Two new hydrothermally synthesised hexamine bridged L–M–L type coordination polymers:...
11
Two new hydrothermally synthesised hexamine bridged L–M–L type coordination polymers: Characterisation and magneto-structural correlation Aurkie Ray a , Joy Chakraborty a , Brajagopal Samanta a , Santarupa Thakurta a , C. Marschner b , M. Salah El Fallah c , Samiran Mitra a, * a Department of Chemistry, Jadavpur University, Kolkata 700 032, West Bengal, India b Institut fu ¨ r Anorganische Chemie, Technische Universita ¨ t Graz, Stremayrgasse 16, A-8010 Graz, Austria c Departamento de Quı ´mica Inorga ´ nica, Universitat de Barcelona, Martı ´ i Franquees, 1-11, 08028 Barcelona, Spain Received 1 March 2007; received in revised form 5 September 2007; accepted 27 September 2007 Available online 10 October 2007 Abstract Two new L–M–L type transition metal coordination polymers [M(C 6 H 12 N 4 )(NCO) 2 (H 2 O) 2 ] n , where M = Co(II) (1) and Ni(II) (2), have been synthesised under controlled hydrothermal condition and characterised spectroscopically and by thermal analyses. Here hmt or hexamethylenetetramine has behaved as a neutral organic bidentate spacer molecule. Both the complexes crystallise in the mono- clinic system as confirmed from single crystal X-ray diffraction studies. Cryomagnetic susceptibility measurements over a wide range of temperature (2–300 K) under 0.5 T magnetic fields show a weak antiferromagnetic interaction of J = 0.65 cm 1 for 1 and 1.6 cm 1 for 2. The values have been given a favorable support by weak covalent and H-bonding interactions between octahedral M(II) metal centers as revealed from X-ray structure determination. The high dimensionality of the structures is probably a manifestation of exten- sively weak covalent interactions and H-bondings. Ó 2007 Elsevier B.V. All rights reserved. Keywords: Hydrothermal; L–M–L type polymers; H-bonding; Co(II); Ni(II); Hmt; Antiferromagnetic 1. Introduction Intense research interest is currently being focused on the crystal engineering of supramolecular architectures organised by coordinate covalent bonds or supramolecular contacts (such as hydrogen bonding, p–p or van der Waal’s interactions) [1]. This field got remarkable growth since the development of important minerals such as zeolites and other nanoporous materials [2–6] with interesting one-, two- or three-dimensional architectural frameworks bear- ing large cavities or channels for their possible applications in separation processes and catalysis [7]. Although supra- molecular polymers have been constructed largely with the use of symmetrically bridging neutral organic spacer ligands (i.e. 2-connectors, trigonal 3-connectors, tetrahe- dral or square-planar 4-connectors) [2,8–12] alone, but their use along with other ligands (e.g. thiocyanate, azide [13–15] or aromatic polycarboxylic acid anions [16–19], etc.) in combination, is emerging nowadays as one of the several popular strategies to increase the dimensionality of polymers. Hexamethylenetetramine (1,3,5,7-tetraazatri- cyclo[3.3.1 2,7 ]decane) or (hmt) can be regarded as a very efficient polydentate organic spacer molecule in this con- text. It can connect through self-assembly with two, three or all four of its tertiary nitrogen atoms to furnish a variety of inorganic coordination polymers bearing interesting topologies involving several transition and non-transition metals [1,20]. Among them, especially the contribution 0020-1693/$ - see front matter Ó 2007 Elsevier B.V. All rights reserved. doi:10.1016/j.ica.2007.09.041 * Corresponding author. Tel.: +91 033 2414 6666x2505; fax: +91 033 2414 6414x6210. E-mail address: [email protected] (S. Mitra). www.elsevier.com/locate/ica Available online at www.sciencedirect.com Inorganica Chimica Acta 361 (2008) 1850–1860
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Two new hydrothermally synthesised hexamine bridged L–M–L type coordination polymers:...

Available online at www.sciencedirect.com
www.elsevier.com/locate/ica
Inorganica Chimica Acta 361 (2008) 1850–1860
Two new hydrothermally synthesised hexamine bridged L–M–Ltype coordination polymers: Characterisation and
magneto-structural correlation
Aurkie Ray a, Joy Chakraborty a, Brajagopal Samanta a, Santarupa Thakurta a,C. Marschner b, M. Salah El Fallah c, Samiran Mitra a,*
a Department of Chemistry, Jadavpur University, Kolkata 700 032, West Bengal, Indiab Institut fur Anorganische Chemie, Technische Universitat Graz, Stremayrgasse 16, A-8010 Graz, Austria
c Departamento de Quımica Inorganica, Universitat de Barcelona, Martı i Franquees, 1-11, 08028 Barcelona, Spain
Received 1 March 2007; received in revised form 5 September 2007; accepted 27 September 2007Available online 10 October 2007
Abstract
Two new L–M–L type transition metal coordination polymers [M(C6H12N4)(NCO)2(H2O)2]n, where M = Co(II) (1) and Ni(II) (2),have been synthesised under controlled hydrothermal condition and characterised spectroscopically and by thermal analyses. Herehmt or hexamethylenetetramine has behaved as a neutral organic bidentate spacer molecule. Both the complexes crystallise in the mono-clinic system as confirmed from single crystal X-ray diffraction studies. Cryomagnetic susceptibility measurements over a wide range oftemperature (2–300 K) under 0.5 T magnetic fields show a weak antiferromagnetic interaction of J = �0.65 cm�1 for 1 and �1.6 cm�1
for 2. The values have been given a favorable support by weak covalent and H-bonding interactions between octahedral M(II) metalcenters as revealed from X-ray structure determination. The high dimensionality of the structures is probably a manifestation of exten-sively weak covalent interactions and H-bondings.� 2007 Elsevier B.V. All rights reserved.
Keywords: Hydrothermal; L–M–L type polymers; H-bonding; Co(II); Ni(II); Hmt; Antiferromagnetic
1. Introduction
Intense research interest is currently being focused onthe crystal engineering of supramolecular architecturesorganised by coordinate covalent bonds or supramolecularcontacts (such as hydrogen bonding, p–p or van der Waal’sinteractions) [1]. This field got remarkable growth since thedevelopment of important minerals such as zeolites andother nanoporous materials [2–6] with interesting one-,two- or three-dimensional architectural frameworks bear-ing large cavities or channels for their possible applicationsin separation processes and catalysis [7]. Although supra-
0020-1693/$ - see front matter � 2007 Elsevier B.V. All rights reserved.
doi:10.1016/j.ica.2007.09.041
* Corresponding author. Tel.: +91 033 2414 6666x2505; fax: +91 0332414 6414x6210.
E-mail address: [email protected] (S. Mitra).
molecular polymers have been constructed largely withthe use of symmetrically bridging neutral organic spacerligands (i.e. 2-connectors, trigonal 3-connectors, tetrahe-dral or square-planar 4-connectors) [2,8–12] alone, buttheir use along with other ligands (e.g. thiocyanate, azide[13–15] or aromatic polycarboxylic acid anions [16–19],etc.) in combination, is emerging nowadays as one of theseveral popular strategies to increase the dimensionalityof polymers. Hexamethylenetetramine (1,3,5,7-tetraazatri-cyclo[3.3.12,7]decane) or (hmt) can be regarded as a veryefficient polydentate organic spacer molecule in this con-text. It can connect through self-assembly with two, threeor all four of its tertiary nitrogen atoms to furnish a varietyof inorganic coordination polymers bearing interestingtopologies involving several transition and non-transitionmetals [1,20]. Among them, especially the contribution

A. Ray et al. / Inorganica Chimica Acta 361 (2008) 1850–1860 1851
from silver(I) is noteworthy in the chemical literature([1,20d,20g] and related references therein). The self-assem-bly of all these metallo-organic frameworks is highly influ-enced by several factors such as solvent system [2,8],template effect [7,11,21], pH of the solutions [7,11], stericparameters of the counter ion [22] and above all theimposed reaction environment. Therefore, more stress isneeded to extend the knowledge of the relevant structuraltypes and establish proper synthetic strategies leading tothe desired species.
In our continuing work [13,20a,20b] on the explorationof the combination effect of different ligands and coligandson transition metals, we have established a feasible routeleading to novel hydrogen bonded frameworks. We havestudied the combination effect of hmt along with cyanateas anionic ligand on Co(II) and Ni(II) metal ions. It success-fully ends to a construction of two new high dimensionalityCo(II) and Ni(II) metallo-organic polymers containinghexamethylenetetramine as a neutral organic spacer.
We report herein the controlled hydrothermal synthesisand systematic characterisation of [MII(C6H12N4)(NCO)2-(H2O)2]n, a L–M–L type interesting polymer, where botha neutral spacer and an anionic ligand have been usedsimultaneously to explore their combination effect on thetransition metal ions i.e. Co(II) and Ni(II). In the poly-meric chain, M(II) is in an octahedral environment assem-bled by a l-(N,N 0) bridging hexamine neutral spacer ligandwhile the cyanate is coordinated as a terminal anionicligand to the metal center. The stability of the system is lar-gely enhanced through O–H� � �N or O–H� � �O inter-chainand C–H� � �O and C–H� � �N intra-chain hydrogen bonds.Although there are ample interesting examples of IPOSsystems involving hmt as a spacer, their systematic charac-terisation, spectral and complete magneto-structural corre-lation studies is not a field which has been much explored,especially for transition metals.
2. Experimental
2.1. Materials
All the chemicals and solvents employed for this synthe-sis were of analytical grade. Cobalt(II) nitrate, hexahy-drate; nickel(II) nitrate, hexahydrate; sodium cyanate andhexamethylenetetramine (hmt) were purchased from E.Merck and used as received without further purification.
2.2. Physical measurements
The Fourier Transform Infrared spectra (4000–400 cm�1) of the complexes were recorded on a Perkin–Elmer Spectrum RX I FT-IR system with solid KBr disc.The electronic spectra were recorded on a Perkin–Elmerk – 40 UV/Vis-spectrometer using HPLC grade methanolas solvent. C, H, N microanalyses were carried out witha Perkin–Elmer 2400 II elemental analyser. The thermalanalyses were done on a Mettler Toledo TGA/SDTA-
851e thermal analyser system in a dynamic atmosphere ofdinitrogen (flow rate: 30 cm3min�1) in a platinum crucibleat a heating rate of 10 �C min�1.
Cryomagnetic susceptibility measurements for the com-plexes were carried out at the Servei de Magnetoquımica ofthe Universitat de Barcelona, Spain with a QuantumDesigned SQUID MPMS-XL susceptometer apparatuswhich works in the range 2–300 K under magnetic fieldof approximately 0.5 T. Diamagnetic corrections were esti-mated from Pascal’s tables.
2.3. Synthesis of the complexes
2.3.1. [Co(hmt)(NCO)2(H2O)2]n (1)The complex was prepared hydrothermally under con-
trolled condition. 0.4365 g (1.5 mmol) Co(NO3)2 Æ 6H2O,0.1982 g (3.05 mmol) NaNCO, 0.2158 g (1.54 mmol) hmtand 15 cm3 of water were loaded into a 23 cm3 Teflon-linedstainless steel vessel, which was then sealed and placed in acomputer programmable furnace. The resulting mixturewas heated at the rate of 100 �C/h up to 150 �C, kept for10 h, then cooled to 120 �C at the rate of 4 �C/h, held foranother 10 h, followed by further cooling at the same rateto room temperature to produce an approximately 80%yield based on cobalt(II), as rosy pink colored needleshaped single crystals. They were later collected bymechanical isolation and washed with water–ethanol (2:1v/v) mixed solvent, and then dried in an oven keeping themat 40 �C for 10 min. Anal. Calc. for [C8H16CoN6O4]: C,30.10; H, 5.05; N, 26.33. Found: C, 30.12; H, 5.04; N,26.36%.
2.3.2. [Ni(hmt)(NCO)2(H2O)2]n (2)
This complex was prepared following the exactly sameprocedure as described above for complex 1 withNi(NO3)2 Æ 6H2O, NaNCO and hexamethylenetetraminetaken in the proportion of 0.4362 g (1.5 mmol), 0.1969 g(3.03 mmol) and 0.2103 g (1.5 mmol), respectively. On slowcooling to ambient temperature pale green needle shapedsingle crystals were obtained with a yield of 87% basedon nickel(II). Suitable crystals were later collected bymechanical isolation and washed with water–ethanol (2:1v/v) mixed solvent, dried in an oven keeping them at40 �C for 10 min. Anal. Calc. for [C8H16NiN6O4]: C,30.13; H, 5.06; N, 26.35. Found: C, 30.16; H, 5.07; N,26.32%.
2.4. Crystal structure determination
The X-ray diffraction experiment was carried out at100 K on a rosy pink needle shaped single crystal(0.34 mm · 0.28 mm · 0.15 mm) of complex 1 and a palegreen needle shaped single crystal (0.21 mm · 0.12 mm ·0.10 mm) of complex 2. The single crystal was mountedon the tip of a glass fibre and was exposed to a graphitemonochromatised Mo Ka radiation (k = 0.71073 A)emerging from a fine-focus sealed tube. The intensity data

1852 A. Ray et al. / Inorganica Chimica Acta 361 (2008) 1850–1860
collection was performed with a Bruker-AXS SMARTCCD diffractometer using the x-scan technique. Standardreflections were measured at fixed intervals during the datacollection to check the stability of the crystal under obser-vation. However, no significant loss of intensity was notedfor both the crystals. A total of 4747 reflections (1258 inde-pendent reflections, Rint = 0.0626) for 1 and 4202 reflec-tions (1071 independent reflections, Rint = 0.0595) for 2
were collected in the range of 2.9� < h < 26.3� for 1 and3.0� < h < 25.0� for 2, applying the boundary conditionI > 2r(I). The data were reduced to F 2
o and corrected formultiscan absorption effects with Bruker SHELXTL [23] andSADABS [24] softwares, respectively. XPREP [25] was usedfor space group assignment, statistics and numericalabsorption corrections. Unit cell refinements were doneusing the Bruker XCANS [25] software. Structure wassolved by direct methods with SHELXS-97 [26] and refinedto convergence on F2 using SHELXL-97 [27]. Maximumand minimum peaks (e A�3) in the final difference Fouriersynthesis were 0.790 and �0.520, 0.56 and �1.02 for 1, 2,respectively. The functions minimised were
Pw½jF oj2�
½jF cj2�2, where w ¼ ½r2ðF 2oÞ þ ð0:0370P Þ2 þ 6:8075P ��1 for
1; w ¼ ½r2ðF 2oÞ þ ð0:0243P Þ2 þ 15:9807P ��1 for 2; with
P = ([jFoj2 + 2jFcj2)/3 in both the cases. All non-hydrogenatoms were refined with anisotropic displacement parame-ters. The hydrogen atoms attached to the carbon wereincluded in the geometric positions and given thermalparameters equivalent to 1.2 times those of the atoms to
Table 1Crystal parameters of 1 and 2
Parameters 1 2
Empirical formula C8H16CoN6O4 C8H16NiN6O4
Formula weight 319.20 318.98Crystal system monoclinic monoclinicSpace group C2/c (No. 15) C2/c (No. 15)a (A) 9.2664(19) 9.2237(18)b (A) 11.160(2) 11.158(2)c (A) 12.510(3) 12.358(3)a (�) 90 90b (�) 107.25(3) 108.16(3)c (�) 90 90V (A3) 1235.5(4) 1208.5(5)Z 2 4Temperature (K) 100(2) 100(2)kMo Ka (A) 0.71073 0.71073Dcalc (g cm�3) 1.716 1.753l (mm�1) 1.411 1.628F(000) 660 664h Range for data collection (�) 2.94–26.33 2.96–24.99Total data 4747 4202Unique data 1258 1071Goodness-of-fit on F2 1.20 1.331Observed data [I > 2r(I)] 1120 997Ra 0.0610 0.0766Rw
b 0.1167 0.1357Rint 0.0626 0.0595Dqmax (e A�3) 0.79 0.555Dqmin (e A�3) �0.518 �1.017
a R =P
(jFo � Fcj)/PjFoj.
b Rw = {P
[w(jFo � Fcj)2]/P
[wjFoj2]}1/2.
which they were attached. All the hydrogen atoms werelocated by difference Fourier synthesis reflections in calcu-lated positions to correspond to standard bond lengths andangles. Finally XCIF [25] was used to produce reports fromstandard CIF files. Selected crystallographic data, experi-mental conditions and some important features of thestructural refinements of both the complexes are summa-rised in Table 1.
3. Results and discussion
Both the complexes were synthesised under controlledhydrothermal condition by continuously varying the hmtconcentration in different sets while keeping others con-stant following the exactly same procedure described inour earlier works [20a,20b]. Surprisingly, it was observedthat the single crystals of the complexes were obtained asthe sole product specifically when the concentration rangereaches metal salt/hmt/NaNCO = 1:1:2.
3.1. Fourier Transform infrared spectra
The solid state Fourier Transform infrared spectra ofboth the complexes were recorded on a high resolutionFT-IR spectrophotometer for the range of 4000–400 cm�1. The spectral region for both the complexes ismore or less similar due to the exactly same coordinationmodes. The entire spectrum region can be split into threedistinctive zones with the characteristic absorptions [28,29].
1. High energy range: Several methylene groups of hexa-methylenetetramine molecules appear at 2900 cm�1
whereas a distinctive broad peak at 3440 cm�1 probablycorresponds to mstr(OH2) for different coordinated watermolecules.
2. Mid energy range: This region showed characteristicabsorption peaks for terminal NCO� groups at2103 cm�1 and 2113 cm�1, respectively, for 1 and 2.
3. Low energy range: The region showed a number of dis-tinctive peaks like, dCH2
: 1453 cm�1 and 1380 cm�1; dOH:1642 cm�1; dCN: 1220 cm�1, 1020 cm�1 and 1005 cm�1.
The peaks obtained in the different spectral regions cor-relate well with the observed values in the case of similarkind of systems found in the literature [20c,20e].
3.2. Electronic spectra
The UV–Vis spectra of both the complexes wererecorded in methanol HPLC grade solvent. Complex 1
shows a few strong and broad absorption bands at 8790,12030, 21000 cm�1, e = 8 L mol�1 cm�1, except m2 (veryweak). The absorption peaks can be tentatively assignedas typical d–d transition bands of an octahedral Co(II)ion [30,31]. The speculated transitions may be due to thefollowing: 4T1g(F)! 4T2g – m1, 4T1g(F)! 4A2g – m2,4T1g(F)! 4T1g(P) – m3.

A. Ray et al. / Inorganica Chimica Acta 361 (2008) 1850–1860 1853
Three distinct bands were observed for 2 supporting anOh Ni(II) ion. Strong and broad absorption bands arefound at 8550, 15540, 25000 cm�1, e = 10 L mol�1 cm�1,except m2 (very weak). Three absorption peaks can be ten-tatively assigned to d–d transitions for an Oh Ni(II) ion:3A2g! 3T2g – m1, 3A2g! 3T1g(F) – m3
2;A2g! 3T1g(P) – m3
[30,31].All the transition bands found for both the complexes
are in well accordance with those found in the literatureand it is seen that the hydrogen bonding network structureposses apparently no effect on electronic absorptionspectrum.
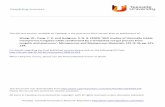
Fig. 1. ORTEP view of 1 with atom numbering scheme. Symmetry cod
Fig. 2. 2D sheet view of 1, showing th
3.3. Description of crystal structures [Co(hmt)(NCO)2-
(H2O)2]n (1) and [Ni(hmt)(NCO)2(H2 O)2]n (2)
The X-ray diffraction analysis reveals that both the com-plexes contain octahedral metal centers coordinated by twoterminal NCO� groups, two water molecules and two neu-tral hexamethylenetetramine molecules (Figs. 1, 4). Thetwo nitrogen atoms [N(3) and its symmetry related coun-terpart N(3**)] of terminal cyanate groups and the twooxygen atoms [O(1) and its symmetry related counterpartO(1**)] of coordinated water molecules, form the equato-rial plane around each metal center [Co(1)–N(3)(NCO�):
e (#) = x, �y, z + 1/2; (*) = �x, y, 1/2 � z; (**) = 1 � x, 1 � y, �z.
e hydrogen-bonding interactions.

Fig. 3. Packing view of 1 along the crystallographic a-axis.
Fig. 4. ORTEP view of 2 with atom numbering scheme. Symmetry code (#) = x, �y, z + 1/2; (*) = �x, y, 1/2 � z; (**) = 1 � x, 1 � y, �z.
1854 A. Ray et al. / Inorganica Chimica Acta 361 (2008) 1850–1860
2.097(3) A; Co(1)–O(1)(H2O): 2.087(3) A; Ni(1)–N(3)(NCO�): 2.022(5) A; Ni(1)–O(1)(H2O): 2.060(4) A].The trans-axial sites are occupied by the two nitrogenatoms [N(1), N(1**)] coming from pendent bridging hmtmolecules to provide each metal center a MIIN2(hmt)N2-(NCO�)O2(H2O) chromophore (Figs. 1, 4). The respective
bond lengths are Co(1)–N(1)(Hmt): 2.328(3) A and Ni(1)–
N(1)(Hmt): 2.262(5) A. The adjacent metal atoms arebridged together by neutral hmt molecules in a l-N,N 0
fashion with the occurrence of a zigzag 1-D infinite poly-meric chain, further carrying into a 2-D net with the helpof several weak inter-chain hydrogen bondings. ORTEPand PLATON [32] drawings of the chain with atom label-ing scheme are shown in Figs. 1, 4. The degrees of distor-

A. Ray et al. / Inorganica Chimica Acta 361 (2008) 1850–1860 1855
tion from the ideal octahedral geometry are reflected incisoid and transoid angles, all resembling the ideal valuesfor an octahedral arrangement. The shortest Co� � �Co dis-tance along the same chain is 6.255(3) A. The shortestNi� � �Ni distance along the same chain is 6.179 A. TheNCO� groups show almost linearity with N–C–O angleof 178.84(2)� in both the complexes. The terminal cyanateligand shows a slight deviation from linearity in theconnection with metal center showing a Co(1)–N(3)–C(5)angle of 164.2(3)� and Ni(1)–N(3)–C(5) angle of165.8(5)�, respectively, for 1 and 2. The hmt molecule con-nects two consecutive metal centers using only two out ofits four possible donor sites to link the adjacent unitstogether. All the water molecules and uncoordinated N-atoms are involved in intermolecular H-bondings (Figs.2, 5) making hmt a good tetrahedral template which isthe foremost requirement factor to establish an orderedsupramolecular structure formed by self-assembly. Eachof the infinite 1-D chains results in a 2-D polymeric net-work (Figs. 3, 6) through pronounced inter-chain O–H� � �N or O–H� � �O hydrogen bonding between the coordi-nated water molecules to the metal atom in one chain andN atom of the hmt in the other chain. Another inter-chainO–H� � �O hydrogen bonding is found between the coordi-nated water molecules of Co(II) or Ni(II) in one chainand the oxygen atoms from terminal cyanate ligands pres-ent in an adjacent chain [20a,20b,20c]. Apart from this,there are also three different types of intra-chain hydrogenbonds present in the system (C–H� � �O, C–H� � �N, etc.).Another noteworthy point is that the water molecules coor-
Fig. 5. 2D sheet view of 2, showing th
dinated to the alternative metal centers along the samechain are not behaving identically in the hydrogen bondingmode to the acceptor sites present in the adjacent chain(Figs. 2, 5). Such motif of inter-chain H-bonding can bedescribed in Etter’s graph set notation as R2
2 (12) [33]. Thesetwo types of hydrogen bonds (inter- and intra-chain) in thesystem give a high degree of thermal stability to the poly-mers and it is clearly reflected through their thermal anal-ysis. We see that the coordinated water molecules are lostat a much higher temperature than what is normallyobserved. A few relevant selected bond distances, bondangles and H-bonding parameters for the complexes aregiven in Tables 2 and 3, respectively.
3.4. Cryomagnetic susceptibility studies
The magnetic properties of complex 1 have been repre-sented by the plot of vMT versus T (vM is the molar mag-netic susceptibility for one Co(II) ion) where T variesfrom 2 K to 300 K (Fig. 7). The value of vMT at 300 K is2.616 cm3 K mol�1, which is larger than that expected forthe spin-only case (vMT = 1.87 cm3 mol�1 K, S = 3/2) indi-cating that an important orbital contribution is involved.The vMT value continuously decreases from room temper-ature to 1.232 cm3 K mol�1 at 2 K. The global feature ischaracteristic of weak antiferromagnetic interactions. ThevM curve is less indicative at room temperature as it startsfrom 0.0087 cm3 mol�1 and increases in a uniform way to0.628 cm3 mol�1 at 2 K. The absence of maxima in thesevM curves may indicate that the possible antiferromagnetic
e hydrogen-bonding interactions.

Fig. 6. Packing view of 2 along the crystallographic a-axis.
Table 2Selected bond lengths and bond angles for 1 and 2
Bond lengths (A) Bond angles (�)
Complex 1
Co(1)–O(1) 2.087(3) Co(1)–N(3)–C(5) 164.20(3)Co(1)–N(1) 2.328(3) N(3)–Co(1)–O(1) 90.75(13)Co(1)–N(3) 2.097(3) N(1)–Co(1)–O(1) 88.24(11)
N(3)–Co(1)–N(3**) 180.00O(1)–Co(1)–O(1**) 180.00(14)N(1)–Co(1)–N(1**) 180.00
Complex 2
Ni(1)–O(1) 2.060(4) Ni(1)–N(3)–C(5) 165.80(5)Ni(1)–N(1) 2.262(5) N(3)–Ni(1)–O(1) 90.90(2)Ni(1)–N(3) 2.022(5) N(1)–Ni(1)–O(1) 92.23(18)
N(3)–Ni(1)–N(3**) 180.00O(1)–Ni(1)–O(1**) 179.99(2)N(1)–Ni(1)–N(1**) 180.00
Translation of symmetry code to equivalent position.** 1 � x, 1 � y, �z.
Table 3Hydrogen bonding parameters for 1 and 2
D–H� � �A d(D–H) (A) d(H� � �A) (A) d(D� � �A) (A) \(DHA) (�)
Complex 1
O(1)–H(1)� � �N(2) 0.74(6) 2.05(5) 2.788(5) 171(5)O(1)–H(2)� � �O(2) 1.10(5) 1.78(5) 2.758(5) 146(4)C(1)–H(1**)� � �O(1) 0.9900 2.5200 2.937(5) 105.00C(3)–H(3**)� � �N(3) 0.9900 2.5400 3.182(6) 123.00C(3)–H(3*)� � �O(1) 0.9900 2.6000 3.158(5) 116.00
Complex 2
O(1)–H(1)� � �N(3) 0.75(10) 2.06(9) 2.781(8) 163(10)O(1)–H(2)� � �O(2) 0.84(8) 1.93(8) 2.761(6) 171(8)C(2)–H(2**)� � �O(1) 0.9900 2.4700 2.874(8) 104.00C(2)–H(2*)� � �O(1) 0.9900 2.4700 2.874(8) 104.00C(3)–H(3**)� � �N(1) 0.9900 2.4700 3.097(7) 121.00
Translation of symmetry code to equivalent position.* �x, y, 1/2 � z.
** 1 � x, 1 � y, �z.
1856 A. Ray et al. / Inorganica Chimica Acta 361 (2008) 1850–1860
coupling is very weak. The analysis of magnetic data forcobalt complexes is usually complicated by the fact thatsingle-ion effects, such as spin–orbit coupling, distortion
from regular stereochemistry, electron delocalisation, crys-tal field mixing of excited states into the ground state, affectthe magnetic properties in addition to possible magnetic

Fig. 7. Plot of vMT vs. T for complex 1.
Fig. 8. Plot of reduced magnetisation M/Nb vs. applied field H at 2 K forcomplex 1.
Fig. 9. Plot of vMT vs. T for complex 2. (Inset shows the plot of reducedmagnetisation M/Nb vs. applied field H at 2 K for complex 2).
A. Ray et al. / Inorganica Chimica Acta 361 (2008) 1850–1860 1857
exchange interactions [34a]. Considering the spin–orbitcoupling due to the 4T1g ground state for octahedral Co(II)complexes [34b,34c,34d,34e,34f], exact calculations forderiving the J parameter from experimental data in allthe temperature ranges are not possible unless for dinuclearcomplexes [35,36]. Other small polynuclear systems canalso be fitted through sophisticated computer programs,based on full diagonalisation methods at low temperatureregion with the effective spin of S 0 = 1/2 [37]. 1-D systemsof Co(II) are frequently associated with anisotropic Isingsystems, and they can be fitted in the low temperature zoneassuming again an effective spin S 0 = 1/2 [38]. Morerecently, Rueff et al. [38c,39], have proposed a phenomeno-logical approach for some low-dimensional Co(II) systemsthat allows to gain an estimate of the strength of the anti-ferromagnetic exchange interactions. They postulated thephenomenological equation as
vMT ¼ A expðE1=kT Þ þ B expðE2=kT Þ ð1Þwhere A + B equals the Curie constant (�2.8–3.4 cm3 mol�1 K for octahedral cobalt(II) ions), and E1,E2 represent the ‘‘activation energies’’ corresponding tothe spin–orbit coupling and the antiferromagnetic ex-change interaction, respectively. The E1/k which is the ef-fect of spin–orbit coupling and site distortion is of theorder of +100 K [38c–41]. Based on Eq. (1), an estimatedJ value for complex 1 has been calculated. The fit valuesfrom this procedure are A + B = 2.85 cm3 mol�1 K, E1/k = 53.22 K and E2/k = 0.47 K, corresponding toJ = �0.94 K (= �0.65 cm�1). This indicates the very weakantiferromagnetic exchange interaction in complex 1. Theattempts have been taken to fit complex 1, assuming thatisolated Co(II) ions with perfect octahedral geometry [34]did not give good fit. Thus, we can conclude that this com-plex behaves magnetically as one-dimensional system,which is weakly coupled by hexamethylenetetramine. Thereduced molar magnetisation (M/Nb) at 2 K per Co(II)reaches 1.82 (Fig. 8). This value is less than that expectedfor an isolated Co(II) ion (2.0–2.5 Nb) [39]. Confrontation
of the overall shape of the plot of 1 with the Brillouin one(solid plot) for one isolated Co(II) with S 0 = 1/2 andg = 4.3 at T = 2 K indicates slower magnetisation whichis consistent with a weak antiferromagnetic interaction.
The variable temperature magnetic properties of thepowdered sample of 2 have been represented as vMT versusT (Fig. 9). At room temperature, the vMT value is1.133 cm3 K mol�1. This value corresponds to one nicke-l(II) ion with two unpaired electrons with g = 2.13. Withdecreasing temperature, the vMT value remains almost con-stant until ca. 50 K and then it decreases very sharply,reaching a minimum of 0.252 cm3 K mol�1. The drop invMT at low temperature indicates the presence of a veryweak antiferromagnetic interaction between the nickel(II)ions. As shown in Fig. 4, the structure of 2 consists ofnickel entities linked by hexamethylenetetramine groupsgiving a one-dimensional zigzag polymeric chain. The

Fig. 10. Representative thermal analysis curve for both the complexes.
1858 A. Ray et al. / Inorganica Chimica Acta 361 (2008) 1850–1860
interactions through the inter-chain hydrogen bonds(Fig. 5) are negligible and only one coupling parameter(J) has been considered to interpret a possible magneticinteraction in 2. Taking into account this consideration,the experimental data were fitted to the Weng equation[40], based upon the spin Hamiltonian H ¼ J
PðSiSiþ1Þ,
where the nickel ion is assumed to be magneticallyisotropic:
v ¼ Ng2b2=KT ½2þ Axþ Bx2�=½3þ Cxþ Dx2 þ Ex3�In which, A = 0.019, B = 0.777, C = 4.346, D = 3.232,E = 5.834, and v = jJj/kT. The J value has been obtainedby minimizing the function: R ¼
PðvMT calc � vMT obsÞ2=P
ðvMT obsÞ2.The best fitting parameters obtained are J = �1.6 cm�1,
g = 2.13, and R = 3.11 · 10�5 indicating a very weakcoupling.
We think that in the case of complexes 1 and 2, we can-not give Haldane gap phenomenon for the complexity ofthe detection of the phenomenon for simple reasons. Inthe literature related to this phenomenon, most systemsstudied were mono-dimensional antiferromagnetic com-plexes with spin S = 1. These complexes exhibit a singletground state separated from the first triplet excited stateby an energy gap Eg (Haldane gap). In other words themagnetic susceptibility decreases to zero at T = 0 in theHaldane gap systems, while the magnetic susceptibility fallsto finite values in the case of the half-odd integer spin sys-tem [42].
In our case, the molar susceptibility vM for the Ni(II)complex tends to infinity at low temperature and not tozero. (see in Fig. 9, vMT is constant and goes to zero at verylow temperatures). It is practically impossible to observethis phenomenon when the exchange parameter J is soweak as in Ni(II) complex (�1.6 cm�1).
Regarding the Co(II) complex, the situation is worse,the spin–orbit coupling which is usually observed in octa-hedral S = 3/2 systems, interferes with the exchange cou-pling J, mainly if J is weak like in the case of the Co(II)complex (J < 1 cm�1). This makes the determination ofthe gap in systems similar to our Co(II) complex difficult.
In the magnetic study, we have not used the Fisher equa-tion in the fitting process, considering that the used equa-tions give better results, mainly in the case of the cobaltcomplex when we tried to determine also the spin orbitcoupling, which is responsible for the observed behaviorin 1. To the best of our knowledge, the Fisher equationgives good result except in S = 5/2 systems (classical onesin general).
The weak antiferromagnetic interaction was confirmedby magnetisation measurements at 2 K up to an externalfield of 5 T. At higher field, the magnetisation in M/Nbunits, indicates a value of 1.19 corresponding to a quasihalf saturated S = 1 system (inset Fig. 9). Comparison ofthe overall shape of the plot with the Brillouin plot (solidplot) for one isolated ion with S = 1 system and g = 2 indi-cates slower magnetisation which is consistent only with a
weak antiferromagnetic interaction. A confrontation withBrillouin curve, as done for 2, has been added in Fig. 8illustrating the presence of antiferromagnetic interactions.
The weak magnetic exchange couplings observed in 1and 2 can be understood considering the hexamethylenetet-ramine molecule which is separating the metal atoms withlarge distance (�6.255 A), leading in this way to almostnegligible coupling.
3.5. Thermal analyses
Thermal analysis was done for both the complexes. Thenature of thermal stability curves found for both the com-plexes are almost similar. So, only one representative curve(complex 1) has been provided in Fig. 10. The curve clearlydepicts the decomposition in three consecutive steps. Theweight losses are approximately 10.35% (203 �C); 15.05%(233 �C) and about 32.37% (234–340 �C) showing threeconsecutive endothermal breaks in DTA trace. Weight lossat 203 �C is very close to the calculated water content fortwo coordinated H2O molecules. So it can be clearly attrib-uted for deaquation. This temperature is rather highercompared to that of similar coordination polymers. It sug-gests the higher thermodynamic stability of the water mol-ecules in the polymeric complex reported herein [43]. Itmay be due to the strong and extensive inter- and intra-chain hydrogen bonding network within the polymeric sys-tem. The weight loss at 233 �C may be correlated to thedecomposition of the CNO�groups. The final step leadsto the formation of a stable residue.
4. Conclusion
In this paper we have reported the synthesis of two newextensively hydrogen bonded coordination polymers[MII(hmt)(NCO)2(H2O)2]n where M = Co(II) and Ni(II)for 1 and 2, respectively, using hexamethylenetetramineas a neutral organic spacer. Cyanate is coordinated as

A. Ray et al. / Inorganica Chimica Acta 361 (2008) 1850–1860 1859
terminal ligand in either case. Both the structures show aninfinite robust 2-D network with several inter- and intra-chain hydrogen bondings which may be a probable causefor the stability of the system. The facts are well supportedby several instrumental analyses and also firmly establishedfrom variable temperature magnetic susceptibility studies.
5. Supplementary material
CCDC 630136 and 630137 contain the supplementarycrystallographic data for 1 and 2. These data can beobtained free of charge from The Cambridge Crystallo-graphic Data Centre via www.ccdc.cam.ac.uk/data_re-quest/cif.
Acknowledgements
Aurkie Ray is thankful to the Department of Scienceand Technology, New Delhi, Government of India, forfinancial support. Joy Chakraborty is especially gratefulto the University Grants Commission, New Delhi, Govern-ment of India, for the award of a Senior Research Fellow-ship to him. S. Thakurta is grateful to the Council ofScientific and Industrial Research, Government of India,for the award of a Junior Research Fellowship to her.
References
[1] S.-L. Zheng, M.-L. Tong, X.-L. Yu, X.-M. Chen, J. Chem. Soc.,Dalton Trans. (2001) 586.
[2] (a) R. Robson, B.F. Abrahms, S.R. Batten, R.W. Gable, B.F.Hoskins, J. Lieu, Supramolecular Architecture, ACS, Washington,DC, 1992, p. 256;(b) J.M. Lehn, Supramolecular Chemistry, VCH, Weinheim, 1985(Chapter 9);(c) G.R. Desiraju, The Crystals as a Supramolecular Entity, vol. 2,John Wiley & Sons, Chichester, UK, 1996;(d) G.R. Desiraju, Acc. Chem. Res. 35 (2002) 565.
[3] C.L. Bowes, G.A. Ozin, Adv. Mater. 8 (1996) 13.[4] P. Feng, X. Bu, G.D. Stucky, Nature 388 (1997) 735.[5] Y. Zhang, M. Nishiura, J. Li, W. Deng, T. Imamoto, Inorg. Chem. 38
(1999) 825.[6] Y. Zhang, J. Li, M. Zhu, Q. Wang, X. Wu, Chem. Lett. (1998)
1803.[7] (a) M.L. Tong, X.-M. Chen, S.W. Ng, Inorg. Chem. Commun. 3
(2002) 436;(b) M.L. Tong, X.-M. Chen, B.-H. Ye, L.-N. Ji, Angew. Chem., Int.Ed. Engl. 38 (1999) 2237;(c) M.L. Tong, H.K. Lee, X.-M. Chen, R.B. Huang, T.C.W. Mak, J.Chem. Soc., Dalton Trans. (1999) 3657.
[8] G.R. Desiraju, Angew. Chem., Int. Ed. Engl. 34 (1995) 2311.[9] O. Ermer, J. Am. Chem. Soc. 110 (1988) 3747.
[10] (a) M.L. Tong, X.-M. Chen, X.-L. Yu, T.C.W. Mak, J. Chem. Soc.,Dalton Trans. (1998) 5;(b) L.R. Macgillicray, R.H. Groeneman, J.L. Atwood, J. Am. Chem.Soc. 120 (1998) 2676;(c) P. Lightfoot, A. Snedden, J. Chem. Soc., Dalton Trans. (1999)3549.
[11] (a) X.-M. Chen, M.L. Tong, Y.-J. Luo, Z.-N. Chen, Aust. J. Chem.49 (1996) 835;(b) M.L. Tong, J.-W. Cai, X.-L. Yu, X.-M. Chen, S.W. Ng, T.C.W.Mak, Aust. J. Chem. 51 (1998) 637;
(c) M.L. Tong, S.-L. Zheng, X.-M. Chen, Polyhedron 19 (2002) 1809;(d) M.L. Tong, B.-H. Ye, J.-W. Cai, X.-M. Chen, S.W. Ng, Inorg.Chem. 37 (1998) 2645.
[12] B.D. Wagner, G.J. Mc Manus, B. Moulton, M.J. Zaworotko, Chem.Commun. (2002) 2176.
[13] N. Mondal, M.K. Saha, S. Mitra, V. Gramlich, J. Chem. Soc., DaltonTrans. (2000) 3218.
[14] S.A. Barnett, A.J. Blake, N.R. Champness, C. Wilson, Chem.Commun. (2002) 1640.
[15] B. Zurowska, J. Mrozinske, M. Julve, F. Lloret, A. Maslezova, W.Sawka-Dobrowolska, Inorg. Chem. 41 (2002) 1771.
[16] F. Wiesbrock, H. Schmidbaur, J. Chem. Soc., Dalton Trans. (2002)3201.
[17] O.R. Evans, W. Lin, Inorg. Chem. 39 (2000) 2189.[18] C. Livage, C. Egger, M. Nogues, G. Ferey, J. Mater. Chem. 8 (1998)
2743.[19] S.O.H. Gutschke, M. Molinier, A.K. Powell, P.T. Wood, Angew.
Chem., Int. Ed. Engl. 36 (1997) 991.[20] (a) J. Chakraborty, B. Samanta, G. Rosair, V. Gramlich, M.S. El
Fallah, J. Ribas, T. Matsushita, S. Mitra, Polyhedron 25 (2006) 3006;(b) J. Chakraborty, B. Samanta, A.S. Batsanov, J. Ribas, M.S. ElFallah, S. Mitra, Struct. Chem. 17 (2006) 401;(c) S. Banerjee, M.G.B. Drew, A. Ghosh, Polyhedron 22 (2003) 2933;(d) M.-L. Tong, S.-L. Zheng, X.-M. Chen, Chem. Commun. (1999)561;(e) Y. Zhang, J. Li, M. Nishiura, T. Imamoto, J. Mol. Struct. 523(2000) 257;(f) S. Wang, M.-L. Hu, S.W. Ng, Acta Crystallogr., Sect. E 58 (2002)m242;(g) S.-L. Zheng, M.-L. Tong, H.-L. Zhu, Y. Fang, X.-M. Chen, J.Chem. Soc., Dalton Trans. (2001) 2049.
[21] (a) T.L. Hennigar, D.C. MacQuarrie, P. Losier, R.D. Rogers, M.J.Zaworotko, Angew. Chem., Int. Ed. Engl. 36 (1997) 972;(b) H. Gudbjartson, K. Biradha, K.M. Poirier, M.J. Zaworotko, J.Am. Chem. Soc. 121 (1999) 2599.
[22] L. Carlucci, G. Ciani, D.M. Proserpio, A. Sironi, J. Am. Chem. Soc.117 (1995) 12861.
[23] G.M. Sheldrick, Programs for the solution and refinement of crystalstructures, SHELXTL ver. 6.12, Bruker AXS P4 Inc., Madison, WI,USA, 1999.
[24] G.M. Sheldrick, Program SADABS: Area-detector Absorption Correc-tion, University of Gottingen, Germany, 1996.
[25] XPREP, XCANS and XCIF are part of the standard software package withBruker Analytical X-ray Systems.
[26] G.M. Sheldrick, SHELXS-97, Program for Crystal Structure Refine-ment, University of Gottingen, Gottingen, Germany, 1990.
[27] G.M. Sheldrick, SHELXL-97, Program for Crystal Structure Refine-ment, University of Gottingen, Gottingen, Germany, 1997.
[28] R.T. Conley, Infrared Spectroscopy, Allyn & Bacon Inc., Boston, 1966.[29] K. Nakamoto, Infrared and Raman Spectra of Inorganic and
Coordination Compounds, Part A and B, fifth ed., John Wiley,1997, 1997.
[30] A.B.P. Lever, Inorganic Electronic Spectroscopy, second ed., Else-vier, New York, 1984.
[31] Y. Zhang, J. Li, Q. Su, G. Zhao, Spectrochim. Acta, Part A 48 (1992)175.
[32] (a) L.J. Farrugia, J. Appl. Crystallogr. 30 (1997) 565;(b) A.L. Spek, Acta Crystallogr., Sect. A 46 (1990) C34.
[33] (a) M.C. Etter, Acc. Chem. Res. 23 (1990) 120;(b) M.C. Etter, J. Phys. Chem. 95 (1991) 4601.
[34] (a) S. Emori, M. Inoue, M. Kubo, Coord. Chem. Rev. 21 (1976) 1;(b) F.E. Mabbs, D.J. Machin, Magnetism and Transition MetalComplexes, Chapman and Hall, London, 1973;(c) J.W. Raebiger, J.L. Manson, R.D. Sommer, U. Geiser, A.L.Rheingold, J.S. Miller, Inorg. Chem. 40 (2001) 2578;(d) E.W. Lee, Y.J. Kim, D.Y. Junag, Inorg. Chem. 41 (2002) 501;(e) B.N. Figgis, M.A. Hitchman, Ligand Field Theory and itsApplications, Wiley-VCH, New York, 2000;

1860 A. Ray et al. / Inorganica Chimica Acta 361 (2008) 1850–1860
(f) D. Armentano, G. de Munno, F. Lloret, M. Julve, Inorg. Chem.38 (1999) 3744;(g) S.J. Retting, R.C. Thompson, J. Trotter, S. Xia, Inorg. Chem. 38(1999) 1360.
[35] G. de Munno, M. Julve, F. Lloret, J. Faus, A. Canneschi, J. Chem.Soc., Dalton Trans. (1994) 1175.
[36] M.E. Lines, J. Chem. Phys. 55 (1971) 2977.[37] MAGPACK Program (a) J.J. Borras-Almenar, J.M. Clemente-Juan, E.
Coronado, B.S. Tsukerblat, Inorg. Chem. 38 (1999) 6081;(b) J.J. Borras-Almenar, J.M. Clemente-Juan, E. Coronado, B.S.Tsukerblat, J. Comput. Chem. 22 (2001) 985.
[38] (a) M.E. Fisher, J. Math. Phys. 4 (1963) 124;(b) S. Angelow, M. Drillon, E. Zhecheva, R. Stoyanova, M. Belaiche,A. Derory, A. Herr, Inorg. Chem. 31 (1992) 1514;(c) J.-M. Rueff, N. Masciocchi, P. Rabu, A. Sironi, A. Skoulios, Eur.J. Inorg. Chem. (2001) 2843.
[39] J.-M. Rueff, N. Masciocchi, P. Rabu, A. Sironi, A. Skoulios, Chem.Eur. J. 8 (2002) 1813, and references therein.
[40] C.Y. Weng, Ph.D. Thesis, Carnegie Institute of Technology,1968.
[41] (a)E.A. Boudreaux, L.N. Mulay (Eds.), Theory and Applications ofMolecular Paramagnetism, John-Wiley, New York, 1976, p. 135(Chapter 3);(b) L.L. Lohr, J.C. Miller, R.R. Sharp, J. Chem. Phys. 111 (1999)10148;(c) S.G. Telfer, T. Sato, R. Kuroda, J. Lefebvre, D.B. Leznoff, Inorg.Chem. 43 (2004) 421.
[42] (a) F.D. Haldane, Phys. Lett. 93A (1983) 464;(b) F.D. Haldane, Phys. Rev. Lett. 50 (1983) 1153;(c) J.P. Renard, M. Verdaguer, L.P. Regnault, W.A.C. Erkelens, J.Rossat-Mignod, W.G. Stirling, Europhys. Lett. 3 (1987) 945;(d) I. Affleck, T. Kennedy, E.H. Lieb, H. Tasaki, Phys. Rev. Lett. 59(1987) 799;(e) M.S. El Fallah, A. Escuer, R. Vicente, X. Solans, M. Font-Bardia,M. Verdaguer, Inorg. Chim. Acta 344 (2003) 133.
[43] J. Xiang, J. He, Y. Yin, D. Li, Inorg. Chem. Commun. 9 (2006) 326.