located in the Actin–binding domain, proved to be the only mutation fully co-segregating with the cardiomyopathic trait in 18 additional family members (of whom 11 clinically affected). ACTN2_Met228Thr was absent in 570 alleles of healthy controls and in 1000 Genomes Project, and was labelled as “Causative” by in silico analysis using PolyPhen-2, as “Deleterious” by SIFT and as “Disease-Causing” by MutationTaster.
Conclusions. A targeted NGS approach allowed the identification of a novel ACTN2 variant associated with mid-apical HCM and juvenile onset of atrial fibrillation, emphasizing the potential of this technique in HCM diagnostic screening.
Word count= 249

CLINICAL PHENOTYPE AND OUTCOME OF
HYPERTROPHIC CARDIOMYOPATHY ASSOCIATED WITH
SARCOMERE THIN FILAMENT GENE MUTATIONS
Raffaele Coppini1, Benedetta Tomberli1, Caroly Ho2, Euan Ashley3, Sharlene Day4, Cecilia Ferrantini1, Francesca Girolami1, Sara Bardi1, Franco Cecchi1,
Corrado Poggesi1, Jil Tardiff5, Iacopo Olivotto1.
1. Centro Interuniversitario di Medicina Molecolare e Biofisica Applicata (CIMMBA), University of Florence, Italy.
2. Referral Center for Cardiomyopathies, Careggi University Hospital, Florence, Italy.
3. Brigham and Women's Hospital, Boston, US. 4. Cardiovascular Medicine, Stanford Medical Center, Stanford, US. 5. University of Michigan Medical Center, Ann Arbor, US. 6. Department of Cellular and Molecular Medicine, University of Arizona,
Tucson, US.

ABSTRACT
Purpose: HCM may be associated with a variety of causal genes. In a distinct subgroup
of patients, HCM is caused by mutations of sarcomere thin filament protein genes,
including cardiac troponin T and I (TNNT2, TNNI3), tropomyosin (TPM1) and cardiac
actin (ACTC). The phenotype and clinical course of thin filament HCM, compared to the
more prevalent thick filament form, is still unresolved. We sought to characterize the
phenotypic correlates and outcome of 84 patients with thin filament HCM, followed for
an average of 5 years, compared to 157 HCM patients with thick filament-‐related
disease. Results. Compared with thick filament patients, patients with thin filament
HCM showed: (1) lesser maximal LV wall thickness values (18±5 vs 24±6 mm, p<0.001),
with larger prevalence of atypical distribution of hypertrophy; (2) higher rate of adverse
events, including cardiovascular death, resuscitated cardiac arrest, appropriate ICD
shock, nonfatal stroke or progression to NYHA class III/IV (annual event rate 4.6% vs
2.7%, p=0.032); (3) higher likelihood of progression towards LV systolic dysfunction
(EF<50%) and/or restrictive diastole, the so-‐called end-‐stage of HCM (30% vs 18%,
p=0.02). Furthermore patients with thin filament HCM had 2.2 fold higher prevalence of
triphasic LV filling pattern (26% vs 12% in thick filament HCM, p<0.001). Conclusions.
HCM related to thin filament mutations is characterized by significant differences in
phenotype and clinical course compared to the more prevalent thick-‐filament HCM,
including higher risk of adverse events related to ventricular arrhythmias as well as
progressive LV dysfunction. Triphasic LV filling is distinctively common in thin filament
HCM, pointing to peculiar molecular determinants of diastolic dysfunction in this genetic
subset

Outcome of Hypertrophic Cardiomyopathy
associated with sarcomere protein gene mutations:
impact of the Implantable Cardioverter-‐Defibrillator
Tomberli B (MD1), Ferrantini C (MD, PhD2), Coppini R (MD, PhD3), Girolami F (BSc5), Pieragnoli P (MD6), Olivotto I (MD4), Padeletti L (MD6), Cecchi F (MD1).
(1) University of Florence, Department of Clinical and Experimental Medicine, Florence, Italy (2) University of Florence, Department of Human Physiology, Florence, Italy (3) University of
Florence, Department of Pharmacology, Florence, Italy (4) Careggi University Hospital, Referral Center for Cardiomyopathies, Florence, Italy (5) Careggi University Hospital, Genetic Unit, Florence, Italy (6) Careggi University Hospital, Electrophysiology Unit, Florence, Italy
Purpose
To date, large clinical studies on hypertrophic cardiomyopathy (HCM) have assessed outcome irrespective of genetic background. However, the large proportion of patients with no detectable sarcomere myofilament gene mutations, possibly including phenocopies, may confound our perception of the the natural history of HCM due to sarcomeric myofilament mutations. We therefore investigated the clinical features and outcome of 250 HCM patients followed 6±3 years after genetic identification of such mutations. The impact of the implantable cardioverter-‐defibrillator (ICD), both in terms of appropriate intervention rates and adverse effects, was specifically assessed.
Results
Overall, 16 pts (6%; incidence 1% per year) died of cardiovascular causes, including progressive heart failure (n=7), SCD (n=5), ischemic stroke (n=1) and other non-‐cardiac diseases (n=3). Of these, 9(5%) occurred in the subset of patents without ICD (group 1) including 3 sudden deaths; while 7 (11%) occurred among pts with ICD (group 2), of whom 3 had prior appropriate interventions. Two of the deaths in group 2 were sudden, occurring despite the device: unfortunately, neither an autopsy nor an ICD interrogation was performed in these pts, and the exact causes of death could not be ascertained. At final evaluation, 6±3 years after genetic testing, 34 pts (14%) were in NYHA class III/IV and 25 (10%) had overt LV systolic dysfunction (LVEF <50%). A total of 53 (21%) pts had received invasive management of LV obstruction by surgical myectomy (n=33) or alcohol septal ablation (n=20). Survival in the two groups was comparable (p=0.15) despite more severe clinical profile and greater prevalence of end-‐stage in group 2. No difference in survival was observed based on the affected gene. Among the 64 pts with ICD, only 7 (11%; 2 in primary and 5 in secondary prevention) experienced appropriate shocks for VT of VF, with an

overall annual incidence of 2%. However, 16 pts (25% of the ICD subset, including 2 with appropriate shocks) experienced device-‐related complications such as inappropriate ICD interventions (n=10; including 4 with electric storms due to lead fracture), infections (n=4) and lead dislocation (n=6).
Conclusions
In HCM patients due to sarcomere myofilament mutations, cardiovascular mortality and sudden cardiac death was low even in the presence of multiple risk factors. End-‐stage progression appeared to be common, supporting the hypothesis of long-‐term disease progression to heart failure. The ICD allowed favourable survival rates in a subset of high risk HCM patients, at the cost of considerable complication rates, even when no appropriate interventions were recorded. Sudden cardiac death may occur even after ICD implantation.

A novel desmoplakin dominant mutation
responsible for Carvajal/Naxos syndrome
identified by exome sequencing
B. Tomberli (1), A. Fornaro (1), S. Bardi (2), F. Torricelli (2), M. Benelli (2), C. Pescucci (2), F. Cecchi (3), F. Girolami (2), I. Olivotto (1)
(1) Careggi University Hospital, Referral Center for Cardiomyopathies, Florence, Italy
(2) Careggi University Hospital, Genetic Unit, Florence, Italy (3) University of Florence, Department of Clinical and Experimental Medicine, Florence, Italy
Purpose
Naxos and Carvajal syndrome are rare forms of recessive Arrhythmogenic Right Ventricular
Cardiomyopathy (ARVC), characterized by ventricle dysplasia/dilated cardiomyopathy and
ventricular arrhythmias, associated with palmoplantar keratoderma and woolly hair.
We report the case of an Italian 37-‐year-‐old woman with a form of ARVC characterized by
phenotypic features overlapping Naxos/Carvajal syndrome, with a dominant model of
inheritance. Whole Exome Sequencing (WES) allowed the identification of a novel mutation in
the desmoplakin gene (DSP), which was previously missed by traditional Sanger sequencing.
Methods
The woman was admitted to the hospital after resuscitated cardiac arrest. Clinical and
instrumental evaluations showed mild biventricular systolic dysfunction, with dilated
ventricles and areas of bulging in the lateral wall of the right ventricle. She also had
palmoplantar keratoderma and family history of juvenile sudden cardiac death (brother, 28
year-‐old). Traditional Sanger sequencing failed to identify any mutations in the genes usually
associated with ARVC (transforming growth factor beta-‐3, ryanodine receptor 2, desmoplakin,
plakophilin-‐2, desmoglein-‐2, desmocollin-‐2 transmembrane protein 43, junction plakoglobin
protein). Whole exome sequencing was therefore performed on the Illumina HiScan SQ

platform. After bioinformatics analysis, data were filtered by frequency, function,
conservation scores and predicted deleteriousness by means in silico 3D modeling.
Prioritization of variants was performed by Gene Prioritization Portal and then cosegregation
was tested in the family.
Results
Family’s pedigree showed a dominant model of inheritance. 65993 variants were individuated
by bioinformatics analysis; after data filtering and integration of filtered results with patient’s
clinical features, 4 potential mutations were selected. Only two of those variants cosegregated
with the disease in the family: c.878A>T, p.Glu293Val in the DSP gene and c.626A>C,
p.Tyr209Ser in the cytochrome c oxidase assembly gene (SCO2). Since the cardiomyopathy
related to SCO2 gene mutations is usually associated with encephalopathy and is fatal in
children, we excluded the causative role of such gene in our family. Therefore, we considered
the DSP-‐Glu293Val variant as disease-‐causing and we hypothesized a modifier role for the
SCO2 variant.
Conclusions
We identified a novel DSP gene variant associated to a rare dominant form of Carvajal/Naxos
syndrome, by Whole Exome Sequencing. Our study confirmed the high efficiency of NGS, both
in terms of accuracy and sensitivity, when compared to traditional sequencing. However, due
to the huge amount of data arising from NGS analysis, bioinformatics management becomes
crucial and traditional genetic rules are still needed to understand and interpret the real
significance of new variants.

PREVALENCE AND CLINICAL SIGNIFICANCE
OF HEART FAILURE SYMPTOMS
IN PATIENTS WITH NONOBSTRUCTIVE HCM
E.J. Rowin, B. Tomberli, A. Arretini, B.J. Maron, S.A. Casey, R. Garberich, M.S. Link, R.H.M.
Chan, E. Appelbaum, J.R. Lesser, J.E. Udelson, M.S. Maron, F. Cecchi, I. Olivotto.
Background. One-third of patients with HCM do not have left ventricular outflow tract (LVOT)
obstruction and the natural history of this subgroup remains undefined. We therefore sought to
evaluate clinical features and outcome of 416 HCM patients, followed for 6.3±2.1 yrs.
Furthermore, we assessed the prevalence of heart failure (HF) symptoms (NYHA class III/IV),
with regard to the presence or absence of LVOT obstruction, in order to evaluate its clinical
implications.
Results. At baseline, LVOT obstruction (≥30 mmHg at rest or following exercise) was present in
436 (64%), while 242 (36%) were non-obstructive. At the end of follow-up, 231 patients (34%)
developed progressive HF symptoms; of these, 201 were obstructive and 30 nonobstructive (87
vs 13%, p<0.001). However, nonobstructive patients who developed HF symptoms, were more
likely to require heart transplant compared to obstructive patients (0.7 vs 2.1%; p=0.02).
Furthermore, nonobstructive HCM patients showed increased risk for end-stage progression
(EF<50%; 8% vs 4% for patients with obstruction; p=0.05). Overall mortality and HCM-related
event rate were comparable in the two subgroups.

Conclusions: Development of HF symptoms in nonobstructive patients, although less common
than in the obstructive group, was associated with higher risk of end-stage progression and the
need of heart transplant. While HF occurring in the context of LVOT obstruction, usually
associated with little or no fibrosis within the LV, is possibly reversible following surgery, HF in
nonosbtructive patients is often subtended by huge LGE and it is usually progressive and
irreversible. This observation suggests the need for close surveillance, even though systolic
function is preserved, and potentially early initiation of antiremodeling treatment.

MYBPC3-‐Glu258Lys RELATED HYPERTROPHIC CARDIOMYOPATHY:
A FOUNDER EFFECT IN TUSCANY
Francesca Girolami BSc (*), Sara Bardi BSc (*), Laura Berti BSc (*),Tommaso Pippucci (°) , Benedetta
Tomberli MD (*), Franco Cecchi MD(*), Francesca Torricelli BSc(*), Iacopo Olivotto MD (*).
(*) Referral Center for Myocardial Diseases, Genetic Diagnostic Unit, Careggi University Hospital,
Florence, Italy;
(°) Medical Genetic Laboratory, Policlinico S. Orsola-‐Malpighi ,University of Bologna, Italy.
ABSTRACT
Background. MYBPC3 is the most common gene involved in HCM. Even if the
majority of mutations identified are novels, it is possible to find some recurrent
pathogenic mutations. A few founding mutations have been defined, and the most
of them are in MYBPC3 gene. The occurrence of a founder effect has never been
reported in Italy so far.
Objectives. The aim of the present study was to describe a founder effect in Tuscany
caused by MYBPC3-‐Glu258Lys mutation.
Methods and Results. A total 635 Italian unrelated index HCM patients underwent
screening for myofilament gene mutations by direct sequencing of 8 genes, including
MYBPC3, MYH7, TNNI2, TNNI3, ACTC, TPM1, MYL2 and MYL3. In 211/635 (33%) was
identified a MYBPC3 mutation and 54/635 (8.5%) consecutive unrelated probands,
all were born in Tuscany, harboured the same MYBPC3 Glu258Lys pathogenic
mutation. Disease haplotypes analysis examined in 4 families using 9 intragenic loci,

showed a unique haplotype extended for a 2Mb genomic region. To confirm these
results haplotype analysis was extended to all index cases and in 100 healthy adult
subjects were born in the same geographical region of Tuscany.
Conclusions. MYPC3-‐Glu258Lys mutation is a founder effect in Tuscany HCM
population. Further studies in family members are currently ongoing, in order to
assess the cardiac phenotype in mutation carriers and potential modifier SNPs or
genes.

SURVIVAL OF CHEMOTHERAPY-‐INDUCED CARDIOMYOPATHY
IN THE LAST TWO DECADES.
Alessandra Fornaro, Gabriele Castelli, Mauro Ciaccheri,
Benedetta Tomberli, Iacopo Olivotto, Gian Franco Gensini.
Background: cardio-‐oncology is an emerging field with an increasing amount of
patients suffering from cardiotoxic effects of antineoplastic drugs. In this context
chemotherapy-‐induced cardiomyopathy (CI-‐CM) is a challenging problem and has
been considered one of the most life-‐threatening conditions so far.
Aim: to analyze the actual survival of a cohort of CI-‐CM pts in comparison to a
population of pts with idiopathic dilated CM (IDCM), both followed at our Center in
the last two decades.
Methods: we consecutively enrolled 62 pts (32,3% male, age 49±14 y) presenting at
our Center with CI-‐CM from 1990 to 2012. Global follow up was 70±62 months. At
baseline mean left ventricular (LV) ejection fraction (EF) was 39.5±10%, mean LV end
diastolic index 32.1±5 mm/m2, mean NYHA class 2.6±1.2. Hypertension was present
in 15%, diabetes in 7% and hypercholesterolemia in 10% of pts; 10% of pts were
smokers. The majority of pts received chemotherapy (mainly anthracyclines) due to
Non-‐Hodgkin’s Lymphoma (64%); 12% were treated because of Hodgkin’s disease,
10% due to breast cancer while a minority of pts were treated because of leukemia,
osteosarcoma, hepatocellular carcinoma and glioblastoma. Twenty-‐three pts
received a previous antineoplastic treatment regimen, while 41% had a history of
mediastinum radiotherapy (RT). 63% of pts showed symptoms of heart failure (HF).
Two or more antineoplastic treatment and RT related complications, beyond LV
dilatation and dysfunction, were evident in 21% of pts. Mean time from last
antineoplastic treatment dose and CI-‐CM diagnosis was 39±64 months with most

cases reporting a late-‐onset cardiotoxicity with LV dilatation/dysfunction occurring
even after 23 years. At the end of follow up 64,5% of pts were receiving ACE-‐
inhibitors, 18% ARBs (ACEI+ARBs: 82%) and 90% betablockers (BB) with a mean dose
of 67%, 75% and 39% of the international HF-‐guidelines recommended dosage,
respectively. Mineral corticoid receptor antagonists (MRA) were administered in
45% of pts; 76% of pts received a combination of ACEI/ARBs plus BB while only 12%
of cases received a combination of ACEI/ARBs+BB+MRA.
Results: twenty-‐five patients (40,3%) reached the combined end-‐point of all cause
death/heart transplantation. Causes of death were refractory HF in 23.8%, cancer-‐
related in 14.3% and other (non cardiac) in 61.9% of pts. Overall survival was 72,6%
at 60 months, 47,7% at 120 months, considerably lower compared to a matched
population of 451 patients with IDCM, enrolled and followed at our Center from
1990 (86,1% and 68,8%, respectively), but far better in respect to data available in
current literature.
Conclusions: CI-‐CM, even when treated with optimal HF therapy, portends a severe
prognosis, especially when compared to IDCM. Our data show anyway an
improvement in survival with respect to data available in current literature.

334

335
9. Acknowledgments

336
"Let us be grateful to people
who make us happy;
they are the charming gardeners
who make our souls blossom"
Marcel Proust

337
9.1 Acknowledgments
Love is all around me
Love for medicine, research and cardiomyopathies
Foremost, I would like to express my sincere gratitude to my supervisor and mentor Dr. Iacopo Olivotto for his continuous support during my Ph.D study, for his patience, motivation, enthusiasm and immense knowledge. He taught me everything I know, from reading an ECG to writing a paper (although I am still working on it!). He has been invaluable on both, academic and personal level. Most importantly, he cares for patients and he showed me the importance of being a good person in order to become a good doctor. His guidance helped me all along the research and writing of this thesis. I could not have asked for a better mentor.
I am also very grateful to Prof. Franco Cecchi for instilling in me a profound sense of love for research and cardiomyopathies. He also gave me the opportunity to start this experience and he helped me understand who I want to be in my life.
Special thanks to Katia Baldini, our professional nurse, who helps us with patients and research, and supports us even during our crazy and over-‐crowded outpatients sessions.
Many thanks to Dr. Francesca Girolami, our passionate geneticist, for the incredible amount of work she did in these years. Without her help none of this would have been possible.
To my collegues and friends, Dr. Gabriele Castelli, Dr. Alessandra Fornaro, Dr. Ceclia Ferrantini and Dr. Raffaele Coppini, for their constant help in clinical and research work.

338
Love for people and family
Last but not least, I would like to thank my entire family, especially those who are no longer here among us. I wish they could have lived a few more years to see me now: married, graduated and PhD. Thanks to every single component of my very big and chaotic family, for the unconditioned trust, great respect and immense love.
Special thanks to Filippo, my soul mate and husband. He has been a true and great supporter and he has unconditionally loved me during my good and bad times. I truly thank him for sticking by my side when I was happy and satisfied, and even when I was irritable and depressed. I want to thank him also for his patience during the innumerable evenings and weekends I spent studying, writing papers and doing research for this thesis.




















